
Atomic-level design of metalloenzyme-like active pockets in metal–organic frameworks for bioinspired catalysis
Weiqing
Xu
a,
Yu
Wu
a,
Wenling
Gu
a,
Dan
Du
 b,
Yuehe
Lin
*b and
Chengzhou
Zhu
*a
b,
Yuehe
Lin
*b and
Chengzhou
Zhu
*a
aNational Key Laboratory of Green Pesticide, International Joint Research Center for Intelligent Biosensing Technology and Health, College of Chemistry, Central China Normal University, Wuhan 430079, P. R. China. E-mail: czzhu@ccnu.edu.cn
bSchool of Mechanical and Materials Engineering, Washington State University, 99164, Pullman, USA. E-mail: yuehe.lin@wsu.edu
First published on 29th November 2023
Abstract
Natural metalloenzymes with astonishing reaction activity and specificity underpin essential life transformations. Nevertheless, enzymes only operate under mild conditions to keep sophisticated structures active, limiting their potential applications. Artificial metalloenzymes that recapitulate the catalytic activity of enzymes can not only circumvent the enzymatic fragility but also bring versatile functions into practice. Among them, metal–organic frameworks (MOFs) featuring diverse and site-isolated metal sites and supramolecular structures have emerged as promising candidates for metalloenzymes to move toward unparalleled properties and behaviour of enzymes. In this review, we systematically summarize the significant advances in MOF-based metalloenzyme mimics with a special emphasis on active pocket engineering at the atomic level, including primary catalytic sites and secondary coordination spheres. Then, the deep understanding of catalytic mechanisms and their advanced applications are discussed. Finally, a perspective on this emerging frontier research is provided to advance bioinspired catalysis.
 Weiqing Xu | Weiqing Xu received her BS Degree (2018) from Jiangxi Agricultural University. She is currently a PhD candidate at Central China Normal University under the supervision of Prof. Chengzhou Zhu. Her research interests are focused on MOF-based nanobiocatalysis and biosensing. |
 Yuehe Lin | Dr Yuehe Lin is a Professor at Washington State University, working on the development of new functional materials and integrated devices for biomedical applications. He has more than 500 peer-reviewed publications with total citations over 80 000 and H-index 145. He has been named among the world's most highly cited researchers every year from 2014 to 2021 by the Web of Science Group. Dr Lin is a fellow of the National Academy of Inventors, the American Association for the Advancement of Science, the Royal Society of Chemistry, the Electrochemical Society, and the American Institute of Medical and Biological Engineering as well as an elected member of the Washington State Academy of Sciences. |
 Chengzhou Zhu | Dr Chengzhou Zhu is currently a full professor at Central China Normal University. He received his PhD degree from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. Since then, he worked as a Humboldt Research Fellow at Dresden University of Technology and then joined Washington State University as an assistant research professor. His scientific interests focus on atomically dispersed materials for nanobiocatalysis and biosensing. He was listed as a highly cited researcher on Clarivate Analytics's lists. He serves as an Associate Editor of Advanced Agrochem, Frontiers in Bioengineering and Biotechnology, Editorial Board Members of Analytica Chimica Acta, Biosensors, and other international journals. He is a Fellow of Royal Society of Chemistry. |
1. Introduction
Metalloenzymatic catalysis has had a transformative impact on industrial production, playing a crucial role in pharmaceutical synthesis, environmental protection, and energy fields.1–3 The sophisticated conformational structure and composition in active pockets endow enzymes with unparalleled reactivity and specificity, leading to efficient chemical transformations.4,5 Although some biological strategies have been honed to engineer catalytic sites to match the increasingly challenging biocatalysis, the operation is very challenging and costly. Additionally, the complexity and vulnerability of enzymatic structures strongly hinder the mechanism exploration and practical industrial applications.6,7 To this end, much effort has been devoted to developing biomimetic nanocatalysts to serve as promising alternatives to metalloenzymes.8–11 Their superior operability and stability not only aid in overcoming the above limitations to meet the needs of practical applications but also bring in opportunities beyond natural enzymes.12–14 It should be noted that the well-defined structure of active centers is conducive to the investigation of the catalytic mechanisms, which in turn deepens the understanding of the nature of enzymatic reactions.Aiming at vividly mimicking metalloenzymes, the construction of artificial metalloenzymes requires essential metal sites and suitable adjacent environments to form active pockets. In this case, metal–organic frameworks (MOFs), which consist of metal nodes and organic linkers forming the ordered periodic networks, are regarded as the ideal candidates, allowing exploration of their enzyme-like performance.15–17 First, the atomically dispersed metal sites and diversified linkers can be easily customized and functionalized to accurately reproduce the primary active sites of metalloenzymes.18 MOF-based biomimetic catalysts possess high-density metal active centers, as well as enable circumvention of the inactivation owing to contact and dimerization of the homogeneous catalysts. Second, the hierarchical architectures and non-covalent interactions in channels endow MOFs with enzyme-like secondary coordination spheres, which are involved in substrate binding, electron/proton transfer, mass transfer, etc. to directly influence the reactivity and selectivity.19,20 Additionally, using the unique photo, electric, and thermal effects of MOFs,21,22 the catalytic behaviour of active sites can be further regulated, which even surpasses the functions of enzymes for realizing challenging chemical transformations.
The rapid development of materials science and the advancement of in situ characterization technologies have fueled the efficient development of MOF-based biomimetic catalysts and the exploration of the underlying mechanisms. Recently, several outstanding reviews presented the progress of MOF-based biocomposites,23–27 while most of them focused on the functions of MOFs as carriers and/or simulating the biocatalytic performance of metalloenzymes by engineering metal sites. Note that the precise design of active pockets at the atomic level, including primary metal sites and secondary coordination spheres, and the merits of heterogeneous catalysts have been overlooked.
Herein, we aim to present state-of-the-art research on the atomic-level design of metalloenzyme-like active pockets over MOFs for efficient bioinspired catalysis in this review (Fig. 1). Drawing inspiration from enzymes, we begin with the design of site-isolated metal ions and various organic linkers to precisely mimic primary active sites. Subsequently, the modulation of secondary coordination spheres in MOFs is emphasized to match the role of enzymatic active pockets and shed light on the fundamental structure–performance relationship. Moreover, the effect of supramolecular architectures and external stimuli on proximal catalysis is systematically discussed. We then will summarize their advanced bioinspired catalytic applications and highlight their advantages over enzymatic catalysis. Finally, a personal outlook on future opportunities and challenges in bioinspired MOF-based catalysis is proposed. We anticipate that this review will contribute to the advancement of artificial catalysis.
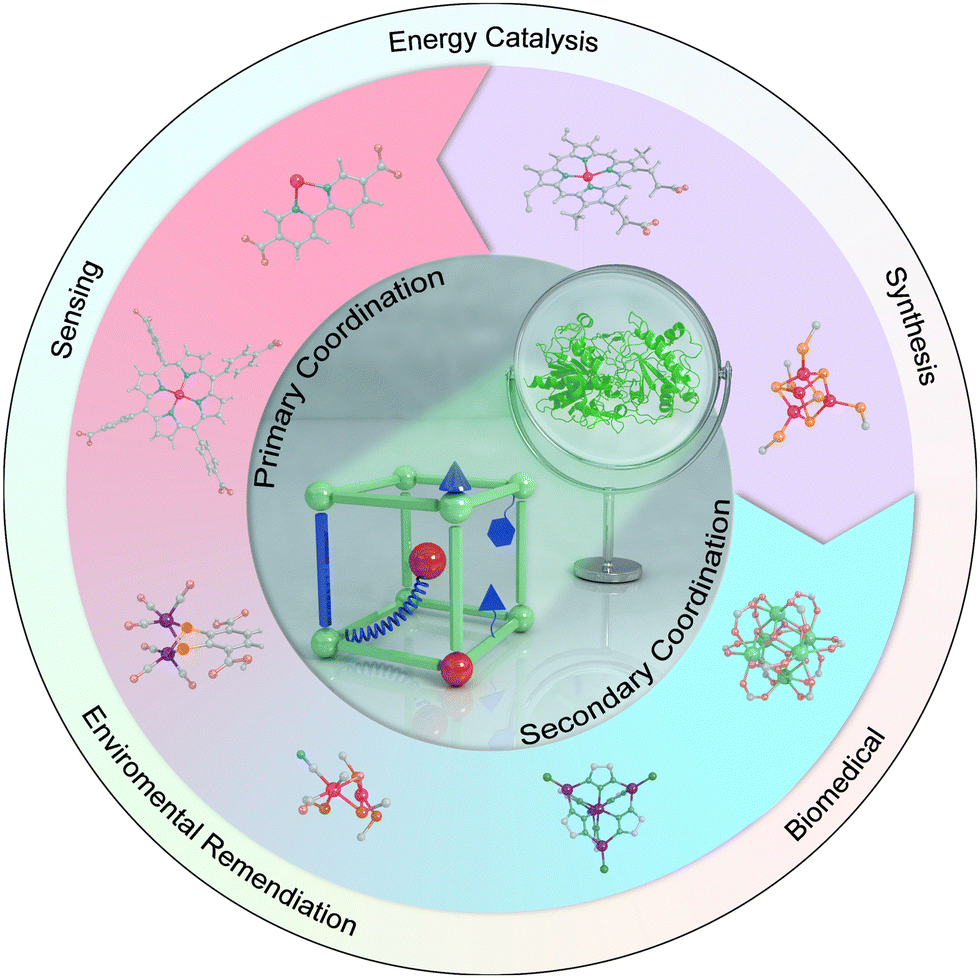 | ||
| Fig. 1 Representative atomic-level design of metalloenzyme-like active pockets over MOFs for various catalytic applications. | ||
2. Designing bioinspired primary metal active sites
In nature, metalloenzymes strongly depend on metal cofactors to drive the chemical transformations necessary for life.28 Each metal ion in enzymes is a natural selection and underpins a specific role in catalysis and function. Of all metalloenzymes, the type of metal cofactors mainly includes monometallic sites (such as heme-containing oxygenase, NiS4-involved dehydrogenases, and Zn–OH center of metallohydrolase) and multimetallic clusters (such as multicopper oxidases, [Fe4S4] and [FeMo] nitrogenase, [NiFe] and [Fe–Fe] hydrogenase, etc.) (Fig. 2). The diverse and spatially isolated metal ions offer MOFs to vividly mimic the catalytic sites at the atomic scale. The construction of enzyme-like active pockets over MOFs is focused on two main building blocks: secondary building units (SBUs) and organic linkers. On the one hand, various metal species and ligands (e.g., TCPP, imidazolate, thiolate, and so on) endow biomimetic MOFs to directly recapitulate the metal coordination structures of metalloenzymes (Fig. 3). On the other hand, the easy tunable building blocks not only allow open sites to anchor single metal sites but also introduce diverse substituents for grafting active centers. The high-density active sites over MOFs provide rich catalytic units with enzyme-like weak ligand field effects for stunning reproduction of catalytic activity. Notably, the design of biomimetic catalysts is not just limited to precisely replicating the metal active units of enzymes within MOFs. Employing other metals such as, but not limited to, transition metals and noble metals, to construct structural analogues of active pockets is capable of replicating the enzymatic catalytic behaviours. It is anticipated to broaden the type of bioinspired catalysts for the development of advanced enzyme mimics. In addition, bioinspired MOFs integrate the merits of molecular and supramolecular catalysts. The porous frameworks with confinement effects protect active sites from aggregation and against external stimulates, endowing MOF-based catalysts with excellent stability. Furthermore, the unique physicochemical properties (photo and electro effects) of MOFs afford biomimetic catalysts with tunable catalytic performance. In this section, the construction of enzyme-like metal cofactors within MOFs, including monometallic sites and multimetallic clusters, and their underlying catalytic mechanisms are introduced.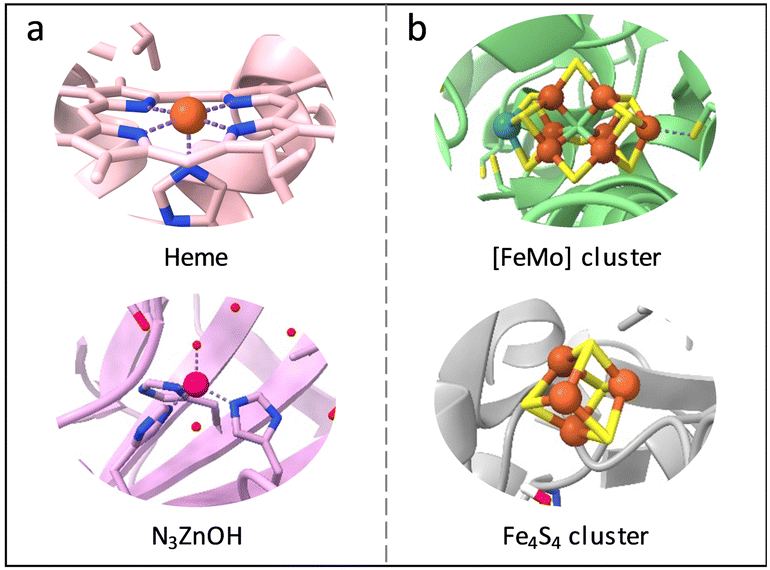 | ||
| Fig. 2 Schematic illustration of the (a) monometallic sites and (b) multimetallic clusters in different metalloenzymes. Orange: Fe, blue: N, purple: Zn, red: O, cyan: Mo, yellow: S. | ||
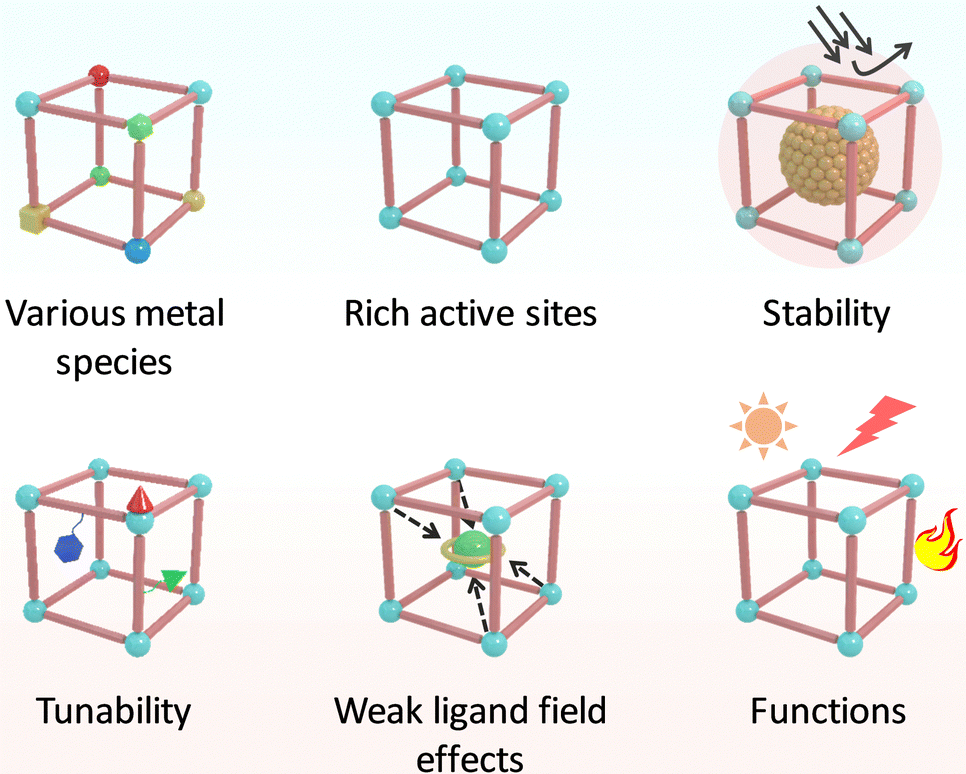 | ||
| Fig. 3 Schematic illustration of the advantages of MOFs in replicating the active center of metalloenzymes. | ||
2.1. Engineering monometallic sites
Heme-containing oxygenases (cytochrome P450, peroxidase (POD)) are a series of ubiquitous metalloenzymes in organisms that catalyze various metabolic reactions for promoting the oxidative transformation of drugs, xenobiotics, and steroids.29 For the active pocket, the iron–porphyrin motif underpins the substrate binding and activation. Specifically, the ground state of Fe-porphyrin is transformed into highly reactive FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species in the presence of O2 or H2O2, affording an efficient oxygenation reaction.30,31 Inspired by this, researchers have developed various molecular analogues of heme.32,33 The proposed metalloporphyrin complexes featuring well-defined coordination structures are beneficial for uncovering the structure–function relationships and deepening understanding of the catalytic mechanism. Nevertheless, due to the lack of confinement effect of enzyme-like supramolecular framework structures, these small molecule complexes can easily contact each other for dimerization and inactivation, as well as suffer from the invasion of extreme stimulates, showing insufficient stability in practice.34 In this regard, porphyrin-based MOFs composed of porphyrins linkers and metal nodes not only inherit the biological functions of porphyrins, including light-harvesting, electron transfer, oxygen transportation, and catalysis,35 but also exhibit spatially isolated sites and good stability to overcome above limits. Zhou's group adopted the Fe-porphyrin-containing tetracarboxylate (Fe-TCPP) ligand coordination with Zr4+ to synthesize a stable three-dimensional (3D) PCN-222(Fe) (Fig. 4a).36 Combined with the open mesoporous channel (3.7 nm), the high-density heme-like sites recapitulate the POD catalytic activity for the efficient oxidization of various substrates with the help of H2O2. Apart from Fe centers, other transition metal (Co,Mn,Cu)-TCPP linkers have been used to construct diversified biomimetic MOFs and show favourable enzyme-like activities.37–39 For example, a Co-TCPP linker and a Cu2(COO)4 paddle-wheel cluster were assembled to create 2D Cu-TCPP(Co) nanosheets.40 Compared with bulk MOFs, the obtained 2D MOFs have a larger surface area, which facilitates the exposure of catalytic sites and accelerates transfer mass for boosted POD-like catalytic performance.
O species in the presence of O2 or H2O2, affording an efficient oxygenation reaction.30,31 Inspired by this, researchers have developed various molecular analogues of heme.32,33 The proposed metalloporphyrin complexes featuring well-defined coordination structures are beneficial for uncovering the structure–function relationships and deepening understanding of the catalytic mechanism. Nevertheless, due to the lack of confinement effect of enzyme-like supramolecular framework structures, these small molecule complexes can easily contact each other for dimerization and inactivation, as well as suffer from the invasion of extreme stimulates, showing insufficient stability in practice.34 In this regard, porphyrin-based MOFs composed of porphyrins linkers and metal nodes not only inherit the biological functions of porphyrins, including light-harvesting, electron transfer, oxygen transportation, and catalysis,35 but also exhibit spatially isolated sites and good stability to overcome above limits. Zhou's group adopted the Fe-porphyrin-containing tetracarboxylate (Fe-TCPP) ligand coordination with Zr4+ to synthesize a stable three-dimensional (3D) PCN-222(Fe) (Fig. 4a).36 Combined with the open mesoporous channel (3.7 nm), the high-density heme-like sites recapitulate the POD catalytic activity for the efficient oxidization of various substrates with the help of H2O2. Apart from Fe centers, other transition metal (Co,Mn,Cu)-TCPP linkers have been used to construct diversified biomimetic MOFs and show favourable enzyme-like activities.37–39 For example, a Co-TCPP linker and a Cu2(COO)4 paddle-wheel cluster were assembled to create 2D Cu-TCPP(Co) nanosheets.40 Compared with bulk MOFs, the obtained 2D MOFs have a larger surface area, which facilitates the exposure of catalytic sites and accelerates transfer mass for boosted POD-like catalytic performance.
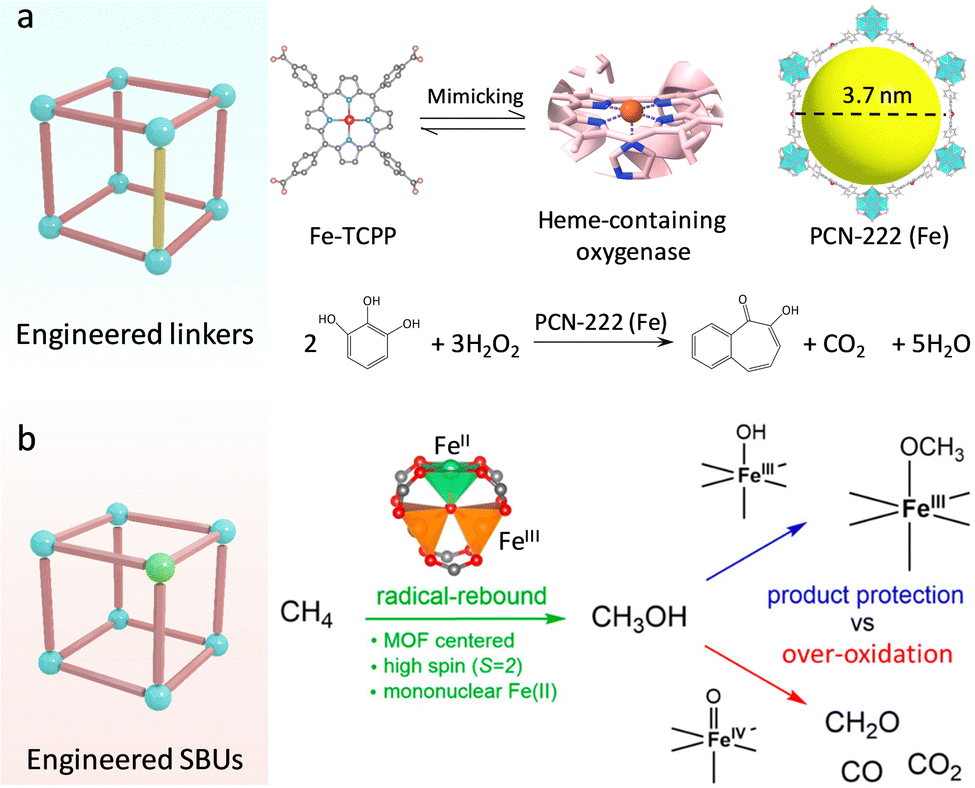 | ||
| Fig. 4 (a) Engineering the linkers over MOFs. Schematic illustration of the building blocks of PCN-222(Fe) to mimic the heme active sites for catalyzing substrate oxidation. (b) Engineering the SBUs over MOFs. Schematic representation of the different catalytic processes for methanol oxidation. Reproduced with permission from ref. 42. Copyright 2021, American Chemistry Society. | ||
In attempts to replicate the skyscraping catalytic ability of enzymes, researchers also focus on designing Fe-containing MOFs, in which Fe sites can be activated to FeIVO and FeIII–OH species for underpinning various important monooxygenase reactions.41 For instance, Bhan's group constructed Fe-based MOFs (MIL-100(Fe)) and created mononuclear FeII sites in the SBUs by using in vacuo thermal treatment (Fig. 4b).42 Similar to nature, the isolated FeII site with a weak ligand field conferred by the organic carboxylate linker presents a high-spin state (S = 2), which can be efficiently converted to FeIVO moieties for C–H bond hydroxylation in methane (CH4). This activation reaction follows the typical radical-rebound mechanism to generate methanol (CH3OH). Notably, the FeIII–OH intermediate can react with CH3OH to form stable FeIII–OCH3 species through the hydrogen atom transfer (HAT) process, inhibiting the overoxidation of CH3OH by FeIVO species. What's more, the proximal zeolites enable dehydration and protection of the diffused CH3OH gas, resulting in enhanced catalytic selectivity. Motivated by this, the researchers directly prepared a mononuclear FeIII–OH site and combined it with redox-active sites and photosensitive units to borrow solar energy for efficient methane photo-oxidation.43 The photosensitizer [RuII(BPy)2(BPyDC)] (Ru-PS) and redox-active polyvanadotungstate [PW9V3O40]6− were in situ immobilized within a porous UiO-67, and then introduced an isolated Fe center on the linker (2,2′-bipyridine-5,5′-dicarboxylicacid, H2BPyDC) by post-synthetic metalation strategy to afford PMOF-RuFe(Cl). Before the catalysis of CH4, the Fe–Cl site was activated to generate highly active FeIII–OH sites by light illumination in an aqueous solution. The photo-reduced PW9V3IV/V with rich electron density can induce the photoreduction of O2 to H2O2via the proton-coupled electron transfer (PCET) process. H2O2 can be readily catalyzed to hydroxyl radicals (˙OH) by FeIII–OH species. Besides, the confined FeIII–OH sites bind with CH4 to form [Fe–OH⋯CH4] intermediates, which are in favour of the coupling with ˙OH to produce CH3OH. In this work, the light-driven oxidation process circumvents the activation conditions of high temperature and pressure. Markedly, light illumination quenches ˙OOH intermediates to suppress the overoxidation of products, realizing 100% selectivity catalysis and an outstanding CH3OH yield of ∼8.81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mmol gcat−1 h−1, which is superior to methane monooxygenase (5.05mmol gcat−1 h−1).
mmol gcat−1 h−1, which is superior to methane monooxygenase (5.05mmol gcat−1 h−1).
Apart from transition metals, a lot of noble metals (e.g., Pt, Ru, Au, etc.) have been reported to simulate the catalytic behaviour of enzymes.44–46 As an example, a single-atom Ru-modified H2BPyDC linker over UiO-67 was reported by our group (Fig. 5a).47 By integrating the photoelectric effect and POD-like activity, the as-prepared UiO-67-Ru realizes a 7.0-fold enhancement in catalytic activity with the help of illumination (Fig. 5b). The UiO-67-Ru system presents lower production of ˙OH than that of the control group (Fig. 5c), which is different from the traditional photo-involved free radical activation process. Further analysis revealed that the photoelectrons can induce the electronic structure redistribution of Ru sites by following the cofactor-mediated electron transfer (ET) process. The excited Ru center facilitates the production of RuO species to perform the high catalytic performance of FeIVO species. Besides, this enzyme-like activation process prevents many side reactions conferred by free radicals, showing good catalytic specificity.
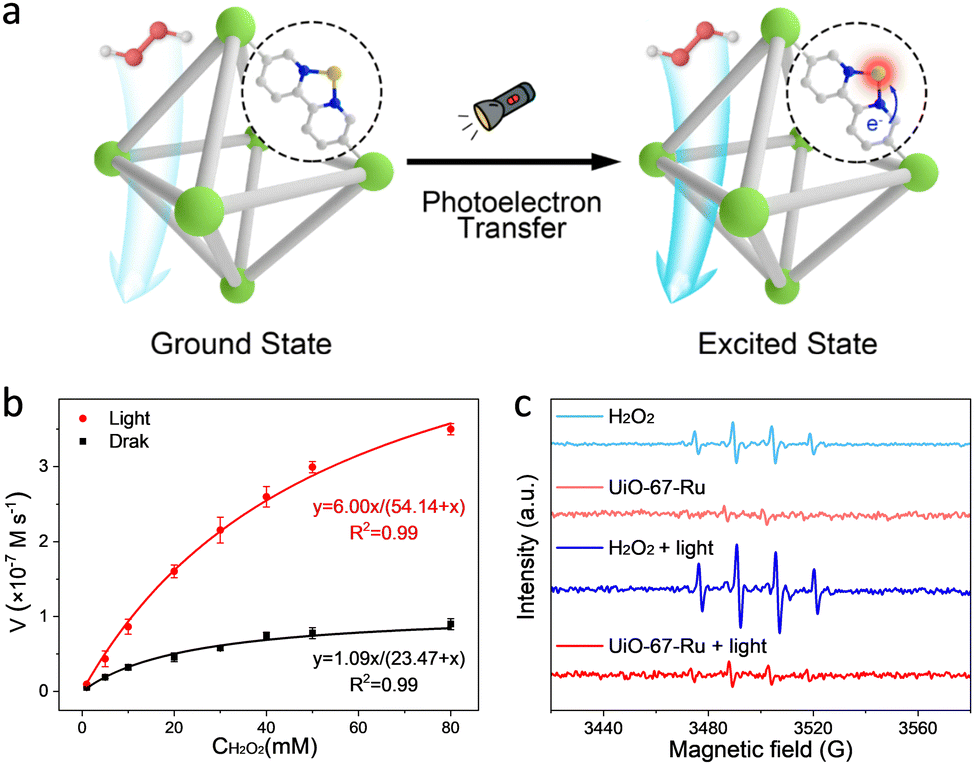 | ||
| Fig. 5 (a) Schematic illustration of the single-atom Ru site on UiO-67 enhancing POD-like activity upon illumination. Green: Zr, red: O, gray: C, bule: N, yellow: Ru, white: H. (b) Kinetic curves of UiO-67-Ru under dark and light conditions. (c) Electron paramagnetic resonance spectra of the UiO-67-Ru–H2O2 system in different conditions. | ||
Dehydrogenases, as another kind of essential oxidoreductases, catalyze substrate oxidation via reducing electron acceptors, except O2.48 The catalysis of dehydrogenases depends on the ET process induced by coenzymes, such as flavin adenine dinucleotide (FAD), nicotinamide adenine dinucleotide (phosphate) (NAD(P)H), and flavin mononucleotide (FMN), endowing reversible transformation between reactants and products. CO dehydrogenase (CODH) and formate dehydrogenase (FDH) are two common dehydrogenases affording carbon dioxide (CO2) fixation and reduction.3,49,50 They have been widely used to deal with the challenge of global warming conferred by the rapidly increased CO2 emissions.51 To mimic the hydrogenation of CODH/FDH, a redox-active nickel bis(dithiolenedibenzoic acid) with [NiS4] cores was designed to replace the tetrathiafulvalene-tetrabenzoate (TTFTB) linker to coordinate with InIII ions to gain (Me2NH2+){InIII–[Ni(C2S2-(C6H4COO)2)2]}·3DMF·1.5H2O (Fig. 6a).52 Experimental investigations show that the unsaturated [NiS4] sites exhibit strong CO2 binding ability for efficient electrocatalytic CO2 reduction reaction (CO2RR) to high-value HCOO− with high selectivity (89.6%) and conversion rate (91.5%). Notably, in addition to mimicking the CODH/FDH catalytic function, the multiple oxidation states of [NiS4] sites enable electrocatalytic dehydrogenation of glucose to glucolactone.53 Based on this, the resultant [Mn2{Ni(C2S2(C6H4COO)2)2}(H2O)2]·2DMF was utilized to construct an electrochemical glucose sensor, exhibiting excellent sensitivity, stability, and repeatability. Furthermore, to investigate the influence of the coordination structure of metal sites on the catalytic properties, a series of CoNx (x = 2, 3, and 4) modified UiO-67 photocatalysts were prepared by Wang et al.54 Under light irradiation, the excited photosensitizer [Ru-PS]* can catalyze CoNx species to the reduced state, serving as key intermediates. Among the three systems, the CoN3-involved system presents faster charge transfer efficiency, achieving an optimized electronic structure in Co sites for improving the CO2RR ability. The CO evolution rate in CoN3 sites is calculated to be 358.6 μmol g−1, which is 1.73 times higher than of CoN2 sites. Different from the mononuclear metal center-mediated CO2 reduction mechanism, a –OH group-involved hydrogenation process was described by Zeng and co-workers.55 A single-atom Pt was assembled onto the nodes of MIL-101(Cr) to obtain Pt1@MIL via the post-synthetic metalation method. The isolated Pt–O cluster with two dangling O moieties was formed by coordinating with the terminal –OH/O2H groups. It is demonstrated that the dangling O atom can adsorb dissociated H atoms to form –OH groups, in which the hydroxy H atom binds with CO2 to form HCOO*. The HCOO* was hydrogenated to HCOOH* for further conversion to CH3OH. Compared with the multinuclear center Ptn@MIL following the traditional hydrogenation mechanism, Pt1@MIL exhibits a 5.6-fold improvement in turnover frequency (TOF = 117 h−1) and higher selectivity (90.3%) for CH3OH.
 | ||
| Fig. 6 (a) Schematic illustration of the ligand structures and the obtained (Me2NH2+){InIII-[Ni(C2S2-(C6H4COO)2)2]}·3DMF·1.5H2O for the electrocatalysis of CO2RR. Gray: C, yellow: S, pink: O, purple: Ni, white: H. (b) Schematic illustration of post-synthetic modification of N3Zn–Cl sites to obtain MFU-4l-(OH) to mimic the active center of CA for reversible CO2 hydration. Reproduced with permission from ref. 58. Copyright 2018, Elsevier. | ||
Carbonic anhydrase (CA), as a ubiquitous zinc-containing metalloenzyme, can catalyze reversible CO2 hydration.56 Therefore, the development of advanced CA mimics is regarded as another efficient strategy for CO2 capture and conversion. The active center consists of one Zn2+, three histidine (His) groups, and a hydroxide (or H2O) to form an N3ZnOH tetrahedral coordination structure, where the strong nucleophilic Zn–OH moiety allows it to bind with CO2. In 2018, Winston's group reported a Zn benzotriazolate MOF (CFA-1-OH) that precisely mimics the N3ZnOH center through a mild ligand exchange and thermal activation procedure.57 By combining the intercluster hydrogen bonding interactions, the obtained Zn–OH site displays an augmented CO2/HCO3− chemisorption, affording trace CO2 capture. Similarly, an MFU-4I-OH was synthesized for efficient CO2 hydration,58 where the pristine N3Zn–Cl centers were converted into N3Zn–OH units with tetrabutylammonium hydroxide via an anion exchange method (Fig. 6b). Experimental studies demonstrated that different from the chemisorption mechanism in CFA-1-OH, the proposed MFU-4I-OH binds CO2 into the Zn–OH bond based on an insertion mechanism. The CO2 adsorption ability of MFU-4I-OH is 3.41 mmol g−1, which is significantly superior to that of MFU-4I (0.86 mmol g−1). Similar to CA, MFU-4I-OH not only catalyzes oxygen atom exchange between H2O and CO2 but also hydrolyzes acetate bonds as well.
Encouraged by the inherent hydrolysis of CA, researchers also leveraged CA mimics to decompose hypertoxic organophosphorus compounds (OPs) that remain deadly threats to humans.59 In 2020, Farha and co-workers described that Zn-based MFU-4I-OH has satisfactory hydrolytic performance toward OPs (Soman, GD) with a half-life (t1/2) of 3 min.60 And then, an array of MFU-4I MOFs with diverse metals (Co, Ni, and Cu) have been also verified with good hydrolytic properties.61 Among them, CuII-MFU-4I presents the highest activity for the degradation of dimethyl (4-nitrophenyl) phosphate (DMNP). After anion modification and thermal activation, the CuII sites are reduced to CuI with stronger binding affinities toward OPs, leading to enhanced catalytic activity. Recently, a Ti-based MFU-4l was constructed by using a transmetallation strategy to replace ZnII with strong Lewis acidic TiIV species.62 To study the effect of coordination environments on the catalytic performance, other five Ti-based MOFs, containing NU-1012-NDC, MIL-125, Ti-MIL-101, MIL-177(LT), and MIL-177(HT), were prepared, which feature different node structures and linkers (such as carboxylate and azolate). As a result, the accessibility of the monometallic TiIV–OH motif is superior to bimetallic and coordinatively saturated sites, endowing Ti-MFU-4l with the best hydrolytic capability toward DMNP. Moreover, the half-life is calculated to be ∼2 min, which is on par with any MOF reported to date.
2.2. Engineering multimetallic clusters
For many metalloenzymes, their extraordinary bioactivities rely on the cooperating catalysis of multiple metal moieties, which perform their duties, including electron transfer, binding and activation of substrates.9 Herein, in this section, we emphasize the regulation of building blocks in MOFs to reproduce the homogeneous and heterogeneous multimetallic clusters of enzymes. Importantly, the synergistic effect between multiple sites will be highlighted to deepen the understanding of catalytic functions.Phosphotriesterase (PTE) serves as another vital metallohydrolase that can catalyze the cleavage of phosphate esters.63 The active center is composed of hydroxyl bridged two Zn2+ ions (Zn–OH–Zn), in which one Zn site binds and activates the PO bond, and the other Zn site combines an –OH group to nucleophilic attack the P atom for cleaving phosphoester bonds.64 Motivated by the structural similarity between M–OH–M building blocks in MOFs and Zn–OH–Zn (Fig. 7a), a lot of related MOFs have been reported as alternatives to PTE for OP hydrolysis. Among them, Zr-based MOFs are one of the most representative PTE mimics,65,66 which show the enzyme-like catalytic mechanism. In 2014, Hupp's group demonstrated that UiO-66 with strong Lewis acidic Zr–OH–Zr centers can hydrolyze DMNP (t1/2 = 45 min) (Fig. 7b).67 However, it should be noted that the actual active sites are only 0.045% because the Zr6 cluster connected to 12 carboxylates has saturated ZrIV sites and bits of surface-defected sites for catalysis. In this regard, NU-1000 featuring four coordinatively unsaturated ZrIV sites in each Zr6 cluster was prepared, resulting in highly efficient degradation of GD and the simulant DMNP.68 Note that the use of the larger 1,3,6,8-tetrakis(p-benzoic acid)-pyrene (H4TBAPy) linker provides the NU-1000 with ultrawide channels (31 Å), which are quite larger than those of UiO-66 (<10 Å) and contribute to phosphate ester molecules to access active sites (Fig. 7c). Benefiting from this, the dehydrated NU-1000 displays superior detoxication ability toward DMNP with a half-life of only 1.5 min and a 100% conversion efficiency at 10 min. Besides enhancing the accessibility, MOF-808 was further prepared, which is connected by the six benzene-1,3,5-tricarboxylate (H3BTC) linkers to construct the Zr6 cluster, leaving six unsaturated sites.69 The appropriate pore size (4.8 to 18 Å) and rich density of active sites owing to the short ligand endow MOF-808 with superb hydrolytic activity (Fig. 7d). The hydrolytic half-life toward DMNP is 30 s, which is the fastest among reported MOFs up to now.
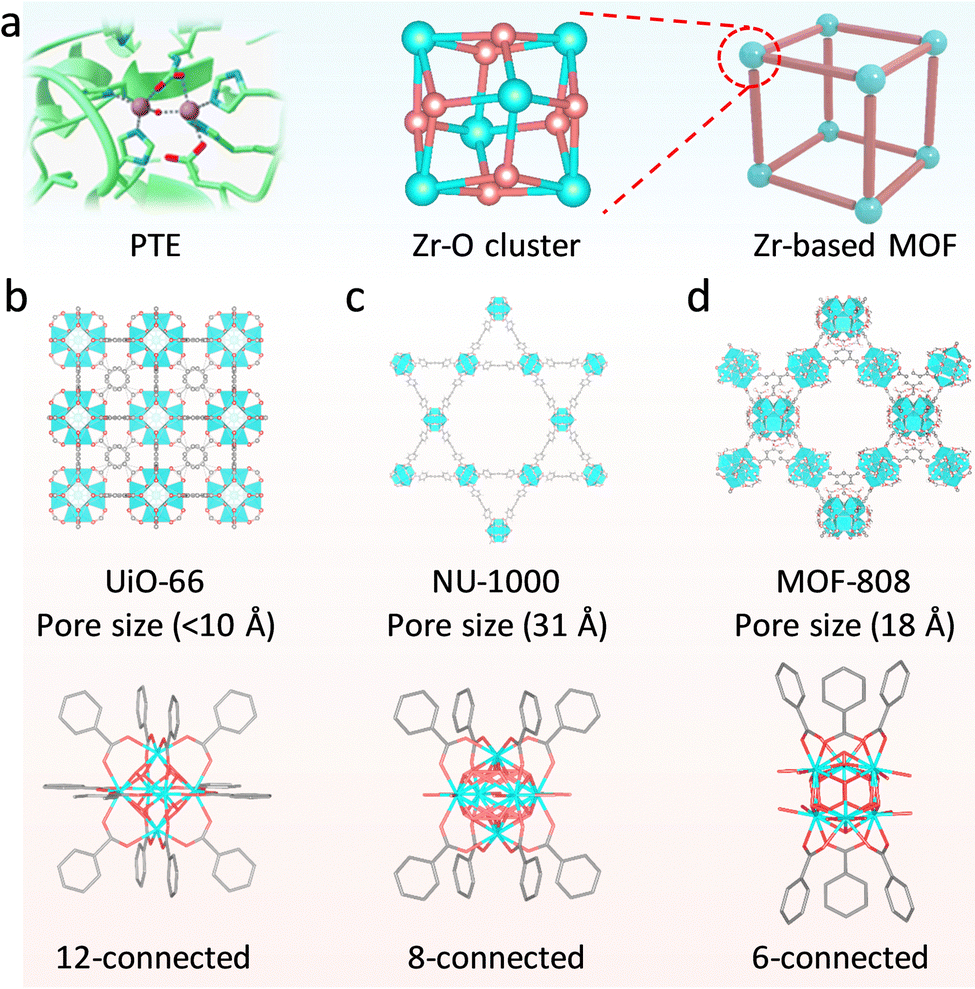 | ||
| Fig. 7 (a) Schematic illustration of active centers of PTE and structure of the Zr–O cluster over Zr-based MOFs. Crystal and the node connectivity structures of (b) UiO-66, (c) NU-1000, and (d) MOF-808. Purple: Zn, bule: N, red: O, light blue: Zr. | ||
Other metal-based MOFs, including hard Lewis acids (Ce and Ti) and intermediate Lewis acids (Cu, Fe, Zn, Mn, and Ni), have been utilized to develop hydrolase mimics. For instance, Wei's group assembled fumaric acid (FMA) and CeIV to construct a Ce-FMA-MOF that mimics the hydrolase.70 The stronger Lewis acidity of CeIV and the shorter length of linkers endow the Ce-FMA-MOF with an efficient and high density of active sites, enabling excellent hydrolyzation of various substrates. Besides, a bicopper-containing ZZU-282 MOF was synthesized to replicate the active center of mushroom tyrosinase,71 which is capable of remarkable degradation of OPs and their simulants (diethoxy-phosphoryl cyanide (DECP)) (t1/2 = 3.5 min) in the presence of H2O2. Furthermore, a series of divalent metal (Fe, Mn, Ni, Cu)-doped Co-based Prussian-blue-type MOF have been realized to decompose p-nitrophenylphosphate.72
It should be pointed out that the irreversible binding between oxohydroxometal sites and degradation products causes the poison of catalysts. To solve this problem, researchers usually introduce the sacrificial base and nucleophilic co-catalyst (N-ethylmorpholine, N-EM) to improve the hydrolytic performance of MOFs. Apart from these, Navarro and co-workers doped a [Mg-(OMe)2(MeOH)2]4 into the SBUs of MOF-808 to tune its native basicity.73 As a result, the basicity of MOF-808@Mg(OMe)2 (pH 7.6) is significantly higher than that of pristine MOF-808 (pH 3.1), leading to efficient hydrolysis of Sarin gas simulant diisopropylfluorophosphate (DIFP) in unbuffered conditions. Assisted by Mg(OMe)2, the TOF of MOF-808 toward DIFP increases from 0.01 min−1 to 0.1 min−1. In addition, a series of heterometallic Ti-based MUV-101(M), [TiIV3M2(μ3-O)(O2CX)6], M = Fe, Co, Ni, and Mg were synthesized by Martí-Gastaldo's group (Fig. 8a).74 Among them, the MUV-101(Fe) displays the highest detoxification capacity toward DIFP without the presence of the buffer solution, while other catalysts show no observable activity (Fig. 8b). This can be attributed to the cooperation of TiIV strong Lewis acid sites and FeIII–OH Brønsted base sites, which provide –OH groups for a nucleophilic attack to facilitate the nucleophilic attack of H2O (Fig. 8c).
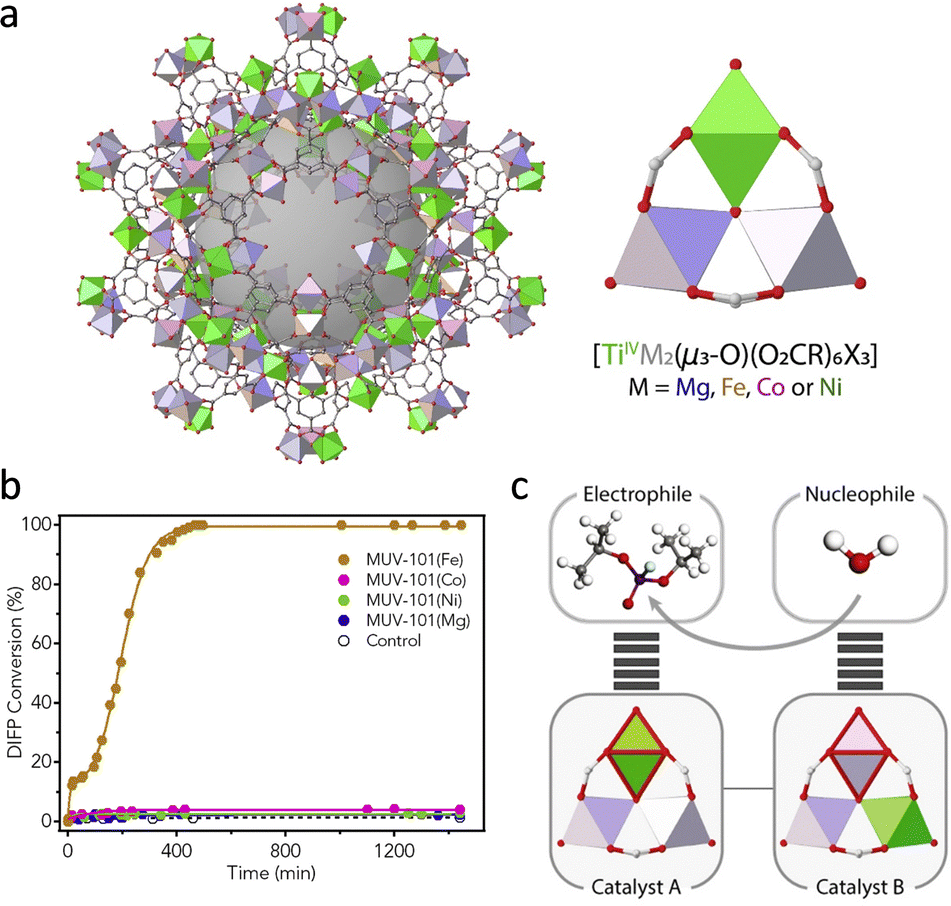 | ||
| Fig. 8 (a) Schematic illustration of the structure of heterometallic MUV-101 and its node structure. (b) The hydrolytic activity of different catalysts. (c) The functions of different metal sites in hydrolysis. Reproduced with permission from ref. 74. Copyright 2020 Elsevier. | ||
Encouraged by the monooxygenase with highly efficient catalytic ability, many MOF-based biomimetic catalysts have been frequently used for various oxidation transformations.15,75 In several natural monooxygenases, such as methane monooxygenase (Fe–Fe),76 tyrosinase,77 and catechol oxidase (Cu–Cu),78 multimetallic cofactors act as co-catalytic sites for O2 activation and oxidization of organic substrates in the active pockets (Fig. 9a). Herein, a bioinspired bicopper-containing Ti-based MOF was reported by Lin's group (Fig. 9b).79 The spatial proximal CuII2(μ2-OH)2 site was formed through LiCH2Si(CH3)3-induced deprotonation of hydroxides in SBUs and the subsequent metallization of Cu(CH3CN)4(BF4). Compared with mononuclear CuI-oxo species, the proposed CuII2(μ2-OH)2 sites display good catalytic performances for a lot of monooxygenation reactions, including hydroxylation, epoxidation, Baeyer–Villiger oxidation, and so on. Computational studies suggested that the rate-limiting step is O2 activation, where O2 binds to the bicopper site in end-on mode to form a stable Cu–O2 adduct. It assists the O–O bond cleavage, affording smaller free energy than mononuclear Cu sites. As a result, the CuII2(μ2-OH)2 sites exhibit more than 17 times higher turnover frequency than that of mononuclear Cu sites. In addition, MOF-818 with trinuclear copper-containing building blocks has been prepared to perform the catalytic behaviour of catechol oxidase for efficient oxidization of o-diphenol to o-quinone with a catalytic activity (Kcat) of 0.383 S−1.80 Note that O2 is reduced to H2O2 rather than H2O, which is different from the natural catalytic process. Additionally, Duan's group prepared a Ce-based MOF with binuclear Ce–O–Ce moieties, which are the structural analogue of bi-iron active centers of monooxygenase for the excellent photooxidation of alkanes, giving a conversion of 54.2% and a good selectivity of 98.4% to produce cyclohexanone.81 Upon light excitation, the binuclear Ce sites are activated to form an oxygen bridge radical by ligand-to-metal charge transfer (LMCT), which is beneficial for the HAT process to generate carbon radicals. Subsequently, the carbon radical combines with photogenerated singlet oxygen to form alkyl peroxy radicals, which are further hydrolyzed into carbonyl adducts conferred by the unsaturated Ce ions. In addition to chemical transformations, the oxygenase-induced oxygen reduction reaction (ORR) motivated great attention to constructing fuel cells and metal–air batteries for high-energy conversions.82,83 As an illustration, a multinuclear Cu center electrocatalyst was reported to simulate the enzyme-like O2 activation mechanism,84 showing eminent ORR property, which is equal to standard 20 wt% Pt/C with good durability.
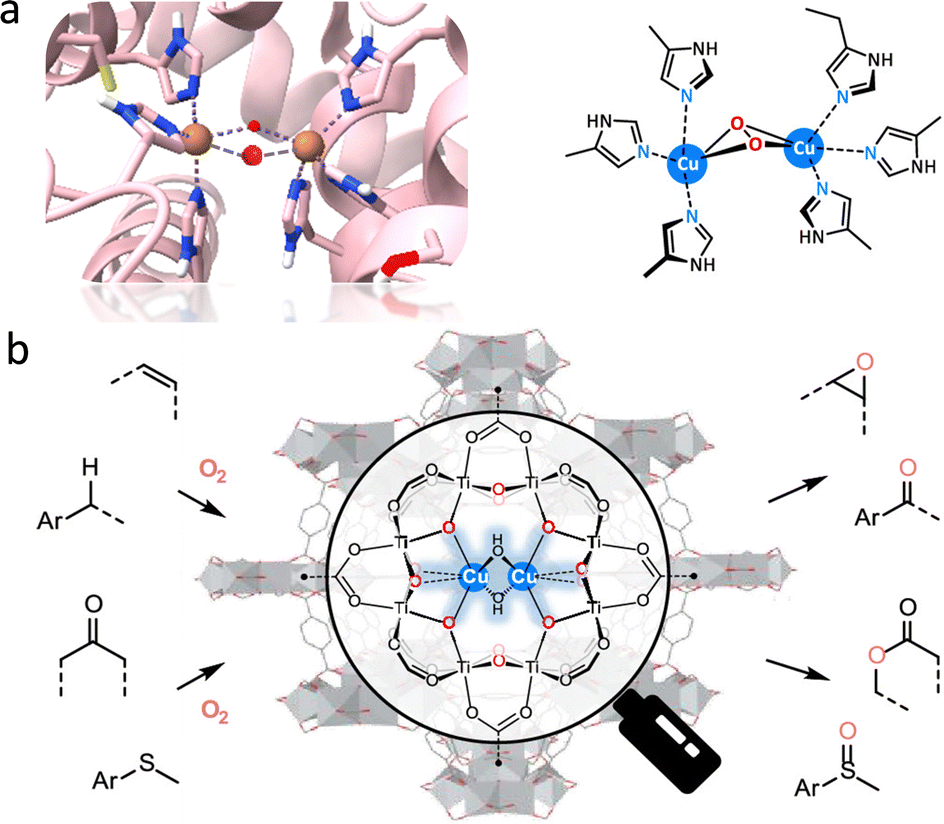 | ||
| Fig. 9 Schematic illustration of the (a) structure of the active center of binuclear copper monooxygenase and (b) grafting CuII2(μ2-OH)2 site within a Ti-based MOF for catalyzing various monooxygenation reactions. Reproduced with permission from ref. 79. Copyright 2021 American Chemistry Society. | ||
With the increasing energy crisis, water splitting-generated H2 with high combustion enthalpy and green energy has inspired extensive interest. In nature, hydrogenases (H2ases) as a kind of common metalloenzyme can catalyze the reversible conversion of H2.85,86 Among them, [FeFe] and [NiFe]–H2ases are two main oxidoreductases that strongly rely on electron transfer from [Fe4S4] clusters to aid H2/H+ binding and activation (Fig. 10a). Although the catalytic activity of [NiFe]–H2ase is slightly lower than that of [FeFe]–H2ase, the tolerability toward O2 is better, affording it to overcome the O2 interference. In an attempt to achieve the superior H2 generation ability of H2ases, researchers have devoted themselves to incorporating a structural analogue of the bimetallic clusters into MOFs for the design of efficient biomimetic catalysts. Ott and co-workers first assembled an organometallic Fe2 complex [FeFe]-(dcbdt)(CO)6 (dcbdt = 1,4-dicarboxylbenzene-2,3-dithiolate) within the robust UiO-66(Zr) by the post-synthesis exchange method.87 The confinement effect of MOFs affords to overcome the labilization and agglomeration of active units. Combined with a photosensitizer, the proposed UiO-[FeFe](dcbdt)(CO)6 realized an enhanced photocatalytic H2 evolution reaction (HER) compared with the organometallic Fe2 complex. The photoexcited ET occurring in the building blocks of MOFs can replicate the function of [Fe4S4] clusters for assistance catalysis. Based on this work, this group further regulated the [FeFe](dcbdt)(CO)6 coordination structure by substituting phosphine groups (PX3, X = Me, Et) for CO groups.88 The catalytic behaviour of the obtained [Fe2(dcbdt)(CO)4(PX3)2] site is more similar to natural [FeFe]–H2ase in comparison with pristine [FeFe](dcbdt)(CO)6. The increased electron density conferred by the phosphine groups facilitates the generation of active intermediates (hydride species), allowing reduction at more negative potentials. As another method, a redox-active linker was introduced into PCN-700 to cooperate with the organometallic Fe2 complex to improve the HER activity (Fig. 10b).89 The organic redox-active naphthalene diimide-based linker as an electron mediator mimics the [Fe4S4] clusters of H2ase to promote ET, enabling good electrocatalytic H2 production ability (400 nmol H2) for 2 h.
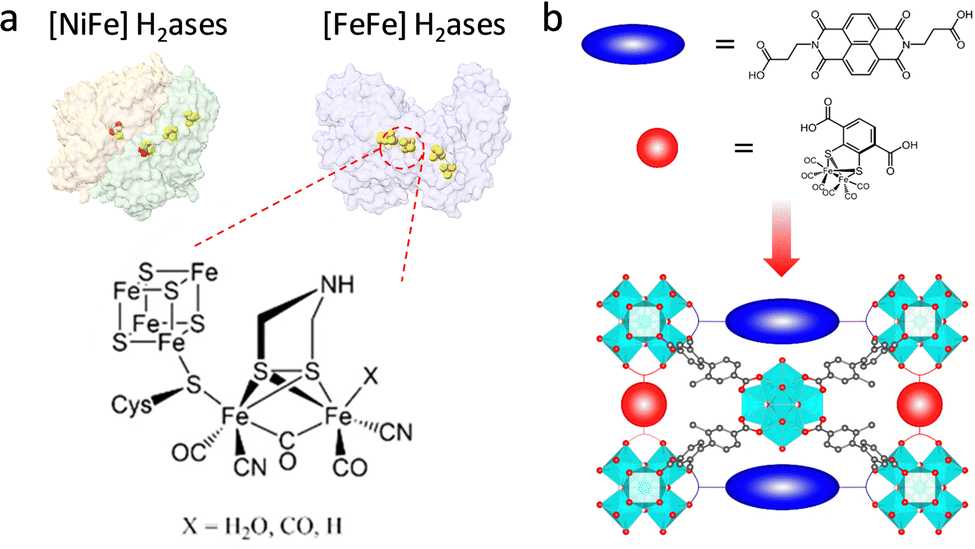 | ||
| Fig. 10 (a) Schematic illustration of two H2ases and the active center of [FeFe] H2ases. (b) Representation of bioinspired PCN-700 with two functional linkers. Reproduced with permission from ref. 89. Copyright 2022, American Chemistry Society. | ||
Similarly, various NiFe bimetallic sites have been built in MOFs, which not only replicate the catalytic behaviour of [NiFe]–H2ases but also show great potential for oxygen evolution reaction (OER), achieving overall water splitting. For instance, Li and co-workers reported a NiFe heterometallic ultrathin 2D MOF with impregnated metal sulfide clusters to obtain NiFe-MS/MOF as a bifunctional HER/OER electrocatalyst.90 Assisted by the MS cluster, the proposed catalyst displays good electrocatalytic water splitting activity, in which the electrocatalytic current density is 50 mA cm−2 at a 1.74 V cell voltage. It can be attributed to the fact that the MS cluster increases the electric conductivity, especially, optimizes the electron structure of active sites to facilitate the absorption of intermediates. Additionally, some of the other metallic clusters are capable of performing the catalytic performance of H2ases. As an illustration, a Ni-based MOF containing [Ni3O16] cluster was synthesized and showed satisfactory photocatalytic HER activity.91 Benefiting from the light-harvesting ability of the H4TBAPy ligand, the NiII–O sites can be activated into NiI sites via the LMCT process. The photoreduction active sites are favourable for the adsorption and activation of H2O, realizing the excellent H2 production rate (5 mmol h−1 g−1). As another example, Tang's group prepared a bimetallic NiCo-MOF-74 electrocatalyst with tunable OER activity by regulating the ratio of Ni to Co species.92 During catalysis, the phase structural transition of MOF from Ni0.5Co0.5(OH)2 to Ni0.5Co0.5OOH0.75 has been obtained by using operando high-resolution transmission electron microscopy imaging. The in situ formed active species (Ni0.5Co0.5OOH0.75) exhibit high OER properties (TOF = 0.86 s−1 at an overpotential of 0.3 V), which is superior to commercial RuO2. This work provides deep insights into the relationship between the structure evolution and catalytic behaviour during the reaction process. It is believed to guide the design of composite catalysts.
Nitrogenase consists of an Fe protein and a MoFe protein, enabling the effective conversion of nitrogen (N2) to bioavailable ammonia (NH3), which involves the biogeochemical nitrogen cycle.93 The [Fe4S4] cluster-containing Fe protein affords ET. As for the MoFe protein, the iron–molybdenum [FeMo] cofactor serves as the catalytic site for binding and reduction of N2, and the P-cluster accepts the electrons from the Fe protein and transfers them to [FeMo]. As for V- and Fe-type nitrogenase, the cofactor is composed of V–Fe and Fe–Fe clusters, respectively. For reproducing the astonishing catalytic activity of nitrogenase, Long et al. constructed a V-based MOF V2Cl2.8(btdd) (H2btdd, bis(1H-1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin) as a structural analogue of the [FeV] cofactor.94 Interestingly, the exposed VII sites back-donate electrons to weak π-acids to efficiently targeting π acidity N2 in the gas mixtures, achieving N2 separation. Moreover, a mixed-valence MIL-53(FeII/FeIII) was prepared to simulate the FeII active sites and high-valence MIII species in nitrogenase, respectively.95 The building linker affords fast ET to in situ form FeII upon photo illumination. The changed ratio of FeII/FeIII leads to the regulation of the catalytic performance. As a result, the optimized FeII/FeIII ratio (1.06:1) exhibits a superior NH3 evolution rate of 306 μmol h−1 g−1, which is nearly 10 folds as high as that of other framework-based catalysts. Apart from the reproduction of metal species of nitrogenase, a bimetallic Zr-Hf-containing UiO-66 was also reported.96 The Zr ions act as catalytic sites, and the Hf species serve as electron buffer tanks to perform the function of Fe protein, enabling a good photocatalytic N2 fixation rate of 116.1 μmol g−1 h−1.
3. Engineering bioinspired secondary coordination spheres
Besides the first coordination sphere of metal sites, the active pocket of metalloenzymes consists of amino acid residues that form a secondary coordination sphere. During catalysis, the spatially adjacent amino acids are involved in substrate activation, which regulates the physicochemical properties of the active sites via non-covalent interactions.97,98 Taking cytochrome P450 as an example, various proximal amino acid residues and solvents combine with iron-coordinated dioxygen to form a hydrogen bond network. Thr252 and anchored H2O facilitate heterolysis of the O–O bond to produce key intermediates through protonation.99 The electron-rich transition states can be stabilized by adjacent cationic residues to reduce the activation energy barrier. The crucial cysteine residue acts as an electron donor, supplying electrons to the iron active center via hydrogen bonding, which contributes to substrate activation. As for metallohydrolase, some amino acids act as proton relays to activate nucleophiles for promoting the cleavage of peptide amides and ester bonds and expulsing the products.100 Additionally, the excellent specificity of enzymes relies on the physicochemical properties of the binding site, including the appropriate shape, chirality, and electronic profile conferred by amino acid residues.101 In general, the secondary coordination spheres play an essential role in encompassing active species production, stabilizing the transition state and conformational structure, promoting the generation of products, and improving selectivity. The crucial functions of amino acid residues have sparked significant interest among researchers in incorporating appropriate analogues into MOFs for building enzyme-like active pockets. Beyond the primary catalytic sites, the modulable and even flexible chemical environments in the secondary coordination spheres afford biomimetic MOF-based catalysts to optimize the catalytic performance through non-covalent forces (Fig. 11). First, engineering co-catalytic units over MOFs can synergistically improve the activation of substrates and the generation of products. Second, the supramolecular chemical properties of MOFs provide a microenvironment akin to enzymes, which can participate in the selection and transport of substrates and the stabilization of intermediates to regulate reactivity and selectivity.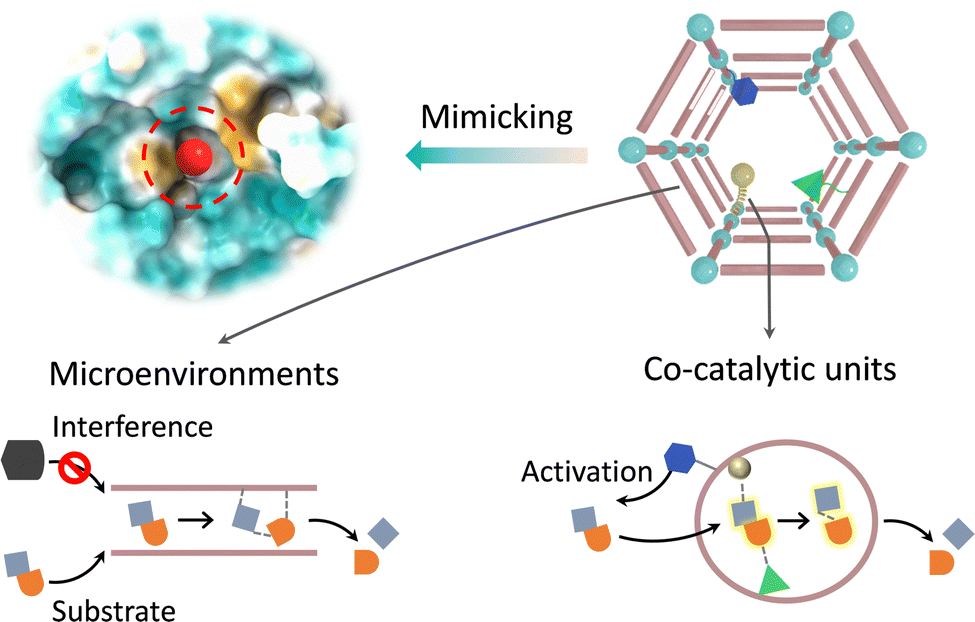 | ||
| Fig. 11 Schematic illustration of building enzyme-like active pockets over MOFs for synergy catalysis. | ||
3.1. Integrating co-catalytic groups
This section focuses on engineering additional catalytic groups that are spatially adjacent to metal sites, such as amino acids, organic molecules, etc., to construct a secondary coordination sphere over MOFs. In addition, we will highlight how these co-catalytic units participate in catalysis, especially in substrate binding, electron transfer, catalyst regeneration, and so on.For recapitulating the proton/electron transfer functions of amino acid residues in enzymes, Zhou's group skilfully developed the photo-responsive Zr-based MOF (PCN-136), which is composed of hexabenzocoronene fragments and Zr–oxo clusters to borrow photoelectron for boosting catalytic activity.102 Upon visible light excitation, photoelectrons are transferred to ZrIV–oxo clusters for reducing ZrIV to ZrIII, which has high reactivity to reduce CO2 to valuable formate, with a yield of 0.88 μmol h−1. As a control, hexaphenylbenzene-containing pbz-MOF-1 was prepared, which has similar metal nodes and topological framework to PCN-136. In comparison with pbz-MOF-1 (yield = 0.29 μmol h−1), PCN-136 shows higher photocatalytic activity because the extended π-conjugated fragments increase the optical absorption performance. What's more, motivated by abundant function groups over MOFs, Wang et al. directly immobilized the CuZn active sites within channels of UiO-66 through absorption and subsequent electroreduction.103 The robust skeleton not only stabilizes the reactive intermediates but also suppresses *H adsorption through the proton transfer process conferred by the pendant Brønsted acidic groups (–COOH). As a consequence, the proposed UiO-CuZn presents impressively excellent electrocatalytic properties and selectivity (95.2%) for the reduction of nitrate to ammonia, with a conversion rate of 97.6%. Moreover, Farha and Hupp's groups verified that –NH2 group modified UiO-66 exhibits a 20-fold improvement in the hydrolysis of OPs compared with the pristine UiO-66.104 The strengthened catalytic activity originates from the –NH2 groups neighboring to Zr active centers, which accelerate the proton transfer to assist the nucleophilic attack. Similarly, incorporating an imidazole derivative, which is analogous to a His residue in PTE, into the cavity of MOF-808 has realized rapid degradation of DMNP in the unbuffered condition, with a half-life time of less than 0.5 min.105
Encouraged by these, Lan et al. integrated various catalytic units to report a MOF-based artificial enzyme, which vividly reproduces the first and second coordinated spheres of active pockets in enzymes for synergy catalysis, containing active metal sites, proximal amino acids, and other cofactors (Fig. 12a).106 The precisely arranged cofactors over the MOF monolayer significantly boost the catalytic activity of metal active sites via the PCET process and hydrogen-bond (H-bond) stabilization. Specifically, for the photocatalytic CO2RR, a photoexcited Ir-PS unit can inject electrons into the adjacent Fe-porphyrin centers driving the transformation of CO2 to CO and CH4. The strong acidic glutamic acid (Gln) serves as a proton source to assist PCET, boosting the CO2RR activity to generate CH4. Notably, urea-based ligand (Ur)-modified Fe-porphyrin presents much higher catalytic efficiency (TOF = 150.7 h−1) and selectivity (over 99%) because the H-bond interaction between the Ur- and Fe-bound CO2, that is (N)H⋯O, stabilizes the reactive intermediates. As for water oxidation (WOR), an Ir-based active metal site was installed into the MOF. During catalysis, Gln can act as an electron mediator to accept electrons to form Gln˙+ through the PCET process. It helps in stabilizing the bound H2O based on H-bond interaction and further oxidizes the H2O molecule into an O–O bond along with HAT. This work skilfully integrated various cofactors to build artificial active pockets and achieve effective chemical transformations, which is expected to provide a promising operating principle for the design of advanced artificial enzymes.
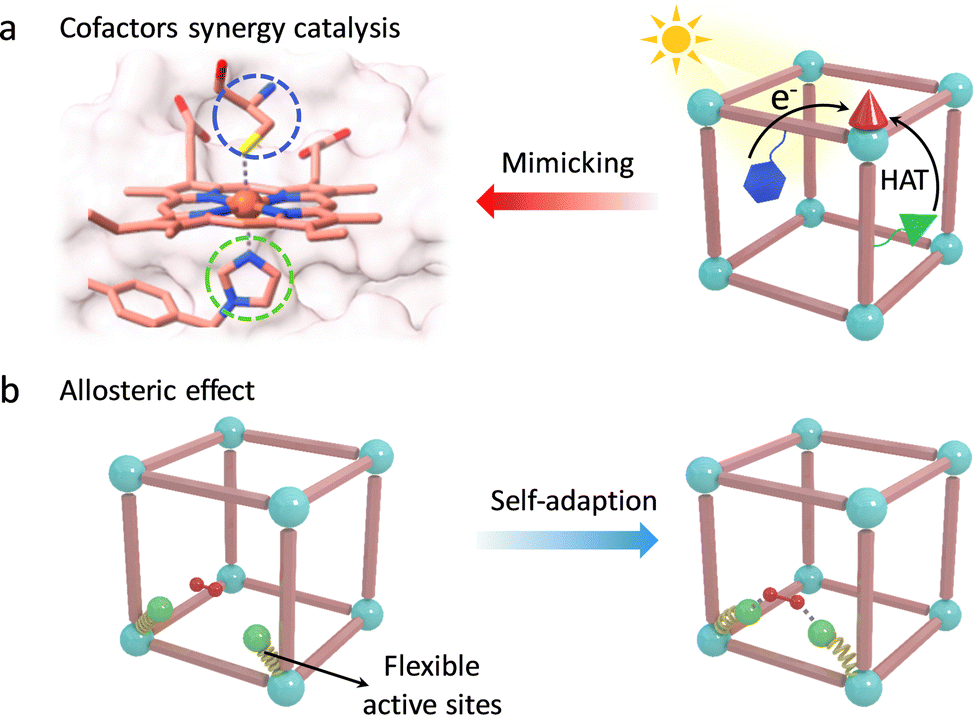 | ||
| Fig. 12 Schematic illustration of (a) the synergy catalysis of various cofactors over MOFs for reproducing enzyme active pockets and (b) the allosteric effect of self-adaptive catalytic units over MOFs. | ||
The conformationally flexible active pocket in enzymes is also a key factor for superior reactivity. The unique allosteric effect of catalytic groups is closely related to substrate binding and transition state stabilization (Fig. 12b). To this end, Tang's group introduced a series of monocarboxylate modulators in UiO-66 to improve the catalytic activity and selectivity of Zr sites.107 During catalysis, formic acid (FC) can change its conformational structure to facilitate the generation of a six-membered ring transition state by strong van der Waals forces. Compared with other linkers, due to the highest freedom of FC, the proposed UiO-66-FC shows the lowest energy barrier for the hydrogenation of alkyl levulinates with a selectivity of 99.3%. Additionally, a bioinspired flexible catalytic site was designed by introducing the variable ethylenediaminetetraacetic acid onto the Zr-oxo clusters and further anchoring dual-metal-site pair (Cu/Ni).108 The obtained MOF-808-CuNi exhibited a dynamic self-adaptive effect to fit the diverse reaction intermediates during the photoreduction CO2 process. Benefited by the stabilized intermediates, MOF-808-CuNi realized a high production rate of CH4 (158.7 μmol g−1 h−1), with fabulous selectivity (99.4%). Similar to these, some other bioinspired catalysts with adaptive frameworks have been reported to reproduce the behaviour of enzymes, including preorganization of substrates, regulation of coordination, and electronic structures of active centers for fitting the intermediates.75,109
To synergistically improve catalysis, a frustrated Lewis pair (FLPs) was immobilized in a Cr-based MOF to afford MIL-101(Cr)–FLP for selective hydrogenation.110 The exposed CrIII sites in the pore wall of MOFs can anchor FLPs, which cooperate with the –OH groups residing at the CrIII sites to preferably interact with the targeted CN bond via an H-bond (N⋯HO) interaction. Benefiting from the activating process, the resultant MOFs achieve a highly selective hydrogenation of imine bonds in α,β-unsaturated imine compounds, rather than a reduction of the CC bond conferred by the homogeneous system. Besides, Tsung et al. immobilized two Ru complexes and engineered the secondary sphere interactions in UiO-66 for the cascade catalysis hydrogenation of CO2 to CH3OH, with a TOF of 9100 h−1.20 The functionalized ammonium groups on the linker of MOFs can work as a general Brønsted acid to reinforce the hydrogenation of CO2 to HCOOH induced by a Ru complex, which may be derived from preventing the production of off-cycle resting species. After the esterification of HCOOH by the Zr–O cluster, the second Ru complex further reduced the formate esters to produce CH3OH. In this work, the synergetic catalysis of multiple active sites in the outer-sphere can achieve challenging reactions. The design principles of this system can be leveraged to develop other attractive catalysts to enhance activity and/or selectivity.
3.2. Tailoring catalytic microenvironments
In this section, we discuss the influence of secondary coordination environments, such as pore sizes, hydrophilicity and hydrophobicity, electronic profiles, and chirality on the catalytic activity of MOFs. | ||
| Fig. 13 Representation of size-selective catalysis of functionalized PCN-700 by engineering size-tailorable linkers. Reproduced with permission from ref. 113. Copyright 2021, American Chemistry Society. | ||
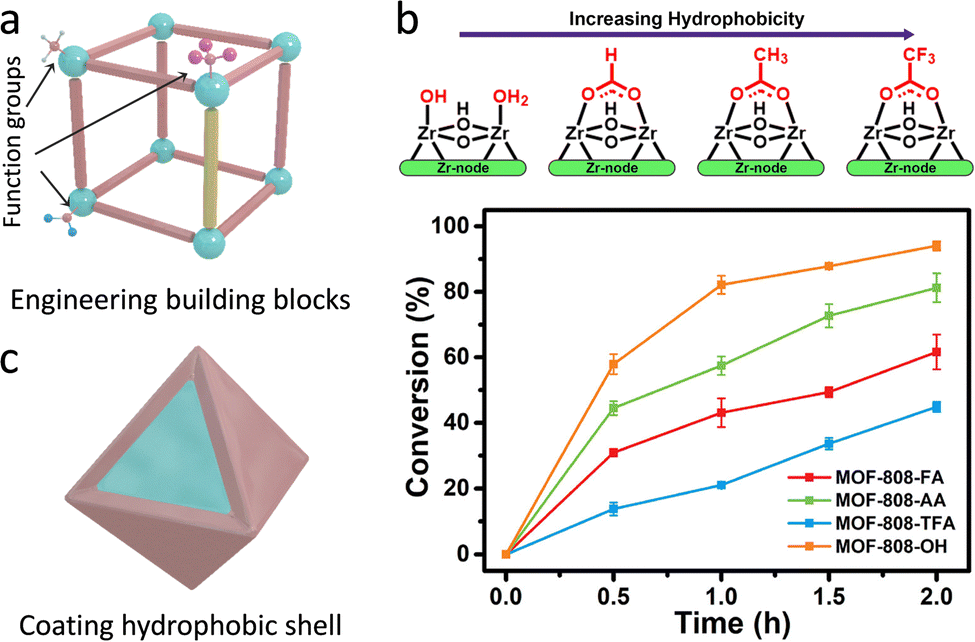 | ||
| Fig. 14 (a) Representation of the engineering building blocks (linkers and nodes) of MOFs. (b) The relationship between the various modulators and the hydrophobicity of MOFs and their corresponding hydrolytic activities. Reproduced with permission from ref. 116. Copyright 2021, American Chemistry Society. (c) Schematic illustration of coating hydrophobic shell for tuning the hydrophilicity and hydrophobicity of MOFs. | ||
Notably, as for some reactions, water may serve as a strong poisoning molecule to bring about the passivation of active sites and quench the active intermediates to suppress catalytic efficiency. Similar to the above-mentioned methods, the hydrophobicity of the pore environment within MOFs can be tuned. Apart from these, coating/growing hydrophobic shells on the surface of hydrophilic MOFs is leveraged to be a useful strategy. For instance, Yu's group utilized a hydrophobic polydimethysiloxane (PDMS) for coating the MOFs (Fig. 14c).117 PDMS-coated MOFs exhibit well-maintained porosity and surface area due to the good permeability of the PDMS layers. Consequently, the active sites are not only accessible to reactants but also have water-repelling features for preventing the entrance and coordination of water molecules, thus retaining the catalytic activity and improving the moisture/water stability. Another example is reported by Li's group.118 The mesoporous covalent organic frameworks (COFs) were fabricated on the surface of NH2-MIL-101(Fe) to form core–shell hybrid materials via a covalent linking process. The synthesized MIL@NTU-COF composite exhibits superior catalytic activity toward styrene oxidation (conversion rate of 32%) to the pristine NH2-MIL-101(Fe) (24%). On the one hand, the hydrophobic NTU-COF shell gathers styrene molecules around the catalytic sites, leading to improved catalytic conversions. On the other hand, the B3O3 ring in NTU-COF as a Lewis acid raises target benzaldehyde generation with a good selectivity of 84% via the radical mechanism path.
O species with the assistance of proximal amino acid groups to afford substrate oxidation. Taken into consideration, researchers functionalized MOFs with different electron-donating/withdrawing substitutes to tune the electronic environment around active sites. For example, Zhou et al. introduced diverse function groups, containing F, Cl, Br, and Et, at the β-position of the Fe-TCPP ligand of PCN-224(Fe).119 Due to the varied electronic properties of the substitutes, the isolated Fe-TCPP center has a tunable chemical environment. Among these, the electron-withdrawing Br group shifts the FeIII/II potential to more positive values for accelerating peroxide decomposition. What's more, halogenation helps to suppress the oxidative degradation of porphyrin. As a result, the obtained Br-PCN-224(Fe) exhibits remarkable activity, selectivity, and stability for the oxidation of 3-methylpentane, with a TOF of 10240 h−1. Similarly, Wei's group prepared a series of functional groups modified Fe-based MOFs and verified that their oxidase-like activities conform to a linear relationship with the Hammett-type structure.120 Apart from the electronic effect, our group further prepared nitro/amino (–NO2/–NH2) groups decorated MIL-101(Fe) and revealed the steric effect of function groups (Fig. 15a).121 The steady-state kinetic experiment demonstrated that the introduction of –NO2 increased the affinity of MIL-101(Fe) toward H2O2, while the opposite effect is obtained in –NH2 functionalization (Fig. 15b). Theoretical studies confirmed that the –NO2 group with strong electron-withdrawing ability tremendously reduces the electron density on the dangling bond of the Fe sites, which promotes the cleavage of H2O2 and reduces the energy change of the rate-determining step. Besides, the orientation of the O–H bond of H2O2 is biased to O of –NO2, which is conducive to breaking the O–O bond to form active intermediates.
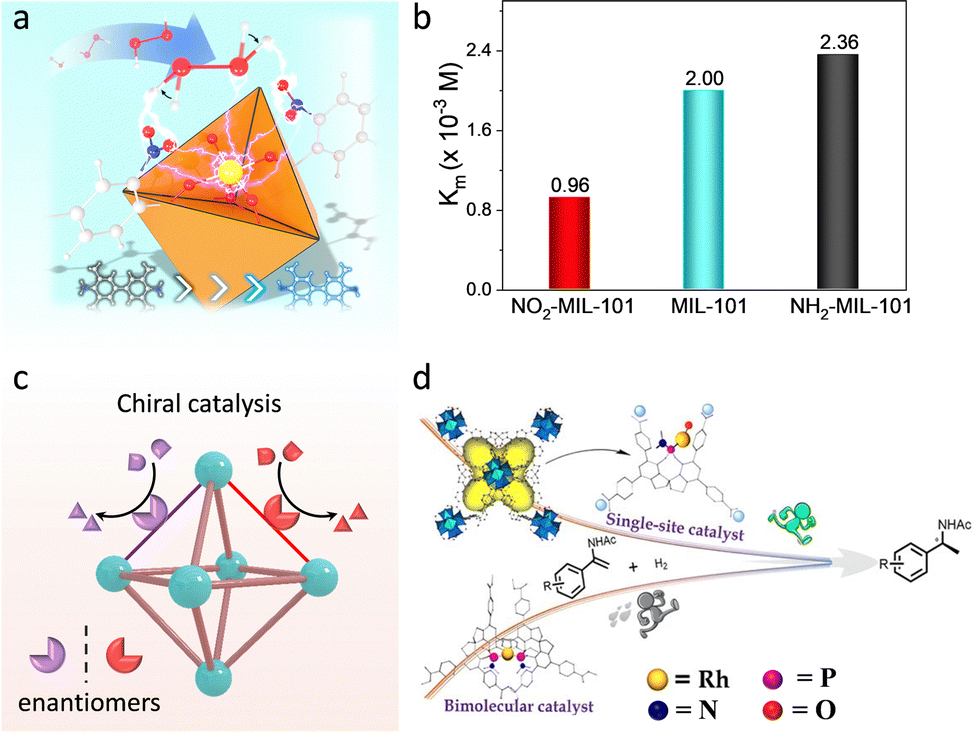 | ||
| Fig. 15 (a) Schematic illustration of electronic and steric effects of over MOFs. Yellow: Fe, red: O, bule: N, gray: C, white: H. (b) Michaelis constant (Km) of different catalysts. (c) Schematic illustration of the structure of chiral MOFs. (d) Representation of catalytic performance of chiral MOF-based catalysts and homogeneous catalysts. Reproduced with permission from ref. 126. Copyright 2022, American Chemistry Society. | ||
In addition to the linker modification method, several defect-engineering strategies have been adopted to tune the electronic environment of the secondary coordination sphere. Yu's group synthesized a defect-containing HKUST-1 via a facile “atomized trimesic acid” method.122 The retardation of the crystal ripening process induced by atomized trimesic acid facilitates the generation of coordinatively unsaturated Cu paddle wheel (CU-CPW) clusters. During electrochemical reconstruction, Cu species in the CU-CPW show a reduced oxidation state compared with the CPW. Benefited by this, the CU-CPW accelerates the proton-coupled multi-electron transfer reaction process, causing enhanced electrocatalytic CO2RR activity, with a total faradaic efficiency (FE) of 85%. Furthermore, a thermal defect-engineering strategy was employed to create CuI/CuII defect pairs in HKUST-1.123 An oxidative decarboxylation occurring at high temperatures can lead to the reduction of CuII/CuII paddle-wheel units. The generated CuI species with rich electrons strengthen the CO binding energy. The defect structure provides extra space for the simultaneous binding of CO and dioxygen, accounting for outstanding low-temperature CO oxidation. Moreover, the linker scission method was adopted to induce lattice strain in MOFs for modulating the electronic structure of metal active sites.124 The monocarboxylic acid ligand was exerted to partially replace the multi-coordinating ligand to form lattice expansion in NiFe-MOFs. The strained NiFe-MOFs show an optimized Ni 3d eg-orbital, which helps the generation of key *OOH intermediate, thus boosting the OER activity. The overpotential is reduced from 320 mV to 230 mV at a current density of 10 mA cm−2.
As mentioned above, the adjustable channel structures of MOFs can provide a steric-hindrance effect to achieve stereoscopic selective catalysis. Importantly, the fabrication of chiral moieties, such as M(salen), biphenol, spinol, and amino acids, to prepare chiral MOF-based catalysts can realize asymmetric transformations (Fig. 15c). Cui's group utilized the dicarboxylic-functionalized chiral Co(salen) linkers to coordinate with square-planar tetrameric [Cd4(O2C)8] units to construct asymmetric catalysts.125 The proposed Co(salen)-based MOFs enable efficient and recyclable resolution of racemic epoxides. In comparison with the homogenous analogues, the heterogeneous catalysts present higher catalytic performance, affording the selectivity at up to 99.5% ee. It derives from the boosted bimetallic cooperative interactions conferred by the confinement effect of MOFs. Later, this group further study the influence of the coordination structure of active sites on the catalytic behaviour.126 A single-site Ru chiral catalyst was prepared by immobilizing the Ru species within the chiral spinol-based ligand of Zr-MOF, where per Ru site features one phosphorous ligand (Fig. 15d). Compared with the homogeneous Rh-biphoshorous catalyst, the Rh-monophosphorus catalyst presents a 5-fold improvement in catalytic activity for asymmetric hydrogenations of enamides and α-dehydroamino acid esters. It can be attributed to the fact that the reduced steric repulsion and the enriched reactants in MOF cavities perfect the accessibility of active sites. What's more, the researchers grafted a chiral amino acid within the pore of MOFs and chelated metal active sites, showing high-activity enantioselective catalysis.127 For instance, our group incorporated chiral His moieties into MOF-808 to construct a bicopper active center.128 The similar active sites to native catechol oxidase not only endow the proposed MOFs to underpin a dehydrogenation of o-diphenols but also beyond natural enzymes for stereoselective catalysis. The study of the mechanism reveals that the stereoselectivity is derived from the binding energy and the steric effect in active pockets. By virtue of this, a MOF-His-Cu-based biosensor was constructed for good enantiorecognition Dopa. Furthermore, a new type of photo-responsive asymmetric MOF-based catalyst was reported by linking the chiral photo redox linkers with varied metal ions.129 As a result, the catalytic property of the asymmetric MOFs is strongly associated with the metal species because of the tunable ET ability between the chiral units and metal nodes. Notably, the chiral catalytic reactions are silent without the help of visible light. This work provides a promising way to develop and tailor heterogeneous asymmetric catalysts.
4. Applications
By virtue of advanced enzyme mimicking, MOF-based biomimetic catalysts as competitive candidates have been employed for reproducing the catalytic function of metalloenzymes in various fields. This section highlights recent achievements and progress in bioinspired systems for energy, synthesis, environmental remediation, sensing, and biomedical applications.4.1. Energy applications
The increasing energy crisis and air pollution have been urging the development of clean and sustainable alternative energy sources to replace traditional fossil fuels. HER, OER, and ORR are three key reactions for many energy processes, including fuel cells, water-splitting, and so on.130,131 Besides, CO2RR is pivotal to mitigating the greenhouse effect through the production of renewable carbon-based fuels and chemical feedstocks.132 These catalyzes of energy relevance are energetically uphill redox processes, which require catalysts to reduce the overpotential requirements.The atomically dispersed redox active sites and semiconductive or even conductive properties afford MOFs to construct bioinspired photocatalysts and electrocatalysts for driving energy-involved catalysis. MOFs with active pockets akin to that of H2ase can efficiently drive H2-relevant reactions (Fig. 16a). As a piece of evidence, Yuan's group integrated a [Fe2S2]-containing complex, which is an analogue of the [FeFe] active sites, and a Ru-photosensitive unit into Zr-MOFs to prepare photocatalysts.133 Due to the confinement effect of MOFs, the distance between the catalytic site and the photosensitizer is close, which helps accelerate the charge transfer for superior photocatalytic HER activity with a total H2 production rate of 32 μmol in 50 h. However, the UiO-MOF and the [FeFe] complex exhibit traces of H2 evolution. The electrocatalytic performance of MOFs is limited by the poor charge transport efficiency and insulating properties. In view of this, considerable efforts are implemented to tailor the linkers for optimizing their orbital overlap and charge delocalization or incorporate redox-active moieties as conduits into MOFs for promoting ET, resulting in enhanced electrocatalytic performance.134 Roy et al. described a new 3D MOF electrocatalyst, UU-100(Co) that exclusively consists of redox-active cobaloxime linkers and zirconium clusters.135 The six-coordinate CoIII catalytic centers in cobaloxime ligands present superior charge transport via a hopping mechanism due to the high porosity and channels of UU-100(Co), showing remarkable HER activity. The H2 evolution is 66 μmol with a faradaic yield of ∼84% and an average TOF of 1650 h−1. More importantly, the structurally fragile molecular cobaloximes stabilized by MOFs endow heterogeneous catalysts with structural integrity over extended periods of catalysis. It overcomes the limitation of the low stability of the homogeneous catalysts. OER is another crucial half-reaction of cater splitting and metal–air batteries.136 Das's group entrapped a mononuclear CoII complex cation into the void space of Co-based MOFs to develop electrocatalysts for outstanding OER.137 Benefited by the confinement effect of MOFs, the synthesized catalyst has satisfying stability even after 1000 cycles. In a follow-up study, a self-supported Co-based MOF nanoarray (CoBDC-Fc-NF) was prepared and introduced carboxyferrocene (Fc) missing linkers to tune the electronic structure of the Co-active centers (Fig. 17a).138 DFT calculation indicated that the extra missing linkers modulate the band gap of CoBDC-Fc, forming new electronic states near the Fermi lever. Besides, the regulated charge distribution of the active sites optimizes the adsorption of intermediates, enabling superior OER performance with a catalytic TOF of 0.034 s−1 at an overpotential of 250 mV (Fig. 17b). Meanwhile, the obtained catalyst exhibits strong durability (Fig. 17c). In virtue of the above-mentioned merits of heterogeneous materials, many biomimetic MOF-based photocatalysts have been reported for underpinning excellent OER.139
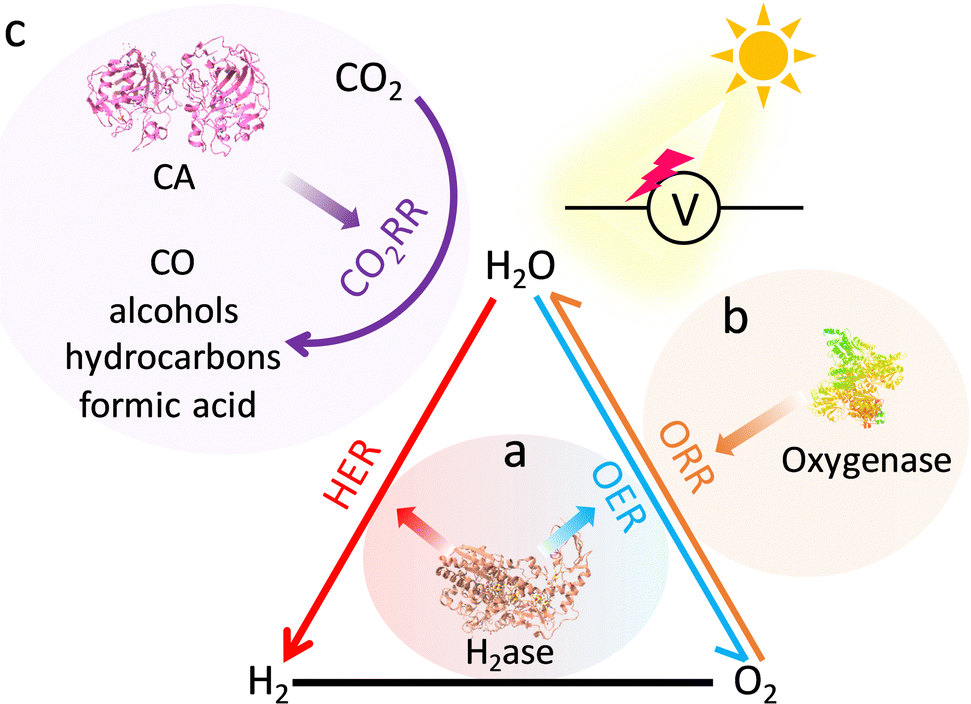 | ||
| Fig. 16 Representative reactions of energy relevance, including (a) water-splitting, (b) ORR, and (c) CO2RR, and their corresponding metalloenzymes. | ||
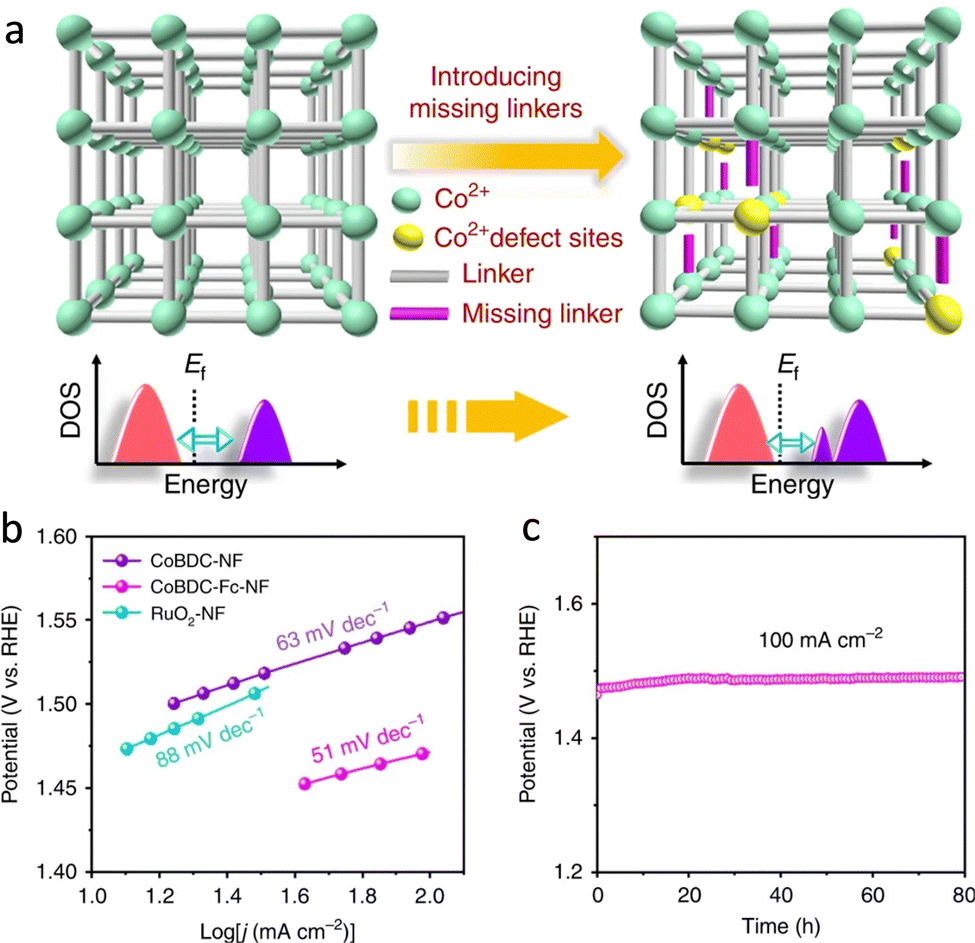 | ||
| Fig. 17 (a) Schematic illustration of the missing linker strategy for regulating electronic structure active sites. (b) Tafel plots of different catalysts. (c) Chronopotentiometry curves of CoBDC-Fc-NF. Reproduced with permission from ref. 138. Copyright 2019, Springer Nature. | ||
In terms of the ORR, the researchers focused on the development of biomimetic MOF-based catalysts with oxygenase-like properties (Fig. 16b).84,140 For optimizing the catalytic activity, researchers focused on engineering the secondary coordination sphere of MOFs to mimic the function of active pockets of metalloenzymes. Typically, an electron-donating 2-methylimidazole was assembled into the Fe-heme linker of MOFs to regulate the electronic properties.141 As a result, the obtained MOFs exhibit quite boosted conductivity and electrocatalytic ORR performance. The introduction of 2-methylimidazole affords a 2.9-time enhancement in the catalytic current. Notably, electrochemical characterization techniques demonstrated that the enhanced activity originates from accelerated kinetics of charge hopping and the rates of proton-involved chemical steps and mass transport. In addition to mimicking enzymes, Miner et al. developed a conductive 2D layered Ni3(HITP)2 MOF (HITP = 2,3,6,7,10,11-hexaiminotriphenylene) with Ni–N4 catalytic center, which shares a similar structure with the long-studied M–Nx ORR electrocatalysts.142 Ni3(HITP)2 has ORR activity and stability competitive with most non-Pt-based electrocatalysts. Given that the transport of ions, protons, and reactants are several key factors in catalysis, Ni3(HITP)2 was further introduced on a gas diffusion electrode. Three orders of magnitude electrocatalytic ORR property was achieved because of the accelerated O2 transport.143
Driving CO2RR to CO, HCO3−, alcohol, and formic acid can store renewable energy in the form of chemical bonds. Hod and co-workers prepared a 2D Zr-MOF containing hemin catalytic sites and modified a cationic functional group on the SUBs for tuning the electrostatic secondary sphere over MOFs.144 Assisted by the extra electrostatic interaction, the Fe-hemin active center shows a 3-fold improvement in the electrocatalytic CO2RR activity and essentially 100% CO selectivity. This can be attributed to the fact that the stabilization of weakly bound CO-intermediate induced by proximal groups allows its rapid release from the surface of catalysts. Besides, a stable OH−-coordinated MOF NNU-15 [Co(OH)2(H2O)2(Co-TIPP)] was developed to electrocatalytically convert CO2 to HCO3−.145 The obtained mononuclear Co–OH sites are structural analogues of Zn–OH in CA (Fig. 16c) and exhibit increased CO2 chemisorption ability for improving the FE(CO) reaching up to 99.2% at −0.6 V along with long-term stability. Moreover, combining the photoactive ReI-containing linkers, a series of Re-UiO-67 photocatalysts were reported.146 The catalytic performance of Re-MOF varies with the distance of active centers. Among these, Re3-UiO-67 with a finely balanced spatial distribution of photoactive sites exhibits the highest CO2-to-CO conversion efficiency. Specifically, integrating with the Ag nanocubes, Re3-UiO-67 achieves a 7-time improvement in CO2RR ability upon illumination. This result stems from the intensified local electric field around Re centers induced by the plasmon effect of Ag species.
4.2. Synthesis
MOF-based bioinspired catalysts with enzyme-like active pockets have been used to perform the function of enzymes for driving organic molecule synthesis and transformations, including C–H bond oxidation, Diels–Alder conversion, asymmetric catalysis, and so on.Inspired by the C–H bond oxidation capacity of monooxygenase, various heme-containing MOFs have been developed for efficient catalytic conversions. As a typical example, Wu's group developed five metalloporphrinic MOFs, consisting of M(Fe or Mn)-TCPP metalloligand, binuclear M(Zn or Cd)2(COO)4 paddle-wheel nodes, and a formate pillar.147 The proposed metalloporphrinic active center enables the reproduction of the function of heme-containing enzymes to selectively oxidize hydrocarbons, including olefins and cyclohexane, and intermolecular aldol reaction of aldehydes and ketones. Very recently, a high-throughput virtual screening workflow was reported to evaluate the catalytic performance of 87 diverse metal (Mn, Fe, Co, Cu)-based MOFs for methane oxidation via density functional theory calculations.148 The computed energetic spans are close to their turnover frequencies, which can be qualitatively comparable to the catalytic activity of MOFs. This work provides a prediction and screening of the catalytic performance of MOFs, which is expected to guide further experimental studies. In addition to the mononuclear active sites, a dicopper-containing MOF-808 was designed by Lin's group, where the imidazole units were installed on the Zr nodes for subsequent metalation with CuI species for recapitulating the catalytic center of methane monooxygenase.149 The resultant catalysts enable highly selective oxidation of CH4 under isothermal conditions at 150 °C. Also, Lercher's group introduced various copper sites into the stable NU-1000 by tuning the loading amount of Cu precursors.150 A study of the mechanism revealed that the formed dinuclear copper oxyl centers display a higher conversion rate (9.7 mmolMeOH molCu−1) and selectivity (90%) for converting CH4 to CH3OH than mononuclear Cu sites (3.3 mmolMeOH molCu−1, selectivity of 70%).
In addition to redox-active properties, various metal species in MOFs with Lewis and Brønsted acidity can also afford organic compound conversions. For example, Feng et al. synthesized a porphyrinic Zr-MOF (PCN-223), where the exposed cationic FeIII-porphyrin centers have a strong affinity for electron-donating species.151 Consequently, PCN-223 is capable of inducing the polarization of aldehyde, realizing a hetero-Diels–Alder reaction between unreactive aldehydes and dienes. Notably, the carboxylate and Zr species over MOFs feature electron-withdrawing ability that can reduce the electron density of Fe centers, thus improving the catalytic activity. In comparison with the homogeneous catalysts, the proposed PCN-223(Fe) shows a higher yield (99%) along with fabulous recyclability. In later work, a heterogeneous Bi-BTC MOF catalyst was synthesized for catalyzing Diels–Alder conversion.152 The Bi3+ species acting as Lewis acid sites are capable of diminishing the Pauli repulsion between the p-electron systems of dienophile and diene to boost the reaction efficiency. Additionally, the BTC linkers can help stabilize the reactive species, enabling a promising yield (92%) of para-xylene from the conversion of 2,5-dimethylfuran and acrylic acid. Moreover, by combining with Brønsted acid sites, Ma's group synthesized a bifunctional Cu-based MOF (JUC-1000).153 The open CuII Lewis sites are customized by –OH and –NH– groups via hydrogen bonding to form Brønsted acid sites and Lewis basic sites (Fig. 18a), which synergistically work as buffer pairs to strikingly elevate the aqueous stability of JUC-1000. The formed acid–base pairs reinforce the activation and interaction of epoxide and CO2 molecules for outstanding cycloaddition reactions, with a yield of 96%.
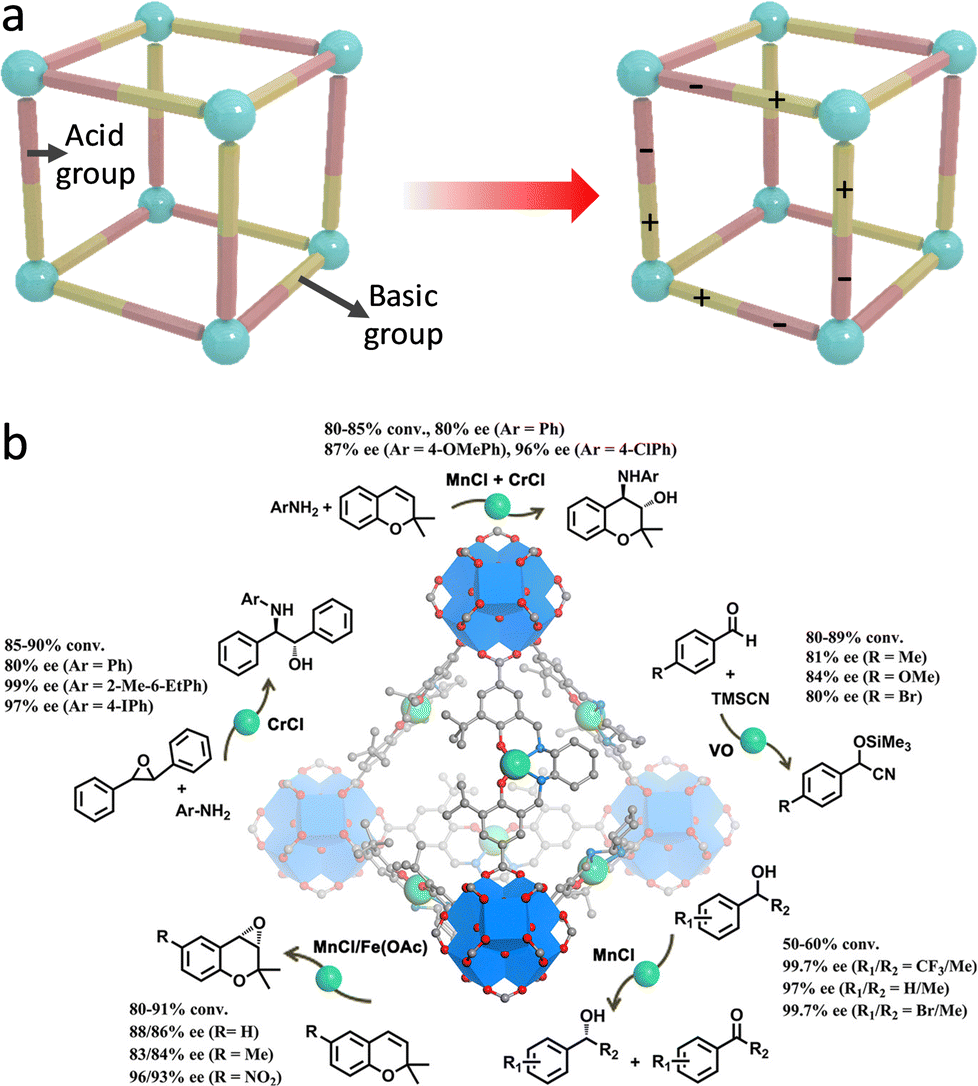 | ||
| Fig. 18 (a) Schematic illustration of integrating Brønsted acid and Lewis basic sites within MOFs. (b) Schematic illustration of chiral MOFs for catalyzing asymmetric organic transformations. Reproduced with permission from ref. 155. Copyright 2018, American Chemistry Society. | ||
Assembling with chiral building blocks, a host of MOF-based catalysts were constructed for asymmetric conversions.154 A case in point, Cui's group exerted the post-synthetic exchange method to incorporate chiral metallosalen linkers into the highly stable Zr-based UiO-68 MOFs to prepare asymmetric catalysts (Fig. 18b).155 The resultant single-metallosalen (Cu, Fe, Cr, V, and Mn) MOFs with high catalytic activity and enantioselectivities for efficient cyanosilylation of aldehydes, oxidative kinetic resolution of secondary alcohols, and ring-opening of epoxides, and aminolysis of stilbene oxide. Furthermore, the construction of mixed-M(salen) linkers over MOFs affords remarkable sequential asymmetric organic reactions. The complete recyclability of heterogeneous catalysts well overcomes the drawbacks of homogeneous catalysts, showing great promise in the pharmaceutical industry. The operating principles of this work are expected to be leveraged to design other robust and versatile heterogeneous chiral catalysts.
4.3. Environmental remediation
Environmental management strategies include not only the above-mentioned reduction of greenhouse gas emissions but also the elimination of threatening pollutants.156 In terms of environmental remediation, many MOF-based catalysts are capable of oxidizing and/or hydrolyzing highly toxic pollutants to low-toxic and even nontoxic small molecule compounds, CO2 and H2O. The oxidative remediation of catalysts depends on their reactive oxygen species (ROS)-producing capacity with the help of oxidants (Fig. 19a). In this regard, Chen and Wang reported a heme-containing Tb-based MOF, which acts like horseradish peroxidase to degrade estrogen endocrine disruptors.157 In the presence of H2O2, the resultant biomimetic MOF-based catalysts afford to produce ˙OH and high-valent Fe–O intermediates, realizing effective pollution elimination. Interestingly, some MOF-based catalysts with inherent photocatalytic ability produce more ROS upon light illumination, thus boosting the degradation results. A case in point, the Au nanorods/Fe-MOF hybrids (Au NRs/Fe-MOFs) integrating surface-enhanced Raman spectroscopy property and photo-enhanced POD-like activity were reported.158 The generated hot electrons on Au NRs excited by localized surface plasmon resonance transfer to Fe-MOFs, which promotes the Fenton reaction and prevents the recombination of electron–hole. The increased ˙OH production endows Au NRs/Fe-MOFs to significantly degrade methylene blue along with pleasurable stability and reproducibility. Based on similar strategies, a series of advanced MOF-based catalysts have been designed for the excellent removal of organic dyes and volatile organic compounds.159,160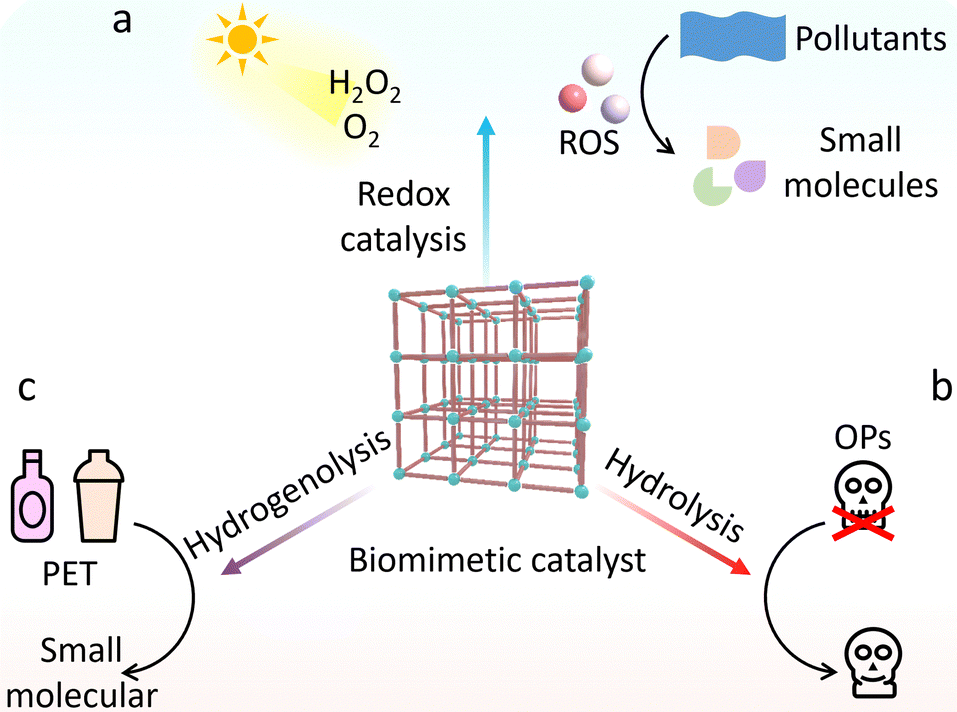 | ||
| Fig. 19 Schematic illustration of the biomimetic catalysts for environmental remediation via (a) redox catalysis, (b) hydrolysis, and (c) hydrogenolysis. | ||
Apart from these, OPs, such as organophosphate pesticides and nerve agents, as a kind of highly toxic pollution can damage, or even kill mankind. By exerting MOFs as the hydrolase mimics, the effective detoxification of OPs has been widely reported (Fig. 19b).161 Therefore, many researchers are devoted to utilizing these MOF-based catalysts to develop nanofibers against OPs.162,163 For example, several MOFs, including UiO-66, UiO-66-NH2, and UiO-67 were prepared and grew on polyamide-6 nanofibers (Fig. 20a).164 The formed MOF-nanofiber thin films with high external surface area and attractive water vapour transport performance are effective for hydrolysis OPs. Compared with UiO-66 and UiO-66-NH2, UiO-67 shows large channels for promoting the diffusion of reactants (Fig. 20b), allowing fast degradation of DMNP and GD with half-life of 7.3 min and 2.3 min, respectively. The development of nanofibers offers an opportunity to design gas filters and smart textile composites with remarkable defensive and detoxification effects against OPs in practice. Moreover, Yao et al. prepared UiO-66-NH2-coated nanofiber membranes by using a polydopamine-mediated strategy.165 Benefited by the desirable properties for filtration and photothermal effect, the obtained UiO-66-NH2-based fabrics allow outstanding degradation of DMNP (t1/2 = 0.5 min) under photo illumination.
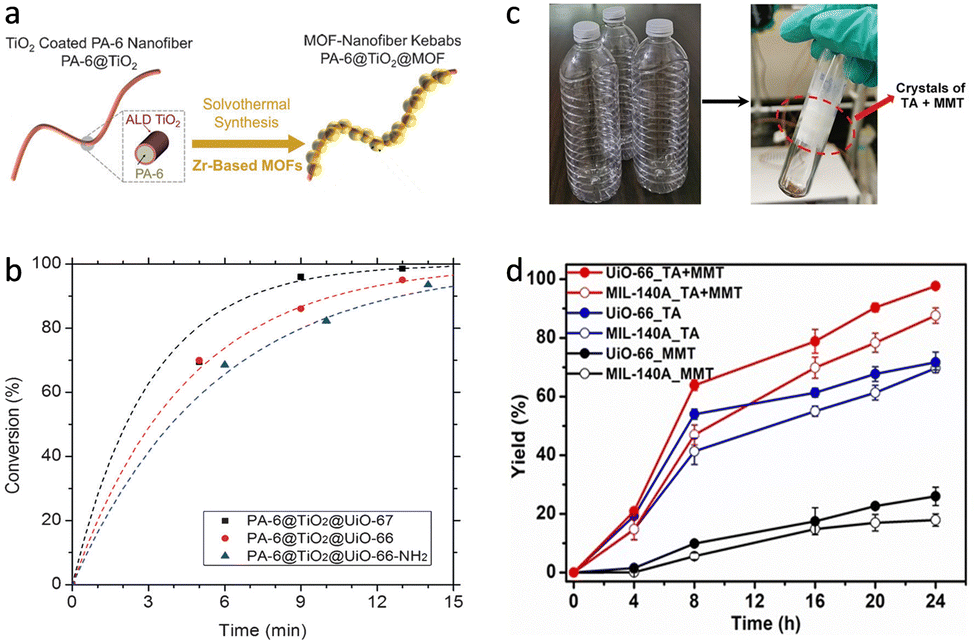 | ||
| Fig. 20 (a) Schematic illustration of synthetic procedure for Zr-MOF-based nanofibers. (b) The conversion rate of GD catalyzed by different catalysts. Reproduced with permission from ref. 164. Copyright 2016, John Wiley and Sons. (c) The picture of waste PET bottles and their degradation product catalyzed by UiO-66. (d) The yield of TA and MMT catalyzed by different catalysts. Reproduced with permission from ref. 169. Copyright 2022, John Wiley and Sons. | ||
The excessive utilization and difficulty of recycling plastics bring about many environmental crises. Polyethylene terephthalate (PET) is one of the most popular plastics worldwide. Many efforts are dedicated to developing hydrolase and their analogous heterogeneous catalysts for depolymerization of PET.166–168 Different from these, Farha's group synthesized UiO-66 to deconstruct PET by using a catalytic hydrogenolysis strategy (Fig. 19c).169 At high temperature (260 °C), UiO-66 can efficiently catalyze the decomposition of PET into terephthalic acid (TA) and mono-methyl terephthalate (MMT) with total yields of 98% within H2 in 24 h (Fig. 20c and d). Owing to its high chemical and thermal stability, UiO-66 shows satisfactory recyclability. This finding is believed to broaden the application of MOF-based catalysts for polymer degradation, leading to a recyclable polymer economy.
4.4. Sensing
Despite enzyme-based sensors with outstanding specificity and signal transformation ability for highly precise and sensitive target analysis,170,171 their inherent vulnerability significantly restrains the widespread applications in practice. By employing biomimetic MOF-based catalysts as substitutes for enzymes, stable and recyclable sensing platforms have achieved good performance.172,173 For example, Willner's group modified mononuclear Cu active sites on the linker of UiO-67 to gain Cu-NMOF, which reproduces the catalytic behaviour of POD for efficient signal transductions.174 In the presence of H2O2, various signal molecules with diverse properties can be oxidized into colorimetric, fluorescent, and chemiluminescent products (Fig. 21a). As a consequence, the resultant biosensor achieved the sensitive detection of H2O2, showing different signal output forms. Furthermore, a cascade glucose oxidase (GOx)-Cu-NMOF sensor was constructed for monitoring glucose. Briefly, GOx converts glucose into H2O2, which subsequently allows signal output catalyzed by Cu-NMOFs. Similar to this cascade catalytic strategy, the detection of some other biomolecules, such as choline, alcohol, lactic acid, etc., has been widely reported.175 Cooperating with natural enzymes for cascade catalysis enables circumvention of the low specificity of biomimetic catalyst-based sensors. In addition, Zhang's group integrated 2D metalloporphyrinic MOF (Cu-TCPP(Fe)) nanosheets and ultrasmall Au NPs, which respectively replicate the catalytic activity of POD and GOx to build a hybrid nanobiomimetic sensing device for glucose analysis.40 The superior stability and recyclability of this system solve the basic challenges in enzyme-based sensors.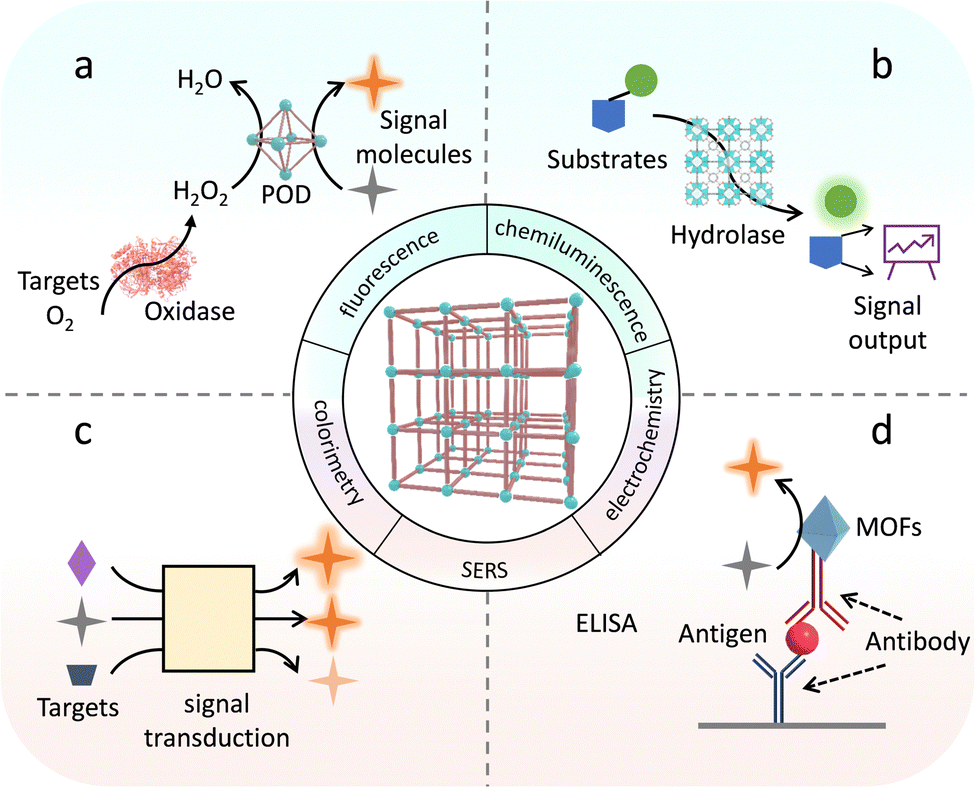 | ||
| Fig. 21 Schematic illustration of MOF-based sensor for various target analyses with different strategies, including (a) oxidation and (b) hydrolysis of substrates, (c) indirect signal transduction, and (d) ELISA. | ||
In addition to redox catalysis, a biomimetic MIP-202(Zr) catalyst was designed to perform the catalytic behaviour of metallohydrolase for the degradation and determination of nerve agents (Fig. 21b).66 The obtained MIP-202(Zr) can catalyze and recognize nerve agents, which was immobilized on a solid-contact fluoride ion-selective electrode (F-ISE) transducer to develop a MIP-202/F-ISE electrochemical sensor chip. The potentiometric detection of an F-containing nerve agent simulant (diisopropylfluorophosphate) was achieved with remarkable stability and biocompatibility in real-world environments. The low cost, sustainability, and scalability of this catalyst afford the on-body sensing application for rapid on-site detection and detoxification of nerve agent threats, expanding the unexploited scope of biomimetic catalysts.
During catalysis, the coexistence of interference molecules, which can react with enzymes or substrates, is bound to change signaling (Fig. 21c). Based on this, various indirect mode sensors have been developed for the analysis of enzyme activity and its inhibitors, small reductive molecules, and metal ions.176–178 For example, our group synthesized a NO2-MIL-101(Fe) possessing good POD-like and cooperated with acetylcholinesterase (AChE) for enzyme activity analysis and OP detection.121 In detail, AChE hydrolyzes acetylthiocholine (ATCh) into reductive mercapto thiocholine (TCh), which can inhibit the oxidative reaction catalyzed by NO2-MIL-101(Fe), resulting in a decrease of the signal output. The reduced signal intensity is positive relative to the AChE activity in the range of 0.2–50 mU mL−1. Furthermore, the inactivation of AChE caused by OPs decreases the generation of TCh and recovers the signaling, realizing sensitive determination of OPs with a limit of detection of 1 ng mL−1.
The enzyme-linked immunosorbent assay (ELISA) method is regarded as a gold standard for bioassays.179–181 Given the recognition units (antibodies or aptamers) with high specificity, the proposed biosensor exhibits robust selectivity. By employing biomimetic MOF-based catalysts as signal transduction units to build an ELISA sensing platform, effective detection of targets, including cells, bacteria, antibiotics, biomacromolecules, and so on, has been realized (Fig. 21d).182–184 For instance, Lin's group immobilized Fe-MOF onto a 2D graphene oxide to prepare efficient POD mimics.183 Due to the large surface area and rich active sites, the obtained catalyst displays good POD-like activity. As a result, the obtained biomimetic ELISA platform achieves highly sensitive detection of woodsmoke exposure biomarkers, with a limit of detection of 0.268 ng mL−1. Interestingly, integrating recognition and catalytic units, chiral MOF-based sensors are capable of distinguishing enantiomers. Zhong et al. modified CoZn bimetallic MOF-74 with D-tartaric acid to prepare a chiral catalyst.185 The metal species as catalytic sites afford the luminol oxidation with the help of H2O2. The H-bond interaction between the D-tartaric acid and chiral amino acids enables control of the catalytic reaction rate for amplification of the signal difference. Benefited by these, the selective recognition of 19 pairs of enantiomer amino acids was realized. The superior chiral recognition ability is hope for chiral catalysis monitoring, food analysis, and clinical diagnosis.
It is evident that the development of advanced sensors strongly depends on the excellent catalytic activity and specificity of biomimetic MOFs, affording efficient signal amplification and selectivity. Combining with enzymes/artificial enzymes and biomimetic MOF-based catalysts for cascade reactions not only widens the target types but also reinforces the performance of sensors owing to synergic catalysis. Importantly, the unique physicochemical properties of nanomaterials endow bioinspired sensors with multi-mode signal output, expanding the range of the applications.
4.5. Biomedical applications
Encouraged by the enzyme-like activity and pleasurable biocompatibility, many biomimetic MOF-based catalysts are effective in performing the catalytic behaviour to modulate the expression of metabolites, resulting in good biomedical applications, including cancer therapy, antibacterial therapy, antioxidation, and neuroprotection.Disrupting the redox balance in the tumor environment, such as increasing ROS production and consuming the reductive species, can efficiently eradicate cancer cells (Fig. 22a). Zhao's group prepared a copper hexacyanoferrate and modified it with a polyethylene glycol protection layer to simultaneously mimic the glutathione (GSH) oxidase and POD properties for cascade amplification chemodynamic therapy.186 The mononuclear CuII active sites are capable of depleting intracellular GSH and producing H2O2. This process is accompanied by the generation of CuI species, which boost the subsequent Fenton-like reaction to convert H2O2 into highly toxic ˙OH, enabling significant tumor-specific therapeutic efficacy. Besides, the porous and large surface areas of MOFs encouraged researchers to immobilize drugs and/or enzymes within biomimetic MOFs for synergy therapy. Note that GOx as a key enzyme not only consumes intracellular glucose and decreases the energy supply, leading to efficient starvation therapy, but also transfers O2 to H2O2 for subsequent chemodynamic therapy.187,188 Ding et al. fabricated Fe-MOFs and immobilized Au NPs and camptothecin to obtain hybrid nanomedicine for chemo/chemodynamic therapies.189 The Au NPs as GOx mimics can consume glucose and produce H2O2, which further converts to ˙OH catalyzed by POD mimics (Fe-MOF). On account of the inhibited cell proliferation and increased production of toxic •OH, the resultant nanomedicine achieves satisfactory anticancer efficacy in vivo.
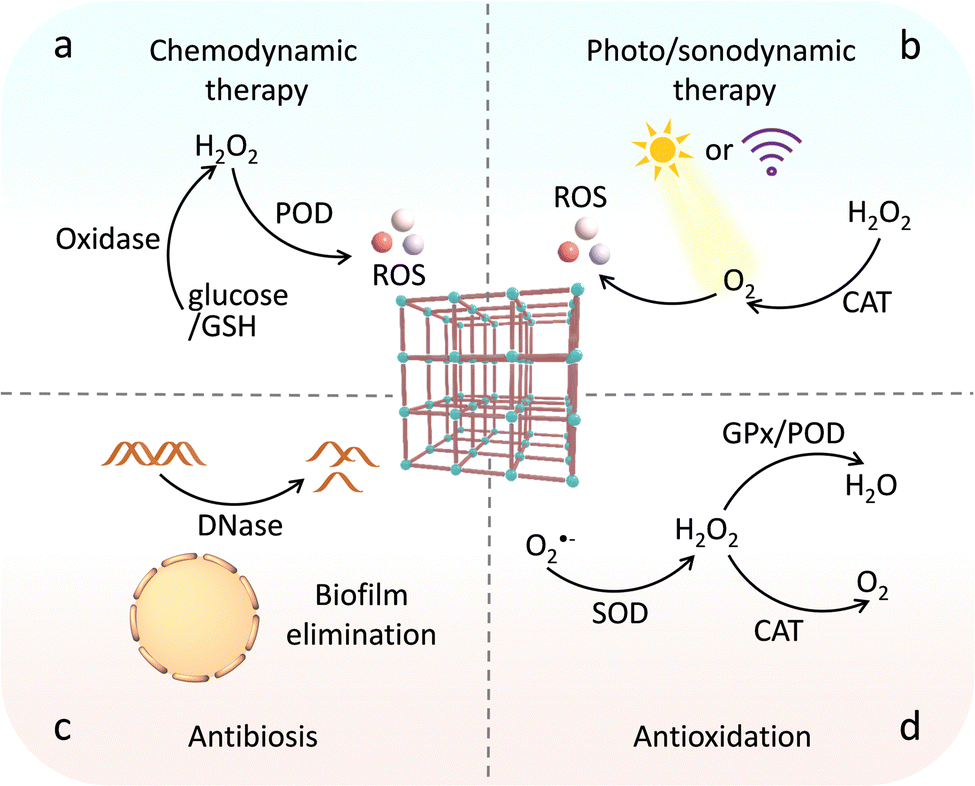 | ||
| Fig. 22 Illustration of biomimetic catalysts for (a) chemodynamic therapy, (b) photo/sonodynamic synergy therapy, (c) antibiosis, and (d) antioxidation by various catalytic reactions. | ||
Cooperating with the photo/sonodynamic effects of nanomaterials, many biomimetic MOF-based catalysts have been used to construct multi-modal synergy therapy systems (Fig. 22b).190,191 Wang et al. developed a CAT mimic by incorporating mononuclear Ru sites into the Mn3[Co(CN)6]2 MOF and integrated chlorin e6 for photodynamic therapy.192 The unsaturated single-atom Ru active sites rapidly decompose the endogenous H2O2 to O2, which overcomes tumor hypoxia and subsequently converts to ROS induced by chlorin e6 for superior cancer treatment. Notably, the high-spin Mn–N6 (S = 5/2) species endow biomimetic catalysts with T1-weighted magnetic resonance imaging ability for tracking therapeutic agents in vivo.
In addition to cancer treatment, the strong oxidative ROS can eradicate bacterial infections for effective antibacterial therapeutics in clinical applications. For example, Qu's group immobilized GOx onto a 2D Cu-TCPP(Fe) nanosheet with POD-like activity.193 The obtained nanocomposites serve as self-activated cascade reagents to generate abundant ˙OH, exhibiting robust antibacterial effects for wound healing. In many bacterial species, extracellular DNA (eDNA) is one of the important components for maintaining biofilm integrity.194 Therefore, this group incorporated CeIV-containing complexes (deoxyribonuclease (DNase) mimics) into Au-modified Fe-MOFs (POD mimics) to prepare dual-modal antibacterial agents for combating biofilms (Fig. 22c).195 The proposed bioinspired catalysts not only afford to hydrolyze eDNA for disrupting biofilms but also avoid the recolonization of bacteria and the recurrence of biofilms. The good antibacterial ability remarkably suppresses inflammatory cell growth, realizing admirable wound healing.
The overproduction of ROS and insufficient antioxidants will inevitably cause oxidative stress, resulting in various diseases, like stroke, Alzheimer's disease, and inflammation.21 In this regard, various biomimetic MOF-based catalysts have been developed to perform the functions of antioxidase, such as catalase (CAT), glutathione POD (GPx), and superoxide dismutase (SOD), for eliminating ROS (Fig. 22d).196 Qu's group synthesized Cu-TCPP MOF nanodots (CTMDs) with SOD- and GPx-like activities, which can act as a powerful enzyme-cooperative platform against several oxidative stresses.197 Based on the bienzyme cascade reaction, the CTMDs can efficiently convert the excess superoxide anion free radical (O2˙−) to H2O2, which can be in situ catalyzed to H2O, avoiding the toxification of O2˙− to H2O2, simultaneously. In an endotoxemia model, the CTMDs are capable of reducing systemic inflammation and mortality. In addition, a multi-copper cluster-containing MOF-818 with good SOD- and CAT-like properties has been used to build an antioxidation system.198 The reshaped oxidative environment restores the diabetic chronic wounds to the proliferation phase, affording effective wound healing.
Apart from the redox-related biomedical applications, our group recently described a biomimetic multifunctional hydrolase mimic for efficient neuroprotection (Fig. 23a).199 Highly toxic OPs can irreversibly inhibit the bioactivity of AChE, causing fatal nerve injury. In this regard, the mononuclear AlIII–OH species were incorporated into the node of MOF-808 to obtain MOF-808-Al for recapitulating the catalytic behaviour of AChE. By leveraging the strong Lewis acidity of AlIII and the high nucleophilic attack capacity of –OH groups, MOF-808-Al exhibits a 2.7-fold increase in activity compared to pristine MOF-808 (Fig. 23b). Importantly, MOF-808-Al with self-defense ability breaks the activity inhibition by OPs due to the intrinsic stability and detoxification effect toward OPs (Fig. 23c). As a result, the proposed AChE mimic is efficient in alleviating apoptosis and neuronal tissue damage. The design principle of this work is expected to develop intelligent bioinspired catalysts for adapting various scenarios of therapeutic applications.
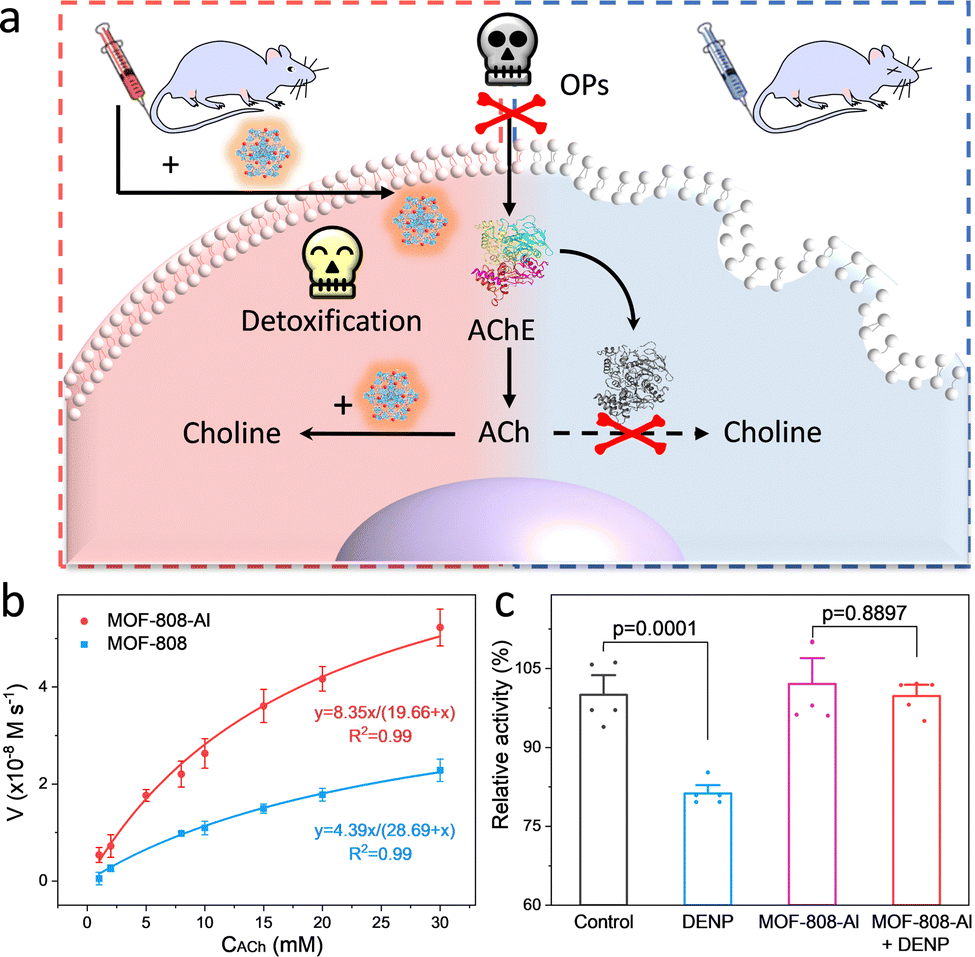 | ||
| Fig. 23 (a) Schematic illustration of neuroprotection by MOF-808-Al. (b) Kinetic curve of MOF-808-Al and MOF-808. (c) The relative activity of different systems for hydrolysis acetylcholine. | ||
Conclusions and perspective
Metalloenzymes possess sophisticated active pockets that enable them to catalyze chemical conversions effectively and selectively under mild conditions. This has sparked considerable interest among researchers in designing active pockets within MOFs to develop bioinspired MOF-based catalysts that replicate the catalytic behaviour of metalloenzymes. On the one hand, diverse and customizable building blocks of MOFs, including linkers and SBUs, allow for the precise reproduction of the primary metal active centers of enzymes at the atomic level. Importantly, the modular nature and supramolecular structures of MOFs provide the opportunity to engineer enzyme-like secondary coordination spheres, which provide non-covalent interaction networks to regulate catalytic performance. On the other hand, compared with homogeneous catalysts, bioinspired MOF-based catalysts exhibit high stability, tolerability, and recyclability due to their robust skeleton structures that prevent the aggregation and leaching of active sites. Additionally, the native physicochemical properties of MOFs broaden the functions of biomimetic catalysts beyond metalloenzymes. Overall, inspired by metalloenzymes, the rational design of innovative bioinspired MOFs not only combines the advantages of enzymes and nanomaterials to expand the scope of applications but also facilitates the exploration of the structure–activity relationship and mechanisms, thus paving a promising path in biomimetic catalysis. The catalytic performance of MOF-based catalysts for various chemical transformations is presented in Table 1. Despite these significant achievements, numerous foreseeable challenges remain in the exploration and utilization of biomimetic catalysts. Some latent research directions are provided personally.| MOF | Active center | Reaction | Activity | Ref. |
|---|---|---|---|---|
| PCN-222(Fe) | Fe-TCPP | Pyrogallol oxidation | K cat = 14 min−1 | 36 |
| MIL-125(Ti) | CuII2(μ2-OH)2 | 1,2-Dichloroethane epoxidation to cyclohexene oxide | Yield = 84%, O2 | 79 |
| MOF-818 | Trinuclear copper centers | Catechol oxidation | K cat = 0.383 s−1 | 80 |
| Ce-MOF | Ce–O–Ce | Alkane oxidation | Conversion = 54.2%, CH3CN, 395 nm LED | 81 |
| PCN-700-BPyDC(Cu) | CuN2 | Alcohol oxidation | Yield = 99%, TEMPO/NMI/CH3CN | 113 |
| MIL@NTU-1 | Fe cluster | Styrene oxidation | Conversion = 32%, CH3CN, at 80 °C | 118 |
| Br-PCN-224(Fe) | Fe-TCPP | 3-Methylpentane oxidation | TOF = 10240 h−1, CH2Cl2 |
119 |
| Metalloporphrinic MOF | Mn-TCPP | Olefins epoxidation | Yield > 99%, PhIO/CH2Cl2 | 147 |
| PMOF-RuFe(Cl) | Fe–OH | Methane oxidation | Productivity = 8.81 ± 0.34mmolg−1h−1, visible light |
43 |
| MOF-808-Bzz-Cu | Cu–O–Cu | Productivity = 71.8 ± 23.4 μmol g−1, 3% steam/He, at 150 °C | 149 | |
| Cu-NU-1000 | Cu–OH–Cu | Productivity = 9.7 mmolMeOH molCu−1, at 150 °C | 150 | |
| JUC-1000 | Cu24 cluster | Cycloaddition | Yield = 96%, TBABr | 153 |
| UiO-666-FC | Zr6 cluster | Alkyl levulinates hydrogenation | Conversion = 99.7%, at 150 °C | 107 |
| Co(salen)-based MOFs | Co(salen) | Racemic epoxide resolution | Conversion = 57%, selectivity = 99.5% ee | 125 |
| Chiral spinol-based Zr-MOF | Rh-biphoshorous | Enamides hydrogenation | Conversion = 99%, H2 (6 MPa), Ph–CH3 | 126 |
| MOF-His-Cu | Cu–O–Cu | o-Diphenols dehydrogenation | K cat = 6.9 × 10−3 min−1 | 128 |
| Ti-MOF | Ti cluster | Aldehydes a-alkylation | Conversion = 98%, THF, > 400 nm xenon lamp | 129 |
| PCN-223(Fe) | Fe-TCPP | Hetero-Diels–Alder reaction | Yield = 99%, AgBF4/toluene, 80 °C | 151 |
| Bi-BTC MOF | Bi cluster | Yield = 92%, DMF, at 160 °C | 152 | |
| MFU-4I | Zn–Cl | CO2 absorption | CO2 capacity = 0.86 mmol g−1 | 58 |
| MFU-4I-OH | Zn–OH | CO2 absorption | CO2 capacity = 3.36 mmol g−1 | 58 |
| CFA-1-OH | CO2 capacity = 2.20 mmol g−1, at 100 °C | 57 | ||
| UiO-66 | Ru complexes | CO2RR | TOF = 9100 h−1, EtOH/DMF, at 70 °C | 20 |
| (Me2NH2+){InIII-[Ni(C2S2-(C6H4COO)2)2]}·3DMF·1.5H2O | NiS4 | FE(HCOO−) = 91.5% | 52 | |
| UiO-Co–N3 | Co–N3 | CO evolution = 358.6 μmol g−1, MeCN/Ru(bpy)3Cl2·6H2O/BIH, > 420 nm xenon lamp | 54 | |
| Pt1@MIL | Pt–O | TOF = 117 h−1, DMF, 32 bar, at 150 °C | 55 | |
| PCN-136 | Zr6 cluster | HCOO− yield = 0.88 μmol h−1, MeCN/TIPA > 420 nm xenon lamp | 102 | |
| pbz-MOF-1 | HCOO− yield = 0.29 μmol h−1, MeCN/TIPA > 420 nm xenon lamp | |||
| MOZ | haem | TOF = 150.7 h−1, DMA/BIH/TFE/Co(bpy)3Cl2, > 300 nm xenon lamp | 106 | |
| MOF-808-CuNi | [CuNi] | Productivity = 158.7 μmol g−1 h−1, [Ru(bpy)3]Cl2·6H2O, MeCN, TEOA, > 420 nm xenon lamp | 108 | |
| CU-CPW | [CuCu] | FE(CH4, C2H4) = 80.5%, 0.1 M KHCO3 | 122 | |
| Zr-BTB@Hemin-TMA | Hemin | CO2RR | FE(CO, H2) = 100%, MeCN/LiClO4, 0.1 M TFE | 144 |
| NNU-15 | Co–OH | CO2RR | FE(CO) = 99.2%, 0.5 M KHCO3 | 145 |
| CuI-MFU-4l | Cu–OH | DMNP degradation | t 1/2 = 2 min | 61 |
| Ni-MFU-4l | Ni–OH | t 1/2 = 44 min | ||
| Co-MFU-4l | Co–OH | t 1/2 = 116 min | ||
| Ti-MFU-4l | Ti–OH | t 1/2 = 2 min | 62 | |
| UiO-66 | Zr–OH–Zr | t 1/2 = 45 min, 0.45 M N-EM | 67 | |
| NU-1000 (dehyd) | t 1/2 = 1.5 min, 0.45 M N-EM | 68 | ||
| MOF-808 | t 1/2 = 0.5 min, 0.45 M N-EM | 69 | ||
| UiO-66-NH2 | t 1/2 = 1 min, 0.45 M N-EM | 104 | ||
| MOF-808-imidazole | t 1/2 = 0.5 min | 105 | ||
| Spirof-MOF | t 1/2 = 1.8 min, 0.45 M N-EM | 111 | ||
| UiO-66-NH2 | t 1/2 = 0.5 min, 0.45 M N-EM, light | 165 | ||
| MFU-4-OH | Zn–OH | GD degradation | t 1/2 = 3 min, 0.45 M N-EM | 60 |
| UiO-67 | Zr–OH–Zr | t 1/2 = 2.3 min, 0.45 M N-EM | 164 | |
| ZZU-282 | Cu–O–Cu | DECP degradation | t 1/2 = 3.5 min | 71 |
| MOF-808@Mg(OMe)2 | MgZr5O2(OH)6 | DIFP degradation | t 1/2 = 6.8 min | 73 |
| MUV-101(Fe) | TiIV and Fe–OH | t 1/2 = 165 min | 74 | |
| UiO-66 | Zr–OH–Zr | PET degradation | Yield = 98%, 1 atm H2, 260 °C | 169 |
| MIL-53(FeII/FeIII) | [FeFe] | Nitrogen fixation | Fixation rate = 306 μmol h−1 g−1 | 95 |
| Zr-Hf bimetallic MOFs | [ZrHf] | Fixation rate = 116.1 μmol g−1 h−1 | 96 | |
| Zr-based MOF | [CuZn] | Nitrate reduction | Conversion = 97.6% | 103 |
| Cu-based coordination polymer | Binuclear Cu | ORR | Onset potential = 840 mV, 0.1 M KOH | 84 |
| PCN-226(Co) | Co-TCPP | Onset potential = 830 mV, 0.1 M KOH | 112 | |
| UiO-66@Hemin-MeIM | Hemin | Onset potential = 124 mV, 0.1 M LiClO4, MeCN | 141 | |
| Ni3(HITP)2 | Ni–N4 | Onset potential = 180 mV, 0.1 M KOH | 142 | |
| Ni3(HITP)2-gas diffusion electrode | Ni–N4 | ORR | Onset potential = 50 mV, 1 M NaCl, 0.3 M NaPi, pH = 7 | 143 |
| NiFe-MS/MOF | [NiFe] | Water splitting | Cell voltage = 1.74 V, current density = 50 mA cm−2, 1 M KOH | 90 |
| PCN-700_NDI_FeFe | [FeFe] | HER | 200 nmol h−1, 0.1 M NaAc, 0.5 M KCl, pH = 5 | 89 |
| Ni-TBAPy | [Ni3O16] | H2 evolution = 5 mmol h−1 g−1, AA, MeOH, > 420 nm xenon lamp | 91 | |
| UiO-MOF-Fe2S2 | Fe2S2 | H2 evolution = 0.64 μmol h−1, AA, pH = 5, > 420 nm xenon lamp | 133 | |
| UU-100(Co) | Co–N4 | TOF = 1650 h−1, DMF, pH = 4 | 135 | |
| Ni0.5Co0.5-MOF-74 | [NiCo] | OER | Onset potential = 270 mV, 1 M KOH | 92 |
| Strained NiFe-MOFs | [NiFe] | Onset potential = 230 mV, 1 M KOH | 124 | |
| CoBDC-Fc-NF | [CoCo] | TOF = 0.034 s−1, 1 M KOH | 138 |
In comparison with natural enzymes, biomimetic MOFs often show relatively low catalytic activity and specificity, which limits their practical applications. These limitations can be attributed to the lack of appropriate active pockets, leading to unsatisfactory ability in binding and activation of reactants. Therefore, the development of advanced biomimetic catalysts requires the rational design of the catalytic environments of metal active centers. On the one hand, tuning primary and higher coordination spheres is believed to optimize the chemical environment of active sites by non-covalent forces to enhance the intrinsic property. On the other hand, drawing inspiration from enzymes to create co-catalytic groups within MOFs is anticipated to cooperate with active sites for promoting substrate activations. However, existing studies predominantly focus on the design of the primary coordination sphere, neglecting the surrounding structures, which may insufficiently activate metal sites and hinder the investigation of catalytic mechanisms. To address this issue, advanced synthesis and characterization techniques are urgently required. The implementation of automated synthesis and machine learning will be leveraged to revolutionize the trial-and-error mode for the design of advanced MOF-based catalysts. Furthermore, a precise design of the pore shape, size, and function over MOFs is capable of reinforcing the chemo-, stereo-, and/or enantioselectivity conferred by the steric-hindrance effect and non-covalent forces. Also, a deep simulation of enzymatic catalysis is anticipated, like constructing flexible active centers to achieve allosteric effects for exceptional catalysis.
Different from metalloenzymes, MOF-based catalysts exhibit unique physicochemical properties, such as photo, thermal, and magnetic response, etc. These properties can be effectively utilized to regulate catalysis for advanced chemical transformations and wide applications. To harness these properties, it is necessary to employ rational modulation strategies that consider the effects on both intrinsic catalysis and physicochemical properties. These multifunctional biomimetic MOF-based catalysts hold promise in utilizing external stimuli for activating catalysts and/or reagents, thereby enabling efficient and sustainable applications. Moreover, the deliberate integration of various active sites within MOFs allows for the creation of catalysts with multiple enzyme-like properties, achieving diverse chemical conversions simultaneously.
Although bioinspired MOF-based catalysts can replicate the metalloenzymatic activities, their catalytic mechanisms are significantly different in most cases. The difference is mainly attributed to their distinct structures. Indeed, the different catalytic mechanism enables enhanced performances and even novel applications. The vast selection of metal elements and organic ligands for MOFs presents both opportunities and challenges. For example, the variable and complex compositions pose significant challenges in investigating and identifying formed intermediates that really work. Employing advanced spectroscopy, microscopy, and crystallography techniques is desirable to precisely characterize the structural information. In particular, the development of in situ characterization methods is of the essence for real-time monitoring of catalysis, allowing for the identification and visualization of reactive intermediates. Meanwhile, given the fact that enzymatic catalysis as one of the advanced nanocatalysis follows the same rules at the nanoscale, deep learning of enzymology is of vital importance to provide theoretical guidance and predictions to the mechanism studies of MOFs. Also, the mechanism of biomimetic catalysts is conducive to exploring native enzymatic reactions and active centers in return.
Considering various application scenarios, there is still much room for innovation in the performance of MOF-based catalysts. First, although various MOFs have been verified to perform the functions of oxidoreductase and hydrolase, other metalloenzymes, like DNAzymes, RNAzymes, isozymes, and so on, have received less attention. Therefore, in addition to improving the catalytic activity, broadening the reactive types of MOFs needs to be investigated in the future. Second, many nanomaterials with inherent limits, like nanotoxicity and restrained solubility, greatly limit their applications in real-life situations. In this regard, modifying MOFs with biocompatible groups and function materials may help alleviate their negative effects. The ultimate goal in applications is to develop more robust and efficient metalloenzyme mimics for underpinning chemical transformations. Combining the existing knowledge and inspiration from nature or other fields is highly desirable to facilitate further research in this area.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of National Natural Science Foundation of China (no. 22074049 and 22004042), the Fundamental Research Funds for the Central Universities (no. CCNU22JC006) and the Program of Introducing Talents of Discipline to Universities of China (111 program, B17019).References
- A. Y. Chen, R. N. Adamek, B. L. Dick, C. V. Credille, C. N. Morrison and S. M. Cohen, Chem. Rev., 2019, 119, 1323–1455 CrossRef CAS.
- A. Cvetkovic, A. L. Menon, M. P. Thorgersen, J. W. Scott, F. L. Poole Ii, F. E. Jenney Jr, W. A. Lancaster, J. L. Praissman, S. Shanmukh, B. J. Vaccaro, S. A. Trauger, E. Kalisiak, J. V. Apon, G. Siuzdak, S. M. Yannone, J. A. Tainer and M. W. W. Adams, Nature, 2010, 466, 779–782 CrossRef CAS PubMed.
- P. S. Adam, G. Borrel and S. Gribaldo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E1166–E1173 CrossRef CAS PubMed.
- C. E. Valdez, Q. A. Smith, M. R. Nechay and A. N. Alexandrova, Acc. Chem. Res., 2014, 47, 3110–3117 CrossRef CAS.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495–3578 CrossRef CAS.
- K.-Y. Wang, J. Zhang, Y.-C. Hsu, H. Lin, Z. Han, J. Pang, Z. Yang, R.-R. Liang, W. Shi and H.-C. Zhou, Chem. Rev., 2023, 123, 5347–5420 CrossRef CAS PubMed.
- T. Heinisch and T. R. Ward, Acc. Chem. Res., 2016, 49, 1711–1721 CrossRef CAS PubMed.
- Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS.
- J.-P. Yuan, Z.-J. Guan, H.-Y. Lin, B. Yan, K.-K. Liu, H.-C. Zhou and Y. Fang, Angew. Chem., Int. Ed., 2023, 62, e202303896 CrossRef PubMed.
- A. Tofoni, F. Tavani, M. Vandone, L. Braglia, E. Borfecchia, P. Ghigna, D. C. Stoian, T. Grell, S. Stolfi, V. Colombo and P. D’Angelo, J. Am. Chem. Soc., 2023, 145, 21040–21052 CrossRef CAS PubMed.
- P. Trogadas, L. Xu and M.-O. Coppens, Angew. Chem., Int. Ed., 2023, e202314446 Search PubMed.
- C. Van Stappen, Y. Deng, Y. Liu, H. Heidari, J.-X. Wang, Y. Zhou, A. P. Ledray and Y. Lu, Chem. Rev., 2022, 122, 11974–12045 CrossRef CAS.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Chem. Soc. Rev., 2013, 42, 8360–8375 RSC.
- F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC.
- J. R. Bour, A. M. Wright, X. He and M. Dincă, Chem. Sci., 2020, 11, 1728–1737 RSC.
- M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed.
- K. Chen and C.-D. Wu, Coord. Chem. Rev., 2019, 378, 445–465 CrossRef CAS.
- W. Xu, L. Jiao, Y. Wu, L. Hu, W. Gu and C. Zhu, Adv. Mater., 2021, 33, 2005172 CrossRef CAS PubMed.
- N. F. Suremann, B. D. McCarthy, W. Gschwind, A. Kumar, B. A. Johnson, L. Hammarström and S. Ott, Chem. Rev., 2023, 123, 6545–6611 CrossRef CAS PubMed.
- T. M. Rayder, A. T. Bensalah, B. Li, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2021, 143, 1630–1640 CrossRef CAS PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- J. Chen, Y. Zhu and S. Kaskel, Angew. Chem., Int. Ed., 2021, 60, 5010–5035 CrossRef CAS PubMed.
- K. J. Erickson, F. Léonard, V. Stavila, M. E. Foster, C. D. Spataru, R. E. Jones, B. M. Foley, P. E. Hopkins, M. D. Allendorf and A. A. Talin, Adv. Mater., 2015, 27, 3453–3459 CrossRef CAS PubMed.
- L. Ma, F. Jiang, X. Fan, L. Wang, C. He, M. Zhou, S. Li, H. Luo, C. Cheng and L. Qiu, Adv. Mater., 2020, 32, 2003065 CrossRef CAS.
- S. Huang, G. Chen and G. Ouyang, Chem. Soc. Rev., 2022, 51, 6824–6863 RSC.
- X. Huang, S. Zhang, Y. Tang, X. Zhang, Y. Bai and H. Pang, Coord. Chem. Rev., 2021, 449, 214216 CrossRef CAS.
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C.-K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef CAS.
- S. H. Lee, D. S. Choi, S. K. Kuk and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 7958–7985 CrossRef CAS PubMed.
- P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed.
- R. Lonsdale, J. Oláh, A. J. Mulholland and J. N. Harvey, J. Am. Chem. Soc., 2011, 133, 15464–15474 CrossRef CAS PubMed.
- M. T. Green, J. H. Dawson and H. B. Gray, Science, 2004, 304, 1653–1656 CrossRef CAS PubMed.
- A. Lombardi, F. Nastri and V. Pavone, Chem. Rev., 2001, 101, 3165–3190 CrossRef CAS.
- C. M. Lemon and M. A. Marletta, Acc. Chem. Res., 2021, 54, 4565–4575 CrossRef CAS PubMed.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- J. M. Park, K.-I. Hong, H. Lee and W.-D. Jang, Acc. Chem. Res., 2021, 54, 2249–2260 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- Y. Wang, M. Zhao, J. Ping, B. Chen, X. Cao, Y. Huang, C. Tan, Q. Ma, S. Wu, Y. Yu, Q. Lu, J. Chen, W. Zhao, Y. Ying and H. Zhang, Adv. Mater., 2016, 28, 4149–4155 CrossRef CAS PubMed.
- Y. Chen, H. Zhong, J. Wang, X. Wan, Y. Li, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 5773–5778 RSC.
- W. Bai, S. Li, J. Ma, W. Cao and J. Zheng, J. Mater. Chem. A, 2019, 7, 9086–9098 RSC.
- Y. Huang, M. Zhao, S. Han, Z. Lai, J. Yang, C. Tan, Q. Ma, Q. Lu, J. Chen, X. Zhang, Z. Zhang, B. Li, B. Chen, Y. Zong and H. Zhang, Adv. Mater., 2017, 29, 1700102 CrossRef PubMed.
- B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Nature, 2016, 536, 317–321 CrossRef CAS PubMed.
- M. C. Simons, S. D. Prinslow, M. Babucci, A. S. Hoffman, J. Hong, J. G. Vitillo, S. R. Bare, B. C. Gates, C. C. Lu, L. Gagliardi and A. Bhan, J. Am. Chem. Soc., 2021, 143, 12165–12174 CrossRef CAS PubMed.
- B. An, Z. Li, Z. Wang, X. Zeng, X. Han, Y. Cheng, A. M. Sheveleva, Z. Zhang, F. Tuna, E. J. L. McInnes, M. D. Frogley, A. J. Ramirez-Cuesta, L. S. Natrajan, C. Wang, W. Lin, S. Yang and M. Schröder, Nat. Mater., 2022, 21, 932–938 CrossRef CAS PubMed.
- F. Li, H. Sun, J. Ren, B. Zhang, X. Hu, C. Fang, J. Lee, H. Gu and D. Ling, Nat. Commun., 2022, 13, 7361 CrossRef CAS PubMed.
- H. Fan, J. Zheng, J. Xie, J. Liu, X. Gao, X. Yan, K. Fan and L. Gao, Adv. Mater., 2023, 2300387 CrossRef PubMed.
- J. Chen, Q. Ma, M. Li, D. Chao, L. Huang, W. Wu, Y. Fang and S. Dong, Nat. Commun., 2021, 12, 3375 CrossRef CAS.
- W. Xu, H. Zhong, Y. Wu, Y. Qin, L. Jiao, M. Sha, R. Su, Y. Tang, L. Zheng, L. Hu, S. Zhang, S. P. Beckman, W. Gu, Y. Yang, S. Guo and C. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220315120 CrossRef CAS.
- E. J. Kim, J. Feng, M. R. Bramlett and P. A. Lindahl, Biochemistry, 2004, 43, 5728–5734 CrossRef CAS.
- S. W. Ragsdale and M. Kumar, Chem. Rev., 1996, 96, 2515–2540 CrossRef CAS.
- V. M. Badiani, C. Casadevall, M. Miller, S. J. Cobb, R. R. Manuel, I. A. C. Pereira and E. Reisner, J. Am. Chem. Soc., 2022, 144, 14207–14216 CrossRef CAS.
- L. Domnik, M. Merrouch, S. Goetzl, J.-H. Jeoung, C. Léger, S. Dementin, V. Fourmond and H. Dobbek, Angew. Chem., Int. Ed., 2017, 56, 15466–15469 CrossRef CAS PubMed.
- Y. Zhou, S. Liu, Y. Gu, G.-H. Wen, J. Ma, J.-L. Zuo and M. Ding, J. Am. Chem. Soc., 2021, 143, 14071–14076 CrossRef CAS PubMed.
- Y. Zhou, Q. Hu, F. Yu, G.-Y. Ran, H.-Y. Wang, N. D. Shepherd, D. M. D’Alessandro, M. Kurmoo and J.-L. Zuo, J. Am. Chem. Soc., 2020, 142, 20313–20317 CrossRef CAS PubMed.
- J. Wang, K. Sun, D. Wang, X. Niu, Z. Lin, S. Wang, W. Yang, J. Huang and H.-L. Jiang, ACS Catal., 2023, 13, 8760–8769 CrossRef CAS.
- Y. Chen, H. Li, W. Zhao, W. Zhang, J. Li, W. Li, X. Zheng, W. Yan, W. Zhang, J. Zhu, R. Si and J. Zeng, Nat. Commun., 2019, 10, 1885 CrossRef PubMed.
- H. Hu, A. Boisson-Dernier, M. Israelsson-Nordström, M. Böhmer, S. Xue, A. Ries, J. Godoski, J. M. Kuhn and J. I. Schroeder, Nat. Cell Biol., 2010, 12, 87–93 CrossRef CAS.
- C. E. Bien, K. K. Chen, S.-C. Chien, B. R. Reiner, L.-C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS PubMed.
- A. M. Wright, Z. Wu, G. Zhang, J. L. Mancuso, R. J. Comito, R. W. Day, C. H. Hendon, J. T. Miller and M. Dincă, Chem, 2018, 4, 2894–2901 CAS.
- F. M. Raushel, Nature, 2011, 469, 310–311 CrossRef CAS PubMed.
- M. R. Mian, T. Islamoglu, U. Afrin, S. Goswami, R. Cao, K. O. Kirlikovali, M. G. Hall, G. W. Peterson and O. K. Farha, Chem. Mater., 2020, 32, 6998–7004 CrossRef CAS.
- M. R. Mian, H. Chen, R. Cao, K. O. Kirlikovali, R. Q. Snurr, T. Islamoglu and O. K. Farha, J. Am. Chem. Soc., 2021, 143, 9893–9900 CrossRef CAS PubMed.
- M. R. Mian, X. Wang, X. Wang, K. O. Kirlikovali, H. Xie, K. Ma, K. M. Fahy, H. Chen, T. Islamoglu, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2023, 145, 7435–7445 CrossRef CAS PubMed.
- C. J. Jackson, J. L. Foo, N. Tokuriki, L. Afriat, P. D. Carr, H. K. Kim, G. Schenk, D. S. Tawfik and D. L. Ollis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21631–21636 CrossRef CAS PubMed.
- S. D. Aubert, Y. Li and F. M. Raushel, Biochemistry, 2004, 43, 5707–5715 CrossRef CAS.
- A. M. Plonka, Q. Wang, W. O. Gordon, A. Balboa, D. Troya, W. Guo, C. H. Sharp, S. D. Senanayake, J. R. Morris, C. L. Hill and A. I. Frenkel, J. Am. Chem. Soc., 2017, 139, 599–602 CrossRef CAS PubMed.
- S. S. Sandhu, Y. G. Kotagiri, P. U. A. I. Fernando I, M. Kalaj, N. Tostado, H. Teymourian, E. M. Alberts, T. L. Thornell, G. R. Jenness, S. P. Harvey, S. M. Cohen, L. C. Moores and J. Wang, J. Am. Chem. Soc., 2021, 143, 18261–18271 CrossRef CAS PubMed.
- M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Angew. Chem., Int. Ed., 2014, 53, 497–501 CrossRef CAS.
- J. E. Mondloch, M. J. Katz, W. C. Isley Iii, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed.
- S.-Y. Moon, Y. Liu, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 6795–6799 CrossRef CAS PubMed.
- S. Li, Z. Zhou, Z. Tie, B. Wang, M. Ye, L. Du, R. Cui, W. Liu, C. Wan, Q. Liu, S. Zhao, Q. Wang, Y. Zhang, S. Zhang, H. Zhang, Y. Du and H. Wei, Nat. Commun., 2022, 13, 827 CrossRef CAS PubMed.
- Q.-Y. Wang, Z.-B. Sun, M. Zhang, S.-N. Zhao, P. Luo, C.-H. Gong, W.-X. Liu and S.-Q. Zang, J. Am. Chem. Soc., 2022, 144, 21046–21055 CrossRef CAS PubMed.
- Y. Seki, H. Tabe and Y. Yamada, J. Phys. Chem. C, 2022, 126, 5564–5574 CrossRef CAS.
- R. Gil-San-Millan, E. López-Maya, A. E. Platero-Prats, V. Torres-Pérez, P. Delgado, A. W. Augustyniak, M. K. Kim, H. W. Lee, S. G. Ryu and J. A. R. Navarro, J. Am. Chem. Soc., 2019, 141, 11801–11805 CrossRef CAS PubMed.
- J. Castells-Gil, N. M. Padial, N. Almora-Barrios, R. Gil-San-Millán, M. Romero-Ángel, V. Torres, I. da Silva, B. C. J. Vieira, J. C. Waerenborgh, J. Jagiello, J. A. R. Navarro, S. Tatay and C. Martí-Gastaldo, Chem, 2020, 6, 3118–3131 CAS.
- Z. Wang, P. Yeary, X. Feng and W. Lin, J. Am. Chem. Soc., 2023, 145, 8647–8655 CAS.
- G. E. Cutsail, III, R. Banerjee, A. Zhou, L. Que Jr., J. D. Lipscomb and S. DeBeer, J. Am. Chem. Soc., 2018, 140, 16807–16820 CrossRef.
- L. M. Mirica, M. Vance, D. J. Rudd, B. Hedman, K. O. Hodgson, E. I. Solomon and T. D. P. Stack, Science, 2005, 308, 1890–1892 CrossRef CAS PubMed.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084–1090 CrossRef CAS.
- X. Feng, Y. Song, J. S. Chen, Z. Xu, S. J. Dunn and W. Lin, J. Am. Chem. Soc., 2021, 143, 1107–1118 CrossRef CAS.
- M. Li, J. Chen, W. Wu, Y. Fang and S. Dong, J. Am. Chem. Soc., 2020, 142, 15569–15574 CrossRef CAS PubMed.
- G. Ji, L. Zhao, Y. Wang, Y. Tang, C. He, S. Liu and C. Duan, ACS Catal., 2022, 12, 7821–7832 CrossRef CAS.
- Y. Liang, J. Wei, X. Qiu and N. Jiao, Chem. Rev., 2018, 118, 4912–4945 CrossRef CAS PubMed.
- N. Zion, A. Friedman, N. Levy and L. Elbaz, Adv. Mater., 2018, 30, 1800406 CrossRef PubMed.
- B. Devi, A. Bhardwaj, D. Gambhir, B. Roy, A. Karmakar, G. Dey, A. Jain, B. Mondal and R. R. Koner, Inorg. Chem., 2022, 61, 15699–15710 CrossRef CAS.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC.
- A. T. Castner, B. A. Johnson, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2021, 143, 7991–7999 CrossRef CAS PubMed.
- M. Zhao, W. Li, J. Li, W. Hu and C. M. Li, Adv. Sci., 2020, 7, 2001965 CrossRef CAS PubMed.
- L. Liu, S. Du, X. Guo, Y. Xiao, Z. Yin, N. Yang, Y. Bao, X. Zhu, S. Jin, Z. Feng and F. Zhang, J. Am. Chem. Soc., 2022, 144, 2747–2754 CrossRef CAS PubMed.
- S. Zhao, C. Tan, C.-T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K.-H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Nat. Energy, 2020, 5, 881–890 CrossRef CAS.
- R. D. Milton and S. D. Minteer, Acc. Chem. Res., 2019, 52, 3351–3360 CrossRef CAS PubMed.
- D. E. Jaramillo, D. A. Reed, H. Z. H. Jiang, J. Oktawiec, M. W. Mara, A. C. Forse, D. J. Lussier, R. A. Murphy, M. Cunningham, V. Colombo, D. K. Shuh, J. A. Reimer and J. R. Long, Nat. Mater., 2020, 19, 517–521 CrossRef CAS PubMed.
- Z. Zhao, D. Yang, H. Ren, K. An, Y. Chen, Z. Zhou, W. Wang and Z. Jiang, Chem. Eng. J., 2020, 400, 125929 CrossRef CAS.
- K. An, H. Ren, D. Yang, Z. Zhao, Y. Gao, Y. Chen, J. Tan, W. Wang and Z. Jiang, Appl. Catal., B, 2021, 292, 120167 CrossRef CAS.
- J. C. Fontecilla-Camps and A. Volbeda, Chem. Rev., 2022, 122, 12110–12131 CrossRef CAS PubMed.
- R. M. Bullock and A. Dey, Chem. Rev., 2022, 122, 11897–11899 CrossRef CAS PubMed.
- K. A. Jesse, S. W. Anferov, K. A. Collins, J. A. Valdez-Moreira, M. E. Czaikowski, A. S. Filatov and J. S. Anderson, J. Am. Chem. Soc., 2021, 143, 18121–18130 CrossRef CAS PubMed.
- G. Schenk, N. Mitić, L. R. Gahan, D. L. Ollis, R. P. McGeary and L. W. Guddat, Acc. Chem. Res., 2012, 45, 1593–1603 CrossRef CAS.
- T. K. Hyster and T. R. Ward, Angew. Chem., Int. Ed., 2016, 55, 7344–7357 CrossRef CAS.
- J.-S. Qin, S. Yuan, L. Zhang, B. Li, D.-Y. Du, N. Huang, W. Guan, H. F. Drake, J. Pang, Y.-Q. Lan, A. Alsalme and H.-C. Zhou, J. Am. Chem. Soc., 2019, 141, 2054–2060 CrossRef CAS PubMed.
- Z. Wang, S. Liu, M. Wang, L. Zhang, Y. Jiang, T. Qian, J. Xiong, C. Yang and C. Yan, ACS Catal., 2023, 13, 9125–9135 CrossRef CAS.
- M. J. Katz, S.-Y. Moon, J. E. Mondloch, M. H. Beyzavi, C. J. Stephenson, J. T. Hupp and O. K. Farha, Chem. Sci., 2015, 6, 2286–2291 RSC.
- H.-B. Luo, A. J. Castro, M. C. Wasson, W. Flores, O. K. Farha and Y. Liu, ACS Catal., 2021, 11, 1424–1429 CrossRef CAS PubMed.
- G. Lan, Y. Fan, W. Shi, E. You, S. S. Veroneau and W. Lin, Nat. Catal., 2022, 5, 1006–1018 CrossRef CAS.
- X. Zhang, C. Yang, P. An, C. Cui, Y. Ma, H. Liu, H. Wang, X. Yan, G. Li and Z. Tang, Sci. Adv., 2022, 8, eadd5678 CrossRef CAS PubMed.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H.-L. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- S. Yuan, L. Zou, H. Li, Y.-P. Chen, J. Qin, Q. Zhang, W. Lu, M. B. Hall and H.-C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 10776–10780 CrossRef CAS.
- Z. Niu, W. Zhang, P. C. Lan, B. Aguila and S. Ma, Angew. Chem., Int. Ed., 2019, 58, 7420–7424 CrossRef CAS.
- H. J. Park, J. K. Jang, S.-Y. Kim, J.-W. Ha, D. Moon, I.-N. Kang, Y.-S. Bae, S. Kim and D.-H. Hwang, Inorg. Chem., 2017, 56, 12098–12101 CrossRef CAS PubMed.
- M. O. Cichocka, Z. Liang, D. Feng, S. Back, S. Siahrostami, X. Wang, L. Samperisi, Y. Sun, H. Xu, N. Hedin, H. Zheng, X. Zou, H.-C. Zhou and Z. Huang, J. Am. Chem. Soc., 2020, 142, 15386–15395 CrossRef CAS PubMed.
- S. Yuan, Y.-P. Chen, J.-S. Qin, W. Lu, L. Zou, Q. Zhang, X. Wang, X. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 8912–8919 CrossRef CAS PubMed.
- R. Wang, K. Shi, J. Liu, R. Q. Snurr and J. T. Hupp, J. Am. Chem. Soc., 2023, 145, 13979–13988 CrossRef CAS.
- Z. Han, Z. Yan, K. Wang, X. Kang, K. Lv, X. Zhang, Z. Zhou, S. Yang, W. Shi and P. Cheng, Sci. China: Chem., 2022, 65, 1088–1093 CrossRef CAS.
- X. Liu, K. O. Kirlikovali, Z. Chen, K. Ma, K. B. Idrees, R. Cao, X. Zhang, T. Islamoglu, Y. Liu and O. K. Farha, Chem. Mater., 2021, 33, 1444–1454 CrossRef CAS.
- W. Zhang, Y. Hu, J. Ge, H.-L. Jiang and S.-H. Yu, J. Am. Chem. Soc., 2014, 136, 16978–16981 CrossRef CAS PubMed.
- M. Cai, Y. Li, Q. Liu, Z. Xue, H. Wang, Y. Fan, K. Zhu, Z. Ke, C.-Y. Su and G. Li, Adv. Sci., 2019, 6, 1802365 CrossRef.
- N. Huang, S. Yuan, H. Drake, X. Yang, J. Pang, J. Qin, J. Li, Y. Zhang, Q. Wang, D. Jiang and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 18590–18597 CrossRef CAS.
- J. Wu, Z. Wang, X. Jin, S. Zhang, T. Li, Y. Zhang, H. Xing, Y. Yu, H. Zhang, X. Gao and H. Wei, Adv. Mater., 2021, 33, 2005024 CrossRef CAS.
- W. Xu, Y. Kang, L. Jiao, Y. Wu, H. Yan, J. Li, W. Gu, W. Song and C. Zhu, Nano-Micro Lett., 2020, 12, 184 CrossRef CAS PubMed.
- W. Zhang, C. Huang, J. Zhu, Q. Zhou, R. Yu, Y. Wang, P. An, J. Zhang, M. Qiu, L. Zhou, L. Mai, Z. Yi and Y. Yu, Angew. Chem., Int. Ed., 2022, 61, e202112116 CrossRef CAS PubMed.
- W. Wang, D. I. Sharapa, A. Chandresh, A. Nefedov, S. Heißler, L. Heinke, F. Studt, Y. Wang and C. Wöll, Angew. Chem., Int. Ed., 2020, 59, 10514–10518 CrossRef CAS PubMed.
- Q. Ji, Y. Kong, C. Wang, H. Tan, H. Duan, W. Hu, G. Li, Y. Lu, N. Li, Y. Wang, J. Tian, Z. Qi, Z. Sun, F. Hu and W. Yan, ACS Catal., 2020, 10, 5691–5697 CrossRef CAS.
- C. Zhu, G. Yuan, X. Chen, Z. Yang and Y. Cui, J. Am. Chem. Soc., 2012, 134, 8058–8061 CrossRef CAS PubMed.
- W. Gong, X. Chen, W. Zhang, K. O. Kirlikovali, B. Nan, Z. Chen, R. Si, Y. Liu, O. K. Farha and Y. Cui, J. Am. Chem. Soc., 2022, 144, 3117–3126 CrossRef CAS PubMed.
- R. Newar, N. Akhtar, N. Antil, A. Kumar, S. Shukla, W. Begum and K. Manna, Angew. Chem., Int. Ed., 2021, 60, 10964–10970 CrossRef CAS PubMed.
- M. Sha, L. Rao, W. Xu, Y. Qin, R. Su, Y. Wu, Q. Fang, H. Wang, X. Cui, L. Zheng, W. Gu and C. Zhu, Nano Lett., 2023, 23, 701–709 CrossRef CAS PubMed.
- Y. Zhang, J. Guo, L. Shi, Y. Zhu, K. Hou, Y. Zheng and Z. Tang, Sci. Adv., 2017, 3, e1701162 CrossRef PubMed.
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- W. Wang, X.-W. Song, Z. Hong, B. Li, Y. Si, C. Ji, K. Su, Y. Tan, Z. Ju, Y. Huang, C.-N. Chen and D. Yuan, Appl. Catal., B, 2019, 258, 117979 CrossRef CAS.
- S. S. Shinde, C. H. Lee, J.-Y. Jung, N. K. Wagh, S.-H. Kim, D.-H. Kim, C. Lin, S. U. Lee and J.-H. Lee, Energy Environ. Sci., 2019, 12, 727–738 RSC.
- S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942–15950 CrossRef CAS.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- P. Manna, J. Debgupta, S. Bose and S. K. Das, Angew. Chem., Int. Ed., 2016, 55, 2425–2430 CrossRef CAS PubMed.
- Z. Xue, K. Liu, Q. Liu, Y. Li, M. Li, C.-Y. Su, N. Ogiwara, H. Kobayashi, H. Kitagawa, M. Liu and G. Li, Nat. Commun., 2019, 10, 5048 CrossRef.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2023, 123, 445–490 CrossRef.
- L. Huang, J. Chen, L. Gan, J. Wang and S. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS.
- I. Liberman, R. Shimoni, R. Ifraemov, I. Rozenberg, C. Singh and I. Hod, J. Am. Chem. Soc., 2020, 142, 1933–1940 CrossRef CAS.
- E. M. Miner, T. Fukushima, D. Sheberla, L. Sun, Y. Surendranath and M. Dincă, Nat. Commun., 2016, 7, 10942 CrossRef CAS.
- R. G. Mariano, O. J. Wahab, J. A. Rabinowitz, J. Oppenheim, T. Chen, P. R. Unwin and M. Dinca, ACS Cent. Sci., 2022, 8, 975–982 CrossRef CAS PubMed.
- R. Shimoni, Z. Shi, S. Binyamin, Y. Yang, I. Liberman, R. Ifraemov, S. Mukhopadhyay, L. Zhang and I. Hod, Angew. Chem., Int. Ed., 2022, 61, e202206085 CrossRef CAS PubMed.
- Q. Huang, Q. Li, J. Liu, Y. R. Wang, R. Wang, L. Z. Dong, Y. H. Xia, J. L. Wang and Y.-Q. Lan, Matter, 2019, 1, 1656–1668 CrossRef.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. D. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
- C. Zou, T. Zhang, M.-H. Xie, L. Yan, G.-Q. Kong, X.-L. Yang, A. Ma and C.-D. Wu, Inorg. Chem., 2013, 52, 3620–3626 CrossRef CAS PubMed.
- H. Adamji, A. Nandy, I. Kevlishvili, Y. Román-Leshkov and H. J. Kulik, J. Am. Chem. Soc., 2023, 145, 14365–14378 CrossRef CAS PubMed.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y.-S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- J. Zheng, J. Ye, M. A. Ortuño, J. L. Fulton, O. Y. Gutiérrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, Y.-P. Chen, J. Park, Z. Wei, Y. Sun, M. Bosch, S. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 17714–17717 CrossRef CAS PubMed.
- J.-Y. Yeh, S. S. Chen, S.-C. Li, C. H. Chen, T. Shishido, D. C. W. Tsang, Y. Yamauchi, Y.-P. Li and K. C. W. Wu, Angew. Chem., Int. Ed., 2021, 60, 624–629 CrossRef CAS PubMed.
- H. He, Q. Sun, W. Gao, J. A. Perman, F. Sun, G. Zhu, B. Aguila, K. Forrest, B. Space and S. Ma, Angew. Chem., Int. Ed., 2018, 57, 4657–4662 CrossRef CAS PubMed.
- Z. Han, K. Wang, M. Wang, T. Sun, J. Xu, H.-C. Zhou, P. Cheng and W. Shi, Chem, 2023, 9, 2561–2572 CAS.
- C. Tan, X. Han, Z. Li, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16229–16236 CrossRef CAS PubMed.
- Y. Wu, W. Xu, L. Jiao, W. Gu, D. Du, L. Hu, Y. Lin and C. Zhu, Chem. Soc. Rev., 2022, 51, 6948–6964 RSC.
- L. Wang and Y. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 8351–8358 CrossRef CAS.
- X. Zhao, T. Yang, D. Wang, N. Zhang, H. Yang, X. Jing, R. Niu, Z. Yang, Y. Xie and L. Meng, Anal. Chem., 2022, 94, 4484–4494 CrossRef CAS.
- H. Zhao, Q. Xia, H. Xing, D. Chen and H. Wang, ACS Sustain, Chem. Eng., 2017, 5, 4449–4456 CAS.
- R. Rao, S. Ma, B. Gao, F. Bi, Y. Chen, Y. Yang, N. Liu, M. Wu and X. Zhang, J. Colloid Interface Sci., 2023, 636, 55–72 CrossRef CAS PubMed.
- Y. Liu, A. J. Howarth, N. A. Vermeulen, S.-Y. Moon, J. T. Hupp and O. K. Farha, Coord. Chem. Rev., 2017, 346, 101–111 CrossRef CAS.
- K. Ma, Y. H. Cheung, K. O. Kirlikovali, H. Xie, K. B. Idrees, X. Wang, T. Islamoglu, J. H. Xin and O. K. Farha, Adv. Mater., 2023, 2300951 CrossRef.
- D. T. Lee, J. Zhao, G. W. Peterson and G. N. Parsons, Chem. Mater., 2017, 29, 4894–4903 CrossRef CAS.
- J. Zhao, D. T. Lee, R. W. Yaga, M. G. Hall, H. F. Barton, I. R. Woodward, C. J. Oldham, H. J. Walls, G. W. Peterson and G. N. Parsons, Angew. Chem., Int. Ed., 2016, 55, 13224–13228 CrossRef CAS PubMed.
- A. Yao, X. Jiao, D. Chen and C. Li, ACS Appl. Mater. Interfaces, 2020, 12, 18437–18445 CrossRef CAS PubMed.
- C.-C. Chen, X. Han, X. Li, P. Jiang, D. Niu, L. Ma, W. Liu, S. Li, Y. Qu, H. Hu, J. Min, Y. Yang, L. Zhang, W. Zeng, J.-W. Huang, L. Dai and R.-T. Guo, Nat. Catal., 2021, 4, 425–430 CrossRef CAS.
- W. Zeng, X. Li, Y. Yang, J. Min, J.-W. Huang, W. Liu, D. Niu, X. Yang, X. Han, L. Zhang, L. Dai, C.-C. Chen and R.-T. Guo, ACS Catal., 2022, 12, 3033–3040 CrossRef CAS.
- S. Zhang, Q. Hu, Y.-X. Zhang, H. Guo, Y. Wu, M. Sun, X. Zhu, J. Zhang, S. Gong, P. Liu and Z. Niu, Nat. Sustainable, 2023, 6, 965–973 CrossRef.
- Y. Wu, X. Wang, K. O. Kirlikovali, X. Gong, A. Atilgan, K. Ma, N. M. Schweitzer, N. C. Gianneschi, Z. Li, X. Zhang and O. K. Farha, Angew. Chem., Int. Ed., 2022, 61, e202117528 CrossRef CAS PubMed.
- C. Ren, X. Wen, J. Mencius and S. Quan, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2101618118 CrossRef CAS PubMed.
- S. Kurbanoglu, C. Erkmen and B. Uslu, TrAC Trend. Anal. Chem., 2020, 124, 115809 CrossRef CAS.
- W. Wang and S. Gunasekaran, TrAC Trend. Anal. Chem., 2020, 126, 115841 CrossRef CAS.
- L. Jiao, W. Xu, Y. Wu, H. Yan, W. Gu, D. Du, Y. Lin and C. Zhu, Chem. Soc. Rev., 2021, 50, 750–765 RSC.
- W.-H. Chen, M. Vázquez-González, A. Kozell, A. Cecconello and I. Willner, Small, 2018, 14, 1703149 CrossRef PubMed.
- S. Li, X. Liu, H. Chai and Y. Huang, TrAC-Trend Anal. Chem., 2018, 105, 391–403 CrossRef CAS.
- H. Li, H. Liu, J. Zhang, Y. Cheng, C. Zhang, X. Fei and Y. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 40716–40725 CrossRef CAS PubMed.
- J.-W. Zhang, H.-T. Zhang, Z.-Y. Du, X. Wang, S.-H. Yu and H.-L. Jiang, Chem. Commun., 2014, 50, 1092–1094 RSC.
- M. Sha, W. Xu, Q. Fang, Y. Wu, W. Gu, C. Zhu and S. Guo, Chem. Catal., 2022, 2, 2552–2589 CrossRef CAS.
- W. Xu, L. Jiao, H. Ye, Z. Guo, Y. Wu, H. Yan, W. Gu, D. Du, Y. Lin and C. Zhu, Biosens. Bioelectron., 2020, 148, 111780 CrossRef CAS PubMed.
- X. Wei, S. Song, W. Song, Y. Wen, W. Xu, Y. Chen, Z. Wu, Y. Qin, L. Jiao, Y. Wu, M. Sha, J. Huang, X. Cai, L. Zheng, L. Hu, W. Gu, M. Eguchi, T. Asahi, Y. Yamauchi and C. Zhu, Chem. Sci., 2022, 13, 13574–13581 RSC.
- Y. Tang, Y. Chen, Y. Wu, W. Xu, Z. Luo, H.-R. Ye, W. Gu, W. Song, S. Guo and C. Zhu, Nano Lett., 2023, 23, 267–275 CrossRef CAS PubMed.
- W.-C. Hu, J. Pang, S. Biswas, K. Wang, C. Wang and X.-H. Xia, Anal. Chem., 2021, 93, 8544–8552 CrossRef CAS PubMed.
- X. Ruan, D. Liu, X. Niu, Y. Wang, C. D. Simpson, N. Cheng, D. Du and Y. Lin, Anal. Chem., 2019, 91, 13847–13854 CrossRef CAS PubMed.
- Y. Li, L. Wang, Z. Cui, S. Liu, S. Wang, J. Ren, Y. Tian, R. Shu, X. Luo, Y. Liao, J. Wang and D. Zhang, Sens. Actuators, B, 2022, 364, 131909 CrossRef CAS.
- Y. Zhong, Y. Chen, L. Chen, Y. Hu, X. Xiao, L. Xia and G. Li, Anal. Chem., 2023, 95, 6971–6979 CrossRef CAS PubMed.
- D. Wang, H. Wu, C. Wang, L. Gu, H. Chen, D. Jana, L. Feng, J. Liu, X. Wang, P. Xu, Z. Guo, Q. Chen and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 3001–3007 CrossRef CAS PubMed.
- L.-H. Fu, C. Qi, J. Lin and P. Huang, Chem. Soc. Rev., 2018, 47, 6454–6472 RSC.
- M. Chang, M. Wang, M. Wang, M. Shu, B. Ding, C. Li, M. Pang, S. Cui, Z. Hou and J. Lin, Adv. Mater., 2019, 31, 1905271 CrossRef CAS PubMed.
- Y. Ding, H. Xu, C. Xu, Z. Tong, S. Zhang, Y. Bai, Y. Chen, Q. Xu, L. Zhou, H. Ding, Z. Sun, S. Yan, Z. Mao and W. Wang, Adv. Sci., 2020, 7, 2001060 CrossRef CAS PubMed.
- F. Yang, J. Dong, Z. Li and Z. Wang, ACS Nano, 2023, 17, 4102–4133 CrossRef CAS PubMed.
- X. Pan, W. Wang, Z. Huang, S. Liu, J. Guo, F. Zhang, H. Yuan, X. Li, F. Liu and H. Liu, Angew. Chem., Int. Ed., 2020, 59, 13557–13561 CrossRef CAS PubMed.
- D. Wang, H. Wu, S. Z. F. Phua, G. Yang, W. Qi Lim, L. Gu, C. Qian, H. Wang, Z. Guo, H. Chen and Y. Zhao, Nat. Commun., 2020, 11, 357 CrossRef CAS PubMed.
- X. Liu, Z. Yan, Y. Zhang, Z. Liu, Y. Sun, J. Ren and X. Qu, ACS Nano, 2019, 13, 5222–5230 CrossRef CAS PubMed.
- Z. Liu, F. Wang, J. Ren and X. Qu, Biomaterials, 2019, 208, 21–31 CrossRef CAS PubMed.
- L. K. Jennings, K. M. Storek, H. E. Ledvina, C. Coulon, L. S. Marmont, I. Sadovskaya, P. R. Secor, B. S. Tseng, M. Scian, A. Filloux, D. J. Wozniak, P. L. Howell and M. R. Parsek, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11353–11358 CrossRef CAS PubMed.
- M. Ghorbani, H. Derakhshankhah, S. Jafari, S. Salatin, M. Dehghanian, M. Falahati and A. Ansari, Nano Today, 2019, 29, 100775 CrossRef CAS.
- L. Zhang, Y. Zhang, Z. Wang, F. Cao, Y. Sang, K. Dong, F. Pu, J. Ren and X. Qu, Mater. Horiz., 2019, 6, 1682–1687 RSC.
- D. Chao, Q. Dong, Z. Yu, D. Qi, M. Li, L. Xu, L. Liu, Y. Fang and S. Dong, J. Am. Chem. Soc., 2022, 144, 23438–23447 CrossRef CAS PubMed.
- W. Xu, X. Cai, Y. Wu, Y. Wen, R. Su, Y. Zhang, Y. Huang, Q. Zheng, L. Hu, X. Cui, L. Zheng, S. Zhang, W. Gu, W. Song, S. Guo and C. Zhu, Nat. Commun., 2023, 14, 6064 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |