
Field-theoretic simulations of random copolymers with structural rigidity†
 *acdef
*acdef
Abstract
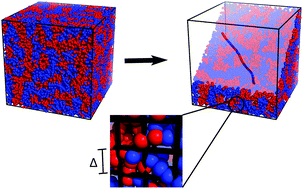
Copolymers play an important role in a range of soft-materials applications and biological phenomena. Prevalent works on block copolymer phase behavior use flexible chain models and incorporate interactions using a mean-field approximation. However, when phase separation takes place on length scales comparable to a few monomers, the structural rigidity of the monomers becomes important. In addition, concentration fluctuations become significant at short length scales, rendering the mean-field approximation invalid. In this work, we use simulation to address the role of finite monomer rigidity and concentration fluctuations in microphase segregation of random copolymers. Using a field-theoretic Monte-Carlo simulation of semiflexible polymers with random chemical sequences, we generate phase diagrams for random copolymers. We find that the melt morphology of random copolymers strongly depends on chain flexibility and chemical sequence correlation. Chemically anti-correlated copolymers undergo first-order phase transitions to local lamellar structures. With increasing degree of chemical correlation, this first-order phase transition is softened, and melts form microphases with irregular shaped domains. Our simulations in the homogeneous phase exhibit agreement with the density–density correlation from mean-field theory. However, conditions near a phase transition result in deviations between simulation and mean-field theory for the density–density correlation and the critical wavemode. Chain rigidity and sequence randomness lead to frustration in the segregated phase, introducing heterogeneity in the resulting morphologies.
 Please wait while we load your content...
Please wait while we load your content...

