
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanocellulose-based hydrogels as versatile materials with interesting functional properties for tissue engineering applications
Arnaud Kamdem
Tamo
 abcd
abcd
aInstitute of Microsystems Engineering IMTEK, University of Freiburg, 79110 Freiburg, Germany. E-mail: arnaudkamdem38@yahoo.com
bFreiburg Center for Interactive Materials and Bioinspired Technologies FIT, University of Freiburg, 79110 Freiburg, Germany
cFreiburg Materials Research Center FMF, University of Freiburg, 79104 Freiburg, Germany
dIngénierie des Matériaux Polymères (IMP), Université Claude Bernard Lyon 1, INSA de Lyon, Université Jean Monnet, CNRS, UMR 5223, 69622 Villeurbanne CEDEX, France
First published on 21st May 2024
Abstract
Tissue engineering has emerged as a remarkable field aiming to restore or replace damaged tissues through the use of biomimetic constructs. Among the diverse materials investigated for this purpose, nanocellulose-based hydrogels have garnered attention due to their intriguing biocompatibility, tunable mechanical properties, and sustainability. Over the past few years, numerous research works have been published focusing on the successful use of nanocellulose-based hydrogels as artificial extracellular matrices for regenerating various types of tissues. The review emphasizes the importance of tissue engineering, highlighting hydrogels as biomimetic scaffolds, and specifically focuses on the role of nanocellulose in composites that mimic the structures, properties, and functions of the native extracellular matrix for regenerating damaged tissues. It also summarizes the types of nanocellulose, as well as their structural, mechanical, and biological properties, and their contributions to enhancing the properties and characteristics of functional hydrogels for tissue engineering of skin, bone, cartilage, heart, nerves and blood vessels. Additionally, recent advancements in the application of nanocellulose-based hydrogels for tissue engineering have been evaluated and documented. The review also addresses the challenges encountered in their fabrication while exploring the potential future prospects of these hydrogel matrices for biomedical applications.
1 Introduction
In the medical field, tissue engineering plays a pivotal role by integrating principles from biology, engineering, and materials science to develop functional biological substitutes that can repair or replace damaged or diseased tissues.1–3 This multidisciplinary approach addresses critical healthcare challenges, offering transformative solutions for disease management, organ transplantation, and regenerative therapies.4 Addressing the persistent shortage of viable donor organs, tissue engineering represents a paradigm shift. By integrating the body regenerative capacities with advanced biocompatible materials, it enables the controlled fabrication of functional tissues and organs in laboratory settings, thus mitigating the risk of immune rejection and offering hope to patients.5,6In recent years, the field of tissue engineering has witnessed a paradigm shift towards the exploration of advanced biomaterials that can emulate the complex and dynamic microenvironment of native tissues.7,8 Biomaterials constitute the foundation of advancements in tissue engineering, offering unique properties that are indispensable for the successful regeneration of damaged or diseased tissues.9 These biomaterials are commonly chosen for their biocompatibility, ensuring a harmonious interaction with living cells and tissues without inducing adverse reactions.1,8 Biomaterials also possess tunable physical and mechanical properties, allowing for the customization of scaffolds to mimic the specific characteristics of the target tissue. Their ability to provide structural support, promote cell adhesion, and guide cellular behavior makes them essential for fabricating three-dimensional frameworks that facilitate tissue engineering.8,10 Furthermore, biomaterials can serve as carriers for biomolecules and cells, enhancing their therapeutic potential. In addition to their biocompatibility, biomaterials are also biodegradable, ensuring a controlled release of encapsulated molecules, aligning with the natural healing process, and eliminating the need for scaffold removal.11–13 While presenting interesting properties and characteristics for their use in tissue engineering, biomaterials are generally combined with other materials and biomolecules to enhance their properties for specific biomedical applications.14,15 This combination approach allows for the fabrication of more complex and functional systems tailored to particular needs, such as the engineering of specific tissues or the customization of medical devices.16,17 The synergy between biomaterials and other components often enhances biocompatibility, mechanical strength, and the ability to induce desired cellular responses.18,19
Among the various types of biomaterials commonly used to design functional constructs for repairing damaged tissues and organs, nanocellulose-based hydrogels have emerged as a promising class of materials, demonstrating remarkable potential for a myriad of applications in tissue engineering.20,21 Nanocellulose, derived from cellulose fibers at the nanoscale level, possesses unique properties that make it an ideal candidate for designing tailored functional constructs in tissue engineering.22,23 Its nanoscale dimensions contribute to a high surface area, allowing for enhanced cellular interactions and promoting cell adhesion and proliferation. Nanocellulose is biocompatible and exhibits remarkable mechanical strength.7,24,25 Moreover, its tunable properties enable the fabrication of hydrogels with customizable stiffness and porosity, providing a platform for mimicking specific tissue microenvironments.26,27 The fibrous nature of nanocellulose also facilitates the formation of intricate 3D structures, closely resembling the architecture of native tissues.28,29 These properties, coupled with its biodegradability, make nanocellulose a versatile biomaterial for creating functional constructs tailored to the unique requirements of tissue engineering.10,21
In the spectrum of recent advancements in tissue engineering, hydrogels play a crucial role.7,8,10 With their unique ability to retain water in a three-dimensional network structure, hydrogels mimic the natural extracellular matrix, creating a nurturing environment for cell growth, proliferation, and differentiation.7,30 Hydrogel versatility in accommodating various cell types and bioactive molecules amplifies their significance, allowing precise engineering for intricate cellular interactions. This biomimetic approach extends to their mechanical properties, tailored to match specific tissues, making hydrogels invaluable for guiding tissue engineering processes.31,32 Additionally, hydrogels address challenges in tissue engineering, playing a fundamental role in controlled drug delivery and scaffold design. They encapsulate therapeutic agents, ensuring targeted release and providing architectural frameworks for tissue engineering.12,33 Furthermore, hydrogels can be infused with bioactive signals to guide cell behavior, 3D directing them towards differentiation and the formation of functional tissues. This process is essential in tissue engineering.
In the field of tissue engineering, integrating nanocellulose into matrices to design composite hydrogels contributes to enhancing the properties of the resulting composites.34–36 Nanocellulose, derived from plant or bacteria sources, possesses exceptional strength, biocompatibility, and versatility, making it an ideal biomimetic reinforcement material. When incorporated into hydrogels, nanocellulose enhances the mechanical stability and structural integrity of the resulting biomaterial hydrogels.8,37,38 Moreover, nanocellulose biocompatibility fosters a conducive environment for cellular interactions in the hydrogel matrix.8,24 Nanocellulose, with its hydroxyl groups (–OH) and other functional groups, can provide an ideal scaffold for cell adhesion, proliferation, and differentiation. These functional groups facilitate hydrogen bonding and electrostatic interactions with cell surface molecules, promoting cellular attachment and signaling cascades essential for tissue regeneration.7,10,30,37 Beyond its contributions to tissue engineering, nanocellulose ability to facilitate controlled drug release makes it invaluable in targeted therapies and regenerative medicine.39–41 By incorporating bioactive molecules into nanocellulose-based hydrogel carriers, scientists could precisely regulate drug release, promising biomimetic responses in a targeted manner.13,42
Extensive research in the literature has highlighted the significant potential of nanocellulose-based hydrogels for repairing diverse types of damaged tissues. Studies demonstrated the versatility of nanocellulose in tissue engineering applications, showcasing its ability to mimic the native extracellular matrix, promote cell adhesion and proliferation, and exhibit excellent biocompatibility. Researchers have successfully utilized nanocellulose hydrogels to address various tissue repair needs, including wound healing, bone, nerve and cardiac engineering as well as cartilage repair.16,17,43,44 The tunable properties of these hydrogels allow for the fabrication of tailored hydrogel constructs with customizable stiffness and porosity, providing an adaptable platform for different tissue microenvironments.22 The review delves into the potential of nanocellulose-reinforced hydrogels for tissue engineering. It initially underscores the pivotal role of hydrogels in this field. Various aspects of nanocellulose are covered, including its types, properties, sources, biocompatibility, and biodegradability. Moreover, it discusses engineering techniques and cross-linking strategies aimed at integrating nanocellulose with other materials to create composite hydrogels. The review further emphasizes the biomimetic properties of nanocellulose-based hydrogels, such as their ability to deliver bioactive molecules, promote cell adhesion and proliferation, and mimic signals from the extracellular matrix. It explores the diverse applications of these hydrogels in bone, cartilage, nerve, vascular, and skin tissue engineering. Additionally, it outlines commonly used characterization methods for assessing the mechanical, structural, and biological attributes of these polymer materials. Recent advancements in utilizing nanocellulose-based hydrogels for tissue engineering are also evaluated and described. Finally, the review addresses the challenges encountered during their fabrication process and explores potential future prospects for these functional hydrogel matrices in biomedical applications.
2 Nanocellulose: properties and types
Nanocellulose, derived from cellulose, exhibits remarkable properties and exists in various forms. It is characterized by its nanoscale dimensions, high aspect ratio, and exceptional mechanical strength. Nanocellulose can be categorized into three main types: cellulose nanocrystals (CNCs), cellulose nanofibrils (CNFs), and bacterial nanocellulose (BNC). CNCs are rigid, rod-like nanoparticles with dimensions on the order of nanometers. CNFs, on the other hand, are long, flexible fibrils with diameters ranging from tens to hundreds of nanometers. BNC is produced by certain bacteria and forms a unique, highly pure form of nanocellulose. Each type offers distinct advantages and applications across various industries.2.1 Exploring nanocellulose: insights into nanocrystals and nanofibrils
Nanocellulose, a versatile and sustainable biomaterial, encompasses a spectrum of types, each with its unique properties and applications. The primary categories of nanocellulose include CNCs, CNFs, and BNC (as illustrated in Fig. 1 and 2a–c). Nanocellulose, can be sourced from both plants and bacteria, each offering distinct properties and extraction techniques.45–47 Plant-based nanocellulose, primarily obtained from natural sources such as wood pulp, cotton, hemp, and agricultural residues, serves as a notable example of sustainable engineering practices.48,49 Wood pulp, derived from various tree species, remains a predominant source, serving as the foundation for producing both CNCs and CNFs. The extraction process involves breaking down the complex cellulose structure into nano-sized entities, resulting in CNCs and CNFs renowned for their exceptional mechanical strength and biocompatibility.50–53 In contrast, BNC represents a cutting-edge approach to nanocellulose production. Cultivated through microbial fermentation, BNC is synthesized by acetic acid bacteria, notably Gluconacetobacter xylinus, using glucose-containing substrates.54 This method yields an ultra-pure and mechanically robust nanocellulose variant, offering distinct advantages such as high purity, mechanical strength, and biocompatibility. BNC stands out as a prime choice for biomedical applications, including tissue engineering and wound healing, owing to its unique properties derived from its bacterial origins.55,56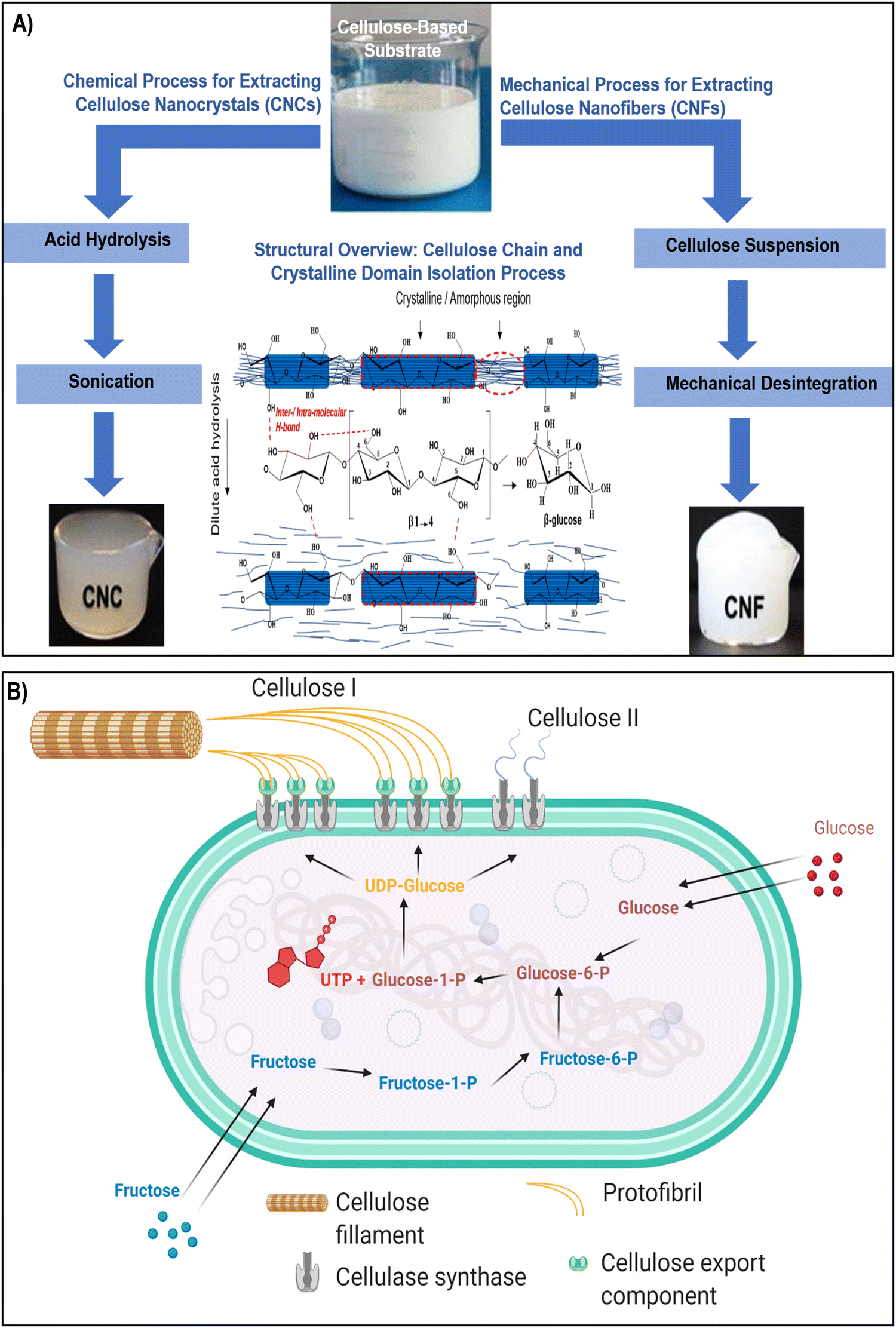 | ||
| Fig. 1 (A) Two established methods for the isolation of CNCs and CNFs involve acid hydrolysis and mechanical disintegration, accompanied by a hypothetical schematic representing the dilute acid pretreatment process aimed at extracting crystalline regions from cellulose amorphous domains. The middle section illustrates the configuration of the cellulose repeating unit, highlighting the β-(1,4) glycosidic linkage influenced by intra/intermolecular hydrogen bonding. Reproduced with permission from ref. 57 Copyright © 2019 Elsevier. (B) Schematic diagram depicting the biosynthesis pathways of bacterial cellulose I and II from glucose and fructose. Reproduced with permission from ref. 46 Copyright © 2021 MDPI. | ||
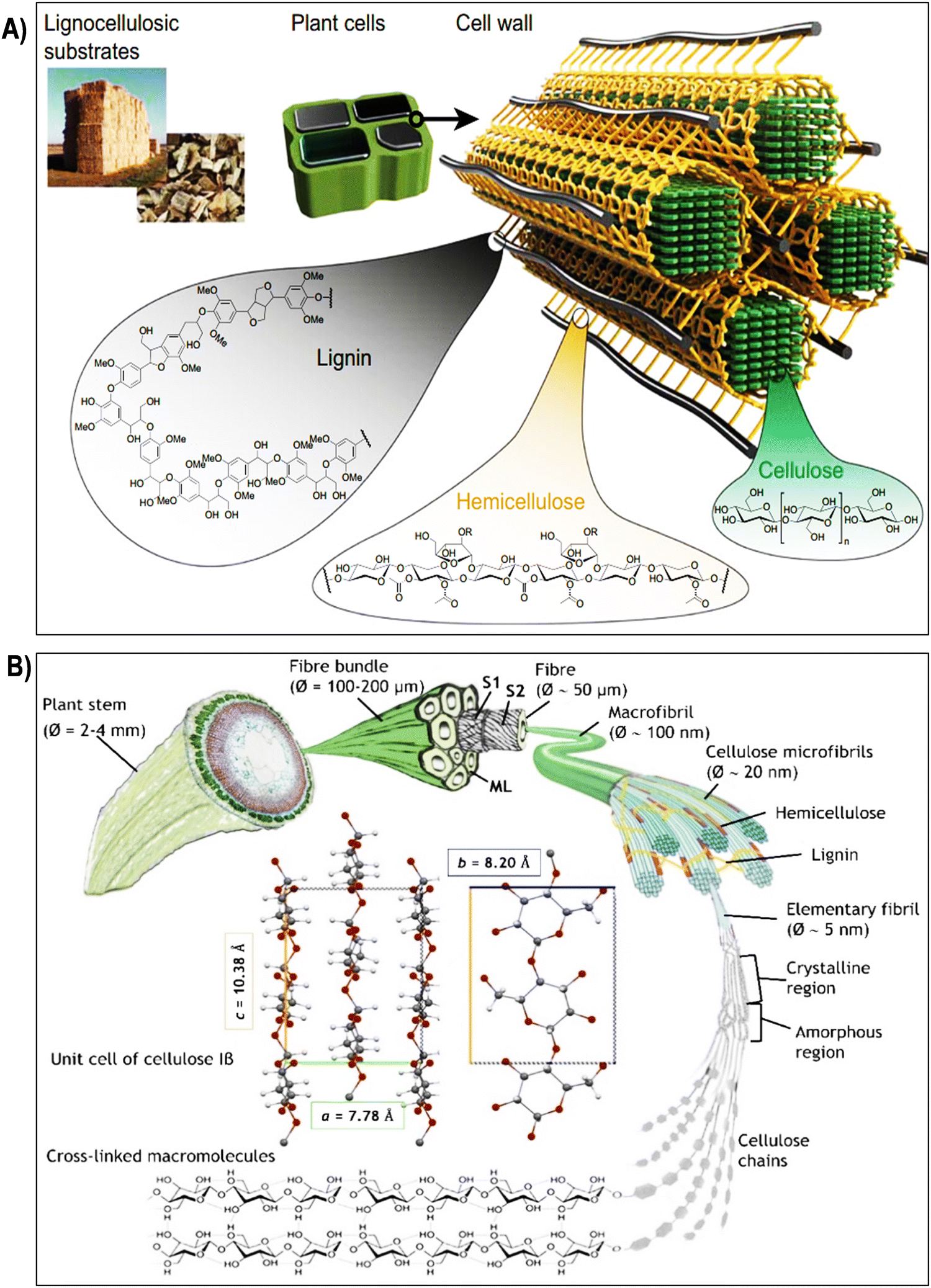 | ||
| Fig. 2 (A) Composition and structure of lignocellulosic plant cell walls. Lignocellulosic plant cell walls primarily consist of cellulose, hemicellulose, and lignin. Reproduced with permission from ref. 58 Copyright © 2020 Springer. (B) Schematic illustration of the hierarchical structure of cellulose, starting from its macroscopic origin in the plant stem (depicted here with flax). Cellulose microfibrils, which form the secondary cell wall of fibers, are surrounded by hemicelluloses, lignin, and pectins, creating an interlocking matrix gel. The unit cell of cellulose Iβ, predominant in flax and cotton, is depicted with its a, b, and c lattice constants. Reproduced with permission from ref. 59 Copyright © 2022 Royal Society of Chemistry. | ||
CNCs are often produced through acid hydrolysis, a chemical process where cellulose-based materials are treated with strong acids, causing the amorphous regions of cellulose to be removed, leaving behind crystalline nanoparticles (Fig. 1A).60,61 CNFs, on the other hand, can be obtained through mechanical disintegration, wherein cellulose-based substrates are subjected to high shear forces using techniques like homogenization or microfluidization. These mechanical forces break down the fibers into nanoscale fibrils, creating a suspension of CNFs.27,55 In contrast, the production of BNC commonly involves specific bacteria like Komagataeibacter xylinus and follows a complex series of biochemical processes (Fig. 1B). In the BNC production pathway, glucose acquired from the surroundings undergoes isomerization to glucose-1-phosphate, subsequently reacting with uridine-5-triphosphate (UTP) to generate uridine diphosphate glucose (UDP-glucose). Catalyzed by cellulose synthase A and activated by cyclic-di-GMP, linear 1,4 glucan chains are synthesized. These chains are then discharged through pores in the bacterial cell wall.46,62 The entire process is regulated by the bacterial cellulose synthesis ABCD operon, where bcsA encodes the catalytic subunit, and bcsB produces a regulatory subunit that binds to c-di-GMP, a crucial step in initiating cellulose production. The exact functions of bcsC and bcsD remain somewhat ambiguous, although it is suggested that bcsC might play a role in the formation of pores in the cell membrane. Following fermentation, the resulting pellicle comprises cellulose, secondary metabolites, microbial biomass, and residual nutrients.46,63 This pellicle can be subjected to purification, ultimately yielding a highly pure crystalline cellulose matrix. Despite its molecular formula being similar to plant-derived cellulose, BNC exhibits distinct physical and chemical properties that render it exceptionally valuable in diverse applications. The production process meticulously follows biochemical pathways, ensuring the synthesis of cellulose in bacterial cultures. Komagataeibacter xylinus produces bacterial cellulose extracellularly in four allomorphic forms, I–IV, with celluloses I and II being the most extensively studied.46,63
The hierarchical structure of cellulose fibers displays an intricate and orderly arrangement across various levels, ranging from the macroscopic organization of the cell wall to the molecular configuration of cellulose chains as illustrated in the Fig. 2 and 3a. At the macroscopic level, cellulose fibers exhibit a well-defined hierarchical structure within the cell wall. This structure comprises layers of cellulose microfibrils embedded in a matrix of hemicelluloses and lignin (Fig. 2A).64 These microfibrils, in turn, consist of bundles of cellulose chains organized in a crystalline lattice. Delving further into the microscopic scale, cellulose microfibrils reveal a highly structured assembly of cellulose molecules. This assembly results in crystalline regions interspersed with less organized, amorphous regions (Fig. 1A and 3b).65 The stability of the crystalline cellulose structure is primarily upheld by hydrogen bonding interactions between adjacent cellulose chains, establishing a robust and secure framework. Cellulose, a complex polysaccharide predominantly found in the cell walls of plants, algae, and select bacteria, boasts a meticulously organized microstructure that underlies its exceptional mechanical strength and adaptability.48,66 Composed of linear chains of glucose molecules linked by β-1,4-glycosidic bonds, cellulose forms intricate crystalline structures due to the regular arrangement of its constituent units. This systematic assembly operates across various scales, with each contributing to the unique properties of cellulose.35,67 At the nanoscale, individual cellulose chains align into extended, flat ribbons, held together by hydrogen bonds, resulting in the formation of robust, inflexible structures known as cellulose microfibrils. These microfibrils, with diameters measuring several nanometers, align themselves into bundles within plant cells, forming a mesh-like network. The hydrogen bonds between these chains provide cohesion and stability, significantly bolstering cellulose tensile strength.68–70 Expanding to a larger scale, cellulose microfibrils come together to create macroscopic units, constituting cellulose fibers. These fibers are bound together by van der Waals forces and additional hydrogen bonds, culminating in the formation of a dense and stable material.71–73 This intricately organized structure, with its interplay of crystalline and amorphous regions, defines the exceptional mechanical integrity of cellulose and serves as the foundation for its diverse applications in various industrial and biological contexts.74,75 Due to the unique arrangement of cellulose microfibrils, cellulose fibers exhibit impressive mechanical properties, including high tensile strength and stiffness. In the context of plant cell walls, cellulose microfibrils are embedded within a matrix of hemicellulose, pectin, and lignin (Fig. 2A). This composite structure provides plants with structural integrity, enabling them to withstand mechanical stress and maintain their shape. The precise arrangement of cellulose microfibrils within this matrix contributes to the rigidity of plant cell walls, allowing plants to grow tall and maintain their form against gravitational forces.8,24,75,76 Plant fibers, depicted in Fig. 2B, comprise elongated cylindrical cells with an outer primary cell wall (S1), an inner secondary cell wall (S2), and a central hollow channel (lumen). S2, rich in microfibrils, primarily consists of crystalline cellulose, representing about 95% of cotton fiber weight and 65–80% of flax fiber weight.59 Other components include waxes, protein, pectate, and minerals in cotton, and hemicellulose, pectins, proteins, and lignin (2–2.5%) in flax. The interaction between cellulose and hemicellulose, along with hemicellulose covalent bonds, influences fiber macro-mechanical behavior. Cellulose molecules form microfibrils through hydrogen bonding, adopting crystalline or semi-crystalline structures with lattice parameters.59–79 Cellulose I occurs naturally in two crystalline forms: Iα and Iβ, with Iβ predominant in flax and cotton, characterized by lattice constants a = 7.80 Å, b = 8.20 Å, and c = 10.38 Å.59,80 Amorphous regions lacking hydrogen bonding are also present. Various treatments target cellulose hydroxyl groups in both ordered and disordered regions, affecting swelling and crystallinity (Fig. 2B).
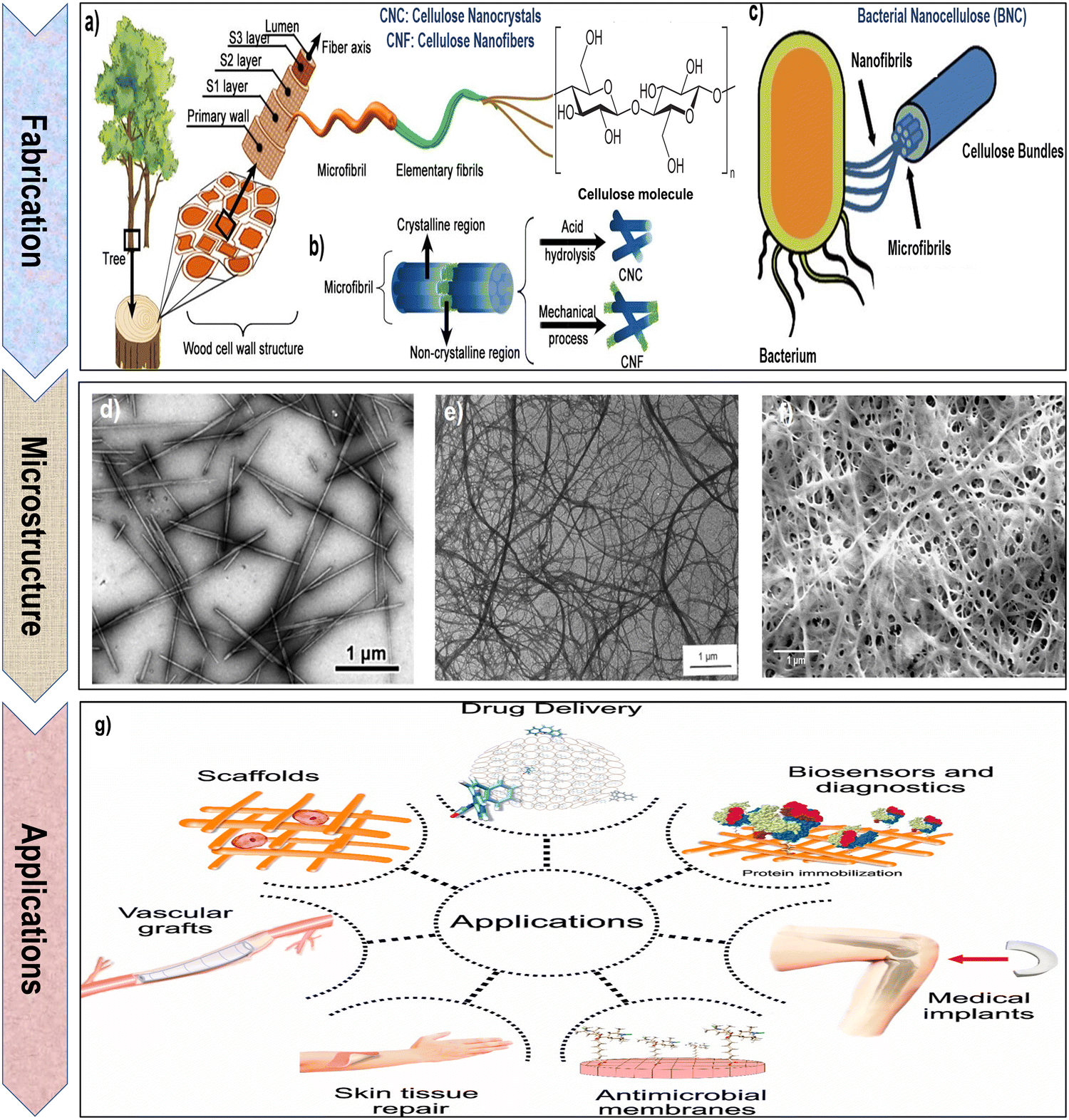 | ||
| Fig. 3 (a) and (b) Schematic representation of the process for extracting cellulose nanocrystals (CNCs) and cellulose nanofibrils (CNFs) from the cell walls of wood and plants. Reproduced with permission from ref. 81 Copyright © 2023 MDPI, as well as (c) bacterial nanocellulose (BNC) from bacteria. Reproduced with permission from ref. 66 Copyright © 2023 Springer. (d) Transmission electron microscopy (TEM) image of CNCs. Reproduced with permission from ref. 35 Copyright © 2012 American Chemical Society. (e) Scanning electron microscopy (SEM) image of CNFs. Reproduced with permission from ref. 82 Copyright © 1997 John Wiley & Sons, Inc. (f) Field emission scanning electron microscopy (FESEM) image of BNC. Reproduced with permission from ref. 83 Copyright © 2020 Springer Nature. (g) Schematic illustration of diverse biomedical applications for nanocellulose. Reproduced with permission from ref. 84 Copyright © 2020 MDPI. | ||
The microstructure of cellulose has significant implications for various industries. In papermaking, the arrangement of cellulose fibers influences the texture, strength, and absorbency of the paper.85,86 Additionally, advancements in nanotechnology have led to the isolation of cellulose nanocrystals and nanofibrils, exploiting the unique microstructure of cellulose for the development of nanocomposites and biomedical materials. These nanoscale cellulose entities exhibit exceptional mechanical properties and have gained attention for their potential applications in a wide range of fields.66,87
The preparation of CNCs involves several advanced methods, each tailored to yield nanocrystals with specific properties for diverse applications. One common approach is acid hydrolysis (Fig. 1A and 3b), where cellulose fibers are treated with strong acids, such as sulfuric acid or hydrochloric acid, under controlled conditions. This process selectively removes the less crystalline or amorphous regions of cellulose, leaving behind highly crystalline CNCs with characteristic rod-like shapes.97 Another method involves enzymatic hydrolysis, wherein enzymes like cellulase break down cellulose fibers into nanocrystals. This technique offers a more environmentally friendly alternative compared to acid hydrolysis and allows precise control over CNC dimensions.98 In addition, mechanical disintegration techniques, such as microfluidization and homogenization, utilize mechanical forces to physically break down cellulose fibers into nanoscale dimensions. These methods assiocated with chemical treatments are efficient in producing CNCs with tunable sizes and morphologies.99 Additionally, chemical oxidation techniques, including TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl)-mediated oxidation, involve the use of specific chemical catalysts to oxidize cellulose, resulting in CNCs with negatively charged surfaces, enhancing their stability in aqueous solutions.100,101 Emerging methods include deep eutectic solvent (DES) treatment, where cellulose fibers are dissolved in a DES and then precipitated to obtain CNCs. This method is environmentally sustainable and produces high-quality CNCs suitable for various applications.102,103 Ultrasonication, which utilizes ultrasound waves to disintegrate cellulose fibers, is another versatile method. It allows for the production of CNCs on both laboratory and industrial scales, with the size and properties of the CNCs controlled by the duration and intensity of ultrasound treatment.104,105 Additionally, methods like steam explosion and combined mechanical-chemical treatments have been explored to enhance the efficiency of CNC extraction, ensuring a more sustainable and economical process.106 These diverse preparation methods empower scientists to obtain CNCs with tailored characteristics, enabling their utilization in advanced materials, nanocomposites, drug delivery systems, and biomedical applications, thus driving innovations in various fields of science and technology (Fig. 3g).
The production of CNFs from plant biomasses or other cellulose sources mainly involves various physical and mechanical methods aimed at breaking down cellulose fibers into nanoscale fibrils. One common technique is mechanical disintegration, where cellulose fibers are subjected to high shear forces using methods such as microfluidization, homogenization, or grinding. These mechanical forces separate the fibers into smaller entities, eventually yielding CNFs.110–112 Microfluidization, in particular, involves forcing cellulose fibers through a narrow channel at high velocities, resulting in effective fibrillation due to shear and cavitation forces.111 Another method is high-pressure homogenization, where cellulose fibers are forced through a narrow gap at high pressures, causing them to shear and fibrillate into nanoscale dimensions.112 Ultrasonication, utilizing ultrasound waves, induces mechanical agitation and cavitation, leading to the disintegration of cellulose fibers into CNFs.104 These methods are advantageous as they do not involve chemical treatments, preserving the natural properties of CNFs. In addition to mechanical approaches, physical methods like cryocrushing have been explored. In this method, cellulose fibers are frozen in liquid nitrogen and then shattered, resulting in fine fibrillation due to the brittle nature of the frozen material. These techniques generate CNFs with high aspect ratios, offering excellent reinforcement potential in various applications.113 Furthermore, combination methods, such as enzymatic pre-treatment and mechanical disintegration, have been employed to enhance the efficiency of CNFs production.8,25,114 Enzymatic treatments, partially break down cellulose fibers, making them more amenable to subsequent mechanical disintegration. These physical and mechanical methods enable the production of CNFs with specific characteristics, including fibril length, width, and surface properties, tailored for diverse applications such as nanocomposites, films, coatings, and biomedical materials.115,116 By optimizing these methods, scientists can obtain CNFs with desired properties, contributing significantly to advancements in nanotechnology and sustainable materials development.
BNC possesses an impressive array of features that elevate its value in industrial and biomedical applications. Its outstanding purity, often reaching 99.9%, ensures it is virtually free from impurities, making it an ideal choice for biomedical applications where material purity is paramount.122 BNC exhibits a degree of polymerization within the range of 4000 to 8000, coupled with a crystallinity spanning from 75% to 80%. The elementary crystallites exhibit a length of 100–150 nm, with a lateral size of 8–10 nm.123–126 BNC plays a crucial role in biomedical engineering due to its unique properties. Its biocompatibility, high porosity, remarkable mechanical strength and water-holding capacity, and capacity to be tailored into diverse forms make it an ideal material for various biomedical applications.16,127,128 From wound dressings to tissue scaffolds, bacterial cellulose serves as a versatile and effective component, contributing to advancements in regenerative medicine and tissue engineering. Additionally, its natural origin and biodegradability align with the growing emphasis on sustainable and eco-friendly practices in biomedical research and applications.54,118,129–132 Moreover, BNC is highly biocompatible and non-toxic, allowing safe direct contact with biological tissues without causing adverse reactions.133,134 Its substantial and high surface area facilitates efficient interactions with various molecules, making it ideal for drug delivery systems.135 The nanofibrous architecture of BNC, characterized by substantial fiber diameters and lengths, offers an extensive surface area-to-volume ratio. With numerous hydroxyl groups and inherent hydrophilicity, BNC exhibits enhanced reactivity, positioning it as a highly suitable material for surface modifications and functionalization.136 Microscopical image of BNC (as highlighted in the Fig. 3f) exhibits a unique, nanofibrous network composed of ultrafine cellulose fibers. These fibers are highly uniform in diameter and appear densely packed, creating a compact and stable material. BNC displays a characteristic mesh-like structure with a high degree of porosity, providing a large surface area for interactions with surrounding environments.83,119 The image showcases the smooth and homogeneous surface of BNC fibers, underscoring their high purity and quality. BNC micrograph often reveals an intricate arrangement of fibers, forming a three-dimensional scaffold with a nanoscale texture.83,119
In summary, nanocellulose, a versatile material with unique properties, holds significant promise for biomedical applications. Its inherent features, such as biocompatibility, high surface area, and tunable mechanical strength, make it an excellent candidate for various biomedical uses. The nanoscale dimensions of nanocellulose allow precise manipulation, facilitating tailored applications in drug delivery, tissue engineering, and wound healing. Moreover, its renewable and sustainable nature aligns with the growing emphasis on eco-friendly biomedical solutions. The combination of these properties positions nanocellulose as a valuable resource for advancing innovations in the biomedical field.55,118,129Fig. 3g emphasizes several biomedical applications of nanocellulose hydrogels.
2.2 Tailoring nanocellulose properties through functionalization
Functionalizing nanocellulose is pivotal for optimizing its properties and expanding its applicability.136,137 Nanocellulose, featuring high surface area, biocompatibility, and mechanical strength, undergoes diverse surface modifications, including oxidation, coupling, coating, etc. These methods tailor its surface chemistry, introducing functionalities like carboxyl, hydroxyl, or amino groups.26 This customization enhances compatibility with other materials, crucial for uniform dispersion in nanocomposites or favorable interactions. In addition, functionalization enables precise control over wettability, adhesion, and reactivity, making nanocellulose versatile for varied applications.138 In biomedicine, this material serves as a foundation for drug delivery, wound dressings, and tissue engineering. Tailoring nanocellulose for targeted drug release or antimicrobial properties enhances its suitability.83,139 In sustainability, functionalized nanocellulose plays a vital role in green technologies, aiding water treatment and reinforcing biodegradable composites.108,140Table 1 provides a comprehensive overview of the different functionalization methods, along with their respective advantages and disadvantages, aiding in the selection of the most suitable method for fabricating functionalized nanocellulose-based hydrogels for tissue engineering applications.| Functionalization methods | Principles | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|
| Esterification | Introduction of ester groups to nanocellulose surface through reaction with carboxylic acid derivatives. | • Enhanced hydrophobicity | • Requires harsh reaction conditions | 141–143 |
| • Improved mechanical properties | • Formation of byproducts | |||
| • Tunable surface chemistry | • Difficulties in controlling degree of substitution | |||
| Etherification | Introduction of ether groups to nanocellulose surface via reaction with alkyl or aryl halides. | • Increased stability against moisture | • Limited reaction selectivity | 144 and 145 |
| • Enhanced mechanical properties | • Formation of heterogeneous products | |||
| • Improved dispersibility | • Moderate to low functionalization efficiency | |||
| Silanization | Coating of nanocellulose with organosilanes to modify surface properties. | • Enhanced hydrophobicity or hydrophilicity | • Requires specific handling and storage conditions for silanes | 146–148 |
| • Improved compatibility with other materials | • Potential toxicity of unreacted silanes | |||
| • Enhanced chemical stability | • May introduce heterogeneous surface coverage | |||
| Sulfonation | Introduction of sulfonate groups onto nanocellulose surface through reaction with sulfonating agents. | • Enhanced water solubility | • Harsh reaction conditions | 149–152 |
| • Improved interaction with polar solvents | • Potential degradation of cellulose structure | |||
| • Increased surface charge | • Formation of side products | |||
| Carboxymethylation | Attachment of carboxymethyl groups to nanocellulose surface using chloroacetic acid or its derivatives. | • Introduction of carboxyl groups for further functionalization | • Requires alkaline conditions | 153–156 |
| • Increased water solubility | • Formation of byproducts | |||
| • Improved dispersibility | • Potential decrease in mechanical properties | |||
| Phosphorylation | Introduction of phosphate groups onto nanocellulose surface through reaction with phosphorus-containing compounds. | • Enhanced bioactivity for tissue regeneration | • Complex synthesis routes may require multiple steps | 157–159 |
| • Improved flame retardancy | • Potential cytotoxicity of reactants | |||
| • Increased surface reactivity | • Potential decrease in mechanical properties | |||
| Polycondensation | Formation of covalent bonds between nanocellulose and polymeric chains through condensation reactions. | • Enhanced mechanical properties of resulting hydrogel | • Complex reaction kinetics | 160 and 161 |
| • Improved stability against environmental factors | • Difficulty in controlling polymer chain length | |||
| • Tunable network structure | • Formation of heterogeneous networks | |||
| Cationization | Introduction of positively charged groups onto nanocellulose surface via reaction with cationic reagents. | • Enhanced interaction with negatively charged molecules (e.g., proteins, cells) | • Potential cytotoxicity of cationic reagents | 162–164 |
| • Improved dispersion in polar solvents | • Non-specific binding to negatively charged species | |||
| • Increased stability in aqueous environments | • Limited control over degree of functionalization | |||
| Coating | Application of a thin layer of functional material onto nanocellulose surface. | • Versatile: can be used to introduce various functionalities | • Limited durability of coating | 165–167 |
| • Controlled deposition of coating material | • Potential delamination | |||
| • Preservation of inherent properties of nanocellulose | • Difficulty in achieving uniform coverage | |||
| Adsorption | Adsorption of functional molecules onto nanocellulose surface via physical interactions. | • Simple and cost-effective method | • Weak bonding between adsorbate and nanocellulose | 168–170 |
| • Minimal alteration of nanocellulose structure | • Difficulty in controlling adsorption efficiency | |||
| • Versatile: wide range of molecules can be adsorbed | • Potential desorption under certain conditions | |||
| Initiation | Introduction of reactive sites onto nanocellulose surface to initiate subsequent polymerization or modification reactions. | • Precise control over location and density of functional groups | • Requires specific initiation agents | 171 and 172 |
| • Compatibility with various polymerization methods | • Limited scalability for large-scale applications | |||
| • Enables grafting of diverse monomers | • Potential decrease in cellulose crystallinity | |||
| Oxidation | Introduction of oxygen-containing functional groups (e.g., hydroxyl, carboxyl) onto nanocellulose surface using oxidizing agents. | • Increased hydrophilicity | • Harsh reaction conditions | 100, 127 and 173 |
| • Enhanced reactivity for further functionalization | • Potential degradation of cellulose structure | |||
| • Improved dispersibility | • Formation of undesired byproducts | |||
In the Fig. 4, the mechanisms behind key chemical modifications of nanocellulose are highlithed.
