
β-Amino amide based covalent adaptable networks with high dimensional stability†
Loc Tan
Nguyen
 a,
Francesca
Portone
ab and
Filip E.
Du Prez
*a
a,
Francesca
Portone
ab and
Filip E.
Du Prez
*a
aPolymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281 S4, 9000 Ghent, Belgium. E-mail: Filip.DuPrez@UGent.be
bDepartment of Chemistry, Life Sciences and Environmental Sustainability and INSTM UdR Parma, University of Parma, Parco Area delle Scienze 17/A, 43124 Parma, Italy
First published on 6th December 2023
Abstract
Herein, we report a scalable synthesis of catalyst-free, covalent adaptable networks (CANs) based on β-amino amides as dynamic linkages. Rheological analysis of their dynamic behaviour shows a remarkably high activation energy of around 300 kJ mol−1. Hence, the obtained elastomers can be (re-)processed at elevated temperatures while possessing high creep resistance in a wide temperature window. Finally, in comparison with the much-studied β-amino ester-based networks, this new generation of CANs incorporating amino amide motifs possess superior hydrolytic resistance under both acidic and basic conditions.
The dramatic increase in plastic pollution has become one of the greatest environmental challenges.1–3 While thermoplastic materials are intrinsically (re-)processable at elevated temperatures, repurposing or recycling the more dimensionally stable thermoset materials, in which polymer chains are covalently cross-linked, is far more cumbersome. Therefore, thermosets typically end up as permanent waste after a single use.4–6 Covalent adaptable networks (CANs) have emerged as a potential solution that can combine the high performance of thermosets and (re)processability of thermoplastics in one single material.7–9 Although CANs are also cross-linked by covalent bonds, the dynamic nature of such bonds endows such materials with the ability to flow with the application of specific stimuli.10–14 On the other hand, reversible linkages within CANs often reduce their dimensional stability and creep is thus observed due to the unfavoured premature exchange under the use conditions.15,16 Suppressing this unwanted deformation at use temperatures therefore remains one of the important challenges in merging academic research on CANs and industrial applications.17–23 Among the different approaches that have been introduced recently to limit the undesired exchange (e.g., protected catalysts, modifying reactive functional groups, etc.), the use of highly endothermic retro-chemistry is one of the strategies showing the greatest potential.24–30 Indeed, by introducing dynamic covalent chemistries that are characterized by a highly endothermic dissociation, the exchange activation energy increases, thereby shifting the exchange away from the application conditions. Hence, an appropriate balance between (re)processability and dimensional stability should be aimed for.15,16,31
Recently, our research group introduced β-amino ester chemistry in the context of dynamic networks.32 In this pioneering study, CANs were prepared from a large range of acrylate and amine building blocks via the aza-Michael addition, in which bonds can exchange without adding an external catalyst, i.e. following an associative or/and a dissociative pathway via transesterification in the presence of hydroxyl groups and retro-Michael reactions, respectively. Subsequently, numerous studies focusing on applying this promising dynamic chemistry platform have been conducted very recently in an effort to fasten the exchange process and thus reduce the stress relaxation time, for example by modifying substituents or employing other neighbouring groups.33–38 On the other hand, polymeric materials including β-amino esters contain hydrolysable esters (Fig. 1) and have therefore often been investigated for their degradability.39–42
 | ||
| Fig. 1 While previous research focused on β-amino esters,32,33,35,37 this study has focused on β-amino amides as dynamic moieties in CANs. | ||
It can therefore be expected that the hydrolytic resistance of the corresponding CANs will be limited to specific matrices and conditions. Therefore, we aimed in this study to replace the ester groups with much more stable amides and thus to develop and investigate β-amino amide-based CANs (Fig. 1). Herein, such CANs were hypothesized to possess a substantial higher activation energy resulting in both very high creep resistance and hydrolytic stability while still being (re)processable, in turn bringing them close to real-life applications.
To investigate the reversibility of a β-amino amide moiety, a model compound was first prepared via the aza-Michael addition between readily available N-isopropylacrylamide and N-methyl butylamine with the formation of the desired β-amino amide compound 1 (1H NMR is reported in Fig. S1†). This was dissolved in different NMR tubes in DMSO-d6 and placed in separate oil baths heated at 180 °C, 160 °C, 140 °C and 120 °C. 1H-NMR spectra were collected before and after heating for 16 h in order to determine the thermally activated dissociation of such a dynamic β-amino amide (Fig. 2). Up to temperatures of 140 °C, there was no significant variation compared to the NMR analysis of the pure model compound 1. In contrast, at 160 °C, an 8% enhancement of the proton signals in the area between 6.24 ppm and 5.51 ppm is clearly visible, indicating the formation of the acrylamide double bonds via the retro-aza-Michael reaction. In addition, the decreasing intensity of proton f relative to Michael adduct 1, as well as the appearance of proton d at 3.98 ppm, confirmed the dissociation. Finally, the increase of the double bond integral value at 180 °C (signals a to c) indicates a faster dissociation at higher temperatures. All these observations confirm the reversibility of the aza-Michael reaction on β-amino amides at temperatures beyond 160 °C.
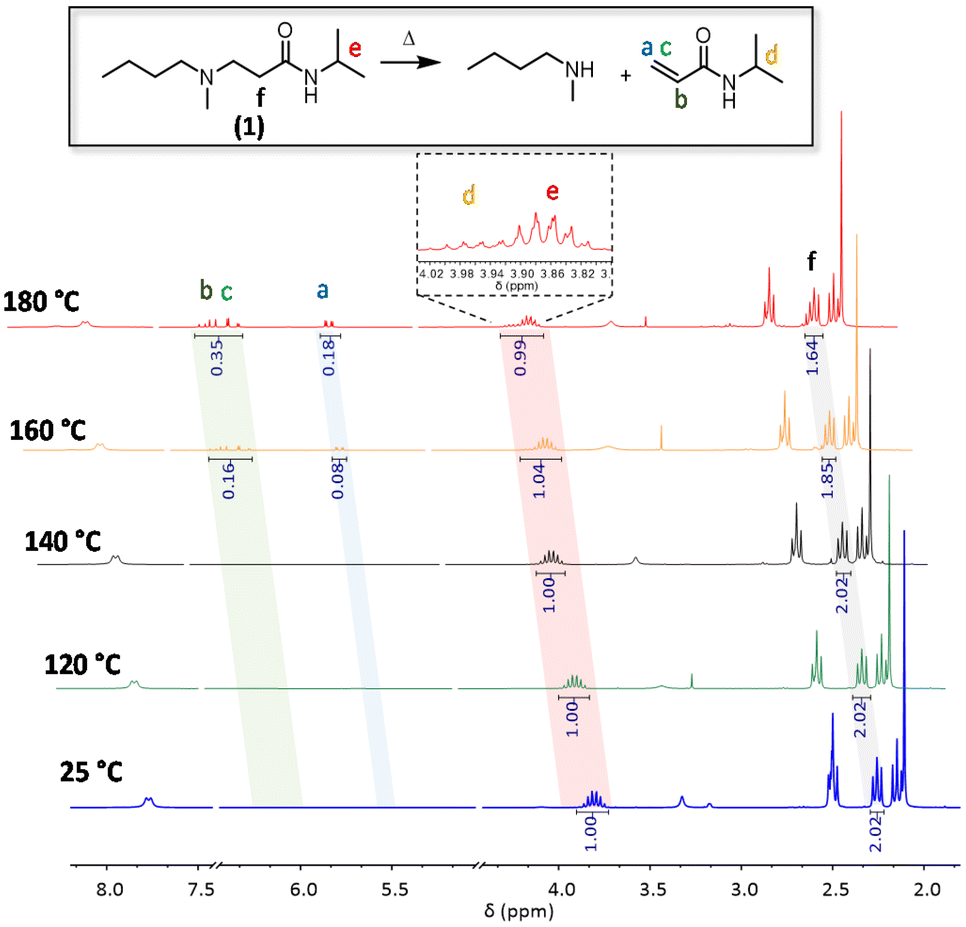 | ||
| Fig. 2 1H NMR stacking spectra of model compound 1 in DMSO-d6 at 25 °C, 120 °C, 140 °C, 160 °C and 180 °C for 16 h. | ||
Generally, when investigating a suitable chemistry for CANs, the scalability of the synthesis pathways should also be considered as a function of the envisaged bulk-scale application areas. Due to the lack of cheap, large-scale available multifunctional acrylamide cross-linkers and the tedious synthesis of acrylamides in general, which typically requires chromatographical purification,43,44 the direct synthesis of β-amino amide networks via aza-Michael addition is not suitable for large-scale synthesis. Therefore, we focused on an earlier reported procedure45 in which crosslinked materials were made in two steps, i.e. the preparation of amino-ester crosslinkers, and then the subsequent amidation reaction with various available amine-containing building blocks (vide infra). Hence, to comprehensively investigate the temperature-dependent nature of the aza-Michael addition and the subsequent amidation reaction, model β-amino-amide compound 2 was prepared by a one-pot reaction between the bulk chemicals methyl acrylate and 2-ethyl hexylamine (Fig. 3a).
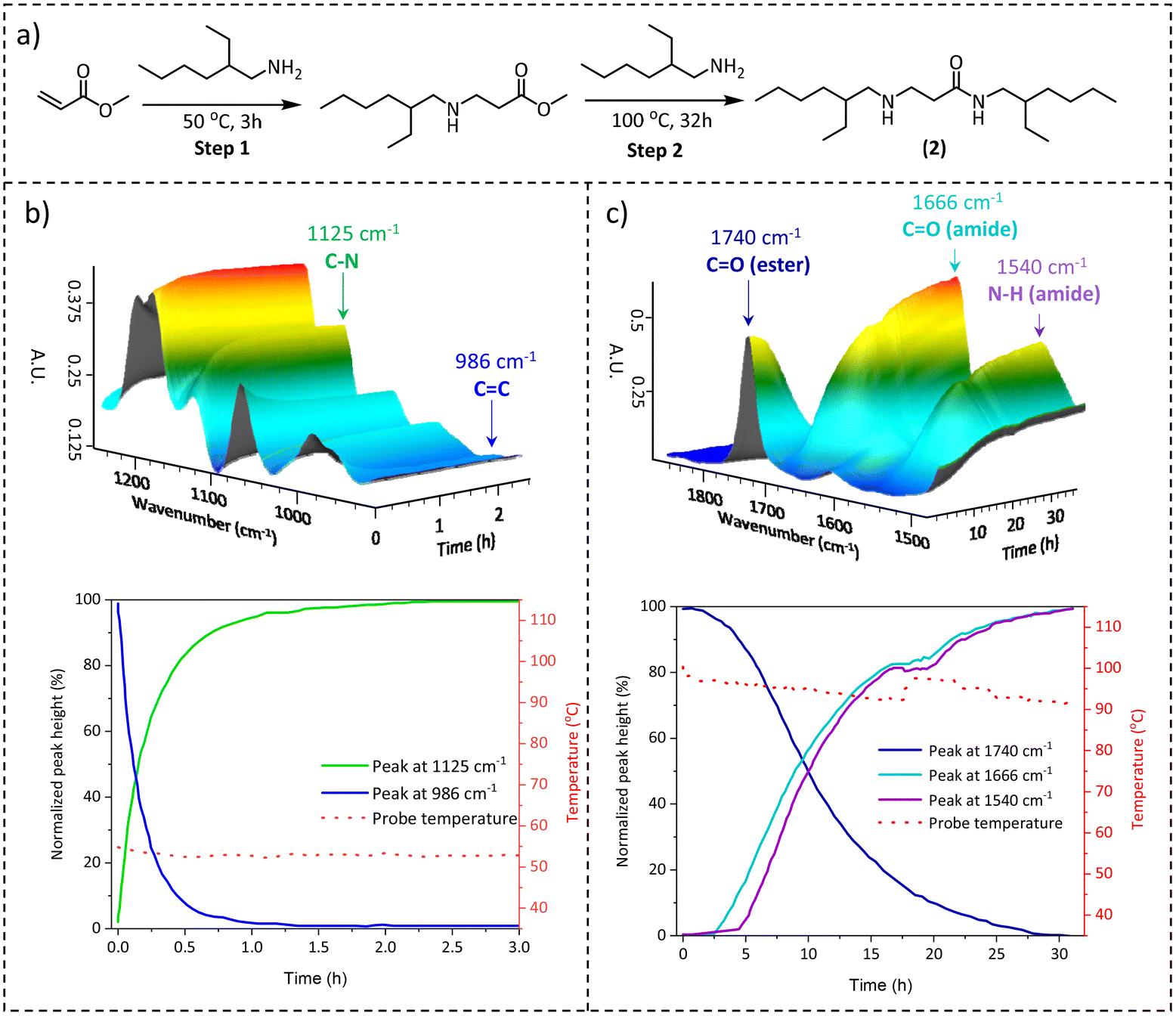 | ||
| Fig. 3 (a) Synthesis of model compound 2, (b) top: three-dimensional plot of time-resolved ATR-FTIR in the region of 1200 cm−1 to 900 cm−1 for the aza-Michael addition step, bottom: plot of the intensity related to the followed peaks versus time. (c) Top: three-dimensional plot of time-resolved ATR-FTIR in the region of 1800 cm−1 to 1300 cm−1 for the amidation reaction, bottom: plot of the intensity related to the followed peak versus time. | ||
The reaction was monitored by performing an online attenuated total reflection Fourier transform infrared (ATR-FTIR) experiment at two temperatures: firstly at 50 °C for the aza-Michael addition and then at 100 °C for the amidation step. In the first step, the progressive disappearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching at 986 cm−1 and the analogous increase of the C–N stretching band at 1125 cm−1 were observed (Fig. 3b top), indicating that the reaction was completed after 2 h (Fig. 3b bottom). The Michael-adduct structure was confirmed by 1H NMR (Fig. S2†), which showed the complete consumption of the methyl acrylate. Next, the amidation step was evaluated using the same technique but increasing the temperature to 100 °C. In this stage, the focus shifted to the stretching band at 1740 cm−1 related to the ester carbonyl moiety, which decreased over time, and to the steady increase of the corresponding carbonyl of the amide bond at 1666 cm−1. At the same time, the bending related to the amide N–H bond at 1540 cm−1 also supported the conversion of esters to amides (Fig. 3c top). The observed FTIR signals reached a plateau after 30 h as depicted in Fig. 3c (bottom), indicating the full conversion of the amino ester to the amino amide functionality. This conclusion was further confirmed by 1H NMR (Fig. S3†). Based on these initial results, as well as those previously reported in the literature,45 we concluded that in the reaction between an acrylate ester and amino derivative at 50 °C, the aza-Michael addition is the only reaction observed, resulting in β-amino esters, which subsequently undergo amidation at 100 °C with the formation of β-amino amides.
C stretching at 986 cm−1 and the analogous increase of the C–N stretching band at 1125 cm−1 were observed (Fig. 3b top), indicating that the reaction was completed after 2 h (Fig. 3b bottom). The Michael-adduct structure was confirmed by 1H NMR (Fig. S2†), which showed the complete consumption of the methyl acrylate. Next, the amidation step was evaluated using the same technique but increasing the temperature to 100 °C. In this stage, the focus shifted to the stretching band at 1740 cm−1 related to the ester carbonyl moiety, which decreased over time, and to the steady increase of the corresponding carbonyl of the amide bond at 1666 cm−1. At the same time, the bending related to the amide N–H bond at 1540 cm−1 also supported the conversion of esters to amides (Fig. 3c top). The observed FTIR signals reached a plateau after 30 h as depicted in Fig. 3c (bottom), indicating the full conversion of the amino ester to the amino amide functionality. This conclusion was further confirmed by 1H NMR (Fig. S3†). Based on these initial results, as well as those previously reported in the literature,45 we concluded that in the reaction between an acrylate ester and amino derivative at 50 °C, the aza-Michael addition is the only reaction observed, resulting in β-amino esters, which subsequently undergo amidation at 100 °C with the formation of β-amino amides.
From the viewpoint of scalability, several covalent adaptable β-amino amide networks (BAA) were prepared by reacting a tetrafunctional cross-linker with β-amino ester moieties, obtained through a straightforward aza-Michael addition (Fig. S4†) and subsequently crosslinked with a series of readily available amino-containing building blocks (Fig. 4a).
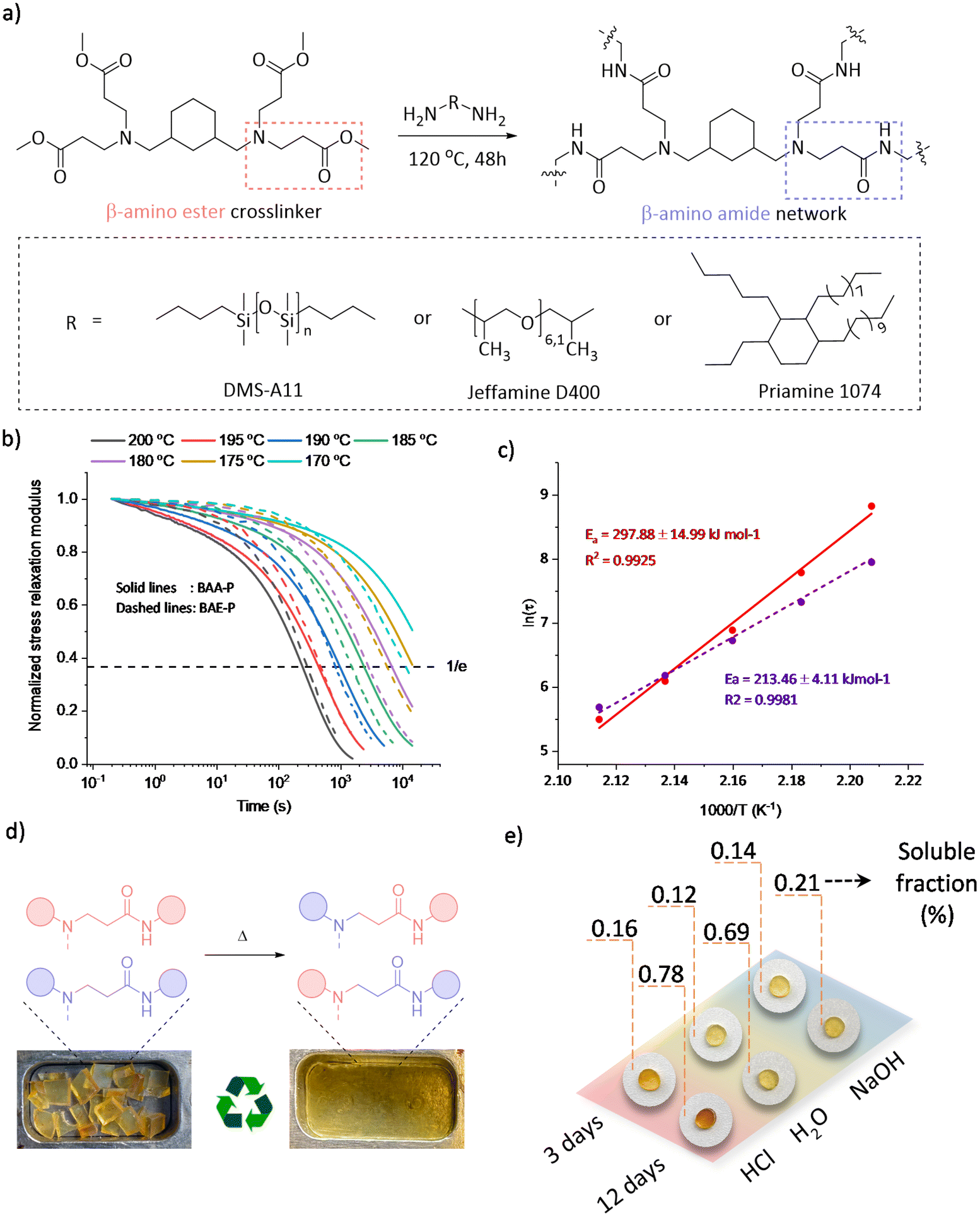 | ||
| Fig. 4 (a) Preparation of β-amino amide containing CANs from a tetrafunctional crosslinker and Priamine 1074 (BAA-P), Jeffamine D400 (BAA-J) or DMS-A11 (BAA-S); (b) normalized stress relaxation curves and (c) corresponding Arrhenius plots of BAA-P (red) and BAE-P (violet) networks; (d) demonstration of (re)processing of BAA-P; (e) appearances and the corresponding soluble fractions of BAA-P samples after the hydrolysis tests under different conditions (1 M HCl, deionized water, 1 M NaOH). | ||
The cross-linking process was followed by ATR-FTIR, in which the disappearance of absorption bands of the ester-carbonyl groups (at 1720 cm−1) was simultaneously observed with the increase of the amide-carbonyl absorbance band at 1665 cm−1, indicating the amidation of the ester to form the targeted amide bonds (Fig. S5†). The overall properties of the obtained materials have been summarized in Table 1. Although BAA-J and BAA-S showed soluble fractions above 10%, associated with a very high swelling ratio (>580%), BAA-P showed a low soluble fraction (6.3%) with a swelling ratio of 350% in THF. Moreover, a wide range of glass transition temperatures (Tg) from −107 to −15 °C were obtained when using different amine building blocks, which affect both the backbone mobility and cross-link density. In addition, thermogravimetric analysis (TGA) showed a high degradation onset temperature (Td5%) ranging between 298 and 325 °C with no significant mass loss (<3.5%) after a period of 2 h at 200 °C under an air atmosphere, revealing sufficient stability for the thermal (re-)processing (vide infra).
| Network | Amine | Swelling ratioa (%) | Soluble fractiona (%) |
T
gDSC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (°C) b (°C) |
T
d-5%c (°C) |
m
iso 200-2 hd (%) |
G′e (MPa) |
|---|---|---|---|---|---|---|---|
| a Swelling ratio and soluble fraction obtained from a four-sample measurement in THF at RT for 24 h. b Determined during the second heating run by DSC with a heating and cooling rate of 10 K min−1. c Determined as an onset temperature for 5% mass loss observed by TGA using a heating rate of 10 K min−1. d Determined mass loss observed with the TGA isothermal mode at 200 °C for 2 h. e Apparent plateau storage modulus determined from frequency sweep measurements at 100 °C and 1 rad s−1. | |||||||
| BAA-P | Priamine | 350 ± 23 | 6.3 ± 0.7 | −15 | 325 | 3.0 | 2.29 |
| BAA-J | Jeffamine | 590 ± 26 | 17.3 ± 3.2 | −25 | 298 | 3.5 | 0.43 |
| BAA-S | PDMS A11 | 582 ± 17 | 13.8 ± 1.5 | −107 | 320 | 3.0 | 0.33 |
| BAA-P R3 | Priamine | 292 ± 19 | 3.3 ± 1.1 | −7 | 326 | — | — |
| BAE-P (Ref) | — | 340 ± 16 | 4.8 ± 2.2 | −43 | 336 | 1.5 | — |
The dynamicity of the selected BAA-P was further examined by rheology with stress relaxation experiments in a temperature range between 200 and 170 °C. A drop in the material's shear storage modulus (G′) upon increasing the temperature was clearly observed in non-normalized stress relaxation curves (Fig. S6,† top), which revealed the decrease in the crosslinking density. Indeed, a frequency sweep experiment confirmed the partial de-cross-linking by showing a drop in the G′ value at higher temperatures, which is another indication of a dissociative exchange of the dynamic amino-amide via reversible (retro) aza-Michael reaction (Fig. S7†). In addition, the elastic plateau modulus in frequency sweep experiments remained almost unchanged at temperatures below 150 °C, yet significantly dropped beyond 160 °C revealing the contribution of the dynamic Michael adduct dissociation beyond that temperature. In Fig. 4b, the normalized stress relaxation curves of BAA-P (solid line) are reported in comparison with the corresponding β-amino ester network BAE-P (dashed line, see the ESI†). A high-temperature dependent dynamic behaviour was observed in which the applied stress could be relaxed completely at elevated temperatures, favouring the (re)processability with relaxation times of around 4 min at 200 °C. In addition, an Arrhenius plot extracted from the characteristic stress relaxation time (τ) was used to derive a high activation energy of 298 kJ mol−1, compared to the value of 213 kJ mol−1 found for BAE-P (Fig. 4c).
Although TGA revealed a degradation onset temperature (Td5%) of 325 °C, time sweep experiments at different temperatures showed a slight increase in the storage modulus at temperatures above 200 °C (220–260 °C), which could be ascribed to the introduction of permanent cross-links as a result of homopolymerisation of the acrylamide groups created at the dissociation stage (Fig. S8†). For this reason, the (re)processing experiments have all been conducted at 180 °C. To demonstrate the (re)processability, BAA-P was cut into small pieces and pressed multiple times by compression moulding at 180 °C under 3 tons for 60 min.
The reprocessing ability of materials with dynamic amino-amide linkages was demonstrated by executing 3 cycles without observing significant changes in the appearance or thermal properties (Fig. 4d and Table 1-BAA-P R3). Although the stress relaxation of the third recycled sample slightly slowed down and thus a higher activation energy was obtained (Fig. S6,† bottom), the chemical structure observed by FTIR remained unchanged during the recycling processes (Fig. S9†). In addition, tensile test results did not reveal significant changes in mechanical properties after 3 cycles of remoulding (Fig. S10†).
Finally, in order to investigate the dimensional stability, a creep recovery experiment was carried out on BAA-P by applying 2 kPa shear stress for 5000 s, followed by a recovery period of 3600 s at temperatures ranging from 50 to 110 °C (Fig. S11†). Despite the low Tg of BAA-P (−15 °C), high creep resistance was observed and the creep rates were below 10−5% s−1 up to 100 °C with remaining strains being less than 0.05% after one hour of recovery. Also, as mentioned earlier, the use of amides instead of esters was expected to improve the chemical stability of the obtained materials. Therefore, hydrolysis tests of BAA-P were carried out for up to 12 days under 4 different conditions: in deionized water, 1 M HCl, and 1 M NaOH at room temperature and in deionized water at 100 °C. All the results presented in Table S1† indicate soluble fractions of less than 1% while the network maintained its structural integrity (Fig. 4e and Fig. S12†). In comparison with the β-amino ester network (Table 1-BAE-P), an overall high hydrolytic resistance was observed for the β-amino amide network. Indeed, BAA-P remained almost unchanged after two days in boiling water whereas BAE-P lost its structural appearance under comparable conditions (Fig. S13†). These results indicate the potential dimensional resistance of such covalent adaptable amino-amide networks under harsh conditions (e.g., marine applications).
Conclusions
In this study, the temperature-dependent reaction between acrylates and amines was first investigated with model studies, in which the aza-Michael addition was exclusively observed at 50 °C, whereas the subsequent amidation (converting esters into amides) efficiently proceeded at 100 °C. Additionally, the dissociation of the resulting dynamic β-amino amide bonds was found to occur at temperatures beyond 160 °C. By exploiting this knowledge, a scalable synthesis of covalent adaptable amino-amide networks using a multifunctional amino-ester cross-linker and various bifunctional amine-containing building blocks was performed. The obtained amino-amide networks showed high thermal stability while rheological measurements revealed a temperature-dependent stress relaxation with a high activation energy of around 300 kJ mol−1, as well as the ability of the networks to be (re)processed multiple times without significant changes in properties. Moreover, the presented materials showed excellent dimensional stability with less than 0.05% of the remaining strain as observed in creep measurements at 100 °C. Finally, in comparison with the corresponding β-amino ester-based material, CANs also presented exceptional hydrolytic stability under acidic, basic and neutral conditions for at least 12 days.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Bernhard De Meyer for technical support, Prof. Alessandro Pedrini, Prof. Enrico Dalcanale, Dr Nezha Badi, Dr Matthieu Soete, Stephan Maes and Niccolò Braidi for fruitful discussions. This project received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and the innovation programme 101021081 (ERC-AdG-2020, CiMaC-project).References
- J.-P. Lange, ACS Sustainable Chem. Eng., 2021, 9, 15722–15738 CrossRef CAS.
- J. Nikiema and Z. Asiedu, Environ. Sci. Pollut. Res., 2022, 29, 24547–24573 CrossRef PubMed.
- S. Engelen, A. A. Wróblewska, K. De Bruycker, R. Aksakal, V. Ladmiral, S. Caillol and F. E. Du Prez, Polym. Chem., 2022, 13, 2665–2673 RSC.
- J. Seay and M. E. Ternes, Clean Technol. Environ. Policy, 2022, 24, 731–738 CrossRef.
- H. Chen, R. Qin, C. L. Chow and D. Lau, Cem. Concr. Compos., 2023, 137, 104922 CrossRef CAS.
- Z. Liu, Z. Fang, N. Zheng, K. Yang, Z. Sun, S. Li, W. Li, J. Wu and T. Xie, Nat. Chem., 2023, 1–7 Search PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- S. Maes, F. Van Lijsebetten, J. M. Winne and F. E. Du Prez, Macromolecules, 2023, 56, 1934–1944 CrossRef CAS.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- F. Van Lijsebetten, J. O. Holloway, J. M. Winne and F. E. Du Prez, Chem. Soc. Rev., 2020, 49, 8425–8438 RSC.
- F. Van Lijsebetten, K. De Bruycker, E. Van Ruymbeke, J. M. Winne and F. E. Du Prez, Chem. Sci., 2022, 13, 12865–12875 RSC.
- V. Zhang, B. Kang, J. V. Accardo and J. A. Kalow, J. Am. Chem. Soc., 2022, 144, 22358–22377 CrossRef CAS PubMed.
- F. Van Lijsebetten, T. Debsharma, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202210405 CrossRef CAS PubMed.
- Y. Liu, Z. Tang, D. Wang, S. Wu and B. Guo, J. Mater. Chem. A, 2019, 7, 26867–26876 RSC.
- Y. You, M. Fu, M. Rong and M. Zhang, Mater. Today Chem., 2022, 23, 100687 CrossRef CAS.
- Y. Ma, H.-Q. Wang, P.-C. Zhao, F.-Z. Wang and C.-H. Li, J. Mater. Chem. A, 2022, 10, 20804–20812 RSC.
- M. A. B. Rusayyis, L. M. Fenimore, N. S. Purwanto and J. M. Torkelson, Polym. Chem., 2023, 14, 3519–3534 RSC.
- C. Cui, X. Chen, L. Ma, Q. Zhong, Z. Li, A. Mariappan, Q. Zhang, Y. Cheng, G. He and X. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 47975–47983 CrossRef CAS PubMed.
- S. K. Schoustra and M. M. Smulders, Macromol. Rapid Commun., 2023, 44, 2200790 CrossRef CAS PubMed.
- L. Zhang, Z. Liu, L. Sun, L. Xiao, Q. Guan and Z. You, Macromolecules, 2021, 54, 4081–4088 CrossRef CAS.
- S. Wang, N. Wang, D. Kai, B. Li, J. Wu, J. C. C. Yeo, X. Xu, J. Zhu, X. J. Loh and N. Hadjichristidis, Nat. Commun., 2023, 14, 1182 CrossRef CAS PubMed.
- F. Van Lijsebetten, K. De Bruycker, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202113872 CrossRef CAS PubMed.
- F. Van Lijsebetten, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 15834–15844 CrossRef CAS PubMed.
- J. Park, H. Y. Song, S. Choi, S.-k. Ahn, K. Hyun and C. B. Kim, J. Mater. Chem. A, 2022, 10, 6475–6480 RSC.
- B. T. Worrell, M. K. McBride, G. B. Lyon, L. M. Cox, C. Wang, S. Mavila, C.-H. Lim, H. M. Coley, C. B. Musgrave and Y. Ding, Nat. Commun., 2018, 9, 2804 CrossRef PubMed.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- F. Van Lijsebetten, K. De Bruycker, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2022, 11, 919–924 CrossRef CAS PubMed.
- S. Engelen, F. Van Lijsebetten, R. Aksakal, J. M. Winne and F. E. Du Prez, Macromolecules, 2023, 56, 7055–7064 CrossRef CAS.
- M. A. B. Rusayyis and J. M. Torkelson, Polym. Chem., 2021, 12, 2760–2771 RSC.
- C. Taplan, M. Guerre and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 9140–9150 CrossRef CAS PubMed.
- G. Lee, H. Y. Song, S. Choi, C. B. Kim, K. Hyun and S.-k. Ahn, Macromolecules, 2022, 55, 10366–10376 CrossRef CAS.
- G. Li, J. Huang, H. S. Soo, Y. Zhao, T. Li, Y. Wang, S. Wang and W. Dong, Eur. Polym. J., 2023, 194, 112165 CrossRef CAS.
- L. Stricker, C. Taplan and F. E. Du Prez, ACS Sustainable Chem. Eng., 2022, 10, 14045–14052 CrossRef CAS.
- D. Jeon, Y. Yoon, D. Kim, G. Lee, S.-k. Ahn, D. Choi and C. B. Kim, Macromolecules, 2023, 56, 697–706 CrossRef CAS.
- D. Berne, G. Coste, R. Morales-Cerrada, M. Boursier, J. Pinaud, V. Ladmiral and S. Caillol, Polym. Chem., 2022, 13, 3806–3814 RSC.
- D. Berne, B. Quienne, S. Caillol, E. Leclerc and V. Ladmiral, J. Mater. Chem. A, 2022, 10, 25085–25097 RSC.
- Y. Al Thaher, S. Latanza, S. Perni and P. Prokopovich, J. Colloid Interface Sci., 2018, 526, 35–42 CrossRef CAS PubMed.
- A. Muralidharan, R. R. McLeod and S. J. Bryant, Adv. Funct. Mater., 2022, 32, 2106509 CrossRef CAS PubMed.
- X. Liu, Z. Zhao, F. Wu, Y. Chen and L. Yin, Adv. Mater., 2022, 34, 2108116 CrossRef CAS PubMed.
- B. G. De Geest, W. Van Camp, F. E. Du Prez, S. C. De Smedt, J. Demeester and W. E. Hennink, Macromol. Rapid Commun., 2008, 29, 1111–1118 CrossRef CAS.
- S. Van Herck, L. Van Hoecke, B. Louage, L. Lybaert, R. De Coen, S. Kasmi, A. P. Esser-Kahn, S. A. David, L. Nuhn and B. Schepens, Bioconjugate Chem., 2017, 29, 748–760 CrossRef PubMed.
- A. Huppertsberg, L. Kaps, Z. Zhong, S. Schmitt, J. Stickdorn, K. Deswarte, F. Combes, C. Czysch, J. De Vrieze and S. Kasmi, J. Am. Chem. Soc., 2021, 143, 9872–9883 CrossRef CAS PubMed.
- C. Yi, J. Zhao, Z. Zhang and J. Zhang, Ind. Eng. Chem. Res., 2017, 56, 13743–13750 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01175e |
| This journal is © The Royal Society of Chemistry 2024 |