
The effect of ionic versus covalent functionalization of polyoxometalate hybrid materials with coordinating subunits on their stability and interaction with DNA†
Daria Nowicka‡
a,
Dawid Marcinkowski‡ a,
Nahir Vadrab,
Martyna Szymańskaa,
Maciej Kubickia,
Giuseppe Consiglioc,
Wojciech Drożdżad,
Artur R. Stefankiewiczad,
Violetta Patroniaka,
Marta Fik-Jaskółka*a and
Adam Gorczyński*a
a,
Nahir Vadrab,
Martyna Szymańskaa,
Maciej Kubickia,
Giuseppe Consiglioc,
Wojciech Drożdżad,
Artur R. Stefankiewiczad,
Violetta Patroniaka,
Marta Fik-Jaskółka*a and
Adam Gorczyński*a
aAdam Mickiewicz University in Poznań, Faculty of Chemistry, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: marta.fik@amu.edu.pl; adam.gorczynski@amu.edu.pl
bUniversidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Química Inorgánica, Analítica y Química Física and CONICET–Universidad de Buenos Aires, Instituto de Química Física de los Materiales, Medio Ambiente y Energía (INQUIMAE), Buenos Aires C1428EGA, Argentina
cUniversità di Catania, Dipartimento di Scienze Chimiche, I-95125 Catania, Italy
dAdam Mickiewicz University in Poznań, Center for Advanced Technology, Uniwersytetu Poznańskiego 10, 61-614 Poznań, Poland
First published on 9th May 2024
Abstract
Inorganic–organic hybrid materials that combine both Polyoxometalates (POMs) and metal ion coordinating subunits (CSUs) represent promising multifunctional materials. Though their individual components are often biologically active, utilization of hybrid materials in bioassays significantly depends on the functionalization method and thus resulting stability of the system. Quite intriguingly, these aspects were very scarcely studied in hybrid materials based on the Wells–Dawson POM (WD POM) scaffold and remain unknown. We chose two model WD POM hybrid systems to establish how the functionalization mode (ionic vs. covalent) affects their stability in biological medium and interaction with nucleic acids. The synthetic scope and limitations of the covalent POM-terpyridine hybrids were demonstrated and compared with the ionic Complex-Decorated Surfactant Encapsulated-Clusters (CD-SECs) hybrids. The nature of POM and CSU binding can be utilized to modulate the stability of the hybrid and the extent of DNA binding. The above systems show potential to behave as model cargo-platforms for potential utilization in medicine and pharmacy.
Introduction
Polyoxometalates (POMs) can be described as a group of transition metals in their highest oxidation state linked together by oxygen atoms with a general formula [MOx]n, where M = Mo, W, V, and Nb, and x = 4–7,1–3 which can form different structures of varying sizes by gradual condensation. Besides, heteroatoms such as P, Si, Al, or S and transition metal ions such as Co, Fe, Ni, Cu, Zn, or Ln may be present, resulting in more stable systems commonly named as archetypical groups as e.g., Keggin, Wells Dawson (WD), Anderson or Preyssler.4 In addition, vacancies in the structure can be generated by controlled hydrolysis (e.g., lacunary POMs), giving new reactive centers that can be linked using electrophiles to form larger aggregates or utilized for postfunctionalization.5,6 Given their anionic characteristics, POMs can also bind different cations,7 and the final architecture and the physicochemical properties of the complex formed depend on the participating components of the complex.8Self-assembly and functionalization processes in POMs are not always straight-forward, and they are highly influenced by small changes in reaction conditions such as pH, temperature or stoichiometry and therefore, it is not always easy to predict the architecture and have a preprogrammed design.4 In the last few years, there have been numerous efforts to find new approaches to control the structure of POMs, some of the reported strategies have pointed to controlling self-assembly using classical approaches (such as trapping of functional metallic cores, molecular nanoparticles, or incorporation of active sites),8 while the latest ones focus on using Machine learning9 and automated chemistry.10
The versatility and variety of properties in these systems have led in the recent years to exploring ways to diversify the physicochemical properties, for example, by generating hybrid inorganic–organic architectures.11 In general terms, hybrid inorganic–organic POMs may be obtained by different approaches such as (1) nanostructurization of small POMs into larger ones;12 (2) covalent linking of organic moieties onto the POM cluster surface;13 (3) non-covalent functionalization (ionic interaction or hydrogen bonding) through the formation of the Surfactant-Encapsulated-Clusters (SECs).13 We have also presented a new pathway (4) to generate a novel family POMs inorganic–organic systems to form Complex-Decorated Surfactant-Encapsulated-Cluster (CD-SEC) materials.14 It is a multilevel functionalization based on the decoration of WD POMs with organic cation surfactant, which could simultaneously act as a metal ion complexing agent allowing for high versatility and tunable functionality. As a result, extensive exploration has been carried out for those systems concerning applications in fields such as catalysis,15,16 electrocatalysis,17,18 hydrogen generation,19 protein crystallography,20 elaboration of functional materials.21–23
Biological applications are also promising in terms of the potential of POMs to modify and enhance interaction with biomacromolecules.24–26 Indeed, a high negative charge accompanied by a large number of oxo-ligands results in POMs that can be treated as water-soluble macroanions, thus representing perfect models for studying the biomacromolecular interactions in polyelectrolyte solutions.27 Iso and heteropolyanions were studied in terms of hydrolytic cleavage of peptides,28 nucleotides,29 and saccharides,30 along with analysis of the POM-biomolecule mechanisms that govern the observed outcomes.31 Consequently, systematic studies on potential applications, such as antiviral,32 antidiabetic,33 antibiotic34 or anticancer35 effects, have been reported. Nevertheless, high toxicity in normal cells and unclear biochemical pathways indicate the need for further studies. Recently recognized organic–inorganic hybrid systems have been shown to enhance the anticancer activity35 and exhibit more effective antibacterial properties than the inorganic analogues.34 Both derivatization with organic moieties via covalent bonds and their encapsulation inside non-toxic organic macromolecules could lead to systems with enhanced biocompatibility, higher stability at physiological pH and lower toxicity.36–39 However, these results are still scarcely understood, and the need to construct such systems and studies is therefore justified.
Herein, we have chosen WD POMs to study due to their ability to undergo a variety of modifications and formation of organic–inorganic hybrids that exhibit potential biological activity. In this work we have studied how the ionic vs. covalent functionalization of the hybrid WD POM based system affects their stability in the presence of DNA and interacts with this biological system. To establish this, two distinct systems were preprogrammed: (i) previously demonstrated CD-SECs; (ii) covalently bound POM-tpy, and chosen, together with the construction algorithms that demonstrate the potential feasibility of such systems, to act as the drug carrier models.
Experimental section
Materials and methods
The metal salts, organic compounds and solvents were supplied by Aldrich and POCH. All chemicals mentioned above were of analytical grade quality and were used as obtained without further purification. The precursor V3POM-TBA TBA5H4[P2W15V3O62] POM was synthesized and characterized following published procedures (K6[α-P2W18O62]·14H2O40 → Na12[α-P2W15O56]·18H2O41 → K8H[P2W15V3O62]·9H2O42 → TBA5H4[P2W15V3O62]).42V3POM + TRIZMA (TBA)4H2[H2NC(CH2O)3P2V3W15O59]·3C3H7NO was synthesized following published procedure.43 All reactions were performed under an argon atmosphere condition unless otherwise stated with magnetic stirring and in the dark to prevent photoreduction of the V5+ containing POM to mixed valence V5+/V−4+ POMs. Ligand Ltpy was obtained according to the previously reported procedure.44 IR spectra were obtained with a PerkinElmer 580 spectrophotometer and peak positions are reported in cm−1. ESI mass spectra were determined in methanolic solution with c = ∼10−4 M using a Waters Micromass ZQ spectrometer. NMR spectra were run on a Varian VNMR-S 400 MHz spectrometer and were calibrated against the residual protonated solvent signals (DMSO-d6, δ 2.50; D2O, δ 3.33) which are given in parts per million. In 31P NMR 25% H3PO4 was used as an external reference. Electronic absorption titrations and stability tests of compounds were performed on JASCO V-770 spectrophotometer equipped with a Peltier PAC-743R at a temperature of 20 °C in 10 × 10 mm quartz cells in the range of 800–200 nm. pH measurements were tested on Mettler Toledo Seven Compact S210 pH meter.
Synthetic procedure of complex K1 [Zn(Ltpy)Cl2]
Ligand Ltpy (100.0 mg, 0.30 mmol) was placed in a 50 ml round-bottom flask and dissolved in the mixture methanol/acetonitrile (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v) (10 ml). The ZnCl2 (40.9 mg, 0.30 mmol) was dissolved in a mixture of methanol/acetonitrile (1:1, v:v) (10 ml). Then the salt solution was added to the ligand and the solution changed colour from transparent to light yellow. The resulting mixture was stirred for 24 hours at room temperature. After this time, the reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in a minimal amount of methanol/acetonitrile (1:1, v:v) (ca. 1 ml) and precipitated by the adding an excess of diethyl ether (approx. 30 ml). The resulting white precipitate was filtered off with suction and dried in a vacuum. Yield: 80.0 mg, 57.0%. ESI-MS(+) m/z (%): 335 (100) [Ltpy + H]+, 433 (30) [ZnLtpyCl]+. Anal. calc.: C, 51.15; H, 4.08; N, 8.95; found: C, 51.05; H, 3.98; N, 9.02%. IR (KBr, cm−1): ν(C–H)ar 3392; νs(CH3) 2449; ν(C
:1, v:v) (10 ml). The ZnCl2 (40.9 mg, 0.30 mmol) was dissolved in a mixture of methanol/acetonitrile (1:1, v:v) (10 ml). Then the salt solution was added to the ligand and the solution changed colour from transparent to light yellow. The resulting mixture was stirred for 24 hours at room temperature. After this time, the reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in a minimal amount of methanol/acetonitrile (1:1, v:v) (ca. 1 ml) and precipitated by the adding an excess of diethyl ether (approx. 30 ml). The resulting white precipitate was filtered off with suction and dried in a vacuum. Yield: 80.0 mg, 57.0%. ESI-MS(+) m/z (%): 335 (100) [Ltpy + H]+, 433 (30) [ZnLtpyCl]+. Anal. calc.: C, 51.15; H, 4.08; N, 8.95; found: C, 51.05; H, 3.98; N, 9.02%. IR (KBr, cm−1): ν(C–H)ar 3392; νs(CH3) 2449; ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1627; ρ(C–H)ar1040; γ(C–H)ar 553. 1H NMR (400 MHz, DMSO-d6): δ = 9.06 (s, 1H), 8.71 (d, J = 7.9 Hz, 1H), 8.17 (t, J = 7.8 Hz, 1H), 7.69 (d, J = 7.7 Hz, 1H), 4.58–4.45 (m, 1H), 3.01 (s, 4H), 1.46 (t, J = 7.1 Hz, 2H). Solubility problems result in a mixture of “open” (metal ion:ligand; 1:1) and “closed” (metal ion:ligand; 1:2) systems.
O) 1627; ρ(C–H)ar1040; γ(C–H)ar 553. 1H NMR (400 MHz, DMSO-d6): δ = 9.06 (s, 1H), 8.71 (d, J = 7.9 Hz, 1H), 8.17 (t, J = 7.8 Hz, 1H), 7.69 (d, J = 7.7 Hz, 1H), 4.58–4.45 (m, 1H), 3.01 (s, 4H), 1.46 (t, J = 7.1 Hz, 2H). Solubility problems result in a mixture of “open” (metal ion:ligand; 1:1) and “closed” (metal ion:ligand; 1:2) systems.
Synthetic procedure of complex K2 [Zn(Ltpy)2](ClO4)2
Ligand Ltpy (100.0 mg, 0.30 mmol) was placed in a 50 ml round-bottom flask and dissolved in the mixture methanol/acetonitrile (1:1, v:v) (10 ml). The Zn(ClO4)2·6H2O (55.9 mg, 0.15 mmol) was dissolved in a mixture of methanol/acetonitrile (1:1, v:v) (10 ml). Then the salt solution was added to the ligand solution. The resulting transparent mixture was stirred for 24 hours at room temperature. After this time, the reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in a minimal amount of methanol/acetonitrile (1:1, v:v) (about 1 ml) and precipitated by adding an excess of diethyl ether (approx. 30 ml). The resulting white precipitate was filtered off with suction and dried in a vacuum. White solid residue was recrystallized by slow diffusion method from diisopropyl ether into methanol/acetonitrile (1:1, v:v) system at lowered temperature. Yield: 105.0 mg, 75.0%. ESI-MS(+) m/z (%): 335 (30) [Ltpy + H]+, 356 (50) [Ltpy + Na]+, 366 (80) [ZnLtpy2]2+, 831 (10) [ZnLtpy2(ClO4)]+. IR (KBr, cm−1): ν(C–H)ar 3104; νs(CH3) 2980, 2931; ν(CO) 1773; ν(CC)ar 1604, 1570, 1478, ν(C–N) 1442, 1372; ν(CN)ar 1269, 1249; ρ(C–H)ar 1139, 1089; γ(C–H)ar 810, 769, 740; γ(ClO) 622. 1H NMR (400 MHz, DMSO-d6): δ = 8.82 ppm (s, 2H), 8.44 ppm (d, 2H), 7.91 ppm (t, 2H), 7.40 ppm (d, 2H), 4.46 ppm (q, 2H), 2.62 ppm (s, 6H), 1.40 ppm (t, 3H).
Synthetic procedure of hybrid H1cov (POM + Ltpy) [V3POM + TRIZMA + Ltpy]TBA6
V3POM + TRIZMA (50 mg, 0.01 mmol) was dissolved in 15 ml of acetonitrile and added to a two necked 50 ml flask equipped with a condenser. Ligand Ltpy (3.2 mg, 0.01 mmol) was dissolved in 5 ml of acetonitrile and was added to solution of V3POM + TRIZMA. The solution changed colour from brown-orange to yellow-green. Then the mixture was heated under reflux for 3 days. The resulting yellow-green precipitate was filtered off with suction, washed with acetonitrile and dried in a vacuum. Yield: 41.5 mg, 78.0%. ESI-MS(−) m/z (%): 716 (20) [Ltpy(V3POM + trizma)]6−. Anal. calc.: C, 25.03; H, 4.25; N, 2.43; found: C, 25.01; H, 4.31; N, 2.38%. IR (KBr, cm−1): ν(N–H) 3446; νs(CH3) 2932, 2872; ν(CO) 1633; ν(C–N) 1465, 1379; ρ(C–H)ar 1086; γ(C–H)ar 952, 910, 817, 528. 1H NMR (400 MHz, DMSO-d6): δ = 8.85 ppm (s, 2H, HL), 8.49 ppm (d, 2H, HL), 7.99 ppm (m, 2H, HL), 7.46 ppm (d, 2H, HL), 5.39 ppm (s, 6H, O-CH2-C), 3.16 ppm (m, 48H, HTBA), 2.65 ppm (s, 6H, HL), 1.57 ppm (p, 48H, HTBA), 1.31 ppm (h, 48H, HTBA), 0.98 ppm (t, 72H, HTBA). 31P NMR (162 MHz, DMSO-d6): δ = −6.65 ppm, −12.70 ppm.
Synthetic procedure of hybrid H2cov (POM + K1) [Zn(V3POM + TRIZMA + Ltpy)Cl2]TBA6
V3POM + TRIZMA (50 mg, 0.01 mmol) was dissolved in 8 ml of acetonitrile and added to two necked 50 ml flask. K1 (4.5 mg, 0.01 mmol) was added to solution of V3POM + TRIZMA. The homogeneous mixture was heated under reflux for 24 hours. After this time clear yellow solution was condensed on rotavapor and diethyl ether was added. The resulting yellow precipitate was centrifuged, washed with diethyl ether and dried in a vacuum. Yield: 50.8 mg, 87.6%. ESI-MS(–) m/z (%): 962 (30) [Ltpy(V3POM + trizma)Cl + 2TBA]5−, 1638 (40) [Zn Ltpy(V3POM + trizma)Cl2 + H + 2TBA]3−. Anal. calc.: C, 25.45; H, 4.15; N, 2.38; found: C, 25.58; H, 4.21; N, 2.31%. IR (KBr, cm−1): ν(N–H) 3436; νs(CH3) 2963, 2873; ν(CO) 1644; ν(C–N) 1473, 1391; ρ(C–H)ar 1079; γ(C–H)ar 952, 900, 804, 737, 514. 1H NMR (400 MHz, DMSO-d6): δ = 8.84 ppm (s, 2H, HL), 8.38 ppm (s, 2H, HL), 7.95 ppm (s, 2H, HL), 7.28 ppm (s, 2H, HL), 5.39 ppm (s, 6H, O-CH2-C), 3.17 ppm (t, 48H, HTBA), 2.59 ppm (s, 6H, HL), 1.56 ppm (p, 48H, HTBA), 1.23 ppm (h, 48H, HTBA), 0.94 ppm (t, 72H, HTBA). 31P NMR (162 MHz, DMSO-d6): δ = −6.65 ppm, −12.70 ppm.
Synthetic procedure of hybrid H3cov (POM + K2) [Zn(V3POM + TRIZMA + Ltpy)2(ClO4)2]TBA(12−x)Hx; x = 11.5
V3POM + TRIZMA (100 mg, 0.02 mmol) was dissolved in 15 ml of acetonitrile and added to two necked 50 ml flask. K2 (7.0 mg, 0.01 mmol) was added to solution of V3POM + TRIZMA. The heterogeneous mixture was heated under reflux for 24 hours. After this time a clear yellow solution was condensed on a rotavapor and placed at 4° deg overnight. The resulting yellow precipitate was filtered off with suction, washed with acetonitrile and dried in a vacuum. Yield: 38.0 mg, 71.7%. ESI-MS(−) m/z (%): 795 (30) [Zn(Ltpy(V3POM + trizma))2ClO4]11−, 1089 (70) [ZnLtpy(V3POM + trizma)]4−. IR (KBr, cm−1): ν(N–H) 3465; νs(CH3) 2952, 2863; ν(CO) 1629; ν(CC)ar 1570; ν(C–N) 1458, 1384; ν(CN)ar 1272; ρ(C–H)ar 1079; γ(C–H)ar 975, 900, 818, 730, 514. 1H NMR (400 MHz, DMSO-d6): 8.85 ppm (s, 4H, HL), 8.50 ppm (d, 4H, HL), 7.99 ppm (m, 4H, HL), 7.48 ppm (d, 4H, HL), 5.40 ppm (s, 6H, O-CH2-C), 3.16 ppm (m, 3H, HTBA), 2.65 ppm (s, 12H, HL), 1.57 ppm (s, 3H, HTBA), 1.30 ppm (p, 3H, HTBA), 0.93 ppm (t, 5H, HTBA). 31P NMR (162 MHz, DMSO-d6): δ = −6.65 ppm, −12.70 ppm.
Synthetic procedure of hybrid H4cov (H1cov + ZnCl2) [Zn(V3POM + TRIZMA + Ltpy)Cl2]H2TBA4
Hybrid H1cov (30 mg, 0.005 mmol) was dissolved in 5 ml of acetonitrile and added to two necked 50 ml flask. Next, solution of ZnCl2 (0.7 mg, 0.005 mmol) was added to solution of hybrid H1cov. The heterogeneous mixture was heated under reflux for 24 hours. After this time the solution changed colour from violet to grey-green. The mixture was condensed on rotavapor and diethyl ether was added. The resulting grey-green precipitate was filtered via suction filtration, washed with diethyl ether and dried in a vacuum. Yield: 21.0 mg, 43.9%. ESI-MS(−) m/z (%): 915 (20) [Ltpy(V3POM + trizma)Cl + H + TBA]5−, 1204 (10) [Ltpy(V3POM + trizma)Cl + H + 2TBA]4−. Anal. calc.: C, 19.53; H, 3.22; N, 2.07; found: C, 19.75; H, 3.29; N, 2.10%. IR (KBr, cm−1): ν(N–H) 3443; νs(CH3) 2960, 2878; ν(CO) 1644; ν(C–N) 1466, 1377; ν(CN)ar 1272; ρ(C–H)ar 1087; γ(C–H)ar 952, 915, 812, 722, 536. 1H NMR (400 MHz, DMSO-d6): δ = 8.85 ppm (s, 2H, HL), 8.48 ppm (d, 2H, HL), 7.96 ppm (m, 2H, HL), 7.45 ppm (d, 2H, HL), 5.39 ppm (s, 6H, O-CH2-C), 3.16 ppm (t, 32H, HTBA), 2.64 ppm (s, 6H, HL), 1.56 ppm (p, 32H, HTBA), 1.32 ppm (h, 32H, HTBA), 0.94 ppm (t, 48H, HTBA). 31P NMR (162 MHz, DMSO-d6): δ = −6.65 ppm, −12.70 ppm.
Synthetic procedure of hybrid H5cov (H1cov + Zn(ClO4)2) [Zn(V3POM + TRIZMA + Ltpy)2(ClO4)2]H12
Hybrid H1cov (30 mg, 0.005 mmol) was dissolved in 5 ml of acetonitrile and added to two necked 50 ml flask. Next, Zn(ClO4)2·6H2O (1.0 mg, 0.0026 mmol) was added to solution of hybrid H1cov. The heterogeneous mixture was heated under reflux for 24 hours. The resulting yellow precipitate was centrifuged, washed with acetonitrile and dried in a vacuum. Yield: 25.8 mg, 98.7%. ESI-MS(−) m/z (%): 575 (10) [Ltpy(V3POM + trizma)(ClO4)3 + H]8−, 604 (30) [Ltpy(V3POM + trizma)(ClO4)3 + TBA]8−. IR (KBr, cm−1): ν(N–H) 3465; νs(CH3) 2982, 2915; ν(CO) 1607; ν(CC)ar 1540; ν(C–N) 1435, 1391; ν(CN)ar 1287; ρ(C–H)ar 1087; γ(C–H)ar 960, 900, 797, 722, 522. 1H NMR (400 MHz, DMSO-d6): 8.85 ppm (s, 4H, HL), 8.48 ppm (d, 4H, HL), 7.97 ppm (m, 4H, HL), 7.45 ppm (d, 4H, HL), 5.39 ppm (s, 12H, O-CH2-C), 2.64 ppm (s, 12H, HL). 31P NMR (162 MHz, DMSO-d6): δ = −6.67 ppm, −12.70 ppm.
Synthetic procedure of hybrid H6cov (POM + Ltpy + ZnCl2) [Zn(V3POM + TRIZMA + Ltpy)Cl2]TBA(6−x)Hx; x = 5.75
V3POM + TRIZMA (50 mg, 0.01 mmol) was dissolved in 8 ml of acetonitrile and added to two necked 50 ml flask. Ltpy (3.2 mg, 0.01 mmol) and ZnCl2 (1.3 mg, 0.01 mmol) were added to solution of V3POM + TRIZMA. The mixture changed colour to dark violet. The heterogeneous mixture was heated under reflux for 24 hours. After 3 hours the mixture turned yellow again. The resulting gray-green precipitate was centrifuged, washed with acetonitrile and dried in a vacuum. Yield: 17.4 mg, 36.4%. ESI-MS(−) m/z (%): 962 (30) [Ltpy(V3POM + trizma)Cl + 2TBA]5−, 1089 (30) [Zn Ltpy(V3POM + trizma)]4−. Anal. calc.: C, 6.48; H, 0.75; N, 1.26; found: C, 6.51; H, 0.80; N, 1.31%. IR (KBr, cm−1): ν(N–H) 3458; ν(C–H)ar 3086; νs(CH3) 2960, 2863; ν(CO) 1636; ν(CC)ar 1577, 1517; ν(C–N) 1428, 1384; ν(CN)ar 1265; ρ(C–H)ar 1071; γ(C–H)ar 960, 908, 812, 737, 522. 1H NMR (400 MHz, DMSO-d6): δ = 8.85 ppm (s, 2H, HL), 8.50 ppm (d, 2H, HL), 7.98 ppm (d, 2H, HL), 7.49 ppm (d, 2H, HL), 5.40 ppm (s, 6H, O-CH2-C), 3.16 ppm (m, 2H, HTBA), 2.66 ppm (s, 6H, HL), 1.57 ppm (p, 2H, HTBA), 1.31 ppm (h, 2H, HTBA), 0.94 ppm (t, 3H, HTBA). 31P NMR (162 MHz, DMSO-d6): δ = −6.65 ppm, −12.70 ppm.
Synthetic procedure of hybrid H7cov (POM + Ltpy + Zn(ClO4)2) [Zn(V3POM + TRIZMA + Ltpy)2(ClO4)2]TBA(12−x)Hx; x = 11.5
V3POM + TRIZMA (50 mg, 0.01 mmol) was dissolved in 8 ml of acetonitrile and added to two necked 50 ml flask. Ltpy (3.2 mg, 0.01 mmol) and Zn(ClO4)2·6H2O (1.8 mg, 0.005 mmol) were added to solution of V3POM + TRIZMA. The heterogeneous mixture was heated under reflux for 24 hours. The resulting yellow precipitate was centrifuged, washed with acetonitrile and dried in a vacuum. Yield: 34.3 mg, 64.7%. ESI-MS(−) m/z (%): 716 (20) [Ltpy(V3POM + trizma)]6−, 732 (20) [Ltpy(V3POM + trizma)ClO4 + H]6−, 772 (40) [Ltpy(V3POM + trizma)ClO4 + TBA]6−, 976 (60) [Ltpy(V3POM + trizma)ClO4 + 2TBA]5−. IR (KBr, cm−1): ν(N–H) 3458; ν(C–H)ar 3086; νs(CH3) 2952, 2863; ν(CO) 1644; ν(CC)ar 1554; ν(C–N) 1428, 1384; ν(CN)ar 1272; ρ(C–H)ar 1094; γ(C–H)ar 960, 923, 812, 752, 581, 522. 1H NMR (400 MHz, DMSO-d6): 8.85 ppm (s, 4H, HL), 8.49 ppm (d, 4H, HL), 7.97 ppm (m, 4H, HL), 7.46 ppm (d, 4H, HL), 5.41 ppm (s, 6H, O-CH2-C), 3.17 ppm (m, 4H, HTBA), 2.64 ppm (s, 12H, HL), 1.58 ppm (m, 4H, HTBA), 1.31 ppm (m, 4H, HTBA), 0.94 ppm (m, 6H, HTBA). 31P NMR (162 MHz, DMSO-d6): δ = −6.65 ppm, −12.70 ppm.
X-ray crystallography
X-ray diffraction data were collected at 100(1) K by the ω-scan technique on Rigaku four-circle XCalibur diffractometer (Eos detector) with graphite-monochromatized MoKα radiation (λ = 0.71073 Å). The data were corrected for Lorentz-polarization and absorption effects.45 The structure was solved with SHELXT46 and refined with the full-matrix least-squares procedure on F2 by SHELXL.47 Non-hydrogen atoms were refined anisotropically (vide infra), hydrogen atoms were placed in the calculated positions and refined as ‘riding model’ with the isotropic displacement parameters set at 1.2 (1.5 for methyl) times the Ueq value for appropriate non-hydrogen atom. The structure has been found disordered, in both ligand molecules the ethyl groups have been refined in two alternative positions, and in both cases the terminal methyl carbon atoms were left isotropic. Some constraints for C–C distances were applied. Additionally, there are regions of diffused electron density in the voids, apparently connected with the disorder solvent molecules. The attempts to model the solvent failed, so it has been refined with the SQUEEZE method.48Crystal data: (C40H38ZnN6O4)2+·2(ClO4), Mr = 931.03, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 12.9401(8) Å, b = 13.1181(8) Å, c = 13.9436(6) Å, α = 68.923(5), β = 80.861(4), γ = 66.577(6), V = 2026.2(2) Å3, Z = 2, dx = 1.523 g·cm−3, F(000) = 958, μ = 0.743 cm−1, 14264 reflection collected, 7131 symmetry independent (Rint = 3.36%), 5905 with I > 2σ(I). Final R[I > 2σ(I)] = 0.0779, wR2[I > 2σ(I)] = 0.1993, R [all reflections] = 0.0928, wR2[all reflections] = 0.2088, S = 1.102, (Δρmax/Δρmin) = 1.45/−0.76 e Å−3. Crystallographic data for the structural analysis has been deposited with the Cambridge Crystallographic Data Centre, CCDC 2078396.†
, a = 12.9401(8) Å, b = 13.1181(8) Å, c = 13.9436(6) Å, α = 68.923(5), β = 80.861(4), γ = 66.577(6), V = 2026.2(2) Å3, Z = 2, dx = 1.523 g·cm−3, F(000) = 958, μ = 0.743 cm−1, 14264 reflection collected, 7131 symmetry independent (Rint = 3.36%), 5905 with I > 2σ(I). Final R[I > 2σ(I)] = 0.0779, wR2[I > 2σ(I)] = 0.1993, R [all reflections] = 0.0928, wR2[all reflections] = 0.2088, S = 1.102, (Δρmax/Δρmin) = 1.45/−0.76 e Å−3. Crystallographic data for the structural analysis has been deposited with the Cambridge Crystallographic Data Centre, CCDC 2078396.†
General for biological activities
CT-DNA, Tris and NaCl were supplied from Merck and used without further purification. CT-DNA was dissolved in Tris Buffer (5 mM Tris HCl, 50 mM NaCl, pH 7.4) prior to use. The CT-DNA solution gave a ratio of UV absorbance at 260 and 280 nm and the value is lower than 1.82:1, indicating that the CT-DNA sample was sufficiently free from protein. CT-DNA concentration per nucleotide was determined from the UV absorbance at 260 nm using the extinction coefficient ε260 = 6600 dm3 mol−1 cm−1. Stock solutions of compounds in DMSO at concentration 2 × 10−3 M were prepared prior to use. All electronic absorption spectra were performed in 10 × 10 mm quartz cells on Jasco V-770 between 800 and 200 nm using solutions ∼2 × 10−5 M with respect to the compound concentration in Tris Buffer (5 mM Tris HCl, 50 mM NaCl, pH 7.4) with 1% DMSO content.
Stability tests
Samples were prepared by diluting aqueous stock solutions (2 mM) of compounds (25 μl) in Tris Buffer (2475 μl; 5 mM Tris HCl, 50 mM NaCl, pH 7.4). UV-Vis absorbance spectra were measured in time intervals until stabilization of spectra has been reached. The obtained data indicate that the compounds remain stable49 and reach equilibrium within 60 minutes.Electronic absorption titration with DNA
The absorbance titrations were performed in a fixed concentration of compounds (20 μM) while gradually increasing the concentration of CT-DNA within the range from 0 to 100 μM. Identical aliquots of CT-DNA stock solution were added to the sample and the blank solutions and a new baseline was recorded before each titration step in order to not observe the bands corresponding to CT-DNA alone in the subsequent spectra. Each sample solution was allowed to equilibrate 5 min before the spectra were recorded. Using the absorption titration data, the binding constant Kb was determined according to the equation:50 [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb (εb − εf), where [DNA] is the concentration of CT-DNA in the base pairs, εa corresponds to the extinction coefficient observed (Aobsd/[M]), εf corresponds to coefficient of free compound, εb is the extinction coefficient of the compound fully bound to CT-DNA, and Kb is the intrinsic binding constant. The Kb value was given by the ratio of slope to intercept in the plot of [DNA]/(ε − εf) versus [DNA].pH driven decomposition of hybrids and electronic absorption titration of H1cov, H2cov, H3cov with DNA in alkaline solutions
Solutions were prepared by adding stock solutions of compounds (20 μM) to MiliQ water (4 ml solution volume). The pH of each of the solutions was measured and the absorption spectra were performed by taking 2.5 ml of the solution. 2 M NaOH (1.5 μl) was then added to the test solutions to adjust the pH to alkaline. The pH of the solutions and the UV spectrum were measured. The next step was to add 100 μM CT-DNA to each sample including reference and the absorption measurement was made after 10 min, 60 min and 24 h.Results and discussion
General rationale
WD POM hybrids with terpyridine-type ligands are known in the literature from both covalent and ionic approaches. The majority of papers focus on the covalent bonding of terpyridine ligands to WD POM, where organosilyl51,52 or organophosphonate53,54 functionalized WD POMs can be post-functionalized with terpyridine ligand-based complexes. In contrast, a distinct minority can be found in the literature showing ionic bonding between WD POM and terpyridine ligand. Among the few articles, the most commonly presented is the binding of a common P2W18 subunit or modified cap WD POM to a pentaerythritol-based metallodendrimer with RuII terpyridine units.55,56Herein, the hybrid systems based on WD POM were obtained via ionic and covalent functionalization to compare if the type of POM-complex interactions affect the stability of the hybrids and whether this can be used in the presence of DNA to demonstrate the possible interactions with a simple biological system. Hybrid materials constructed via ionic interactions were prepared using WD POM and tridentate acyl-hydrazone N2O ligand LN2O with NR4+ cationic moiety and was reported by us in 202014 (Scheme 1b). The simplicity of the system lies in the fact that WD counterpart is non-functionalized, easily available and its highly negative charge is responsible for interactions with ligand LN2O or its complexes, ultimately forming (CD)-SEC materials. Please note that it was previously demonstrated in the literature that introduction of the cationic moiety in the acyl-hydrazone group can influence the binding propensity of the system to the DNA.57 These hybrid materials termed H1ion–H3ion are studied for their interactions with DNA (see Section: interaction of ionic hybrids with DNA). Herein, we constructed new class of WD hybrid materials based on the inorganic WD POM and 6,6′′-dimethyl-2,2′:6′,2′′-terpyridine-type ligand Ltpy, which is based on the covalent functionalization. Tris(hydroxymethyl)aminomethane (TRIZMA) was chosen as the anchoring group, given its wide and successful utilization in the POM chemistry.58
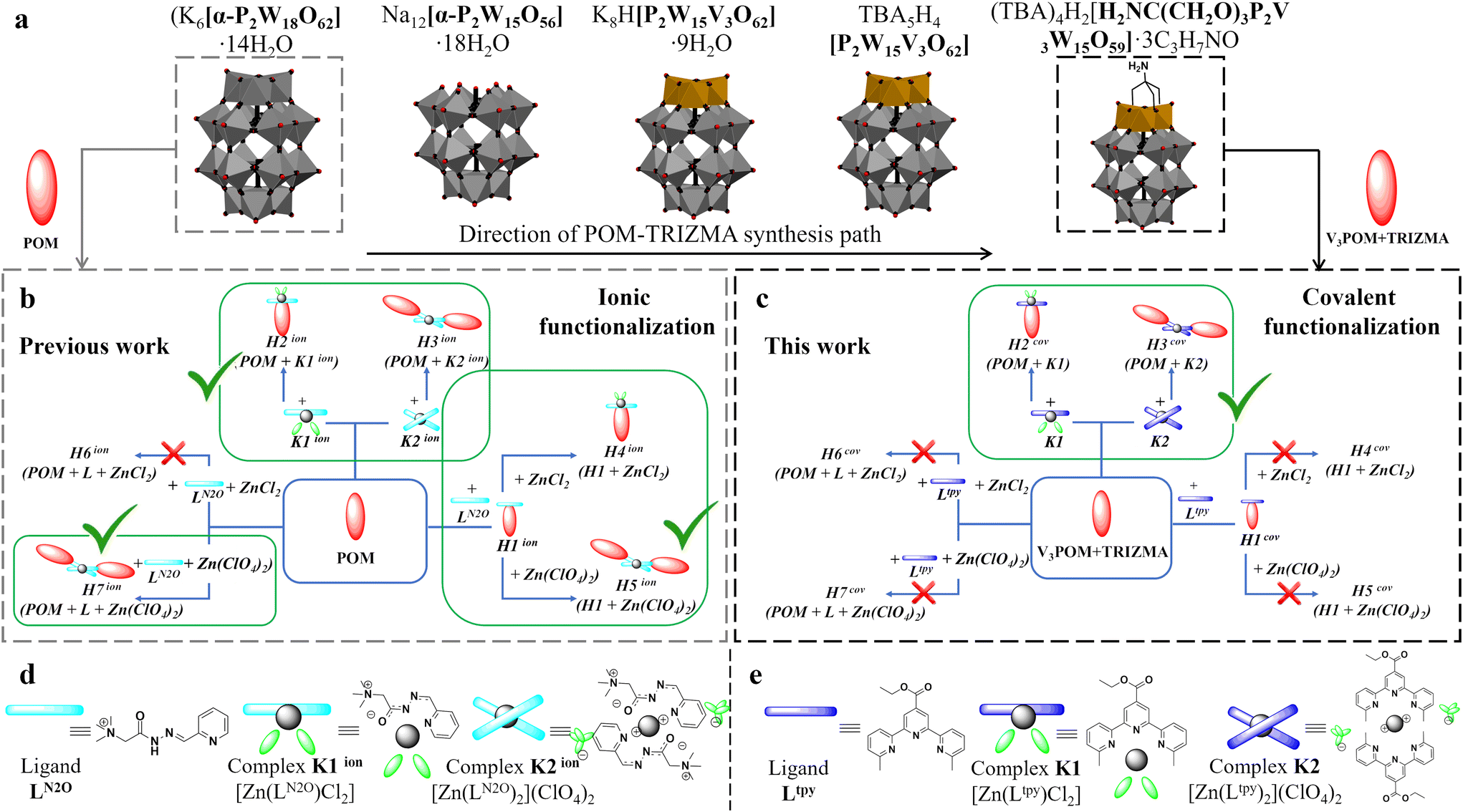 | ||
| Scheme 1 (a) Scheme of synthesis path of WD POM in their polyhedral representations; schematic representation of hybrids constructed via (b) ionic14 and (c) covalent interactions; schematic representation structures of (d) ligand LN2O and complexes K1ion [Zn(LN2O)Cl2], K2ion [Zn(LN2O)2](ClO4)2, respectively; and (e) ligand Ltpy [C20H19N3O2] and complexes K1 [Zn(Ltpy)Cl2], K2 [Zn(Ltpy)2](ClO4)2, respectively. | ||
Onto the TRIZMA-modified mono-substituted WD POM (V3POM + TRIZMA), Ltpy ligand was chemically grafted through the ester group in the p-position of the central tpy N-heterocyclic ring (Scheme 1c). We have previously demonstrated that the methylated version of the tpy ligand can efficiently form bimetallic [Ag2L2]2+ helicates, which demonstrated cytotoxic character towards chosen cancer cell lines,59 hence we envisaged that the formed POM covalent hybrids H1cov–H3cov have the potential to exhibit related properties and therefore interaction with DNA were studied and compared with H1ion–H3ion materials. Please note that irrespectively of the chosen type of connection (ionic or covalent), H1 denotes POM-ligand hybrid, whereas H2 and H3 its complex with zinc(II) chloride and perchlorate, respectively.
Synthetic considerations for substrates
The synthetic pathway was divided into independent groups: preparation of TRIZMA-functionalized Wells–Dawson system V3POM + TRIZMA (Scheme 1a), synthesis of ligand Ltpy and its two zinc(II) complexes K1 [Zn(Ltpy)Cl2], K2 [Zn(Ltpy)2](ClO4)2 (Scheme 1e). After preparing the precursors, the synthesis of the seven hybrid materials H1cov–H7cov was carried out using different routes (Scheme 1c). Details, including synthetic procedures and characterization are included in the Experimental section and ESI,† respectively.The general procedure (Scheme 1a) involves selective lacunarization of the parent K6[P2W18O62] WD POM and subsequent implementation of the vanadium cap, which renders the system prone to selective integration of the TRIZMA linker. Such amino-functionalized POM allows one to graft the tpy ligand in a robust, covalent manner through the amide bond, which was demonstrated to be feasible approach.60,61
Ligand Ltpy was obtained according to the slightly modified reported procedure44 via the Stille coupling protocol (Scheme S1†) and was used for complexation reactions with ZnCl2 and Zn(ClO4)2, thus allowing formation of complexes K1 [Zn(Ltpy)Cl2] and K2 [Zn(Ltpy)2](ClO4)2, respectively. In both cases, the reaction was conducted in a mixture of methanol/acetonitrile (1:1, v:v) for 24 hours at room temperature. The purity of the compound and the repeatability of the desired product were confirmed by ESI-MS, elementary analysis, FT-IR, 1H NMR and 31P NMR spectroscopy are described in detail in the Experimental section and ESI.† X-ray crystal structure of complex K2 is described in Section: X-ray crystal characterization of complex K2.
X-ray crystal characterization of complex K2
Fig. 1a shows the perspective view of the [Zn(Ltpy)2]2+ cation in structure K2. The ZnII ion is 6-coordinated, by six nitrogen atoms – three from each ligand molecule. The coordination might be described as distorted octahedral, with the Zn–N distances within the range 2.054(5) Å–2.311(4) Å, and the three largest.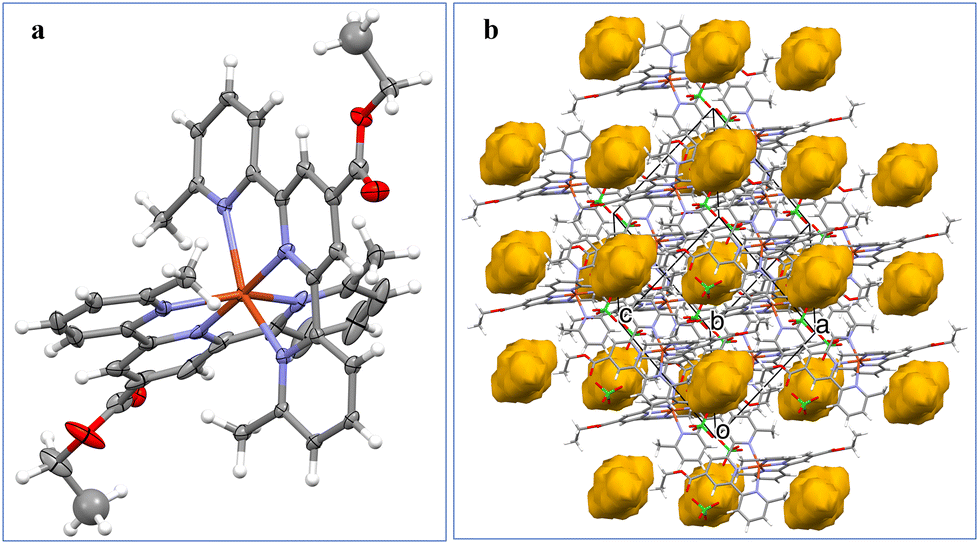 | ||
| Fig. 1 (a) Perspective view of the dicationic complex K2 [Zn(Ltpy)2]2+; (b) the ellipsoids are drawn at the 30% probability level, hydrogen atoms are represented by spheres of arbitrary radii. | ||
N–Zn–N angles equal 176.17(17)°, 151.07(19)°, and 151.63(15)°. The crystal architecture is determined mainly be electrostatic interactions between charged species; there are voids filled with diffused electron density (Fig. 1b).
Synthesis and characterization of hybrid materials
The obtained precursors, both V3POM + TRIZMA, ligand Ltpy and Zn(II) complexes K1 and K2, were reacted to obtain three distinct families of hybrid materials H1cov–H3cov (H1cov – POM with grafted ligand; H2cov – POM with grafted complex K1; H3cov – POM with grafted complex K2) that can be potentially formed via seven different routes – depicted in Scheme 1c. Since we observed in our previous study14 that not every route is feasible for the construction of ionic assemblies H1ion–H3ion, the same approach needs to be performed for the covalent class. Please note that for clarity purposes, each process was termed as H1cov–H7cov, where H1cov–H3cov denote reactions of POM with ligand Ltpy or its complexes; H4cov and H5cov denote reaction of H1cov with metal salts (so called step-by-step approach) and H6cov and H7cov denote one-pot reaction of all substrates (POM, Ltpy and metal ion salts).Synthesis of H1cov–H3cov hybrids
Synthesis of hybrids of H1cov–H3cov through direct reaction of amino-functionalized WD POM synthesis with Ltpy (H1cov) or its zinc complexes K1 (H2cov) and K2 (H3cov) was performed through heating of substrates under reflux and successfully confirmed by a series of analytical methods (ESI-MS, elementary analysis, FT-IR, 1H NMR spectroscopy, 31P NMR spectroscopy) and is described in detail in the Experimental section and ESI.† Comparison of 1H NMR spectra of respectivesubstrates and their products (H1cov – Fig. S24 and S25; H2cov – Fig. S26 and S27; H3cov – Fig. S28 and S29†) shows changes that are consistent with covalent bonding of the organic counterparts with the inorganic POM core (see also Fig. 2 of the aromatic region). FT-IR spectra (Fig. S4–S6†) also confirm the conducted reactions via the NH and CO frequency shifts but importantly, together with 31P NMR (Fig. S40–S42†) confirms that the WD POM retained its structural integrity. ESI-MS analyses also prove formation of the desired products. Interestingly, while the sole complex K1 shows the 1H NMR signature of the solution speciation between the 1:1 M:L [Zn(Ltpy)Cl2] and the 1:2 M:L [Zn(Ltpy)2]2+ complexes – which is also observed in the literature for other tpy systems62 – the H2cov hybrid does not exhibit such a character. The nBu4N+ cations inherent to the negatively charged POM species are also observed in the hybrid materials and integrated accordingly (6 for H1cov and H2cov, 12 for H3cov hybrid). These were demonstrated to be inter-exchangeable,63 which provides additional means for future functionalization.
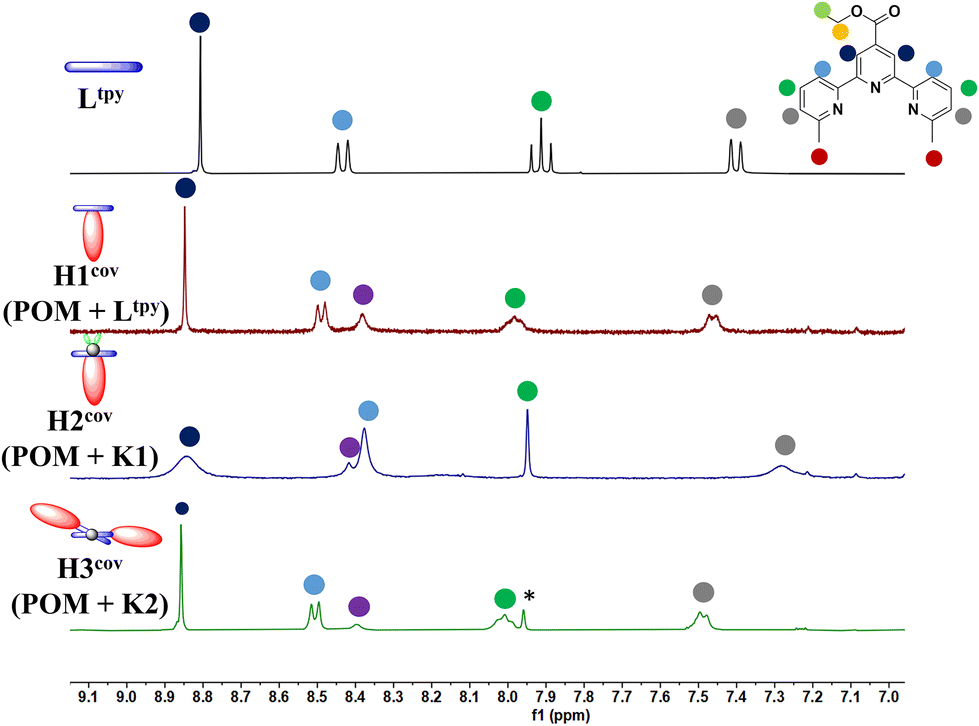 | ||
| Fig. 2 Comparison of aromatic region of 1H NMR spectra of Ltpy and hybrids: H1cov, H2cov and H3cov in d6-DMSO. Counterions are omitted for clarity. | ||
Synthesis of H4cov and H5cov hybrids
Synthesis of H4cov and H5cov hybrids was anticipated to form analogues of H2cov and H3cov hybrids respectively, attempted from H1cov hybrid i.e. system with Ltpy ligand covalently grafted onto the WD POM. While changes indicative of the molecular transformations were observed during the synthetic procedure, the isolation and characterization of hybrids (H4cov – Fig. S7, S18, S30, S31 and S43; H5cov – Fig. S8, S19, S32, S33 and S44†) showed that ZnCl2 and Zn(ClO4)2 salts are not readily coordinated in the tpy pocket, but lead to partial hydrolysis resulting in the exchange of nBu4N+ counteractions for the acidic protons – partial in the case of H4cov (2H+ and 4nBu4N+ remain ionically bound to the POM hybrid) and complete in the case of H5cov (12H+ and no nBu4N+ cations). What needs to be noted is the fact that in both cases the covalently bound tpy ligand remained grafted on the POM cluster and the polyoxometalate remained its plenary character. This is interesting, given the fact that in the presence of ionic hybrids,14 we did observe the possibility of retrieving H2ion and H3ion analogues via this synthetic route to the certain extent (H3ion perchlorates being more stable than the H2ion chloride analogues) and that H1ion hybrid was ultimately the most stable system.Synthesis of H6cov and H7cov hybrids
Synthesis of H6cov and H7cov hybrids was expected to form analogues of H2cov and H3cov hybrids, respectively, attempted in the form of the one-pot reaction i.e. from which means combination of both POM, ligand Ltpy and appropriate Zn(II) salt in one vessel. Isolation and characterization of products (H6cov – Fig. S9, S20, S34, S35 and S45; H7cov – Fig. S10, S21, S36, S37 and S46†) and comparison with previously synthesized samples (Fig. S38, S39 and S47†) allowed us to establish that covalent grafting of the tpy ligand was successful (per analogy to the H1cov hybrid) and zinc(II) cations did not coordinate to the N3 tridentate pocket but resulted in an almost complete counterion exchange from nBu4N+ cations to the acidic protons (H6cov – TBA(6−x)Hx; x = 5.75; H7cov – TBA(12−x)Hx; x = 11.5). This further confirms that in order for the covalent WD hybrids to retain Zn(II) ions in their molecular composition, one has to use the preformed complexes in the form of K1 or K2 compounds. As in previous instances, integrity of the WD POM counterpart was confirmed via 31P NMR (Fig. S47†). The remarkable stability of the H1cov and H1ion (previously demonstrated14) hybrids combined with excellent water solubility led us to focus on the domain of bioassays.Biological activities – general considerations
Polyoxometalates,64,65 organic multidentate ligands and their Zn(II) complexes57,66,67 were demonstrated in the literature to exhibit promising biological properties. Utilization of such materials in bioassays significantly depends on the functionalization method and stability of the system, yet these aspects were very scarcely studied in hybrid materials based on WD POM scaffold and remain unknown, and only relatively recently such a niche was highlighted upon the fact that organic functionalization of the POM inorganic clusters alleviates some of the drawbacks that are inherent to the sole POM within the context of biotoxicity.24 One of the primary cellular targets of drugs in cancer cells is the DNA present in the nuclei. Owing to the large size and the negative charge of both unfunctionalized POMs and DNA assemblies they are expected to rather repulse than interact between each other. It was however demonstrated that functionalization of POM clusters may reverse this trend or at least provide an alternative interaction path. For instance, Wang and co-workers68 have reported cyclopentadienyltitanium substituted polyoxotungstate [CoW11O39(CpTi)]7− (Cp = η5-C5H5) that is able to cleave the supercoiled DNA macrostructure. In addition, Dianat et al.69 showed K6H[CoW11O39CpZr], K6H[CoW11O39CpTi] and K7H2[CoW11O39CpFe] Keggin-type POMs, that tend to interact with the phosphate backbone of the DNA via electrostatic interactions.Herein, we aimed to elucidate the affinity of the hybrid compounds (ionic Hxion vs. covalent Hxcov) toward CT-DNA (deoxyribonucleic acid sodium salt from calf thymus), a model reagent used for routine DNA studies.70 Since the electrostatic character of Hxion–H1 (neutral), H2 (anionic) and H3 (anionic) is structure dependent, the interplay between them and DNA scaffold may be dissimilar to those previously described. At the same time, covalent functionalization of the POM hybrid can also be anticipated to help, especially since we did demonstrate previously that the silver(I) helical complexes of the related tpy ligand bind nucleic acids.59 Both types of hybrids reach equilibrium within ca. 60 min and remain stable in the buffered medium (Fig. S48 and S49†).49 Therefore, in order to probe the interactions between the synthesized hybrid materials H1ion, H2ion, H3ion, H1cov, H2cov, H3cov and also the corresponding complexes with the nucleic acid, a solution of the corresponding compound was titrated with CT-DNA samples.
Interaction of ionic hybrids with DNA
Significant changes in the shape of the spectra were observed for a series of CD-SEC hybrids H2ion and H3ion, contrary to the H1ion system, which remained unchanged (Fig. 3 and Fig. S50†). In particular, the MLCT bands at 345 nm for H2ion and 339 nm for H3ion characteristic for the CD-SEC entities disappeared. Simultaneously, new bands, peculiar for H1ion, at 257 nm and 290 nm appear. We surmise that this means that H2ion and H3ion undergo a structural transformation to H1ion in the presence of DNA (compare Fig. 3) with the binding constant Kb of 2.5 × 105 M−1 (Fig. S50†) through decomplexation mechanism. Further experiments confirmed that the substrate complexes K1ion and K2ion, in fact, interact with DNA what is manifested by the moderately high binding constants of Kb = 6.7 × 104 and 1.2 × 105 M−1, respectively (Fig. S51†). In case of both complexes, a hypsochromic shifts of ca. 10 nm of ILCT bands (ca. 295 nm) were observed. Changes in the intensity and position of the ligand-derived bands may suggest electrostatic interaction with CT-DNA. On the other hand, isosbestic points at ca. 305 nm as well as hypochromism of MLCT bands at ca. 345 nm are present and, together with the reported Kb values, indicate binding to DNA, possibly, by intercalation.71,72 This indicates that the H2ion and H3ion are able to rearrange themselves to H1ion. It also highlights the stability of the H1ion in a cellular-like environment, making it a potential drug delivery platform.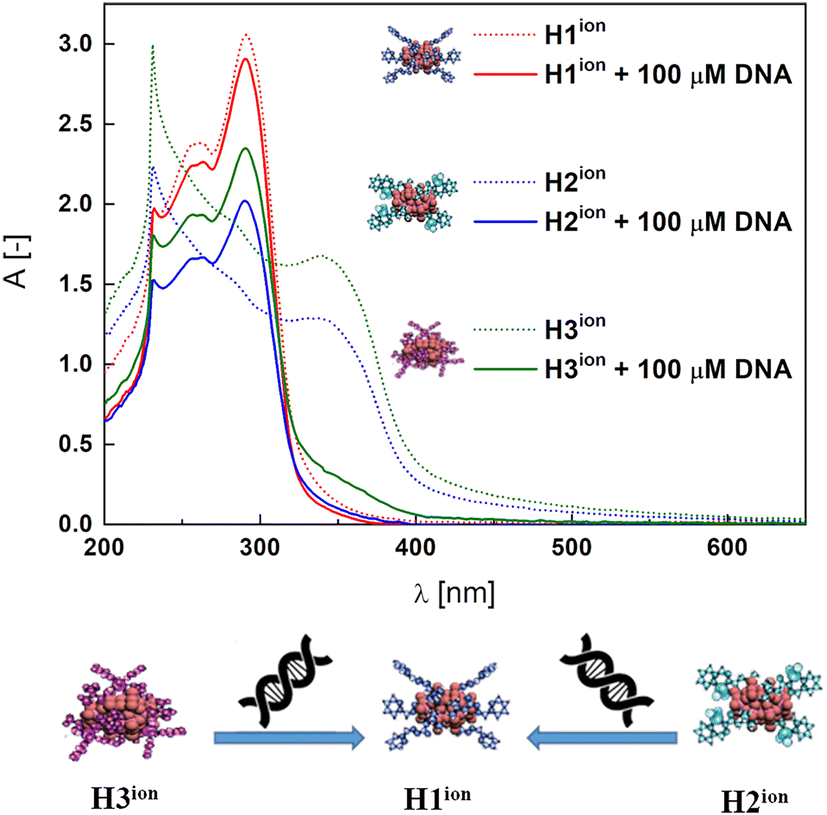 | ||
| Fig. 3 H1ion, H2ion and H3ion alone and the effect of CT-DNA addition. | ||
Potential drugs such as platinum complexes73 bound in SEC system as CD-SEC systems may be released in the presence of cellular DNA and then interact with its backbone via covalent binding thus inhibiting the cells vital processes as replication and transcription.
Interaction of covalent hybrids with DNA
It should be noted that in order to gain a deeper insight into the nature of the counterions present in the system and the changes associated to the POM transformations (Scheme 1a), a much larger number of substrates were tested for stability – including the V3-capped WD POM with varying countercations (K+ (V3POM-K8), nBu4N+ (V3POM-TBA), nBu4N+ functionalized with TRIZMA (V3POM + trizma)), including the Ltpy ligand and its K2 complex (K1 is very poorly soluble in aqueous media). It occurred that functionalization of the WD POM with TRIZMA renders the system more stable that its non-substituted counterparts V3POM-K8 and V3POM-TBA (Fig. S49†).The covalently linked H1cov–H3cov hybrids did not bind to the DNA scaffold (Fig. S51†). However, the substrate ligand Ltpy and K2 complex easily bound to DNA what is reflected in the high binding constants, respectively, 2.6 × 106 M−1 and 1.1 × 105 M−1 (Fig. 4 and Fig. S52†).
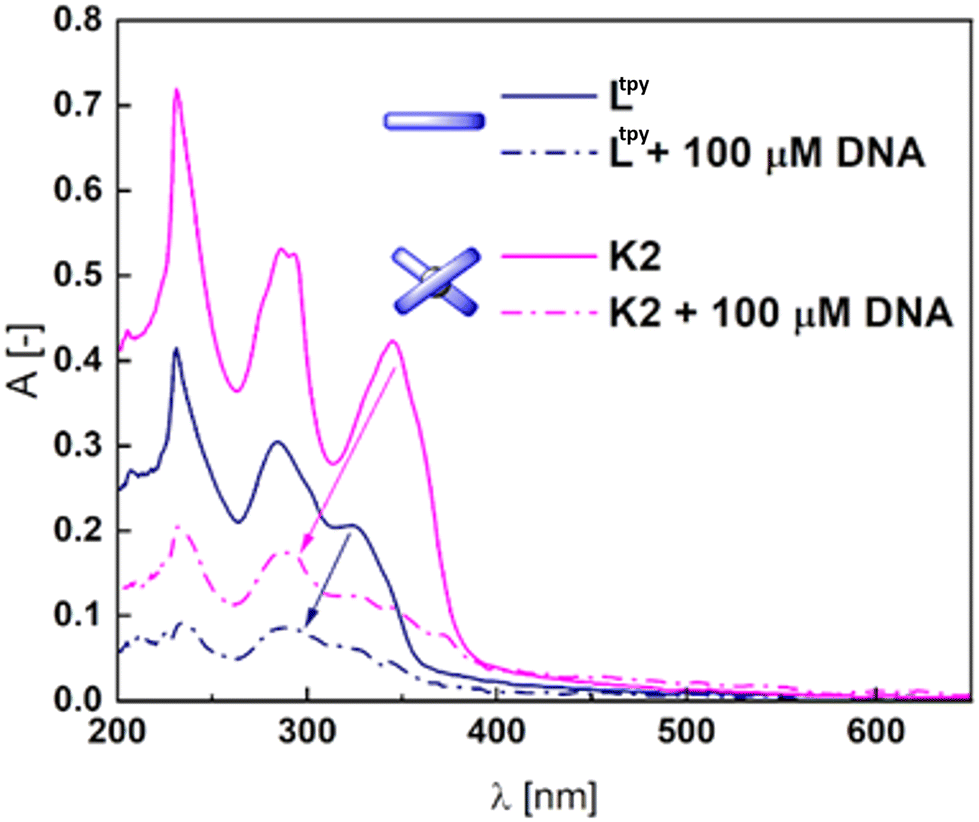 | ||
| Fig. 4 Ltpy and K2 alone and the effect of CT-DNA addition. Arrows indicate the trend of changes. | ||
Addition of DNA to both compounds caused significant hypochromic changes in the spectra implying the intercalation.71,72 K1 complex is insoluble in buffer with DMSO, therefore the Kb was not calculated. However, its derivatization with POM increased its solubility (Fig. S52†) and the structure similarity to Ltpy and K2 allows to surmise it could interfere with the DNA after the exchange of monovalent ligands. The stability of H1cov–H3cov hybrids in acidic (cf. synthesis) and normal cellular pH (7.4) opens a path to a controlled release of the ligand and complexes in basic pH in the duodenum and small intestine (pH ∼ 8). Basification of the acidic solutions of H1cov–H3cov (ca. 5.9) to pH ∼ 8.2–8.6 led to some morphological changes within 1 h in the UV spectra indicating slow decomposition of the hybrids in the POM region63 (Fig. 5 and Fig. S53†). Then, to the partially decomposed covalent hybrids, the DNA was added and some subtle changes in the band's intensity were detected (Fig. 5 and Fig. S53†). One can observe that the H2cov is more readily decomposed than H3cov probably due to the insolubility of K1 and the lower stability of the complexes of 1:1 ligand:metal ratio in comparison to 2:1 complexes as K2. We find it as evidence for the facilitation of the DNA binding process, possibly, due to the detaching of the POM octahedrons.63,74 Changes are less abrupt that in the case of Hxion hybrids (Fig. 3).
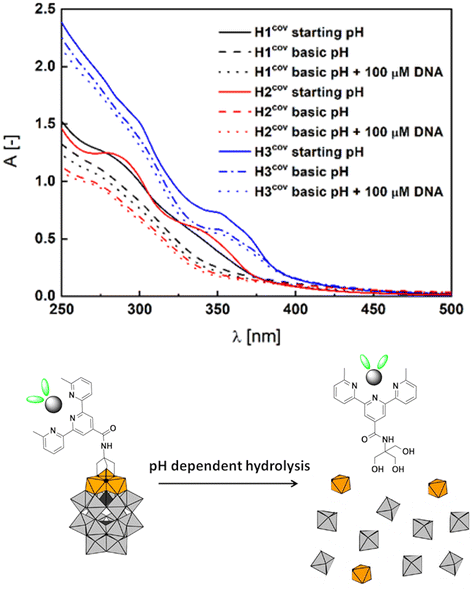 | ||
| Fig. 5 H1cov, H2cov and H3cov alone in miliQ water (starting pH ca. 5.9) and the effect of basification (up to pH ∼ 8.6) and CT-DNA addition. Scheme below – schematic illustration using the example of H2cov of pH dependent hydrolysis leading to the breakdown into basic inorganic building subunits. | ||
This means that depending on the character of the chosen hybrid (covalent vs. ionic) one can envisage those systems to become cargo-loading drug candidates, with functionalization mode ultimately affecting the stability and eventual pharmacotherapeutic effect – which was demonstrated for the anchored organic ligand/complex (interaction constant Kb = 2.5 × 105 M−1, insets in Fig. S50†). The ultimate mechanism is however different; for Hxion hybrids, the ionic interactions make it very fast system, where POM remains intact in its H1ion form and the disturbance of ionic interactions results in cargo-release of the appended molecules; for Hxcov hybrids the POM hydrolyses through the pH dependent mechanisms, therefore ligand/complex is released together with disintegration of WD POM into the basic inorganic building subunits (Fig. 4).
Conclusions
In summary, we have presented the effect of various synthetic methods on the synthesis of three different families of H1cov–H3cov hybrid materials. The first synthetic method involved the reaction of POM with the ligand Ltpy or its complexes: K1 or K2; the second synthetic method involved the reaction of H1cov with metal salts (step-by-step approach), and the third involved the reaction between POM, Ltpy and metal ion salts (one-pot approach). As a result, the desired hybrid materials were obtained using the first synthetic method and we have demonstrated for the first time how hybrid materials based on Wells–Dawson Polyoxometalates interact with DNA and how their functionalization mode (ionic/covalent) modulates the stability of the hybrid and the extent of DNA binding. This was done by comparing the ionic Hxion hybrids synthesized previously14 and newly prepared covalent Hxcov hybrids. Protocols for the latter ones were also developed, giving therefore different means for their functionalization and demonstrating which molecular parts of the hybrids are amenable to further synthetic changes. Specifically, the acidic character of the zinc(II) salts is responsible for part of those transformations.In the presence of DNA, the synthesized ionic and covalent hybrids undergo reorganizations, which in both instances lead to the release of the bound ligands/complexes, nonetheless the ultimate mechanism is different. For Hxion ionic hybrids, fast elimination of the coordinated zinc(II) ions/ligands from H2ion and H3ion hybrids is observed, concomitant with transformation to the H1ion hybrid due to the significant stability of the latter one. For Hxcov covalent hybrids, slow pH mediated hydrolysis of the polyoxometalate was observed, which leads to release of the grafted ligand/complex to the surrounding aqueous medium. High binding constant of terpyridine ligand and its complex [Zn(Ltpy)2](ClO4)2 to the DNA was also demonstrated. This overall shows that the WD POM has the potential to act as the cargo-loading system, which we anticipate to study further.
Author contributions
Daria Nowicka: investigation, methodology, formal analysis, data curation, visualization, writing – review & editing, funding acquisition. Dawid Marcinkowski: investigation, methodology, formal analysis, data curation, visualization, writing – review & editing, funding acquisition. Nahir Vadra: investigation, methodology, formal analysis, data curation, visualization, writing – review & editing. Martyna Szymańska: investigation, methodology, formal analysis, data curation, visualization, writing – review & editing. Maciej Kubicki: formal analysis, investigation, data curation, writing – review & editing, visualization. Giuseppe Consiglio: conceptualization, visualization, writing – review & editing. Wojciech Drożdż: investigation, formal analysis, data curation, writing – review & editing. Artur R. Stefankiewicz: resources, visualization, writing – review & editing. Violetta Patroniak: supervision, resources, writing – review & editing, funding acquisition. Marta Fik-Jaskółka: investigation, conceptualization, methodology, writing – review & editing, project administration, supervision, resources, funding acquisition. Adam Gorczyński: conceptualization, methodology, writing – review & editing, project administration, supervision, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We express gratitude to Dr Aleksandra Bocian for help in synthesis of ionic hybrids H1ion–H7ion. This work was supported by the National Science Centre, Poland (grant numbers UMO-2022/45/N/ST4/00632 (D. N.), UMO-2022/45/N/ST4/00344 (D. M.), UMO-2019/32/C/ST4/00565 (W. D.), UMO-2022/47/B/ST4/02310 (V. P.)). Daria Nowicka is a scholarship holder of the Adam Mickiewicz University in Poznan Foundation for the academic year 2023/2024. Adam Gorczyński is a scholarship holder of the Polish Ministry of Education and Science for outstanding young scientists. MFJ: the work was supported by IDUB-UAM (project no. 038/04/NŚ/0023). This work was also supported by the University of Catania, PIACERI 2020/2022, Linea di Intervento 2.References
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- S. Lentink, D. E. Salazar Marcano, M. A. Moussawi and T. N. Parac-Vogt, Angew. Chem., Int. Ed., 2023, 62, e202303817 CrossRef PubMed.
- X.-K. Lian, H.-B. Chen, Y.-D. Lin, X.-X. Li and S.-T. Zheng, Coord. Chem. Rev., 2023, 497, 215440 CrossRef CAS.
- L. Vilà-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 17054 CrossRef.
- L. Cronin, Ann. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2004, 100, 323–383 RSC.
- L.-L. Liu, L. Wang, X.-Y. Xiao, P. Yang, J. Zhao and U. Kortz, Coord. Chem. Rev., 2024, 506, 215687 CrossRef CAS.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596–612 CrossRef CAS PubMed.
- H. N. Miras, J. Yan, D.-L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403–7430 RSC.
- V. Duros, J. Grizou, W. Xuan, Z. Hosni, D.-L. Long, H. N. Miras and L. Cronin, Angew. Chem., Int. Ed., 2017, 56, 10815–10820 CrossRef CAS PubMed.
- D. S. Salley, G. A. Keenan, D. L. Long, N. L. Bell and L. Cronin, ACS Cent. Sci., 2020, 6, 1587–1593 CrossRef CAS PubMed.
- A. V. Anyushin, S. Vanhaecht and T. N. Parac-Vogt, Inorg. Chem., 2020, 59, 10146–10152 CrossRef CAS PubMed.
- Q. Liu and X. Wang, Matter, 2020, 2, 816–841 CrossRef.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- A. Bocian, W. Drożdż, M. Szymańska, J. Lewandowski, M. Fik-Jaskółka, A. Gorczyński, V. Patroniak and A. R. Stefankiewicz, Nanoscale, 2020, 12, 4743–4750 RSC.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- S. Li, Y. Ma, Y. Zhao, R. Liu, Y. Zhao, X. Dai, N. Ma, C. Streb and X. Chen, Angew. Chem., Int. Ed., 2023, 62, e202314999 CrossRef CAS PubMed.
- M. Yang, X. Wang, C. J. Gómez-García, Z. Jin, J. Xin, X. Cao, H. Ma, H. Pang, L. Tan, G. Yang and Y. Kan, Adv. Funct. Mater., 2023, 33, 2214495 CrossRef CAS.
- X. Chen, C. Ma, Z. Tan, X. Wang, X. Qian, X. Zhang, J. Tian, S. Yan and M. Shao, Chem. Eng. J., 2022, 433, 134504 CrossRef CAS.
- J.-J. Chen, M. D. Symes and L. Cronin, Nat. Chem., 2018, 10, 1042–1047 CrossRef CAS PubMed.
- A. Bijelic and A. Rompel, Acc. Chem. Res., 2017, 50, 1441–1448 CrossRef CAS PubMed.
- Y. Martinetto, B. Pégot, C. Roch-Marchal, B. Cottyn-Boitte and S. Floquet, Eur. J. Inorg. Chem., 2020, 2020, 228–247 CrossRef CAS.
- S.-M. Wang, J. Hwang and E. Kim, J. Mater. Chem. C, 2019, 7, 7828–7850 RSC.
- U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315–2327 CrossRef CAS.
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327–358 CrossRef CAS PubMed.
- B. Hasenknopf, Front. Biosci., 2005, 10, 275–287 CrossRef CAS PubMed.
- M. Arefian, M. Mirzaei, H. Eshtiagh-Hosseini and A. Frontera, Dalton Trans., 2017, 46, 6812–6829 RSC.
- T. Liu, Langmuir, 2010, 26, 9202–9213 CrossRef CAS PubMed.
- P. Gao, Y. Wu and L. Wu, Soft Matter, 2016, 12, 8464–8479 RSC.
- S. Vanhaecht, G. Absillis and T. N. Parac-Vogt, Dalton Trans., 2012, 41, 10028–10034 RSC.
- K.-i. Shimizu, H. Furukawa, N. Kobayashi, Y. Itaya and A. Satsuma, Green Chem., 2009, 11, 1627–1632 RSC.
- J. Zhang, Trends in Polyoxometalates Research, 2015 Search PubMed.
- A. Flütsch, T. Schroeder, M. G. Grütter and G. R. Patzke, Bioorg. Med. Chem. Lett., 2011, 21, 1162–1166 CrossRef PubMed.
- K. Nomiya, H. Torii, T. Hasegawa, Y. Nemoto, K. Nomura, K. Hashino, M. Uchida, Y. Kato, K. Shimizu and M. Oda, J. Inorg. Biochem., 2001, 86, 657–667 CrossRef CAS PubMed.
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169 RSC.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980–2999 CrossRef CAS PubMed.
- L. Fu, H. Gao, M. Yan, S. Li, X. Li, Z. Dai and S. Liu, Small, 2015, 11, 2938–2945 CrossRef CAS PubMed.
- D. Li, P. Yin and T. Liu, Dalton Trans., 2012, 41, 2853–2861 RSC.
- X. Wang, J. Liu and M. Pope, Dalton Trans., 2003, 957–960 RSC.
- S. K. Petrovskii, E. V. Grachova and K. Y. Monakhov, Chem. Sci., 2024, 15, 4202–4221 RSC.
- R. Contant and A. Tézé, Inorg. Chem., 1985, 24, 4610–4614 CrossRef CAS.
- B. J. Hornstein and R. G. Finke, Inorg. Chem., 2002, 41, 2720–2730 CrossRef CAS PubMed.
- R. G. Finke, B. Rapko, R. J. Saxton and P. J. Domaille, J. Am. Chem. Soc., 1986, 108, 2947–2960 CrossRef CAS.
- C. P. Pradeep, D.-L. Long, G. N. Newton, Y.-F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4388–4391 CrossRef CAS PubMed.
- G. Ulrich, S. Bedel, C. Picard and P. Tisnès, Tetrahedron Lett., 2001, 42, 6113–6115 CrossRef CAS.
- CrysAlis PRO (Version 1.171.38.41), 2015.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- J. Skiba, T. Bernaś, D. Trzybiński, K. Woźniak, G. Ferraro, D. Marasco, A. Merlino, M. Z. Shafikov, R. Czerwieniec and K. Kowalski, Molecules, 2017, 22, 809 CrossRef PubMed.
- L. Shivakumar, K. Shivaprasad and H. D. Revanasiddappa, Spectrochim. Acta, Part A, 2012, 97, 659–666 CrossRef CAS PubMed.
- G. Izzet, B. Abécassis, D. Brouri, M. Piot, B. Matt, S. A. Serapian, C. Bo and A. Proust, J. Am. Chem. Soc., 2016, 138, 5093–5099 CrossRef CAS PubMed.
- B. Matt, C. Coudret, C. Viala, D. Jouvenot, F. Loiseau, G. Izzet and A. Proust, Inorg. Chem., 2011, 50, 7761–7768 CrossRef CAS PubMed.
- E. Hampson, J. M. Cameron, S. Amin, J. Kyo, J. A. Watts, H. Oshio and G. N. Newton, Angew. Chem., 2019, 131, 18449–18453 CrossRef.
- E. Hampson, J. M. Cameron, J. A. Watts and G. N. Newton, Chem. Commun., 2020, 56, 8237–8240 RSC.
- M. Zynek, M. Serantoni, S. Beloshapkin, E. Dempsey and T. McCormac, Electroanalysis, 2007, 19, 681–689 CrossRef CAS.
- R. I. Nooney, D. Thirunavukkarasu, Y. Chen, R. Josephs and A. E. Ostafin, Chem. Mater., 2002, 14, 4721–4728 CrossRef CAS.
- W. Drożdż, A. Walczak, Y. Bessin, V. Gervais, X.-Y. Cao, J.-M. Lehn, S. Ulrich and A. R. Stefankiewicz, Chem. – Eur. J., 2018, 24, 10802–10811 CrossRef PubMed.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- M. A. Fik, A. Gorczyński, M. Kubicki, Z. Hnatejko, A. Fedoruk-Wyszomirska, E. Wyszko, M. Giel-Pietraszuk and V. Patroniak, Eur. J. Med. Chem., 2014, 86, 456–468 CrossRef CAS PubMed.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- M. Najafi, ChemCatChem, 2023, 15, e202201045 CrossRef CAS.
- M. Wałęsa-Chorab, A. R. Stefankiewicz, D. Ciesielski, Z. Hnatejko, M. Kubicki, J. Kłak, M. J. Korabik and V. Patroniak, Polyhedron, 2011, 30, 730–737 CrossRef.
- D. E. Salazar Marcano, S. Lentink, M. A. Moussawi and T. N. Parac-Vogt, Inorg. Chem., 2021, 60, 10215–10226 CrossRef CAS PubMed.
- N. I. Gumerova and A. Rompel, Inorg. Chem., 2021, 60, 6109–6114 CrossRef CAS PubMed.
- L. S. Van Rompuy and T. N. Parac-Vogt, Curr. Opin. Biotechnol., 2019, 58, 92–99 CrossRef CAS PubMed.
- W. Drożdż, Y. Bessin, V. Gervais, X. Y. Cao, J. M. Lehn, A. R. Stefankiewicz and S. Ulrich, Chem. – Eur. J., 2018, 24, 1518–1521 CrossRef PubMed.
- F. Nachon, X. Brazzolotto, J. Dias, C. Courageux, W. Drożdż, X.-Y. Cao, A. R. Stefankiewicz and J.-M. Lehn, ChemBioChem, 2022, 23, e202200456 CrossRef CAS PubMed.
- X. Wang, J. Liu, J. Li, Y. Yang, J. Liu, B. Li and M. T. Pope, J. Inorg. Biochem., 2003, 94, 279–284 CrossRef CAS PubMed.
- S. Dianat, A. K. Bordbar, S. Tangestaninejad, B. Yadollahi, S. H. Zarkesh-Esfahani and P. Habibi, J. Photochem. Photobiol., B, 2013, 124, 27–33 CrossRef CAS PubMed.
- M. Szymańska, M. Kubicki, G. N. Roviello, G. Consiglio, M. A. Fik-Jaskółka and V. Patroniak, Dalton Trans., 2022, 51, 15648–15658 RSC.
- S. Tabassum, S. Amir, F. Arjmand, C. Pettinari, F. Marchetti, N. Masciocchi, G. Lupidi and R. Pettinari, Eur. J. Med. Chem., 2013, 60, 216–232 CrossRef CAS PubMed.
- A. A. Almaqwashi, T. Paramanathan, I. Rouzina and M. C. Williams, Nucleic Acids Res., 2016, 44, 3971–3988 CrossRef CAS PubMed.
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- N. I. Gumerova and A. Rompel, Chem. Soc. Rev., 2020, 49, 7568–7601 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic analysis of synthesized hybrids and chosen biological spectra. CCDC 2078396. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00965g |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |