
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand engineering of circularly polarized luminescence inversion and enhancement for chiral Ag6 nanoclusters†
Hui
Chen
,
Xiangyang
Zhang
,
Kuo
Fu
,
Xuejuan
Wang
and
Guofeng
Liu
 *
*
School of Chemical Science and Engineering, Advanced Research Institute, Tongji University, Shanghai, 200092, P. R. China. E-mail: liuguofeng@tongji.edu.cn
First published on 14th May 2024
Abstract
Coinage metal nanoclusters usually show bright emission, which is useful for obtaining circularly polarized luminescence (CPL) after modifying with chiral organic ligands. However, it remains challenging to understand the chiral transfer and amplification mechanisms and achieve CPL inversion without changing the absolute configuration of the chiral ligands. Here, we present four pairs of six-core silver nanoclusters protected by the chiral thiazinthione- and oxazinthione-based ligands, showing S⋯S interaction-enhanced luminescence quantum yield (QY) and supramolecular chirality inversion induced by the peripheral methyl and phenyl groups in both ground and excited states.
 Guofeng Liu | Guofeng Liu received his MSc degree from East China University of Science and Technology under the supervision of Prof. He Tian and obtained his PhD degree at Shanghai Jiao Tong University under the supervision of Prof. Chuanliang Feng. After postdoctoral research in Prof. Yanli Zhao's lab at Nanyang Technological University, he joined the School of Chemical Science and Engineering at Tongji University as a faculty member and is also selected as a professor of special appointment (Eastern Scholar) by Shanghai Institutions of Higher Learning. His research mainly focuses on dynamic chemical self-assembly systems and chiral supramolecular materials. |
1. Introduction
Metal nanoclusters are ultra-small nanoparticles with diameters ranging from 1 to 3 nm, typically consisting of tens to hundreds of metal atoms.1,2 Compared with other nanoparticles like carbon dots3 and semiconductor quantum dots,4 nanoclusters have atomically precise structures with a discrete energy level like the small organic molecules, and their functions can be finely tailored by means of metal atom exchanging/doping and ligand-modifying engineering.5,6 In the nanoclusters modified with different organic ligands, organic ligands make them exhibit excellent physical and chemical properties, such as photoluminescence,7 catalytic activity,8 pharmacokinetic parameters,9,10 and supramolecular chirality.11 Therefore, metal nanoclusters with controlled functionality through ligand-modifying engineering have shown great potential for application in the fields of optoelectronic devices, catalysis, and biomedicine.12–16Recently, nanomaterials with excellent circularly polarized luminescence (CPL) have attracted much attention due to their fascinating chiroptical performances and great potential for application in advanced information processing, chiral optoelectronic devices, and chiral biosensing.17–29 An ideal CPL-active material usually needs to have both high photoluminescence quantum yield (QY) and large luminescence asymmetry factor (glum).30,31 Thus, chiral nanoclusters became an excellent candidate for constructing CPL-active materials with both high QY and tuneable supramolecular chirality.32,33 In general, the chirality of nanoclusters mainly originates from the following three chiral sources: (a) intrinsic chirality of the inorganic metal cores with asymmetrical stacking, (b) twisted arrangement of achiral ligands on the surface of the cluster core, and (c) induced chirality from the chiral ligand on the surface of the cluster.34–37 Chiral induction from organic ligands is the most convenient method for the preparation of chiral metal nanoclusters since it is easy to control the chirality by tuning the R/S-configuration of chiral ligands and endowing the obtained nanocluster with amplified chirality performances especially for the CPL emissive intensity.38 In addition, based on the atomically precise structures, the structure–function relationship of nanoclusters can be better established by regulating the luminescent factors, including wavelength, QY, and glum through ligand engineering.39–43 However, due to the complexity and dynamics of metal coordination, as well as the abundant interactions between the metal core and peripheral ligand, it is hard to predict the handedness of nanoclusters, especially for the understanding of chirality inversion in both ground and excited states. Thus, it is necessary to reveal the chirality transfer and amplification mechanisms in the complex arrangements and assembly behaviours of nanoclusters by ligand-modifying engineering.
Herein, we synthesized four pairs of chiral Ag6L6 nanoclusters through ligand engineering involving thiazithione and oxazinthione (Fig. 1). These nanoclusters exhibited excellent photoluminescence properties in both the solid and solution states, achieving a maximum luminescence QY of 28.19% and the largest glum at −2.77 × 10−3. Interestingly, the nanoclusters protected by a thiazidethione ligand showed a higher QY than those modified with oxazinthione, which is attributed to the weak S⋯S interactions from adjacent cluster units in the thiazidethione-protected ones. In addition, the opposite arrangement of methyl and phenyl groups of chiral ligands was revealed in the single crystals of clusters protected by both thiazithione- and oxazinthione-based ligands, showing opposite helical senses of cluster chains and inverted CD/CPL performances. The obtained chiral Ag6L6 nanoclusters exhibited CPL inversion and enhanced QY by means of ligand engineering, which would pave a way to develop CPL-active nanoclusters for applications in chiral sensors, optoelectronic engineering and asymmetric catalysis.
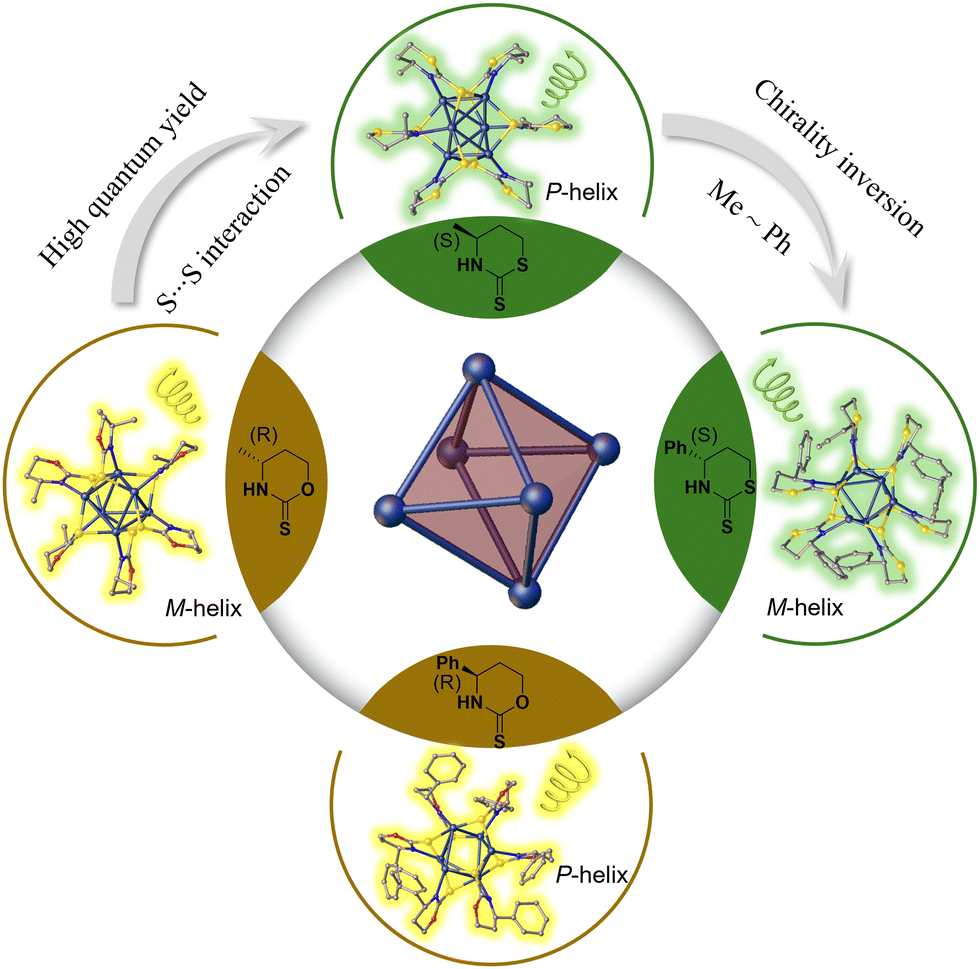 | ||
| Fig. 1 Ligand engineering to achieve CPL enhancement and chirality inversion for silver nanoclusters protected by chiral thiazinthione- and oxazinthione-derived ligands. | ||
2. Experimental
2.1 Materials
(S)-3-amino-3-phenylpropan-1-ol, (R)-3-amino-3-phenylpropan-1-ol, chlorosulphonic acid, carbon disulfide potassium hydroxide, triethylamine, (S)-3-aminobutan-1-ol, (R)-3-aminobutan-1-ol, 30% hydrogen peroxide and silver acetate were purchased from Shanghai Titan Scientific Co., Ltd. The organic solvents were purchased from Shanghai Titan Scientific Co., Ltd, and used without further purification.2.2 Synthesis of (S)-4-phenyl-1,3-thiazinane-2-thione (S-SPh)
Chlorosulphonic acid (1.0 mL, 14.5 mmol) was added dropwise to a solution of (S)-3-amino-3-phenylpropan-1-ol (2.0 mL, 13.8 mmol) in CH2Cl2 (20 mL) under a constant flow of N2 at 0 °C. The reaction mixture was stirred for 15 h at RT, after which the solvent was removed in vacuo to afford a white solid. The white solid was triturated with MeOH and dried under reduced pressure to afford 2.32 g of (S)-3-amino-3-phenylpropyl hydrogen sulfate (10.0 mmol, 72%), which was pure enough, as confirmed by 1H NMR spectroscopy and required no further purification. Then, CS2 (0.72 mL, 12 mmol) was added dropwise to (S)-3-amino-3-phenylpropyl hydrogen sulfate (2.32 g, 10.0 mmol) under N2 at RT. A solution of KOH (1.23 g, 22 mmol in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O/EtOH 20 mL) was added dropwise to the reaction mixture at 0 °C. The resulting solution was then heated to reflux and stirred for 2 h, after which it was further stirred overnight at RT. Then, it was cooled in an ice-water bath. The white precipitate was then filtered and rinsed with cold water. It was dissolved in CH2Cl2, dried over anhydrous Na2SO4, and the solvent was removed in vacuo to obtain a white solid. The crude product was purified by recrystallization in CH2Cl2/hexane to yield (S)-4-phenyl-1,3-thiazinane-2-thione (S-SPh, 1.8 g, 62% for two steps). Physical state: white solid. TLC: Rf = 0.32 (PE/EtOAc = 1:1). 1H NMR (400 MHz, DMSO-d6) δ 10.62 (s, 1H), 7.41 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 7.22 (d, J = 7.3 Hz, 2H), 4.82 (d, 1H), 2.89–2.78 (m, 1H), 2.66–2.54 (m, 1H), 2.26–2.12 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 193.21, 141.38, 129.08, 128.02, 126.60, 56.66, 27.94, 26.65.
:1 H2O/EtOH 20 mL) was added dropwise to the reaction mixture at 0 °C. The resulting solution was then heated to reflux and stirred for 2 h, after which it was further stirred overnight at RT. Then, it was cooled in an ice-water bath. The white precipitate was then filtered and rinsed with cold water. It was dissolved in CH2Cl2, dried over anhydrous Na2SO4, and the solvent was removed in vacuo to obtain a white solid. The crude product was purified by recrystallization in CH2Cl2/hexane to yield (S)-4-phenyl-1,3-thiazinane-2-thione (S-SPh, 1.8 g, 62% for two steps). Physical state: white solid. TLC: Rf = 0.32 (PE/EtOAc = 1:1). 1H NMR (400 MHz, DMSO-d6) δ 10.62 (s, 1H), 7.41 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 7.22 (d, J = 7.3 Hz, 2H), 4.82 (d, 1H), 2.89–2.78 (m, 1H), 2.66–2.54 (m, 1H), 2.26–2.12 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 193.21, 141.38, 129.08, 128.02, 126.60, 56.66, 27.94, 26.65.
2.3 Synthesis of (R)-4-phenyl-1,3-thiazinane-2-thione (R-SPh)
Chlorosulphonic acid (0.5 mL, 7 mmol) was added dropwise to a solution of (R)-3-amino-3-phenylpropan-1-ol (1 g, 6.6 mmol) in CH2Cl2 (15 mL) under a constant flow of N2 at 0 °C. The reaction mixture was stirred for 15 h at RT after which the solvent was removed in vacuo to afford a white solid. The white solid was triturated with MeOH and dried under reduced pressure to afford 1.04 g of (R)-3-amino-3-phenylpropyl hydrogen sulfate (4.5 mmol, 68%), which was pure enough, as confirmed by 1H NMR spectroscopy and required no further purification. Then, CS2 (0.4 mL, 5.3 mmol) was added dropwise to (R)-3-amino-3-phenylpropyl hydrogen sulfate (1.04 g, 4.4 mmol) under N2 at RT. A solution of KOH (560 mg, 10 mmol in 1:1 H2O/EtOH, 20 mL) was added dropwise to the reaction mixture at 0 °C. The resulting solution was then heated to reflux and stirred for 2 h, after which it was further stirred overnight at RT. Then, it was cooled in an ice-water bath. The white precipitate was filtered and rinsed with cold water. It was dissolved in CH2Cl2, dried over anhydrous Na2SO4, and the solvent was removed in vacuo to obtain a white solid. The crude product was purified by recrystallization in CH2Cl2/hexanes to obtain (R)-4-phenyl-1,3-thiazinane-2-thione (R-SPh, 820 mg, 60% over two steps). Physical state: white solid. TLC: Rf = 0.32 (PE/EtOAc = 1:1). 1H NMR (600 MHz, DMSO-d6) δ 10.63 (s, 1H), 7.41 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.2 Hz, 1H), 7.22 (d, J = 7.9 Hz, 2H), 4.82 (s, 1H), 2.87–2.82 (m, 1H), 2.60 (td, J = 12.4, 11.5, 3.7 Hz, 1H), 2.25–2.17 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 193.20, 141.38, 129.07, 128.02, 126.60, 56.66, 27.94, 26.65.
2.4 Synthesis of (S)-4-methyl-1,3-thiazinane-2-thione (S-SMe)
Chlorosulphonic acid (0.7 mL, 10.5 mmol) was added dropwise to a solution of (R)-3-aminobutan-1-ol (890 mg, 10 mmol) in CH2Cl2 (25 mL) under a constant flow of N2 at 0 °C. The reaction mixture was stirred for 15 h at RT, after which the solvent was removed in vacuo to afford a white solid. The white solid was triturated with MeOH and dried under reduced pressure to afford 1.18 g of (S)-3-aminobutyl hydrogen sulfate (7 mmol, 70%), which was pure enough, as confirmed by 1H NMR spectroscopy and required no further purification. Then, CS2 (0.63 mL, 10.5 mmol) was added dropwise to (R)-3-aminobutyl hydrogen sulfate (1.18 g, 7 mmol) under N2 at RT. A solution of KOH (840 mg, 15 mmol in 1:1 H2O/EtOH, 25 mL) was added dropwise to the reaction mixture at 0 °C. The resulting solution was then heated to reflux and stirred for 2 h, after which it was further stirred overnight at RT. Then, it was cooled in an ice-water bath. The white precipitate was filtered and rinsed with cold water. It was dissolved in CH2Cl2, dried over anhydrous Na2SO4, and the solvent was removed in vacuo to obtain a white solid. The crude product was purified by recrystallization in CH2Cl2/hexanes to yield (S)-4-methyl-1,3-thiazinane-2-thione (S-SMe, 970 mg, 66% over two steps). TLC: Rf = 0.40 (PE/EtOAc = 1:1). Physical state: white solid. TLC: Rf = 0.40 (PE/EtOAc = 1:1). 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, 1H), 3.56–3.48 (m, 1H), 3.01 (ddd, J = 13.3, 9.9, 3.7 Hz, 1H), 2.94–2.86 (m, 1H), 2.12–2.05 (m, 1H), 1.71–1.64 (m, 1H), 1.20 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.30, 50.21, 28.41, 27.78, 20.79.
2.5 Synthesis of (R)-4-methyl-1,3-thiazinane-2-thione (R-SMe)
Chlorosulphonic acid (2.1 mL, 31.5 mmol) was added dropwise to a solution of (R)-3-aminobutan-1-ol (2.67 g, 30 mmol) in CH2Cl2 (100 mL) under a constant flow of N2 at 0 °C. The reaction mixture was stirred for 15 h at RT, after which the solvent was removed in vacuo to afford a white solid. The white solid was triturated with MeOH and dried under reduced pressure to afford 3.8 g of (R)-3-aminobutyl hydrogen sulfate (22.5 mmol, 75%), which was pure enough as confirmed by 1H NMR spectroscopy and required no further purification. Then, CS2 (1.63 mL, 27 mmol) was added dropwise to (R)-3-aminobutyl hydrogen sulfate (3.8 g, 22.5 mmol) under N2 at RT. A solution of KOH (2.8 g, 50 mmol in 1:1 H2O/EtOH, 100 mL) was added dropwise to the reaction mixture at 0 °C. The resulting solution was then heated to reflux and stirred for 2 h, after which it was further stirred overnight at RT. Then, it was cooled in an ice-water bath. The white precipitate was filtered and rinsed with cold water. It was dissolved in CH2Cl2, dried over anhydrous Na2SO4, and the solvent was removed in vacuo to obtain a white solid. The crude product was purified by recrystallization in CH2Cl2/hexanes to yield (R)-4-methyl-1,3-thiazinane-2-thione (R-SMe, 3.17 g, 72% over two steps). TLC: Rf = 0.40 (PE/EtOAc = 1:1). 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 3.56–3.48 (m, 1H), 3.01 (ddd, J = 13.1, 9.9, 3.7 Hz, 1H), 2.93–2.86 (m, 1H), 2.12–2.05 (m, 1H), 1.72–1.62 (m, 1H), 1.20 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 192.28, 50.21, 28.42, 27.77, 20.79.
2.6 Synthesis of (S)-4-phenyl-1,3-oxazinane-2-thione (S-OPh)
Triethylamine (0.92 mL, 6.6 mmol) was added to a solution of (S)-3-amino-3-phenylpropan-1-ol (1.0 g, 6.6 mmol) in methanol (20 mL) at 0 °C, and carbon disulfide (0.4 mL, 6.6 mmol) was added dropwise. The solution was stirred at room temperature for 30 min. Hydrogen peroxide (30%, 0.43 mL, 8 mmol) was then added and stirred for 2 h. Methanol was removed by reduced pressure, and the resulting mixture was diluted with CH2Cl2 (100 mL) and washed with H2O (2 × 50 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness under vacuum. The crude product was purified by recrystallization in CH2Cl2/hexanes to yield (S)-4-phenyl-1,3-oxazinane-2-thione (S-OPh, 1.04 g, 82%). Physical state: white solid. TLC: Rf = 0.38 (PE/EtOAc = 1:1). 1H NMR (400 MHz, DMSO-d6) δ 9.95 (s, 1H), 7.40 (t, J = 7.4 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 7.26 (d, J = 7.3 Hz, 2H), 4.64 (td, J = 5.8, 2.5 Hz, 1H), 4.32–4.26 (m, 1H), 4.13–4.07 (m, 1H), 2.33–2.25 (m, 1H), 1.96–1.88 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 186.49, 141.79, 129.02, 128.07, 126.69, 65.78, 53.99, 28.79.
2.7 Synthesis of (R)-4-phenyl-1,3-oxazinane-2-thione (R-OPh)
Triethylamine (0.56 mL, 4 mmol) was added to a solution of (R)-3-amino-3-phenylpropan-1-ol (605 mg, 4 mmol) in methanol (10 mL) at 0 °C, and carbon disulfide (0.24 mL, 4 mmol) was added dropwise. Then the solution was stirred at room temperature for 30 min. Hydrogen peroxide (30%, 0.26 mL, 8 mmol) was then added and stirred for 2 h. Methanol was removed by reduced pressure, and the resulting mixture was diluted with CH2Cl2 (100 mL) and washed with H2O (2 × 50 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and then concentrated to dryness under vacuum. The crude product was purified by recrystallization in CH2Cl2/hexanes to yield (R)-4-phenyl-1,3-oxazinane-2-thione (R-OPh, 586 mg, 76%). Physical state: white solid. TLC: Rf = 0.38 (PE/EtOAc = 1:1). 1H NMR (400 MHz, DMSO-d6) δ 9.95 (s, 1H), 7.40 (t, J = 7.4 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 7.26 (d, J = 7.1 Hz, 2H), 4.64 (td, J = 5.8, 2.5 Hz, 1H), 4.33–4.26 (m, 1H), 4.13–4.07 (m, 1H), 2.33–2.25 (m, 1H), 1.96–1.88 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 186.49, 141.79, 129.02, 128.07, 126.69, 65.78, 53.99, 28.79.
2.8 Synthesis of (S)-4-methyl-1,3-oxazinane-2-thione (S-OMe)
Triethylamine (1.8 mL, 13 mmol) was added to a solution of (S)-3-aminobutan-1-ol (1.16 g, 13 mmol) in methanol (30 mL) at 0 °C, and carbon disulfide (0.78 mL, 13 mmol) was added dropwise. The solution was stirred at room temperature for 30 min. Hydrogen peroxide (30%, 0.85 mL, 26 mmol) was then added and stirred for 2 h. Methanol was removed by reduced pressure, and the resulting mixture was diluted with CH2Cl2 (100 mL) and washed with H2O (2 × 50 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and then concentrated to dryness under vacuum. The crude product was purified by recrystallization in CH2Cl2/hexane to yield (S)-4-methyl-1,3-oxazinane-2-thione (S-OMe, 936 mg, 55%). Physical state: white solid. TLC: Rf = 0.30 (PE/EtOAc = 1:1). 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H), 4.30–4.25 (m, 1H), 4.20 (td, J = 11.2, 10.2, 3.2 Hz, 1H), 3.47 (q, J = 6.5 Hz, 1H), 2.05–1.98 (m, 1H), 1.62–1.54 (m, 1H), 1.13 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 185.68, 66.83, 46.86, 27.68, 21.17.
2.9 Synthesis of (R)-4-methyl-1,3-thiazinane-2-thione (R-OMe)
Triethylamine (1.8 mL, 13 mmol) was added to a solution of (R)-3-aminobutan-1-ol (1.16 g, 13 mmol) in methanol (30 mL) at 0 °C, and carbon disulfide (0.78 mL, 13 mmol) was added dropwise. Then the solution was stirred at room temperature for 30 min. Hydrogen peroxide (30%, 0.85 mL, 26 mmol) was then added and stirred for 2 h. Methanol was removed by reduced pressure, and the resulting mixture was diluted with CH2Cl2 (100 mL) and then was washed with H2O (2 × 50 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and then concentrated to dryness under vacuum. The crude product was purified by recrystallization in CH2Cl2/hexane to yield (R)-4-methyl-1,3-oxazinane-2-thione (R-OMe, 1.02 g, 60%). Physical state: white solid. TLC: Rf = 0.30 (PE/EtOAc = 1:1). 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H), 4.31–4.24 (m, 1H), 4.20 (td, J = 11.2, 10.3, 3.2 Hz, 1H), 3.52–3.42 (m, 1H), 2.06–1.97 (m, 1H), 1.58 (dtd, J = 13.5, 9.0, 4.0 Hz, 1H), 1.14 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 185.67, 66.83, 46.85, 27.67, 21.17.
2.10 General synthesis of Ag clusters
S/R-OPh (97 mg, 0.5 mmol) and silver acetate (84 mg, 0.5 mmol) were dissolved in THF (3 mL) and stirred overnight at room temperature. The resulting yellow precipitate was filtered to obtain 91 mg of the crude product. The crude product was divided into four parts and dissolved separately in a mixed solvent of CH2Cl2/hexane (v/v, 6:1) to form a saturated solution. The solution was then allowed to evaporate slowly at RT for 5 days, yielding yellow block crystals of the S/R-AgOPh cluster (62 mg, yield: 74.2% based on Ag). The synthesis procedures of other clusters were the same as described above, with yields of 78.6% for S/R-AgSMe, 69.1% for S/R-AgOMe, and 58% for S/R-AgSPh.
2.11 Preparation of single-crystals of silver clusters
A 20 mg sample of the (S)-AgSMe cluster was dissolved in a 3.60 mL mixed solvent of DCM and n-hexane (v/v, 5:1), yielding bulk crystals after standing at room temperature and undergoing solvent evaporation for 3–4 days. Similarly, the single crystals of the (R)-AgSMe, (S/R)-AgOMe, and R-AgOPh clusters were obtained using the same solvent evaporation method for 3–4 days to facilitate single-crystal X-ray diffraction (SCXRD) characterization.
2.12 Characterization
NMR spectra were recorded on a Bruker AVANCE III HD 400 MHz and 600 MHz Instrument at ambient temperature. Circular dichroism (CD) spectra were obtained using a JASCO J-810 CD spectrometer with a bandwidth of 1.0 nm, a scanning speed of 500 nm min−1, and a data integration time of 1 s. The CD spectra of the samples were recorded in the UV/Vis region (250–450 nm) using a 0.1 mm quartz cuvette. Fluorescence spectra were measured with an F-4500 fluorescence spectrophotometer. CPL measurements were recorded on a JASCO CPL-300 spectrometer, with an Ex and Em slit width of 3000 μm, a scanning speed of 500 nm min−1, and a data integration time of 1 s. The CPL spectra of the gels were measured using a 0.1 mm quartz cuvette. Single-crystal data were collected from a shock-cooled single crystal at 100(2) K on a Bruker D8 VENTURE dual-wavelength Mo three-circle diffractometer with a microfocus sealed X-ray tube using mirror optics as a monochromator and a Bruker PHOTON II detector. QYs were measured using an Edinburgh FLS1000 equipped with a xenon lamp.3. Results and discussion
According to the literature,44,45 chiral thiazinthione- and oxazinthione-derived ligands such as (S/R)-SMe, (S/R)-OMe, (S/R)-OMe, (S/R)-SPh, and (S/R)-SPh were synthesized from (S/R)-3-aminobutin-1-ol and (S/R)-3-phenylpropion-1-ol, respectively (Fig. 2a). Their chemical structures were fully characterized and confirmed by 1H and 13C NMR measurements (see the ESI†). Then, the corresponding enantiomeric silver clusters of (S/R)-AgSMe, (S/R)-AgOMe, (S/R)-AgOPh and (S/R)-AgOPh were successfully prepared by the reaction of AgOAc with the above chiral ligands in THF (Fig. 2b and c). To better understand the composition and three-dimensional structure of chiral silver clusters, the single crystals of (S/R)-AgSMe, (S/R)-AgOMe, R-AgOPh and S-AgSPh were carefully cultured from the mixed solvent of DCM and n-hexane (v/v, 5:1) through the solvent evaporation method (Tables S1–S6, ESI†).
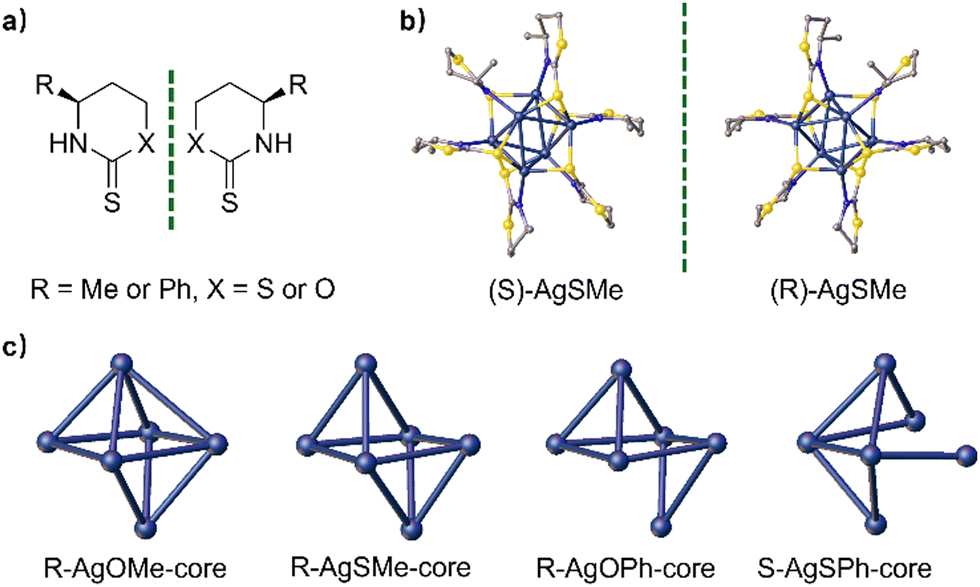 | ||
| Fig. 2 (a) Chemical structures of the four pairs of chiral 1,3-thiazinane-2-thione- and 1,3-oxazinane-2-thione-based ligand compounds modified with methyl and phenyl moieties, (b) structures of (S)-AgSMe (left) and (R)-AgSMe (right) in the single crystal, and (c) core structures of (R)-AgOMe, (R)-AgSMe, (R)-AgOPh, and (S)-AgSPh in a single crystal. | ||
Single-crystal X-ray diffraction (SCXRD) analysis showed that S/R-AgSMe, R-AgOMe and R-AgOPh crystallized in the orthorhombic chiral space group of P212121. S-AgOMe crystallized in the trigonal chiral space group of R-3, while S-AgSPh crystallized in the tetragonal chiral space group of P41. The central nuclei of the four pairs of clusters are made up of 6 silver atoms, forming a twisted octahedral configuration through Ag–Ag metallophilic interactions. The degree of distortion varied depending on the chiral ligand utilized. As depicted in Fig. 2b, six (S/R)-SMe ligands are bonded to each silver cluster nucleus via Ag–S and Ag–NH bonds, arranged alternately in a twisted mode around the periphery of each Ag6 nanocluster core. The resulting single crystals of S-AgSMe and R-AgSMe exhibit perfect mirror-image symmetry. Interestingly, considering the single crystals of R-AgOMe, R-AgSMe, R-AgOPh, and S-AgSPh as examples, the cluster cores of AgOMe, AgSMe and AgOPh hold 11, 10, 9, and 8-number Ag–Ag metallophilic interactions, respectively, indicating increasingly twisted configurations. The gradual decrease in the number of Ag–Ag metallophilic interactions in the cluster cores of AgOMe, AgSMe, AgOPh, and AgSPh strongly suggests that both the atomic radius of S/O and the steric hindrance of the methyl/phenyl groups play crucial roles in determining the degree of distortion in the obtained cluster nucleus. These factors greatly influenced the chirality transfer of the distorted Ag6 nanocluster backbone (Fig. 2c).
The photophysical properties were first explored in detail. As shown in Fig. 3a and Fig. S1–S4 (ESI†), the four enantiomeric Ag6L6 clusters, AgSPh, AgSMe, AgOPh, and AgOMe, exhibited bright photoluminescence at 534 nm, 530 nm, 542 nm, and 565 nm, respectively. It is evident that the silver clusters, protected by the oxazine cyclothione ligands, showed a significantly redshifted emission wavelength compared to those protected by the thiazide cyclothione ligands, as also evident from the yellow and green emission colour observed in Fig. 3b. On the other hand, the QYs of the R-type clusters were measured. As shown in Fig. 3b, the QYs of R-AgSMe, R-AgSPh, R-AgOMe, and R-AgOPh were 28.19%, 5.81%, 14.23%, and 5.07%, respectively (Fig. S5–S8, ESI†). Previous studies have indicated that the surface rigidification of ligands can indeed enhance the emission intensity of clusters.46,47 In reality, there are also cases where the opposite effect occurs,48–51 the metal core structure of clusters can induce changes in the emission of the cluster itself. The volume of the surface ligands can affect the size of the cluster core and its metallophilic interactions, thereby affecting the emission intensity of the cluster. This means that the silver clusters protected by the methyl-modified ligand showed much higher emission intensity than those protected by phenyl-modified ones, as the exchange of methyl and phenyl significantly influenced the Ag–Ag distance in the cluster core, as seen in Table S7 (ESI†). Furthermore, silver clusters protected by thiazide cyclothione ligands showed noticeably greater emission intensities than those modified by oxazine cyclothione ligands. To gain further insight into the emission mechanism of the clusters, we conducted DFT calculations using the CAM−B3LYP/6-31G*method, as depicted in Fig. 3c. The simulation results show that the energy gap between the HOMO and LUMO in the S-atom containing ligand-based clusters was significantly higher than that in the O-atom containing ligand-based clusters, corresponding to the redshifted emission observed in the AgOPh and AgOMe-based clusters mentioned above (Fig. 3c).
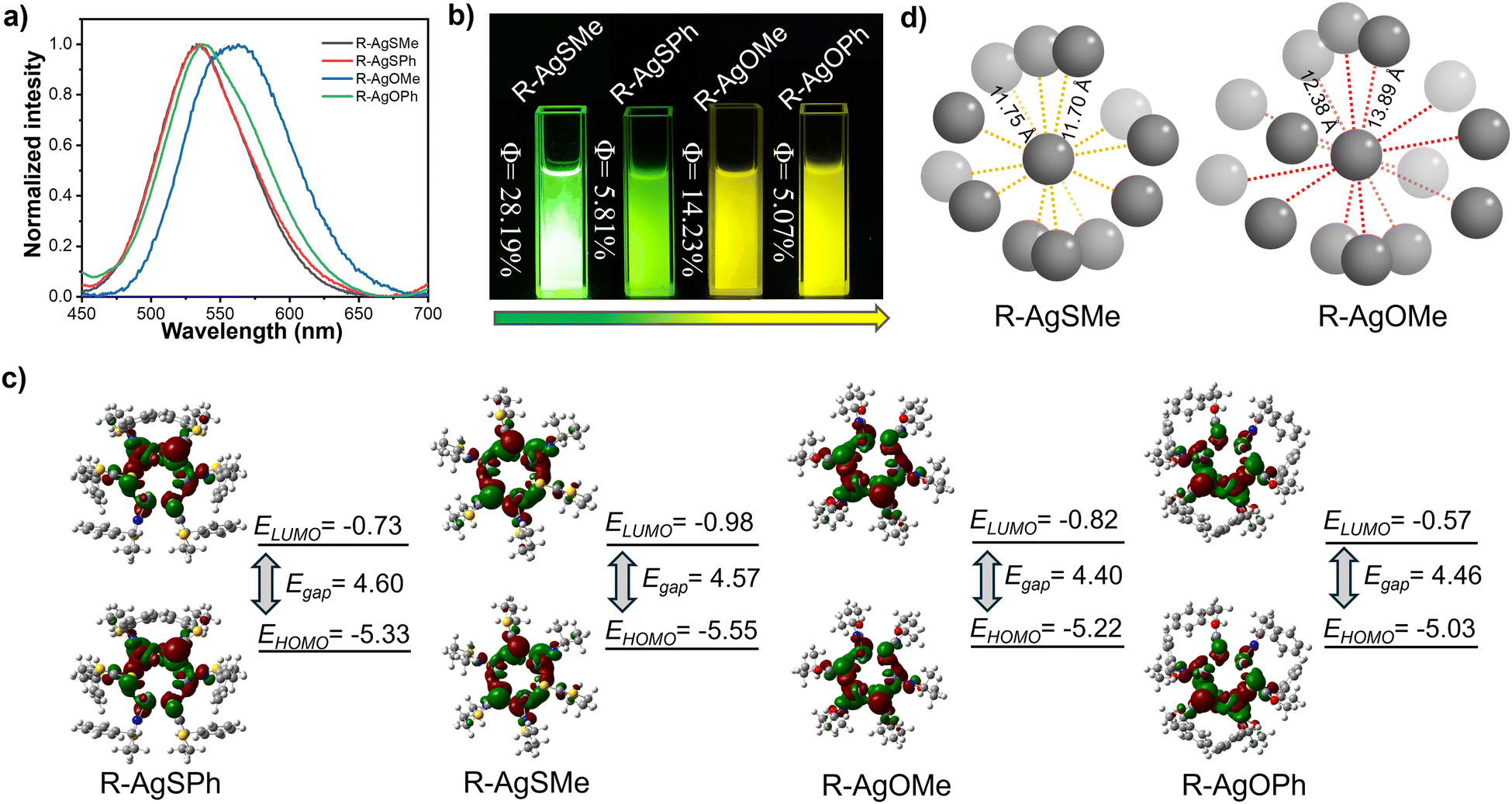 | ||
| Fig. 3 (a) Photoluminescence (PL) spectra of R-type chiral silver nanoclusters R-AgSMe, R-AgSPh, R-AgOMe, and R-AgOPh. (b) Photoimages of R-AgSMe, R-AgSPh, R-AgOMe, and R-AgOPh clusters under 365 nm light. (c) DFT calculations of the LUMO/HOMO energy gap among the four R-type clusters R-AgSMe, R-AgSPh, R-AgOMe, and R-AgOPh. (d) Schematic diagram of R-AgSMe and R-AgOMe cluster stacking in the single crystal. | ||
At the same time, these clusters exhibited aggregation-induced luminescence properties (Fig. S9, ESI†). After carefully studying the crystal structures of R-AgSMe and R-AgOMe, we found that the packing mode of R-AgSMe clusters was much more compact than the packing mode of R-AgOMe-based clusters (Fig. 3d, Fig. S10 and S11, ESI†). Compared to 12.38–13.89 Å measured in the R-AgOMe cluster-based single crystal, the distance between the adjacent clusters of R-AgSMe was measured to be 11.70–11.75 Å. This is because there are S⋯S interactions formed in the adjacent clusters protected by the thiazide cyclothione ligands. These interactions resulted in a tighter packing and suppressed the rotational vibration of the single cluster, thereby increasing the energy of its radiative transition and enhancing the emissive intensity (Table S8, ESI†).52 These emission properties of various R-type Ag6L6 clusters have also been demonstrated through detailed investigations of the S-type Ag6L6 clusters described in Fig. S1–S4 (ESI†).
To gain a deeper understanding of the photophysical properties of the chiral clusters, we measured the CD signals of the four pairs of clusters and their corresponding ligands in a CH2Cl2 solution. In comparison to the weak CD absorption peaks of ligands observed at ∼250 nm for R/S-SMe and ∼290 nm for R/S-OMe, R/S-SPh, and R/S-OPh(Fig. S12 (ESI†)), the four pair of clusters, namely AgSMe, AgSPh, AgOMe, and AgOPh, showed strong CD absorption peaks at 303 nm, 285 nm, 290 nm, and 258 nm, respectively (Fig. 4a–d). Interestingly, in comparison to the positive CD absorption peaks of R-SMe and R-SPh displayed in Fig. S12 (ESI†), the corresponding clusters of R-AgSMe showed a positive CD effect at ∼350 nm and a negative CD absorption peak at ∼300 nm (Fig. 4a). Conversely, R-AgSPh displayed a negative CD absorption peak at ∼350 nm and a positive CD absorption peak at ∼300 nm (Fig. 4c). This CD inversion was further demonstrated by the S-type clusters of S-AgSMe and S-AgSPh (Fig. 4a and c). It seems that the clusters modified with phenyl-containing ligands have stronger CD signals than those modified with methyl groups. Additionally, the obtained chiral silver clusters exhibited inverted CD absorption despite the methyl/phenyl-containing chiral ligands having the same R/S configuration. The ligand-induced CD inversion was further demonstrated in the clusters of 1,3-oxazinane-2-thione-based ligands, such as R/S-AgOMe and R/S-AgOPh systems. As displayed in Fig. 4b and d, the R-AgOMe cluster showed negative and positive CD effects at 350 nm and 290 nm, respectively. In contrast, the cluster of R-AgOPh showed positive and negative CD effects at 350 nm and 295 nm, respectively (and vice versa).
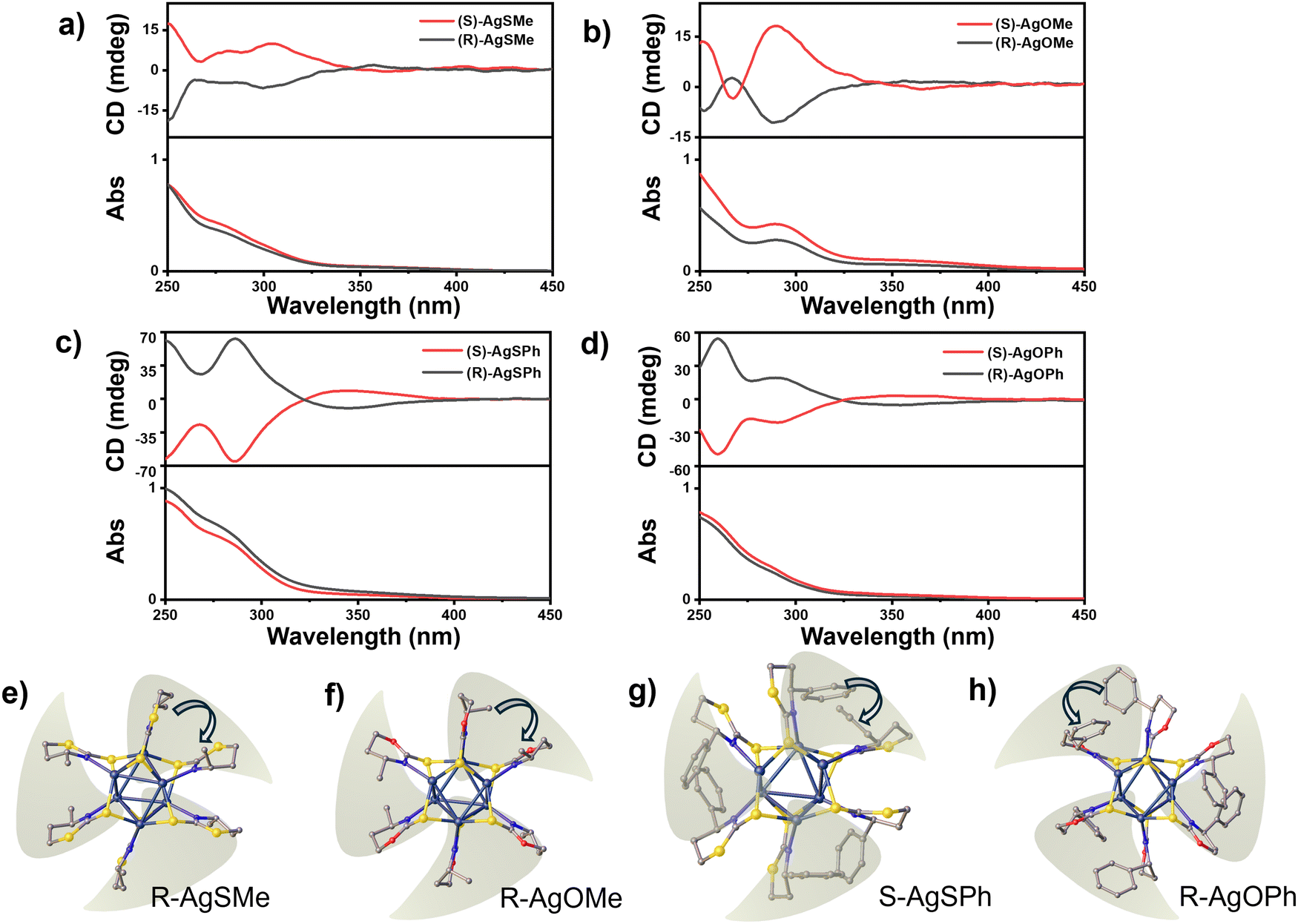 | ||
| Fig. 4 (a) CD spectra of S-AgSMe and R-AgSMe in DCM solution, (b) CD spectra of S-AgOMe and R-AgOMe in DCM solution, (c) CD spectra of S-AgSPh and R-AgSPh in DCM solution, (d) CD spectra of S-AgOPh and R-AgOPh in DCM solution, (e)–(h) single-crystal structures of (e) R-AgSMe, (f) R-AgOMe, (g) S-AgSPh, and (h) R-AgOPh. The concentration of cluster was fixed at 1.0 × 10−5 mol L−1. | ||
To better understand the ligand-induced chirality inversion in these clusters, a detailed investigation was conducted on single crystal structures. It was found that the ligand-induced CD inversion of the cluster resulted from the opposite arrangement of peripheral ligands. Taking R-type AgSMe, AgOMe, AgOPh, and S-AgSPh clusters as examples, the ligands are arranged alternately on the nuclear periphery. When the three chosen clusters were fixed in the same direction, the two adjacent methyl and phenyl groups in the packing chains of R-AgSMe, R-AgOMe, and S-AgSPh are arranged in a clockwise direction (Fig. 4e–g). In contrast, the two adjacent phenyl groups in the chain of R-AgOPh are arranged in a counter-clockwise direction (Fig. 4h). Therefore, the opposite arrangements of peripheral ligands could have a significant impact on the CD signal and even lead to chirality inversion of clusters.
Based on the effective chirality transmission and bright photoluminescence performance of Ag6 clusters, CPL investigations were conducted accordingly. As shown in Fig. 5a and b, the clusters AgSMe, AgSPh, AgOMe, and AgOPh displayed decent CPL emissions at 525 nm, 533 nm, 568 nm, and 563 nm, respectively, in ethanol or methanol suspensions. Compared with the relatively low glum of the clusters (Fig. S13–S16, ESI†), the S-AgSPh cluster had a maximum glum value of 2.77 × 10−3 (Fig. 5c). Intriguingly, supramolecular chirality inversion has also been observed in CPL investigations. As described in Fig. 5a, the R-AgSMe cluster displayed a right-handed CPL at 525 nm, while the R-AgSPh cluster displayed a left-handed CPL at 533 nm (and vice versa). CPL inversion was in good agreement with CD inversion, as shown in Fig. 4a and c. This CPL inversion was further demonstrated by the S-AgOMe and S-AgOPh cluster systems (Fig. 5b), although the CPL intensity of the R-AgOPh cluster was too weak to be detected. This means that if the side chain group of the ligand is methyl, both R-AgSMe and R-AgOMe clusters show negative CPL signals. However, both R-AgSPh and R-AgOPh showed positive CPL signals (Fig. 5a and b). Compared with the previously reported side group-induced supramolecular chirality inversion in the assembly hydrogel system,53,54 the side-chain group of the ligand-induced supramolecular chirality inversion could also be achieved in a chiral nanocluster system.
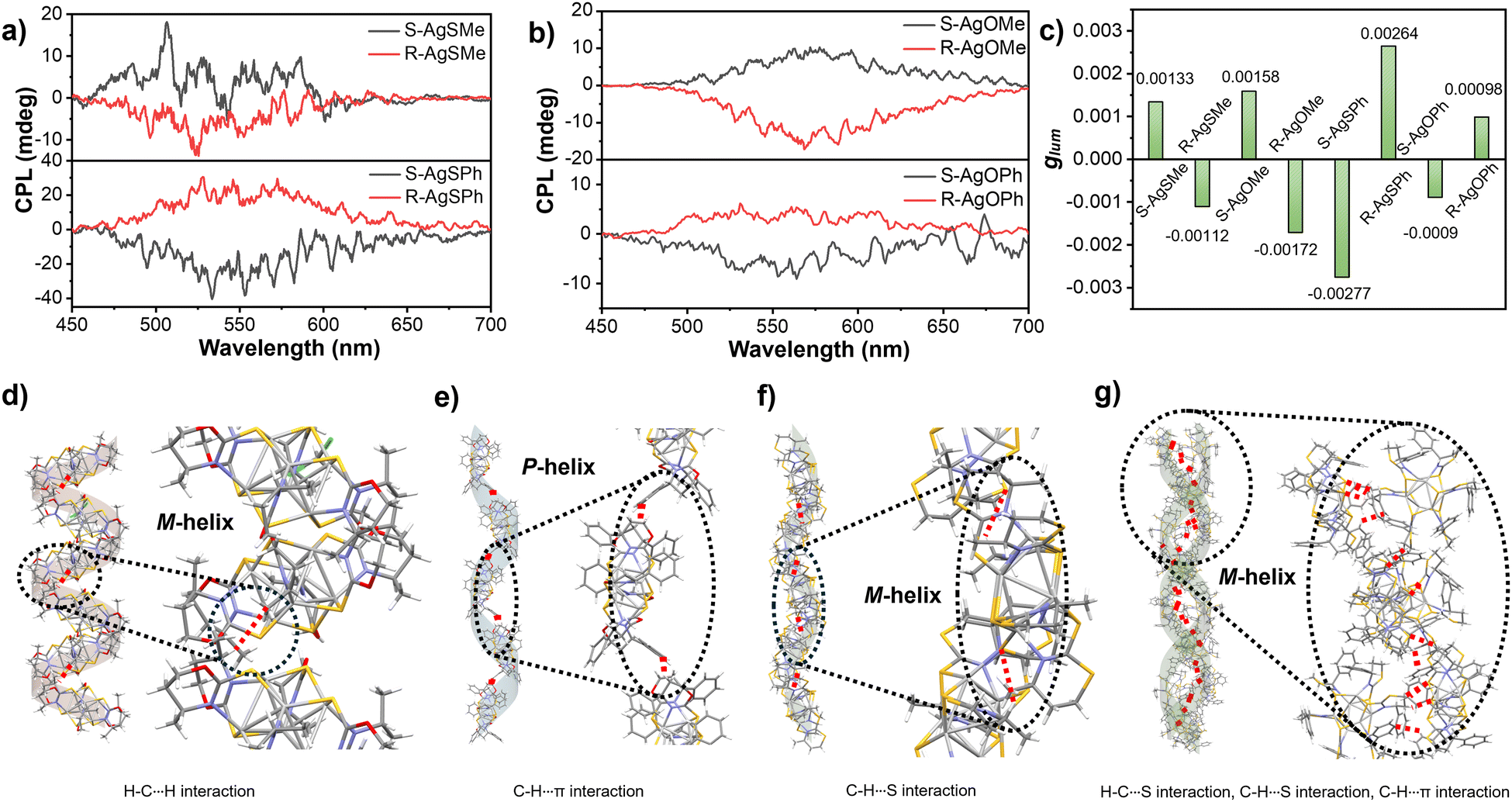 | ||
| Fig. 5 (a) CPL spectra of R/S-AgSMe and R/S-AgSPh in EtOH suspension, (b) CPL spectra of R/S-AgOMe in EtOH suspension and R/S-AgOPh in MeOH suspension, (c) glum factors of various clusters detected in (a) and (b), (d) H–C···H interaction-driven left-handed (M) helical stacking of the R-AgOMe cluster in a single-crystal structure, (e) C–H···π interaction-driven right-handed (P) helical stacking of the R-AgOPh cluster in a single-crystal structure, (f) C–H···S interaction-driven left-handed (M) helical stacking of the R-AgSMe cluster in a single-crystal structure, and (g) H–C⋯S interaction-, H–C⋯S interaction- and C–H⋯π interaction-driven left-handed (M) double helix of the S-AgSPh cluster in the single-crystal structure. | ||
Ligand-induced CPL inversion of nanoclusters could also provide insight into the stacking mode of the single-crystal structure of various clusters. Taking the R-AgOMe and R-AgOPh clusters as examples, except for the different orientations of the methyl and phenyl substituents shown in Fig. 4f and h, the stacking mode and the main driving forces in the single crystals of R-AgOMe and R-AgOPh clusters are very different. As clearly shown in Fig. 5d, the R-AgOMe cluster was stacked in a left-handed (M-type) helix, mainly driven by the H–C···H interaction between the adjacent cluster units. However, for the R-AgOPh cluster shown in Fig. 5e, the main driving force was the C–H···π interaction, which promoted the adjacent cluster units to pack in a right-handed (P) helical mode. This suggests that CPL inversion might originate from the different helical stacking modes of the cluster units between R-AgOMe and R-AgOPh since the left-helical stacking of the R-AgSMe cluster driven by the C–H···S interaction (Fig. 5f) also exhibited a negative CPL emission (Fig. 5a). The S-AgSPh packed in a left (M) helical mode was driven by the H–C⋯S, H–C⋯S and C–H⋯π interactions between the adjacent cluster units (Fig. 5g). Different helical senses of the cluster chains are clearly observed in Fig. S17 (ESI†). Additionally, for the R-AgSMe cluster, apart from the C–H···S interaction driving the helical stacking of the cluster unit in the left-handed chain, there were S⋯S interactions in the multi-layer stacking of chains (Fig. S18, ESI†). Therefore, we speculated that both the absolute R/S configuration of the peripheral substituents of methyl/phenyl and the stacking mode of the cluster units collectively determine the final CPL performance and induce CPL and CD inversion.
4. Conclusions
In summary, we synthesized four pairs of thiazithione- and oxazinthione ligand-protected chiral Ag6L6 nanoclusters with O/S-dependent emissive performances and found methyl/phenyl-directed chirality inversion using both CD and CPL measurements. First, the chiral silver clusters protected by a methyl-modified ligand showed significantly higher emission intensity than those protected by phenyl-modified ligands, which is attributed to the shorter Ag–Ag bonds in the cluster core. Additionally, silver clusters protected by thiazide cyclothione exhibited obviously greater emission intensity than the clusters modified by the oxazine cyclothione ligands, owing to the S···S interactions between the adjacent thiazithione-protected clusters. Secondly, the exchange of methyl/phenyl induced CD/CPL inversion in Ag6L6 nanoclusters, as evidenced in both thiazithione- and oxazinthione-modified clusters. This inversion was attributed to the different interactions between adjacent cluster units in the assembled cluster chains with opposite helical senses.Overall, the ligands played an important role in both the photoluminescence properties, such as emissive wavelength and QY, as well as supramolecular chirality performances like CD and CPL inversion of Ag6L6 nanoclusters. Through the analysis of PL, CD and CPL spectra, as well as the single-crystal structure, we gained insights into the intrinsic chiroptical properties of silver nanoclusters at the atom-precise level. This also sheds light on the mechanism of chirality transmission and regulation at the supramolecular scale, which benefits the development of CPL-active nanoclusters with precise chirality modulation in chiral sensors, optoelectronic engineering and even asymmetric catalysis.
Author contributions
Hui Chen: investigation, methodology, data curation, DFT-calculation, and writing the original draft. Xiangyang Zhang: data curation and formal analysis. Kuo Fu: data curation and formal analysis. Xuejuan Wang: resources and formal analysis. Guofeng Liu: conceptualization, writing – review & editing, and supervision. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the National Natural Science Foundation of China (NSFC) (No. 22101208), the Fundamental Research Funds for the Central Universities, and the Program for Professors of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning.Notes and references
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed
.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed
- Y. Zhang, J. Wang, L. Wang, R. Fu, L. Sui, H. Song, Y. Hu and S. Y. Lu, Adv. Mater., 2023, 35, 2302536 CrossRef CAS PubMed
- L. Lv, J. Li, Y. Wang, Y. Shu and X. Peng, J. Am. Chem. Soc., 2020, 142, 19926–19935 CrossRef CAS PubMed
- T. Omoda, S. Takano and T. Tsukuda, Small, 2021, 17, 2001439 CrossRef CAS PubMed
- Z. Liu, L. Luo and R. Jin, Adv. Mater., 2024, 36, 2309073 CrossRef CAS PubMed
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed
- H. Tang, Q. Li, W. Yan and X. Jiang, Angew. Chem., Int. Ed., 2021, 60, 13829–13834 CrossRef CAS PubMed
- T. Yang, J. Zhang, Y. Yu and T. Sun, Curr. Med. Chem., 2021, 28, 6990–7005 CrossRef CAS PubMed
- T. Jia, Z.-J. Guan, C. Zhang, X.-Z. Zhu, Y.-X. Chen, Q. Zhang, Y. Yang and D. Sun, J. Am. Chem. Soc., 2023, 145, 10355–10363 CrossRef CAS PubMed
- X. Wang, B. Yin, L. Jiang, C. Yang, Y. Liu, G. Zou, S. Chen and M. Zhu, Science, 2023, 381, 784–790 CrossRef CAS PubMed
- Y.-M. Wang, F.-Q. Yan, Q.-Y. Wang, C.-X. Du, L.-Y. Wang, B. Li, S. Wang and S.-Q. Zang, Nat. Commun., 2024, 15, 1843 CrossRef CAS PubMed
- J.-Q. Wang, R.-L. He, W.-D. Liu, Q.-Y. Feng, Y.-E. Zhang, C.-Y. Liu, J.-X. Ge and Q.-M. Wang, J. Am. Chem. Soc., 2023, 145, 12255–12263 CrossRef CAS PubMed
- X. Song, W. Zhu, X. Ge, R. Li, S. Li, X. Chen, J. Song, J. Xie, X. Chen and H. Yang, Angew. Chem., Int. Ed., 2021, 60, 1306–1312 CrossRef CAS PubMed
- Y. Shi, Z. Wu, M. Qi, C. Liu, W. Dong, W. Sun, X. Wang, F. Jiang, Y. Zhong, D. Nan, Y. Zhang, C. Li, L. Wang and X. Bai, Adv. Mater., 2023, 35, 2310529 Search PubMed
- K. Fu and G. Liu, ACS Nano, 2024, 18, 2279–2289 CrossRef CAS PubMed
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef
- K. Staszak, K. Wieszczycka, V. Marturano and B. Tylkowski, Coord. Chem. Rev., 2019, 397, 76–90 CrossRef CAS
- G. Liu, M. G. Humphrey, C. Zhang and Y. Zhao, Chem. Soc. Rev., 2023, 52, 4443–4487 RSC
- S. Zheng and G. Liu, CCS Chem., 2024, 6 DOI:10.31635/ccschem.024.202303722
- S. Sun, X. Li, C. Xu, Y. Li, Y. Wu, B. L. Feringa, H. Tian and X. Ma, Natl. Sci. Rev., 2023, 10, nwad072 CrossRef PubMed
- L. Zhou, J. Song, Z. He, Y. Liu, P. Jiang, T. Li and X. Ma, Angew. Chem., Int. Ed., 2024, 63, e202403773 CrossRef CAS PubMed
- R. Liu, Z. Feng, X. Yan, Y. Lv, J. Wei, J. Hao and Z. Yang, J. Am. Chem. Soc., 2023, 145, 17274–17283 CrossRef CAS PubMed
- C. Ren, W. Sun, T. Zhao, C. Li, C. Jiang and P. Duan, Angew. Chem., Int. Ed., 2023, 62, e202315136 CrossRef CAS PubMed
- T. Zhao, D. Meng, Z. Hu, W. Sun, Y. Ji, X. Wu and P. Duan, Nat. Commun., 2023, 14, 81 CrossRef CAS PubMed
- J. Song, H. Xiao, B. Zhang, L. Qu, X. Zhou, P. Hu, Z.-X. Xu and H. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202302011 CrossRef CAS PubMed
- J. Song, H. Xiao, L. Fang, L. Fang, L. Qu, X. Zhou, Z.-X. Xu, C. Yang and H. Xiang, J. Am. Chem. Soc., 2022, 144, 2233–2244 CrossRef CAS PubMed
- B. Li, Y. Li, M. H.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 21676–21684 CrossRef CAS PubMed
- T. Zhao, J. Han, P. Duan and M. Liu, Acc. Chem. Res., 2020, 53, 1279–1292 CrossRef CAS PubMed
- Y.-J. Liu, Y. Liu and S.-Q. Zang, Angew. Chem., Int. Ed., 2023, 62, e202311572 CrossRef CAS PubMed
- J. Lu, B. Shao, R.-W. Huang, L. Gutiérrez-Arzaluz, S. Chen, Z. Han, J. Yin, H. Zhu, S. Dayneko, M. N. Hedhili, X. Song, P. Yuan, C. Dong, R. Zhou, M. I. Saidaminov, S.-Q. Zang, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2024, 146, 4144–4152 CrossRef CAS PubMed
- X.-H. Ma, Y. Si, J.-H. Hu, X.-Y. Dong, G. Xie, F. Pan, Y.-L. Wei, S.-Q. Zang and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 25874–25886 CrossRef CAS PubMed
- Y. Zhu, H. Wang, K. Wan, J. Guo, C. He, Y. Yu, L. Zhao, Y. Zhang, J. Lv, L. Shi, R. Jin, X. Zhang, X. Shi and Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 9059–9063 CrossRef CAS PubMed
- Y. Sang, Q. Zhu, X. Zhou, Y. Jiang, L. Zhang and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202215867 CrossRef CAS PubMed
- Z. Han, X.-Y. Dong, P. Luo, S. Li, Z.-Y. Wang, S.-Q. Zang and T. C. W. Mak, Sci. Adv., 2020, 6, eaay0107 CrossRef CAS PubMed
- H. Wu, X. He, B. Yang, C.-C. Li and L. Zhao, Angew. Chem., Int. Ed., 2021, 60, 1535–1539 CrossRef CAS PubMed
- S. Knoppe and T. Bürgi, Acc. Chem. Res., 2014, 47, 1318–1326 CrossRef CAS PubMed
- X. Wei, Y. Lv, H. Shen, H. Li, X. Kang, H. Yu and M. Zhu, Aggregate, 2023, 4, e246 CrossRef CAS
- G. Li, H. Abroshan, C. Liu, S. Zhuo, Z. Li, Y. Xie, H. J. Kim, N. L. Rosi and R. Jin, ACS Nano, 2016, 10, 7998–8005 CrossRef CAS PubMed
- Y. Zhong, J. Zhang, T. Li, W. Xu, Q. Yao, M. Lu, X. Bai, Z. Wu, J. Xie and Y. Zhang, Nat. Commun., 2023, 14, 658 CrossRef CAS PubMed
- J. Zhao, A. Ziarati, A. Rosspeintner, Y. Wang and T. Bürgi, Chem. Sci., 2023, 14, 7665–7674 RSC
- M. He, H. Sun, L. Zhu and G. Liu, Chem. Mater., 2024, 36, 2508–2519 CrossRef CAS
- N. Amir, M. Motonishi, M. Fujita, Y. Miyashita, K. Fujisawa and K.-i Okamoto, Eur. J. Inorg. Chem., 2006, 1041–1049 CrossRef CAS
- R. L. Willer, C. G. Campbell and R. F. Storey, J. Heterocycl. Chem., 2012, 49, 421–423 CrossRef CAS
- E. Khatun, A. Ghosh, P. Chakraborty, P. Singh, M. Bodiuzzaman, P. Ganesan, G. Nataranjan, J. Ghosh, S. K. Pal and T. Pradeep, Nanoscale, 2018, 10, 20033–20042 RSC
- S. Ito, S. Takano and T. Tsukuda, J. Phys. Chem. Lett., 2019, 10, 6892–6896 CrossRef CAS PubMed
- Y. Zeng, S. Havenridge, M. Gharib, A. Baksi, K. L. D. M. Weerawardene, A. R. Ziefuß, C. Strelow, C. Rehbock, A. Mews, S. Barcikowski, M. M. Kappes, W. J. Parak, C. M. Aikens and I. Chakraborty, J. Am. Chem. Soc., 2021, 143, 9405–9414 CrossRef CAS PubMed
- Z. Han, X.-Y. Dong, P. Luo, S. Li, Z.-Y. Wang, S.-Q. Zang and T. C. W. Mak, Sci. Adv., 2020, 6, eaay0107 CrossRef CAS PubMed
- S. Mukherjee, P. Chandrashekar, I. E. Aby, S. Mittal, A. Varghese, B. Pathak and S. Mandal, J. Phys. Chem. Lett., 2023, 14, 8548–8554 CrossRef CAS PubMed
- Y. Chen, M. Zhou, Q. Li, H. Gronlund and R. Jin, Chem. Sci., 2020, 11, 8176–8183 RSC
- Y. Zeng, S. Havenridge, M. Gharib, A. Baksi, K. L. Dimuthu, M. Weerawardene, A. R. Ziefuß, C. Strelow, C. Rehbock, A. Mews, S. Barcikowski, M. M. Kappes, W. J. Parak, C. M. Aikens and I. Chakraborty, J. Am. Chem. Soc., 2021, 143, 9405–9414 CrossRef CAS PubMed
- G. Liu, X. Li, J. Sheng, P.-Z. Li, W. K. Ong, S. Z. Fiona Phua, H. Ågren, L. Zhu and Y. Zhao, ACS Nano, 2017, 11, 11880–11889 CrossRef CAS PubMed
- G. Liu, C. Zhou, W. L. Teo, C. Qian and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 9366–9372 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2342764–2342768 and 2351225. For ESI and crystallographic data in CIF or other electronic format, see DOI: https://doi.org/10.1039/d4tc01280a |
| This journal is © The Royal Society of Chemistry 2024 |