
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic-assisted formulation of cell membrane-camouflaged anisotropic nanostructures†
Kenry
 abc
abc
aDepartment of Pharmacology and Toxicology, R. Ken Coit College of Pharmacy, University of Arizona, Tucson, AZ 85721, USA. E-mail: kenry@arizona.edu
bUniversity of Arizona Cancer Center, University of Arizona, Tucson, AZ 85721, USA
cBIO5 Institute, University of Arizona, Tucson, AZ 85721, USA
First published on 18th March 2024
Abstract
Anisotropic gold (Au) nanostructures have been widely explored for various nanomedicine applications. While these nanomaterials have shown great promise for disease theranostics, particularly for cancer diagnosis and treatment, the utilization and clinical translation of anisotropic Au nanostructures have been limited by their high phagocytic uptake and clearance and low cancer targeting specificity. Numerous efforts have thus been made toward mitigating these challenges. Many conventional strategies, however, rely on all-synthetic materials, involve complex chemical processes, or have low product throughput and reproducibility. Herein, by integrating cell membrane coating and microfluidic technologies, a high-throughput bioinspired approach for synthesizing biomimetic anisotropic Au nanostructures with minimized phagocytic uptake and improved cancer cell targeting is reported. Through continuous hydrodynamic flow focusing, mixing, and sonication, Au nanostructures are encapsulated within the macrophage and cancer cell membrane vesicles effectively. The fabricated nanostructures are uniform and highly stable in serum. Importantly, the macrophage membrane vesicle-encapsulated Au nanostructures can be preferentially internalized by breast cancer cells, but not by macrophages. Overall, this study has demonstrated the feasibility of employing an integrated microfluidic-sonication technique to formulate uniform and highly stable biomimetic anisotropic nanostructures for enhanced cancer theranostic applications.
1. Introduction
Nanomaterials of various compositions and shapes have been actively developed over the years for different theranostic and nanomedicine applications.1–12 Among these, anisotropic gold (Au) nanostructures, such as Au nanocages, nanorods, and nanostars, stand out because of their distinct physicochemical properties which may be leveraged for biomedical imaging, biological sensing, and disease phototherapies.13–17 While anisotropic Au nanostructures have demonstrated tremendous potential for biomedical applications, the utilization and clinical translation of these nanomaterials are still hampered by numerous obstacles.18,19 One of the most common challenges associated with the nanomedicine applications of synthetic nanomaterials like Au nanostructures is their relatively low theranostic efficacy, which is predominantly influenced by their poor delivery and targeting efficacy. To date, the delivery and targeting of theranostic nanomaterials to disease sites, particularly cancer sites, have been realized through either passive or active strategies or a combination of both.20,21 Passive targeting leverages an enhanced permeation and retention effect of nanomaterials, in which the nanomaterials permeate through the leaky tumor vasculature to accumulate passively at the tumor sites.22,23 Active targeting, in contrast, relies on the surface modification of nanomaterials with certain targeting moieties (e.g., antibodies, aptamers, and peptides) to improve targeting specificity to disease sites.24,25 Unfortunately, a previous work reported that regardless of the targeting modes, both strategies still have a relatively poor targeting efficacy, which is less than 5%.26While many factors contribute to the low delivery and targeting efficacy of nanomaterials,27–29 one of the most significant factors is the recognition of exogenous nanomaterials by the immune system and the uptake and clearance of these nanomaterials from blood circulation by the mononuclear phagocyte system.30,31 Numerous approaches have been explored to overcome this challenge. These include the formulation of nanomaterials with specific shapes or surface charges, surface modification of nanomaterials with a hydrophilic polyethylene glycol (PEG) layer, or a combination of both.32–35 Nevertheless, increasing studies have reported that PEGylation of nanomaterials induces an accelerated blood clearance phenomenon, where repeated systemic administrations of PEGylated nanomaterials cause their rapid clearance from the circulatory system.36,37 To address this issue, bioinspired and biomimetic strategies, notably coating of nanomaterials with natural cellular components, such as membranes from erythrocytes, leukocytes, platelets, macrophages, and cancer cells, have been increasingly explored as alternatives.38–40 The cell membrane coating technique has been shown to endow the modified nanomaterials with biological properties and functionalities similar to those of the source cells from which the membranes are derived, such as immune evasion capability, prolonged in vivo residence time, and enhanced homologous tumor targeting.41–46 For instance, nanomaterials camouflaged with membranes derived from erythrocytes have been reported to display improved immune evasion characteristics.42–44 Separately, polymeric nanoparticles coated with cancer cell membranes have been demonstrated to exhibit much higher cancer targeting specificity than their bare uncoated counterparts.45,47,48 Consequently, the cell membrane coating technique has emerged as an attractive option to generate biomimetic nanostructures with potentially reduced phagocytic uptake and enhanced tumor targeting ability.
The formulation of cell membrane-coated nanostructures typically involves the synthesis of nanostructures, preparation of cell membrane vesicles, and coating of the membrane vesicles on nanostructures, which until now, has been largely achieved through two approaches, i.e., sequential mechanical extrusion and ultrasonication.38,39 While these approaches can assemble membrane vesicles around nanomaterials to form core–shell nanostructures, the methods suffer from several drawbacks, notably laborious and time-consuming formulation processes, low product throughput, and substantial batch-to-batch variations. Furthermore, most of the studies that employed these methods have focused on isotropic nanomaterials. Less attention has been placed on the coating of cell membranes on anisotropic nanostructures. As such, active efforts have been geared toward developing more effective methods capable of formulating highly uniform cell-mimicking nanomaterials in high yield. One such strategy that has gained traction recently is the microfluidic-based method.49,50 As opposed to other nanomaterial formulation strategies, the microfluidic-based method offers numerous advantages, including a highly controlled and automated process, precise manipulation of the sample volume and mixing rate, shorter formulation time, higher throughput, ease of scale-up, and improved product quality, consistency, and reproducibility.51–53 Specifically for the formulation of cell membrane-coated nanomaterials, recent studies have demonstrated that, in contrast to the typical batch synthesis strategies, for comparable coating quality and product size, the microfluidic-facilitated approaches can significantly accelerate the formulation process from hours down to milliseconds to seconds.49,50 Moreover, combining the hydrodynamic forces within the microfluidic channels with external force fields may enhance the applicability of microfluidic platforms to assemble a wider variety of cell membrane-coated nanostructures. As shown in previous reports,50,54,55 a higher microscale mixing efficiency can be achieved by integrating microhydrodynamic mixing with external forces like sonication. This, in turn, may be beneficial for realizing a more uniform and reproducible cell membrane coating on nanomaterials.
Motivated by this, herein, a one-stage continuous flow microfluidic platform was developed to formulate highly stable biomimetic cell membrane vesicle-coated Au nanostructures. Microfluidic hydrodynamic flow focusing was combined with sonication to realize uniform coating of membrane vesicles derived from macrophages and cancer cells onto anisotropic Au nanostructures. These biomimetic nanostructures were highly stable and experienced minimal aggregation in serum. In particular, the macrophage membrane vesicle-encapsulated Au (MMV-Au) nanostructures were minimally taken up by macrophages but preferentially internalized by breast cancer cells. Overall, this work demonstrates the feasibility of leveraging microfluidics to realize high-throughput and uniform synthesis of cell membrane-camouflaged anisotropic nanostructures for a range of disease theranostic and nanomedicine applications.
2. Results and discussion
Microfluidic-mediated assembly and characterization of cell membrane-camouflaged Au nanostructures
The one-stage continuous flow microfluidic platform used for the formulation of cell membrane vesicle-encapsulated anisotropic nanostructures was designed using a computer-aided design software (Fig. 1). Architecturally, the microfluidic device consists of three inlet channels, a serpentine-shaped mixing region, and an outlet channel. The nanomaterial cores would be introduced into the middle inlet channel, while the cell membrane vesicles would be introduced into the two side inlet channels. These two distinct components would interact in the mixing region and undergo hydrodynamic focusing and mixing under a continuous flow condition. Sonication would be employed throughout the synthesis process to enhance the mixing efficiency and eventually, the membrane vesicle-encapsulated Au nanostructures would be collected from the outlet channel.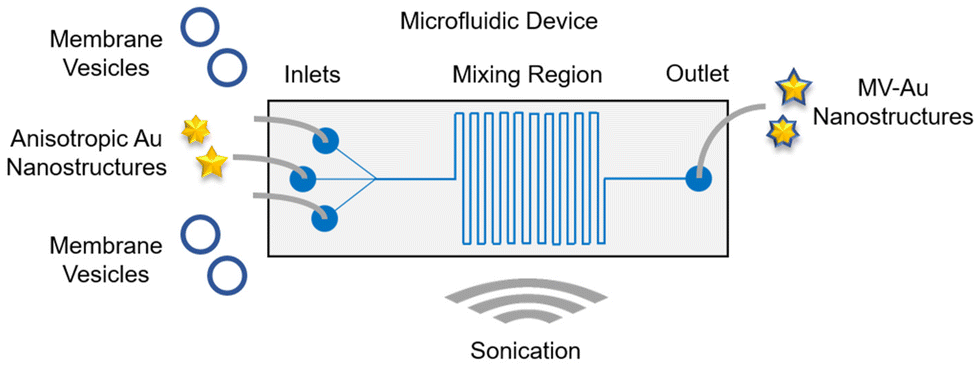 | ||
| Fig. 1 Schematic of the microfluidic device used for the synthesis of the cell membrane vesicle-encapsulated anisotropic Au nanostructures. The device consists of three inlet ports and an outlet port connected by a serpentine-shaped mixing region. It was immersed in a bath sonicator throughout the nanostructure synthesis process. | ||
After designing the microfluidic device, it was then fabricated in polydimethylsiloxane (PDMS) through a soft lithography process and bonded to a glass slide (Fig. 2a). Next, two anisotropic Au nanostructures with different tip-to-core ratios were synthesized and characterized. Au nanostructures with a lower tip-to-core ratio, termed here as Au nanonuggets (AuNNs), were synthesized using an ascorbic acid-mediated process. On the other hand, a 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer-facilitated process was employed to prepare Au nanostructures with a higher tip-to-core ratio, termed here as Au nanostars (AuNSs). Characterization of the morphology of AuNNs and AuNSs using transmission electron microscopy (TEM) showed that Au nanostructures with varying tip-to-core ratios were successfully prepared (Fig. 2b, bare row). Both Au nanostructures possessed an average hydrodynamic size of around 70 nm (Fig. 2c) and a negative zeta potential from −40 to −45 mV (Fig. 2d). Besides, AuNNs exhibited an absorbance peak at around 585 nm, while that of AuNSs centered at roughly 765 nm (Fig. S1†).
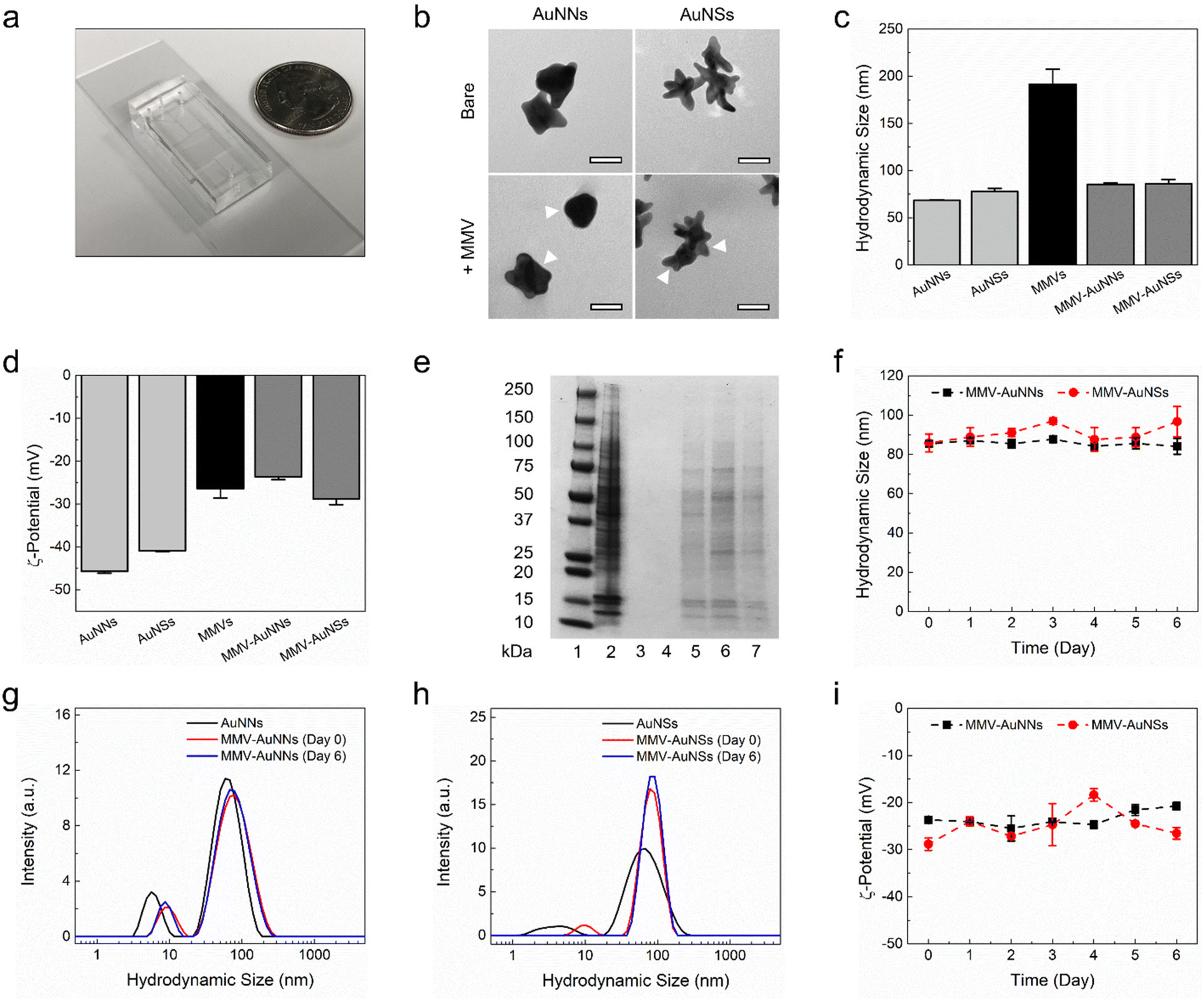 | ||
| Fig. 2 Physicochemical characterization of the macrophage membrane vesicle-encapsulated Au nanostructures. (a) Photograph of the fabricated microfluidic device. (b) Surface morphology, (c) hydrodynamic size, (d) zeta potential, and (e) protein expression of the bare and macrophage membrane vesicle-encapsulated Au (MMV-Au) nanostructures. Scale bars are 40 nm. The white arrows indicate the presence of macrophage membrane coating. Lanes 1–7 of the polyacrylamide gel represent the protein marker (lane 1), cell lysate (lane 2), AuNNs (lane 3), AuNSs (lane 4), MMVs (lane 5), MMV-AuNNs (lane 6), and MMV-AuNSs (lane 7). (f–i) Stability of the MMV-Au nanostructures based on the variations of: (f) hydrodynamic size, (g and h) size distribution, and (i) zeta potential over time. n = 3, mean ± standard deviation. | ||
Following the synthesis and characterization of the bare anisotropic Au nanostructures, MMVs were prepared for the microfluidic-assisted formulation of membrane vesicle-coated AuNNs and AuNSs, i.e., MMV-AuNNs and MMV-AuNSs, respectively. The membrane vesicles from macrophages were specifically selected in this study due to their outstanding reticuloendothelial system evasion and tumor homing capability. Characterization of MMVs revealed that they had an average hydrodynamic size of about 200 nm and a zeta potential of roughly −26 mV (Fig. 2c and d). These membrane vesicles were next introduced into the two side inlet channels of the microfluidic device at a flow rate of 14 mL h−1, while either AuNNs or AuNSs were simultaneously injected into the middle inlet channel at a flow rate of 7 mL h−1. After going through hydrodynamic focusing and mixing under a continuous flow condition in the presence of sonication, MMV-AuNNs or MMV-AuNSs were eventually formed and collected from the outlet channel. TEM evaluation of the surface morphology of the resulting biomimetic Au nanostructures showed the existence of thin membrane layers wrapping AuNNs and AuNSs (as indicated by the white arrows), essentially forming core–shell nanostructures (Fig. 2b, +MMV row). These nanostructures possessed an average hydrodynamic size of approximately 85 nm (Fig. 2c) and a zeta potential between −25 and −30 mV (Fig. 2d), which was close to that of MMVs. Characterization of the membrane protein expression of both MMV-AuNNs and MMV-AuNSs revealed that most of the membrane proteins of MMVs were still retained on the biomimetic nanostructures (Fig. 2e). Finally, evaluations of the evolution of hydrodynamic size (Fig. 2f), size distribution (Fig. 2g and h), and zeta potential (Fig. 2i) over time confirmed that both MMV-AuNNs and MMV-AuNSs were highly stable over at least six days, where minimal variations were observed.
To further evaluate the performance of the developed microfluidic platform, the standard sequential physical extrusion method was employed to coat the anisotropic Au nanostructures with MMVs, which were then compared against those obtained using the microfluidic-sonication process (Fig. S2–S4†). The formation of core–shell nanostructures could be clearly seen based on TEM imaging (Fig. S2†). The average hydrodynamic size of the extrusion-obtained MMV-AuNNs and MMV-AuNSs was about 105 nm (Fig. S3†), while their average zeta potential was approximately −32 mV, which was close to that of the MMVs used in the extrusion process (Fig. S4†). This suggests that the biomimetic nanostructures acquired through the microfluidic-sonication formulation process and the sequential mechanical extrusion process had comparable size and surface charge. Nevertheless, it is noteworthy that the microfluidic-assisted formulation method has a much higher product throughput and is more automated, controlled, and less laborious than the physical extrusion approach.
Next, the feasibility of utilizing the continuous flow microfluidic device to coat cancer cell-derived membrane vesicles (CCMVs) on AuNNs and AuNSs was assessed (Fig. S5–S11†). The CCMV-Au nanostructures were synthesized and characterized similarly to the MMV-Au nanostructures. Both CCMV-AuNNs and CCMV-AuNSs had core–shell structures (Fig. S5†), an average hydrodynamic size of between 100 and 110 nm (Fig. S6†), a negative zeta potential of around −25 mV (Fig. S7†), and membrane protein expressions similar to that of CCMVs (Fig. S8†). The CCMV-Au nanostructures also displayed excellent stability in terms of the hydrodynamic size (Fig. S9†), size distribution (Fig. S10†), and zeta potential (Fig. S11†) over at least six days. As a comparison, sequential physical extrusion was employed to coat CCMVs onto AuNNs and AuNSs and their properties were found to be similar to those of the microfluidic-acquired CCMV Au nanostructures in terms of the surface morphology, (Fig. S12†), hydrodynamic size (Fig. S13†), and zeta potential (Fig. S14†).
Altogether, the obtained experimental data have demonstrated the feasibility of employing the developed microfluidic-sonication platform to process and coat different cell membrane vesicles onto anisotropic nanomaterials for the formulation of highly stable biomimetic core–shell nanostructures.
Interactions of biomimetic Au nanostructures with serum
Most of the theranostic and nanomedicine applications of nanomaterials require their introduction into the body, which will expose them to a plethora of biomolecules in the physiological system. The nanomaterials may then interact almost immediately with biomolecules, leading to non-specific biomolecule adsorption on the nanomaterial surfaces.56–58 Consequently, the integrity, properties, and functionalities of the nanomaterials may be altered. In fact, many studies have shown that biomolecule adsorption and protein corona formation around nanomaterials may influence their uptake by mammalian cells.59,60 As such, it is essential to elucidate the interactions between the biomolecules in serum and the Au nanostructures and how these interactions will affect their cellular uptake, particularly by immune and cancer cells.Motivated by this, after formulating MMV-AuNNs and characterizing their physicochemical properties, their interactions with the biomolecules in serum were examined. Specifically, the recruitment of non-specific biomolecules in cell culture medium by MMV-AuNNs and how this would affect their stability in terms of the hydrodynamic size and zeta potential were assessed. The surface morphology of the bare and MMV-encapsulated AuNNs in the absence and presence of serum was first qualitatively characterized through TEM imaging (Fig. 3a). It is evident that the bare AuNNs aggregated substantially in serum, which led to a significant increase in their size. Conversely, although MMV-AuNNs also aggregated in the presence of serum, this aggregation appeared less substantial, as evidenced from the morphology of MMV-AuNNs in serum. Quantitative analysis of the hydrodynamic size of AuNNs and MMV-AuNNs over time confirmed the increased size of both nanostructures in serum (Fig. 3b). It is important to note that while the size of these nanostructures increased in serum, the extent of size change was different (Fig. 3d). In particular, the percentage change of the hydrodynamic size of AuNNs was consistently higher than that of MMV-AuNNs over the observed period of 6 h. The difference in size variation was the largest immediately after the introduction of nanostructures into serum, where AuNNs experienced close to 50% size change while MMV-AuNNs had less than 10% size change. At 6 h, the percentage change of the size of AuNNs was almost twice that of MMV-AuNNs. The zeta potential of AuNNs also varied significantly in the presence of serum throughout the observation period (Fig. 3c). In fact, the average zeta potential of the bare AuNNs increased from about −41 mV to −24 mV in serum. Intriguingly, certain level of zeta potential variation was also observed from MMV-AuNNs Specifically, AuNNs in serum experienced approximately 40% change in their zeta potential, while the percentage change in the zeta potential of MMV-AuNNs under the same condition was consistently less than 10% (Fig. 3e).
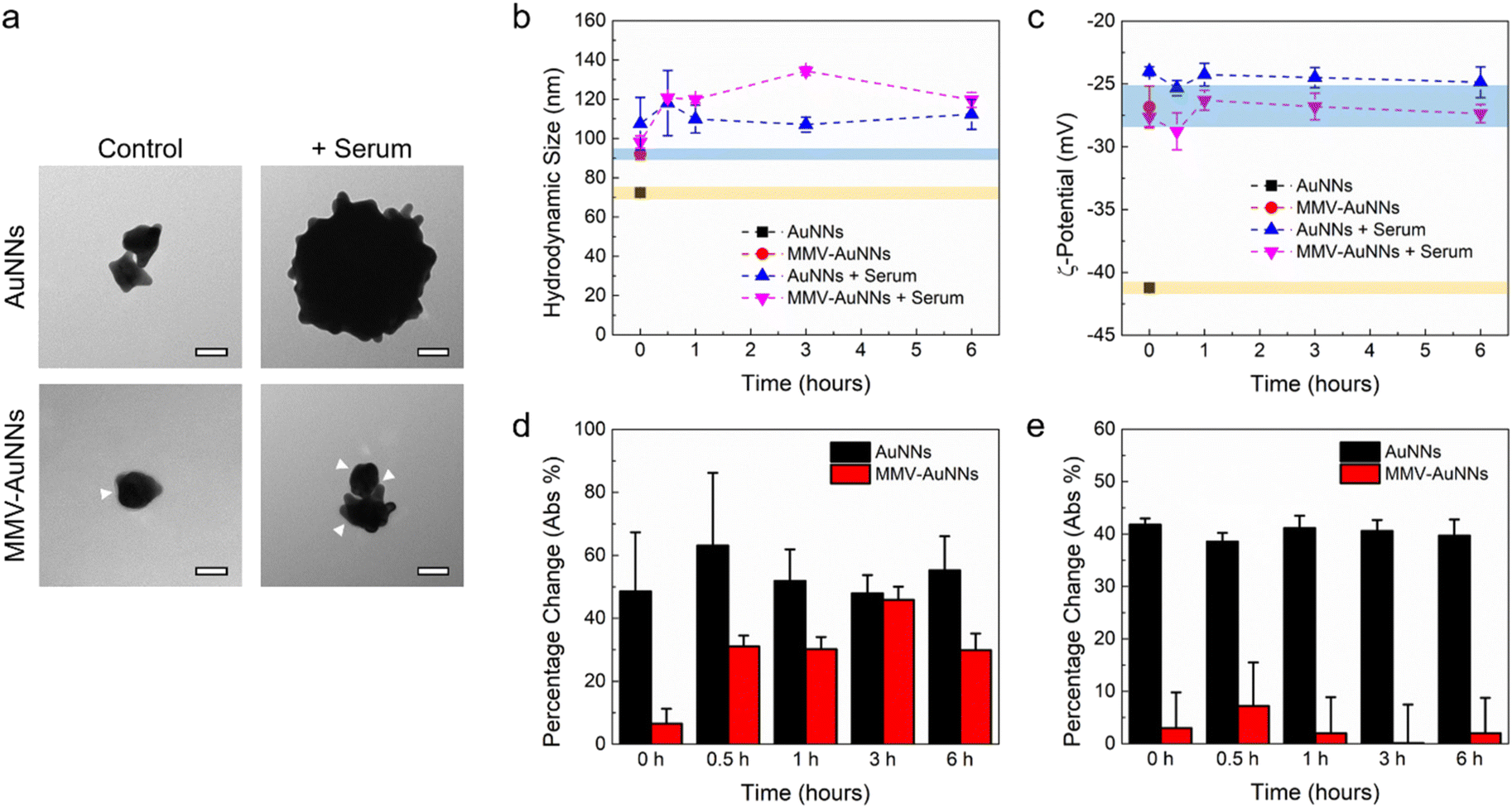 | ||
| Fig. 3 Interactions of the macrophage membrane vesicle-encapsulated Au nanostructures with serum. (a) Representative TEM images showing the morphology of the bare and MMV-Au nanostructures in the presence of serum. Scale bars are 40 nm. The white arrows indicate the presence of macrophage membrane coating. (b) Hydrodynamic size and (c) zeta potential evolution of the bare and MMV-Au nanostructures in the absence and presence of serum over time (i.e., 0, 0.5, 1, 3, and 6 h). The yellow and blue strips indicate the properties of the bare and MMV-Au nanostructures, respectively, in the absence of serum. (d and e) Percentage change of: (d) hydrodynamic size and (e) zeta potential of the bare and MMV-Au nanostructures in the presence of serum with respect to controls. | ||
Overall, these findings suggest that the MMV-camouflaged AuNNs possessed better stability in serum than the bare AuNNs in terms of changes in their surface morphology, hydrodynamic size, and zeta potential, which might facilitate MMV-AuNNs to avoid immune uptake and clearance.
Immune evasion and cancer cell uptake of biomimetic Au nanostructures
One of the key criteria in realizing theranostic nanostructures with improved delivery and targeting efficacy is a sufficiently long circulation lifetime for accumulation at the target sites,61–63 which depends predominantly on the ability of the nanostructures to avoid immune uptake and rapid body clearance.64,65 Because of this, after verifying the lower non-specific biomolecule adsorption on MMV-AuNNs than on the bare AuNNs, the phagocytic uptake of MMV-AuNNs was investigated. Specifically, RAW 264.7 macrophages were incubated with AuNNs and MMV-AuNNs and their macrophage uptake and cellular effects were examined. Bright-field microscopy images obtained after treating macrophages with both Au nanostructures over 12 h revealed a lower uptake of MMV-AuNNs by macrophages than the bare AuNNs (Fig. 4a and Fig. S15†). Indeed, semi-quantitative analysis of the bright-field images showed that MMV-AuNNs consistently exhibited a lower macrophage uptake than their bare counterparts (Fig. 4b). This difference in macrophage uptake was evident as early as 3 h after both Au nanostructures were introduced into the macrophage cultures. Separately, the cell spreading areas and circularity, which are typically used to evaluate cellular health and proliferation,66 of the AuNN- and MMV-AuNN-treated macrophages over time were similar (Fig. 4c and d). On the same note, the difference in cell solidity, which measures the density of a cell, of both nanostructure-treated macrophages was negligible (Fig. S16†). This suggests the minimal effect of MMV-AuNNs on the spreading and proliferation of macrophages.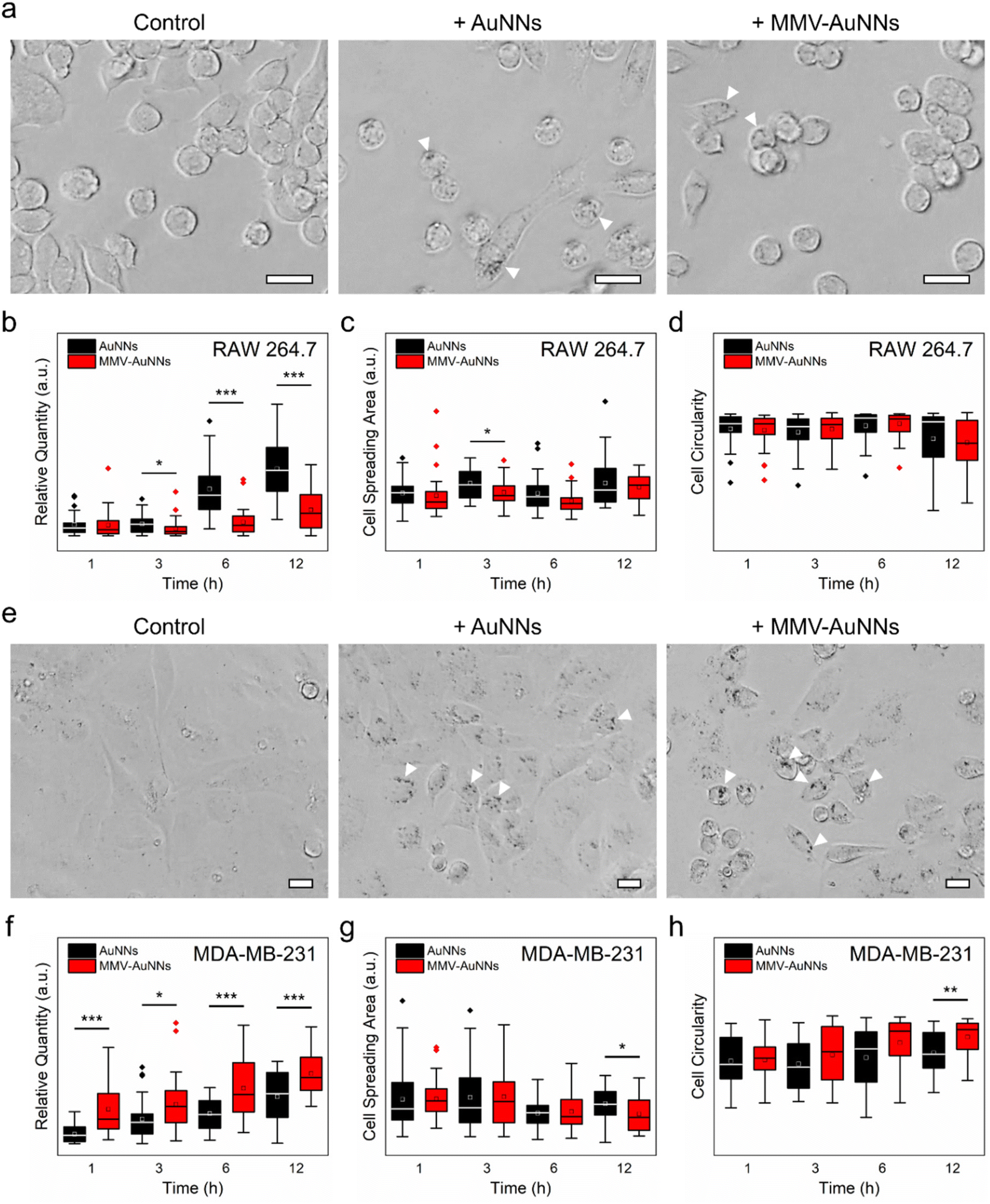 | ||
| Fig. 4 Immune evasion and cancer cell uptake of the macrophage membrane vesicle-encapsulated Au nanostructures. (a and e) Representative bright-field images of the uptake of AuNNs and MMV-AuNNs by (a) RAW 264.7 macrophages and (e) MDA-MB-231 breast cancer cells after 12 h of incubation. The white arrows on the bright-field images indicate the presence of Au nanostructures. All scale bars are 20 μm. (b and f) Corresponding semi-quantitative estimation of the uptake of different Au nanostructures by (b) macrophages and (f) breast cancer cells over time. (c and g) Spreading areas and (d and h) circularity of (c and d) macrophages and (g and h) breast cancer cells after different Au nanostructure treatments over time. n = 30 cells. * p < 0.05; ** p < 0.01; *** p < 0.001 based on either the parametric student's t test or non-parametric Mann–Whitney test. | ||
In addition to having a reduced immune uptake and clearance, theranostic nanostructures need to be able to target specific cancer cells and induce preferential uptake by these cells to realize the intended theranostic effects.67,68 To this end, after establishing the more pronounced immune evasion ability of MMV-AuNNs as compared to the bare AuNNs, the uptake of MMV-AuNNs by cancer cells was investigated. Here, the triple-negative MDA-MB-231 breast cancer cells were selected as the model cancer cells. These cells were first incubated with both AuNNs and MMV-AuNNs for 1, 3, 6, and 12 h and the cellular uptake of these nanostructures was then evaluated. Bright-field microscopy images showed that both Au nanostructures could be taken up easily by MDA-MB-231 breast cancer cells over time (Fig. 4e and Fig. S17†). Nevertheless, it appeared that MMV-AuNNs could be internalized more easily by the cancer cells as compared to the bare AuNNs. This was verified through semi-quantitative analysis of the acquired bright-field images, where the higher cellular uptake of MMV-AuNNs occurred as early as 1 h after the introduction of Au nanostructures (Fig. 4f). This indicates the preferential uptake of the biomimetic Au nanostructures by breast cancer cells. Analysis of cell spreading areas and circularity after nanostructure treatments up to 6 h revealed that both Au nanostructures appeared to induce negligible influence on the health and proliferation of breast cancer cells (Fig. 4g and h). However, at 12 h post-nanostructure treatments, as compared to the AuNN-treated breast cancer cells, those incubated with MMV-AuNNs experienced a significant decrease in spreading areas and a substantial increase in cell circularity. This suggests that MMV-AuNNs might alter the morphological integrity of breast cancer cells more effectively than AuNNs. Furthermore, evaluation of cell solidity at various time points uncovered a significant difference in the distribution of the solidity of the nanostructure-treated breast cancer cells, suggesting that MMV-AuNNs elicited a more pronounced effect on the density of breast cancer cells than the bare AuNNs (Fig. S18†).
Altogether, the immune evasion capability and preferential cancer cell uptake of MMV-AuNNs could be leveraged to potentially enhance the cancer targeting of anisotropic Au nanostructures for various cancer theranostic applications.
3. Conclusion
In summary, a one-step continuous flow microfluidic-sonication method was developed for the formulation of cell membrane vesicle-encapsulated anisotropic Au nanostructures. The encapsulation of Au nanostructures within the macrophage and cancer cell membrane vesicles was realized through the combined effect of hydrodynamic flow focusing, mixing, and sonication. The formulated biomimetic Au nanostructures displayed core–shell structures with a hydrodynamic size of <100 nm, a negative zeta potential, and excellent stability in serum over time. These biomimetic nanostructures also exhibited immune evasion property, where their uptake by macrophages was significantly less than that of their bare counterparts. Importantly, as compared to the uncoated Au nanostructures, the membrane-camouflaged Au nanostructures could be preferentially internalized by breast cancer cells due to the higher cancer targeting specificity of the biomimetic nanostructures. Altogether, it is anticipated that this study will further expand the library of approaches available for the synthesis of cell membrane-camouflaged nanostructures for theranostic and nanomedicine applications.4. Methods
Design and fabrication of the microfluidic device
The microfluidic device used for the synthesis of cell membrane vesicle-coated Au nanostructures consists of three inlet channels, a mixing region, and an outlet channel for collecting the final products. The total length of the microfluidic device from the middle inlet port to the outlet port is 3 cm. All microchannels have a uniform height of 50 μm. Each inlet channel has a width of 100 μm, while the mixing region and outlet channel have a width of 300 μm. The microfluidic device was fabricated in polydimethylsiloxane (PDMS) based on a soft lithography process.Synthesis of Au nanostructures
AuNNs with a lower tip-to-core ratio were prepared based on a seed-mediated process.69 In brief, 2 mL of 25 mM gold(III) chloride (MilliporeSigma, USA) solution was diluted in 200 mL of deionized water. Next, 6 mL of ice-cold 100 mM NaBH4 (MilliporeSigma, USA) solution was introduced into the mixture under continuous stirring for 30 s, followed by further dilution of the mixture in deionized water to a volume of 1 L. The resulting Au seeds were stored at room temperature for further use. 2 mL of 400 mM gold(III) chloride (MilliporeSigma, USA) aqueous solution and 7 mL of Au seeds were introduced into 1.8 L of 200 mM ascorbic acid (MilliporeSigma, USA) aqueous solution at 4 °C under turbulent mixing. The resulting mixture was subsequently centrifuged at 4000g and 0 °C for 20 min. After the removal of the supernatant, the liquid pellets were collected in a 15 mL dialysis cassette (MWCO 20000, Thermo Fisher Scientific Inc., USA) and subjected to dialysis for 3 days. The acquired AuNNs were then characterized and used for subsequent experiments.A 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer-driven process was used to prepare AuNSs with a higher tip-to-core size ratio.70 In brief, 10 mL of 20 mM aqueous solution of gold(III) chloride (MilliporeSigma, USA) was introduced into a 1 L solution of 200 mM HEPES buffer pH 7.2 under continuous stirring. After the stirring was stopped, the mixture was stored at room temperature overnight. The acquired AuNS solution was then centrifuged at 4000g and 0 °C for 20 min. After the removal of the supernatant, the liquid pellets were collected in a 15 mL dialysis cassette (MWCO 20000, Thermo Fisher Scientific Inc., USA) and subjected to dialysis for 3 days. The acquired AuNSs were then characterized and used for subsequent experiments.
Cell lines and culture
RAW 264.7 murine macrophages and MDA-MB-231 human breast cancer cells were purchased from American Type Culture Collection (ATCC). All cells were grown and maintained in T75 flasks containing Dulbecco's modified Eagle's medium (DMEM) (Corning Inc., USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific Inc., USA) and 1% antibiotic–antimycotic (Gibco, Thermo Fisher Scientific Inc., USA). All cells were cultured in a 37 °C humidified incubator supplemented with 5% CO2.Preparation of cell membrane vesicles
After growing to confluency in the culture flasks, the cells were detached using 0.25% trypsin–ethylenediaminetetraacetic acid (EDTA) (Gibco, Thermo Fisher Scientific Inc., USA). The cell suspensions were then centrifuged at 1500 rpm for 5 min to remove the supernatants, followed by washing with phosphate-buffered saline (PBS) 1× (MilliporeSigma, USA). The collected cell pellets were then resuspended in a hypotonic lysis buffer comprising 20 mM Tris–HCl pH 7.5 (Teknova Inc., USA), 10 mM KCl (Teknova Inc., USA), 2 mM MgCl2 (New England Biolabs Inc., USA), and 1 EDTA-free mini protease inhibitor tablet (Roche Diagnostics GmbH, Germany) per 10 mL of solution. The cell suspensions were subjected to 5 cycles of freeze–thaw treatment, followed by centrifugation at 700g for 10 min to remove cell debris. The supernatants were then collected and subjected to centrifugation at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min. After the removal of the supernatants, the pellets containing cell membrane materials were then stored immediately at −80 °C for future use or suspended in PBS 1× for subsequent experiments. To generate membrane vesicles, the suspended membrane materials were sonicated for 4 min or physically extruded through a 200 nm polycarbonate membrane (Avanti Polar Lipids Inc., USA) for 13 passes using a mini-extruder (Avanti Polar Lipids Inc., USA).
000g for 30 min. After the removal of the supernatants, the pellets containing cell membrane materials were then stored immediately at −80 °C for future use or suspended in PBS 1× for subsequent experiments. To generate membrane vesicles, the suspended membrane materials were sonicated for 4 min or physically extruded through a 200 nm polycarbonate membrane (Avanti Polar Lipids Inc., USA) for 13 passes using a mini-extruder (Avanti Polar Lipids Inc., USA).
Synthesis of cell membrane vesicle-encapsulated Au nanostructures
Au nanostructure and cell membrane vesicle solutions were first loaded into syringes (BD, USA) installed on syringe pumps (Harvard Apparatus, USA and KD Scientific Inc., USA) and connected with tubing (ID 0.02 inch and OD 0.06 inch) (Tygon®, Saint-Gobain Performance Plastics Corp., USA) to the appropriate ports of the microfluidic device. The device was immersed in an ultrasonic bath (CPX1800, Thermo Fisher Scientific Inc., USA) operating throughout the synthesis process. The flow rate of the middle inlet channel where Au nanostructures were introduced was set at 7 mL h−1, while the flow rate of the two side inlet channels where cell membrane vesicles were injected was set at 14 mL h−1. The synthesized products were collected from the outlet channel and centrifuged at 2000g for 20 min. After the removal of the supernatants, the pellets containing membrane vesicle-coated nanostructures were resuspended in deionized water to appropriate concentrations for characterization and subsequent experiments. For comparison, cell membrane vesicle-encapsulated Au nanostructures were also prepared through sequential physical extrusion. In brief, the Au nanostructures and cell membrane vesicles were mixed and then co-extruded through a 200 nm polycarbonate membrane (Avanti Polar Lipids Inc., USA) for 21 passes using a mini-extruder (Avanti Polar Lipids Inc., USA). The processed samples were then centrifuged and the collected pellets were resuspended in deionized water.Characterization of Au nanostructure properties
A UV-vis spectrophotometer (UV-2600, Shimadzu Scientific Instruments Inc., USA) was used to measure the UV-vis absorbance of the Au nanostructures. Transmission electron microscopy (TEM) (JEOL USA, Inc., USA) was used to characterize the surface morphology of the bare and membrane vesicle-coated Au nanostructures. Their hydrodynamic size and zeta potential were characterized using dynamic light scattering (DLS) (Malvern Panalytical Ltd, USA). The protein expressions of cell membrane vesicles and membrane vesicle-coated Au nanostructures were visualized using one-dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Briefly, each sample was mixed with Laemmli sample buffer 2× (Bio-Rad Laboratories Inc., USA) and heated at 100 °C for 7 min. 10 μL of each mixture was then loaded into 4–20% MP TGX polyacrylamide gel (Bio-Rad Laboratories Inc., USA) installed in an electrophoresis cell (Bio-Rad Laboratories Inc., USA), which was then operated at constant 200 V for 40 min in SDS running buffer 1× (Bio-Rad Laboratories Inc., USA). The polyacrylamide gel was then stained with Coomassie Brilliant Blue R-250 staining solution (Bio-Rad Laboratories Inc., USA) for 2 h and washed with destaining solution (Bio-Rad Laboratories Inc., USA) overnight to visualize the protein expressions of different Au nanostructures.Evaluation of the stability of Au nanostructures in serum
25 μL of 0.02 nM of each Au nanostructure solution was mixed with 25 μL of complete cell culture medium (DMEM (Corning Inc., USA) supplemented with 10% FBS (Gibco, Thermo Fisher Scientific Inc., USA) and 1% antibiotic–antimycotic (Gibco, Thermo Fisher Scientific Inc., USA)). The different mixtures were incubated at room temperature over varying durations (i.e., 0, 0.5, 1, 3, and 6 h). The Au nanostructure stability in serum was assessed according to changes in the nanostructure morphology using TEM (JEOL USA Inc., USA) and the hydrodynamic size and zeta potential using DLS (Malvern Panalytical Ltd, USA).Imaging and semi-quantitative analysis of the cellular uptake and effects of Au nanostructures
10000 cells in 100 μL of medium were first cultured in 96-well plates (Falcon, Corning Inc., USA) overnight. The culture medium was then replaced with the solutions of bare or membrane vesicle-coated Au nanostructures diluted in fresh medium. The uptake of different Au nanostructures by macrophages and breast cancer cells was visualized using inverted bright-field microscopy (Olympus CKX41, Olympus Corp., Japan) after incubating the cells with Au nanostructures for 1, 3, 6, and 12 h. Semi-quantitative evaluations of the cellular uptake of Au nanostructures, cell spreading areas, cell circularity, and cell solidity were performed using ImageJ software (National Institutes of Health, USA). A total of 30 cells were randomly selected from bright-field images for each semi-quantitative evaluation.
Statistical analysis
The experimental data were presented as the average ± standard deviation. GraphPad Prism 9.1 (GraphPad Software Inc., USA) was used to analyze all data. The normality of data was first evaluated based on the Shapiro–Wilk normality test. The data that were normally distributed were analyzed using the parametric student's t test. On the other hand, the non-parametric Mann–Whitney test was used to analyze the data that were not normally distributed. The difference between compared groups was considered statistically significant for *p < 0.05, **p < 0.01, and ***p < 0.001.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Kenry would like to acknowledge Dr Bohdan Andreiuk for his gift of Au nanostructures and the funding support from Dana-Farber Cancer Institute. Kenry would also like to especially acknowledge the dedicated mentorship of Dr Moritz F. Kircher, the late Head of the Kircher Lab and Chair of the Department of Imaging, Dana-Farber Cancer Institute/Harvard Medical School. Finally, the departmental start-up fund of Kenry from the Department of Pharmacology and Toxicology, R. Ken Coit College of Pharmacy, University of Arizona, is gratefully acknowledged.References
- J. Xie, G. Liu, H. S. Eden, H. Ai and X. Chen, Acc. Chem. Res., 2011, 44, 883–892 CrossRef CAS PubMed.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed.
- R. Bardhan, S. Lal, A. Joshi and N. J. Halas, Acc. Chem. Res., 2011, 44, 936–946 CrossRef CAS PubMed.
- Kenry, A. Geldert, Z. Lai, Y. Huang, P. Yu, C. Tan, Z. Liu, H. Zhang and C. T. Lim, Small, 2017, 13, 1601925 CrossRef PubMed.
- S. M. A. Fateminia, L. Kacenauskaite, C.-J. Zhang, S. Ma, Kenry, P. N. Manghnani, J. Chen, S. Xu, F. Hu, B. Xu, B. W. Laursen and B. Liu, Small, 2018, 14, 1803325 CrossRef PubMed.
- G. Liao, F. He, Q. Li, L. Zhong, R. Zhao, H. Che, H. Gao and B. Fang, Prog. Mater. Sci., 2020, 112, 100666 CrossRef CAS.
- Kenry, T. Yeo, D. T. She, M. H. Nai, V. L. Marcelo Valerio, Y. Pan, E. Middha, C. T. Lim and B. Liu, ACS Nano, 2021, 15, 17412–17425 CrossRef CAS.
- G. Liao, L. Zhang, C. Li, S.-Y. Liu, B. Fang and H. Yang, Matter, 2022, 5, 3341–3374 CrossRef CAS.
- L. Zhang, G. Oudeng, F. Wen and G. Liao, Biomater. Res., 2022, 26, 61 CrossRef CAS PubMed.
- Kenry, L. Sun, T. Yeo, E. Middha, Y. Gao, C. T. Lim, S. Watanabe and B. Liu, Small, 2022, 18, 2203285 CrossRef CAS PubMed.
- Z. Mo, Q. Li, K. Zhao, Q. Xu, H. Hu, X. Chen, Y. Luo, B. Chi, L. Liu, X. Fang, G. Liao, Z. Xu, J. Wang and S. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 10001–10014 CrossRef CAS PubMed.
- Kenry and B. Liu, Chem, 2023, 9, 2078–2094 CAS.
- Y. Xia, W. Li, C. M. Cobley, J. Chen, X. Xia, Q. Zhang, M. Yang, E. C. Cho and P. K. Brown, Acc. Chem. Res., 2011, 44, 914–924 CrossRef CAS PubMed.
- N. Li, P. Zhao and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 1756–1789 CrossRef CAS PubMed.
- J. E. Ortiz-Castillo, R. C. Gallo-Villanueva, M. J. Madou and V. H. Perez-Gonzalez, Coord. Chem. Rev., 2020, 425, 213489 CrossRef CAS.
- L. Fabris, J. Phys. Chem. C, 2020, 124, 26540–26553 CrossRef CAS.
- J. Zheng, X. Cheng, H. Zhang, X. Bai, R. Ai, L. Shao and J. Wang, Chem. Rev., 2021, 121, 13342–13453 CrossRef CAS.
- H. He, L. Liu, E. E. Morin, M. Liu and A. Schwendeman, Acc. Chem. Res., 2019, 52, 2445–2461 CrossRef CAS PubMed.
- M. A. Younis, H. M. Tawfeek, A. A. H. Abdellatif, J. A. Abdel-Aleem and H. Harashima, Adv. Drug Delivery Rev., 2022, 181, 114083 CrossRef CAS.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- T. D. Clemons, R. Singh, A. Sorolla, N. Chaudhari, A. Hubbard and K. S. Iyer, Langmuir, 2018, 34, 15343–15349 CrossRef CAS PubMed.
- Y. Shi, R. van der Meel, X. Chen and T. Lammers, Theranostics, 2020, 10, 7921–7924 CrossRef PubMed.
- H. Kang, S. Rho, W. R. Stiles, S. Hu, Y. Baek, D. W. Hwang, S. Kashiwagi, M. S. Kim and H. S. Choi, Adv. Healthcare Mater., 2020, 9, 1901223 CrossRef CAS.
- A. K. Pearce and R. K. O'Reilly, Bioconjugate Chem., 2019, 30, 2300–2311 CrossRef CAS.
- Z. R. Goddard, M. J. Marín, D. A. Russell and M. Searcey, Chem. Soc. Rev., 2020, 49, 8774–8789 RSC.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- H. Meng, W. Leong, K. W. Leong, C. Chen and Y. Zhao, Biomaterials, 2018, 174, 41–53 CrossRef CAS PubMed.
- J. Jia, Z. Wang, T. Yue, G. Su, C. Teng and B. Yan, Chem. Res. Toxicol., 2020, 33, 1055–1060 Search PubMed.
- Y. Liu, Z. Wang, Y. Liu, G. Zhu, O. Jacobson, X. Fu, R. Bai, X. Lin, N. Lu, X. Yang, W. Fan, J. Song, Z. Wang, G. Yu, F. Zhang, H. Kalish, G. Niu, Z. Nie and X. Chen, ACS Nano, 2017, 11, 10539–10548 CrossRef CAS PubMed.
- J. G. Cronin, N. Jones, C. A. Thornton, G. J. S. Jenkins, S. H. Doak and M. J. D. Clift, Chem. Res. Toxicol., 2020, 33, 1061–1073 Search PubMed.
- H. Hadji and K. Bouchemal, J. Controlled Release, 2022, 342, 93–110 CrossRef CAS PubMed.
- Y. Qie, H. Yuan, C. A. von Roemeling, Y. Chen, X. Liu, K. D. Shih, J. A. Knight, H. W. Tun, R. E. Wharen, W. Jiang and B. Y. S. Kim, Sci. Rep., 2016, 6, 26269 CrossRef CAS PubMed.
- L. Sanchez, Y. Yi and Y. Yu, Nanoscale, 2017, 9, 288–297 RSC.
- Kenry, T. Yeo, P. N. Manghnani, E. Middha, Y. Pan, H. Chen, C. T. Lim and B. Liu, ACS Nano, 2020, 14, 4509–4522 CrossRef CAS PubMed.
- R. Saadati, S. Dadashzadeh, Z. Abbasian and H. Soleimanjahi, Pharm. Res., 2013, 30, 985–995 CrossRef CAS PubMed.
- M. Liu, Y. Chu, H. Liu, Y. Su, Q. Zhang, J. Jiao, M. Liu, J. Ding, M. Liu, Y. Hu, Y. Dai, R. Zhang, X. Liu, Y. Deng and Y. Song, Mol. Pharm., 2020, 17, 1059–1070 CrossRef CAS PubMed.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2018, 30, 1706759 CrossRef PubMed.
- Y. Liu, J. Luo, X. Chen, W. Liu and T. Chen, Nano-Micro Lett., 2019, 11, 100 CrossRef CAS PubMed.
- S. Zou, B. Wang, C. Wang, Q. Wang and L. Zhang, Nanomedicine, 2020, 15, 625–641 CrossRef CAS PubMed.
- C. M. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- J.-G. Piao, L. Wang, F. Gao, Y.-Z. You, Y. Xiong and L. Yang, ACS Nano, 2014, 8, 10414–10425 CrossRef CAS PubMed.
- L. Rao, L.-L. Bu, J.-H. Xu, B. Cai, G.-T. Yu, X. Yu, Z. He, Q. Huang, A. Li, S.-S. Guo, W.-F. Zhang, W. Liu, Z.-J. Sun, H. Wang, T.-H. Wang and X.-Z. Zhao, Small, 2015, 11, 6225–6236 CrossRef CAS PubMed.
- X. Hao, H. Zhang, R. Liu, J. Che, D. Zhang, J. Liang and L. Sun, Mater. Today Adv., 2022, 16, 100294 CrossRef CAS.
- R. H. Fang, C.-M. J. Hu, B. T. Luk, W. Gao, J. A. Copp, Y. Tai, D. E. O'Connor and L. Zhang, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS.
- Z. Chen, P. Zhao, Z. Luo, M. Zheng, H. Tian, P. Gong, G. Gao, H. Pan, L. Liu, A. Ma, H. Cui, Y. Ma and L. Cai, ACS Nano, 2016, 10, 10049–10057 CrossRef CAS PubMed.
- L. Rao, G.-T. Yu, Q.-F. Meng, L.-L. Bu, R. Tian, L.-S. Lin, H. Deng, W. Yang, M. Zan, J. Ding, A. Li, H. Xiao, Z.-J. Sun, W. Liu and X. Chen, Adv. Funct. Mater., 2019, 29, 1905671 CrossRef CAS.
- S. Zhu, Z. Li, D. Zheng, Y. Yu, J. Xiang, X. Ma, D. Xu, J. Qiu, Z. Yang, Z. Wang, J. Li, H. Sun, W. Chen, X. Meng, Y. Lu and Q. Ren, Mol. Ther. – Oncolytics, 2023, 28, 182–196 CrossRef CAS.
- L. Rao, B. Cai, L.-L. Bu, Q.-Q. Liao, S.-S. Guo, X.-Z. Zhao, W.-F. Dong and W. Liu, ACS Nano, 2017, 11, 3496–3505 CrossRef CAS PubMed.
- C. Liu, W. Zhang, Y. Li, J. Chang, F. Tian, F. Zhao, Y. Ma and J. Sun, Nano Lett., 2019, 19, 7836–7844 CrossRef CAS PubMed.
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Nano Lett., 2008, 8, 2906–2912 CrossRef CAS PubMed.
- N. M. Belliveau, J. Huft, P. J. C. Lin, S. Chen, A. K. K. Leung, T. J. Leaver, A. W. Wild, J. B. Lee, R. J. Taylor, Y. K. Tam, C. L. Hansen and P. R. Cullis, Mol. Ther. – Nucleic Acids, 2012, 1, e37 CrossRef PubMed.
- J. Ma, S. M.-Y. Lee, C. Yi and C.-W. Li, Lab Chip, 2017, 17, 209–226 RSC.
- T. C. Marentis, B. Kusler, G. G. Yaralioglu, S. Liu, E. O. Hæggström and B. T. Khuri-Yakub, Ultrasound Med. Biol., 2005, 31, 1265–1277 CrossRef PubMed.
- Z. Cao and C. Lu, Anal. Chem., 2016, 88, 1965–1972 CrossRef CAS PubMed.
- Kenry, K. P. Loh and C. T. Lim, Small, 2015, 11, 5105–5117 CrossRef CAS PubMed.
- Kenry, K. P. Loh and C. T. Lim, Nanoscale, 2016, 8, 9425–9441 RSC.
- Kenry, A. Geldert, Y. Liu, K. P. Loh and C. T. Lim, NPG Asia Mater., 2017, 9, e422 CrossRef CAS.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailänder, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
- A. Aliyandi, C. Reker-Smit, R. Bron, I. S. Zuhorn and A. Salvati, ACS Biomater. Sci. Eng., 2021, 7, 5573–5584 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS.
- J. Bourquin, A. Milosevic, D. Hauser, R. Lehner, F. Blank, A. Petri-Fink and B. Rothen-Rutishauser, Adv. Mater., 2018, 30, 1704307 CrossRef PubMed.
- H. B. Haroon, A. C. Hunter, Z. S. Farhangrazi and S. M. Moghimi, Adv. Drug Delivery Rev., 2022, 188, 114396 CrossRef CAS PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS PubMed.
- K. M. Tsoi, S. A. MacParland, X.-Z. Ma, V. N. Spetzler, J. Echeverri, B. Ouyang, S. M. Fadel, E. A. Sykes, N. Goldaracena, J. M. Kaths, J. B. Conneely, B. A. Alman, M. Selzner, M. A. Ostrowski, O. A. Adeyi, A. Zilman, I. D. McGilvray and W. C. W. Chan, Nat. Mater., 2016, 15, 1212–1221 CrossRef CAS PubMed.
- Kenry, P. K. Chaudhuri, K. P. Loh and C. T. Lim, ACS Nano, 2016, 10, 3424–3434 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef CAS PubMed.
- M. A. Wall, S. Harmsen, S. Pal, L. Zhang, G. Arianna, J. R. Lombardi, C. M. Drain and M. F. Kircher, Adv. Mater., 2017, 29, 1605622 CrossRef PubMed.
- K. Chandra, K. S. B. Culver, S. E. Werner, R. C. Lee and T. W. Odom, Chem. Mater., 2016, 28, 6763–6769 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00415a |
| This journal is © The Royal Society of Chemistry 2024 |