
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The efficacy of Pb, As(V) and Sb(III) removal by biochar is determined by solution chemistry†
Sampriti
Chaudhuri
 ab,
Gabriel
Sigmund
*ac,
Naresh
Kumar
d,
Thorsten
Hüffer
a,
Andreas
Mautner
ef and
Thilo
Hofmann
*a
ab,
Gabriel
Sigmund
*ac,
Naresh
Kumar
d,
Thorsten
Hüffer
a,
Andreas
Mautner
ef and
Thilo
Hofmann
*a
aDepartment of Environmental Geosciences, Centre for Microbiology and Environmental Systems Science, University of Vienna, Josef-Holaubek Platz 2, 1090 Vienna, Austria. E-mail: thilo.hofmann@univie.ac.at
bDoctoral School in Microbiology and Environmental Science, University of Vienna, 1090 Vienna, Austria
cEnvironmental Technology, Wageningen University, P.O. Box 17, 6700 AA Wageningen, The Netherlands. E-mail: gabriel.sigmund@wur.nl
dSoil Chemistry, Wageningen University, 6708 PB Wageningen, The Netherlands
eInstitute of Materials Chemistry and Research, Polymer and Composite Engineering (PaCE) Group, Faculty of Chemistry, University of Vienna, Währinger Strasse 42, 1090, Vienna, Austria
fInstitute of Environmental Biotechnology, IFA-Tulln, University of Natural Resources & Life Sciences (BOKU), Konrad-Lorenz-Straße 20, 3430, Tulln an der Donau, Austria
First published on 12th February 2024
Abstract
Biochars (BC) are cost-effective and sustainable sorbents to clean up waters polluted with metal(loid)s. Understanding the influence of water chemistry is critical in identifying processes that limit metal(loid) removal. To address this, we investigated the removal of lead [Pb], arsenate [As(V)], and antimonite [Sb(III)] using BC in the presence of various solution constituents. A design of experiments approach was used to investigate sorption for each metal(loid)-BC setup (Pb with a straw BC, As(V) with charred wood-dolomite and Sb(III) with a steam-activated wood BC) with twenty-five different background solutions varying in calcium (Ca), natural organic matter (NOM), phosphorus (P), and iron [Fe(III)] content. Background solution composition affected removal of Pb (29 to 100%) more strongly than that of As(V) (37 to 92%) and Sb(III) (20 to 70%), with the selected BC at the metal(loid) concentrations studied. Pb removal was associated with Fe(III)–NOM–Ca organo-mineral phases for solutions containing Fe(III), NOM and Ca. As(V) sorption was enhanced by Ca due to cation-bridging and reducing the competition for sorption sites by NOM and P in high NOM and/or P containing solutions. Sb(III) sorption was hindered by oxidation to Sb(V) through redox active moieties in the BC in all solutions. Sb(III) removal decreased in the presence of high Fe(III), because Fe(III)/Fe(III)–NOM phases blocked accessibility to sorption sites in the highly porous BC, and/or due to enhanced oxidation of Sb(III) to the more mobile (but less toxic) Sb(V). Ideally, the design of BC sorbents for the removal of metal(loid)s from contaminated waters should a priori consider complex solution compositions.
Water impactThe influence of dissolved and particulate solution constituents on contaminant removal by biochar remains poorly understood. To address this, a design of experiments approach was used to determine lead, arsenate, and antimonite removal by biochars from solutions of varying chemical composition. This study explored the mechanisms involved and highlights the need to test biochars in target water matrices to fairly assess their contaminant removal efficacy for a given application. |
1. Introduction
Biochars (BC) have been used to successfully treat contaminated water, sediments, and soils. These materials can be tailored favourably to remove a diverse suite of contaminants by combining high porosity and specific surface area with surface functionality and reactivity.1 Hydrophobic organic contaminants are well immobilised by activated carbon and high temperature biochars.2 However, for inorganic contaminants, i.e. metals and metalloids referred to as “metal(loid)s”, the extent and mechanism of immobilisation differ largely, depending on BC properties and the type of contaminant.3,4Lead (Pb), arsenic (As), and antimony (Sb) are commonly detected metal(loid)s in contaminated water from mining and industrial activities. While Pb exists predominantly in the cationic form at pH < 8, prevalent forms of As and Sb are mostly oxyanionic or neutral, depending on the redox conditions. Their environmental speciation (e.g., positively or negatively charged) and redox state (e.g., Sb(III)/Sb(V) and As(III)/As(V)), influence the potential immobilisation of each of these metals(loid)s by BC. For example, Pb(II) can be removed by predominantly negatively charged BC through cation-exchange processes, complexation with surface functional groups, and precipitation–adsorption mechanisms.5 In contrast, anionic species of As and Sb are removed to a lesser extent, due to electrostatic repulsion and the lack of positively charged binding sites on the BC.6,7 Relevant factors for the removal of metal(loid)s in oxic waters include, but are not limited to, pH, dissolved calcium (Ca) and phosphorus (P), along with natural organic matter (NOM) and iron (Fe). pH is crucial in determining the dominant species of a particular metal(loid) in water. For example, at pH > 8 the Pb speciation shifts from cationic forms (Pb2+, PbOH+) towards the neutral Pb(OH)2. An even higher pH (>10) causes the negatively charged Pb(OH)3− to form, decreasing Pb retention by negatively charged sorbents.8 Ca, which is ubiquitously present, can increase the removal of anionic As(V) by bridging negatively charged moieties between As and BC, or screening negative charges of BC.9 P belongs to the same group of elements (group 15) as Sb and As and can compete with them for sorption sites. Contrastingly, P can promote the removal of Pb from solution through formation of Pb-phosphate precipitates.10 NOM can hinder metal(loid) removal by complexing the target ions and competing for, or blocking sorption sites on BC.11 Hydrolysed Fe(III) phases are omnipresent in the environment and can trigger multiple mechanisms that affect the removal of targeted metal(loid)s from water. These include (i) production of new sorption sites on the BC,12 (ii) association of targeted metal(loid)s to Fe(III) phases that can sorb to the BC surface or, conversely be mobilised through colloidal transport of these Fe(III) phases,13 and (iii) redox reactions which may produce more mobile species of particular metal(loid)s.14
Previously, we demonstrated that the simultaneous existence of high concentrations of Ca and/or chloride in NOM-rich solutions can compensate the inhibitory effects that NOM has on contaminant removal by a BC.11 Similarly, the inhibitory role of P in As sorption by BC can be mitigated by the concurrent presence of Fe(III) phases that may complex with P.15 If NOM is complexed with Ca, the amount of NOM available for Pb complexation will be lower, facilitating increased Pb removal by BC. The co-existence of NOM and Fe(III) leads to a new co-precipitated or adsorbed organo-mineral phase. Studies report conflicting results for the mobilisation and remobilisation of metal(loid)s by such phases.16 The complex role of Fe(III)–NOM phases/composites in sorbent performance, and the changing reactivity of these phases with P and Ca has been investigated by few studies and thorough investigation is warranted.17–20 How this complexity will affect metal(loid) sorption by BC in waters of diverse ionic composition remains unknown. The complexity and variety in solution composition in the environment necessitates the study of metal(loid) sorption by BC from the perspective of combined factors rather than a single factor.21
The overall objective of this study was to understand the influence of key dissolved and particulate solution constituents on metal(loid) sorption to allow for better sorbent design and/or selection. The specific objectives were (i) investigating sorption of three metal(loid)s with different geochemical behaviour to three specific BC, i.e., Pb to a straw BC, As(V) to co-pyrolyzed wood and dolomite, and Sb(III) to a steam activated wood BC, and (ii) analysing the influence of combinations of key solution constituents, i.e., Ca, P, NOM and Fe(III), on sorption. We hypothesise that specific combinations of these solution constituents strongly alter sorption to BC. To account for the large variety of possible combinations of environmental factors, we used a central composite design approach. Therein we conducted single-metal(loid) batch sorption experiments under oxic conditions for a total of twenty-five different solutions covering a wide range of background solution compositions.
2. Materials and methods
2.1 Materials
Ultrapure water from an Elga PURELAB Chorus 3 water purification system (ELGA LabWater, UK) was used to prepare all solutions. Calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), sodium phosphate dibasic dihydrate (Na2HPO4·2H2O), iron(III) nitrate nonahydrydrate (Fe(NO3)3·9H2O), lead nitrate (Pb(NO3)2), sodium hydrogen arsenate heptahydrate (Na2HAsO4·7H2O), potassium antimonyl tartrate trihydrate (K2[Sb2(C4H2O6)2]·3H2O), and potassium hexahydroxoantimonate (KSb(OH)6) of analytical grade or higher were purchased from Merck (Germany). NOM was extracted from Pahokee Peat bulk material purchased from the International Humic Substances Society (IHSS-2BS103P) following previously described protocols.11 The dissolved organic carbon (DOC) content of the extract was measured (Shimadzu TOC-L series, Japan) and found to be 1200 mg DOC L−1. A portion of the extract was freeze dried for carbon (C), nitrogen (N), hydrogen (H), and sulfur (S) analysis (Elementar VarioMacro, Elementar Analysensysteme GmbH, Germany). Biochar produced from miscanthus straw pellets pyrolyzed at 550 °C (MSP550), purchased from UK Biochar Research Centre (UKBRC, Edinburg, UK), was used for Pb removal experiments. Wood biomass and dolomite (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40) pyrolyzed at 850 °C (BCD) (University of Bologna, Italy) was used for As(V) removal experiments, and a steam activated wood char pyrolyzed at 900 °C (SAWC) (Ithaka Institute, Switzerland) was used for Sb(III) removal experiments. The optimal material selection for each metal(loid) out of 12 biochars is explained in Section 2 of the ESI.† The selected materials were washed with ultrapure water multiple times and vacuum dried before use to minimise BC derived changes to water chemistry. Details on the characterisation of the washed materials are provided in Sections 2.4 and 7 of the ESI.†
:40) pyrolyzed at 850 °C (BCD) (University of Bologna, Italy) was used for As(V) removal experiments, and a steam activated wood char pyrolyzed at 900 °C (SAWC) (Ithaka Institute, Switzerland) was used for Sb(III) removal experiments. The optimal material selection for each metal(loid) out of 12 biochars is explained in Section 2 of the ESI.† The selected materials were washed with ultrapure water multiple times and vacuum dried before use to minimise BC derived changes to water chemistry. Details on the characterisation of the washed materials are provided in Sections 2.4 and 7 of the ESI.†
2.2 Central composite design
To assess singular as well as combined effects of Ca, P, NOM, and Fe(III) phases on sorption of the selected metal(loid)s, a central composite design was generated for those four factors using the Minitab Statistical Software. pH was not selected as a factor, given that BC have strong buffering capacities and tend to increase the pH of surrounding solutions towards alkaline. Each factor was set at 3 levels (−1, 0, +1), with α = 2 (distance of axial points from the centre point). These included 16 cube points, 7 central points (identical), and 8 axial points. Accordingly, in total, 25 distinctly different background solutions were used. The axial points accounted for the end most (highest and lowest) values of each factor, while keeping the others at the ‘medium’ level. Table 1 summarises the levels of the different factors used. Table S1 in the ESI† shows the entire test matrix used. To ensure that sorption to Fe(III) phases would not overshadow BC as the sorbents of interests, Fe(III) was kept at concentrations below 6.0 mg L−1. The values for the other factors cover a more diverse range that can be encountered in surface and pore waters in soils and sediments.22| Factor mg L−1 | Levels | ||||
|---|---|---|---|---|---|
| −α (very low) | −1 (low) | 0 (medium) | +1 (high) | +α (very high) | |
| Ca | 0.4 | 48.5 | 96.6 | 144.7 | 192.8 |
| P | 0.01 | 1.51 | 3.01 | 4.51 | 6.01 |
| NOM as DOC | 0.01 | 5.01 | 10.01 | 15.01 | 20.01 |
| Fe(III) | 0.01 | 1.51 | 3.01 | 4.51 | 6.01 |
2.3 Sorption experiments
Stock solutions of 1000 mg Pb L−1, 1000 mg As(V) L−1, and 100 mg Sb(III) L−1 were freshly prepared by dissolving their corresponding salts (Pb(NO3)2, Na2HAsO4·7H2O, and K2[Sb2(C4H2O6)2]·3H2O respectively) in ultrapure water. Aqueous phase metal(loid) concentrations in contaminated areas can vary considerably. For example, 0.02 to 2.62 mg L−1 Pb, 0.07 to 15.7 mg L−1 As and 0.02 to 2.06 mg L−1 Sb concentrations was found in contaminated water close to recycling plants and legacy mining areas.23,24 Up to 7080 mg kg−1 of Pb, 410 mg kg−1 of As, and 99.4 mg kg−1 of Sb has been recorded in soils and sediments across Europe.22 Across 9 contaminated soils collected from across Austria, we found median concentrations of 170 mg kg−1 of Pb, 15 mg kg−1 of As, and 9 mg kg−1 of Sb (data not shown). In the current study, Pb, As(V), and Sb(III) were spiked at 1 mg L−1, 0.5 mg L−1, and 0.1 mg L−1, respectively. For Pb sorption experiments, MSP550 was weighed into 25 mL polystyrene tubes with subsequent addition of ultrapure water at a solid:liquid ratio of 1:1000. All four factors of Ca, NOM, P, and Fe(III) were spiked into the experimental solutions in that order from stock solutions of 1 M Ca(NO3)2·4H2O, Pahokee Peat NOM extract (1200 mg DOC L−1), 50 mM Na2HPO4·2H2O, and 10 mM Fe(NO3)3·9H2O (pH ∼ 1.0), according to the test matrix (Table S1†). After every spike, the tubes were shaken by hand to homogenise. Finally, Pb from the stock solution was added followed by equilibration for 48 hours in the dark at 150 rpm on a horizontal shaker. The pH of selected solutions (axial or end most points in the test matrix) was measured at the end of the experiments. The solutions were then filtered through 0.45 μm cellulose acetate filters (Sartorius, Germany) and acidified (∼1% wt HNO3) for subsequent analysis of Pb, Ca, Fe, and P by inductively coupled plasma optical emission spectroscopy (ICP-OES, Agilent 5100 Agilent Technologies, USA). The same experimental protocol and experimental test matrix were used for As(V) with BCD and Sb(III) with SAWC, using solid:liquid ratios of 1:2000 and 1:800, respectively. The solid:liquid ratios were optimised for each metal(loid) such that (i) at least 40% removal was achieved for >50% of the solutions, and (ii) quantification of the metal(loid)s was still possible after sorption. To minimise bias, experiments were randomised within a test matrix and conducted in duplicates. Additional control experiments without BC were set up for selected combinations. Sorption coefficients (Kd) and removal (%) were calculated based on the amount eliminated from the aqueous phase as described in Section 3 of the ESI.†
For Sb(III) experiments, certain solution conditions with and without SAWC were selected to determine the aqueous phase speciation of Sb at the end of the experiments and to check the stability of Sb(III) under oxic conditions. Sb(III) and Sb(V) speciation was analysed by high performance liquid chromatography (Agilent 1200 series, Agilent Technologies, USA) hyphenated to an inductively coupled plasma mass spectrometer (Agilent 8800 ICP-MS, Agilent Technologies, USA) following the protocol of Zheng et al.25 Details on sample preparation and instrumentation are provided in Section 4 of the ESI.† Additionally, four randomised points from the test matrix were selected for Sb(V) sorption experiments using KSb(OH)6.
2.4 Material characterisation and solid phase analysis
The specific surface area (SSA), pore volume (PV), persistent free radicals (PFR) from electron spin resonance spectroscopy, elemental composition, ash content, carbon phase speciation, and X-ray diffractograms (XRD) of the materials were analysed, and morphology (using scanning electron microscope) investigated as described in Section 7 of the ESI.† Some of the end most points in the design matrix were scaled up at the same solid:liquid ratio, such that at least 200 mg of solids were available for solid phase analysis, i.e., XRD, energy dispersive X-ray spectroscopy (EDX), and micro-X-ray fluorescence (μ-XRF). These samples were repeatedly suspended and washed with 200 mL of ultrapure water at least thrice to eliminate loosely attached phases and salts, while filtering through a vacuum filtration unit.
3. Results and discussion
3.1 Selection of sorbents and sorption mechanisms involved
The Kd of the metal(loid)s with 12 biochars are shown in Fig. S1.† Pb showed the greatest affinity towards SAWC and MSP550. As(V) sorbed only to BCD which, in contrast to the other BC, has also positive surface charges owing to the presence of charred dolomite.26 Sb(III) exhibited negligible affinity towards almost all BC other than BCD and SAWC. MSP550, BCD, and SAWC were chosen as sorbents for Pb, As(V), and Sb(III), respectively. A different well-performing BC was selected for each of the metal(loid)s to cover a broad variety of materials in the study (un-activated, a mineral-BC composite and a BC physically activated by steam). When compared to other BC reported in literature,27 MSP550 had a specific surface area and pore volume lower than 63% and 90% of the BC, respectively. BCD had a specific surface area and a pore volume greater than 70% and 80% of the BC, respectively. SAWC had a specific surface area and pore volume greater than all the BC reported in the cited study. Comparing H/OC ratios, MSP550 was more aromatic than 70% of 2640 BC screened in a recent meta-analysis, BCD was more aromatic than 42%, and SAWC was more aromatic than 98%.28Kd with the selected sorbents in ultrapure water for Pb, As(V), and Sb(III) were 60000 L kg−1, 75000 L kg−1, and 570 L kg−1, respectively (Fig. S1†).
Removal of Pb by MSP550 likely involved complexation with surface functional groups (carboxyl), Pb–π bonding, along with P/Si interactions, based on previous attenuated total reflection fourier transformation infrared spectroscopic investigations on Pb-loaded MSP550 by Soria et al.29 Precipitation and/or chelation with P/Si phases was also observed by others.10,30,31 In our study, XRD analysis of pristine and Pb loaded MSP550 did not show the formation of any new crystalline phase (Fig. S7†), but EDX spectra of MSP550 revealed the presence of appreciable amounts of Si and P at the BC surface (Fig. S11†).
Removal of As(V) using BCD can occur by precipitation as arsenic carbonates or calcium arsenates.26 XRD analysis of BCD revealed the presence of calcite (CaCO3) and brucite (Mg(OH)2) phases (Fig. S7†), and 1.8% of BCD was composed of inorganic C as carbonate (Table 2). The mechanism of As(V) sorption by BCD likely involved electrostatic interactions. The point of zero charge for charred/calcined dolomite has been reported at 10–11.1.26,32 As(V), existing as an oxyanionic species migrated to localised positive charges (pH of BCD solutions: 9.5–9.8) and thereafter was bound to BCD through inner-sphere surface complexes (corner-sharing with Ca and Mg atoms).33–35
| SSA | PV | C | H | N | S | O | Ash | OC | IC | O/OC | H/OC | PFR | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MSP550 | 14 | 0.02 | 76 | 2.7 | 0.5 | 0.2 | 10.5 | 10.2 | 81.4 | 0.11 | 0.10 | 0.39 | 1.72 × 1019 |
| BCD | 150 | 0.10 | 35 | 1.6 | 0.7 | 0.2 | 59.4 | 29.4 | 1.8 | 0.65 | 6.81 × 1014 | ||
| SAWC | 850 | 0.57 | 77 | 0.9 | 0.7 | 0.5 | 11.7 | 9.5 | 72.3 | 15.6 | 0.12 | 0.15 | n.d. |
A specific driving mechanism for Sb(III) sorption by SAWC was not identified. However SAWC's high specific surface area (850 m2 g−1) and porosity (0.57 cm3 g−1), resulting from steam activation, probably increased the physical entrapment of Sb(III) molecules, compared to the other tested BC.36 XRD pattern of SAWC showed broad diffraction peaks, which are characteristics of an amorphous carbonaceous structure (Fig. S7†). A sharp peak at 2θ of 26.6° indicated the growth of a crystalline graphitic phase which is a result of partial devolatilization from steam activation.37,38 This high degree of graphitisation indicates that Sb–π-interactions could occur.39 EDX spectra of SAWC samples revealed the presence of Al and Si, and traces of Mg (Fig. S11†), suggesting traces of phyllosilicate-like constituents, which can drive Sb(III) sorption through inner-sphere complex formation.40
3.2 Pb removal was more strongly affected by water chemistry compared to As(V) and Sb(III)
The removal of metal(loid)s varied substantially in the presence of other ions and phases (Fig. 1), compared to ultrapure water (Fig. S1†). Across different water matrices, the variation in metal(loid) removal by the respective BC followed the order: Pb (29–100%) > As(V) (37–92%) > Sb(III) (20–70%). Sorption coefficients normalised to the sorbent mass (Kd) for Pb, As(V), and Sb(III) stretched across 3.0, 1.5, and 1.0 orders of magnitude, respectively (Fig. 1a). Sorption coefficients normalised to the specific surface area (KSSA) resulted in similar trends, highlighting that the specific surface area and available sorption sites alone were not driving these differences in variation (Fig. 1b). The trends imply that in the given metal(loid)-BC systems, the removal of Pb by MSP550 was most strongly affected by the solution composition.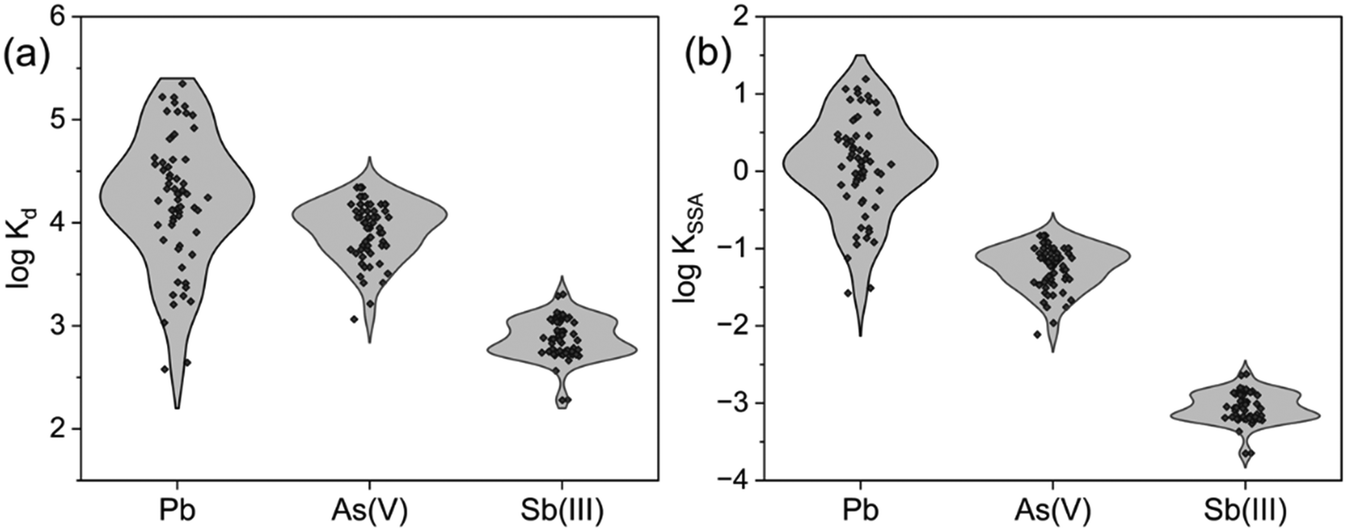 | ||
| Fig. 1 Violin plots depicting variations in (a) logKd and (b) logKSSA for Pb, As(V), and Sb(III) across different ionic composition in the experimental test matrix. Kd units: L kg−1; KSSA units: L m−2. | ||
The pH of the solutions at the end of the experiments for Pb ranged from 6.7 to 8.1 (MSP550), for As(V) from 9.5 to 9.8 (BCD) and for Sb(III) from 7.2 to 7.8 (SAWC). At such pH, Fe(III), introduced as Fe3+ ions (acidic spike), forms poorly ordered polymeric phases of Fe(III)(oxyhydr)oxide, such as ferrihydrite,41,42 which can constitute an additional sorbent phase for metal(loid)s. This phase can sorb to the BC or form organo-mineral phases with NOM by a co-precipitation mechanism.19,43,44 Both P and Ca can change the formation and reactivity of these phases. The effects of singular and combined factors on the removal of targeted metal(loid)s by the respective BC will be discussed in detail in the following sections.
3.3 Ca improved Pb removal by MSP550 in NOM containing solutions
At the pH range in the Pb-MSP550 experiments (6.7 to 8.1), Pb exists primarily as a combination of (i) positively charged Pb2+ and PbOH+, (ii) organic matter complexed Pb, and (iii) a minimal proportion (1–2%) complexed to P, as estimated from equilibrium calculations performed in Visual MINTEQ 3.1. Pb exhibits affinity primarily for N containing groups in NOM, specifically amine groups such as aminoquinone, amino sugars, and amino acids, and phenolic groups, but additionally can complex with carboxylate groups.45,46 The Pahokee Peat NOM extract used in all experiments contained 43.1% C, 3.8% N, 0.9% S, and 3.9% H by weight. Considering 20–30% of total N to be amino acid derived N,47,48 Pb would be in excess to N binding sites and thus partially also complex with O sites. In the absence of NOM, 99–100% of Pb was removed (logKd > 5.3). Increasing NOM concentrations to 5 mg DOC L−1, decreased the removal of Pb to 95% ± 5% (n = 16). At NOM concentrations of 10 or 15 mg DOC L−1 combined with low Ca concentrations of 0.4 or 48.5 mg Ca L−1, the removal of Pb fell below 80% (logKd < 3.5) (Fig. 2). Even at high NOM levels (≥10 mg DOC L−1), as soon as Ca levels were high (>48.5 mg Ca L−1), Pb removal increased above 80%. Ca could have competed with Pb for binding sites in NOM, thereby increasing the removal of free Pb2+ by MSP550. However, since Pb has a high affinity for specific ligands in NOM46,49 and Ca undergoes more non-specific binding with NOM,50 this would not have been driving the increased Pb removal in Ca and NOM containing solutions in our experiments.
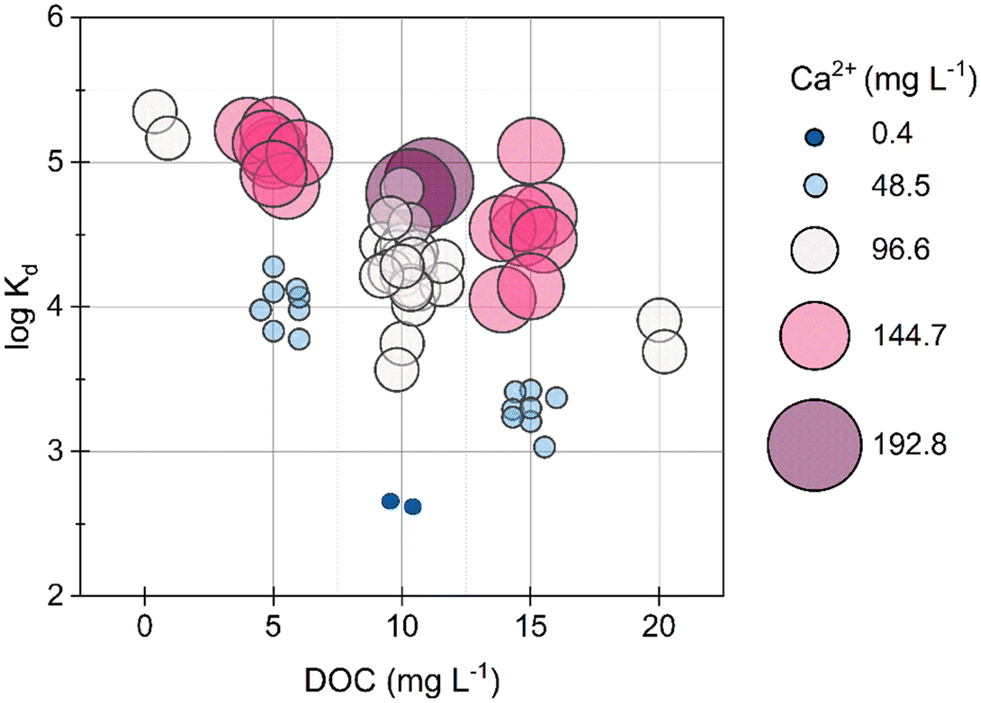 | ||
| Fig. 2 Multivariable bubble plots of logarithmic sorption coefficients of Pb with DOC (as a measure of NOM content). Increasing bubble size and colour gradient shifting from deep blue to purple indicates increasing Ca concentrations. | ||
The removal of total Fe with Pb across samples correlated well (Fig. 3). In solutions containing Fe(III) and NOM, Fe(III)–NOM phases are formed which are known to exhibit a strong affinity towards cations such as Pb.43,44 Increasing Ca concentrations give rise to a micrometric network of NOM and Fe(III) associated NOM.17 These phases can co-precipitate or interact with the surface of MSP550, as was confirmed from the EDX spectra of the thoroughly washed and dried MSP550 solids (Fig. S11†) i.e., elevated levels of Ca and the presence of Fe were observed on MSP550 when excess Ca was present in solution.
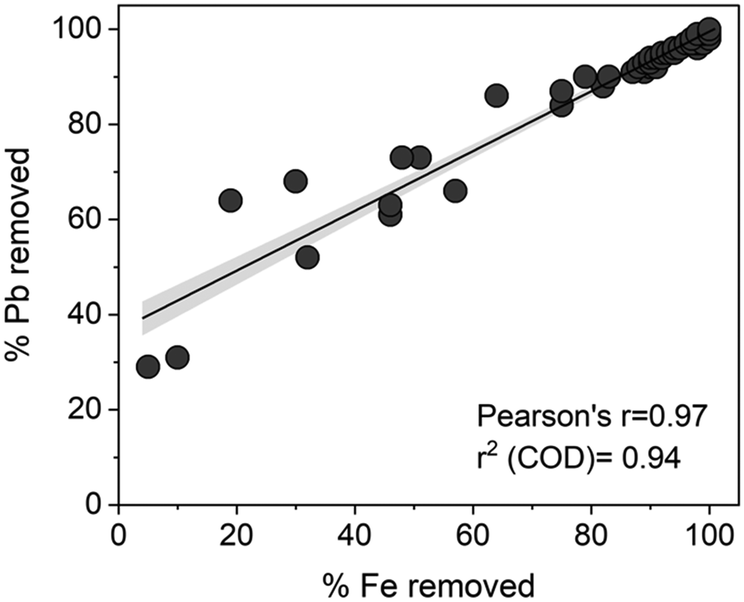 | ||
| Fig. 3 Scatterplot showing correlation between Pb and total Fe removal. 95% confidence band included, with linear regression line. The concurrent presence of Fe(III) and NOM gives rise to Fe(III)–NOM complexes which can stabilise Fe(III) in solution at the studied environmentally relevant pH.51 | ||
μ-XRF maps showed Fe deposition on MSP550 even in the absence of Ca (Fig. S12b†), but clusters of Fe and Ca were denser once the Ca concentration was increased to 192.8 mg Ca L−1 (Fig. S12c†). The simultaneous removal of Pb and total Fe was especially enhanced at high Ca concentrations. This further suggests that Pb was embedded in a network of organo-mineral phases which facilitated its removal by MSP550. Pb removal at the highest concentrations of Ca in the experimental test matrix (axial points in the central composite design) was higher (98%/logKd 4.7) than in Ca-free solutions (30%/logKd 2.6) at the same Fe(III), NOM, and P conditions. This highlights that Fe(III)–NOM–Ca phases were more active in Pb removal than Fe(III)–NOM phases. At the highest condition of Fe(III) with 6.0 mg Fe(III) L−1, Pb removal was stronger (97.3%/logKd 4.6) than in Fe(III)-free solutions (81%/logKd 3.7) under the same Ca, NOM, and P conditions. This highlights that the contribution of Fe(III)–NOM–Ca phases surpassed NOM–Ca phases in Pb removal.
Thus, interactions among Pb, Ca, Fe(III), and NOM collectively facilitate Pb removal by MSP550. Therein, removal of Pb by a BC such as MSP550 is highest when NOM levels are low, or a combination of high levels of NOM and Ca exist.
3.4 Ca increased As(V) removal by BCD
The variation in As(V) removal was lower than that of Pb, suggesting an overall high affinity of As(V) to BCD, less influenced by the solution composition. 90% of the solutions showed >80% As(V) removal (Kd > 5000 L kg−1; logKd > 3.7) contained at least 96.6 mg Ca L−1, suggesting that the primary factor affecting As(V) sorption was Ca (Fig. 4). Ca was consistently depleted from solutions in the As(V)-BCD experiments compared to the other metal(loid)-BC experiments (Fig. S5†). At the end most conditions in the experimental test matrix, EDX spectra and μ-XRF maps revealed more Ca clusters on BCD at 192.8 mg Ca L−1 compared to the Ca-free system (Fig. S11 and S13†) under the same conditions of Fe(III), NOM, and P. Further, 90% As(V) was removed with 192.8 mg Ca L−1 (Kd: 18571 L kg−1), whereas only 41% As(V) was removed in the Ca-free system (Kd: 1397 L kg−1). At the pH of As(V)-BCD experiments (pH: 9.5–9.8), As(V) exists primarily as the arsenate anion (HAsO42−). The influence of Ca on As removal was thus likely caused by (i) cation bridging between arsenate anions and oxygen containing sites, and (ii) partial screening of negative surface charges over BCD.9,52
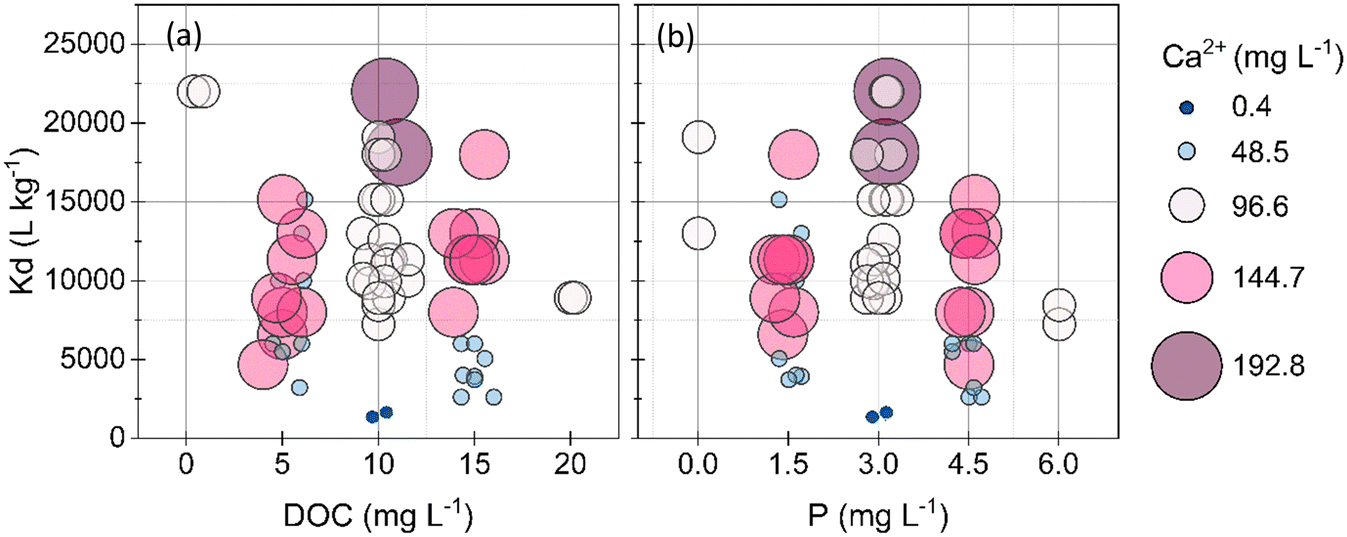 | ||
| Fig. 4 Multivariable bubble plots of sorption coefficients of As(V) with (a) DOC (as a measure of NOM content), and (b) P. Increasing bubble size and colour gradient shifting from deep blue to purple indicates increasing ca concentrations. | ||
At the highest investigated concentrations of NOM (20.0 mg DOC L−1) and P (6.0 mg P L−1), BCD still sorbed up to 83% of As(V), indicating that NOM and P did not strongly inhibit As(V) sorption. However, an inhibitory role of NOM on As(V) sorption by BCD was observed only at low Ca (48.5 mg Ca L−1) coupled with low Fe(III) (1.5 mg Fe(III) L−1) concentrations. Under these low concentrations of Ca and Fe(III), in solutions with 5.0 mg DOC L−1 BCD removed 75% to 86% of As(V) (Kd: 6000 to 12571 L kg−1), whereas in solutions with 15.0 mg DOC L−1, BCD only removed 57% to 66% As(V) (Kd: 2615 to 3857 L kg−1). Under low concentrations of Ca and Fe(III), P also inhibited As(V) sorption. In these solutions with 1.5 mg P L−1, BCD removed 66% to 86% of As(V) (Kd: 3857 to 12571 L kg−1) and in solutions with 4.5 mg P L−1, BCD removed 57% to 75% of As(V) (Kd: 2615 to 6000 L kg−1). Bridging with Ca or Fe cations is the primary means of As(V) binding to NOM, and direct binding only secondary.53–55 The lower removal of As(V) in the presence of NOM with low Ca and Fe(III) concentrations, can thus be explained by negatively charged humic and fulvic acids competing for positively charged sorption sites in BCD, rather than As(V) complexation with NOM.56,57
Phosphate anions were effectively removed by BCD, similarly to arsenate anions. In 94% of the solutions, P removal was >95% (Fig. S6†). μ-XRF elemental maps of BCD at the end of experiments showed an increase of P compared to pristine BCD (Fig. S13†). Other than at low Ca concentrations, excess P (6.0 mg P L−1) did not hinder As(V) removal. The feedstock for BCD contained 40% dolomite and XRD analysis revealed the dominating presence of calcite and brucite in the sorbent matrix, which is likely to drive the efficient removal of As(V) and P anions. Sorption sites were therefore available in excess compared to As(V) and P anions in the current study. In highly P rich systems, where As(V) to P molar ratios are lower than our current value of 1:29, competition effects can be more drastic.58,59 Lack of competition effects could also be ascribed to P precipitation as Ca–P phases (such as hydroxyapatite and calcium phosphate)60 at the high pH of BCD solutions (∼9.5–9.8) that drove P depletion from solution independently. The density of P on BCD at high Ca concentrations was higher than at low Ca concentrations (Fig. S13†), but Ca–P phases were too low in concentration (<1% by mass) to be detected by XRD and/or may have had low crystallinity for our experimental duration.
3.5 Oxidation to Sb(V) limits Sb(III) removal by SAWC
The Sb(III) redox state in the oxic experiments was ensured by introducing Sb(III) as a tartrate complex, which is an anionic dimer upon dissociation in water.61 Sb(III) oxidation solely by O2 is slow,62 but active substances such as Fe(III)–NOM can accelerate Sb(III) oxidation.63 In this study, SAWC oxidised Sb(III) to the more mobile Sb(V) in all solutions tested, which explains the generally low removal of Sb(III) (2.3 < logKd < 3.3). The BC related oxidation of Sb is illustrated in Fig. 5 which is a representative example of measured HPLC-ICP-MS chromatograms of the aqueous phase with and without SAWC. Low Sb(V) sorption affinity was independently confirmed from an additional sorption experiment with Sb(V), wherein Sb(V) was not sorbed by SAWC at all (Table S3†). Other studies have also observed limited interactions between BC and Sb(V) compared to Sb(III).39,64
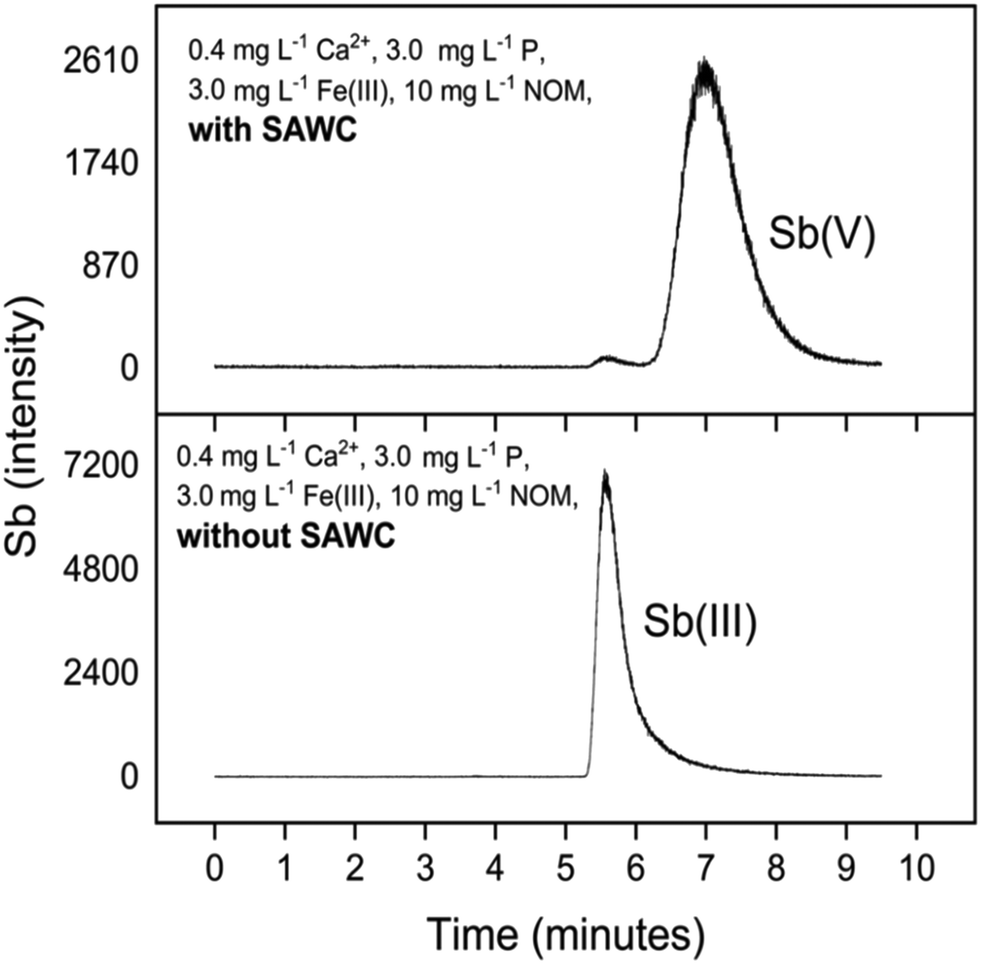 | ||
| Fig. 5 HPLC-ICP-MS chromatograms of Sb(III)/(V) (complexed to citrate as a stabilising agent) at the end of sorption experiments with SAWC, and control experiments without SAWC under the same solution matrix. | ||
Oxidizing moieties such as solid phase PFR and quinoid groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in BC can promote Sb(III) oxidation, thereby hindering Sb removal. As an example, from electron spin resonance spectroscopy, MSP550 had high concentrations of PFR (1.72 × 1019 spins per g) (Table 2) that could oxidise Sb(III), explaining its negligible Sb(III) sorption affinity. Out of all 12 BC screened in the preliminary experiments, except BCD, only SAWC exhibited an affinity for Sb(III) (Fig. S1†). PFR were not detectable in SAWC, but Sb(III) oxidation still occurred (Fig. 5), albeit to a lower extent than with the other BC. SAWC was pyrolyzed at 900 °C, wherein the thermal reorganisation of organic structures lowers the PFR content,65 making quinoid moieties the likely driver for Sb(III) oxidation.66–68 These results corroborate findings by Zhong et al.69,70 who demonstrated that electron accepting moieties i.e., PFR and quinoid groups in rice husk biochar were able to oxidise As(III) to As(V). Therefore, BC induced oxidation of metal(loid)s such as Sb(III) can limit their sorption by BC.
O) in BC can promote Sb(III) oxidation, thereby hindering Sb removal. As an example, from electron spin resonance spectroscopy, MSP550 had high concentrations of PFR (1.72 × 1019 spins per g) (Table 2) that could oxidise Sb(III), explaining its negligible Sb(III) sorption affinity. Out of all 12 BC screened in the preliminary experiments, except BCD, only SAWC exhibited an affinity for Sb(III) (Fig. S1†). PFR were not detectable in SAWC, but Sb(III) oxidation still occurred (Fig. 5), albeit to a lower extent than with the other BC. SAWC was pyrolyzed at 900 °C, wherein the thermal reorganisation of organic structures lowers the PFR content,65 making quinoid moieties the likely driver for Sb(III) oxidation.66–68 These results corroborate findings by Zhong et al.69,70 who demonstrated that electron accepting moieties i.e., PFR and quinoid groups in rice husk biochar were able to oxidise As(III) to As(V). Therefore, BC induced oxidation of metal(loid)s such as Sb(III) can limit their sorption by BC.
The variability in Sb(III) removal was the least compared to Pb and As(V), with Kd extending across only one order of magnitude and never exceeding logKd 3.3. Other than the strong influence of Fe(III) (Fig. S3†), no singular or combined effect of Ca, P, and NOM on Sb(III) sorption by SAWC was evident.
Increasing Fe(III) concentrations in solutions decreased Sb(III) sorption. Although we were unable to conclusively determine the driving mechanism behind this phenomenon, from EDX, μ-XRF, and HPLC-ICP-MS analyses we were able to identify two likely mechanisms. Compared to Fe(III)-free solutions, we observed spatially densely distributed Fe on SAWC upon contact with 6.0 mg Fe(III) L−1 solutions (Fig. S14†). Both EDX and μ-XRF spectra semi-quantitatively verified the increased surface Fe content (Fig. S11 and S14†). Fe(III)(oxyhydr)oxide phases formed in situ could block pores in SAWC, thereby decreasing the accessible pore space within the BC. The porosity assisted sorption of Sb(III) molecules at sites within the BC pore network could thus be hindered in the presence of Fe(III). The inhibitory effect of Fe(III) on Sb(III) removal can also be linked to changes in Sb redox state. Up to 22% Sb(V) was formed in control experiments at 6.0 mg Fe(III) L−1 without SAWC, as observed from HPLC-ICP-MS analysis of the solutions after 48 hours in the dark (Fig. S4†). Oxic systems containing Fe(III)(oxyhydr)oxides with NOM can catalyse Sb(III) oxidation processes.14,71 Increased Sb(V) formation mediated by adsorption–oxidation processes via Fe(III)(oxyhydr)oxide phases could thereby decrease the sorption of Sb by SAWC. Our results underline that in addition to BC, Fe(III) phases in solution are capable of catalysing oxidation processes and decreasing Sb retention by BC.
4. Conclusions
This study investigated the combined effects of Ca, NOM, P, and Fe(III) on metal(loid) removal from oxic water by BC using a central composite design. We found that Fe(III)–NOM–Ca phases bound to BC are an effective sorbent for Pb and the simultaneous presence of Ca, Fe(III), and NOM can promote the removal of Pb, even in waters having higher NOM concentrations (10–20 mg DOC L−1).A calcareous BC, such as the charred biomass and dolomite blend that was used in this study, can be an effective sorbent for anionic As(V) under diverse solution conditions. Competition by other anions such as phosphate, humics, or fulvics limits As(V) sorption only when Ca and/or Fe(III) concentrations are low.
Sorption of Sb(III) to conventional BC is minimal, and a BC with superior specific surface area and microporosity can be used in water with low levels of Sb(III) contamination (<100 μg L−1) and low levels of Fe(III). Fe(III) hindered Sb(III) sorption, which may be either due to (i) reduced accessibility of sorption sites in the microporous BC by Fe(III)/Fe(III)–NOM, and/or (ii) Fe(III) mediated oxidation to the more mobile Sb(V). Most BC, even with low amounts of PFR, oxidise Sb(III) to the more mobile Sb(V) because of redox active moieties such as quinoid groups on the BC surface. Since Sb(V) is less toxic than Sb(III), this can be a treatment strategy for less contaminated waters. However, natural redox equilibrium processes are dynamic and mobile Sb(V) may reach an anoxic region or interact with a reductant and be reduced to the more toxic Sb(III) again.
Interactions of dissolved and particulate solution constituents can strongly influence sorbent performance. The sorption of metal(loid)s by BC strongly depends on the ionic composition of the solution. For best performance, variable solution compositions and ideally a tailored optimisation for the targeted water matrix need to be included at the sorbent selection or development stage. We advocate for the development of a comprehensive knowledge database allowing an informed material selection based on complex solution compositions.
Abbreviations
| BC | Biochar |
| MSP550 | Miscanthus straw pellets pyrolyzed at 550 °C |
| BCD | Charred biomass dolomite composite |
| SAWC | Steam activated wood char |
| Pb | Lead |
| As | Arsenic |
| Sb | Antimony |
| Ca | Calcium |
| P | Phosphorus |
| NOM | Natural organic matter |
| DOC | Dissolved organic carbon |
| Fe | Iron |
| C | Carbon |
| H | Hydrogen |
| N | Nitrogen |
| S | Sulphur |
| O | Oxygen |
| SSA | Specific surface area |
| PV | Pore volume |
| PFR | Persistent free radicals |
| XRD | X-ray diffraction |
| EDX | Energy dispersive X-ray spectroscopy |
| μ-XRF | Micro-X-ray fluorescence |
| K d | Sorption coefficient normalised to sorbent mass |
| K SSA | Sorption coefficient normalised to specific surface area |
Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors wish to thank Carlotta Carlini from University of Bologna, Department of Physics and Astronomy for providing the BCD material, and the Ithaka Institute for carbon strategies for providing the SAWC material. The authors would like to acknowledge University of Vienna's Facility for Nanostructure Research for scanning electron microscope access, and Marc Pignitter from the University of Vienna's Faculty of Chemistry for electron spin resonance spectroscopy access. This research was funded by the Bundesministerium für Klimaschutz, Umwelt, Energie, Mobilität, Innovation und Technologie (BMK), management by Kommunalkredit Public Consulting GmbH (grant number B820017).References
- L. Beesley, E. Moreno-Jiménez, J. L. Gomez-Eyles, E. Harris, B. Robinson and T. Sizmur, Environ. Pollut., 2011, 159, 3269–3282 CrossRef CAS PubMed.
- J. J. Pignatello, W. A. Mitch and W. Xu, Environ. Sci. Technol., 2017, 51, 8893–8908 CrossRef CAS PubMed.
- X. Zhu, X. Wang and Y. S. Ok, J. Hazard. Mater., 2019, 378, 120727 CrossRef CAS PubMed.
- B. Qiu, X. Tao, H. Wang, W. Li, X. Ding and H. Chu, J. Anal. Appl. Pyrolysis, 2021, 155 Search PubMed.
- H. Lu, W. Zhang, Y. Yang, X. Huang, S. Wang and R. Qiu, Water Res., 2012, 46, 854–862 CrossRef CAS PubMed.
- S. S. A. Alkurdi, I. Herath, J. Bundschuh, R. A. Al-Juboori, M. Vithanage and D. Mohan, Environ. Int., 2019, 127, 52–69 CrossRef CAS PubMed.
- L. Wang, J. Wang, Z. Wang, C. He, W. Lyu, W. Yan and L. Yang, Chem. Eng. J., 2018, 354, 623–632 CrossRef CAS.
- X. Yang, S. Yang, S. Yang, J. Hu, X. Tan and X. Wang, Chem. Eng. J., 2011, 168, 86–93 CrossRef CAS.
- E. Smith, R. Naidu and A. M. Alston, J. Environ. Qual., 2002, 31, 557–563 CAS.
- S. Cairns, S. Chaudhuri, G. Sigmund, I. Robertson, N. Hawkins, T. Dunlop and T. Hofmann, Environ. Technol. Innovation, 2021, 24, 101961 CrossRef CAS.
- S. Chaudhuri, G. Sigmund, S. E. Bone, N. Kumar and T. Hofmann, Environ. Sci. Technol., 2022, 56, 11354–11362 CrossRef CAS PubMed.
- A. Kappler, M. L. Wuestner, A. Ruecker, J. Harter, M. Halama and S. Behrens, Environ. Sci. Technol. Lett., 2014, 1, 339–344 CrossRef CAS.
- E. Neubauer, F. V. D. Kammer and T. Hofmann, Water Res., 2013, 47, 2757–2769 CrossRef CAS PubMed.
- N. Belzile, Y. W. Chen and Z. Wang, Chem. Geol., 2001, 174, 379–387 CrossRef CAS.
- P. Wilfert, P. S. Kumar, L. Korving, G. J. Witkamp and M. C. M. Van Loosdrecht, Environ. Sci. Technol., 2015, 49, 9400–9414 CrossRef CAS PubMed.
- Y. Bao, N. S. Bolan, J. Lai, Y. Wang, X. Jin, M. B. Kirkham, X. Wu, Z. Fang, Y. Zhang and H. Wang, Crit. Rev. Environ. Sci. Technol., 2022, 52, 4016–4037 CrossRef CAS.
- A. Beauvois, D. Vantelon, J. Jestin, C. Rivard, M. Bouhnik-Le Coz, A. Dupont, V. Briois, T. Bizien, A. Sorrentino, B. Wu, M. S. Appavou, E. Lotfi-Kalahroodi, A. C. Pierson-Wickmann, M. Davranche and Environ Sci, NANO, 2020, 7, 2833–2849 CAS.
- A. Beauvois, D. Vantelon, J. Jestin, M. Bouhnik-Le Coz, C. Catrouillet, V. Briois, T. Bizien and M. Davranche, J. Hazard. Mater., 2021, 404, 124127 CrossRef CAS PubMed.
- R. Mikutta, D. Lorenz, G. Guggenberger, L. Haumaier and A. Freund, Geochim. Cosmochim. Acta, 2014, 144, 258–276 CrossRef CAS.
- J. Yan, T. Jiang, Y. Yao, S. Lu, Q. Wang and S. Wei, J. Environ. Sci., 2016, 42, 152–162 CrossRef CAS PubMed.
- D. Sedlak, Environ. Sci. Technol., 2018, 52, 3327–3328 CrossRef CAS PubMed.
- R. Salminen, M. J. Batista, M. Bidovec, A. Demetriades, B. De Vivo, W. De Vos, M. Duris, A. Gilucis, V. Gregorauskiene, J. Halamić and P. Heitzmann, Geochemical atlas of Europe, part 1, background information, methodology and maps, 2005 Search PubMed.
- S. Mykolenko, V. Liedienov, M. Kharytonov, N. Makieieva, T. Kuliush, I. Queralt, E. Marguí, M. Hidalgo, G. Pardini and M. Gispert, Environ. Pollut., 2018, 237, 569–580 CrossRef CAS PubMed.
- T. Lomaglio, N. Hattab-Hambli, A. Bret, F. Miard, D. Trupiano, G. S. Scippa, M. Motelica-Heino, S. Bourgerie and D. Morabito, J. Geochem. Explor., 2017, 182, 138–148 CrossRef CAS.
- J. Zheng, A. Iijima and N. Furuta, J. Anal. At. Spectrom., 2001, 16, 812–818 RSC.
- Y. Salameh, A. B. Albadarin, S. Allen, G. Walker and M. N. M. Ahmad, Chem. Eng. J., 2015, 259, 663–671 CrossRef CAS.
- H. Li, X. Dong, E. B. da Silva, L. M. de Oliveira, Y. Chen and L. Q. Ma, Chemosphere, 2017, 178, 466–478 CrossRef CAS PubMed.
- J. A. Ippolito, L. Cui, C. Kammann, N. Wrage-Mönnig, J. M. Estavillo, T. Fuertes-Mendizabal, M. L. Cayuela, G. Sigua, J. Novak, K. Spokas and N. Borchard, Biochar, 2020, 2, 421–438 CrossRef.
- R. I. Soria, S. A. Rolfe, M. P. Betancourth and S. F. Thornton, Heliyon, 2020, 6(11), E05388 CrossRef CAS PubMed.
- Y. Fan, H. Wang, L. Deng, Y. Wang, D. Kang, C. Li and H. Chen, Environ. Res., 2020, 191, 110030 CrossRef CAS PubMed.
- H. Lu, W. Zhang, Y. Yang, X. Huang, S. Wang and R. Qiu, Water Res., 2012, 46, 854–862 CrossRef CAS PubMed.
- F. L. Braghiroli, I. L. Calugaru, C. Gonzalez-Merchan, C. M. Neculita, H. Bouafif and A. Koubaa, Miner. Eng., 2020, 151, 106310 CrossRef CAS.
- V. G. Alexandratos, E. J. Elzinga and R. J. Reeder, Geochim. Cosmochim. Acta, 2007, 71, 4172–4187 CrossRef CAS.
- X. Ou, X. Liu, W. Liu, W. Rong, J. Li and Z. Lin, Environ. Sci.: Nano, 2018, 5, 2570–2578 RSC.
- S. Tresintsi, K. Simeonidis, M. Katsikini, E. C. Paloura, G. Bantsis and M. Mitrakas, J. Hazard. Mater., 2014, 265, 217–225 CrossRef CAS PubMed.
- T. Sizmur, T. Fresno, G. Akgül, H. Frost and E. Moreno-Jiménez, Bioresour. Technol., 2017, 246, 34–47 CrossRef CAS PubMed.
- A. I. Osman, C. Farrell, A. H. Al-Muhtaseb, J. Harrison and D. W. Rooney, Sci. Rep., 2020, 10, 1–13 CrossRef PubMed.
- A. U. Rajapaksha, S. S. Chen, D. C. W. Tsang, M. Zhang, M. Vithanage, S. Mandal, B. Gao, N. S. Bolan and Y. S. Ok, Chemosphere, 2016, 148, 276–291 CrossRef CAS PubMed.
- X. Cui, Q. Ni, Q. Lin, K. Y. Khan, T. Li, M. B. Khan, Z. He and X. Yang, Environ. Pollut., 2017, 229, 394–402 CrossRef CAS PubMed.
- A. G. Ilgen and T. P. Trainor, Environ. Sci. Technol., 2012, 46, 843–851 CrossRef CAS PubMed.
- U. Schwertmann, J. Friedl and H. Stanjek, J. Colloid Interface Sci., 1999, 209, 215–223 CrossRef CAS PubMed.
- J. S. Weatherill, K. Morris, P. Bots, T. M. Stawski, A. Janssen, L. Abrahamsen, R. Blackham and S. Shaw, Environ. Sci. Technol., 2016, 50, 9333–9342 CrossRef CAS PubMed.
- H. Du, Q. Huang, M. Lei and B. Tie, ACS Earth Space Chem, 2018, 2, 556–564 CrossRef CAS.
- Z. Zhao, L. Yao, J. Li, X. Ma, L. Han, Z. Lin and S. Guan, Environ. Sci. Pollut. Res., 2022, 29, 21561–21575 CrossRef CAS PubMed.
- J. Adusei-Gyamfi, B. Ouddane, L. Rietveld, J. P. Cornard and J. Criquet, Water Res., 2019, 160, 130–147 CrossRef CAS PubMed.
- S. E. Cabaniss, Environ. Sci. Technol., 2011, 45, 3202–3209 CrossRef CAS PubMed.
- K. A. Thorn and L. G. Cox, Org. Geochem., 2009, 40, 484–499 CrossRef CAS.
- A. Vairavamurthy and S. Wang, Environ. Sci. Technol., 2002, 36, 3050–3056 CrossRef CAS PubMed.
- S. E. Cabaniss, Environ. Sci. Technol., 2009, 43, 2838–2844 CrossRef CAS PubMed.
- J. P. Pinheiro, A. M. Mota and M. F. Benedetti, Environ. Sci. Technol., 1999, 33, 3398–3404 CrossRef CAS.
- T. Karlsson and P. Persson, Chem. Geol., 2012, 322–323, 19–27 CrossRef CAS.
- S. Klitzke and F. Lang, J. Environ. Qual., 2009, 38, 933–939 CrossRef CAS PubMed.
- A. Aftabtalab, J. Rinklebe, S. M. Shaheen, N. K. Niazi, E. Moreno-Jiménez, J. Schaller and K. H. Knorr, Chemosphere, 2022, 286, 131790 CrossRef CAS PubMed.
- V. Lenoble, D. H. Dang, M. Loustau Cazalet, S. Mounier, H. R. Pfeifer and C. Garnier, Talanta, 2015, 134, 530–537 CrossRef CAS PubMed.
- P. Sharma, J. Ofner and A. Kappler, Environ. Sci. Technol., 2010, 44, 4479–4485 CrossRef CAS PubMed.
- G. Cornelissen and Ö. Gustafsson, Environ. Pollut., 2006, 141(3), 526–531 CrossRef CAS PubMed.
- A. D. Redman, D. L. Macalady and D. Ahmann, Environ. Sci. Technol., 2002, 36, 2889–2896 CrossRef CAS PubMed.
- Md. A. Rahman, D. Lamb, M. M. Rahman, M. M. Bahar and P. Sanderson, ACS Omega, 2022, 7, 101–117 CrossRef CAS PubMed.
- K. Yoon, D.-W. Cho, D. C. W. Tsang, N. Bolan, J. Rinklebe and H. Song, Bioresour. Technol., 2017, 246, 69–75 CrossRef CAS PubMed.
- H. R. Sindelar, M. T. Brown and T. H. Boyer, Chemosphere, 2015, 138, 218–224 CrossRef CAS PubMed.
- X. Li, T. Reich, M. Kersten and C. Jing, Environ. Sci. Technol., 2019, 53, 5221–5229 CrossRef CAS PubMed.
- A. K. Leuz and C. A. Johnson, Geochim. Cosmochim. Acta, 2005, 69, 1165–1172 CrossRef CAS.
- Y. Wang, L. Kong, M. He, W. Ouyang, C. Lin and X. Liu, ACS Earth Space Chem., 2020, 4, 661–671 CrossRef CAS.
- M. Vithanage, A. U. Rajapaksha, M. Ahmad, M. Uchimiya, X. Dou, D. S. Alessi and Y. S. Ok, J. Environ. Manage., 2015, 151, 443–449 CrossRef CAS PubMed.
- J. Yang, B. Pan, H. Li, S. Liao, D. Zhang, M. Wu and B. Xing, Environ. Sci. Technol., 2016, 50, 694–700 CrossRef CAS PubMed.
- H. Chen, Y. Gao, J. Li, C. Sun, B. Sarkar, A. Bhatnagar, N. Bolan, X. Yang, J. Meng, Z. Liu, H. Hou, J. W. C. Wong, D. Hou, W. Chen and H. Wang, Biochar, 2022, 4(1), 37 CrossRef CAS.
- L. Klüpfel, M. Keiluweit, M. Kleber and M. Sander, Environ. Sci. Technol., 2014, 48, 5601–5611 CrossRef PubMed.
- Y. Zhang, X. Xu, P. Zhang, L. Zhao, H. Qiu and X. Cao, Chemosphere, 2019, 232, 273–280 CrossRef CAS PubMed.
- D. Zhong, Z. Zhao, Y. Jiang, X. Yang, L. Wang, J. Chen, C. Y. Guan, Y. Zhang, D. C. W. Tsang and J. C. Crittenden, Water Res., 2020, 183, 116106 CrossRef CAS PubMed.
- D. Zhong, Y. Jiang, Z. Zhao, L. Wang, J. Chen, S. Ren, Z. Liu, Y. Zhang, D. C. W. Tsang and J. C. Crittenden, Environ. Sci. Technol., 2019, 53, 9034–9044 CrossRef CAS PubMed.
- A. K. Leuz, H. Mönch and C. A. Johnson, Environ. Sci. Technol., 2006, 40, 7277–7282 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ew00726j |
| This journal is © The Royal Society of Chemistry 2024 |