Surface modification of mesostructured cellular foam to enhance hydrogen storage in binary THF/H2 clathrate hydrate†
Received
22nd January 2024
, Accepted 12th April 2024
First published on 15th April 2024
Abstract
This study introduces solid-state tuning of a mesostructured cellular foam (MCF) to enhance hydrogen (H2) storage in clathrate hydrates. Grafting of promoter-like molecules (e.g., tetrahydrofuran) at the internal surface of the MCF resulted in a substantial improvement in the kinetics of formation of binary H2-THF clathrate hydrate. Identification of the confined hydrate as sII clathrate hydrate and enclathration of H2 in its small cages was performed using XRD and high-pressure 1H NMR spectroscopy respectively. Experimental findings show that modified MCF materials exhibit a ∼1.3 times higher H2 storage capacity as compared to non-modified MCF under the same conditions (7 MPa, 265 K, 100% pore volume saturation with a 5.56 mol% THF solution). The enhancement in H2 storage is attributed to the hydrophobicity originating from grafting organic molecules onto pristine MCF, thereby influencing water interactions and fostering an environment conducive to H2 enclathration. Gas uptake curves indicate an optimal tuning point for higher H2 storage, favoring a lower density of carbon per nm2. Furthermore, a direct correlation emerges between higher driving forces and increased H2 storage capacity, culminating at 0.52 wt% (46.77 mmoles of H2 per mole of H2O and 39.78% water-to-hydrate conversions) at 262 K for the modified MCF material with fewer carbons per nm2. Notably, the substantial H2 storage capacity achieved without energy-intensive processes underscores solid-state tuning's potential for H2 storage in the synthesized hydrates. This study evaluated two distinct kinetic models to describe hydrate growth in MCF. The multistage kinetic model showed better predictive capabilities for experimental data and maintained a low average absolute deviation. This research provides valuable insights into augmenting H2 storage capabilities and holds promising implications for future advancements.
1. Introduction
2022 was a critical year for the European Union, both in terms of changing the traditional energy import routes and hastening its energy transition towards low-emission alternative fuels. This complements the recommendation of the 27th United Nations Climate Change Conference to prioritize decarbonization.1 Hydrogen (H2) is a clean energy carrier with a >200% higher energy density than methane (CH4) (H2 ∼120 MJ Kg−1; CH4: ∼50 MJ Kg−1). With a predicted global H2 demand of 95 million tonnes by 2030, H2 is considered to be a crucial pillar of the future energy system, especially for industrial sectors.2,3 Despite H2's potential, its safety and processability (i.e. storage and transportation) remain major challenges limiting the development of novel H2 technologies and delaying the deployment of an H2 economy.4 Chemi- and physi-sorption based H2 storage has often been touted as an alternative to conventional storage of molecular H2 in high-pressure tanks or via liquefaction, which comes with a variety of drawbacks.5,6 While sorption based storage can exhibit superior gravimetric and/or volumetric H2 storage densities, many materials potentially exceed the DOE target of 5.6 wt% H2 for mobile applications.7–9 H2 release is often energy intensive and not fully reversible. To confront slow kinetics, poor reversibility, cycling instability, and thermal management issues, substantial R&D resources are committed to the field of materials science.6,10
Gas hydrates or clathrate hydrates are crystalline materials formed under favorable thermodynamic conditions when suitable guest (H2 in this work) molecules are incorporated in the cages of 3-dimensional hydrogen bonded solid water (H2O) structures.11 Because of its renewable and environmentally friendly nature as the principal storage medium is H2O, clathrate based H2 storage technology has regained significant momentum.12,13 Minimal temperature or pressure swings outside the hydrate stability zone enable swift recovery of the stored molecular H2 without losing H2O or promoter molecules.14–16 Recently, gas recovery from clathrate hydrate was even demonstrated without melting the solid water phase, further improving the energy balance of the process.17 Pure H2 clathrates entered the focus of attention when Mao et al.15 reported that classical sII hydrate structures allow to encapsulate respectively two and four hydrogen molecules in each of its sixteen dodecahedra (small) and eight hexakaidecahedron (large) cages. In this scheme, complete occupancy would deliver a total H2 storage capacity of 5 wt%. Neutron diffraction analysis by Lokshin et al.,18 however, refuted the claims of double occupancy of H2 in small cages. Regardless of this occupancy debate, the exceedingly high pressures (between 0.75 and 3.1 GPa) required to stabilize pure H2 clathrate hydrate in the bulk remain a major factor limiting deployment of bulk H2 clathrate hydrate in H2 storage and transportation applications. To resolve this aspect and make H2 clathrates industrially viable, binary clathrate hydrates incorporating both H2 and promoter molecules have been put forward.13
Stoichiometric concentrations of thermodynamic promoters (e.g. tetrahydrofuran (THF); 5.56 mol%) can stabilize binary promoter-gas sII clathrate hydrate structures at pressures as low as 5 MPa and 279.6 K, albeit at the expense of storage capacity for the gas.12,13,16,19,20 Promoter molecules typically occupy a significant fraction of larger clathrate cages, indeed leaving mostly only small cages open for gas enclathration. In an attempt to enable a higher H2 storage density, various studies have proposed strategies for reducing the concentrations of these thermodynamic promoters below the stoichiometric concentration.21–24 These strategies were, however, contested by claims that the H2 storage capacity at reduced pressure and elevated temperature is dependent on the promoter concentration. Optimization of the promoter concentration would therefore not allow the storage capacity to be enhanced.25–28
While much attention has been focused on cage occupancies and determining overall H2 storage capacities with different thermodynamic promoters (THF being cited as the best promoter thus far),13,19,29 the constraints on enclathration kinetics and the mass transfer rate have largely been overlooked, causing further impediments to foster this technology. While mechanical mixing can potentially assist in enhancing its kinetics, gas enclathration is typically a slow process due to insufficient contact between gas and liquid phases27,30,31 (in this case between H2 and H2O + promoter molecules). Mixing on top is an energy-intensive operation as the viscosity of the system increases with increasing conversion.32–34 As an alternative to mechanical mixing of the bulk gas–liquid system, pre-synthesized pure promoter molecules, including THF, have been made to come into contact with H2 gas at high-pressure to convert the pure promoter-molecule clathrate hydrate into binary clathrate hydrate. Melting of the binary clathrate hydrate to a liquid phase upon gas release however renders this strategy unsuitable for continuous cycles.27,35–38 Enhancing the kinetics of synthesizing a binary H2 hydrate from a bulk aqueous phase under mild thermodynamic conditions in a process incorporating recycling of the H2O and promoter molecules therefore remains a major challenge.
A limited number of publications report the option to accelerate the kinetics of binary clathrate hydrate formation by incorporating porous materials in an unstirred reactor.25,35,39–45 This paves the way for further research on quiescent and porous systems that could propel clathrate-based H2 technology to new frontiers. While the literature clearly demonstrates enhanced growth of clathrate hydrate with increasing the surface-to-volume ratio, the process is affected by multiple factors including surface chemistry, pore morphology, and wettability.27,30 Pore size and network morphology not only affect the gas–liquid contact, but together with the surface chemistry of the pores, they also profoundly impact the local organization of H2O,46 thereby directly affecting clathrate hydrate nucleation. Molecular simulations by Bai et al.47 also show that gas hydrate nucleation occurs more easily on less hydrophilic surfaces. This is supported experimentally by J. Wang et al.48 who reported the induction time of gas clathrate hydrate formation to be more than 8 times shorter in hydrophobic materials than in hydrophilic materials. Hydrophobic nano-confinement indeed induces a reorganization of the hydrogen bonding structure of H2O, towards a more ice-like tetrahedrally bonded organization which appears to promote clathrate hydrate nucleation.39,42,46,49–60
Given the advantages of hydrophobic surfaces in gas hydrate nucleation, the present work attempts to design and synthesize porous materials enabling the enclathration kinetics of binary H2 clathrate hydrates to be boosted. Consequently, this study focuses on silica-based mesoporous materials that have been comprehensively designed in terms of pore networks and with pore diameters exceeding 17.3 Å.11 Such pore size should allow the accommodation of an sII clathrate hydrate unit cell, while functionalization with a promoter molecule (THF) could assist in stabilizing H2 clathrate hydrate and potentially allow its concentration in the aqueous phase to be reduced. The chosen silica material, specifically mesostructured cellular foam (MCF), offers several advantages due to its relatively large pores in the mesoporous region, which, in turn, facilitate network accessibility. As THF today still is the best promoter molecule for binary H2 clathrate hydrate formation, the MCF pore surface was functionalized with tetrahydro furfuryloxypropyl triethoxysilane, a THF-like molecule that can be grafted on silica surfaces while retaining high mobility due to its long ether chain. To evaluate if the grafted THF-like molecules assist in perturbing the H2O structure and lowering the nucleation barrier, binary H2-THF clathrate hydrate formation was studied as a function of temperature (268 K, 265 K, and 262 K; 7 MPa initial pressure) in grafted and pristine MCF foams, evaluating kinetics as well as H2 storage capacity.
2. Experimental methods
2.1. Materials
Table 1 lists all the chemicals used in this work and all of them were used without any purification. H2 (99.99% purity) was supplied by Air Liquide Benelux Industries and for the liquid phase deionized H2O was used.
Table 1 Description of chemicals
| Chemical name |
Purity |
Source |
| P123 (EO20PO70EO20) |
Average Mn ∼5800 |
Sigma-Aldrich |
| Ammonium fluoride (NH4F) |
95% |
| 1, 3, 5-trimethylbenzene (mesitylene) |
99% |
| Tetraethylorthosilicate (TEOS) |
99% |
| Toluene |
99% |
| Tetrahydro furfuryloxypropyl triethoxysilane |
97% |
2.2. Synthesis and characterization methods
The MCF silica material was synthesized via a previously reported template approach.61 In a typical experimental procedure, the triblock copolymer P123 (EO20PO70EO20) surfactant was dissolved in a solution of HCl acidified H2O and stirred overnight. Next, appropriate amounts of etching agent ammonium fluoride (NH4F) and swelling agent 1, 3, 5-trimethylbenzene (mesitylene) were added and the mixture was equilibrated at 308–313 K for 1 h while stirring. Next, tetraethylorthosilicate (TEOS) was added dropwise to the stirred solution over a timeframe of 30 minutes (approximately 1 mL min−1). Subsequently, the newly formed white precipitate was aged at 313 K for 20 h while stirring. The synthesis was finalized by transferring the white precipitate to an autoclave and subjecting it to hydrothermal treatment for 24 h at 373 K. Afterwards, the solid was recovered by filtering, subsequently rinsed with distilled H2O, and then dried. The molar ratio of the precursors used in the synthesis was as follows: 1 TEOS: 5.87 HCl: 194 H2O: 0.017 P123: 0.031 NH4F: 0.815 1, 3, 5-trymethilbenzene. Before using it in the subsequent experiments, the dry material was calcined at 550 °C for 6 hours, raising the temperature from 295 to 823 K at 1 K per minute. The resulting product was denoted as MCF (n-f) and will be referred to as such throughout this manuscript.
After the synthesis, the MCF silica material underwent surface modification using tetrahydro furfuryloxypropyl triethoxysilane, drawing inspiration from a previously documented approach.62 Prior to the procedure, each batch of 1.2 g of the MCF material was subjected to overnight drying at 473 K under atmospheric pressure. Subsequently the dried batches were introduced in a glovebox under an argon atmosphere and each batch was suspended in 30 ml toluene. Subsequently a mixture of toluene and tetrahydro furfuryloxypropyl triethoxysilane (0.002 moles for f-1 and 0.006 moles for f-2) was added to the stirred suspension. The resulting mixture was sealed and continuously stirred for 72 h at 353 K. The resulting solid phase was isolated by filtration and thoroughly rinsed with toluene to eliminate any remaining unreacted triethoxysilane. Finally, the material was dried overnight at 373 K. The resulting products were denoted as MCF (f-1) and MCF (f-2) and will be referred to as such throughout this manuscript.
The material's porosity was evaluated using N2-sorption, conducted on an automated Quantachrome Quadrasorb system at 77 K. Before analysis, the samples underwent degassing at 393 K for 16 h. In situ diffuse reflectance infrared FT (DRIFT) measurements were performed using a Nicolet 6700 Fourier transform IR (FT-IR) spectrometer. To prepare the samples for analysis, they were diluted in KBr (2 wt%) and heated under vacuum to 393 K. Following a 30-min equilibration time and keeping the temperature constant, 100 scans were performed in the region of 4000–500 cm−1 with a resolution of 4 cm−1. Thermogravimetric analyses (TGA) were conducted using a Mettler Toledo TGA/DSC 3+ star system, in cups of 150 μL and 80 mL min−1 flow of pure O2. The TGA results were cross-referenced with the N2-sorption analysis (BET surface area) to estimate the surface functional group density of the modifier molecule. This methodology was previously validated and documented in the existing literature.63,64 The analysis of the TGA-profile considered the mass loss between 423 K and 1073 K, assuming that toluene, used as a solvent and rinsing agent in the post-synthesis modification protocol, had evaporated below this temperature.
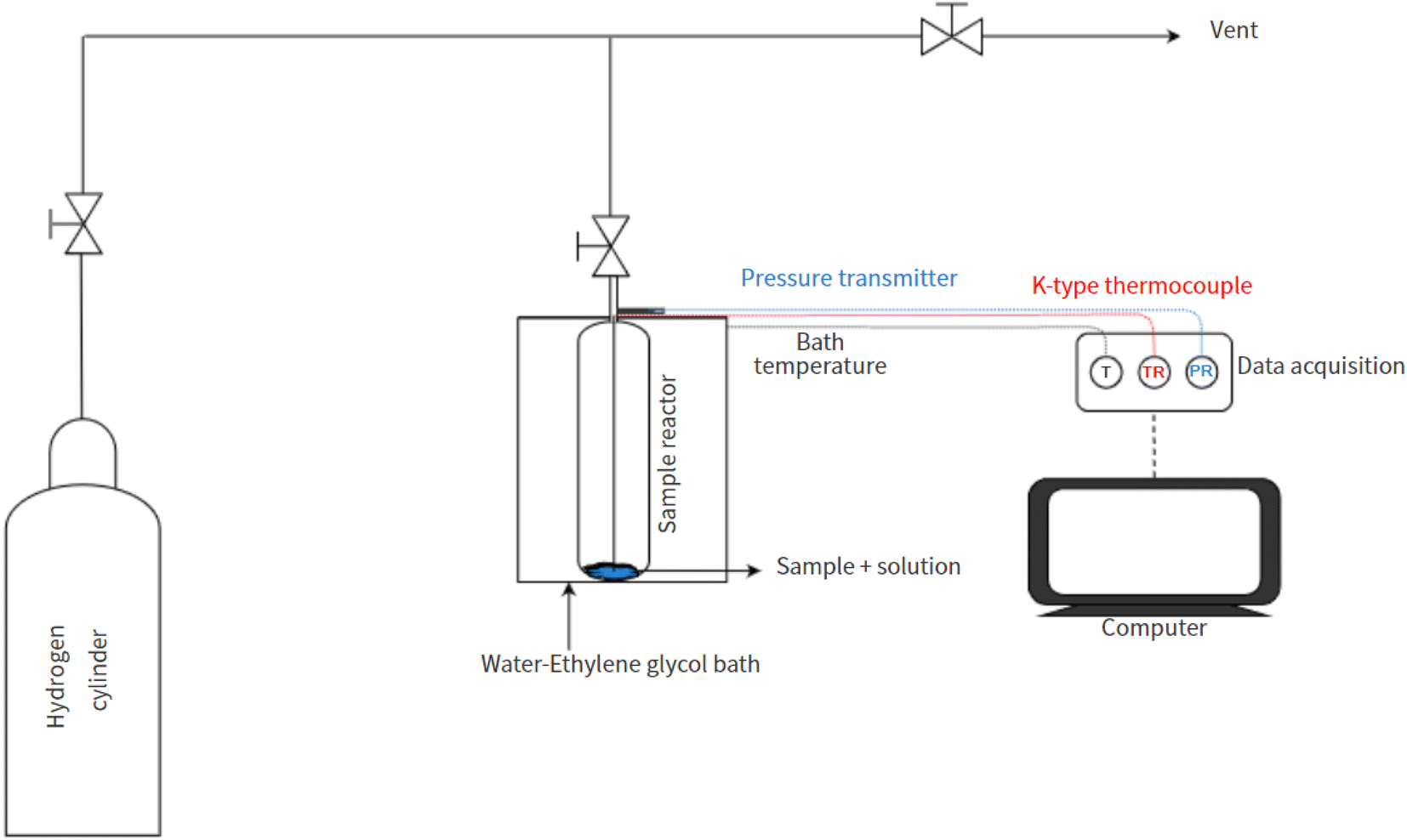
2.3. Experimental setup and procedure
The schematic layout of the experimental setup employed in this work is presented in Fig. 1. The binary H2-THF hydrate formation tests were performed in a Swagelok stainless steel cylindrical reactor (316L-50DF4-150; designed pressure: 34.4 MPa) with an effective inner volume of 150 cm3. The temperature within the reactor was kept cold and stable (measured using a K-type thermocouple, ± 0.1 K; Testo SE & Co. KGaA) by immersing it in a circulating water–ethylene glycol bath (CORIO CP-1000F, JULABO GmbH, stability: ± 0.03 K). The pressure in the reactor was monitored every second using a pressure transmitter acquired from KELLER AG für Druckmesstechnik (PAA3X-30 MPa; range of 0–30 MPa absolute, with ± 0.01% FS accuracy).
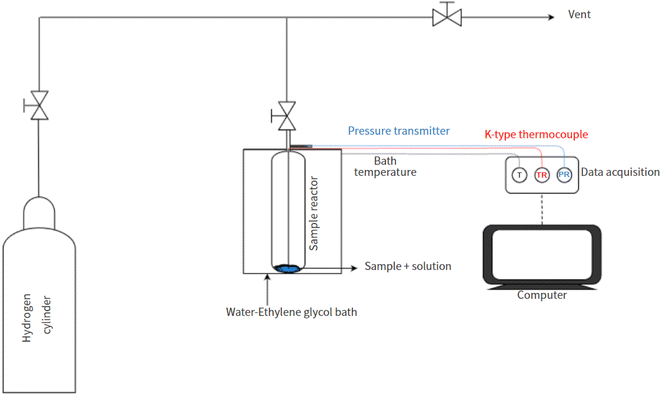 |
| | Fig. 1 The schematic of the experimental setup. | |
All experiments were performed in accordance with the previously reported standard procedure.45,64,65 In summary, the reactor was thoroughly cleaned with H2O and dried to remove contaminants/impurities before commencing any experiment. To carry out the H2 uptake experiments, 5.56 mol% THF solution was added to 0.5 g of the synthesized material (dried overnight at 343 K) based on 100% pore volume saturation. To remove any atmospheric gases the reactor was subsequently subjected to at least 10 rapid pressurization (0.2 MPa) and depressurization (0.12 MPa) cycles with H2 gas. The reactor was then immersed in a water–ethylene glycol bath at 298 K before gradual pressurization with H2 gas to 7 MPa. Sufficient time was allowed for the system to stabilize under these thermodynamic conditions before cooling the reactor to the experimental temperature. The maximal H2 storage capacity of the binary H2-THF clathrate hydrate was considered to have been attained when no considerable pressure reduction (0.02 ± 1 × 10−4 MPa in 30 min) was observed. Each experiment in this study was performed in triplicate.
The amount of H2 gas consumed during hydrate formation at any given real-time was quantified using the compressibility factor equation of state as shown in eqn (1):
| | 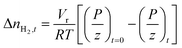 | (1) |
with Δ
nH2,t being the moles of H
2 gas consumed at time
t;
Vr is the gas-phase volume in the reactor measured using the helium expansion method;
65R is the ideal gas constant;
T and
P respectively are the temperature and pressure within the reactor and
z is the compressibility of H
2 gas calculated using the Lemmon–Huber–Leachman correlation.
66
The normalized H2 gas uptake (NGt) and the percentage of H2O to hydrate conversion were quantified using eqn (2) and eqn (3) and 4, respectively.
| | 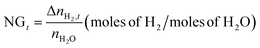 | (2) |
| | 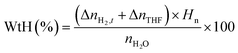 | (3) |
| | 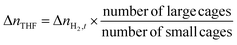 | (4) |
with
nH2O representing the moles of H
2O introduced into the reactor; Δ
nTHF is the moles of THF consumed for hydrate formation, assuming that THF solely occupies the hexakaidecahedron cages of the expected sII clathrate hydrate, and
Hn is the hydration number, which is considered to be 5.67 in order to comply with previously reported binary H
2-THF sII clathrate hydrate studies.
45,67
The volumetric gas storage capacity was determined using eqn (5),19
| | 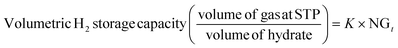 | (5) |
| | 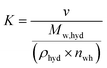 | (6) |
| | | Mw,hyd = (136 × 18.01) + (8 × 72.11) + (16 × 2.016) | (7) |
| | 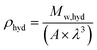 | (8) |
where the proportionality coefficient
K is defined as shown in
eqn (6),
v is the gas volume under STP conditions (22.4 cm
3 mmol
−1 of gas),
Mw,hyd is the molecular weight (g mol
−1) of sII clathrate hydrate considering the thermodynamic promoter used in this study (THF) and is calculated as shown in
eqn (7),
nwh is the mole of H
2O per mole of sII clathrate hydrate (
i.e. 136), and
ρhyd (g cm
−3) is the hydrate density, calculated using
eqn (8), with
A the Avogadro constant (6.023 × 10
23 mol
−1)
68 and
λ the sII hydrate lattice parameter which is 17.145 Å when THF is used as a promoter and pressurized with H
2 gas in the bulk.
69
The rate of H2 hydrate formation was determined by applying a discrete first-order forward difference approach, as shown in eqn (9), where Δt (1 min) is the time difference between two observations.
| | 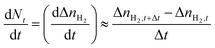 | (9) |
2.4. Characterization of the confined clathrate hydrate phase
The crystal structure of the clathrate hydrate phase formed in an MCF (f-1) host material mixed with an amount of 5.56 mol% THF solution corresponding to 100% of its pore volume, and subsequently pressurized to 7 MPa with H2 gas and cooled to 265 K, was established by transmission X-ray diffraction using a B STOE STADI P combi diffractometer with a focusing Ge(111) monochromator (CuKα1 radiation, λ = 0.154 nm) in a high throughput set-up in transmission geometry. Samples were collected from the reactor, ground in a mortar filled with liquid nitrogen (LN2) and finally transferred to a sample holder which had been submerged in LN2 up until the transfer. The metal sample holder was retrofitted with a Kapton® bottom film to withstand the low LN2 temperature (77 K) required for the stability of the clathrate hydrate phase at atmospheric pressure. PXRD data were recorded using a 140° curved image plate position sensitive detector (IP PSD) from −17.5 to 62.5° 2θ. To account for reflections originating from the MCF host material, a diffraction pattern of the dry MCF (f-1) material was recorded as a reference. X-ray diffraction experiments at liquid nitrogen (LN2) temperature (77 K) and atmospheric pressure were performed in a metal sample holder with a Kapton® bottom film. The low temperature ensured the stability of the clathrate hydrate phase under these conditions. Before XRD analysis, the H2-THF clathrate hydrate samples were first ground in a mortar filled with LN2 and then transferred to the metal sample holder which had been submerged in LN2 up until the transfer.
Incorporation of H2 in 512 cages of the sII clathrate hydrate lattice was observed using in situ high-pressure 1H nuclear magnetic resonance (NMR) spectroscopy at 800 MHz. 1H NMR spectra were acquired using a static 5 mm BBO probe head operating at a Larmor frequency of 801.25 MHz and a Bruker Avance Neo console. The temperature was regulated using a BCU II unit. In order to minimize the 1H background signal of the host material and facilitate identification of all components in spectra, the MCF (f-1) material was rinsed 3 times with D2O and subsequently dried at 363 K under vacuum. Approximately 30 mg of dried D-exchanged MCF (f-1) sample was packed in a 5 mm high-pressure NMR cell built according to the design by Houlleberghs et al.70 before adding 60 μL of a 5.56 mol% THF-D2O mixture to the system. The tube was pressurized to 7 MPa with H2 gas before transferring to a pre-cooled (265 K) NMR probe head and allowing the system to equilibrate for 10 h. Following shimming, 48 transients were recorded for each 1H NMR spectrum using a 30° pulse with 18 kHz RF strength and a repetition delay of 10 s. Chemical shift referencing was carried out with respect to TMS, using ethylbenzene (10% in chloroform-d) as a secondary reference with δ (1H) = 1.22 ppm. Spectral decomposition was performed using dmFit software using Gaussian lineshapes.71
3. H2-THF binary hydrate kinetics
The kinetic regime of post-induction time H2 gas uptake was analyzed using the widely adopted Johnson–Mehl–Avrami–Kolmogorow (JMAK) model,72–80 as shown in eqn (10). This model was established to describe the kinetics of phase change under isothermal/isotropic conditions with a linear growth rate and random nucleation.| | | αWtH = 1 − exp(−k(t)n) | (10) |
where αWtH is water-to-hydrate conversion at time t, k is the crystallization rate constant and n is the Avrami exponent.
However, in a later phase of hydrate growth, the kinetics of hydrate crystallization is often impeded due to the poor diffusion of guest gas (H2) or H2O through the hydrate layer.81,82 Considering this, the kinetics of the experimental data were also analyzed by adopting Hay's two-stage crystallization kinetic model,83 as given in eqn (11), which can represent both the initial hydrate growth based on the JMAK model (first term in eqn (11)) and the hydrate growth owing to diffusion (second term in eqn (11)),
| | | αWtH = αP(1 − exp(−kPtn))(1 + (kst0.5)) | (11) |
where
αWtH is the water-to-hydrate conversion at time
t and
αP is the water-to-hydrate conversion at the end of the reaction-controlled primary crystallization process.
kP and
ks are the rate constants for reaction-controlled primary and diffusion-controlled secondary crystallization growth, respectively.
n is the Avrami exponent. Additional details about the analysis are provided in the Results and discussion section.
The efficacy of the kinetic models in predicting water-to-hydrate conversion is evaluated using average absolute deviation (AAD) as shown in eqn (12),
| | 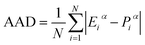 | (12) |
N is the number of datasets and
Eiα and
Piα are experimental and predicted water-to-hydrate conversion respectively.
4. Results and discussion
4.1. Host material characterization
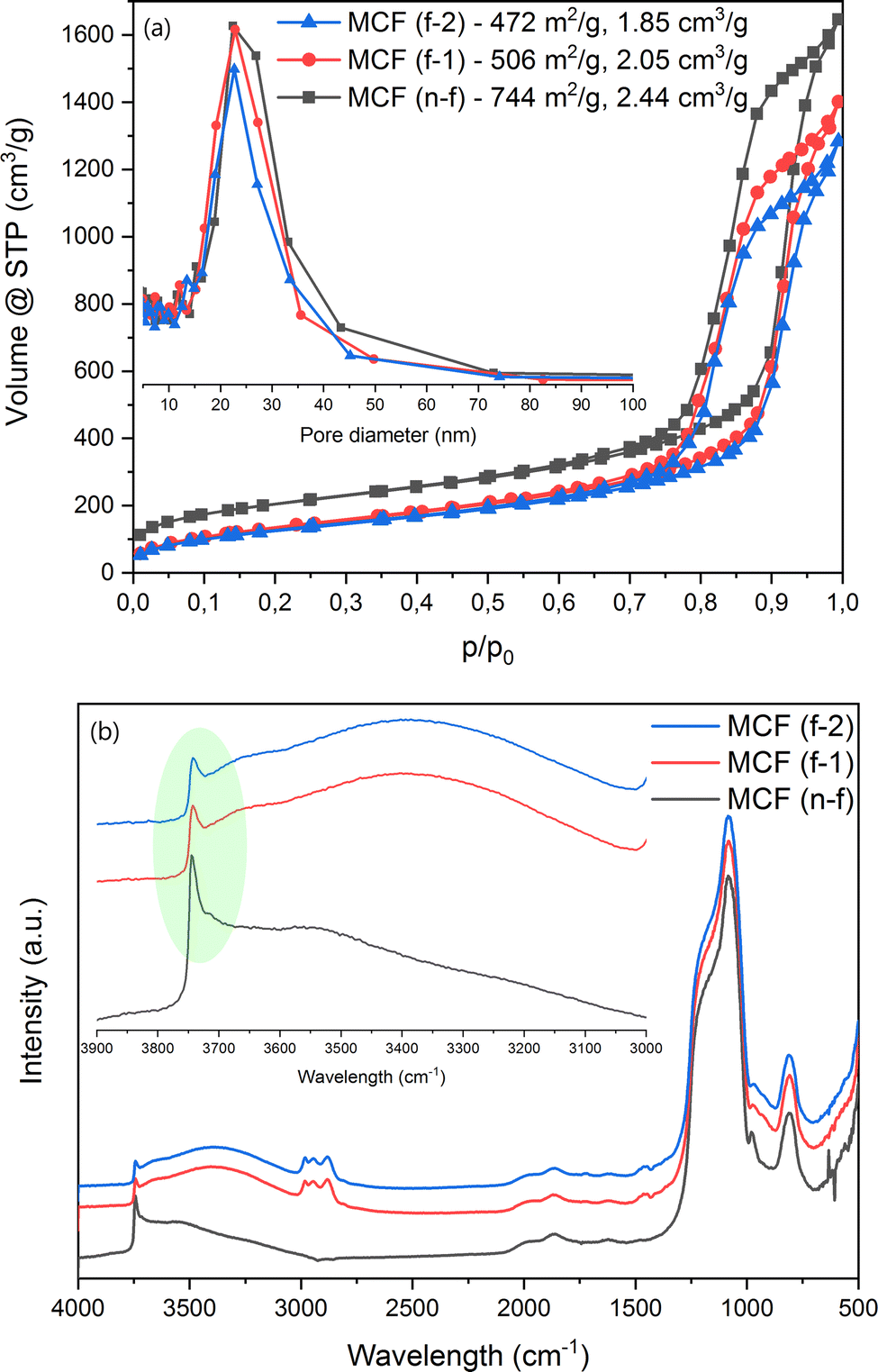
N2-sorption (Fig. 2a) proved the successful formation of the MCF (n-f) material. The presence of the swelling agent pushed for the formation of considerably larger pores as compared to a standard SBA-15 material.84 The maximum size of the ultra-big mesopores determined by the BJH method is approx. 22 nm, while the BET surface area and total pore volume reach 744 m2 g−1 and 2.44 cm3 g−1, respectively. Following the procedure of post-synthesis modification, a decrease in both surface area and total pore volume is observed (Fig. 2a), serving as an initial confirmation of the successful grafting process. The most important band observed during the in situ DRIFT spectral analysis refers to the presence of silanol groups on the surface, localized at around 3745 cm−1 (Fig. 2b). The decreased intensity of this band after the grafting procedure suggests the chemical attachment of the modifier molecule. In other words, fewer Si–OH groups are present on the surface of the material after the modification procedure takes place, which directly implies that these groups are involved in the surface reactions. Next, the features belonging to the silica material are present in all the samples, while the bands at around 2981, 2944, 2881 and 1456 cm−1 are related to the CHx groups of the modifier.62,85
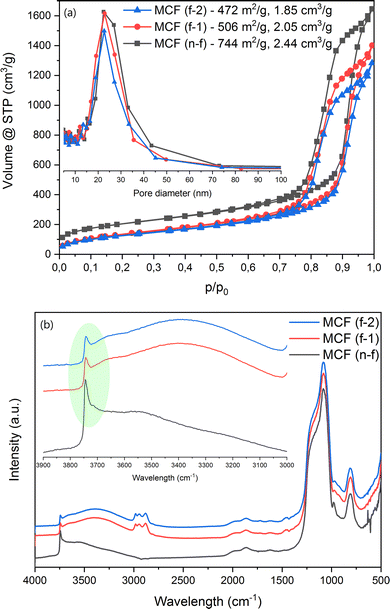 |
| | Fig. 2 (a: top) N2 sorption and (b: bottom) in situ DRIFT analyses of the materials. | |
Thermogravimetric analysis (Fig. S1†) showed a consistent mass loss above 423 K for the modified MCF materials (12–13%). It is clear that increasing the amount of the modifier did not achieve a considerably higher modification degree. This might be connected to the surface becoming saturated with the modifier molecule, leading to steric interactions that restrict the chemical grafting process. The carbon content exhibits variation between the two modified samples, resulting in an approximately 7% increase in the density of the groups on the surface of MCF (f-2), totaling 0.75 g nm−2 compared to MCF (f-1). To conclude here, all the results presented above suggest a successful modification of the MCF material with the THF-ending molecule.
4.2. Characterization of the clathrate hydrate phase
The identity of clathrate hydrate was confirmed to be sII clathrate hydrate using XRD analysis of the product obtained with MCF (f-1) as a porous host. Next to the sII clathrate hydrate also hexagonal ice was found (Fig. S2†). At present it cannot be distinguished if the impurity was formed in the reactor or by condensation and subsequent freezing of moisture from the air during the XRD analysis.
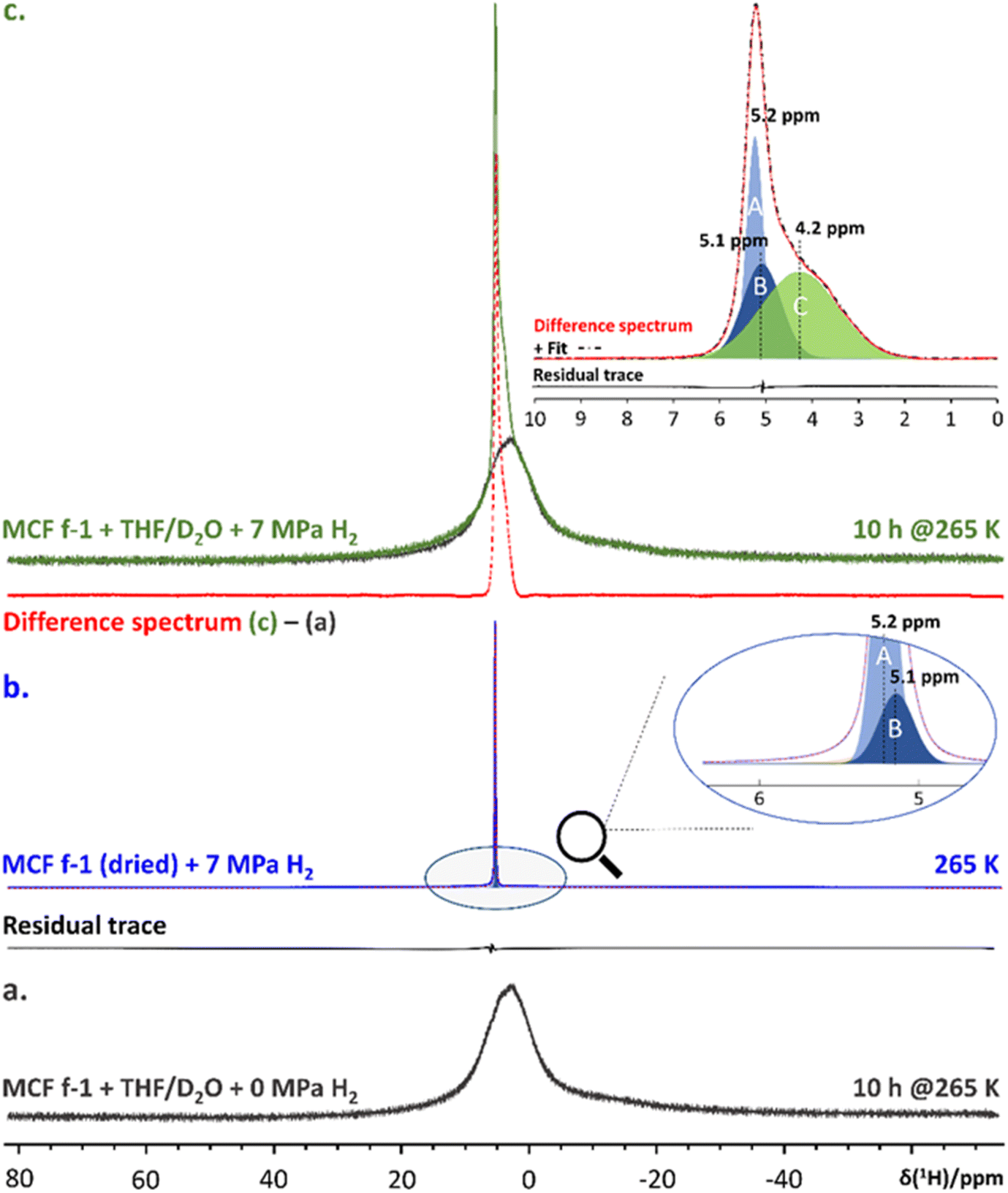
Enclathration of H2 in 512 cages of the sII hydrate structure was demonstrated using in situ high-pressure 1H NMR spectroscopy (Fig. 3). Subtracting the 1H NMR spectrum of pure THF sII clathrate hydrate confined in MCF (f-1) (Fig. 3a) from the spectrum of the binary H2-THF clathrate hydrate formed at the same temperature in the presence of 7 MPa of H2 pressure (Fig. 3c) yielded a difference spectrum containing the resonance of gaseous H2 and H2 occupying the small cage of the clathrate hydrate (Fig. 3c, inset). To identify the signature of gaseous H2 and enable assignment, the 1H NMR spectrum of a dry MCF (f-1) material pressurized with 7 MPa H2 gas was measured (Fig. 3b). Decomposition of this spectrum revealed two resonances at 5.1 and 5.2 ppm respectively, which are associated with gaseous H2 in the presence of the host material. Decomposition of the difference spectrum between confined binary H2-THF clathrate hydrate and confined pure sII THF clathrate hydrate revealed the presence of 3 spectral components (Fig. 3c, inset), two of which (A and B) were also present in the spectrum of the dry host material pressurized with H2 gas. The third resonance (C) at 4.2 ppm can therefore unequivocally be assigned to H2 molecules residing in the 512 cages of the sII hydrate structure. A similar resonance was reported by Lee et al.21 in their pioneering work on binary sII H2-THF clathrate hydrate.
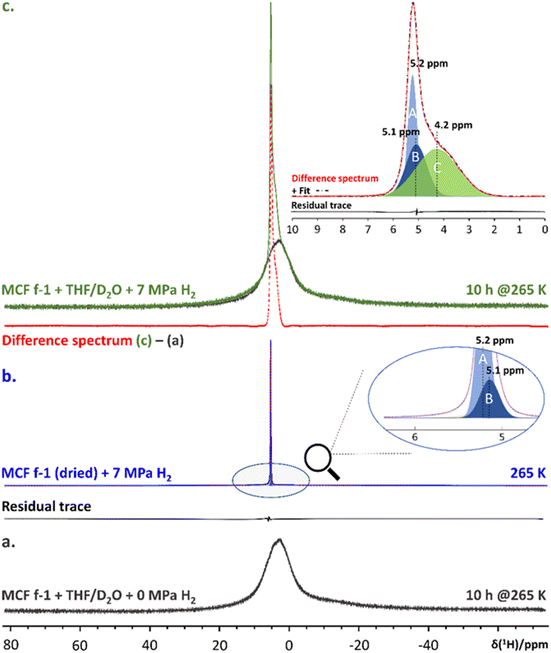 |
| | Fig. 3 (a). 1H NMR spectrum of MCF (f-1) + 5.56 mole% THF/D2O + 0 MPa H2 measured after equilibration for 10 hours at 265 K (black trace). (b). 1H NMR spectrum of D2O-exchanged and subsequently dried MCF (f-1) pressurized with 7 MPa H2 recorded at 265 K (blue trace). (Inset): zoom-in highlighting the most relevant spectral components, i.e., ‘A’ and ‘B’, and their corresponding chemical shifts. The added residual trace reflects the accuracy of the modelled fit (dotted red trace). (c). 1H NMR spectrum of MCF (f-1) + 5.56 mole% THF/D2O + 7 MPa H2 after 10 hours at 265 K (green trace). The red trace below shows the 1H NMR spectrum obtained from computing the difference between spectra (c) and (a). (Inset): spectral decomposition of the resulting difference spectrum. The different components, i.e., A, B and C, are color coded in accordance with previous decompositions. The residual trace reflects the accuracy of the modelled fit (dashed/dotted black trace). | |
4.3. Effect of functionalized porous materials on H2 storage
The storage capacities of H2 in the binary H2-THF clathrate hydrate were investigated using an unstirred reactor filled with ≈0.5 g of porous material and 5.56 mole% of aqueous THF solution. All the experiments were conducted at 265 K with an initial H2 gas pressure of 7 MPa and all systems were saturated with a stoichiometric solution (5.56 mol%), corresponding to 100% of pore volume saturation of each material. The host material consisting of hydrophilic silica-based materials (MCF) and the same material grafted with THF-like functional groups (hydrophobic) were used in the experiments to evaluate the impact of the grafting on hydrate formation rates and H2 storage capacities. The primary distinctions between materials are their pore volumes and the number of carbons per nm2 as a result of functionalization. Table 2 summarizes the list of experimental conditions, H2 storage capacities and other analyzed outcomes from this work. Note that porous media can influence the phase equilibrium of hydrate formation and thus can serve as thermodynamic inhibitors, shifting the phase boundaries to significantly higher pressures and lower temperatures compared to bulk systems. Such effects are expected to become increasingly important as the pore size decreases and the hydrophilicity increases.49,86,87 To assess the occurrence of such effects in our study, a comparative phase equilibrium analysis of binary H2-THF clathrate hydrates in the MCF (f-1) material and in the bulk system was performed. As can be seen from Fig. S3,† the presence of a porous host indeed induces a small shift in the phase diagram (up to 4 K at the lowest experimental temperature), but overall its effect is limited given the large mesopore size of 25 nm and the more hydrophobic nature of the grafted tetrahydro furfuryloxypropyl triethoxysilane functional groups. This small thermodynamic penalty does however not outweigh the observed kinetic promotion, where dispersion of the aqueous phase throughout the modified porous matrix coupled with favourable pore wall–water interactions enables larger amounts of clathrate hydrate to be formed under non-stirred conditions in hours rather than days as compared to non-confined, bulk systems under similar experimental conditions.
Table 2 Summary of experimental observation for H2/THF(5.56 mol%)/H2O hydrate in THF-like functionalized and non-functionalized silica based porous materials under different thermodynamic conditionsa
| Porous system |
P (MPa) |
T (K) |
NGt (mmoles of H2 per mole of H2O) |
WtH (%) |
q
H2
w (wt%) |
q
H2
A (wt%) |
q
H2
T (wt%) |
Volumetric H2 storage (v/v) |
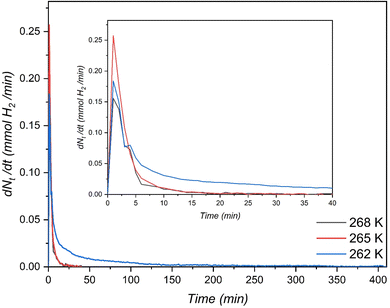
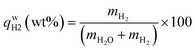 , , 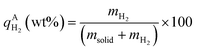 , , 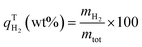 , mH2 = mass of H2, mH2O = mass of H2O, msolid = mass of solid, and mtotal = mass of H2O + mass of solid + mass of H2. , mH2 = mass of H2, mH2O = mass of H2O, msolid = mass of solid, and mtotal = mass of H2O + mass of solid + mass of H2.
|
| MCF(n-f) |
7 |
265 |
13.53 |
11.50 |
0.15 |
0.30 |
0.10 |
13.58 |
| MCF(f-2) |
7 |
265 |
16.15 |
13.73 |
0.18 |
0.27 |
0.11 |
16.21 |
| MCF(f-1) |
7 |
265 |
18 |
15.31 |
0.21 |
0.34 |
0.13 |
18.06 |
| MCF(f-1) |
7 |
268 |
14.06 |
11.96 |
0.16 |
0.27 |
0.1 |
14.12 |
| MCF(f-1) |
7 |
262 |
46.77 |
39.78 |
0.52 |
0.88 |
0.33 |
46.95 |
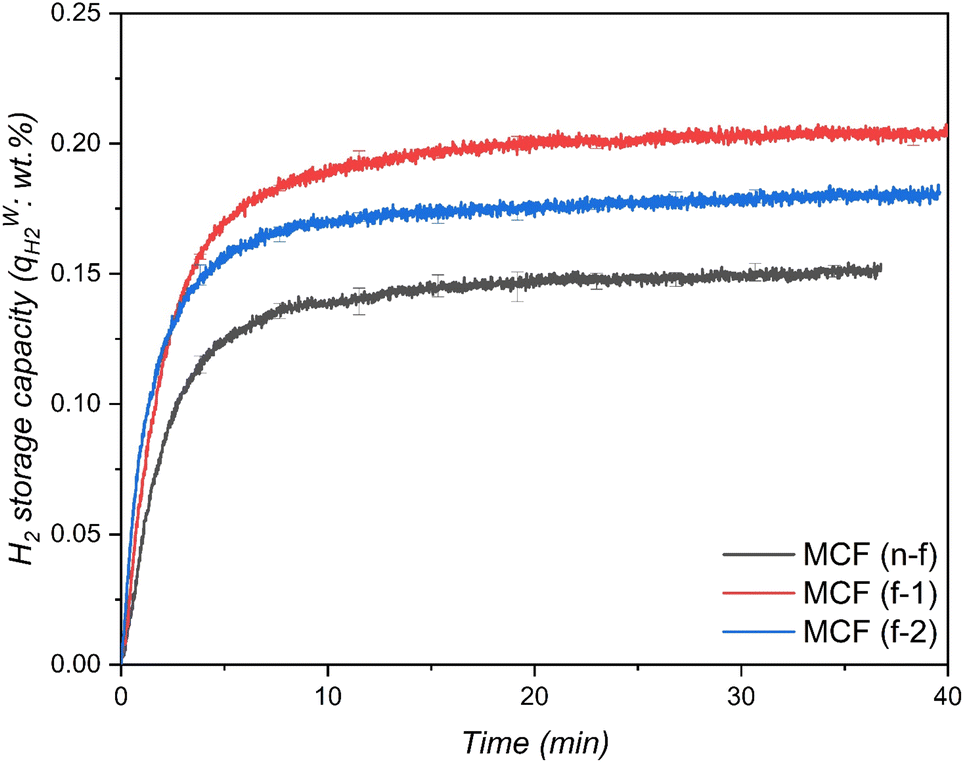
Fig. 4 shows the amount of H2 enclathrated during binary H2-THF hydrate formation in MCF (f-1 and f-2) hydrophobic and MCF (n-f) hydrophilic silica based porous materials. Time zero in this figure corresponds to the commencement of hydrate formation observed from the first temperature spike due to the exothermic nature of the process and all three gas uptake curves also demonstrate the consistency of hydrate formation with a minor standard deviation. As shown in Fig. 4, all three materials exhibit rapid hydrate formation before tapering off to a steady state, and the characteristic time t90, defined as the time required for the system to reach 90% of the maximum obtained gas uptake, was less than 7.5 min for all the systems, implying that this parameter could be a critical criterion in evaluating the kinetic performance of gas uptake as well as hydrate formation. It can also be seen from the figure that the THF-like functionalized materials MCF (f-1 and f-2) showed an excellent promoting effect on H2 storage compared to non-functionalized material MCF (n-f) despite having a pore volume ∼16% and 24% lower than MCF (n-f), respectively. For instance, MCF (f-1), at 265 K, attained a maximum H2 storage capacity (qH2w) of 0.21 wt%, which corresponds to ∼15.31% of water-to-hydrate conversion and 18 mmol H2 per mol H2O, while MCF (n-f) attained only 0.15 wt% and ∼11.50% of water-to-hydrate conversion which is 28.6% and 24.8% less than MCF (f-1), respectively. It is also evident from the gas uptake curves that there also exists an optimal amount of tuning THF-like moieties for enhancing the H2 storage, where lowering the number of carbons per nm2 showed a better gas uptake compared to MCF (f-2) having ∼7% higher carbons per nm2 (which is relatively more hydrophobic than MCF (f-1)) and MCF (n-f) which is a completely hydrophilic material. It is also important to mention that in order to corroborate the suggestions from Lee et al.,21 THF-like functionalized and non-functionalized materials were also tested with 0.1 and 0.3 mol% THF solution; however, no clathrate formation was observed at 265 K for a period of 6 h. The induced hydrophobicity generated by the organic molecules grafted on the surface of the MCF directly affects the interaction of the material with water. In this regard, as already proved before, the organic groups are not allowing the water to interact very strongly with the surface of the SiO2 material and provide a favorable environment for stabilizing clathrate cages and promoting the encapsulation of hydrogen molecules within those cages.54,57 On top of this, one can assume that the enhanced mobility induced by the ether (containing 5 atoms) linker may allow the THF-ending group to have an influence on the water structure disruption, lowering nucleation barrier.88
 |
| | Fig. 4 H2 storage capacity in THF-like functionalized MCF (f-1 and f-2) hydrophobic and non-functionalized MCF (n-f) hydrophilic porous materials at 265 K with an initial pressure of 7 MPa. | |
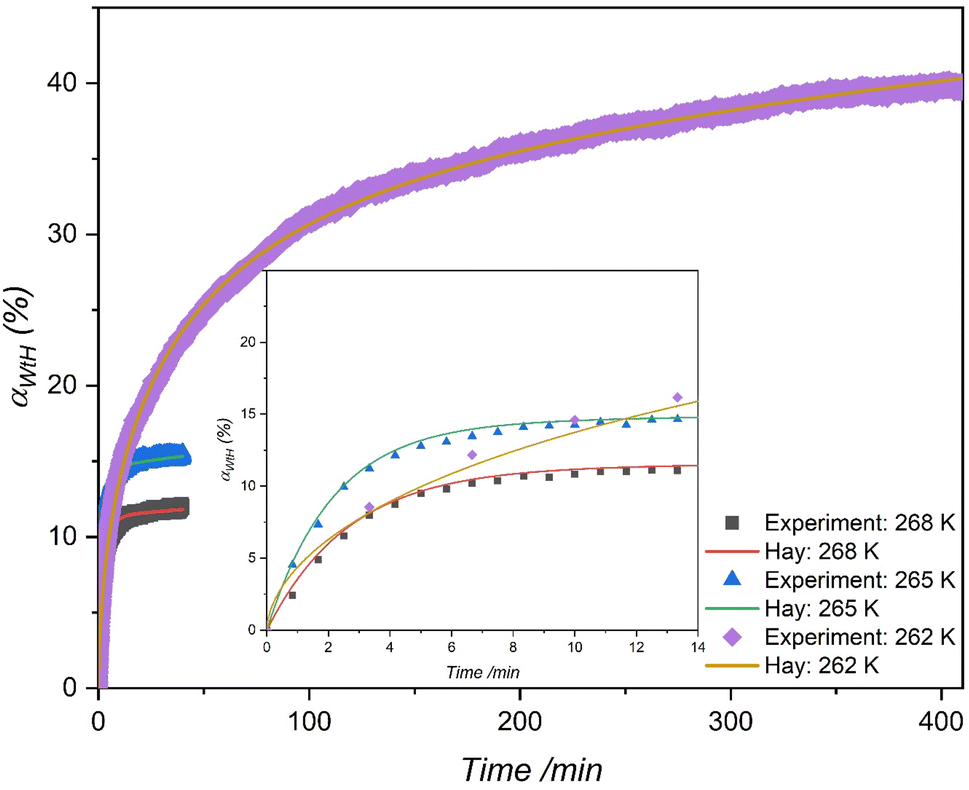
4.4. Effect of temperature on H2 storage
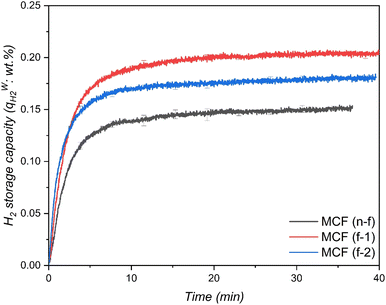
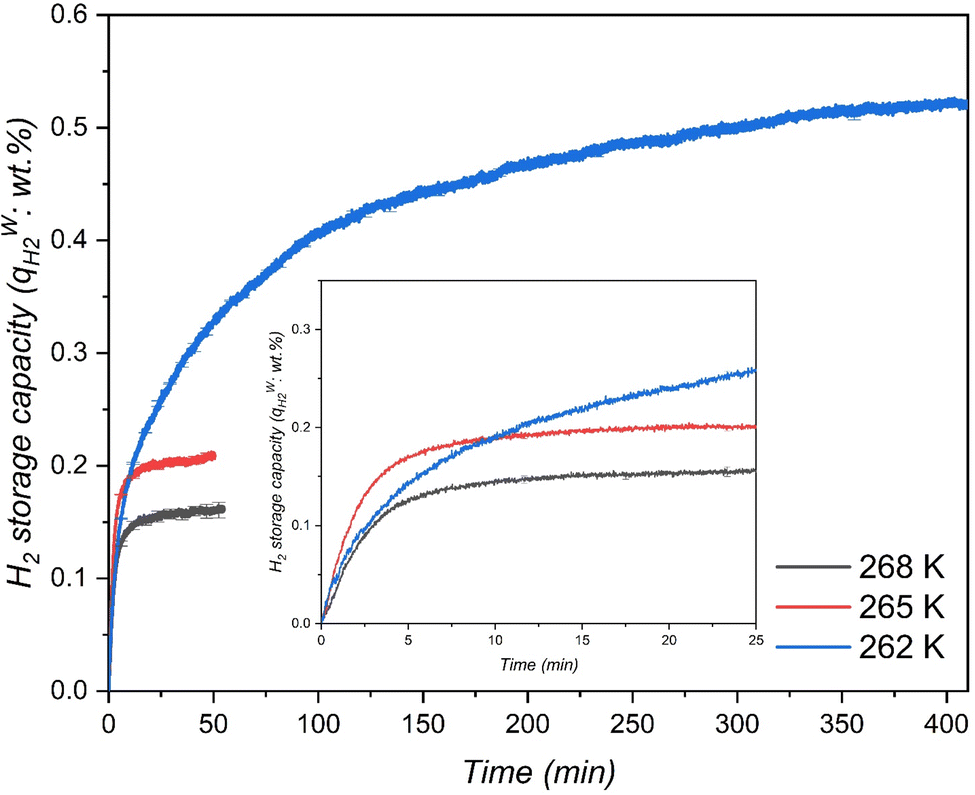
The material that demonstrated higher H2 uptake at 265 K was further investigated at two different temperatures to determine its H2 storage capacities. Typically, lower temperatures for hydrate formation demonstrate higher driving force at constant pressure indicating improved gas enclathration. However, few studies89,90 reported that the gas storage capacities increased with an increase in temperature, implying that lowering the driving force enhances gas storage capacity. In this perspective, two different temperatures, one lower than 265 K and one higher than 265 K were chosen to investigate the effect of the hydrate formation rate and H2 enclathration. The corresponding experiments were performed at 262 K and 268 K with an initial H2 gas pressure of 7 MPa and both the systems were saturated with 5.56 mol% of THF solution, which corresponded to 100% of pore-volume saturation. Fig. 5 exhibits H2 storage capacity of the systems at 268 K, 265 K and 262 K. As can be seen, the temperature has a significant impact on the hydrate formation rate and H2 storage capacities, where a decrease in temperature from 268 K to 262 K resulted in a substantial increase in H2 enclathration, attaining a maximum storage capacity of ∼0.52 wt% at the lowest temperature. These findings are consistent with Tian and Zhang simulation work,91 which indicated that the maximal H2 storage capacity in a binary H2-THF hydrate can be obtained at 260 K at constant pressure. Similarly, results from Liu et al.92 also support our findings by demonstrating that THF hydrate formation diminishes with increasing temperature, which consequently affects H2 enclathration in the small cages (512).
 |
| | Fig. 5 H2 storage capacity in the THF-like functionalized MCF (f-1) porous material at three different temperatures with an initial pressure of 7 MPa. | |
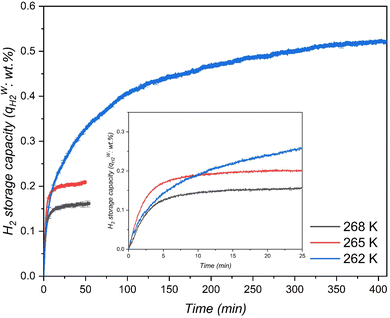
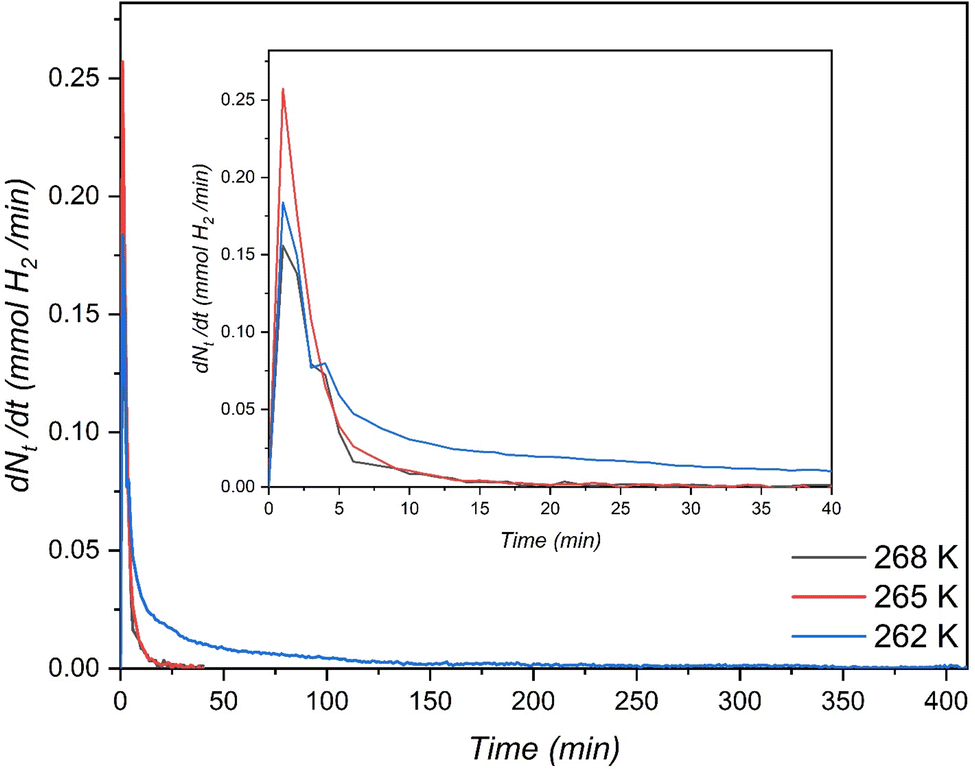
Meanwhile, the material MCF (f-1) also showed higher H2 storage capacity than H2/THF hydrate systems and relatively similar storage capacities compared to H2/1,3-dioxolane systems using stirring reactors (Table 3). Nevertheless, it is worth mentioning that the pressures used in these stirring systems are at least 75% higher than the pressures used in this work. This indicates that the surface grafted THF-like functionalized porous materials show better mass transfer performance for a static/unstirred H2 storage system compared to stirring systems (which are energy intensive). Nevertheless, at the end of hydrate growth reaching a steady state the total H2 storage capacity at 262 K was 2.47 and 3.25 times higher than that in the case of 265 K and 268 K respectively, which corresponds to 39.78% water-to-hydrate conversion and 46.77 mmol H2 per mol H2O. Fig. 6 also displays rapid hydrate growth in the first 2 min at all the temperatures, where the initial rate of hydrate formation increased with decreasing temperature from 268 K to 265 K and then dropped down quickly to near-zero after 30 min. However, lowering the temperature to 262 K showed a slight decrease in the initial rate of hydrate formation (which is evident from Fig. 5 (inset)) compared to 265 K, followed by a very gradual reduction, and reaching near-zero at about 350 min, indicating diffusion-controlled hydrate growth. Further explanation of hydrate growth kinetics is presented in the next section.
Table 3 H2 storage capacity in sII hydrates below 15 MPa using promoters (stoichiometric conc.)
| System |
P (MPa) |
T (K) |
H2 storage capacity (wt%) |
Occupancy of 512 cagesa (%) |
Ref. |
|
Estimated based on two H2 molecules in a single small cage.
|
| This work |
7 |
268 |
0.16 |
6.08 |
— |
| 7 |
265 |
0.21 |
7.97 |
| 7 |
262 |
0.52 |
19.84 |
| Powdered THF hydrate |
6.7 |
270 |
0.36 |
13.63 |
27
|
| 12.2 |
0.46 |
17.70 |
| 6.2 |
0.38 |
14.56 |
| 12.1 |
0.55 |
21.13 |
| 12.4 |
0.32 |
12.28 |
| Powdered THF hydrates |
10.7 |
277.15 |
0.19 |
7.23 |
38
|
| 11.4 |
0.26 |
9.90 |
| Powdered THF hydrates |
6.5 |
269.5 |
0.23 |
8.75 |
99
|
| 6.5 |
269.5 |
0.23 |
8.75 |
| 6.5 |
269.5 |
0.20 |
7.61 |
| 8.4 |
269.5 |
0.25 |
9.52 |
| 6.5 |
269.5 |
0.23 |
8.75 |
| 3.6 |
269.5 |
0.19 |
7.23 |
| 6.5 |
266.7 |
0.26 |
9.90 |
| 6.5 |
269.5 |
0.23 |
8.75 |
| 6.5 |
275.1 |
0.21 |
7.99 |
| THF-bulk solution/stirring |
7.0 |
278.2 |
0.14 |
5.25 |
67
|
| 5.0 |
278.2 |
0.12 |
4.40 |
| 5.0 |
278.2 |
0.13 |
4.76 |
| 5.0 |
278.2 |
0.10 |
3.92 |
| Powdered THF hydrate |
5.0 |
265.1 |
0.18 |
6.85 |
100
|
| 5.0 |
269 |
0.16 |
6.10 |
| 5.0 |
273.2 |
0.15 |
5.71 |
| Powdered DXN hydrate |
12.0 |
233 |
0.4 |
15.25 |
101
|
| THF-bulk solution/stirring |
13 |
279.2 |
0.183 |
6.96 |
102
|
| DIOX-bulk solution/stirring |
12.3 |
271.15 |
0.216 |
8.22 |
20
|
| THF in porous media |
11.6 |
270 |
0.4 |
15.25 |
41
|
| ECP-bulk solution |
12.2 |
273.25 |
0.310 |
11.81 |
37
|
 |
| | Fig. 6 Rate of H2-THF binary hydrate formation in the THF-like functionalized (f-1) porous material at three different temperatures with an initial pressure of 7 MPa. | |
It could be intriguing to compare the findings of various research groups endeavoring to enhance H2 storage in clathrate hydrates. Nevertheless, a direct comparison of the absolute values pertaining to the hydrate growth rate, maximum gas stored capacity, and other parameters obtained from different research laboratories might not be a reliable way to ascertain which one leapfrogged the other as the experimental procedures, reactor setups, and methodologies employed by different groups could significantly influence the results, which could also be a major contributing factor to the observed differences in kinetics of hydrate formation, possibly involving mass transfer limitations. However, it is exemplary to make a comparison between the studies focusing on H2 storage capacity in the binary H2-THF clathrate hydrate, which is presented in Table 3.
Scientifically, it is well-established that under extremely high pressures, H2 can form sII hydrate, reaching 5.6 wt%, with both small (512) and large cages (51264) accommodating two and five H2 molecules, respectively.15,93 However, when THF (a thermodynamic promoter) is introduced in stoichiometric concentrations, the H2 molecules can only occupy small cages, while large cages are left for THF. Consequently, the overall storage capacity decreases to 1.3 wt% and 2.5 wt% when one and two H2 molecules occupy a small cage, respectively. From the data presented in Table 3, it is evident that in a H2-promoter (stoichiometric conc.) H2O system under various thermodynamic conditions, the maximum H2 storage capacity falls below 1.3 wt%. This indicates that either the fractional H2 occupancy in the small cages is less than 1 or not all small (512) cages are filled under those specific conditions. However, this limitation can be overcome by increasing the pressure of the system, which leads to an enhancement in H2 storage capacity. Furthermore, Table 3 demonstrates that by employing solid-state tuning of the material, it is possible to improve the H2 storage capacity significantly even at relatively low pressures when compared to values reported in the literature. While powder-THF hydrate exhibits better H2 enclathration, it is essential to note that the fine THF hydrate particles revert to a liquid phase upon melting. This characteristic makes them unsuitable for continuous cycles of H2 storage and release, thereby limiting their practical applicability in certain situations.
4.5. Kinetic analysis
In this study, the formation kinetics of hydrates at different temperatures were investigated using two distinct kinetic models, as mentioned earlier. The predicted kinetic curves of hydrate formation from the JMAK model73 (eqn (10)) are depicted in Fig. S4,† and the corresponding values of the rate constant (k) and the Avrami kinetic exponent (n) were determined and are presented in Table S1.† The Avrami kinetic exponent (n) is commonly used to interpret the growth geometry and the degree of crystallization during hydrate formation in a porous medium.94 The value of the Avrami exponent (n) can provide insights into the growth dimension of hydrate crystals. A value of n = 3 indicates instantaneous three-dimensional growth from a spherical nucleus, n = 2 suggests instantaneous two-dimensional growth, and n = 1 signifies one-dimensional growth. However, interestingly, the regressed exponent values (n) obtained for the entire growth period, as shown in Table S1,† are consistently less than 1 for all conditions. This indicates that the hydrate crystals experienced instantaneous one-dimensional growth, followed by a decrease in hydrate nucleation and growth rates. This decrease in growth rates could be attributed to the presence of mass transfer resistance in the system. Similar low Avrami exponent values for hydrate growth were also reported by other researchers.77,95,96 Conclusively, the predictive power of the JMAK model revealed limitations in evaluating the hydrate growth at both the beginning or later stages of the growth kinetics.
The limitations of single-step kinetic models have led to a preference for using an integrated multistep model, which offers a more accurate representation of hydrate kinetics. To achieve this, the model proposed by Hay83 has been adopted and adapted for evaluating the hydrate growth kinetics at both the reaction-controlled stage (primary growth) and the diffusion-controlled stage (secondary growth). Hay's model, in a more detailed version, was previously described in their earlier work on crystallization kinetics.97,98 This integrated approach allows for a more comprehensive understanding of the hydrate formation process by considering the distinct contributions of both primary and secondary growth stages. By incorporating these stages into the model, a more accurate depiction of the overall hydrate kinetics can be obtained, improving our insights into the complex nature of hydrate formation. As indicated in eqn (11), the total water-to-hydrate conversion (αWtH) can be categorized into two components: the water-to-hydrate conversion in the primary growth stage and the secondary growth stage, as shown in eqn (13).
| | | αWtH = αWtH,P + αWtH,S | (13) |
Here,
αWtH,P, the primary crystal growth stage, attributed to the rapid nucleation and hydrate growth was analyzed using the JMAK model (
eqn (14)), assuming that the primary growth stage ends before the onset of the diffusion-controlled regime
i.e., the secondary growth stage (
αP).
| | | αWtH = αP(1 − exp(−kptn)) | (14) |
where
kp is rate constant for primary crystallization growth and
n is the Avrami exponent.
Eqn (14) can also be rewritten by applying a double logarithm as shown in
eqn (15).
| | ln(−ln(1−αWtH,P/αP)) = n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(t) + ln(kp) ln(t) + ln(kp) | (15) |
In this analysis, exponent
n was set to 1 assuming one-dimensional growth and the value of the adjustable parameter,
αP, was chosen from the best linear fit to
eqn (15) based on the coefficient of determination (
R2) (for MCF (f-1) at 265 K: Fig. S5
†). Finally, the corresponding value of ln(
kp) was obtained from the intercept. The water-to-hydrate conversion in the secondary hydrate growth stage is defined as shown in
eqn (16).| | | αWtH,S = αWtH,P(kst0.5) | (16) |
By substituting
eqn (16) and
eqn (14) into (
eqn (13)), the complete kinetic model for the process is derived, as described in (
eqn (11)). The kinetic rate constant governing the secondary growth or crystallization stage was determined by plotting

against
t0.5 and calculating the slope of the resulting linear fit. These rate constants and
αP, obtained from this study, are summarized in
Table 4 and the predicted kinetic curves of hydrate formation from the Hay model are depicted in
Fig. 7.
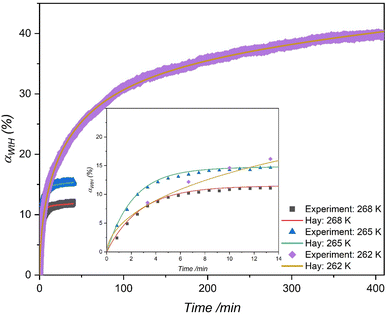 |
| | Fig. 7 Comparing the multistep kinetic model (Hay)83 to experimental data for water-to-hydrate conversion (%) in the THF-like functionalized (f-1) porous material at three different temperatures with an initial pressure of 7 MPa. | |
Table 4 reveals that the secondary kinetic rate constant (ks) decreases to a lower value after the initial growth stage, regardless of the driving force applied. This reduction can be attributed to slower conversion rates due to mass transfer resistance within these porous media, particularly upon reaching a certain water-to-hydrate conversion level. Furthermore, the multistage kinetic model consistently demonstrated superior predictive capabilities for hydrate kinetics compared to the JMAK model, as evident from the AAD values. Moreover, the multistage model allows for an insightful examination of the contributions of primary and secondary-stage hydrate growth to the overall water-to-hydrate conversion. For instance, at 262 K, as shown in Fig. S6(c)†, the secondary stage contributes to around 30% of the overall water-to-hydrate conversion. This suggests that the growth of hydrates, equally driven by primary and diffusion-controlled stages, significantly influenced the slow rate of hydrate formation from inception, leading to relatively low-rate constants at very low temperatures. These findings highlight the significance of the multistage kinetic model in understanding hydrate formation dynamics.
Table 4 Regressed values of rate constants (kp and ks), from the Hay83 model
| System |
T (K) |
Multistep kinetic model |
AAD |
|
k
p (min−1) |
k
s (min−1) |
| MCF (f-1) |
268 |
0.38 |
0.012 |
0.16 |
| 265 |
0.48 |
0.015 |
0.18 |
| 262 |
0.17 |
0.022 |
0.23 |
4.6. Clathrate hydrate formation pathway
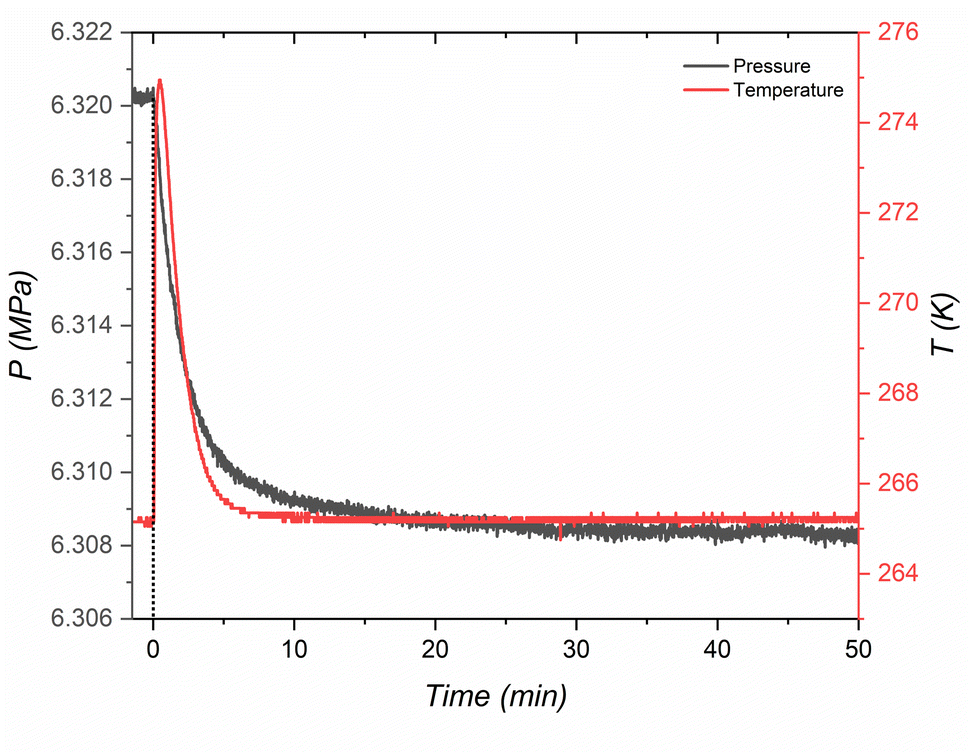
The kinetic model conclusively indicates that the formation pathway for H2-THF hydrate in porous media involves the concurrent occurrence of primary hydrate growth and diffusion-controlled secondary growth. Unlike sequential pathways where a separate THF hydrate forms first followed by H2 enclathration, primary growth here involves the direct formation of H2-THF hydrate (as can be seen from Fig. 8, where a single sharp exothermic peak representing hydrate nucleation coincides with a significant pressure drop, signifying simultaneous hydrate formation and H2 enclathration occurring at the same time). This initial stage is characterized by rapid hydrate growth within the pore space, where the growing hydrate crystal makes it progressively more challenging for H2 molecules to reach the growing crystal front creating a diffusion limitation. Despite the diffusion limitations, the inherent high diffusivity of H2 within clathrate hydrates allows for continued crystal growth until steric hindrance (impingement) occurs. This initial burst of growth (as observed from relatively high kP) corresponds to primary crystallization, while the interplay between continued growth and the increasing influence of diffusion limitations (ks < kP) reflects the concept of secondary growth.
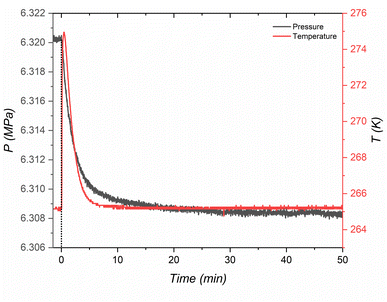 |
| | Fig. 8 A single sharp exothermic peak representing hydrate nucleation coinciding with a significant pressure drop inferring simultaneous THF hydrate formation and H2 enclathration for MCF (f-1) at 265 K. | |
5. Conclusion
This study introduces a new strategy to expedite H2 storage via clathrate hydrates by solid-state tuning of silica-based porous materials (MCF) by grafting promoter-like molecules (THF), leading to a notable enhancement in H2 enclathration kinetics and storage capacities within a binary H2-THF hydrate, as compared to conventional bulk stirring systems. The performance of the materials was evaluated under the conditions of 100% pore volume saturation with a stoichiometric concentration of THF solution (5.56 mol%), at 7 MPa in a range of different temperatures (262 K, 265 K, and 268 K) using a volumetric approach. The findings revealed that the modified MFC materials (MCF (f-1) and (f-2)) can enhance the overall H2 storage capacity compared to the non-modified material (MCF (n-f)). This enhancement could potentially be attributed to the induced hydrophobicity achieved by grafting organic molecules onto the surface of MCF (n-f), which directly influenced the interaction of the material with H2O, creating a conducive milieu for stabilizing clathrate cages and promoting the encapsulation of H2 within these cages. Additionally, the mobility engendered by the ether linker is hypothesized to enable the THF-ending group to function as a promoter, perturbing the water structure and lowering the nucleation barrier. MCF (f-1) and MCF (f-2) achieved a maximum H2 gas uptake of 0.21 wt% and 0.18 wt%, respectively, as opposed to MCF (n-f) which reached 0.15 wt% at 265 K. It is also evident from the gas uptake curves that there also exists an optimal amount of tuning the material that enhances the H2 storage, where a lower number of carbons per nm2 showed a better gas uptake. Furthermore, the results also show that the H2 storage capacity increases with an increase in driving force (i.e., a decrease in temperature), where a maximum storage capacity of 0.52 wt% corresponding to 46.77 mmoles of H2 per mole of H2O and 39.78 water-to-hydrate conversions was achieved at 262 K in MCF (f-1). An intriguing aspect of these results is the substantial H2 storage capacity observed without necessitating supplementary energy-intensive procedures such as agitation or crystal grinding. This underscores the potential of solid-state tuning of porous materials as promoters for H2 storage within artificially synthesized hydrates. Additionally, this work also employed two distinct models for kinetic analysis of hydrate formation and growth in MCF (f-1) at all driving forces. Notably, the multistage kinetic model exhibited better predictive capabilities for the experimental data (water-to-hydrate conversion), with a commendably low average absolute deviation (AAD). Conclusively, it is also recommended to conduct a comprehensive molecular-level investigation to glean a more profound comprehension of the solid-state tuning effects on the supporting materials in enhancing H2 storage capacities. Additionally, these insights can contribute to the refinement and upscaling of clathrate-based H2 storage processes, thereby paving the way for further technological advancements.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work received funding from the European Research Council (ERC) under grant agreement no. 834134 (WATUSO). The authors acknowledge VLAIO for Moonshot funding (ARCLATH, no. HBC.2019.0110, ARCLATH2, no. HBC.2021.0254). NMRCoRe is supported by the Flemish Government as an international research infrastructure (I001321N) and acknowledges infrastructure support by Department EWI via the Hermes Fund (AH.2016.134) and by the Hercules Foundation (AKUL/13/21). M. H. acknowledges FWO Vlaanderen for an FWO-SB fellowship.
References
-
UNFCC, Action Taken by the Conference of the Parties Serving as the Meeting of the Parties to the Kyoto Protocol at its Seventeenth Session, Egypt, 2023, https://newsroom.unfccc.int/sites/default/files/resource/cmp2022_09_add1_adv.pdf Search PubMed.
-
E. Tzimas, et al., Hydrogen Storage: State-Of-The-Art and Future Perspective, Netherlands, 2003 Search PubMed.
-
IEA, Net Zero by 2050, Paris, 2001, https://www.iea.org/reports/net-zero-by-2050.
-
IEA, Hydrogen, Paris, 2022, https://www.iea.org/reports/hydrogen.
-
B. Zohuri, Cryogenics and Liquid Hydrogen Storage, Hydrogen Energy, Springer, 2018, pp. 121–139 Search PubMed.
-
H. W. Langmi, et al., Chapter 13 – Hydrogen storage, in Electrochemical Power Sources: Fundamentals, Systems, and Applications, ed. T. Smolinka, J. Garche, Elsevier, 2022, pp. 455–486 Search PubMed.
- S. K. Dewangan,
et al., A comprehensive review of the prospects for future hydrogen storage in materials-application and outstanding issues, Int. J. Energy Res., 2022, 46, 16150–16177 CrossRef CAS.
- R. Moradi and K. M. Groth, Hydrogen storage and delivery: Review of the state of the art technologies and risk and reliability analysis, Int. J. Hydrogen Energy, 2019, 44, 12254–12269 CrossRef CAS.
- Y. Huang,
et al., A review of high density solid hydrogen storage materials by pyrolysis for promising mobile applications, Ind. Eng. Chem. Res., 2021, 60, 2737–2771 CrossRef CAS.
- H. Q. Nguyen and B. Shabani, Review of metal hydride hydrogen storage thermal management for use in the fuel cell systems, Int. J. Hydrogen Energy, 2021, 46, 31699–31726 CrossRef CAS.
-
E. D. Sloan and C. A. Koh, Clathrate Hydrates of Natural Gases, Taylor & Francis-CRC Press, Boca Raton, FL, 3rd edn, 2008 Search PubMed.
- H. P. Veluswamy,
et al., Hydrogen storage in clathrate hydrates: Current state of the art and future directions, Appl. Energy, 2014, 122, 112–132 CrossRef CAS.
- A. Gupta,
et al., Hydrogen clathrates: Next generation hydrogen storage materials, Energy Storage Mater., 2021, 41, 69–107 CrossRef.
- Y. A. Dyadin,
et al., Clathrate hydrates of hydrogen and neon, Mendeleev Commun., 1999, 5, 209–210 CrossRef.
- W. L. Mao,
et al., Hydrogen clusters in clathrate hydrate, Science, 2002, 297, 2247–2249 CrossRef CAS PubMed.
- L. J. Florusse,
et al., Stable low-pressure hydrogen clusters stored in a binary clathrate hydrate, Science, 2004, 306, 469–471 CrossRef CAS PubMed.
- E. J. Beckwée,
et al., Enabling hydrate-based methane storage under mild operating conditions by periodic mesoporous organosilica nanotubes, Heliyon, 2023, 9, e17662 CrossRef PubMed.
- K. A. Lokshin,
et al., Structure and dynamics of hydrogen molecules in the novel clathrate hydrate by high pressure neutron diffraction, Phys. Rev. Lett., 2004, 93, 125503 CrossRef PubMed.
- T. Saikia,
et al., Hydrogen hydrate promoters for gas storage—A review, Energies, 2023, 16, 2667 CrossRef CAS.
- Y. Zhang,
et al., Hydrogen storage as clathrate hydrates in the presence of 1,3-dioxolane as a dual-function promoter, J. Chem. Eng., 2022, 427, 131771 CrossRef CAS.
- H. Lee,
et al., Tuning clathrate hydrates for hydrogen storage, Nature, 2005, 434, 743–746 CrossRef CAS PubMed.
- T. Sugahara,
et al., Increasing hydrogen storage capacity using tetrahydrofuran, J. Am. Chem. Soc., 2009, 131, 14616–14617 CrossRef CAS PubMed.
- D.-Y. Kim,
et al., Tuning clathrate hydrates: Application to hydrogen storage, Catal. Today, 2007, 120, 257–261 CrossRef CAS.
- D.-Y. Kim,
et al., Critical guest concentration and complete tuning pattern appearing in the binary clathrate hydrates, J. Am. Chem. Soc., 2006, 128, 15360–15361 CrossRef CAS PubMed.
- R. Anderson,
et al., Phase relations and binary clathrate hydrate formation in the system H2-THF-H2O, Langmuir, 2007, 23, 3440–3444 CrossRef CAS PubMed.
- S. Hashimoto,
et al., Thermodynamic Stability of H2 + tetrahydrofuran mixed gas hydrate in nonstoichiometric aqueous solutions, J. Chem. Eng. Data, 2007, 52, 517–520 CrossRef CAS.
- T. A. Strobel,
et al., Molecular hydrogen storage in binary THF−H2 clathrate hydrates, J. Phys. Chem. B, 2006, 110, 17121–17125 CrossRef CAS PubMed.
- N. I. Papadimitriou,
et al., Evaluation of the hydrogen-storage capacity of pure H2 and binary H2-THF hydrates with monte
carlo simulations, J. Phys. Chem. C, 2008, 112, 10294–10302 CrossRef CAS.
- H. P. Veluswamy,
et al., Influence of cationic and non-ionic surfactants on the kinetics of mixed hydrogen/tetrahydrofuran hydrates, Chem. Eng. Sci., 2015, 132, 186–199 CrossRef CAS.
- A. Lokshin and Y. Zhao, Fast synthesis method and phase diagram of hydrogen clathrate hydrate, Appl. Phys. Lett., 2006, 88, 131909 CrossRef.
- S. Alavi and J. A. Ripmeester, Hydrogen-gas migration through clathrate hydrate cages, Angew. Chem., Int. Ed., 2007, 46, 6102–6105 CrossRef CAS PubMed.
- P. Englezos, Nucleation and growth of gas hydrate crystals in relation to kinetic inhibition, Rev. Inst. Fr. Pet., 1996, 51, 789 CrossRef CAS.
- F. Rossi,
et al., Investigation on a novel reactor for gas hydrate production, Appl. Energy, 2012, 99, 167–172 CrossRef CAS.
- W. Lee,
et al., Rapid formation of hydrogen-enriched hydrocarbon gas hydrates under static conditions, ACS Sustainable Chem. Eng., 2021, 9, 8414–8424 CrossRef CAS.
- A. Talyzin, Feasibility of H2–THF–H2O clathrate hydrates for hydrogen storage applications, Int. J. Hydrogen Energy, 2008, 33, 111–115 CrossRef CAS.
- T. Tsuda,
et al., Storage capacity of hydrogen in tetrahydrothiophene and furan clathrate hydrates, Chem. Eng. Sci., 2009, 64, 4150–4154 CrossRef CAS.
- S. Chen,
et al., Rapid and high hydrogen storage in epoxycyclopentane hydrate at moderate pressure, Energy, 2023, 268, 126638 CrossRef CAS.
- K. Ogata,
et al., Storage capacity of hydrogen in tetrahydrofuran hydrate, Chem. Eng. Sci., 2008, 63, 5714–5718 CrossRef CAS.
- J. Farrando-Perez,
et al., Rapid and efficient hydrogen clathrate hydrate formation in confined nanospace, Nature, 2022, 13, 5953 Search PubMed.
- D. Saha and S. Deng, Accelerated Formation of THF-H2 Clathrate Hydrate in Porous Media, Langmuir, 2010, 26, 8414–8418 CrossRef CAS PubMed.
- F. Su,
et al., Rapid and reversible hydrogen storage in clathrate hydrates using emulsion-templated polymers, Adv. Mater., 2008, 20, 2663–2666 CrossRef CAS PubMed.
- D. Saha and S. Deng, Enhanced hydrogen adsorption in ordered mesoporous carbon through clathrate formation, Int. J. Hydrogen Energy, 2009, 34, 8583–8588 CrossRef CAS.
- F. Su,
et al., Reversible hydrogen storage in hydrogel clathrate hydrates, Adv. Mater., 2009, 21, 2382–2386 CrossRef CAS.
- E. J. Beckwée,
et al., Structure I methane hydrate confined in C8-grafted SBA-15: A highly efficient storage system enabling ultrafast methane loading and unloading, Appl. Energy, 2024, 353, 122120 CrossRef.
- G. Watson,
et al., Engineering of hollow periodic mesoporous organosilica nanorods for augmented hydrogen clathrate formation, J. Mater. Chem. A, 2023, 11, 26265–26276 RSC.
- E. Breynaert,
et al., Water as a tuneable solvent: a perspective, Chem. Soc. Rev., 2020, 49, 2557–2569 RSC.
- D. Bai,
et al., How properties of solid surfaces modulate the nucleation of gas hydrate, Sci. Rep., 2015, 5, 12747 CrossRef CAS PubMed.
- J. L. Wang,
et al., Use of hydrophobic particles as kinetic promoters for gas hydrate formation, J. Chem. Eng. Data, 2015, 60, 383–388 CrossRef CAS.
- L. Wang,
et al., A review of the effect of porous media on gas hydrate formation, ACS Omega, 2022, 7, 33666–33679 CrossRef CAS PubMed.
- N. N. Nguyen and A. V. Nguyen, “Nanoreactors” for boosting gas hydrate formation toward energy storage applications, ACS Nano, 2022, 16, 11504–11515 CrossRef CAS PubMed.
- H. Li and L. Wang, Hydrophobized particles can accelerate nucleation of clathrate hydrates, Fuel, 2015, 140, 440–445 CrossRef CAS.
- J. Miyawaki,
et al., Macroscopic evidence of enhanced formation of methane nanohydrates in hydrophobic nanospaces, J. Phys. Chem. B, 1998, 102, 2187–2192 CrossRef CAS.
- N. N. Nguyen and A. V. Nguyen, Hydrophobic effect on gas hydrate formation in the presence of additives, Energy Fuels, 2017, 31, 10311–10323 CrossRef CAS.
- N. N. Nguyen,
et al., Interfacial gas enrichment at hydrophobic surfaces and the origin of promotion of gas hydrate formation by hydrophobic solid particles, J. Phys. Chem. C, 2017, 121, 3830–3840 CrossRef CAS.
- N. N. Nguyen,
et al., Critical Review on Gas Hydrate Formation at Solid Surfaces and in Confined Spaces-Why and How Does Interfacial Regime Matter?, Energy Fuels, 2020, 34, 6751–6760 CrossRef CAS.
- Z. Li and R.-H. Yoon, Thermodynamics of hydrophobic interaction between silica surfaces coated with octadecyltrichlorosilane, J. Colloid Interface Sci., 2013, 392, 369–375 CrossRef CAS PubMed.
- M. E. Casco,
et al., Experimental evidence of confined methane hydrate in hydrophilic and hydrophobic model carbons, J. Phys. Chem. C, 2019, 123, 24071–24079 CrossRef CAS.
- M. E. Casco,
et al., Methane hydrate formation in confined nanospace can surpass nature, Nat. Commun., 2015, 6, 6432 CrossRef CAS PubMed.
- M. S. P. Sansom and P. C. Biggin, Water at the nanoscale, Nature, 2001, 414, 157–159 CrossRef PubMed.
- P. G. M. Mileo,
et al., Interfacial study of clathrates confined in reversed silica pores, J. Mater. Chem. A, 2021, 9, 21835–21844 RSC.
- V. Meynen,
et al., Verified syntheses of mesoporous materials, Microporous Mesoporous Mater., 2009, 125, 170–223 CrossRef CAS.
- T. Yasmin and K. Müller, Synthesis and characterization of surface modified SBA-15 silica materials and their application in chromatography, ACS Sustainable Chem. Eng., 2011, 1218, 6464–6475 CAS.
- J. G. V. Dijck,
et al., The interaction of water with organophosphonic acid surface modified titania: An in-depth in-situ DRIFT study, Surf. Interfaces, 2020, 21, 100710 CrossRef.
- N. B. Kummamuru,
et al., Accelerated methane storage in clathrate hydrates using mesoporous (Organo-) silica materials, Fuel, 2023, 354, 129403 CrossRef CAS.
- N. B. Kummamuru,
et al., Experimental investigation of methane hydrate formation in the presence of metallic packing, Fuel, 2022, 323, 124269 CrossRef CAS.
- E. W. Lemmon,
et al., Revised standardized equation for hydrogen gas densities for fuel consumption applications, J. Res. Natl. Inst. Stand. Technol., 2008, 113, 341–350 CrossRef CAS PubMed.
- H. P. Veluswamy and P. Linga, Macroscopic kinetics of hydrate formation of mixed hydrates of hydrogen/tetrahydrofuran for hydrogen storage, Int. J. Hydrogen Energy, 2013, 38, 4587–4596 CrossRef CAS.
-
NIST Standard Reference Database, Fundamental Physical Constants: Avogadro Constant, U. S. A, 2018 Search PubMed.
- T. Kawamura,
et al., Enclathration of hydrogen by organic-compound clathrate hydrates, Chem. Eng. Sci., 2011, 66, 2417–2420 CrossRef CAS.
- M. Houlleberghs,
et al., Building a Cost-Efficient High-Pressure Cell for Online High-Field NMR and MRI Using Standard Static Probe Heads: An In Situ Demonstration on Clathrate Hydrate Formation, Anal. Chem., 2023, 95, 16936–16942 CrossRef CAS PubMed.
- D. Massiot,
et al., Modelling one- and two-dimensional solid-state NMR spectra, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- M. Avrami, Kinetics of phase change. i general theory, J. Chem. Phys., 1939, 7, 1103 CrossRef CAS.
- M. Avrami, Kinetics of phase change. ii transformation-time relations for random distribution of nuclei, J. Chem. Phys., 1940, 8, 212 CrossRef CAS.
- M. Fanfoni and M. Tomellini, The Johnson-Mehl-Avrami-Kohnogorov model: A brief review, Il Nuovo Cimento D, 1998, 20, 1171–1182 CrossRef.
- A. Kumar,
et al., Crystallization kinetics for carbon dioxide gas hydrate in fixed bed and stirred tank reactor, Korean J. Chem. Eng., 2016, 33, 1922–1930 CrossRef CAS.
- I. L. Moudrakovski,
et al., Nucleation and growth of hydrates on ice surfaces: new insights from 129xe nmr experiments with hyperpolarized xenon, J. Phys. Chem. B, 2001, 105, 12338–12347 CrossRef CAS.
- M. Luzi,
et al., Systematic kinetic studies on mixed gas hydrates by Raman spectroscopy and powder X-ray diffraction, J. Chem. Thermodyn., 2012, 48, 28–35 CrossRef CAS.
- P. S. R. Prasad,
et al., Enhanced methane gas storage in the form of hydrates: role of the confined water molecules in silica powders, RSC Adv., 2020, 10, 17795–17804 RSC.
- A. Nambiar,
et al., CO2 capture using the clathrate hydrate process employing cellulose foam as a porous media, Can. J. Chem., 2015, 93, 1–7 CrossRef.
- J. Zhao,
et al., Combined replacement and depressurization methane hydrate recovery method, Energy Explor. Exploit., 2016, 34, 129–139 CrossRef CAS.
- J. Zhao,
et al., Growth kinetics and gas diffusion in formation of gas hydrates from ice, J. Phys. Chem. C, 2020, 124, 12999–13007 CrossRef CAS.
- H. Liang,
et al., Characterizing mass-transfer mechanism during gas hydrate formation from water droplets, Chem. Eng. J., 2022, 428, 132626 CrossRef CAS.
- J. N. Hay, Secondary crystallization kinetics, Polym. Cryst., 2018, 1, e10007 Search PubMed.
- O. Aktas,
et al., Structural variations of MCF and SBA-15-like mesoporous materials as a result of differences in synthesis solution pH, Mater. Chem. Phys., 2011, 131, 151–159 CrossRef CAS.
- X.-M. Wang,
et al., Direct synthesis and characterization of phenyl-functionalized SBA-15, Appl. Surf. Sci., 2008, 254, 3753–3757 CrossRef CAS.
- C. Yu,
et al., Study on the dissociation characteristics of methane hydrate in clayey silts, Chem. Eng. Sci., 2022, 252, 117508 CrossRef CAS.
-
R. Anderson, et al., Gas hydrate growth and dissociation in narrow pore networks: capillary inhibition and hysteresis phenomena, Sediment-Hosted Gas Hydrates: New Insights on Natural and Synthetic Systems, The Geological Society, London, 2009 Search PubMed.
- T. Yagasaki,
et al., Mechanism of Slow Crystal Growth of Tetrahydrofuran Clathrate Hydrate, J. Phys. Chem. C, 2016, 120, 3305–3313 CrossRef CAS.
- K. Inkong,
et al., Effects of temperature and pressure on the methane hydrate formation with the presence of tetrahydrofuran (THF) as a promoter in an unstirred tank reactor, Fuel, 2019, 255, 115705 CrossRef CAS.
- D. W. Kang,
et al., Confined tetrahydrofuran in a superabsorbent polymer for sustainable methane storage in clathrate hydrates, Chem. Eng. J., 2021, 411, 128512 CrossRef CAS.
- H. Tian and Z. Zhang, Revealing the Growth of H2 + THF Binary Hydrate through Molecular Simulations, Energy Fuels, 2020, 34, 15004–15010 CrossRef CAS.
- B. Liu,
et al., Effect of the Temperature and Tetrahydrofuran (THF) Concentration on THF Hydrate Formation in Aqueous Solution, Energy Fuels, 2022, 36, 10642–10651 CrossRef CAS.
- R. Ma,
et al., Molecular Insights into Cage Occupancy of Hydrogen Hydrate: A Computational Study, Processes, 2019, 7, 699 CrossRef CAS.
-
V. V. Koryakina and E. Y. Shitz, Condensed Matter and Interphases 22 (Usage of the Kolmogorov−Johnson−Mehl−Avrami Model for the Study of the Kinetics of the Formation of Natural Gas Hydrate in Inverse Oil Emulsions), 2020, 327–335 Search PubMed.
- R. Susilo,
et al., Methane conversion rate into structure H hydrate crystals from Ic, AIChE J., 2007, 53, 2451–2460 CrossRef CAS.
- B. L. L. D. Silva,
et al., Sodium alginate polymer as a kinetic inhibitor of methane hydrate formation, J. Mater. Res. Technol., 2021, 12, 1999–2010 CrossRef CAS.
- A. A. Aziz,
et al., The effect of a secondary process on polymer crystallization kinetics – 3. Co-poly (lactic acid), Eur. Polym. J., 2017, 94, 311–321 CrossRef CAS.
- K. Phillipson,
et al., The effect of a secondary process on crystallization kinetics – Poly (ε-caprolactone) revisited, Eur. Polym. J., 2016, 84, 708–714 CrossRef CAS.
- Y. Nagai,
et al., Binary hydrogen–tetrahydrofuran clathrate hydrate formation kinetics and models, AIChE J., 2008, 54, 3007–3016 CrossRef CAS.
- H. Yoshioka,
et al., Decomposition Kinetics and Recycle of Binary Hydrogen-Tetrahydrofuran Clathrate Hydrate, AIChE J., 2011, 57, 265–272 CrossRef CAS.
- J.-H. Yoon,
et al., Spectroscopic identification, thermodynamic stability and molecular composition of hydrogen and 1,4-dioxane binary clathrate hydrate, J. Phys. Chem. Solids, 2008, 69, 1432–1435 CrossRef CAS.
- H. P. Veluswamy,
et al., Clathrate hydrates for hydrogen storage: The impact of tetrahydrofuran, tetra-n-butylammonium bromide and cyclopentane as promoters on the macroscopic kinetics, Int. J. Hydrogen Energy, 2014, 39, 16234–16243 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d,
Johan
Martens
d,
Johan
Martens
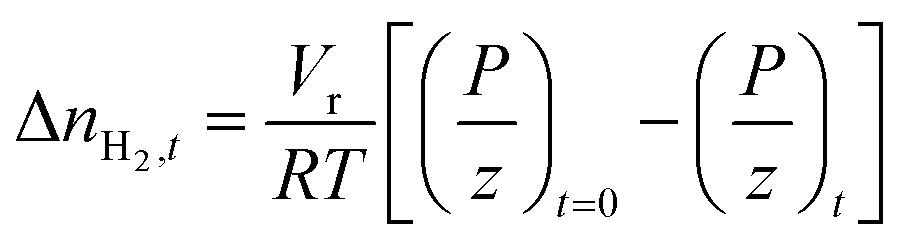
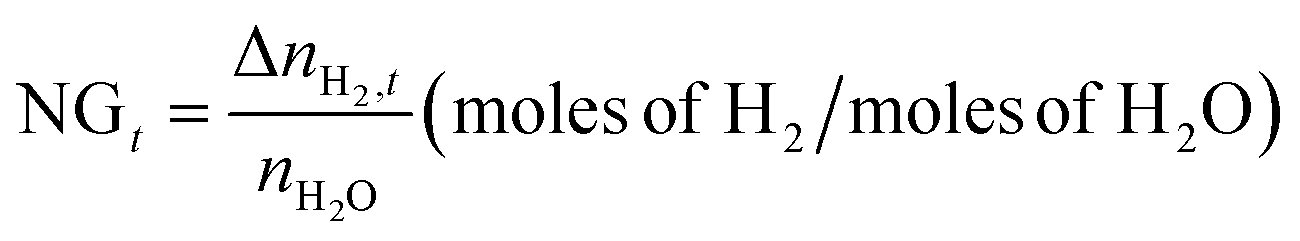
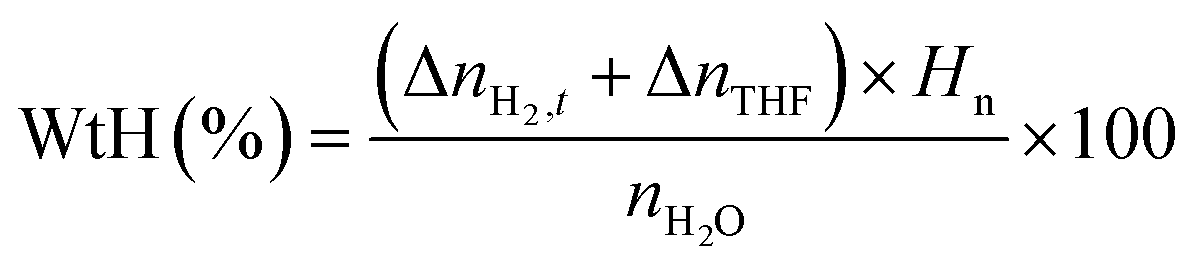
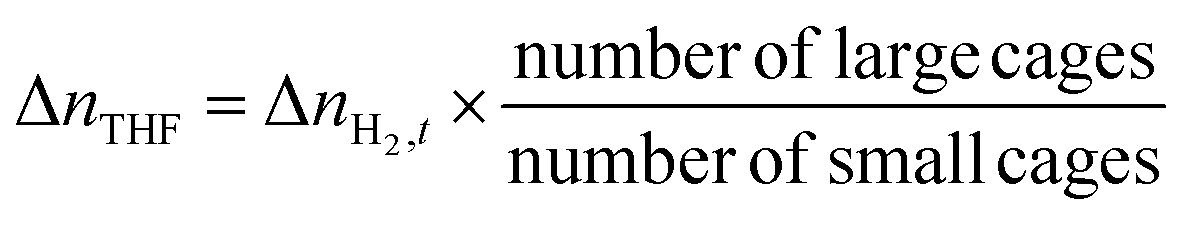
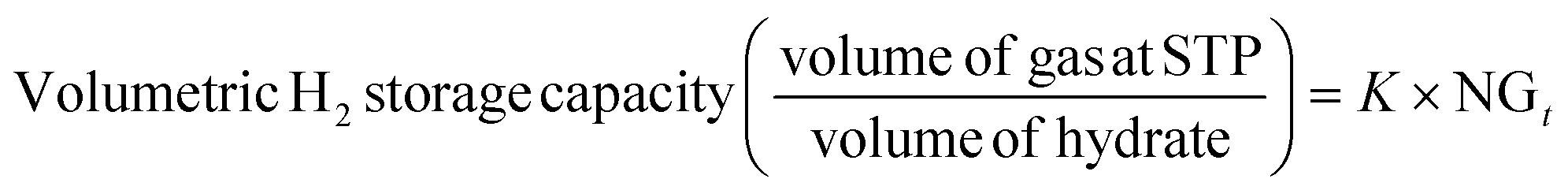
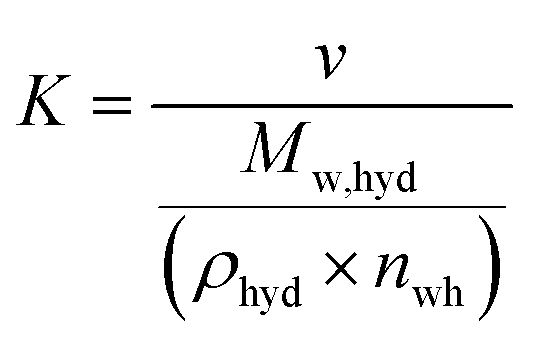
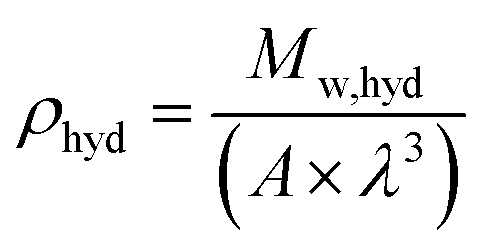
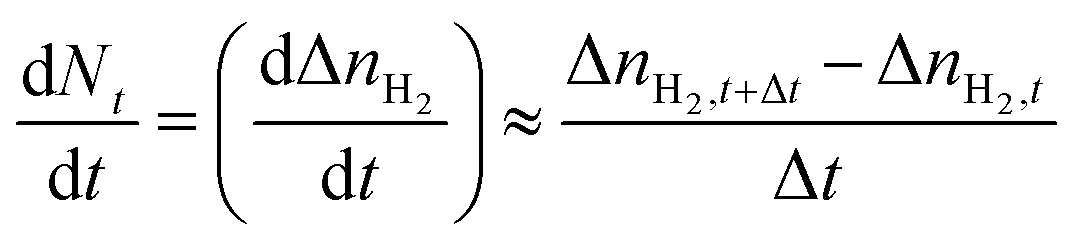
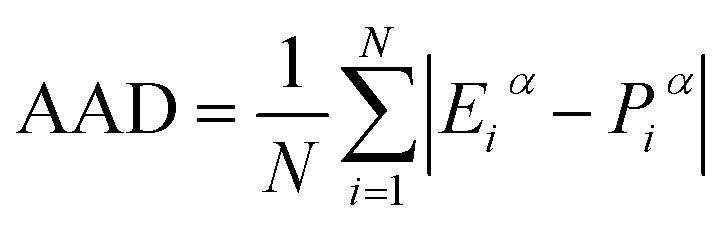
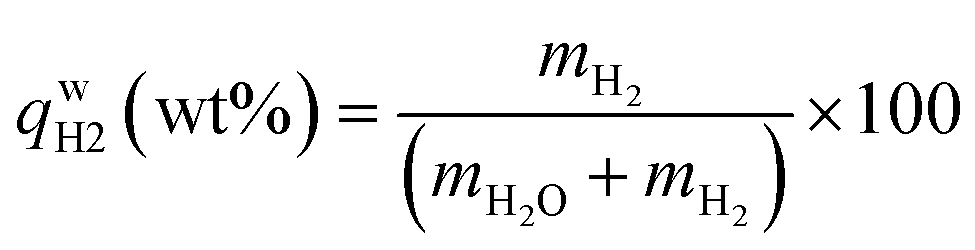 ,
, 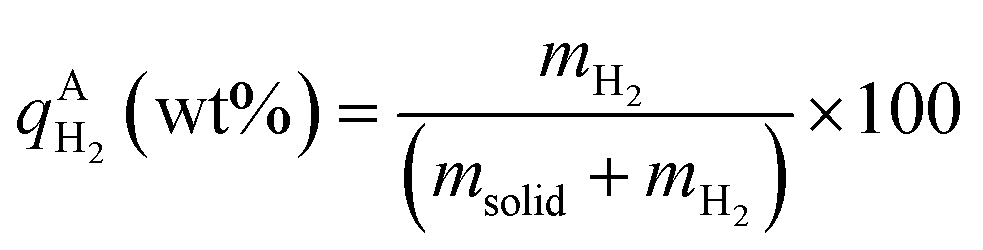 ,
, 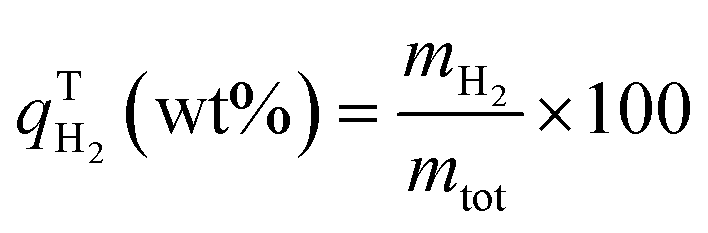 , mH2 = mass of H2, mH2O = mass of H2O, msolid = mass of solid, and mtotal = mass of H2O + mass of solid + mass of H2.
, mH2 = mass of H2, mH2O = mass of H2O, msolid = mass of solid, and mtotal = mass of H2O + mass of solid + mass of H2.
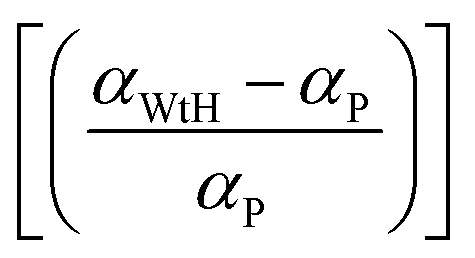 against t0.5 and calculating the slope of the resulting linear fit. These rate constants and αP, obtained from this study, are summarized in Table 4 and the predicted kinetic curves of hydrate formation from the Hay model are depicted in Fig. 7.
against t0.5 and calculating the slope of the resulting linear fit. These rate constants and αP, obtained from this study, are summarized in Table 4 and the predicted kinetic curves of hydrate formation from the Hay model are depicted in Fig. 7.