
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Latest technological advances and insights into capture and removal of hydrogen sulfide: a critical review
Muhammad Syahir
Aminuddin
 *ab,
Mohamad Azmi
Bustam
ab and
Khairiraihanna
Johari
*ac
*ab,
Mohamad Azmi
Bustam
ab and
Khairiraihanna
Johari
*ac
aDepartment of Chemical Engineering, Universiti Teknologi PETRONAS, 32610 Bandar Seri Iskandar, Perak, Malaysia. E-mail: khairiraihana.j@utp.edu.my
bCentre of Research in Ionic Liquids (CORIL), Universiti Teknologi PETRONAS, 32610 Bandar Seri Iskandar, Perak, Malaysia
cCO2 Research Centre (CO2RES), Universiti Teknologi PETRONAS, 32610 Bandar Seri Iskandar, Perak, Malaysia
First published on 7th March 2024
Abstract
Hydrogen sulfide is an extremely toxic, poisonous and flammable gas often found in natural gas streams and crude oil reservoirs. Due to its hazardous and corrosive nature, it must be effectively removed to protect human health and for economic reasons. To overcome these issues, various technologies and methods have been implemented for efficient capture of H2S. This work presents a comprehensive review of various up-to-date technologies and materials such as ionic liquids, deep eutectic solvents, carbon-based adsorbents, zeolites, metal organic frameworks, membranes and composite materials. Furthermore, an in-depth discussion for each technology and class of material is also included. Besides, potential opportunities and limitations are also identified to further enhance the development in future research. By evaluating eco-friendly and cost-effective techniques, our work contributes to reducing harmful emission of H2S, protecting air quality and promoting cleaner industries. Our work aligns with the UN's sustainable development goals, specifically SDG 7 (affordable and clean energy) and SDG 13 (climate action), by advocating for a cleaner and more sustainable energy sector, as well as SDG 9 (industry, innovation, and infrastructure) through innovative solutions for cleaner industrial processes.
Sustainability spotlightTo minimize the impact of global climate change and environmental degradation, it is of paramount importance to address the urgent need to capture and remove hydrogen sulfide, an extremely toxic and pollutant gas. Our review paper provides a comprehensive analysis of cutting-edge methods that promise sustainable solutions for this critical issue. By evaluating eco-friendly and cost-effective techniques, our work contributes to reducing harmful emission of H2S, protecting air quality and promoting cleaner industries. Our work aligns with the UN's sustainable development goals, specifically SDG 7 (affordable and clean energy) and SDG 13 (climate action), by advocating for a cleaner and more sustainable energy sector, as well as SDG 9 (industry, innovation, and infrastructure) through innovative solutions for cleaner industrial processes. |
1. Introduction
Hydrogen sulfide (H2S) conversion and capture has presented a persistent economic and environmental challenge throughout the last century.1 H2S is a noxious, foul-smelling, and toxic substance that frequently contaminates crucial fuel gases.2 It is essential to eliminate this compound from these streams due to both economic and safety concerns. The natural tendency of H2S to create acidic solutions when combined with water leads to corrosive damage in equipment and pipelines.3 Additionally, the presence of H2S in fuel gases diminishes the heating value4 and causes catalyst poisoning.4 Notably, the combustion of H2S generates sulfur dioxide and other harmful sulfur oxides, which contribute to acid rain.5 Furthermore, as indicated in Table 1, H2S is a toxic gas that poses hazards even at low concentrations.6,7 Prolonged exposure to around 5 ppm causes irritation of the eyes and respiratory system, while concentrations of 1000–2000 ppm result in immediate fatality. Hence, it is crucial to control and limit H2S emissions to enhance worldwide atmospheric chemistry and improve overall life quality.8| Concentration (ppm) | Acute health symptoms |
|---|---|
| 0.0001–0.0003 | Standard ambient levels |
| 0.01–1.5 | Minimum limit where the distinctive smell of rotten egg becomes noticeable |
| 2–5 | Extended exposure might lead to headaches, nausea and insomnia |
| 20 | May cause fatigue, headache, appetite loss, forgetfulness, irritability and nausea |
| 50–100 | Irritation to respiratory tract and slight conjunctivitis “gas eyes” after 1 hour |
| Might lead to loss of appetite and digestive upset | |
| 100–150 | Eye irritation, coughing and loss of smell within 2 to 15 minutes |
| Altered breathing and sleepiness after 15 to 30 minutes | |
| Sore throat after 1 hour | |
| The symptom severity increases steadily over several hours | |
| Death possibility after 48 hours | |
| 200–300 | Evident conjunctivitis and irritation of breathing passages after 60 minutes |
| Extended exposure may cause pulmonary edema to occur | |
| 500–700 | Staggering, loss of coordination and collapse after 5 minutes |
| Serious eye damage in 30 minutes | |
| Death within 30 to 60 minutes | |
| 700–1000 | Death within several minutes due to respiratory paralysis |
| Instant “knockdown” or collapse within a few breaths | |
| 1000–2000 | Almost instant death |
Consequently, to utilize various fuel gases for generating energy or chemical manufacturing, it becomes necessary to purify them by eliminating and/or converting acid gases such as H2S and CO2. The acceptable level of H2S in a gas stream relies on the specific intended use and local regulations. For instance, in the United States and Denmark, pipeline gas must contain less than 4 ppm of H2S,9 whereas fuel cell and reformer applications typically need lower than 1 ppm of H2S.10,11 The techniques for H2S capture can be categorized into several classes such as absorption, adsorption, chemical conversion, membrane separation and cryogenic distillation. This extensive review focuses on the latest developments in each of these technologies, with a particular emphasis on advancements made within the past 5 to 15 years. Each technique has its own sets of advantages and disadvantages influenced by various aspects, which will be elaborated upon in the following sections. The summary of technology selection for this review is presented in Fig. 1.
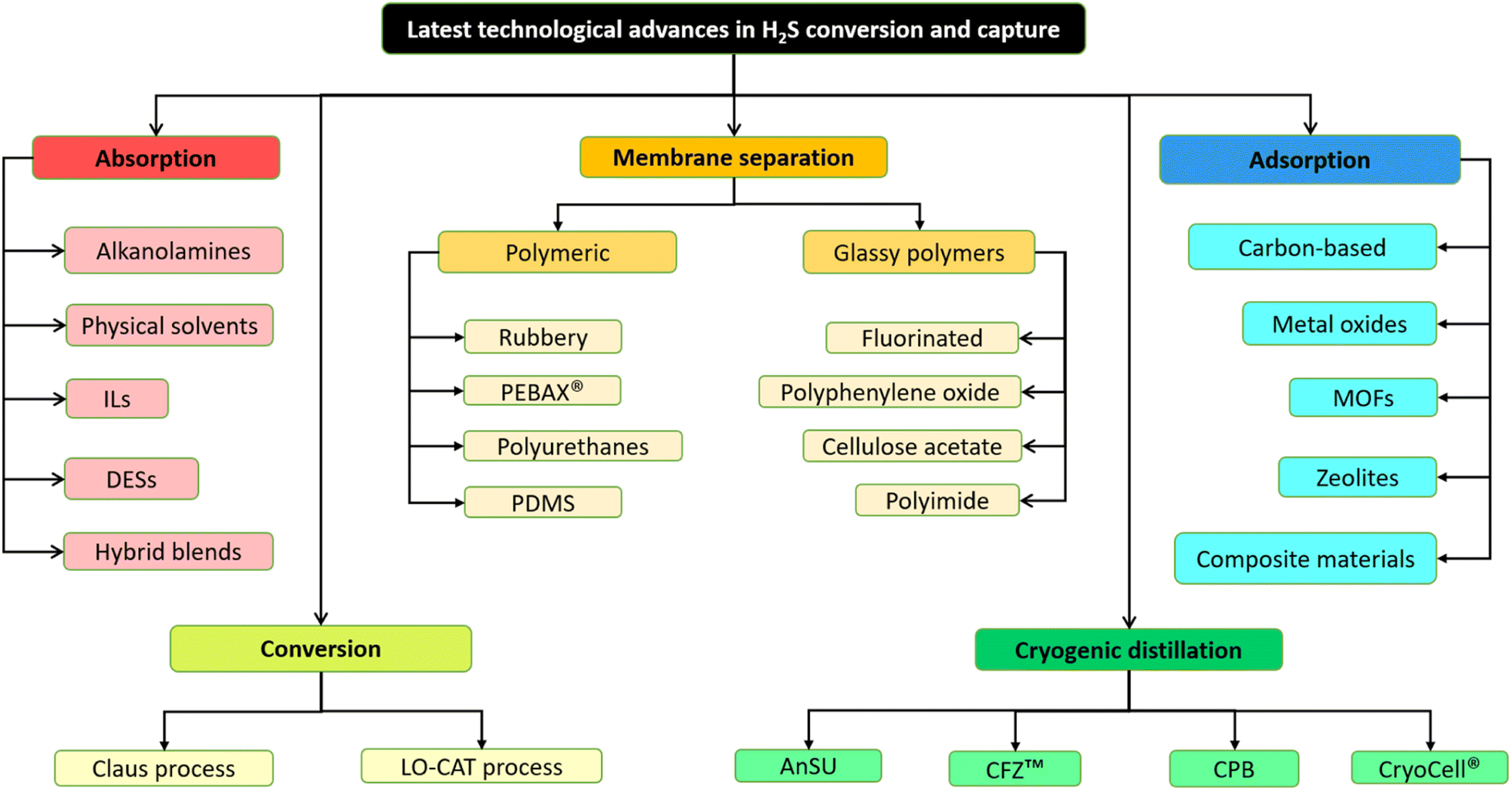 | ||
| Fig. 1 The summary of technology selection for H2S removal and capture. | ||
2. Technologies for H2S capture and removal
2.1. Absorption
Absorption is among the most established technologies in the oil and gas industry for removal of acid gases such as H2S and CO2 from natural gas streams using a liquid solvent.12 H2S absorption involves transfer of H2S from the gas feed into the physical or chemical solvent in the packed or plated conventional absorption columns. In general, the reaction mechanisms for the absorption process can be divided into two categories such as chemical and physical absorption, based on the interaction strength between the solvents and H2S.Chemical absorption involves strong chemical bonds that are difficult to break, whereas physical absorption involves weak intermolecular forces that are easier to overcome. Typically, chemical absorption operates at low pressure, while physical absorption requires high pressure to operate optimally. As a result, physical solvents are preferable as compared to chemical solvents under the conditions where the feed gas contains high H2S concentrations and partial pressure. Physical solvent systems are identical to chemical solvent systems but are dependent on the gas solubility in the solvent instead of reaction stoichiometry.
The solubility of acid gases such as H2S and CO2 is heavily reliant on the gas partial pressure and reaction temperature of the system. Higher partial pressures lead to higher gas solubility. In contrast, low temperatures lead to higher gas solubility. However, in general, temperature is not as critical as pressure. Moreover, the solvents can be regenerated by adjusting the pressure and temperature.13 Absorption methods can be classified into five major classes such as alkanolamines, physical solvents, ionic liquids (ILs), deep eutectic solvents (DESs) and hybrid blends. Table 2 provides the summary of recent studies on H2S absorption using the different classes of solvents stated beforehand.
| Solvents | Feed gas | Operating conditions and H2S partial pressure | Solubility/loading (mol mol−1) | H2S removal efficiency | Reference |
|---|---|---|---|---|---|
| Alkanolamines | |||||
| 30–50 wt% MDEA + 80 wt% MEA | Simulated coke oven gas | Temperature: 30–50 °C, partial pressure: 0.003–0.005 bar | 0.022–0.096 | 93.6–95.9% (6 sieve trays) | 14 |
| 3.5–11.6 wt% MEA | Gases: H2S and CH4 | Temperature: 40–60 °C, partial pressure: 0–0.45 bar | −0.1–1.0 | n/a | 15 |
| 19.0–46.3 wt% MDEA | Partial pressure: 0.01–0.5 bar | −0.05–0.85 | n/a | 16 | |
| 5.6 wt% MEA + 26.3 wt% MDEA + 3–7 wt% PZ | Partial pressure: 0.04–0.55 bar | −0.09–0.95 | n/a | ||
| 20 wt% MDEA | Gases: H2S and N2 | Temperature: 5–40 °C, partial pressure: 0.0003–0.01 bar | 0.015–0.25 | 100% | |
| 20 wt% MDEA + 80 wt% MEG | Temperature: 5 °C, partial pressure: 0.0003–0.01 bar | 0.01–0.09 | 77% | ||
| 20 wt% MDEA + 80 wt% TEG | 0.006–0.040 | 17% | |||
| 20 wt% t-BDEA | 0.009–0.407 | n/a | |||
| 20 wt% DIPA | 0.012–0.185 | n/a | |||
| 20 wt% TEA | 0.013–0.165 | n/a | |||
| 20–50 wt% DEAE-EO | Gases: H2S and N2 | Temperature: 5–40 °C, partial pressure: 0.005–0.01 bar | 0.094–0.416 | 100% | |
| 20 wt% DEAE-EO + 80 wt% MEG | Temperature: 5 °C, partial pressure: 0.01 bar | 0.280 | 76% | ||
| 20 wt% DEAE-EO + 80 wt% TEG | 0.073 | 21% | |||
| 20–50 wt% 3DEA-1P | Gases: H2S and N2 | Temperature: 5–40 °C, partial pressure: 0.005–0.01 bar | 0.061–0.355 | 100% | |
| 20 wt% 3DEA-1P + 80 wt% MEG | Temperature: 5 °C, partial pressure: 0.01 bar | 0.193 | 57% | ||
| 20 wt% 3DEA-1P + 80 wt% TEG | 0.080 | 24% | |||
| MDEA + PZ | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppmv H2S and remaining CO2 000 ppmv H2S and remaining CO2 |
Temperature: 30–45 °C, partial pressure: 1 bar | n/a | 100% | 17 |
| 10–30 wt% TBE | 15 mol% H2S and 85 mol% CO2 | Temperature: 25–45 °C, partial pressure: n/a | n/a | 50–94% | 18 and 19 |
| 10–30 wt% AMP | n/a | 65–95% | |||
|
|||||
| ILs | |||||
| [N2224]2[maleate] | Gas: H2S | Temperature: 40 °C, partial pressure: 1.1 bar | 1.43 | n/a | 20 |
| [N2224]2[maleate] + maleic acid (1:1) |
Partial pressure: 1.18 bar | 1.30 | n/a | ||
| [BDMAEEH][NTf2] + MAA (1:1) |
0.8 | n/a | |||
| [BDMAEEH][MAA] | 1.0 | n/a | |||
| [BDMAEEH][NTf2] | Partial pressure: 1.1 bar | 0.4 | n/a | ||
| [DBNH][1,2,4-triaz] | Gas: H2S | Temperature: 25 °C, partial pressure: 1 bar | 1.4 | n/a | 21 |
| Temperature: 40 °C, partial pressure: 0.012–0.019 bar | 0.253–1.198 | n/a | |||
| Temperature: 60 °C, partial pressure: 1 bar | 1.0 | n/a | |||
| [DBNH][1,2,3-triaz] | Gas: H2S | Temperature: 60 °C, partial pressure: 0.016–1.089 bar | 0.287–1.081 | n/a | |
| [DBUH][1,2,4-triaz] | Partial pressure: 0.02–1.03 bar | 0.447–1.174 | n/a | ||
| [DBUH][1,2,3-triaz] | Partial pressure: 0.02–1.03 bar | 0.412–1.046 | n/a | ||
| [DBNH][Im] | Gas: H2S | Temperature: 25 °C, partial pressure: 0.1 bar | 1.18 | n/a | 22 |
| Partial pressure: 0.01–1 bar | 0.63–1.36 | n/a | |||
| Temperature: 40 °C, partial pressure: 0.1 bar | 0.95 | n/a | |||
| Partial pressure: 0.01–1 bar | 0.40–1.26 | n/a | |||
| Temperature: 60 °C, partial pressure: 0.01–1 bar | 0.27–1.08 | n/a | |||
| [DBUH][Im] | Gas: H2S | Temperature: 40 °C, partial pressure: 0.1–1 bar | 0.98–1.26 | n/a | |
| [DBNH][Pyr] | 0.99–1.31 | n/a | |||
| [DBUH][Pyr] | 0.95–1.17 | n/a | |||
| [TMGH][PhO] | Gas: H2S | Temperature: 25 °C, partial pressure: 0.1 bar | 0.69 | n/a | 23 |
| Partial pressure: 0.01–1 bar | 0.38–0.97 | n/a | |||
| Temperature: 40 °C, partial pressure: 0.1 bar | 0.56 | n/a | |||
| Partial pressure: 0.01–1 bar | 0.20–0.85 | n/a | |||
| Temperature: 60 °C, partial pressure: 0.1 bar | 0.47 | n/a | |||
| Partial pressure: 0.01–1 bar | 0.08–0.78 | n/a | |||
| [DBUH][PhO] | Temperature: 40 °C, partial pressure: 0.1–1 bar | 0.6–0.8 | n/a | ||
| [Hmim][PhO] | 0.59–0.84 | n/a | |||
| [P4444][PhO] | 0.62–0.88 | n/a | |||
| 40–70 wt% [BDMAEE][Ac] | Temperature: 25 °C, partial pressure: 1 bar | 0.950–1.044 | n/a | 24 | |
| [NEMH][Bu] | Temperature: 25 °C, partial pressure: 0.035–1.056 bar | 0.012–0.156 | n/a | 25 | |
| Temperature: 35 °C, partial pressure: 0.056–1.07 bar | 0.008–0.114 | n/a | |||
| Temperature: 45 °C, partial pressure: 0.03–1.05 bar | 0.007–0.098 | n/a | |||
| [NEMH][Ac] | Temperature: 25 °C, partial pressure: 0.04–1.06 bar | 0.009–0.127 | n/a | ||
| [NEMH][Pro] | 0.011–0.151 | n/a | |||
| [NEMH][MoAc] | 0.010–0.142 | n/a | |||
| [TEAH][Bu] | Partial pressure: 0.04–1.1 bar | 0.009–0.135 | n/a | ||
| [TEAH][MoAc] | Partial pressure: 0.06–1.02 bar | 0.010–0.138 | n/a | ||
| [C4Py][BF4] | Gas: H2S | Temperature: 40 °C, partial pressure: 1 bar | 0.07 | n/a | 26 |
| [C4Py][SCN] | Partial pressure: 1 bar | 0.1 | n/a | ||
| [Emim][BF4] | Temperature: 25 °C, partial pressure: 0.003–14 bar | 0–1.46 | n/a | 27 | |
| Temperature: 40 °C, partial pressure: 0.003–18.4 bar | 0–1.26 | n/a | |||
| Temperature: 60 °C, partial pressure: 0.003–18.2 bar | 0–0.674 | n/a | |||
| Temperature: 80 °C, partial pressure: 0.003–17.6 bar | 0–0.425 | n/a | |||
|
|||||
| DESs | |||||
| ChCl-urea (1:1.5) |
Temperature: 40 °C, partial pressure: 0.11–2.02 bar | 0.0037–0.055 | n/a | 28 | |
| Temperature: 50 °C, partial pressure: 0.11–2.01 bar | 0.0025–0.046 | n/a | |||
| Temperature: 60 °C, partial pressure: 0.10–2.02 bar | 0.0021–0.037 | n/a | |||
| Temperature: 80 °C, partial pressure: 0.1–1.98 bar | 0.0014–0.023 | n/a | |||
| ChCl-urea (1:2) |
Temperature: 40–80 °C, partial pressure: 0.10–2.02 bar | 0.0015–0.046 | n/a | ||
| ChCl-urea (1:2.5) |
Temperature: 40–80 °C, partial pressure: 0.10–2.01 bar | 0.0011–0.035 | n/a | ||
| TBAB-ProH (1:1) |
Gas: H2S | Temperature: 25 °C, partial pressure: 2.45–4.96 bar | 0.40–0.60 | n/a | 29 |
| TBAB-AcH (1:1) |
Partial pressure: 2.25–5.11 bar | 0.32–0.59 | n/a | ||
| TBAB-ForH (1:1) |
Partial pressure: 1.78–5.06 bar | 0.21–0.55 | n/a | ||
| ChCl-ProH (1:2) |
Partial pressure: 1.84–5.40 bar | 0.13–0.44 | n/a | ||
| ChCl-AcH (1:2) |
Partial pressure: 2.49–5.22 bar | 0.16–0.33 | n/a | ||
| ChCl-ForH (1:2) |
Partial pressure: 2.62–5.10 bar | 0.12–0.25 | n/a | ||
| [C4-TMHDA][Cl]-Im (1:2) |
Gases: H2S, CH4, and CO2 | Temperature: 30 °C, partial pressure: 1 bar | 0.99 | n/a | 30 |
| [C4-TMPDA][Cl]-Im (1:2) |
0.48 | n/a | |||
| [C4-TMEDA][Cl]-Im (1:2) |
0.36 | n/a | |||
| [C4-TMEDA][Cl]-1,2,4-triaz (1:2) |
0.27 | n/a | |||
| [C4-TMEDA][Cl]-1,2,3-triaz (1:2) |
0.24 | n/a | |||
| [C4-TMHDA][Cl]-Im (1:2) |
Temperature: 40 °C, partial pressure: 1 bar | 0.71 | n/a | ||
| [C1-TMHDA][Ac]-MDEA (1:2) |
Gases: H2S, CH4, and CO2 | Temperature: 40 °C, partial pressure: 1 bar | 1.44 | n/a | 31 |
| [C1-TMHDA][Ac]-pyrol (1:2) |
1.17 | n/a | |||
| [C1-TMHDA][Ac]-AA (1:2) |
1.09 | n/a | |||
| [C1-TMHDA][Ac]-Im (1:2) |
1.02 | n/a | |||
|
|||||
| Hybrid blends | |||||
| 30–50 wt% MDEA + 2.5–7.5 wt% [N1111][Arg] | Simulated coke oven gas | Temperature: 30–50 °C, partial pressure: 0.003–0.005 bar | 0.039–0.143 | 100% (requires 4 sieve trays) | 32 |
| 30–50 wt% MDEA + 2.5–7.5 wt% [N1111][Gly] | Simulated coke oven gas | Temperature: 30–50 °C, partial pressure: 0.003–0.005 bar | 0.035–0.98 | 100% (requires 6 sieve trays) | 14 |
| 30–50 wt% DIPA + 5–50 wt% [Bmim][Ac] | 30 mol% CO2 and H2S | Temperature: 50–75 °C, partial pressure: 0.46–6.68 bar | 0.010–0.153 | n/a | 33 |
|
|||||
| Benchmarks | |||||
| Dimethyl sulfoxide (DMSO) | Gas: H2S | Temperature: 25 °C, partial pressure: 1 bar | 0.123 | n/a | 34 |
| N-methyl-2-pyrrolidone (NMP) | Gas: H2S | Temperature: 25 °C, partial pressure: 1 bar | 0.013 | n/a | 35 |
| Sulfolane | Gas: H2S | Temperature: 25 °C, partial pressure: 1 bar | 0.072 | n/a | 36 |
| Temperature: 40 °C, partial pressure: 1 bar | 0.043 | n/a | |||
| 50 wt% MDEA | Gas: H2S | Temperature: 25 °C, partial pressure: 1 bar | 0.944 | n/a | 37 |
| Temperature: 40 °C, partial pressure: 0.1–1 bar | 0.3–0.85 | n/a | |||
| Gases: CH4 and H2S | Temperature: 50 °C, partial pressure: 0.026–1.78 bar | 0.084–0.775 | n/a | 38 | |
| [Bmim][PF6] | Gas: H2S | Temperature: 25–130 °C, partial pressure: 0.69–96.30 bar | 0.016–0.875 | n/a | 39 |
| Methanol | Gas: H2S | Temperature: 25 °C, partial pressure: 1 bar | 0.027 | n/a | 40 |
| Partial pressure: 14 bar | 0.8 | n/a | 41 | ||
| [TEGDME] | Gas: H2S | Temperature: 40 °C, partial pressure: 1 bar | 0.162 | n/a | 42 |
Besides, there are also a large number of conventional solvents available for acid gas absorptive removal such as ADIP, ADEG. OASE, FLEXSORB, GAS/SPEC and UCARSOL series. Privately owned amine processes of low energy by oil companies such as Shell, i.e., ADIP and Sulfinol;49 and ExxonMobil, i.e., FLEXSORB™SE50 and OASE®sulfexx™51 were able to selectively remove H2S in the presence of CO2 commercially. The long-established hot potassium carbonate (HPC) process was pioneered by Benson and Field, before being licensed by Universal Oil Products (UOP) as the UOP Benfield™ process.52 On the other hand, the CATACARB® process which utilizes enhanced HPC technology is commercially proven, with over 150 plants in 30 different countries worldwide facilitating a variety of applications such as natural gas, ammonia, hydrogen and ethylene oxide.52
In chemical solvent processes, the acid gas feed enters the absorption columns at high pressure between 5 and 205 bar and low temperature between 35 and 50 °C, while the solvent enters the regenerator at high temperature between 115 and 130 °C and low pressure between 1.4 and 1.7 bar.53 Generally, primary amines (MEA) and secondary amines (DEA) are more reactive towards acid gas, whereas tertiary amines (MDEA) and sterically hindered amines (AMP) are less reactive but possess higher selectivity towards H2S.
MEA is the most widely used alkanolamine for removal of acid gas due to certain factors such as low cost, low hydrocarbon solubility, high reactivity and ease of recovery. However, MEA suffers from various drawbacks such as strong affinity towards CO2, zero selectivity towards H2S, stable carbamate formation, and highly volatility, corrosivity and degradability, thus limiting its absorption potential.54,55 Similarly, DEA has advantages comparable to those of MEA but is less corrosive and has higher vapor pressure.56 MDEA surpasses both DEA and MEA in performance and efficiency due to its high loading capacity, high selectivity towards H2S, low vapor pressure, low corrosivity and high efficiency. Conversely, when MEA and DEA react with CO2, they form stable carbamates, resulting in a reduced hydrolysis rate to bicarbonate, consequently leading to low absorption of H2S into the amines.57 In contrast, AMP reacts with CO2 to form unstable carbamates, whereas MDEA does not form any carbamates, making them more favorable choices compared to MEA and DEA.58
Among these solvents, a combination of MEA and DEA is commonly used in the sweetening process of natural gas streams. Industrially, aqueous MDEA is mixed with primary or secondary amines such as 2-tertiarybutylamino-2-ethoxyethanol (TBEE), AMP, DGA, MEA, DEA and DIPA to obtain desirable performance and effects. Primary or secondary amines were used as they have higher reaction rates, whereas MDEA has a higher equilibrium capacity. It is also common that three different types of solvents were mixed to achieve the desired target. The blended solvents offer a combined effect of a high reaction rate and high equilibrium capacity. However, these multi-solvent systems involve very complex chemical reactions causing the solvents to be hard to regenerate. Moreover, the gas stripping process at elevated temperatures is an energy intensive process, leading to solvent losses due to thermal degradation and volatilization.59
Abdulrahman and Sebastine60 investigated the best solvent for simultaneous removal of acid gases among MEA, DEA and MDEA. They suggested that 35 wt% DEA is the best and most cost-effective solvent for removal of both CO2 and H2S gases. Another study by Mandal et al.61 revealed that MDEA performs slightly better as compared to AMP in H2S absorption. Lu et al.53 discovered that the performance of an aqueous blend consisting of 1.5 kmol m−2 of MDEA and 1 kmol m−2 of TBEE is far higher than that of 2.5 kmol m−2 of MDEA alone for selective removal of H2S. The authors also deduced that both primary and secondary sterically hindered amines, i.e., TBEE and AMP, exhibited comparable characteristics to tertiary amines such as MDEA. Moreover, Li et al.19 and Du et al.19 investigated the performance of AMP and 2-tert-butylamino ethanol (TBE) for selective removal of H2S from a feed gas that contains 15% H2S and 85% CO2. AMP has a higher performance and selectivity towards H2S at low amine concentrations, whereas TBE performs better at high amine concentrations and displayed slightly higher selectivity towards H2S as compared to AMP. Nevertheless, both AMP and TMP have a similar removal efficiency for H2S.
An activator is typically added to the aqueous amine blend as an additive to enhance the base amine performance. Piperazine (PZ) is an example of a commonly used activator in amine blends because of its high resistance to oxidative and thermal decomposition.62 Sheng et al.63 and Lin et al.64 recorded a higher reaction rate of PZ with CO2 as compared to that of other conventional solvents such as MEA, DEA, MDEA and AMP. Nguyen et al.65 and Dash et al.66 reported that the blends of amines and PZ possess lower volatility than the amines alone as the blends typically form non-ideal solutions. On the other hand, Yunhai et al.15 evaluated the performance of MEA, MDEA, a mixture of MEA and MDEA and a mixture of MEA, MDEA and PZ for H2S capture from natural gas streams. They reported that the performance of MEA was the highest followed by the mixture of MEA, MDEA and PZ, the mixture of MEA and MDEA and MDEA, respectively. The results proved that PZ could improve the loading capacity of H2S but possesses lower selectivity towards H2S in the presence of CO2. Meanwhile, Zhan et al.17 investigated an aqueous blend of MDEA and PZ for H2S absorption under different conditions. It was observed that PZ has a higher reaction rate with CO2 as compared to MDEA. In addition, it was also noticed that a higher concentration of PZ has led to higher CO2 removal but a lower H2S removal efficiency. Similarly, Haghtalab and Izadi67 also recorded a lower H2S loading capacity with the addition of PZ to each aqueous solution of MDEA and DIPA, respectively.
Lee et al.48 performed simultaneous absorption and desorption of H2S and CO2 from a feed gas containing 50 ppmv H2S, 35% CH4, and 15% CO2 with the remainder being N2 using a total of 11 aqueous blends containing 4.5 wt% MDEA and 5 wt% activator. The activators used are AMP, PZ, bis(3-aminopropyl)amine (APA), diethylenetriamine, tetraethylenepentamine, 1-dimethylamino-2-propanol, 2-amino-1-butanol, 5-amino-1-pentanol, dibutylamine, N-propylethylenediamine and 1,4-diaminobutane. PZ was found to be the best activator for CO2 absorption but poor for H2S absorption. On the other hand, APA demonstrated excellent performance for H2S absorption but does not show any selectivity towards H2S when CO2 is present.
On the other hand, Zhan et al.17 discovered that when the MDEA concentration rose above 1.68 mol L−1 in the aqueous blend of MDEA and PZ, the removal efficiency of CO2 and H2S declined sharply. Fu et al.68 and Foo et al.69 also stated that higher concentration of MDEA led to an increase in viscosity, which resulted in lower diffusivity of amine molecules in the solvent. Consequently, it led to lower capacity and absorption rate of the amine solvents. Similarly, Tian et al.70 recorded a decreasing performance of MEA and DEA blends as the concentration of MDEA increases at low partial pressure of H2S. Thus, it can be deduced that there should be an optimal concentration of MDEA that could lead to the highest performance in any solvent containing MDEA. The viscosity of the solvent also has a great impact on CO2 absorption as compared to H2S absorption. Generally, H2S absorption is restricted by the gas mass transfer, whereas the absorption of CO2 is restricted by reaction kinetics and liquid mass transfer.53,61,71
Alkanolamines are extensively utilized for acid gas removal via absorption methods due to their efficiency and versatility. Despite their wide applications in the natural gas industry, alkanolamine-based processes suffer from several drawbacks such as alkanolamine degradation, losses due to volatility, water presence in outlet gas and being energy intensive.72 These limitations cause alkanolamine-based processes to be uneconomic and unsustainable, which has led to limited adoption outside the natural gas industry.
There are several processes that employ physical solvents for H2S absorption such as Selexol, Rectisol, Purisol, Fluor, Genosorb, Morphysorb, and Coastal AGR II processes. Selexol, Genosorb and Coastal AGR II use dimethyl ethers in polyethylene glycol (PEG), Rectisol uses methanol, Fluor uses propylene carbonate, Morphysorb uses N-formylmorpholine and morpholine derivatives, whereas Purisol process uses sulfolane and N-methyl-2-pyrrolidone (NMP) as solvents, respectively.13,74 Among the listed processes, Rectisol and Selexol are the most outstanding due to their capability of acid gas deep removal and reducing the concentration of COS and H2S to 0.01 ppm and 1 ppm, respectively. This makes Rectisol suitable for chemical synthesis applications which require less than 1 ppm of sulfur compounds. However, Rectisol is an energy-intensive process and uneconomic as the recovery process of the methanol solvent used needs a high cooling duty and large volume of water for washing. In addition, the methanol used is also highly volatile and has a low selectivity towards H2S over CO2. In contrast, the Selexol process exhibits exceptional selectivity for H2S, low vapor pressure, and demonstrates high thermal and chemical stability. The only downside to the Selexol process is its high viscosity at low temperature, which contributes to lower efficiency and mass transfer rates.
By comparing all conventional physical solvents, Purisol demonstrated the highest selectivity towards H2S in the presence of CO2, making it highly viable for selective removal of H2S. In addition, Purisol also possess a higher vapor pressure compared to Fluor and Selexol. Nevertheless, similar to Rectisol, Purisol needs water for washing in order to recover the spent solvents. Fluor is suitable for CO2 bulk removal from feed gas containing a low content of H2S with minimum loss of solvents. Moreover, Morphysorb is also excellent for selective removal of H2S but suffers from high volatility.75 The performance of physical solvent processes in H2S absorption can be improved by increasing the gas pressure, as the solubility of H2S in the solvent is directly proportional to its gas phase partial pressure.
Recently, nanofluid application for gas absorption has gained increasing attention. Nano-sized particles such as alumina, silica, graphene oxide and carbon nanotubes (CNTs) were mixed with solvents such as MEA, DEA and MDEA76 to improve the mass transfer coefficient in three-phase systems which consist of gas, liquid and solid phases. The gas adsorption by nanoparticles aids in sustaining the flow of gas from areas of high concentration to areas of low concentration, a phenomenon commonly referred to as the shuttle effect. The collision between the gas–liquid interface and the nanoparticles causes the diffusion boundary layer to become thinner and inhibits bubble coalescence, resulting in higher contact surface area between the gas and liquid interface. The performance of Fe-5MEA-DES systems for H2S removal was quite promising; nevertheless, the H2S removal efficiency dropped to below 50% after the third regeneration cycle.77 This showed the need to find better ILs which can maintain high desulfurization performance and H2S removal efficiency after multiple regeneration cycles.
In general, ILs consist of bulky, nonsymmetrical organic cations such as imidazolium, pyrrolidinium, pyridinium, ammonium or phosphonium and various different inorganic or organic anions such as chloride and tetrafluoroborate anions. Compared to conventional solvents, ILs have many fascinating properties that make them unique. Generally, ILs are colorless liquids possessing relatively high viscosities. Under ambient conditions, ILs exhibit very low vapor pressures contributing to their effective non-volatile nature. Due to their non-volatility, ILs have been advertised as green solvents.83 Additionally, ILs are excellent solvents for a wide spectrum of organic, inorganic and polymeric materials and are immiscible with numerous organic solvents. The wide usage of ILs in various extraction processes as organic solvent replacements is considered a hot and interesting research topic.84 The fact that ILs are not flammable and volatile makes them highly desirable for safer process development. Moreover, IL physicochemical properties such as hydrophobicity, polarity, viscocity and others can be tuned85 by interchanging their constituent cations and anions. ILs are often regarded as “designer solvents” due to their tunable nature, which increases their potential uses.86 Generally, ILs can be further divided into two major classes, ordinary ILs and task-specific ILs.
Among the first to study H2S absorption using ILs, Jou and Mather39 prepared [Bmim][PF6] and classified it as a physical absorbent. They also deduced that ILs could be used for bulk removal of acid gas at high pressures. Pomelli et al.41 investigated the H2S solubility in [Bmim][NTf2] and recorded a low correlation between H2S solubility and Kamlett–Taft parameters. They deduced that the strength of anion interactions with H2S is similar to that of hydrogen bonds and the H2S solubility in ILs is less affected by the cations. Furthermore, the capacity and regenerability of any particular IL can be tuned by changing the anions of the IL. Wang et al.26 found out that pyridinium-based ILs performed excellently for selective absorption of H2S over CO2 due to the presence of hydrogen protons in H2S molecules. The solubility of H2S and CO2 was also found to have increased as the length of alkyl chains of cations increased. The solubility of H2S increases in the following order of [BF4], [C4Py], [C4Py][NO3], [C4Py][SCN], [C6Py][SCN] and [C8Py][SCN], respectively. By comparing the anions with similar cations, [SCN]− displayed the highest H2S over CO2 selectivity value of 8.99 at 30 °C. which is far higher than that of imidazolium-based ILs. On the other hand, Jalili et al.27 reported that [Emim][BF4] possesses similar absorption capacity for H2S to that of other [BF4]-based ILs and similar selectivity to other types of ILs such as [Emim][eFAP],87, [C4mim][PF6],88, [C8mim][NTf2]89 and [C8mim][PF6].90 Nevertheless, conventional ILs act as physical absorbents and exhibit reduced absorption capacities for H2S and CO2 under low to moderate pressures.
To mitigate current deficiencies of ordinary ILs, new subcategories of ILs are introduced known as task-specific ionic liquids (TSILs). TSILs are specifically designed for a particular purpose of reaction such as for H2S absorption applications.91 Typically, TSILs are prepared by tethering one or more functionalized groups that could improve their physicochemical properties.92 As a result, their chemical interactions with H2S can be tailored to improve the absorption capacity of TSILs, exceeding that of ordinary ILs. Huang et al.93 synthesized 1-alkyl-3-methylimidazolium carboxylates as TSILs for H2S absorption. The TSILs exhibited a higher solubility of H2S than ordinary ILs, reaching up to 0.6 mol mol−1 at room temperature and pressure. Nonetheless, the H2S selectivity over CO2 is unsatisfactorily low reaching only 1.0 at 30 °C. Consequently, Huang et al.94 also synthesized dual Lewis based-ILs (DLB-ILs) for selective H2S capture over CO2. [N2224][IMA] exhibited a H2S solubility of 0.85 mol mol−1 and selectivity of 10 for H2S over CO2 at standard temperature and pressure. Despite their advantages, DLB-ILs arevery viscous with over 2500 cP at room temperature and require complex synthesis procedures.
Furthermore, Huang et al.95 prepared protic ionic liquids (PILs) by combining alkanolamines such as MDEA and DMEA with acetic and formic acids for selective removal of H2S over CO2. The synthesized PILs displayed a high selectivity of 8.9 to 19.5, and absorption capacities of 0.04 to 0.16 at 30 °C and 1 bar.96,97 Additionally, they are also less viscous as compared to the common ILs. Zhao et al.25 synthesized ten carboxylate-based PILs and recorded low viscosities of 4.3 cP at 25 °C and high absorption capacities of H2S. However, these PILs are not thermally stable as most of them can be easily decomposed below 50 °C, making them unsuitable for regeneration and H2S removal at higher temperatures. Similarly, Huang et al. prepared hydrophobic PILs functionalized with tertiary amines for selective removal of H2S. The results showed that these PILs are highly selective towards H2S with minimum absorption of CO2. Consequently, Huang et al.98 attempted to create hydrophilic PILs by replacing the tertiary amines with carboxylate groups. They observed that the solubilities of the hydrophilic PILs increases in the following order from [BMEE][Ac] to [TMEDA][Ac], [TMPDA][Ac] and [BDMAEE][Ac], respectively. As the concentration of PILs and temperature increased, the solubilities of H2S and CO2 were also found to have increased. Moreover, these PILs can be diluted by mixing with water.99 Despite having a high H2S absorption capacity of 1.044 mol mol−1, aqueous PILs have a low selectivity of 1 to 2 only.
Huang et al.23 also studied the selectivity and solubility of phenolic ILs for selective removal of polar gases, e.g., CO2 and H2S over non-polar gases, e.g., CH4. They found out that the ILs are more soluble towards polar gases as compared to non-polar gases, leading to higher selectivity of H2S and CO2 over CH4.100 In addition, phenolic ILs also exhibited a decent absorption capacity of 0.6 mol mol−1 at 0.1 bar, which increases slightly to 0.85 mol mol−1 at 1 bar. Among all phenolic ILs, [TMGH][PhO] demonstrated the highest selectivity of 9.4 for H2S absorption over CO2. Nevertheless, phenolic ILs possess several drawbacks such as the toxic nature of phenolic compounds and high viscosities between 125.7 and 435.1 cP at 30 °C. Zhang et al.21 studied four azole-based PILs for simultaneous removal of CO2 and H2S at 30 °C and 1 bar. [DBNH][1,2,4-triaz] has a high potential of becoming an excellent absorbent for H2S due to its high absorption capacity of 1.2 mol mol−1 and a rather low viscosity of 42.6 cP at 40 °C. On the other hand, Xiong et al.22 prepared four superbase PILs (SPILs) such as [DBUH][Im], [DBUH][Pyr], [DBNH][Im] and [DBNH][Pyr] for acid gas removal from natural gas.101 SPILs possess low viscosity at 40 °C and demonstrated a remarkable H2S absorption capacity of 6.81 mol kg−1 and excellent selectivity of H2S and CO2 over CH4, respectively. However, their low thermal decomposition temperatures of around 80 °C hinder their regeneration capabilities and absorption at high temperatures. For example, [DBNH][Im] lost 25% of its original absorption capacity after being regenerated four times.
Most of the ILs reported so far did not undergo a rigorous selection process to select the best IL for the H2S absorption process but mostly rely on the trial-and-error method.102 This is not only ineffective but failed to identify better possible combinations of anions and cations. In addition, the selection process via experimental methods is not realistic considering the huge amount of time and costs needed for completion.103 Thus, a preliminary screening to identify desirable physical and thermodynamics properties is necessary to narrow down the selection of ILs from a vast array of possible ILs. One of the most effective ways of finding the most optimal ILs is by using a predictive model based on computational chemistry such as conductor-like screening model for real solvents (COSMO-RS). Mortazavi-Manesh et al.104 used a COSMO-RS approach105,106 and the Peng–Robinson equation of states107 to screen 425 ILs for selective removal of H2S over CO2. From their findings, cations such as PMG, TMG and N4111 and anions such as CH3SO4, NO3 and BF4 were suggested as the best IL candidates. Zhao et al.108 also utilized COSMO-RS to screen over 10000 ILs but not for absorption of H2S. Santiago et al.109 integrated Aspen Plus with COSMO-RS to select from over 700 ILs with absorption process simulation using the selected ILs. The authors identified [Emim][DCN] as the IL with the highest absorption performance. By using similar methods, Lemus et al.110 found out that hybrid blends of 75 wt% [Emim][DCN] and 25 wt% [Bmim][Ac] were able to recover over 98% of H2S at 10 bar.
Apart from IL screening, several researchers compared industrial processes with IL-based processes to study the potential commercialization of ILs in industrial applications. Kazmi et al.111 performed comparisons of imidazolium-based ILs with aqueous MDEA for simultaneous removal of acid gas from natural gas at a high pressure of 68 bar and 30 °C. The activation energies of ILsbased processes and MDEA were determined to be around 3981 kW and 18619 kW, respectively. A huge reduction in activation energy of the IL-based processes was found to significantly reduce thermal energy usage by 78.6% and total annual cost by 59.8%. Additionally, the ILs are recoverable by passing through flash drums while removing the absorbed CO2 and H2S completely. However, the maximum absorption capacity of the ILs was not determined, making the performance hard to assess realistically. Wang et al.112 and Yang et al.113 compared the Rectisol process with [Bmim][NTf2] for simultaneous removal of acid gas from syngas. They discovered that the process utilizing ILs is far better in terms of solvent recovery, cooling energy consumption and CO2 capture. Based on a review by Haider et al.,114 there is promising potential for applications of ILs in acid gas removal at lower cost as compared to the conventional methods.
It is very clear that ILs have huge potential as both CO2 and H2S absorbents due to their remarkable properties. The only drawbacks hindering them from being used in industries are the scalability costs and the commercialization of processes based on ILs due to their high costs of production, complex synthesis procedures, high viscosity, toxicity, etc. High viscosity is the main limitation of ILs,115 as it limits the gas–liquid mass transfer and decreases the diffusion rates of gases across the liquid. As a result, the process requires a longer reaction time and larger size of the absorption column. Furthermore, high viscosity requires high pumping power and initial cost of storage for the solvent. Thus, it is necessary to find solutions to lower the viscosity of ILs. One such way is by mixing ILs with low-cost solvents to reduce the viscosity of ILs significantly and efficiently.116 Other concerns regarding ILs are their high toxicity and poor regeneration capability that lead to additional losses as the spent ILs could not be recovered and will lead to environmental pollution. These issues can be tackled by further investigating and designing ILs that could function not only as solvents, but also as catalysts that can be regenerated multiple times without needing to be discarded. Finding an ideal balance between viscosity, regeneration capability and ease of synthesis should definitely be research goal to further enhance the development of ILs for adoption in the oil and gas industry.
Liu et al.28 prepared a series of DESs using choline chloride (ChCl) and urea in various ratios, such as 1.5, 2 and 2.5 for the concurrent removal of H2S, CH4 and CO2. They found that these DESs undergo physical absorption and as the ratios between ChCl and urea decreases, the solubility of H2S also decreases. They also discovered that selectivity of H2S and CO2 over CH4 is easily tunable by changing the ChCl and urea ratios. The DES is also rather stable thermally with a decomposition temperature of 176.85 °C. Wu et al.29 prepared two types of DESs by mixing tetrabutylammonium bromide (TBAB) and ChCl with carboxylic acids (CA) for H2S capture. Both DESs were acting as physical absorbents with similar trends. TBAB-CA performed better as compared to ChCl-CA due to strong hydrogen bond interactions in ChCl-CA contributing to its lower free volume for absorption. The carboxylic acid-based DESs also outperformed other reported DESs28 and many ordinary ILs.
Similar to TSILs, DESs can also be functionalized to create more chances of chemisorption to increase the selectivity and absorption capacity. The functionalized DESs are known as task-specific deep eutectic solvents (TSDESs). Shi et al.30 developed five TSDESs of quaternary ammonium salts with tertiary amines and azoles. Among them, only [C4-TMEDA][Cl]-1,2,3-triaz has a viscosity below 100 cP at 40 °C making them unfavorable for industrial applications. Despite this, these TSDESs showed a high selectivity towards H2S ranging from 5.2 to 12.1. Nonetheless, [C4-TMHDA][Cl]-[Im] displayed the highest absorption capacity of 0.996 mol mol−1 at room temperature and pressure. Moreover, [C4-TMHDA][Cl]-[Im] also can be regenerated five times while retaining 92% of its capacity at 69 °C and 0.1 bar. Shi et al.31 also prepared a series of TSDESs containing chemical dual sites such as acetate and ternary amine anions to form dual chemisorption sites with H2S. [C1-TMHDA][Ac]-[MDEA] exhibited the highest absorption capacity for H2S followed by [C1-TMHDA][Ac]-[Pyrol], [C1-TMHDA][Ac]-[AA] and [C1-TMHDA][Ac]-[Im], respectively. These TSDESs also displayed a high selectivity for H2S over CO2 ranging from 6.9 to 9.3 and can be regenerated up to six times while maintaining their absorption capacity of 92%. Furthermore, [C1-TMHDA][Ac]-[Pyrol] and [C1-TMHDA][Ac]-[Im] possess viscosities lower than 75 cP at 40 °C, increasing their potential for industrial applications.
Another method to evaluate the interactions between DESs and H2S molecules is by computational methods such as ab initio calculations, and molecular dynamics (MD), Monte Carlo (MC) and COSMO-RS simulations. These methods could prove useful in cases where there is a lack of experimental data. Karibayev et al.125 applied ab initio calculations and MD simulations to evaluate the interactions in four DESs made up of TBAB, tetrabutylammonium chloride (TBAC), caprolactam (CPL), ChCl, urea, MEA and methyltriphenylphosphonium bromide (MTPB) to form TBAB-CPL, TBAC-CPL, ChCl-urea and MTPB-MEA. The authors discovered that the TBAB-CPL DES demonstrated stronger affinity towards H2S over CH4, due to stronger interactions between CPL and H2S as compared to CH4. As a result, TBAB-CPL can selectively remove H2S efficiently from CH4. However, these DESs require a high pressure of 10 bar to operate optimally. Salehi et al.126 predicted the solubilities of H2S, CO2, H2 and N2 in ChCl-ethylene glycol and ChCl-urea DESs by MC simulations. Their results showed that both DESs are highly soluble in H2S followed by CO2, CH4, CO, H2 and N2. On the other hand, Stupek et al.127 predicted the thermodynamic properties of 23 cost-effective DESs for biogas upgrading using COSMO-RS. ChCl-urea and ChCl-oxalic acid were found to be the best DESs to remove CO2, H2S and siloxanes effectively.
DESs are an interesting class of solvents with high potential to replace traditional solvents in various applications both from economics and sustainability viewpoints. Nonetheless, the applications of DESs in H2S capture are still at the grassroots level and require further research to address issues such as high viscosities124 and low H2S absorption capacities.128 Overcoming these limitations would make DESs highly viable for industrial adoption. TSDES development has demonstrated that chemisorption can be initiated to achieve greater absorption capacity and reduced viscosity. TSDESs have been applied for removal of other contaminant gases but are yet to be explored for H2S absorption.129–131 Therefore, more studies should be carried out in this area to further enhance the potential of TSDESs in the near future.
Despite their significant potential for H2S removal applications, DESs are showing concerning signs of toxicity and ecotoxicity. Recent studies by Jung et al.132 have revealed that ChCl-based DESs with urea contain ammonia, which contributes to their toxic effects. Similarly, Lomba et al.122 also concluded that the individual toxicity of the initial components cannot serve as a predictive factor of DES ecotoxicity. Furthermore, Hayyan et al.133 discovered that the cytotoxicity of DESs was much higher than that of their individual components such as ChCl and glycerine, indicating that their toxicological behavior is different. These findings highlight the importance of assessing the safety and environmental impact of DESs before their widespread adoption. Further research is crucial to thoroughly investigate the potential risks associated with DES usage, particularly focusing on safety and ecotoxicity, in order to ensure their responsible and sustainable application in H2S removal processes.
The limitations of most ILs such as high viscosity and cost can be overcome by blending the ILs with more affordable solvents, i.e., physical solvents and alkanolamines, to decrease the viscosity of ILs and enhance mass transfer. Several researchers14,32,70 studied the effect of additional activators such as [N1111][Arg], [N1111][Gly] and MEA within 7.5 to 30 wt% on the performance of MDEA solution for H2S removal at low partial pressure. All activators were observed to enhance the MDEA solution capacity for absorbing H2S due to the increased availability of interaction sites. [N1111][Arg] recorded the highest removal efficiency and capacity followed by [N1111][Gly] and MEA, respectively. On the other hand, Afsharpour and Hagthalab33 prepared a hybrid blend of [Bmim][Ac] with DIPA for simultaneous removal of CO2 and H2S. They noticed that both solvents are highly selective towards H2S, thus leading to higher solubilities of H2S at high pressures. Moreover, Lemus et al.110 discovered that a hybrid blend of 75 wt% TEGDME and 25 wt% [Bmim][Ac] displayed an excellent removal performance of H2S due to low viscosity of TEGDME and high absorption capacity of [Bmim][Ac].
Chemicals not adhering to green chemistry principles can cause significant negative environmental impacts throughout their lifecycle, from production to disposal. Many conventional solvents like chlorinated solvents (e.g., dichloromethane and chloroform), aromatic solvents (e.g., benzene and toluene), and volatile organic compounds (VOCs) contribute to air pollution,135 soil and water contamination,136 and harm wildlife.137 Long-term exposure to certain solvents can lead to health risks such as respiratory issues, neurological disorders, and cancer.138 Some toxic solvents can severely impact ecosystems, leading to soil and water contamination, disrupting ecosystems, and harming aquatic life. Improper disposal of solvents containing heavy metals can lead to environmental contamination, affecting soil quality and agricultural productivity. Highly toxic metals such as mercury and lead can pose serious health risks to humans, especially through food and water contamination.139
Therefore, adopting sustainable and environmentally friendly alternatives is crucial. Green solvents140–142 such as ILs and DESs offer advantages such as enhanced selectivity, improved absorption capacity, reduced environmental impact, and alignment with green chemistry principles, which aim to minimize hazardous substance use and generation, thereby reducing environmental pollution and protecting human health and ecosystems. A summary of conventional H2S removal absorption processes is provided in Table 3.
| Conventional technology | Feed gas composition | Operating conditions | Types of solvent used | H2S removal efficiency | Reference |
|---|---|---|---|---|---|
| Selexol | 44 mol% CO2, 0.6 mol% H2S, and 55.4 mol% CH4 | Temperature: −9.4 to 38 °C, pressure: 20–120 bar | Dimethyl ether of polyethylene glycol (DMPEG) | 90% | 143 |
| Rectisol | Gas ratio: 6–7 mol% CO2: 1 mol% H2S | Temperature: −34 to −74 °C, pressure: 40–78 bar | Methanol | 70% | 144–146 |
| Purisol | 8 mol% H2S, 18 mol% CO2, 70 mol% CH4, and 4 mol% N2 | Temperature: −15 °C, pressure: 32–35 bar | N-Methyl-2-pyrrolidone (NMP) | 94% | 74 |
| ADIP | 0.5 mol% H2S, 5.5 mol% CO2, and 94 mol% CH4 | Temperature: 100–150 °C, pressure: 4.1–24.8 bar | DIPA | 75% | 147 |
| Sulfinol | 15 mol% H2S, 6 mol% CO2, 57.69 mol% CH4, 6.24 mol% C2H6, 7.57 mol% C3H8, and 7.5 mol% N2 | Temperature: 57–66 °C, pressure: 49.3–91.7 bar | Sulfolane® (tetra-hydrothiophene dioxide) and DIPA | 70% | 148 |
| FLEXSORB™SE | 1.8 mol% H2S, 6.7 mol% CO2, and 91.5 mol% CH4 | Temperature: 32 °C, pressure: 64.5 bar | MDEA | 67% | 50 and 149 |
| OASE®sulfexx™ | 2 mol% H2S, 10 mol% CO2, and 88 mol% CH4 | Temperature: 45 °C, pressure: 64.5 bar | MDEA | 81% | 51 and 150 |
| UOP Benfield™ | 2.65 mol% H2S, 11.5 mol% CO2, and 85.85 mol% CH4 | Temperature: 110–130 °C, pressure: 20.7 bar | Hot potassium carbonate, K2CO3 | 93.7% | 151 |
| CATACARB® | 2.1 mol% H2S, 6.7 mol% CO2, and 91.2 mol% CH4 | Temperature: 153–204 °C, pressure: 20.7–56.5 bar | Hot potassium carbonate, K2CO3 | 38.7% | 151 |
2.2 Adsorption
Adsorption is a process in which the molecules of a fluid are adhered to a solid surface.152 The molecules that adhere to the surface are referred to as adsorbates, whereas the surface where the molecules accumulate is the adsorbent. Adsorption can be divided into two different classes such as physisorption and chemisorption.153,154 Physisorption, also known as physical adsorption, is a rather weak interaction where the molecules of adsorbates were adhered to the surface of the adsorbent by van der Waals forces,i.e., dipole–dipole interactions and London dispersion forces.155 Physisorption involves rather low energy and gets stronger at lower temperatures. Furthermore, physisorption is a reversible process, which means that the adsorbates can be desorbed easily from the adsorbent surface by increasing the temperature or reducing the pressure. On the other hand, chemisorption or also known as chemical adsorption is a stronger interaction as compared to physisorption due to the formation of bonds such as covalent or ionic bonds between the adsorbate molecules and the adsorbent surface.156 Generally, chemisorption requires an activation energy which is specific to both the adsorbate and the adsorbent. Unlike physisorption, chemisorption is mostly irreversible and needs high temperatures and drastic conditions to desorb the molecules from the adsorbent.In general, gas adsorption by solid adsorbents was governed by five different mass transport mechanism stages such as external, pore, film, intraparticle and surface adhesive diffusion.157 The process of H2S adsorption from a gas stream is an exothermic process and occurs under dry conditions without requiring an extremely high temperature to operate. There are various adsorbent materials commonly used for large scale H2S removal, including carbon-based adsorbents, zeolites, metal oxides, metal organic frameworks (MOFs) and composite materials. These materials were used mainly because of their high removal capacity, good selectivity and thermal and mechanical stability.158,159 The removal efficiency of H2S is significantly affected by the adsorbent properties such as porosity and surface chemistry; and operating parameters such as mixture gas concentration, temperature, gas hourly space velocity and relative humidity.160
| NaOH + H2S → NaHS + H2O | (2.1) |
| 2NaOH + 2H2S → Na2S + 2H2O | (2.2) |
| NaHS + 0.5O2 → S + NaOH | (2.3) |
| Na2S + 0.5O2 + H2O → S + 2NaOH | (2.4) |
| 2NaOH + H2SO4 → Na2SO4 + 2H2O | (2.5) |
The exothermic reaction releases heat that may damage the adsorptive sites. H2S oxidation is reported to be influenced by surface chemistry and local pH. An acidic carbon surface will result in higher yield of water-soluble products, but at the same time lower the total sorption capacity. When the adsorption performance of H2S using impregnated ACs with various alkaline solutions such as K2CO3, Na2CO3, KOH and NaOH was compared, NaOH-activated carbons outperformed the other alkaline solutions, despite the drawbacks mentioned earlier.165 Recently, Ahmadi et al.166 developed an N–S rich jute-derived nanoporous carbon activated by KOH; which possesses excellent gas adsorption capacity and high cycling performance. Meanwhile, Wang et al.167 synthesized Cu-impregnated peanut shell-based AC for H2S adsorption in an enclosed area, which displayed good regenerability with a 90% removal rate. A ZnO–MgO-based AC adsorbent investigated by Yang et al.168 exhibited an impressive adsorption capability of 96.5 mg g−1 under dry conditions. Nevertheless, the regeneration capability of the adsorbent was not studied.
Yang et al.169 also developed a ZnFe2O4-based AC adsorbent which demonstrated a 122.5 mg g−1 breakthrough capacity with an optimum loading of 10%, able to be regenerated three times with small capacity reduction after being treated thermally at 500 °C. Similarly, Yuan et al.170 synthesized leftover rice-based ACs that displayed a 12.11 mg g−1 breakthrough capacity with 50 wt% loading. On the other hand, Pan et al.171 prepared 2D nanostructures of CaO/CH carbonized at a temperature of 700 °C and above. CaO/CH-700 displayed the highest alkalinity, and a breakthrough adsorption capacity of 9.1 g g−1. Nonetheless, CaO/CH-700 displayed a very low regeneration capability of lower than 0.2 g g−1 as the reaction between H2S and CaO was irreversible.
In the last few years, N-rich carbon-based adsorbents are being developed as better alternatives to metal oxides, metal-doped carbons and caustic compounds for H2S removal due to their high sustainability, safety and efficiency. Chen et al.172 prepared N-rich porous carbons that shows higher breakthrough capacities as the CN/CS ratio increases from 0.5 to 1, which become lower after the ratio increases to 2. Furthermore, Fakhraie et al.173 synthesized KOH-activated high N-doped ACs (HNAC) at 800 °C over 1 h. Among the synthesized adsorbents, HNAC-802 was proven to have the best adsorption capacity at 316.35 mg g−1 with a 2.17 selectivity for H2S/CO2. On the other hand, Yu et al.174 used ZIF-8 to develop N-rich mesoporous carbon nanosheets (N-MCNS). Remarkably, N-MCNS have outperformed those in previous studies from Chen et al.172 and Fakhraie et al.173 by exhibiting an excellent adsorption capacity of 510 mg g−1 and a large surface area of 1937 m2 g−1. On the other hand, Wang et al.175 integrated polyethyleneimine into MCNS to form MCNS-PEI with different loading percentages ranging from 0 to 65 wt%. MCNS-PEI 25 displayed a high breakthrough sulfur capacity of 13.68 mmol g−1 and regenerability without any performance drop within 6 cycles. Xu et al.176 acquired N-doped porous carbons impregnated with KOH by treating them thermally with ammonia. The N-doped carbons managed to achieve over 85% conversion and 80% selectivity towards sulfur formation for 1 vol% of H2S in the feed gas.
There are various kinds of functionalizations for synthesized carbon-based adsorbents such as impregnation, direct integration, heteroatom doping, or deposition–precipitation that has been studied up to now. Moreover, Ou et al.177 synthesized granular ACs to remove H2S with concentrations of 932 to 2060 ppm at 25 °C. The breakthrough capacity of the granulated ACs was found to be 745–1293 mg g−1 for low concentration (932–1560 ppm) and even lower capacity of 615–703 mg g−1 at high concentration (1920–2060 ppm). However, no regeneration study of the adsorbent was conducted. Recently, Sawalha et al.178 developed biochar from waste biomass materials such as eucalyptus barks, almond shells and used coffee grains to remove H2S from biogas with an average concentration of 970 ppm. The adsorption capacity, breakthrough time and removal efficiency were observed to increase in the order of used coffee grains, almond shells and eucalyptus barks. Eucalyptus barks exhibited a capacity of 490 mg g−1, which was the highest among all three but performed lower than the granular ACs in a study by Ou et al.177 at the same temperature. Recently, Qi Dong et al.179 developed a polyethylene polyamine impregnated carbon catalyst, exhibiting outstanding performance with a H2S adsorption capacity of 1.58 g H2S/g-catalyst at 30 °C. This achievement is credited to its optimal steric hindrance and enhanced stability during the H2S adsorption–desorption cycle.
The development of carbon-based adsorbents that meets certain criteria such as cost-effectiveness, regenerability, sustainability and high capacity is in great demand nowadays. As a matter of fact, some of the adsorbents are capable of removing H2S at low temperatures. Therefore, further research should be carried out to increase the regenerability of the adsorbents as most of the reactions involving carbon-based adsorbents are irreversible. More emphasis should also be placed on developing N-doped or metal oxide-doped porous carbons that are economic, sustainable and easy to synthesize. Despite the high-cost requirements to perform the functionalization of these adsorbents, it is noteworthy that their adsorption capacity and regeneration capability could be significantly improved making them excellent alternatives for removal of H2S.
Aside from this, the deployment of metal oxide adsorbents in industrial applications for H2S capture such as iron oxide (FeO) adsorbents and mixed metal oxide-based adsorbents is getting recognition due to their capability of reducing H2S concentration to 10 ppm while avoiding heat loss at high temperature.185,186 FeO-based adsorbents such as SULFATREAT and SELECT FAMILY were developed by Schlumberger,187 while mixed-metal oxide adsorbents such as CuO/ZnO-based adsorbents, i.e., PURISTAR® and alumina-based adsorbents, i.e., Selexsorb® were developed by BASF. H2S adsorption on a metal oxide reaches 95% even at high temperature and resulted in the formation of a solid or liquid phase containing sulfur and an adsorbent metal. Nonetheless, various studies have been conducted to harness their exceptional performance for H2S capture. Hassankiadeh et al.188 proposed the use of molybdenum oxide (MoO2) nanoparticles for H2S capture. Despite possessing a remarkable adsorptive capacity of 0.081 g of H2S/g (non-spherical) and 0.074 g of H2S/g (spherical) at 85 °C,188 the adsorbent requires a high pressure of 16 bar with a low initial concentration of 43 ppm H2S to operate optimally.
On the other hand, Orojlou et al.189 developed nanocomposites such as NiO/TiO2, CoO/TiO2 and CuO/TiO2 for H2S capture at a very high temperature of 480 °C. The best nanocomposite at 480 °C was found to be CoO/TiO2. However, the performance of these nanocomposites began to drop significantly at a temperature of 400 °C, indicating that these nanocomposites are not capable of operating optimally at low temperatures. A similar study was conducted by Pan et al.190 who recorded a better performance of NiO/TiO2 at 500 °C. Furthermore, Orojlou et al.189 discovered that these nanocomposites can only be regenerated once before deteriorating in performance. Furthermore, Kim et al.191 developed a Mn2O3/Fe2O3 nanocomposite to remove H2S at room temperature. The adsorption capacity recorded was 11.97 mg g−1 which decreases as the flow rate of the feed gas increases. Regenerability performance of these nanocomposites should be further researched to improve their regeneration capability.
Wu et al.192 prepared double metal oxides (DMOs) containing Zn and Fe with different molar ratios from 1:1 to 5:1 for H2S capture at an elevated temperature ranging from 450 °C to 700 °C. DMO-5 exhibited the best performance at 550 °C with a breakthrough capacity of 250 mg g−1 and breakthrough time of 321 min. The Zn–Fe-based adsorbents can be fully regenerated at a temperature between 600 °C and 650 °C in the presence of 2 to 4% oxygen. On the other hand, Ahn et al.193 synthesized acid mine drainage sludge (AMDS) which contains various types of metal oxides, and 56.6% of the sludge is iron oxide. AMDS demonstrated a breakthrough capacity of 8361 mg g−1 for removal of low concentration H2S ranging from 110 ppmv to 126 ppmv.
Metal oxide adsorbents generally possess excellent adsorption capacities for H2S. Nevertheless, they suffer from irreversible chemisorption, which leads to high replacement costs, and the spent adsorbents have to be disposed of. Another disadvantage of metal oxide adsorbents is the high temperature requirement to operate optimally, and a further increase in temperature might also lead to sintering of the adsorbents.194 Furthermore, unsupported metal oxides may deteriorate with time due to rapid sintering and fail to be regenerated at high temperatures. In addition, metal oxide adsorbents also experience issues such as spalling and sublimation. The applications of metal oxide adsorbents is restricted by several factors, including low thermal and chemical stability at high temperatures, as well as limited surface areas at low temperatures.
Ozekmekci et al.201 determined that zeolite 13X (Na-X) and its derivatives have the highest performance in removing S compounds. Furthermore, Bareschino et al.202 also investigated zeolite 13X for H2S removal with concentrations ranging from 50 to 100 ppm. They discovered that H2S adsorption is higher with the presence of water in the system and changes from physisorption to chemisorption at the end of the reaction. Similarly, Barelli et al.203 synthesized a Cu-replaced 13X (Cu-Ex 13X) zeolite for removal of H2S from biogas with concentrations between 200 and 1000 ppm. Cu-Ex 13X has a breakthrough time of 126 min and adsorption capacity of 11.46 mg g−1. By comparison with the study carried out by Bareschino et al.,202 it is noticeable that the presence of Cu2+ ions has led to an efficient adsorption process by the Cu-Ex 13X zeolite. Moreover, a higher H2S concentration in the feed gas has led to a lower breakthrough time and adsorption capacity. On the other hand, Bahraminia et al.204 prepared a nanozeolite of AgNaA for H2S removal and reported a breakthrough time of 310 min and 33.24 mg g−1 capacity to obtain 1 ppmv H2S at the outlet. The reported performance is far lower than that of commercial 4A and the pure NaA nanozeolite. Nonetheless, the AgNaA nanozeolite performed better than Cu-Ex 13X. Formation of water inside the zeolite indicated that H2S chemisorption occurred and led to higher performance. Additionally, the AgNaA zeolite can be regenerated with a 5% reduction of adsorption capacity for every cycle, probably because of the water loss during regeneration at high temperatures.
Currently, zeolite-based adsorbents are gaining worldwide attention for industrial adoption and being rigorously studied for H2S capture purposes owing to their substantial surface areas and porosity. Jafari et al.205 studied the effect of magnetite nanoparticle addition on the performances of Y and ZSM-5 zeolite substrates for removal of up to 120 ppm of H2S at relatively high temperatures between 100 and 300 °C. Zeolite Y doped with 5% magnetite was found to have the highest adsorption capacity of 69.92 mg g−1 as the doping process has improved the specific surface area and increased the number of micropores and mesopores. Meanwhile, Florent and Bandosz206,207 synthesized a new biosolid-based adsorbent incorporated with pluronic surfactant F127 for H2S removal from air. The surfactant-added adsorbent displayed a 250% increase in H2S removal efficiency compared to the adsorbent without surfactant addition. The surfactant addition increased the mesopore volume and carbon content, leading to higher catalytic center dispersion and carbon structural order which resulted in the conversion of H2S into elemental sulfur.
Yan et al.208 screened 95 different types of silica zeolites for removal of toxic gases such as H2S, NO, NO2, NH3, SO2 and CO using grand canonical ensemble Monte Carlo simulations. According to their prediction results, the top 10 adsorbents for H2S removal showed a higher capacity towards NH3 as compared to H2S. Zeolites AFY, MER and PAU are found to be the best H2S adsorbents with loading capacities of 7.8, 5.4 and 5.4 mmol g−1, respectively. For adsorption that involves all gases, zeolite RWY showed an outstanding performance with a H2S adsorption capacity of 17.74 mmol g−1. Similarly, Song et al.206 discovered that the CHA zeolite displayed the best adsorption capacity and decent H2S selectivity, whereas LTA and FAU zeolites exhibited excellent H2S selectivity but suffered from poor adsorption capacities. Georgiadis et al.209 used an industrial molecular sieve with a ratio of 0.97 between Si and Al that resembles the LTA zeolite. The molecular sieve exhibited an adsorption capacity of 193.59 mg g−1 for 10000 ppmv of H2S and can be regenerated up to 15 times without any significant loss in its capacity at 200 °C. The adsorption capacity was also found to decrease to 164.5 mg g−1 after H2S concentration increases to 30000 ppmv.
One of the major disadvantages of zeolites is their high temperature requirement for regeneration, which hinders them from being widely implemented in industrial applications, especially for H2S adsorption. Among all classes of commercial adsorbents, only zeolites are able to be regenerated. Another drawback commonly faced by zeolites is low selectivity towards H2S in the presence of other gases, i.e., CO2, SO2 and NH3. There are a few ways to boost the development of zeolites for H2S adsorption such as by implementing more feasibility studies of the newly developed zeolites and changing the research approach to focus on developing new structures and frameworks.
000 m2 g−1.211 In addition, MOFs are thermally and chemically stable, besides having a high tunability of their pore size.212 Due to their remarkable properties, MOFs are excellent candidates for gas storage and separation applications.213 MOFs are widely used as gas storage media for H2 and CH4 for clean energy production and as high-capacity adsorbents for separation of various gases, i.e., NH3, H2S, SO2 and CO2.
Gupta et al.214 developed Cu-based MOFs such as CuBTC, CuBDC and CuBDC-N for H2S capture at 25 °C. It was observed that CuBDC with the smallest ratio of Cu+ to Cu2+ ions possesses the best adsorption capacity of 105.6 mg g−1. Gupta et al.215 also prepared Cu(BDC)0.5(BDC-NH2)0.5 and noticed that a higher adsorption capacity of 128.4 mg g−1 was recorded for 500 ppm of H2S inlet. The presence of moisture has appeared to affect the MOFs adversely by reducing their performance. The authors have attempted a methanol and UV-aided regeneration process but are unable to fully regenerate the MOF, with only 9.9 mg g−1 of adsorption capacity being recorded after three regeneration cycles. Further characterization conducted confirmed that the UV-regeneration method has damaged the internal structure of the studied MOF. On the other hand, Zarate et al.216 used MIL-53(Al)-TDC to remove 5 vol% of H2S in the inlet gas under ambient conditions and reported an extremely high adsorption capacity of 618 mg g−1. Additionally, MIL-53(Al)-TDC can be regenerated up to 5 times in a temperature range of 65 to 200 °C, without losing any capacity.
Similarly, Flores et al.217 developed MFM-300(Sc) for removal of 10 vol% of H2S in the feed gas and recorded a high adsorption capacity of 565 mg g−1 at room temperature. Nevertheless, the MFM-300(Sc) zeolite was unable to be regenerated completely as the adsorption capacity dropped to 344 mg g−1 after being regenerated once. The capacity reduction was attributed to the irreversible chemisorption and formation of polysulfides from H2S molecules. Grape et al.218 prepared Bi2O(H2O)2(C14H2O8)·H2O (SU-101) to remove 4.3 vol% of H2S from the feed gas under room temperature conditions. The adsorption capacity recorded was 543.6 mg g−1, almost similar to that of MFM-300(Sc) despite having smaller surface area. SU-101 also exhibited a notable decrease in capacity after the initial regeneration cycle, attributed to an irreversible chemisorption process. Even though both MFM-300(Sc) and SU-101 could not be regenerated, they have huge potential in the MOF-based Li–S battery industry219 due to their capability of strong polysulfides formation and high stability.
Ngo and Chiang220 compared H2S removal performance using zeolite, calcined dolomite, and activated carbon in a hot gas cleaning system at 250 °C, with simulated synthesis gas containing 200 ppm of H2S. The results showed that activated carbon exhibited the highest removal efficiency and adsorption capacity, followed by calcined dolomite and zeolite. The adsorption capacities were 5.88 mg S g−1 for activated carbon, 3.16 mg S g−1 for calcined dolomite, and 2.22 mg S g−1 for zeolite. Calcined dolomite displayed poor adsorption–regeneration ability due to H2S deactivation, while zeolite and activated carbon were fully regenerated at 350 °C over four cycles.
Zeolitic imidazolate frameworks (ZIFs) are a unique subclass of MOFs which possess structural similarity to zeolites with the presence of imidazolate-based ligands.221 In ZIFs, the imidazolate ligands coordinate with metal ions forming a high porosity crystalline network. Consequently, ZIFs possess both added advantages of MOFs and zeolites.222 In general, ZIFs are more stable chemically and thermally as compared to MOFs, and more porous as compared to zeolites. Their unique features have led to much research for potential applications in various fields, especially for CO2 capture and storage. Nevertheless, similar applications for H2S removal are still limited. On the other hand, Jameh et al.223 developed a zeolitic imidazolate framework (ZIF-8) functionalized with ethylenediamine (ED) for effective removal of H2S from natural gas streams. The ED-functionalized ZIF-8 showed remarkable improvements in terms of breakthrough time and adsorption capacities compared to the pure ZIF-8 adsorbent. Nevertheless, the adsorption performance after regeneration was not reported. Another study by Liu et al.224 reported the usage of ZIF-67 for H2S removal. Despite having high removal efficiencies, ZIF-67 was facing structural collapse after the reaction, and no data on the adsorption capacity were reported.
In general, MOFs suffer from structural collapse in the presence of certain chemicals and gases, especially H2S. Commonly used metals such as Fe, Zn and Cu were reported for H2S adsorption but were unstable. Meanwhile, metals such as Zr, V, Al, Ni. Ti and Mg displayed strong reactivity towards H2S but were still rarely used.225,226 Moreover, the stability of ZIFs is also affected by the linkers and their functionalization.227 Additionally, the high cost of MOFs hinders their adoption in industrial applications. To mitigate the structural collapse in MOFs, higher emphasis should be given for designing linkers and metal nodes with higher stability, developing post-synthetic stabilization procedures and mixing the MOFs with metals that could enhance their stability. Besides, researchers should also focus on developing MOFs at much more affordable prices. Despite their limitations, MOFs have huge potential in H2S adsorption applications and there is still much to explore and uncover in this regard.
Gupta et al.232 developed Zn-MOFs/ZnO nanocomposites such as ZnBDC/N/ZnO, ZnBDC/ZnO and ZNBTC/ZnO for H2S adsorption at 25 °C. ZnBTC/ZnO demonstrated a 14.2 mg g−1 capacity followed by ZnBDC/ZnO and ZnBDC-N/ZnO. Zn-HKUST1 performed lower than the nanocomposites indicating that the presence of ZnO in the MOFs is crucial for excellent adsorption of H2S. The nanocomposites could only be regenerated twice by using methanol and UV-based methods before showing a major decline in their adsorption capacity. Similarly, Wu et al.233 prepared nanocomposites of ZnO mounted on MCM-41 and MCM-48. The optimum temperatures for ZnO/MCM-41 and ZnO/MCM-48 to achieve the highest performance were 500 °C and 600 °C, respectively. It was observed that the addition of ZnO into the nanocomposites provided more active sites for adsorption to occur, thus increasing the reaction rate of the adsorbents. Nevertheless, metal oxides are prone to conversion into metal sulfides which are larger in size, causing the pore structure to expand and might cause structural collapse. Similarly, Lin et al.234 synthesized a series of cuprous chloride/copper chloride hydroxide composites (CCCCHs) and recorded a high H2S adsorption capacity of 259.5 mg g−1. Upon reaction with H2S, the CCH surface undergoes decomposition, generating CuCl2·2H2O and reducing Cu2+ to Cu+, leading to the oxidation of H2S to elemental sulfur and sulfate.
Okonkwo et al.235–237 studied SBA-15 based nanocomposites of sterically hindered and unhindered amines for H2S removal under humid and dry conditions. Only (tertbutylaminopropyl)methoxysilanes (TBAPS) and (N,N-dimethylaminopropyl)trimethoxysilane (DMAPS) were selected to study the effect of humidity on H2S adsorption performance in a feed gas containing 1 vol% of H2S. Under humid conditions with 45% relative humidity, DMAPS/SBA-15 displayed a higher adsorption capacity as compared to TBAPS/SBA-15. In addition, TBAPS/SBA-15 was found to be the most selective towards H2S in the presence of other gases such as CO2 and CH4.237 After desorption at 120 °C, the capacity of both materials decreased sharply as the desorption process could not be completed under dry conditions. However, when subjected to desorption at 80 °C under humid conditions, both DMAPS/SBA-15 and TBAPS/SBA-15 displayed a minor decline in performance in the first cycle and it remained constant for the second and third cycles.237 This probably occurred because the chemisorption bond was likely reversible under humid conditions. As a result, this could lead to further research for reversing the chemisorption bonds under such conditions. Nonetheless, the regeneration process under humid conditions demands extra energy and water resources, potentially making it economically impractical.
Generally, composite materials could offer a versatile and effective approach for H2S adsorption applications due to the added advantages of the combined materials. By tailoring their composition and structures in order to be adapted for specific process requirements, researchers can design composite materials that are durable, cost-effective and highly efficient for specific H2S removal applications. Nevertheless, it is rather challenging to select the right combination of materials and modifications to enhance the overall performance due to limited adjustability of the main adsorbent component and lack of understanding of the reaction mechanisms for each component within the composite materials.
There are a few advantages of the adsorption process such as low cost, energy-efficiency, easy operation and simple design besides having a high temperature variation.185,186,188 However, there are various drawbacks associated with the adsorption process such as high-pressure steam requirement to desorb high molecular weight pollutants and a gas stream pre-filter to prevent any particles from clogging the adsorbent bed.238 In general, the adsorption process requires high temperature and pressure to operate optimally.188 Moreover, the capacity of adsorbents gradually deteriorates as the number of cycles increases, and a steam or vacuum source is required to regenerate the adsorbent. Nonetheless, some adsorbents are non-regenerable, resulting in a huge volume of sulfide wasted. In addition, the product recovery might require a special expensive distillation or extraction. The spent absorbent is often considered as hazardous waste as some contaminants might undergo an exothermic reaction with the adsorbent leading to explosion. The existing conventional technologies and recent research findings on H2S capture through adsorption are summarized in Table 4.
| Adsorbent | Adsorption conditions | Initial concentration of H2S | Breakthrough time, concentration of H2S and capacity | BET surface area (m2 g−1) | Reference |
|---|---|---|---|---|---|
| Conventional adsorbents | |||||
| SULFATREAT | Temperature: 100 °C, pressure: 20–120 bar | Pure | Time:1–2 min, capacity: 150 mg g−1 | n/a | 13 |
| SELECT FAMILY | Temperature: −34 to −74 °C, pressure: 40–78 bar | n/a | n/a | 13 | |
| PURISTAR® | Temperature: −15 °C, pressure: 32–35 bar | n/a | n/a | 74 | |
| Selexsorb® | Temperature: 100–150 °C, pressure: 4.1–24.8 bar | Capacity: 8 mg g−1 | n/a | 13 | |
|
|||||
| Carbon-based adsorbents | |||||
| AC | Gases present: H2S, O2, N2, and H2O, temperature: 30 °C | 850 mg m−3 | Concentration: 0.1 ppmv, capacity: 3.4 mg g−1 | 1120 | 168 |
| Mg–Ac | Capacity: 32.7 mg g−1 | 366 | |||
| Zn–Ac | Capacity: 38.5 mg g−1 | 769 | |||
| Mg0.2Zn0.8-AC | Capacity: 113.4 mg g−1 | 653 | |||
| Cu–Ac | Gases present: H2S, O2, N2, and H2O, temperature: 30 °C | 100 ppmv | Concentration: saturated, capacity: 129.2 mg g−1 | 559 | 239 |
| Zn–Ac | Capacity: 30.9 mg g−1 | n/a | |||
| Cu0.5Zn0.5-AC | Capacity: 118 mg g−1 | 570 | |||
| COF | Gases present: H2S, CO2, O2, and N2, temperature: 25 °C, pressure: 1 bar | 970 ppm | Concentration: saturated, capacity: 22 mg g−1 | n/a | 178 |
| ALM | Capacity: 230 mg g−1 | ||||
| EUC | Capacity: 460 mg g−1 | ||||
| GAC | Gases present: H2S, CO2, CH4, and O2, temperature: 20–25 °C, pressure: 1 bar | 932–2350 ppm | Concentration: 100 ppm, capacity: 615–1293 mg g−1 | 177 | |
| Alkaline graphene aerogel | Gases present: H2S, O2, and N2, temperature: 30 °C | 1000 ppmv | Concentration: 50 ppm, capacity: 3190 mg g−1 | 40 | 240 |
| CaCO3/CH-600 | Gas present: H2S | 1000 ppmv | Concentration: 50 ppmv | 22 | 171 |
| Temperature: 30 °C | Capacity: 99 mg g−1 | ||||
| CaO/CH-700 | Capacity: 9100 mg g−1 | 162 | |||
| CaO/CH-800 | Capacity: 4400 mg g−1 | 203 | |||
| CaO/CH-900 | Capacity: 2380 mg g−1 | 312 | |||
| N-rich mesoporous carbon nanosheets, NMCNS-0-5 | Gases present: H2S, N2, O2, and H2O | 1000 ppm | Concentration: 250 ppm | 1389 | 174 |
| Temperature: 25 °C, pressure: 1 bar | Capacity: 48 mg g−1 | ||||
| (NaCO3)0.2/NMCNS-0-5 | Capacity: 65 mg g−1 | n/a | |||
| NMCNS-50-8 | Capacity: 510 mg g−1 | 1937 | |||
| (NaCO3)0.2/NMCNS-50-8 | Capacity: 1370 mg g−1 | 983 | |||
| KOH-activated, Cu-impregnated peanut shell derived-ACs | Temperature: 25–110 °C, humidity: 60% | 200–1000 ppm | Capacity: 34.4–97.6 mg g−1 | 1326–1523 | 167 |
| Cu-modified AC fiber (ACF) | Temperature: 25–110 °C, mass: 0.5 g | 30 ppm | Capacity: 22.4–75.9 mg g−1 | 980.8–1768.9 | 241 |
| CuSO4, ZnAc2, KOH, KI, Na2CO3-modified coconut shell-derived AC (CAC) | Temperature: 25 °C, pressure: 1 bar, mass: 32–155 g, flowrate: 1.5–5.5 L min−1 | 1000–5000 ppm | Time: 63–68 min, capacity: 0.673–1.83 mg g−1 | 39.8–901.6 | 242 |
| ZnAc2-CAC | Gases present: H2S, N2, temperature: 25 °C, pressure: 1 bar | 5000 ppmv | Concentration: 5–10 ppmv, capacity: 2.37 mg g−1 | 620.6 | 243 |
| Thermally treated biowaste residues | Temperature: 25 °C, mass: 26.7–168.8 g, flowrate: 1 L min−1 | 1700 ppm | Capacity: 3.1–171.8 mg g−1 | 3–919 | 244 |
| Rice-based ACs, RBC-500 | Gases present: H2S, O2, N2, and H2O, temperature: 25 °C | 300 ppm | Concentration: 270 ppm, capacity: 12.11 mg g−1 | 2.76 | 170 |
| 50 wt% ZnFe2O4-loaded biochar, RZF-500-1:1 | Capacity: 228.29 mg g−1 | 1065 | |||
| Maize cob waste (MCW)-based ACs | Temperature: 25 °C, pressure: 1 bar, mass: 0.5 g, particle size: 1–3 mm, flowrate: 400 cm3 min−1 | 1100–1800 ppm | Time:11–629 s, capacity: 0,25–15.5 mg g−1 | 8–820 | 245 |
| BAX-1520 (commercial AC) | Gases present: H2S, O2, N2, and H2O, temperature: 25 °C, pressure: 1 bar | 1000 ppm | Concentration: 100 ppm, capacity: 5.6 mg g−1 | 2158 | 169 |
| 10 wt% ZnFe2O4-BAX-1520 | Capacity: 122.5 mg g−1 | 1403 | |||
| Porous carbons (PC) | Gases present: H2S, O2, N2, and H2O, temperature: 30 °C | 1000 ppmv | Concentration: 50 ppmv, capacity: 12.5 mg g−1 | 1340 | 172 |
| N-rich carbons | Capacity: 19.5 mg g−1 | 187 | |||
| N-rich PC (0.5) | Capacity: 119.1 mg g−1 | 2459 | |||
| N-rich PC (1) | Capacity: 426.2 mg g−1 | 1839 | |||
| N-rich PC (1.5) | Capacity: 334.5 mg g−1 | 1274 | |||
| N-rich PC (2) | Capacity: 267.2 mg g−1 | 1048 | |||
| N-rich hierarchical PC, NHPC-802 | Gas present: H2S, temperature: 30 °C, pressure: 1–10 bar | Pure | Concentration: saturated, capacity: 284.1 mg g−1 (1 bar), 669.7 mg g−1 (10 bar) | 1544 | 173 |
| NHPC-812 | Capacity: 316.5 mg g−1 (1 bar), 613.8 mg g−1 (10 bar) | 1477 | |||
| AN-OMC-700 | Gases present: H2S, O2, and N2, temperature: 25 °C, pressure: 1 bar | 5000 ppm | Concentration: saturated, capacity: 276.21 mg g−1 | 1538 | 246 |
| N-OMCS-700 | Gases present: H2S, O2, and N2, temperature: 0 °C, pressure: 1 bar | Capacity: 456.94 mg g−1 | 1575 | 247 | |
| N-OMCS-800 | Capacity: 371.69 mg g−1 | 1201 | |||
| N-OMCS-900 | Capacity: 303.49 mg g−1 | 1202 | |||
| N-PCNF-1/2-700 | Gases present: H2S, O2, N2, and H2O, temperature: 25 °C, pressure: 1 bar, humidity: 70% | 1000 ppm | Concentration: 50 ppm, capacity: 1490 mg g−1 | 1279 | 248 |
| N-PCNF-1/2-800 | Capacity: 1840 mg g−1 | 1308 | |||
| 40 wt% N-PCNF-1/2-800 | Capacity: 3340 mg g−1 | n/a | |||
| N-MCNS-PEI-25 | Gases present: H2S, O2, and N2, temperature: 25 °C | 1000 ppm | Concentration: 50 ppm, capacity: 466.23 mg g−1 | 193 | 175 |
| Gases present: H2S, CO2, O2, and N2, temperature: 25 °C | Capacity: 436.82 mg g−1 | ||||
| D-glucose and D-fructose-based PC | Temperature: 25 °C, pressure: 1 bar, mass: 100 mg, flowrate: 0.2 L min−1 | 20 ppm | Time:18–68.2 min, capacity: 6.32–25.7 mmol g−1 | 229–3217 | 249 |
| ZnO-supported N-modified ACs | Temperature: 25 °C, pressure: 1 bar, flowrate: 100 mL min−1 | 400 ppm | Time: 105–345 min, capacity: 24.2–62.5 mg S g−1 | 495–816 | 250 |
| Jute-derived NPC | Temperature: 25 °C, pressure: 1–10 bar (pure H2S), 35 bar (mixed gas), mass: 1 g | Pure and mixed with He (20:80 vol% H2S/He) |
Capacity: 6.45–19.10 mmol g−1 (1 bar), 12.8–32.6 mmol g−1 (10 bar), 16.2–45 mmol g−1 (35 bar) | 1065–2580 | 166 |
| KOH-modified waste oil fly ash derived ACs | Temperature: 1–50 °C, pressure: 1.38–3.45 bar, mass: 1 g, flowrate: 0.2–0.8 L min−1 | 50 and 100 ppm | Time: 25–120 min, capacity: 3.5–8.5 mg g−1 | 35.24 | 251 |
|
|||||
| Metal oxides | |||||
| ZnO | Gases present: H2S, CO2, CO, H2, N2, and H2O, temperature: 400–600 °C | 100 ppmv | Concentration: 20 ppmv, capacity: 10 mg g−1 (400 °C) | 2–3 | 190 |
| 28 wt% Ni–ZnO | Capacity: 210 mg g−1 (400 °C), 178 mg g−1 (500 °C), 117 mg g−1 (600 °C) | ||||
| 28 wt% Co–ZnO | Capacity: 184 mg g−1 (400 °C), 177 mg g−1 (400 °C), 145 mg g−1 (400 °C) | ||||
| 28 wt% Cr–ZnO | Capacity: 48 mg g−1 (400 °C) | ||||
| 28 wt% Fe–ZnO | Capacity: 101 mg g−1 (400 °C) | ||||
| CuO/TiO2 | Gases present: H2S and N2, temperature: 400–480 °C | 2000 ppm | Concentration: saturated, capacity: 40–300 mg g−1 | 44.34 | 189 |
| CoO/TiO2 | Capacity: 100–410 mg g−1 | 45.92 | |||
| NiO/TiO2 | Capacity: 100–580 mg g−1 | 52.77 | |||
| Mn2O3/Fe2O3 | Gas present: H2S, temperature: 25 °C, pressure: 1 bar | 500 ppm | Concentration: 400 ppm, capacity: 11.97 mg g−1 | 6.19 | 191 |
| CaO–Fe2O3 | Gases present: H2S, CO2, NH3, and H2, temperature: 850 °C | 230 ppmv | n/a | 1.50 | 252 |
| ZnFe2O4 | Gases present: H2S, O2, N2, and H2O, temperature: 25 °C, pressure: 1 bar | 1000 ppm | Concentration: 100 ppm, capacity: 1.6 mg g−1 | 16 | 169 |
| MoO2 | Temperature: 65–89 °C, pressure: 10–19 bar, Mass: 1 g | 38–73 ppm | Time: 78–85 min, capacity: 0.03–0.08 g S g−1 | 48–65 | 188 |
| Acid mine drainage sludge (AMDS) | Gases present: H2S and N2, temperature: 25 °C, pressure: 1 bar | 110–126 ppmv | Concentration: 5.5–6.3 ppmv, capacity: 8361 mg g−1 | 155.65 | 193 |
| 1000 ppmv | Concentration: 50 ppmv, capacity: 0.23–0.31 mg g−1 | 155.65 | |||
| ZnFe-DMO-5 | Gases present: H2S, CO2, CO, N2, and H2, temperature: 550 °C | 2000 ppmv | Concentration: 100 ppmv, capacity: 250 mg g−1 | 27 | 192 |
| NaMnO-D | Gases present: H2S, N2, and H2O, temperature: 25 °C, pressure: 1 bar | 500 ppm | Concentration: 400 ppm, capacity: 439.2 mg g−1 | 1.77 | 253 |
| NaMnO-T | Capacity: 818.7 mg g−1 | 2.56 | |||
| NaCo0.7O2.4-D | Gases present: H2S and N2, temperature: 25 °C | 500 ppm | Concentration: 10 ppm, capacity: 154.6 mg g−1 | 1.15 | 254 |
| NaCo1.1O3.3-T | Capacity: 168.2 mg g−1 | 1.90 | |||
|
|||||
| Zeolites | |||||
| NaA | Gases present: H2S and N2, temperature: 25 °C, pressure: 1 bar | 15 ppmv | Concentration: 1 ppmv, capacity: 13.95 mg g−1 | 263.35 | 204 |
| AgNaA | Capacity: 33.24 mg g−1 | 201.79 | |||
| NaX | 110–126 ppmv | Concentration: 5.5–6.3 ppmv, capacity: 277.86 mg g−1 | 515 | 193 | |
| FeX | Capacity: 10.01 mg g−1 | 350 | |||
| Magnetite-supported Y and ZSM-5 zeolites | Temperature: 100, 200, and 300 °C, pressure: 1 bar, mass: 0.5 g, flowrate: 1 L min−1 | 30, 60, 90 and 120 ppm | Time: 120–330 min, capacity: 14.7–69.9 mg g−1 | 332.6–550.1 | 205 |
| Industrial molecular sieve (IMS) | Gases: H2S and Ar, temperature: 25 °C, pressure: 1 bar | 200 and 10000 ppmv |
Concentration: saturated, capacity: 32 mg g−1 (200 ppmv), 194 mg g−1 (10000 ppmv) |
590 | 209 |
|
|||||
| MOFs | |||||
| UiO-66 | Gases: H2S and N2, temperature: 30–50 °C | 4000 ppm | Concentration: saturated, capacity: 50–80 mg g−1 | 1351 | 255 |
| 4500 ppm | Concentration: 2250 ppm, capacity: 51.1–95.4 mg g−1 | 256 | |||
| UiO-66-NH2 | Gases: H2S and N2, temperature: 25 °C, pressure: 1 bar | 10 ppmv | Concentration: 1 ppmv, saturated, capacity: 0.04 mg g−1 (1 ppmv), 0.07 mg g−1 (saturated) | 963 | 257 |
| MOF-5 | Capacity: 1.2 mg g−1 (1 ppmv), 4.97 mg g−1 (saturated) | 424 | |||
| MOF-199 | Capacity: 40 mg g−1 (1 ppmv), 69 mg g−1 (saturated) | 725 | |||
| CuBDC | Gases: H2S, N2, and H2O, temperature: 25 °C | 500 ppm | Concentration: 400 ppm, capacity: 105.6 mg g−1 | 218 | |
| CuBDC-N | Capacity: 1.3 mg g−1 | 38 | |||
| CuBTC | Capacity: 27.1 mg g−1 | 317 | |||
| Gases: H2S and N2, temperature: 25 °C | 500 ppm | Concentration: saturated, capacity: 77.1 mg g−1 | 231.9 | 258 | |
| Gases: H2S and N2, temperature: 25 °C, pressure: 1 bar | 99.6 ppm | Concentration: 5 ppmv, capacity: 17.1–56.3 mg g−1 | 434–1380 | 259 | |
| Cu(BDC)0.5(BDC-NH2)0.5 | Gases: H2S and N2, temperature: 25 °C | 500 ppm | Concentration: 50 ppm, capacity: 128.4 mg g−1 | 19.4 | 215 |
| MFM-300(In) | Gases: H2S and N2, temperature: 25 °C, pressure: 1 bar | 10 vol% | Concentration: saturated, capacity: 310 mg g−1 | 1060 | 217 |
| MFM-300(Sc) | Capacity: 564 mg g−1 | 1360 | |||
| MIL-53(Al)-TDC | Gases: H2S and N2, temperature: 30 °C, pressure: 1 bar | 5 vol% | Concentration: saturated, capacity: 606–654 mg g−1 | 1150 | 216 |
| ZIF-8 | Gases: H2S, CO2, CH4, and He, temperature: 25 °C, pressure: 2–10 bar | 3 vol% | Time: 190–400 s, concentration: 1500 ppmv, capacity: 3299 mg g−1 | 412–1389 | 223 |
| Bi2O(H2O)2(C14H2O8)·nH2O | Gases: H2S and N2, temperature: 25 °C, pressure: 1 bar | 4.3 vol% | Concentration: saturated, capacity: 543.58 mg g−1 | 412 | 218 |
|
|||||
| Porous organic polymers | |||||
| Hierarchical porous polymers (HPP) | Gas: H2S, temperature: 25 °C, pressure: 1 bar | Pure | Concentration: saturated, capacity: 94 mg g−1 | 1605 | 260 |
| N-HPP-pyridine | Capacity: 170 mg g−1 | 792 | |||
| N-HPP-bipyridine | Capacity: 188 mg g−1 | 1350 | |||
| N-HPP-3-aminophenol | Capacity: 153 mg g−1 | 1229 | |||
| N-HPP-HMTA | Capacity: 198 mg g−1 | 1397 | |||
| N-HPP-p-phenylenediamine | Capacity: 177 mg g−1 | 1186 | |||
| Pluronic® F127-modified biosolid-derived | Temperature: 25 °C, volume: 2 cm3, particle size: 425–600 μm | 1000 ppm | Time: 120–325 min, capacity: 89.1–221.2 mg g−1 | 110–180 | 207 |
| CBAP-1-EDA | Gases: H2S and N2, temperature: 25 °C, pressure: 1 bar | 10 ppmv | Concentration: 1 ppmv, saturated, capacity: 0.033 mg g−1 (1 ppmv), 0.12 mg g−1 (saturated) | 672 | 257 |
| CBAP-1-DETA | Capacity: 0.026 mg g−1 (1 ppmv), 0.1 mg g−1 (saturated) | 667 | |||
|
|||||
| Composite materials | |||||
| ZnBDC/ZnO | Gases: H2S and N2, temperature: 25 °C | 500 ppm | Concentration: 400 ppm, capacity: 13.6 mg g−1 (moist), 10.6 mg g−1 (dry) | 12.4 | 232 |
| ZnBDC-N/ZnO | Capacity: 9.4 mg g−1 (moist), 7.9 mg g−1 (dry) | 15.0 | |||
| ZnBTC/ZnO | Capacity: 7.8 mg g−1 (moist), 14.3 mg g−1 (dry) | 20.6 | |||
| ZnO/MCM-41 | Temperature: 50, 175, and 300 °C | 5000 ppm | Time: 119–850 min | 254–739 | 261 |
| 20 wt% ZnO/MCM-41 | Gases: H2S, N2, and H2O, temperature: 25 °C | 500 mg m−3 | Concentration: 1.5 mg m−3, capacity: 54.9 mg g−1 | 686 | 262 |
| 30 wt% ZnO/MCM-41 | Gases: H2S, CO2, CO, H2, and N2, temperature: 500 °C | 2000 ppmv | Concentration: 100 ppmv, capacity: 84 mg g−1 | 459 | 233 |
| 20 wt% ZnO/SBA-15 | Gases: H2S, N2, and H2O, temperature: 25 °C | 500 mg m−3 | Concentration: 1.5 mg m−3, capacity: 41 mg g−1 | 213 | 262 |
| 30 wt% ZnO/MCM-48 | Capacity: 53.2 mg g−1 | 323 | |||
| 30 wt% ZnO/MCM-48 | Gases: H2S, CO2, CO, H2, and N2, temperature: 500 °C | 2000 ppmv | Concentration: 100 ppmv, capacity: 54 mg g−1 | 459 | 233 |
| ZnO/SiO2 | Gases: H2S and N2, temperature: 30 °C | 850 mg m−3 | Concentration: 0.15 mg m−3, capacity: 28.6–108.9 mg g−1 | 106.3–168.5 | 263 |
| Co3O4/SiO2 | Capacity: 114.3 mg g−1 | n/a | |||
| CuO/SiO2 | Capacity: 145.6 mg g−1 | n/a | |||
| Gases: H2S and N2, temperature: 150 °C, pressure: 1 bar | 100 ppm | Concentration: 5 ppm, capacity: 32–363 mg g−1 | 13.44–175.75 | 264 | |
| HKUST-1/GO-PEI | Gases: H2S and N2, temperature: 25 °C, pressure: 1 bar | 99.6 ppm | Concentration: 5 ppmv, capacity: 30.7–55.6 mg g−1 | 56–489 | 259 |
| SBA-15/PEI-25 | Gases: H2S, N2, and O2, temperature: 25 °C, pressure: 1 bar | 1000 ppm | Concentration: 50 ppm, capacity: 26.6 mg g−1 | 495 | |
| UiO-66/GO | Gases: H2S and N2, temperature: 30–50 °C | 4500 ppm | Concentration: 2250 ppm, capacity: 115.9–296.5 mg g−1 | 1002–1432 | 256 |
| 1 wt% TiO2/UiO-66 | 4000 ppm | Concentration: saturated, capacity: 180–210 mg g−1 | 1171 | 255 | |
| 3 wt% TiO2/UiO-66 | Capacity: 140–180 mg g−1 | 986 | |||
| 5 wt% TiO2/UiO-66 | Capacity: 80–130 mg g−1 | 652 | |||
| 5 wt% TiO2/GO | Gases: H2S and N2, temperature: 220 °C, pressure: 1 bar | 4400 ppm | Concentration: 2200 ppm, capacity: 200 mg g−1 | 154 | 265 |
| 5 wt% N–TiO2/GO | Capacity: 250 mg g−1 | 145 | |||
2.3 Chemical conversion
There are two major conventional technologies for H2S removal by chemical conversion into elemental sulfur such as Claus and LO-CAT processes.The Claus process was further improved to enhance the overall sulfur recovery up to 97% of inlet sulfur.268 The feed gases for the majority of Claus process applications typically originate from absorption units, and they primarily consist of H2S, H2O, and CO2 as major components, along with minor components such as NH3, N2 and hydrocarbons.269 By utilizing amine extraction, H2S is absorbed from the absorption unit gas stream and channeled into the Claus unit. The furnace and waste heat boiler are usually considered as a single unit. The process of H2S conversion in Claus technology involves two main steps such as thermal oxidation and catalytic reaction. The two-step process is described using eqn (2.7) and (2.8); meanwhile, eqn (2.9) is the overall reaction of the Claus process.182
| H2S + 1.5O2 → H2O + SO2 | (2.7) |
| 2H2S + SO2 → 3S + 2H2O | (2.8) |
| 2H2S + O2 → 2S + 2H2O | (2.9) |
The Claus process combines multiple thermal and catalytic steps.270 One third of the feed gas containing H2S is burned in a furnace with oxygen from air to give sufficient SO2 at 823 K in the thermal step as in eqn (2.7). In the presence of a catalyst based on alumina- or titanium dioxide, the remaining H2S undergoes further reaction with SO2 to produce water and elemental sulfur in the catalytic steps as described in eqn (2.8). However, since O2 in eqn (2.9) is limited, H2S is not fully converted.271 Hence, the treatment of tail gas is required to comply with the environmental regulations.
Nearly all refinery plants require above 99% sulfur recovery from SRUs to meet the strict SOX emission laws. The Claus process is only able to attain 97% of sulfur recovery even with 3 catalytic reactors. Since H2S is not converted fully in the Claus process, its tail gas requires an additional sulfur recovery unit for tail gas treatment to meet environmental regulations regardless of the plant capacity. Three main methods of tail gas treatment that are available at the commercial scale are sub dew point, selective or direct catalytic oxidation and reduction–absorption.272 The sub dew point process uses a special alumina-based catalyst to process H2S and SO2 in the tail gas from the Claus reactor at or below the sulfur dew point in a liquid system to produce more elemental sulfur at a low temperature. Nevertheless, the catalyst is vulnerable to deactivation due to deposition of liquid elemental sulfur on its surface and requires occasional replacements.182
The advantages of the Claus process are it is convenient for a high percentage of H2S conversion and a well proven process technology for desulfurization. The disadvantages of the Claus process are the process is not economical and might not operate after H2S concentration falls below 20%. In addition to this, the catalyst tends to deactivate because of liquid elemental sulfur deposition on its surface. The presence of heavy hydrocarbons will lead to catalyst fouling and deactivation, resulting in a lower quality sulfur product.273 Besides, the Claus process is an energy intensive process and requires a significant investment in capital equipment, and high operating costs due to the need for high-temperature combustion and the use of a catalyst.274 There are also safety concerns as the process requires careful handling of high-temperature and high-pressure gases, which can pose safety risks if not managed properly. Last but not least, the process has a limited operating range and may not be effective for gas streams with low H2S concentrations or high levels of other impurities.275
The LO-CAT technology was first used commercially in 1980. Commencing with the oil and gas industry, the LO-CAT process has been persistently upgraded and modified from time to time to allow for extended usage in other industrial segments and markets such as petrochemicals, metals, water and wastewater treatment and last but not least, CO2-based products such as food and beverages. More recently, alternative and conventional energy sources such as stranded offshore gas, shale gas, gasification syngas and biogas have also successfully utilized LO-CAT technology for sulfur recovery.276 The LO-CAT process involves an exclusive liquid redox catalyst that converts H2S to elemental sulfur by carrying out direct oxidation of H2S.
The LO-CAT process uses iron(III) chelate catalytic solution as a catalyst reagent to promote the overall reaction.276 In the absorber, the sulfide ions are oxidized by the iron ions. Eqn (2.10) and (2.11) represent H2S absorption into the aqueous, chelated iron (Fe) solution and its subsequent ionization. Eqn (2.12) represents the S−2 ion oxidation to elemental sulfur and reduction of Fe3+ to Fe2+. In the oxidizer, the reduced iron ions are regenerated by oxidation in dissolved oxygen for reuse as expressed in eqn (2.13). The function of organic chelating agents is to inhibit the precipitation of ferric hydroxide or ferrous sulfide. The Fe+2/Fe+3 reactions are very rapid so minimum excess S2− ions are carried over into the oxidizer. Eqn (2.12) and (2.14) occur very fast. Hence, the unwanted byproduct, thiosulfate salts are not formed by the side reaction. Eqn (2.13) and (2.14) represent the oxygen absorption from surrounding air into the aqueous solution followed by Fe2+ to Fe3+ oxidation. However, eqn (2.10) and (2.13) are quite slow and become the rate controlling steps in a LO-CAT process.276
| H2S + 0.5O2 → H2O + S | (2.10) |
| H2S → 2H+ + S−2 | (2.11) |
| Fe−3 + S−2 → 2Fe+2 + S | (2.12) |
| 2Fe+2 + 0.5O2 + H2O → 2Fe+3 + 2OH− | (2.13) |
| 2S−2 + 1.5O2 → S2O3−2 | (2.14) |
Also, excess dissolved O2 is limited by the presence of Fe+2 ions in the regenerated solution. The presence of high partial pressures of CO2 in the process gas stream leads to pH reduction of the LO-CAT solution. Thus, a buffer solution such as ammonium, sodium or potassium carbonate is added. Since the reaction of H2S absorption is very fast compared with CO2 absorption, high selectivity of H2S removal can be attained. Fe was chosen for the LO-CAT process mainly because it is safe to operate and inexpensive, even though there are many metals that are able to perform similar functions. The iron(III) chelate catalytic solution does not participate in the reaction. Its role is just to retain Fe ions in solution because both Fe2+ and Fe3+ ions are not very stable and soluble in aqueous solutions.276,277 Usually at low concentrations, Fe will form precipitates as either ferrous sulfide, FeS or ferric hydroxide, Fe(OH)3. The chelating agents are organic compounds that wrap around the Fe ions to prevent them from precipitating and holding them in solution throughout the operation.
LO-CAT has been developed into an adaptable processing technology for treating gas streams containing any percentage of H2S. Among the advantages of LO-CAT include the ability to treat both aerobic and anaerobic streams of gases, high efficiencies of H2S removal and production of only safe products and byproducts. Elemental sulfur recovered by the LO-CAT technology is totally different from sulfur produced by other processes. Compared to the sulfur generated by other methods, LO-CAT sulfur particle sizes are finer, within a range of 8 to 45 microns.278
There are a few advantages of the LO-CAT process such as easily obtainable and cheap catalysts besides being stable at any pH, low catalyst consumption and tolerating CO2, NH3 and other gas contaminants. The LO-CAT process almost ensures complete H2S removal from acid gas streams with low H2S content. The process operates under mild reaction conditions and does not require high temperatures or pressures, resulting in lower energy consumption compared to other processes.279 However, there are also a few disadvantages associated with the LO-CAT process such as the catalyst reagent used is slightly corrosive, and therefore, equipment cannot be fabricated from carbon steel. Besides, the process has a limited operating range, and its performance can be affected by variations in operating conditions such as gas composition and temperature.280 Furthermore, the degradation of chelated iron and the formation of sulfur-oxo-acid salts reduce the concentration of iron(III) chelate in the solution, hence limiting the sulfur production to 1050 kg h−1.281 Additionally, the LO-CAT catalyst needs to be periodically regenerated, which requires additional equipment and chemicals. Furthermore, the LO-CAT catalyst is proprietary, which can limit the availability and increase the cost of the catalyst.280
2.4 Membrane separation
Membrane separation is an industrially established technology in various industrial processes and has several advantages over other technologies such as absorption and adsorption due to its high modularity, large surface area of the membrane per unit module, smaller footprint and simple operation.282 In addition, membrane separation can optimize the gas separation processes by lowering the energy requirement in the presence of pressurized gas, reducing the size of equipment and capital costs and improving the process safety, and it does not require a complex system to operate.283 Besides, H2S removal can be accomplished effectively via membrane separation at low cost at both offshore and remote areas.284 Nonetheless, current research and development of membranes were focusing mainly on CO2 removal instead of H2S. There was a higher emphasis on membrane development for various industrial gases such as light hydrocarbons, CO2, N2, O2 and H2 based on review papers to date.285–288 Only a few reviews289,290 reported the development of polymeric membranes for H2S removal due to high purification requirements (<4 ppm) and the toxic nature of the gas. Essentially, the membrane separation process uses a physical porous barrier where selective gas transport via permeation occurs due to transmembrane pressure. The gas separation of these membranes is influenced by the permeability–selectivity relationship while the gas permeability is controlled by the solution–diffusion mechanism. In recent years, membrane materials have been engineered to cater to the need for higher performance, efficiency and economy by optimizing several criteria such as selectivity, permeability, and chemical and mechanical stability.291The application of membrane separation technology is mostly overshadowed by polymeric membranes as inorganic membranes are expensive and difficult to manufacture.292 The only disadvantage of polymeric membranes as compared to inorganic membranes is their inability to operate under high temperature conditions. Nonetheless, polymeric membranes have proven to be one of the most successful membranes in industrial applications for H2S separation due to their high chemical stability, ability to operate under harsh conditions and very efficient pretreatment for other technologies to purify streams containing high level of H2S.291 Such polymeric membranes include cellulose acetate-based membranes, i.e., Schlumberger Cynara®293 and UOP Separex™293 and polyimide-based membranes, i.e., Air Liquide MEDAL™.294 In general, polymeric membranes are used for natural gas sweetening processes which contains 10–20% or higher of H2S concentration and are driven by the selectivity of H2S over CO2 and CH4, respectively. Despite having similar operating conditions to biogas separation, the performances of these membranes for H2S removal from biogas are rarely reported. This is due to the membrane material failure to withstand acidic properties of H2S in the gas streams and since they are only capable of removing very low concentrations of H2S (<1%) efficiently.
Membrane-based gas separation operates through a pressure-driven mechanism as the gases flow through a thin selective membrane capable of separating different chemicals species. The process is driven by the pressure difference from high to low pressure across the membrane which allows the gas molecules to pass through the membrane layer. The rate of gas permeation is highly dependent on diffusivity, membrane thickness, partitioning of chemical species in the feed-membrane phase and the interactions between the molecules and the membranes. A higher gas solubility and diffusivity rate will result in a higher flow rate of gases passing through the membrane at a certain membrane thickness and pressure gradient.
