
Wildfire plume ageing in the Photochemical Large Aerosol Chamber (PHOTO-LAC)†
Hendryk
Czech
 *a,
Olga
Popovicheva
*b,
Dmitriy G.
Chernov
c,
Alexander
Kozlov
d,
Eric
Schneider
ae,
Vladimir P.
Shmargunov
c,
Maxime
Sueur
fg,
Christopher P.
Rüger
ae,
Carlos
Afonso
fg,
Viktor
Uzhegov
c,
Valerii S.
Kozlov
c,
Mikhail V.
Panchenko
c and
Ralf
Zimmermann
ae
*a,
Olga
Popovicheva
*b,
Dmitriy G.
Chernov
c,
Alexander
Kozlov
d,
Eric
Schneider
ae,
Vladimir P.
Shmargunov
c,
Maxime
Sueur
fg,
Christopher P.
Rüger
ae,
Carlos
Afonso
fg,
Viktor
Uzhegov
c,
Valerii S.
Kozlov
c,
Mikhail V.
Panchenko
c and
Ralf
Zimmermann
ae
aDepartment of Analytical and Technical Chemistry, Chair of Analytical Chemistry, Joint Mass Spectrometry Centre (JMSC), University of Rostock, 18059, Rostock, Germany. E-mail: hendryk.czech@uni-rostock.de
bSkobeltsyn Institute of Nuclear Physics, Lomonosov Moscow State University, 119991, Moscow, Russia. E-mail: olga.popovicheva@gmail.com
cV. E. Zuev Institute of Atmospheric Optics, Siberian Branch of the Russian Academy of Sciences, 634055, Tomsk, Russia
dVoevodsky Institute of Chemical Kinetics and Combustion, Siberian Branch of the Russian Academy of Sciences, 630090, Novosibirsk, Russia
eDepartment Life, Light & Matter (LLM), University of Rostock, 18059, Rostock, Germany
fNormandie Université, UNIROUEN, INSA Rouen, CNRS, COBRA, 76000, Rouen, France
gInternational Joint Laboratory – iC2MC: Complex Matrices Molecular Characterization, 76700, Harfleur, France
First published on 24th October 2023
Abstract
Plumes from wildfires are transported over large distances from remote to populated areas and threaten sensitive ecosystems. Dense wildfire plumes are processed by atmospheric oxidants and complex multiphase chemistry, differing from processes at typical ambient concentrations. For studying dense biomass burning plume chemistry in the laboratory, we establish a Photochemical Large Aerosol Chamber (PHOTO-LAC) being the world's largest aerosol chamber with a volume of 1800 m3 and provide its figures of merit. While the photolysis rate of NO2 (jNO2) is comparable to that of other chambers, the PHOTO-LAC and its associated low surface-to-volume ratio lead to exceptionally low losses of particles to the walls. Photochemical ageing of toluene under high-NOx conditions induces substantial formation of secondary organic aerosols (SOAs) and brown carbon (BrC). Several individual nitrophenolic compounds could be detected by high resolution mass spectrometry, demonstrating similar photochemistry to other environmental chambers. Biomass burning aerosols are generated from pine wood and debris under flaming and smouldering combustion conditions and subsequently aged under photochemical and dark ageing conditions, thus resembling day- and night-time atmospheric chemistry. In the unprecedented long ageing with alternating photochemical and dark ageing conditions, the temporal evolution of particulate matter and its chemical composition is shown by ultra-high resolution mass spectrometry. Due to the spacious cavity, the PHOTO-LAC may be used for applications requiring large amounts of particulate matter, such as comprehensive chemical aerosol characterisation or cell exposures under submersed conditions.
Environmental significanceWildfires are a significant source of airborne particulate matter (PM), which has consequences for ecosystems, climate and human health. When wildfires occur in remote places, biomass burning plumes undergo physical and chemical transformation in the atmosphere under alternating day- and night-time conditions during long-range transport before arrival at more densely populated regions. Plume ageing significantly differs from atmospheric processing at typical ambient levels because of optical plume thickness and different oxidant availability. The Photochemical Large Aerosol Chamber (PHOTO-LAC) was established on the basis of a LAC and provides a platform for systematic studies of wildfire plume ageing under well-defined UV light and dark exposure conditions. Large-scale PM sampling may be potentially linked to physico-chemical properties with its toxicological characteristics. |
1. Introduction
Biomass burning is a substantial contributor to ambient particulate matter (PM) and releases between 26 and 70 Tg of organic aerosols and about 6 Tg of black carbon (BC) or elemental carbon (EC) per year into the atmosphere.1,2 It involves both anthropogenic and natural sources, spanning a range of emission from residential heating in small-scale combustion appliances to open burning in wildfires. Wildfires may appear as a global phenomenon from subpolar areas to demi-deserts, in dry regions and wetlands, which may naturally occur by lightning during the so-called fire seasons.3 However, in the past few decades, anthropogenic activities and climate change have promoted wildfires by means of a higher frequency of abundance and larger areas burned as well as lengthened fire seasons.4,5Wildfires in Siberia impact the climate-relevant species, including organic and BC aerosols, and cause pronounced pollution events.6 Plumes of Siberian wildfires spread over vast territories, threaten sensitive ecosystems7,8 and deteriorate the air quality in Europe and East Asia.9,10
The release of products from incomplete combustion during wildfires is driven by the fire propagation, combustion conditions, composition of fuel and meteorological conditions; thus the resulting emissions are highly variable. Fuels involved in wildfires span the range from dry and weathered plant foliage to wood with a high moisture content of approximately 50% by mass, which separates itself from residential heating by dry logwood.
Burning conditions may be divided into smouldering and flaming. The smouldering process is driven by the heat released from heterogeneous oxidation of a solid fuel on its surface by oxygen in the atmosphere. Flaming combustion appears at higher temperatures than smouldering, so the solid fuel is volatilised first and subsequently oxidised in the gas phase. Small radicals in the flame may combine and form aromatic rings, which further build up to a disordered graphitic microstructure in soot particles.11 Slow and low-temperature smouldering fires yield substantially higher emissions of organic matter and produce weakly light-absorbing organic carbon (brown carbon, BrC), while flaming combustion contributes to BC.12 Moreover, flaming combustion conditions promote the conversion of fuel-nitrogen to nitrous gases (NOx), while during smouldering fuel-nitrogen is released as nitrogen-containing organic compounds.13
When wildfires appear close to populated regions, they may rapidly elevate the short-term exposure to respirable fine particulate matter (PM2.5), which is associated with increased mortality as well as respiratory and cardiovascular diseases.14,15 Wildfire-related PM2.5 has been found to be more relevant for respiratory health than other sources,16 but fuel composition and combustion conditions triggered different mutagenicity and lung toxicity observed in in vivo models.17–19
It is well known that biomass burning aerosols are rapidly transformed in the atmosphere (“atmospheric ageing”) and increase in organic matter by oxidative gas-to-particle conversion, which is called secondary organic aerosol (SOA) formation.20 However, in dense biomass burning plumes, the ambient level of hydroxyl radicals, the main atmospheric oxidant during daytime, decreases so other oxidants may increase in relative importance. Due to the high optical depth of plumes, the formation of nitrate radicals (·NO3) in a plume centre is enhanced, despite the high susceptibility of ·NO3 towards photolysis by visible UV light. Consequently, atmospheric ageing of plumes involves spatial variations, which may lead to concentration gradients or spatially different optical properties21–23 that may not be represented in biomass burning ageing studies using lower concentrations in environmental chambers24,25 or oxidation flow reactors.26,27 Furthermore, plume chemistry enhances the production of ozone28 with consequences for air quality in urban areas when VOC-rich wildfire plumes are mixed with NOx-rich urban air29 or affect stratospheric ozone depletion.30
In epidemiological as well as toxicological studies, SOAs in general, SOAs from individual biomass burning-related SOA precursors and aged wood combustion aerosols have been linked to increased mortality and several biological effects in in vitro exposures.31–36 Moreover, atmospheric long-range transported wildfire smoke was associated with increases in cardiopulmonary morbidity and mortality37 Therefore, systematic laboratory studies on the effect of atmospheric ageing on biomass burning plumes may help to understand potential health effects from long-range transport of wildfire emissions.
A Large Aerosol Chamber (LAC) with a total volume of approximately 1800 m3 has been established for studies of optical properties and chemical composition of fresh and dark aged biomass burning under defined conditions, limiting the impact of soil, wind, and unpredictable combustion phase.12,38–40 The analyses by Popovicheva & Kozlov (2020)41 have revealed the influence of combustion temperature on the scattering and light absorption of pine and debris smoke. While a high-temperature open flaming regime mostly emits strongly light-absorbing soot, low-temperature smouldering generates weakly light-absorbing particles.
With a LAC modification to a PHOTO-LAC, we aim to bridge the gap between atmospheric processing of wildfire plumes and laboratory-ageing of biomass burning aerosols under day–night environmental conditions corresponding to high plume concentration levels. In the following, we introduce photochemistry in the LAC, provide the figures of merit for environmental chambers, such as wall loss of particles and gases as well as the photolysis rate of NO2, and describe unprecedented long chamber-ageing under alternating photochemical and dark ageing conditions.
2. Materials and methods
2.1. Large Aerosol Chamber (LAC)
The Large Aerosol Chamber (LAC) located in the Institute of Atmospheric Optics, Siberian Branch, Russian Academy of Sciences (Tomsk), comprises a cigar-like, approximately cylindrical shape, with a height of 10 m, a length of 25 m, an approximate volume of 1800 m3 and an inner surface of 942 m2. A scheme of the LAC with instrumentation and a photograph is shown in Fig. 1. The resulting ratio of inner surface to volume of ∼0.52 m−1 is beneficial to minimise particle losses, so aerosol studies over 48 h with acceptable wall losses are possible.40 The inner walls of the LAC are made of steel, which may affect aerosol chemistry.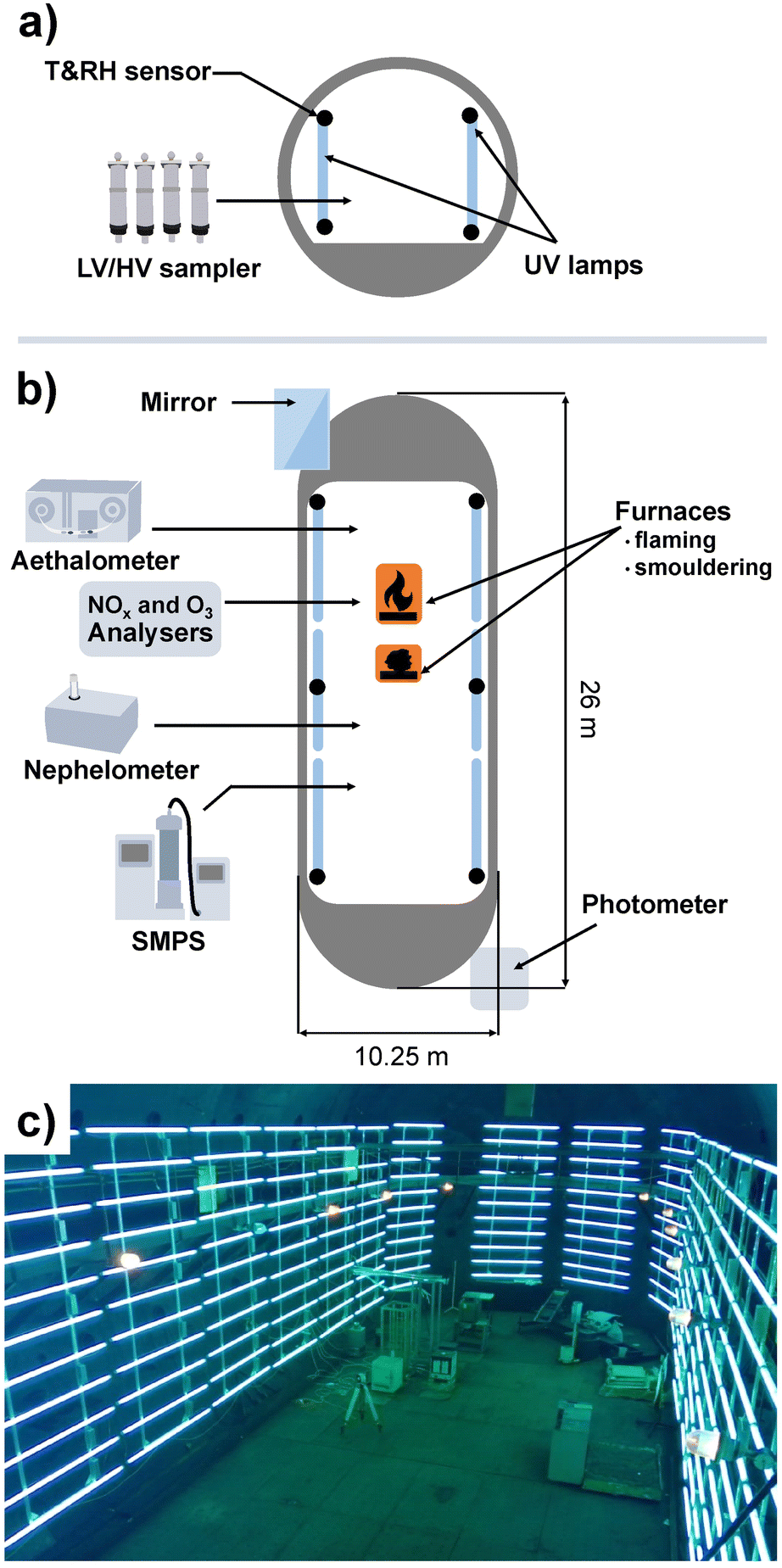 | ||
| Fig. 1 Scheme of the PHOTO-LAC from (a) longitudinal and (b) plan views with equipment for aerosol generation and measurements and (c) a photograph. UV lamps and ovens are located inside the PHOTO-LAC, whereas analytical instrumentation is placed in a laboratory aside and connected to the PHOTO-LAC cavity by stainless steel tubing. | ||
Ambient air is used to fill the chamber and therefore determine the initial temperature, humidity and aerosol composition in an experiment. However, when the LAC is closed, ambient temperature has no effect on the temperature inside. Two furnaces are located inside the LAC with combustion temperatures of ∼400 °C for smouldering and ∼700 °C for open flaming,12 which leads to aerosol concentration cancelling out the effect of background air composition. About 100 to 400 g of biomass is typically used for small-scale fires. Temperature and relative humidity are measured using 12 sensors uniformly distributed in the LAC at two levels: 0.5 m and 4 m from the floor. Via a sampling line in the centre of the LAC with 25 and 40 mm diameter at 2 m height, PM is collected by low and high-volume sampling for further chemical analysis.
For the characterisation of the LAC homogeneity, the LAC was equipped with a modernised path photometer of atmospheric spectral transmittance, operating in the wavelength (λ) range from 0.45 to 3.9 μm.42,43 The photometer of atmospheric spectral transmittance also monitors the radiation extinction for particles with a radius r > λ/5 μm.44 Measurements of aerosol extinction in the IR spectral range at a wavelength λ = 3.9 μm provide information on the content of particles with a radius r > 0.8 μm (coarse particles). The difference between the measured aerosol extinction in the visible (at λ = 0.55 μm) and IR (at λ = 3.9 μm) spectral ranges allows the determination of the variation of the optical properties of submicron particles in the size range 0.1 < r < 0.8 μm. Comparing the path measurements of atmospheric spectral transmittance with the scattering coefficients measured with a nephelometer in a local volume, the homogeneity of the optical characteristics of smoke aerosols throughout the chamber volume can be estimated.43
2.2. Modification of the LAC to PHOTO-LAC
In order to facilitate atmospheric processing of aerosols under daytime conditions, the LAC was further equipped with 198 UV lamps (length: 2 m; diameter: 4 cm; lamp wattage: 180 W; JW Sales GmbH, Brand Division Cosmedico) inside, which were mounted vertically distributed over both sides of the LAC cavity (PHOTO-LAC) (Fig. 1). It is possible to turn on all UV lamps (100% UV intensity) or only on the left or the right long side of the PHOTO-LAC (50% UV intensity). With a final surface-to-volume ratio of 0.55 m−1, the additional surface from the UV lamps had only a minor effect on the overall inner surface of the PHOTO-LAC. Compared to traditional environmental chambers, the PHOTO-LAC inherently features lower particle wall losses, but the placement of the UV lamps inside the PHOTO-LAC does not allow temperature control during photochemical ageing.Irradiance from the 198 UV lamps was measured by using a spectroradiometer (TKA-ΠKM, TKA LLC) with a bandwidth of 1 nm over the wavelength range from 200 to 1100 nm. The spectrum covers the atmospherically important region of approximately 300 to 400 nm, thereby initialising fundamental processes, including the photolysis of NO2 and O3, and partially resembles the solar spectrum on the 37° sun-facing tilted surface of the Earth (ASTM G173-03 Reference Spectra) (Fig. 2). Similar to UV lamps used in other chambers, the emission spectrum has a broad maximum at around 350 nm and a narrower peak at 365 nm.45 Additionally, distinct emission peaks in the visible UV range appear at 405, 436, 545, and 578 nm. In order to generate different atmospheric ageing scenarios and to promote the formation of hydroxyl radicals (·OH), the PHOTO-LAC was equipped with a humidifier (Par-Tuman LLC) and an ozone generator (Groza-60M; Absolut LLC).
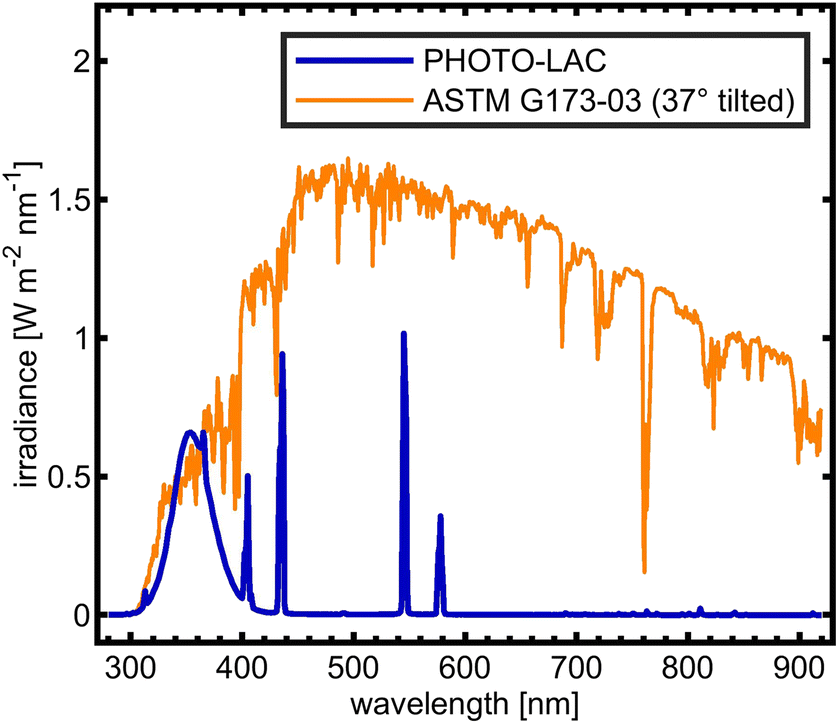 | ||
| Fig. 2 UV lamp emission inside the PHOTO-LAC and solar spectrum (ASTM G173-03). | ||
2.3. Instrumentation
Online aerosol particle measurements were conducted with a scanning mobility particle sizer (SMPS), a custom-made aethalometer 
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46 and a multi-angle multi-wavelength nephelometer APSN-2.47 By sampling from the centre of the PHOTO-LAC through a stainless steel tube (10 mm inner diameter), the SMPS (Grimm #5.403; Grimm Aerosol Technik GmbH & Co. KG) provided particle number concentrations and was operated with a time resolution of 3.5 min. The total particle number concentration was obtained by integration of the particle size distribution from 11.1 nm to 1083.3 nm.
46 and a multi-angle multi-wavelength nephelometer APSN-2.47 By sampling from the centre of the PHOTO-LAC through a stainless steel tube (10 mm inner diameter), the SMPS (Grimm #5.403; Grimm Aerosol Technik GmbH & Co. KG) provided particle number concentrations and was operated with a time resolution of 3.5 min. The total particle number concentration was obtained by integration of the particle size distribution from 11.1 nm to 1083.3 nm.
The method for analysing the spectral absorption of aerosols is based on measuring the attenuation of radiation passing through a filter during continuous sampling of aerosols from the atmosphere. With laser diodes at 460, 530 and 630 nm, the aethalometer determines the attenuation ATNλ of the light transmittance T by PM sampled on a filter tape from which the attenuation coefficient bATN,i for a time interval Δt can be retrieved at individual wavelengths λ:
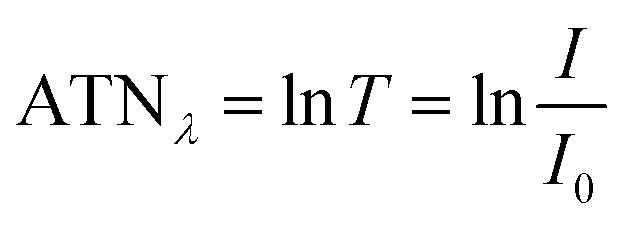 | (1) |
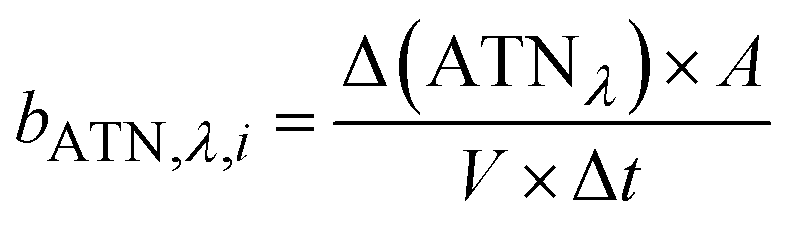 | (2) |
| babs,λ ≈ bATN,λ | (3) |
The spectral dependence of babs over λ can be described by
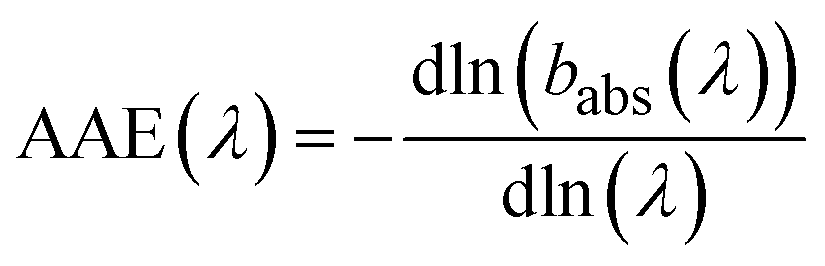 | (4) |
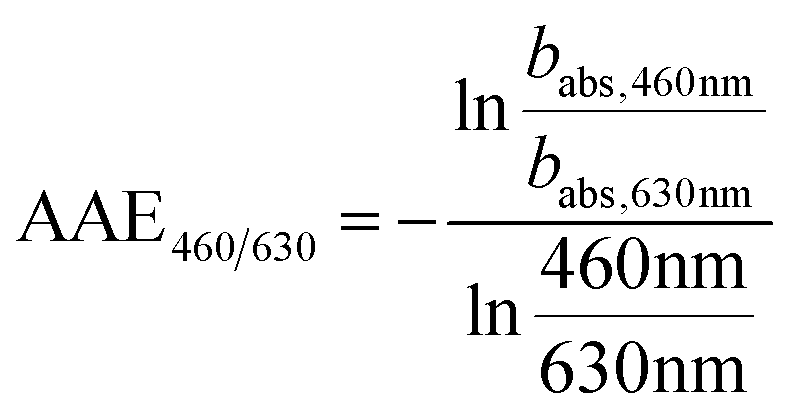 | (5) |
A transmit/receive system and a mirror reflector of the path meter are set on opposite sides of the chamber to control the homogeneity of optical characteristics throughout the entire chamber volume. They are installed behind quartz windows where the measuring path runs along the major axis of the chamber. The transmit/receive system and a mirror reflector are spaced by 26 m, and the total path length L is 52 m. The transmittance meter operates at 12 spectral wavelengths in the range 0.45–3.91 μm.
The total radiation extinction coefficients βEXT(λ) are determined from T through the Bouguer equation:
| lnT(λ) = −βEXT(λ) × L | (6) |
The directional scattering coefficients βSCT(λ,φ) are measured simultaneously with the total extinction coefficients βEXT(λ) with the APSN-2 in the wavelength range λ from 0.46 to 0.63 μm and in the range of scattering angles φ from 25 to 165°. βSCT(λ,φ) measured at a wavelength λ of 0.53 μm at a scattering angle φ of 45° is proportional to the integral scattering coefficient and related to the submicron PM fraction of the smoke aerosol as
| PM (μg m−3) = 2.4 × 103 μg m−3 × βSCT(45°) | (7) |
A SOA from toluene photooxidation was analysed using a Velos Orbitrap (Thermo Scientific, Bremen, Germany) with electrospray ionisation in negative ionization mode (ESI-) with a direct-infusion ion source electrospray setup using a spray voltage of 4.2/3.5 kV and a syringe flow rate of 7 μL min. Data were collected in the range of 90 to 600 Da, resulting in a resolving power of >100000 at m/z 400 and mass accuracy below 3 ppm. MS/MS data were acquired by isolation of selected m/z values in a window of 1 Da by ion trapping and subsequent collision-induced dissociation (CID) at voltages optimised for each selected mass range.
BB PM filter samples were analysed by Fourier-transform Ion Cyclotron Resonance Mass Spectrometry (FT-ICR MS) using a solariX 2XR 12T (Paracell™) instrument (Bruker Daltonik, Bremen, Germany). The instrument was equipped with a krypton lamp (10.6/10.0 eV) for atmospheric pressure photoionisation (APPI). The ion source was operated in positive mode and includes a vaporiser unit set to 350 °C and a syringe flow rate of 600 μL h−1. In total 100 transients were acquired and added up for each analysis. Transients were digitalised using 4 M data points and a mass range from m/z 92.1 to 1000, leading to a transient length of 1.0486 s. The time of flight to the ICR cell was set to 0.6 ms and ions were accumulated for 150 ms. The resolving power m/z 400 and mass accuracy were >300000 and below 1 ppm, respectively.
The exported mass spectra were processed by using self-written MATLAB algorithms and routines combined in a graphical user interface named CERES Processing. After careful investigation and taking into account attribution boundaries from the literature, the following restrictions were deployed for elemental composition assignment for ESI-Orbitrap-MS in the range of 90 to 600 Da: CcHhNnOo; 4 ≤ c ≤ 50, 2 ≤ h ≤ 100, n ≤ 4, and o ≤ 15, with a maximum error of 3 ppm. Additional restrictions were applied for the H-to-C ratio (H:C): 0.4–2.4, O-to-C ratio (O:C): 0–1.4 and double bond equivalents (DBE) 0–20.
For APPI-FT-ICR-MS the following attribution boundaries were applied: CcHhNnOo; 4 ≤ c ≤ 100, 2 ≤ h ≤ 200, n ≤ 3, o ≤ 20, and s ≤ 1, with a maximum error of 1 ppm. Additional restrictions were applied for the H:C: 0.4–2.4, O:C: 0–1.4 and double bond equivalents (DBE) 0–30.
In order to address the variability of the APPI-HRMS analysis, a photochemically-aged BB aerosol from smouldering combustion (referring to BB06 in Table 2) was analysed in three repetitions. Due to a large percentage of peaks with a signal-to-noise ratio just above the threshold of six, one measurement gave 20% fewer detected peaks. However, the variation of the intensive properties of the aerosol, such as elemental ratios O:C and H:C, was minor and did not affect the interpretation of the results (Table S1†).
For the determination of toluene wall losses, gas phase samples were collected on adsorbent tubes (Orbo 32, Supelco) at 0.3 L min−1 for 90 min each over a total experimental duration of 24 h. Adsorbent tubes were extracted with 1.5 mL of carbon disulphide (extra pure, Acros Organics) and analysed by gas chromatography single-quadrupole mass spectrometry (GC 6890/MS 5973; type, Agilent) with a temperature programme of 40 °C (10 min) to 230 °C (10 °C min−1) and quantified by external calibration.
2.4. PHOTO-LAC characterisation
With these model aerosol particles, the particle number and PM mass losses were determined. The loss of particle number concentration (PN) in cm−3 over time t was fitted according to
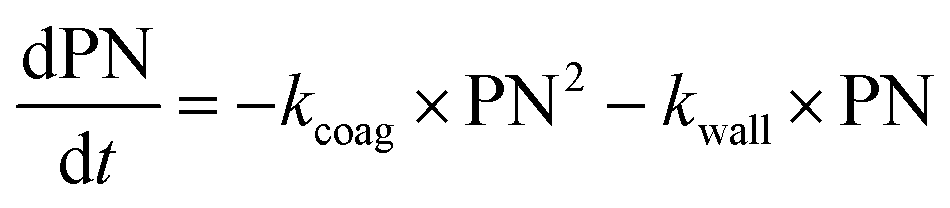 | (8) |
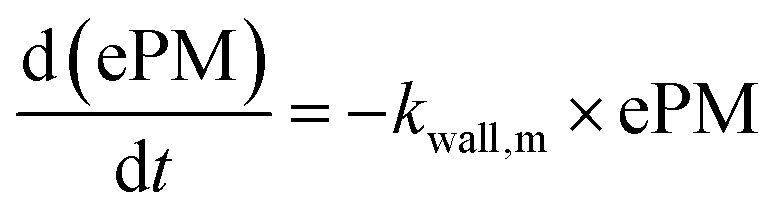 | (9) |
The VOC wall loss was determined with toluene, which is an abundant VOC in biomass burning aerosols.13 First, 11 g of toluene was evaporated in a crucible at 150 °C. Shortly after complete evaporation toluene was sampled on adsorbent tubes, and then after 2, 4, 19, and 21 h and analysed by GC/MS.
| Cu + 4HNO3 → Cu2+ + 2NO3− + 2H2O + 2NO2 | (10) |
| 3Cu + 8HNO3 → 3Cu2+ + 6NO3− + 4H2O + 2NO | (11) |
In the atmosphere, NO2 is photolysed by UV light, generating O(3P), which subsequently converts O2 to O3 according to
| NO2 + hν (<420 nm) → NO + O(3P) | (12) |
| O(3P) + O2 + M → O3 + M | (13) |
| O3 + hν (<335 nm) → O2 + O(1D) | (14) |
| O(1D) + H2O → 2·OH | (15) |
Finally, the NO2 photolysis rate j(NO2) denotes the ratio between the product of O3, NO and kO3,NO (1.8 × 10−14 s cm−3) with the remaining concentration of NO2
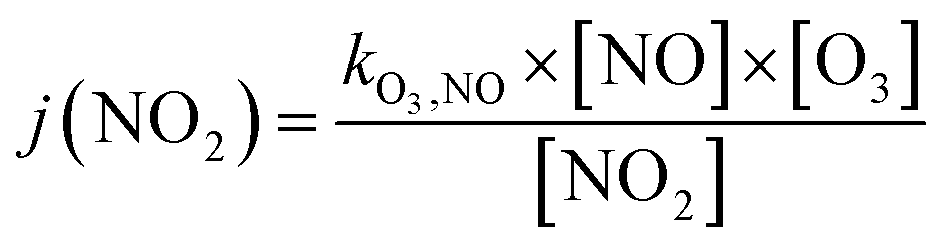 | (16) |
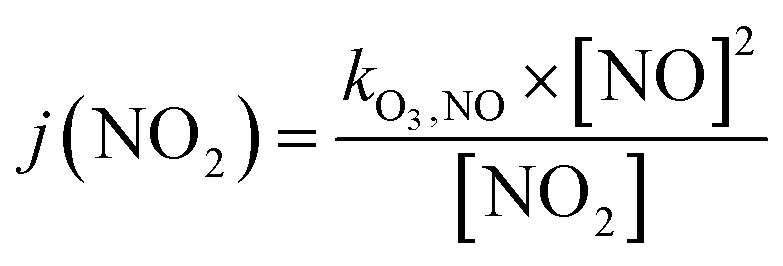 | (17) |
| Exp. | [tol]0a ppb | [NO2]0 [ppb] | [O3]0 [ppb] | RH0 [%] | j(NO2) [min−1] | t UV [h] | [ΔSOA]endb [μg m−3] | ΔmSOA,maxc [mg] |
|---|---|---|---|---|---|---|---|---|
| a Theoretical concentration of toluene from an evaporated toluene mass of 10 to 11 g. b From the nephelometer assuming a particle density of 1.5 g cm−3 and wall loss corrected. c Uncorrected maximum ePM mass in the PHOTO-LAC. | ||||||||
| tol1 | 1500 | 800 | 125 | 63 | 0.17 | 4.0 | 780 | 1320 |
| tol2 | 1500 | 110 | <1 | 57 | 0.17 | 2.0 | 210 | 350 |
| tol3 | 1600 | 1000 | 250 | 57 | 0.34 | 4.1 | 640 | 1000 |
| tol4 | 1500 | 320 | 150 | 69 | 0.34 | 3.5 | 1100 | 1730 |
| Exp. | Combustion | Ageing sequence | [NO2]0 [ppb] | [O3]0 [ppb] | t ageing [h] | [ΔePM]0b [mg m−3] | [ΔePM]endc [mg m−3] |
|---|---|---|---|---|---|---|---|
| a Unreliable for NO2 detection due to high VOC, and NO2 estimated from NO assuming full conversion to NO2 by O3. b Conversion to emission factors in g kg−1 by a factor of 4.5 m3 kg−1. c Wall loss-corrected. | |||||||
| BB1 | Flaming | Dark | 92 | 117 | 4 | 2.9 | 3.1 |
| BB2 | Smouldering | Dark | 33 | 34 | 4 | 15.7 | 16.7 |
| BB3 | Flaming | Photo-dark | 181 | 55 | 3/2.5 | 2.3 | 2.1 |
| BB4 | Flaming | Photo-dark | 153 | 76 | 2.5/2 | 2.2 | 2.0 |
| BB5 | Flaming | Photo-dark | 140 | 260 | 2/2 | 1.8 | 1.5 |
| BB6 | Smouldering | Photo-dark | 25a | 34 | 3/2 | 16.6 | 14.1 |
| BB7 | Flaming | Photo-dark-photo | 131 | 115 | 7.5/13/10 | 3.6 | 4.3 |
| BB8 | Smouldering | Photo-dark-photo | 34a | n.a. | 8.5/13.5/10 | 11.0 | 17.8 |
For initial dark ageing conditions (BB1 and BB2), the addition of ozone defines the experimental start. NO2 in the presence of O3 is partially converted into ˙NO3, which is the main oxidant during nighttime52 and important in ageing of optically dense biomass burning plumes during daytime.53 The dark ageing conditions of the flaming biomass burning aerosol resemble the experimental conditions of Kodros et al. (2022),54 but at higher biomass burning aerosol concentrations. Assuming that the majority of O3 reacts with NO2, substantial generation of ˙NO3 may be expected, particularly for the biomass burning aerosol generated under flaming conditions.
For photochemical ageing, UV lamps were turned on for 3 h (BB3–BB6) to 9 h (BB7 and BB8) after addition of O3. Due to the absence of an additional radical source through photolysis, like HONO or H2O2, the production of OH radicals proceeds from O3viaeqn (14) and (15), while the photolysis of NO2 (eqn (12)) and subsequent reaction of O(3P) with O2 (eqn (13)) reflate ozone production. In order to simulate further atmospheric plume processing after sunset, the aged biomass burning plume remained in the PHOTO-LAC for approximately the same duration as UV lamps were turned on, accounting for different rate constants of the main atmospheric oxidants ˙OH, O3, and ˙NO3 together with UV light toward airborne organic compounds.51 In two long ageing experiments (BB7 and BB8), photochemical ageing was repeated after dark ageing with a total experimental duration of approximately 30 h from biomass ignition to the end.
3. Results and discussion
3.1. Temperature and humidity distribution
The mean temperature and relative humidity are evaluated from the 12 sensors distributed inside the PHOTO-LAC. Owing to the large volume and cylindrical shape of the chamber, measurements at individual spots may deviate from the mean from the 12 sensors, particularly when heat or humidity is introduced from a point source (furnace or humidifier, respectively) or when only 50% of the UV lamps are turned on. During approximately one day of stable conditions, the 95% confidence interval (CI) around the mean temperature accounts for ±0.15 K. When the furnace was operated to vaporise toluene, the average temperature increased by 0.002 K min−1 associated with a larger 95% CI of ±0.25 K. In parallel, the mean RH decreased by 0.002 ppm with a 95% CI around the mean RH of ±1.1 pp. In an overnight experiment with a total duration of 16 h, the average temperature decreased from (24.8 ± 0.2) °C to (24.1 ± 0.1) °C and RH from (60.1 ± 1.3)% to (59.4 ± 2.1)%.When the PHOTO-LAC was operated with UV lamps on, larger changes in temperature and RH were observed. During 120 h with 50% of UV lamps turned on and RH of 57% (tol2), the average temperature and RH increased by 0.03 K min−1 and 0.1 ppm with average 95% CIs of 3.5 K and 10.8 pp around the mean. Therefore, the temperature increased from 20.4 °C to 23.8 °C following the empirical equation
| θ(t) = 0.323 °C min−1 × t0.513 + 20.2 °C | (18) |
For a longer experiment of 250 min and 100% UV lamp intensity (tol3), the increase in temperature can be described by
| θ(t) = 0.521 °C min−1 × t0.542 + 16.6 °C | (19) |
3.2. Homogeneity of the biomass burning smoke distribution
The aerosol extinction coefficients βEXT(0.53) at the path and local-volume scattering coefficients βSCT(0.53, 45°) during smouldering and flaming were measured to investigate the spatial homogeneity of the aerosol inside the PHOTO-LAC (Fig. 3a). Three hours after the burning of mixed pine wood and debris under smouldering and after two hours under flaming conditions, βEXT(0.53) at the path and βSCT(0.53, 45°) in the local volume demonstrate the synchronous aerosol dynamics. Thus, three hours after the burning, a homogeneously distributed smoke is formed in the PHOTO-LAC, which demonstrates the consistency of variation of the aerosol optical parameters throughout the chamber cavity and allows the comparability of measurements conducted at different locations inside the PHOTO-LAC.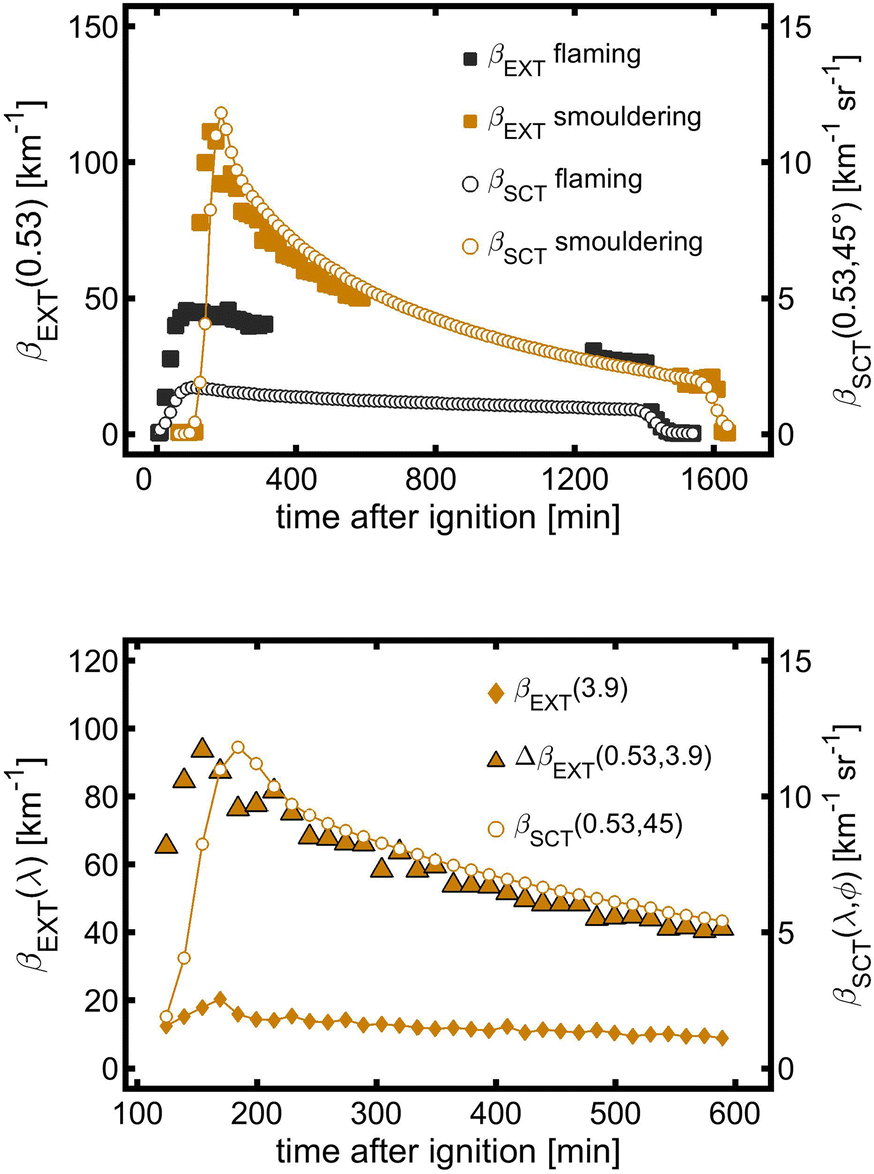 | ||
| Fig. 3 (Top) Evolution of the extinction coefficient βEXT at 0.53 μm and the directional scattering coefficient βSCT at 0.53 μm and 45° of flaming and smouldering aerosols without addition of atmospheric oxidants under dark conditions. (Bottom) Evolution of βEXT at 3.9 μm, difference in βEXT at 0.53 μm and 3.9 μm, and βSCT at 0.53 μm and 45° of the smouldering aerosol for comparison. | ||
Between 200 and 600 min after ignition, βSCT(0.53, 45°) obtained from the local volume and the difference in βEXT between 0.53 μm and 3.9 μm (ΔβEXT) gradually decreased (Fig. 3b) with parallel time trends, reflecting the evolution of the submicron aerosol during the residence of the smouldering BB aerosol. After peaking, βEXT(3.9) decreases by 30% during 30 min of residence, hence indicating a substantial loss of the coarse particle fraction from the size spectrum. Subsequently, the concentration of the coarse aerosol reduces synchronously with the submicron component. The aerosol optical properties in the PHOTO-LAC become homogeneously distributed for the coarse particle fraction 3.5 hours after the biomass ignition. Overall, these estimates support the interpretation of optical data obtained at the centre and integrally throughout the PHOTO-LAC cavity as well as the consistency of variations of different particle sizes collected by the sampling system.
3.3. Wall losses of particles, toluene and reactive gases
Wall losses of particles by means of number and mass concentration were determined for BCP-SOA particles during 6 h and 17 h of ageing, respectively (Fig. 4). Results from fitting eqn (8) demonstrate that particle number losses were significantly caused by coagulation with a kcoag,PN of (2.54 ± 0.1) × 10−5 cm3 h−1. Despite a mean loss of particles to the wall with a kwall,PN of 0.0112 h−1, the 95% confidence interval (−0.0843 h−1 to 0.0538 h−1) of kwall,PN indicates that kwall,PN is not significantly different from zero. The half-life t1/2 of a particle with respect to the wall loss can be estimated from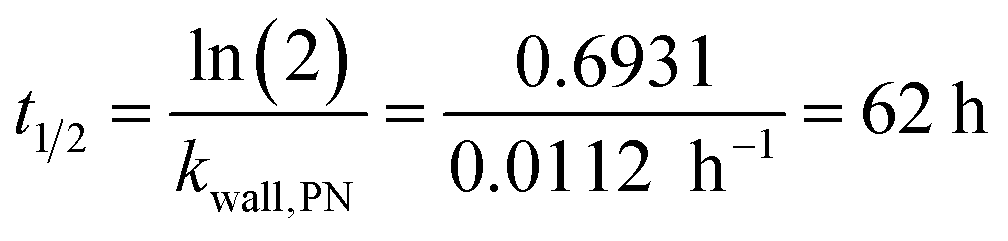 | (20) |
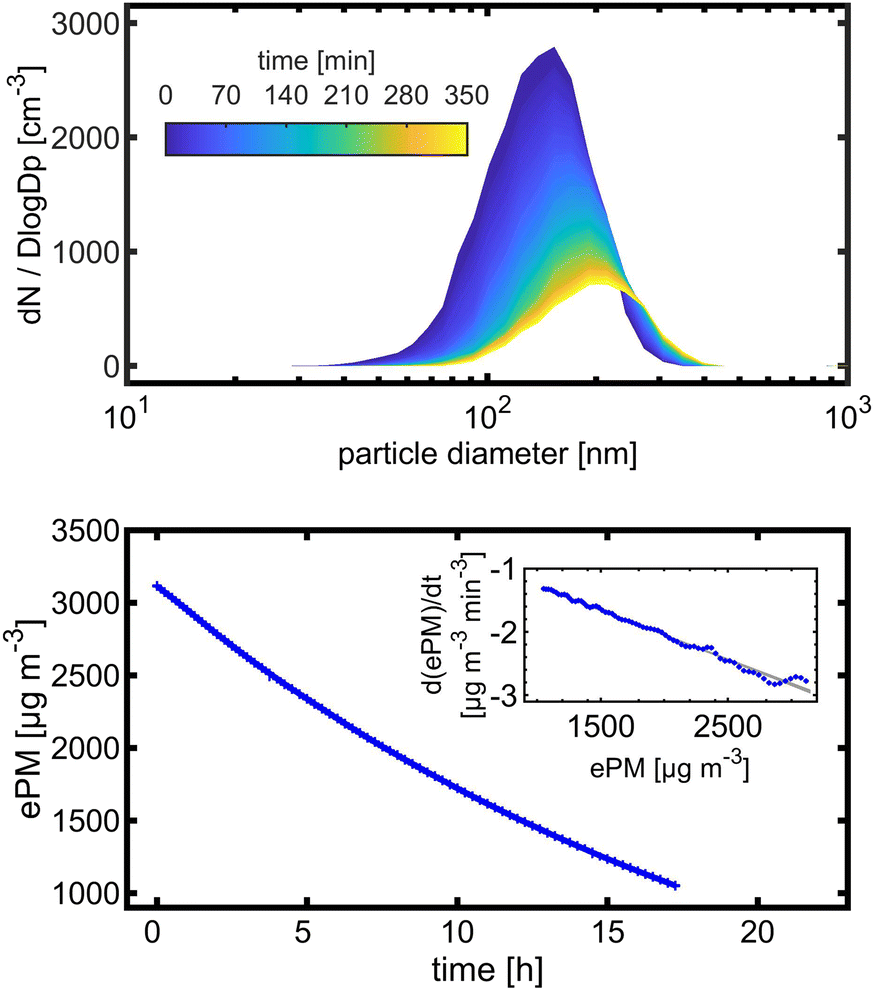 | ||
| Fig. 4 (Top) Change of BCP-SOA number size distribution over time with a kwall,PN of 0.0112 h−1 (95% CI: −0.0843 h−1 to 0.0538 h−1). (Bottom) Wall loss of BCP-SOA mass over time with a wall loss coefficient kwall,PM of 0.0468 h−1 (95% CI: 0.0480 to 0.0457); inset shows the first derivative of ePM according to time t with linear fit in blue and the non-simultaneous functional 95% confidence band in grey. | ||
The dimensions of the PHOTO-LAC and more specifically the low surface-to-volume ratio substantially increases the particle lifetime. Leskinen et al. (2015)45 and Wang 2014 (ref. 55) compiled the rates of particle losses to the wall with the corresponding half-lives ranging from 2.5 to 17 h for chambers of volumes from 12 to 270 m2. Hence, the particle half-life in the PHOTO-LAC exceeds the particle half-lives on environmental chambers of medium size by a factor >3, prolonging the experimental duration and thus longer ageing times.
The rate of wall losses and particle coagulation varies by orders of magnitude for individual particle size bins (Fig. S2†). Therefore, direct comparisons between wall loss experiments with a polydisperse aerosol in different environmental chambers are less conclusive45 and often the kwall,PN values of 100 nm particles are compared. With 0.135 h−1, kwall,PN (100 nm) is one order of magnitude higher than for the total PN concentrations. However, the 95% confidence interval does not indicate that kwall,PN (100 nm) is significantly different from zero, equal to total PN. Overall, the low wall loss rate of particles and similar particle size distribution of the BCP-SOA to the BB aerosol (Fig. S3†) emphasise the benefit of the large dimension of the PHOTO-LAC and give evidence for the applicability of a loss function to combustion experiments, respectively.
In an experiment with BCP-SOA over 17 h, a wall loss rate of kwall,PM of 0.0468 h−1 (95% CI: 0.0480 to 0.0457) was measured by fitting PM mass concentrations derived from nephelometer measurements to a first-order kinetic model (Fig. 4). Larger particles with a major contribution to the total PM mass concentration, but low contribution to PN, got lost to the inner wall of the PHOTO-LAC. The corresponding PM mass concentration half-life of 14.8 h underlines the suitability of the PHOTO-LAC for long lasting aerosol ageing experiments.
The organic matter of the freshly generated BB aerosol is not yet in gas-particle equilibrium, and consequently can affect the particle losses in the PHOTO-LAC. Since smouldering aerosols are rich in organic material compared to flaming combustion aerosols, which contain approximately the same amount of elemental and organic carbon,40 larger losses from evaporating organic particle constituents are expected for smouldering BB aerosols. On that account, flaming and smouldering BB aerosols were generated and left inside the PHOTO-LAC without addition of atmospheric oxidants or light over approximately 24 h. As expected, the combustion of 400 g of biomass (300 g of wood and 100 g of debris) under smouldering conditions generated almost one order of magnitude higher PM mass concentration than under flaming (Fig. S4†).
Flaming aerosol particles were lost to the walls with 0.0440 h−1 (95% CI: 0.0411 to 0.0455 h−1), similar to particles of BCP-SOA. In contrast, the rate for smouldering aerosol particles was 0.0747 h−1 (95% CI: 0.0727 to 0.0767 h−1), likely because of evaporation of volatile and semi-volatile compounds from fresh organic aerosols. These losses coincide with the results of 0.043 h−1 for flaming and 0.065 h−1 for smouldering from Kalogridis et al. (2018)40 based on the EC tracer method. Moreover, a similar wall loss rate was derived from babs from aethalometer measurements at 530 nm of (0.0392 ± 0.0034) h−1 for flaming and of (0.0406 ± 0.0045) h−1 for smouldering conditions (Fig. S4†), which are similar to the wall loss rate for BCP-SOA. Hence, all experiments on characterising the wall losses of particles gave consistent results and demonstrated that the large dimension of the PHOTO-LAC substantially reduce the wall losses of particles.
Losses of organic vapours in the Teflon chamber experiment were recognised as a significant bias for modelling SOA formation from organic precursors.56 On that account, toluene was injected into the PHOTO-LAC filled with ambient air without addition of any oxidant or UV light for one day. A significant decrease in toluene by 40% was found as toluene may be absorbed on the inner walls despites its high volatility. From first-order kinetics, a wall loss rate kwall,tol of (0.0235 ± 0.0188) h−1 was obtained, corresponding to a toluene half-life of 29.5 h (Fig. S5†). However, due to the low number of toluene samples collected, determining the wall loss rate is associated with large uncertainties, spanning the range of toluene half-lives from 16.4 to 147 h at 95% confidence. Nevertheless, the average wall loss rate of toluene is comparable to a 24 m3 fluorinated ethylene propylene chamber.57
Higher loss rates of (0.0039 ± 0.0085) min−1 and (0.0212 ± 0.0022) min−1 were observed for the reactive gases NO2 and O3 with the corresponding half-lives of 3 h and 33 min, respectively, which is two orders of magnitude distant for that of environmental chambers consisting of Teflon and operated at aerosol concentrations close to ambient air.55 Both reactive species are drivers of atmospheric chemistry and particularly for the generation of hydroxyl radicals, and hence their higher losses compared to particles likely affect the chemical composition of aerosols inside the PHOTO-LAC over time. The inner walls of the PHOTO-LAC contain a thin layer of particles from the BB aerosol, including soot and several BB-related organic species, which are known to react efficiently with ozone and NO2 on surfaces,58–61 so both adsorption on or reactive losses to walls are possible processes to explain the decay in the concentrations of NO2 and O3. However, ambient air containing approximately 104 particles per cm3, which was used to fill the PHOTO-LAC, provides a surface area in the same order of magnitude as the inner surface of the PHOTO-LAC. Therefore, it cannot be completely ruled out to which extent the losses of NO2 and O3 are caused by the inner walls or a consequence of moderately high levels of background particles. An overview on the wall loss rates of PN, ePM BCP-SOA, ePM flaming BB, ePM smouldering BB, toluene and NO2 and O3 expressed as half-lives is summarised in Table 3.
| Gas or particle quantity | Average | 95%-CIlow | 95%-CIup |
|---|---|---|---|
| PN | 62 h | 31 h | ∞ |
| PN (100 nm) | 5.13 h | 1.96 h | ∞ |
| ePM BCP-SOA | 14.8 h | 14.4 h | 15.2 h |
| ePM flaming BB | 15.8 h | 15.2 h | 16.9 h |
| ePM smouldering BB | 9.3 h | 9.0 h | 9.5 h |
| Toluene | 29.5 h | 16.4 h | 147 h |
| NO2 | 3.0 h | 2.5 h | 3.8 h |
| O3 | 33 min | 30 min | 37 min |
3.4. UV lamp emission spectrum and NO2 photolysis rate
The radiation from the 198 UV lamps inside the PHOTO-LAC covers the wavelength range from approximately 300 to 450 nm, which may initiate fundamental chemical processes in the atmosphere, such as the photolysis of NO2. At full UV lamp power, the NO2 photolysis rate j(NO2) accounts for (0.35 ± 0.01) min−1, which appears well within the range of other environmental chambers.45 Moreover, UV lamps on the opposite long side of the PHOTO-LAC may be turned on individually for a lower total light intensity. Therefore, j(NO2) for both sides account for approximately half of j(NO2) at full UV light power with a j(NO2)left of (0.18 ± 0.01) min−1 and a j(NO2)right of (0.19 ± 0.01) min−1. When all UV lamps are turned on, aerosols inside the PHOTO-LAC receive an integrated irradiance of 24 W m−2, which is close to the ground level of UV solar radiation in Siberian summer.43In addition to the photolysis of NO2 with subsequent formation of O3, the photolysis of the latter one is essential for the generation of OH radicals. For that, the wavelength region between approximately 300 and 320 nm shows significant quantum yields for the photodissociation of O3,62 which is covered by the UV lamps with an integrated irradiance of 0.7 W m−2 and may be favourable compared to blacklight emission sources of narrow bandwidths in the UV-A region. Furthermore, the photolysis of carbonyls, such as acetone and formaldehyde being the major VOC emission constituents in BB aerosol emissions,2 appearing at wavelengths below 360 nm, is well-covered and denotes an additional source of OH radicals with atmospheric relevance.62 For larger carbonyls, such as methyl vinyl ketone or methacrolein, light of the UV-A range is absorbed and may lead to photoisomerisation and the formation of, e.g., unsaturated alcohols.63 Also visible UV light may induce photochemical reactions in soot particles,64 or photolysis of α-diketones, such as 2,5-pentadione,65 which is less represented by the UV lamp emission spectrum and may favour specific reactions due to its narrow visible UV emission bands around 436, 545 and 578 nm.
3.5. SOA formation from toluene photooxidation
Toluene refers to a common SOA precursor, which has been widely investigated as a proxy for anthropogenic VOCs66,67 and frequently used for the performance characterization of environmental chambers.45 The study design includes four toluene photooxidation experiments under similar initial toluene concentrations and relative humidity, but different initial ratios of VOC/NOx and NO2/O3 and times of irradiance. A few minutes after UV lamps were turned on, particles formed by nucleation can be observed with an initial diameter mode of 24.7 nm, which increases to 82.8 nm over 4 h of photochemical ageing (Fig. 5a) by condensation of oxidised vapours, thus creating typical “banana-type” nucleation events.68 In all four experiments, substantial SOA formation was observed with a broad range from 210 to 1100 μg m−3, indicating that lower NO2/O3 and more intense exposure to UV lights increase SOA concentrations (Fig. 5b). For experiment tol1 (Table 1), the concentrations of initial and wall loss-corrected aged toluene ([tol]0 and [tol]aged) could be determined by GC/MS to estimate the OH exposure (OHexp) according to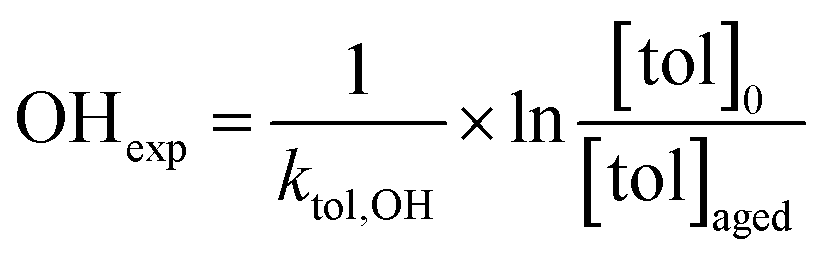 | (21) |
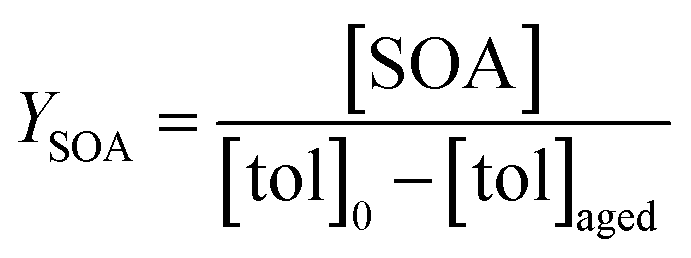 | (22) |
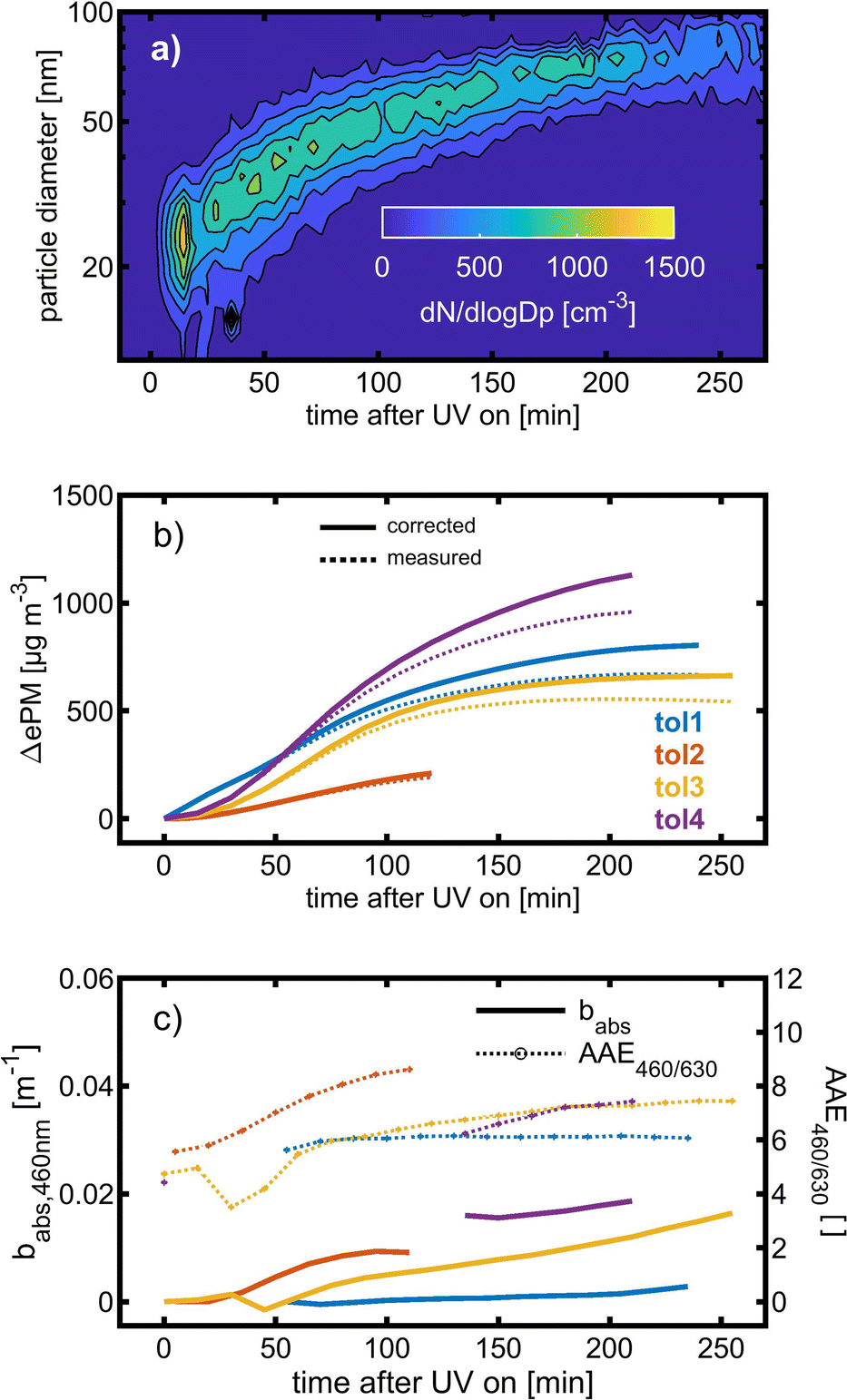 | ||
| Fig. 5 SOA formation from toluene photooxidation (Table 1): (a) new particle formation and growth for experiment tol4 (uncorrected), (b) background-corrected equivalent particle mass concentration (ΔePM), and (c) absorption coefficient (babs,460nm) together with the Angström absorption exponent at 460 and 630 nm (AAE460/630). | ||
The optical properties, related to the chemical composition of the individual toluene-derived SOAs, were analysed over the course of photochemical ageing by using a custom-made aethalometer. The formation light absorbing carbon, here assumed to consist predominantly of BrC and described by the light absorption coefficient babs, from photooxidation of toluene compares to the trend of the ePM concentration and shows an increasing trend in each of the four experiments (Fig. 5c). The same observation was made for AAE460/630 except experiment tol1, showing a plateau forAAE460/630 at 6.1. However, the largest AAE460/630 was observed for experiment tol2, in which only a negligible initial ozone concentration was present. Consequently, these conditions generate the most 460 nm-light-absorbing toluene-SOA among all experiments related to the formed mass of ePM (Table 1).
BrC formation from toluene photooxidation under high NOx conditions is well-known72 and coherent with the observed trends of AAE460/630. The enhanced absorption in the lower visible UV range was attributed to nitrogen-containing species, particularly to nitrophenolic species.73 However, the absorption properties of such brown carbon species are affected by the pH of the liquid particle phase,72 which is unknown in this study, but biases a direct comparison to previous studies.
The molecular composition of the toluene-derived SOA was investigated by high-resolution mass spectrometry. Qualitatively, 10 out of 11 BrC species detected by HPLC-UV/vis-(−)ESI-HRMS in previous high NOx toluene photooxidation chamber experiments73 could be identified in at least one experiment of this study by -ESI-HRMS with direct infusion (Fig. 6). In all toluene photooxidation experiments, peak (8) detected at m/z 168.0303 denoted the base peak with the sum formula C7H6NO4, pointing towards isomers of deprotonated methyl-nitro-catechol. For the second highest peak at m/z 154.0147 with the sum formula C6H4NO4−, collision-induced dissociation (CID) was performed to examine isomers of nitro-catechol (Fig. S7†). The most intense fragment was observed at m/z 124.0163 with the sum formula C6H4O3−, originating from a neutral loss of NO, which is characteristic of 4-nitro-catechol. Other neutral losses, like H2O and CHO2, indicate the presence of other isomers. For example, neutral losses of H2O are favoured for nitrophenolic species having the nitro group in the ortho position to a hydroxyl group.74 This is in line with Lin et al. (2015)73 who detected only a single peak in the HPLC chromatogram for the m/z trace corresponding to neutral C6H5NO4, but could resolve co-eluting 3- and 4-nitrocatechol based on UV absorption.
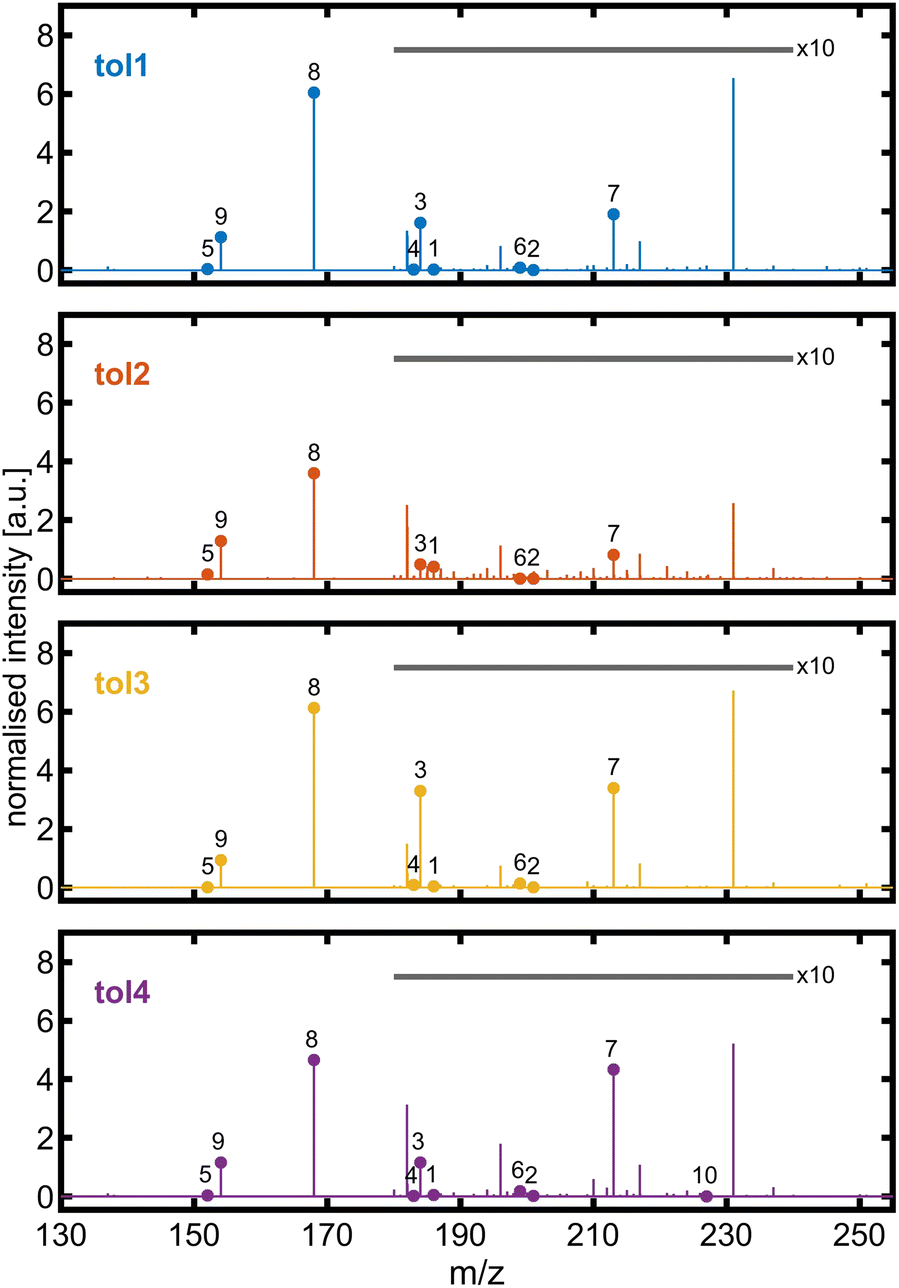 | ||
| Fig. 6 ESI mass spectra of toluene-SOA with major brown carbon species identified in high-NOx chamber experiments by the HPLC-UV/vis-(−)ESI-Orbitrap from Lin et al. (2015):73 (1) C7H8NO5−, (2) C6H5N2O6−, (3) C7H6NO5−, (4) C6H3N2O5−, (5) C7H6NO3−, (6) C6H3N2O6−, (7) C7H5N2O6−, (8) C7H6NO4−, (9) C6H4NO4−, and (10) C7H3N2O7−. The intensity of peaks in the m/z range from 180 to 240 is 10-fold magnified. The unlabelled peak at m/z 231.026 belongs to the sum formula C7H7N2O7− and is supposed to be non-aromatic. | ||
The formation of nitroaromatics from aromatic hydrocarbon in photooxidation proceeds via the addition of an OH radical on the aromatic ring. Subsequently, the OH-toluene adduct reacts with NO2 and forms 3-nitrotoluene. For the detected nitrophenolic species, the OH-toluene adduct may first react with O2 to cresol, which may further react with methylcatechol and finally with methylnitrocatechol or directly from cresol to methylnitrocatechol as later-generation oxidation products.72 Additionally, an intense peak in the ESI mass spectrum was observed at m/z 231.0256 with a sum formula of C7H7N2O7−, which is likely a non-aromatic SOA compound due to its low double bond equivalent.73 Alternatively, the OH attack on toluene starts at its methyl group, forming a benzyl radical, which reacts with benzaldehyde as a first-generation oxidation product. In this study, several sum formulae of multifunctional species detected by LC-APCI-HRMS75 could be found in the presented toluene photooxidation experiments despite the differences in ionisation selectivity between (−)ESI and APCI and the optimised range of m/z for the Orbitrap mass spectrometer. Furthermore, the later-generation oxidation products after ring-opening, dihydroxy-oxobutanoic and -pentanoic acid, which were established as markers for toluene SOAs,76 could also be detected at m/z 133.0142 (C4H5O5−) and 147.0298 (C5H7O5−), respectively, in all toluene photooxidation experiments of this study. Overall, these photooxidation experiments with an established SOA precursor demonstrate that the PHOTO-LAC is capable of generating expected compound classes and marker substances, sensitive to changes in oxidation conditions and the abundance of reactive gases.
3.6. Ageing of biomass burning aerosols
Eight plume ageing experiments were conducted with biomass burning aerosols generated under flaming and smouldering conditions (Table 2). While the smouldering biomass burning aerosol is organic-rich and releases significant amounts of organic nitrogen species, the flaming biomass burning aerosol contains equally distributed organic and elemental carbon with higher concentrations of NOx formed from the combustion of the same biofuel. Generally, the concentrations of ePM from smouldering combustion exceed those from flaming combustion conditions. Short durations of dark (BB1 and BB2) or sequential photochemical and dark ageing of biomass burning plumes (BB3–BB6) were used to demonstrate differences in quantitative aerosol evolution and optical properties compared to conventional chamber experiments. Finally, two unprecedented long ageing experiments (BB7 and BB8) over >24 h with alternating photochemical and dark ageing conditions are presented to simulate the fate of dense wildfire plumes during long-range transport.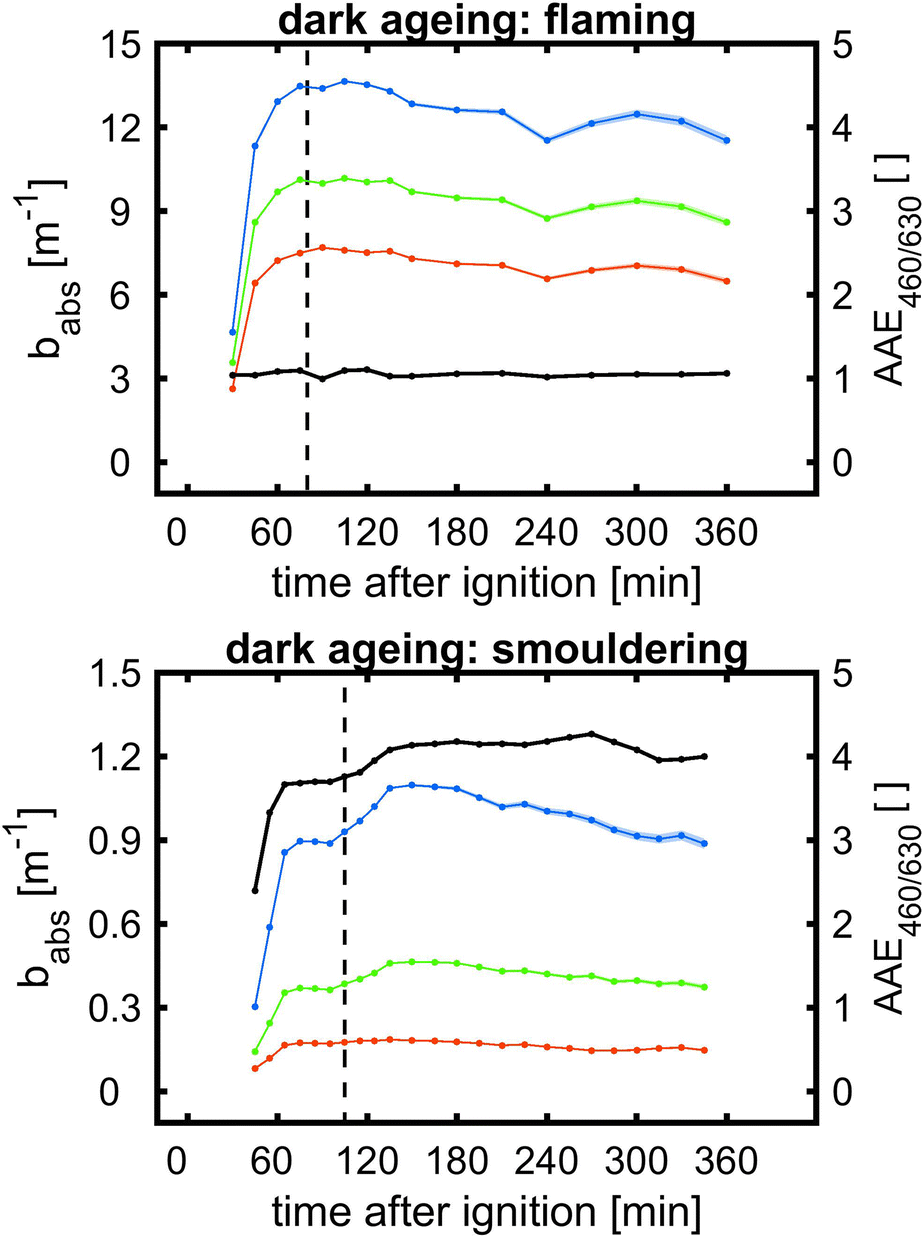 | ||
| Fig. 7 Temporal evolution of wall loss-corrected absorption coefficient babs (blue: 460 nm; green: 530 nm; red: 630 nm) and Angström absorption exponent (AAE460/630; black) from 460 nm and 630 nm during short dark ageing for flaming (top) and smouldering (bottom) biomass burning aerosols. Vertical dashed line denotes the addition of ozone to initiate ˙NO3 formation and shaded area refers to the 95% CI. | ||
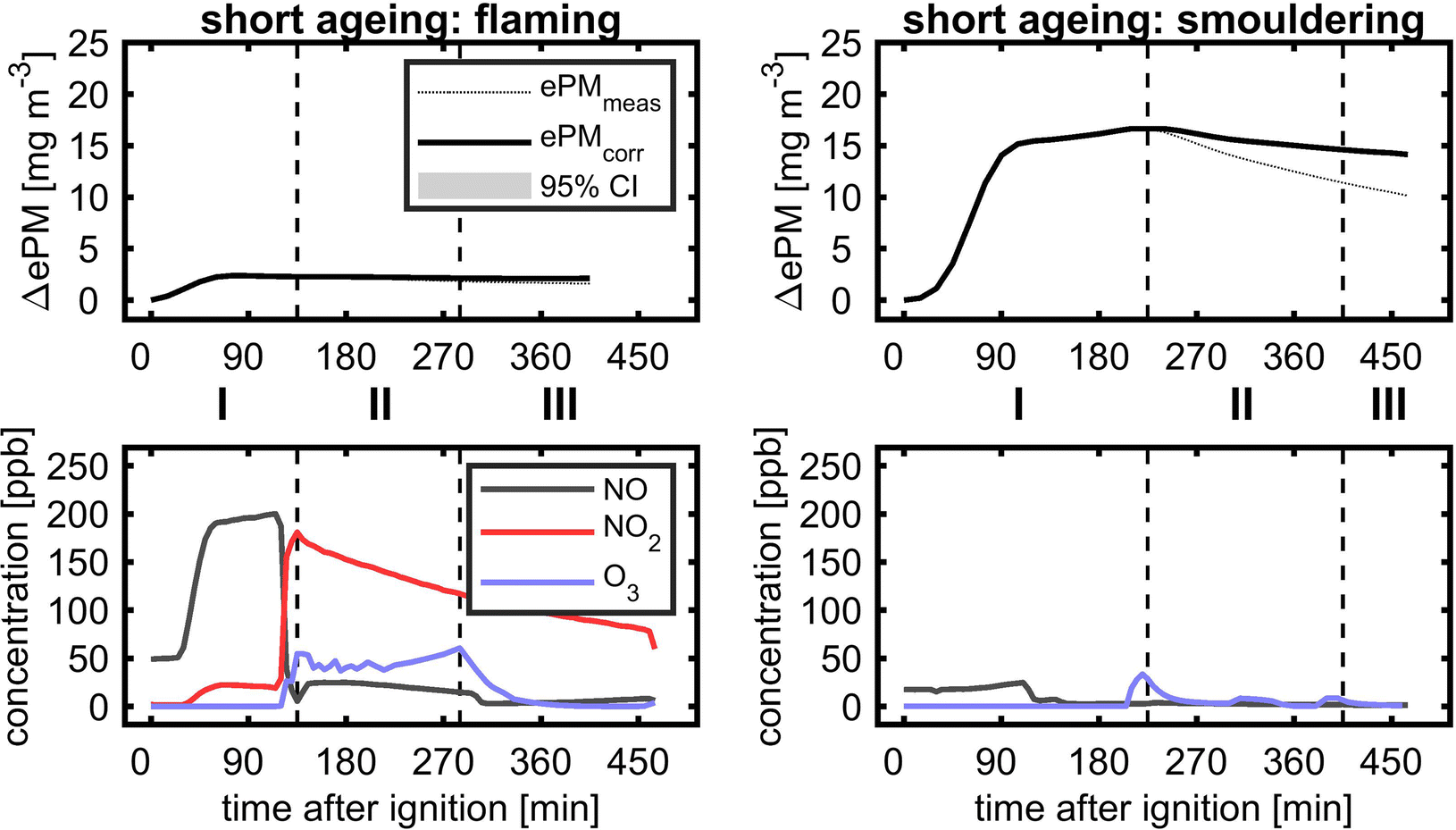 | ||
| Fig. 8 Short dark ageing of flaming (left) and smouldering BB aerosols (right) with the temporal evolution of equivalent particle mass (ePM; top) and gases NO, NO2 and O3 (bottom) for (I) the generation of the BB aerosol and ozone addition, (II) photochemical ageing with UV lights turned on and (III) dark ageing. For smouldering conditions, no NO2 data are available. | ||
The ozone addition, before UV lamps were turned on, already increased the ePM of smouldering BB to a small extent. However, during photochemical and consecutive dark ageing, ePM slightly decreased, so the wall loss corrected ePM values of fresh and photochemically dark aged at the end of the experiment are the same. BB plumes of high aerosol concentrations age by dilution-driven evaporation and recondensation,79,80 but in these experiments the dilution, mainly caused by aerosol wall losses of less than 30%, was too low. Therefore, the observed evolution of flaming and smouldering BB ePM with missing substantial formation of secondary aerosols is likely a consequence of a lower ratio of oxidants to reactive SOA precursors, so measurable SOAs might require higher exposures to atmospheric oxidants.
3.6.3.1 Evolution of PM and reactive gases. In long-ageing experiments with consecutive photochemical and dark ageing, the diurnal cycle during summertime with longer day- and shorter night-time was followed by the experimental procedure with a total ageing duration of 28 h (Fig. 9). Similar to the short ageing described in the previous section, ePM slightly decreased after UV lights were turned on for both flaming and smouldering BB aerosols. However, during the subsequent dark ageing, the wall loss corrected concentration of ePM stabilised and reincreased to the fresh ePM level for flaming and even exceed the fresh ePM level for the smouldering BB aerosol. After turning on UV lights for a second time, ePM increased further, but with a lower slope than during dark ageing. At the end of the ageing experiment after 28 h, a net gain in ePM of 16% for flaming and 35% for smouldering was obtained. Low-temperature pyrolysis under smouldering combustion conditions produces higher yields of VOCs and likely more SOA precursors,13 which may explain the higher SOA formation compared to the flaming BB aerosol. The concentrations of reactive gases showed the same profile as during the short ageing experiment, but approached the lower ppb range. During the second photochemical ageing, the concentrations of the BB aerosol and reactive gases were generally lower than at the beginning of the experiment. However, UV lights still induced substantial ozone formation to a level of approximately 25 ppb.
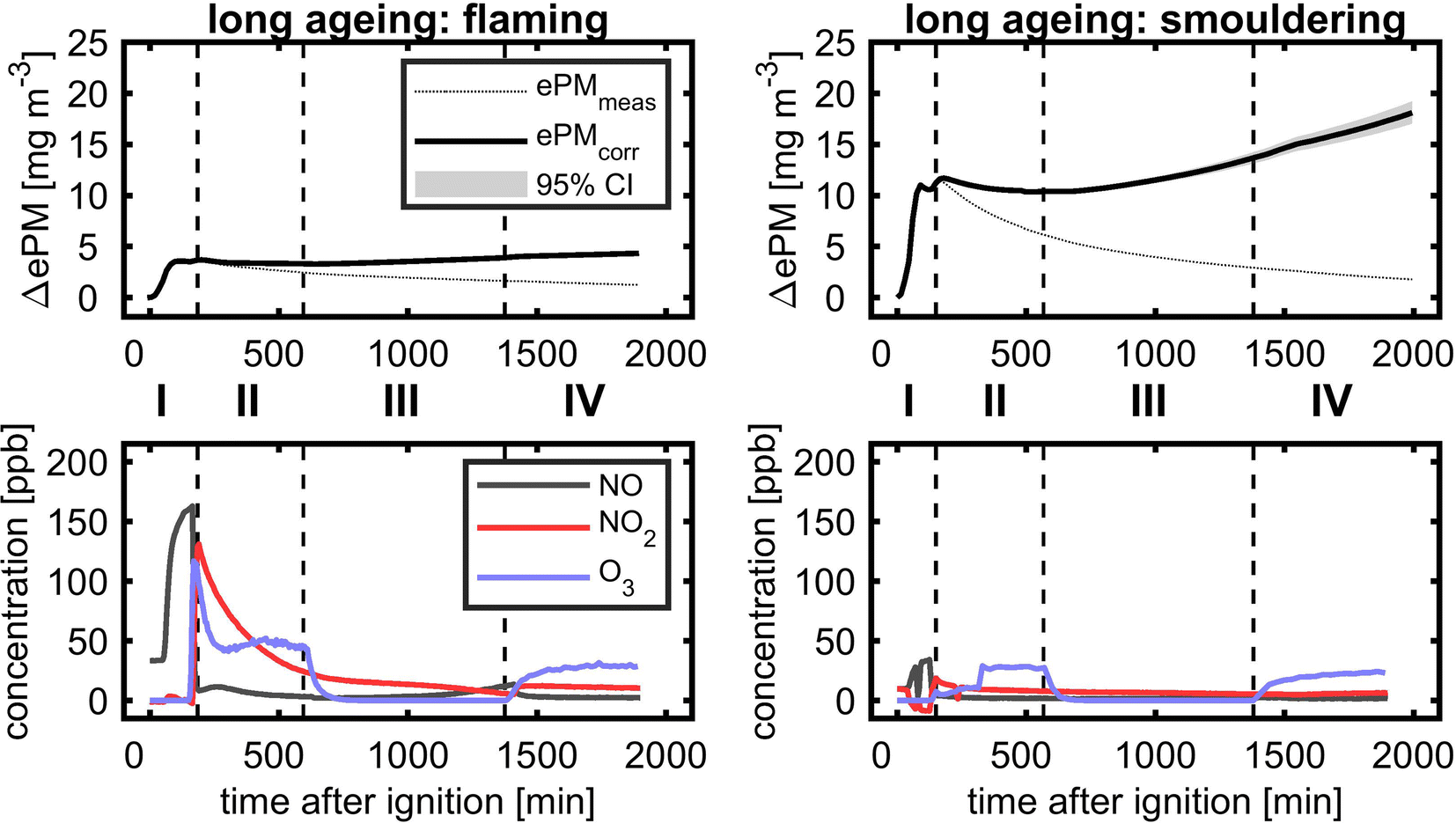 | ||
| Fig. 9 Long ageing of flaming (left) and smouldering BB aerosols (right) with the temporal evolution of equivalent particle mass (ePM; top) and gases NO, NO2 and O3 (bottom) for (I) the generation of the BB aerosol and ozone addition shortly before transition to (II) first photochemical ageing, (III) dark ageing, and (IV) second photochemical ageing. | ||
3.6.3.2 Evolution of chemical PM composition. The chemical composition of the BB aerosol was investigated after combustion, first photochemical ageing, dark ageing, and second photochemical ageing by FT-ICR-MS using direct infusion APPI. In each sample, approximately 2000 to 5500 peaks could be detected and assigned to unsaturated hydrocarbons and oxygenated hydrocarbons as well as nitrogen- and sulphur-containing compounds with a hydrocarbon backbone based on their sum formula. Furthermore, the sum formulae were used to calculate intensive chemical properties including elemental ratios O
:C and H:C, the modified aromaticity index (AImod)81 and the molar ratio of organic matter to organic carbon (OM:OC).
In both fresh BB aerosols, the sum formula of particularly coniferous biomass burning markers could be detected, such as C18H18 (retene), C10H10O3 (coniferyl aldehyde) or C20H28O2 (dehydroabietic acid), as well as several sum formulae of polycyclic aromatic hydrocarbons (PAH), such as C14H10 (phenanthrene and anthracene), C16H10 (pyrene and fluoranthene), C18H12 (e.g. benz(a)anthracene and chrysene) and C20H12 (e.g. benz(a)pyrene and benzo(b)fluoranthene). In agreement with previous studies, smouldering combustion led to higher emissions of PAHs, PAH derivatives and phenolic species than flaming combustion conditions12,40,82 and a generally more complex mass spectrum.
Long ageing in the PHOTO-LAC induced chemical changes for both flaming and smouldering BB aerosols under all ageing conditions. With each ageing step of photochemical or dark ageing, new sum formulae along the entire m/z range were detected which were not present in the sample collected before (Fig. 10, highlighted in red). The most significant changes were observed after the second photochemical ageing of the flaming BB aerosol, while in the aged smouldering BB aerosol, the stepwise addition of new sum formulae was more evenly distributed between the dark and two photochemical ageing conditions. Smog chamber ageing is inherently affected by changing ratios of oxidants to BB aerosol due to wall losses of PM, vapours and reactive gases. The experiments of this study were conducted at high aerosol concentrations (see Section 3.5.4), which separates itself from smog chamber ageing at near-ambient aerosol concentrations. At the end of the long ageing experiments, only 25% of the flaming and 9.5% of smouldering BB ePM from the beginning of the experiment remained in the PHOTO-LAC (Fig. 9). Hence, in the second photochemical ageing there was likely a higher oxidant to aerosol ratio in the PHOTO-LAC than during the first one, which consequently caused a higher conversion of organic aerosol constituents.
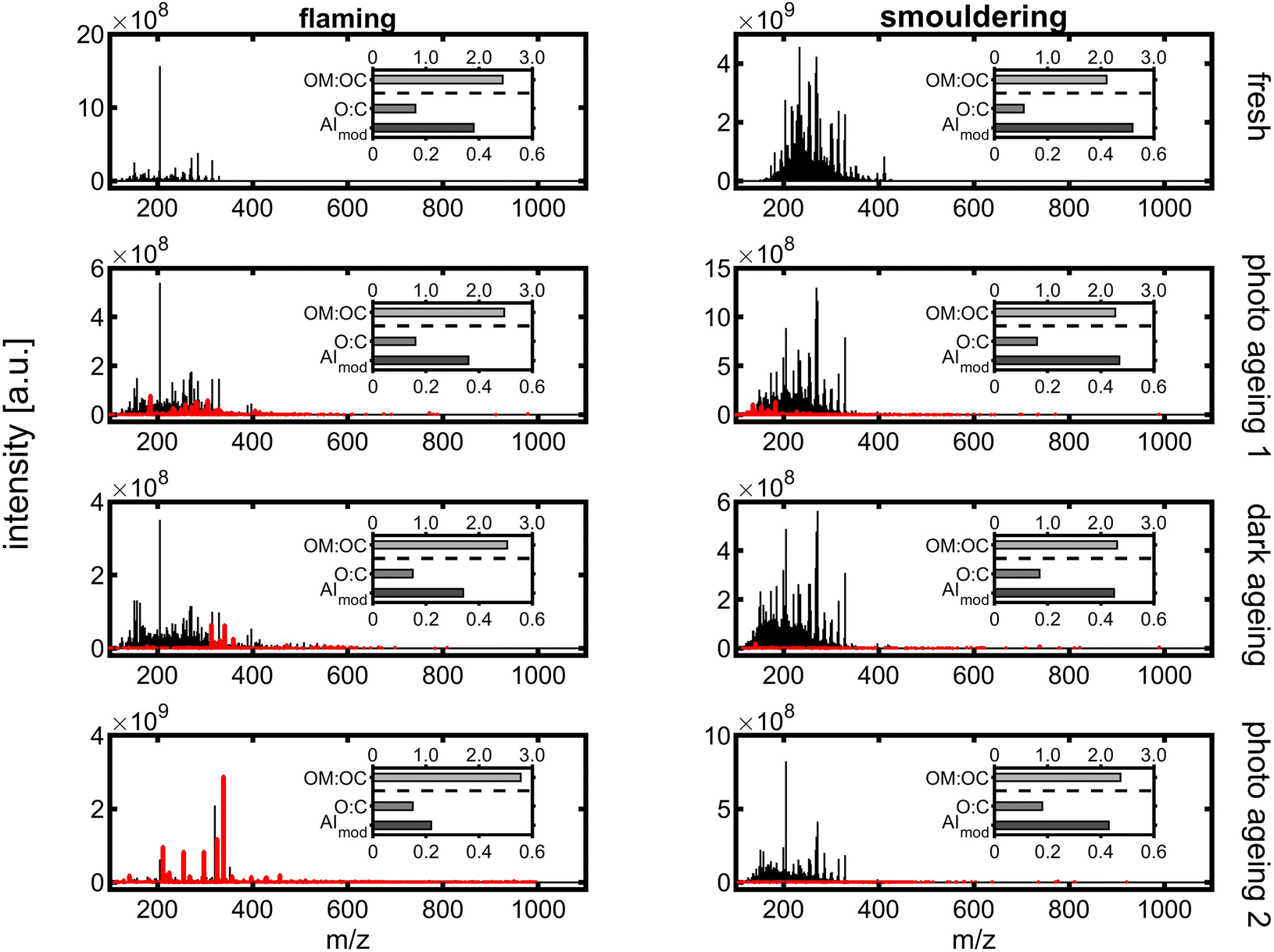 | ||
| Fig. 10 APPI-FT-ICR mass spectra from long ageing of flaming (left) and smouldering BB aerosols (right). Peaks in red denote sum formulae not detected in the BB sample before. | ||
On a peak number basis, 35% and 28% of detected peaks were found in all four samples of flaming and smouldering BB aerosols, respectively, indicating the survival of a substantial part of organic aerosol constituents or, in the case of oxygenated species, the generation of secondary compounds similar to primary ones. In contrast, 37% of the peaks in the smouldering BB aerosol were exclusively detected in the fresh emissions, which was only found for 2% of the peaks in the flaming BB aerosol (Fig. S9†). Apart from large numbers of common peaks in the repeated photochemically aged flaming BB aerosol and fresh smouldering BB aerosol, also the patterns of intersecting peaks in flaming and smouldering were similar (Pearson correlation coefficient of 0.96).
For each individual peak, the exact mass enables the calculation of the corresponding sum formula. The BB aerosol is known to contain substantial fractions of aromatic compounds, which is also reflected by an AImod of 0.38 for flaming and 0.52 for the smouldering BB aerosol (Fig. 10), being larger than the AImod of lignin of 0.33.81 Aromatic compounds have an AImod of ≥0.5, while condensed aromatic compounds have an AImod of ≥0.67. During ageing, AImod continuously decreases for both flaming and smouldering BB aerosols to finally 0.22 and 0.43, respectively, as oxidation reactions may cause ring-opening and aromatic aerosol constituents lose aromaticity. The largest difference in AImod could be observed after the first photochemical ageing for flaming, but after the second photochemical ageing for the smouldering BB aerosol. A constant increase could be observed for O:C in the smouldering BB aerosol (0.12 to 0.18) as typical for atmospheric ageing. However, in the flaming BB aerosol O:C remains at 0.156 ± 0.020, which is close to the 95% confidence interval of three replicate analyses of ±0.014. On the one hand, aromatic compounds are degraded, and thus oxidation products might not be accessible anymore by APPI constraining the O:C. On the other hand, some oxidation reactions, such as the conversion of an alcohol to a carbonyl, do not involve a change in O:C. However, the carbon backbone of both BB aerosols is increasingly functionalised, indicated by increasing OM:OC from 2.4 to 2.8 for flaming and from 2.1 to 2.4 for the smouldering BB aerosol, which agrees with high resolution mass spectrometry analysis of the photochemically aged smouldering biomass burning aerosol.83 Moreover, both BB aerosols show a slight increase in H:C during ageing in contrast to often declining H:C, indicating a significant role of particle phase reactions, such as hydrolysis84 or photooxidation of large condensed aromatics.85
To illustrate the chemistry in the PHOTO-LAC on a molecular level, the evolution of individual sum formulae, associated with organic BB aerosol constituents, was investigated (Fig. 11). For example, polycyclic aromatic hydrocarbons (PAHs), alkylated PAHs (alk-PAHs) and oxygenated PAHs (oxy-PAHs) appear in fresh emissions of the BB aerosol.86–88 Particularly, alkylated phenanthrenes, such as retene or pimanthrene, are used as markers for the combustion of coniferous plants because they are formed by thermal conversion of resin acids. The relative intensities of peaks with sum formulae C18H12 (chrysene and benz(a)anthracene), C20H12 (e.g. benzo(a)pyrene and benzo(k)fluoranthene), C18H18 (C4-phenanthrenes such as retene), and C16H10O (hydroxy-pyrene and hydroxyl-fluoranthene) notably decreased over all periods of photochemical ageing for both flaming and smouldering BB aerosols, demonstrating their photochemical and dark degradation in the PHOTO-LAC. However, retene slightly reincreased after the second photochemical ageing period, perhaps because of volatilisation from the inner walls of the PHOTO-LAC by increasing temperature. In contrast to PAHs and alk-PAHs, oxy-PAHs were both degraded and formed by atmospheric ageing,89 and hence the relative abundances of hydroxy-pyrene reasonably changed to a smaller extent than that of PAHs and alk-PAHs. Other oxygenated aerosol particle constituents reveal temporally different concentration profiles and steadily increase or peak after the first photochemical ageing or after dark ageing. For example, C10H6O2 (naphthoquinone), formed from the oxidation of naphthalene,90 shows its maximum after the first photochemical ageing for both flaming and smouldering BB aerosols. However, many oxygenated species detected by the APPI FT-ICR-MS peak in relative concentrations for the flaming aerosol, but steadily increase during ageing of the smouldering BB aerosol. Due to the higher aerosol concentrations generated by smouldering combustion and consequently a lower oxidant to aerosol ratio, ageing seems to proceed less intensely. This effect is illustrated by the temporal evolution of, e.g., C8H8O4 (vanillic acid), C7H7NO3 (nitrocresol) and C12H8O4 (naphthalaldehydic acid). Particularly the latter one was identified as an early-generation oxidation product, peaking within less than 2 days of equivalent photochemical ageing.27
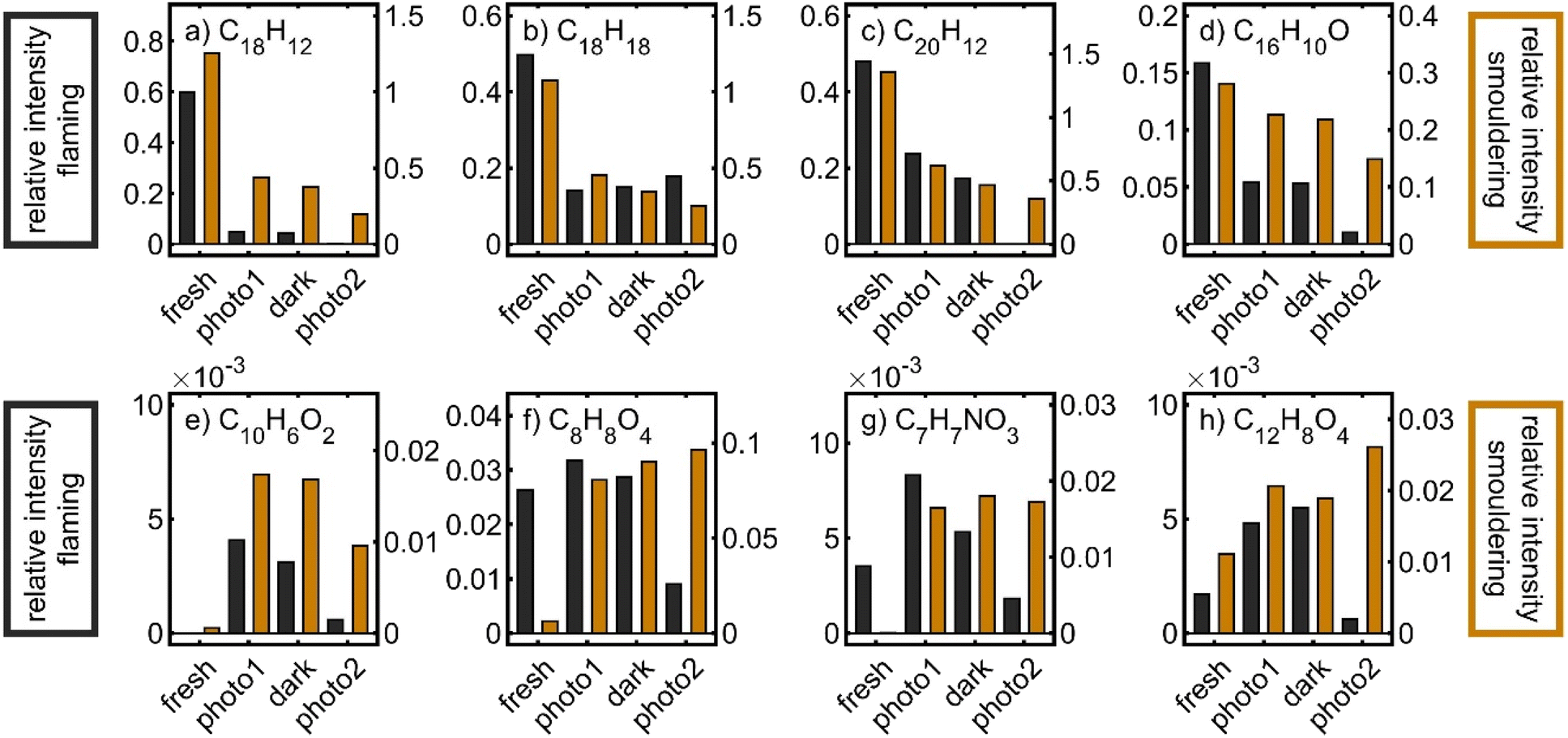 | ||
| Fig. 11 Relative abundance of sum formulae corresponding to the tentative structures (a) benz(a)anthracene/chrysen, (b) retene, (c) benz(a)pyrene, (d) hydroxy-pyrene, (e) naphthalenedione, (f) vanillic acid, (g) nitrocresol, and (h) naphthaldehydic acid, sampled after the generation of the BB aerosol (fresh), first photochemical ageing (photo1), dark ageing (dark), and second photochemical ageing (photo2). | ||
The degradation kinetics of PAHs are known to be largely dependent on the composition and morphology of the particle core.61 For example, soot particles may convert NO2 into HONO, which is a source of OH radicals or act as photosensitiser.64,91 Furthermore, SOA coatings may preserve fresh aerosol species from heterogeneous oxidation by shielding, especially in thick plumes.80,92 Overall, a quantitative assessment of the degradation and formation of aerosol species in the BB plume cannot be made, but the relative trends of decreasing, peaking and increasing peaks of known primary and secondary aerosol species demonstrate the occurrence of fundamental atmospheric reactions in the PHOTO-LAC on a molecular level.
3.7. Opportunities and outlook for aerosol characterisation and toxicity testing
At the end of the long ageing experiments, 72% of flaming and 90% of the smouldering BB aerosol was lost to the walls. However, when starting with 400 g of mixed pine biomass, the PHOTO-LAC chamber still contains more than 1 mg m−3 of ePM. Owing to the large volume, high-volume sampling of PM can be conducted on a filter over an appropriate time to conduct subsequent cell exposure to the aged BB aerosol under submersed conditions. For example, despite a concentration of 6 mg m3 of SOA from α-pinene ozonolysis in an oxidation flow reactor (OFR), the applied flow rate of 8 L min−1 required 18.5 h of continuous sample collection for 20 mg PM.93 In the PHOTO-LAC facility, stainless steel tubing of 10 mm inner diameter was installed at the axial centre of the cavity at 2 m height, so high-volume samples with 20 mg filter load can be collected within 30 min. Hence, sufficient material for cell exposure and comprehensive chemical aerosol characterisation can be provided and sampling artefacts such as a blow-off of semi-volatile species is largely avoided.A general limitation of ageing in environmental chambers is the maximum achievable photochemical age equivalent of approximately 2 days, which is thus lower than the lifetime of aerosol particles in the troposphere up to two weeks. This may be overcome by an OFR, exposing the aerosol to higher levels of atmospheric oxidants.94 Principally, PM of a higher photochemical age equivalent can be generated and sampled in a reasonable time using the high-volume OFR “Photochemical Emission Aging flow tube Reactor” (PEAR), which can be operated at >100 L min−1,95 and would shorten the sampling time of 20 mg from 6 mg m−3 in the OFR by one order of magnitude. However, OFR aerosol processing differs from atmospheric ageing, for example because of high oxidant exposure and short residence time on the minute scale, which prevent oxidised vapours from condensation, but promote multiple oxidation vapours in the gas phase. Alternatively to the batch mode, the PHOTO-LAC may be used in flow-through mode, such as the SAPHIR chamber,96 at lower concentrations, combining the benefits of a high-volume OFR and a high-volume smog chamber. Venting of the PHOTO-LAC with current technical equipment has to be conducted for approximately one hour to reach ambient PM and NOx concentrations, thus enabling continuous operation with sufficient time for gas-particle equilibrium, leading to opportunities for future experiments.
4. Conclusion
A Large Aerosol Chamber (LAC) with a volume of 1800 m3 was equipped with 198 UV lamps becoming a PHOTO-LAC to enable studies on the atmospheric fate of optically dense biomass burning plumes, associated with wildfire emission ageing. The figures of merit were provided for the PHOTO-LAC including wall losses, induction of photochemistry and ability to generate SOAs. While the photolysis rate of NO2 and SOA formation rate from toluene were comparable to those of other environmental chambers, the wall loss rates of particles were significantly lower, but the wall losses of reactive gases were higher and the temperature increases during photochemical ageing due to the placement of the UV lamps inside the PHOTO-LAC cavity. Moreover, the chemical composition of toluene-derived SOAs agrees fairly well with results from small-scale chamber ageing experiments under similar experimental conditions.The low wall losses of PM allow the accomplishment of long ageing experiments and finally to study the atmospheric fate of long-range transported biomass burning plumes from wildfire under laboratory conditions. With a total experimental duration of ≈30 h, the BB aerosol could be exposed to alternating day- and night-time chemical ageing. Therefore, chemical changes in the aerosol particle composition could be revealed by ultra-high resolution mass spectrometry.
In an ageing experiment with the PHOTO-LAC, more than 1 g of fresh or aged BB aerosol can be generated and collected on the filter material. Hence, the PHOTO-LAC gives the opportunity for parallel cell exposures and comprehensive chemical aerosol particle analysis towards a deeper understanding of health effects from long-range transported wildfire plumes.
Author contributions
HC: conceptualisation, formal analysis, investigation, writing – original draft, visualization, supervision, and project administration; OP: conceptualisation, investigation, resources, data curation, writing – original draft, supervision, project administration, and funding acquisition; DGC: investigation, validation, and visualisation; AK: conceptualisation, methodology, investigation, resources, data curation, and supervision; ES. methodology, investigation, and writing – review and editing; VPS: investigation; MS: investigation, validation, and writing – review and editing; CPR: methodology, software, resources, writing – review and editing, supervision, and project administration; CA: resources and supervision; VU: investigation; VSK: methodology and investigation; MVP: resources and funding acquisition; RZ: conceptualisation, resources, writing – review and editing, project administration, and funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Russian Foundation for Basic Research (RFBR) and the German Research Foundation (DFG) under the grants 20-55-12001 and ZI 764/24-1. RSF project 19-77-3004-Π funding is appreciated for support of developing the custom-made aethalometer methodology. Long-term work on the development of the large aerosol chamber and the creation of optical instruments used in experiments was carried out within the framework of the State task for IAO SB RAS No. 121031500342-0. The modernisation of the LAC ventilation and control system was carried out using the equipment of the Atmosfera Shared Use Centre with partial financial support from the Ministry of Education and Science of Russia (contract No. 075-15-2021-661). Research of fundamental photochemistry on the combustion aerosol was performed according to the Development program of the Interdisciplinary Scientific and Educational School of M. V. Lomonosov Moscow State University « Future Planet and Global Environmental Change » with a purpose of comprehensive evaluation of wildfire impact on aerosol pollution in the global atmosphere. Access to the CNRS research infrastructure infranalytics (FR2054) is gratefully acknowledged.References
- M. O. Andreae and D. Rosenfeld, Aerosol–cloud–precipitation interactions. Part 1. The nature and sources of cloud-active aerosols, Earth-Sci. Rev., 2008, 89, 13–41 CrossRef.
- M. O. Andreae, Emission of trace gases and aerosols from biomass burning – an updated assessment, Atmos. Chem. Phys., 2019, 19, 8523–8546 CrossRef CAS.
- M. D. Flannigan, M. A. Krawchuk, W. J. de Groot, B. M. Wotton and L. M. Gowman, Implications of changing climate for global wildland fire, Int. J. Wildland Fire, 2009, 18, 483 CrossRef.
- W. M. Jolly, M. A. Cochrane, P. H. Freeborn, Z. A. Holden, T. J. Brown, G. J. Williamson and D. M. J. S. Bowman, Climate-induced variations in global wildfire danger from 1979 to 2013, Nat. Commun., 2015, 6, 7537 CrossRef CAS PubMed.
- D. M. J. S. Bowman, C. A. Kolden, J. T. Abatzoglou, F. H. Johnston, G. R. van der Werf and M. Flannigan, Vegetation fires in the Anthropocene, Nat. Rev. Earth Environ., 2020, 1, 500–515 CrossRef.
- V. S. Kozlov, E. Yausheva, S. A. Terpugova, M. V. Panchenko, D. G. Chernov and V. P. Shmargunov, Optical–microphysical properties of smoke haze from Siberian forest fires in summer 2012, Int. J. Remote Sens., 2014, 35, 5722–5741 Search PubMed.
- O. B. Popovicheva, N. Evangeliou, V. O. Kobelev, M. A. Chichaeva, K. Eleftheriadis, A. Gregorič and N. S. Kasimov, Siberian Arctic black carbon: gas flaring and wildfire impact, Atmos. Chem. Phys., 2022, 22, 5983–6000 CrossRef CAS.
- O. Popovicheva, E. Molozhnikova, S. Nasonov, V. Potemkin, I. Penner, M. Klemasheva, I. Marinaite, L. Golobokova, S. Vratolis, K. Eleftheriadis and T. Khodzher, Industrial and wildfire aerosol pollution over world heritage Lake Baikal, J. Environ. Sci., 2021, 107, 49–64 CrossRef PubMed.
- E. Diapouli, O. Popovicheva, M. Kistler, S. Vratolis, N. Persiantseva, M. Timofeev, A. Kasper-Giebl and K. Eleftheriadis, Physicochemical characterization of aged biomass burning aerosol after long-range transport to Greece from large scale wildfires in Russia and surrounding regions, Summer 2010, Atmos. Environ., 2014, 96, 393–404 CrossRef CAS.
- K. Ikeda and H. Tanimoto, Exceedances of air quality standard level of PM 2.5 in Japan caused by Siberian wildfires, Environ. Res. Lett., 2015, 10, 105001 CrossRef.
- M. Santoso, E. G. Christensen, J. Yang and G. Rein, Review of the Transition From Smouldering to Flaming Combustion in Wildfires, Front. Mech. Eng., 2019, 5, 49 CrossRef.
- O. Popovicheva, V. S. Kozlov, R. F. Rhakimov, V. P. Shmargunov, E. D. Kireeva, N. M. Persiantseva, M. A. Timofeev, G. Engling, K. Eleftheriadis, E. Diapouli, M. V. Panchenko, R. Zimmermann and J. Schnelle-Kreis, Optical-Microphysical and Physical-Chemical Characteristics of Siberian Biomass Burning: Experiments in Aerosol Chamber, Atmos. Oceanic Opt., 2016, 29, 492–500 CrossRef CAS.
- S. K. Akagi, R. J. Yokelson, C. Wiedinmyer, M. J. Alvarado, J. S. Reid, T. Karl, J. D. Crounse and P. O. Wennberg, Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys., 2011, 11, 4039–4072 CrossRef CAS.
- G. Chen, Y. Guo, X. Yue, S. Tong, A. Gasparrini, M. L. Bell, B. Armstrong, J. Schwartz, J. J. K. Jaakkola, A. Zanobetti, E. Lavigne, P. H. Nascimento Saldiva, H. Kan, D. Royé, A. Milojevic, A. Overcenco, A. Urban, A. Schneider, A. Entezari, A. M. Vicedo-Cabrera, A. Zeka, A. Tobias, B. Nunes, B. Alahmad, B. Forsberg, S.-C. Pan, C. Íñiguez, C. Ameling, C. de La Cruz Valencia, C. Åström, D. Houthuijs, D. van Dung, E. Samoli, F. Mayvaneh, F. Sera, G. Carrasco-Escobar, Y. Lei, H. Orru, H. Kim, I.-H. Holobaca, J. Kyselý, J. P. Teixeira, J. Madureira, K. Katsouyanni, M. Hurtado-Díaz, M. Maasikmets, M. S. Ragettli, M. Hashizume, M. Stafoggia, M. Pascal, M. Scortichini, M. de Sousa Zanotti Stagliorio Coêlho, N. Valdés Ortega, N. R. I. Ryti, N. Scovronick, P. Matus, P. Goodman, R. M. Garland, R. Abrutzky, S. O. Garcia, S. Rao, S. Fratianni, T. N. Dang, V. Colistro, V. Huber, W. Lee, X. Seposo, Y. Honda, Y. L. Guo, T. Ye, W. Yu, M. J. Abramson, J. M. Samet and S. Li, Mortality risk attributable to wildfire-related PM2·5 pollution: a global time series study in 749 locations, Lancet Planet. Health, 2021, 5, e579–e587 CrossRef PubMed.
- C. E. Reid, M. Brauer, F. H. Johnston, M. Jerrett, J. R. Balmes and C. T. Elliott, Critical Review of Health Impacts of Wildfire Smoke Exposure, Environ. Health Perspect., 2016, 124, 1334–1343 CrossRef PubMed.
- R. Aguilera, T. Corringham, A. Gershunov and T. Benmarhnia, Wildfire smoke impacts respiratory health more than fine particles from other sources: observational evidence from Southern California, Nat. Commun., 2021, 12, 1493 CrossRef CAS PubMed.
- Y. H. Kim, S. H. Warren, Q. T. Krantz, C. King, R. Jaskot, W. T. Preston, B. J. George, M. D. Hays, M. S. Landis, M. Higuchi, D. M. DeMarini and M. I. Gilmour, Mutagenicity and Lung Toxicity of Smoldering vs. Flaming Emissions from Various Biomass Fuels: Implications for Health Effects from Wildland Fires, Environ. Health Perspect., 2018, 126, 17011 CrossRef PubMed.
- Y. H. Kim, C. King, T. Krantz, M. M. Hargrove, I. J. George, J. McGee, L. Copeland, M. D. Hays, M. S. Landis, M. Higuchi, S. H. Gavett and M. I. Gilmour, The role of fuel type and combustion phase on the toxicity of biomass smoke following inhalation exposure in mice, Arch. Toxicol., 2019, 93, 1501–1513 CrossRef CAS PubMed.
- L. E. Koval, C. K. Carberry, Y. H. Kim, E. McDermott, H. Hartwell, I. Jaspers, M. I. Gilmour and J. E. Rager, Wildfire Variable Toxicity: Identifying Biomass Smoke Exposure Groupings through Transcriptomic Similarity Scoring, Environ. Sci. Technol., 2022, 56, 17131–17142 CrossRef CAS PubMed.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- Z. C. J. Decker, K. J. Zarzana, M. Coggon, K.-E. Min, I. Pollack, T. B. Ryerson, J. Peischl, P. Edwards, W. P. Dubé, M. Z. Markovic, J. M. Roberts, P. R. Veres, M. Graus, C. Warneke, J. de Gouw, L. E. Hatch, K. C. Barsanti and S. S. Brown, Nighttime Chemical Transformation in Biomass Burning Plumes: A Box Model Analysis Initialized with Aircraft Observations, Environ. Sci. Technol., 2019, 53, 2529–2538 CrossRef CAS PubMed.
- A. L. Hodshire, A. Akherati, M. J. Alvarado, B. Brown-Steiner, S. H. Jathar, J. L. Jimenez, S. M. Kreidenweis, C. R. Lonsdale, T. B. Onasch, A. M. Ortega and J. R. Pierce, Aging Effects on Biomass Burning Aerosol Mass and Composition: A Critical Review of Field and Laboratory Studies, Environ. Sci. Technol., 2019, 53, 10007–10022 CrossRef CAS.
- J. E. Lee, M. K. Dubey, A. C. Aiken, P. Chylek and C. M. Carrico, Optical and Chemical Analysis of Absorption Enhancement by Mixed Carbonaceous Aerosols in the 2019 Woodbury, AZ, Fire Plume, J. Geophys. Res.: Atmos., 2020, 125(15), e2020JD032399 CrossRef CAS.
- M. F. Heringa, P. F. DeCarlo, R. Chirico, T. Tritscher, J. Dommen, E. Weingartner, R. Richter, G. Wehrle, A. S. H. Prévôt and U. Baltensperger, Investigations of primary and secondary particulate matter of different wood combustion appliances with a high-resolution time-of-flight aerosol mass spectrometer, Atmos. Chem. Phys., 2011, 11, 5945–5957 CrossRef CAS.
- P. Tiitta, A. Leskinen, L. Hao, P. Yli-Pirilä, M. Kortelainen, J. Grigonyte, J. Tissari, H. Lamberg, A. Hartikainen, K. Kuuspalo, A.-M. Kortelainen, A. Virtanen, K. E. J. Lehtinen, M. Komppula, S. Pieber, A. S. H. Prévôt, T. B. Onasch, D. R. Worsnop, H. Czech, R. Zimmermann, J. Jokiniemi and O. Sippula, Transformation of logwood combustion emissions in a smog chamber, Atmos. Chem. Phys., 2016, 16, 13251–13269 CrossRef CAS.
- E. A. Bruns, M. Krapf, J. Orasche, Y. Huang, R. Zimmermann, L. Drinovec, G. Močnik, I. El Haddad, J. G. Slowik, J. Dommen, U. Baltensperger and A. S. H. Prévôt, Characterization of primary and secondary wood combustion products generated under different burner loads, Atmos. Chem. Phys., 2015, 15, 2825–2841 CrossRef CAS.
- A. Hartikainen, P. Tiitta, M. Ihalainen, P. Yli-Pirilä, J. Orasche, H. Czech, M. Kortelainen, H. Lamberg, H. Suhonen, H. Koponen, L. Hao, R. Zimmermann, J. Jokiniemi, J. Tissari and O. Sippula, Photochemical transformation of residential wood combustion emissions: dependence of organic aerosol composition on OH exposure, Atmos. Chem. Phys., 2020, 20, 6357–6378 CrossRef CAS.
- D. A. Jaffe and N. L. Wigder, Ozone production from wildfires: A critical review, Atmos. Environ., 2012, 51, 1–10 CrossRef CAS.
- L. Xu, J. D. Crounse, K. T. Vasquez, H. Allen, P. O. Wennberg, I. Bourgeouis, S. S. Brown, P. Campuzano-Jost, M. M. Coggon, J. H. Crawford, J. P. DiGangi, G. S. Diskin, A. Fried, E. M. Gargulinski, J. B. Gilman, G. I. Gkatzelis, H. Guo, J. W. Hair, S. R. Hall, H. A. Halliday, T. F. Hansco, R. A. Hannun, C. D. Holmes, L. G. Huey, J. L. Jimenez, A. Lamplugh, Y. R. Lee, J. Liao, J. Lindaas, J. A. Neuman, J. B. Nowak, J. Peischl, D. A. Peterson, F. Piel, D. Richter, P. S. Rickly, M. A. Robinson, A. W. Rollins, T. B. Ryerson, K. Sekimoto, V. Selimovic, T. Shingler, A. J. Soja, J. M. St. Clair, D. J. Tanner, K. Ullmann, P. R. Veres, J. Walega, C. Warneke, R. A. Washenfelder, P. Weibring, A. Wisthaler, G. M. Wolfe, C. Womack and R. J. Yokelson, Ozone chemistry in western U.S. wildfire plumes, Sci. Adv., 2021, 7, eabl3648 CrossRef CAS PubMed.
- P. Voosen, High-flying wildfire smoke poses potential threat to ozone layer, Science, 2021, 374, 921–922 CrossRef CAS PubMed.
- A. C. Rohr, The health significance of gas- and particle-phase terpene oxidation products: a review, Environ. Int., 2013, 60, 145–162 CrossRef CAS PubMed.
- H. Tong, P. S. J. Lakey, A. M. Arangio, J. Socorro, C. J. Kampf, T. Berkemeier, W. H. Brune, U. Pöschl and M. Shiraiwa, Reactive oxygen species formed in aqueous mixtures of secondary organic aerosols and mineral dust influencing cloud chemistry and public health in the Anthropocene, Faraday Discuss., 2017, 200, 251–270 RSC.
- M. Krapf, L. Künzi, S. Allenbach, E. A. Bruns, I. Gavarini, I. El-Haddad, J. G. Slowik, A. S. H. Prévôt, L. Drinovec, G. Močnik, L. Dümbgen, M. Salathe, N. Baumlin, C. Sioutas, U. Baltensperger, J. Dommen and M. Geiser, Wood combustion particles induce adverse effects to normal and diseased airway epithelia, Environ. Sci.: Processes Impacts, 2017, 19, 538–548 RSC.
- H. O. T. Pye, C. K. Ward-Caviness, B. N. Murphy, K. W. Appel and K. M. Seltzer, Secondary organic aerosol association with cardiorespiratory disease mortality in the United States, Nat. Commun., 2021, 12, 7215 CrossRef CAS PubMed.
- S. Offer, E. Hartner, S. Di Bucchianico, C. Bisig, S. Bauer, J. Pantzke, E. J. Zimmermann, X. Cao, S. Binder, E. Kuhn, A. Huber, S. Jeong, U. Käfer, P. Martens, A. Mesceriakovas, J. Bendl, R. Brejcha, A. Buchholz, D. Gat, T. Hohaus, N. Rastak, G. Jakobi, M. Kalberer, T. Kanashova, Y. Hu, C. Ogris, A. Marsico, F. Theis, M. Pardo, T. Gröger, S. Oeder, J. Orasche, A. Paul, T. Ziehm, Z.-H. Zhang, T. Adam, O. Sippula, M. Sklorz, J. Schnelle-Kreis, H. Czech, A. Kiendler-Scharr, Y. Rudich and R. Zimmermann, Effect of Atmospheric Aging on Soot Particle Toxicity in Lung Cell Models at the Air-Liquid Interface: Differential Toxicological Impacts of Biogenic and Anthropogenic Secondary Organic Aerosols (SOAs), Environ. Health Perspect., 2022, 130, 27003–27021 CrossRef CAS PubMed.
- M. Pardo, S. Offer, E. Hartner, S. Di Bucchianico, C. Bisig, S. Bauer, J. Pantzke, E. J. Zimmermann, X. Cao, S. Binder, E. Kuhn, A. Huber, S. Jeong, U. Käfer, E. Schneider, A. Mesceriakovas, J. Bendl, R. Brejcha, A. Buchholz, D. Gat, T. Hohaus, N. Rastak, E. Karg, G. Jakobi, M. Kalberer, T. Kanashova, Y. Hu, C. Ogris, A. Marsico, F. Theis, T. Shalit, T. Gröger, C. P. Rüger, S. Oeder, J. Orasche, A. Paul, T. Ziehm, Z.-H. Zhang, T. Adam, O. Sippula, M. Sklorz, J. Schnelle-Kreis, H. Czech, A. Kiendler-Scharr, R. Zimmermann and Y. Rudich, Exposure to naphthalene and β-pinene-derived secondary organic aerosol induced divergent changes in transcript levels of BEAS-2B cells, Environ. Int., 2022, 166, 107366 CrossRef CAS PubMed.
- S. Magzamen, R. W. Gan, J. Liu, K. O'Dell, B. Ford, K. Berg, K. Bol, A. Wilson, E. V. Fischer and J. R. Pierce, Differential Cardiopulmonary Health Impacts of Local and Long-Range Transport of Wildfire Smoke, Geohealth, 2021, 5, e2020GH000330 CrossRef PubMed.
- V. S. Kozlov and M. V. Panchenko, Investigation of optical characteristics and particle-size distribution of wood-smoke aerosols, Combust. Explos. Shock Waves, 1996, 32, 577–586 CrossRef.
- R. F. Rakhimov, R. V. Makienko and V. P. Shmargunov, Variations of the Optical Constants and Size Spectra of Smoke Aerosols Produced during the Thermal Decomposition of Different Types of Wooden Materials, Atmos. Oceanic Opt., 2010, 23, 64–374 Search PubMed.
- A.-C. Kalogridis, O. B. Popovicheva, G. Engling, E. Diapouli, K. Kawamura, E. Tachibana, K. Ono, V. S. Kozlov and K. Eleftheriadis, Smoke aerosol chemistry and aging of Siberian biomass burning emissions in a large aerosol chamber, Atmos. Environ., 2018, 185, 15–28 CrossRef CAS.
- O. Popovicheva and V. Kozlov, in Proceedings of SPIE - 26th International Symposium on Atmospheric and Ocean Optics, ed. G. G. Matvienko and O. A. Romanovskii, 2020, p. 252 Search PubMed.
- V. N. Uzhegov, A. P. Rostov and Y. A. Pkhalagov, Automated path photometer, Opt. Atmos. Okeana, 2013, 26, 590–594 Search PubMed.
- V. S. Kozlov, I. B. Konovalov, V. N. Uzhegov, D. G. Chernov, V. V. Pol’kin, P. N. Zenkova, E. P. Yausheva, V. P. Shmargunov and S. N. Dubtsov, in 26th International Symposium on Atmospheric and Ocean Optics, Atmospheric Physics, ed. G. G. Matvienko and O. A. Romanovskii, SPIE, 2020, p. 189 Search PubMed.
- G. M. Krekov and R. F. Rakhimov, Optical - Radar Model of Continental Aerosol, Nauka, Novosibirsk, 1982 Search PubMed.
- A. Leskinen, P. Yli-Pirilä, K. Kuuspalo, O. Sippula, P. Jalava, M.-R. Hirvonen, J. Jokiniemi, A. Virtanen, M. Komppula and K. E. J. Lehtinen, Characterization and testing of a new environmental chamber, Atmos. Meas. Tech., 2015, 8, 2267–2278 CrossRef.
- V. S. Kozlov, V. Shmargunov and M. Panchenko, Modified aethalometer for monitoring of Black Carbon concentration in atmospheric aerosol and technique for correction of the spot loading effect, Proc. SPIE 10035, 22nd International Symposium on Atmospheric and Ocean Optics: Atmospheric Physics, 2016 Search PubMed.
- R. F. Rakhimov, V. S. Kozlov, M. V. Panchenko, A. G. Tumakov and V. P. Shmargunov, Properties of Atmospheric Aerosol in Smoke Plumes from Forest Fires according to Spectronephelometer Measurements, Atmos. Oceanic Opt., 2014, 27, 275–282 CrossRef.
- V. Kozlov, M. Panchenko and E. Yausheva, Mass fraction of black carbon in submicron aerosol as an indicator of influence of smoke from remote forest fires in Siberia, Atmos. Environ., 2008, 42, 2611–2620 CrossRef CAS.
- M. Jaoui, S. Leungsakul and R. M. Kamens, Gas and Particle Products Distribution from the Reaction of beta-Caryophyllene with Ozone, J. Atmos. Chem., 2003, 45, 261–287 CrossRef CAS.
- V. Pospisilova, F. D. Lopez-Hilfiker, D. M. Bell, I. El Haddad, C. Mohr, W. Huang, L. Heikkinen, M. Xiao, J. Dommen, A. S. H. Prévôt, U. Baltensperger and J. G. Slowik, On the fate of oxygenated organic molecules in atmospheric aerosol particles, Sci. Adv., 2020, 6, 1–11 Search PubMed.
- R. Atkinson, Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 2000, 34, 2063–2101 CrossRef CAS.
- S. S. Brown and J. Stutz, Nighttime radical observations and chemistry, Chem. Soc. Rev., 2012, 41, 6405–6447 RSC.
- Z. C. J. Decker, M. A. Robinson, K. C. Barsanti, I. Bourgeois, M. M. Coggon, J. P. DiGangi, G. S. Diskin, F. M. Flocke, A. Franchin, C. D. Fredrickson, G. I. Gkatzelis, S. R. Hall, H. Halliday, C. D. Holmes, L. G. Huey, Y. R. Lee, J. Lindaas, A. M. Middlebrook, D. D. Montzka, R. Moore, J. A. Neuman, J. B. Nowak, B. B. Palm, J. Peischl, F. Piel, P. S. Rickly, A. W. Rollins, T. B. Ryerson, R. H. Schwantes, K. Sekimoto, L. Thornhill, J. A. Thornton, G. S. Tyndall, K. Ullmann, P. van Rooy, P. R. Veres, C. Warneke, R. A. Washenfelder, A. J. Weinheimer, E. Wiggins, E. Winstead, A. Wisthaler, C. Womack and S. S. Brown, Nighttime and daytime dark oxidation chemistry in wildfire plumes: an observation and model analysis of FIREX-AQ aircraft data, Atmos. Chem. Phys., 2021, 21, 16293–16317 CrossRef CAS.
- J. K. Kodros, C. Kaltsonoudis, M. Paglione, K. Florou, S. Jorga, C. Vasilakopoulou, M. Cirtog, M. Cazaunau, B. Picquet-Varrault, A. Nenes and S. N. Pandis, Secondary aerosol formation during the dark oxidation of residential biomass burning emissions, Environ. Sci.: Atmos., 2022, 2, 1221–1236 CAS.
- X. Wang, T. Liu, F. Bernard, X. Ding, S. Wen, Y. Zhang, Z. Zhang, Q. He, S. Lü, J. Chen, S. Saunders and J. Yu, Design and characterization of a smog chamber for studying gas-phase chemical mechanisms and aerosol formation, Atmos. Meas. Tech., 2014, 7, 301–313 CrossRef CAS.
- X. Zhang, C. D. Cappa, S. H. Jathar, R. C. McVay, J. J. Ensberg, M. J. Kleeman and J. H. Seinfeld, Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, Proc. Natl. Acad. Sci. U.S.A., 2014, 111, 5802–5807 CrossRef CAS PubMed.
- J. H. Seinfeld, Effect of vapor wall loss in laboratory chambers on yields of secondary organic aerosols, Final Report to the California Air Resources Board. Contract 13-321, California Air Resources Board and the California Environmental Protection Agency, California Institute of Technology, Pasadena, CA 91125, 2015 Search PubMed.
- J. McCabe and J. P. D. Abbatt, Heterogeneous Loss of Gas-Phase Ozone on n -Hexane Soot Surfaces: Similar Kinetics to Loss on Other Chemically Unsaturated Solid Surfaces, J. Phys. Chem. C, 2009, 113, 2120–2127 CrossRef CAS.
- A. F. Khalizov, M. Cruz-Quinones and R. Zhang, Heterogeneous Reaction of NO2 on Fresh and Coated Soot Surfaces, J. Phys. Chem. A, 2010, 114, 7516–7524 CrossRef CAS PubMed.
- D. A. Knopf, S. M. Forrester and J. H. Slade, Heterogeneous oxidation kinetics of organic biomass burning aerosol surrogates by O3, NO2, N2O5, and NO3, Phys. Chem. Chem. Phys., 2011, 13, 21050–21062 RSC.
- I. J. Keyte, R. M. Harrison and G. Lammel, Chemical reactivity and long-range transport potential of polycyclic aromatic hydrocarbons-a review, Chem. Soc. Rev., 2013, 42, 9333–9391 RSC.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics. From Air Pollution to Climate Change, John Wiley & Sons, Ltd, Hoboken, New Jersey, 3rd edn, 2016 Search PubMed.
- S. So, U. Wille and G. Da Silva, Photoisomerization of Methyl Vinyl Ketone and Methacrolein in the Troposphere: A Theoretical Investigation of Ground-State Reaction Pathways, ACS Earth Space Chem., 2018, 2, 753–763 CrossRef CAS.
- M. Li, F. Bao, Y. Zhang, W. Song, C. Chen and J. Zhao, Role of elemental carbon in the photochemical aging of soot, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 7717–7722 CrossRef CAS PubMed.
- H. Bouzidi, C. Fittschen, P. Coddeville and A. Tomas, Photolysis of 2,3-pentanedione and 2,3-hexanedione: Kinetics, quantum yields, and product study in a simulation chamber, Atmos. Environ., 2014, 82, 250–257 CrossRef CAS.
- L. Hildebrandt, N. M. Donahue and S. N. Pandis, High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 2009, 9, 693–733 CrossRef.
- Y. Li, J. Zhao, Y. Wang, J. H. Seinfeld and R. Zhang, Multigeneration Production of Secondary Organic Aerosol from Toluene Photooxidation, Environ. Sci. Technol., 2021, 55, 8592–8603 CrossRef CAS PubMed.
- J. Joutsensaari, M. Ozon, T. Nieminen, S. Mikkonen, T. Lähivaara, S. Decesari, M. C. Facchini, A. Laaksonen and K. E. J. Lehtinen, Identification of new particle formation events with deep learning, Atmos. Chem. Phys., 2018, 18, 9597–9615 CrossRef CAS.
- P. Barmet, J. Dommen, P. F. DeCarlo, T. Tritscher, A. P. Praplan, S. M. Platt, A. S. H. Prévôt, N. M. Donahue and U. Baltensperger, OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 2012, 5, 647–656 CrossRef CAS.
- J. R. Odum, T. Hoffmann, F. Bowman, D. Collins, R. C. Flagan and J. H. Seinfeld, Gas/Particle Partitioning and Secondary Organic Aerosol Yields, Environ. Sci. Technol., 1996, 30, 2580–2585 CrossRef CAS.
- N. L. Ng, J. H. Kroll, A. W. H. Chan, P. S. Chhabra, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 2007, 7, 4085–4126 Search PubMed.
- Z. Yang, L. Du, Y. Li and X. Ge, Secondary organic aerosol formation from monocyclic aromatic hydrocarbons: insights from laboratory studies, Environ. Sci.: Processes Impacts, 2022, 24, 351–379 RSC.
- P. Lin, J. Liu, J. E. Shilling, S. M. Kathmann, J. Laskin and A. Laskin, Molecular characterization of brown carbon (BrC) chromophores in secondary organic aerosol generated from photo-oxidation of toluene, Phys. Chem. Chem. Phys., 2015, 17, 23312–23325 RSC.
- Z. Kitanovski, I. Grgić, F. Yasmeen, M. Claeys and A. Cusak, Development of a liquid chromatographic method based on ultraviolet-visible and electrospray ionization mass spectrometric detection for the identification of nitrocatechols and related tracers in biomass burning atmospheric organic aerosol, Rapid Commun. Mass Spectrom., 2012, 26, 793–804 CrossRef CAS PubMed.
- K. Sato, S. Hatakeyama and T. Imamura, Secondary organic aerosol formation during the photooxidation of toluene: NOx dependence of chemical composition, J. Phys. Chem. A, 2007, 111, 9796–9808 CrossRef CAS PubMed.
- A. Fushimi, D. Nakajima, A. Furuyama, G. Suzuki, T. Ito, K. Sato, Y. Fujitani, Y. Kondo, A. Yoshino, S. Ramasamy, J. J. Schauer, P. Fu, Y. Takahashi, K. Saitoh, S. Saito and A. Takami, Source contributions to multiple toxic potentials of atmospheric organic aerosols, Sci. Total Environ., 2021, 773, 145614 CrossRef CAS PubMed.
- Y. Kuang, J. Shang and Q. Chen, Effect of ozone aging on light absorption and fluorescence of brown carbon in soot particles: The important role of polycyclic aromatic hydrocarbons, J. Hazard. Mater., 2021, 413, 125406 CrossRef CAS PubMed.
- D. Liu, S. Li, D. Hu, S. Kong, Y. Cheng, Y. Wu, S. Ding, K. Hu, S. Zheng, Q. Yan, H. Zheng, D. Zhao, P. Tian, J. Ye, M. Huang and D. Ding, Evolution of Aerosol Optical Properties from Wood Smoke in Real Atmosphere Influenced by Burning Phase and Solar Radiation, Environ. Sci. Technol., 2021, 55, 5677–5688 CrossRef CAS PubMed.
- A. L. Hodshire, E. Rammarine, A. Akherati, M. J. Alvarado, D. K. Farmer, S. H. Jathar, S. M. Kreidenweis, C. R. Lonsdale, T. B. Onasch, S. R. Springston, J. Wang, Y. Wang, L. I. Kleinman, A. J. Sedlacek III and J. R. Pierce, Dilution impacts on smoke aging: evidence in Biomass Burning Observation Project (BBOP) data, Atmos. Chem. Phys., 2021, 21, 6839–6855 CrossRef CAS.
- B. B. Palm, Q. Peng, C. D. Fredrickson, B. H. Lee, L. A. Garofalo, M. A. Pothier, S. M. Kreidenweis, D. K. Farmer, R. P. Pokhrel, Y. Shen, S. M. Murphy, W. Permar, L. Hu, T. L. Campos, S. R. Hall, K. Ullmann, X. Zhang, F. Flocke, E. V. Fischer and J. A. Thornton, Quantification of organic aerosol and brown carbon evolution in fresh wildfire plumes, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29469–29477 CrossRef CAS PubMed.
- B. P. Koch and T. Dittmar, From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter, Rapid Commun. Mass Spectrom., 2006, 20, 926–932 CrossRef CAS.
- A. V. Semenova, Y. A. Zavgorodnyaya, M. A. Chichaeva, V. S. Kozlov and O. B. Popovicheva, Chemical Composition and Toxicity of Siberian Biomass Burning in the Large Aerosol Chamber (Tomsk), Atmos. Oceanic Opt., 2022, 35, S38–S47 CrossRef CAS.
- K. Atwi, S. N. Wilson, A. Mondal, R. C. Edenfield, K. M. Symosko Crow, O. El Hajj, c. Perrie, C. K. Glenn, C. A. Easley IV, H. Handa and R. Saleh, Differential response of human lung epithelial cells to particulate matter in fresh and photochemically aged biomass-burning smoke, Atmos. Environ., 2022, 271, 118929 CrossRef CAS.
- C. L. Heald, J. H. Kroll, J. L. Jimenez, K. S. Docherty, P. F. DeCarlo, A. C. Aiken, Q. Chen, S. T. Martin, D. K. Farmer and P. Artaxo, A simplified description of the evolution of organic aerosol composition in the atmosphere, Geophys. Res. Lett., 2010, 37, 8350 Search PubMed.
- C. P. Ward, R. L. Sleighter, P. G. Hatcher and R. M. Cory, Insights into the complete and partial photooxidation of black carbon in surface waters, Environ. Sci.: Processes Impacts, 2014, 16, 721–731 RSC.
- Y. Liang, C. N. Jen, R. J. Weber, P. K. Misztal and A. H. Goldstein, Chemical composition of PM2.5 in October 2017 Northern California wildfire plumes, Atmos. Chem. Phys., 2021, 21, 5719–5737 CrossRef CAS.
- R. Avagyan, R. Nyström, R. Lindgren, C. Boman and R. Westerholm, Particulate hydroxy-PAH emissions from a residential wood log stove using different fuels and burning conditions, Atmos. Environ., 2016, 140, 1–9 CrossRef CAS.
- T. Miersch, H. Czech, A. Hartikainen, M. Ihalainen, J. Orasche, G. Abbaszade, J. Tissari, T. Streibel, J. Jokiniemi, O. Sippula and R. Zimmermann, Impact of photochemical ageing on Polycyclic Aromatic Hydrocarbons (PAH) and oxygenated PAH (Oxy-PAH/OH-PAH) in logwood stove emissions, Sci. Total Environ., 2019, 686, 382–392 CrossRef CAS PubMed.
- C. Walgraeve, K. Demeestere, J. Dewulf, R. Zimmermann and H. van Langenhove, Oxygenated polycyclic aromatic hydrocarbons in atmospheric particulate matter: Molecular characterization and occurrence, Atmos. Environ., 2010, 44, 1831–1846 CrossRef CAS.
- E. Hartner, A. Paul, U. Käfer, H. Czech, T. Hohaus, T. Gröger, M. Sklorz, G. Jakobi, J. Orasche, S. Jeong, R. Brejcha, T. Ziehm, Z.-H. Zhang, J. Schnelle-Kreis, T. Adam, Y. Rudich, A. Kiendler-Scharr and R. Zimmermann, On the Complementarity and Informative Value of Different Electron Ionization Mass Spectrometric Techniques for the Chemical Analysis of Secondary Organic Aerosols, ACS Earth Space Chem., 2022, 6, 1358–1374 CrossRef CAS.
- M. E. Monge, B. D'Anna, L. Mazri, A. Giroir-Fendler, M. Ammann, D. J. Donaldson and C. George, Light changes the atmospheric reactivity of soot, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 6605–6609 CrossRef CAS PubMed.
- M. Shrivastava, S. Lou, A. Zelenyuk, R. C. Easter, R. A. Corley, B. D. Thrall, P. J. Rasch, J. D. Fast, S. L. Massey Simonich, H. Shen and S. Tao, Global long-range transport and lung cancer risk from polycyclic aromatic hydrocarbons shielded by coatings of organic aerosol, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 1246–1251 CrossRef CAS PubMed.
- F. Khan, K. Kwapiszewska, Y. Zhang, Y. Chen, A. T. Lambe, A. Kołodziejczyk, N. Jalal, K. Rudzinski, A. Martínez-Romero, R. C. Fry, J. D. Surratt and R. Szmigielski, Toxicological Responses of α-Pinene-Derived Secondary Organic Aerosol and Its Molecular Tracers in Human Lung Cell Lines, Chem. Res. Toxicol., 2021, 34, 817–832 Search PubMed.
- Z. Peng and J. L. Jimenez, Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev., 2020, 49, 2570–2616 RSC.
- M. Ihalainen, P. Tiitta, H. Czech, P. Yli-Pirilä, A. Hartikainen, M. Kortelainen, J. Tissari, B. Stengel, M. Sklorz, H. Suhonen, H. Lamberg, A. Leskinen, A. Kiendler-Scharr, H. Harndorf, R. Zimmermann, J. Jokiniemi and O. Sippula, A novel high-volume Photochemical Emission Aging flow tube Reactor (PEAR), Aerosol Sci. Technol., 2019, 53, 276–294 CrossRef CAS.
- Z. Tan, H. Fuchs, A. Hofzumahaus, W. J. Bloss, B. Bohn, C. Cho, T. Hohaus, F. Holland, C. Lakshmisha, L. Liu, P. S. Monks, A. Novelli, D. Niether, R. Rohrer, R. Tillmann, T. S. E. Valkenburg, V. Vardhan, A. Kiendler-Scharr, A. Wahner and R. Sommariva, Seasonal variation in nitryl chloride and its relation to gas-phase precursors during the JULIAC campaign in Germany, Atmos. Chem. Phys., 2022, 22, 13137–13152 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00280b |
| This journal is © The Royal Society of Chemistry 2024 |