
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
UV-light crosslinked photocatalytic polymer gels for batch and continuous flow reactions†
Sarah
Freeburne
a and
Christian W.
Pester
 *ab
*ab
aDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: pester@psu.edu
bDepartment of Materials Science and Engineering, Department of Chemistry, The Pennsylvania State University, University Park, PA 16802, USA
First published on 10th June 2024
Abstract
Using visible light to drive reactions is a growing field due to its potential to provide efficient pathways towards chemical syntheses. In contrast to adding photocatalysts to the reaction mixtures as small molecules, heterogeneous photocatalytic approaches provide the ability to facilitate recovery and reuse after a reaction. Herein, we describe a heterogeneous polymer gel-based photocatalyst comprised of a crosslinked terpolymer. We use an activated ester approach to substitute in an amine-functionalized Eosin Y dye while also providing benzophenone-based moieties which afford for UV-crosslinking via C,H-insertion chemistry. The resulting Eosin Y-gels were analyzed for their ability to oxidize thioanisole and their longevity in subsequent recycles for this reaction was analyzed at various light intensities. We interrogated how the incorporation of additional crosslinkers influences photocatalytic properties. Finally, the utility of Eosin Y-gels in continuous packed bed flow photoreactors was investigated, showing good long-term stability against photobleaching and high yields for extended reaction times.
Introduction
The ability to turn photons into chemical energy has gathered increasing scientific interest in recent years.1 Beyond inorganic photocatalysts, such as titania, both transition metal-based and organic photocatalysts can perform various organic (both bond forming and breaking) reactions under irradiation of light2–4 of ultraviolet (UV)5,6 to visible,7,8 all the way to (near) infrared (NIR) wavelengths.9–11 Similarly, polymer chemistry has been significantly impacted by this area,12 where photons can be used in free radical13–15 and reversible deactivation radical polymerizations (RDRPs),16–18 step growth polymerizations,19 post polymerization modifications,19,20 or depolymerization reactions.21,22 Limitations in organic photocatalysis are the often high cost of catalysts and catalyst contamination of products. Heterogeneous photocatalysis offers a viable alternative to allow post-synthetic separation and reuse of catalyst materials. To this end, immobilizing photocatalysts on solid supports or incorporating them into networks has shown promise. Examples include photocatalysts that have been immobilized on inorganic nanoparticles,23 microparticles,24–26 organic substrates (such as cotton),27,28 or incorporated into hybrid (e.g., metal organic frameworks)29 or organic polymer networks and polymer nanoparticles.30,31For heterogeneous photocatalysts in particular, organic dyes such as Eosin Y32 or Rose Bengal33 have become increasingly popular for their wide range of applicability and their relatively low cost. Eosin Y is widely studied in a variety of chemical reactions, including for its ability to produce singlet oxygen34–37 and for hydrogen atom transfer reactions.38 Eosin Y also operates as RDRP photocatalyst for atom transfer radical polymerization (ATRP)39,40 or photoinduced electron/energy transfer reversible addition–fragmentation chain-transfer (PET-RAFT) polymerization.41 Another emerging and highly relevant application for Eosin Y is for the degradation of polymers and for depolymerization reactions.42
As will be discussed, Eosin Y and Rose Bengal can be readily modified for heterogenization. Examples of Eosin Y-based heterogeneous photocatalysts include cotton threads (Eosin Y@cotton),43 Eosin Y attached to poly(urethane)-based scaffolds,44 interpenetrating polymer networks (IPNs) comprised of poly(ethylene glycol) diglycidyl ether and 3-(diethylamino)propylamine, and 1,2-bis(2-aminoethoxy)ethane as a crosslinker,45 as well as Eosin-Y functionalized Amberlite IRA 900 chloride resin.46 Each system offers means for recovery through removal of the catalyst by simple means such as using filter paper. This adds to the ease of use and recovery of the catalyst. However, these systems may either require involved syntheses, as exemplified by the Eosin Y-IPN, or are limited in their shapes and geometries, such as the cotton thread scaffolds or the amberlite resin supports.
Herein, we synthesize and study a photo-crosslinked photocatalytic gel. The polymer network is comprised of a ternary copolymer of ethyl methacrylate (EtMA), 4-benzoylphenyl methacrylate (BPMA), and pentafluorophenyl methacrylate (PFPMA). This terpolymer provides capabilities to be crosslinked under UV light through C,H insertion crosslinking (CHic) reactions,47–50 introducing solvent-stability and the ability to recover the photocatalyst gel post-reaction, as well as modularity opportunities to substitute the PFPMA51 with various amine-functionalized photocatalysts. After synthesizing the desired photocatalytic Eosin Y-based gels, we study their performance in an oxidation reaction both in batch as well as in continuous flow.
Results and discussion
Free radical polymerization was used to synthesize a photo-cross linkable terpolymer of ethyl methacrylate (EtMA), 4-benzoylphenyl methacrylate (BPMA), and pentafluorophenyl methacrylate (PFPMA) (see Fig. 1) by using azobisisobutyronitrile (AIBN) as a radical initiator. BPMA was selected as the crosslinking agent via C,H-insertion chemistry (CHic), and is expected to form crosslinks between the individual polymer chains. Because the rate constant for hydrogen abstraction in CHic chemistry is faster for secondary protons than primary protons,49,52 we chose EtMA as a suitable basis for the bulk of the copolymer. PFPMA was chosen to afford substitution with amine-functionalized photocatalysts.53 A similar approach was demonstrated, for example, by Cai and co-workers to engineer polymeric coatings on chitin microspheres as RDRP photocatalysts.54 While our work focuses on Eosin Y, this modular active ester-based approach provides ample opportunity to functionalize these terpolymers with any amine-functionalized photocatalyst. The molecular weight of the resulting P(EtMA-co-BPMA-co-PFPMA) copolymer was measured at Mn = 56.7 ± 9.1 kg mol−1 (Đ = 2.03 ± 0.16) by gel permeation chromatography (GPC; see Fig. S1 & S2†). The variation reported as standard deviations in the molecular weight and dispersity are the result of multiple batches of polymer being synthesized over the course of this study. 1H NMR spectroscopy was used to estimate the copolymer composition as 88.0 mol% EtMA, 6.2 mol% BPMA, and 5.8 mol% PFPMA, which was in reasonable agreement with the molar monomer feed ratio of [EtMA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[BPMA]:[PFPMA] = 92.5:5.0:2.5. Some of the discrepancy can be accounted for by the incomplete conversion of EtMA at the end of the reaction. Incorporation of PFPMA was estimated through 19F NMR spectroscopy of the purified polymer, exhibiting peaks at δ = −149.9 ppm, −151.6 ppm, −157.2 ppm, and −162.1 ppm, respectively in deuterated chloroform (CDCl3, see Fig. S3†).
:[BPMA]:[PFPMA] = 92.5:5.0:2.5. Some of the discrepancy can be accounted for by the incomplete conversion of EtMA at the end of the reaction. Incorporation of PFPMA was estimated through 19F NMR spectroscopy of the purified polymer, exhibiting peaks at δ = −149.9 ppm, −151.6 ppm, −157.2 ppm, and −162.1 ppm, respectively in deuterated chloroform (CDCl3, see Fig. S3†).
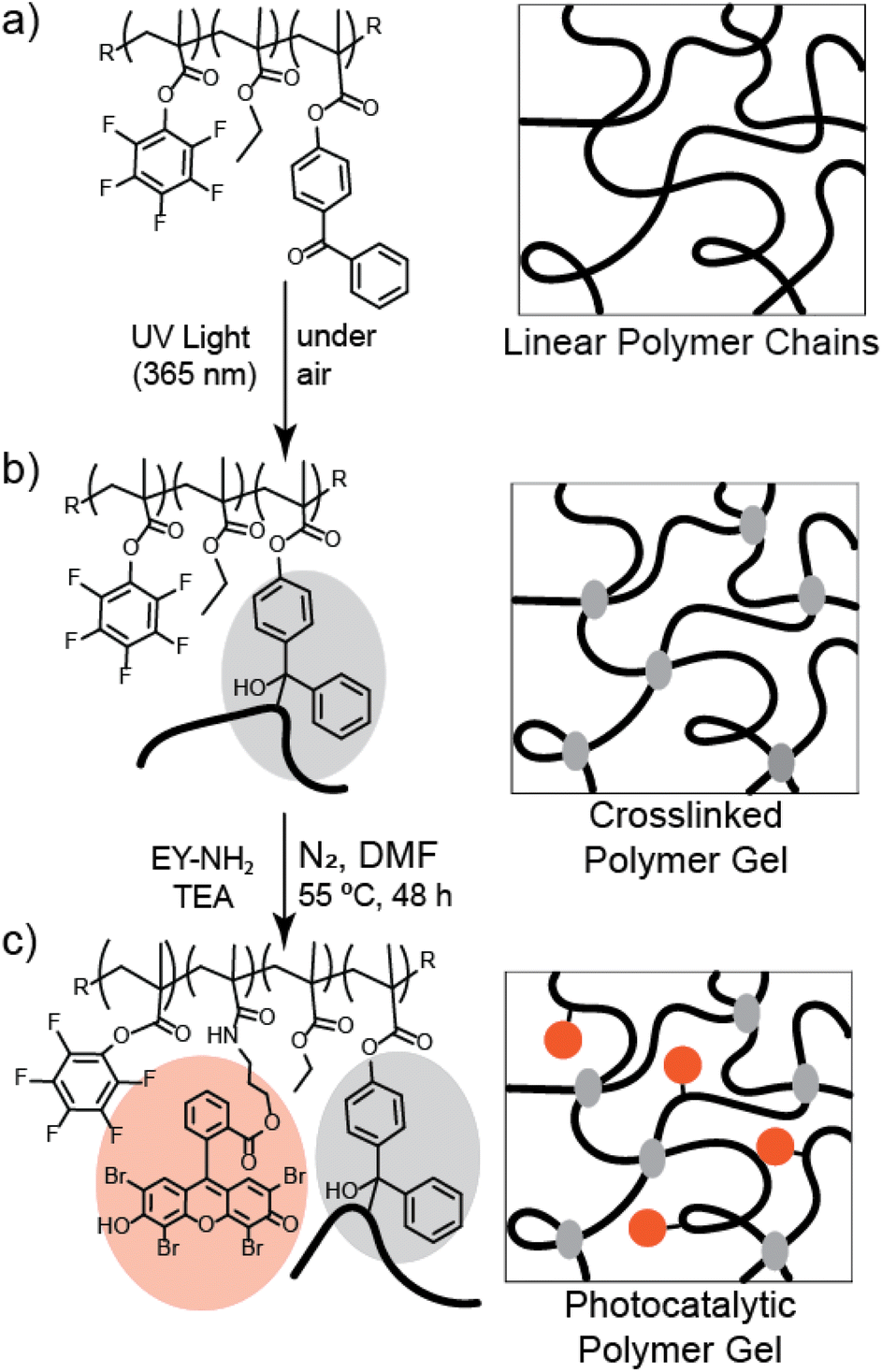 | ||
| Fig. 1 (a) The chemical structure of the synthesized P(EtMA-co-BPMA-co-PFPMA) copolymer. R groups represent the variety of chain ends produced from free radical polymerization. (b) The terpolymer was treated with UV light (λmax = 365 nm) to crosslink the polymer via C,H-insertion chemistry. (c) Treatment with a primary amine-bearing photocatalyst (Eosin Y; EY-NH2) in presence of triethylamine (TEA) substituted the pentafluorophenyl moieties to yield the final photocatalytic crosslinked network. | ||
To create a crosslinked polymer network, the P(EtMA-co-BPMA-co-PFPMA) terpolymer was exposed to UV light (λmax = 365 nm, 4.5 mW cm−2) for two hours. This activated C,H-insertion (CHic) crosslinking through the BPMA functionalities (see Fig. 1b), wherein the UV light results in a ketyl group and a hydrogen is abstracted from a nearby hydrocarbon, and the radical carbons form a bond creating a tertiary carbon.55 Immersion in acetone was used to purify the product and confirm that gel formation was successful, and the polymer network remained insoluble. The gel was functionalized with Eosin Y photocatalysts by substituting the pentafluorophenyl functionalities using activated ester chemistry51 with an amine-functionalized Eosin Y (EY-NH2) in dimethyl formamide (DMF) and the presence of triethyl amine (TEA; see Fig. 1). We estimated the incorporation of Eosin Y into the gel with UV/Vis spectroscopy of aliquots of the substitution reaction (see Fig. S4†) as roughly 2.5 mg of Eosin Y per 130 mg of EY-gel. Variation of substitution reaction times did not conclusively appear to alter the performance of the gel (see Fig. S5†), and 19F NMR of the linear polymer before and after substitution did not appear to show significant changes in substitution efficiency (see Fig. S6†). We hypothesize that the retention of the PFPMA monomers despite the substitution reaction is the result of the lower efficiency of substitution associated with PFPMA compared to pentafluorophenyl acrylate (PFPA).56 Another possible reason could be steric hinderance that may arise from the BPMA monomers and the reactivity ratios of PFPMA and BPMA.
The oxidation of thioanisole to methyl phenyl sulfoxide was selected as a model reaction to investigate the photocatalytic properties of the synthesized heterogeneous photocatalyst (see Fig. 2a and b). This reaction is based on photo-generation of reactive singlet oxygen (1O2), which then proceeds to oxidize the sulfur atom in thioanisole.57 Others have studied this reaction using scaffolds for Eosin Y (such as cotton threads, Amberlite Resins, polyurethane foams, or chitin spheres).28,44,46,54 Rose bengal-based materials have also been studied on chitosan, and yields of 92% of thioanisole in 6 hours of blue light irradiation were obtained.58
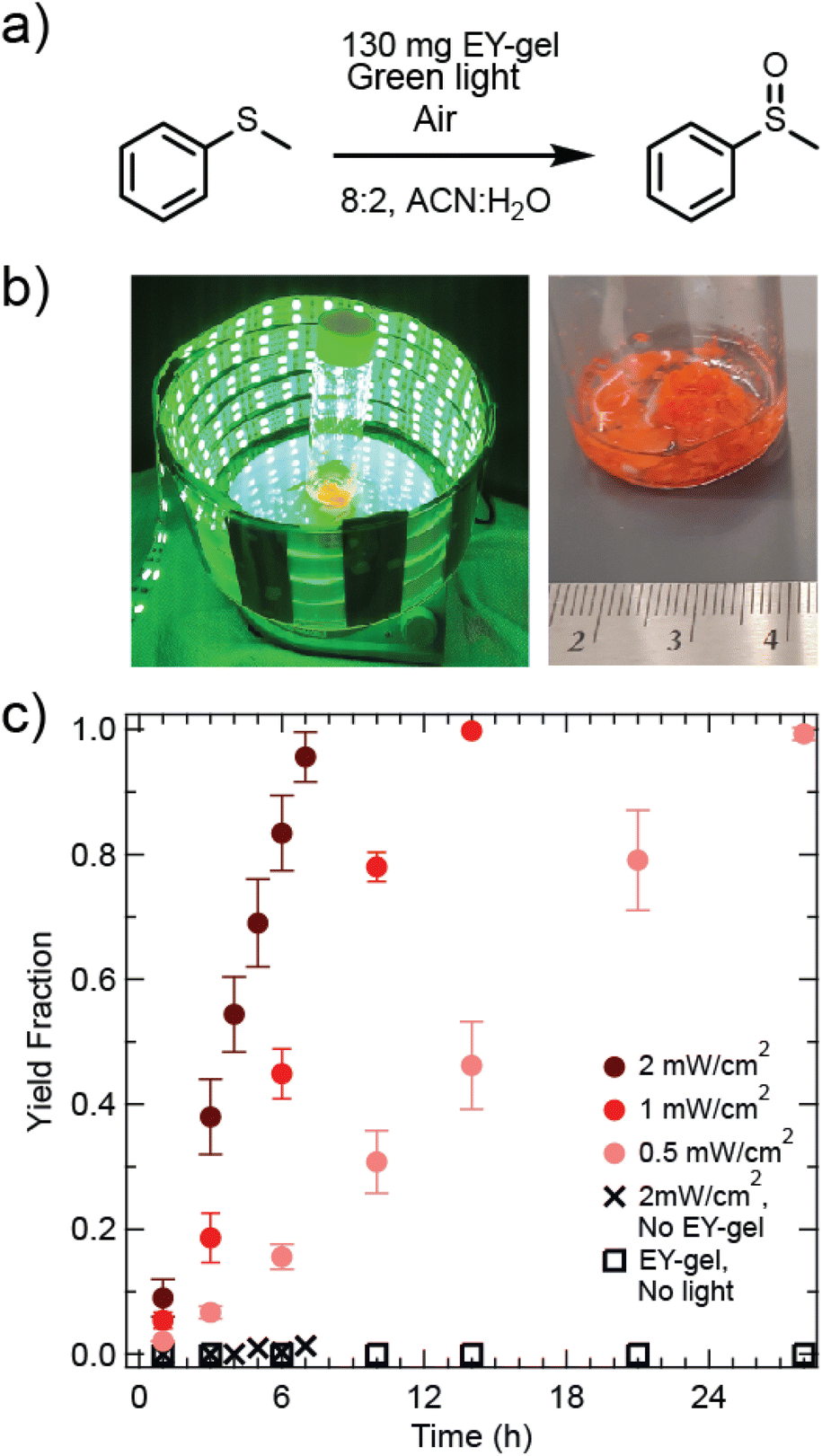 | ||
| Fig. 2 (a) Reaction schematic for oxidation of thioanisole. (b) Photographs of the light bath reaction setup at 1 mW cm−2 (left), and the EY-gel in reaction solution (right). (c) Yield fraction as a function of reaction time for the oxidation of thioanisole (0.67 mmol) using 130 mg of EY-gel under varied intensities of green light irradiation (λmax = 520 nm) at room temperature. | ||
A volumetric ratio of 8:2 acetonitrile to water for the solvent choice for this reaction was based off of optimizations performed by Sridhar et al.59 and Chu et al.43 It is hypothesized that this mixture may combine the benefits that acetonitrile results in a relatively longer excited state lifetime of singlet oxygen60 and it is thought that the addition of water aids in the prevention of catalyst agglomeration for some catalyst types.61 Studies were then performed to determine reactor size and head space were to provide sufficient oxygen supply for the progression of the reaction (see section 7 of the ESI and Fig. S7a†).
Fig. 2c shows kinetic studies of EY-gel catalyzed oxidation of thioanisole as a function of light intensity and monitored by 1H NMR. Yields were determined by relating the increase in the methyl phenyl sulfoxide methyl group signal at δ = 2.71 ppm (3H, CH3) with the decreasing methyl group in thioanisole (δ = 2.47 ppm; 3H, CH3) while using the CDCl3 solvent peak (δ(CHCl3) = 7.26 ppm) as a reference (see Fig. S8†). A comparison of different gel loadings of 60 mg, 130 mg, and 200 mg showed that 130 mg resulted in the fastest rate of conversion (see Fig. S9†). Next, light intensities were modulated, and the highest light intensity (2 mW cm−2) resulted in the fastest conversion of thioanisole, reaching near completion after 7 hours of reaction time. In contrast, the lowest examined light intensity (0.5 mW cm−2) resulted in the slowest rate of conversion, reaching 98% yield within 28 hours of irradiation. Assuming a pseudo-0th order rate law, apparent reaction rates (kapp) were determined as kapp = 0.024 mmol h−1, 0.049 mmol h−1, and 0.091 mmol h−1 for 0.5 mW cm−2, 1 mW cm−2, 2 mW cm−2 irradiance, respectively. This linear relationship between light intensity and reaction rate matches theoretical expectations for the range of studied light intensities.62 This suggests that the activation of Eosin Y photocatalysts is a potential limiting factor under these conditions. A control experiment performed under 2 mW cm−2 green light exposure with no photocatalyst showed negligible yield under these conditions. Another control experiment was performed using the same loading of EY-gel, but without exposure to light and also revealed negligible yield (see Fig. 2c). Overall, results from our materials suggest faster kinetics than were observed by Chu et al., who performed this reaction using Eosin Y coupled to cotton thread (EY@cotton) catalysts and found 45% product yield after 28 hours (green light, λmax = 530 nm, 0.45 mW cm−2) even with air being bubbled through the system as an additional O2 supply.28 For a comparison of this catalyst's performance compared to other similar systems based off of Eosin Y, the reader is referred to a summary table prepared by Peng et al.63
The degree of crosslinking was investigated to further understand the impact of the benzophenone groups on the gel's photocatalytic performance. Riga et al. found that increased amounts of benzophenone groups at 15 mol% resulted in higher amounts of gel compared to 5 mol% on silica wafers when controlled for the same input of UV light energy for crosslinking.64 To investigate the effects of the benzophenone incorporation, three polymers of 5 mol%, 10 mol%, and 15 mol% target BPMA incorporations were synthesized, while maintaining the same 2.5 mol% target PFPMA content. Fig. S10–13† compile NMR and GPC analyses for the resulting materials. The experimentally determined EtMA:BPMA ratios were: 16.7:1 (expected 18.5:1 for 5 mol%), 5:1 (expected 8.75:1 for 10 mol%), and 3.8:1 (expected 5.5:1 for 15 mol%), indicating a slightly faster BPMA incorporation rate than EtMA during the polymerization process. The crosslinked polymers were investigated for their swelling ratios in acetone to gain insight on the degrees of crosslinking. The crosslinked 5 mol% BPMA copolymer exhibited the lowest swelling ratio 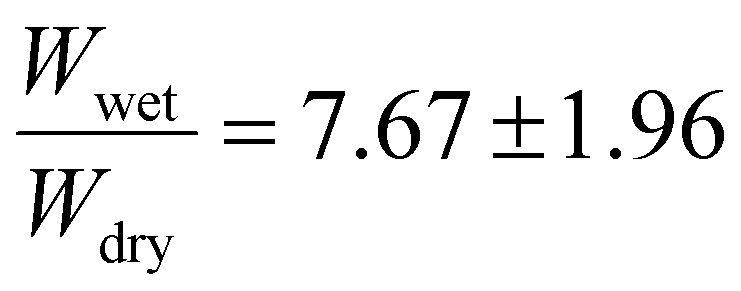 . The 10 mol% target BPMA incorporation showed a higher swelling ratio of
. The 10 mol% target BPMA incorporation showed a higher swelling ratio of 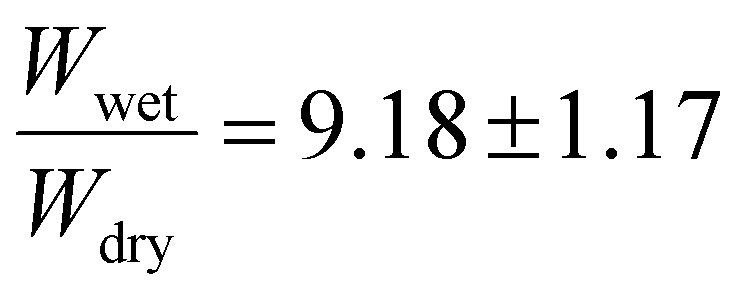 . Finally, the 15 mol% target incorporation resulted in a swelling ratio of
. Finally, the 15 mol% target incorporation resulted in a swelling ratio of 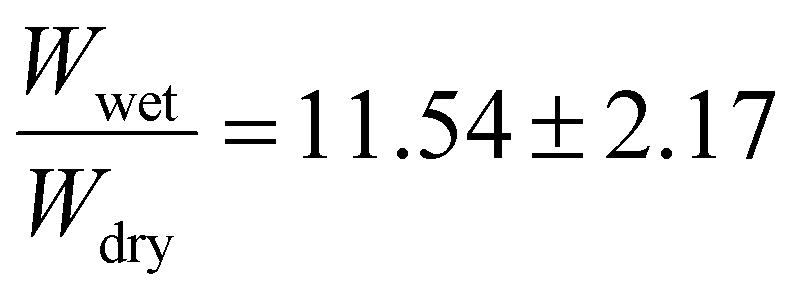 (see Table S1†). The variations in standard deviations are likely the result of uneven penetration of UV light during the crosslinking process because of the amorphous shapes and differing sizes of polymer clusters used. The trend of increasing swelling ratios with increasing incorporations of benzophenone may likely be explained by the expected decrease in UV light penetration depth with increasing amounts of benzophenone. While the exact penetration depth in this system is uncertain, different studies by Riga et al.64 and Liu et al.,65 who studied different polymers with benzophenone groups, each found similar thicknesses on the order of roughly 150–200 nm after UV light crosslinking their polymers onto silicon wafers. Beer–Lambert absorption effects in these types of applications have been estimated to result on the order of 15–30% UV light intensity declines.50,65 While the exact number of BPMA groups activated by the UV treatment is uncertain, Christensen et al. found in their studies that the crosslinking efficiency with PMMA can reach 70%.50
(see Table S1†). The variations in standard deviations are likely the result of uneven penetration of UV light during the crosslinking process because of the amorphous shapes and differing sizes of polymer clusters used. The trend of increasing swelling ratios with increasing incorporations of benzophenone may likely be explained by the expected decrease in UV light penetration depth with increasing amounts of benzophenone. While the exact penetration depth in this system is uncertain, different studies by Riga et al.64 and Liu et al.,65 who studied different polymers with benzophenone groups, each found similar thicknesses on the order of roughly 150–200 nm after UV light crosslinking their polymers onto silicon wafers. Beer–Lambert absorption effects in these types of applications have been estimated to result on the order of 15–30% UV light intensity declines.50,65 While the exact number of BPMA groups activated by the UV treatment is uncertain, Christensen et al. found in their studies that the crosslinking efficiency with PMMA can reach 70%.50
Fig. 3 shows that varying the molar incorporation of BPMA did not appear to significantly alter photocatalytic reaction rates. The slight trend of decreased rates with increased BPMA loadings is reflective of the expected increased proportion of mass being BPMA in the higher loading gels. Nonetheless, there does not appear to be a benefit to increasing the amount of BPMA in the EY-gel photocatalysts, and therefore the expected increases in swelling associated did not appear to considerably improve performance.
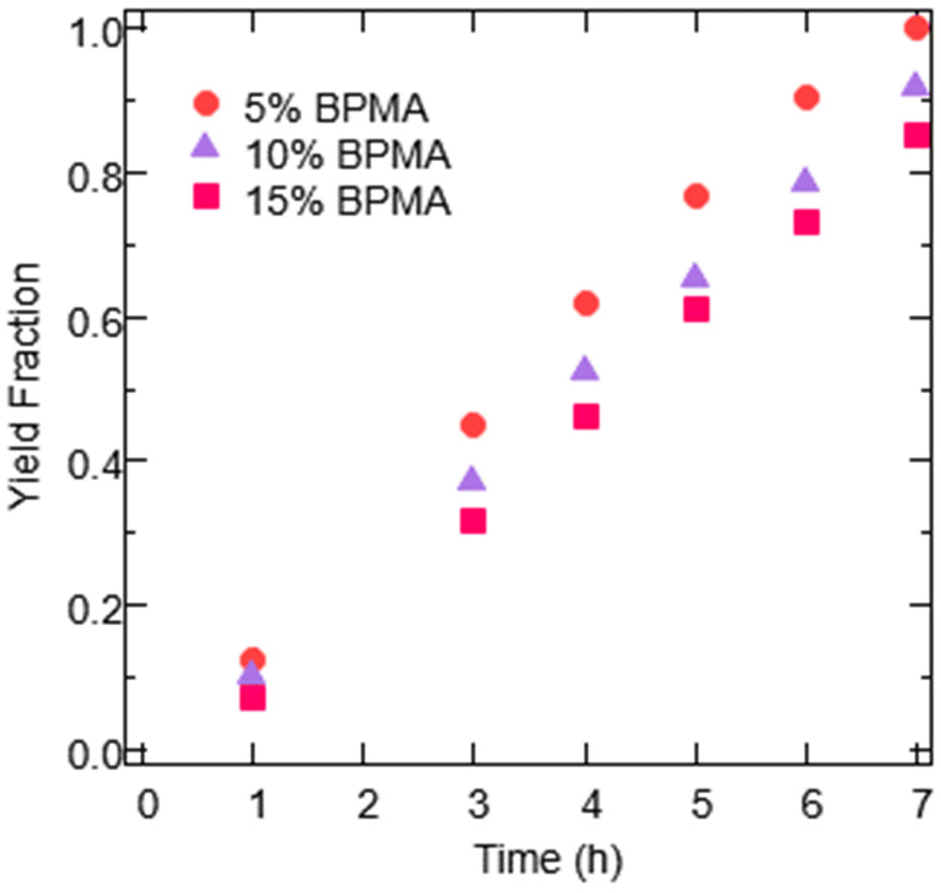 | ||
| Fig. 3 EY-gel photocatalysts evaluated with different target incorporations of BPMA, with each percentage representing the molar feed ratio used in the free radical polymerization to synthesize the materials. Data shown are product yield as a function of reaction time for EY-gels being evaluated for the oxidation of thioanisole (130 mg of EY-gel in 2 mL of an 8:2 ACN/H2O solvent mixture and 0.67 mmol of thioanisole), with the 5% BPMA sample being the best performing set from Fig. 2. Light intensity was constant at 2 mW cm−2 green LED exposure. | ||
One benefit of heterogeneous photocatalysis is the potential for recycling the catalyst materials, since the gels largely retained the characteristic orange color after use (see Fig. S14†), it was hypothesized that the gels could be recycled. To investigate this, we first determined the stability of EY-gels with respect to leaching of potentially physiosorbed Eosin Y from the network, first by testing the gel for retention of Eosin Y under heating in a good solvent, and next by measuring leaching. To provide further evidence for the covalent incorporation of EY-NH2, 100 mg of gel was subjected to heating in a 40 mL vial of DMF at 80 °C and 100 °C. While some pink color was observed in the surrounding liquid after removal of the gel, the gel retained its color and subsequent immersion in DMF at room temperature showed no additional leaching (see Fig. S15†).
To measure the leaching amounts observed from immersion in reaction solvent, dried EY-gel (130 mg) was placed into a 40 mL vial, which was subsequently charged with 3 mL of an 8:2 v/v acetonitrile/DI water mixture. The EY-gel was left to stir in the dark overnight and the concentration of EY-NH2 in solution was measured via UV/vis spectroscopy. Fig. S16† shows representative UV/vis data and a calibration curve which was used to determine the concentration of EY-NH2 through the linear relationship between absorbance at λ = 531 nm and concentration. From this data, we determined that approximately 9.5 μg mL−1 of EY-NH2 was leached from the 130 mg EY-gel. Through monitoring EY-NH2 concentration decline during the incorporation process into the gel, we estimated that about 2.5 mg of EY-NH2 was incorporated per 130 mg of gel polymer, which suggests that the observed leaching amounts are negligible compared to the Eosin Y incorporated into the gel. While low EY-NH2 concentrations are effective even at 20.5 μg mL−1 in catalyzing reactions (see Fig. S17†), the observed rate overall remains slower than the reaction rate produced by the EY-gel. The solution of EY-NH2 (20.5 μg mL−1) appeared to rapidly decrease in conversion rate after approximately 5 hours at 2 mW cm−2 intensity, which is likely a result of photobleaching.
Furthermore, the effects of leaching were further reduced by allowing time for swelling to reach equilibrium, by pre-soaking the gels for at least 16 hours in the ACN:H2O reaction solvent before use, and the solvent was replaced before use in a photocatalytic reaction if leaching was observed. If the gels are not pre-soaked, and instead are used directly after adding solvent, tests showed slightly higher rates of conversion, showing an average of about 15% higher yields (see Fig. S18†), likely as a result of leaching. While this suggests leaching can play a role in the reaction rate, it reinforces that leaching plays a lesser role than the gel. In contrast, the EY-gel catalyst reached quantitative thioanisole conversion after 7 hours. This provides evidence that the EY-gel performance is not solely explained by catalyst leaching.
After investigating leaching, the recyclability of the EY-gel was then investigated. To test the performance of the EY-gels in recycle reactions, the same photocatalytic material was re-used for multiple reaction cycles (see Fig. 4). Between runs, the EY-gels were cleaned by soaking overnight in acetone and rinsed an additional three times with acetone. An aliquot was taken before beginning the recycling tests was to ensure that residual product had been sufficiently removed before beginning a new test. Only minor (0–3 mol%) amounts of residual product from previous reactions were present upon recycling.
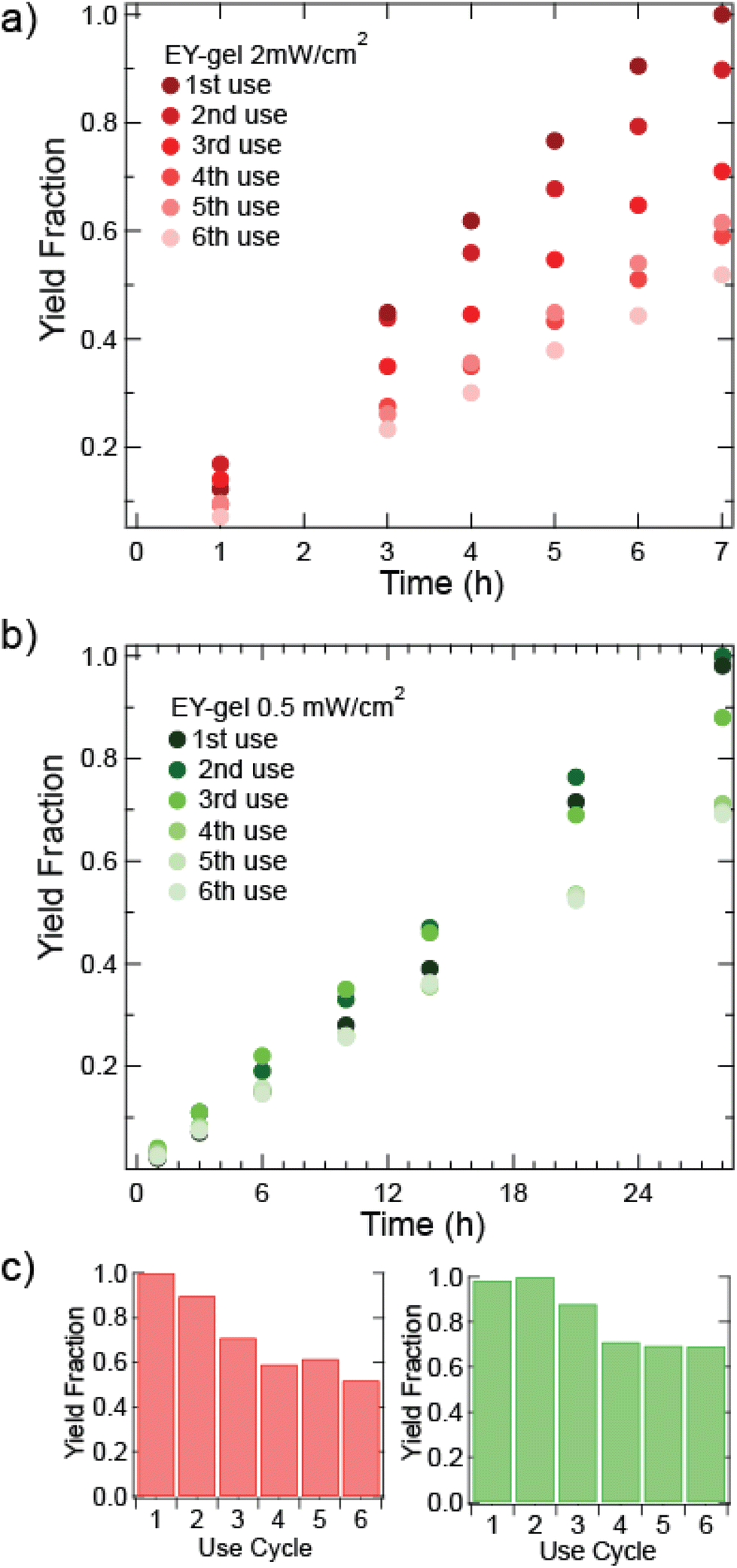 | ||
| Fig. 4 Recycling kinetics for the oxidation of thioanisole (0.67 mmol) in 2 mL of 8:2 ACN:H2O v/v solvent of 130 mg of EY-gel at 2 mW cm−2 (a) 2 mW cm−2 kinetics of each use cycle. (b) 0.5 mW cm−2 kinetics of each use cycle. (c) Bar graph summaries of the final yields (left) at 2 mW cm−2 and (right) 0.5 mW cm−2 intensities. Each reaction employed λmax = 520 nm Green LED light exposure at room temperature. Each use relates to the same EY-gel being recovered and used an additional time in the same reaction. Gels were cleaned through soaking in acetone and further rinses in acetone before reuse. | ||
For heterogeneous photocatalysts, we hypothesized that lower light intensity would result in a slower decrease in rate of thioanisole oxidation and increase catalyst longevity. While increased photon flux generally increases reaction rates, significant photobleaching and loss of catalytic activity can occur. Fig. 4 shows recycling kinetics of EY-gel at a higher (2 mW cm−2) and lower (0.5 mW cm−2) light intensities. For reactions under 2 mW cm−2 green light exposure, a slight visible change in color intensity of the EY-gels could be observed after their first use (see Fig. S14†). In the subsequent recycling experiments, a decrease in product yield to about 90% was observed for the second use of the EY-gel. The yield further decreased to approximately 52% yield on the 6th recycle (see Fig. 4a). An apparent rate of kapp = 0.091 mmol h−1 for the generation of methyl phenyl sulfoxide was determined for the first EY-gel use at 2 mW cm−2 (see above). This rate decreases to kapp = 0.050 mmol h−1 after the 6th use. This decline in rate of nearly 45% suggests photobleaching (deactivation of the photocatalyst material) occurred with the 42 hours total of light exposure. For the decreased irradiance of 0.5 mW cm−2, the EY-gels remained more stable (see Fig. 4b). After a total of 168 hours of irradiation, the yield decreased to about 69% by 28 hours of reaction time. In contrast to higher light intensity recycling experiments, the reaction rate showed a slower decline, from kapp = 0.022 mmol h−1 for the first use to kapp = 0.017 mmol h−1 during the 6th use. This represents a decline in the rate of reaction of 24%. Design considerations may favor the more stable catalyst as opposed to faster reaction rates. For instance, while the reaction took longer under 0.5 mW cm−2 light exposure, a total of 15% more product was generated. Continuous synthetic processes, like flow photochemistry, may benefit from this increased stability of the EY-gels to achieve reliable yields over increased durations of use.
Fig. 5 demonstrates the successful utility of the EY-gels as heterogeneous photocatalysts in continuous flow. A 1.5 m continuous flow packed bed reactor was constructed using #13 AWG Teflon tubing (Tef Cap Industries Inc.) wrapped around a glass beaker and placed into a light bath (see section 12 and Fig. S19 of the ESI† for more details). Teflon tubing was selected both for its inertness towards many organic solvents and reactants, as well as for its oxygen permeability.66 Considering the relevance of O2 for the thioanisole oxidation reaction, this was expected to allow for more oxygen delivery into the tubing.67,68 Because the diffusion of oxygen through the tube walls (0.3 mm thickness) is unlikely to provide similar amounts of O2 as an open vial, a decreased concentration of thioanisole (0.08 M in ACN/H2O) was used compared to the batch system. Alternative strategies to maximize yield could include the addition of a compressed air feed line and the construction of a droplet flow reactor.69 The reactor was packed with both approximately 100 mg of EY-gel and glass beads (SiO2, diameter 212–300 μm). The glass beads served to space out the EY-gel, to avoid clogging, and to improve static mixing. Previous work by others have demonstrated that the inclusion of transparent and curved material in continuous flow reactors can result in slight increases in the duration of time photons spend in the reactor, thus increasing yield.70 For example, Herbrik et al. found that the inclusion of glass beads increased the yield in their slurry flow reactor.71
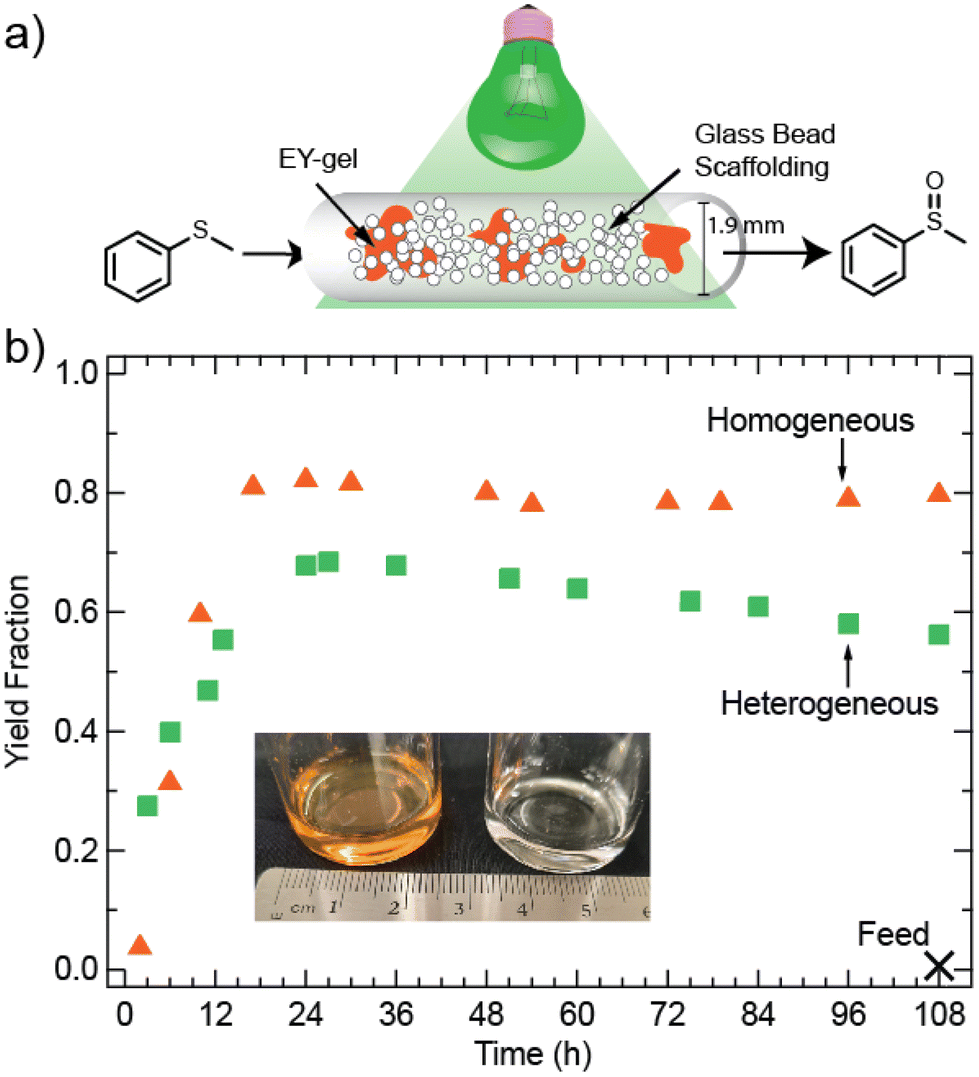 | ||
| Fig. 5 (a) Schematic of continuous flow reactor comprised of Teflon tubing filled with glass beads and EY-gel photocatalyst. (b) Yield fraction as a function of time for the oxidation of thioanisole in a packed bed continuous flow reactor for homogenous and heterogeneous photocatalysis using Eosin Y and EY-gel, respectively. (inset) A sample of product outflow was collected into a vial without any additional processing after leaving the reactor tubing, showing a clear and colorless liquid (right) compared to the homogeneous system (left) which contains coloration from Eosin Y. | ||
Fig. 5b shows a yield of about 69% after the expected residence time. The measured free volume (volume excluding the packed materials) of the reactor was about 1.96 mL, therefore the expected residence time at a flow rate of 2.1 μL min−1 was determined to be 15.6 hours. Many literature studies on immobilized heterogeneous photocatalysts in flow have focused on only a few residence times and long-term stabilities are not yet understood. Examples include Eosin Y@Merrifield Resin beads (reactor volume = 1.95 mL, flow rate ∼16 μL min−1)71 or tetraphenylporphyrin@cotton (reactor volume = 1.16 mL, flow rate = 1.93 μL min−1).72 For this study, we extended the reaction time to gain an understanding of the stability of the photocatalyst in continuous operation. For both systems, an aliquot from the feed system was taken to ensure no product was seen in the syringe after 108 hours of flow. Once the expected residence time has passed, the described EY-gel flow photoreactor produces consistent results without the need for researcher intervention or maintenance. A linear approximation starting at the 27-hour time-point and up until 108 hours of total reaction time suggests a decline in yield at a rate of 0.15% per hour (R2 = 0.99), perhaps resulting from photobleaching. In comparison, a homogeneous mixture of Eosin Y (304 μg mL−1) and thioanisole at the same flow rate produced a product yield of about 80% after steady state was reached. The homogeneous photocatalytic system does not display a drop in yield with time. However, because EY is continuously passing through the reactor, this approach becomes increasingly expensive at scale. The resulting product outflow (collected at the final 12 hours) is also notably orange in color, resulting from photocatalyst contamination (see Fig. 5c). Based on the loaded amount of homogeneous EY, the outflow has a concentration of 304 μg mL−1 of EY. In contrast, the product output from the heterogeneous EY-gel approach (collected in the final 12 hours of the experiment) appears nearly colorless. A comparison of UV/vis absorbances suggests only 0.94 μg mL−1 of EY-NH2 in the output stream (see Fig. S20†), which is significantly less than the homogeneous photocatalytic approach, and suggests negligible leaching at the end of the reaction period studied.
Lastly, a comparison of the turnover number (TON) (calculated as the moles of product divided by the moles of catalyst73) and turnover frequency (TOF) (TON divided by time) values can be made (see Table S2†). In batch, the TON and TOF values for Eosin Y were calculated to 712 and 142, respectively. This indicates a relatively high efficiency of homogeneous Eosin Y in the thioanisole oxidation reaction. The EY-gel TON and TOF values in batch conditions were estimated to 180 and 26, respectively, which are relatively high compared to other heterogeneous catalysts based on Eosin Y for this reaction, as summarized by Peng et al.63 In flow, the Eosin Y TON and TOF values were calculated to 142 and 9, showing lower efficiency as a result of the limitations on oxygen diffusion into the tubing. The EY-gel in flow by comparison was estimated to have TON and TOF values of 229 and 15, respectively. This implies an increase in the efficiency of the use of photocatalyst because the EY-gel in flow remains inside the reactor for the duration of the experiment, while the homogeneous Eosin Y is continuously added into the reactor tubing from the feed syringe with the solvent and reactant. Overall, this highlights the benefits of heterogeneous photocatalysis in continuous flow.
Conclusions
In this work we synthesized a bi-functional polymer that can crosslink through pendant 4-benzoylphenyl methacrylate (BPMA) groups and is also able to be functionalized with Eosin Y photocatalysts through pentafluorophenyl methacrylate groups. The polymer was crosslinked under UV light to form an insoluble macroscopic gel and was then functionalized with Eosin Y to produce an EY-gel heterogeneous photocatalyst. The EY-gel was then studied under different light intensities, BPMA inclusions, and both in batch and continuous flow conditions to evaluate its efficacy in the oxidation of thioanisole as a model reaction. Findings indicated that the EY-gels exposed to lower light intensities could withstand longer reaction cycles with lower rates of photocatalytic decline while higher light intensities resulted in faster production kinetics, but also faster rates of photocatalyst degradation. Different incorporations of BPMA were investigated, and no additional benefit was observed from increasing molar feed BPMA amounts from 5 mol% to 15 mol%. Finally, the EY-gel was packed into Teflon tubing and was demonstrated to be an effective photocatalyst for the oxidation of thioanisole with a single feed line delivered through a syringe pump. The resulting product collected was shown to have vastly reduced the amount of Eosin Y found in the product collected when compared to a homogeneous system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge NSF CBET Award # 2143628 for financial support. The authors would like to thank Dr Gina Noh for UV/Vis access, Dr Christy George for assistance with 19F NMR experiments, and Sierra Yost and Dr Bryan Vogt for helpful discussions on monomer selection.References
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- O. Tokode, R. Prabhu, L. A. Lawton and P. K. J. Robertson, Handb. Environ. Chem., 2015, 35, 159–179 Search PubMed.
- G. Goti, K. Manal, J. Sivaguru and L. Dell’amico, Nat. Chem., 2024, 16, 684–692 CrossRef CAS PubMed.
- A. B. Djurišić, Y. He and A. M. C. Ng, APL Mater., 2020, 8, 30903 CrossRef.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- Z. Xia, B. Shi, W. Zhu, Y. Xiao, C. Lü, Z. Xia, B. Shi, W. Zhu, Y. Xiao and C. Lü, Adv. Funct. Mater., 2022, 32, 2207655 CrossRef CAS.
- Y. Sang, H. Liu and A. Umar, ChemCatChem, 2015, 7, 559–573 CrossRef CAS.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- J. H. Back, Y. Kwon, J. C. Roldao, Y. Yu, H. J. Kim, J. Gierschner, W. Lee and M. S. Kwon, Green Chem., 2020, 22, 8289–8297 RSC.
- M. V. Encinas, A. M. Rufs, S. G. Bertolotti and C. M. Previtali, Polymer, 2009, 50, 2762–2767 CrossRef CAS.
- G. Zhang, I. Y. Song, K. H. Ahn, T. Park and W. Choi, Macromolecules, 2011, 44, 7594–7599 CrossRef CAS.
- S. Freeburne, B. Hunter, K. Bell and C. W. Pester, ChemPhotoChem, 2023, 7, e202300090 CrossRef CAS.
- N. Zivic, M. Bouzrati-Zerelli, A. Kermagoret, F. Dumur, J.-P. Fouassier, G. Didier and J. Lalevée, ChemCatChem, 2016, 8, 1617–1631 CrossRef CAS.
- C. Wu, N. Corrigan, C. H. Lim, K. Jung, J. Zhu, G. Miyake, J. Xu and C. Boyer, Macromolecules, 2018, 52, 236–248 CrossRef PubMed.
- J. Xu and C. Boyer, Macromolecules, 2015, 48, 520–529 CrossRef CAS.
- D. J. Lunn, E. H. Discekici, J. Read de Alaniz, W. R. Gutekunst and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2903–2914 CrossRef CAS.
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. De Alwis Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Polym. Chem., 2022, 14, 253–258 RSC.
- K. Parkatzidis, N. P. Truong, K. Matyjaszewski and A. Anastasaki, J. Am. Chem. Soc., 2023, 145, 21146–21151 CrossRef CAS PubMed.
- S. Shanmugam, S. Xu, N. N. M. Adnan and C. Boyer, Macromolecules, 2018, 51, 779–790 CrossRef CAS.
- K. Bell, Y. Guo, S. Barker, S. H. Kim and C. W. Pester, Polym. Chem., 2023, 14, 2662–2669 RSC.
- K. Bell, S. Freeburne, M. Fromel, H. J. Oh and C. W. Pester, J. Polym. Sci., 2021, 59, 2844–2853 CrossRef CAS.
- K. Bell, S. Freeburne, A. Wolford and C. W. Pester, Polym. Chem., 2022, 116, 6120 RSC.
- Y. Chu, N. Corrigan, C. Wu, C. Boyer and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15245–15253 CrossRef CAS.
- Y. Chu, Z. Huang, R. Liu, C. Boyer and J. Xu, ChemPhotoChem, 2020, 4, 5201–5208 CrossRef CAS.
- L. Zhao, Z. Du, G. Ji, Y. Wang, W. Cai, C. He and C. Duan, Inorg. Chem., 2021, 61, 7256–7265 CrossRef PubMed.
- X. Yu, Z. Yang, B. Qiu, S. Guo, P. Yang, B. Yu, H. Zhang, Y. Zhao, X. Yang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 632–636 CrossRef CAS PubMed.
- J. Heuer, T. Kuckhoff, R. Li, K. Landfester and C. T. J. Ferguson, ACS Appl. Mater. Interfaces, 2022, 15, 2891–2900 CrossRef PubMed.
- D. P. Haria and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- S. Sharma and A. Sharma, Org. Biomol. Chem., 2019, 17, 4384–4405 RSC.
- L. V. Lutkus, S. S. Rickenbach and T. M. McCormick, J. Photochem. Photobiol., A, 2019, 378, 131–135 CrossRef CAS.
- F. Amat-Guerri, M. M. C. López-González, R. Martínez-Utrilla and R. Sastre, J. Photochem. Photobiol., A, 1990, 53, 199–210 CrossRef CAS.
- Ritu, C. Sharma, S. Kumar and N. Jain, Org. Biomol. Chem., 2020, 18, 2921–2928 RSC.
- J. J. Lessard, G. M. Scheutz, A. B. Korpusik, R. A. Olson, C. A. Figg and B. S. Sumerlin, Polym. Chem., 2021, 12, 2205–2209 RSC.
- X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. Da Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed.
- M. Atilla Tasdelen, M. Ciftci, Y. Yagci, M. A. Tasdelen, M. Ciftci and Y. Yagci, Macromol. Chem. Phys., 2012, 213, 1391–1396 CrossRef.
- G. Szczepaniak, J. Jeong, K. Kapil, S. Dadashi-Silab, S. S. Yerneni, P. Ratajczyk, S. Lathwal, D. J. Schild, S. R. Das and K. Matyjaszewski, Chem. Sci., 2022, 13, 11540–11550 RSC.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- K. Parkatzidis, H. S. Wang and A. Anastasaki, Angew. Chem., Int. Ed., 2024, 63, e202402436 CrossRef CAS PubMed.
- Y. Chu, Z. Huang, R. Liu, C. Boyer and J. Xu, ChemPhotoChem, 2020, 4, 5201–5208 CrossRef CAS.
- H. Peng, T. Romero, P. Bertani and V. Ritleng, Catalysts, 2023, 13, 589 CrossRef CAS.
- X. Li, Y. C. Zhang, S. Ye, X. R. Zhang and T. Cai, J. Mater. Chem. A, 2020, 8, 25363–25370 RSC.
- A. Sridhar, R. Rangasamy and M. Selvaraj, New J. Chem., 2019, 43, 17974–17979 RSC.
- B. Photophores, B. G. Dorman and G. D. Prestwich, Biochemistry, 1994, 33, 5661–5673 CrossRef PubMed.
- G. Dormán, H. Nakamura, A. Pulsipher and G. D. Prestwich, Chem. Rev., 2016, 116, 15284–15398 CrossRef PubMed.
- L. Giering, M. Berger and C. Steel, J. Am. Chem. Soc., 1974, 96, 953–958 CrossRef CAS.
- S. K. Christensen, M. C. Chiappelli and R. C. Hayward, Macromolecules, 2012, 45, 5237–5246 CrossRef CAS.
- K. A. Günay, P. Theato and H. A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- S. K. Christensen, M. C. Chiappelli and R. C. Hayward, Macromolecules, 2012, 45, 5237–5246 CrossRef CAS.
- M. Eberhardt and P. Théato, Macromol. Rapid Commun., 2005, 26, 1488–1493 CrossRef CAS.
- W. L. Guo, Y. Zhou, B. Duan, W. F. Wei, C. Chen, X. Li and T. Cai, Chem. Eng. J., 2022, 429, 132120 CrossRef CAS.
- G. Rydzek, P. Schaaf, J. C. Voegel, L. Jierry and F. Boulmedais, Soft Matter, 2012, 8, 9738–9755 RSC.
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS.
- C. Ye, Y. Zhang, A. Ding, Y. Hu and H. Guo, Sci. Rep., 2018, 8, 1–6 Search PubMed.
- W. Chen, D. Gu, T. Zhou and X. Peng, Dyes Pigm., 2023, 210, 111042 CrossRef CAS.
- A. Sridhar, R. Rangasamy and M. Selvaraj, New J. Chem., 2019, 43, 17974–17979 RSC.
- M. Bregnhøj, M. Westberg, F. Jensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2016, 18, 22946–22961 RSC.
- T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654–1663 CrossRef.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2021, 122, 2752–2906 CrossRef PubMed.
- H. Peng, T. Romero, P. Bertani and V. Ritleng, Catalysts, 2023, 13, 589 CrossRef CAS.
- E. K. Riga, J. S. Saar, R. Erath, M. Hechenbichler and K. Lienkamp, Polymers, 2017, 9, 686 CrossRef PubMed.
- Q. Liu, A. T. Shaver, Y. Chen, G. Miller, D. R. Paul, J. S. Riffle, J. E. McGrath and B. D. Freeman, Polymer, 2016, 87, 202–214 CrossRef CAS.
- G. E. Everitt, E. C. Potter and R. G. Thompson, J. Appl. Chem., 1965, 15, 398–402 CrossRef CAS.
- T. R. Holm, G. K. George and M. J. Barcelona, Groundwater Monit. Rem., 1988, 8, 83–89 CrossRef CAS.
- F. G. Roper, J. Chromatogr. Sci., 1971, 9, 697–699 CrossRef CAS.
- L. J. Pan, J. W. Tu, H. T. Ma, Y. J. Yang, Z. Q. Tian, D. W. Pang and Z. L. Zhang, Lab Chip, 2017, 18, 41–56 RSC.
- L. Zheng, H. Xue, W. K. Wong, H. Cao, J. Wu and S. A. Khan, React. Chem. Eng., 2020, 5, 1058–1063 RSC.
- F. Herbrik, S. Rossi, M. Sanz, A. Puglisi and M. Benaglia, ChemCatChem, 2022, 14, e202200461 CrossRef CAS.
- Y. Chu, N. Corrigan, C. Wu, C. Boyer and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15245–15253 CrossRef CAS.
- T. Gensch, M. J. James, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 2296–2306 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical reagents/materials, instrumentation and methods, reaction procedures, gel permeation chromatography (GPC) data of polymers used, nuclear magnetic resonance (NMR) of polymers used and thioanisole oxidations, additional kinetic experiments for different amounts of EY-gel, UV/vis calibration curve data, continuous Flow reactor set-up. See DOI: https://doi.org/10.1039/d4py00313f |
| This journal is © The Royal Society of Chemistry 2024 |