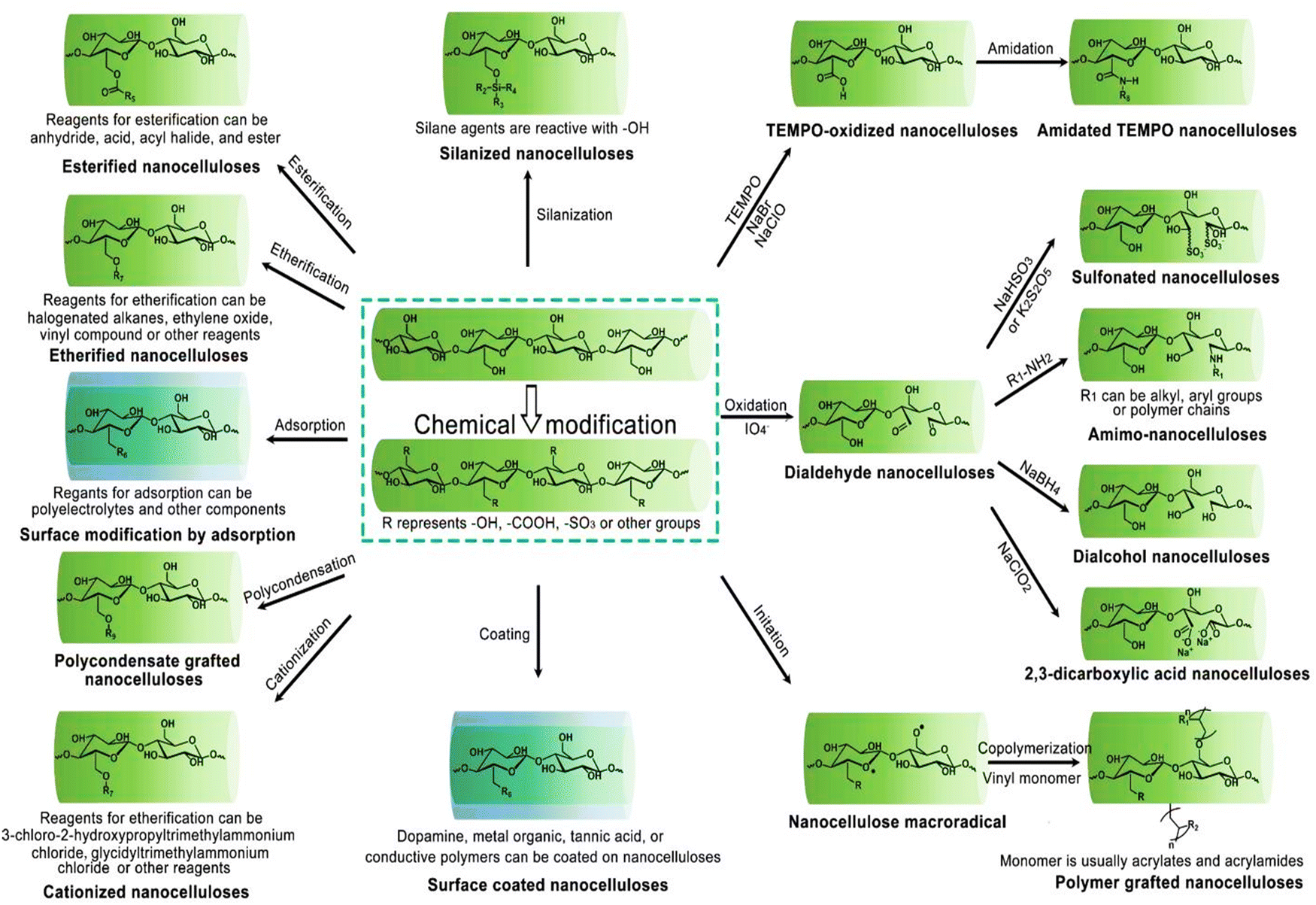 | ||
| Fig. 4 Overview of customizing nanocellulose functionality. Reproduced with permission from ref. 81 Copyright © 2023 MDPI. | ||
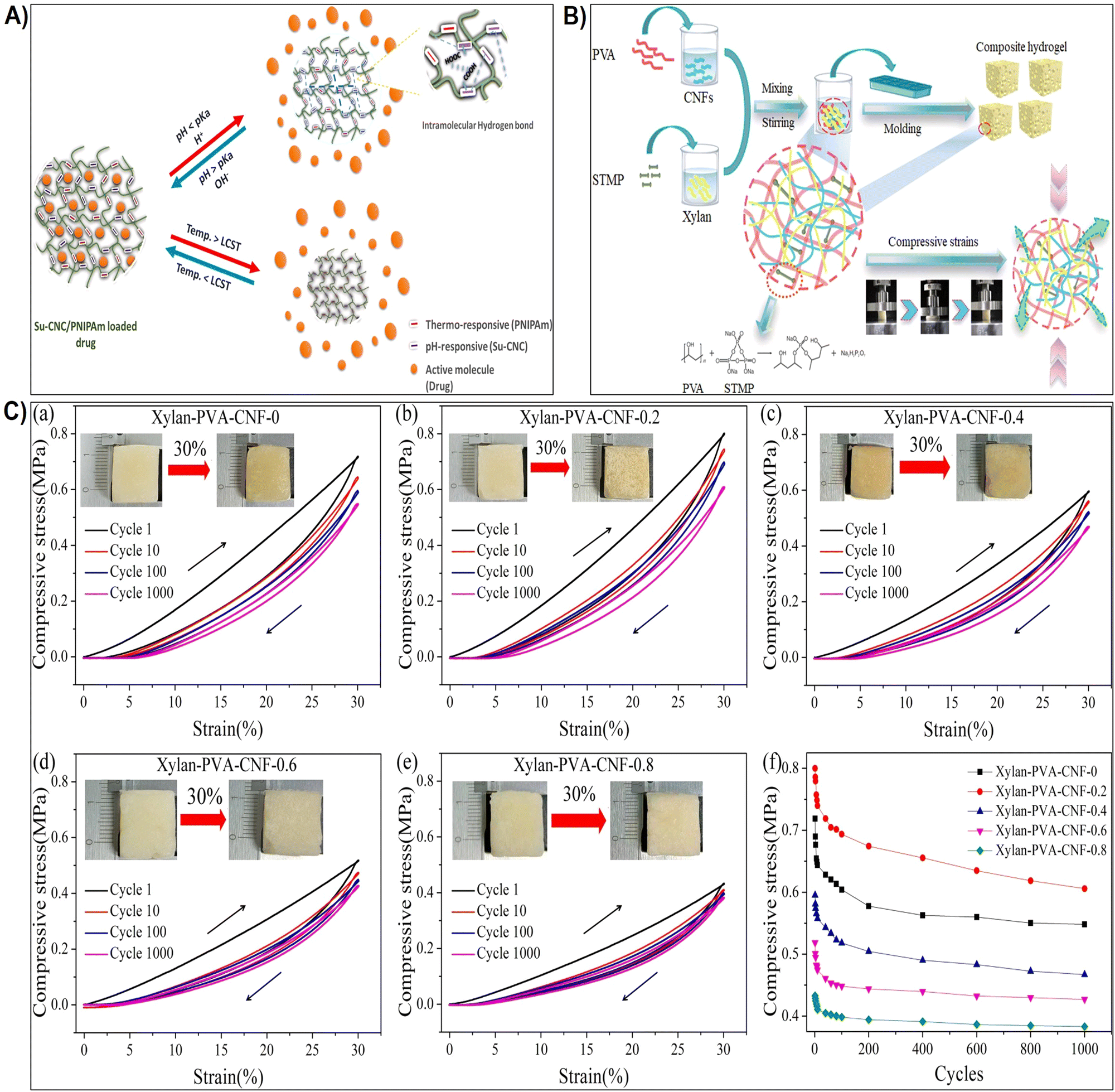 | ||
| Fig. 5 (A) Schematic representation illustrating the influence of pH and thermo-responsiveness on the drug release. Reproduced with permission from ref. 176 Copyright © 2022 Elsevier. (B) Schematic depiction of the hydrogel preparation process and the proposed behavior of the hydrogel under compression. Reproduced with permission from ref. 177 Copyright © 2023 Elsevier. (C) (a)–(e) Strength-strain curves of xylan/PVA/CNF hydrogels at maximum strains of 30% and after 1000 cycles of loading–unloading; (f) maximum strength plotted against the number of compressive test cycles. Reproduced with permission from ref. 177 Copyright © 2023 Elsevier. | ||
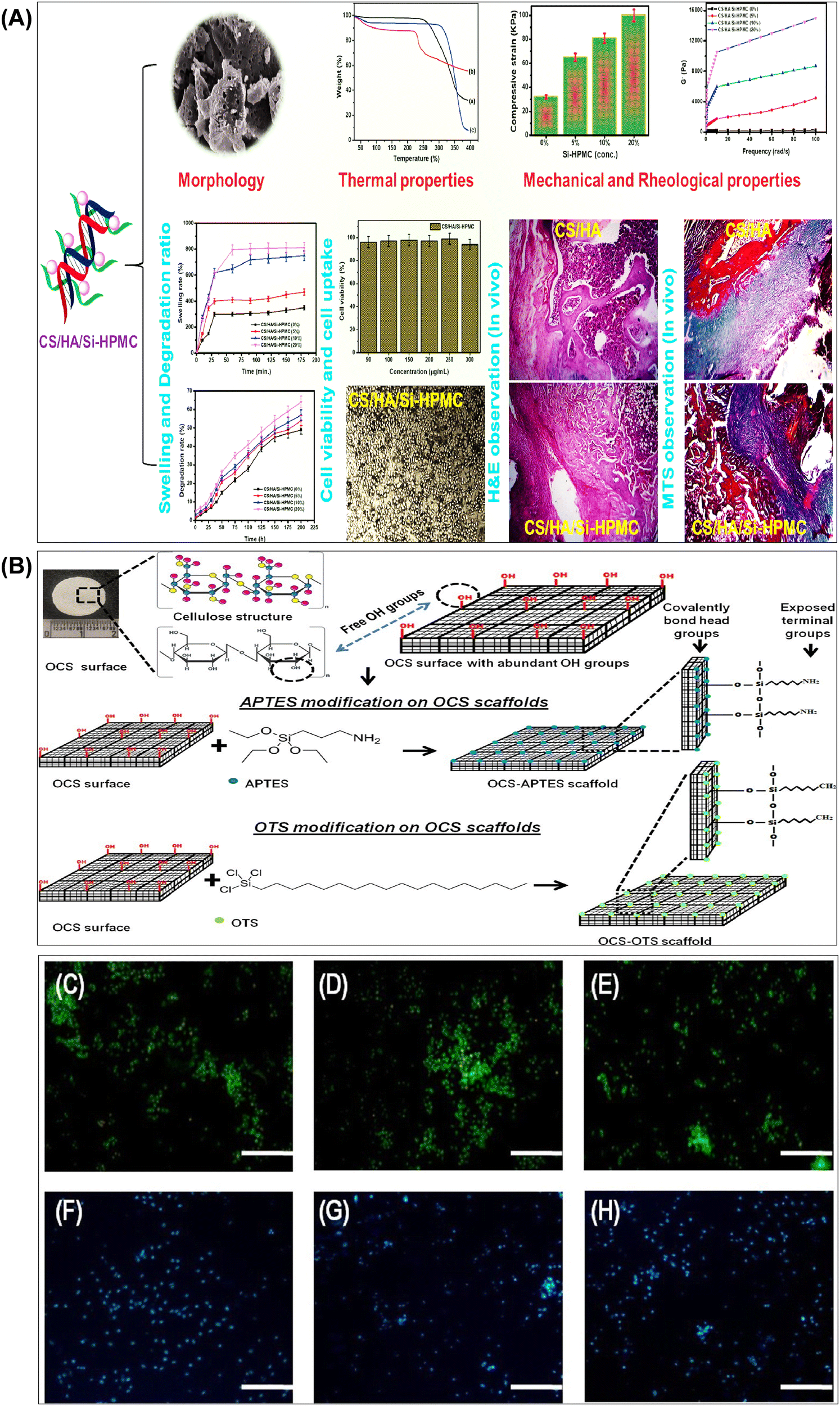 | ||
| Fig. 6 (A) Summary of results obtained from the investigation of the structural and biological properties of chitosan/hyaluronic acid hydrogel reinforced with silanized-hydroxypropyl methylcellulose as an injectable interpenetrating network for cartilage tissue engineering. Reproduced with permission from ref. 148 Copyright © 2021 Taylor & Francis. (B) Schematic representation of surface functionalization of OCS scaffolds using APTES and OTS via chemical grafting to form self-assembled monolayers. Representative images of (C) OCS scaffold, (D) OCS-APTES scaffold, and (E) OCS-OTS scaffold. Cell colonization assessed by Hoechst staining in (F) OCS scaffold, (G) OCS-APTES scaffold, and (H) OCS-OTS scaffold (scale bar: C–H, 100 μm). Reproduced with permission from ref. 182 Copyright © 2022 American Chemical Society. | ||
However, it is worth noting that there are notable differences between sulfonation and sulfatation of nanocellulose.185,186 Sulfonation typically involves the reaction of nanocellulose with a sulfonating agent under specific conditions to introduce sulfate groups onto the surface. This process may require harsh reaction conditions and can lead to the degradation of cellulose chains if not carefully controlled.186 This process results in a high density of sulfate groups distributed along the cellulose chains, imparting a strong negative charge to the nanocellulose surface. This modification enhances the hydrophilicity of nanocellulose and introduces ion-exchange properties, making it useful in various applications.186,187 On the other hand, sulfatation involves the reaction of nanocellulose with sulfate-containing reagents or precursors, such as sulfur trioxide or sulfuric acid esters. Sulfatation reactions are often milder and can be conducted under more controlled conditions, reducing the risk of cellulose degradation.188,189 Additionally, sulfatation may offer more flexibility in terms of the degree of substitution and the distribution of sulfate groups on the nanocellulose surface. Unlike sulfonation, sulfatation primarily targets the hydroxyl groups at the C6 position of the glucose units in cellulose, leading to the formation of sulfate esters (–OSO3H) at this position.188,190 This selective modification results in a lower density of sulfate groups compared to sulfonation but still imparts a negative charge to the nanocellulose surface. Sulfatation also enhances the hydrophilicity of nanocellulose and introduces ion-exchange properties, similar to sulfonation.186,188,190
When exposed to water or physiological fluids, these hydrogels absorb and retain a significant amount of liquid, forming a gel-like structure.187 This characteristic is particularly advantageous in wound healing applications, where maintaining a moist environment around the wound is crucial for optimal healing. The hydrogel ability to absorb and retain moisture helps create a conducive environment for tissue engineering, accelerates wound healing, and minimizes the risk of infection.149,191 For instance, Fan et al.184 synthesized carboxymethyl cellulose sulfates by reacting carboxymethyl cellulose with N(SO3Na)3, formed from sodium bisulfite and sodium nitrite in water. Reaction conditions influenced the degree of substitution (DS), assessed via barium sulfate nephelometry.184 Anticoagulant activity of carboxymethyl cellulose sulfates with varied DS, concentration, and molecular weights was studied through activated partial thromboplastin time, thrombin time, and prothrombin time. Their impact on wound healing was assessed by wound closure rate and histological analysis. Results demonstrated enhanced anticoagulant activity and accelerated wound healing with carboxymethyl cellulose sulfates treatment.184 In addition, sulfonated cellulose-based hydrogels were widely used for tissue engineering applications.150,192,193 Dixit et al.193 fabricated an injectable interpenetrating hydrogel system to mimic the extracellular matrix (ECM) and promote the differentiation of stem cells into chondrocytes (Fig. 7A). The hydrogel replicated the gradual stiffening and growth factor presentation of natural ECM, employing silk fibroin for stiffening and sulfated-carboxymethyl cellulose (s-CMC) for growth factor presentation.193 Combined with tyraminated-carboxymethyl cellulose (t-CMC) and crosslinked with HRP/H2O2, the hydrogel provided a soft environment initially, promoting chondrogenic differentiation, which gradually stiffened over time for joint support (Fig. 7A). The presence of s-CMC facilitated the prolonged presentation of growth factors like TGF-β, inducing chondrogenic differentiation of stem cells and deposition of cartilage ECM components. This hydrogel system served as a reservoir of biological cues for cartilage regeneration while offering mechanical support (Fig. 7A).193 Furthermore, Bhutada et al.150 developed electrospun poly(hydroxybutyrate)/gelatin fibers with anionic sulfated carboxymethylcellulose to immobilize growth factors, mimicking natural electrostatic interactions in the extracellular matrix. This fibrous scaffold effectively bound cationic molecules, remaining cytocompatible and stable in morphology and function.150 Transforming growth factor-β1 remained immobilized for at least 4 weeks with minimal release (3%). Fibroblast growth factor-2 and connective tissue growth factor, when immobilized, induced proliferation and fibrogenic differentiation of mesenchymal stem cells from infrapatellar fat pad, comparable to or better than free growth factors. These findings highlighted the potential of sCMC conjugated PG fibers in tissue engineering applications.150 In addition, Huang et al.192 assessed cellulose sulfate suitability as a scaffold for cartilage tissue engineering. The cellulose sulfate mimicked chondroitin sulfate C (CSC), a natural glycosaminoglycan, in sulfation levels and distribution (Fig. 7B). This partially sulfated cellulose (pSC) was integrated into gelatin construct using electrospinning.192 Scaffold characteristics, including fiber morphology, stability in water, growth factor interaction, and support for mesenchymal stem cell (MSC) chondrogenesis in vitro, were analyzed. All scaffolds maintained stability with micron-sized fibers.192 Increasing pSC concentration correlated with higher levels of transforming growth factor-beta 3 on scaffolds. The scaffold with the highest pSC concentration facilitated enhanced MSC chondrogenesis, demonstrated by increased collagen type II production and expression of cartilage-specific genes (Fig. 7B). These findings underscored pSC sulfate potential as a scaffold for cartilage tissue engineering.192 In drug delivery, sulfonation imparts nanocellulose with unique features such as increased drug loading capacity and controlled release behavior. The hydrophilic nature of sulphonated nanocellulose allows for efficient encapsulation of water-soluble drugs, while its negatively charged surface facilitates the binding of positively charged drugs. This modification enables precise control over drug release kinetics, contributing to the development of drug delivery systems with improved therapeutic efficacy and reduced side effects.194 Su et al.195 fabricated polyelectrolyte complexes (PEC) microcapsules, comprising sodium cellulose sulfate-chitosan hydrochloride (sample 1), along with variants patched using sodium tripolyphosphate (sample 2), sodium pyrophosphate (sample 3), and sodium hexametaphosphate (sample 4), all fabricated under mild conditions. The microcapsules exhibited excellent drug loading capacity and encapsulation efficiency (max. 66.9 ± 4.6% and 74.2 ± 5.1%).195In vitro release studies indicated that samples 2 and 3 had a higher cumulative drug release rate of 5-aminosalicylic acid and released completely within 12 hours. The drug release mechanisms were identified as mainly diffusion-controlled for samples 1 and 3, while samples 2 and 4 followed a non-Fickian transport mechanism. These findings suggested that PEC microcapsules, consolidated by polyphosphates, held promise as drug delivery vehicles with sustained release profiles.195
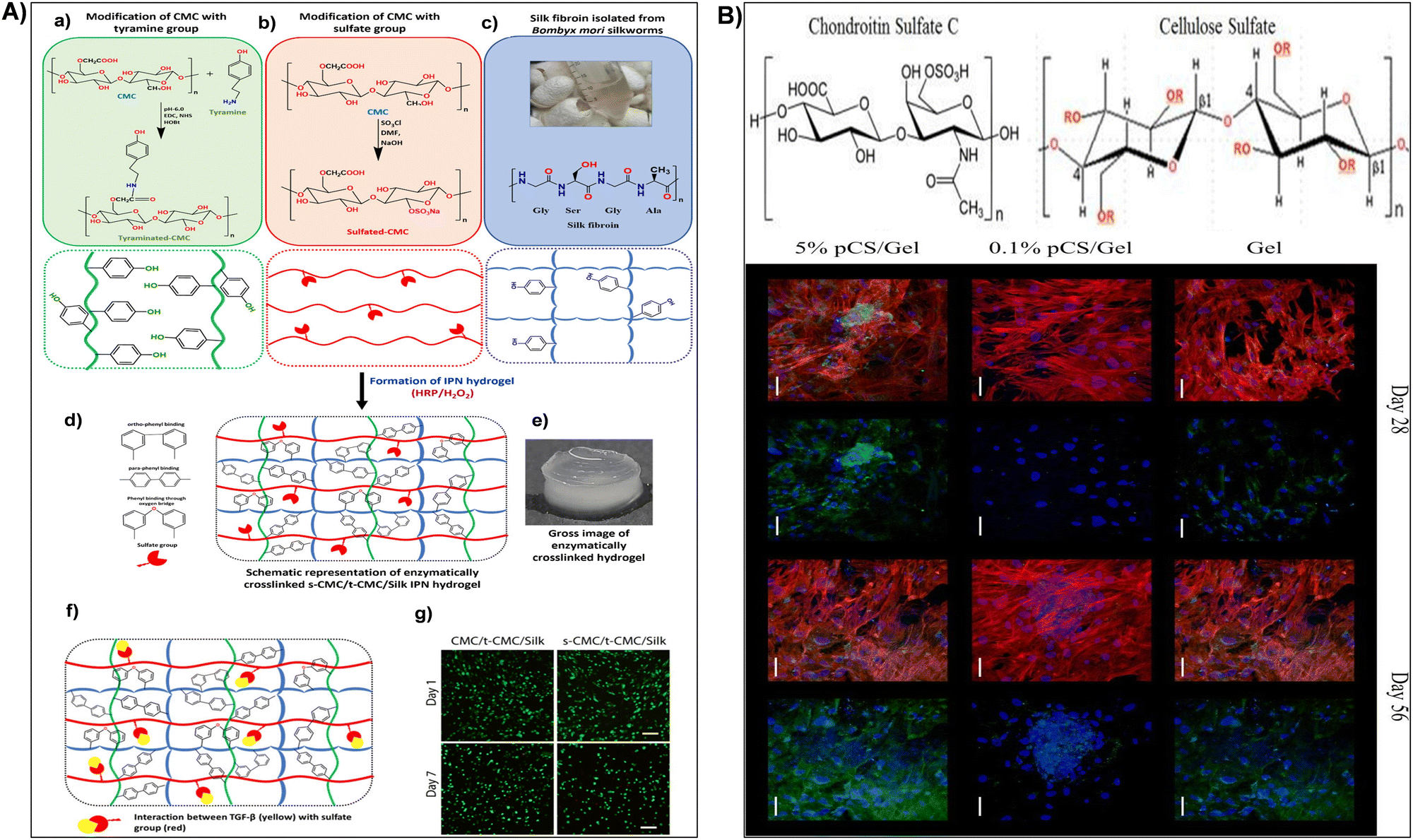 | ||
| Fig. 7 (A) (a)–(e) Illustration of interpenetrating network (IPN) hydrogel formation using tyraminated-CMC (t-CMC), sulfated-CMC (s-CMC), and silk fibroin via enzymatic crosslinking (HRP/H2O2). (a) Chemical modification of CMC to t-CMC (upper box) and schematic representation of t-CMC polymeric chains (lower box). (b) Chemical modification of CMC to s-CMC (upper box) and schematic representation of s-CMC polymeric chains (lower box). (c) Isolation of silk fibroin from Bombyx mori silkworms (upper box) and schematic representation showing tyrosine residues on silk fibroin (lower box). (d) Enzymatically crosslinked polymeric network of s-CMC/t-CMC/silk IPN hydrogel. (e) Macroscopic image of enzymatically crosslinked s-CMC/t-CMC/silk IPN hydrogel. Reproduced with permission from ref. 193 Copyright © 2024 Royal Society of Chemistry. (B) (a) Structure of chondroitin sulfate C (CSC) or chondroitin-6-sulfate (a) and cellulose sulfate (b) where R = –SO3Na or –H depending on degree of sulfation. (b) Confocal microscopy images of cells on Gel, 0.1% pSC/Gel, and 5% pSC/Gel in CCM at day 28 and 56. Red indicates F-actin, blue indicates the nucleus, and green indicates collagen type II. Merged image showing all three stains is on the left and the same image showing nucleus (blue) and collagen type II (green) staining is on the right for each group at each time point. Magnification = 40×. Scale bar = 50 μm. Reproduced with permission from ref. 192 Copyright © 2017 Mary Ann Liebert, Inc. | ||
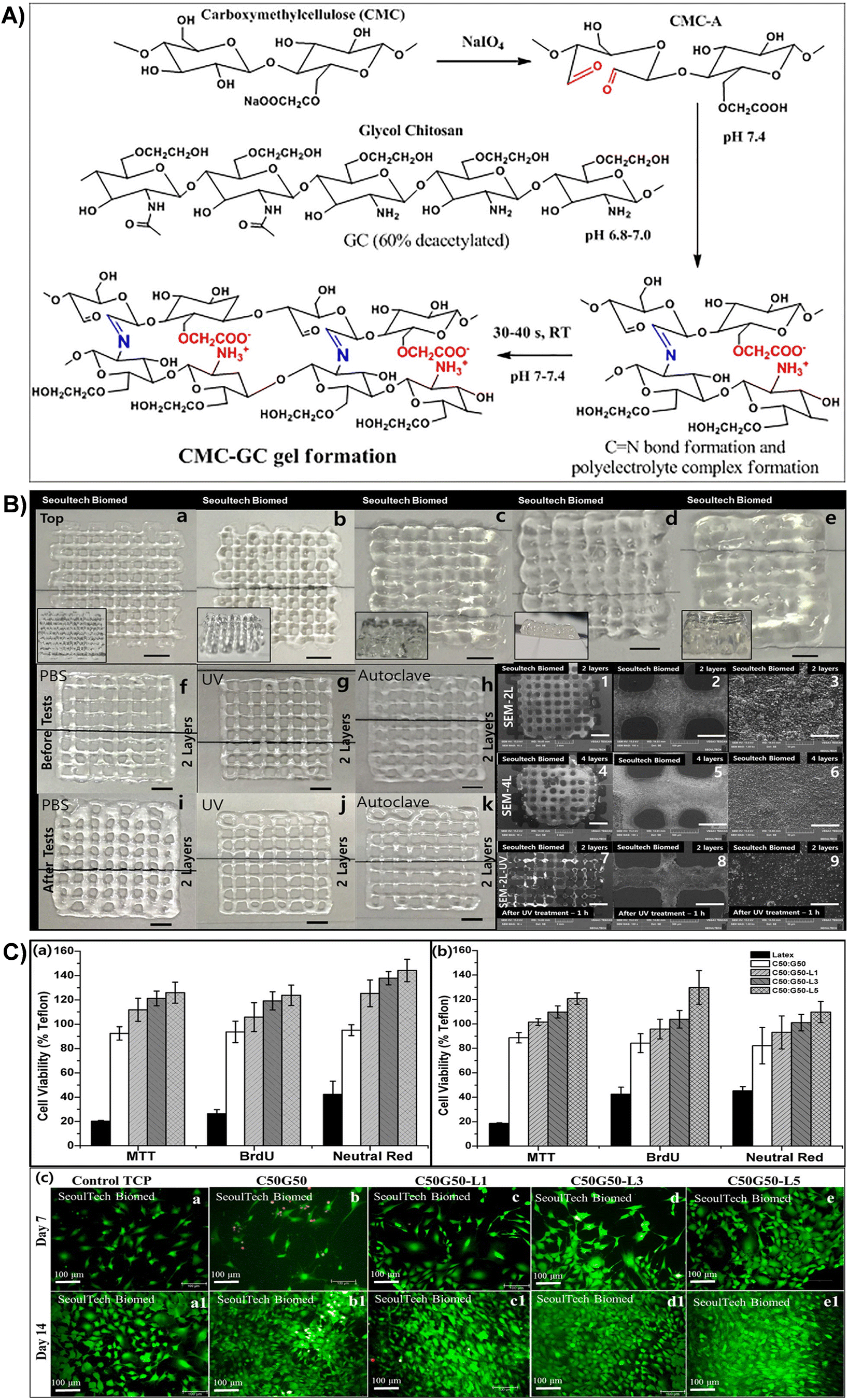 | ||
| Fig. 8 (A) Schematic representation of gel formation via the Schiff base reaction between the aldehyde group of CMC-A and the amine group of GC, resulting in imine bond formation and polyelectrolyte complex formation. (B) 3D printed C50G50 gel lattice structures with stability tests: top views of samples with (a)–(e) 2, 4, 6, 8, and 16 layers (scale bar: 2 mm). Side views provided as inserts. Stability tests: digital images of 2-layer structures before (f)–(h) and after (i)–(k) exposure to PBS, UV irradiation, and autoclaving. SEM images of 2-(1–3) and 4-layer structures (4–6) at various magnifications [scale bar: (1,4) – 2 mm; (2,5) – 500 μm; (3,6) – 50 μm]. SEM images of 2-layer structure samples after UV treatment (1 h) (7–9) (scale bar: (7) – 2 mm; (8) – 500 μm; (9) – 50 μm). (C) Viability assays of MC3T3 and BM MSC cells using MTT, BrdU, and Neutral red assays for different lactoferrin-incorporated CMC-GC gels. Live/dead assay images of lactoferrin-incorporated CMC-GC gels seeded with MC3T3 cells at days 7 and 14. Reproduced with permission from ref. 201 Copyright © 2020 Elsevier. | ||
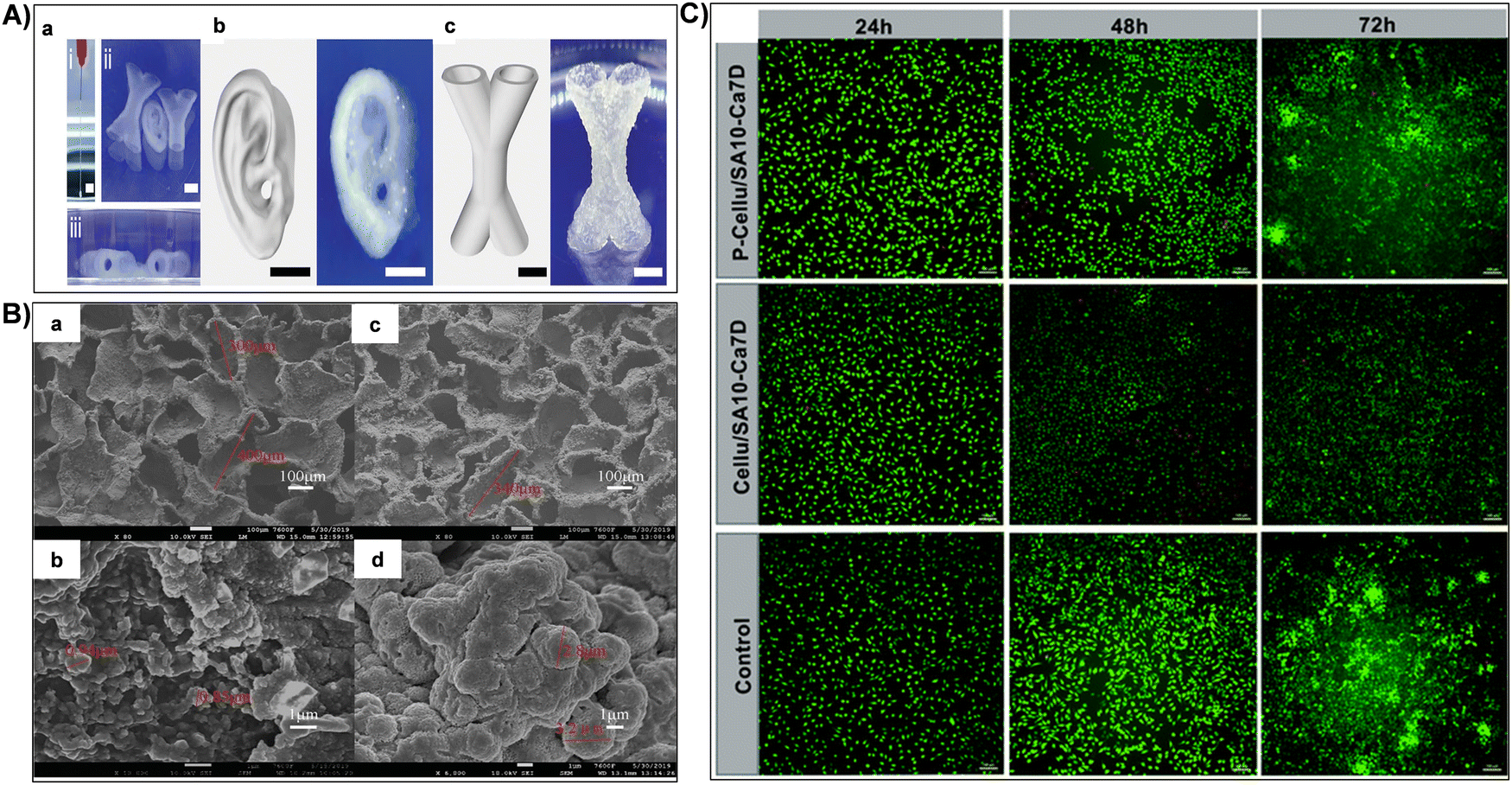 | ||
| Fig. 9 (A) (i) Continuous extrusion of composite inks demonstrating structural integrity post-extrusion. Representative images from (ii) frontal and (iii) lateral views of printed structures immersed in PBS after 7 days. (b) and (c) Representative images of printed centimeter-scale hydrogels featuring complex structures (human ear and X-shaped hollow tubes). Reproduced with permission from ref. 212 Copyright © 2024 John Wiley & Sons, Inc. (B) SEM images showcasing the surface of Cellu/SA10-Ca7D (a) and (b) and P-Cellu/SA10-Ca7D (c) and (d): (a) and (c) low magnification; (b) and (d) high magnification. Reproduced with permission from ref. 158 Copyright © 2021 Royal Society of Chemistry. (C) Live/dead staining of L929 cells at 24 h, 48 h, and 72 h for Cellu/SA10-Ca7D and P-Cellu/SA10-Ca7D. Scale bar: 100 μm. Reproduced with permission from ref. 158 Copyright © 2021 Royal Society of Chemistry. | ||
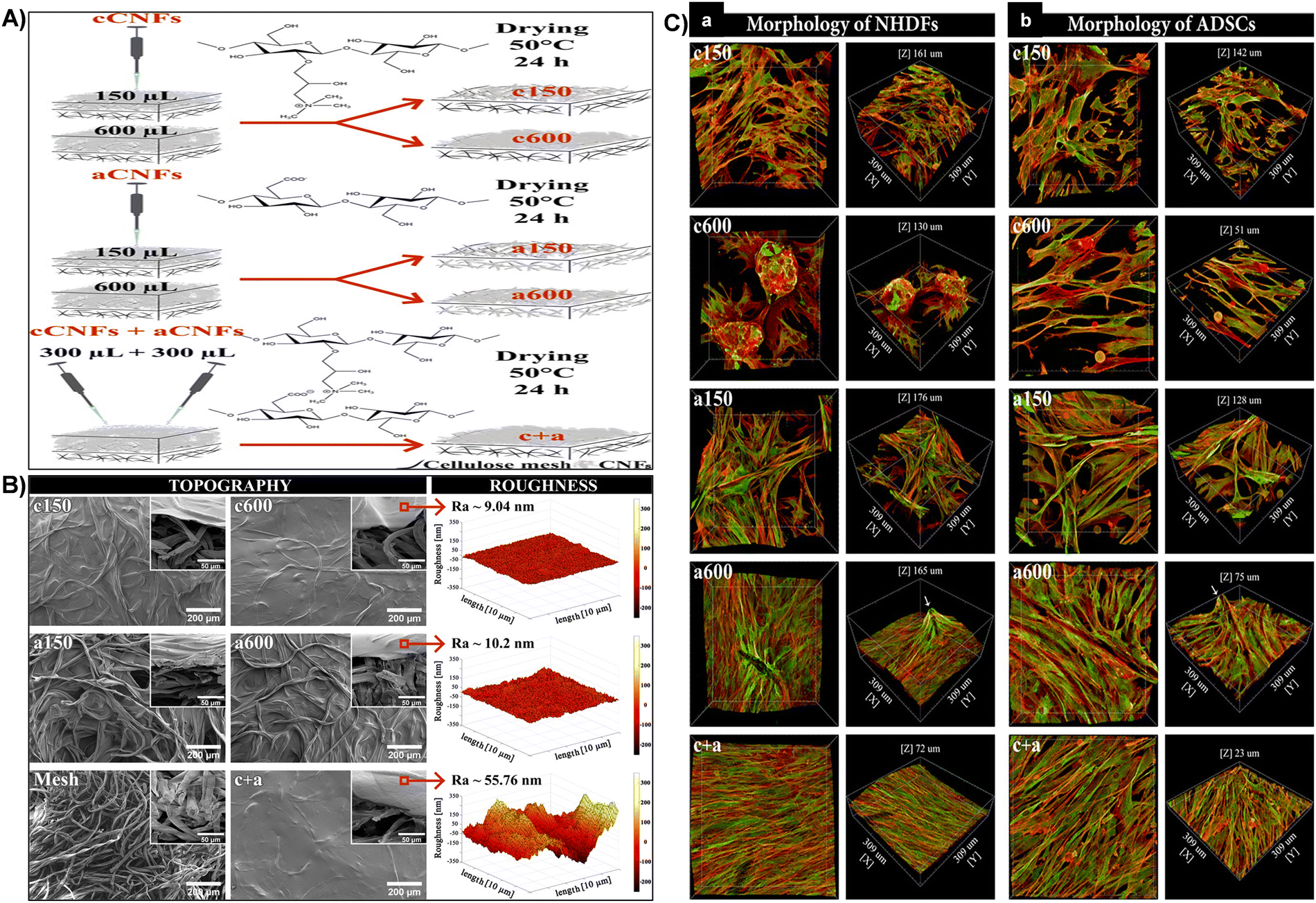 | ||
| Fig. 10 (A) Schematic representation of the process for fabricating diverse coating topographies on cellulose meshes using cCNF and aCNF solutions. (B) Surface topography and roughness. SEM images of c150, c600, a150, a600, and c + aCNF-coated and uncoated meshes (front and side views, inset). AFM measured roughness of c600, a600, and c + aCNF-coated meshes (right). (C) Cellular morphology of NHDFs (A) and ADSCs (B) influenced by CNF-coated mesh topography after 7 days of cultivation. 3D projection microscopy images showing cells on CNF-coated meshes. F-actin of cell cytoskeleton stained in red, vinculin in green. Confocal microscope, 40× objective magnification. Reproduced with permission from ref. 165 Copyright © 2021 American Chemical Society. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mass ratio). The size and aldehyde content of oxidized NC samples were assessed to understand their impact on hydrogel properties.260 Crosslinked hydrogels were characterized by field emission scanning electron microscopy (FESEM), swelling ability, Fourier transform infrared spectroscopy (FTIR), compression tests, thermal stability, and cell culture conditions. Oxidized-MFC hydrogel improved mechanical stability and swelling behavior but lacked stability in cell conditions due to low aldehyde content. Conversely, oxidized CNF and CNC formed suitable hydrogels for cell adhesion and MSC proliferation in 3D spheroids, with PO-CNF/chitosan hydrogel exhibiting antibacterial activity and MSC proliferation.260 Furthermore, Abouzeid et al.259 designed three-dimensional printed scaffolds by partially cross-linking TEMPO-oxidized cellulose nanofibril/alginate hydrogel with calcium ions, maintaining shape and filament integrity. Post-printing, full cross-linking with calcium ions enhanced hydrogel rigidity and long-term stability.259 Rheological properties, including thixotropic behavior and viscosity recovery, were studied, revealing improved recovery with cellulose nanofibrils. Mineralization with simulated body fluid confirmed hydroxyapatite nucleation on the scaffold, indicating biomimetic properties. Compressive strength analysis demonstrated the scaffold's suitability for bone tissue engineering. The composite scaffold showed superior properties compared to pure alginate, suggesting promise for 3D printing applications in bone regeneration.259
:5 mass ratio). The size and aldehyde content of oxidized NC samples were assessed to understand their impact on hydrogel properties.260 Crosslinked hydrogels were characterized by field emission scanning electron microscopy (FESEM), swelling ability, Fourier transform infrared spectroscopy (FTIR), compression tests, thermal stability, and cell culture conditions. Oxidized-MFC hydrogel improved mechanical stability and swelling behavior but lacked stability in cell conditions due to low aldehyde content. Conversely, oxidized CNF and CNC formed suitable hydrogels for cell adhesion and MSC proliferation in 3D spheroids, with PO-CNF/chitosan hydrogel exhibiting antibacterial activity and MSC proliferation.260 Furthermore, Abouzeid et al.259 designed three-dimensional printed scaffolds by partially cross-linking TEMPO-oxidized cellulose nanofibril/alginate hydrogel with calcium ions, maintaining shape and filament integrity. Post-printing, full cross-linking with calcium ions enhanced hydrogel rigidity and long-term stability.259 Rheological properties, including thixotropic behavior and viscosity recovery, were studied, revealing improved recovery with cellulose nanofibrils. Mineralization with simulated body fluid confirmed hydroxyapatite nucleation on the scaffold, indicating biomimetic properties. Compressive strength analysis demonstrated the scaffold's suitability for bone tissue engineering. The composite scaffold showed superior properties compared to pure alginate, suggesting promise for 3D printing applications in bone regeneration.259
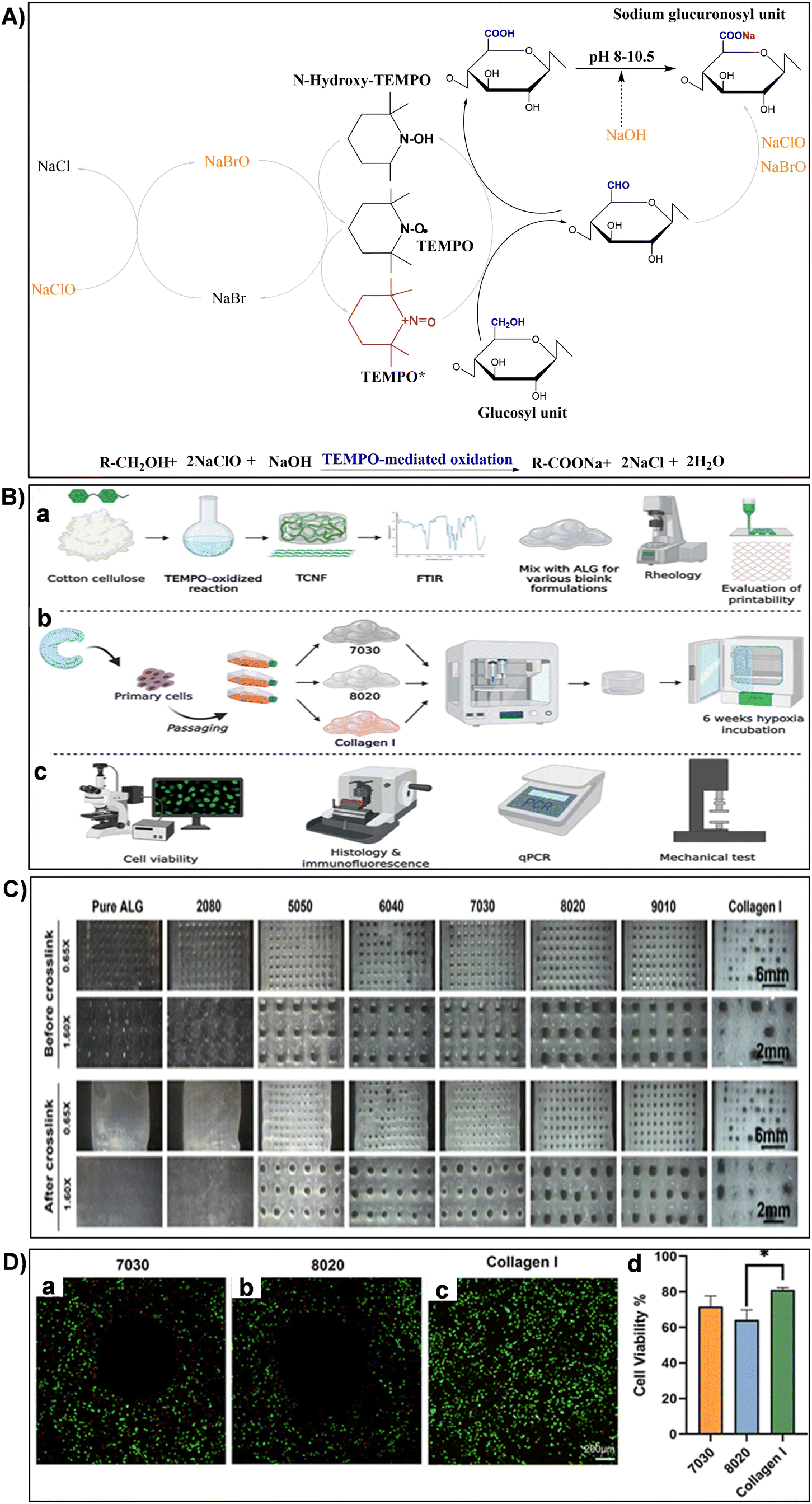 | ||
| Fig. 11 (A) Illustration of TEMPO-mediated oxidation mechanism converting cellulose primary hydroxy groups to sodium C6-carboxylate groups using TEMPO/NaBr/NaClO in water at pH 10. Reproduced with permission from ref. 251 Copyright © 2018 Elsevier. (B) Experimental design schematic: (a) biomaterial formation and characterization, (b) engineered tissue formation, and (c) evaluation of engineered tissues. Reproduced with permission from ref. 173 Copyright © 2021 Frontiers. (C) Printed mesh structures utilizing various TCNF/ALG precursor formulations. Reproduced with permission from ref. 173 Copyright © 2021 Frontiers. (D) Live/dead images of (a) 7030, (b) 8020, and (c) COL bioinks, where live cells appear green and dead cells red. (d) Quantitative cell viability analysis of live/dead assay images. Reproduced with permission from ref. 173 Copyright © 2021 Frontiers. | ||
2.3 Structural and mechanical properties of nanocellulose
Investigating the structural and mechanical characteristics of nanocellulose is crucial in the field of advanced materials research. Originating from plant or bacterial sources, nanocellulose displays a distinctive hierarchical structure at the nanoscale, which provides exceptional mechanical strength, flexibility, and biocompatibility.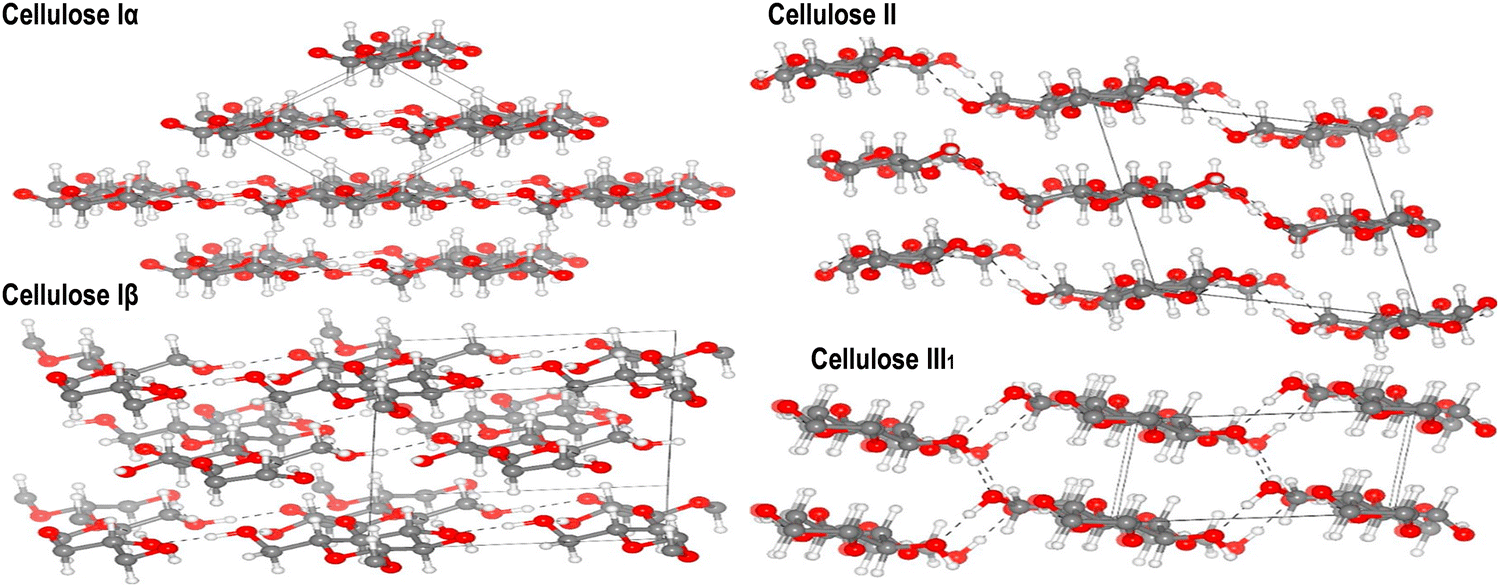 | ||
| Fig. 12 Refined crystal structures of cellulose allomorphs Iα, Iβ, II, and III1 are shown, with carbon, oxygen, and hydrogen atoms depicted as gray, red, and white spheres, respectively. Hydrogen bonds are indicated by dashed lines. Reproduced with permission from ref. 269 Copyright © 2020 Elsevier. | ||
Understanding the characteristics of the primary crystalline allomorph is pivotal for tailoring nanocellulose to specific applications. CNCs primarily adopt the cellulose I crystalline allomorph. In this form, cellulose chains are organized in a parallel arrangement with a repeating unit structure.270 The crystalline nature of CNCs contributes to their exceptional strength, stiffness, and thermal stability. The high aspect ratio and surface area of CNCs make them ideal reinforcements in nanocomposites, where they enhance mechanical properties.60,110,271 CNFs can exhibit a combination of cellulose I and cellulose II crystalline forms, depending on the processing method. The mechanical treatment used during CNF production can induce a transformation from cellulose I to cellulose II.272,273 The resulting crystalline structure impacts CNF properties such as flexibility, porosity, and surface area. This versatility allows CNFs to be tailored for diverse applications, including paper manufacturing, biocompatible materials, and reinforcing agents in composites.8,24,108 BNC typically adopts the cellulose I crystalline allomorph, resembling the crystalline structure found in plant-derived cellulose.274 BNC is produced by bacterial fermentation, leading to a highly pure and crystalline nanocellulose structure. The complex network of BNC fibers contributes to its remarkable mechanical properties, high purity and biocompatibility.54,130 BNC found applications in biomedicine owing to its resemblance to the extracellular matrix and its ability to support cell growth.56,118
Crystallinity, a fundamental feature of nanocellulose can profoundly influence its mechanical, thermal, and chemical behaviors. This property quantifies the proportion of crystalline regions within the material, indicating the ordered arrangement of cellulose chains. For CNCs, a remarkable feature is their high degree of crystallinity, often surpassing 80%, contributing to their remarkable strength and stiffness.60,275 This remarkable crystallinity is a result of their small size and the thorough isolation process. During isolation, CNCs are selectively derived from the most crystalline regions of cellulose, emphasizing their ordered structure and reinforcing their mechanical strength.110,113 For CNFs, their crystallinity remains noteworthy, generally ranging from 60% to 80%. This range signifies a substantial degree of ordered cellulose chains within CNFs.276,277 However, CNFs typically exhibit a lower crystallinity compared to CNFs. The structure of CNFs is characterized by a network of intertwined cellulose chains, resulting in a less ordered arrangement. The lower crystallinity contributes to the flexibility and deformability of CNFs, making them suitable for applications requiring enhanced flexibility and toughness. The amorphous regions within CNFs contribute to their ability to disperse more readily in certain matrices, enhancing their compatibility in composite materials.87,278 In addition, the BNC variant is notably captivating, showcasing crystallinity levels typically falling within the range of 70% to 80%. This characteristic is complemented by a high degree of polymerization, spanning from 7000 to 16000 glucose residues.139,279 The crystallinity of BNC is attributed to the biosynthesis process by bacteria, which leads to a highly ordered arrangement of cellulose chains. BNC crystallinity is profoundly affected by the intricate interplay between genetic factors and environmental parameters, shaping its unique structure.117,280 The crystalline structure of BNC is characterized by a well-organized network of cellulose nanofibers, forming a three-dimensional matrix. This high degree of crystallinity results in BNC possessing remarkable tensile strength, elasticity, and water-holding capacity. Its unique combination of strength, biocompatibility, and controllable porosity makes BNC an appealing choice for advancing research and innovation in regenerative medicine, tissue engineering, and drug delivery systems.128,281
Specific gravity, expressed as the ratio of a substance density to that of a reference material, commonly water, serves as a foundational property delineating material weightiness. Regarding nanocellulose, this parameter highlights the lightweight nature that distinguishes these materials. More precisely, the specific gravity of nanocellulose materials consistently falls within a confined range, typically ranging from 1.3 to 1.6 g cm−3.282–284 This specific range underscores the inherently low-density characteristic of nanocellulose, emphasizing its exceptionally lightweight quality.282,283,285 The low specific gravity of nanocellulose materials is a result of their unique structural arrangement at the nanoscale. CNCs, CNFs, BNC possess highly organized nanostructures characterized by their intricate networks of cellulose chains. These structures are adept at minimizing the overall mass while maximizing the volume, contributing to the exceptionally low specific gravity values observed.286,287 The lightweight nature of nanocellulose is crucial in applications requiring weight reduction, particularly in aerospace, automotive, and construction industries. In these sectors, material weight significantly impacts fuel efficiency, maneuverability, and structural integrity. Nanocellulose low specific gravity makes it an attractive option, allowing for reduced component weight without sacrificing strength or durability.87,288 Moreover, in biomedical applications such as drug delivery systems and tissue engineering scaffolds, the lightweight nature of nanocellulose is essential. This characteristic minimizes the burden on biological systems while providing necessary support and functionality. Thus, nanocellulose emerges as a valuable material driving advancements in medical technologies.21,289
One of the key metrics used to evaluate the mechanical performance of nanocellulose is the elastic modulus, which measures its resistance to deformation under an applied load.70,291–293 This property is profoundly influenced by the degree of crystallinity and the alignment of the nanofibrils within the material. The elastic modulus of nanocellulose can vary significantly, ranging from a few GPa to over 100 GPa, depending on factors such as cellulose source, processing methods, and nanocellulose dimensions and types.91,294 For instance, CNFs exhibit anisotropic physical properties, with the elastic modulus reported to be about 150 GPa in the longitudinal direction and about 18–50 GPa in the transverse direction for highly crystalline CNF.70,295 On the other hand, CNCs, generally obtained by acid hydrolysis of cellulose fibers, have high crystallinity and an elastic modulus reported to be between 100 and 150 GPa.293,296 Furthermore, atomic force microscopy was employed in a study to determine the elastic modulus of BNC. The findings revealed a constant value of 78 ± 17 GPa over a fiber diameter range of 27–88 nm.292 These characteristics position nanocellulose within the range of certain engineering reinforcement materials, highlighting its remarkable rigidity and structural integrity.8,24,25 The high elastic modulus of nanocellulose is attributed to its well-defined crystalline structure and the efficient load-bearing capability of individual nanofibers or nanocrystals. This ability underscores the potential of nanocellulose for applications demanding superior mechanical strength, including reinforcing materials in advanced composites, manufacturing lightweight yet robust structural components, and developing innovative products in fields such as aerospace, automotive engineering, and bioengineering.290,297,298
Tensile and compressive strengths are also essential mechanical properties that define the ability of nanocellulose materials to withstand various mechanical loads. These features can significantly vary depending on parameters such as cellulose types, sources, the isolation techniques, crystallinity, and the size of individual nanofibers or nanocrystals.86,299 CNCs are characterized by their remarkable mechanical properties. Their theoretical tensile strength reported ranges from 7.5 to 7.7 GPa, making them one of the strongest naturally occurring materials. Typically, the aspect ratio of CNCs, isolated from cellulosic materials like cotton, sisal, flax, and jute through acid hydrolysis, falls between 12 and 50.300,301 Moreover, CNCs exhibit remarkable stiffness, with a Young's modulus ranging from 100 to 170 GPa, showcasing their ability to reinforce composites significantly.88,89,302 On the other hand, CNFs boast high tensile strength, reaching up to 10 GPa, making them among the strongest natural fibers and highlighting their exceptional mechanical performance. CNFs characterized by high crystallinity (approximately 88%) exhibit notable strength, measuring approximately 1.6–7.7 GPa, along with a stiffness of around 150 GPa.70,108,303 CNFs also exhibit a remarkable aspect ratio, ranging from 10 to 250, highlighting their slender and elongated structure.304 The excellent mechanical properties of the nanofibers stem from the highly crystalline structure of native cellulose and the high aspect ratio achievable through nanofiber processing. Additionally, BNC in dry state exhibits remarkable mechanical strength, with tensile strength values ranging from 200 to 300 MPa and Young's modulus ranging from 15 to 35 GPa.305 Mechanical attributes of cellulose have been extensively scrutinized through experimental and theoretical investigations, utilizing techniques such as X-ray diffraction, Raman spectroscopy, and atomic force microscopy.86 Purely crystalline nanocellulose exhibits a crystalline structure. Similar to the modulus of elasticity, the tensile strength of nanocellulose is influenced by its anisotropic crystalline arrangement. Molecular modeling predicted a tensile strength ranging from 5 to 7 GPa along the chain direction, while the transverse strength is markedly lower at 0.3 to 0.9 GPa.306 Computational predictions aligned well with experimental findings from sonication-induced fragmentation, reporting tensile strengths of 1.6 to 3 GPa for wood cellulose nanofibrils and 3–6 GPa for highly crystalline tunicate cellulose nanofibrils.307 This underscores the cellulose crystal substantial stiffness and strength, with pronounced anisotropy at this level of structural hierarchy. Tensile strength, representing the maximum tensile stress a material can endure without fracturing, is a crucial parameter for materials used in applications requiring resistance to stretching or pulling forces.308 This property is advantageous for mimicking load-bearing tissues like tendons and ligaments. The aligned and entangled nanofibrils within nanocellulose contribute to its superior tensile strength.186 Conversely, compressive strength indicates a material capacity to withstand loads that push it together, compressing it. Compressive strength is vital for applications where tissues undergo compression, such as in cartilage and bone engineering.309–311 Nanocellulose, with its porous structure and ability to form hydrogels, can provide a supportive environment for cells and promote tissue engineering in compression-loaded regions.290 In addition, cellulose, with its inherent strength and versatility, serves as an excellent candidate for reinforcement strategies in composite materials.108,147 By combining cellulose with other matrices, a synergistic effect can be achieved, enhancing the overall compressive strength of the composite. This strategy is particularly valuable in fields like construction, where the reinforced materials can contribute to the development of high-performance and durable structures.312,313 The utilization of cellulose in composite materials opens up possibilities for innovative solutions across various industries, showcasing its potential as a sustainable and robust reinforcement agent. For instance, a recent study investigated the impact of incorporating nanocrystalline cellulose into cement mortars at varying concentrations (0.5%, 1.0%, and 1.5% by weight of cement).314 The research assessed physical and mechanical properties, frost resistance, salt resistance, and microstructure. Results indicated that higher nanocellulose content correlates with reduced weight loss in frost and salt tests. The addition of nanocrystalline cellulose led to a notable improvement in compressive strength (27.6%) and flexural strength (10.9%). Mortars with 1.5% nanocellulose exhibited a remarkable 98% enhancement in frost resistance after 50 freezing and thawing cycles.314 Furthermore, the orientation and alignment of cellulose nanofibers or nanocrystals within cellulose-based hydrogels play a pivotal role in determining their tensile and compressive properties. When these nanomaterials are strategically oriented, it enhances the overall mechanical strength of the hydrogel. In the case of tensile properties, the aligned cellulose structures allow for better load distribution along the direction of force, resulting in improved tensile strength.8,25 Similarly, in compression, the organized arrangement of cellulose chains contributes to heightened resistance against compressive forces. This controlled orientation at the nanoscale level imparts superior mechanical performance to cellulose-based hydrogels, making them promising materials for applications requiring robustness and structural integrity.50,86 In a recent study, two composite hydrogels, featuring interpenetrating polymer networks, were fabricated by initiating free-radical polymerization of acrylamide within preformed physical networks of regenerated plant cellulose (PC) or bacterial cellulose (BC), both swollen in the reactive solution.315 The mechanical responses of these hydrogels were investigated under compressive deformations of varying amplitudes. Results from compression tests revealed distinct mechanical behaviors between PC- and BC-based hydrogels. While both withstood single compressions up to 80% amplitude, cyclic compressions exposed a notable increase in stiffness for BC-based hydrogels beyond 60% deformation. This phenomenon suggested structural reorganization, potentially influenced by stress-induced reorientation of BC microfibrils.315
In addition to the previously mentioned mechanical attributes of nanocellulose-based materials, another significant property is viscoelasticity. This distinctive characteristic encapsulates the unique behavior of nanocellulose materials, combining both viscous and elastic properties.243,316,317 This dual nature is pivotal for applications demanding materials to deform under stress and revert to their original shape upon stress removal, underpinning their versatility in various industrial sectors.316,317 The viscoelastic properties of nanocellulose are meticulously tuned by several key parameters inclusding concentration, processing conditions and nanocellulose types.318,319 The concentration of nanocellulose within a material can significantly influence its viscoelastic behavior. Higher concentrations often lead to increased viscosity, affecting the material flow and deformation properties.320 For instance, in a recent investigation, a noteworthy enhancement in viscoelastic properties was observed by augmenting the concentration of CNCs within CNCs/collagen hydrogels. The study demonstrated a substantial increase in the rate of stress relaxation (τ1/2) and creep (γ1/2), along with heightened values of storage modulus (G′) and loss modulus (G′′). The τ1/2 values for the hydrogels exhibited a range from approximately 29.2 to 261 seconds, with the frequency of the strain sweep measuring G′ and G′′ set at 1 Hz. Furthermore, within the linear viscoelastic region, the G′ values of the hydrogels ranged from 10 to 30 kPa.320 Moreover, Jiang et al.321 developed a robust hydrogel system using dialdehyde cellulose nanocrystals (DAC) and gelatin (GEL) as bio-ink for 3D printing. To prepare the composite hydrogel, 4% CNC or 4% DAC suspension was blended with 8% GEL solution at a volume ratio of 1:1, yielding 4:8-CNC/GEL or 4:8-DAC/GEL hydrogel. The behavior of 4:8-DAC/GEL hydrogel was investigated under different conditions to optimize printability and structural integrity.321 Rheological analysis revealed GEL and 4:8-CNC/GEL samples had Tsol–gel values of 23.8 and 30.7 °C, respectively (Fig. 13a). However, 4:8-DAC/GEL showed no intersection of G′ and G′′ from 3 to 50 °C. While GEL and 4:8-CNC/GEL hydrogels were unsuitable for tissue engineering due to low Tsol–gel values, 4:8-DAC/GEL exhibited promising results, with continuous improvement of gel structure, reaching a storage modulus (G′) of 5297.23 Pa after 4 hours (Fig. 13b).321 The viscosity of 4:8-DAC/GEL at 1, 2, 3 and 4 h was 5.71, 14.06, 29.11 and 152.98 Pa s, respectively (Fig. 13c). The hydrogel displayed shear-thinning behavior, with viscosity decreasing from 17.60 to 10.62 Pa s as shear rate increased from 20 to 60 s−1 (Fig. 13d). 3D printed scaffolds exhibited exceptional architectural precision and fidelity, thanks to precisely tailored printing parameters and the hydrogel intrinsic characteristics (Fig. 13e–j).321
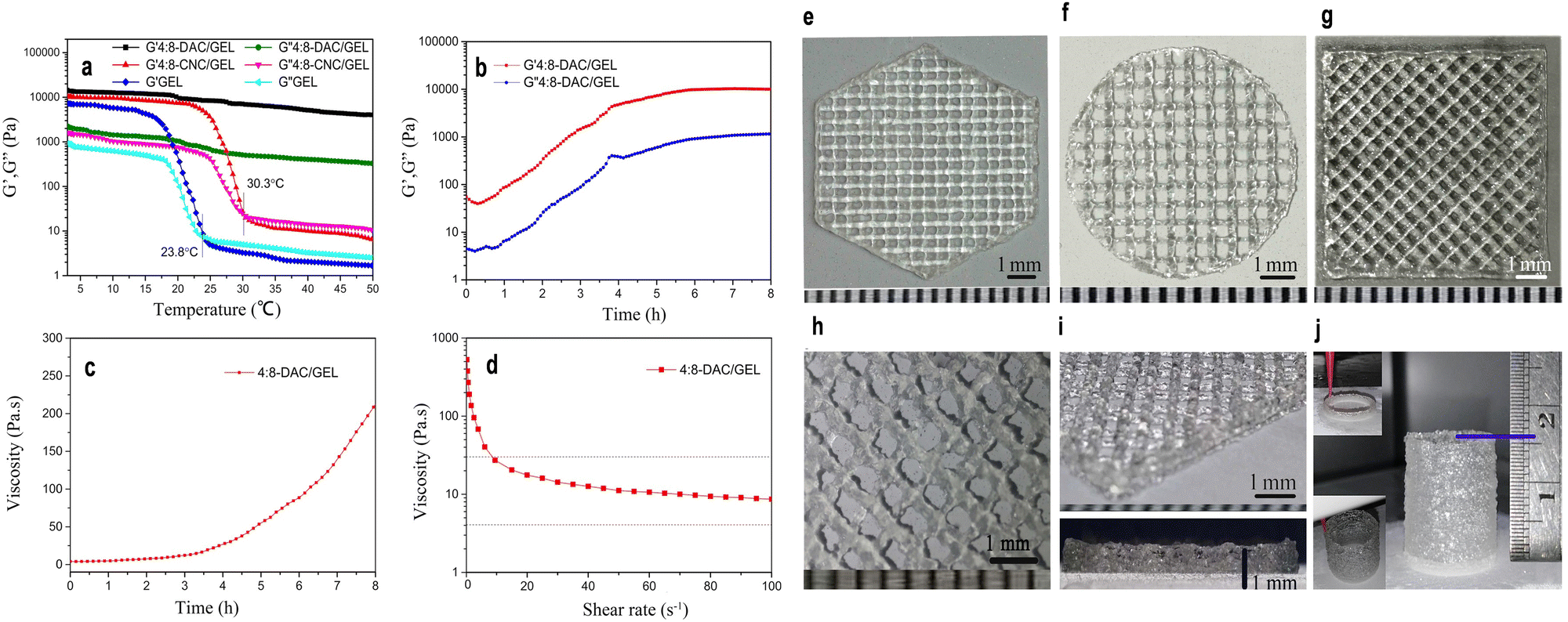 | ||
| Fig. 13 (a) Graphical representation of the changes in storage modulus (G′) and loss modulus (G′′) observed in GEL, 4:8-CNC/GEL, and 4:8-DAC/GEL hydrogels as the temperature increases, showcasing their viscoelastic behavior. (b) Plot illustrating alterations in storage modulus (G′) and loss modulus (G′′) of the 4:8-DAC/GEL hydrogel concerning increasing incubation time, indicating the evolving mechanical properties during gelation. (c) Graph depicting variations in viscosity of the 4:8-DAC/GEL hydrogel over time, illustrating the changes in flow behavior with increasing incubation duration. (d) Relationship between viscosity and shear rate for the 4:8-DAC/GEL hydrogel, demonstrating its shear-thinning behavior, crucial for extrusion-based 3D printing processes. (e)–(g) Images of 4:8-DAC/GEL scaffolds fabricated in distinct geometric shapes, including regular hexagons, circles, and squares, highlighting the versatility in design possibilities. (h) Magnified photograph showcasing a detailed section of the scaffold, emphasizing its intricate structure and fidelity. (i) Oblique and side-view images offering detailed perspectives of the 4:8-DAC/GEL scaffold architecture, highlighting its intricate and well-defined nature. (j) Tubular construct printed utilizing the 4:8-DAC/GEL hydrogel, underscoring its potential for creating complex and functional tissue engineering constructs. Reproduced with permission from ref. 321 Copyright © 2018 Springer. | ||
In addition, the processing conditions, including temperature, pressure, and shear rate during the production process, play a crucial role in determining the viscoelastic response of nanocellulose-based materials.322 Controlled processing conditions are essential for achieving desired mechanical properties. Variation in these conditions can result in materials with tailored viscoelastic characteristics suitable for specific applications. The viscoelastic properties of nanocellulose materials are also intricately influenced by temperature and shear rate, both playing pivotal roles in defining the material behavior under mechanical stress. Temperature affects the mobility and interactions of nanocellulose chains in the material, thereby altering its rheological responses. As temperature increases, the material may exhibit changes in viscosity, storage modulus (G′), and loss modulus (G′′). Higher temperatures often lead to increased molecular mobility and reduced material stiffness, affecting both the elastic and viscous components of the composite response to mechanical stress. For example, research study conducted by Hassan et al.323 demonstrated that varying temperatures induced changes in the mechanical properties of nanocellulose-based composites. The study investigated the impact of crosslinking with 1, 3, and 5% (w/w) citric acid in starch/cellulose composites on viscoelastic properties using dynamic mechanical thermal analysis from room temperature to 200 °C.323 The storage modulus (E′) of the starch/cellulose composites exhibited an initial increase with the rising concentration of citric acid up to 5% (w/w). However, further increments in citric acid concentration resulted in a subsequent decrease in the storage modulus. Across all samples, an elevation in temperature led to a consistent reduction in the storage modulus, indicating a progressive decline in stiffness.323 Notably, the control starch/cellulose composite foam demonstrated the lowest stiffness at all temperatures, while the starch/cellulose composite foam crosslinked with 5% (w/w) citric acid exhibited the highest storage modulus (E′). This trend aligned with the observed flexural moduli, emphasizing the temperature-dependent effect on the mechanical properties of the cellulose-based composites.323 In addition, the viscoelastic properties of nanocellulose materials can also be significantly influenced by shear rate, a crucial parameter dictating the response of the material to mechanical forces. Shear rate refers to the rate at which adjacent layers of the material move relative to each other under applied shear stress.324 In nanocellulose suspensions, varying shear rates can induce distinct rheological behaviors. At low shear rates, nanocellulose materials may exhibit more solid-like properties, with increased resistance to flow and pronounced elastic characteristics.8,325 This behavior is attributed to the entanglement and alignment of nanocellulose entities under slower shear conditions. As shear rates escalate, nanocellulose structures may undergo alignment and disentanglement more rapidly, resulting in a transition to a more fluid-like response characterized by decreased viscosity and enhanced flow.7,8,325 Futhermore, different types of nanocellulose, such as CNCs, CNFs, and BNC, exhibit distinct viscoelastic behaviors. CNCs, due to their rod-like shape and high aspect ratio, often contribute to materials with higher stiffness and viscosity.326 CNFs, with their fibrillar structure, offer improved deformability, strength and flexibility.327 BNC, characterized by its nanofibrous network, possesses unique viscoelastic properties, making it suitable for biomedical applications.118
3. Engineering biomimetic nanocellulose-reinforced hydrogels
Engineering biomimetic nanocellulose-reinforced hydrogels involves mimicking the intricate structure and properties of natural cellulose to fabricate advanced materials for various applications. By integrating nanocellulose into hydrogel matrices, scientists aim to enhance mechanical strength, biocompatibility, and bioactivity. These biomimetic hydrogels hold significant promise in tissue engineering, offering tailored scaffolds that closely mimic the extracellular matrix of living tissues.7,8,24,25,52,328 Through precise control of nanocellulose dispersion and hydrogel crosslinking, these materials can replicate the hierarchical organization and mechanical resilience found in biological tissues, paving the way for innovative biomedical solutions.20,3293.1 Physical and chemical technologies for processing nanocellulose-based hydrogels
The incorporation of nanocellulose into diverse matrices requires precise and innovative techniques to harness its unique properties effectively. Physical and chemical methods provide exceptional control and versatility in integrating nanocellulose into composite matrices, rendering them pivotal in a broad spectrum of applications.26,227,330,331 The selection of a method for preparing nanocellulose-based hydrogels heavily depends on the intended application, balancing advantages and challenges (Table 2). Physical methods like solvent casting and mechanical mixing offer simplicity and scalability but may compromise hydrogel quality due to the use of potentially toxic solvents or limited control over nanocellulose distribution, respectively.332–334 Advanced techniques like freeze-casting and electrospinning, while beneficial for creating structured and porous hydrogels suitable for applications like tissue engineering, face challenges like the need for specialized equipment and the potential use of harmful solvents.51,53,330,332 3D printing excels in customizability and precision in hydrogel structuring but is limited by equipment cost and material constraints.29,328,335 On the chemical side, methods like surface modification and functionalization improve hydrogel stability and functionality, albeit at the cost of potentially complex synthesis and batch variability.43,335,336 Cross-linking offers enhanced mechanical properties and stability; however, toxicity of cross-linkers remains a concern.29,337 Polymer blending allows tailoring of hydrogel properties through the integration of various polymers, though compatibility issues may arise.338,339 Incorporation of functionalized nanoparticles can impart specific advantageous properties but may introduce complexity in synthesis and concerns about uniformity and cytotoxicity.331,340,341 Finally, the layer-by-layer (LbL) assembly provides precision in hydrogel design but suffers from scalability issues and lengthy processing.166,342| Methods | Description | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|
| Physical methods | ||||
| Solvent casting | Casting nanocellulose in a solvent to form gel. | • Simple and cost-effective method. | • May involve the use of toxic solvents. | 332, 333, 343 and 344 |
| • Allows for control over hydrogel thickness and porosity. | • Limited control over pore size and distribution. | |||
| • Suitable for large-scale production | • May result in low mechanical strength of hydrogels. | |||
| Mechanical mixing | Dispersion of nanocellulose within a polymer matrix through mechanical agitation, such as stirring or high-shear mixing. | • Requires minimal equipment. | • Limited control over the orientation and distribution of nanocellulose. | 217, 334 and 345 |
| • Can be easily scaled up for production. | • May lead to uneven mechanical properties of the hydrogel. | |||
| • Facilitates homogeneous dispersion of nanocellulose within the hydrogel matrix. | ||||
| Freeze-casting | Freeze-casting, also known as ice-templating, involves the controlled freezing of a nanocellulose-based suspension followed by sublimation of the ice crystals to create a porous structure. | • Enables the fabrication of highly porous structures with aligned pores. | • Requires specialized equipment for controlled freezing. | 330 and 346 |
| • Allows for the control of pore size and morphology. | • May lead to structural collapse during drying – Time-consuming process. | |||
| • Preserves the hierarchical structure of nanocellulose within the hydrogel matrix. | ||||
| Electrospinning | Nanocellulose fibers are electrostatically spun into a gel matrix. | • Produces nanofibrous hydrogel scaffolds with high surface area and porosity. | • Requires high voltage for electrospinning setup. | 109, 347 and 348 |
| • Offers tunable fiber diameter and orientation. | • May involve the use of toxic solvents. | |||
| • Suitable for tissue engineering applications. | • Limited scalability for large-scale production. | |||
| 3D printing | Nanocellulose-based inks are deposited layer by layer to fabricate complex 3D hydrogel structures. | • Offers precise control over the architecture and geometry of hydrogel constructs. | • Requires specialized 3D printing equipment. | 8, 29, 349 and 350 |
| • Allows for the incorporation of multiple materials for functionalization. | • Limited availability of biocompatible printing materials. | |||
| • Enables patient-specific customization in biomedical applications. | • May involve post-processing steps for cross-linking and sterilization. | |||
| Chemical methods | ||||
| Nanocellulose surface modification and functionalization | Chemical modification of nanocellulose surface to introduce desired functionalities and enhance its properties. | • Enhances the compatibility and stability of nanocellulose within the hydrogel matrix. | • May involve complex synthesis procedures. | 137, 221 and 238 |
| • Enables the introduction of functional groups for targeted applications. | • Requires optimization to avoid cytotoxicity or alteration of native properties of nanocellulose. | |||
| • Potential for batch-to-batch variability. | ||||
| Cross-linking approaches | Chemical cross-linking agents (e.g., glutaraldehyde, epoxides, isocyanates) are used to form covalent bonds between nanocellulose chains to stabilize the hydrogel structure and improve its mechanical strength and stability. | • Improves the mechanical strength and stability of nanocellulose-based hydrogels. | • Some cross-linkers may be toxic or environmentally unfriendly. | 194, 331, 351 and 352 |
| • Offers control over cross-link density and swelling behavior. | • Optimization required to achieve desired mechanical properties without compromising biocompatibility. | |||
| • Enhances biocompatibility and resistance to enzymatic degradation. | • May lead to non-uniform cross-linking within the hydrogel matrix. | |||
| Polymer blending | Nanocellulose is blended with other polymers (such as polyethylene glycol, polyvinyl alcohol, or polyacrylic acid to form a composite hydrogel. | • Enables the incorporation of various polymers to modify the properties of nanocellulose-based hydrogels. | • Requires compatibility between nanocellulose and polymer matrices. | 24, 25, 43, 158, 341 and 353 |
| • Offers tunable mechanical, thermal, and swelling properties. | • May lead to phase separation or poor interfacial adhesion. | |||
| • Facilitates the creation of multifunctional hydrogel composites. | • Optimization needed to achieve desired synergistic effects. | |||
| Nanoparticle-functionalized nanocellulose hydrogels | Incorporation of functionalized nanoparticles (e.g., silica, metal oxides, quantun dots) into nanocellulose hydrogels. | • Allows for the incorporation of functionalized nanoparticles to impart specific properties to the hydrogel. | • Synthesis and functionalization of nanoparticles may be complex and costly. | 331, 340, 341 and 354 |
| • Enhances mechanical strength, thermal stability, or responsiveness to external stimuli. | • Requires optimization to ensure uniform dispersion within the hydrogel matrix. | |||
| • Offers potential for multifunctional applications. | • May introduce additional processing steps and potential cytotoxicity concerns. | |||
| Layer-by-layer (LbL) Assembly | Alternating deposition of nanocellulose and oppositely charged polyelectrolytes onto a substrate, followed by cross-linking or drying to stabilize the multilayer structure. | • Provides precise control over the layer thickness and composition of nanocellulose-based hydrogels. | • Requires multiple deposition steps, leading to increased processing time. | 166, 342 and 355 |
| • Allows for the incorporation of bioactive molecules, nanoparticles, or polymers in a step-wise manner. | • May involve the use of toxic or environmentally harmful chemicals. | |||
| • Offers tailored properties for specific applications. | • Limited scalability for large-area coatings or thick hydrogel constructs. | |||
| • Optimization needed to achieve desired layer architecture and stability. | ||||
3.1.1.1 Solvent casting. It is a fundamental technique in the fabrication of nanocellulose-based hydrogels for tissue engineering applications. In this process, nanocellulose is dissolved in a compatible solvent, forming a solution. The solution is then cast into specific molds or shapes and allowed to evaporate, leading to the formation of a hydrogel structure. During the evaporation process, nanocellulose particles become uniformly dispersed throughout the matrix, creating a nanocellulose-infused hydrogel.344,356 The resulting cellulose-based hydrogel retains the morphology of the original solution and can be further crosslinked or modified to achieve desired properties such as mechanical strength, porosity, and biocompatibility.221,269,351 This method offers several advantages. Firstly, the uniform dispersion of nanocellulose within the hydrogel ensures consistent mechanical properties, enhancing the overall strength and stability of the composite material.290 Additionally, the presence of nanocellulose enhances the barrier functions of the hydrogel, making it suitable for applications where controlled substance release or protection against external agents is required.333,344,357 In the literature, cellulose composites prepared using solvent casting have demonstrated improved mechanical properties for tissue engineering applications.358,359 Utilizing favorable properties of cellulose and its derivatives, Pooyan et al.358 developed a fully cellulose-based nanohybrid material by dispersing CNCs within a cellulose acetate propionate (CAP) matrix to fabricate a 3D percolating network, targeting small diameter vascular graft scaffolds in tissue engineering. CNCs from microcrystalline cellulose were dispersed in acetone to ensure uniform distribution.358 Thin films were produced via solvent casting, exhibiting excellent mechanical performance at body temperature, with CNCs enhancing strength and rigidity even at low concentrations (0.2 wt%), surpassing CAP alone.358 In a subsequent study, CNC alignment within the CAP matrix using a weak external magnetic field (0.3 T) significantly improved mechanical and thermal properties up to 3 wt% nanofiller concentration.359 Magnetic alignment enhanced filler interactions, dispersion, and filler-matrix contact. The composites showed a controlled porous structure conducive to cell seeding and proliferation, with aligned CNCs potentially guiding cell growth.359 Furthermore, Ten et al.360 developed bacterial polyester poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) reinforced with cellulose nanowhiskers (CNW) at concentrations of 1–5 wt% via solvent casting. CNW, derived from microcrystalline cellulose (MCC) through sulfuric acid hydrolysis, enhanced PHBV crystallization and mechanical properties.360 Evaluation through polarized optical microscopy (POM), differential scanning calorimetry (DSC), dynamic mechanical analysis (DMA), and tensile and bulge tests revealed CNW effectiveness as a nucleation agent, increasing PHBV tensile strength, Young's modulus, and toughness. DMA showed enhanced storage modulus and restrained molecular mobility near CNW surfaces, indicating improved mechanical properties due to strong interphase interactions.360
3.1.1.2 Mechanical mixing. Mechanical mixing stands as a crucial technique in the fabrication of nanocellulose-based hydrogels. This method involves the application of high shear forces to achieve the uniform dispersion of nanocellulose within a host material, including polymers and composites.8,10 The significance of mechanical mixing lies in its ability to ensure homogeneity, scalability, and the development of materials with enhanced mechanical properties. In tissue engineering, hydrogels function as scaffold materials providing structural support and a conducive environment for cell growth and tissue engineering.8 By incorporating nanocellulose into matrix through mechanical mixing to fabricate hydroges, several advantages can be achieved. The high shear forces applied during the mixing process disperse the nanocellulose evenly, preventing clumping or agglomeration. This uniform distribution enhances the structural integrity of the hydrogel and promotes cell adhesion, proliferation, and differentiation.8,10,361 A significant benefit of mechanical mixing in the fabrication of nanocellulose-based hydrogels lies in the enhancement of their strength, flexibility, and durability. By incorporating nanocellulose, which inherently possesses high tensile strength and stiffness, into the hydrogel matrix through mechanical mixing, the resulting material gains increased robustness and resilience, enabling it to withstand mechanical stresses more effectively.362,363
3.1.1.3 Freeze-casting. It is a technique that takes advantage of controlled freezing to organize nanocellulose chains in a structured manner. By subjecting a nanocellulose suspension to carefully controlled freezing conditions, the water within the solution forms ice crystals, guiding the alignment of the cellulose chains.364,365 This unique process results in materials with hierarchical porosity, offering a sponge-like structure where interconnected pores at multiple size scales create a network, resembling natural tissues.366,367 The porous structure of nanocellulose-based constructs is highly desirable for tissue engineering applications. The aligned porosity fosters cell adhesion and proliferation, promoting cellular activities as well as the exchange of nutrients and waste, which are vital for the proper functioning of artificial constructs.23,290 Furthermore, this technology has been utilized in the development of advanced filtration membranes, where the precisely controlled porosity enhances filtration efficiency.332
3.1.1.4 Electrospinning. This technology showed great potential in fabricating cellulose-based hydrogels for tissue engineering applications.347,348 In this process, a polymer solution containing nanocellulose is subjected to a high voltage electrical field, causing the solution to form a charged jet. As the jet travels towards a collector, solvent evaporation and polymer solidification occur, resulting in the formation of ultrafine nanofibers with diameters ranging from tens to hundreds of nanometers.51,368 Electrospun cellulose-based hydrogels have been extensively used for tissue engineering.227,347,348,369 Recently, uniaxially aligned cellulose nanofibers with embedded cellulose nanocrystals (CNCs) were produced via electrospinning, using a rotating drum as the collector. Scanning electron microscope images confirmed the alignment of most nanofibers.52 Incorporating CNCs improved the uniformity and orientation of electrospun cellulose/CNCs nanocomposite nanofibers (ECCNN), enhancing their physical properties significantly. ECCNN nonwovens exhibited significantly enhanced physical properties, with 20% CNC loading increasing tensile strength and elastic modulus by 101.7% and 171.6%, respectively. ECCNN demonstrated biocompatibility in MTT tests and supported cell proliferation and organization, making them promising scaffold materials for tissue engineering.52 In addition, these materials found applications in wound healing. In another study, a bilayer electrospun scaffold was fabricated, consisting of regenerated cellulose (RC)/quaternized chitosan (CS) as the primary layer and collagen/hyaluronic acid as the second layer. Both layers were chemically crosslinked with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC)/N-hydroxysuccinimide (NHS).369 Cell adhesion, proliferation, and viability of electrospun scaffolds evaluated using L929 fibroblast cells showed increased spreading after 3 days compared to 1 day (Fig. 14A). Both layers underwent crosslinking, resulting in increased ultimate tensile strength (2.29 MPa for the bilayer scaffold compared to 1.82 MPa for the RC scaffold) (Fig. 14B).369In vitro studies with fibroblasts revealed enhanced gene expression of PDGF, VEGF-A, and COL1, indicating angiogenesis potential. Moreover, antimicrobial, antioxidant, and protease inhibitory activities were observed, demonstrating the bilayer scaffold potential in wound healing (Fig. 14C).369 Moreover, in a recent investigation, core–shell fibers composed of poly(lactic acid) (PLA) as the core and cellulose acetate (CA) as the shell were fabricated through coaxial electrospinning. These fibers exhibited superior mechanical strength and biocompatibility compared to individual PLA and CA fibers and blend PLA/CA fibers.370 Specifically, optimal core-to-shell flow rates produced defect-free fibers with high ultimate tensile strength (19.53 ± 1.68 MPa) and stiffness (0.62 ± 0.09 GPa), matching native cancellous bone properties.370 Enhanced mechanical strength resulted from higher PLA–core weight fraction and improved crystallinity. In vitro studies with human fetal osteoblasts confirmed improved cell attachment, proliferation, and alkaline phosphatase activity on these core–shell fibers, promising their use for bone tissue engineering.370
 | ||
| Fig. 14 (A) SEM images depicting cell-adhered electrospun scaffolds. RC: (a1) and (a3); RC/quaternized CS: (b1) and (b3); RC/quaternized CS-collagen: (c1) and (c3); RC/quaternized CS-collagen/HA: (d1) and (d3); EDC/NHS crosslinked RC/quaternized CS-collagen/HA: (e1) and (e3). Numbers denote culture time in days. (B) Mechanical properties shown by stress–strain curves of the electrospun scaffolds. (C) FDA-stained electrospun scaffolds after 3 days of culture with L929 cells: (a) RC; (b) RC/quaternized CS; (c) RC/quaternized CS-collagen; (d) RC/quaternized CS-collagen/HA scaffolds. Bilayer scaffolds were crosslinked. Reproduced with permission from ref. 369 Copyright © 2024 Elsevier. | ||
3.1.1.5 3D printing. 3D printing stands at the forefront of tissue engineering, offering unparalleled precision in scaffold design and fabrication. Through the layer-by-layer deposition of nanocellulose-based inks, this cutting-edge technology enables the manufacturing of intricate three-dimensional structures with remarkable control over geometry and internal architecture.29,335,371 In tissue engineering, 3D-printed cellulose-based scaffolds played pivotal roles as biomimetic platforms offering promising and functional supports for cell growth and proliferation.8,10,130,328,338 For instance, researchers have utilized nanocellulose-based hydrogels in 3D printing to fabricate scaffolds that closely resemble the native tissue environment.34,349 These biomimetic hydrogels possessed excellent biocompatibility and mechanical properties. By precisely controlling the printing parameters, such as layer thickness and filament alignment, scientists engineered cellulose-based hydrogel scaffolds tailored to specific tissues like bone,34 cartilage,328,372 or skin.371 Kamdem et al.8 developed 3D printed hydrogel constructs using chitosan (CHI) and cellulose nanofibers (CNFs) (Fig. 15). The unique mechanical properties of native CNFs provided environmentally friendly high-performance composites. CHI served as a biocompatible matrix, while CNFs reinforced the hydrogel.8 By means of extrusion-based printing functional hydrogel scaffolds were achieved with optimized concentrations of CHI and CNFs, exhibiting good mechanical performance (Young's modulus 3.0 MPa, stress at break 1.5 MPa, and strain at break 75%), anisotropic microstructure, and favorable biological response (Fig. 15).8 Furthermore, 3D-printed hybrid biodegradable hydrogels, comprising cellulose nanocrystals, alginate and gelatin (CNC/Alg/Gel), were recently developed for bone tissue engineering. These scaffolds aimed to support cell proliferation, adhesion, nutrient exchange, and matrix mineralization.373 They exhibited superior mechanical strength compared to pure polymer scaffolds. Assessment through various assays demonstrated enhanced cell proliferation, adhesion, and osteogenic differentiation on 1% CNC/Alg/Gel scaffolds. Moreover, increased expression of osteogenic-specific genes and improved bone formation in calvaria critical-sized defects model highlighted their potential for bone regeneration.373
 | ||
| Fig. 15 (Left) 3D printed chitosan (CHI)/cellulose nanofiber (CNF) hydrogel scaffolds measuring 30 × 30 mm2 with an interfilament distance of 0.85 mm. The scale bars in the top image represent 11.5 mm. (Right) Live/dead assay confocal laser scanning microscopy (CLSM) images after culturing NIH/3T3 fibroblast cells in 3D printed CHI or CHI/CNF hydrogel composite scaffolds. Live cells are indicated by green fluorescence, while dead cells are indicated by red fluorescence. Scale bars in the CLSM images represent 500 μm. Three-dimensional frames (3 days) scale: CHI2) x × y × z: 2.3 × 2.3 × 0.075 mm3; CHI2/CNF0.4) x × y × z: 2.3 × 2.3 × 0.50 mm3. Reproduced with permission from ref. 8 Copyright © 2021 MDPI. | ||
Nanocellulose-based hydrogels have emerged as promising scaffolds for tissue regeneration due to their biocompatibility, biodegradability, mechanical robustness, and ability to mimic the ECM of native tissues.22,374 One crucial aspect in the fabrication of nanocellulose-based hydrogels is the development of suitable inks for their precise deposition onto substrates, especially for fabricating complex structures with high fidelity.8,29,338 Nanocellulose-based inks represent a class of printable materials with unique rheological, mechanical, and chemical properties that make them well-suited for 3D printing.328,371 These inks typically consist of nanocellulose fibrils dispersed in a solvent or matrix, along with additives such as cross-linkers,29 thickeners,133,371,375 and bioactive agents373 to enhance their printability and functionality. The key properties of nanocellulose-based inks influencing printability include viscosity, shear thinning behavior, surface tension, solid content, and compatibility with printing technologies.376,377 The viscosity of nanocellulose-based inks plays a crucial role in their printability, affecting extrusion, droplet formation, and deposition onto substrates. Generally, these inks exhibit shear-thinning behavior, wherein their viscosity decreases under applied shear stress, facilitating smooth flow through printing nozzles and precise deposition onto target surfaces.378,379 The rheological properties of nanocellulose-based inks can be tailored by adjusting factors such as nanocellulose concentration, solvent composition, and the presence of rheology modifiers, enabling control over printing parameters and structural fidelity of fabricated hydrogels.378,379 In addition, surface tension influences the wetting behavior of inks on substrate surfaces, affecting droplet spreading, adhesion, and resolution during printing. Nanocellulose-based inks with optimized surface tension properties exhibit improved wetting on various substrates, including hydrophobic and hydrophilic surfaces, ensuring uniform deposition and adhesion of printed layers.380 Surface modification techniques, such as plasma treatment or chemical functionalization, can be employed to tailor the surface properties of substrates, enhancing ink-substrate interactions and printability.380,381 Furthermore, the solid content of nanocellulose-based inks dictates the mechanical properties and gelation kinetics of printed hydrogels, influencing their structural integrity and stability post-printing.8 Higher solid content formulations result in stiffer hydrogels with enhanced mechanical strength, while lower solid content formulations may exhibit faster gelation kinetics and improved printability. Optimizing the solid content of inks is crucial to achieve a balance between printability, structural fidelity, and post-printing properties of fabricated hydrogels for tissue engineering applications.8,382 Nanocellulose-based inks can be employed in various printing technologies, including extrusion-based printing,8 inkjet printing,224 and digital light processing,383 each offering distinct advantages in terms of resolution, speed, and scalability for fabricating hydrogels with tailored architectures. Extrusion-based printing, for instance, enables layer-by-layer deposition of inks to create complex 3D structures,384 while inkjet printing allows for precise droplet placement with high resolution.385 Selecting an appropriate printing technology depends on the desired resolution, throughput, and material properties required for specific tissue engineering applications.335,386 The printability of nanocellulose-based inks has enabled the fabrication of hydrogel scaffolds with intricate architectures and controlled porosity, mimicking the native ECM of various tissues such as cartilage,34,328 bone,133 skin,206,371 and blood vessels.387 These scaffolds served as platforms for cell adhesion, proliferation, and differentiation, promoting tissue regeneration and repair in vitro and in vivo.8,10,133,328
3.1.2.1 Nanocellulose surface modification and functionalization. Nanocellulose functionalization plays a pivotal role in enhancing its compatibility and performance in various applications, particularly in the biomedical field. Through techniques like carboxymethylation, phosphorylation, and silanization, specific chemical groups are introduced onto the nanocellulose surface. This functionalization enables tailored interactions with different materials, including polymers and metals, ensuring uniform dispersion and robust interfacial bonding.26,137,388 In biomedical applications, nanocellulose-based hydrogels, modified and functionalized using these techniques, find extensive use. For instance, carboxymethylated nanocellulose hydrogels were employed in wound dressings due to their excellent water absorption properties.206,389 Furthermore, carboxymethylation of cellulose has garnered interest for its role in constructing adhesive hydrogels with potential in chronic diabetic wound repair. A mussel-inspired hydrogel, composed of catechol modified carboxymethyl cellulose (CMC-DA) and tannic acid, demonstrated rapid shape adaptability, wet adhesion, and antioxidant properties.390 The hydrogel, with injectability and self-healing ability, adhered strongly to tissues (45.9 kPa) and retained adhesion even on wet surfaces, with adhesion strength remaining at 14.9 kPa. It offered efficient wound healing, hemostasis, and antibacterial effects, showing promise for diabetic wound management.390 Moreover, phosphorylated nanocellulose hydrogels served as drug delivery carriers, offering controlled release mechanisms for pharmaceuticals.159 In tissue engineering, phosphorylation of cellulose received attention due to its ability to enhance bone cell differentiation and calcification. In this context, surface-phosphorylated cellulose nanofibers (P-CNFs) were developed as eco-friendly scaffolds for bone tissue engineering.211 These P-CNFs, with adjustable phosphate group content, showed improved zeta-potential and formed stable, uniform scaffolds. Notably, osteoblasts cultured on P-CNFs exhibited better adherence, proliferation, and osteogenic differentiation compared to those on native CNFs, particularly at optimal phosphate levels. This research underscored the potential of bio-based P-CNFs in simulating bone components and regulating osteoblast activity.211 Similarly, in another study, a novel three-dimensional composite scaffold was successfully fabricated for bone tissue repair using a strategy that incorporated cellulose as a backbone and alginate to expand pore size.209 The cellulose underwent bisphosphonate modification through oxidation with sodium periodate and a Schiff-base reaction with sodium alendronate, followed by biomimetic mineralization.209 The resultant scaffolds were porous and exhibited a chemical structure and morphology that closely mimicked natural bone, including crystallite size and Ca/P ratio. The phosphorylated scaffolds showed no cytotoxicity and supported excellent cell attachment, highlighting their potential for bone tissue engineering applications.209 In addition, silanized nanocellulose hydrogels found applications in tissue engineering, providing scaffolds that supported cell growth and proliferation.146,148 In a study, Vinatier et al.146 developed an injectable and self-setting hydrogel consisting of hydroxypropyl methylcellulose grafted with silanol groups (Si-HPMC) for cartilage repair. The authors tested its cytocompatibility and its ability to maintain a chondrocyte-specific phenotype. Primary chondrocytes from rabbit and human cell lines were cultured within Si-HPMC.146 Results confirmed that the hydrogel supported chondrocyte viability, proliferation, and allowed growth while maintaining the cell ability to produce key cartilage components such as type II collagen and sulfated glycosaminoglycans. This suggests that Si-HPMC is a promising scaffold for the three-dimensional culture and transfer of chondrocytes in cartilage tissue engineering.146
3.1.2.2 Chemical cross-linking approaches. These methods have revolutionized the field of nanocellulose-based materials, particularly in the development of advanced hydrogels tailored for biomedical applications. By employing cross-linkers such as epoxides, isocyanates, and multifunctional acrylates, covalent bonds are formed in nanocellulose-based hydrogels, creating robust networks that significantly enhance the material structural integrity and mechanical properties.20,138,357 A noteworthy utilization of cross-linked nanocellulose hydrogels resides in the generation of biocompatible scaffolds. These scaffolds function as three-dimensional matrices, closely emulating the natural extracellular matrix.210,337,391 This emulation facilitates cell adhesion, growth, and proliferation, proving conducive for the engineering of compromised or diseased tissues. Such applications render these scaffolds indispensable in the advancement of functional organs and tissues.29,331 Furthermore, the cross-linking of nanocellulose to fabricate hydrogels enables the mechanical reinforcement of the resulting hydrogels, enhancing their suitability for biomedical applications. Porous structures with controlled pore size, mechanical robustness, and biocompatibility are crucial for tissue engineering.323,392 A recent investigation proposed a novel approach using bacterial cellulose (BC) and eco-friendly additives for enhanced properties. Glucose, vanillin, and citric acid served as non-toxic cross-linkers, while γ-aminopropyltriethoxysilane was used to modify cellulose surface OH groups.393 Modified BC sponges exhibited 80–90% open porosity, with cross-linked glucose and citric acid variants showing 150% and 120% higher strength-to-weight ratios. In vitro assessments confirmed biocompatibility and lack of inflammatory response.393 Moreover, responsive hydrogels, representing an innovative application, exhibit sensitivity to external stimuli such as pH, temperature, or specific biomolecules. The introduction of cross-linking in nanocellulose-based hydrogels enhances their responsiveness, positioning them as optimal candidates for controlled drug delivery systems. These intelligent hydrogels demonstrated the capability to release therapeutic agents in response to physiological cues, ensuring precision and targeted drug delivery.394,395 For instance, in the field of cancer therapy, responsive nanocellulose-based hydrogels exhibited the capacity to selectively release chemotherapy drugs within tumor sites, thereby minimizing damage to healthy tissues and improving treatment efficacy.396,397 Moreover, cross-linked nanocellulose-based hydrogels have found utility in the fabrication of robust coatings for medical devices. These coatings have elevated the biocompatibility of implants and devices, consequently reducing the risk of adverse reactions within the living organisms.398,399 Illustratively, orthopedic implants derived from nanocellulose hydrogels have demonstrated heightened integration with surrounding tissues, thereby reducing the likelihood of implant rejection and promoting enhanced in vivo tissue recovery.400–402 In a study, to overcome the biological inertness of pristine bacterial cellulose (BC) for bone regeneration, a strategy was devised to crosslink short-cut BC nanofibers with Ca2+, yielding a macroporous BC scaffold (MPBC@Ca) with enhanced bioactivity.402 This scaffold exhibited improved structural stability and facilitated hydroxyapatite (HAp) deposition, mimicking the bone tissue extracellular matrix microenvironment. Hydroxyapatite-deposited MPBC@Ca scaffolds (HAp-MPBC@Ca) demonstrated excellent cytocompatibility and enhanced osteogenic differentiation of stem cells in vitro.402In vivo tests confirmed the scaffold osteoinductivity and ability to accelerate cranial bone tissue regeneration, suggesting its potential for repairing large cranial bone defects. This study highlights the importance of crosslinking cellulose for creating biomimetic scaffolds with enhanced bioactivity and regenerative potential.402 Furthermore, cross-linked cellulose hydrogels have been successfully used for tendon tissue engineering. For instance, fibers based on alginate (Alg) and hydroxyethyl cellulose (HEC) were developed via wet-spinning to mimic the fibrous ECM sheath. Blends of 1% Alg and 4% HEC (25
:75, 50:50, 75:25) were cross-linked with CaCl2 and glutaraldehyde to enhance physical and mechanical properties.403 Characterization revealed molecular interactions and favorable mechanical strength comparable to collagenous fibers. Lower HEC concentrations exhibited good degradability and mechanical features, while increased cross-linking altered mechanical behavior. The fibers demonstrated excellent biocompatibility, tenocyte proliferation, and migration, suggesting their potential as tendon substitutes.403 Additionally, the versatility of cross-linked nanocellulose hydrogels has allowed for their modification with bioactive molecules. For instance, the incorporation of growth factors or antimicrobial agents within the hydrogel structure has augmented their therapeutic potential. In applications related to wound healing, cross-linked nanocellulose hydrogels have been loaded with growth factors, accelerating tissue engineering and proving valuable in the management of chronic wounds.336 In a research work, carboxymethyl chitosan (CMCS)/hydroxyethyl cellulose (HEC) hydrogel films were developed to address challenges in fibroblast growth factor 2 (FGF-2) delivery for wound healing. These films, with a porous structure and high swelling capacity, facilitated controlled release of FGF-2, enhancing its bioavailability.404 They promoted cell proliferation, protected FGF-2 from degradation, and sustained its release. In animal burn treatment, FGF-2-loaded hydrogel films accelerated wound healing, stimulating re-epithelialization, granulation tissue formation, and angiogenesis, while reducing hypertrophic scarring. This innovative approach suggests CMCS/HEC hydrogel films as promising wound dressings, potentially revolutionizing burn treatment with enhanced FGF-2 delivery and wound healing outcomes.404
3.1.2.3 Polymer blending. This technique represent advanced methods for integrating nanocellulose into various polymeric matrices, yielding advanced materials with exceptional properties.405,406 By blending nanocellulose with polymers, such as biodegradable poly(lactic acid) (PLA)407 or biocompatible polyethylene glycol (PEG),408 researchers have developed hybrid materials that combined the unique attributes of nanocellulose with the desirable properties of the host polymers. In biomedical applications, nanocellulose-based hydrogels were recognized as prominent examples of these advanced materials.21,329 Hydrogels are 3D networks that can hold large amounts of water while maintaining their structural integrity. Nanocellulose high surface area and excellent water-absorption capacity made it an ideal candidate for hydrogel formulations.409 When incorporated into hydrogel matrices through blending, nanocellulose imparted mechanical strength, and responsiveness to the resulting hydrogels.7,8,24 One notable application of composite nanocellulose-based hydrogels is in tissue engineering. These hydrogels served as scaffolds that mimic the natural extracellular matrix, providing a supportive environment for cells to adhere, proliferate, and differentiate.20,29 The mechanical properties of nanocellulose hydrogels can be tailored to match specific tissues, making them invaluable for engineering functional tissues. Moreover, the biocompatibility of nanocellulose ensures that these hydrogels are well-tolerated by living tissues, reducing the risk of adverse reactions.7 Bacterial cellulose (BC) for instance holds promise for wound healing, but its limited malleability hinders applications. Blending with non-cellulosic polysaccharides like dextran (D) and xyloglucan (XG) could enhance wound healing and flexibility.410 A study introduced a novel in situ method to create BC-based hybrid hydrogels containing dextran (BC–D) and xyloglucan–dextran (BC–XG–D), with unique mechanical properties. Structural analysis revealed dextran forming micron-sized particles bound loosely to cellulose fibers.410 Xyloglucan acted as a lubricant, reducing Young's modulus to 4 MPa and increasing maximum tensile strain to 53%. BC–XG–D hydrogels exhibited improved wound healing and skin maturation in animal models. These findings advanced functional BC-based materials for wound healing.410 In another study, an injectable hydrogel mimicking the bone microenvironment and supporting bone regeneration was developed and characterized. The hydrogel, comprising chitosan (CS) and cellulose nanofibers/nanocrystals (CNFs/CNCs), exhibited enhanced mechanical properties. Fig. 16A(a) and (b) highlights schematic illustration of interactions in composite hydrogels prepared in this study.411 Cellulose nanocrystals (CNCs) notably improved gelation kinetics (from ∼24 s to 7 s) and mechanical strength (from ∼28 kPa to ∼379 kPa) of the chitosan hydrogel. Scanning electron microscopy imaging confirmed the CNCs incorporation into the chitosan network, resulting in a densely crosslinked structure.411 Fourier-transform infrared spectroscopy analysis revealed increased hydrogen bonding, enhancing mechanical reinforcement. FTIR also confirmed chemical interactions between chemical groups of chitosan and those of cellulose (Fig. 16A(c)–(f)). The hydrogel was biocompatible, promoting high cell viability, and altered cell morphology, making it a promising biomaterial for bone regeneration (Fig. 16B).411 In drug delivery, nanocellulose-based hydrogels offer unique advantages. Their high water content enables the encapsulation and controlled release of drugs,159,395 proteins,412 or growth factors.404 Researchers achieved precise control over drug release rates by modulating the composition and structure of the hydrogels and enhancing their properties through blending nanocellulose with other polymers, making them suitable for long-term therapeutic applications.13,329 Furthermore, nanocellulose-based hydrogels can find utility in wound healing and regenerative medicine. They can be loaded with antimicrobial agents to prevent infections and accelerate wound closure.337,413 Recently, poly(vinyl alcohol)/cellulose acetate (PVA/CA) films were fabricated using a novel method blending principles of solvent casting and phase inversion. Films with varying PVA/CA ratios were produced for flexibility. Dopamine was employed to anchor the antimicrobial peptide (AMP) LL37 onto the films.414 LL37-modified films exhibited potent antibacterial activity against Staphylococcus aureus, Staphylococcus epidermidis, and Escherichia coli, with approximately 75% inhibition for Staphylococcus aureus, 85% for Staphylococcus epidermidis, and 60% for Escherichia coli, irrespective of PVA/CA ratio.414 Presence of albumin reduced bacterial inhibition due to binding of protein molecules to antimicrobial agents. LL37-treated films also accelerated clotting time (≈10 min), surpassing vancomycin and bare surfaces, indicating effective activation of the coagulation cascade.414
 | ||
Fig. 16 (A) Chemical interactions within hydrogel systems. (a) Chemical structures and pKa values of hydrogel compositions: chitosan (CS), β-glycerophosphate (BGP), hydroxyethyl cellulose (HEC), and CNCs. (b) Hydrogel formation mechanism comparison between CS and CS-CNC networks. (c) FTIR spectra of various CS hydrogel formulations in deuterium oxide, with inset showing absorbance related to intermolecular hydrogen bonding. (d) Overlay FTIR spectra from inset of c demonstrating increased absorbance at 3400 cm−1 in CS-CNC hydrogels compared to CS hydrogels in deuterium oxide. (e) Overlay FTIR spectra of different CS hydrogel formulations in water, indicating absorbance at 2360 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch resulting from interactions between glyoxal molecules from HEC and chitosan amine groups. (f) FTIR assignments of various chitosan hydrogel formulations in deuterium oxide (D2O). (B) In vitro cell-based assays of encapsulated cells in hydrogels: (a) Z-stacking confocal images of MC3T3-E1 cells encapsulated in different hydrogel formulations, with live cells stained green and dead cells stained red. (b) Morphology of MC3T3-E1 cells encapsulated in different hydrogel formulations, stained for cytoskeletons and nuclei. (c) Percentage cell proliferation on day 7 compared to day 1 in each hydrogel formulation using cell counter ImageJ analysis. Reproduced with permission from ref. 411 Copyright © 2020 Elsevier. N stretch resulting from interactions between glyoxal molecules from HEC and chitosan amine groups. (f) FTIR assignments of various chitosan hydrogel formulations in deuterium oxide (D2O). (B) In vitro cell-based assays of encapsulated cells in hydrogels: (a) Z-stacking confocal images of MC3T3-E1 cells encapsulated in different hydrogel formulations, with live cells stained green and dead cells stained red. (b) Morphology of MC3T3-E1 cells encapsulated in different hydrogel formulations, stained for cytoskeletons and nuclei. (c) Percentage cell proliferation on day 7 compared to day 1 in each hydrogel formulation using cell counter ImageJ analysis. Reproduced with permission from ref. 411 Copyright © 2020 Elsevier. | ||
3.1.2.4 Nanoparticle-functionalized nanocellulose hydrogels. This represents a groundbreaking avenue in the field of advanced materials, particularly in biomedical applications. Serving as a versatile platform, nanocellulose offers a stable foundation for the functionalization by diverse nanoparticles, facilitating the creation of innovative nanocomposites with multifaceted functionalities.415,416 One of the key applications lies in targeted drug delivery systems. Nanocellulose, with its high surface area and biocompatibility, can be modified to carry therapeutic agents, such as anticancer drugs417–419 or antibiotics.159,420 By attaching drug-loaded onto nanocellulose surfaces, these nanocomposites can navigate the body biological barriers more effectively, delivering medications directly to specific cells or tissues. This targeted approach minimizes side effects and enhances the overall efficacy of treatments.175,421 Recently, two morphological structures, blending polylactic acid (PLA) with nanocellulose (NC) and self-assembled copolymer (M-PLA-co-NC), were synthesized with Fe3O4 NPs incorporated into both. These magnetic nanocomposites served as carriers for anti-cancer drugs 5-fluorouracil (5-FU) and curcumin (CUR).417 Both carriers showed similar 5-FU loading, but M-PLA-co-NC doubled CUR loading compared to PLA/M-NC, indicating copolymeric micelle superiority. M-PLA-co-NC/5-FU/CUR exhibited slower release and higher antibacterial and antitumor efficacy than PLA/M-NC/5-FU/CUR, suggesting it as a promising nanomedicine for bacterial infections and cancer treatment.417 Another study investigated pH-responsive drug release from various cellulose forms: micro-, nano-, and functionalized cellulose. Nanocrystalline cellulose (NCC) was derived from microcrystalline cellulose (MCC) via sulfuric acid hydrolysis (Fig. 17A(a)).422 Selective oxidation introduced aldehyde groups at carbon-2 and carbon-3 positions, yielding di-aldehyde nanocellulose (DANC). Chemical, spectroscopic and cristallographic analyses confirmed the conversion (Fig. 17A(b) and (c)). DANC exhibited higher drug binding capacity (200.8 mg g−1) than NCC (138.3 mg g−1) and MCC (120.2 mg g−1).422 Drug release increased with pH from 2.5 to 8.5. At pH 2.5, slow release occurred, while equilibrium (84.8% drug release) was reached within 10 minutes at pH 8.5 (Fig. 17B). Higuchi and Korsmeyer–Peppas kinetic models accurately described drug release, suggesting potential for pH-responsive drug delivery applications with functionalized cellulose.422 In addition, nanocellulose-based hydrogels incorporating functionalized nanoparticles found extensive use in biomedical imaging. Quantum dots, for instance, can be conjugated with nanocellulose to create luminescent probes. These probes emit specific wavelengths of light when exposed to external energy sources, enabling highly sensitive and precise imaging of biological structures.340,341 Such imaging techniques are invaluable in diagnosing diseases, monitoring cellular activities, and understanding complex biological processes.340,341 In addition, nanoparticle-functionalized nanocellulose hydrogels are emerging as promising materials for tissue engineering applications. By incorporating nanoparticles such as bioactive glass nanoparticles423 or metal oxides424 into nanocellulose-based hydrogels, researchers aim to enhance their mechanical properties, biocompatibility, and bioactivity. These hybrid materials offer a versatile platform for controlling cell behavior, promoting tissue regeneration, and delivering therapeutic agents. In a recent study, blend polymers comprising natural polymers were investigated for their mechanical and biological properties. Composite scaffolds of bacterial cellulose (BC)/silk fibroin (SF) with bioactive glass nanoparticles (BGNPs) were developed to promote osteogenesis in human adipose-derived stem cells (hASCs).423 SEM analysis revealed spherical BGNPs with sizes ranging from 15 to 30 nm. Incorporating BC and BGNPs reduced pore diameter (as shown by SEM images of composites (Fig. 17(C)), increased compressive strength, and modulus of SF scaffolds. Flow cytometry validated hASC surface markers. Adipogenic and osteogenic differentiation were confirmed by Alizarin Red S and Oil Red O staining, showing calcium deposition and lipid vacuole formation after 21 days (Fig. 17D(a) and (b)).423 Treatment with osteogenic medium for 21 days led to calcified nodule formation, indicating differentiation into bone cells (Fig. 17D(a)). Conversely, cells cultured in regular medium did not form mineral nodules (Fig. 17D(c)). Quantitative analysis revealed a significant increase in calcified nodule concentration with osteogenic differentiation medium (Fig. 17D(d)), highlighting its efficacy in promoting hASC differentiation into bone cells. Gene expression studies indicated osteogenic potential. Enhanced cell adhesion, viability, and differentiation in BC/SF and BC/SF/BGNPs scaffolds demonstrated their therapeutic potential for bone defects.423 In another study, Ghanbari et al.424 fabricated a 3D nanocomposite scaffold was developed using green-synthesized magnesium oxide nanoparticles (MgONPs) and bacterial cellulose (BC) nanofibers for bone regeneration. Camellia sinensis extract was utilized for MgONP synthesis.424 The scaffold showed improved mechanical strength and porosity due to MgONPs, enhancing osteoblast proliferation and osteogenic gene expression. The MgONP-BC scaffold demonstrated higher calcium deposition and alkaline phosphatase activity compared to pure BC, indicating enhanced osteogenic potential. This study highlights the benefits of blending cellulose with MgONPs for creating biocompatible scaffolds with promising therapeutic potential in bone tissue engineering.424 Moreover, in another study, a biomimetic Ag/bacterial cellulose/hydroxyapatite (Ag/BC@HAp) hydrogel mesh was designed for temporary cranioplasty with guided bone regeneration.425 The hydrogel featured a double-sided functionalized structure, with one layer dense and covered with Ag nanoparticles, while the other was porous and anchored with HAp through mineralization for varying durations.425 This design endowed the hydrogel with distinguished antibacterial activities and guided bone regeneration effects, while maintaining biocompatibility with preosteoblasts. In vivo experiments showed optimal guided bone regeneration with 7-day mineralization, while the hydrogel exhibited low swelling and strong mechanical strength, ensuring protection of soft brain tissues without increasing intracranial pressure.425
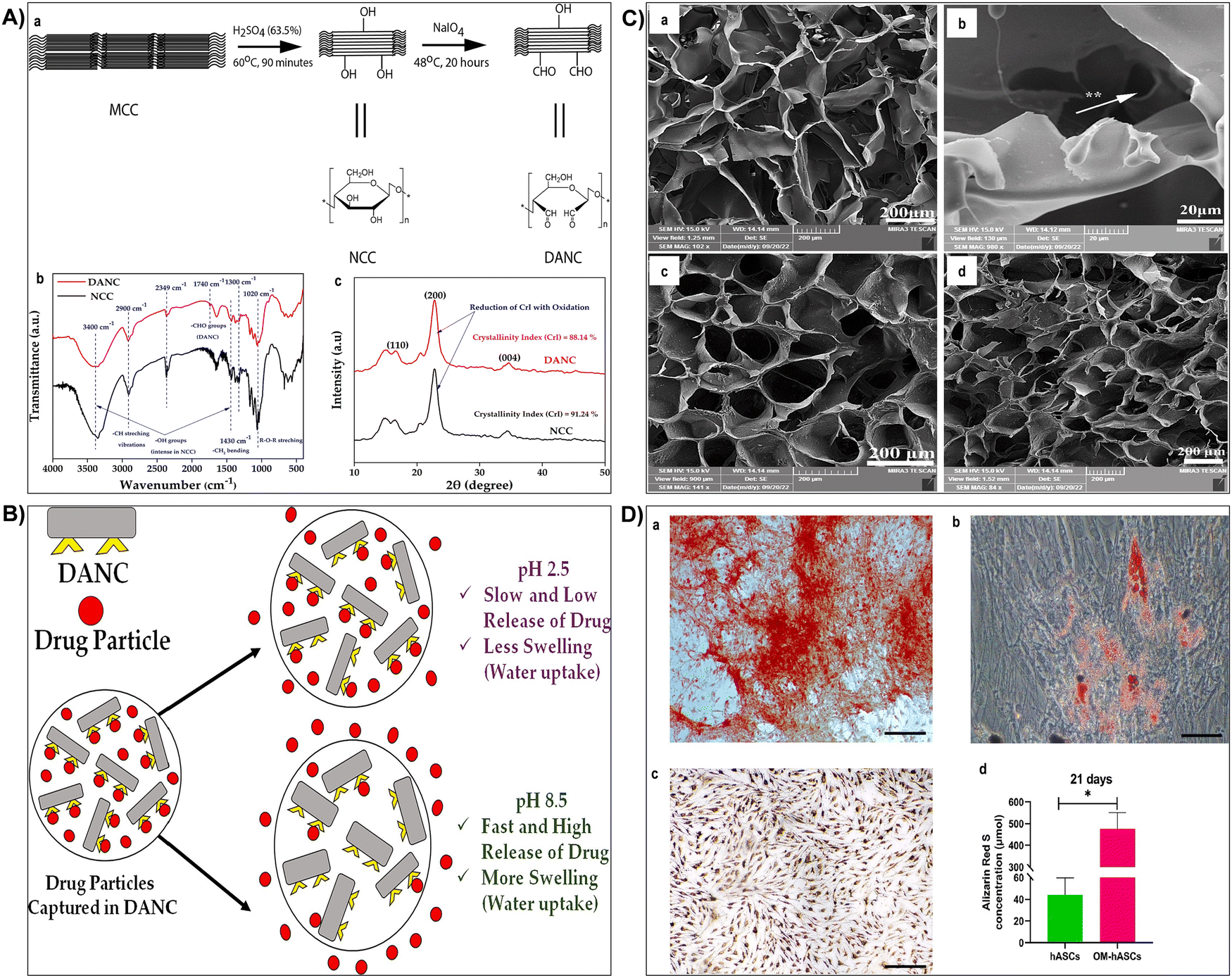 | ||
| Fig. 17 (A) (a) Synthesis scheme and structures of DANC from MCC via NCC. Characteristic peaks in FTIR spectra (b) and characteristic plane in XRD analysis (c) of NCC and DANC. Reproduced with permission from ref. 422 Copyright © 2024 Elsevier (B) release of ciprofloxacin hydrochloride from DANC matrix at pH 2.5 and pH 8.5. Reproduced with permission from ref. 422 Copyright © 2024 Elsevier (C) SEM images showing composite scaffold morphology: (a) SF scaffold (scale bar: 200 μm), (b) interconnected pores in SF scaffolds (scale bar: 20 μm), (c) BC/SF composite scaffolds (scale bar: 200 μm), (d) BC/SF/BGNPs composite scaffolds (scale bar: 200 μm). Reproduced with permission from ref. 423 Copyright © 2024 Elsevier (D) osteogenic and adipogenic differentiation assay of hASCs over 21 days: (a) mineralized nodules stained with Alizarin Red S after 21 days, (b) lipid vacuoles stained with Oil Red after 21 days, (c) hASCs cultured in osteogenic differentiation media and stained with ARS after 21 days, (d) quantitative analysis of ARS staining on hASCs after 21 days (n = 4/group; scale bar = 200 μm). OM-hASCs refers to hASCs cultured in osteogenic differentiation media. Reproduced with permission from ref. 423 Copyright © 2024 Elsevier. | ||
3.1.2.5 Layer-by-layer (LbL) assembly. LbL assembly is a remarkable technique that has gained significant attention for the fabrication of nanocellulose-based hydrogels, particularly for biomedical applications.342,355 In this method, nanocellulose is meticulously deposited in alternating layers with other polyelectrolytes, creating a structurally controlled and multifunctional composite material. The driving force behind this assembly lies in electrostatic interactions between the positively and negatively charged components, ensuring the precise arrangement of layers.342,426,427 LbL assembly empowers researchers to design and engineer nanocellulose-based hydrogels with tailored properties. By choosing specific polyelectrolytes and controlling the number of layers, the resulting hydrogel mechanical, chemical, and biological characteristics can be precisely customized. This level of control is especially advantageous for biomedical applications, where the hydrogel must meet specific requirements, such as tissue compatibility, drug delivery, or cell adhesion.428,429 One of the remarkable aspects of LbL assembly is its ability to control the film thickness with exceptional precision. This feature is pivotal in biomedical applications, where the hydrogel thickness can influence factors like drug release rates, cellular interactions, or barrier properties. Researchers can fine-tune the films thickness by simply adjusting the number of layers deposited, ensuring the hydrogel aligns with the intended application.430,431 Nanocellulose-based hydrogels produced through LbL assembly have shown immense promise in various biomedical applications. For example, they can be engineered to serve as drug delivery carriers, with the controlled release of therapeutic agents over time.355 In tissue engineering, these hydrogels can provide structural support for cell growth and engineering, mimicking the extracellular matrix.432 Recently, a magnetically-guided oxygen-propelled CoPt/gold nanosheet motor (NSM) was fabricated as an active self-propelled platform for anti-cancer doxorubicin drug (DOX) delivery to cancer cells. Microcapsules, serving as drug carriers, were fabricated using layer-by-layer (LbL) coating of chitosan and carboxymethyl cellulose layers, with gold and magnetite nanoparticles incorporated.433 Two sorts of nanoparticles (NPs) including magnetite (Fe3O4) and gold (Au) NPs were incorporated into the microcapsule shells by the LbL procedure for different functions. DOX loading efficiency reached 77%. NSMs successfully delivered DOX-loaded magnetic multilayer microcapsules to target cancer cells via external magnetic guidance.433 Under near-infrared irradiation, gold nanoparticles induced rapid drug release. Cytotoxicity studies demonstrated minimal side effects of NSMs and multilayer microcapsules across all tested concentrations.433 Moreover, they have been explored in wound dressings to enhance the healing process, benefiting from their biocompatibility and tunable properties. The multilayered nanocellulose-based hydrogels produced through LbL assembly exhibited an inherent biological compatibility, making them suitable for biomedical applications.434 The precisely designed layers can promote cell adhesion, proliferation, and tissue integration. In addition, their porous structure allows for the efficient diffusion of nutrients and oxygen, contributing to improved biocompatibility.355,432,434 A layer-by-layer (LbL) transdermal patch was crafted using bacterial cellulose (BC) via a Schiff base reaction. Ethylene diamine-modified carboxymethylated BC and aldehyde-modified pectin were assembled through this method. The presence of imine bonds facilitated self-healing upon scratching in pH 7.4 buffer solution, as observed through optical and atomic force microscopy.434 Field-emission scanning electron microscopy confirmed the assembly formation. Water swelling and deswelling tests demonstrated efficient water retention. Incorporation of silver nanoparticles (AgNPs) was verified, exhibiting antimicrobial activity against Staphylococcus aureus and Escherichia coli. The patch cytotoxicity and wound healing properties were assessed on NIH 3T3 fibroblast and A549 epithelial cell lines, showing positive effects.434
3.2 Crosslinking strategies for enhancing mechanical properties of nanocellulose based-hydrogels
Fabricating robust and mechanically stable hydrogels is crucial in tissue engineering applications to ensure the structural integrity and long-term functionality of implanted constructs.435–437 Biomimetic hydrogels reinforced with nanocellulose offer a promising avenue for tissue engineering due to their biological and tunable properties. To enhance the mechanical stability of these hydrogels, various crosslinking strategies are commonly employed, each tailored to meet specific requirements and applications.140,438 The chemical and structural properties of a hydrogel significantly influence the crosslinking mechanism, which can be categorized into physical (non-covalent) and chemical (covalent) bonding, as shown in Fig. 18A. In hydrogels, physical crosslinking is often complemented by chemical crosslinking, as physical bonds alone are insufficient to maintain hydrogel integrity. Specific crosslinking techniques are illustrated in Fig. 18B.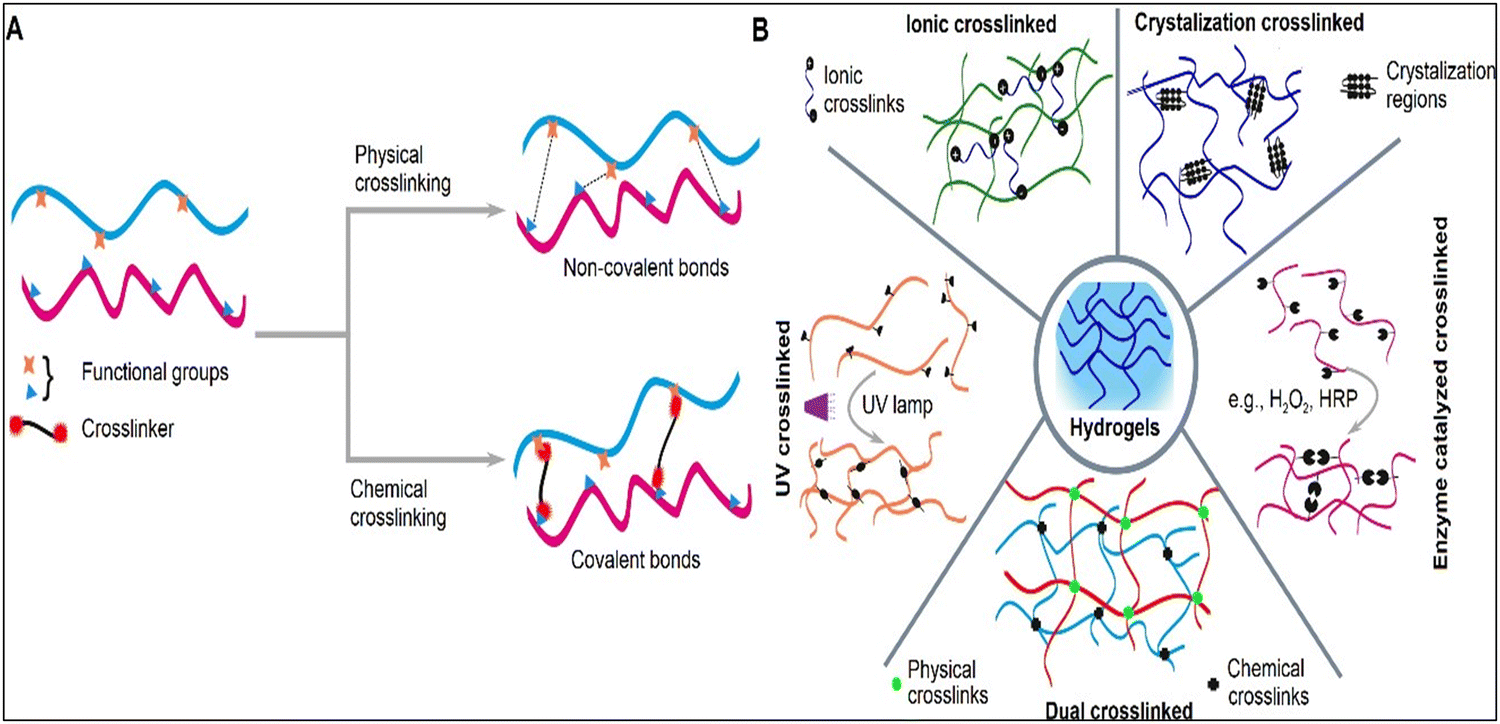 | ||
| Fig. 18 Illustration depicting (A) the influence of physical and chemical crosslinking on bond formation and (B) various examples of distinct crosslinking techniques. Reproduced with permission from ref. 438 Copyright © 2021 MDPI. | ||
Table 3 provides an overview of various cross-linking methods commonly used for nanocellulose-based hydrogel fabrication, highlighting their principles, advantages, disadvantages, and properties of the resulting hydrogels. Depending on specific application requirements, the choice of cross-linking method can be tailored to achieve desired properties and performances.
| Cross-linking methods | Principles | Advantages | Disadvantages | Properties and features of hydrogels | Ref. |
|---|---|---|---|---|---|
| Chemical cross-linking | Chemical reactions between functional groups on nanocellulose and cross-linking agents. | • High cross-linking density can be achieved. | • May involve toxic chemicals. | • High mechanical strength. | 351 and 439 |
| • Tunable mechanical properties. | • Potential alteration of nanocellulose properties. | • Improved stability and durability. | |||
| • Enhanced stability. | • Tunable swelling behavior. | ||||
| • Potential toxicity concerns depending on the cross-linking agents used. | |||||
| Physical cross-linking | Entanglement or physical interactions such as hydrogen bonding or hydrophobic interactions between nanocellulose chains. | • Simple and environmentally friendly. | • Lower mechanical strength compared to chemical cross-linking. | • Moderate mechanical strength. | 392 and 440 |
| • No chemical agents required. | • Limited stability. | • Responsive to environmental stimuli (e.g., temperature, pH). | |||
| • Biocompatible and suitable for biomedical applications. | |||||
| • May undergo reversible gel–sol transitions. | |||||
| Ionic cross-linking | Interaction between oppositely charged ions, often involving polyelectrolytes and counterions. | • Mild conditions and simple preparation. | • Limited stability in certain environments. | • Variable mechanical properties depending on ionic interactions. | 10 and 72 |
| • Can achieve tunable mechanical properties. | • Sensitivity to pH and ionic strength variations. | • Responsive to environmental changes, such as pH and ionic strength. | |||
| • Suitable for controlled release applications due to ion exchange capabilities. | |||||
| UV cross-linking | Initiation of cross-linking reactions by ultraviolet (UV) radiation-induced radicals. | • Rapid cross-linking process. | • Requires photoinitiators which may have toxicity concerns. | • Quick gelation and cross-linking. | 441 and 442 |
| • Precise spatial and temporal control. | • Limited penetration depth of UV light. | • Possibility of achieving spatially controlled cross-linking. | |||
| • High mechanical strength and stability. | |||||
| • Applicable for patterning and 3D printing of hydrogels. | |||||
| Enzymatic cross-linking | Enzyme-mediated formation of cross-links between nanocellulose chains. | • Biocompatible and environmentally friendly. | • Relatively slower reaction kinetics. | • Biocompatible and suitable for biomedical applications. | 244, 439 and 443 |
| • High specificity and mild reaction conditions. | • Limited availability of suitable enzymes. | • Controlled and precise cross-linking. | |||
| • Retention of native nanocellulose properties. | |||||
| • Potential for functionalization through enzyme engineering. | |||||
| Doubly cross-linked hydrogels | Combination of two or more cross-linking methods to enhance hydrogel properties. | • Synergistic combination of advantages from different cross-linking methods. | • Complex synthesis process. | • Superior mechanical properties compared to single cross-linking methods. | 444 and 445 |
| • Enhanced mechanical strength and stability. | • May require optimization of cross-linking conditions. | • Enhanced stability and durability. | |||
| • Tunable properties by adjusting the ratio of cross-linking methods. | |||||
| • Suitable for applications requiring high mechanical strength and stability. | |||||
4 Nanocellulose-based hydrogels for tissue engineering applications
Nanocellulose-based hydrogels have revolutionized the field of tissue engineering and regenerative medicine, owing to their exceptional versatility and biocompatibility.21,29,30,51,401 In this section, the latest applications of these hydrogels in regenerating different biological tissues are described and discussed. The recent progress in employing cellulose-based hydrogels for tissue engineering applications is outlined in Table 4. Nanocellulose is frequently combined with other biomaterials and polymers to fabricate customized hydrogels tailored to meet the specific challenges presented by different tissue types. By encapsulating biological cues and incorporating diverse functionalities, these hydrogels demonstrate mechanical strength and biological activities essential for effective tissue engineering. Vital tissues in living organisms, such as skin, cartilage, bone, cardiac muscle, nerves, and vasculature, play critical roles in their structural integrity, function, and overall well-being.20,49,335 However, injuries, diseases, or congenital conditions can compromise the integrity of these tissues, leading to pain, reduced mobility, impaired organ function, or life-threatening situations. Consequently, the imperative to repair, regenerate, or replace damaged tissues has driven significant advancements in regenerative medicine.485–487 Nanocellulose hydrogels have emerged as remarkable materials, offering interesting properties that facilitate tissue repair. Fig. 19 summarizes key properties and various tissue engineering applications of nanocellulose-based hydrogels.| Regenerated tissues | Hydrogel composition | Results | Ref. |
|---|---|---|---|
| Skin | Bacterial cellulose/acrylic acid hydrogel | • In vitro tests confirmed hydrogel supported keratinocytes and fibroblasts attachment. | 488 |
| • Hydrogel maintained cell viability and facilitated cell transfer without excess migration. | |||
| • In vivo studies showed hydrogel alone and with cells accelerated wound healing. | |||
| • Hydrogel with cells outperformed hydrogel alone in healing, shown by gross appearance. | |||
| • Masson's trichrome staining highlighted better performance of hydrogel with cells. | |||
| Skin | Chloramphenicol (CAP) loaded 2,3 dialdehyde cellulose (DABC) hydrogel membranes | • Bacterial cellulose transformed into DABC using sodium metaperiodate oxidation. | 489 |
| • DABC membranes released 99–99.5% of CAP within 24 hours, no burst effect. | |||
| • Both CAP-loaded BC and DABC showed prolonged antibacterial activity for 3 days. | |||
| • CAP-loaded DABC enhanced fibroblast adhesion and proliferation versus CAP-loaded BC. | |||
| • Fibroblast support confirmed by MTT tests. | |||
| Skin | Bacterial cellulose/polyacrylamide hydrogel microparticles | • Microparticles showed cytocompatibility with L929 cells. | 490 |
| • Non-irritating in dermal irritation tests. | |||
| • Hydrogel improved wound contraction and epithelialization. | |||
| • Accelerated fibroblast proliferation; resulted in complete wound repair. | |||
| • Confirmed biocompatibility and effectiveness as wound dressing material. | |||
| Skin | Carboxymethyl cellulose/hydroxyethyl cellulose/LAPONITE®/acetylated distarch phosphate/berberine | • The biocomposite effectively controlled oxidative stress and inflammation in chronic wounds. | 491 |
| • It reduced keratinocyte hyperproliferation by 25%, aiding wound healing. | |||
| • Berberine and hydrogel synergy notably enhanced material properties. | |||
| • The biocomposite is highly promising for dermatological applications, promoting wound healing. | |||
| Skin | A series of hydrogels (CMC-ADH/PEG-FBA) was developed by chemically linking carboxymethyl cellulose-graft-adipic dihydrazide (CMC-ADH) with 4-formylbenzoic acid-terminated poly(ethylene glycol) (PEG-FBA) | • The hydrogels sustained drug release and showed effective antibacterial properties in vitro. | 413 |
| • In vivo studies confirmed their hemostatic and wound-healing effects. | |||
| • They accelerated wound healing and exhibited superior hemostatic effects. | |||
| • The hydrogels adhered well to irregular wounds and enhanced angiogenic capabilities. | |||
| • Their performance attributes include stopping bleeding, creating a protected wound environment, releasing ciprofloxacin, reducing inflammation, and promoting tissue engineering. | |||
| Skin | Boronic acid was grafted onto carboxyethyl cellulose (CMC) to create boronic acid grafted CMC (CMC-BA). Subsequently, a self-healing hydrogel was produced by crosslinking with polyvinyl alcohol (PVA) through dynamic boronic ester bonds. | • The CMC-BA/PVA hydrogel displayed excellent biocompatibility and degradability. | 492 |
| • It was susceptible to cellulase both in vitro and in vivo. | |||
| • Rapid formation of hydrogel enabled localized injection, suitable for irregular wounds. | |||
| • In vivo experiments demonstrated airtight adhesion, reducing blood loss. | |||
| • The hydrogel acted as a drug carrier, minimizing acute in vivo toxicity. | |||
| • It preserved the antitumor efficacy of doxorubicin through controlled release. | |||
| Skin | Hydrogel fabricated using TEMPO-oxidized bacterial cellulose (TBC) combined with tannic acid-modified metal–organic framework (TA@ZIF-8 [TZ]), and loaded with MXene (TBC-TA@ZIF-8-MXene [TTZM]). | • The hydrogel exhibited strong antibacterial effects and sped up wound healing. | 493 |
| • Under near-infrared light, Zn2+ and MXene agents synergistically inhibited bacterial metabolism. | |||
| • TTZM hydrogel stimulated blood vessel growth and regulated inflammation for wound healing. | |||
| • In bacteria-infected wounds, NIR-responsive TTZM hydrogel accelerated closure and eliminated infections. | |||
| Skin | A bilayer material was prepared by electrospinning a hybrid of PVA and bacterial cellulose (BC NFs) atop a highly interconnected porous 3D gelatin-PVA hydrogel obtained by freeze-drying. | • FTIR confirmed cross-linking of layers, SEM showed porous structure, random and aligned NFs. | 494 |
| • Water contact angle indicated a hydrophilic surface for the bilayer material. | |||
| • Swelling analysis indicated high swelling, while degradation analysis showed good stability. | |||
| • The bilayer material released Ag-sulfadiazine in a sustained and controlled manner, exhibiting antibacterial activity. | |||
| • In vitro studies on fibroblasts and human embryonic kidneys showed desirable cell viability and adhesion. | |||
| • The synergistic effect of NFs and hydrogel resulted in a potential wound dressing material. | |||
| Skin | Multimodal antibacterial cellulose wound dressing (MACD) with photothermal therapy (PTT) was developed by graft polymerizing an imidazolium ionic liquid. monomer containing an iron complex anion structure. | • The developed hydrogels showed remarkable antibacterial properties. | 495 |
| • Ionic liquids and quaternary ammonium salts contributed to photothermal conversion ability. | |||
| • Hydrogel dressings exhibited 99.57% antibacterial efficacy against S. aureus. | |||
| • They also demonstrated 99.16% efficacy against E. coli. | |||
| • Minimal hemolysis (<5%) and high cell viability (>85%) were observed. | |||
| • In vivo experiments confirmed accelerated wound healing. | |||
| Skin | Bacterial cellulose (BC) dressings incorporating ε-polylysine (ε-PL), cross-linked with a biocompatible and mussel-inspired polydopamine (PDA). | • BC membranes were coated with PDA and treated with ε-PL. | 496 |
| • Modified membranes exhibited robust antibacterial properties in vitro and in vivo. | |||
| • Membranes showed excellent hemocompatibility and cytocompatibility. | |||
| • Functionalized membranes significantly improved wound healing in rats. | |||
| • Complete wound closure occurred with functionalized membranes. | |||
| • Histological analysis showed thicker, smoother skin with modified membranes. | |||
| Skin | Water-responsive hybrid hydrogels based on carboxymethyl cellulose (CMC) and poly(vinyl alcohol) (PVA) using citric acid (CA) as the crosslinking agent were developed. | • Hydrogels were designed with flexible swelling properties through molecular mass and degree of functionalization. | 497 |
| • Swelling degrees ranged from 100% to 5000%, with gel fraction from 40% to 80%. | |||
| • Highly absorbent hydrogels were produced, influenced by CA crosslinker concentration and presence of PVA. | |||
| • Crosslinking mechanism involved ester bonds formed between CA, CMC, and PVA polymers. | |||
| • Hydrogels presented hydrophilicity, permeability and structural features dependent on crosslinking and composition. | |||
| • Hydrogels showed cytocompatibility with in vitro cell viability responses over 90%, promising for skin repair. | |||
| Cartilage | Hydrogel composite comprising cross-linked thiolated chitosan (TCS) and carboxymethyl cellulose (CMC) (TCS/CMC). | • Rheological analysis confirmed TCS/CMC hydrogels strength exceeding 2.5 kPa elastic modulus. | 498 |
| • Crosslinking impacted network distribution, surface morphology, pore size, gelation time, and degradation. | |||
| • Hydrogels maintained 90% weight and shape for 21 days, releasing TGF-β1. | |||
| • In vitro experiments showed accelerated BMSC growth with TGF-β1-loaded hydrogels. | |||
| • In vivo rat knee studies for 8 weeks demonstrated hydrogel effectiveness in cartilage repair. | |||
| Cartilage | Injectable hydrogels based on chitosan (CS), cellulose nanocrystals (CNCs) and pectin were designed | • Injectable hydrogels of CS, CNCs, and pectin exhibited significant internal structure. | 499 |
| • Pectin and CS-based hydrogels mimicked ECM, but their low mechanical strength limited cartilage repair. | |||
| • CNCs were added to reinforce natural polysaccharide-based hydrogels, aiming to enhance mechanical properties. | |||
| • Varying weight ratios of CNCs improved network structure, reduced swelling, and enhanced degradation resistance. | |||
| • CNC incorporation increased chondrocyte proliferation, suggesting potential for cartilage tissue regeneration. | |||
| Cartilage | Double-network microcrystalline cellulose hydrogels | • A chemical-physical multi-step cross-linking strategy was used to fabricate hydrogels. | 500 |
| • Hydrogels biomimetic macroporous shape and high water content suited chondrocyte adhesion and proliferation. | |||
| • Hydrogels met cartilage tissue engineering scaffold requirements with high compressive strength (4.53 MPa). | |||
| • In vitro findings showed hydrogels enhanced cell adhesion and proliferation safely. | |||
| • Hydrogels demonstrated stability, slow degradation, and strong histocompatibility in vivo implantation. | |||
| Cartilage | Azide and alkyne functional groups were incorporated into citric acid-modified hydroxyethyl cellulose (HEC) utilizing a bioorthogonal click chemistry approach. | • Lyophilized samples exhibited interconnected microstructure suitable for cartilage tissue engineering. | 391 |
| • Crosslinking improved scaffold stability. | |||
| • The click sample retained high water uptake (∼650% swelling degree). | |||
| • Click sample showed Young's modulus ∼10 MPa, tensile strength ∼0.43 MPa, resembling natural cartilage. | |||
| • In vitro assays confirmed click sample biocompatibility, chondrogenic potential, and bioorthogonal characteristics. | |||
| Cartilage | Composite hydrogels comprising methacrylated gelatin (GelMA) and bacterial cellulose (BC) were successfully fabricated. | • The morphology of GelMA/BC hydrogels was analyzed with SEM. | 501 |
| • Hydrogels displayed interconnected porous structure, pore sizes decreased with BC content. | |||
| • Characterization included swelling experiments, XRD, TGA, rheology, and compressive tests. | |||
| • Composite hydrogels showed enhanced mechanical properties compared to pure GelMA. | |||
| • Preliminary biocompatibility assessment with chondrocytes showed cell proliferation and phenotype maintenance. | |||
| Cartilage | Bacterial cellulose-chitosan (BCC) and alginate-gelatin (AG) were synergistically utilized to fabricate 3D hydrogel scaffolds. | • Composite scaffolds featured robust 3D architecture with average pore diameters. | 16 |
| • BCC–AG scaffolds exhibited substantial compressive strength, stability, and biocompatibility. | |||
| • Mesenchymal stem cells (hMSCs) mixed with hydrogel, cultured in chondrogenic medium. | |||
| • Hydrogel scaffold supported cell proliferation, leading to glycosaminoglycan and collagen accumulation at 6.26% and 6.71% (w/w), respectively, after 42 days of incubation. | |||
| • BCC–AG scaffolds show potential for cartilage tissue engineering applications. | |||
| Cartilage | Alginate dialdehyde–gelatin (ADA–GEL)/nanofibrillated cellulose (NFC) hydrogel | • Nanofibrillated cellulose (NFC) introduced into ADA–GEL matrix to improve physicochemical properties. | 502 |
| • Fourier transform infrared spectra show no chemical interaction between NFC and ADA–GEL. | |||
| • Scanning electron microscopy reveals NFC fibers embedded in hydrogel matrix, confirming fiber-reinforced composite. | |||
| • NFC-reinforced ADA–GEL (AG-N) composite hydrogels exhibited increased stiffness, up to 19.6 ± 3.0 kPa. | |||
| • Cell viability, proliferation, and chondrogenic differentiation assessed, suggesting potential osteochondral plug application. | |||
| Cartilage | Poly(vinyl alcohol) grafted glycidyl methacrylate/cellulose nanofiber (PVA-g-GMA/CNF) injectable hydrogels were developed. | • PVA-g-GMA lacked mechanical properties and cell proliferation. | 503 |
| • Cellulose nanofibers (CNF) were chosen as reinforcement to enhance properties. | |||
| • UV curing formed hydrogels with interconnected microporosity and pore size diameters of 3–68 μm. | |||
| • Hydrogels exhibited high physicochemical properties: gel fraction (81–82%), porosity (83–94%), water content (73–87%), and swelling (272–652%). | |||
| • Increasing CNF concentration improved water content, swelling, compressive strength, and modulus while reducing porosity. | |||
| • 0.7% CNF hydrogels showed superior cell proliferation, indicating potential for meniscus tissue engineering. | |||
| Bone | New hydrogel scaffolds were developed for bone tissue engineering, incorporating inorganic calcium in the form of calcium phosphate, both alone and in combination with CaCO3, within bacterial cellulose (BC) matrices. | • Fourier transform infrared and thermogravimetric analyses confirmed BC and inorganic calcium presence. | 504 |
| • Scanning electron microscopy revealed porous structures ranging from 50 to 200 μm. | |||
| • Swelling studies showed notable capacity in calcium phosphate-filled and calcium phosphate & CaCO3-filled scaffolds. | |||
| • Compressive strength of calcium-filled scaffolds ranged from 0.24 to 0.60 MPa, comparable to trabecular bone. | |||
| • Significant cell viability persisted for up to 72, 120, and 168 hours. | |||
| Bone | Cellulose hydrogels were crosslinked using citric acid. | • Cellulose hydrogels were crosslinked using non-toxic citric acid. | 505 |
| • Citric acid induced –COOH functional groups, enhancing hydrophilicity and roughness. | |||
| • Physiochemical, morphological, and mechanical analyses were conducted before and after crosslinking. | |||
| • Human mesenchymal stem cell responses were influenced by surface chemistry and roughness. | |||
| Bone | Well-defined porous silk fibroin/cellulose hydrogels were successfully designed. | • Cellulose enhanced mechanical properties and stiffness of silk fibroin (SF) material. | 506 |
| • Atmospheric humidity regenerated cellulose framework, followed by SF assembly. | |||
| • Hydrogels exhibited β-sheet conformation in surface-near layers, decreasing in deeper layers. | |||
| • Cellulose framework boosted mechanical properties; SF matrix supported cell growth. | |||
| • MC3T3-E1 cells on SF/cellulose hydrogels showed accelerated osteocyte differentiation. | |||
| Bone | Nanocomposite scaffold of semi-interpenetrating network cellulose-graft-polyacrylamide/nano-hydroxyapatite was synthesized using free radical polymerization. | • Scaffolds fabricated via freeze-drying with compressive strength of 4.80 MPa. | 507 |
| • Elastic modulus measured at 0.29 GPa under optimal conditions. | |||
| • Scaffolds exhibited 47.37% porosity. | |||
| • Apatite-forming ability assessed by immersion in simulated body fluid (SBF). | |||
| • Apatite particles observed on scaffold surface after 14 days in SBF. | |||
| Bone | Cellulose nanofiber (CNF)-reinforced oxidized alginate (OSA)/gelatin (Gel) hydrogel | • OSA/Gel/CNF sample displayed notable compressive modulus (up to 361.3 kPa). | 508 |
| • Gelation time ensured excellent injectability (∼150 s). | |||
| • Self-healing efficiency reached up to 92%, allowing dynamic adjustments and personalized therapies. | |||
| • OSA/Gel/CNF hydrogel exhibited excellent biomineralization (Ca/P ratio ∼1.69). | |||
| • Enhanced preosteoblast cell (MC3T3-E1) viability, proliferation, and osteogenic differentiation observed (over 96%). | |||
| Bone | Citric acid-cross-linked carboxymethyl cellulose (C3CA) scaffolds were fabricated using freeze-drying | • Citric acid-cross-linked carboxymethyl cellulose scaffolds were developed via freeze-drying at −20, −40, or −80 °C. | 509 |
| • Scaffold pore sizes differed, with −20 °C scaffolds having larger pores (74 ± 4 μm) compared to −40 and −80 °C. | |||
| • Saos-2 osteoblast cell line proliferation and differentiation were supported, with significant matrix mineralization observed in −40 °C scaffolds. | |||
| • Subcutaneous implantation in rats showed matrix formation as early as 14 days in −40 °C scaffolds, compared to 28 days in −20 and −80 °C. | |||
| Heart | Electroconductive, thermosensitive, and injectable hydrogel composed of chitosan, pluronic, and gold-decorated cellulose nanofibers was developed. | • The hydrogels possessed porous structures with open pore channels ranging from 50 to 200 μm. | 510 |
| • Shear rate sweep measurements demonstrated reversible sol-to-gel phase transitions within 5 minutes. | |||
| • These hydrogels exhibited shear-thinning behavior, with a shear modulus ranging from 1 to 12 kPa. | |||
| • Electrical conductivity studies revealed a conductance of 6 × 10−2 S m−1 at 75 mM Au concentration. | |||
| • In vitro cytocompatibility assays with H9C2 cells showed high biocompatibility and strong cell adhesion. | |||
| • The developed hydrogel showed significant potential as injectable biomaterials in cardiac tissue engineering. | |||
| Heart | Cellulose acetate (CA)/regenerated cellulose (RC) hydrogel scaffolds were developed. | • CA and RC surfaces promoted cardiac cell growth, enhanced cell connectivity, and improved electrical functionality. | 511 |
| • CA scaffolds provided optical clarity and non-autofluorescent properties, enabling accurate optical measurements in cell constructs. | |||
| • These scaffolds were moldable to intricate shapes, crucial for tissue engineering design. | |||
| • Cellulose scaffolds offered excellent adhesion, nanoscale molding capabilities, and controlled biodegradability for in vitro cardiac construct growth. | |||
| Heart | Carboxymethyl cellulose–alginate interpenetrating hydroxy ethyl methacrylate crosslinked polyvinyl alcohol reinforced hybrid hydrogel templates | • Two superabsorbent hydrogels developed using interpenetration chemistry displayed superior physiochemical features. | 512 |
| • In vitro degradation showed acceptable weight composure and pH changes for both hydrogels. | |||
| • Both hydrogels demonstrated hemocompatibility and biocompatibility. | |||
| • Hydrogels facilitated anterograde and retrograde migration of H9c2 cells. | |||
| • Coculture with swine epicardial adipose tissue cells and cardiac fibroblasts showed synchronous cell growth. | |||
| • Hydrogels containing H9c2 cells promoted extracellular matrix production. | |||
| Heart | TEMPO-oxidized cellulose nanofibrils (TOCNs) and sulfonated carbon nanotubes (SCNTs) used to prepare TOCN–SCNT composite hydrogels. | • TOCN–SCNT hydrogels showed conductivity between 5.2 × 10−6 to 6.2 × 10−2 S cm−1 by adjusting SCNT content from 0 to 5 wt%. | 513 |
| • 1 wt% SCNT incorporation promoted adhesive cell growth and proliferation. | |||
| • Hydrogel exhibited higher Connexin 43 and cardiac troponin-T proteins, suggesting myocardial tissue engineering scaffold potential. | |||
| Nerve | Cellulose nanofibers (CNFs)/polypyrrole (PPy) | • CNFs were electrospun from cellulose acetate. | 53 |
| • CNFs were surface-modified via in situ PPy polymerization. | |||
| • PPy particles adhered to nanofiber surface, increasing conductivity 105-fold. | |||
| • No cytotoxic effects observed in vitro. | |||
| • SH-SY5Y human neuroblastoma cells exhibited enhanced adhesion on PPy-coated CNFs. | |||
| • Cell viability sustained for up to 15 days of differentiation. | |||
| • Cells adopted neuron-like morphology on PPy-coated CNFs. | |||
| Nerve | Cellulose acetate (CA)/poly(ε-caprolactone) (PCL)/graphene oxide (GO) | • A novel electroactive biocompatible nanofibrous scaffold consisting of CA, PCL and GO was prepared using electrospinning. | 514 |
| • An optimum sample containing 0.5 wt% of GO exhibited a diameter of 280 nm. | |||
| • The electrical conductivity of the sample was approximately 75 μS m−1. | |||
| • Chemical analysis showed no reaction during the distribution of GO within the nanofibers. | |||
| • The optimized sample displayed a water contact angle of 39 and a tensile strength of 2.5 MPa. | |||
| • Electrical activity, assessed by cyclic voltammetry, was higher in the presence of GO. | |||
| • Cell viability, adhesion, and differentiation were significantly enhanced in the presence of GO. | |||
| Nerve | Nanoporous cellulose gels (NCG) were coated with PPy nanoparticles, creating electroconductive materials. | • The NCG/PPy composite hydrogels, turned into aerogels using supercritical CO2 drying, demonstrated remarkable properties. | 515 |
| • They had a density of 0.41–0.53 g cm−3 and nitrogen adsorption surface areas ranging from 264 to 303 m2 g−1. | |||
| • Additionally, they showed robust mechanical strength and electrical conductivity up to 0.08 S cm−1. | |||
| • In vitro studies indicated improved adhesion and proliferation of PC12 cells. | |||
| • Electrical stimulation led to PC12 cells attaching and extending longer neurites. | |||
| Nerve | Chitosan–cellulose nanofiber (CS–CNF) composite self-healing hydrogels | • Incorporating cellulose nanofibers (CNFs) into chitosan (CS) improved hydrogel properties. | 516 |
| • CNF-modified hydrogels showed enhanced self-healing, dynamic bonding, and strain sensitivity. | |||
| • Neural stem cells in CS–CNF hydrogels exhibited improved oxygen metabolism and differentiation. | |||
| • Metabolic changes in hydrogels aligned with neural stem cell differentiation. | |||
| • Optimized CS–CNF hydrogel with 0.09 wt% CNFs showed 50% better neural engineering. | |||
| • Improved hydrogel outperformed pristine CS hydrogel in zebrafish brain injury model. | |||
| Vasculature | Functional bacterial cellulose (BC) hydrogel incorporating silver-loaded zeolitic imidazolate framework-8 (ZIF-8) antibacterial agent. | • The biopolymer dressing exceeded 1 MPa in tensile strength. | 517 |
| • In vitro tests showed antibacterial efficacy, with 0.85% survival for E. coli and 0.39% for S. aureus. | |||
| • Cell culture experiments demonstrated excellent biocompatibility and notable angiogenic potential. | |||
| • In vivo studies on rats exhibited remarkable wound healing and skin re-epithelialization. | |||
| • This study introduced a high-performing dressing with potent antibacterial properties. | |||
| Vasculature | Multifunctional hydrogel (CCS/CCHO-Ag) was developed by incorporating silver (Ag) into catechol-modified chitosan (CCS) and aldehyde-modified cellulose nanocrystals (CCHOs). | • Cellulose nanocrystals (CNCs) in liquid crystal phase enhanced vascularization. | 518 |
| • CCS/CCHO-Ag hydrogels had in situ formation, satisfactory strength, controlled Ag release. | |||
| • Hydrogels exhibited antibacterial properties and biocompatibility. | |||
| • They seamlessly covered and adhered to irregular wounds, reducing reinjury risk. | |||
| • CCS/CCHO-Ag hydrogels promoted neovascularization and tissue engineering. | |||
| • In vitro and in vivo experiments confirmed their effectiveness. | |||
| Vaculature | Regenerative cellulose hydrogel matrix and photo-polymerized poly(hydroxyethyl methacrylate) filler | • Biomimetic approach inspired by native blood vessels enabled fabrication of cellulose-based small-diameter artificial blood vessels (SDABV). | 519 |
| • Gradient distribution of poly(hydroxyethyl methacrylate) in cellulose network created composite gel tube mimicking native blood vessels. | |||
| • Cellulose-based SDABV hydrogels exhibited similar mechanical performance to native counterparts: tensile strength (1.4 MPa), strain (93%), Young's modulus (2.98 MPa), toughness (1040 kJ m−3). | |||
| • Materials demonstrated excellent suture retention, burst pressure, in vitro biocompatibility, low fibrinogen adsorption, and platelet adhesion. | |||
| • In vivo experiments on rabbits confirmed material safety with no blood vessel blockages or inflammatory responses after three months. | |||
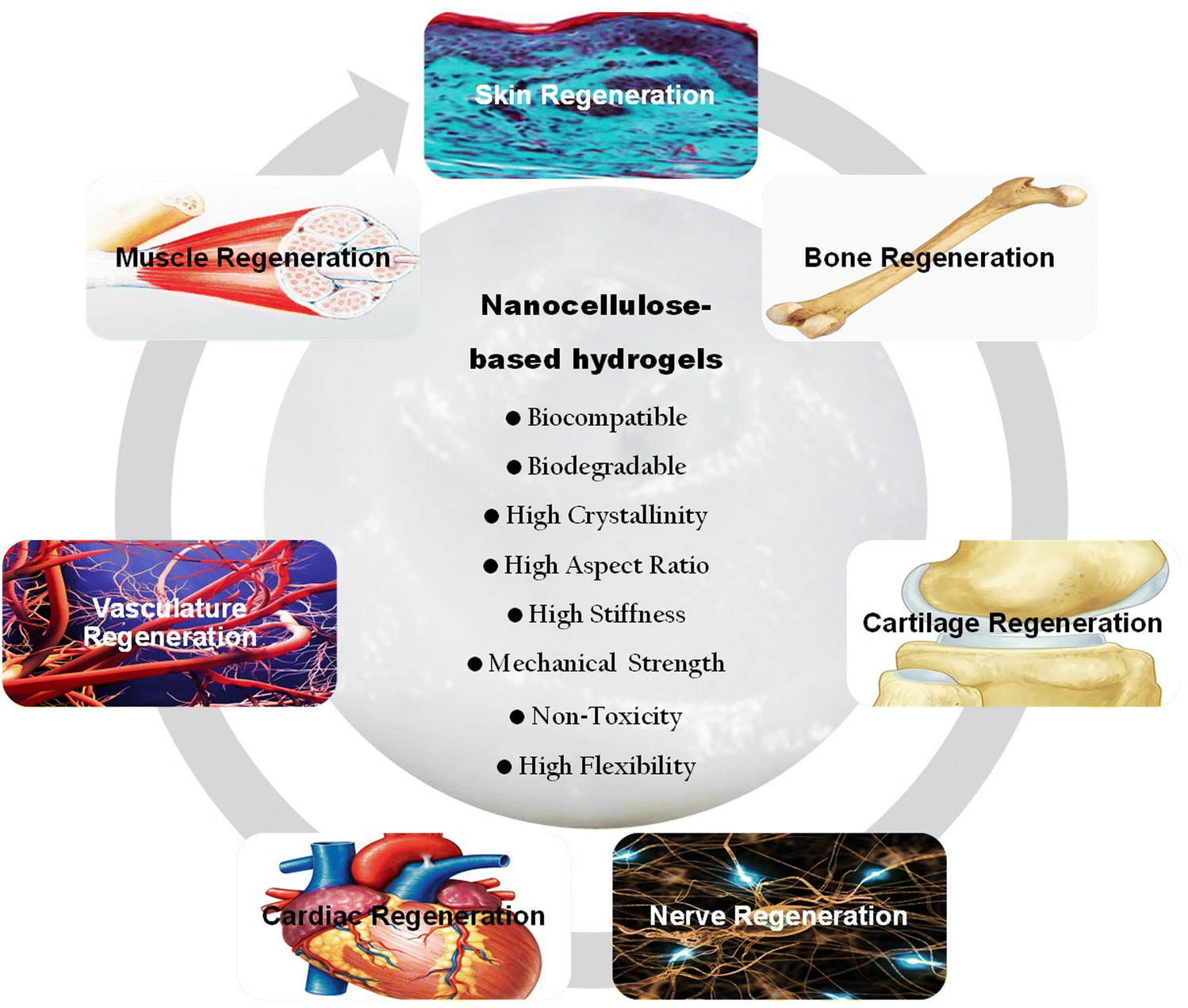 | ||
| Fig. 19 Key properties and tissue engineering applications of nanocellulose-based hydrogels. | ||
As highlighted in Table 4, the utilization of nanocellulose-based hydrogels highlights a significant advancement in the field of tissue engineering. Through meticulous design and innovative processing techniques, these hydrogels have demonstrated remarkable mechanical and biological properties essential for the engineering of various tissues, including skin, cartilage, bone, heart, and nerve, among others. The data in Table 4 provides a comprehensive overview of nanocellulose-based hydrogel compositions, processing methods, and the achieved mechanical and biological outcomes. These findings underscore the promising potential of nanocellulose-based hydrogels in revolutionizing tissue engineering therapies, offering tailored solutions for diverse clinical applications. The use of nanocellulose-based hydrogels represents a significant step forward in the pursuit of innovative and effective strategies to repair and regenerate damaged tissues, ultimately improving the quality of life for patients in need of such treatments.
4.1 Skin tissue engineering
Skin acts as a barrier protecting the body from pathogens and regulating temperature. Skin injuries, whether due to wounds, burns, or chronic ulcers, can lead to severe complications. Nanocellulose hydrogels provide a biocompatible and supportive scaffold, promoting cell adhesion, proliferation, and wound healing. Their high water content maintains a moist environment, enhancing tissue engineering and minimizing scarring.207,520 For instance, in a recent study, a biocompatible and biodegradable hydrogel system was developed by combining curcumin (Cur) solubilization with pluronic F-127, polyvinyl alcohol (PVA) gelation, and porosity enhancement by TEMPO-oxidized cellulose nanofiber (TOCN). This physically crosslinked TOCN-PVA-Cur hydrogel, prepared via freeze-thawing, effectively released Cur, enhancing wound healing. In vitro studies demonstrated the uptake of curcumin by fibroblast cells. Application of TOCN-PVA-Cur to rat full-thickness skin wounds increased wound closure, stimulated neoepidermis and granulation tissue formation, and encouraged collagen fiber accumulation, highlighting the hydrogel potential for promoting natural wound healing (Fig. 20A and B).521 In addition to the compelling findings presented in Table 4 regarding the use of nanocellulose-based hydrogels as functional matrices for repairing skin tissues, it's worth noting that certain microbial infections can damage skin tissue. In this regard, cellulose hydrogels have also demonstrated intriguing antibacterial properties and proven biocompatibility, thus supporting the regeneration of skin tissues.219,517,522 In a recent study conducted by Hu et al.,522 ε-polylysine (ε-PL), known for its biocompatibility and antibacterial properties, was integrated into a cellulose/γ-polyglutamic acid (γ-PGA) hydrogel, creating a double-network hydrogel termed CGLH (Fig. 20C).522 Its elastic modulus increased from 0.097 ± 0.015 MPa to 0.441 ± 0.096 MPa, and swelling ratio from 382.7 ± 24.3% to 611.2 ± 8.6%. CGLH showed promising results in preclinical models, demonstrating biocompatibility and antibacterial activity, aiding in healing infected and critical-size wounds within 12 days. It also enhanced collagen synthesis, vascularization and cell proliferation.522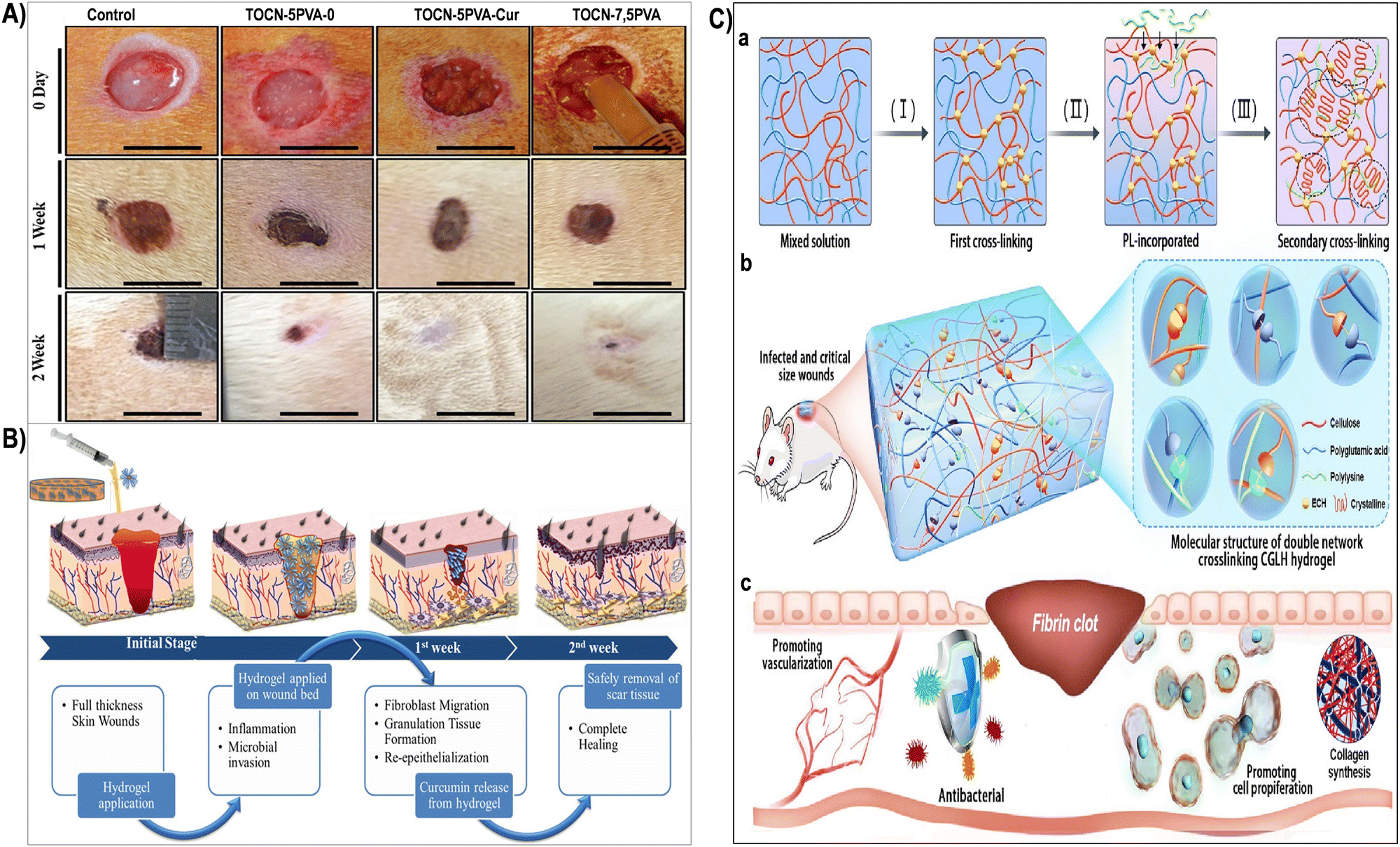 | ||
| Fig. 20 Wound healing stages are illustrated (A), showing enhanced neoepidermis formation using TOCN-5PVA-0, TOCN-5PVA-Cur and TOCN-7.5PVA-Cur hydrogel samples (B). Reproduced with permission from ref. 521 Copyright © 2020 Elsevier. (C) Schematic depiction of the preparation process and biomedical utilization of double-network CGLH. (a) Sequential chemical (primary) and physical (secondary) crosslinking led to CGLH formation. (b) Emphasizing the biomolecular structure and crosslinking sites within CGLH. (c) Illustrating CGLH capability to expedite wound healing by eradicating bacteria and enhancing vascularization, cell proliferation, and collagen deposition. Reproduced with permission from ref. 522 Copyright © 2023 Elsevier. | ||
Additionally, cellulose-based hydrogels have shown promising outcomes in skin tissue regeneration. Nanocellulose-based hydrogels have been thoroughly assessed in murine models, examining wound healing kinetics, inflammatory responses, and tissue remodeling. Metrics including wound closure rates, collagen deposition, and angiogenesis are meticulously quantified, providing valuable insights into the hydrogel effectiveness in advancing skin tissue engineering.523 A recent investigation showcased the expedited healing of full-thickness wounds through the application of bacterial cellulose-based multilayer composites. This innovative strategy involved employing a wound dressing composed of bacterial cellulose (BC) hydrogel, gelatin/alginate (Gel/Alg) hydrogel, and polycaprolactone (PCL) nanofibers loaded with ciprofloxacin (CIP) to facilitate in vivo healing.524 The developed dressings exhibited notable water absorption and an adequate water vapor transmission rate within the initial week, confirming their effective wound exudate absorption. The drug-containing dressing demonstrated a significant release of CIP in the first four hours, followed by a gradual reduction and stabilization from hours 4 to 24, persisting until day 9.524 This controlled CIP release effectively inhibited both positive and negative bacterial strains while ensuring the normal morphology and metabolic activity of fibroblasts. Additionally, in vivo experiments showcased the remarkable efficacy of CIP-loaded multilayer dressings in accelerating full-thickness wound healing within a 14-day timeframe. The dressings not only reduced inflammation but also stimulated re-epithelialization and enhanced skin engineering.524 In an another study, bacterial cellulose nanocrystals (BCNCs) were employed to reinforce regenerated chitin (RC) fibers, forming BCNC/RC filaments. Mechanical assessments revealed a significant enhancement in the strength of the BCNC/RC filament compared to RC. A yarn of 30 BCNC-loaded fibers exhibited satisfactory mechanical performance with a knot-pull tensile strength of 9.8 ± 0.6 N.525In vivo experiments on mice demonstrated that suturing with BCNC/RC yarn promoted wound healing without adverse effects. Animal models were divided into four groups: a negative control (Group I), a positive control with commercial polyamide (PA) sutures (Group II), and two novel sutures twisted and coated BCNC/RC yarn (Group III) and twisted but uncoated BCNC/RC sutures (Group IV). Images showing the wound healing process are highlighted in Fig. 21A. Histopathology analysis revealed complete tissue morphology in Groups II, III, and IV at three and ten days post-surgery, contrasting with the unsutured Group-I mice (Fig. 21B). No decay or inflammatory lesions were observed, and collagen around the suture increased over an extended recovery period. BCNC/RC sutures promoted effective wound healing without adverse effects, comparable to commercial PA sutures after 10 days (Fig. 21B).525
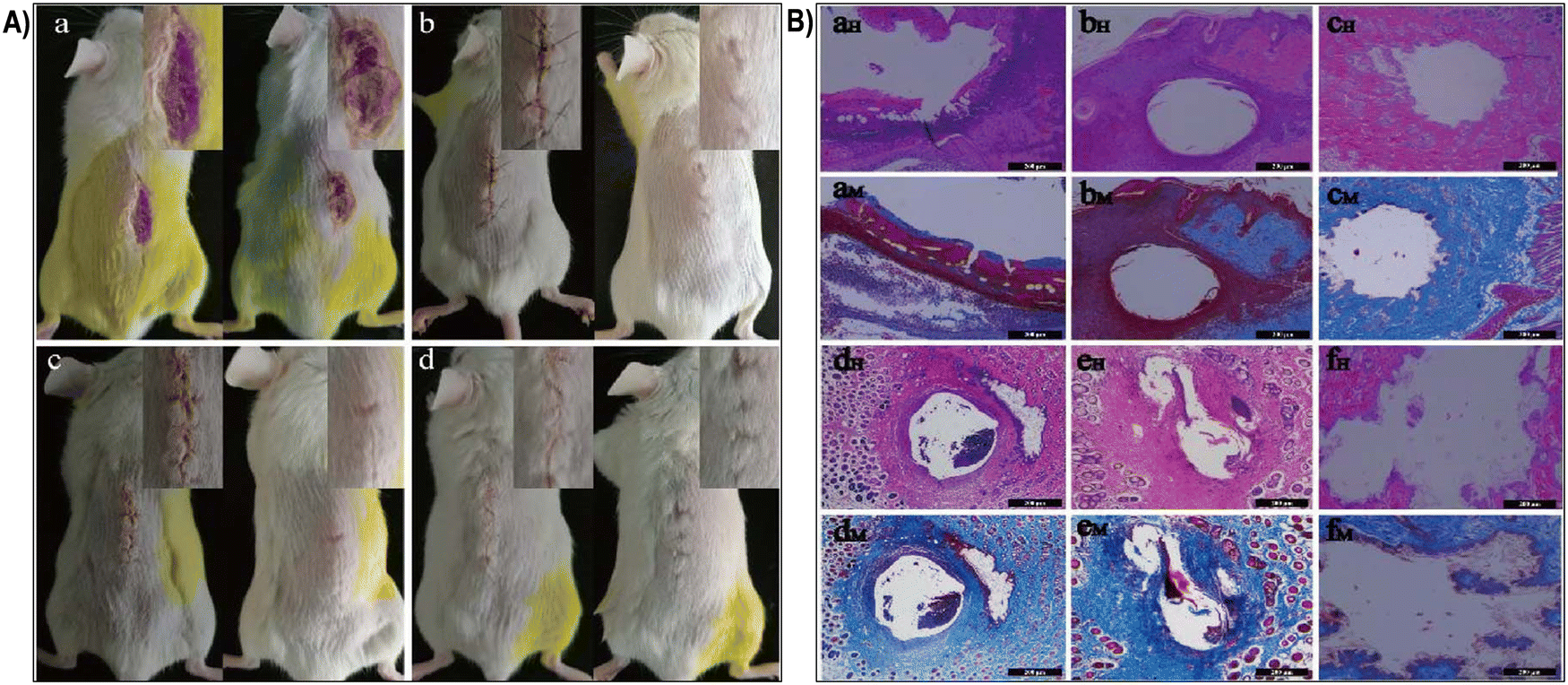 | ||
| Fig. 21 (A) Wound healing process images after 3 days (left) and 10 days (right) for (a) suture-less mice (Group-I), (b) mice with polyamide sutures (Group-II), (c) mice sutured with coated BCNC-10 mL/RC yarns (Group-III), and (d) mice sutured with uncoated BCNC-10 mL/RC yarns (Group-IV). Insets show enlarged views of the wound area. (B) Histological analysis with HE staining (aH) –(fH) and Masson's trichrome staining (aM)–(fM) for Group-I at 10 days (aH) and (aM), Group-II at 3 days (bH) and (bM), Group-II at 10 days (cH) and (cM), Group-III at 3 days (dH) and (dM), Group-III at 10 days (eH) and (eM), and Group-IV at 10 days (fH) and (fM). Scale bars: 200 μm. Reproduced with permission from ref. 525 Copyright © 2018 Elsevier. | ||
4.2 Cartilage tissue engineering
Cartilage, crucial for joint mobility, can deteriorate due to injuries or degenerative diseases, causing pain and impaired movement. Nanocellulose hydrogels, when combined with chondrocytes or stem cells, could mimic the natural cartilage matrix. These hydrogels provide mechanical support, facilitate cell adhesion, and offer sustained release of growth factors, aiding in cartilage engineering and mitigating joint-related discomfort.16,391 For example, 3D hierarchical porous bacterial cellulose (BC)/decellularized cartilage extracellular matrix (DCECM) scaffolds, mimicking cartilage microenvironment, were fabricated.526 A schematic of the experimental design used this work is shown in the Fig. 22A. These scaffolds demonstrated exceptional mechanical strength, water absorbency, and shape-memory properties. Enhanced cell adhesion and proliferation were observed in the fabricated scaffolds (Fig. 22B). In vitro and in vivo studies showed successful neocartilage tissue engineering with natural appearance and cartilage-specific lacuna formation (Fig. 22C). The BC/DCECM scaffolds led to superior repair outcomes, forming hyaline cartilage-like tissue within defect sites (Fig. 22D). This study marked significant progress in utilizing BC for cartilage tissue engineering.526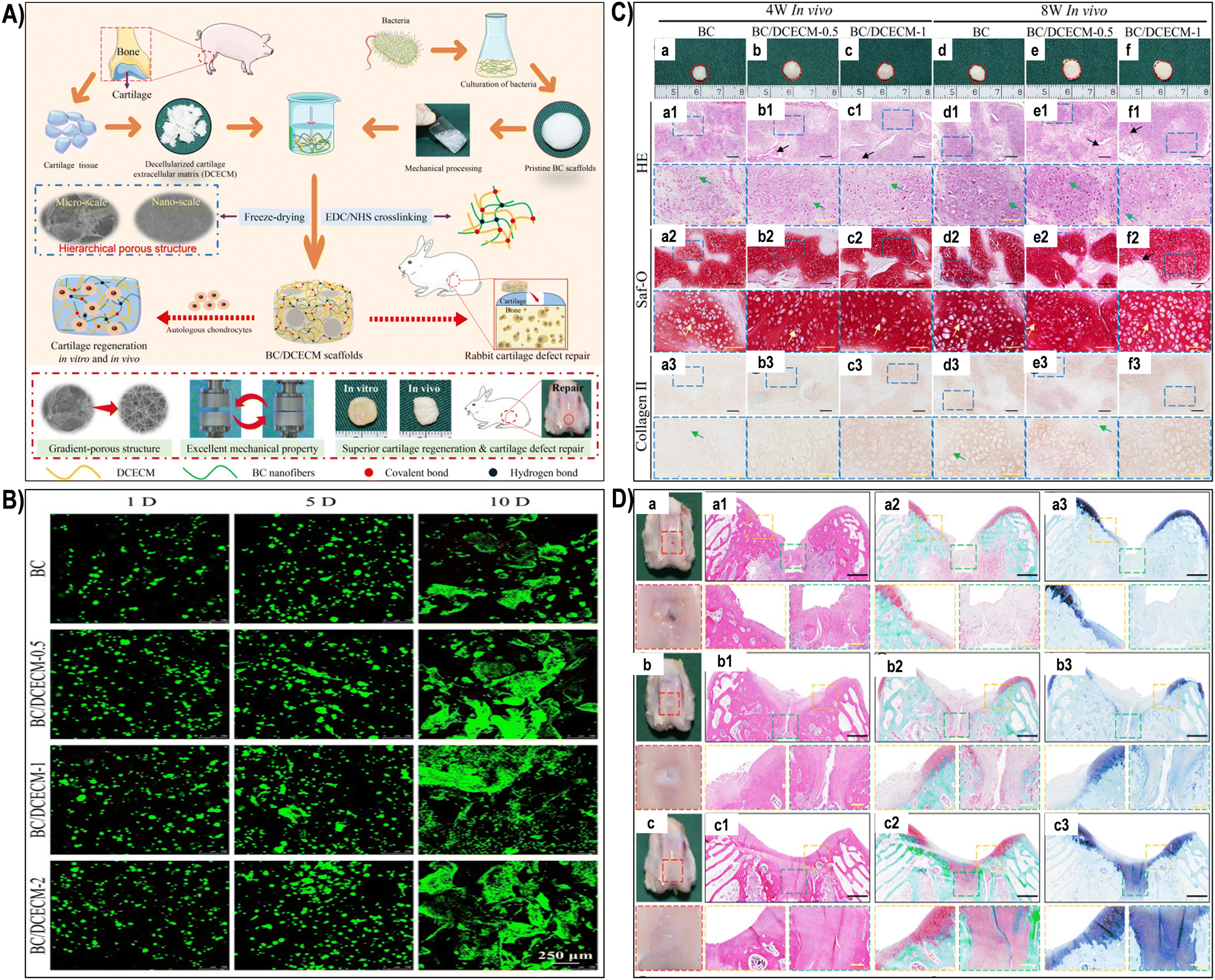 | ||
| Fig. 22 (A) Illustration of the fabrication process for 3D hierarchical porous scaffolds mimicking cartilage microenvironment for tissue engineering applications. (B) Live/dead staining of chondrocytes after 1, 5, and 10 days of culture. (C) Gross, histological, and immunohistological views of cell-scaffold constructs after 4 and 8 weeks in vivo. Gross images show engineered constructs at 4 and 8 weeks (a)–(f). Histological stains include HE for cells and extracellular matrix (a1)–(f1), Saf-O for GAG (a2)–(f2), and Collagen II for type II collagen (a3)–(f3). (D) In vivo assessment of scaffold therapeutic efficacy for cartilage defects at 8 weeks post-surgery. Macroscopic images of defects (a: BC control; b: BC/DCECM-0.5; c: BC/DCECM-1). Histological stains depict regenerated tissues (a1)–(c1), (a2)–(c2), (a3)–(c3). Scale bars: black, 200 μm; yellow, 100 μm. Reproduced with permission from ref. 526 Copyright © 2021 Elsevier. | ||
Moreover, nanocellulose-based hydrogels have showcased significant potential for in vivo cartilage tissue regeneration. Studies utilizing small animal models, such as mice and rabbits, have explored the effectiveness of these hydrogels in repairing cartilage defects. Current treatments for cartilage damage, such as autologous chondrocyte implantation (ACI) and microfracture surgery, often pose challenges, leading researchers to explore innovative solutions. In a recent study, researchers developed a novel hydrogel by oxidizing methylcellulose with sodium periodate, yielding dialdehyde methylcellulose (DAC). This DAC was then combined with succinyl-chitosan (SUC) to form an in situ DAC-SUC hydrogel, characterized by its stiffness, elasticity, and porosity as observed through advanced microscopy and rheological analysis. Notably, the DAC-SUC13 hydrogel exhibited excellent compatibility with cells, particularly bone marrow mesenchymal stem cells (BM-pMSCs), showcasing their chondrogenic potential.527 To assess the hydrogel efficacy, researchers created osteochondral defects in porcine knee joints, injecting BM-pMSC/DAC-SUC hydrogel after joint irrigation with sterile isotonic saline (Fig. 23A). After a 6-month observation period, the defect site treated with BM-pMSC/DAC-SUC hydrogel showed significant engineering, characterized by distinct reddish-white tissue filling the defect area, contrasting sharply with the irregular and depressed defect sites in the sham group (Fig. 23B). Moreover, histological analysis revealed that the DAC-SUC13 hydrogel acted as an efficient stem cell carrier, stimulating the synthesis of glycosaminoglycans and type II collagen in porcine knee osteochondral defects.527
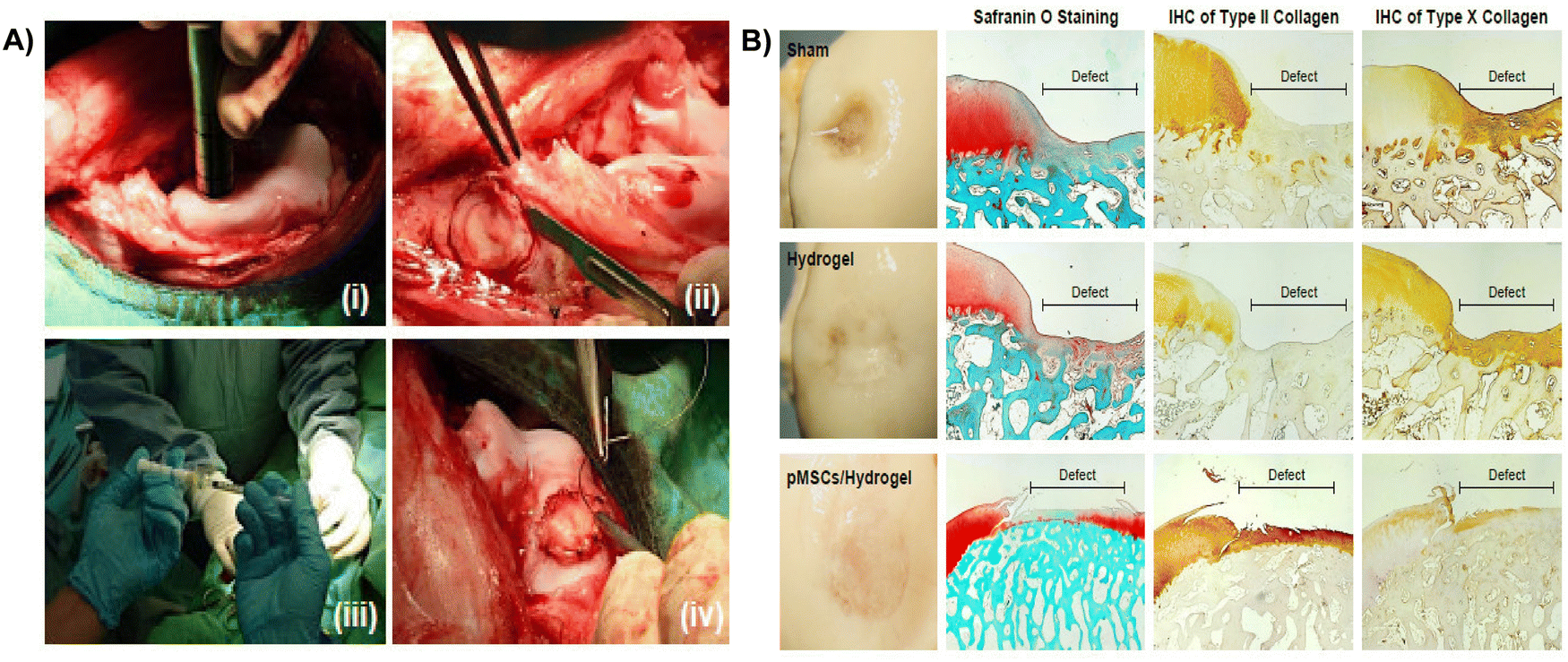 | ||
| Fig. 23 (A) Surgical procedure: (i) OCD generated using an electric drill; (ii) 6.5 mm × 5 mm OCD area obtained; (iii) pMSC-hydrogel scaffold implanted into OCD; (iv) patella relocated, and the wound sutured in layers. (B) Histological evaluation: microscopic views and histological sections of repaired sites at 6 months post-implantation. Macroscopic image of femoral condyle with pMSC in hydrogel implants. Paraffin-embedded sections stained with Safranin O, type II collagen, and type X collagen. The detailed examination provided insights into the regenerative potential of the implanted pMSC-hydrogel constructs. Reproduced with permission from ref. 527 Copyright © 2022 MDPI. | ||
4.3 Bone tissue engineering
Bone provides structural support, protects organs, and maintains mineral balance. Bone injuries resulting from trauma or diseases like osteoporosis require effective engineering to maintain skeletal integrity.528 Nanocellulose hydrogels, combined with osteogenic factors and nanomaterials, can create a conducive environment for osteoblast differentiation and bone tissue engineering. These hydrogels aid in bone defect repair and enhance overall skeletal stability.210,506 In a recent work, bacterial cellulose (BC) was treated with TEMPO-mediated oxidation (TO-BC) and maleic acid (MA-BC) to fabricate BC dispersions for bone engineering scaffolds (Fig. 24A). Both MA-BC and TO-BC-based 3D scaffolds exhibited distinct pore structures, with MA-BC showing regular, loose, and well-arranged lamellar patterns (Fig. 24B).529 MA-BC, combined with gelatin, demonstrated enhanced rheological properties and compression modulus for 3D printing, promoting osteogenic marker gene expression and mineralized nodule formation in vitro. Moreover, MA-BC-based gelatin scaffolds exhibited superior rat calvaria engineering, displaying higher bone mineral density and trabecular thickness in vivo, making them promising for biomedical applications.529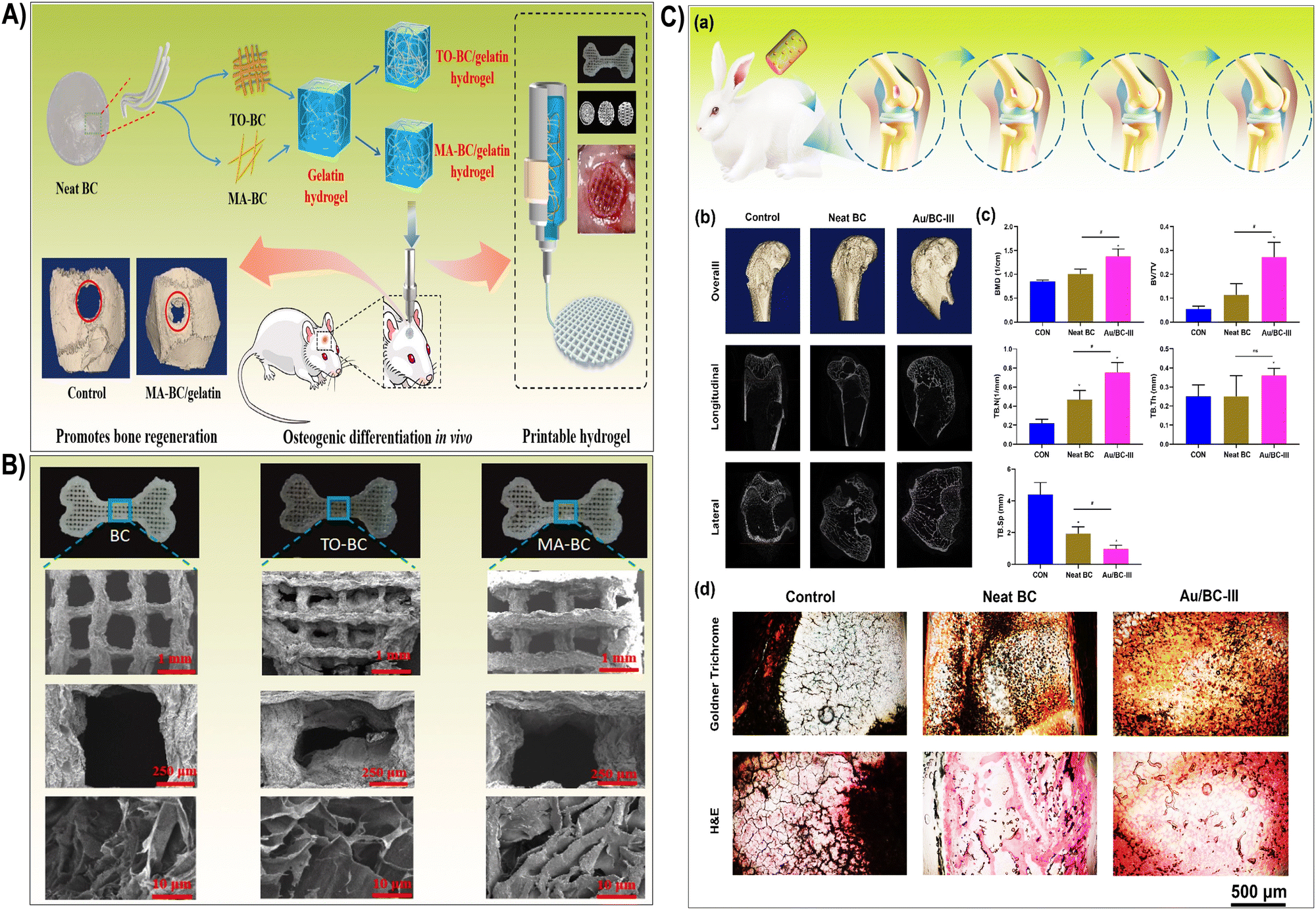 | ||
| Fig. 24 (A) Schematic depicting the preparation of printable bacterial cellulose-gelatin gel for in vivo osteogenic engineering. (B) Scanning electron microscopy images showcasing printed scaffolds. Reproduced with permission from ref. 529 Copyright © 2021 Elsevier. (C) In vivo osteogenic assessment of Au/BC hydrogels. (a) Illustration outlining the experimental procedure. (b) 3D micro-CT reconstructions and cross-sectional images of the femoral condyle treated with neat BC and Au/BC-III hydrogels, respectively. (c) Quantitative evaluation of the regenerated area using micro-CT parameters: BMD, BV/TV, TB·N, TB·Sp, and TB·Th. (d) Staining images with Goldner Trichrome and H&E, depicting bone defects implanted with neat BC and Au/BC-III hydrogels, respectively. Asterisks denote statistically significant differences between the control and experimental groups (*p < 0.05; **p < 0.01; ****p < 0.001). Reproduced with permission from ref. 400 Copyright © 2023 Elsevier. | ||
In vivo evaluation of nanocellulose-based hydrogels is pivotal, particularly in the field of bone and cartilage engineering. Animals serve as indispensable models for assessing the efficacy and safety of these hydrogels, serving as a bridge between laboratory investigations and clinical applications. In a recent investigation, cellulose nanofibrils (CNF) were employed in scaffold fabrication, leveraging their biocompatibility and capacity to form robust 3D porous networks from aqueous suspensions. A method enhancing their bioactivity for bone formation resulted in highly interconnected 3D networks.44 These materials exhibited optimal morphological and mechanical features, fostering hydroxyapatite formation and releasing essential ions crucial for in vivo bone repair. The scaffold porosity and roughness supported various cellular functions, and the released ions influence gene expression associated with cell differentiation. The controlled release of ions played a pivotal role in stimulating the production of bone morphogenetic protein 2 (BMP-2) from cells within the fractured area, expediting in vivo bone repair. Comprehensive biocompatibility assessments revealed no adverse effects on vital organs, such as the liver and kidneys. These findings signified a significant stride toward the facile development of advanced, high-performance CNF-based scaffolds tailored for bone tissue engineering applications.44 In addition, cutting-edge imaging technologies such as micro-computed tomography (micro-CT) and magnetic resonance imaging (MRI), have been strategically employed for non-invasive visualization of bone engineering progression in animal models subjected to nanocellulose-based hydrogels. These techniques provide quantitative assessments of critical parameters such as bone volume, density, and microstructure, delivering valuable insights into the hydrogel efficacy in facilitating bone healing.530 In a notable study,400 bacterial cellulose (BC) was combined with gold (Au) nanoparticles (GNPs) through in situ fermentation, with BC possessing attributes like biocompatibility, high porosity, robust mechanical strength, and a large surface area. GNPs are known for their remarkable osteogenic differentiation capability. Based on cellular assays, the Au/BC-III sample, which had the highest GNP content, exhibited superior in vitro osteogenic differentiation potential.400 It was subsequently chosen for in vivo bone engineering evaluation in a rabbit femoral defect model (Fig. 24C(a)). After 2 months, femurs were harvested and evaluated using micro-computed tomography (micro-CT) to assess structural changes. The 3D-reconstruction and cross-sectional images of femurs (Fig. 24C(b)) revealed significant bone defects in the control and neat BC hydrogel groups.400 Conversely, the femurs treated with Au/BC-III hydrogel showed more newly formed bone in the defect areas, indicating its capacity for in vivo bone engineering. Quantitative assessments, including bone mineral density (BMD), bone volume fraction (BV/TV), and bone trabecular parameters (TB·N, TB·Th, and TB·Sp), further characterized the differences in osseous tissue formation within the defect areas (Fig. 24C(c)). BMD in the defect areas treated with neat BC hydrogel and Au/BC-III hydrogels increased by 1.19-fold and 1.62-fold, respectively, compared to the control group, emphasizing the role of BC hydrogel as a platform for osteogenic cell adhesion and mineralization. Furthermore, treatment with Au/BC-III hydrogel led to significant increases in BV/TV, TB·N, and TB·Th, while reducing TB·Sp when compared to the control group and the neat BC hydrogel group. These results confirmed that Au/BC-III, due to the inclusion of GNPs, had a positive impact on in vivo bone engineering. Histological analysis using Masson-Goldner's trichrome staining (Fig. 24C(d)) revealed a substantial empty area in the newly formed osteoid region of the control group. In contrast, the groups with BC-based hydrogel implants displayed more newly formed bone adhering to the bone surface. Remarkably, the osteoid in the Au/BC-III group exhibited alignment and compactness, closely resembling healthy bone tissue. Similar trends were observed in H&E staining images, collectively highlighting Au/BC-III as the most effective in enhancing bone formation within the defect areas.400
Furthermore, in the pursuit of attaining optimal mechanical and biological characteristics in engineered cellulose-based hydrogels for bone tissue regeneration, it is common practice to integrate various components such as bioactive glasses and ceramics into cellulose inks, thereby crafting composite materials.44,531 Ceramics are inorganic, non-metallic materials characterized by their high melting points, hardness, and brittleness. In tissue engineering, ceramics like hydroxyapatite (HA) and tricalcium phosphate (TCP) are favored due to their similarity to the mineral component of bone, facilitating integration with native tissue. They provide a scaffold for cell attachment, proliferation, and differentiation, promoting bone regeneration.532,533 For instance, cellulose scaffolds incorporated with nano- or micro-hydroxyapatite (nHA or μHA) were created through cellulose regeneration and mechanical immobilization of inorganic particles, followed by freeze-drying. Micro-computed tomography revealed highly interconnected porous structures in both scaffolds, with mean pore diameters of 490 ± 94 μm for cellulose/nHA and 540 ± 132 μm for cellulose/μHA.533In vitro and in vivo studies were conducted, with osteoblastic cell cultures and a rabbit calvarial defect model, respectively. Results showed enhanced cell adhesion, metabolic activity, and osteoblastic gene expression in cellulose/nHA scaffolds compared to cellulose/μHA and the control. Furthermore, greater mineralized tissue formation was observed in defects loaded with cellulose/nHA, indicating promising potential for bone regeneration applications.533 In another study, Matinfar et al.534 fabricated porous scaffolds of chitosan (CS) and carboxymethyl cellulose (CMC), strengthened with whisker-like biphasic and triphasic calcium phosphate fibers via freeze drying. They evaluated the effects of CMC addition, fiber type, and content on mechanical, physicochemical, and biological properties.534 The synthesized fibers, containing hydroxyapatite (HA), monetite, β-tricalcium phosphate, and calcium pyrophosphate, were characterized. Composite scaffolds showed high porosity (61–75%) and interconnected pores (35–200 μm). CMC addition significantly improved mechanical properties (up to 150%) without affecting water uptake or biocompatibility. Both fiber types enhanced in vitro cell proliferation, attachment, and mineralization, with triphasic fibers showing superior reinforcement and cell viability. Scaffold reinforcement with 50 wt% triphasic fibers demonstrated superior mechanical and biological properties, suggesting their potential in bone tissue engineering, albeit requiring further mechanical enhancement.534
Bioactive glasses, on the other hand, possess a unique ability to bond with host tissue through the formation of a hydroxycarbonate apatite layer on their surface when in contact with bodily fluids. This bioactivity triggers cellular responses, such as enhanced osteogenic differentiation and angiogenesis, crucial for bone regeneration.535,536 In bone tissue engineering, various types of bioactive glasses are utilized, including silicate-based glasses, phosphate-based glasses, and borate-based glasses. Silicate-based glasses, such as 45S5 Bioglass, are among the most extensively studied and are known for their ability to promote osteogenesis and bond to bone tissue. Phosphate-based glasses offer enhanced bioactivity and degradation rates, while borate-based glasses exhibit excellent mechanical properties and bioactivity, making them suitable for load-bearing applications.535,537 In the study conducted by Li et al.538 3D bioactive glass/lignocellulose (BG/cellulose) composite scaffolds were successfully produced using 3D printing. The scaffold physical structure, morphology, mechanical properties, hydroxyapatite growth, and cell response were analyzed. Scanning electron microscopy revealed uniform macropores and rough surfaces in the BG/cellulose scaffolds. They demonstrated superior mechanical strength compared to pure cellulose scaffolds, meeting human trabecular bone strength requirements.538 BG enhanced hydroxyapatite formation, confirmed by mineralization in simulated body fluid (SBF). The scaffolds exhibited low cytotoxicity to human bone marrow mesenchymal stem cells (hBMSCs), with maximum cell viability observed at a cellulose to BG weight ratio of 2. These findings suggested promising applications for 3D-printed BG/cellulose scaffolds in bone tissue engineering.538 Additionally, surface functionalization of orthopedic implants was extensively explored to enhance bone-to-implant contact and expedite bone healing. In the study conducted by Chen et al.,167 a hybrid coating, comprising 45S5 bioactive glass (BG) individually enveloped and linked with fibrous cellulose nanocrystals was applied onto 316L stainless steel via one-step electrophoretic deposition.167 Zeta-potential and scanning electron microscopy clarified the codeposition mechanism. In vitro assessment in simulated body fluid showcased rapid mineralization of BG particles on the coating, forming a hydroxyapatite layer within 0.5 days. Comparative trials and characterization elucidated mineralization dynamics. Cell culture trials with MC3T3-E1 cells demonstrated enhanced cell behavior and matrix mineralization, affirming CNC role in augmenting BG bioactivity for improved bone implant coatings.167
4.4 Cardiac tissue engineering
Heart muscle damage, frequently resulting from myocardial infarctions, has profound implications for cardiac function. Nanocellulose hydrogels, when laden with cardiac-specific cells and frequently supplemented with growth factors, play a pivotal role in orchestrating the engineering of myocardial tissue. These hydrogels serve a dual purpose in this context. Firstly, they offer essential mechanical support to the damaged cardiac tissue, helping to structurally reinforce the compromised areas of the heart. Secondly, the hydrogels can create a microenvironment conducive to the survival and proliferation of cardiac cells, fostering the engineering process. By providing mechanical support and enhancing cell survival, nanocellulose-based hydrogels actively contribute to the recovery of heart function post-injury.511,539 This approach holds promise in addressing the challenges associated with myocardial damage, potentially offering innovative solutions for cardiac tissue engineering and functional recovery. In a specific study, an electroconductive cardiac patch was developed using bacterial cellulose (BC) infused with polypyrrole (Ppy) nanoparticles to mimic the natural myocardial environment. For assessing mechanical properties and cell experiments, BC materials prepared with Py concentrations of 1–10 mM were selected, denoted as BP1 (1 mM), BP3 (3 mM), BP5 (5 mM), and BP10 (10 mM).540 The properties of BC-Ppy composites were adjustable by varying precursor concentration. BC films changed from transparent white to opaque yellow-black with increasing monomer concentration. Scaffold thickness increased with Ppy functionalization, reaching ∼140 μm at 100 mM Py. Flexibility persisted up to 10 mM Py, but higher concentrations resulted in fragility (Fig. 25A).540 Comprehensive experiments involving cardiac fibroblasts and H9c2 cells highlighted the exceptional biocompatibility of BC-Ppy composites, enhancing cell viability and attachment, and fostering favorable cardiomyoblast morphology (Fig. 25B). Biochemical analyses unveiled diverse cardiomyocyte phenotypes and maturation levels in H9c2 cells, correlated with Ppy content. Furthermore, the BC-Ppy composites induced partial differentiation of H9c2 cells towards a cardiomyocyte-like phenotype.540 In addition, Ma et al.539 developed photocrosslinkable composite hydrogels mNCC-MeGel (mNG) by integrating TEMPO-modified nanocrystalline cellulose (mNCC) into methacrylated gelatin (MeGel) backbone. Encapsulated human adipose-derived mesenchymal stem cells (HADMSC) exhibited valve interstitial cell phenotypes over 14 days in both normal and osteogenic media.539 Compared to MeGel, mNG reduced αSMA expression and increased vimentin and aggrecan expression, supporting a quiescent fibroblastic phenotype. Under osteogenic conditions, mNG lowered expression of osteogenic genes, resisting calcification. Bioprinted tall tubular structures with sustained cell viability demonstrated mNG potential for tissue-engineered heart valves.539 In another study, Hou et al.541 synthesized crosslinked TEMPO-oxidized cellulose nanofibers (TOCN), polymerized with pyrrole monomer, yielding polypyrrole nanocellulose composite hydrogel scaffolds for myocardial tissue engineering (Fig. 25C). The hydrogel displayed mechanical properties (50–75 kPa) and electrical conductivity (5 × 10−5–1 × 10−3 S cm−1) akin to natural myocardial tissue, with high biocompatibility (relative growth rate >90%) and non-cytotoxicity.541 Cells cultured on TOCN-PPy scaffold exhibited increased expression of myocardial-specific proteins like connexin 43 and cardiac troponin T, suggesting its potential in myocardial tissue engineering (Fig. 25D).541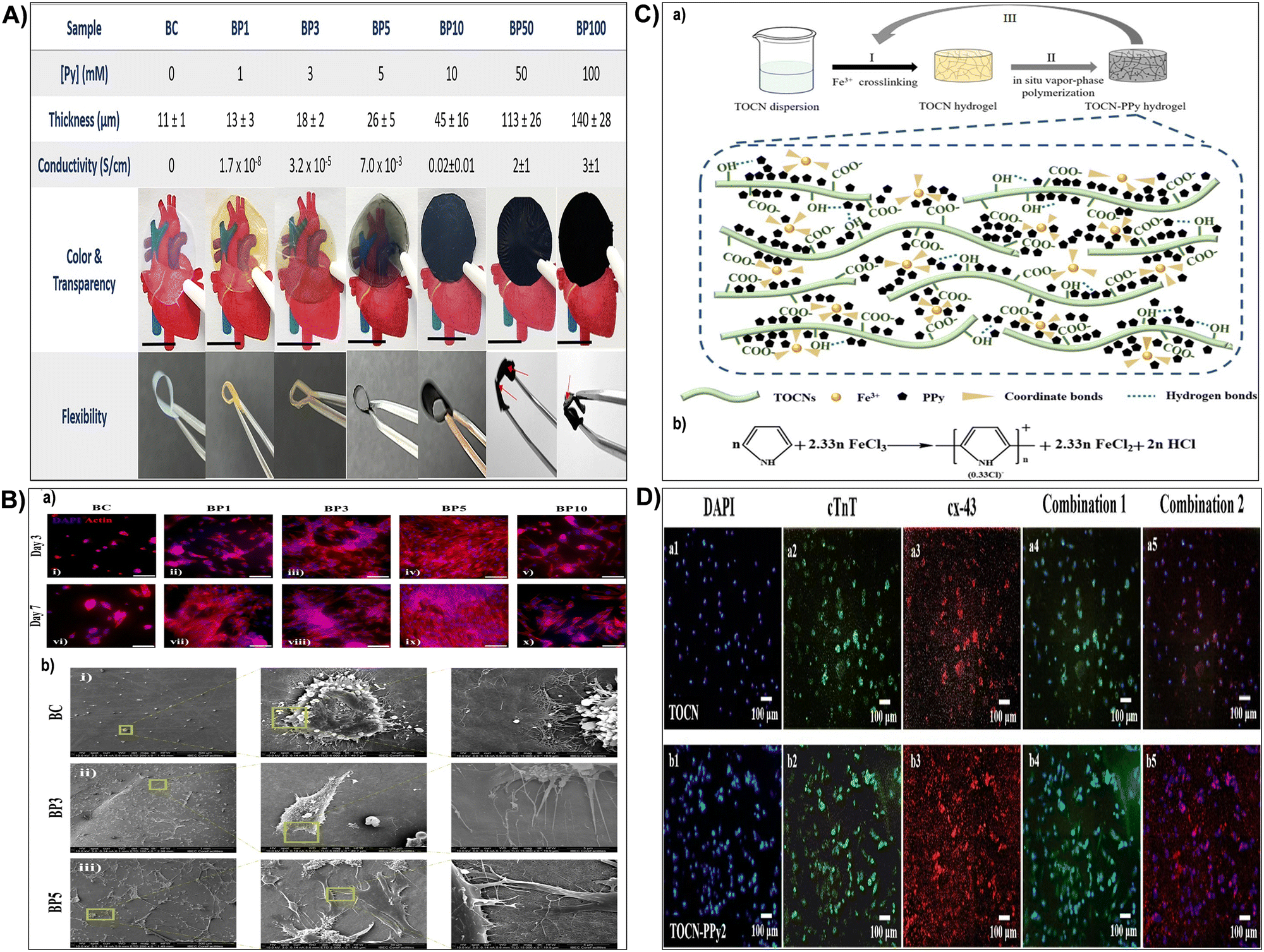 | ||
| Fig. 25 (A) Impact of Py concentration on scaffold properties. Bacterial nanocellulose (BC) and bacterial nanocellulose-polypyrrole nanocomposites (BC-Ppy) at varying pyrrole (Py) concentrations and their property alterations. Scale bars: 1 cm. Reproduced with permission from ref. 540 Copyright © 2023 American Chemical Society. (B) H9c2 cell attachment to BC and BC-Ppy scaffolds. (a) Fluorescence microscopy images showing H9c2 cell populations on nanocellulose composites after 3 and 7 days of culture. Scale bars: 100 μm. (b) SEM images illustrating H9c2 attachment and morphology after 7 days of culture on BC, BP3, and BP5 materials. Reproduced with permission from ref. 540 Copyright © 2023 American Chemical Society. (C) Preparation process of nano-cellulose/polypyrrole conductive hydrogel. (a) Sequential cross-linking of TOCN dispersion with Fe3+ ions, followed by vapor-phase pyrrole monomer polymerization to form TOCN-PPy1 composite hydrogel, repeated to generate TOCN-PPy2 and TOCN-PPy3 composite hydrogels. (b) Schematic representation of pyrrole polymerization with FeCl3. Reproduced with permission from ref. 541 Copyright © 2024 Springer. (D) Immunofluorescent staining of cells on pure TOCN and TOCN-PPy2 hydrogels. DAPI (a1) and (b1), cTnT protein (a2) and (b2), cx-43 protein (a3) and (b3), (a4) combination diagram of a1 and a2, (a5) combination diagram of a1 and a3, (b4) combination diagram of b1 and b2, (b5) combination diagram of b1 and b3. Reproduced with permission from ref. 541 Copyright © 2024 Springer. | ||
4.5 Nerve tissue engineering
In the context of nerve injuries or neurological disorders, which can lead to sensory or motor impairments, nanocellulose hydrogels can serve as promising scaffolds to repair damaged nerve tissues.388,516,542 These hydrogels serve a dual purpose, offering both physical and biochemical cues that foster an environment conducive to guided nerve engineering. They facilitate controlled delivery of neurotrophic factors, supporting and enhancing axonal growth while establishing a supportive matrix for functional reinnervation.543,544 This multifaceted approach facilitates nerve tissue repair, contributing to improved functional recovery. Additionally, the neurotrophic factors delivered by nanocellulose hydrogels are crucial signaling molecules that promote the survival, growth, and differentiation of neurons.516,542 Recently, Chang et al.544 sought to improve in vitro and in vivo survival and neuronal differentiation of otic neuronal progenitors (ONPs) by creating a stem cell niche. They used 3D human embryonic stem cells (hESC)-derived ONP spheroids with a nanofibrillar cellulose hydrogel and sustained-release neurotrophic factor delivery.544 Results showed transplanted hESC-derived ONP spheroids survived, differentiated into otic neuronal lineages, and extended neurites towards the cochlea bony wall after 90 days, without immunosuppressants. This robust protocol established viable in vivo transplantation of hESC-derived ONPs to the inner ear, potentially enabling further integration and functional auditory neuron generation.544 In addition, as illustrated by several recent research studies presented in Table 4 focused on the use of nanocellulose-based hydrogels for neural tissue engineering, nanocellulose-based hydrogels have emerged as promising matrices for neural tissue engineering due to their remarkable biocompatibility and tunable properties.388,514 Nanocellulose, derived from renewable sources, possesses inherent biocompatibility and interesting mechanical properties, making it suitable for a wide range of biomedical applications. Its exceptional mechanical properties provide hydrogels with robust performances, offering physical cues that influence and guide the growth of cells in hydrogels during regenration process of neural tissues.514,515 Moreover, the nanostructured morphology of nanocellulose provides a significantly high surface area for cell adhesion and proliferation, which is crucial for supporting neural cell growth and differentiation.516,542 One significant advancement in this field lies in the ability to functionalize nanocellulose hydrogels with electroconductive polymers, enabling the development of electroconductive nanocellulose hydrogels. By incorporating electroconductive polymers like polypyrrole53,515 or graphene514 into nanocellulose matrices, these hydrogels gain the capacity to facilitate electrical signal propagation, effectively mimicking the natural electroconductive environment of neural tissues. This electroconductivity is vital for neural tissue engineering, as it allows neural cells to communicate effectively and propagate electrical signals necessary for proper tissue function.53,514,5154.6 Vascular tissue engineering
The vascular system, comprising arteries, veins, and capillaries, is indispensable for delivering oxygen and nutrients to tissues throughout the body.545–547 Disruptions in vascular function, such as those caused by atherosclerosis, can lead to critical conditions like heart attacks and strokes due to impaired blood flow.548 In addressing these challenges, the use of nanocellulose hydrogels in vascular tissue engineering offers a promising solution. These hydrogels are engineered to mimic the structural and mechanical properties of native blood vessels, making them ideal candidates for developing vascular grafts.517,518 The utilization of nanocellulose hydrogels for blood vessel fabrication presents a compelling avenue for addressing critical challenges in vascular tissue regeneration. Each approach outlined in the reviewed research studies highlighted in Table 4 offers unique contributions and insights into the potential applications of nanocellulose-based materials in this context. Firstly, the functional bacterial cellulose hydrogel incorporating a silver-loaded antibacterial agent demonstrated exceptional mechanical strength exceeding 1 MPa, alongside potent antibacterial efficacy against common pathogens such as E. coli and S. aureus.517 Moreover, cell culture experiments underscored the remarkable biocompatibility and angiogenic potential of this dressing, while in vivo studies on rats showcased its efficacy in promoting wound healing and skin re-epithelialization, highlighting its clinical relevance.517 Similarly, the multifunctional hydrogel incorporating silver into catechol-modified chitosan and aldehyde-modified cellulose nanocrystals exhibited enhanced vascularization, controlled silver release, and seamless coverage of irregular wounds, highlighting its versatility and efficacy in promoting neovascularization and tissue engineering.518 Lastly, the biomimetic approach employed in fabricating cellulose-based small-diameter artificial blood vessels showcased impressive mechanical performance comparable to native vessels, coupled with excellent biocompatibility and safety profiles validated through comprehensive in vitro and in vivo experiments.519 Overall, these findings collectively underscore the significant potential of nanocellulose hydrogels in advancing vascular tissue engineering by offering robust mechanical properties, antibacterial efficacy, and enhanced tissue regeneration capabilities, thereby paving the way for innovative therapeutic interventions in tissue repair.5 Conclusion, challenges and future propects
Biomimetic nanocellulose-based hydrogels play a pivotal role in tissue engineering. These advanced materials are designed to mimic the functions and architecture of native tissues and are characterized by their exceptional biocompatibility, robust mechanical strength, and customizable biofunctionalities. The unique attributes of cellulose-based hydrogels render them exceptionally well-suited for a broad spectrum of tissue engineering applications, offering promising solutions for the field of regenerative medicine.However, nanocellulose-based hydrogels, while gaining significant attention for their biocompatibility, biodegradability, and tunable mechanical strength, face several challenges that hinder their widespread biomedical applications. Achieving optimal mechanical strength, which matches that of native tissues, remains elusive due to variations in fabrication methods, crosslinking techniques and processing conditions, which significantly impact their structural integrity and mechanical properties. Moreover, ensuring biological compatibility and biofunctionality is critical. Although nanocellulose possesses inherent biocompatibility, the inclusion of modifications and additives during the fabrication process of nanocellulose-based hydrogels can disrupt its interactions with cells and tissues. This disruption can have significant effects on cell viability, proliferation, and differentiation in the hydrogel matrix, which are essential factors for successful tissue regeneration. Additionally, nanocellulose-based hydrogels should degrade at a rate compatible with tissue regeneration while maintaining structural integrity during the healing process. Balancing biodegradation and stability is challenging and requires precise control over hydrogel composition, crosslinking density, and degradation kinetics. Rapid degradation may lead to premature loss of mechanical support, while slow degradation can impede tissue remodeling and integration. On the practical side, scaling up the production of nanocellulose-based hydrogels to meet clinical demands in a cost-effective manner involves complex processing techniques and significant investment in infrastructure and equipment, with the cost of raw materials and purification processes adding to the overall expense. The immunogenicity and potential inflammatory responses, especially in vivo, also need careful assessment as variations in nanocellulose sources, purification methods, and surface modifications can influence immunological properties of resulting hydrogels. Addressing potential immune reactions is crucial to ensure the long-term safety and efficacy of these hydrogels.
Despite the challenges faced, ongoing research endeavors aim to surmount existing limitations and propel the utilization of nanocellulose-based hydrogels for tissue engineering forward. This includes the development of multifunctional hydrogels tailored to possess enhanced mechanical strength, controlled biodegradation, and bioactive functionalities. Integration of bioactive molecules and therapeutic agents into the hydrogel matrix can stimulate specific cellular responses, fostering tissue regeneration. Additionally, exploration of advanced fabrication techniques such as 3D bioprinting and electrospinning offers precise control over hydrogel architecture and properties, enabling customizable scaffold designs for complex tissue regeneration. Looking ahead, the utilization of nanocellulose-based hydrogels in organ-on-chips presents promising prospects for biomedical applications. Organ-on-chip technology seeks to replicate human organ structure and function in microfluidic devices, facilitating physiologically relevant models for drug screening and disease modeling. Nanocellulose hydrogels offer advantages including biocompatibility, tunable mechanical properties, and support for cell growth and differentiation, enhancing biomimicry of the microenvironment in organ-on-chip systems. Future research directions may involve optimizing nanocellulose-based hydrogel formulations for specific organ models and integrating advanced fabrication techniques to create complex organ systems. Efforts to enhance hydrogel functionality through biofunctionalization and biochemical cue incorporation could further enhance their relevance for organ-on-chip applications, ultimately advancing biomedical research and improving human health outcomes.
Author contributions
Arnaud Kamdem Tamo: conceptualization, methodology, investigation, data curation, supervision, funding acquisition, writing – original draft, writing – review & editing.Data availability
No data was used for the research described in the article.Conflicts of interest
The author declares there is no conflict of interest. The author declares that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.Acknowledgements
The German Academic Exchange Service (DAAD) is acknowledged for financial support.References
- M. M. Farag, J. Mater. Sci., 2023, 58, 527–558 CrossRef CAS.
- F. Han, J. Wang, L. Ding, Y. Hu, W. Li, Z. Yuan, Q. Guo, C. Zhu, L. Yu, H. Wang, Z. Zhao, L. Jia, J. Li, Y. Yu, W. Zhang, G. Chu, S. Chen and B. Li, Front. Bioeng. Biotechnol., 2020, 8, 83 CrossRef PubMed.
- S. Verma, V. Khanna, S. Kumar and S. Kumar, ACS Omega, 2023, 8, 47322–47339 CrossRef CAS PubMed.
- J. M. Colazo, B. C. Evans, A. F. Farinas, S. Al-Kassis, C. L. Duvall and W. P. Thayer, Tissue Eng., Part B, 2019, 25, 259–290 CrossRef PubMed.
- C. Mandrycky, K. Phong and Y. Zheng, MRS Commun., 2017, 7, 332–347 CrossRef CAS PubMed.
- D. Shopova, A. Yaneva, D. Bakova, A. Mihaylova, P. Kasnakova, M. Hristozova, Y. Sbirkov, V. Sarafian and M. Semerdzhieva, Bioengineering, 2023, 10, 287 CrossRef PubMed.
- I. Doench, M. Torres-Ramos, A. Montembault, P. Nunes De Oliveira, C. Halimi, E. Viguier, L. Heux, R. Siadous, R. Thiré and A. Osorio-Madrazo, Polymers, 2018, 10, 1202 CrossRef PubMed.
- A. Kamdem Tamo, I. Doench, L. Walter, A. Montembault, G. Sudre, L. David, A. Morales-Helguera, M. Selig, B. Rolauffs, A. Bernstein, D. Hoenders, A. Walther and A. Osorio-Madrazo, Polymers, 2021, 13, 1663 CrossRef PubMed.
- M. Brovold, J. I. Almeida, I. Pla-Palacín, P. Sainz-Arnal, N. Sánchez-Romero, J. J. Rivas, H. Almeida, P. R. Dachary, T. Serrano-Aulló, S. Soker and P. M. Baptista, in Novel Biomaterials for Regenerative Medicine, ed. H. J. Chun, K. Park, C.-H. Kim and G. Khang, Springer Singapore, Singapore, 2018, vol. 1077, pp. 421–449 Search PubMed.
- A. K. Tamo, T. A. Tran, I. Doench, S. Jahangir, A. Lall, L. David, C. Peniche-Covas, A. Walther and A. Osorio-Madrazo, Materials, 2022, 15, 6039 CrossRef CAS PubMed.
- A. Sood, A. Gupta and G. Agrawal, Carbohydr. Polym. Technol. Appl., 2021, 2, 100067 CAS.
- N. Desai, D. Rana, S. Salave, R. Gupta, P. Patel, B. Karunakaran, A. Sharma, J. Giri, D. Benival and N. Kommineni, Pharmaceutics, 2023, 15, 1313 CrossRef CAS PubMed.
- S. Das, B. Ghosh and K. Sarkar, Sens. Int., 2022, 3, 100135 CrossRef.
- A. E. Eldeeb, S. Salah and N. A. Elkasabgy, AAPS PharmSciTech, 2022, 23, 267 CrossRef PubMed.
- D. S. Kohane and R. Langer, Pediatr. Res., 2008, 63, 487–491 CrossRef CAS PubMed.
- P. P. Phatchayawat, A. Khamkeaw, S. Yodmuang and M. Phisalaphong, Biochem. Eng. J., 2022, 184, 108476 CrossRef CAS.
- P.-H. Chen, H.-C. Liao, S.-H. Hsu, R.-S. Chen, M.-C. Wu, Y.-F. Yang, C.-C. Wu, M.-H. Chen and W.-F. Su, RSC Adv., 2015, 5, 6932–6939 RSC.
- D. M. Alshangiti, T. K. El-damhougy, A. Zaher, M. Madani and M. Mohamady Ghobashy, RSC Adv., 2023, 13, 35251–35291 RSC.
- R. Li, Y. Zhao, Z. Zheng, Y. Liu, S. Song, L. Song, J. Ren, J. Dong and P. Wang, RSC Adv., 2023, 13, 7153–7167 RSC.
- C. Chen, Y. Xi and Y. Weng, Polymers, 2022, 14, 3335 CrossRef CAS PubMed.
- R. Curvello, V. S. Raghuwanshi and G. Garnier, Adv. Colloid Interface Sci., 2019, 267, 47–61 CrossRef CAS PubMed.
- H. P. S. A. Khalil, F. Jummaat, E. B. Yahya, N. G. Olaiya, A. S. Adnan, M. Abdat, N. A. M. Nasir, A. S. Halim, U. S. U. Kumar, R. Bairwan and A. B. Suriani, Polymers, 2020, 12, 2043 CrossRef PubMed.
- F. V. Ferreira, C. G. Otoni, K. J. De France, H. S. Barud, L. M. F. Lona, E. D. Cranston and O. J. Rojas, Mater. Today, 2020, 37, 126–141 CrossRef CAS.
- I. Doench, T. Tran, L. David, A. Montembault, E. Viguier, C. Gorzelanny, G. Sudre, T. Cachon, M. Louback-Mohamed, N. Horbelt, C. Peniche-Covas and A. Osorio-Madrazo, Biomimetics, 2019, 4, 19 CrossRef CAS PubMed.
- S. Marquez-Bravo, I. Doench, P. Molina, F. E. Bentley, A. K. Tamo, R. Passieux, F. Lossada, L. David and A. Osorio-Madrazo, Polymers, 2021, 13, 1563 CrossRef CAS PubMed.
- S. Tortorella, V. Vetri Buratti, M. Maturi, L. Sambri, M. Comes Franchini and E. Locatelli, IJN, 2020, 15, 9909–9937 CrossRef CAS PubMed.
- F. Rol, M. N. Belgacem, A. Gandini and J. Bras, Prog. Polym. Sci., 2019, 88, 241–264 CrossRef CAS.
- Z. Wei, C. Wu, R. Li, D. Yu and Q. Ding, Cellulose, 2021, 28, 7497–7520 CrossRef CAS.
- M. Monfared, D. Mawad, J. Rnjak-Kovacina and M. H. Stenzel, J. Mater. Chem. B, 2021, 9, 6163–6175 RSC.
- D. K. Patel, S. D. Dutta, W.-C. Shin, K. Ganguly and K.-T. Lim, RSC Adv., 2021, 11, 7466–7478 RSC.
- A. Kantaros and T. Ganetsos, IJMS, 2023, 24, 15748 CrossRef CAS PubMed.
- E. Merotto, P. G. Pavan and M. Piccoli, Biomedicines, 2023, 11, 1742 CrossRef CAS PubMed.
- A. Lall, A. Kamdem Tamo, I. Doench, L. David, P. Nunes De Oliveira, C. Gorzelanny and A. Osorio-Madrazo, IJMS, 2020, 21, 5602 CrossRef CAS PubMed.
- L. Lin, S. Jiang, J. Yang, J. Qiu, X. Jiao, X. Yue, X. Ke, G. Yang and L. Zhang, IJB, 2022, 9, 637 CrossRef PubMed.
- A. Osorio-Madrazo, M. Eder, M. Rueggeberg, J. K. Pandey, M. J. Harrington, Y. Nishiyama, J.-L. Putaux, C. Rochas and I. Burgert, Biomacromolecules, 2012, 13, 850–856 CrossRef CAS PubMed.
- X. He and Q. Lu, Carbohydr. Polym., 2023, 301, 120351 CrossRef CAS PubMed.
- A. Kamdem Tamo, I. Doench, A. Morales Helguera, D. Hoenders, A. Walther and A. O. Madrazo, Polymers, 2020, 12, 1522 CrossRef PubMed.
- W. Chimpibul, T. Nakaji-Hirabayashi, X. Yuan and K. Matsumura, J. Mater. Chem. B, 2020, 8, 7904–7913 RSC.
- T. Agarwal, S. N. G. H. Narayana, K. Pal, K. Pramanik, S. Giri and I. Banerjee, Int. J. Biol. Macromol., 2015, 75, 409–417 CrossRef CAS PubMed.
- M. G. Bekaroğlu, Y. İşçi and S. İşçi, Mater. Sci. Eng., C, 2017, 78, 847–853 CrossRef PubMed.
- L. Ning, C. You, Y. Zhang, X. Li and F. Wang, Composites Commun., 2021, 26, 100739 CrossRef.
- M. Y. Leong, Y. L. Kong, M. Y. Harun, C. Y. Looi and W. F. Wong, Carbohydr. Res., 2023, 532, 108899 CrossRef CAS PubMed.
- V. Durairaj, P. Li, T. Liljeström, N. Wester, J. Etula, I. Leppänen, Y. Ge, K. S. Kontturi, T. Tammelin, T. Laurila and J. Koskinen, ACS Appl. Nano Mater., 2021, 4, 5842–5853 CrossRef CAS.
- F. V. Ferreira, L. P. Souza, T. M. M. Martins, J. H. Lopes, B. D. Mattos, M. Mariano, I. F. Pinheiro, T. M. Valverde, S. Livi, J. A. Camilli, A. M. Goes, R. F. Gouveia, L. M. F. Lona and O. J. Rojas, Nanoscale, 2019, 11, 19842–19849 RSC.
- M. Nagalakshmaiah, M. Rajinipriya, S. Afrin, M. A. Ansari, M. Asad and Z. Karim, in Bio-based Polymers and Nanocomposites, ed. M. L. Sanyang and M. Jawaid, Springer International Publishing, Cham, 2019, pp. 49–65 Search PubMed.
- S. Swingler, A. Gupta, H. Gibson, M. Kowalczuk, W. Heaselgrave and I. Radecka, Polymers, 2021, 13, 412 CrossRef CAS PubMed.
- S. Yu, J. Sun, Y. Shi, Q. Wang, J. Wu and J. Liu, Environ. Sci. Ecotechnol., 2021, 5, 100077 CrossRef CAS PubMed.
- M. N. Norizan, S. S. Shazleen, A. H. Alias, F. A. Sabaruddin, M. R. M. Asyraf, E. S. Zainudin, N. Abdullah, M. S. Samsudin, S. H. Kamarudin and M. N. F. Norrrahim, Nanomaterials, 2022, 12, 3483 CrossRef CAS PubMed.
- T. V. Patil, D. K. Patel, S. D. Dutta, K. Ganguly, T. S. Santra and K.-T. Lim, Bioactive Mater., 2022, 9, 566–589 CrossRef CAS PubMed.
- W. Yan, J. Liu, X. Zheng, J. Zhang and K. Tang, e-Polymers, 2023, 23, 20230010 CrossRef CAS.
- D. K. Patel, S. D. Dutta, J. Hexiu, K. Ganguly and K.-T. Lim, Int. J. Biol. Macromol., 2020, 162, 1429–1441 CrossRef CAS PubMed.
- X. He, Q. Xiao, C. Lu, Y. Wang, X. Zhang, J. Zhao, W. Zhang, X. Zhang and Y. Deng, Biomacromolecules, 2014, 15, 618–627 CrossRef CAS PubMed.
- J. Thunberg, T. Kalogeropoulos, V. Kuzmenko, D. Hägg, S. Johannesson, G. Westman and P. Gatenholm, Cellulose, 2015, 22, 1459–1467 CrossRef CAS.
- K. Liu and J. M. Catchmark, Carbohydr. Polym., 2019, 219, 12–20 CrossRef CAS PubMed.
- A. Ghilan, R. Nicu, D. E. Ciolacu and F. Ciolacu, Materials, 2023, 16, 4447 CrossRef CAS PubMed.
- M. Moniri, A. Boroumand Moghaddam, S. Azizi, R. Abdul Rahim, A. Bin Ariff, W. Zuhainis Saad, M. Navaderi and R. Mohamad, Nanomaterials, 2017, 7, 257 CrossRef PubMed.
- P. Tayeb and A. H. Tayeb, Carbohydr. Polym., 2019, 224, 115149 CrossRef CAS PubMed.
- S. Brethauer, R. L. Shahab and M. H. Studer, Appl. Microbiol. Biotechnol., 2020, 104, 5201–5212 CrossRef CAS PubMed.
- A. Dal Fovo, J. Striova, D. Quintero Balbas, S. Mattana, N. Tacconi, R. Cicchi and R. Fontana, RSC Adv., 2022, 12, 26744–26752 RSC.
- J. Shojaeiarani, D. S. Bajwa and S. Chanda, Composites, Part C, 2021, 5, 100164 CAS.
- S. L. Leong, S. I. X. Tiong, S. P. Siva, F. Ahamed, C.-H. Chan, C. L. Lee, I. M. L. Chew and Y. K. Ho, J. Environ. Chem. Eng., 2022, 10, 108145 CrossRef CAS.
- P. V. Krasteva, J. Bernal-Bayard, L. Travier, F. A. Martin, P.-A. Kaminski, G. Karimova, R. Fronzes and J.-M. Ghigo, Nat. Commun., 2017, 8, 2065 CrossRef PubMed.
- W. D. Jang, T. Y. Kim, H. U. Kim, W. Y. Shim, J. Y. Ryu, J. H. Park and S. Y. Lee, Biotechnol. Bioeng., 2019, 116, 3372–3381 CrossRef CAS PubMed.
- L. J. Gibson, J. R. Soc., Interface, 2012, 9, 2749–2766 CrossRef CAS PubMed.
- L. Solhi, V. Guccini, K. Heise, I. Solala, E. Niinivaara, W. Xu, K. Mihhels, M. Kröger, Z. Meng, J. Wohlert, H. Tao, E. D. Cranston and E. Kontturi, Chem. Rev., 2023, 123, 1925–2015 CrossRef CAS PubMed.
- Y. Qi, Y. Guo, A. A. Liza, G. Yang, M. H. Sipponen, J. Guo and H. Li, Cellulose, 2023, 30, 4115–4147 CrossRef CAS.
- R. Tarrahi, A. Khataee, A. Karimi and Y. Yoon, Chemosphere, 2022, 288, 132529 CrossRef CAS PubMed.
- C. T. Brett, International Review of Cytology, Elsevier, 2000, vol. 199, pp. 161–199 Search PubMed.
- T. I. Baskin, Protoplasma, 2001, 215, 150–171 CrossRef CAS PubMed.
- S. Iwamoto, W. Kai, A. Isogai and T. Iwata, Biomacromolecules, 2009, 10, 2571–2576 CrossRef CAS PubMed.
- Y. Nishiyama, J. Wood Sci., 2009, 55, 241–249 CrossRef CAS.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- J. L. Fredricks, A. M. Jimenez, P. Grandgeorge, R. Meidl, E. Law, J. Fan and E. Roumeli, J. Polym. Sci., 2023, 61, 2585–2632 CrossRef CAS.
- S. Amine, A. Montembault, M. Fumagalli, A. Osorio-Madrazo and L. David, Polymers, 2021, 13, 2023 CrossRef CAS PubMed.
- P. Samyn and A. Osorio-Madrazo, in Handbook of Nanofibers, ed. A. Barhoum, M. Bechelany and A. S. H. Makhlouf, Springer International Publishing, Cham, 2019, pp. 287–321 Search PubMed.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- L. I. Mikhalovska, V. M. Gun’ko, A. A. Rugal, O. I. Oranska, Y. I. Gornikov, C. Morvan, N. Follain, C. Domas, E. M. Pakhlov and S. V. Mikhalovsky, RSC Adv., 2012, 2, 2032 RSC.
- N. Zhang, S. Li, L. Xiong, Y. Hong and Y. Chen, Modelling Simul. Mater. Sci. Eng, 2015, 23, 085010 CrossRef.
- U. P. Agarwal, Molecules, 2019, 24, 1659 CrossRef PubMed.
- P. Langan, N. Sukumar, Y. Nishiyama and H. Chanzy, Cellulose, 2005, 12, 551–562 CrossRef CAS.
- Y. Zhang, W. Deng, M. Wu, M. Rahmaninia, C. Xu and B. Li, Nanomaterials, 2023, 13, 1489 CrossRef CAS PubMed.
- A. Dufresne, J.-Y. Cavaill and M. R. Vignon, J. Appl. Polym. Sci., 1997, 64, 1185–1194 CrossRef CAS.
- D. Abol-Fotouh, M. A. Hassan, H. Shokry, A. Roig, M. S. Azab and A. E.-H. B. Kashyout, Sci. Rep., 2020, 10, 3491 CrossRef CAS PubMed.
- H. P. S. Abdul Khalil, A. S. Adnan, E. B. Yahya, N. G. Olaiya, S. Safrida, Md. S. Hossain, V. Balakrishnan, D. A. Gopakumar, C. K. Abdullah, A. A. Oyekanmi and D. Pasquini, Polymers, 2020, 12, 1759 CrossRef PubMed.
- M. Zhao, L. Robertsén, L. Wågberg and T. Pettersson, Cellulose, 2022, 29, 2617–2632 CrossRef CAS.
- M. Jakob, A. R. Mahendran, W. Gindl-Altmutter, P. Bliem, J. Konnerth, U. Müller and S. Veigel, Prog. Mater. Sci., 2022, 125, 100916 CrossRef CAS.
- D. Trache, A. F. Tarchoun, M. Derradji, T. S. Hamidon, N. Masruchin, N. Brosse and M. H. Hussin, Front. Chem., 2020, 8, 392 CrossRef CAS PubMed.
- J. George and S. N. Sabapathi, Nanotechnol., Sci. Appl., 2015, 2015(8), 45–54 CrossRef PubMed.
- P. Mali and A. P. Sherje, Carbohydr. Polym., 2022, 275, 118668 CrossRef CAS PubMed.
- O. M. Vanderfleet, M. S. Reid, J. Bras, L. Heux, J. Godoy-Vargas, M. K. R. Panga and E. D. Cranston, Cellulose, 2019, 26, 507–528 CrossRef CAS.
- P. G. Gan, S. T. Sam, M. F. B. Abdullah and M. F. Omar, J. Appl. Polym. Sci., 2020, 137, 48544 CrossRef CAS.
- R. Nasseri, C. P. Deutschman, L. Han, M. A. Pope and K. C. Tam, Mater. Today Adv., 2020, 5, 100055 CrossRef.
- P. Liu, X. Guo, F. Nan, Y. Duan and J. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 3085–3092 CrossRef CAS PubMed.
- F. D’Acierno, W. Y. Hamad, C. A. Michal and M. J. MacLachlan, Biomacromolecules, 2020, 21, 3374–3386 CrossRef PubMed.
- J. Mao, A. Osorio-Madrazo and M.-P. Laborie, Cellulose, 2013, 20, 1829–1840 CrossRef CAS.
- H.-M. Ng, L. T. Sin, T.-T. Tee, S.-T. Bee, D. Hui, C.-Y. Low and A. R. Rahmat, Composites, Part B, 2015, 75, 176–200 CrossRef CAS.
- S. Soleimani, A. Heydari and M. Fattahi, Starch Stärke, 2022, 74, 2200159 CrossRef CAS.
- T. Yang, X. Li, N. Xu, Y. Guo, G. Liu and J. Zhao, Bioresour. Bioprocess., 2023, 10, 42 CrossRef PubMed.
- C. M. Pacheco, C. Bustos A and G. Reyes, J. Dispersion Sci. Technol., 2020, 41, 1731–1741 CrossRef CAS.
- X. Hu, M. Song, S. Li, Y. Chu, W. Zhang and Z. Deng, Chemosphere, 2023, 343, 140212 CrossRef CAS PubMed.
- Y. Zhou, T. Saito, L. Bergström and A. Isogai, Biomacromolecules, 2018, 19, 633–639 CrossRef CAS PubMed.
- Y. Xu, Y. Xu, W. Deng, H. Chen and J. Xiong, Int. J. Biol. Macromol., 2023, 246, 125604 CrossRef CAS PubMed.
- L. Douard, J. Bras, T. Encinas and M. N. Belgacem, Carbohydr. Polym., 2021, 252, 117136 CrossRef CAS PubMed.
- S. Zhu, H. Sun, T. Mu, Q. Li and A. Richel, Food Chem., 2023, 403, 134496 CrossRef CAS PubMed.
- Z. Pang, P. Wang and C. Dong, Cellulose, 2018, 25, 7053–7064 CrossRef CAS.
- D. Z. Haddis, M. Chae, J. Asomaning and D. C. Bressler, Carbohydr. Polym., 2024, 323, 121460 CrossRef CAS PubMed.
- N. Carter, I. Grant, M. Dewey, M. Bourque and D. J. Neivandt, Front. Nanotechnol., 2021, 3, 729743 CrossRef.
- P. Xi, F. Quan, Y. Sun and Y. Jiang, Composites, Part B, 2022, 242, 110078 CrossRef CAS.
- M. Mohammadalipour, S. Karbasi, T. Behzad, Z. Mohammadalipour and M. Zamani, Int. J. Biol. Macromol., 2022, 220, 1402–1414 CrossRef CAS PubMed.
- S. Widiarto, E. Pramono, Suharso, A. Rochliadi and I. M. Arcana, Fibers, 2019, 7, 44 CrossRef CAS.
- C. Carneiro Pessan, J. Silva Bernardes, S. H. P. Bettini and E. R. Leite, Carbohydr. Polym., 2023, 304, 120505 CrossRef CAS PubMed.
- C. Yao, F. Li, T. Chen and Y. Tang, Ind. Crops Prod., 2023, 206, 117575 CrossRef CAS.
- P. Nehra and R. P. Chauhan, Iran. Polym. J., 2022, 31, 771–778 CrossRef CAS.
- X. Liu, Y. Jiang, C. Qin, S. Yang, X. Song, S. Wang and K. Li, Cellulose, 2018, 25, 7065–7078 CrossRef CAS.
- B. S. Santucci, J. Bras, M. N. Belgacem, A. A. D. S. Curvelo and M. T. B. Pimenta, Ind. Crops Prod., 2016, 91, 238–248 CrossRef CAS.
- Y. Chen, Y. He, D. Fan, Y. Han, G. Li and S. Wang, J. Nanomater., 2017, 2017, 1–12 Search PubMed.
- M. Gama, P. Gatenholm and D. Klemm, Bacterial NanoCellulose: A Sophisticated Multifunctional Material, CRC Press, Boca Raton, 1st edn, 2016 Search PubMed.
- C. Sharma and N. K. Bhardwaj, Mater. Sci. Eng., C, 2019, 104, 109963 CrossRef CAS PubMed.
- P. Lv, Y. Yao, D. Li, H. Zhou, M. A. Naeem, Q. Feng, J. Huang, Y. Cai and Q. Wei, Carbohydr. Polym., 2017, 172, 93–101 CrossRef CAS PubMed.
- D. Lahiri, M. Nag, B. Dutta, A. Dey, T. Sarkar, S. Pati, H. A. Edinur, Z. Abdul Kari, N. H. Mohd Noor and R. R. Ray, IJMS, 2021, 22, 12984 CrossRef CAS PubMed.
- C. Zhong, Front. Bioeng. Biotechnol., 2020, 8, 605374 CrossRef PubMed.
- K. Qiu and A. N. Netravali, Polym. Rev., 2014, 54, 598–626 Search PubMed.
- D. Klemm, B. Heublein, H. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- D. Klemm, D. Schumann, F. Kramer, N. Heßler, M. Hornung, H.-P. Schmauder and S. Marsch, in Polysaccharides II, ed. D. Klemm, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, vol. 205, pp. 49–96 Search PubMed.
- D. Klemm, D. Schumann, F. Kramer, N. Heßler, D. Koth and B. Sultanova, Macromol. Symp., 2009, 280, 60–71 CrossRef CAS.
- M. Ioelovich, Fabrication and Self-Assembly of Nanobiomaterials, Elsevier, 2016, pp. 243–288 Search PubMed.
- H. Luo, G. Xiong, D. Hu, K. Ren, F. Yao, Y. Zhu, C. Gao and Y. Wan, Mater. Chem. Phys., 2013, 143, 373–379 CrossRef CAS.
- N. Halib, I. Ahmad, M. Grassi and G. Grassi, Int. J. Pharm., 2019, 566, 631–640 CrossRef CAS PubMed.
- G. F. Picheth, C. L. Pirich, M. R. Sierakowski, M. A. Woehl, C. N. Sakakibara, C. F. De Souza, A. A. Martin, R. Da Silva and R. A. De Freitas, Int. J. Biol. Macromol., 2017, 104, 97–106 Search PubMed.
- A. Retegi, N. Gabilondo, C. Peña, R. Zuluaga, C. Castro, P. Gañan, K. De La Caba and I. Mondragon, Cellulose, 2010, 17, 661–669 CrossRef CAS.
- H. Suryanto, M. Muhajir, T. A. Sutrisno, Mudjiono, N. Zakia and U. Yanuhar, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 515, 012053 CAS.
- A. G. N. Sofiah, J. Pasupuleti, M. Samykano, K. Kadirgama, S. P. Koh, S. K. Tiong, A. K. Pandey, C. T. Yaw and S. K. Natarajan, Polymers, 2023, 15, 3044 CrossRef CAS.
- D. Aki, S. Ulag, S. Unal, M. Sengor, N. Ekren, C.-C. Lin, H. Yılmazer, C. B. Ustundag, D. M. Kalaskar and O. Gunduz, Mater. Des., 2020, 196, 109094 CrossRef CAS.
- M. Vielreicher, D. Kralisch, S. Völkl, F. Sternal, A. Arkudas and O. Friedrich, Sci. Rep., 2018, 8, 9401 CrossRef PubMed.
- M. M. Abeer, M. C. I. Mohd Amin and C. Martin, J. Pharm. Pharmacol., 2014, 66, 1047–1061 CrossRef CAS.
- S. M. Choi and E. J. Shin, Nanomaterials, 2020, 10, 406 Search PubMed.
- Y. Shi, H. Jiao, J. Sun, X. Lu, S. Yu, L. Cheng, Q. Wang, H. Liu, S. Biranje, J. Wang and J. Liu, Carbohydr. Polym., 2022, 285, 119208 Search PubMed.
- C. Yadav, J.-M. Lee, P. Mohanty, X. Li and W.-D. Jang, Nanoscale, 2023, 15, 15108–15145 RSC.
- H. Seddiqi, E. Oliaei, H. Honarkar, J. Jin, L. C. Geonzon, R. G. Bacabac and J. Klein-Nulend, Cellulose, 2021, 28, 1893–1931 Search PubMed.
- M. Akter, M. Bhattacharjee, A. K. Dhar, F. B. A. Rahman, S. Haque, T. U. Rashid and S. M. F. Kabir, Gels, 2021, 7, 30 CrossRef CAS PubMed.
- L. Cellante, R. Costa, I. Monaco, G. Cenacchi and E. Locatelli, New J. Chem., 2018, 42, 5237–5242 Search PubMed.
- R. Alimohammadzadeh, A. A. Rafi, L. Goclik, C.-W. Tai and A. Cordova, Carbohydr. Polym. Technol. Appl., 2022, 3, 100205 Search PubMed.
- Y. Wang, X. Wang, Y. Xie and K. Zhang, Cellulose, 2018, 25, 3703–3731 Search PubMed.
- T. Heinze, O. A. El Seoud and A. Koschella, Cellulose Derivatives, Springer International Publishing, Cham, 2018, pp. 429–477 Search PubMed.
- J. You, X. Zhang, Q. Mi, J. Zhang, J. Wu and J. Zhang, Cellulose, 2022, 29, 9583–9596 Search PubMed.
- C. Vinatier, D. Magne, P. Weiss, C. Trojani, N. Rochet, G. F. Carle, C. Vignes-Colombeix, C. Chadjichristos, P. Galera, G. Daculsi and J. Guicheux, Biomaterials, 2005, 26, 6643–6651 Search PubMed.
- K. Jin, Y. Tang, X. Zhu and Y. Zhou, Int. J. Biol. Macromol., 2020, 162, 1109–1117 Search PubMed.
- M. Hu, J. Yang and J. Xu, Drug Delivery, 2021, 28, 607–619 Search PubMed.
- I. Rocha, N. Ferraz, A. Mihranyan, M. Strømme and J. Lindh, Cellulose, 2018, 25, 1899–1910 CrossRef CAS.
- S. S. Bhutada, M. Sriram and D. S. Katti, Carbohydr. Polym., 2021, 268, 118256 CrossRef CAS PubMed.
- K. Min and G. Tae, Biomater. Res., 2023, 27, 28 CrossRef CAS PubMed.
- J. H. Jordan, M. W. Easson and B. D. Condon, Nanomaterials, 2019, 9, 1232 Search PubMed.
- Y. S. Abdullaevich, Y. K. Ergashovich, S. A. Abdukhalilovich and G. I. Shavkat O’G’Li, Polym. Eng. Sci., 2022, 62, 677–686 Search PubMed.
- G. Juncu, A. Stoica-Guzun, M. Stroescu, G. Isopencu and S. I. Jinga, Int. J. Pharm., 2016, 510, 485–492 Search PubMed.
- G. Mosconi, M. L. Formica, S. D. Palma and R. Rojas, New J. Chem., 2024, 48, 406–415 Search PubMed.
- R. K. Singh and A. K. Singh, Waste Biomass Valor, 2013, 4, 129–137 Search PubMed.
- M. K. Patoary, S. R. Islam, A. Farooq, M. A. Rashid, S. Sarker, Md. Y. Hossain, M. A. N. Rakib, M. Al-Amin and L. Liu, Ind. Crops Prod., 2023, 201, 116965 CrossRef CAS.
- L. Zha, Y. Zheng, J. Che and Y. Xiao, New J. Chem., 2021, 45, 22354–22360 Search PubMed.
- S. Wu, Y. Gong, S. Liu, Y. Pei and X. Luo, Carbohydr. Polym., 2021, 254, 117421 Search PubMed.
- G. M. A. Khan, M. A. Haque, M. Terano and M. S. Alam, J. Appl. Polym. Sci., 2014, 131, 40139 Search PubMed.
- M. H. Rahaman, M. A. Haque, M. A. Rahman, M. M. Rana, M. M. Parvez and S. M. N. Alam, Reactions, 2022, 3, 213–223 CrossRef CAS.
- M. Spaic, D. P. Small, J. R. Cook and W. Wan, Cellulose, 2014, 21, 1529–1540 Search PubMed.
- J. C. Courtenay, J. G. Filgueiras, E. R. deAzevedo, Y. Jin, K. J. Edler, R. I. Sharma and J. L. Scott, J. Mater. Chem. B, 2019, 7, 53–64 RSC.
- C. Warwar Damouny, P. Martin, G. Vasilyev, R. Vilensky, R. Fadul, I. Redenski, S. Srouji and E. Zussman, Biomacromolecules, 2022, 23, 3222–3234 Search PubMed.
- J. Pajorova, A. Skogberg, D. Hadraba, A. Broz, M. Travnickova, M. Zikmundova, M. Honkanen, M. Hannula, P. Lahtinen, M. Tomkova, L. Bacakova and P. Kallio, Biomacromolecules, 2020, 21, 4857–4870 Search PubMed.
- N. Forsman, A. Lozhechnikova, A. Khakalo, L.-S. Johansson, J. Vartiainen and M. Österberg, Carbohydr. Polym., 2017, 173, 392–402 CrossRef CAS PubMed.
- Q. Chen, R. P. Garcia, J. Munoz, U. Pérez De Larraya, N. Garmendia, Q. Yao and A. R. Boccaccini, ACS Appl. Mater. Interfaces, 2015, 7, 24715–24725 Search PubMed.
- A. Yamaguchi, H. Nakayama, Y. Morita, H. Sakamoto, T. Kitamura, M. Hashimoto and S. Suye, ACS Omega, 2020, 5, 18826–18830 Search PubMed.
- S. Lombardo and W. Thielemans, Cellulose, 2019, 26, 249–279 Search PubMed.
- K. Mazeau and L. Charlier, Cellulose, 2012, 19, 337–349 CrossRef CAS.
- J. R. G. Navarro, J. Rostami, A. Ahlinder, J. B. Mietner, D. Bernin, B. Saake and U. Edlund, Biomacromolecules, 2020, 21, 1952–1961 Search PubMed.
- Z. Zhang, G. Sèbe, Y. Hou, J. Wang, J. Huang and G. Zhou, J Appl. Polym. Sci., 2021, 138, 51458 CrossRef CAS.
- X. Lan, Z. Ma, A. R. A. Szojka, M. Kunze, A. Mulet-Sierra, M. J. Vyhlidal, Y. Boluk and A. B. Adesida, Front. Bioeng. Biotechnol., 2021, 9, 766399 CrossRef PubMed.
- R. Sharma, K. H. Putera, M. M. Banaszak Holl, G. Garnier and V. S. Haritos, Int. J. Biol. Macromol., 2024, 254, 127972 CrossRef CAS PubMed.
- M. Mujtaba, A. Negi, A. W. T. King, M. Zare and J. Kuncova-Kallio, Curr. Opin. Biomed. Eng., 2023, 28, 100475 CrossRef CAS.
- H. E. Emam and T. I. Shaheen, Carbohydr. Polym., 2022, 278, 118925 CrossRef CAS PubMed.
- W. Fang, T. Song, L. Wang, T. Han, Z. Xiang and O. J. Rojas, Carbohydr. Polym., 2023, 308, 120663 CrossRef CAS PubMed.
- Y. Xie, Y. Pan and P. Cai, Ind. Crops Prod., 2022, 176, 114381 CrossRef CAS.
- B. P. Frank, D. P. Durkin, E. R. Caudill, L. Zhu, D. H. White, M. L. Curry, J. A. Pedersen and D. H. Fairbrother, ACS Appl. Nano Mater., 2018, 1, 7025–7038 CrossRef CAS.
- J. Jo, H. Kim, S.-Y. Jeong, C. Park, H. S. Hwang and B. Koo, Nanomaterials, 2021, 11, 1542 CrossRef CAS PubMed.
- C. Boyer, G. Réthoré, P. Weiss, C. d’Arros, J. Lesoeur, C. Vinatier, B. Halgand, O. Geffroy, M. Fusellier, G. Vaillant, P. Roy, O. Gauthier and J. Guicheux, Front. Bioeng. Biotechnol., 2020, 8, 23 CrossRef PubMed.
- B. Mahendiran, S. Muthusamy, G. Janani, B. B. Mandal, S. Rajendran and G. S. Krishnakumar, ACS Biomater. Sci. Eng., 2022, 8, 2000–2015 CrossRef CAS PubMed.
- J. Luo, N. Semenikhin, H. Chang, R. J. Moon and S. Kumar, Carbohydr. Polym., 2018, 181, 247–255 CrossRef CAS PubMed.
- L. Fan, X. Zhou, P. Wu, W. Xie, H. Zheng, W. Tan, S. Liu and Q. Li, Int. J. Biol. Macromol., 2014, 66, 245–253 CrossRef CAS PubMed.
- A. Randhawa, S. D. Dutta, K. Ganguly, T. V. Patil, D. K. Patel and K.-T. Lim, Applied Sciences, 2022, 12, 7090 CrossRef CAS.
- B. Thomas, M. C. Raj, K. B. Athira, M. H. Rubiyah, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Chem. Rev., 2018, 118, 11575–11625 CrossRef CAS PubMed.
- S. Thiangtham, J. Runt and H. Manuspiya, Carbohydr. Polym., 2019, 208, 314–322 CrossRef CAS PubMed.
- W. Li, Y. Xue, M. He, J. Yan, L. A. Lucia, J. Chen, J. Yu and G. Yang, Nanomaterials, 2021, 11, 2778 CrossRef CAS PubMed.
- A. S. Kazachenko, N. Yu Vasilieva, Y. D. Berezhnaya, O. Yu Fetisova, V. S. Borovkova, Y. N. Malyar, I. G. Sudakova, V. V. Sychev, N. Issaoui, M. A. Lutoshkin and A. A. Karacharov, Polymers, 2023, 15, 1116 Search PubMed.
- Z. Qin, L. Ji, X. Yin, L. Zhu, Q. Lin and J. Qin, Carbohydr. Polym., 2014, 101, 947–953 CrossRef CAS PubMed.
- A. Adewuyi, C. A. Otuechere, O. L. Adebayo, C. Anazodo and F. V. Pereira, Chem.-Biol. Interact., 2018, 284, 56–68 CrossRef CAS PubMed.
- G. Portocarrero Huang, R. Menezes, R. Vincent, W. Hammond, L. Rizio, G. Collins and T. L. Arinzeh, Tissue Eng., Part A, 2017, 23, 1011–1021 CrossRef CAS PubMed.
- A. Dixit, A. Mahajan, R. Saxena, S. Chakraborty and D. S. Katti, Biomater. Sci., 2024, 12, 2067–2085 Search PubMed.
- A. Ashames, F. Pervaiz, M. Al-Tabakha, K. Khalid, N. Hassan, H. Shoukat, M. Buabeid and G. Murtaza, J. Saudi Chem. Soc., 2022, 26, 101541 CrossRef CAS.
- T. Su, Q.-X. Wu, Y. Chen, J. Zhao, X.-D. Cheng and J. Chen, Int. J. Pharm., 2019, 555, 291–302 CrossRef CAS PubMed.
- H. Kono, E. Tsukamoto and K. Tajima, ACS Omega, 2021, 6, 34107–34114 Search PubMed.
- W. Klunklin, K. Jantanasakulwong, Y. Phimolsiripol, N. Leksawasdi, P. Seesuriyachan, T. Chaiyaso, C. Insomphun, S. Phongthai, P. Jantrawut, S. R. Sommano, W. Punyodom, A. Reungsang, T. M. P. Ngo and P. Rachtanapun, Polymers, 2020, 13, 81 CrossRef PubMed.
- R. Ramakrishnan, J. T. Kim, S. Roy and A. Jayakumar, Int. J. Biol. Macromol., 2024, 259, 129194 Search PubMed.
- N. Ninan, M. Muthiah, I.-K. Park, A. Elain, S. Thomas and Y. Grohens, Carbohydr. Polym., 2013, 98, 877–885 Search PubMed.
- J. Liuyun, L. Yubao and X. Chengdong, J. Biomed. Sci., 2009, 16, 65 CrossRef PubMed.
- G. Janarthanan, H. N. Tran, E. Cha, C. Lee, D. Das and I. Noh, Mater. Sci. Eng., C, 2020, 113, 111008 CrossRef CAS PubMed.
- J. Namkaew, P. Laowpanitchakorn, N. Sawaddee, S. Jirajessada, S. Honsawek and S. Yodmuang, Molecules, 2021, 26, 578 CrossRef CAS PubMed.
- S. Parvaneh, M. Pourmadadi, M. Abdouss, S. A. Pourmousavi, F. Yazdian, A. Rahdar and A. M. Díez-Pascual, Int. J. Biol. Macromol., 2023, 241, 124566 Search PubMed.
- S. Javanbakht and A. Shaabani, Int. J. Biol. Macromol., 2019, 133, 21–29 Search PubMed.
- J. Wei, S. Jia, L. Zhang, Y. Zhou, Y. Lv, X. Zhang and Z. Shao, J. Appl. Polym. Sci., 2021, 138, 50092 CrossRef CAS.
- A. Rees, L. C. Powell, G. Chinga-Carrasco, D. T. Gethin, K. Syverud, K. E. Hill and D. W. Thomas, BioMed Res. Int., 2015, 2015, 1–7 Search PubMed.
- P. Khadivar, S. Khajeniazi and A. Karimi, J. Mater. Res. Technol., 2022, 19, 3966–3979 Search PubMed.
- I. Ali, A. Rizwan, T. T. Vu, S.-H. Jo, C.-W. Oh, Y. H. Kim, S.-H. Park and K. T. Lim, Int. J. Biol. Macromol., 2024, 260, 129549 Search PubMed.
- Y. Zheng, L. Wang, X. Bai, Y. Xiao and J. Che, Colloids Surf., A, 2022, 635, 127958 Search PubMed.
- D. A. Osorio, B. E. J. Lee, J. M. Kwiecien, X. Wang, I. Shahid, A. L. Hurley, E. D. Cranston and K. Grandfield, Acta Biomater., 2019, 87, 152–165 Search PubMed.
- Q. Liu, Q. Li, M. Hatakeyama and T. Kitaoka, Int. J. Biol. Macromol., 2023, 253, 126842 CrossRef CAS PubMed.
- Q. Wang, Ö. Karadas, J. M. Rosenholm, C. Xu, T. Näreoja and X. Wang, Adv Funct Mater., 2024, 2400431 CrossRef.
- T. Gullinkala and I. Escobar, J. Membr. Sci., 2010, 360, 155–164 Search PubMed.
- Q. Wang, X. Chen, S. Zeng, P. Chen, Y. Xu, W. Nie, R. Xia and Y. Zhou, Int. J. Biol. Macromol., 2023, 240, 124515 CrossRef CAS PubMed.
- W. He, Z. Zhang, Y. Zheng, S. Qiao, Y. Xie, Y. Sun, K. Qiao, Z. Feng, X. Wang and J. Wang, J Biomed. Mater. Res, 2020, 108, 1086–1098 CrossRef CAS PubMed.
- S. Andra, S. K. Balu, J. Jeevanandam, M. Muthalagu and M. K. Danquah, Cellulose, 2021, 28, 5895–5910 CrossRef CAS.
- P. Willberg-Keyriläinen, P. Pitkänen, J. Hulkko, M. Asikainen and H. Setälä, Heliyon, 2019, 5, e01349 Search PubMed.
- B. J. Boese and R. R. Breaker, Nucleic Acids Res., 2007, 35, 6378–6388 CrossRef CAS PubMed.
- M. Horue, M. L. Cacicedo, M. A. Fernandez, B. Rodenak-Kladniew, R. M. Torres Sánchez and G. R. Castro, Mater. Sci. Eng., C, 2020, 116, 111152 CrossRef CAS PubMed.
- H. Kim, S. Han, K. Choi, J. Lee, S.-H. Lee, Y.-W. Won and K. S. Lim, Biotechnol. Bioproc. E, 2023, 28, 83–90 Search PubMed.
- G. Xu, R. Nigmatullin, T. T. Koev, Y. Z. Khimyak, I. P. Bond and S. J. Eichhorn, ACS Appl. Mater. Interfaces, 2022, 14, 12722–12733 Search PubMed.
- M. Yusefi, K. Shameli, M. S. Lee-Kiun, S.-Y. Teow, H. Moeini, R. R. Ali, P. Kia, C. J. Jie and N. H. Abdullah, Int. J. Biol. Macromol., 2023, 233, 123388 Search PubMed.
- L. A. Goetz, B. Jalvo, R. Rosal and A. P. Mathew, J. Membr. Sci., 2016, 510, 238–248 Search PubMed.
- F. Hoeng, J. Bras, E. Gicquel, G. Krosnicki and A. Denneulin, RSC Adv., 2017, 7, 15372–15381 Search PubMed.
- M.-H. Song, T. P. T. Pham and Y.-S. Yun, Sci. Rep., 2020, 10, 13905 Search PubMed.
- A. Kumar, K. Dixit and N. Sinha, Ceramics International, 2023, 49, 17639–17649 Search PubMed.
- X. Zhang, C. Wang, M. Liao, L. Dai, Y. Tang, H. Zhang, P. Coates, F. Sefat, L. Zheng, J. Song, Z. Zheng, D. Zhao, M. Yang, W. Zhang and P. Ji, Carbohydr. Polym., 2019, 213, 27–38 Search PubMed.
- F. L. Hatton, E. Malmström and A. Carlmark, Eur. Polym. J., 2015, 65, 325–339 Search PubMed.
- I. D. A. A. Fernandes, G. M. Maciel, I. S. Ribeiro, A. C. Pedro, D. G. Bortolini, V. R. Ribeiro, L. Barros and C. W. I. Haminiuk, Int. J. Biol. Macromol., 2023, 240, 124349 Search PubMed.
- I. Spiridon, N. Anghel, M. V. Dinu, S. Vlad, A. Bele, B. I. Ciubotaru, L. Verestiuc and D. Pamfil, Polymers, 2020, 12, 1191 Search PubMed.
- M. E. Mahmoud, R. M. El-Sharkawy and G. A. A. Ibrahim, J. Mol. Liq., 2022, 368, 120676 CrossRef CAS.
- A. Gennari, R. Simon, E. V. Benvenutti, S. Nicolodi, G. Renard, J. M. Chies, G. Volpato and C. F. Volken De Souza, Int. J. Biol. Macromol., 2024, 256, 128418 CrossRef CAS PubMed.
- N. B. Haro-Mares, J. C. Meza-Contreras, F. A. López-Dellamary, A. Richaud, F. Méndez, B. G. Curiel-Olague, G. Buntkowsky and R. Manríquez-González, Surf. Interfaces, 2022, 35, 102412 CrossRef CAS.
- Y. Chen, X. Zhang and X. Luo, Colloids Surf., B, 2023, 223, 113184 CrossRef CAS PubMed.
- N. O. Gomes, E. Carrilho, S. A. S. Machado and L. F. Sgobbi, Electrochim. Acta, 2020, 349, 136341 CrossRef CAS.
- K. Neubauerova, M. C. C. G. Carneiro, L. R. Rodrigues, F. T. C. Moreira and M. G. F. Sales, Sens. Bio-Sens. Res., 2020, 29, 100368 Search PubMed.
- L. Ning, Y. Jia, X. Zhao, R. Tang, F. Wang and C. You, Int. J. Biol. Macromol., 2022, 222, 1500–1510 Search PubMed.
- Y. Y. Khine, R. Batchelor, R. Raveendran and M. H. Stenzel, Macromol. Rapid Commun., 2020, 41, 1900499 Search PubMed.
- L. Ji, F. Zhang, L. Zhu and J. Jiang, Int. J. Biol. Macromol., 2021, 170, 459–468 Search PubMed.
- Z. Cui, J. Lin, C. Zhan, J. Wu, S. Shen, J. Si and Q. Wang, J. Biomater. Sci., Polym. Ed., 2020, 31, 561–577 CrossRef CAS PubMed.
- Y. Yin, X. Tian, X. Jiang and P. Zhu, Ind. Crops Prod., 2022, 188, 115575 CrossRef CAS.
- F. Lin, X. Lu, Z. Wang, Q. Lu, G. Lin, B. Huang and B. Lu, Cellulose, 2019, 26, 1825–1839 CrossRef CAS.
- R. Blažic, K. Marušić and E. Vidović, Gels, 2023, 9, 94 Search PubMed.
- Y. Zhong, J. Wang, Z. Yuan, Y. Wang, Z. Xi, L. Li, Z. Liu and X. Guo, Colloids Surf., B, 2019, 179, 462–469 Search PubMed.
- E. Toshikj, A. Tarbuk, K. Grgić, B. Mangovska and I. Jordanov, Cellulose, 2019, 26, 777–794 CrossRef CAS.
- J. Wang, X. Han, W. Wu, X. Wang, L. Ding, Y. Wang, S. Li, J. Hu, W. Yang, C. Zhang and S. Jiang, Int. J. Biol. Macromol., 2023, 231, 123343 CrossRef CAS PubMed.
- T. Nypelö, B. Berke, S. Spirk and J. A. Sirviö, Carbohydr. Polym., 2021, 252, 117105 CrossRef PubMed.
- U.-J. Kim, S. Kuga, M. Wada, T. Okano and T. Kondo, Biomacromolecules, 2000, 1, 488–492 CrossRef CAS PubMed.
- I. Nikolits, S. Radwan, F. Liebner, W. Dietrich, D. Egger, F. Chariyev-Prinz and C. Kasper, ACS Appl. Bio Mater., 2023, 6, 543–551 CrossRef CAS PubMed.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- A. Isogai and L. Bergström, Curr. Opin. Green Sustainable Chem., 2018, 12, 15–21 Search PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- P. Sharma, V. K. Pal, H. Kaur and S. Roy, Biomacromolecules, 2022, 23, 2496–2511 CrossRef CAS PubMed.
- H. Yin, P. Song, X. Chen, Q. Huang and H. Huang, Int. J. Biol. Macromol., 2022, 221, 1606–1617 CrossRef CAS PubMed.
- C.-N. Wu, S.-C. Fuh, S.-P. Lin, Y.-Y. Lin, H.-Y. Chen, J.-M. Liu and K.-C. Cheng, Biomacromolecules, 2018, 19, 544–554 CrossRef CAS PubMed.
- D. Shao, Q. Gao, Y. Sheng, S. Li and Y. Kong, Int. J. Biol. Macromol., 2022, 202, 37–45 CrossRef CAS PubMed.
- F. Xie, P. De Wever, P. Fardim and G. Van Den Mooter, Molecules, 2021, 26, 1030 CrossRef CAS PubMed.
- A. T. Thodikayil, A. Yadav, P. Hariprasad and S. Saha, Int. J. Biol. Macromol., 2024, 254, 127604 CrossRef CAS.
- R. E. Abouzeid, R. Khiari, D. Beneventi and A. Dufresne, Biomacromolecules, 2018, 19, 4442–4452 CrossRef CAS PubMed.
- M. A. Mariño, K. Oyarce, C. Tobar, R. S. Del Río, M. G. Paredes, P. Pavez, M. Sarabia, A. Amoroso, J. L. Concha, J. Norambuena-Contreras, G. C. Barjas and J. Castaño, Cellulose, 2024, 31, 363–379 CrossRef.
- D. Srivastava, J. Ahopelto and A. J. Karttunen, Molecules, 2022, 27, 6240 Search PubMed.
- M. Wohlert, T. Benselfelt, L. Wågberg, I. Furó, L. A. Berglund and J. Wohlert, Cellulose, 2022, 29, 1–23 CrossRef CAS.
- R. K. Mishra, S. K. Ha, K. Verma and S. K. Tiwari, J. Sci.: Adv. Mater. Dev., 2018, 3, 263–288 Search PubMed.
- A. L. Barnette, C. Lee, L. C. Bradley, E. P. Schreiner, Y. B. Park, H. Shin, D. J. Cosgrove, S. Park and S. H. Kim, Carbohydr. Polym., 2012, 89, 802–809 CrossRef CAS PubMed.
- P. C. S. Faria-Tischer, C. A. Tischer, L. Heux, S. Le Denmat, C. Picart, M.-R. Sierakowski and J.-L. Putaux, Mater. Sci. Eng., C, 2015, 51, 167–173 Search PubMed.
- Z. Ling, S. Chen, X. Zhang, K. Takabe and F. Xu, Sci. Rep., 2017, 7, 10230 CrossRef PubMed.
- Q. Wu, J. Xu, S. Zhu, Y. Kuang, B. Wang and W. Gao, Carbohydr. Polym., 2020, 249, 116827 Search PubMed.
- P. Zugenmaier, Prog. Polym. Sci., 2001, 26, 1341–1417 CrossRef CAS.
- D. Srivastava, M. S. Kuklin, J. Ahopelto and A. J. Karttunen, Carbohydr. Polym., 2020, 243, 116440 CrossRef CAS PubMed.
- P. Chen, Y. Ogawa, Y. Nishiyama, A. E. Ismail and K. Mazeau, Cellulose, 2018, 25, 4345–4355 CrossRef CAS.
- S. A. Kedzior, J. O. Zoppe, R. M. Berry and E. D. Cranston, Curr. Opin. Solid State Mater. Sci., 2019, 23, 74–91 Search PubMed.
- Y. Yue, J. Han, G. Han, Q. Zhang, A. D. French and Q. Wu, Carbohydr. Polym., 2015, 133, 438–447 CrossRef CAS PubMed.
- H. Wang, S. Li, T. Wu, X. Wang, X. Cheng and D. Li, Polymers, 2019, 11, 153 CrossRef PubMed.
- M. Wada, T. Okano and J. Sugiyama, J. Wood Sci., 2001, 47, 124–128 Search PubMed.
- X. Wang, C. H. Chang, J. Jiang, Q. Liu, Y. Liao, J. Lu, L. Li, X. Liu, J. Kim, A. Ahmed, A. E. Nel and T. Xia, Small, 2019, 15, 1901642 CrossRef CAS PubMed.
- G. Kandhola, A. Djioleu, K. Rajan, N. Labbé, J. Sakon, D. J. Carrier and J.-W. Kim, Bioresour. Bioprocess., 2020, 7, 19 CrossRef.
- K. J. Nagarajan, N. R. Ramanujam, M. R. Sanjay, S. Siengchin, B. Surya Rajan, K. Sathick Basha, P. Madhu and G. R. Raghav, Polym. Compos., 2021, 42, 1588–1630 CrossRef CAS.
- R. K. Mishra, A. Sabu and S. K. Tiwari, J. Saudi Chem. Soc., 2018, 22, 949–978 CrossRef CAS.
- B. Anwar, B. Bundjali, Y. Sunarya and I. M. Arcana, Fibers Polym., 2021, 22, 1228–1236 Search PubMed.
- S. Manan, M. W. Ullah, M. Ul-Islam, Z. Shi, M. Gauthier and G. Yang, Prog. Mater. Sci., 2022, 129, 100972 CrossRef CAS.
- J. M. Dugan, J. E. Gough and S. J. Eichhorn, Nanomedicine, 2013, 8, 287–298 CrossRef CAS PubMed.
- K. Daicho, K. Kobayashi, S. Fujisawa and T. Saito, Biomacromolecules, 2020, 21, 939–945 CrossRef CAS PubMed.
- M. Ioelovich, Polymers, 2021, 13, 4313 CrossRef CAS PubMed.
- P. Phanthong, P. Reubroycharoen, X. Hao, G. Xu, A. Abudula and G. Guan, Carbon Resour. Convers., 2018, 1, 32–43 CrossRef.
- F. Fortess, Text. Res. J., 1949, 19, 23–35 CrossRef CAS.
- T. Kuga, N. Sunagawa and K. Igarashi, Cellulose, 2022, 29, 2999–3015 CrossRef CAS PubMed.
- J. R. Olson, C. J. Jourdain and R. J. Rousseau, Can. J. For. Res., 1985, 15, 393–396 CrossRef CAS.
- M. Y. Khalid, A. Al Rashid, Z. U. Arif, W. Ahmed and H. Arshad, J. Mater. Res. Technol., 2021, 14, 2601–2623 CrossRef CAS.
- Y. Feng, H. Cölfen and R. Xiong, J. Mater. Chem. B, 2023, 11, 5321–5349 RSC.
- F. V. Ferreira, A. G. Souza, R. Ajdary, L. P. De Souza, J. H. Lopes, D. S. Correa, G. Siqueira, H. S. Barud, D. D. S. Rosa, L. H. C. Mattoso and O. J. Rojas, Bioactive Mater., 2023, 29, 151–176 CrossRef CAS PubMed.
- L. Geng, Y. Shi, W. Fang, K. Jiang, P. Fan and Y. Zhang, Mater. Today Commun., 2023, 35, 106414 CrossRef CAS.
- G. Guhados, W. Wan and J. L. Hutter, Langmuir, 2005, 21, 6642–6646 CrossRef CAS PubMed.
- A. Šturcová, G. R. Davies and S. J. Eichhorn, Biomacromolecules, 2005, 6, 1055–1061 CrossRef PubMed.
- B. Joseph, V. K. Sagarika, C. Sabu, N. Kalarikkal and S. Thomas, J. Bioresour. Bioprod., 2020, 5, 223–237 CrossRef CAS.
- R. R. Lahiji, X. Xu, R. Reifenberger, A. Raman, A. Rudie and R. J. Moon, Langmuir, 2010, 26, 4480–4488 CrossRef CAS PubMed.
- R. Rusli and S. J. Eichhorn, Appl. Phys. Lett., 2008, 93, 033111 CrossRef.
- N. Lin and A. Dufresne, Eur. Polym. J., 2014, 59, 302–325 CrossRef CAS.
- A. Sugawara, T.-A. Asoh, Y. Takashima, A. Harada and H. Uyama, Polym. Degrad. Stab., 2020, 177, 109157 CrossRef CAS.
- A. A. B. Omran, A. A. B. A. Mohammed, S. M. Sapuan, R. A. Ilyas, M. R. M. Asyraf, S. S. Rahimian Koloor and M. Petrů, Polymers, 2021, 13, 231 CrossRef CAS PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941 RSC.
- P. Panchal, E. Ogunsona and T. Mekonnen, Processes, 2018, 7, 10 CrossRef.
- B. L. Pelegrini, F. Ré, M. M. De Oliveira, T. Fernandes, J. H. De Oliveira, A. G. Oliveira Junior, E. M. Girotto, C. V. Nakamura, A. R. Sampaio, A. Valim and M. M. De Souza Lima, Macro Mater. Eng., 2019, 304, 1900092 CrossRef.
- R. T. Olsson, M. A. S. Azizi Samir, G. Salazar-Alvarez, L. Belova, V. Ström, L. A. Berglund, O. Ikkala, J. Nogués and U. W. Gedde, Nat. Nanotechnol., 2010, 5, 584–588 CrossRef CAS PubMed.
- P. Raj, A. Mayahi, P. Lahtinen, S. Varanasi, G. Garnier, D. Martin and W. Batchelor, Cellulose, 2016, 23, 3051–3064 CrossRef CAS.
- P. Gatenholm and D. Klemm, MRS Bull., 2010, 35, 208–213 CrossRef CAS.
- X. Wu, R. J. Moon and A. Martini, Cellulose, 2014, 21, 2233–2245 CrossRef CAS.
- T. Saito, R. Kuramae, J. Wohlert, L. A. Berglund and A. Isogai, Biomacromolecules, 2013, 14, 248–253 CrossRef CAS PubMed.
- T. Pal, S. Pramanik, K. D. Verma, S. Z. Naqvi, P. K. Manna and K. K. Kar, Handbook of Fly Ash, Elsevier, 2022, pp. 243–270 Search PubMed.
- D. Marković, B. Ćetenović, A. Vuković, V. Jokanović and T. Marković, Nanobiomaterials in Dentistry, Elsevier, 2016, pp. 269–307 Search PubMed.
- A. J. Kerin, M. R. Wisnom and M. A. Adams, Proc. Inst. Mech. Eng. H, 1998, 212, 273–280 CrossRef CAS PubMed.
- Z. L. Li, Q. Lu, J. R. Honiball, S. H. Wan, K. W. Yeung and K. M. Cheung, J. Biomed. Mater. Res., 2023, 111, 1888–1902 CrossRef CAS PubMed.
- A. Guo, Z. Sun, N. Sathitsuksanoh and H. Feng, Nanomaterials, 2020, 10, 2476 CrossRef CAS PubMed.
- M. A. Aziz, M. Zubair and M. Saleem, Case Studies Construction Mater., 2021, 15, e00761 CrossRef.
- D. Barnat-Hunek, M. Grzegorczyk-Frańczak, M. Szymańska-Chargot and G. Łagód, Polymers, 2019, 11, 2088 CrossRef PubMed.
- A. L. Buyanov, I. V. Gofman and N. N. Saprykina, J. Mech. Behav. Biomed. Mater., 2019, 100, 103385 CrossRef CAS PubMed.
- E. B. Heggset, B. L. Strand, K. W. Sundby, S. Simon, G. Chinga-Carrasco and K. Syverud, Cellulose, 2019, 26, 581–595 CrossRef CAS.
- M. Kim, S. Kim, N. Han, S. Lee and H. Kim, Carbohydr. Polym., 2023, 300, 120218 CrossRef CAS PubMed.
- Z. Yu, J. Zhan, H. Wang, H. Zheng, J. Xie and X. Wang, J. Phys.: Conf. Ser., 2020, 1653, 012059 CrossRef.
- M. Sobanwa, T. J. Foster and N. J. Watson, Food Hydrocolloids, 2020, 101, 105460 CrossRef CAS.
- D. Liu, H. Zhang, X. Dong, L. Sang and M. Qi, Front. Bioeng. Biotechnol., 2022, 10, 959409 CrossRef PubMed.
- Y. Jiang, J. Zhou, Z. Yang, D. Liu, X. Xv, G. Zhao, H. Shi and Q. Zhang, J. Mater. Sci., 2018, 53, 11883–11900 CrossRef CAS.
- M. A. Hubbe, P. Tayeb, M. Joyce, P. Tyagi, M. Kehoe, K. Dimic-Misic and L. Pal, BioRes, 2017, 12, 9556–9661 CAS.
- M. M. Hassan, N. Tucker and M. J. Le Guen, Carbohydr. Polym., 2020, 230, 115675 CrossRef CAS PubMed.
- T. Tadros, in Encyclopedia of Colloid and Interface Science, ed. T. Tadros, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 1020–1042 Search PubMed.
- S.-A. Jin and R. J. Spontak, Adv. Ind. Eng. Polym. Res., 2023, 6, 356–381 CAS.
- V. Hynninen, J. Patrakka and Nonappa, Materials, 2021, 14, 5137 CrossRef CAS PubMed.
- M. E. Lamm, K. Li, J. Qian, L. Wang, N. Lavoine, R. Newman, D. J. Gardner, T. Li, L. Hu, A. J. Ragauskas, H. Tekinalp, V. Kunc and S. Ozcan, Adv. Mater., 2021, 33, 2005538 CrossRef CAS PubMed.
- K. Markstedt, A. Mantas, I. Tournier, H. Martínez Ávila, D. Hägg and P. Gatenholm, Biomacromolecules, 2015, 16, 1489–1496 CrossRef CAS PubMed.
- Y. Ai, Z. Lin, W. Zhao, M. Cui, W. Qi, R. Huang and R. Su, J. Mater. Chem. B, 2023, 11, 7004–7023 RSC.
- X. Dong, X. Guo, Q. Liu, Y. Zhao, H. Qi and W. Zhai, Adv. Funct. Mater., 2022, 32, 2203610 CrossRef CAS.
- N. E. Zander, H. Dong, J. Steele and J. T. Grant, ACS Appl. Mater. Interfaces, 2014, 6, 18502–18510 CrossRef CAS PubMed.
- S. Ma, M. Zhang, B. Yang, S. Song, J. Nie and P. Lu, BioRes, 2018, 13, 5894–5908 CAS.
- F. V. Borbolla-Jiménez, S. I. Peña-Corona, S. J. Farah, M. T. Jiménez-Valdés, E. Pineda-Pérez, A. Romero-Montero, M. L. Del Prado-Audelo, S. A. Bernal-Chávez, J. J. Magaña and G. Leyva-Gómez, Pharmaceutics, 2023, 15, 1914 CrossRef PubMed.
- J. C. Muñoz-García, K. R. Corbin, H. Hussain, V. Gabrielli, T. Koev, D. Iuga, A. N. Round, D. Mikkelsen, P. A. Gunning, F. J. Warren and Y. Z. Khimyak, Biomacromolecules, 2019, 20, 4180–4190 CrossRef PubMed.
- J. Li, C. Wu, P. K. Chu and M. Gelinsky, Mater. Sci. Eng., R, 2020, 140, 100543 CrossRef.
- J. Liu, Y. Shi, L. Cheng, J. Sun, S. Yu, X. Lu, S. Biranje, W. Xu, X. Zhang, J. Song, Q. Wang, W. Han and Z. Zhang, Cellulose, 2021, 28, 5643–5656 CrossRef CAS.
- S. Ilkar Erdagi, F. Asabuwa Ngwabebhoh and U. Yildiz, Int. J. Biol. Macromol., 2020, 149, 651–663 CrossRef CAS.
- P. Maturavongsadit, L. K. Narayanan, P. Chansoria, R. Shirwaiker and S. R. Benhabbour, ACS Appl. Bio Mater., 2021, 4, 2342–2353 CrossRef CAS PubMed.
- L. Berglund, P. Squinca, Y. Baş, E. Zattarin, D. Aili, J. Rakar, J. Junker, A. Starkenberg, M. Diamanti, P. Sivlér, M. Skog and K. Oksman, Biomacromolecules, 2023, 24, 2264–2277 CrossRef CAS PubMed.
- C. Chang, J. Peng, L. Zhang and D.-W. Pang, J. Mater. Chem., 2009, 19, 7771 RSC.
- A. M. L. Piloto, D. S. M. Ribeiro, S. S. M. Rodrigues, J. L. M. Santos, P. Sampaio and M. G. F. Sales, Microchim. Acta, 2022, 189, 134 CrossRef CAS PubMed.
- H. Luo, J. Dong, F. Yao, Z. Yang, W. Li, J. Wang, X. Xu, J. Hu and Y. Wan, Nano-Micro Lett., 2018, 10, 42 CrossRef PubMed.
- L. Li, Z. Lin, X. Yang, Z. Wan and S. Cui, Sci. Bull., 2009, 54, 1622–1625 CrossRef CAS.
- H. Najaf Zadeh, T. Huber, V. Nock, C. Fee and D. Clucas, Bioengineering, 2020, 7, 58 CrossRef PubMed.
- E. Campodoni, M. Montanari, S. M. Dozio, E. B. Heggset, S. Panseri, M. Montesi, A. Tampieri, K. Syverud and M. Sandri, Nanomaterials, 2020, 10, 1219 CrossRef CAS PubMed.
- L. Orgéas, S. Gupta, F. Martoïa and P. J. J. Dumont, Mater. Des., 2022, 223, 111201 CrossRef.
- J. Joy, J. Pereira, R. Aid-Launais, G. Pavon-Djavid, A. R. Ray, D. Letourneur, A. Meddahi-Pellé and B. Gupta, Int. J. Biol. Macromol., 2018, 107, 1922–1935 CrossRef CAS PubMed.
- H. Samadian, S. Zamiri, A. Ehterami, S. Farzamfar, A. Vaez, H. Khastar, M. Alam, A. Ai, H. Derakhshankhah, Z. Allahyari, A. Goodarzi and M. Salehi, Sci. Rep., 2020, 10, 8312 CrossRef CAS PubMed.
- M. Bordoni, E. Karabulut, V. Kuzmenko, V. Fantini, O. Pansarasa, C. Cereda and P. Gatenholm, Cells, 2020, 9, 682 CrossRef CAS PubMed.
- L. Diaz-Gomez, I. Gonzalez-Prada, R. Millan, A. Da Silva-Candal, A. Bugallo-Casal, F. Campos, A. Concheiro and C. Alvarez-Lorenzo, Carbohydr. Polym., 2022, 278, 118924 CrossRef CAS PubMed.
- J. Batta-Mpouma, G. Kandhola, J. Sakon and J.-W. Kim, Biomacromolecules, 2022, 23, 4085–4096 CrossRef CAS PubMed.
- A. Sannino, S. Pappadà, M. Madaghiele, A. Maffezzoli, L. Ambrosio and L. Nicolais, Polymer, 2005, 46, 11206–11212 CrossRef CAS.
- R. Vel, A. Bhatt, A. Priyanka, A. Gauthaman, V. Anilkumar, A. S. Safeena and S. Ranjith, Mater. Today Commun., 2022, 33, 104335 CrossRef CAS.
- R. Xing, L. Ning, L. Li, L. He, H. Lin, C. You and F. Wang, J. Drug Delivery Sci. Technol., 2023, 86, 104654 CrossRef CAS.
- V. Mohanta, G. Madras and S. Patil, ACS Appl. Mater. Interfaces, 2014, 6, 20093–20101 CrossRef CAS PubMed.
- X. Huang, Y. Wang, Y. Wang and L. Yang, Molecules, 2023, 28, 1301 CrossRef CAS PubMed.
- C. Wang, J. Bai, P. Tian, R. Xie, Z. Duan, Q. Lv and Y. Tao, Front. Bioeng. Biotechnol., 2021, 9, 732513 CrossRef PubMed.
- P. Pooyan, R. Tannenbaum and H. Garmestani, J. Mech. Behav. Biomed. Mater., 2012, 7, 50–59 CrossRef CAS PubMed.
- P. Pooyan, I. T. Kim, K. I. Jacob, R. Tannenbaum and H. Garmestani, Polymer, 2013, 54, 2105–2114 CrossRef CAS.
- E. Ten, J. Turtle, D. Bahr, L. Jiang and M. Wolcott, Polymer, 2010, 51, 2652–2660 CrossRef CAS.
- P. Zou, J. Yao, Y.-N. Cui, T. Zhao, J. Che, M. Yang, Z. Li and C. Gao, Gels, 2022, 8, 364 CrossRef CAS PubMed.
- J. Han, T. Lei and Q. Wu, Cellulose, 2013, 20, 2947–2958 CrossRef CAS.
- K. Hou, Y. Li, Y. Liu, R. Zhang, B. S. Hsiao and M. Zhu, Polymer, 2017, 123, 55–64 CrossRef CAS.
- J. Hu, Y. Wu, Q. Yang, Q. Zhou, L. Hui, Z. Liu, F. Xu and D. Ding, Carbohydr. Polym., 2022, 275, 118697 CrossRef CAS PubMed.
- S. Mandin, S. Moreau, M. Talantikite, B. Novalès, J.-E. Maigret, B. Cathala and C. Moreau, Gels, 2021, 7, 5 CrossRef CAS PubMed.
- Y. Zhou, S. Fu, Y. Pu, S. Pan, M. V. Levit and A. J. Ragauskas, RSC Adv., 2013, 3, 19272 RSC.
- R. Dash, Y. Li and A. J. Ragauskas, Carbohydr. Polym., 2012, 88, 789–792 CrossRef CAS.
- S. Rahmani, Z. Khoubi-Arani, S. Mohammadzadeh-Komuleh and M. Maroufkhani, in Handbook of Nanocelluloses, ed. A. Barhoum, Springer International Publishing, Cham, 2022, pp. 263–296 Search PubMed.
- J. Adhikari, S. Dasgupta, P. Das, D. A. Gouripriya, A. Barui, P. Basak, M. Ghosh and P. Saha, Int. J. Biol. Macromol., 2024, 261, 129661 CrossRef CAS PubMed.
- M. F. Abdullah, A. Andriyana, F. Muhamad and B. C. Ang, J. Polym. Res., 2023, 30, 257 CrossRef CAS.
- N. S. Lameirinhas, M. C. Teixeira, J. P. F. Carvalho, B. F. A. Valente, R. J. B. Pinto, H. Oliveira, J. L. Luís, L. Pires, J. M. Oliveira, C. Vilela and C. S. R. Freire, Int. J. Biol. Macromol., 2023, 229, 849–860 CrossRef CAS PubMed.
- K. Yan, X. Zhang, Y. Liu, J. Cheng, C. Zhai, K. Shen, W. Liang and W. Fan, Mater. Des., 2023, 225, 111531 CrossRef CAS.
- S. D. Dutta, J. Hexiu, D. K. Patel, K. Ganguly and K.-T. Lim, Int. J. Biol. Macromol., 2021, 167, 644–658 CrossRef CAS PubMed.
- X. Wang, Q. Wang and C. Xu, Bioengineering, 2020, 7, 40 CrossRef CAS PubMed.
- M. P. Sekar, H. Budharaju, S. Sethuraman and D. Sundaramurthi, SLAS Technol., 2023, 28, 183–198 CrossRef CAS PubMed.
- H. Baniasadi, R. T. Polez, E. Kimiaei, Z. Madani, O. J. Rojas, M. Österberg and J. Seppälä, Int. J. Biol. Macromol., 2021, 192, 1098–1107 CrossRef CAS PubMed.
- S. Shin, S. Park, M. Park, E. Jeong, K. Na, H. J. Youn and J. Hyun, BioResources, 2017, 12, 2941–2954 CrossRef CAS.
- N. Ashammakhi, S. Ahadian, C. Xu, H. Montazerian, H. Ko, R. Nasiri, N. Barros and A. Khademhosseini, Mater. Today Bio, 2019, 1, 100008 CrossRef CAS PubMed.
- H. Baniasadi, R. Ajdary, J. Trifol, O. J. Rojas and J. Seppälä, Carbohydr. Polym., 2021, 266, 118114 CrossRef CAS PubMed.
- R. Prathapan, B. A. Glatz, A. K. Ghosh, S. Michel, A. Fery, G. Garnier and R. F. Tabor, Langmuir, 2019, 35, 7155–7160 CrossRef CAS PubMed.
- B. He, S. Yang, Z. Qin, B. Wen and C. Zhang, Sci. Rep., 2017, 7, 11841 CrossRef PubMed.
- A. I. Cernencu, A. Lungu, I.-C. Stancu, A. Serafim, E. Heggset, K. Syverud and H. Iovu, Carbohydr. Polym., 2019, 220, 12–21 CrossRef CAS PubMed.
- D. Cafiso, A. A. Septevani, C. Noè, T. Schiller, C. F. Pirri, I. Roppolo and A. Chiappone, Sustainable Mater. Technol., 2022, 32, e00444 CrossRef CAS.
- A. Sadeghianmaryan, S. Naghieh, H. Alizadeh Sardroud, Z. Yazdanpanah, Y. Afzal Soltani, J. Sernaglia and X. Chen, Int. J. Biol. Macromol., 2020, 164, 3179–3192 CrossRef CAS PubMed.
- X. Li, B. Liu, B. Pei, J. Chen, D. Zhou, J. Peng, X. Zhang, W. Jia and T. Xu, Chem. Rev., 2020, 120, 10793–10833 CrossRef CAS PubMed.
- Z. Xie, M. Gao, A. O. Lobo and T. J. Webster, Polymers, 2020, 12, 1717 CrossRef CAS PubMed.
- Y.-L. Tsai, P. Theato, C.-F. Huang and S. Hsu, Appl. Mater. Today, 2020, 20, 100778 CrossRef.
- S. C. Pandanaboina, A. B. RanguMagar, K. D. Sharma, B. P. Chhetri, C. M. Parnell, J. Y. Xie, M. Srivatsan and A. Ghosh, JFB, 2021, 12, 64 CrossRef CAS PubMed.
- R. R. Palem, B. J. Kim, I. Baek, H. Choi, M. Suneetha, G. Shimoga and S.-H. Lee, Carbohydr. Polym., 2024, 334, 122020 CrossRef CAS PubMed.
- H. Xie, G. Shi, R. Wang, X. Jiang, Q. Chen, A. Yu and A. Lu, Carbohydr. Polym., 2024, 334, 122014 CrossRef CAS PubMed.
- M. Nouri-Felekori, N. Nezafati, M. Moraveji, S. Hesaraki and T. Ramezani, Int. J. Biol. Macromol., 2021, 183, 2030–2043 CrossRef CAS PubMed.
- S. Palantöken, K. Bethke, V. Zivanovic, G. Kalinka, J. Kneipp and K. Rademann, J. Appl. Polym. Sci., 2020, 137, 48380 CrossRef.
- A. N. Frone, D. M. Panaitescu, C. A. Nicolae, A. R. Gabor, R. Trusca, A. Casarica, P. O. Stanescu, D. D. Baciu and A. Salageanu, Mater. Sci. Eng., C, 2020, 110, 110740 CrossRef CAS PubMed.
- S. Dutta, P. Samanta and D. Dhara, Int. J. Biol. Macromol., 2016, 87, 92–100 CrossRef CAS PubMed.
- N. Chen, H. Wang, C. Ling, W. Vermerris, B. Wang and Z. Tong, Carbohydr. Polym., 2019, 225, 115207 CrossRef CAS PubMed.
- X. Jiang, F. Zeng, X. Yang, C. Jian, L. Zhang, A. Yu and A. Lu, Acta Biomater., 2022, 141, 102–113 CrossRef CAS PubMed.
- I. Ali, M. Gulfam, S.-H. Jo, J.-W. Seo, A. Rizwan, S.-H. Park and K. T. Lim, Int. J. Biol. Macromol., 2022, 219, 109–120 CrossRef CAS PubMed.
- R. Ajdary, G. Reyes, J. Kuula, E. Raussi-Lehto, T. S. Mikkola, E. Kankuri and O. J. Rojas, ACS Polym. Au, 2022, 2, 97–107 CrossRef CAS PubMed.
- J. Tie, H. Chai, Z. Mao, L. Zhang, Y. Zhong, X. Sui and H. Xu, Carbohydr. Polym., 2021, 273, 118600 CrossRef CAS PubMed.
- C. Huang, Q. Ye, J. Dong, L. Li, M. Wang, Y. Zhang, Y. Zhang, X. Wang, P. Wang and Q. Jiang, Smart Mater. Med., 2023, 4, 1–14 CrossRef.
- M. Suneetha, H. Kim and S. S. Han, Int. J. Biol. Macromol., 2024, 256, 128364 CrossRef CAS PubMed.
- X. Xun, Y. Li, M. Ni, Y. Xu, J. Li, D. Zhang, G. Chen, H. Ao, H. Luo, Y. Wan and T. Yu, Composites, Part B, 2024, 275, 111277 CrossRef CAS.
- M. Hojabri, T. Tayebi, M. Kasravi, A. Aghdaee, A. Ahmadi, R. Mazloomnejad, R. Tarasi, A. Shaabani, S. Bahrami and H. Niknejad, Int. J. Biol. Macromol., 2023, 240, 124492 CrossRef CAS PubMed.
- K.-T. Le, C.-T. Nguyen, T.-D. Lac, L.-G. T. Nguyen, T. L. Tran and H. Tran-Van, J. Drug Delivery Sci. Technol., 2023, 82, 104318 CrossRef CAS.
- M. P. Arrieta, E. Fortunati, N. Burgos, M. A. Peltzer, J. López and L. Peponi, Multifunctional Polymeric Nanocomposites Based on Cellulosic Reinforcements, Elsevier, 2016, pp. 205–252 Search PubMed.
- S. Kalia, A. Dufresne, B. M. Cherian, B. S. Kaith, L. Avérous, J. Njuguna and E. Nassiopoulos, Int. J. Polym. Sci., 2011, 2011, 1–35 Search PubMed.
- A. A. Singh, M. E. Genovese, G. Mancini, L. Marini and A. Athanassiou, ACS Sustainable Chem. Eng., 2020, 8, 4128–4136 CrossRef CAS.
- S. Sundararajan, A. B. Samui and P. S. Kulkarni, Sol. Energy, 2017, 144, 32–39 CrossRef CAS.
- L. Del Valle, A. Díaz and J. Puiggalí, Gels, 2017, 3, 27 CrossRef PubMed.
- S.-Q. Chen, Q. Liao, O. W. Meldrum, L. Guo, K. Wang, S. Zhang, Y. Liu, X. Chen, J. Zhu and L. Li, Carbohydr. Polym., 2023, 321, 121268 CrossRef CAS PubMed.
- P. Maturavongsadit, G. Paravyan, R. Shrivastava and S. R. Benhabbour, Materialia, 2020, 12, 100681 CrossRef CAS.
- F. Coelho, M. Cavicchioli, S. S. Specian, R. M. Scarel-Caminaga, L. D. A. Penteado, A. I. D. Medeiros, S. J. D. L. Ribeiro and T. S. D. O. Capote, PLoS One, 2019, 14, e0221286 CrossRef CAS PubMed.
- L. Li, L. Wang, X. Luan, Y. Pang, K. Zhang, Y. Cheng, Z. Ji and J. Pang, Carbohydr. Polym., 2023, 320, 121235 CrossRef CAS PubMed.
- H. P. Felgueiras, M. A. Teixeira, T. D. Tavares, N. C. Homem, A. Zille and M. T. P. Amorim, J. Appl. Polym. Sci., 2020, 137, 48626 CrossRef CAS.
- H. Dong, J. F. Snyder, D. T. Tran and J. L. Leadore, Carbohydr. Polym., 2013, 95, 760–767 CrossRef CAS PubMed.
- C. Palo-Nieto, A. Blasi-Romero, C. Sandström, D. Balgoma, M. Hedeland, M. Strømme and N. Ferraz, Mater. Adv., 2023, 4, 1555–1565 RSC.
- E. A. Bakr, M. Gaber, D. R. Saad and N. Salahuddin, Int. J. Biol. Macromol., 2023, 230, 123315 CrossRef CAS PubMed.
- J.-H. Seo, S. Y. Lee, C. Hwang, M. Yang, J. Lee, S.-H. Lee and H.-J. Cho, Int. J. Biol. Macromol., 2020, 162, 798–809 CrossRef CAS PubMed.
- N. S. V. Capanema, I. C. Carvalho, A. A. P. Mansur, S. M. Carvalho, A. P. Lage and H. S. Mansur, ACS Appl. Nano Mater., 2019, 2, 7393–7408 CrossRef CAS.
- E. Tamahkar, Chem. Pap., 2021, 75, 3979–3987 CrossRef CAS.
- T. M. S. U. Gunathilake, Y. C. Ching, H. Uyama, N. D. Hai and C. H. Chuah, Cellulose, 2022, 29, 1821–1840 CrossRef CAS PubMed.
- H. Roy, K. Hasan Parvej, M. Mozammal Hosen, Md Shahinoor Islam and S. H. Firoz, Arabian J. Chem., 2024, 17, 105763 CrossRef CAS.
- B. Niknafs, M. Meskaraf-asadabadi, K. Hamdi and E. Ghanbari, Int. J. Biol. Macromol., 2024, 266, 131167 CrossRef CAS PubMed.
- E. Ghanbari, M. Khazaei, A. Mehdipour, A. Khoshfeterat and B. Niknafs, Cell J., 2023, 25, 483–495 Search PubMed.
- X. Yang, J. Huang, C. Chen, L. Zhou, H. Ren and D. Sun, ACS Appl. Mater. Interfaces, 2023, 15, 10506–10519 CrossRef CAS PubMed.
- F. Yu, H. Shi, K. Wang, H. Li and L. Peng, Int. J. Biol. Macromol., 2022, 222, 1238–1249 CrossRef CAS PubMed.
- S. Li, D. Wang, H. Xiao, H. Zhang, S. Cao, L. Chen, Y. Ni and L. Huang, Carbohydr. Polym., 2021, 255, 117352 CrossRef CAS PubMed.
- A. Díez-Pascual and A. Rahdar, Nanomaterials, 2022, 12, 949 CrossRef PubMed.
- G. Liu, Z. Ding, Q. Yuan, H. Xie and Z. Gu, Front. Chem., 2018, 6, 439 CrossRef CAS PubMed.
- W. Yuan, G.-M. Weng, J. Lipton, C. M. Li, P. R. Van Tassel and A. D. Taylor, Adv. Colloid Interface Sci., 2020, 282, 102200 CrossRef CAS PubMed.
- A. Díez-Pascual and P. Shuttleworth, Materials, 2014, 7, 7472–7512 CrossRef PubMed.
- W. Li, X. Li, W. Li, T. Wang, X. Li, S. Pan and H. Deng, Eur. Polym. J., 2012, 48, 1846–1853 CrossRef CAS.
- S. Kariminia, M. Shamsipur and K. Mansouri, J. Mater. Chem. B, 2024, 12, 176–186 RSC.
- M. Khamrai, S. L. Banerjee, S. Paul, A. K. Ghosh, P. Sarkar and P. P. Kundu, ACS Appl. Bio Mater., 2021, 4, 428–440 CrossRef CAS PubMed.
- F. Zhang, N. Zhang, H.-X. Meng, H.-X. Liu, Y.-Q. Lu, C.-M. Liu, Z.-M. Zhang, K.-Y. Qu and N.-P. Huang, ACS Biomater. Sci. Eng., 2019, 5, 3022–3031 CrossRef CAS PubMed.
- S. Choy, D. V. Lam, S.-M. Lee and D. S. Hwang, ACS Appl. Mater. Interfaces, 2019, 11, 38440–38447 CrossRef CAS PubMed.
- H.-W. Kang, S. J. Lee, I. K. Ko, C. Kengla, J. J. Yoo and A. Atala, Nat. Biotechnol., 2016, 34, 312–319 CrossRef CAS PubMed.
- P. Bhattacharjee and M. Ahearne, Pharmaceutics, 2021, 13, 319 CrossRef CAS PubMed.
- A. Sommer, P. Dederko-Kantowicz, H. Staroszczyk, S. Sommer and M. Michalec, IJMS, 2021, 22, 3346 CrossRef CAS PubMed.
- D. Fu, Y. Xie, L. Zhou, L. Zhang, T. Zheng and J. Shen, Carbohydr. Polym., 2024, 325, 121572 CrossRef CAS PubMed.
- M. Uva, M. Tambasco, G. Grassi, I. Corsi, G. Protano and A. Atrei, J. Environ. Chem. Eng., 2017, 5, 3632–3639 CrossRef CAS.
- R. Cruz-Medina, D. A. Ayala-Hernández, A. Vega-Rios, E. I. López-Martínez, M. E. Mendoza-Duarte, A. Estrada-Monje and E. A. Zaragoza-Contreras, Polymers, 2021, 13, 2342 CrossRef CAS.
- L. Wang, W. Zhao, Y. Zhao, W. Li, G. Wang and Q. Zhang, Theranostics, 2023, 13, 673–684 CrossRef CAS.
- D. Zhao, J. Huang, Y. Zhong, K. Li, L. Zhang and J. Cai, Adv Funct Materials, 2016, 26, 6279–6287 CrossRef CAS.
- M. Xeroudaki, M. Rafat, P. Moustardas, A. Mukwaya, S. Tabe, M. Bellisario, B. Peebo and N. Lagali, Acta Biomater., 2023, 172, 234–248 CrossRef CAS.
- S. R. S. Veloso, A. G. Azevedo, P. F. Teixeira and C. B. P. Fernandes, Gels, 2023, 9, 574 CrossRef CAS.
- F. Doustdar, A. Olad and M. Ghorbani, Int. J. Biol. Macromol., 2022, 208, 912–924 CrossRef CAS PubMed.
- Z. Rao, Y. Dong, J. Liu, X. Zheng, Y. Pei and K. Tang, Int. J. Biol. Macromol., 2022, 222, 3155–3167 CrossRef CAS PubMed.
- P. C. Nath, S. Debnath, M. Sharma, K. Sridhar, P. K. Nayak and B. S. Inbaraj, Foods, 2023, 12, 350 CrossRef CAS.
- A. K. Singh, P. Itkor and Y. S. Lee, Gels, 2023, 9, 433 CrossRef CAS PubMed.
- H. Nasution, H. Harahap, N. F. Dalimunthe, M. H. S. Ginting, M. Jaafar, O. O. H. Tan, H. K. Aruan and A. L. Herfananda, Gels, 2022, 8, 568 CrossRef CAS PubMed.
- A. A. D. N. Pomari, T. L. D. A. Montanheiro, C. P. De Siqueira, R. S. Silva, D. B. Tada and A. P. Lemes, J. Compos. Sci., 2019, 3, 84 CrossRef.
- F. J. Burpo, J. L. Palmer, A. N. Mitropoulos, E. A. Nagelli, L. A. Morris, M. Y. Ryu and J. K. Wickiser, JoVE, 2019, 59176 Search PubMed.
- Y. Jia, M. Huo, H. Huang, W. Fu, Y. Wang, J. Zhang and S. Jia, Proc. Inst. Mech. Eng., Part N, 2015, 229, 41–48 CAS.
- M. Hajiabbas, I. Alemzadeh, M. Vossoughi and A. Shamloo, Chem. Eng. Commun., 2021, 208, 976–992 CrossRef CAS.
- D.-J. Lim, IJMS, 2022, 23, 5444 CrossRef CAS PubMed.
- J. Tang, Y. Li, Y. Song, X. Wu, G. Yu and K. C. Tam, Carbohydr. Polym., 2020, 244, 116512 CrossRef CAS PubMed.
- A. Azarniya, E. Tamjid, N. Eslahi and A. Simchi, Int. J. Biol. Macromol., 2019, 134, 280–289 CrossRef CAS PubMed.
- F. Xu, C. Dawson, M. Lamb, E. Mueller, E. Stefanek, M. Akbari and T. Hoare, Front. Bioeng. Biotechnol., 2022, 10, 849831 CrossRef.
- Y. Zeng, C. Huang, D. Duan, A. Lou, Y. Guo, T. Xiao, J. Wei, S. Liu, Z. Wang, Q. Yang, L. Zhou, Z. Wu and L. Wang, Acta Biomater., 2022, 153, 108–123 CrossRef CAS.
- G. G. De Lima, B. D. Ferreira, M. Matos, B. L. Pereira, M. J. D. Nugent, F. A. Hansel and W. L. E. Magalhães, Carbohydr. Polym., 2020, 245, 116612 CrossRef CAS PubMed.
- M. Bustamante-Torres, D. Romero-Fierro, B. Arcentales-Vera, K. Palomino, H. Magaña and E. Bucio, Gels, 2021, 7, 182 CrossRef CAS.
- H. Kang, R. Liu and Y. Huang, Macromol. Chem. Phys., 2016, 217, 1322–1334 CrossRef CAS.
- X. Zhang, X. Ma, T. Hou, K. Guo, J. Yin, Z. Wang, L. Shu, M. He and J. Yao, Angew. Chem., 2019, 131, 7444–7448 CrossRef.
- C. Wu, J. Li, Y. Zhang, X. Li, S. Wang and D. Li, ChemSusChem, 2023, 16, e202300518 CrossRef CAS PubMed.
- M. Ghasemi, M. Tsianou and P. Alexandridis, Bioresour. Technol., 2017, 228, 330–338 CrossRef CAS PubMed.
- B. Lindman, B. Medronho, L. Alves, C. Costa, H. Edlund and M. Norgren, Phys. Chem. Chem. Phys., 2017, 19, 23704–23718 RSC.
- D. E. Ciolacu and D. M. Suflet, Biomass as Renewable Raw Material to Obtain Bioproducts of High-Tech Value, Elsevier, 2018, pp. 401–439 Search PubMed.
- C. Xie, G. Liu, L. Wang, Q. Yang, F. Liao, X. Yang, B. Xiao and L. Duan, Pharmaceutics, 2024, 16, 430 CrossRef CAS PubMed.
- Z. Xie, B.-L. Hu, R.-W. Li and Q. Zhang, ACS Omega, 2021, 6, 9319–9333 CrossRef CAS PubMed.
- T. Kopač, A. Ručigaj and M. Krajnc, Int. J. Biol. Macromol., 2020, 159, 557–569 CrossRef PubMed.
- Y. Dong, S. Zhao, W. Lu, N. Chen, D. Zhu and Y. Li, RSC Adv., 2021, 11, 10794–10803 RSC.
- O. Haske-Cornelius, S. Bischof, B. Beer, M. Jimenez Bartolome, E. Olatunde Olakanmi, M. Mokoba, G. M. Guebitz and G. S. Nyanhongo, Eur. Polym. J., 2019, 120, 109201 CrossRef CAS.
- S. Zhao, Z. Chen, Y. Dong, W. Lu and D. Zhu, Gels, 2022, 8, 146 CrossRef CAS PubMed.
- X. Zhang, W. Wang, Y. Wang, Y. Wang, X. Wang, G. Gao, G. Chen and A. Liu, Food Hydrocolloids, 2018, 84, 1–8 CrossRef CAS.
- W. Song, J. Ko, Y. H. Choi and N. S. Hwang, APL Bioengineering, 2021, 5, 021502 CrossRef CAS PubMed.
- X. Xue, Y. Hu, S. Wang, X. Chen, Y. Jiang and J. Su, Bioactive Mater., 2022, 12, 327–339 CrossRef CAS PubMed.
- R. Naranjo-Alcazar, S. Bendix, T. Groth and G. Gallego Ferrer, Gels, 2023, 9, 230 CrossRef CAS PubMed.
- L. Trachsel, C. Johnbosco, T. Lang, E. M. Benetti and M. Zenobi-Wong, Biomacromolecules, 2019, 20, 4502–4511 CrossRef CAS.
- L. Zhang, C. Wan, J. Su, C. Zhang, S. Wei, W. Tian, X. Liu, W. Cheng, X. Li, X. Li, X. Guo, K.-T. Yong and Y. Wu, Mater. Des., 2022, 215, 110464 CrossRef CAS.
- L. Li, J. Guo, C. Kang and H. Song, Polymers, 2023, 15, 1765 CrossRef CAS PubMed.
- K. J. De France, K. J. W. Chan, E. D. Cranston and T. Hoare, Biomacromolecules, 2016, 17, 649–660 CrossRef CAS PubMed.
- T. Zhang, T. Zuo, D. Hu and C. Chang, ACS Appl. Mater. Interfaces, 2017, 9, 24230–24237 CrossRef CAS.
- H. Xu, Y. Liu, Y. Xie, E. Zhu, Z. Shi, Q. Yang and C. Xiong, Cellulose, 2019, 26, 8645–8654 CrossRef CAS.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Materials, 2019, 12, 3323 CrossRef CAS PubMed.
- P. Bertsch, M. Diba, D. J. Mooney and S. C. G. Leeuwenburgh, Chem. Rev., 2023, 123, 834–873 CrossRef CAS.
- S. A. Eming, P. Martin and M. Tomic-Canic, Sci. Transl. Med., 2014, 6 DOI:10.1126/scitranslmed.3009337.
- E. Y. X. Loh, N. Mohamad, M. B. Fauzi, M. H. Ng, S. F. Ng and M. C. I. Mohd Amin, Sci. Rep., 2018, 8, 2875 CrossRef.
- N. T. Laçin, Int. J. Biol. Macromol., 2014, 67, 22–27 CrossRef PubMed.
- M. Pandey, N. Mohamad, W.-L. Low, C. Martin and M. C. I. Mohd Amin, Drug Delivery Transl. Res., 2017, 7, 89–99 CrossRef CAS PubMed.
- S. Cometa, C. Licini, M. A. Bonifacio, P. Mastrorilli, M. Mattioli-Belmonte and E. De Giglio, Carbohydr. Polym., 2022, 283, 119145 CrossRef CAS PubMed.
- K. Zhang, D. Wu, L. Chang, W. Duan, Y. Wang, W. Li and J. Qin, Int. J. Biol. Macromol., 2023, 230, 123294 CrossRef CAS PubMed.
- F. Cheng, X. Yi, J. Dai, Z. Fan, J. He, Y. Huang and H. Li, Cell Rep. Phys. Sci., 2023, 4, 101619 CrossRef CAS.
- R. Khan, M. U. Aslam Khan, G. M. Stojanović, A. Javed, S. Haider and S. I. Abd Razak, ACS Omega, 2024, 9, 6527–6536 CrossRef CAS PubMed.
- W. Song, T. Xu, L. Qian, S. Zhang, C. Wang, Y. Zhao, Z. He, V. Nica and Z. Miao, Int. J. Biol. Macromol., 2023, 244, 125353 CrossRef CAS PubMed.
- F. Wahid, X.-J. Zhao, X.-Q. Zhao, X.-F. Ma, N. Xue, X.-Z. Liu, F.-P. Wang, S.-R. Jia and C. Zhong, ACS Appl. Mater. Interfaces, 2021, 13, 32716–32728 CrossRef CAS.
- N. S. V. Capanema, A. A. P. Mansur, I. C. Carvalho, S. M. Carvalho and H. S. Mansur, Gels, 2023, 9, 166 CrossRef CAS.
- Z. Zhang, S. Lin, Y. Yan, X. You and H. Ye, J. Biomater. Sci., Polym. Ed., 2021, 32, 2402–2422 CrossRef CAS.
- M. Ghorbani, L. Roshangar and J. Soleimani Rad, Eur. Polym. J., 2020, 130, 109697 CrossRef CAS.
- X. Yu, X. Li, L. Kan, P. Pan, X. Wang, W. Liu and J. Zhang, Int. J. Biol. Macromol., 2023, 238, 124113 CrossRef CAS PubMed.
- L. Gu, T. Li, X. Song, X. Yang, S. Li, L. Chen, P. Liu, X. Gong, C. Chen and L. Sun, Regener. Biomater., 2020, 7, 195–202 CrossRef CAS PubMed.
- S. Chayanun, A. A. Soufivand, J. Faber, S. Budday, B. Lohwongwatana and A. R. Boccaccini, Adv. Eng. Mater., 2024, 26, 2300641 CrossRef CAS.
- J. Sinna, R. Jeencham, P. Mueangkhot, S. Sophon, P. Noralak, R. Raksapakdee, P. Numpaisal and Y. Ruksakulpiwat, Polymers, 2023, 15, 4230 CrossRef CAS PubMed.
- P. Basu, N. Saha and P. Saha, Int. J. Polym. Mater. Polym. Biomater., 2019, 68, 134–144 CrossRef CAS.
- M. G. Raucci, M. A. Alvarez-Perez, C. Demitri, D. Giugliano, V. De Benedictis, A. Sannino and L. Ambrosio, J. Biomed. Mater. Res., 2015, 103, 2045–2056 CrossRef CAS PubMed.
- D. Burger, M. Beaumont, T. Rosenau and Y. Tamada, Molecules, 2020, 25, 5097 CrossRef CAS PubMed.
- S. Saber-Samandari, S. Saber-Samandari, S. Kiyazar, J. Aghazadeh and A. Sadeghi, Int. J. Biol. Macromol., 2016, 86, 434–442 CrossRef CAS PubMed.
- S. Cui, S. Zhang and S. Coseri, Carbohydr. Polym., 2023, 300, 120243 CrossRef CAS PubMed.
- G. Priya, B. Madhan, U. Narendrakumar, R. V. Suresh Kumar and I. Manjubala, ACS Omega, 2021, 6, 1246–1253 CrossRef CAS PubMed.
- H. Tohidi, N. Maleki-Jirsaraei, A. Simchi, F. Mohandes, Z. Emami, L. Fassina, F. Naro, B. Conti and F. Barbagallo, Materials, 2022, 15, 5122 CrossRef CAS PubMed.
- E. Entcheva, H. Bien, L. Yin, C.-Y. Chung, M. Farrell and Y. Kostov, Biomaterials, 2004, 25, 5753–5762 CrossRef CAS PubMed.
- S. Sedighim, Y. Chen, C. Xu, R. Mohindra, H. Liu, D. K. Agrawal and F. G. Thankam, Biotechnol. Bioeng., 2023, 120, 819–835 CrossRef CAS PubMed.
- C. Sun, Y. Xie, H. Zhu, X. Zheng, R. Hou, Z. Shi, J. Li and Q. Yang, Biomacromolecules, 2023, 24, 5989–5997 CrossRef CAS PubMed.
- E. Ahmadi, P. Harirchi, P. Zahedi, B. Bakhshandeh, S. Zolfagharian and A. Khatibi, Fibers Polym., 2023, 24, 2293–2303 CrossRef CAS.
- Z. Shi, H. Gao, J. Feng, B. Ding, X. Cao, S. Kuga, Y. Wang, L. Zhang and J. Cai, Angew. Chem., Int. Ed., 2014, 53, 5380–5384 CrossRef CAS PubMed.
- K.-C. Cheng, C.-F. Huang, Y. Wei and S. Hsu, NPG Asia Mater., 2019, 11, 25 CrossRef CAS.
- L. Deng, Y. Huang, S. Chen, Z. Han, Z. Han, M. Jin, X. Qu, B. Wang, H. Wang and S. Gu, Carbohydr. Polym., 2023, 308, 120647 CrossRef CAS PubMed.
- H. Lu, X. Li, M. Zhang, C. Xu, W. Li and L. Wan, Front. Bioeng. Biotechnol., 2022, 10, 876936 CrossRef PubMed.
- M. Tian, J. Shuai, B. A. Bishop, W. Zhang, J. Chen and X. Wang, Chem. Eng. J., 2023, 476, 146751 CrossRef CAS.
- T. Sultana, M. Hossain, S. Rahaman, Y. S. Kim, J.-G. Gwon and B.-T. Lee, Carbohydr. Polym., 2021, 272, 118482 CrossRef CAS PubMed.
- A. A. Shefa, T. Sultana, M. K. Park, S. Y. Lee, J.-G. Gwon and B.-T. Lee, Mater. Des., 2020, 186, 108313 CrossRef CAS.
- W. Hu, Z. Chen, X. Chen, K. Feng, T. Hu, B. Huang, J. Tang, G. Wang, S. Liu, G. Yang and Z. Wang, Carbohydr. Polym., 2023, 319, 121193 CrossRef CAS PubMed.
- G. Yang, Z. Zhang, K. Liu, X. Ji, P. Fatehi and J. Chen, J. Nanobiotechnol., 2022, 20, 312 CrossRef CAS PubMed.
- H. Mohaghegh, Z. Assadi, A. Derakhshan and E. Masaeli, J. Pharm. Sci., 2023, 113, 754–763 CrossRef PubMed.
- H. Wu, G. R. Williams, J. Wu, J. Wu, S. Niu, H. Li, H. Wang and L. Zhu, Carbohydr. Polym., 2018, 180, 304–313 CrossRef CAS PubMed.
- Y. Li, X. Xun, Y. Xu, A. Zhan, E. Gao, F. Yu, Y. Wang, H. Luo and C. Yang, Carbohydr. Polym., 2022, 276, 118790 CrossRef CAS PubMed.
- Y.-C. Chen, H.-J. Liao, Y.-M. Hsu, Y.-S. Shen and C.-H. Chang, Polymers, 2022, 14, 1474 CrossRef CAS PubMed.
- G. Osterhoff, E. F. Morgan, S. J. Shefelbine, L. Karim, L. M. McNamara and P. Augat, Injury, 2016, 47, S11–S20 CrossRef PubMed.
- X. Wang, S. Tang, S. Chai, P. Wang, J. Qin, W. Pei, H. Bian, Q. Jiang and C. Huang, Carbohydr. Polym., 2021, 270, 118342 CrossRef CAS PubMed.
- P. Wang and X. Wang, Eng. Regener., 2022, 3, 440–452 Search PubMed.
- C. Ao, Y. Niu, X. Zhang, X. He, W. Zhang and C. Lu, Int. J. Biol. Macromol., 2017, 97, 568–573 CrossRef CAS.
- S. Odabas, J. Bioact. Compat. Polym., 2016, 31, 411–422 CrossRef CAS.
- P. Daugela, M. Pranskunas, G. Juodzbalys, J. Liesiene, O. Baniukaitiene, A. Afonso and P. Sousa Gomes, J. Tissue Eng. Regener. Med., 2018, 12, 1195–1208 CrossRef CAS.
- M. Matinfar, A. S. Mesgar and Z. Mohammadi, Mater. Sci. Eng., C, 2019, 100, 341–353 CrossRef CAS.
- M. Vallet-Regi and A. J. Salinas, Mater. Today Bio, 2021, 11, 100121 CrossRef CAS PubMed.
- S. Simorgh, N. Alasvand, M. Khodadadi, F. Ghobadi, M. Malekzadeh Kebria, P. Brouki Milan, S. Kargozar, F. Baino, A. Mobasheri and M. Mozafari, Methods, 2022, 208, 75–91 CrossRef CAS PubMed.
- M. N. Rahaman, D. E. Day, B. Sonny Bal, Q. Fu, S. B. Jung, L. F. Bonewald and A. P. Tomsia, Acta Biomater., 2011, 7, 2355–2373 CrossRef CAS PubMed.
- L. Li, P. Lu, Y. Liu, J. Yang and S. Li, Polymers, 2023, 15, 2226 CrossRef CAS PubMed.
- N. Ma, D. Y. Cheung and J. T. Butcher, J. Biomed. Mater. Res., 2022, 110, 76–91 CrossRef CAS PubMed.
- S. Y. Srinivasan, M. Cler, O. Zapata-Arteaga, B. Dörling, M. Campoy-Quiles, E. Martínez, E. Engel, S. Pérez-Amodio and A. Laromaine, ACS Appl. Bio Mater., 2023, 6, 2860–2874 CrossRef CAS PubMed.
- R. Hou, Y. Xie, R. Song, J. Bao, Z. Shi, C. Xiong and Q. Yang, Cellulose, 2024, 31, 4247–4262 CrossRef CAS.
- V. Kuzmenko, E. Karabulut, E. Pernevik, P. Enoksson and P. Gatenholm, Carbohydr. Polym., 2018, 189, 22–30 CrossRef CAS PubMed.
- S. Gnavi, B. Fornasari, C. Tonda-Turo, R. Laurano, M. Zanetti, G. Ciardelli and S. Geuna, IJMS, 2015, 16, 12925–12942 CrossRef CAS PubMed.
- H.-T. Chang, R. A. Heuer, A. M. Oleksijew, K. S. Coots, C. B. Roque, K. T. Nella, T. L. McGuire and A. J. Matsuoka, Acta Biomater., 2020, 108, 111–127 CrossRef CAS PubMed.
- R. J. Dilley and W. A. Morrison, Int. J. Biochem. Cell Biol., 2014, 56, 38–46 CrossRef CAS PubMed.
- L. M. Miller and A. Gal, Pathologic Basis of Veterinary Disease, Elsevier, 2017, pp. 561–616.e1 Search PubMed.
- Y. Lee, S. I. Ahn and Y. Kim, Encyclopedia of Biomedical Engineering, Elsevier, 2019, pp. 384–393 Search PubMed.
- P. Libby, J. E. Buring, L. Badimon, G. K. Hansson, J. Deanfield, M. S. Bittencourt, L. Tokgözoğlu and E. F. Lewis, Nat. Rev. Dis. Primers, 2019, 5, 56 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |