
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycosyl oxazolines serve as active donors for iterative synthesis of type I oligosaccharides†
Nitish Verma abc,
Zhijay Tua,
Septila Renataacd and
Chun-Hung Lin*ace
abc,
Zhijay Tua,
Septila Renataacd and
Chun-Hung Lin*ace
aInstitute of Biological Chemistry, Academia Sinica, No. 128, Academia Road, Section 2, Nan-kang, Taipei 11529, Taiwan. E-mail: chunhung@gate.sinica.edu.tw
bDepartment of Chemistry, National Tsing Hua University, Taiwan
cChemical Biology and Molecular Biophysics, Taiwan International Graduate Program, Academia Sinica, Taiwan
dInstitute of Bioinformatics and Structural Biology, College of Life Science, National Tsing Hua University, Taiwan
eDepartment of Chemistry and Institute of Biochemical Sciences, National Taiwan University, Taiwan
First published on 6th September 2024
Abstract
Synthesis of Galβ1 → 3GlcNAc-repeating saccharides is limited mainly by the formation of less-reactive oxazolines. We herein report an expeditious approach that requires trichloroacetyloxazolines as reactive glycosyl donors. Using only two disaccharide building blocks, the iterative oxazoline formation and glycosylation synthesized hexa- and octasaccharides with overall yields of 47% and 26% in four and six steps, respectively.
Type I LacNAc (Galβ1 → 3GlcNAc)-repeating oligosaccharides are a unique structural epitope on the surface of several cancer cell types. For instance, they occur on the surface of colon cancer cells1–4 and exist as the backbone of Lewis a—Lewis a and Lewis b—Lewis a antigens.5,6 They also serve as ligands for cancer-related proteins, such as galectin-37 and mannose-binding protein.4 Efficient preparation of these glycans is necessary to study their biological potential.
Few efforts, however, have been made to carry out the synthesis of these type I oligomers.7–9 Thioglycoside-based synthesis is commonly used to prepare a variety of glycans, but this approach has a few longstanding challenges.10–13 For example, an aglycon transfer usually occurs if the reactivities of the thioglycoside acceptor and donor are not significantly different.10,12,14 When glucosamine-derived donors are used for glycosylation, amide- and carbamate-based groups are usually used to protect the amino group at C2 and at the same time to establish stereoselective glycosylations. Nevertheless, the formation of oxazoline is often observed,15–17 which prevents further elongation.
We recently reported the synthesis of type I oligosaccharides by chemoselective glycosylations.13 The threshold of the reactivity difference between thioglycoside donors and acceptors is critical for obtaining optimal yields. However, it is not trivial to measure the reactivity of saccharides, especially long glycans. Glycosylation often leads to severe aglycon transfer from a shorter acceptor thioglycoside to a longer donor thioglycoside, resulting in acceptor decomposition/hydrolysis and donor regeneration. This results in moderate to low yields.10–12 To develop an efficient method for the rapid assembly of type I oligosaccharides, an ideal approach involves using as few glycosyl donors and acceptors as possible, and not measuring the reactivities of these thioglycosides.
Although trichloroacetyl- and trifluoroacetyloxazolines have previously been used as donors for glycosylation, they are either electronically or sterically disarmed.18–22 Meanwhile, acceptors that have previously been used contain orthogonal protecting groups (e.g., OTBDPS or OBn) at the anomeric position,20,21 requiring extra steps at a later stage to extend the glycan chains. Here we describe an iterative approach that uses reactive trichloroacetyloxazolines as donors to react with thioglycoside acceptors. Because thioglycoside activation was used for oxazoline formation only, there was no aglycon transfer during glycosylation. The product contained an anomeric STol and could be readily converted to another oxazoline donor. The iterative oxazoline formation and glycosylation were performed to give type I LacNAc octasaccharide with a yield of 26% (within six steps) and with exclusive β-stereoselectivity.
Our synthetic endeavour began with the conversion of thioglycoside disaccharide 1 to oxazoline 2 (Table S1, ESI†). First, Ph2SO-based preactivation was examined in the presence of 2,4,6-tri-t-butylpyrimidine (TTBP) as the base.23 Although 2 was obtained with an excellent yield (90%, entry 1, Table S1, ESI†), a trace amount of 1 remained, which was observed by crude 1H NMR. Increasing the amount of activator (Ph2SO and Tf2O) to 0.65 equiv. led to quantitative formation of 2 (100%, entry 2). Additionally, 1 was activated in the absence of TTBP to form 2 with a 93% yield (entry 3) with no starting material left. Next, preactivation using NIS and TMSOTf (entry 4) was completed within 15 min with clean formation of oxazoline 2, with the characteristic doublet signal of H1 at 5.86 ppm in the 1H NMR spectrum of the crude reaction mixture (Fig. S1, ESI†).24 In addition, the signals of H1 and H2′ (of the adjacent Gal) appeared to be clearly observed in 5–6 ppm in the 1H NMR spectra (Fig. S1, ESI†). With oxazoline 2 available as the donor, the corresponding glycosylation was studied by screening different promoters. The combination of NHTCA with benzylidene protection on GlcNAc–based donors (e.g., 2b, Scheme S1, ESI†) leads to the formation of a less-reactive oxazoline.15–17 In contrast, oxazoline 2, having armed-type (OBn) protecting groups at O4- and O6-positions in GlcNTCA, is presumably more reactive. We synthesized and used oxazolines 2 and 2b for a systematic comparison and to couple with cyclohexanol to study the glycosylation in the presence of various promoters.
In the presence of Brønsted acids (0.2 equiv.) such as TfOH and Tf2NH, oxazoline 2 coupled with cyclohexanol for 1 h to result in good yields (entries 1 and 2, Table S2, ESI†). The use of TMSOTf as the promoter at −50 °C (entry 3) resulted in an excellent yield (94%). Among the various metal triflates examined, no reaction happened with mild Lewis acids, such as Cu(OTf)2 and Sm(OTf)3 (entries 4 and 7). Most of the starting material in these cases remained unreacted, and a trace of product was observed (<5%, based on TLC analysis). In fact, raising the temperature from −50 to −20 °C was not useful for the reaction of Cu(OTf)2 (entry 5). The reaction catalyzed by In(OTf)3 was found to be slightly more reactive (24%, entry 6). Interestingly, the reactions of Sn(OTf)2 and Hf(OTf)4 provided good yields (entries 8 and 9). Comparing metal chlorides, FeCl3 appeared to be better than AlCl3 and AuCl3 (entries 10–12) despite incomplete reaction (entry 12).
Furthermore, when using oxazoline 2b for the glycosylations, reaction time was prolonged to 2 h as a result of the lower reactivity (Table S2, ESI†). The reactions of 2b displayed a similar trend to those of 2, albeit with lower yields (entries 13–19), suggesting that oxazoline 2 appears to be an armed donor and thus qualifies as a suitable donor for further study. Meanwhile, considering the cost of Lewis acid promoters and glycosylation yield, we decided to proceed further with TMSOTf as a promoter.
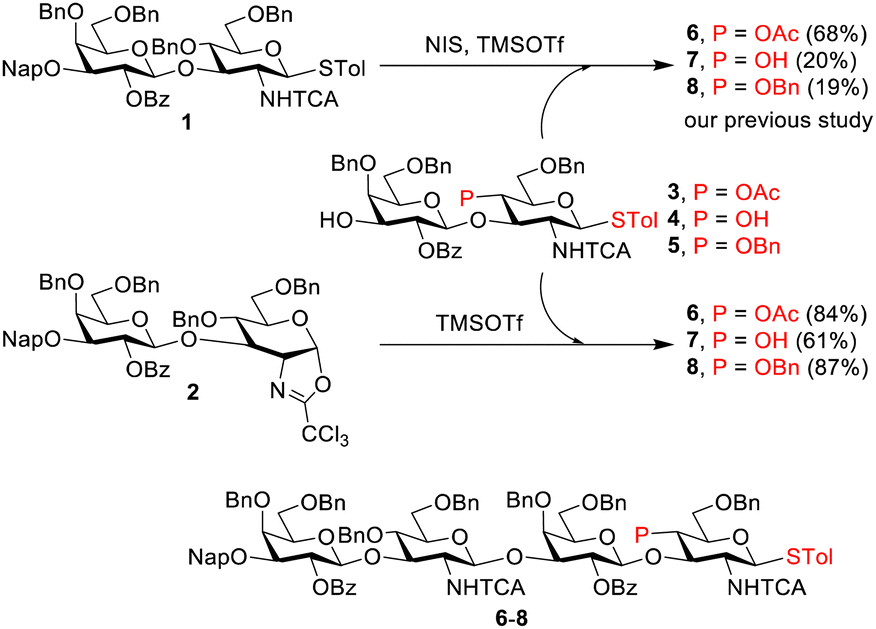
Under optimized conditions, pure oxazoline 2 was subjected to glycosylation with disaccharide acceptors 3–5 (Scheme 1). In the previous chemoselective glycosylations between thioglycoside donor 1 and acceptors 3–5, low to moderate yields of tetrasaccharide products (19–68%) were observed,13 because of an insufficient reactivity difference between the donor and acceptor. To solve this problem, we decided to explore using oxazoline 2 as the donor for glycosylation. As oxazoline can be selectively activated with TMSOTf as the promoter in the absence of thiophilic reagents, the activation of acceptor STol cannot occur or can be minimized.
 | ||
| Scheme 1 [2 + 2] Glycosylations of donors 1 and 2 with disaccharide acceptors 3–5. | ||
Oxazoline 2 reacted cleanly with acceptor 3, leading to an excellent yield of tetrasaccharide 6 (84%, Scheme 1; also see entry 1, Table S3, ESI†). The reactions with armed acceptors (4 and 5) also provided better yields (entries 2–9) than the use of donor 1, suggesting that the usage of an oxazoline donor indeed prevented aglycon transfer. Moderate yields (55% and 61%, entries 2 and 3) were obtained for the reaction of 2 with acceptor 4, owing to the formation of an undesired product. Therefore, optimization was continued with acceptor 5. Decreasing the amount of 2 to 1.1 equiv. marginally decreased the yield for 8 to 70% (compare entries 4 and 5) in conjunction with unreacted 5. Hence, the reaction time was doubled to 2 h, leading to an improved yield (8, 87%, entry 6). An increase in glycosylation temperature from −60 °C to −50 °C led to no change in yield (compare entries 6 and 8). Moreover, the yield was decreased by a negligible amount to 84% by either lowering the reaction temperature to −70 °C (comparing entries 6 and 7) or decreasing the promoter amount to 0.1 equiv. (entry 9).
In addition, we generated 2 in situ from 1 and then coupled it with acceptor 5 without purification (Table S4, ESI†). The glycosidation was initiated by adding promoters at a low temperature and was quenched with the addition of Et3N, followed by workup. The NIS/TMSOTf activation method was found to provide higher yields (70–74% over two steps, entries 2 and 3, Table S4, ESI†) than the Ph2SO/Tf2O activation method (58%, entry 1). It is noteworthy that tetrasaccharide product 8 is equipped with STol at the reducing end, allowing further activation to oxazoline 9 for the next round of glycosylation.
Several conditions were examined to convert 8 into oxazoline 9 (Table S5, ESI†). First, when applying the Ph2SO/Tf2O-based condition, most of the starting material remained intact, even with prolonged stirring. Another 0.65 equiv. of Tf2O was thus added to the reaction mixture after 3.75 h. The reaction was quenched another 2 h later to give oxazoline 9 with a 45% yield (entry 1, Table S5, ESI†), in conjunction with several tailing spots based on subsequent TLC. The NIS/TMSOTf activation (entries 2–9) appeared to be better than the Ph2SO-based activation. The activation in the presence of NIS (1.2 equiv.)/TMSOTf (0.2 equiv.)(see entry 4, Table S1, ESI†) produced 9 at a low yield (entry 2) that was accompanied by many inseparable side products, as indicated by TLC analysis. Reducing the amount of TMSOTf to 0.1 equiv. led to an encouraging 55% yield (entry 3). When the activation was performed at −60 °C with an increase in the reaction time to 1.5 h, 9 was produced with an 87% yield (entry 4), as determined by clean crude 1H NMR of the reaction mixture (Fig. S1, ESI†). Further lowering the temperature to −70 °C decreased the yield of 9 to 63% (entry 5). The yield was further improved to 76% after modifying several factors, including the amount of molecular sieves (entry 5 vs. 6), reaction time (entry 6 vs. 7) and the concentration of 8 (entry 7 vs. 8). Finally, the gradual increase in temperature from −70 to −50 °C generated a yield of 82% (entry 9).
Furthermore, oxazoline 9 was coupled with acceptor 5 through [4 + 2] glycosylation, yielding hexasaccharide 10 with a 77% yield (Scheme 2). [2 + 4] Glycosylation was also examined between disaccharide oxazoline 2 and tetrasaccharide acceptor 8a (Scheme 2), resulting in 10 with a 76% yield, with concomitant recovery of 2 (25% relative to the initial amount of 2). These conditions were then used to improve the synthesis of hexasaccharide 15 that was previously obtained with a 52% yield by chemoselective glycosylation of 14 with 8 (Scheme S2, ESI†).13 Tetrasaccharide 8 (1.1 equiv.) was first activated to oxazoline 9 under the optimized condition (1.3 equiv. of NIS and 0.1 equiv. of TMSOTf at −60 °C). Without purification, the crude 9 was coupled with 14 (1.0 equiv.) in the presence of TMSOTf (0.2 equiv.) for 3.5 h to obtain hexasaccharide 15 with a 65% yield over two steps.
 | ||
| Scheme 2 Synthesis of hexasaccharide 10 by [4 + 2] and [2 + 4] glycosylation by using oxazolines 9 and 2, respectively. | ||
Having STol on the reducing end, hexasaccharide 10 was activated by NIS/TMSOTf at −70 °C. The reaction was sluggish to produce oxazoline 11 with a 50% yield (entry 1, Table S6, ESI†) in addition to recovered 10 (9% relative to the initial amount). An increase in the reaction time was not useful (entry 2). Hence, the activation was performed at −50 °C to obtain 11 with a 67% yield, according to 1H NMR analysis of the crude product (entry 3).
We then investigated the synthesis of type I hexa- and octasaccharides in an iterative manner. Because oxazoline formation had been optimized, the resulting product is presumably ready for subsequent glycosylations, without the need of a chromatographic step. Furthermore, because glycosylation with oxazoline as the donor uses TMSOTf as the promoter, the reaction product is still a thioglycoside that can be further activated to form an elongated oxazoline donor by the mere addition of NIS. The activation and glycosylation procedure can thus be iteratively performed to yield a final desired saccharide.
1 was first activated to oxazoline 2 by using the condition in entry 4 of Table S1, followed by simple workup (see the ESI† for the details), as shown in step I in Scheme 3. Crude oxazoline 2 was then coupled with acceptor 5 by [2 + 2] glycosylation in the presence of TMSOTf (0.1 equiv.) at −50 °C, producing tetrasaccharide 8 (step II), which was further activated in situ by addition of NIS (1.1 equiv.) at −60 °C (step III). We did not add any TMSOTf at this stage because step II used 0.1 equiv. of TMSOTf and the resulting mixture was directly subjected to step III without either quenching or workup. After formation of oxazoline 9 and subsequent quenching and workup, crude 9 was coupled with 5 via [4 + 2] glycosylation in the presence of TMSOTf (0.14 equiv., step IV). The product, hexasaccharide 10, was obtained at a 47% isolated yield over four steps (i.e., >80% for each step).
 | ||
| Scheme 3 Synthesis of type I hexa- and octasaccharides 10 and 12, respectively, by [2 + 2 + 2] and [2 + 2 + 2 + 2] iterative oxazoline formation (blue arrows) and glycosylations (red arrows). | ||
Likewise, 10 was further activated in situ by addition of NIS (1 equiv.) at −50 °C to produce oxazoline 11 (step V). After quenching and workup, crude oxazoline 11 was coupled with 5 through [6 + 2] glycosylation (step VI) with a gradual increase in temperature from −60 to −40 °C over 5.5 h. The desired octasaccharide (12) was purified, resulting in a 26% yield in six steps. It is critical to raise the temperature and increase the amount of TMSOTf for the glycosylation step when long oxazoline donors are used. For example, [2 + 2] glycosylation was performed at −50 °C with 0.1 equiv. of TMSOTf, as compared with [4 + 2] (−60 to −50 °C, 0.2 equiv. of TMSOTf) and [6 + 2] (−60 to −40 °C, 0.48 equiv. of TMSOTf w.r.t. oxazoline 11) glycosylations. In particular, TMSOTf has to be carefully added at a low temperature, but the reaction needs to be carried out at a higher temperature, especially when a longer oxazoline is used. As a consequence, we recommend gradually increasing the temperature after the addition of TMSOTf at an initial lower temperature.
In summary, we have demonstrated the usage of reactive oxazolines as glycosyl donors to solve the existing problem of aglycon transfer when thioglycoside-based glycosylation is carried out. The whole procedure used only two disaccharide building blocks, 1 and 5 (the latter of which was derived from the former in one step), without measuring the relative reactivity of any donors or acceptors. The iterative oxazoline formation and glycosylation represent an economic, rapid, and efficient synthesis. Using NIS and TMSOTf only, type I LacNAc hexa- and octasaccharides (10 and 12) were synthesized with 47% (over four steps) and 26% (six steps) overall yields, respectively. Starting from 1, 12 was obtained in seven steps (16% total yield) with two column chromatography–based purifications, a process that was completed within 18 h.
This work was supported by Academia Sinica (AS-GC-110-MD04, AS-GC-110-04) and National Science and Technology Council (112-2113-M-001-010), Taiwan. We thank Academia Sinica High-Field NMR Center, and NMR facility of the Institute Biological Chemistry, Academia Sinica, Taiwan for technical support.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- M. Terada, K.-H. Khoo, R. Inoue, C.-I. Chen, K. Yamada, H. Sakaguchi, N. Kadowaki, B. Y. Ma, S. Oka, T. Kawasaki and N. Kawasaki, J. Biol. Chem., 2005, 280, 10897–10913 CrossRef CAS PubMed.
- Y.-Y. Fan, S.-Y. Yu, H. Ito, A. Kameyama, T. Sato, C.-H. Lin, L.-C. Yu, H. Narimatsu and K.-H. Khoo, J. Biol. Chem., 2008, 283, 16455–16468 CrossRef CAS PubMed.
- C.-H. Lin, Y.-Y. Fan, Y.-Y. Chen, S.-H. Wang, C.-I. Chen, L.-C. Yu and K.-H. Khoo, Glycobiology, 2009, 19, 418–427 CrossRef CAS.
- N. Kawasaki and T. Kawasaki, Trends Glycosci. Glycotechnol., 2010, 22, 141–151 CrossRef CAS.
- M. R. Stroud, S. B. Levery, E. D. Nudelman, M. E. Salyan, J. A. Towell, C. E. Roberts, M. Watanabe and S. Hakomori, J. Biol. Chem., 1991, 266, 8439–8446 CrossRef CAS PubMed.
- N. Kawasaki, C.-W. Lin, R. Inoue, K.-H. Khoo, N. Kawasaki, B.-Y. Ma, S. Oka, M. Ishiguro, T. Sawada, H. Ishida, T. Hashimoto and T. Kawasaki, Glycobiology, 2009, 19, 437–450 CrossRef CAS PubMed.
- T. Fischoder, D. Laaf, C. Dey and L. Elling, Molecules, 2017, 22, 1320 CrossRef PubMed.
- M. Henze, S. Schmidtke, N. Hoffmann, H. Steffens, J. Pietruszka and L. Elling, ChemCatChem, 2015, 7, 3131–3139 CrossRef CAS.
- D. Kobayashi, A. Ueki, T. Yamaji, K. Nagao, A. Imamura, H. Ando, M. Kiso and H. Ishida, Molecules, 2016, 21, 614 CrossRef PubMed.
- C. Arboe Jennum, T. Hauch Fenger, L. M. Bruun and R. Madsen, Eur. J. Org. Chem., 2014, 3232–3241 CrossRef CAS.
- Z. Tu, P.-K. Liu, M.-C. Wu and C.-H. Lin, Isr. J. Chem., 2015, 55, 325–335 CrossRef CAS.
- Z. Li and J. C. Gildersleeve, J. Am. Chem. Soc., 2006, 128, 11612–11619 CrossRef CAS.
- N. Verma, Z. Tu, M.-S. Lu, S.-H. Liu, S. Renata, R. Phang, P.-K. Liu, B. Ghosh and C.-H. Lin, J. Org. Chem., 2021, 86, 892–916 CrossRef CAS PubMed.
- Z. Tu, H.-W. Hsieh, C.-M. Tsai, C.-W. Hsu, S.-G. Wang, K.-J. Wu, K.-I. Lin and C.-H. Lin, Chem. – Asian J., 2013, 8, 1536–1550 CrossRef CAS PubMed.
- J. Dinkelaar, J. D. Codee, L. J. van den Bos, H. S. Overkleeft and G. A. van der Marel, J. Org. Chem., 2007, 72, 5737–5742 CrossRef CAS PubMed.
- X. Lu, M. N. Kamat, L. Huang and X. Huang, J. Org. Chem., 2009, 74, 7608–7617 CrossRef CAS PubMed.
- X. Lu and P. Kovac, J. Org. Chem., 2016, 81, 6374–6394 CrossRef CAS.
- S. Nakabayashi, C. D. Warren and R. W. Jeanloz, Carbohydr. Res., 1986, 150, C7–C10 CrossRef CAS PubMed.
- M. Colon, M. M. Staveski and J. T. Davis, Tetrahedron Lett., 1991, 32, 4447–4450 CrossRef CAS.
- G. Blatter, J. M. Beau and J. C. Jacquinet, Carbohydr. Res., 1994, 260, 189–202 CrossRef CAS PubMed.
- G. Despras, A. Alix, D. Urban, B. Vauzeilles and J. M. Beau, Angew. Chem., Int. Ed., 2014, 53, 11912–11916 CrossRef CAS PubMed.
- T. J. Donohoe, J. G. Logan and D. D. Laffan, Org. Lett., 2003, 5, 4995–4998 CrossRef CAS PubMed.
- J. D. C. Codée, R. E. J. N. Litjens, R. den Heeten, H. S. Overkleeft, J. H. van Boom and G. A. van der Marel, Org. Lett., 2003, 5, 1519–1522 CrossRef.
- The 1H NMR signal at 5.86 ppm (d, J = 7.3 Hz, 1H, H−1) was used to identify the formation of oxazoline 2 in the crude reaction mixture. For the purpose of quantitation, there was an extra addition of dimethyl terephthalate to the mixture as the internal standard because of the distinguishable signal at 8.1 ppm (s, 4H, Ar-H). Please see the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of oxazolines 2 and 2b, glycosylation of oxazoline donors with cyclohexanol with different promoters, glycosylation of in situ-generated oxazoline 2 with acceptor 5, improved synthesis of hexasaccharide 15 by coupling of 9 and 14, synthesis of hexasaccharide oxazoline 11, and NMR spectra of new compounds. See DOI: https://doi.org/10.1039/d4cc03247k |
| This journal is © The Royal Society of Chemistry 2024 |