The key factors contributing to the performance of polymeric membranes in gas separation are permeability and selectivity. Permeability is defined as the rate at which a chemical species penetrates through a membrane, whereas selectivity is the membrane’s ability to effectively select the desired product with high purity and recovery. According to Robeson,295 there is an inverse relationship between permeability and selectivity. A higher permeability will lead to a lower selectivity and vice versa. To achieve the optimal separation conditions and highest performance using polymeric membranes, a reasonable balance between both parameters must be obtained. Additionally, high durability with high chemical and thermal stability must be present in polymeric membranes to withstand harsh operating conditions during application.
Rubbery polymers have low glass transition temperatures, are flexible under ambient conditions and rely mainly on solubility for transport across rubbery membranes,300 thus making them favorable for H2S removal from CH4 which is influenced by solubility.301 Furthermore, the performance of a polyethylene glycol (PEG)-based membrane302 can be tuned by changing the crosslinking density and polymer crystallinity. Membrane rigidification was found to be effective in resisting the strong effect of plasticization and improving the overall diffusion in a solubility-controlled membrane, contributing to outstanding H2S/CH4 and CO2/CH4 selectivity of higher than 110 and 60, respectively.302 Nevertheless, polydimethylsiloxane (PDMS) was found to be the most permeable rubbery polymer compared to the rest of the polymeric membranes.303
According to Robb,312 the H2S presence could significantly increase the elasticity of PDMS membranes leading to a very high permeability of 10000 Barrer for H2S as compared to CO2 (3250 Barrer) and CH4 (900 Barrer). Besides, the permeability of the PDMS membrane is also observed to be very high in acidic compounds, with a selectivity of 3 and 10 for H2S/CO2 and H2S/CH4, respectively. However, no plasticization effect was observed as the permeability coefficients remain unchanged despite the increase in feed pressure. Several authors313,314 discovered that a PDMS membrane is more permeable in H2S compared to CO2. Meanwhile, Bhide et al.313 determined that the permeability of the PDMS membrane is 1000 Barrer at 25 °C, whereas Merkel et al.314 obtained a value of 5100 Barrer at 23 °C.
A similar observation was also reported by Merket and Toy. A study revealed that the applied PDMS coating layer on the crosslinked 6FDA-DAM/DABA polyimide hollow fiber membranes successfully enhanced membrane performance. The selectivity of H2S to CH4 was in the range of 22 to 29 under demanding sour gas feed conditions, such as a 20% H2S concentration and very high pressures ranging from 34.47 to 48.26 bar.315 It was anticipated that the PDMS coating layer assisted in sealing and leveraging the morphological defects present in the crosslinked polyimide fibers, which could potentially worsen under such harsh conditions.
Based on a comparison between different types of fluorinated and non-fluorinated polymers, Merkel and Toy323 found out that both Cytop and Teflon membranes were capable of absorbing more CO2 than H2S. These results also indicate that fluorinated polymers are more plasticization-resistant as compared to non-fluorinated polymers as they are highly resistant to chemical degradation which allows them to withstand acidic conditions without a decrease in performance. On the other hand, perfluorosulfonic acid (PFSA) such as Aquivion and Nafion exhibited a glassy behavior under dry conditions and increasing permeability under humid conditions. Several authors324–326 reported that the PFSA membranes are showing glassy behavior under dry conditions, the gas permeation is dependent on the solution–diffusion and their performance is affected by the kinetic diameter of the reactants. However, PFSA membranes were found to have a low permeability which makes them undesirable for gas separation purposes under dry conditions.327 Nonetheless, PFSA membranes demonstrated outstanding performances and strong affinity towards polar compounds when being used under humid conditions. By increasing the relative humidity of the feed gas at 35 °C, PFSA membranes exhibited increasing permeability from 32 to 370 Barrer and from 40 to 250 Barrer for H2S and CO2, respectively.328
Other polyimide membranes that are commonly used are semi-fluorinated modified polyimides such as 6FDA, 6F-PAI-1, 6FDA-HAB and 6FDA-DAM:DABA. These polymers are proven to have a high selectivity towards H2S, and CO2 as compared to CH4 with a selectivity range of 10 to 16 for H2S/CH4 and 30 to 60 for CO2/CH4.307,332,333 Kraftschik et al.334 discovered that the plasticization effect of the 6FDA-DAM:DABA membrane can be minimized by thermal treatment when tested with binary mixture gases of H2S and CH4, and also ternary mixture gases of H2S, CO2 and CH4. They found out that upon treatment, the permeability of H2S has increased to 100 Barrer and the interactions between the reactant and polymers have increased the selectivity of membranes. On the other hand, Vaughn and Koros307 investigated the performances of 6FDA and 6F-PAI-1 membranes for H2S removal and discovered a selectivity of 10 for H2S/CH4.
In addition, Baker344 discovered that the permeability of H2S in cellulose acetate membranes become lower at higher relative humidity due to the hydrophilic nature of the membranes which lose their rigidity as they began to absorb water from the surroundings.339 Due to these limitations, their application at the industrial level has been restricted to operating only in the presence of a little water in the feed gas to maintain a high level of permeability and selectivity. To improve the separation performance and mechanical resistance of the cellulose acetate membrane, various studies have been conducted to develop a better membrane by functionalizing the surface with specific groups such as vinyl methoxysilanes. The modified cellulose acetate membrane not only showed similar selectivity to the unmodified membrane but a significantly improved permeability up to 200 Barrer from 8.7 Barrer originally.342
On the other hand, Liu et al.345 investigated the plasticization effect of CTA hollow fibers on the removal of acid gas from a feed stream containing a mixture of gases of H2S, CO2, CH4, C2H6 and C3H8 at 35 °C. The CTA hollow fibers demonstrated a remarkably higher permeability towards H2S and CO2 as the feed gas pressure increases. As pressure increased from 5 to 30 bar, the permeability rose from 80 GPU to 140 and 120 GPU for H2S and CO2, respectively. This happened due to the competitive absorption between H2S and CO2 and also the plasticization effect of CTA hollow fibers at high temperature. Furthermore, Liu et al.345 concluded that the plasticization effect of acid gases and high separation efficiency contributed to high selective removal of H2S in the presence of CO2.
| Membrane | Feed composition | Membrane separation conditions | H2S permeability (mol m−2 s−1 Pa−1) | H2S/CO2 selectivity | H2S/CH4 selectivity | Reference |
|---|---|---|---|---|---|---|
| PEBAX 3533 SA00 | 1.3 mol% H2S, 27.9 mol% CO2, and 70.8 mol% CH4 | Temperature: 35 °C, pressure: 10 bar | 2.97 × 10−13 | 3.65 | 21 | 309 |
| PEBAX 4033 SA00 | 1.04 × 10−13 | 3.7 | 24 | |||
| PEBAX 6333 SA00 | 1.26 × 10−14 | 5.11 | 20 | |||
| PEBAX 7233 SA00 | 2.54 × 10−15 | 1.85 | 15 | |||
| PEBAX MX 1041 | 5.85 × 10−14 | 4.41 | 49 | |||
| PEBAX MX 1657 | 8.3 × 10−14 | 3.59 | 50.6 | |||
| PEBAX MX 1074 | 1.85 × 10−13 | 4.53 | 54 | |||
| 12.5 mol% H2S, 18.1 mol% CO2, and 69.4 mol% CH4 | 2.33 × 10−13 | 4.48 | 50.4 | |||
| PEBAX SA01 MV3000 | Pure | Temperature: 35 °C, pressure: 1.17 bar | 1.75 × 10−13 | 4.87 | 50.48 | 307 |
| Pressure: 2 bar | 2.1 × 10−13 | 5.83 | 60.43 | |||
| Pressure: 2.76 bar | 2.25 × 10−13 | 5.83 | 67.32 | |||
| Pressure: 3.45 bar | 2.42 × 10−13 | 6.49 | 64.94 | |||
| PDMS | Pure | Temperature: 35 °C, pressure: 1.1 bar | 1.68 × 10−12 | 3.08 | 10 | 314 |
| n/a | 1.8 | 6.5 | 348 | |||
| PDMS + PEG + PSf (glycerol) | Temperature: 25 °C, pressure: 4.4 bar | n/a | 5.7 | 140 | 317 | |
| Cytop | 15 mol% H2S and 10.5 mol% CO2 | Temperature: 37 °C, pressure: 1 bar | n/a | n/a | n/a | 314 |
| Silica-rich CHA-type zeolite | 0.3 mol% H2S, 2.13 mol% CO2, and 97.57 mol% CH4 | Temperature: 25 °C, pressure: 4 bar | 1.70 × 10−8 | n/a | 3.24 | 349 |
| Amine-based CNT butylamine, sec-butylamine. dodecylamine, and octadecylamine | 0.5, 1.2, and 2.5% (6000, 12000, 2 and 5000 ppm of H2S, remaining gas is CH4) |
Temperature: 30 °C | n/a | : | 350 | |
| Pressure: 1–5 bar | (6.2–7.6) × 10−7 | 7–8.4 | ||||
| (3.2–4.1) × 10−7 | 3.9–4.8 | |||||
| (4.7–5.7) × 10−7 | 5.5–7 | |||||
| (2.9–3.6) × 10−7 | 3.5–4.5 | |||||
| Cellulose acetate (CA) | 20 mol% H2S, 20 mol% CO2, and 60 mol% CH4 | Temperature: 35 °C, pressure: 34 bar | 2.91 × 10−15 | 1.01 | 30.5 | 342 |
| Pressure: 35 bar | n/a | n/a | 30 | |||
| Pressure: 48 bar | 1.33 × 10−14 | 1.44 | 27.5 | |||
| 6 mol% H2S, 29 mol% CO2, and 65 mol% CH4 | Pressure: 10.8 bar | n/a | 0.88 | 19 | 309 | |
| Modified CA | 20 mol% H2S, 20 mol% CO2, and 60 mol% CH4 | Temperature: 35 °C, pressure: 34 bar | 6.82 × 10−14 | 1.58 | 34.3 | 342 |
| Pressure: 48 bar | 6.36 × 10−14 | 1.40 | 27.5 | |||
| CTA | 1000 ppm H2S and remaining N2 | Temperature: 25–80 °C, pressure: 1–9 bar | (1.57–3.41) × 10−15 | n/a | n/a | 351 |
| CTA hollow fiber | 20 mol% H2S, 5 mol% CO2, 3 mol% C2H6, 3 mol% C2H8, 100–300 ppm toluene, and remaining CH4 | Temperature: 35, 50 °C, pressure: 6.9–31.3 bar | 4.69 × 10−14 | n/a | 28 | 345 |
| 6FDA-DAM. 6FDA-DAM/DABA (3:2) |
(i) 0.5 mol% H2S, 20 mol% CO2, and 79.5 mol% CH4 | Temperature: 35 °C, pressure: 7–46 bar | (6.93–36.4) × 10−14, (7.63–29.3) × 10−15 | n/a | 14–38.7, 18.8–49.1 | 301 |
| (ii) 5 mol% H2S, 45 mol% CO2, and 50 mol% CH4 | ||||||
| (iii) 20 mol% H2S, 20 mol% CO2, and 60 mol% CH4 | ||||||
| 6FDA-DAM/6FpDA (1:3), 6FDA-DAM/CARDO (1:3), 6FDA-DAM/ABL-21 (1:3), 6FDA-DAM/6FDA-CARDO (1:1) |
20 mol% H2S, 10 mol% CO2, 57–59 mol% CH4, 1–3 mol% C2H6, and 10 mol% N2 | Temperature: 22 °C, pressure: 24,35, 46 bar | (1.76–2.28) × 10−14, (7.83–8) × 10−15, (1.05–1.1) × 10−14, (3.85–6.03) × 10−14 | n/a | 19.7–21.2, 15.2–15.9, 15.2–15.9, 23–25 | 352 |
| Crosslinked telechelic PEG | 5 mol% H2S, 3 mol% CO2, and 92 mol% CH4 | Temperature: 25 °C, pressure: 55.2 bar | (0.027–8.37) × 10−15 | n/a | 65–116 | 302 |
| Vinyl-added polynorbornene (VAPNB) membranes | (i) 5 mol% H2S, 3 mol% CO2, and 92 mol% CH4 | Temperature: 25 °C, pressure: 55.2 bar | 3-Gas mixture: (1.51–2.01) × 10−12 | n/a | 6–45 | 353 |
| (ii) 20 mol% H2S, 10 mol% CO2, 57 mol% CH4, 3 mol% C2H6, and 10 mol% N2 | 5-Gas mixture: (2.01–2.34) × 10−12 | 9–40 | ||||
| 6FDA-HAB-DAM | 1000 ppm H2S and remaining N2 | Temperature: 25, 35, 55, 75 °C, pressure: 2–9 bar | (5.19–5.69) × 10−14 | n/a | n/a | 354 |
| 6FDA-HAB-DAM-DABA | (4.35–5.02) × 10−14 | |||||
| 6FDA-DAM:DABA (3:2) |
10 mol% H2S, 20 mol% CO2, and 70 mol% CH4 | Temperature: 35 °C, pressure: 6.7 bar | 3.88 × 10−15 | 0.21 | 9.0 | 334 |
| Pressure: 20.8 bar | 5.15 × 10−15 | n/a | 10.3 | |||
| Pressure: 41.3 bar | 7.26 × 10−15 | n/a | 13.2 | |||
| Pressure: 62 bar | 9.77 × 10−15 | n/a | 15.6 | |||
| Pure | Pressure: 2 bar | 1.0 × 10−14 | 0.29 | 10 | ||
| 6F-PAI 1 | Pure | Temperature: 35 °C, pressure: 4.5 bar | 2.07 × 10−15 | 0.19 | 8.1 | 355 |
| 6F-PAI 2 | 1.0 × 10−15 | 0.21 | 10.3 | |||
| 6F-PAI 3 | 1.67 × 10−15 | 0.23 | 10.9 | |||
| PPO | 0.4 mol% H2S and 99.96 mol% CH4 | Temperature: 22 °C, pressure: 4.5 bar | n/a | n/a | 3.1 | 339 |
| PPOP | Pure | Temperature: 30 °C, pressure: 2.1 bar | 4.01 × 10−15 | 2.5 | 10 | 356 |
| Polyester urethane urea (PEUU) + Teflon | 3 mol% H2S, 5.4 mol% CO2, and 91.6 mol% CH4 | Temperature: 35 °C, pressure: 10 bar | n/a | 3.79 | 43 | 357 |
| Pressure: 30 bar | n/a | 2.72 | 27 | |||
| Temperature: 55 °C, pressure: 10 bar | n/a | 3.91 | 43 | |||
| Pressure: 30 bar | n/a | 1.9 | 12 | |||
| PPO hollow fiber, PEUU | 101, 198, 401, 968, 3048, and 5008 ppm H2S in CH4, and real natural gas concentration: 3360 ppm H2S | Temperature: 22, 40 °C, pressure: 3.45, 5.17, 6.89 bar | PPO:(0.5–2.44) × 10−14 | n/a | 1.34–4.1 | 358 |
| PEUU: 1.67 × 10−15 | 3.25–3.43 | |||||
| PPG-PEU (PU1) | 1.3 mol% H2S, 27.9 mol% CO2, and 70.8 mol% CH4 | Temperature: 35 °C, pressure: 10.8 bar | 8.0 × 10−14 | 3.0 | 21 | 309 |
| 12.5 mol% H2S, 18.1 mol% CO2, and 69.4 mol% CH4 | 6.12 × 10−14 | 4.28 | 22.6 | |||
| PPG-PEUU (PU2) | 1.3 mol% H2S, 27.9 mol% CO2, and 70.8 mol% CH4 | 2.05 × 10−13 | 3.1 | 19 | ||
| 12.5 mol% H2S, 18.1 mol% CO2, and 69.4 mol% CH4 | 2.07 × 10−13 | 3.17 | 18 | |||
| PEG-PEU (PU3) | 1.3 mol% H2S, 27.9 mol% CO2, and 70.8 mol% CH4 | 9.07 × 10−14 | 4.6 | 58 | ||
| 12.5 mol% H2S, 18.1 mol% CO2, and 69.4 mol% CH4 | 9.37 × 10−14 | 4.5 | 54.9 | |||
| PEG-PEUU (PU4) | 1.3 mol% H2S, 27.9 mol% CO2, and 70.8 mol% CH4 | 6.66 × 10−14 | 4.45 | 74 | ||
| 12.5 mol% H2S, 18.1 mol% CO2, and 69.4 mol% CH4 | 7.46 × 10−14 | 4.39 | 66 | |||
| PVDC | Pure | Temperature: 30 °C, pressure: 0.93 bar | n/a | n/a | n/a | 356 |
| PVDF + [Bmim][BF4] | Pure | Temperature: 35–65 °C, pressure: 4 bar | (0.54–3.68) × 10−13 | n/a | 130–260 | 359 and 360 |
| PVDF + [Bmim][BF4] | Temperature: 40 °C, pressure: 1 bar | 1.36 × 10−12 | 3.7 | 36.6 | 361 | |
| PVDF + [Bmim][PF6] | 6.38 × 10−13 | 3.7 | 29.8 | |||
| PVDF + [Bmim][AC] | 2.44 × 10−12 | 11.7 | 136 | |||
| PVDF + [Bmim][NTf2] | 8.7 × 10−13 | 2.1 | 19.8 | |||
| PVDF + [Bmim][CF3SO3] | 1.44 × 10−12 | 4.0 | 50.7 | |||
| PVTMS | Pure | n/a | 1.17 × 10−13 | n/a | 1.59 | 362 |
| PVTFA | Pure | Temperature: 30 °C, pressure: 1 bar | n/a | n/a | n/a | 363 |
| PTBP | Pressure: 2.1 bar | 5.35 × 10−15 | 0.94 | 9.4 | 356 | |
| PDTBP | 6.69 × 10−15 | 0.74 | 4 | |||
| PVBTAF | 10.5 mol% H2S and 89.5 mol% CH4 | Temperature: 30 °C, pressure: 1.2 bar | n/a | n/a | 7000 | 364 |
| 10 mol% CO2 | Pressure: 1.1 bar | n/a | 10.8 | 3300 | ||
| 10.3 mol% H2S | Pressure: 8.5 bar | n/a | 8.7 | 2100 | ||
| 79.7 mol% CH4 | Pressure: 5 bar | n/a | n/a | 950 | ||
| Nafion NE111 | Pure | Temperature: 20 °C, pressure: 1 bar | 1.87 × 10−13 | n/a | n/a | 365 |
| Nafion NE117 | 7.56 × 10−13 | n/a | n/a | |||
| Aquivion® | 10 mol% H2S and 90 mol% CH4 | Temperature: 35 °C, pressure: 1 bar | 1.07 × 10−14 | 0.53 | 12 | 328 |
| Pressure: 2 bar | 1.26 × 10−13 | 0.93 | 28 | |||
| PBI composites | 5.3 mol% H2S, 5.1 mol% CO2, and 89.6 mol% CH4 | Temperature: 50 °C, pressure: 1.2 bar | 2.72 × 10−13 | 7.5 | 140 | 366 |
| TEGMC:6FDA-DAM/DABA (3:2) with/without PDMS |
(i) 5 mol% H2S, 45 mol% CO2, and 50 mol% CH4 | Temperature: 35 °C, pressure: 6.9–48.3 bar | (3.18–6.7) × 10−15 | n/a | With PDMS: 20–29 | 315 |
| (ii) 20 mol% H2S, 20 mol% CO2, and 60 mol% CH4 | Without PDMS: 12–20 |
2.5 Cryogenic distillation
The distillation process has been used since the 19th and 20th centuries, specifically for separation of alcohol applications.367 Frank Sherwood Taylor368 pioneered the distillation concept in 1945 using laboratory apparatus such as condensing apparatus, a vessel and a receiver. In 1953, John M. Chambers369 further developed the idea by creating a structured distillation column for the purification of fermented alcohols by taking into consideration heat input and optimum reflux ratio factors. Since then, distillation technology has improved significantly and is well-suited for harsh operating conditions such as low and high temperatures, high pressure as well as vacuum conditions. A distillation column is widely used for separation of two or more components from the feed gas based on different boiling points and volatility of the respective components.370 The component with a lower boiling point left the column as the top product, whereas the component with a higher boiling point remained as a liquid as the bottom product.Cryogenic distillation technology is a process used to separate different components of a gas mixture by cooling the mixture to very low temperatures and then distilling it.371 Cryogenic distillation technology is one of the most efficient methods used for the simultaneous removal of CO2 and H2S from natural gas streams.372 Commercially, cryogenic separation is widely utilized for CO2 separation to fulfill the pipeline requirements. There are a few advancements in cryogenic separation technology being implemented currently such as anti-sublimation unit (AnSU), controlled free zone (CFZ), cryogenic packed bed (CPB) and CryoCell®.
The advantage of the AnSU method is high purity of CO2 which can be captured without the presence of any contaminants.376 Clodic et al.375 discovered that AnSU consumed a high level of energy for the removal of low CO2 concentration and the energy consumption decreases as the concentration of CO2 increases. On the other hand, Schach et al.377 investigated and made a comparison between AnSU and the adsorption method in terms of energy consumption using ASPEN HYSYS simulation for CO2 removal at 90% efficiency. The results showed that the energy consumption of AnSU is far lower at 178 MW as compared to the adsorption method using MEA at 208 MW.
Despite having high efficiency of CO2 removal, AnSU has several drawbacks such as high costs for regular maintenance for the compression systems and high capital costs for AnSU since it requires five stages of separation mechanisms. Furthermore, the formation of a CO2 frost layer on the surface of the heat exchanger affects its efficiency and thus, a better material is required for the heat exchanger with higher mechanical stress tolerance and thermal conductivity.378 Moreover, simultaneous removal of CO2 and H2S using AnSU has not yet been investigated.
CFZ™ technology is regarded as economically viable due to its capability of handling various compositions of H2S and CO2 in the feed stream. The discharge of both top and bottom products at relatively high pressure helps to minimize the recompression cost for delivery and reinjection of acid gas back to the reservoir. The acid gas injection (AGI) method is usually used to discard the separated H2S and CO2 from the feed gas. By combining the CFZ™ and AGI approaches, the geological sequestration of CO2 could be facilitated using the high-pressure liquid stream released from the bottom column.384 Furthermore, Northrop et al.149 finalized the CDP of CFZ™ technology and concluded that the technology is ready for scale-up and commercialization for sour gas treatment up to 327 m3 s−1. However, CFZ™ application for simultaneous removal of H2S and CO2 is facing drawbacks as the presence of H2S inhibits the freezing of CO2.
One of the major advantages of this process is its ability to simultaneously remove H2S, CO2 and water from the sour gas based on their respective sublimation and dew points.378 Additionally, the CPB process does not require chemical solvents and high pressure to operate, which in turn could save cost as the chemical solvents require regular replacement, which could lead to a higher capital cost.387 The crystallization of H2S, CO2 and water only requires the cold energy provided by the packed bed materials without requiring high pressure.387 Furthermore, the methane produced by the CPB process has a higher purity as compared to that produced by the PSA process. Ali et al.388 compared the energy requirements between the CPB and conventional methods for the removal of 70% CO2. The authors found that CPB consumed lower energy at 810 kJ kg−1 of CO2 as compared to the conventional cryogenic process which used 1472 kJ kg−1 of CO2. On the other hand, Turnier et al.389 concluded that the energy consumption by the CPB method was much lower at 2.9 MJ kg−1 of CH4 as compared to the pressure swing adsorption (PSA) which used 3.7 MJ kg−1 of CH4.
However, the CPB process faces several drawbacks such as limited availability of commercial thermal insulators that can maintain a very low temperature required by the process, loss of cold energy to the surroundings and high energy consumption for simultaneous removal of H2S and CO2.390 The reason behind high energy requirement for simultaneous removal of H2S and CO2 is the requirement to reach the H2S dew point at −150 °C. To achieve high removal efficiency of H2S, the process requires long hours of operation at extremely low temperatures and leads to high costs of operation. Therefore, to minimize losses during the purification process, it is recommended to have a proper heat integration system between the LNG production and air separation unit.391
The initial step of the CryoCell® process involves the treatment of feed gas, which has a high CO2 content, by reducing its moisture level to as low as 5 ppm through a dehydration process.388 After this, the dehydrated feed gas is cooled to a temperature above the freezing point of CO2 at a constant pressure, condensing the gas mixture into a liquid phase. Subsequently, the liquid mixture undergoes expansion through a Joule–Thomson valve, while maintaining a constant enthalpy that induces a phase change in CO2, into liquid, solid, and vapor phases which is later separated by a three-phase separator.395 It is very critical to maintain a low CO2 content in the vapor phase and high CO2 level in the liquid product in the CryoCell® process. The solid CO2 product will then be melted and mixed with the liquid product.
Cool Energy Ltd. collaborated with Shell Global Solutions and developed a commercial demonstration plant (CDP) in 2006 to investigate the viability of CryoCell® technology.392 The CDP of CryoCell® was designed to process 60 mol% of CO2 feed gas with the pressure ranging from 55 to 65 bar and feed flowrate between 600 and 1300 kg h−1. The separator was designed to operate at 12, 16 and 19 bar whereas the reboiler was kept between −50 and −60 °C. The results from CDP showed that CryoCell® technology can remove up to 81% of CO2 from natural gas. On the other hand, Hart and Gnanendran392 performed a comparison between the CryoCell® and amine process for treatment of feed gas containing 20 mol% and 30 mol% of CO2 using Aspen HYSYS simulation. Their results indicated that CryoCell® required a lower heat duty as low as 0.1 MW, as compared to the amine process which required between 19 and 35 MW. Nonetheless, the simulation data also revealed that the CryoCell® process requires more compression between 4.3 and 7.0 MW, as compared to the amine process which only requires 1.9 to 3.8 MW at higher CO2 content in the feed gas.392
Based on initial testing through CDP and simulation data, it is expected that this technology can be commercialized and has huge potential for treatment of natural gas involving high CO2 content and geo-sequestration of CO2 for storage. Furthermore, Amin et al.393 stated that the CryoCell® process can handle a diverse range of contaminants including H2S and heavy hydrocarbons. Nevertheless, limited applications of CryoCell® technology for H2S removal were reported. Besides, the CryoCell® process also faces several drawbacks such as uncontrollable freezing of CO2 and poor handling of solids formed. Therefore, more research should be carried out for H2S removal applications by benefiting from their progress for CO2 purification.
In general, cryogenic distillation can achieve very high levels of sulfur recovery, up to 99% or higher, which means that almost all of the H2S in the natural gas stream can be separated and recovered.396 However, H2S purification is an energy-intensive process and requires greater cooling duty to cool down H2S to below its boiling point. As a result, H2S removal by cryogenic separation is rarely reported as of now. In H2S removal, the process starts with the compression of the gas stream to a high pressure and then cooling to a very low temperature below the boiling point of H2S, typically around −50 °C.397 This causes the H2S gas to condense and form a liquid, which can be removed from the gas stream via the distillation method. The process requires a significant amount of energy to cool the natural gas stream to very low temperatures necessary for the H2S to condense.398 This leads to high operating costs, especially for large-scale industrial applications.
In addition, cryogenic distillation requires complex and expensive specialized equipment with high maintenance costs such as refrigeration units, distillation columns and heat exchangers.371 Furthermore, there are also safety concerns regarding cryogenic distillation as the process involves handling extremely cold materials, which can be hazardous if not properly managed. There is also a risk of leaks or equipment failure, which could result in the release of toxic gases or other safety hazards. Besides, the effectiveness of cryogenic distillation for H2S is limited by the feed gas composition. High levels of other impurities such as CO2 and N2 will interfere with the separation process, reducing its efficiency and increasing the operation costs. Last but not least, the process also has a significant environmental impact due to the high energy consumption and the potential for leaks or release of toxic gases.371
3. Status, opportunities and challenges
There are various methods and materials being developed specifically for the purpose of H2S capture from mainly gaseous streams. This emphasizes the significance and urgency of addressing this problem. The suitability of any method to a specific application is highly dependent on various factors such as cost, operating conditions, initial concentration of H2S, gas volume, space and weight limitations, outlet specification and technological readiness level (TRL). Table 6 provides a general comparison between various techniques for H2S removal. However, the general comparison used cannot be applied specifically for every material used under each technique category. For example, a technology with TRL of 9 is mainly because of its wide industrial application such as the wide usage of alkanolamines as absorbents. However, the same absorption method by using ionic liquids or deep eutectic solvents would only have a TRL of 2–4. Comparatively, an absorbent material with a TRL of 4 is more likely to be used for industrial application as compared to a membrane material with the same level of TRL. This is because the absorption technique has a higher TRL generally than membrane separation technology for selective removal of H2S. This highlights the significance of the TRL as a systematic approach to assess the maturity level of a particular material and in technology development for any application, and the TRL is also crucial to determine the ease of its integration into the current industrial environment.| Conventional technologies | Absorption | Adsorption | Conversion | Membrane separation | Cryogenic distillation | |
|---|---|---|---|---|---|---|
| Claus process | LO-CAT process | |||||
| Operating principle | Absorption of H2S in a liquid solvent | Adsorption of H2S onto a solid surface | Conversion of H2S to elemental sulfur via chemical reactions | Catalytic oxidation of H2S to elemental sulfur on a proprietary catalyst | Separation of H2S from a gas stream by a semi-permeable membrane | Separation of H2S from a gas stream by condensation and distillation |
| Reaction involved | Absorptive oxidation reaction | Adsorption reaction | Thermal oxidation and catalytic reaction | Liquid catalytic oxidation reaction | No chemical reaction involved | No chemical reaction involved |
| Phase separation | Gas–liquid phase | Gas–solid phase | Gas–liquid phase | Gas–liquid and liquid–solid phase | Gas phase | Gas–liquid phase |
| Sulfur recovery | 92.8–94.8% of H2S conversion | ≤95% of H2S conversion | 95–97% of H2S conversion | ≥99% of H2S conversion | 0% (pre-treatment step before sulfur recovery) | ≥99% of H2S conversion |
| Advantages | • High removal efficiency | • Easily regenerated | • High removal efficiency | • Low energy consumption | • Simple process | • High purity separation |
| • Simple process | • Designed for high selectivity | • Produces elemental sulfur as a byproduct | • High removal efficiency | • No chemicals required | • Low energy consumption for high flow rates | |
| • Low capital cost | • Proven technology | • Produces no waste products | • Compact design | • Proven technology | ||
| • Can handle large gas volumes | • Produces elemental sulfur as a byproduct | • Can handle high gas volumes | ||||
| • Compact design | ||||||
| Disadvantages | • High solvent consumption | • Requires high temperature and pressure conditions | • High capital and operating costs | • Limited to low H2S concentrations | • Limited to low H2S concentrations | • High capital and operating costs |
| • Requires high temperature and pressure conditions | • Requires frequent replacement or regeneration of the adsorbent | • Complex process | • Limited operating range | • May require additional treatment for high H2S concentrations | • Requires high pressure and low temperature conditions | |
| • Requires regeneration of solvent | • Potential for bed fouling | • Requires additional equipment for sulfur recovery | • Requires careful temperature control and a proprietary catalyst | • Limited by membrane selectivity | • High energy consumption | |
| • Potential solvent degradation | • Limited capacity | • Requires high temperature and pressure conditions | • Require additional treatment for high H2S concentrations | • Potential for membrane fouling | • Requires specialized equipment and expertise | |
| • Can be affected by other gases in the feed stream | • Formation of sulfur-oxo-acid salts that could lead to catalyst poisoning and degradation | • High capital cost | • Can be affected by other gases in the feed stream | |||
| • Spent absorbent is considered as hazardous waste as some contaminants might undergo exothermic reaction with the adsorbent leading to explosion | • Requires high temperature and pressure conditions | |||||
| Capital costs | Medium to high | Medium to high | High | High | Medium to high | High |
| Operating costs | Medium to high | Medium to high | High | High | Medium | High |
| TRL | 9 | 9 | 9 | 9 | 6 | 6 |
The determination of the TRL for a certain technology is carried out based on the economics and sustainability of a certain technology. The most widely used materials currently are activated carbons, aqueous amines, metal oxides, zeolites and chelated iron solution (LO-CAT process). Despite their disadvantages in certain criteria, most of these materials have been used in industrial applications for many decades. Nevertheless, low-cost and sustainable alternatives are yet to be discovered. ILs are widely known as sustainable solvents, but this was proven to be an overstatement for most conventional ILs. Protic ILs and DESs have shown potential as simpler, more sustainable and cheaper options as compared to the conventional ILs, but they are still more costly than amines. Over the recent years, MOFs have made a significant impact on adsorption applications. Despite comparable performances of MOFs to zeolites, MOFs suffer from high material costs as compared to zeolites and amines. Functionalized carbonaceous materials have shown promising development for H2S removal ability, but still require further studies to explore various potential functionalization techniques. Generally, most of the adsorbents were facing regeneration failure due to irreversible chemical reactions on the surface. Membrane separation technology presents numerous benefits and possibilities due to its lightweight and compact characteristics, making it well-suited for applications in remote areas and small-scale settings. However, they are not economical as standalone applications as they are mostly effective when being used together with other techniques such as adsorption or absorption where the main purpose of the membrane is to reduce the feed flowrates before entering the separation columns and helping to reduce the column size and energy requirements. Cryogenic distillation has huge potential for H2S capture applications. Nevertheless, most of the available methods focus mostly on CO2 capture and rarely focus on H2S. Advancement of this particular technology for H2S capture would be beneficial in terms of technical, economic and sustainability factors.
By taking into consideration all the provided information, a general comparison between technologies for H2S capture could be represented clearly as shown in Table 6. The selection of a suitable technology to be used is highly dependent on the operational requirements and conditions of the specific process. For small-scale applications (associated gas, offshore, biogas, etc.), there is a requirement for processes with smaller footprints. For the required sulfur production between 0.5 and 20 tons per day, liquid redox processes such as the LO-CAT process is the best option. As the total gas flowrate got higher, a combination of H2S removal methods which involves membranes and chemical solutions followed by catalytic adsorbents (impregnated ACs and metal oxides) may be adopted. As the H2S partial pressure increases and the required sulfur production is above 20 tons per day, the absorption method is more suitable economically, possibly combined with the membrane separation method. For the operating pressures between 1 and 25 bar, chemical solvents are a viable option. When the operating pressure is between 15 and 40 bar, hybrid solvents are a better option as compared to chemical solvents. As the operating pressure exceeds 30 bar, especially at a very high pressure of 50 bar or even higher, physical solvents are probably a better option as compared to the latter. If there is an additional requirement for other products such as methane or other light gases, then cryogenic distillation could be the most effective economically compared to other methods. Generally, the adsorption method is mostly economic for removal of a low concentration of H2S between 10 and 20 ppm as currently, most of the spent adsorbents cannot be regenerated and need to be disposed.
Recent development of hybrid and composite materials such as functionalized MOFs, supported IL membranes, functionalized mesoporous silica or activated carbons, and mixed matrix membranes based on zeolites are excellent examples of such development. However, these materials were mostly being developed for specific CO2 removal applications and their use for H2S capture is still at the grassroots level. It is also predicted that the development of a new generation of membranes, adsorbents, functionalized ILs and DESs will become the major focal points of H2S capture research in the near future. The usage of computational tools to design the molecules and prediction of their physicochemical properties for IL and DES application such as a conductor-like screening model for real solvents (COSMO-RS) and process modeling is also expected to gain more interest and become a driving force in designing materials that are excellent in various criteria which includes economic, functionality and sustainability aspects.
The exploration of challenges and the way forward in industrial applications of H2S removal, with a specific focus on large-scale adoption, deployment, and medium-scale application, is essential for advancing effective strategies and technologies in this domain. This involves addressing issues such as scalability, integration into existing infrastructure, cost-effectiveness, regulatory compliance, and ensuring optimal performance across diverse operational settings. Scalability remains a critical consideration, ensuring that solutions can be adapted to meet varying demands across different scales of operation. Integration into existing infrastructure is vital for seamless implementation, while cost-effectiveness is essential for widespread adoption. Regulatory compliance underscores the need for solutions that meet environmental standards. Operational performance across diverse settings, from energy production to wastewater treatment, requires robust technologies capable of consistent and reliable H2S removal. Understanding these challenges and potential pathways forward is crucial for enhancing the efficiency, reliability, and sustainability of H2S removal processes in various industrial contexts.
The cost of the material also plays a significant role in determining the suitability of materials for H2S capture application. Usually, the cost of any material decreases as its TRL increases which eventually lead to a larger production of that specific material. The present state of lab-scale development discussed so far is still far from transcending them to a higher level of TRLs. Most of the experimental studies conducted so far have not considered the influence of realistic feed gas compositions on their performances. The presence of impurities in the feed gas such as CO2 and water are recognized to impact the efficiency of H2S removal. This work also addresses a clear gap between science and engineering. Almost all adsorbents and absorbents reported the performance of material in terms of breakthrough capacity or equilibrium, mostly focusing on maximizing their performance. Nonetheless, these parameters are insufficient to provide comprehensive data on the overall performance of the process. Many other critical process parameters have been overlooked such as selectivity, regenerability, reaction kinetics, purity and activation energy which could provide better understanding of the overall process and its cost of H2S capture. Additionally, the specific challenges that need to be addressed by each class of materials are further elaborated in their individual sections to further accelerate the development of these materials.
4. E-factors
In the context of green chemistry, e-factors399 refer to the environmental impact of chemical processes,400 specifically the efficiency of resource utilization and the generation of waste. Green chemistry aims to design chemical products and processes that minimize environmental pollution, reduce resource consumption, and promote sustainability.401,402 E-factors serve as quantitative indicators of the environmental performance of chemical processes, providing insights into their sustainability and potential for improvement. The e-factor is defined as the ratio of the mass of waste generated by a chemical process to the mass of the desired product. It quantifies the efficiency of a chemical process in terms of waste generation, with lower e-factors indicating more sustainable practices. The e-factor can be expressed using the following equation: | (4.1) |
E-factors are used to evaluate the environmental impact of chemical processes and guide the development of greener alternatives. By assessing the amount of waste generated per unit of product, researchers and industrial players can identify opportunities to minimize waste and optimize resource utilization. The evaluation of e-factors relies on waste definition, encompassing non-recoverable starting materials, solvents, catalysts and undesired side products from reactions. Smaller e-factor means closer to zero waste. Similarly, the e-factor can be used to estimate the total waste generated by using the following expression:
| Amount of waste = e-factor × amount of product | (4.2) |
Comparing the latest technological advances in H2S removal, such as absorption, adsorption, conversion, membrane separation, and cryogenic distillation, regenerable solvents like ILs emerge as the most promising option with the lowest e-factors and highest efficiency for H2S removal applications. This is because regenerable ILs produce minimal or zero waste as compared to their desired products.81,403,404 Their high regenerability contributes to efficient H2S removal, enabling multiple uses over a certain period. Emphasizing the high regenerability of materials used in other methods is essential to minimize e-factors. The high regenerability of materials leads to lower e-factors as it allows for the repeated use of the same materials in a chemical process.405 When materials can be regenerated and reused multiple times, there is less need to produce new materials, resulting in reduced waste generation per unit mass of the desired product. This efficient recyclability of materials decreases the overall amount of waste generated during the process, thereby lowering the e-factor. Lower e-factors indicate higher efficiency in a chemical process, signifying less waste generated per unit mass of the desired product. In other words, a lower e-factor suggests that the process produces less waste relative to the amount of product obtained, aligning with sustainability goals and efficient resource utilization.
In regard to recyclability, ILs are well-known for their potential recyclability406,407 due to their unique properties, such as low vapor pressure408 and high stability.409 In general, ILs can be recovered through processes like solvent regeneration or extraction. Plus, ILs are generally considered to be less prone to leaching compared to traditional solvents due to their low vapor pressure, non-volatile nature and high chemical stability. However ILs still face the risk of contamination and should be monitored periodically, especially in long-term applications. Additionally, ILs can undergo degradation or deactivation over time,410 leading to reduced efficiency in H2S removal. However, proper selection of ILs and optimization of operating conditions can mitigate this issue. Some of the ILs were also reported to be non-regenerative and cannot be recycled.411 These could lead to several drawbacks such as increased chemical consumption, waste generation, and higher operational costs. Additionally, disposal of used ILs may pose environmental concerns.
To date, e-factors are often overlooked in previous studies and should be given higher emphasis in future work. E-factors, which measure the mass of waste generated per unit mass of desired product, provide an excellent means to assess the efficiency of a chemical process by considering the amount of waste generated such as used solvents, reagents and catalysts, relative to the production amount of the desired product. While e-factors provide valuable insights into the environmental performance of chemical processes, challenges remain in their practical implementation and interpretation. Factors such as the definition of waste, the inclusion of energy consumption, and the consideration of life cycle impacts can influence e-factor calculations and their relevance to sustainability. In conclusion, e-factors play a crucial role in green chemistry by quantifying the environmental impact of chemical processes and guiding the development of more sustainable alternatives. By reducing waste generation, optimizing resource utilization, and promoting innovative synthesis strategies, green chemistry contributes to the transition towards a more sustainable and environmentally conscious chemical industry. As a result, a more holistic approach must be used to assess the properties of each material and their respective performances in H2S capture.
5. Conclusion
Effective capture and removal of H2S is very crucial to avert harmful consequences upon exposure to human beings, the environment and production facilities. To date, there are various conventional methods being implemented for H2S capture on the commercial scale such as absorption, adsorption, conversion, membrane separation and cryogenic distillation. However, there is still huge room for improvements and optimizations for these processes, to achieve high efficiency and significant reduction in overall costs. Adsorption via metal oxide adsorbents is by far the most established method for fine H2S removal. Meanwhile, absorption and membrane separation methods are primarily applicable for the bulk removal of acid gases, with alkanolamine solvents and polymeric membranes being the dominant choices for this purpose.Highly regenerable solvents such as ILs emerge as highly promising candidates for H2S removal, offering the potential for efficient and sustainable processes with a low environmental impact. Their high regenerability enables multiple uses over time, contributing to lower e-factors by minimizing waste generation per unit mass of the desired product. The unique properties of ILs, such as low vapor pressure, high stability, and strong solvent power, inherently make them less prone to leaching compared to traditional solvents, further enhancing their appeal for environmentally conscious applications. However, challenges such as degradation or deactivation over time and the potential non-regenerability of some ILs highlight the importance of careful selection, optimization, and monitoring to ensure their continued effectiveness and sustainability. Addressing these challenges is crucial to fully capitalize on the potential of ILs in H2S removal while also minimizing environmental risks and maximizing resource utilization.
Much on-going research is still conducted to develop novel materials by functionalization and molecular design to enhance their performance, whilst at the same time, being cost effective and environmentally friendly. Even though H2S capture can be performed simultaneously with CO2 efficiently using highly established carbon capture technologies with a high TRL, the selective removal of H2S from CO2 still requires deeper research. This would pave the road for further research on the recovery and separation of H2S into valuable end-products such as elemental sulfur, hydrogen, mercaptan and other sulfur-containing materials which are highly lucrative for a circular economy.
Author contributions
Muhammad Syahir Aminuddin: conceptualization, writing – original draft, review & editing, visualization. Mohamad Azmi Bustam: supervision, Khairiraihanna Johari: supervision, writing – review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the funding for research work from the Malaysian Ministry of Higher Education (MOHE) under the Fundamental Research Grant Scheme (FRGS 015MA0-010) and GA Scheme from Universiti Teknologi PETRONAS (UTP).Notes and references
- A. G. Georgiadis, N. Charisiou, I. V. Yentekakis and M. A. Goula, Materials, 2020, 13, 3640 Search PubMed.
- S. Batterman, A. Grant-Alfieri and S.-H. Seo, Crit. Rev. Toxicol., 2023, 53, 244–295 Search PubMed.
- L. Zhang, Y.-Y. Qiu, K. R. Sharma, T. Shi, Y. Song, J. Sun, Z. Liang, Z. Yuan and F. Jiang, Water Res., 2023, 120046 Search PubMed.
- N. N. Zulkefli, L. S. Mathuray Veeran, A. M. I. Noor Azam, M. S. Masdar and W. N. R. Wan Isahak, Materials, 2022, 15, 5409 Search PubMed.
- D. S. Rathore, P. Singh, C. Misra, C. Chandel, S. Bugalia and K. S. Gupta, Environ. Monit. Assess., 2023, 195, 1–12 Search PubMed.
- O. S. a. H. Administration, Safety and Health Topics/Hydrogen Sulfide, https://www.osha.gov/SLTC/hydrogensulfide/hazards.html, (accessed 30th January, 2023) Search PubMed.
- M. D. Group, The Dangers of Hydrogen Sulfide Exposure, https://www.mdgbio.com/news/the-dangers-of-hydrogen-sulfide-exposure/, (accessed 18th February, 2023) Search PubMed.
- C. Secco, M. E. K. Fuziki, A. M. Tusset and G. G. Lenzi, Energies, 2023, 16, 1759 Search PubMed.
- S. Mokhatab, W. A. Poe and J. Y. Mak, Handbook of Natural Gas Transmission and Processing: Principles and Practices, Gulf Professional Publishing, 2018 Search PubMed.
- R. Mohtadi, W.-K. Lee, S. Cowan, J. Van Zee and M. Murthy, Electrochem. Solid-State Lett., 2003, 6, A272 Search PubMed.
- Z. Du, C. Liu, J. Zhai, X. Guo, Y. Xiong, W. Su and G. He, Catalysts, 2021, 11, 393 Search PubMed.
- K. Gonzalez, L. Boyer, D. Almoucachar, B. Poulain, E. Cloarec, C. Magnon and F. de Meyer, Chem. Eng. J., 2023, 451, 138948 Search PubMed.
- M. I. Stewart, in Surface Production Operations, ed. M. I. Stewart, Gulf Professional Publishing, Boston, 3rd edn, 2014, vol. 2, pp. 433–539 Search PubMed.
- X. Tian, L. Wang and D. Fu, Energy Fuels, 2019, 33, 8413–8422 Search PubMed.
- S. Yunhai, L. Shan, L. Wei, L. Dong, G. Remy and D. JianPeng, Int. J. Ind. Chem., 2016, 7, 297–307 Search PubMed.
- U. Shoukat, D. D. Pinto and H. K. Knuutila, Processes, 2019, 7, 160 Search PubMed.
- J. Zhan, B. Wang, L. Zhang, B.-C. Sun, J. Fu, G.-W. Chu and H. Zou, Ind. Eng. Chem. Res., 2020, 59, 8295–8303 Search PubMed.
- L. Du, H. Li, L. Li, J. Xu and Y. Li, Pet. Sci. Technol., 2019, 37, 56–60 Search PubMed.
- H. Li, L. Li, J. Xu and Y. Li, Pet. Sci. Technol., 2019, 37, 1825–1829 Search PubMed.
- X. Zhang, W. Xiong, M. Shi, Y. Wu and X. Hu, Chem. Eng. J., 2021, 408, 127866 Search PubMed.
- X. Zhang, W. Xiong, L. Peng, Y. Wu and X. Hu, AIChE J., 2020, 66, e16936 Search PubMed.
- W. Xiong, M. Shi, L. Peng, X. Zhang, X. Hu and Y. Wu, Sep. Purif. Technol., 2021, 263, 118417 Search PubMed.
- K. Huang, X.-M. Zhang, L.-S. Zhou, D.-J. Tao and J.-P. Fan, Chem. Eng. Sci., 2017, 173, 253–263 Search PubMed.
- K. Huang, J.-Y. Zhang, X.-B. Hu and Y.-T. Wu, Energy Fuels, 2017, 31, 14060–14069 Search PubMed.
- T. Zhao, P. Li, X. Feng, X. Hu and Y. Wu, J. Mol. Liq., 2018, 266, 806–813 Search PubMed.
- X. Wang, S. Zeng, J. Wang, D. Shang, X. Zhang, J. Liu and Y. Zhang, Ind. Eng. Chem. Res., 2018, 57, 1284–1293 Search PubMed.
- A. H. Jalili, M. Shokouhi, G. Maurer, A. T. Zoghi, J. S. Ahari and K. Forsat, J. Chem. Thermodyn., 2019, 131, 544–556 Search PubMed.
- F. Liu, W. Chen, J. Mi, J. Y. Zhang, X. Kan, F. Y. Zhong, K. Huang, A. M. Zheng and L. Jiang, AIChE J., 2019, 65, e16574 Search PubMed.
- H. Wu, M. Shen, X. Chen, G. Yu, A. A. Abdeltawab and S. M. Yakout, Sep. Purif. Technol., 2019, 224, 281–289 Search PubMed.
- M. Shi, W. Xiong, Z. Tu, X. Zhang, X. Hu and Y. Wu, Sep. Purif. Technol., 2021, 276, 119357 Search PubMed.
- M. Shi, W. Xiong, X. Zhang, J. Ji, X. Hu, Z. Tu and Y. Wu, Sep. Purif. Technol., 2022, 283, 120167 Search PubMed.
- X. Tian, L. Wang, P. Zhang, D. Fu and Z. Wang, Environ. Sci. Pollut. Res., 2021, 28, 5822–5832 Search PubMed.
- A. Afsharpour and A. Haghtalab, Int. J. Greenhouse Gas Control, 2017, 58, 71–80 Search PubMed.
- H. Renon, J. Y. Lenoir and P. Renault, J. Chem. Eng. Data, 1971, 16, 340–342 Search PubMed.
- F. Murrieta-Guevara and A. Trejo Rodriguez, J. Chem. Eng. Data, 1984, 29, 456–460 Search PubMed.
- F.-Y. Jou, R. Deshmukh, F. Otto and A. Mather, Fluid Phase Equilib., 1990, 56, 313–324 Search PubMed.
- F. Y. Jou, A. E. Mather and F. D. Otto, Ind. Eng. Chem. Process Des. Dev., 1982, 21, 539–544 Search PubMed.
- E. Skylogianni, I. Mundal, D. D. Pinto, C. Coquelet and H. K. Knuutila, Fluid Phase Equilib., 2020, 511, 112498 Search PubMed.
- F.-Y. Jou and A. E. Mather, Int. J. Thermophys., 2007, 28, 490–495 Search PubMed.
- K. Fischer, J. Chen, M. Petri and J. Gmehling, AIChE J., 2002, 48, 887–893 Search PubMed.
- C. S. Pomelli, C. Chiappe, A. Vidis, G. Laurenczy and P. J. Dyson, J. Phys. Chem. B, 2007, 111, 13014–13019 Search PubMed.
- S. F. Sciamanna and S. Lynn, Ind. Eng. Chem. Res., 1988, 27, 492–499 Search PubMed.
- J. Xue, C. Yang, J. Fu, J. He and J. Li, Processes, 2022, 10, 1795 Search PubMed.
- H. M. Polat, F. de Meyer, C. Houriez, C. Coquelet, O. A. Moultos and T. J. Vlugt, Fluid Phase Equilib., 2023, 564, 113587 Search PubMed.
- J. Mustafa, A. H. Al-Marzouqi, N. Ghasem, M. H. El-Naas and B. Van der Bruggen, Desalination, 2023, 548, 116263 Search PubMed.
- N. Agarwal, L. Cao Nhien and M. Lee, Energies, 2022, 15, 6817 Search PubMed.
- L. Li, J. Chem. Eng. Jpn., 2022, 55, 15–21 Search PubMed.
- W. Y. Lee, S. Y. Park, K. B. Lee and S. C. Nam, Energy Fuels, 2020, 34, 1992–2000 Search PubMed.
- S. Energy, Natural Gas Purification, https://www.shell.com/business-customers/catalysts-technologies/licensed-technologies/gas-processing/natural-gas-purification.html, (accessed 7th March, 2023) Search PubMed.
- E. M. Corporation, Selective H2S removal (FLEXSORB™), https://www.exxonmobilchemical.com/en/catalysts-and-technology-licensing/gas-treating/flexsorb, (accessed 20th March, 2023) Search PubMed.
- E. M. Corporation, Selective H2S removal (OASE® sulfexx™), https://www.exxonmobilchemical.com/en/catalysts-and-technology-licensing/gas-treating/oase-sulfexx, (accessed 13th March, 2023) Search PubMed.
- K. H. Smith, N. J. Nicholas and G. W. Stevens, in Absorption-Based Post-Combustion Capture of Carbon Dioxide, ed. P. H. M. Feron, Woodhead Publishing, 2016, pp. 145–166, DOI:10.1016/B978-0-08-100514-9.00007-X.
- J.-G. Lu, Y.-F. Zheng and D.-L. He, Sep. Purif. Technol., 2006, 52, 209–217 Search PubMed.
- N. Upreti, S. Mohan and P. D. Vaidya, Chem. Eng. Commun., 2023, 1–16 Search PubMed.
- A. Haghtalab and M. Dehghani Tafti, Ind. Eng. Chem. Res., 2007, 46, 6053–6060 Search PubMed.
- A. A. Abd, M. R. Othman, Z. Helwani and J. Kim, Energy, 2023, 272, 127060 Search PubMed.
- N. Haimour and O. C. Sandall, Chem. Eng. Commun., 1987, 59, 85–93 Search PubMed.
- A. Vrachnos, G. Kontogeorgis and E. Voutsas, Ind. Eng. Chem. Res., 2006, 45, 5148–5154 Search PubMed.
- P. Pal and F. Banat, J. Nat. Gas Sci. Eng., 2016, 29, 479–487 Search PubMed.
- R. Abdulrahman and I. Sebastine, J. Nat. Gas Sci. Eng., 2013, 14, 116–120 Search PubMed.
- B. P. Mandal, A. Biswas and S. Bandyopadhyay, Sep. Purif. Technol., 2004, 35, 191–202 Search PubMed.
- M. Zamani, M. Shokouhi, H. Fatoorehchi and M. Vahidi, J. Solution Chem., 2023 DOI:10.1007/s10953-023-01342-8.
- M. Sheng, C. Xie, X. Zeng, B. Sun, L. Zhang, G. Chu, Y. Luo, J.-F. Chen and H. Zou, Fuel, 2018, 234, 1518–1527 Search PubMed.
- C.-C. Lin, Y.-H. Lin and C.-S. Tan, J. Hazard. Mater., 2010, 175, 344–351 Search PubMed.
- T. Nguyen, M. Hilliard and G. T. Rochelle, Int. J. Greenhouse Gas Control, 2010, 4, 707–715 Search PubMed.
- S. K. Dash, A. N. Samanta and S. S. Bandyopadhyay, Fluid Phase Equilib., 2011, 307, 166–174 Search PubMed.
- A. Haghtalab and A. Izadi, Fluid Phase Equilib., 2014, 375, 181–190 Search PubMed.
- D. Fu, P. Zhang and C. Mi, Energy, 2016, 101, 288–295 Search PubMed.
- C. Foo, C. Leo, R. Aramesh, M. Aroua, N. Aghamohammadi, M. Shafeeyan and A. Shamiri, J. Mol. Liq., 2015, 209, 596–602 Search PubMed.
- X. Tian, L. Wang, D. Fu and C. Li, Energy Fuels, 2018, 33, 629–635 Search PubMed.
- X. Rozanska, A. Valtz, M. Riva, C. Coquelet, E. Wimmer, K. Gonzalez-Tovar and F. de Meyer, Ind. Eng. Chem. Res., 2023, 62, 11480–11490 Search PubMed.
- C. Chiappe and C. S. Pomelli, in Ionic Liquids II, Springer, 2017, pp. 265–289 Search PubMed.
- Ludwig's Applied Process Design for Chemical and Petrochemical Plants, ed. A. K. Coker, Gulf Professional Publishing, Boston, 4th edn, 2010, pp. 679–686, DOI:10.1016/B978-0-7506-8366-1.10021-0.
- A. L. Kohl and R. B. Nielsen, in Gas Purification, ed. A. L. Kohl and R. B. Nielsen, Gulf Professional Publishing, Houston, 5th edn, 1997, pp. 1187–1237, DOI:10.1016/B978-088415220-0/50014-8.
- B. Burr and L. Lyddon, A comparison of physical solvents for acid gas removal, in Gas Processors’ Association Convention, Grapevine, TX, 2008 Search PubMed.
- X. Liu, B. Wang, X. Dong, Y. Qiu and Q. Meng, J. Hazard. Mater., 2021, 419, 126394 Search PubMed.
- X. Liu, B. Wang, Y. Qiu, X. Dong, Y. Song, Q. Meng and M. Li, Environ. Sci. Pollut. Res., 2021, 28, 38026–38033 Search PubMed.
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 Search PubMed.
- X. Zhao, H. Xing, R. Li, Q. Yang, B. Su and Q. Ren, Prog. Chem., 2011, 23, 2258 Search PubMed.
- M. G. Cowan, M. Masuda, W. M. McDanel, Y. Kohno, D. L. Gin and R. D. Noble, J. Membr. Sci., 2016, 498, 408–413 Search PubMed.
- M. S. Aminuddin, M. A. B. Khalil and B. Abdullah, RSC Adv., 2022, 12, 11906–11912 Search PubMed.
- P. K. Mohapatra, Dalton Trans., 2017, 46, 1730–1747 Search PubMed.
- Z. Wang, S. He, V. Nguyen and K. E. Riley, Eng. Sci., 2020, 11, 3–18 Search PubMed.
- I. V. Vorotyntsev, A. A. Atlaskin, M. M. Trubyanov, A. N. Petukhov, O. R. Gumerova, A. I. Akhmetshina and V. M. Vorotyntsev, Desalin. Water Treat., 2017, 75, 305–313 Search PubMed.
- M. Han and R. M. Espinosa-Marzal, ACS Appl. Mater. Interfaces, 2019, 11, 33465–33477 Search PubMed.
- T. Koishi, J. Phys. Chem. B, 2018, 122, 12342–12350 Search PubMed.
- A. H. Jalili, M. Shokouhi, G. Maurer and M. Hosseini-Jenab, J. Chem. Thermodyn., 2013, 67, 55–62 Search PubMed.
- M. B. Shiflett and A. Yokozeki, Fluid Phase Equilib., 2010, 294, 105–113 Search PubMed.
- A. H. Jalili, M. Safavi, C. Ghotbi, A. Mehdizadeh, M. Hosseini-Jenab and V. Taghikhani, J. Phys. Chem. B, 2012, 116, 2758–2774 Search PubMed.
- M. Safavi, C. Ghotbi, V. Taghikhani, A. H. Jalili and A. Mehdizadeh, J. Chem. Thermodyn., 2013, 65, 220–232 Search PubMed.
- B. A. Neto, A. A. Lapis and R. Y. Souza, Encyclopedia of Ionic Liquids, 2023, pp. 1273–1283 Search PubMed.
- M. Nisar, H. Y. Gondal, S. Munir, Z. M. Cheema, S. A. Alhussain and M. E. Zaki, J. Saudi Chem. Soc., 2023, 27, 101687 Search PubMed.
- K. Huang, D. N. Cai, Y. L. Chen, Y. T. Wu, X. B. Hu and Z. B. Zhang, AIChE J., 2013, 59, 2227–2235 Search PubMed.
- K. Huang, D. N. Cai, Y. L. Chen, Y. T. Wu, X. B. Hu and Z. B. Zhang, ChemPlusChem, 2014, 79, 241–249 Search PubMed.
- K. Huang, X. M. Zhang, Y. Xu, Y. T. Wu, X. B. Hu and Y. Xu, AIChE J., 2014, 60, 4232–4240 Search PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 Search PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2015, 115, 11379–11448 Search PubMed.
- K. Huang, X. M. Zhang, X. B. Hu and Y. T. Wu, AIChE J., 2016, 62, 4480–4490 Search PubMed.
- C. F. Patzschke, J. Zhang, P. S. Fennell and J. M. Trusler, J. Chem. Eng. Data, 2017, 62, 2075–2083 Search PubMed.
- L.-Y. Wang, Y.-L. Xu, Z.-D. Li, Y.-N. Wei and J.-P. Wei, Energy Fuels, 2018, 32, 10–23 Search PubMed.
- C. Wang, H. Luo, D. E. Jiang, H. Li and S. Dai, Angew. Chem., 2010, 122, 6114–6117 Search PubMed.
- A. A. Sulaimon, P. I. Murungi and D. F. B. Mohshim, Pet. Sci. Technol., 2023, 41, 257–301 Search PubMed.
- M. Huo, X. Peng, J. Zhao, Q. Ma, R. Cai, C. Deng, B. Liu, C. Sun and G. Chen, Int. J. Hydrogen Energy, 2023, 48(85), 33173–33185 Search PubMed.
- S. Mortazavi-Manesh, M. A. Satyro and R. A. Marriott, AIChE J., 2013, 59, 2993–3005 Search PubMed.
- L. S. Tomaník, L. R. Rulíšek and P. Slavíček, J. Chem. Theory Comput., 2023, 19, 1014–1022 Search PubMed.
- M. S. Aminuddin, Z. Man, M. A. Bustam Khalil and B. Abdullah, E3S Web Conf., 2021, 287, 02003 Search PubMed.
- M. Yazdani, E. Salehi, S. Zilabi and G. Nikravesh, J. Chem. Thermodyn., 2023, 107092 Search PubMed.
- Y. Zhao, R. Gani, R. M. Afzal, X. Zhang and S. Zhang, AIChE J., 2017, 63, 1353–1367 Search PubMed.
- R. Santiago, J. Lemus, A. X. Outomuro, J. Bedia and J. Palomar, Sep. Purif. Technol., 2020, 233, 116050 Search PubMed.
- J. Lemus, R. Santiago, D. Hospital-Benito, T. Welton, J. P. Hallett and J. Palomar, ACS Sustain. Chem. Eng., 2021, 9, 2080–2088 Search PubMed.
- B. Kazmi, J. Haider, M. A. Qyyum, S. Saeed, M. R. Kazmi and M. Lee, Int. J. Greenhouse Gas Control, 2019, 87, 89–99 Search PubMed.
- Y. Wang, X. Liu, A. Kraslawski, J. Gao and P. Cui, J. Cleaner Prod., 2019, 213, 480–490 Search PubMed.
- S. Yang, Y. Qian and S. Yang, Ind. Eng. Chem. Res., 2016, 55, 6186–6193 Search PubMed.
- J. Haider, M. A. Qyyum, A. Riaz, A. Naquash, B. Kazmi, M. Yasin, A.-S. Nizami, M. Byun, M. Lee and H. Lim, Fuel, 2022, 314, 123064 Search PubMed.
- J. Jacquemin, P. Husson, A. A. Padua and V. Majer, Green Chem., 2006, 8, 172–180 Search PubMed.
- J. Li, Z. Dai, M. Usman, Z. Qi and L. Deng, Int. J. Greenhouse Gas Control, 2016, 45, 207–215 Search PubMed.
- L. I. Tomé, V. Baião, W. da Silva and C. M. Brett, Appl. Mater. Today, 2018, 10, 30–50 Search PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustain. Chem. Eng., 2014, 2, 1063–1071 Search PubMed.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich and B. W. Doherty, Chem. Rev., 2020, 121, 1232–1285 Search PubMed.
- D. O. Abranches, M. A. Martins, L. P. Silva, N. Schaeffer, S. P. Pinho and J. A. Coutinho, Chem. Commun., 2019, 55, 10253–10256 Search PubMed.
- Y. Liu, J. B. Friesen, J. B. McAlpine, D. C. Lankin, S.-N. Chen and G. F. Pauli, J. Nat. Prod., 2018, 81, 679–690 Search PubMed.
- L. Lomba, M. P. Ribate, E. Sangüesa, J. Concha, M. P. Garralaga, D. Errazquin, C. B. García and B. Giner, Appl. Sci., 2021, 11, 10061 Search PubMed.
- S. Sarmad, J. P. Mikkola and X. Ji, ChemSusChem, 2017, 10, 324–352 Search PubMed.
- G. García, S. Aparicio, R. Ullah and M. Atilhan, Energy Fuels, 2015, 29, 2616–2644 Search PubMed.
- M. Karibayev and D. Shah, Energy Fuels, 2020, 34, 9894–9902 Search PubMed.
- H. S. Salehi, R. Hens, O. A. Moultos and T. J. Vlugt, J. Mol. Liq., 2020, 316, 113729 Search PubMed.
- E. Słupek, P. Makoś and J. Gębicki, Energies, 2020, 13, 3379 Search PubMed.
- K. D. O. Vigier, G. Chatel and F. Jérôme, ChemCatChem, 2015, 7, 1250–1260 Search PubMed.
- D. Deng, X. Liu and B. Gao, Ind. Eng. Chem. Res., 2017, 56, 13850–13856 Search PubMed.
- Y. Chen, B. Jiang, H. Dou, L. Zhang, X. Tantai, Y. Sun and H. Zhang, Energy Fuels, 2018, 32, 10737–10744 Search PubMed.
- Y. Gu, Y. Hou, S. Ren, Y. Sun and W. Wu, ACS Omega, 2020, 5, 6809–6816 Search PubMed.
- D. Jung, J. B. Jung, S. Kang, K. Li, I. Hwang, J. H. Jeong, H. S. Kim and J. Lee, Green Chem., 2021, 23, 1300–1311 Search PubMed.
- M. Hayyan, M. A. Hashim, A. Hayyan, M. A. Al-Saadi, I. M. AlNashef, M. E. Mirghani and O. K. Saheed, Chemosphere, 2013, 90, 2193–2195 Search PubMed.
- M. Xiao, H. Liu, H. Gao, W. Olson and Z. Liang, Appl. Energy, 2019, 235, 311–319 Search PubMed.
- S. J. Kumar, S. R. Prasad, R. Banerjee, D. K. Agarwal, K. S. Kulkarni and K. Ramesh, Chem. Cent. J., 2017, 11, 1–7 Search PubMed.
- P. L. McCarty, In Situ Remediation of Chlorinated Solvent Plumes, 2010, pp. 1–28 Search PubMed.
- W. W. Anku, M. A. Mamo and P. P. Govender, Phenolic Compounds-Natural Sources, Importance and Applications, 2017, pp. 419–443 Search PubMed.
- M. Saygun, A. Ekici, N. B. Muluk, A. Çakmak, T. Pinar, E. Dağ and M. Ekici, Clinical and Investigative Medicine, 2012, pp. E190–E205 Search PubMed.
- M. Jaishankar, T. Tseten, N. Anbalagan, B. B. Mathew and K. N. Beeregowda, Interdiscip. Toxicol., 2014, 7, 60 Search PubMed.
- H. Vanda, Y. Dai, E. G. Wilson, R. Verpoorte and Y. H. Choi, C. R. Chim., 2018, 21, 628–638 Search PubMed.
- Y. Chen and T. Mu, Green Chem. Sci., 2021, 2, 174–186 Search PubMed.
- R. Malolan, K. P. Gopinath, D.-V. N. Vo, R. S. Jayaraman, S. Adithya, P. S. Ajay and J. Arun, Environ. Chem. Lett., 2021, 19, 1001–1023 Search PubMed.
- B. Miller, in Fossil Fuel Emissions Control Technologies, ed. B. Miller, Butterworth-Heinemann, 2015, pp. 367–438, DOI:10.1016/B978-0-12-801566-7.00008-7.
- D. A. Bell, B. F. Towler and M. Fan, in Coal Gasification and Its Applications, ed. D. A. Bell, B. F. Towler and M. Fan, William Andrew Publishing, Boston, 2011, pp. 113–136, DOI:10.1016/B978-0-8155-2049-8.10006-3.
- A. K. Coker, in Ludwig's Applied Process Design for Chemical and Petrochemical Plants, ed. A. K. Coker, Gulf Professional Publishing, Boston, 4th edn, 2010, pp. 269–344, DOI:10.1016/B978-0-7506-8366-1.10011-8.
- B. G. Miller, in Clean Coal Engineering Technology, ed. B. G. Miller, Butterworth-Heinemann, 2nd edn, 2017, pp. 261–308, DOI:10.1016/B978-0-12-811365-3.00006-5.
- A. L. Kohl and R. B. Nielsen, in Gas Purification, ed. A. L. Kohl and R. B. Nielsen, Gulf Professional Publishing, Houston, 5th edn, 1997, pp. 40–186, DOI:10.1016/B978-088415220-0/50002-1.
- L. A. Pellegrini, M. Gilardi, F. Giudici and E. Spatolisano, Energies, 2021, 14 DOI:10.3390/en14206687.
- S. Northrop, J. Seagraves, S. Ramkumar and T. Cullinane, ExxonMobil's Experience with Sour Gas Treating and Acid Gas Handling, in Abu Dhabi International Petroleum Exhibition & Conference, OnePetro, 2019 Search PubMed.
- J. Seagraves and G. Vorberg, OASE® sulfexxTM : The next generation of super selective solvents, in GPA Europe Autumn Conference, Royal Ascot, Gas Processors Association Europe, 2020, https://www.gpaeurope.com Search PubMed.
- A. L. Kohl and R. B. Nielsen, in Gas Purification, ed. A. L. Kohl and R. B. Nielsen, Gulf Professional Publishing, Houston, 5th edn, 1997, pp. 330–414, DOI:10.1016/B978-088415220-0/50005-7.
- S. S. Hosseini and J. F. Denayer, J. Environ. Chem. Eng., 2022, 10, 107483 Search PubMed.
- O. D. Agboola and N. U. Benson, Front. Environ. Sci., 2021, 9, 167 Search PubMed.
- M. Atif, H. Z. Haider, R. Bongiovanni, M. Fayyaz, T. Razzaq and S. Gul, Surf. Interfaces, 2022, 31, 102080 Search PubMed.
- H. Tang, J. Tao, A. Ruzsinszky and J. P. Perdew, J. Phys. Chem. C, 2019, 123, 13748–13757 Search PubMed.
- S. Saini, J. Halldin Stenlid and F. Abild-Pedersen, npj Comput. Mater., 2022, 8, 163 Search PubMed.
- S. N. Kudahi, A. R. Noorpoor and N. M. Mahmoodi, J. CO2 Util., 2017, 21, 17–29 Search PubMed.
- K.-J. Ko, H. Kim, Y.-H. Cho, K.-M. Kim and C.-H. Lee, Sep. Purif. Technol., 2023, 305, 122539 Search PubMed.
- N. N. Zulkefli, A. M. I. Noor Azam, M. S. Masdar and W. N. R. W. Isahak, Materials, 2023, 16, 462 Search PubMed.
- N. Le-Minh, E. C. Sivret, A. Shammay and R. M. Stuetz, Crit. Rev. Environ. Sci. Technol., 2018, 48, 341–375 Search PubMed.
- H. Yildiz, H. Gülşen, Ö. Şahin, O. Baytar and S. Kutluay, Int. J. Phytorem., 2023, 1–13 Search PubMed.
- S. E. Manahan, Environmental Chemistry, CRC Press, 2017 Search PubMed.
- J. Gopalan, A. Buthiyappan and A. A. A. Raman, J. Ind. Eng. Chem., 2022, 113, 72–95 Search PubMed.
- Y. Wang, R. Cui, H. Jiang, M. Bai, K. Lin, M. Zhang and L. Ren, Processes, 2022, 10, 2016 Search PubMed.
- J.-H. Tsai, H.-M. Chiang, G.-Y. Huang and H.-L. Chiang, J. Hazard. Mater., 2008, 154, 1183–1191 Search PubMed.
- R. Ahmadi, M. S. Alivand, N. H. M. H. Tehrani, M. Ardjmand, A. Rashidi, M. Rafizadeh, A. Seif, F. Mollakazemi, Z. Noorpoor and J. Rudd, Chem. Eng. J., 2021, 415, 129076 Search PubMed.
- S. Wang, H. Nam and H. Nam, J. Environ. Chem. Eng., 2020, 8, 103683 Search PubMed.
- C. Yang, Y. Wang, H. Fan, G. de Falco, S. Yang, J. Shangguan and T. J. Bandosz, Appl. Catal., B, 2020, 266, 118674 Search PubMed.
- C. Yang, M. Florent, G. de Falco, H. Fan and T. J. Bandosz, Chem. Eng. J., 2020, 394, 124906 Search PubMed.
- Y. Yuan, L. Huang, T. C. Zhang, L. Ouyang and S. Yuan, Sep. Purif. Technol., 2021, 279, 119686 Search PubMed.
- Y. Pan, M. Chen, Z. Su, K. Wu, Y. Zhang and D. Long, Appl. Catal., B, 2021, 280, 119444 Search PubMed.
- L. Chen, J. Yuan, T. Li, X. Jiang, S. Ma, W. Cen and W. Jiang, Sci. Total Environ., 2021, 768, 144452 Search PubMed.
- S. Fakhraie, H. R. Rajabi, A. Rashidi, Y. Orooji, E. Ghasemy, A. S. Zeraati, R. Rahighi and A. Mirhashemi, Appl. Surf. Sci., 2021, 559, 149892 Search PubMed.
- Z. Yu, X. Wang, Y.-N. Hou, X. Pan, Z. Zhao and J. Qiu, Carbon, 2017, 117, 376–382 Search PubMed.
- J. Wang, C. Ke, X. Jia, C. Ma, X. Liu, W. Qiao and L. Ling, Appl. Catal., B, 2021, 283, 119650 Search PubMed.
- C. Xu, J. Chen, S. Li, Q. Gu, D. Wang, C. Jiang and Y. Liu, J. Hazard. Mater., 2021, 403, 123806 Search PubMed.
- H. Ou, M. Fang, M. Chou, H. Chang and T. Shiao, J. Air Waste Manage. Assoc., 2020, 70, 641–648 Search PubMed.
- H. Sawalha, M. Maghalseh, J. Qutaina, K. Junaidi and E. R. Rene, Bioengineered, 2020, 11, 607–618 Search PubMed.
- Q. Dong, J. Wang, C. Ma, Y. Wu, W. Qiao and L. Ling, Sep. Purif. Technol., 2024, 330, 125152 Search PubMed.
- Y. Feng, J. Lu, J. Wang, J. Mi, M. Zhang, M. Ge, Y. Li, Z. Zhang and W. Wang, J. Cleaner Prod., 2020, 273, 123080 Search PubMed.
- P. R. Westmoreland and D. P. Harrison, Environ. Sci. Technol., 1976, 10, 659–661 Search PubMed.
- X. Zhang, Y. Tang, S. Qu, J. Da and Z. Hao, ACS Catal., 2015, 5, 1053–1067 Search PubMed.
- L. R. Pahalagedara, A. S. Poyraz, W. Song, C.-H. Kuo, M. N. Pahalagedara, Y.-T. Meng and S. L. Suib, Chem. Mater., 2014, 26, 6613–6621 Search PubMed.
- S. Cheah, D. L. Carpenter and K. A. Magrini-Bair, Energy Fuels, 2009, 23, 5291–5307 Search PubMed.
- S. Sureshkumar, S. Rajakumari and R. Manonmani, J. Iran. Chem. Soc., 2023, 1–16 Search PubMed.
- A. Marikutsa, A. A. Dobrovolskii, M. N. Rumyantseva, A. A. Mikhaylov, A. G. Medvedev, O. Lev and P. V. Prikhodchenko, J. Alloys Compd., 2023, 169141 Search PubMed.
- Schlumberger, H2S Removal Adsorbents, https://www.slb.com/-/media/files/mi/brochure/h2s-removal-absorbents-br.ashx, (accessed 28th February, 2023) Search PubMed.
- M. N. Hassankiadeh and A. Hallajisani, J. Pet. Sci. Eng., 2020, 190, 107131 Search PubMed.
- S. H. Orojlou, B. Zargar and S. Rastegarzadeh, J. Nat. Gas Sci. Eng., 2018, 59, 363–373 Search PubMed.
- Z. Pan, W. P. Chan, W. Da Oh, A. Veksha, A. Giannis, S. Kumaran, O. Tamilselvam, J. Lei, D. K. B. Mohamed and H. Wang, Fuel Process. Technol., 2020, 201, 106344 Search PubMed.
- S. Kim, N. K. Gupta, J. Bae and K. S. Kim, J. Environ. Chem. Eng., 2021, 9, 105216 Search PubMed.
- M. Wu, E. Guo, Q. Li, J. Mi and H. Fan, Chem. Eng. J., 2020, 389, 123750 Search PubMed.
- Y. Ahn, K. Pandi, M. Lee and J. Choi, J. Cleaner Prod., 2020, 272, 122849 Search PubMed.
- K. Morioka, S. Inaba, M. Shimizu, K. Ano and T. Sugiyama, ISIJ Int., 2000, 40, 280–285 Search PubMed.
- C. J. Heard, L. Grajciar, F. Uhlík, M. Shamzhy, M. Opanasenko, J. Čejka and P. Nachtigall, Adv. Mater., 2020, 32, 2003264 Search PubMed.
- J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 Search PubMed.
- M. Król, Crystals, 2020, 10, 622 Search PubMed.
- M. W. Ackley, S. U. Rege and H. Saxena, Microporous Mesoporous Mater., 2003, 61, 25–42 Search PubMed.
- M. G. Rimoli, M. R. Rabaioli, D. Melisi, A. Curcio, S. Mondello, R. Mirabelli and E. Abignente, Journal of Biomedical Materials Research Part A: An Official Journal of the Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials, 2008, vol. 87, pp. , pp. 156–164 Search PubMed.
- Y. Li and J. Yu, Nat. Rev. Mater., 2021, 6, 1156–1174 Search PubMed.
- M. Ozekmekci, G. Salkic and M. F. Fellah, Fuel Process. Technol., 2015, 139, 49–60 Search PubMed.
- P. Bareschino, E. Mancusi, A. Forgione and F. Pepe, Chem. Eng. Sci., 2020, 223, 115744 Search PubMed.
- L. Barelli, G. Bidini, L. Micoli, E. Sisani and M. Turco, Energy, 2018, 160, 44–53 Search PubMed.
- S. Bahraminia, M. Anbia and E. Koohsaryan, Int. J. Hydrogen Energy, 2020, 45, 31027–31040 Search PubMed.
- M. Jafari, R. Zendehdel, A. Rafieepour, M. Nakhaei Pour, H. Irvani and S. Khodakarim, Int. J. Environ. Sci. Technol., 2020, 17, 187–194 Search PubMed.
- L. Song, X. Du, Y. Chen, Z. Yang, J. Ran, G. Yang, Q. Shi and Z. Xue, Microporous Mesoporous Mater., 2021, 328, 111495 Search PubMed.
- M. Florent and T. J. Bandosz, Chem. Eng. J., 2020, 401, 125986 Search PubMed.
- Z. Yan, S. Tang, X. Zhou, L. Yang, X. Xiao, H. Chen, Y. Qin and W. Sun, Chin. J. Chem. Eng., 2019, 27, 174–181 Search PubMed.
- A. G. Georgiadis, N. D. Charisiou, S. Gaber, K. Polychronopoulou, I. V. Yentekakis and M. A. Goula, ACS Omega, 2021, 6, 14774–14787 Search PubMed.
- S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 Search PubMed.
- E. Sharmin and F. Zafar, in Metal–Organic Frameworks, IntechOpen, 2016 Search PubMed.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 Search PubMed.
- Q. Liu, C. Bian, S. Ming, L. Guo, S. Zhang, L. Pang, P. Liu, Z. Chen and T. Li, Appl. Catal., A, 2020, 607, 117865 Search PubMed.
- N. K. Gupta, S. Kim, J. Bae and K. S. Kim, RSC Adv., 2021, 11, 4890–4900 Search PubMed.
- N. K. Gupta, S. Kim, J. Bae and K. S. Kim, Chem. Eng. J., 2021, 411, 128536 Search PubMed.
- J. A. Zárate, E. Sánchez-González, T. Jurado-Vázquez, A. Gutiérrez-Alejandre, E. González-Zamora, I. Castillo, G. Maurin and I. A. Ibarra, Chem. Commun., 2019, 55, 3049–3052 Search PubMed.
- J. G. Flores, J. A. Zárate-Colín, E. Sanchez-Gonzalez, J. R. Valenzuela, A. Gutierrez-Alejandre, J. Ramirez, V. Jancik, J. Aguilar-Pliego, M. C. Zorrilla and H. A. Lara-García, ACS Appl. Mater. Interfaces, 2020, 12, 18885–18892 Search PubMed.
- E. S. Grape, J. G. Flores, T. Hidalgo, E. Martínez-Ahumada, A. Gutiérrez-Alejandre, A. Hautier, D. R. Williams, M. O'Keeffe, L. Öhrström and T. Willhammar, J. Am. Chem. Soc., 2020, 142, 16795–16804 Search PubMed.
- J. Xu, T. Lawson, H. Fan, D. Su and G. Wang, Adv. Energy Mater., 2018, 8, 1702607 Search PubMed.
- T. N. L. T. Ngo and K.-Y. Chiang, J. Environ. Chem. Eng., 2023, 11, 109592 Search PubMed.
- Y. Liu, H. Cheng, M. Cheng, Z. Liu, D. Huang, G. Zhang, B. Shao, Q. Liang, S. Luo and T. Wu, Chem. Eng. J., 2021, 417, 127914 Search PubMed.
- J. O. Ighalo, S. Rangabhashiyam, C. A. Adeyanju, S. Ogunniyi, A. G. Adeniyi and C. A. Igwegbe, J. Ind. Eng. Chem., 2022, 105, 34–48 Search PubMed.
- A. A. Jameh, T. Mohammadi, O. Bakhtiari and M. Mahdyarfar, J. Environ. Chem. Eng., 2019, 7, 103058 Search PubMed.
- X. Liu, B. Wang, J. Cheng, Q. Meng, Y. Song and M. Li, Sep. Purif. Technol., 2020, 250, 117300 Search PubMed.
- L. Feng, K.-Y. Wang, G. S. Day, M. R. Ryder and H.-C. Zhou, Chem. Rev., 2020, 120, 13087–13133 Search PubMed.
- P. Lyu and G. Maurin, ACS Appl. Mater. Interfaces, 2021, 13, 4813–4822 Search PubMed.
- A. J. Rieth, A. M. Wright and M. Dincă, Nat. Rev. Mater., 2019, 4, 708–725 Search PubMed.
- T. W. Clyne and D. Hull, An Introduction to Composite Materials, Cambridge University Press, 2019 Search PubMed.
- B. Zimmerli, M. Strub, F. Jeger, O. Stadler and A. Lussi, Composite materials: composition, properties and clinical applications. A literature review, Schweizer Monatsschrift fur Zahnmedizin= Revue mensuelle suisse d’odonto-stomatologie= Rivista mensile svizzera di odontologia e stomatologia, 2010, vol. 120, pp. 972–986 Search PubMed.
- K. N. Keya, N. A. Kona, F. A. Koly, K. M. Maraz, M. N. Islam and R. A. Khan, Mater. Eng. Res., 2019, 1, 69–85 Search PubMed.
- D. K. Rajak, D. D. Pagar, R. Kumar and C. I. Pruncu, J. Mater. Res. Technol., 2019, 8, 6354–6374 Search PubMed.
- N. K. Gupta, J. Bae, S. Kim and K. S. Kim, Chemosphere, 2021, 274, 129789 Search PubMed.
- M. Wu, Q. Li, X. Wang and J. Mi, Energy Fuels, 2021, 35, 2456–2467 Search PubMed.
- B. Lin, H. Chen, W. Wei, J. Zhang, M. Wu, W. Li, W. Zhu, Y. Zhang and Y. Wang, Fuel, 2024, 358, 130318 Search PubMed.
- C. N. Okonkwo, C. Okolie, A. Sujan, G. Zhu and C. W. Jones, Energy Fuels, 2018, 32, 6926–6933 Search PubMed.
- C. N. Okonkwo, J. J. Lee, A. De Vylder, Y. Chiang, J. W. Thybaut and C. W. Jones, Chem. Eng. J., 2020, 379, 122349 Search PubMed.
- C. N. Okonkwo, H. Fang, D. S. Sholl, J. E. Leisen and C. W. Jones, ACS Sustain. Chem. Eng., 2020, 8, 10102–10114 Search PubMed.
- S. Kasulla, S. Malik, S. Zafar and A. Saraf, Int. J. Trend Sci. Res. Dev., 2021, 5, 857–863 Search PubMed.
- S. Cimino, L. Lisi, A. Erto, F. Deorsola, G. de Falco, F. Montagnaro and M. Balsamo, Microporous Mesoporous Mater., 2020, 295, 109949 Search PubMed.
- Y. Pan, M. Chen, M. Hu, M. Tian, Y. Zhang and D. Long, Appl. Catal., B, 2020, 262, 118266 Search PubMed.
- Y. Wang, P. Ning, R. Zhao, K. Li, C. Wang, X. Sun, X. Song and Q. Lin, Front. Environ. Sci. Eng., 2021, 15, 1–10 Search PubMed.
- N. N. Zulkefli, M. S. Masdar, W. N. R. Wan Isahak, J. Md Jahim, S. A. Md Rejab and C. Chien Lye, PLoS One, 2019, 14, e0211713 Search PubMed.
- N. N. Zulkefli, M. S. Masdar, W. N. R. Wan Isahak, S. N. H. Abu Bakar, H. Abu Hasan and N. Mohd Sofian, Catalysts, 2021, 11, 545 Search PubMed.
- V. Gasquet, B. Kim, L. Sigot and H. Benbelkacem, Waste Biomass Valorization, 2020, 11, 5363–5373 Search PubMed.
- E. Surra, M. C. Nogueira, M. Bernardo, N. Lapa, I. Esteves and I. Fonseca, Waste Manage., 2019, 94, 136–145 Search PubMed.
- S. Liang, J. Mi, F. Liu, Y. Zheng, Y. Xiao, Y. Cao and L. Jiang, Chem. Eng. Sci., 2020, 221, 115714 Search PubMed.
- X. Kan, X. Chen, W. Chen, J. Mi, J.-Y. Zhang, F. Liu, A. Zheng, K. Huang, L. Shen and C. Au, ACS Sustain. Chem. Eng., 2019, 7, 7609–7618 Search PubMed.
- M. Sun, X. Wang, X. Pan, L. Liu, Y. Li, Z. Zhao and J. Qiu, Fuel Process. Technol., 2019, 191, 121–128 Search PubMed.
- S. A. Nicolae, P. Á. Szilágyi and M. M. Titirici, Carbon, 2020, 169, 193–204 Search PubMed.
- C. Yang, S. Yang, H. Fan, Y. Wang and J. Shangguan, J. Colloid Interface Sci., 2019, 555, 548–557 Search PubMed.
- Z. Aslam, I. A. Hussein, R. A. Shawabkeh, M. A. Parvez, W. Ahmad and Ihsanullah, J. Air Waste Manage. Assoc., 2019, 69, 246–257 Search PubMed.
- F. Dashtestani, M. Nusheh, V. Siriwongrungson, J. Hongrapipat, V. Materic and S. Pang, Fuel, 2021, 295, 120586 Search PubMed.
- N. K. Gupta, J. Bae and K. S. Kim, Chem. Eng. J., 2022, 427, 130909 Search PubMed.
- N. K. Gupta, J. Bae and K. S. Kim, Sci. Rep., 2021, 11, 14740 Search PubMed.
- M. Daraee, R. Saeedirad and A. Rashidi, J. Solid State Chem., 2019, 278, 120866 Search PubMed.
- M. Daraee, E. Ghasemy and A. Rashidi, J. Environ. Chem. Eng., 2020, 8, 104351 Search PubMed.
- M.-H. Lee, K. Vikrant, S. A. Younis, J. E. Szulejko and K.-H. Kim, J. Cleaner Prod., 2020, 250, 119486 Search PubMed.
- N. K. Gupta, J. Bae and K. S. Kim, ACS Omega, 2021, 6, 25631–25641 Search PubMed.
- N. Bhoria, G. Basina, J. Pokhrel, K. S. K. Reddy, S. Anastasiou, V. V. Balasubramanian, Y. F. AlWahedi and G. N. Karanikolos, J. Hazard. Mater., 2020, 394, 122565 Search PubMed.
- J. Mi, F. Liu, W. Chen, X. Chen, L. Shen, Y. Cao, C. Au, K. Huang, A. Zheng and L. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 29950–29959 Search PubMed.
- N. Hazrati, M. Abdouss, A. Vahid, A. Miran Beigi and A. Mohammadalizadeh, Int. J. Environ. Sci. Technol., 2014, 11, 997–1006 Search PubMed.
- Q. Geng, L.-J. Wang, C. Yang, H.-Y. Zhang, Y.-R. Zhao, H.-L. Fan and C. Huo, Fuel Process. Technol., 2019, 185, 26–37 Search PubMed.
- C. Yang, J. Kou, H. Fan, Z. Tian, W. Kong and J. Shangguan, Langmuir, 2019, 35, 7759–7768 Search PubMed.
- G. Basina, D. A. Gaber, S. Al Yafei, V. Tzitzios, S. A. Gaber, I. Ismail, B. V. Vaithilingam, K. Polychronopoulou, S. Al Hashimi and Y. Al Wahedi, Chem. Eng. J., 2020, 398, 125585 Search PubMed.
- M. Daraee, E. Ghasemy and A. Rashidi, J. Environ. Chem. Eng., 2020, 8, 103836 Search PubMed.
- Q. Li, X. Liu, L. Du, B. Bai, Z. Fang, M. Jing and X. Li, Energy Procedia, 2013, 37, 2505–2510 Search PubMed.
- M. J. Ledoux, C. Pham-Huu, N. Keller, J.-B. Nougayrède, S. Savin-Poncet and J. Bousquet, Catal. Today, 2000, 61, 157–163 Search PubMed.
- F. Tari, M. Shekarriz, S. Zarrinpashne and A. Ruzbehani, J. Nat. Gas Sci. Eng., 2018, 59, 124–135 Search PubMed.
- H. Xu, D. Zhang, F. Wu, X. Wei and J. Zhang, Fuel, 2018, 225, 104–110 Search PubMed.
- A. P. Reverberi, J. J. Klemeš, P. S. Varbanov and B. Fabiano, J. Cleaner Prod., 2016, 136, 72–80 Search PubMed.
- M. Sekhavatjou, R. Moradi, A. A. Hosseini and H. A. Taghinia, Int. J. Environ. Res., 2014, 8, 273–278 Search PubMed.
- B. Lal, D. B. Pal, C. Nayak, A. K. Gupta and A. Singh, J. Inst. Eng. (India): Ser. E, 2022, 103(2), 357–363 Search PubMed.
- B. Lal, D. B. Pal, C. Nayak, A. K. Gupta and A. Singh, J. Inst. Eng. (India): Ser. E, 2022, 103, 357–363 Search PubMed.
- H. R. Mahdipoor and H. Ganji, Petroleum & Coal, 2022, vol. 64, p. 1011 Search PubMed.
- B. Salomatov, Z. Azimova and S. Gaybullaev, Eur. J. Innov. Nonform. Edu., 2022, 2, 51–57 Search PubMed.
- W. Rouleau and J. Watson, LO-CAT: A Flexible Hydrogen Sulphide Removal Process, (accessed 3rd March, 2023) Search PubMed.
- L. Hardison and D. Kamshow, New developments in the LO-CAT hydrogen sulfide oxidation process, in 40th Annual laurance reid gas conditioning conference, 1991 Search PubMed.
- A. Bassani, G. Bozzano, C. Pirola, C. Frau, A. Pettinau, E. Maggio, E. Ranzi and F. Manenti, J. Sustain. Dev. Energy Water Environ. Syst., 2017, 6, 210–226 Search PubMed.
- K. H. Al Hatmi, A. d. N. Al Mashrafi and M. S. Shaikh, An Innovative and Well-Demonstrated Approach Toward Achieving Zero-Flaring, in ADIPEC, OnePetro, 2022 Search PubMed.
- K. Gaj and K. Cichuta, Energies, 2023, 16, 100 Search PubMed.
- M. J. Goodwin, O. M. Musa and J. W. Steed, Energy Fuels, 2015, 29, 4667–4682 Search PubMed.
- S. P. Nunes and K.-V. Peinemann, Membrane Technology, Wiley Online Library, 2001 Search PubMed.
- S. N. Mithra and S. Ahankari, Mater. Today Sustain., 2022, 19, 100191 Search PubMed.
- X. He, I. Kumakiri and M. Hillestad, Sep. Purif. Technol., 2020, 247, 116993 Search PubMed.
- Y. Shi, B. Liang, R.-B. Lin, C. Zhang and B. Chen, Trends Chem., 2020, 2, 254–269 Search PubMed.
- R. S. K. Valappil, N. Ghasem and M. Al-Marzouqi, J. Ind. Eng. Chem., 2021, 98, 103–129 Search PubMed.
- Y. Han and W. W. Ho, J. Membr. Sci., 2021, 628, 119244 Search PubMed.
- W. F. Yong and H. Zhang, Prog. Mater. Sci., 2021, 116, 100713 Search PubMed.
- Y. Alqaheem, A. Alomair, M. Vinoba and A. Pérez, Int. J. Polym. Sci., 2017, 2017, 4250927 Search PubMed.
- G. George, N. Bhoria, S. AlHallaq, A. Abdala and V. Mittal, Sep. Purif. Technol., 2016, 158, 333–356 Search PubMed.
- X. He, Energy Sustain. Soc., 2018, 8, 1–14 Search PubMed.
- F. Sabbagh and B. S. Kim, J. Controlled Release, 2022, 341, 132–146 Search PubMed.
- U. W. Siagian, A. Raksajati, N. F. Himma, K. Khoiruddin and I. Wenten, J. Nat. Gas Sci. Eng., 2019, 67, 172–195 Search PubMed.
- J. R. Klaehn, C. J. Orme and D. M. Ginosar, Sep. Sci. Technol., 2022, 1–11 Search PubMed.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 Search PubMed.
- L. Ansaloni and L. Deng, in Recent Developments in Polymer Macro, Micro and Nano Blends, Elsevier, 2017, pp. 163–206 Search PubMed.
- D. J. Jasim, T. J. Mohammed and M. F. Abid, Eng. Technol. J., 2022, 40, 441–450 Search PubMed.
- B. Wilks and M. E. Rezac, J. Appl. Polym. Sci., 2002, 85, 2436–2444 Search PubMed.
- M. Farnam, H. Bin Mukhtar and A. Bin Mohd Shariff, ChemBioEng Rev., 2021, 8, 90–109 Search PubMed.
- M. Klepić, K. Setničková, M. Lanč, M. Žák, P. Izák, M. Dendisová, A. Fuoco, J. C. Jansen and K. Friess, J. Membr. Sci., 2020, 597, 117623 Search PubMed.
- Y. Liu, Z. Liu, G. Liu, W. Qiu, N. Bhuwania, D. Chinn and W. J. Koros, J. Membr. Sci., 2020, 593, 117430 Search PubMed.
- D. J. Harrigan, J. A. Lawrence III, H. W. Reid, J. B. Rivers, J. T. O'Brien, S. A. Sharber and B. J. Sundell, J. Membr. Sci., 2020, 602, 117947 Search PubMed.
- Y. Yampolskii, L. Starannikova, N. Belov, M. Bermeshev, M. Gringolts and E. Finkelshtein, J. Membr. Sci., 2014, 453, 532–545 Search PubMed.
- M. Isanejad, N. Azizi and T. Mohammadi, J. Appl. Polym. Sci., 2017, 134 DOI:10.1002/app.44531.
- E. G. Estahbanati, M. Omidkhah and A. E. Amooghin, J. Ind. Eng. Chem., 2017, 51, 77–89 Search PubMed.
- K. Amo, R. Baker, V. Helm, T. Hofmann, K. Lokhandwala, I. Pinnau, M. Ringer, T. Su, L. Toy and J. Wijmans, Low-Quality Natural Gas Sulfur Removal/Recovery, Federal Energy Technology Center Morgantown (FETC-MGN), Morgantown, WV …, 1998 Search PubMed.
- J. T. Vaughn and W. J. Koros, J. Membr. Sci., 2014, 465, 107–116 Search PubMed.
- A. P. Isfahani, M. Sadeghi, A. H. S. Dehaghani and M. A. Aravand, J. Ind. Eng. Chem., 2016, 44, 67–72 Search PubMed.
- G. Chatterjee, A. Houde and S. Stern, J. Membr. Sci., 1997, 135, 99–106 Search PubMed.
- Z. Si, H. Wu, P. Qin and B. Van der Bruggen, Sep. Purif. Technol., 2022, 121612 Search PubMed.
- C. A. Scholes, G. W. Stevens and S. E. Kentish, J. Membr. Sci., 2010, 350, 189–199 Search PubMed.
- W. Robb, Ann. N. Y. Acad. Sci., 1968, 146, 119–137 Search PubMed.
- B. Bhide and S. A. Stern, J. Membr. Sci., 1993, 81, 209–237 Search PubMed.
- T. Merkel, R. Gupta, B. Turk and B. Freeman, J. Membr. Sci., 2001, 191, 85–94 Search PubMed.
- V. P. Babu, B. E. Kraftschik and W. J. Koros, J. Membr. Sci., 2018, 558, 94–105 Search PubMed.
- L. M. Robeson, Q. Liu, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2015, 476, 421–431 Search PubMed.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 Search PubMed.
- Y. Yampolskii, Macromolecules, 2012, 45, 3298–3311 Search PubMed.
- Z.-X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 Search PubMed.
- V. F. Cardoso, D. M. Correia, C. Ribeiro, M. M. Fernandes and S. Lanceros-Méndez, Polymers, 2018, 10, 161 Search PubMed.
- B. Ameduri and H. Sawada, Fluorinated Polymers: Volume 1: Synthesis, Properties, Processing and Simulation, Royal Society of Chemistry, 2016 Search PubMed.
- D. J. Kim, M. J. Jo and S. Y. Nam, J. Ind. Eng. Chem., 2015, 21, 36–52 Search PubMed.
- T. C. Merkel and L. G. Toy, Macromolecules, 2006, 39, 7591–7600 Search PubMed.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 Search PubMed.
- L. Olivieri, H. Aboukeila, M. G. Baschetti, D. Pizzi, L. Merlo and G. C. Sarti, J. Membr. Sci., 2017, 542, 367–377 Search PubMed.
- M. De Angelis, S. Lodge, M. G. Baschetti, G. Sarti, F. Doghieri, A. Sanguineti and P. Fossati, Desalination, 2006, 193, 398–404 Search PubMed.
- J. S. Chiou and D. R. Paul, Ind. Eng. Chem. Res., 1988, 27, 2161–2164 Search PubMed.
- V. Signorini, M. G. Baschetti, D. Pizzi and L. Merlo, J. Membr. Sci., 2021, 640, 119809 Search PubMed.
- H. Ohya, V. Kudryavsev and S. I. Semenova, Polyimide Membranes: Applications, Fabrications and Properties, Routledge, 2022 Search PubMed.
- R. Castro-Muñoz, V. Martin-Gil, M. Z. Ahmad and V. Fíla, Chem. Eng. Commun., 2018, 205, 161–196 Search PubMed.
- C. A. Scholes, G. Q. Chen, W. X. Tao, J. Bacus, C. Anderson, G. W. Stevens and S. E. Kentish, Energy Procedia, 2011, 4, 681–687 Search PubMed.
- J. Hao, P. Rice and S. Stern, J. Membr. Sci., 2002, 209, 177–206 Search PubMed.
- J. Hao, P. Rice and S. Stern, J. Membr. Sci., 2008, 320, 108–122 Search PubMed.
- B. Kraftschik, W. J. Koros, J. Johnson and O. Karvan, J. Membr. Sci., 2013, 428, 608–619 Search PubMed.
- P. Goel, P. Mandal, E. Bhuvanesh, V. K. Shahi and S. Chattopadhyay, Sep. Purif. Technol., 2021, 255, 117730 Search PubMed.
- S. Willdorf-Cohen, A. N. Mondal, D. R. Dekel and C. E. Diesendruck, J. Mater. Chem. A, 2018, 6, 22234–22239 Search PubMed.
- M. P. Chenar, M. Soltanieh, T. Matsuura, A. Tabe-Mohammadi and C. Feng, Sep. Purif. Technol., 2006, 51, 359–366 Search PubMed.
- M. Aguilar-Vega and D. Paul, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 1577–1589 Search PubMed.
- M. P. Chenar, H. Savoji, M. Soltanieh, T. Matsuura and S. Tabe, Korean J. Chem. Eng., 2011, 28, 902–913 Search PubMed.
- S. Fischer, K. Thümmler, B. Volkert, K. Hettrich, I. Schmidt and K. Fischer, Properties and applications of cellulose acetate, in Macromolecular symposia, Wiley Online Library, 2008, vol. 262(1), pp. 89–96 Search PubMed.
- A. Moghadassi, Z. Rajabi, S. Hosseini and M. Mohammadi, J. Ind. Eng. Chem., 2014, 20, 1050–1060 Search PubMed.
- C. S. Achoundong, N. Bhuwania, S. K. Burgess, O. Karvan, J. R. Johnson and W. J. Koros, Macromolecules, 2013, 46, 5584–5594 Search PubMed.
- A. Houde, B. Krishnakumar, S. Charati and S. Stern, J. Appl. Polym. Sci., 1996, 62, 2181–2192 Search PubMed.
- R. W. Baker, Membrane Technology and Applications, John Wiley & Sons, 2012 Search PubMed.
- Y. Liu, Z. Liu, A. Morisato, N. Bhuwania, D. Chinn and W. J. Koros, J. Membr. Sci., 2020, 601, 117910 Search PubMed.
- H. Lu, L. Liu, S. Kanehashi, C. Scholes and S. Kentish, J. Membr. Sci., 2018, 555, 362–368 Search PubMed.
- M. Shahbaz, N. Rashid, J. Saleem, H. Mackey, G. McKay and T. Al-Ansari, Fuel, 2023, 332, 126220 Search PubMed.
- B. Bhide, A. Voskericyan and S. Stern, J. Membr. Sci., 1998, 140, 27–49 Search PubMed.
- H. Maghsoudi and M. Soltanieh, J. Membr. Sci., 2014, 470, 159–165 Search PubMed.
- N. Gilani, J. T. Daryan, A. Rashidi and M. R. Omidkhah, Appl. Surf. Sci., 2012, 258, 4819–4825 Search PubMed.
- H. Lu, S. Kanehashi, C. Scholes and S. Kentish, J. Membr. Sci., 2017, 539, 432–440 Search PubMed.
- G. O. Yahaya, A. Hayek, A. Alsamah, Y. A. Shalabi, M. M. B. Sultan and R. H. Alhajry, Sep. Purif. Technol., 2021, 272, 118897 Search PubMed.
- J. A. Lawrence III, D. J. Harrigan, C. R. Maroon, S. A. Sharber, B. K. Long and B. J. Sundell, J. Membr. Sci., 2020, 616, 118569 Search PubMed.
- C. A. Scholes, G. Dong, J. S. Kim, H. J. Jo, J. Lee and Y. M. Lee, Sep. Purif. Technol., 2017, 179, 449–454 Search PubMed.
- J. Vaughn and W. Koros, Macromolecules, 2012, 45, 7036–7049 Search PubMed.
- C. J. Orme, J. R. Klaehn and F. F. Stewart, J. Membr. Sci., 2004, 238, 47–55 Search PubMed.
- T. Mohammadi, M. T. Moghadam, M. Saeidi and M. Mahdyarfar, Ind. Eng. Chem. Res., 2008, 47, 7361–7367 Search PubMed.
- S. Niknejad, H. Savoji, M. Pourafshari Chenar and M. Soltanieh, Int. J. Environ. Sci. Technol., 2017, 14, 375–384 Search PubMed.
- Y.-I. Park, B.-S. Kim, Y.-H. Byun, S.-H. Lee, E.-W. Lee and J.-M. Lee, Desalination, 2009, 236, 342–348 Search PubMed.
- P. J. Carvalho and J. A. Coutinho, Energy Environ. Sci., 2011, 4, 4614–4619 Search PubMed.
- X. Zhang, Z. Tu, H. Li, K. Huang, X. Hu, Y. Wu and D. R. MacFarlane, J. Membr. Sci., 2017, 543, 282–287 Search PubMed.
- O. Malykh, A. Y. Golub and V. Teplyakov, Adv. Colloid Interface Sci., 2011, 164, 89–99 Search PubMed.
- W. Heilman, V. Tammela, J. Meyer, V. Stannett and M. Szwarc, Ind. Eng. Chem., 1956, 48, 821–824 Search PubMed.
- R. Quinn and D. Laciak, J. Membr. Sci., 1997, 131, 49–60 Search PubMed.
- J. Pellegrino and P. Giarratano, Gas Separation Using Ion Exchange Membranes for Producing Hydrogen from Synthesis Gas. Quarterly Report 22 Covering the Period October 1, 1991–December 31, 1991, National Inst. of Standards and Technology, Boulder, CO (United States …), 1992 Search PubMed.
- R. Quinn, J. Appleby and G. Pez, Sep. Sci. Technol., 2002, 37, 627–638 Search PubMed.
- D. A. Nicol, Whisky and Other Spirits, 2022, pp. 247–270 Search PubMed.
- F. S. Taylor, Ann. Sci., 1945, 5, 185–202 Search PubMed.
- J. M. Chambers, Alcohol distillation process, US pat., US2647078A, 1953 Search PubMed.
- J. H. Cho and B. Hariharan, On the efficacy of knowledge distillation, in Proceedings of the IEEE/CVF international conference on computer vision, 2019, pp. 4794–4802 Search PubMed.
- T. He, Z. Liu, H. Son, T. Gundersen and W. Lin, J. Cleaner Prod., 2023, 383, 135264 Search PubMed.
- T. N. A. Tengku Hassan, A. M. Shariff, M. M. i. Mohd Pauzi, M. S. Khidzir and A. Surmi, Molecules, 2022, 27, 1424 Search PubMed.
- D. Clodic and M. Younes, A new method for CO2 capture: frosting CO2 at atmospheric pressure, in Greenhouse gas control technologies-6th international conference, Elsevier, 2003, pp. 155–160 Search PubMed.
- D. Berstad, R. Anantharaman and P. Nekså, Int. J. Refrig., 2013, 36, 1403–1416 Search PubMed.
- D. Clodic, R. El Hitti, M. Younes, A. Bill and F. Casier, CO2 capture by anti-sublimation Thermo-economic process evaluation, in 4th annual conference on carbon capture and sequestration, Citeseer, 2005, pp. 2–5 Search PubMed.
- X. Pan, D. Clodic and J. Toubassy, Greenhouse Gases: Sci. Technol., 2013, 3, 8–20 Search PubMed.
- M.-O. Schach, B. Oyarzún, H. Schramm, R. Schneider and J.-U. Repke, Energy Procedia, 2011, 4, 1403–1410 Search PubMed.
- C. Song, Q. Liu, S. Deng, H. Li and Y. Kitamura, Renewable Sustainable Energy Rev., 2019, 101, 265–278 Search PubMed.
- R. Haut, R. Denton and E. Thomas, SPE Prod. Eng., 1989, 4, 265–271 Search PubMed.
- B. Kelley, J. Valencia, P. Northrop and C. Mart, Energy Procedia, 2011, 4, 824–829 Search PubMed.
- T. E. Rufford, S. Smart, G. C. Watson, B. Graham, J. Boxall, J. D. Da Costa and E. May, J. Pet. Sci. Eng., 2012, 94, 123–154 Search PubMed.
- J. Nichols, B. Friedman, A. Nold, S. McCutcheon and A. Goethe, Processing technologies for CO2 rich gas, in Proceedings of the 88th Annual Convention of the Gas Processors Association, San Antonio, TX, USA, 2009, pp. 8–11 Search PubMed.
- P. S. Northrop and J. A. Valencia, Energy Procedia, 2009, 1, 171–177 Search PubMed.
- J. A. Valencia, S. D. Kelman, A. K. Nagavarapu and D. W. Maher, The controlled freeze zone technology for the commercialization of sour gas resources, in IPTC 2014: International Petroleum Technology Conference, European Association of Geoscientists & Engineers, 2014, pp. cp-395-00180 Search PubMed.
- H. Tan, Z. Ding and N. Wen, Appl. Therm. Eng., 2022, 214, 118903 Search PubMed.
- C. Font-Palma, D. Cann and C. Udemu, C, 2021, 7, 58 Search PubMed.
- M. Tuinier, M. van Sint Annaland and J. Kuipers, Int. J. Greenhouse Gas Control, 2011, 5, 694–701 Search PubMed.
- A. Ali, K. Maqsood, N. Syahera, A. B. Shariff and S. Ganguly, Chem. Eng. Technol., 2014, 37, 1675–1685 Search PubMed.
- M. J. Tuinier and M. van Sint Annaland, Ind. Eng. Chem. Res., 2012, 51, 5552–5558 Search PubMed.
- M. Tuinier, M. van Sint Annaland, G. J. Kramer and J. Kuipers, Chem. Eng. Sci., 2010, 65, 114–119 Search PubMed.
- Q. Fu, C. Song, Y. Liu, M. Ishizuka and A. Tsutsumi, Energy Procedia, 2014, 61, 1673–1676 Search PubMed.
- A. Hart and N. Gnanendran, Energy Procedia, 2009, 1, 697–706 Search PubMed.
- R. Amin, A. T. Jackson and T. Kennaird, The Cryocell: an advanced gas sweetening technology, International Petroleum Technology Conference, 2005, IPTC-10106-MS Search PubMed.
- S. Iglauer, C. Pentland and A. Busch, Water Resour. Res., 2015, 51, 729–774 Search PubMed.
- J. Park, S. Yoon, S.-Y. Oh, Y. Kim and J.-K. Kim, Energy, 2021, 214, 118844 Search PubMed.
- H. Ababneh, A. AlNouss, I. A. Karimi and S. A. Al-Muhtaseb, Energies, 2022, 15, 5286 Search PubMed.
- H. Li, R. Zhang, T. Wang, X. Sun, C. Hou, R. Xu, Y. Wu and Z. Tang, Carbon Capture Sci. Technol., 2022, 3, 100012 Search PubMed.
- D. Haldar, N. Bhattacharjee, A. M. Shabbirahmed, G. S. Anisha, A. K. Patel, J.-S. Chang, C.-D. Dong and R. R. Singhania, Biomass Bioenergy, 2023, 173, 106804 Search PubMed.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 Search PubMed.
- M. Becker, S. Lütz and K. Rosenthal, Molecules, 2021, 26, 573 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 Search PubMed.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 Search PubMed.
- H. Zhang, L. Chen, Y. Chen and Z. Wang, Braz. J. Chem. Eng., 2023, 1–35 Search PubMed.
- F. Li, A. Laaksonen, X. Zhang and X. Ji, Ind. Eng. Chem. Res., 2022, 61, 2643–2671 Search PubMed.
- B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952–10958 Search PubMed.
- B. Wu, W. Liu, Y. Zhang and H. Wang, Chem.–Eur. J., 2009, 15, 1804–1810 Search PubMed.
- S. I. Abu-Eishah, Ionic Liquids-Classes and Properties, 2011, pp. 239–272 Search PubMed.
- S. Ravula, N. E. Larm, M. A. Mottaleb, M. P. Heitz and G. A. Baker, ChemEngineering, 2019, 3, 42 Search PubMed.
- E. Maria Siedlecka, M. Czerwicka, S. Stolte and P. Stepnowski, Curr. Org. Chem., 2011, 15, 1974–1991 Search PubMed.
- A. S. Berenblyum, E. A. Katsman and Y. Z. Karasev, Appl. Catal., A, 2006, 315, 128–134 Search PubMed.
- R. Abro, A. A. Abdeltawab, S. S. Al-Deyab, G. Yu, A. B. Qazi, S. Gao and X. Chen, RSC Adv., 2014, 4, 35302–35317 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |