
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContrasted redox-dependent structural control on Fe isotope fractionation during its adsorption onto and assimilation by heterotrophic soil bacteria†
Aridane G.
González
 *a,
Franck
Poitrasson
b,
Felix
Jiménez-Villacorta
c,
Liudmila S.
Shirokova
bd and
Oleg S.
Pokrovsky
be
*a,
Franck
Poitrasson
b,
Felix
Jiménez-Villacorta
c,
Liudmila S.
Shirokova
bd and
Oleg S.
Pokrovsky
be
aInstituto de Oceanografía y Cambio Global, IOCAG, Universidad de Las Palmas de Gran Canaria, ULPGC, Spain. E-mail: aridane.gonzalez@ulpgc.es; Tel: +34 928 45 44 51
bGéosciences Environnement Toulouse (GET), CNRS UMR 5563, UPS-IRD-CNES 14-16, Avenue Edouard Belin, 31400, Toulouse, France
cESS Bilbao, Parque Tecnológico Bizkaia, Nave 201, 48170 Zamudio, Spain
dN.P. Laverov Federal Center for Integrated Arctic Research (FCIArctic), Russian Academy of Sciences, Arkhangelsk, Russia
eBIO-GEO-CLIM Laboratory, National Research Tomsk State University, Tomsk, Russia
First published on 7th December 2023
Abstract
Despite the importance of structural control on metal stable isotope fractionation in inorganic and abiotic systems, the link between metal structural changes and related isotopic fractionation during reactions with organic surfaces and live cells remains poorly established. We conducted reversible adsorption of Fe(II) and Fe(III) on the surface of exopolysaccharide (EPS)-rich and EPS-poor Pseudomonas aureofaciens, and we allowed Fe intracellular uptake by growing cells. We analyzed the Fe isotopic composition of the remaining fluid and cell biomass, and compared the isotopic fractionation during adsorption and assimilation reaction with relative changes in Fe structural status between aqueous solution and bacterial cells, based on available and newly collected X-ray absorption spectroscopy (XAS) observations. Iron(III) adsorption onto P. aureofaciens at 2.8 ≤ pH ≤ 6.0 produced an enrichment of the cell surface in heavier isotopes with Δ57Fecell-solution ranging from +0.7 to +2.1‰, without a link to pH in EPS-rich cultures. In contrast, the magnitude of isotopic fractionation increased with pH in EPS-poor cultures. Iron(II) adsorption produced an even larger enrichment of the cell surface in heavier isotopes, by up to 3.2‰, tentatively linked to Fe(III) hydroxide precipitation. Intracellular assimilation of Fe(II) favored heavier isotopes and led to Δ57Fecell-solution of +0.8‰. In addition, Fe(III) cellular uptake produced an enrichment of the bacterial biomass in lighter isotopes with Δ57Fecell-solution of −1‰. The XAS analyses demonstrated the dominance of Fe(III)–phosphate complexes both at the cell surface and in the cell interior. We suggest that heavier isotope enrichment of the cell surface relative to the aqueous solution is due to strong Fe(III)–phosphoryl surface complexes and Fe complexation to ligands responsible for metal transfer from the surface to the inner cell. In case of Fe(II) adsorption or assimilation, its partial oxidation within the cell compartments may lead to cell enrichment in heavier isotopes. In contrast, loss of symmetry of assimilated Fe(III) relative to the aqueous Fe3+ ion and longer bonds of intracellular ions relative to aqueous Fe(III)–citrate or hydroxo-complexes could produce an enrichment of cells in lighter isotopes. The versatile nature of Fe(II) and Fe(III) fractionation without a distinct effect of pH and surface exopolysaccharide coverage suggests that, in natural soil and sedimentary environments, Fe isotope fractionation during interaction with heterotrophic bacteria will be primarily governed by Fe complexation with DOM and Fe redox status in the soil pore water.
Environmental significanceThis investigation provides valuable insights into the mechanisms governing Fe isotope fractionation in soil-fluid bacterial systems. Isotopic partitioning during the adsorption of Fe(II) and Fe(III) by a common soil bacterium is dominated by Fe complexation with DOM. The adsorption of Fe(III) results in an enrichment of heavier isotopes on the cell surface that is attributed to the stronger and more symmetrical complexation of Fe(III) with surface phosphoryl moieties compared to hydroxyl complexes in the aqueous solution. In natural environments, the δ57Fe values in solution or microbial biomass may vary from approximately −1 to +3‰, depending on the Fe redox status in soil porewater, and this can occur rapidly over short time scales, independent of pH and the presence of microbial EPS. |
1. Introduction
Iron (Fe) is the fourth most abundant element in the Earth's crust and it is crucial for most living organisms because of its implication in a number of metabolic processes.1–5 Besides, Fe is a limiting factor of primary productivity in many aquatic systems6,7 because of very low free-ion concentration and low solubility of its hydroxides.8 Due to the high abundance of Fe in soil systems,9 versatile Fe redox status,10–12 strong complexation with natural organic matter,13 and the ability of Fe isotopes to track sources and processes, Fe isotope fractionation in natural Earth-surface systems has been at the forefront of isotopic studies for the past two decades.14,15Iron has four stable isotopes (54Fe, 56Fe, 57Fe, and 58Fe), known to fractionate mass-dependently during abiotic and bacterially mediated processes in low temperature environments.16–19 Iron isotope fractionation during Fe interaction with bacteria has received significant attention, mostly with regard to anaerobic phototrophic Fe-oxidizing,20 aerobic neutrophilic Fe-oxidizing,21 and heterotrophic Fe-reducing bacteria.22 Most of these studies demonstrated significant isotopic fractionation occurring during Fe assimilation by the cells and the production of bacterially induced Fe biominerals. For example, Croal et al.20 investigated the Fe(II) isotopic fractionation in anaerobic conditions caused by the photoautotrophic bacteria of genus Thiodictyon. The hydrous ferric oxide (HFO) metabolic products yielded δ57Fe values with heavier isotopic composition than the initial Fe(II), with a fractionation factor (Δ57FeFe(III)–oxides–Fe(II)aq.) of 2.2‰. Crosby et al.23 reported that the Fe dissimilatory reduction promoted by Geobacter sulfurreducens and Shewanella putrefaciens strains caused Fe isotopic fractionation of approximately 2.2‰, with final Fe(II) species enriched in light isotopes. Beard et al.,24 Wiesli et al.25 and Johnson et al.26,27 showed that Fe(II)aq. produced by the dissimilatory reduction exhibited lower 57Fe/54Fe ratios when compared to the initial ferrihydrite substrates, with a fractionation factor (Δ57FeFe(III)–oxide–Fe(II)aq.) ranging from 1.9 to 4‰.
Bacterial adsorption and assimilation of metals, in particular Fe, play an essential role in overall metal biogeochemical cycling in soils and waters.4,28–30 It is known that, during equilibrium isotope fractionation processes, heavier Fe isotopes are preferentially adsorbed onto solid surfaces.23,31–33 Generally, the binding affinity of metals on bacteria cell walls and their exopolymer substances (EPS) is controlled by proton-active surface functional groups such as carboxyl, hydroxyl, sulfhydryl, phosphoryl and amine moieties that undergo deprotonation and bind metal ions to form stable ligand–metal surface complexes.30,34–40 However, while the main molecular mechanisms controlling metal adsorption onto bacterial surfaces and metal assimilation into microbial cells are identified, this is not so for metal isotope–bacteria reaction. Rarely have researchers combined structural study of metal speciation in the cell or solution and its isotope fractionation measurements and this was mostly applied to Zn and Cu isotopes.38,41–44 To our knowledge, the combined XAS and Fe isotope approach on bacteria was used only twice so far. Mulholland et al.32 investigated the Fe isotope fractionation during the interaction of aqueous Fe with three species of cyanobacteria (Gloeocapsa sp., Synehococcus sp., and Planthothrix sp.) conducted in batch experiments using two distinct Fe oxidation states (Fe(II) and Fe(III)). In these cultures, the structural status of adsorbed Fe was characterized in a concomitant in situ X-ray Absorption Spectroscopy (XAS) study.40 It was demonstrated that in the presence of surface organic ligands, the oxidation of divalent iron occurs, but the polymerization of Fe(III) oxy(hydr)oxides is partially inhibited. As a result, the adsorbed iron stays in the form of both Fe–O–Fe polymers and individual Fe atoms which are attached to phosphoryl moieties. We also showed that the presence of EPS in solution reduces metal-cell surface binding capacity and enhances Fe polymerization in the form of Fe(III) oxy(hydr)oxides at the bacterial surface. The isotopic results showed a systematic enrichment in heavy Fe isotopes upon iron adsorption onto bacterial cell surfaces, and this was particularly strong when Fe oxidation was involved in the process.32 Subsequently, Swanner et al.21 expanded on this Fe isotopic study of Fe oxidation by Synechoccus sp. and focused the XAS investigation on the characterization of the Fe–oxide minerals produced.
Upon assimilation inside the cell organelles, metals, including Fe, can be subjected to redox reactions such as a reduction to form S-bearing proteins, complexation with strong ligands in the cytoplasm45–47 or scavenging in the form of insoluble carbonates, phosphates and oxalates.48 Any single reaction involving metals inside the cell has its specific equilibrium isotopic fractionation, in addition to the kinetic isotope effect which can operate in each reaction of intracellular Fe transformation. This may set serious limitations on the capacity to interpret the isotopic fractionation measured between the aqueous solutions and intracellular assimilated metals, even though the average structural status of metal can be determined. In contrast, adsorption processes are typically fast, reversible and rarely include more than 2–3 major binding sites, thus allowing a more straightforward assessment of the structural control on metal isotope fractionation. Considering these intrinsic limitations and the information available at the present time for Fe and other metal interaction with microorganisms, the goal of the current investigation was to test the hypothesis that both Fe(II) and Fe(III) adsorption onto the cell surface brings about preferential removal of the heavier isotopes from solution, whereas cell growth in Fe(II) or Fe(III)-bearing media leads to cell enrichment in lighter Fe isotopes inside the cells. In other words, adsorption of Fe can favor heavier isotopes and incorporation might enrich the cells with lighter Fe isotopes. Relative to previous work where we combined structural (XAS) and isotopic measurements during Fe adsorption on phytoplankton cells,32,40 here we further intended to test the impact of bulk Fe structural status assessed by synchrotron-based spectroscopy (oxidation state, coordination number, nature of atomic neighbors) on Fe isotopic fractionation between the bacteria and aqueous solution, both at the cell surface and inside the cells. For this, we used this time a well-known soil heterotrophic bacterium, Pseudomonas aureofaciens. Examination of the link between the speciation of surface- and intracellularly bound Fe and Fe isotope fractionation between the cell and aqueous solution was conducted considering, in particular, the effect of (i) Fe redox state; (ii) solution pH, and (iii) the presence or not of microbial cell exometabolites. By encompassing a wide variety of soil and aquatic environments, we anticipate that the results can be used to better constrain factors controlling Fe isotopic composition of soil or sediment porewaters and surface waters in the presence of heterotrophic bacteria.
2. Materials and methods
2.1. Heterotrophic bacterial culture
The bacterial strain P. aureofaciens CNMN PsB-03 was obtained from the Laboratory of Plant Mineral Nutrition and Hydric Regime (Institute of Genetics and Plant Physiology, Moldovan Academy of Sciences, Chisinau, Moldova). It was isolated from temperate organic-rich soil and selected due to its capacity of producing EPS on a sucrose-containing medium.49 The strain of P. aureofaciens was maintained at 4 °C in a liquid succinic acid (SA) medium,50 and cultured either in the sucrose-peptone (SP) media,51 thus promoting rich EPS synthesis, or in the SA media with poor EPS production. Cultivation was performed at 25 °C during 48–72 h with continuous shaking. The qualitative monosaccharide composition of the EPS produced is different depending on the culture media: the SP-media yields the EPS composed of 76% fructose, 11% glucose and <10% of other sugars, whereas in the SA media, polysaccharides contain 50% glucose, 22% fructose and 14% mannose and other sugars.52Experiments were performed for non-rinsed and rinsed bacterial biomass that produced two sub-samples for both EPS-rich and EPS-poor samples. In the first case, the bacteria were collected from the culture without rinsing, using centrifugation for 20 min at 8000 g. Therefore, for non-rinsed biomass, the EPS content of the cell wall is expected to remain intact. The rinsed biomass was produced by repeated (3 times) centrifugation in an inert electrolyte solution (0.01 M NaNO3). Repetitive rinsing of biomass in this electrolyte solution should lead to the removal of part of the EPS from the cell surface. For EPS-rich (SP-media) and EPS-poor (SA-media) cultures, 28% and 6% of the total biomass were removed during centrifugation, respectively. The biomass concentration is given in gwet l−1 (after centrifugation for 20 min at 8000 g). The conversion factor of wet to dry biomass is equal to 3.6 and 5.0 for EPS-rich and EPS-poor cultures, respectively.
2.2. Adsorption experiments
Prior to the adsorption experiments, the biomass was collected via centrifugation and rinsed with 0.01 M NaNO3 solution three times via centrifugation. The last supernatant was considered as the reference solution used for control experiments. The biomass of P. aureofaciens was fixed to 4 gwet l−1 for all the experiments. The pH was adjusted by adding aliquots of 0.01–0.1 M NaOH or HNO3. Adsorption experiments were carried out in 0.01 M NaNO3 under darkness at 25 ± 0.2 °C and continuously shaken bacteria suspension. The initial concentrations of Fe(II)–ammonium sulfate or Fe(III) chloride (Sigma) solutions were 20 and 3.3 mg l−1, respectively (Table ESI-1†). Divalent Fe was added to thoroughly rinse bacterial biomass and maintained in 0.01 M NaNO3 at pH of 4.9 under continuous nitrogen bubbling and stirring. Iron(III) adsorption was studied on live bacteria as a function of pH (2.8 to 6.0) and with 3 hours of exposure time. Bacteria-free control experiments with Fe(II) and Fe(III) did not yield significant (i.e., >5–10%) dissolved (<0.45 μm) Fe concentration decrease during an exposure time comparable to that of the adsorption experiments with bacteria. This indicated a lack of Fe adsorption on reactor cell walls, Fe(II) oxidation and particulate Fe(III) precipitation.After sampling, experimental solutions were filtered through 0.45 μm acetate cellulose filters, acidified with ultrapure NaNO3 and stored at 5 °C pending analysis. The reversibility of adsorption was tested following the method developed by Fowle and Fein.53 A homogeneous parent bacteria suspension with Fe(III) was adjusted to pH ∼ 5, under which 100% of Fe was adsorbed onto bacteria. After 3 hours of interaction, aliquots of this parent suspension solution were collected and adjusted to sequentially lower pH values (to 3 and then to 1.5). The reaction vessels were equilibrated at new pH values for 3 hours and sampled for dissolved Fe. The “desorbed” metal supernatant concentration was used to calculate the level of irreversibly “incorporated” metal. This amount ranged between 5 and 15% of the initially added Fe, which suggests an equilibrium adsorption process with a relatively small quantity of Fe(III) penetrating inside the cell under our experimental adsorption conditions (not shown). For the reversibility of Fe(II) adsorption and oxidation, a similar technique yielded 100 ± 10% reversibility. The average experimental reproducibility of metal adsorption yield, calculated based on 2 to 3 replicates at identical conditions, was between 5 and 10%, as assessed in a series of separate adsorption experiments. For these reasons, the replicates were not measured for isotopic ratios.
2.3. Iron assimilation by living cells
Iron incorporation into the cells during microbial growth experiments was performed over 2–3 days after 2 days of cell culturing. This period corresponded to maximal cell viability at the exponential to the beginning of the stationary growth state,39,40 and was consistent with previous studies on metal isotope fractionation during metal assimilation by bacterial cultures.38,42 Iron was added in the EDTA-free nutrient medium in the form of either Fe(II) ammonium sulfate, Fe(II) lactate, Fe(III) citrate, or FeCl3. Total aqueous Fe concentration ranged from 2 to 100 mg l−1. Other nutrient metals (Cu, Zn, Mn, Co, Ni, Mo) were present in trace amounts required for bacteria growth, with concentrations of 1–2 orders of magnitude lower than that of Fe. The main organic ligands controlling Fe speciation in organic-rich nutrient media of P. aureofaciens, in addition to cell exometabolites, are soluble proteins, phosphate, and carboxylates which are capable of complexing Fe(II) and Fe(III) in solution. By analogy with Cu2+ (ref. 38) and according to the Fe-binding capacities, thermodynamic calculations using visual Minteq software demonstrate that >99% of total dissolved Fe is complexed with phosphate and peptone in SP-media and with phosphate and succinate in SA-media.For all Fe assimilation experiments during bacterial growth, prior to the analysis, the cells collected via centrifugation were rinsed in 0.01 M Fe-free NaNO3 aqueous solution and then in a 0.01 M EDTA solution for 10 min, in order to remove Fe that was reversibly adsorbed on surface envelopes (e.g., Hudson and Morel;29 Knauer et al.;54 Le Faucheur et al.55). We also performed additional washing in inert electrolyte solution of the same ionic strength as that of the growth media to ensure complete removal of all the EDTA traces from the wet pellet. In this work, surface adsorbed external (EDTA-removable) and incorporated (non EDTA-removable) iron fractions are operationally defined entities.56,57 The experimental reproducibility of metal assimilation degree, calculated based on 3 replicates under identical conditions, ranged between 5 and 15%.
The oxidation of some Fe(II) in solutions could not be excluded because the experiments were not conducted in a glove box. To quantitatively account for this possibility, abiotic reference experiments were performed in bacterial supernatant solution containing a DOC concentration comparable with that of culture experiments. These experiments demonstrated the absence, within the experimental reversibility of ±10%, of the Fe dissolved (<0.45 μm) concentration decrease under the conditions where most of the assimilation occurred. This suggests that the fraction of oxidized iron during the experiments remained lower than the experimental uncertainty of 10%, if it ever occurred.
2.4. Chemical analyses
The analysis of aqueous iron concentration was carried out using a flame atomic absorption spectrometer (PerkinElmer 5100 PC) with an uncertainty of ±2% and a detection limit of 0.05 mg l−1. For all experiments, sterile de-ionized water (Milli-Q, 18 MΩ) purged with CO2 by N2 bubbling was used. The amount of Fe adsorbed onto the biomass in each vial was calculated by subtracting the concentration of Fe in the filtrate at the end of the experiment from the initial quantity of metal added in the suspension.2.5. Iron isotopic measurements
During sample preparation for isotopic analyses, we followed the methodology of bacterial cell decomposition elaborated by Mulholland et al.32 The filtered (<0.45 μm) solutions were evaporated at 80 °C prior to their digestion. The initial salts were digested and dissolved through a double-step addition of 2 ml of bi-distilled 6 M HCl with overnight reaction on a hotplate at 80 °C. The biomasses, the evaporated experimental solutions, and the experimental analytical blanks were processed using a multi-step acid attack on a Teflon hot plate in an individual evaporation hood (class A 100) of the clean room, using Merck Suprapur H2O2 (30%), bidistilled 6 M HCl, Merck Suprapur conc. HF, bi-distilled conc. HNO3, and de-ionized water (Milli-Q, 18 MΩ). The digested samples did not produce any solid residue. Iron purification was processed through anionic exchange chromatography in HCl medium using 0.5 ml of Bio Rad AG1 X4 resin, 200–400 mesh (ref. 58) to remove all matrix elements. Iron was purified using thermoretractable Teflon columns with an internal diameter of 4 mm, as described by Poitrasson et al.59 The resins were conditioned using 6 M HCl prior to sample loading in 0.5 ml of 6 M HCl. The matrix species were eluted in 3 ml of the same acid and, subsequently, Fe was quantitatively eluted with 2 ml of 0.05 M HCl. The purified Fe solutions were evaporated at 120 °C and re-diluted in 1 to 5 ml of 0.05 M HCl, depending on the amount of Fe available, before sample analyses.Iron isotope measurements were performed using a Thermo Electron Neptune MC-ICPMS (Multi-Collector Inductively Coupled Plasma Mass Spectrometer) as described by Poitrasson and Freydier.60 This protocol allows mass-bias correction through a combination of Ni-doping and sample-standard-bracketing approaches. Depending on the available Fe and the MC-ICPMS sensitivity, the samples were analyzed using a medium resolution entry slit and the SIS (Stable Introduction System) consisting of a tandem quartz glass spray chamber (cyclone + standard Scott double pass) coupled with a low flow PFA nebulizer. Iron isotope results (Table 1) are reported using the delta notation relative to the IRMM-14 iron isotopic reference material, expressed in ‰ (per thousand) for 56Fe/54Fe and 57Fe/54Fe ratios as:
| δ56Fe = [(56Fe/54Fesample)/(56Fe/54FeIRMM14) − 1]103 | (1) |
| δ57Fe = [(57Fe/54Fesample)/(57Fe/54FeIRMM14) − 1]103 | (2) |
| Name | Type of sample | pH | δ57Fe | 2SE | δ56Fe | 2SE | Δ57Fe (‰) | 2SE |
|---|---|---|---|---|---|---|---|---|
| a Recalculated from δ57Fe. | ||||||||
| Adsorption experiments (3 h) | ||||||||
| EPS-rich-2 | Biomass | 4.0 | 0.678 | 0.047 | 0.432 | 0.061 | 0.74 | 0.14 |
| Solution | −0.062 | 0.129 | −0.062 | 0.317 | ||||
| EPS-poor-3 | Biomass | 4.0 | 0.963 | 0.040 | 0.670 | 0.099 | 0.89 | 0.10 |
| Solution | 0.084 | 0.091 | 0.005 | 0.045 | ||||
| EPS-rich-4 | Biomass | 5.5 | 0.466 | 0.044 | 0.279 | 0.090 | 0.81 | 0.11 |
| Solution | −0.347 | 0.098 | −0.250 | 0.129 | ||||
| EPS-poor-5 | Biomass | 6.0 | 0.747 | 0.148 | 0.515 | 0.093 | 1.96 | 0.22 |
| Solution | −1.212 | 0.163 | −0.790 | 0.108 | ||||
| EPS-poor-1 | Biomass | 2.0 | 0.198 | 0.108 | 0.129 | 0.124 | 0.72 | 0.15 |
| Solution | −0.525 | 0.111 | −0.419 | 0.076 | ||||
| EPS-poor-2 | Biomass | 2.8 | 0.199 | 0.034 | 0.122 | 0.068 | 0.78 | 0.13 |
| Solution | −0.578 | 0.123 | −0.407 | 0.152 | ||||
| EPS-rich-3 | Solution | 4.6 | −0.680 | 0.132 | −0.503 | 0.136 | 1.29 | 0.30 |
| EPS-poor-Fe2 | Solution | 4.9 | −0.176 | 0.191 | −0.135 | 0.121 | 3.16 | 1.60 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||
| Assimilation experiments (48 h) | ||||||||
| EPS-rich-4 | Solution | 7.2 | 0.558 | 0.058 | 0.363 | 0.177 | −1.10 | 0.46 |
| EPS-rich-5 | Biomass | 7.1 | 1.003 | 0.132 | 0.653 | 0.124 | 0.77 | 0.32 |
| Solution | 0.236 | 0.291 | 0.208 | 0.119 | ||||
| EPS-poor-6 | Biomass | 7.5 | 0.996 | 0.107 | 0.709 | 0.128 | 0.83 | 0.66 |
| EPS-poor-8 | Biomass | 7.1 | 0.749 | 0.068 | 0.490a | 0.088 | −0.99 | 0.24 |
| EPS-rich-6 | Biomass | 7.2 | 0.172 | 0.097 | 0.127 | 0.063 | −1.01 | 0.51 |
| EPS-rich-7 | Biomass | 7.2 | −0.392 | 0.095 | −0.256 | 0.056 | −1.12 | 0.23 |
| EPS-poor-Fe2-2 | Biomass | 7.8 | 0.749 | 0.119 | 0.492 | 0.093 | −0.04 | 0.19 |
|
||||||||
| Starting stock solutions | ||||||||
| FeCl3 | Stock | — | 0.55 | 0.179 | 0.349 | 0.045 | — | — |
| Fe(II)–ammonium sulfate | Stock | 0.188 | 0.068 | 0.104 | 0.066 | — | — | |
| Fe(III)–citrate | Stock | — | 0.295 | 0.098 | 0.197 | 0.065 | — | — |
| Fe(II)–lactate | Stock | — | 0.788 | 0.142 | 0.537 | 0.106 | — | — |
| BCR482 | Stock | — | −0.002 | 0.109 | −0.028 | 0.034 | — | — |
The validation of the analyses was performed by comparing repeated measurements of our in-house hematite standard from Milhas, Pyrénées, every 6 samples (see Poitrasson and Freydier60). The δ57Fe value obtained by pooling 18 individual analyses obtained in the present study by group of 6 was 0.753 ± 0.032‰ (2SD), consistent with the values previously reported.31,33,60–62 We also analyzed the European lichen reference material BCR 482 and obtained δ57Fe = −0.00 ± 0.11‰ (2SE; Table 1), which is undistinguishable from the previously reported value of −0.03 ± 0.15‰ by Aebischer et al.63
2.6. X-ray absorption spectroscopy for adsorbed and assimilated iron
X-ray absorption spectroscopy (XAS) experiments of Fe-bearing solids (standards) and freeze-dried cells, including XANES and EXAFS, were carried out at the SpLine-BM25A (Spanish CRG beamline) of the European Synchrotron Radiation Facilities (ESRF). The spectra were collected in transmission and fluorescence mode as a function of the Fe concentrations at the Fe K-edge (∼7.112 keV) over the energy range 6.9–7.9 keV. The energy was selected using a Si(111) double-crystal monochromator and a 5 μm iron metal foil was used as a reference; the energy of the first inflection point in the XANES spectrum was set to 7112 eV. The spectra of the bacteria were collected in fluorescence mode with a 13-element Si(Li) detector (e2v Instruments). Iron(III) standard compounds (citrate, phosphate, goethite and alginate) and Fe(II) references (lactate, L-ascorbate, oxalate, gluconate and phosphate) were measured in transmission mode. The incident beam was monitored with a N2-filled gas ionization chamber. XANES spectra were analyzed using linear combination fitting considering several footprint components in agreement with the local environment and geometry around Fe atoms (hydroxide as goethite, carboxylate as ferric citrate and phosphoryl as Fe(III) phosphate). EXAFS signals were analyzed up to a maximum k value of 12 Å−1. Multiple scans of 60 min per scan acquisition time were recorded at different spots of the sample to further avoid beam induced reactions. Data treatment and analysis have been performed using the ATHENA and VIPER software, using a weighted k3·c(k) signal with a window ranging from 2.5 to around 12 Å−1. Fourier transforms of the spectra were filtered in the 1–2.3 Å range to isolate contribution from the first coordination sphere. All the biomass samples were analyzed at 5–10 K using a liquid-helium cryostat because the low temperature offers great benefits such as decreasing the thermal disorder, and minimizing the damage in terms of redox sensitive elements. All biomass samples for XAS analysis were prepared on-site at the European Molecular Biology Laboratory (EMBL) in the vicinity of the ESRF and processed at the beamline within several hours after the end of the freeze-drying procedure. This greatly reduced the possibility of sample degradation, and allowed achievement of the highest Fe concentration for XAS spectra acquisition without increasing the level of initial Fe loading.2.7. Calculation of isotopic fractionation in adsorption and assimilation experiments
The experimental conditions of Fe adsorption and assimilation assays are listed in Table ESI-1.† It presents the data for both biomass and solutions analyzed in this work. For samples from the batch adsorption and assimilation experiments performed in a closed system, the isotopic signature of the bacterial cells (δ57Fecell) was calculated from the mass balance equation using the isotopic ratio in the aqueous phase (δ57Fesolution), the proportion of adsorbed Fe (A, %), and the isotopic composition of the initial solution (δ57Feinitial):| δ57Fecell = (100 × δ57Feinitial − (100 − A) × δ57Fesolution)/A | (3) |
Similarly, in case if only bacterial cell Fe isotope measurements were performed, the isotopic signature of the fluid phase (δ57Fesolution) was calculated via:
| δ57Fesolution = (100 × δ57Feinitial − A × δ57Fecell)/(100 − A) | (4) |
In this work, we define the isotopic fractionation factor between Fe in solution and Fe adsorbed onto or assimilated by bacteria as:
| Δ57Fecell-solution = δ57Fecell − δ57Fesolution | (5) |
The values of isotopic signature (δ57Fe and δ56Fe) and isotopic fractionation factor (Δ57Fecell-solution) and those of the starting products are listed in Table 1. In the adsorption experiments, 6 pairs of samples (cell biomass and corresponding aqueous solution) were processed for isotopic analyses. This allowed straightforward calculation of Δ57Fecell-solution using eqn (4) (Table 1). In these experiments, we also calculated δ57Fecell using eqn (3); the values of Δ57Fecell-solution obtained from such indirect calculation agreed within propagated analytical uncertainties with directly measured Δ57Fecell-solution values for 4 out of the 6 experiments. For the remaining two experiments (EPS-poor-1 and 2), it is possible the proportion of adsorbed Fe on the cell surfaces is only known with an uncertainty larger than our 3% estimate. With this caveat in mind, we computed δ57Fecell using eqn (3) when only δ57Feaqueous was available (exp. EPS-rich-3 and EPS-poor-Fe2 in Table 1). Similarly, in the assimilation experiments, coupling of cell and solution measurements in EPS-rich-5 samples yielded direct Δ57Fecell-solution (+0.767 ± 0.32‰ by eqn (4)), which is similar within uncertainty to the calculated value (+0.739 ± 0.25‰ by eqn (3) and (4)). This allowed using other measured δ57Fecell to calculate δ57Fesolution and Δ57Fecell-solutionviaeqn (4) and (5), respectively.
We also obtained δ56Fe values, though slightly less precisely than δ57Fe values on a per amu basis. Given that the relationships between δ56Fe and δ57Fe of both adsorbed and assimilated samples plot on a single mass fractionation line (Fig. ESI-1†), only δ57Fe values are discussed in this paper. The resulting linear regression for δ57Fe vs. δ56Fe (Fig. ESI-1†) was:
| δ57Fe = (0.016 ± 0.009) + (1.456 ± 0.022) × δ56Fe (R2 = 0.996) | (6) |
This is consistent with δ57Fe being equal to ∼3/2 of δ56Fe. Given the findings reported by Amor et al.64 on mass-independent Fe isotope fractionation during magnetotactic bacteria metabolism, we searched for such mass independent Fe isotope fractionation on our experimental products to see whether such an effect could also occur with other type of bacteria. However, we could not detect any significant mass independent effect outside 0.1‰ uncertainty.
3. Results
3.1. Isotope fractionation during Fe adsorption onto and assimilation by the bacteria
In case of Fe3+ adsorption onto EPS-rich and EPS-poor cells (see Table ESI-1†), the δ57Fecell was always higher than δ57Fesolution (Table 1). However, for the two sets of experiments having the same pH range (close to 4 and 4.6–4.9, respectively), the Δ57Fecell-solution is slightly higher for EPS-poor cultures compared to the EPS-rich ones, although the difference is comparable to uncertainties. Iron(III) adsorption onto P. aureofaciens at 2.8 ≤ pH ≤ 6.0 produced an enrichment of the cell surface in the heavier isotope with Δ57Fecell-solution ranging from +0.72 to +2.0‰ (Table 1 and Fig. 1). The EPS-poor culture exhibited a possible impact of pH on the isotopic offset during adsorption, which increased from +0.72‰ at pH of 2 to +1.96‰ at pH of 6 (Table 1 and Fig. 1).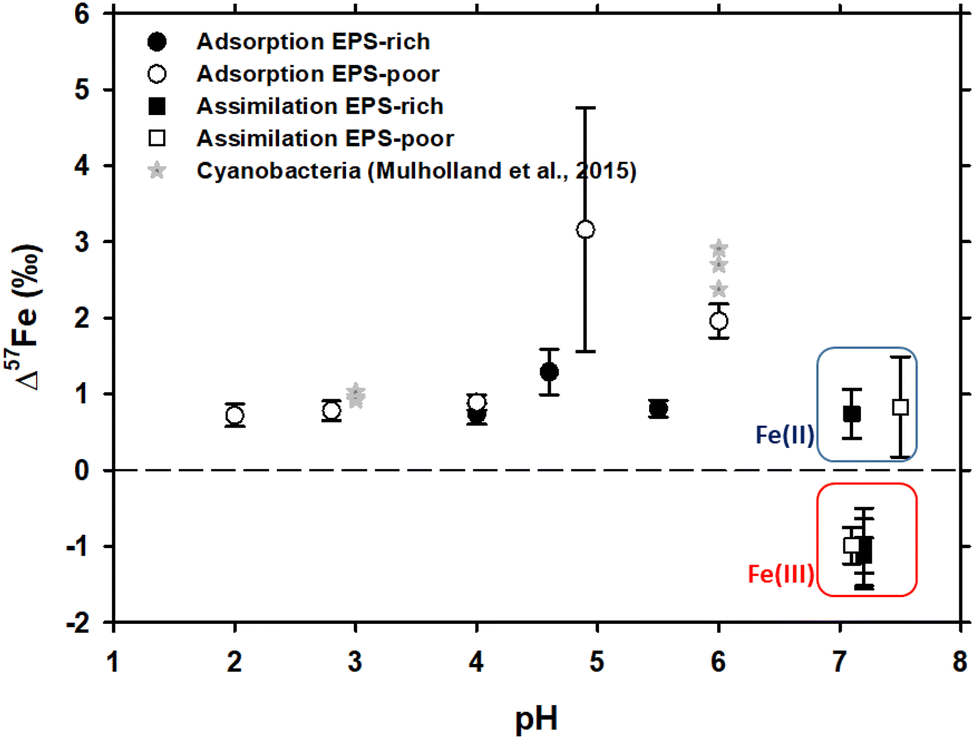 | ||
| Fig. 1 δ57Fe values of the initial solution calculated by the mass balance between cell and solution Fe concentrations and isotopic compositions. Asterisks represent the data on Fe isotope fractionation during adsorption onto cyanobacteria, as reported by Mulholland et al., 2015.32 Note that the isotopic offset during assimilation is strongly dependent on the initial Fe redox status – negative in case of Fe(III) and positive in case of Fe(II) as distinguished by red and blue rectangles, respectively. | ||
The isotope fractionation induced by Fe(II) and Fe(III) assimilation during bacterial growth demonstrated the following features. The EPS-poor-8 cells exhibited higher δ57Fecell values of assimilated Fe compared to those of the EPS-rich-6 ones for the same pH range (Table 1), even though the starting Fe(III)–citrate of the former was isotopically lighter than the starting FeCl3 of the latter (Table 1). This feature of Fe(III) assimilation is not confirmed for the case of Fe(II) assimilation since the δ57Fecell values in EPS-poor-6 and EPS-rich-5 experiments were undistinguishable (Table 1). Intracellular assimilation of Fe(II) led to preferential uptake of heavier isotopes producing Δ57Fecell-solution of +0.77 and +0.83‰ for EPS-rich-5 and EPS-poor-6 experiments. In contrast, intracellular uptake of Fe(III) yielded an enrichment of the cell in lighter isotopes with Δ57Fecell-solution close to −1‰ as calculated from EPS-poor-8 and EPS-rich-4,6,7 experiments (Table 1). It is noteworthy that the Fe(III) uptake from both initial FeCl3 and Fe–citrate salts yielded similar isotopic offsets (Tables ESI-1† and 1). Considering the propagated uncertainties (see Table 1), the magnitude of isotopic offset (Δ57Fecell-solution) in the EPS-rich cells (from −1.01 to −1.12‰) was not distinguishable from that in the EPS-poor cells (Δ57Fecell-solution = −0.99‰) for the experiments using Fe(III) salts (Tables ESI-1† and 1).
3.2. XAS characterization of Fe bound to soil bacteria
The structural characterization of Fe adsorbed onto P. aureofaciens was based on XAS (EXAFS and XANES) analysis of samples as was also described in previous work.40 These results are directly used for testing structural control on adsorbed Fe(III) and Fe(II) from aqueous solution as described in Section 4.1 below. The intracellularly assimilated Fe was not characterized previously and newly obtained results are presented below.The XANES spectra of assimilated Fe–bacteria samples are illustrated in Fig. 2 and Table 2. When comparing the samples with reference spectra, the analysis clearly shows that the unique organic component in most of the samples (and main component in all cases) is Fe(III)–phosphate, corresponding to Fe binding to the phosphoryl group. The relative proportion of Fe(III)–phosphate and goethite was estimated from linear combination fits (LCF analysis) of XANES spectra and using Fe(III) reference compounds. The choice of the best corresponding standard for each spectrum was made using automated combinational fitting (Athena software).
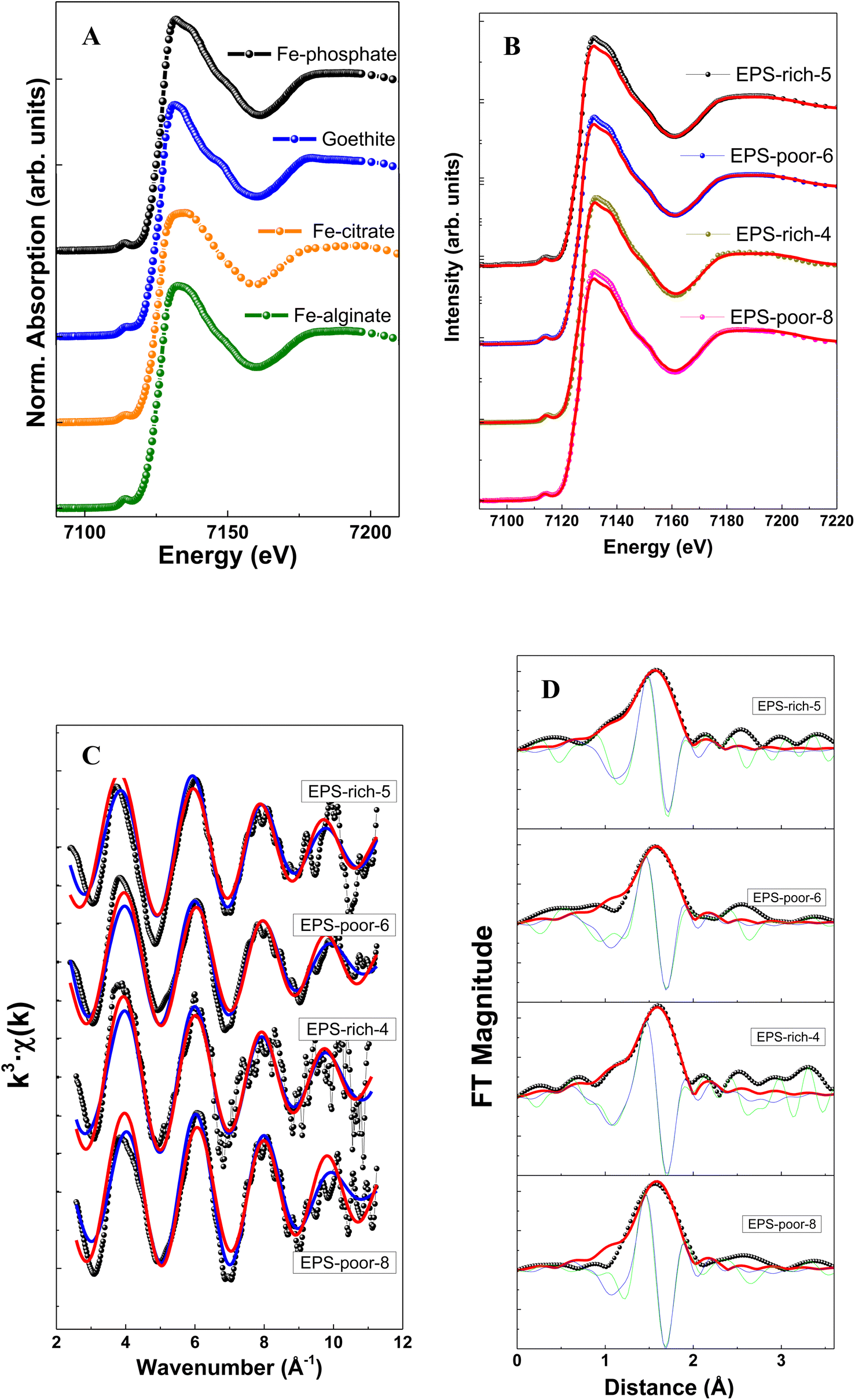 | ||
| Fig. 2 Fe K-edge XANES spectra at the Fe K-edge energies (A) and (B), presenting Fe K-edge oscillations (C) and Fourier transform magnitudes (D) of the Fe adsorbed on cells. | ||
| Sample | pH | Fe(III) phosphate (%) | Goethite (%) | R (Å) (±0.05 Å) | N (atom) (±0.5) | σ 2 (Å2) (±0.001 Å2) | R-factor |
|---|---|---|---|---|---|---|---|
| EPS-rich-5 | 7.1 | 95 | 5 | 2.07 | 5.9 | 0.007 | 0.08 |
| EPS-poor-6 | 7.8 | 100 | 2.07 | 5.1 | 0.006 | 0.13 | |
| EPS-rich-4 | 7.2 | 100 | 2.07 | 6.0 | 0.007 | 0.05 | |
| EPS-poor-8 | 7.1 | 100 | 2.06 | 5.6 | 0.005 | 0.15 |
The EXAFS results allowed extracting the first shell of Fe (Table 2) which exhibited similar features among samples (Fig. 2). Note that the modeling of EXAFS spectra in biological samples is very difficult due to distorted geometries and the presence of different neighbors, like O, N and S, light elements with similar weak cross section values, in the nearest metal coordination shell. In this sense, XANES results may complement the EXAFS via identifying the ligands using the edge features.
The filtered Fourier transform curves (EXAFS signals filtered for the first coordination sphere) were fitted using Fe–O bonds. The inelastic loss factor was estimated/set, based on references and the EPS-rich-4 sample, as S02 = 0.87. Results show a coordination number of around 6 in all cases. This is in good agreement with an octahedral geometry of 6 oxygen atoms surrounding Fe cations, with a Fe–O distance of ∼2.07 Å. We could not quantify the relative contribution of Fe(III)–phosphoryl or carboxyl complexes that are typical in samples of microorganisms,38,65,66 although the samples showed an important contribution of Fe(III)–phosphate like groups in the XANES analysis (peak at ∼1.9–2.0 Å−1). Overall, the EXAFS spectra confirmed the XANES results.
4. Discussion
4.1. Heavier isotope enrichment of cells during Fe(II) and Fe(III) adsorption
The adsorption of Fe onto P. aureofaciens allowed quantifying the adsorption constants and surface binding site concentrations. The total amount of proton-binding sites was two times higher in the EPS-rich cells compared to the EPS-poor ones (5.5 and 2.6 mmol gwet−1), whereas the concentration of phosphoryl-like groups was higher in the EPS-poor cells and the concentration of carboxyl-like groups was higher in the EPS-rich cells. The adsorption of Fe(III) started at acidic pH (∼1.5) and increased with the pH, following the “universal pH-edge” for metals.67,68 González et al.40 provided chemical characterization of Fe3+ adsorbed onto soil bacteria by X-ray absorption spectroscopy (XAS), and demonstrated that Fe(III)–phosphate was the predominant Fe-binding compound on the cell surfaces of the EPS-poor cultures, at pH from 2.0 to 6.0. In addition, the goethite-like component was present in the EPS-rich cells (pH from 3.0 to 4.6). Therefore, EXAFS spectra supported the XANES results.The present study demonstrated that, during Fe(III) adsorption, the effect of pH was strongly pronounced for the EPS-poor culture (Δ57Fecell-solution increased from +0.72‰ at pH = 2.0 to +1.96‰ at pH = 6.0; Table 1 and Fig. 1). The pH effect could not be tested for the EPS-rich culture, for which our two experiments yielded significantly different Δ57Fecell-solution values (+0.74 ± 0.14‰ and +1.29 ± 0.30‰; Table 1 and Fig. 1) at similar 4 ≤ pH ≤ 4.6. The exact reasons for these differences between these EPS-rich and EPS-poor cultures in the circumneutral pH range (Fig. 1) are not clear. It can be hypothesized that, in the case of EPS-poor strain, the phosphoryl moieties of the cell wall are not protected from solution and adsorption occurs essentially on these high affinity sites. This could produce stronger fractionation at higher pH, where phosphorylated groups are mostly deprotonated. However, this seems to be inconsistent with XAS-based bulk speciation of Fe at the cell surface, because the surface-bound Fe(III) was dominated by Fe–phosphoryl ligands, regardless of pH and the presence or absence of the EPS. At the same time, one should keep in mind that XAS treatment provides dominant (ca. 90%) speciation of an element. The remaining 10% are not detectable by spectroscopy but it is not excluded that they can be responsible for observed isotope fractionation, provided that the isotopic offsets of reactions with these minor compounds strongly exceed those of the major binding constituents of Fe. Recently, Oleinikova et al.69 studied the adsorption of organo-ferric colloids onto P. aureofaciens and demonstrated an enrichment in δ57Fe (+0.4‰) of cell surfaces compared to the remaining solution. Consistent with the results of Gonzalez et al.40 and of the present study, this could be explained by the dominance of Fe(III)-complexes with phosphoryl groups on cell surfaces. Therefore, the only possible explanation for the observed difference in isotopic offset between the aqueous solution and cell biomass of EPS-rich an EPS-poor cultures is the shielding effect of carboxylate-rich EPS on aqueous Fe ions. These rather inert EPS layers can prevent direct interaction and binding of Fe2+(aq.) and Fe3+(aq.) with the strong phosphoryl moieties of the external cell membrane.
Globally, the XAS analyses of adsorbed Fe demonstrated a dominance of Fe(III)–phosphate complexes at the cell surface regardless of the identity of strain and the chemical composition of solution. Therefore, we suggest that the heavier isotope enrichment of the cell surface relative to aqueous solution is due to strong Fe(III)–phosphoryl surface complexes compared to FeOH2+, Fe(OH)2+ and Fe(III)–organic complexes in aqueous solution. Presumably, these phosphoryl surface complexes exhibit shorter bond lengths and higher symmetry than aqueous Fe(III) inorganic species (FeOH2+, Fe(OH)2+, FeCl2+) and Fe(III)–EPS complexes. Note that the isotopic measurements of Fe(III) adsorption rule out the possibility of Fe(OH)3 precipitation at the cell surface, because the observed isotopic offset (Δ57Fecell-solution = +0.70 to +2.1‰) is opposite to the sign of the Fe3+(aq.) → Fe(OH)3(solid) reaction (Δ57FeFe(III)hydroxide–Fe3+(aq.) from −0.2 to −1.5‰), with more fractionated values at slower reaction rates.70
In contrast, in case of Fe(II) adsorption on EPS-poor cultures (Δ57Fecell-solution of +3.16‰ measured in this study; EPS-poor-F2; Tables ESI-1† and 1, and Fig. 1), in addition to surface complexation with stronger than aqueous ligand, Fe(II) partial oxidation at the cell surface may lead to overall cell enrichment in heavier isotopes. Indeed, it is known that the Fe(II)aq. → Fe(III)–hydroxide (solid) reaction exhibits a Δ57Fesolid-solution of +2.3 ± 0.3‰,16,19,20 although an isotopic offset as high as +4.8‰ was also reported in case of hematite precipitation.71
4.2. Fe assimilation by bacterial cells
Intracellular assimilation of Fe(II) enriched the cells in heavier isotopes producing undistinguishable Δ57Fecell-solution of +0.77 ± 0.02‰ and +0.83 ± 0.08‰ for EPS-rich and EPS-poor cultures, respectively (Tables ESI-1† and 1). Two possible mechanisms can be responsible for this enrichment. First, partial oxidation of Fe(II) on the cell surface (not removed by EDTA treatment) or inside the cells, favorable at circumneutral pH of nutrient media (7.2–7.5), which can yield sizable enrichment in heavier isotopes at the surface, as follows from Δ57Fesolid-solution of +2.3 ± 0.3‰ for the Fe(II)aq. → Fe(III)–hydroxide (solid).16,19,20 However, in order for this effect to be pronounced, at least 30% of Fe(II) should be transformed into Fe(III) (15–20% in the case where the isotopic offset of Fe2+ oxidation equals +4.8‰ as reported by ref. 71). Second, higher symmetry and shorter bonds of Fe(II) in the cells compared to Fe(II)–lactate solutions may be responsible for cell enrichment in heavy isotopes relative to the nutrient media. Unfortunately, we could not resolve the 2nd neighbor symmetry in this type of sample due to the relatively low Fe concentration in the nutrient solution.In contrast to Fe(II), cellular uptake of Fe(III) produced an enrichment of the cell biomass in lighter isotopes with Δ57Fecell-solution around −1‰, regardless of the presence or not of EPS and the form of Fe(III) in the nutrient media (chloride or citrate; Tables ESI-1† and 1, and Fig. 1). Interestingly, the long-term bacterial uptake of Fe(III) oxy(hydr)oxide colloids and organic complexes by P. aureofaciens produced Δ57Fecell-solution of −0.7‰,69 which is similar to the results of the present study. Iron can be involved in a number of both extra- and intra-cellular redox reactions, under strong control of organic binding ligands. For example, Fe(II) can be oxidized and Fe(III) can be reduced within the cells,47 where both inorganic insoluble Fe, inorganic soluble Fe and organically complexed soluble Fe can be adsorbed onto and transported into the cells. In addition, Fe can be adsorbed onto cell surfaces prior to being reduced to Fe(II) or to being incorporated.4 The preference of cell intracellular compartments with lighter Fe isotopes may be controlled by two factors. First, Fe(III) in the nutrient media can be reduced to Fe(II). However, the lack of XAS confirmation of this redox transformation and also lack of information on Fe(II) speciation in cell compartments cannot support this possibility. Another mechanism of lighter isotope enrichment of cell biomass could be a loss of symmetry of assimilated Fe(III) relative to octahedrally coordinated aqueous Fe3+ ions and longer bonds of intracellular ions relative to aqueous Fe(III)–citrate or hydroxo-complexes.
Overall, although the obtained results are fully consistent with available data on Fe isotope fractionation during adsorption and redox transformation, we could not evidence a straightforward structural control on Fe isotope fractionation between aqueous solution and soil bacteria. The versatile nature of Fe(II) and Fe(III) fractionation without the distinct effect of pH and surface exopolysaccharide coverage suggests that, in soil environments, the nature of microorganisms (presence or not of EPS) which adsorb and assimilate Fe and solution pH (between 4 and 6) exhibit a subordinate effect on Fe isotopic signature in the microbial biomass. As such, Fe isotope fractionations during its interaction with heterotrophic bacteria, mineral or organic sediments will be primarily governed by Fe complexation with DOM and Fe redox state in soil or sediment porewater.72,73
5. Conclusions
With the aim of assessing the role of heterotrophic bacteria in Fe isotope fractionation in aqueous solutions, we measured the isotopic partitioning during Fe(II) and Fe(III) adsorption onto and assimilation by a common soil bacterium. We notably compared the obtained isotopic offsets between the fluid and the biomass with Fe speciation in the nutrient media and Fe chemical status on the cell surface and in the cell interior.Adsorption of Fe(III) led to cell surface enrichment in heavier isotopes, tentatively linked to the stronger and more symmetrical complex of Fe(III) with surface phosphoryl moieties compared to hydroxyl complexes of Fe(III) in aqueous solution. This was especially pronounced for the EPS-poor culture in circumneutral solutions, when the binding sites of the cell membrane were not protected by the carboxylates of EPS. Adsorption of Fe(II) could yield some Fe(III) hydroxides at the surface, which produced even stronger enrichment of cells (by up to 3.2‰) in heavier isotopes.
Iron(III) assimilation from nutrient media produced ca. −1‰ cell enrichment in lighter isotopes. This isotopic offset was similar for EPS-rich and EPS-poor cultures and Fe(III) citrate or Fe(III) chloride in the initial nutrient media. In contrast, Fe(II) uptake by bacteria yielded an enrichment of cells by ca. +0.8‰ in heavier isotopes, following the formation of Fe(III) hydroxides or other minerals inside the cells or due to formation of stronger bonds with phosphoryl moieties in cell organelles compared to nutrient media.
Taken together, the results obtained indicate that the mechanisms controlling Fe isotope fractionation in the soil fluid–bacterial system are better constrained. In natural settings, the δ57Fe in solution or microbial biomass may vary from ca. −1 to +3‰ depending primarily on the Fe redox status in soil and sediment porewater and these changes may occur over a short time scale (hours to days) regardless of pH and the presence or not of microbial extracellular substances.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. E. Emnova (Moldova Academy of Science) is thanked for providing the P. aureofaciens strain. F. P. is funded by CNRS. O. P. acknowledges the support from the TSU Development Programme («Priority-2030»).References
- K. H. Nealson and D. Saffarini, Iron and manganese in anaerobic respiration: environmental significance, physiology, and regulation, Annu. Rev. Microbiol., 1994, 48, 311–344 CrossRef CAS PubMed
.
- D. R. Lovley, Microbial Fe(III) reduction in subsurface environments, FEMS Microbiol. Rev., 1997, 20, 305–313, DOI:10.1111/j.1574-6976.1997.tb00316.x
-
B. Thamdrup, in Advances in microbial ecology, Springer, 2000, pp. 41–84 Search PubMed
- F. M. M. Morel, A. B. Kustka and Y. Shaked, The role of unchelated Fe in the iron nutrition of phytoplankton, Limnol. Oceanogr., 2008, 53, 400–404, DOI:10.4319/lo.2008.53.1.0400
-
R. M. Cornell and U. Schwertmann, The iron oxides: structure, properties, reactions, occurrences and uses, John Wiley & Sons, 2003 Search PubMed
- J. H. Martin, K. H. Coale, K. S. Johnson, S. E. Fitzwater, R. M. Gordon, S. J. Tanner, C. N. Hunter, V. A. Elrod, J. L. Nowickl, T. L. Coley, R. T. Barber, S. Lindley, A. J. Watson, K. Van Scoy, C. S. Law, M. I. Liddicoat, R. Ling, T. Stanton, J. Stockel, C. Collins, A. Anderson, R. Bidigare, M. Ondrusek, M. Latasa, F. J. Millero, K. Lee, W. Yao, J. Z. Zhang, G. Friederich, C. Sakamoto, F. Chavez, K. Buck, Z. Kolber, R. Greene, P. Falkowski, S. W. Chisholm, F. Hoge, R. Swift, J. Yungel, S. Turner, P. Nightingale, A. Hatton, P. Liss and N. W. Tindale, Testing the iron hypothesis in ecosystems of the equatorial Pacific Ocean, Nature, 1994, 371, 123–129, DOI:10.1038/371123a0
- K. H. Coale, K. S. Johnson, S. E. Fitzwater, R. M. Gordon, S. Tanner, F. P. Chavez, L. Ferioli, C. Sakamoto, P. Rogers, F. Millero, P. Steinberg, P. Nightingale, D. Cooper, W. P. Cochlan, M. R. Landry, J. Constantinou, G. Rollwagen, A. Trasvina and R. Kudela, A massive phytoplankton bloom induced by an ecosystem-scale iron fertilization experiment in the equatorial Pacific Ocean, Nature, 1996, 383, 495, DOI:10.1038/383495a0
- X. Liu and F. J. Millero, The solubility of iron in seawater, Mar. Chem., 2002, 77, 43–54, DOI:10.1016/S0304-4203(01)00074-3
-
E. Murad and W. R. Fischer, in Iron in soils and clay minerals, Springer, 1988, pp. 1–18 Search PubMed
- F. J. Millero, S. Sotolongo and M. Izaguirre, The oxidation kinetics of Fe(II) in seawater, Geochim. Cosmochim. Acta, 1987, 51, 793–801, DOI:10.1016/0016-7037(87)90093-7
- J. M. Santana-Casiano, M. González-Dávila and F. J. Millero, Oxidation of nanomolar levels of Fe(II) with oxygen in natural waters, Environ. Sci. Technol., 2005, 39, 2073–2079, DOI:10.1021/es049748y
- A. G. González, J. M. Santana-Casiano, N. Pérez and M. González-Dávila, Oxidation of Fe(II) in natural waters at high nutrient concentrations, Environ. Sci. Technol., 2010, 44, 8095–8101, DOI:10.1021/es1009218
- E. L. Rue and K. W. Bruland, Complexation of iron(III) by natural organic ligands in the Central North Pacific as determined by a new competitive ligand equilibration/adsorptive cathodic stripping, Mar. Chem., 1995, 50, 117–138, DOI:10.1016/0304-4203(95)00031-L
- B. Wu, W. Amelung, Y. Xing, R. Bol and A. E. Berns, Iron cycling and isotope fractionation in terrestrial ecosystems, Earth-Sci. Rev., 2019, 190, 323–352, DOI:10.1016/j.earscirev.2018.12.012
-
C. Johnson, B. Beard and S. Weyer, Iron geochemistry: an isotopic perspective, Springer, 2020 Search PubMed
- T. D. Bullen, A. F. White, C. W. Childs, D. V Vivit and M. S. Schulz, Demonstration of significant abiotic iron isotope fractionation in nature, Geology, 2001, 29, 699–702, DOI:10.1130/0091-7613(2001)029<0699:DOSAII>2.0.CO;2
- A. D. Anbar, Molybdenum stable isotopes: observations, interpretations and directions, Rev. Mineral. Geochem., 2004, 55, 429–454, DOI:10.2138/gsrmg.55.1.429
- A. Gronstal, V. Pearson, A. Kappler, C. Dooris, M. Anand, F. Poitrasson, T. P. Kee and C. S. Cockell, Laboratory experiments on the weathering of iron meteorites and carbonaceous chondrites by iron-oxidizing bacteria, Meteorit. Planet. Sci., 2009, 44, 233–247, DOI:10.1111/j.1945-5100.2009.tb00731.x
- B. L. Beard, R. M. Handler, M. M. Scherer, L. Wu, A. D. Czaja, A. Heimann and C. M. Johnson, Iron isotope fractionation between aqueous ferrous iron and goethite, Earth Planet. Sci. Lett., 2010, 295, 241–250, DOI:10.1016/j.epsl.2010.04.006
- L. R. Croal, C. M. Johnson, B. L. Beard and D. K. Newman, Iron isotope fractionation by Fe(II)-oxidizing photoautotrophic bacteria, Geochim. Cosmochim. Acta, 2004, 68, 1227–1242, DOI:10.1016/j.gca.2003.09.011
- E. D. Swanner, T. Bayer, W. Wu, L. Hao, M. Obst, A. Sundman, J. M. Byrne, F. M. Michel, I. C. Kleinhanns, A. Kappler and R. Schoenberg, Iron isotope fractionation during Fe(II) oxidation mediated by the oxygen-producing marine cyanobacterium Synechococcus PCC 7002, Environ. Sci. Technol., 2017, 51, 4897–4906, DOI:10.1021/acs.est.6b05833
- G. A. Icopini, A. D. Anbar, S. S. Ruebush, M. Tien and S. L. Brantley, Iron isotope fractionation during microbial reduction of iron: the importance of adsorption, Geology, 2004, 32, 205–208, DOI:10.1130/G20184.1
- H. A. Crosby, C. M. Johnson, E. E. Roden and B. L. Beard, Coupled Fe(II)-Fe(III) electron and atom exchange as a mechanism for Fe isotope fractionation during dissimilatory iron oxide reduction, Environ. Sci. Technol., 2005, 39, 6698–6704, DOI:10.1021/es0505346
- B. L. Beard, C. M. Johnson, L. Cox, H. Sun, K. H. Nealson and C. Aguilar, Iron isotope biosignatures, Science, 1999, 285, 1889–1892, DOI:10.1126/science.285.5435.1889
- R. A. Wiesli, B. L. Beard and C. M. Johnson, Experimental determination of Fe isotope fractionation between aqueous Fe(II), siderite and ‘green rust’ in abiotic systems, Chem. Geol., 2004, 211, 343–362, DOI:10.1016/j.chemgeo.2004.07.002
- C. M. Johnson, B. L. Beard, E. E. Roden, D. K. Newman and K. H. Nealson, Isotopic constraints on biogeochemical cycling of Fe, Rev. Mineral. Geochem., 2004, 55, 359–408, DOI:10.2138/gsrmg.55.1.359
- C. M. Johnson, E. E. Roden, S. A. Welch and B. L. Beard, Experimental constraints on Fe isotope fractionation during magnetite and Fe carbonate formation coupled to dissimilatory hydrous ferric oxide reduction, Geochim. Cosmochim. Acta, 2005, 69, 963–993, DOI:10.1016/j.gca.2004.06.043
- K. O. Konhauser, W. S. Fyfe, F. G. Ferris and T. J. Beveridge, Metal sorption and mineral precipitation by bacteria in two Amazonian river systems: Rio Solimoes and Rio Negro, Brazil, Geology, 1993, 21, 1103–1106, DOI:10.1130/0091-7613(1993)021<1103:MSAMPB>2.3.CO;2
- R. J. M. Hudson and F. M. M. Morel, Distinguishing between extra-and intracellular iron in marine phytoplankton, Limnol. Oceanogr., 1989, 34, 1113–1120, DOI:10.4319/lo.1989.34.6.1113
- N. Yee, L. G. Benning, V. R. Phoenix and F. G. Ferris, Characterization
of metal−cyanobacteria sorption reactions: a combined macroscopic and infrared spectroscopic investigation, Environ. Sci. Technol., 2004, 38, 775–782, DOI:10.1021/es0346680
- N. Teutsch, U. Von Gunten, D. Porcelli, O. A. Cirpka and A. N. Halliday, Adsorption as a cause for iron isotope fractionation in reduced groundwater, Geochim. Cosmochim. Acta, 2005, 69, 4175–4185, DOI:10.1016/j.gca.2005.04.007
- D. S. Mulholland, F. Poitrasson, L. S. Shirokova, A. G. González, O. S. Pokrovsky, G. R. Boaventura and L. C. Vieira, Iron isotope fractionation during Fe(II) and Fe(III) adsorption on cyanobacteria, Chem. Geol., 2015, 400, 24–33, DOI:10.1016/j.chemgeo.2015.01.017
- K. Dideriksen, J. A. Baker and S. L. S. Stipp, Equilibrium Fe isotope fractionation between inorganic aqueous Fe(III) and the siderophore complex, Fe(III)-desferrioxamine B, Earth Planet. Sci. Lett., 2008, 269, 280–290, DOI:10.1016/j.epsl.2008.02.022
- J. B. Fein, C. J. Daughney, N. Yee and T. A. Davis, A chemical equilibrium model of metal adsorption onto bacterial surfaces, Geochim. Cosmochim. Acta, 1997, 61, 3319–3328, DOI:10.1016/S0016-7037(97)00166-X
- C. J. Daughney, J. B. Fein and N. Yee, A comparison of the thermodynamics of metal adsorption onto two common bacteria, Chem. Geol., 1998, 144, 161–176, DOI:10.1016/S0009-2541(97)00124-1
- O. S. Pokrovsky, R. E. Martinez, S. V. Golubev, E. I. Kompantseva and L. S. Shirokova, Adsorption of metals and protons on Gloeocapsa sp. cyanobacteria: a surface speciation approach, Appl. Geochem., 2008, 23, 2574–2588, DOI:10.1016/j.apgeochem.2008.05.007
- O. S. Pokrovsky, G. S. Pokrovski and A. Feurtet-Mazel, A structural study of cadmium interaction with aquatic microorganisms, Environ. Sci. Technol., 2008, 42, 5527–5533, DOI:10.1021/es800521a
- O. S. Pokrovsky, J. Viers, E. E. Emnova, E. I. Kompantseva and R. Freydier, Copper isotope fractionation during its interaction with soil and aquatic microorganisms and metal oxy(hydr)oxides: possible structural control, Geochim. Cosmochim. Acta, 2008, 72, 1742–1757, DOI:10.1016/j.gca.2008.01.018
- A. G. González, L. S. Shirokova, O. S. Pokrovsky, E. E. Emnova, R. E. Martínez, J. M. Santana-Casiano, M. González-Dávila and G. S. Pokrovski, Adsorption of copper on Pseudomonas aureofaciens: protective role of surface exopolysaccharides, J. Colloid Interface Sci., 2010, 350, 305–314, DOI:10.1016/j.jcis.2010.06.020
- A. G. González, O. S. S. Pokrovsky, F. Jiménez-Villacorta, L. S. Shirokova, J. M. Santana-Casiano, M. González-Dávila and E. E. Emnova, Iron adsorption onto soil and aquatic bacteria: XAS structural study, Chem. Geol., 2014, 372, 32–45, DOI:10.1016/j.chemgeo.2014.02.013
- A. Coutaud, M. Meheut, J. Viers, J.-L. Rols and O. S. Pokrovsky, Zn isotope fractionation during interaction with phototrophic biofilm, Chem. Geol., 2014, 390, 46–60, DOI:10.1016/j.chemgeo.2014.10.004
- M. Coutaud, M. Méheut, P. Glatzel, G. S. Pokrovski, J. Viers, J.-L. Rols and O. S. Pokrovsky, Small changes in Cu redox state and speciation generate large isotope fractionation during adsorption and incorporation of Cu by a phototrophic biofilm, Geochim. Cosmochim. Acta, 2018, 220, 1–18, DOI:10.1016/j.gca.2017.09.018
- F. C. A. Kafantaris and D. M. Borrok, Zinc isotope fractionation during surface adsorption and intracellular incorporation by bacteria, Chem. Geol., 2014, 366, 42–51, DOI:10.1016/j.chemgeo.2013.12.007
- J. H. Jamieson-Hanes, H. K. Shrimpton, H. Veeramani, C. J. Ptacek, A. Lanzirotti, M. Newville and D. W. Blowes, Evaluating zinc isotope fractionation under sulfate reducing conditions using a flow-through cell and in situ XAS analysis, Geochim. Cosmochim. Acta, 2017, 203, 1–14, DOI:10.1016/j.gca.2016.12.034
- F. C. Fang, E. R. Frawley, T. Tapscott and A. Vázquez-Torres, Bacterial stress responses during host infection, Cell Host Microbe, 2016, 20, 133–143, DOI:10.1016/j.chom.2016.07.009
- D. J. R. Lane, A. M. Merlot, M.-H. Huang, D.-H. Bae, P. J. Jansson, S. Sahni, D. S. Kalinowski and D. R. Richardson, Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1130–1144, DOI:10.1016/j.bbamcr.2015.01.021
-
A. Marchetti and M. T. Maldonado, Iron, The Physiology of Microalgae, ed. M. A. Borowitzka, J. Beardall and J. A. RavenSpringer International Publishing, Cham, 2016, pp. 233–279 Search PubMed
- G. M. Gadd, Metals, minerals and microbes: geomicrobiology and bioremediation, Microbiology, 2010, 156, 609–643, DOI:10.1099/mic.0.037143-0
-
E. E. Emnova, R. III, D. A. Dascualinc, S. Toma, O. Gojnetschi and I. Seniscovscaia, Soil invertase and levansucrase activities under soil water stress, elevated copper level, and soybean seed inoculation with the levan-producing bacteria Pseudomonas aureofaciens, 2005, vol. 1–3, pp. 128–136 Search PubMed
- J. M. Meyer and M. A. Abdallah, The fluorescent pigment of Pseudomonas fluorescens: biosynthesis, purification and physicochemical properties, J. Gen. Microbiol., 1978, 107, 319–328, DOI:10.1099/00221287-107-2-319
-
U. Behrens and M. Ringpfeil, in Bericht über das Jahr 1963, Springer, 1964, pp. 420–429 Search PubMed
- E. E. Emnova, L. D. Varbanets, V. N. Vasiliev, A. G. Ciocarlan, O. S. Brovarskaia, N. J. Caunova, O. G. Ganea and S. I. Toma, Properties of exopolysaccharides from rhizospheric fluorescent bacteria of Pseudomonas genus, Bulletin of Moldovan Academy of Sciences Life Sciences, 2007, 1(310), 14–20 Search PubMed
- D. A. Fowle and J. B. Fein, Experimental measurements of the reversibility of metal–bacteria adsorption reactions, Chem. Geol., 2000, 168, 27–36, DOI:10.1016/S0009-2541(00)00188-1
- K. Knauer, R. Behra and L. Sigg, Adsorption and uptake of copper by the green alga Scenedesmus subspicatus (chlorophyta), J. Phycol., 1997, 33, 596–601, DOI:10.1111/j.0022-3646.1997.00596.x
- S. Le Faucheur, R. Behra and L. Sigg, Thiol and metal contents in periphyton exposed to elevated copper and zinc concentrations: a field and microcosm study, Environ. Sci. Technol., 2005, 39, 8099–8107, DOI:10.1021/es050303z
- M. Ma, W. Zhu, Z. Wang and G. J. Witkamp, Accumulation, assimilation and growth inhibition of copper on freshwater alga (Scenedesmus subspicatus 86.81 SAG) in the presence of EDTA and fulvic acid, Aquat. Toxicol., 2003, 63, 221–228, DOI:10.1016/S0166-445X(02)00179-0
- K. A. C. De Schamphelaere, J. L. Stauber, K. L. Wilde, S. J. Markich, P. L. Brown, N. M. Franklin, N. M. Creighton and C. R. Janssen, Toward a biotic ligand model for freshwater green algae: surface-bound and internal copper are better predictors of toxicity than free Cu2+-ion activity when pH is varied, Environ. Sci. Technol., 2005, 39, 2067–2072, DOI:10.1021/es049256l
- F. W. E. Strelow, Improved separation of iron from copper and other elements by anion-exchange chromatography on a 4% cross-linked resin with high concentrations of hydrochloric acid, Talanta, 1980, 27, 727–732, DOI:10.1016/0039-9140(80)80166-4
- F. Poitrasson, A. N. Halliday, D.-C. Lee, S. Levasseur and N. Teutsch, Iron isotope differences between Earth, Moon, Mars and Vesta as possible records of contrasted accretion mechanisms, Earth Planet. Sci. Lett., 2004, 223, 253–266, DOI:10.1016/j.epsl.2004.04.032
- F. Poitrasson and R. Freydier, Heavy iron isotope composition of granites determined by high resolution MC-ICP-MS, Chem. Geol., 2005, 222, 132–147, DOI:10.1016/j.chemgeo.2005.07.005
- K. Dideriksen, J. A. Baker and S. L. S. Stipp, Fe isotope fractionation between inorganic aqueous Fe(III) and a Fe siderophore complex, Mineral. Mag., 2008, 72, 313–316, DOI:10.1180/minmag.2008.072.1.313
- M. A. Fehr, P. S. Andersson, U. Hålenius and C.-M. Mörth, Iron isotope variations in holocene sediments of the Gotland Deep, Baltic Sea, Geochim. Cosmochim. Acta, 2008, 72, 807–826, DOI:10.1016/j.gca.2007.11.033
- S. Aebischer, C. Cloquet, J. Carignan, C. Maurice and R. Pienitz, Disruption of the geochemical metal cycle during mining: multiple isotope studies of lake sediments from Schefferville, subarctic Québec, Chem. Geol., 2015, 412, 167–178, DOI:10.1016/j.chemgeo.2015.07.028
- M. Amor, V. Busigny, P. Louvat, A. Gélabert, P. Cartigny, M. Durand-Dubief, G. Ona-Nguema, E. Alphandéry, I. Chebbi and F. Guyot, Mass-dependent and -independent signature of Fe isotopes in magnetotactic bacteria, Science, 2016, 352(6286), 705–708, DOI:10.1126/science.aad7632
- M. I. Boyanov, S. D. Kelly, K. M. Kemner, B. A. Bunker, J. B. Fein and D. A. Fowle, Adsorption of cadmium to Bacillus subtilis bacterial cell walls: a pH-dependent X-ray absorption fine structure spectroscopy study, Geochim. Cosmochim. Acta, 2003, 67, 3299–3311, DOI:10.1016/S0016-7037(02)01343-1
- O. S. Pokrovsky, G. S. Pokrovski, A. Gélabert, J. Schott and A. Boudou, Speciation of Zn associated with diatoms using X-ray absorption spectroscopy, Environ. Sci. Technol., 2005, 39, 4490–4498, DOI:10.1021/es0480419
- A. G. González and O. S. Pokrovsky, Metal adsorption on mosses: toward a universal adsorption model, J. Colloid Interface Sci., 2014, 415, 169–178, DOI:10.1016/j.jcis.2013.10.028
- N. Yee and J. Fein, Cd adsorption onto bacterial surfaces: a universal adsorption edge?, Geochim. Cosmochim. Acta, 2001, 65, 2037–2042, DOI:10.1016/S0016-7037(01)00587-7
- O. V. Oleinikova, F. Poitrasson, O. Y. Drozdova, L. S. Shirokova, S. A. Lapitskiy and O. S. Pokrovsky, Iron Isotope Fractionation during Bio-and Photodegradation of Organoferric Colloids in Boreal Humic Waters, Environ. Sci. Technol., 2019, 53, 11183–11194, DOI:10.1021/acs.est.9b02797
- N. Balci, T. D. Bullen, K. Witte-Lien, W. C. Shanks, M. Motelica and K. W. Mandernack, Iron isotope fractionation during microbially stimulated Fe(II) oxidation and Fe(III) precipitation, Geochim. Cosmochim. Acta, 2006, 70, 622–639, DOI:10.1016/j.gca.2005.09.025
- L. Wu, B. L. Beard, E. E. Roden and C. M. Johnson, Stable Iron Isotope Fractionation between Aqueous Fe(II) and Hydrous Ferric Oxide, Environ. Sci. Technol., 2011, 1847–1852, DOI:10.1021/es103171x
- W. B. Homoky, S. Severmann, R. A. Mills, P. J. Statham and G. R. Fones, Pore-fluid Fe isotopes reflect the extent of benthic Fe redox recycling: evidence from continental shelf and deep-sea sediments, Geology, 2009, 37(8), 751–754 CrossRef CAS
- J. K. Klar, W. B. Homoky, P. J. Statham, A. J. Birchill, E. L. Harris, E. M. S. Woodward, B. Silburn, M. J. Cooper, R. H. James, D. P. Connelly, F. Chever, A. Lichtschlag and C. Graves, Stability of dissolved and soluble Fe(II) in shelf sediment pore waters and release to an oxic water column, Biogeochemistry, 2017, 135, 49–67 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00332a |
| This journal is © The Royal Society of Chemistry 2024 |