
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomaterial engineering strategies for B cell immunity modulations
Ali
Zareein†
ab,
Mina
Mahmoudi†
ab,
Shruti Sunil
Jadhav
 a,
Joel
Wilmore
c and
Yaoying
Wu
*abc
a,
Joel
Wilmore
c and
Yaoying
Wu
*abc
aDepartment of Biomedical Engineering, Syracuse University, Syracuse, NY, USA. E-mail: ywu294@syr.edu; Fax: +315 443 4891; Tel: +315 443 4891
bThe BioInspired Institute for Material and Living Systems, Syracuse University, Syracuse, NY, USA
cDepartment of Microbiology & Immunology, SUNY Upstate Medical University, Syracuse, NY, USA
First published on 23rd February 2024
Abstract
B cell immunity has a penetrating effect on human health and diseases. Therapeutics aiming to modulate B cell immunity have achieved remarkable success in combating infections, autoimmunity, and malignancies. However, current treatments still face significant limitations in generating effective long-lasting therapeutic B cell responses for many conditions. As the understanding of B cell biology has deepened in recent years, clearer regulation networks for B cell differentiation and antibody production have emerged, presenting opportunities to overcome current difficulties and realize the full therapeutic potential of B cell immunity. Biomaterial platforms have been developed to leverage these emerging concepts to augment therapeutic humoral immunity by facilitating immunogenic reagent trafficking, regulating T cell responses, and modulating the immune microenvironment. Moreover, biomaterial engineering tools have also advanced our understanding of B cell biology, further expediting the development of novel therapeutics. In this review, we will introduce the general concept of B cell immunobiology and highlight key biomaterial engineering strategies in the areas including B cell targeted antigen delivery, sustained B cell antigen delivery, antigen engineering, T cell help optimization, and B cell suppression. We will also discuss our perspective on future biomaterial engineering opportunities to leverage humoral immunity for therapeutics.
 Ali Zareein | Ali Zareein is a PhD student at Syracuse University. He obtained his Bachelor degree in Biomedical Engineering from Islamic Azad University. His current research focuses on the development of biomaterials tailored to interact with immune cells, with the aim of enhancing antibody responses for diverse therapeutic applications. |
 Mina Mahmoudi | Mina Mahmoudi is a PhD student in the Department of Biomedical and Chemical Engineering at Syracuse University. She obtained her BS from Islamic Azad University and her MS in medical microbiology from Ilam University of Medical Science. Mina's work seeks to advance our understanding of antibody responses. |
 Shruti Sunil Jadhav | Shruti Sunil Jadhav is an MS student in Biotechnology at Syracuse University. She obtained her Bachelor of Technology degree in Biotechnology from DY Patil University, Navi Mumbai. Her research is focused on biomaterial-mediated delivery. |
 Joel Wilmore | Joel Wilmore is an Assistant Professor in the Microbiology and Immunology Department at SUNY Upstate Medical University. He received his BS in Biotechnology from Rochester Institute of Technology and his PhD from SUNY Upstate Medical University. After completing his postdoctoral training at the University of Pennsylvania, he started his independent research group focused on the interaction of the gut microbiome and IgA plasma cell responses. |
 Yaoying Wu | Yaoying Wu is an Assistant Professor of Biomedical Engineering at Syracuse University. He received his BS in Materials Science and Engineering from Tianjin University, MS in Polymer Chemistry from Beijing University of Chemical Technology, and PhD in Chemistry from the University of Minnesota, Twin Cities, before completing his postdoctoral training at Duke University. His research team develops B cell modulation strategies for disease prevention and treatment. |
1. Introduction
B cells and the antibodies they produce profoundly impact many aspects of human health, such as infection,1 inflammation,2,3 autoimmunity,4,5 and malignancies.6,7 Antibodies are crucial for the neutralization and opsonization of invading pathogens during infections,8–11 and are responsible for immune-surveillance against early carcinogenesis.12,13 Mucosal antibodies also play pivotal roles in regulating the gut microbiome and maintaining homeostasis.14 Tumor-infiltrating B cells regulate the tumor microenvironment through cytokine secretion, antigen presentation, tumor-associated antibody production, and tumor-associated tertiary lymphoid structures (TLSs) formation.6,7,15–19 B cells also drive autoimmunity by producing autoreactive antibodies and promoting the differentiation of pathogenic T cells.3,20,21 Because of these central functions of B cells in many human diseases and conditions, modulating B cell responses has been an effective target for therapeutics.Biomaterial engineering strategies have been examined extensively for a broad range of therapeutic and diagnostic purposes, including the targeted delivery of antigens or immunomodulatory biologics to specific tissues or cell types,22–24 the temporally controlled release of biologics,25,26 local immune niches for therapeutics or diagnosis,27–31 and the promotion of tissue regeneration and immune tolerance.32–35 Leveraging biomaterial platforms for modulating B cell immunity has already shown significant progress in preclinical and clinical research, with SARS-CoV-2 lipid nanoparticle (LNP) mRNA vaccine as the most prominent research achievement in this field.36,37 Current B cell modulation efforts are focused on tackling several significant therapeutic challenges utilizing a variety of biomaterials engineering strategies, including the elicitation of broadly neutralizing antibodies (bnAbs),38 the generation of mucosal immunity,39 the promotion of anti-tumor B cell responses,40 and the suppression of allergy- or autoimmune-associated B cells.41,42
In this review, we aim to highlight recent biomaterial strategies for B cell modulation in different therapeutic applications, with a focus on engineering innovations. We also provide a brief overview of B cell immune responses and introduce several factors impacting B cell responses that biomaterial engineers need to consider when designing therapeutic strategies. Lastly, we will share our perspective on future biomaterial engineering opportunities for different diseases.
2. Overview of B cell responses
B cells respond in a multi-step process that involves cellular and molecular regulations at various stages ranging from the initial antigen presentations to the eventual B cell differentiation. This complex regulatory network provides engineering opportunities for modulating B cell responses at various stages. Herein, we briefly summarize the cellular dynamics of B cell responses, and direct the reader to recent advances for more comprehensive information.B cell antigen acquisition is one of the first engineering opportunities to promote B cell responses. B cells primarily encounter cognate antigens in secondary lymphoid organs (SLOs), i.e. lymph nodes (LNs), spleens, and Peyer's patches43 (Fig. 1A). Antigen size significantly influences antigen trafficking. Low molecular weight soluble antigens (<70 kDa) or particles with a diameter of around 5 nm can diffuse into the B cell follicles directly through pores in the subcapsular sinus region.44–46 However, large antigens, such as immune complexes and viral particles, rely on intermediate cells to be transported to follicular B cells. Different specialized resident cells play this intermediate role in different SLOs depending on the routes of antigen exposure. In LNs, it is the subcapsular sinus macrophages (SSM; CD169+) that capture antigens from the incoming lymph and mediate antigen presentation to the follicular B cells47–50 (Fig. 1B). In the spleen, the responsibility of antigen transport is shared by several types of cell, including marginal zone B cells (MZB; IgM+IgDlo)51 and marginal metallophilic macrophages.52,53 The process of antigen presentation to B cells in Peyer's patches is not well understood, but the antigens are shown to be predominantly acquired by modified epithelial cells (M cells) via transcytosis at the follicle-associated epithelium.54 The antigen acquisition is receptor-mediated and often depends on the molecular features of the antigens. For example, SSM cells express various receptors for antigen retention, including macrophage receptor 1 (MAC1) for engaging complement component 3 (C3) deposited cell surface antigens,48 dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN; also known as CD209) for retaining glycosylated antigens,55 and CD169 for recognizing sialylated pathogens.49
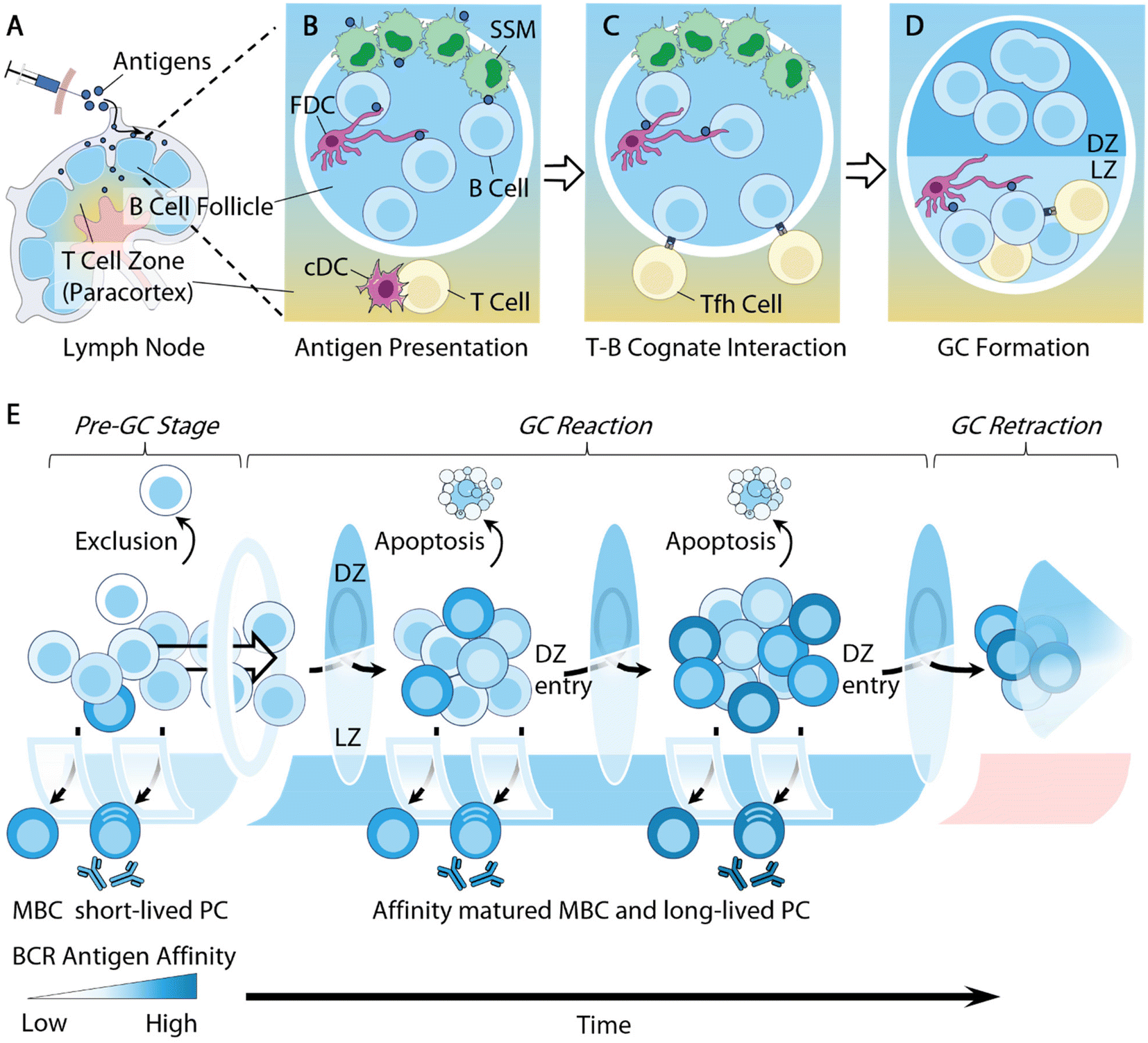 | ||
| Fig. 1 Overview of B cell responses. (A) Antigens accumulate in dLN following administration. (B) Antigen presentations. Opsonized antigens are acquired by subcapsular sinus macrophages (SSM) from the subcapsular sinus and handed off to follicular dendritic cells (FDC) in the B cell follicle; native CD4+ T cells receive antigen presentation by conventional dendritic cells (cDC) at the paracortex and differentiate into early follicular T helper (Tfh) cells. (C) Antigen-stimulated B cells migrate toward the T–B border to seek cognate interactions with Tfh cells. (D) B cells that received sufficient T cell help spontaneously organize into germinal center (GC) structures that are separated into two distinct components: the light zone (LZ) is composed of LZ B cells, Tfh cells and FDC; the dark zone (DZ) is mainly composed of DZ B cells that undergo somatic hypermutations (SHMs). (E) An overview of the dynamics of GC affinity selection. Prior to GC entry, B cells with a low antigen affinity are excluded from GC entry; relatively high affinity B cells differentiate extrafollicularly into short-lived plasma cells (PCs) and memory B cells (MBCs). Cells with intermediate antigen affinity are often selected to enter GC reactions. GC B cells go through rounds of mutation, selection, and expansion. During the GC reactions, some high-affinity B cells exit as MBCs and long-lived PCs, while low-affinity B cells undergo apoptosis. Other B cells potentially enter into the next rounds of mutation and selection. Eventually the GC retracts and the GC B cell population dwindles. | ||
Follicular dendritic cells (FDCs), a type of stromal cell in B cell follicles, are another type of intermediate cell for antigen presentation to B cells. They are responsible for retaining antigens using complement receptors, allowing for B cells to test the antigen affinity of their B cell receptors (BCRs)48,50,51,56 (Fig. 1C). Additionally, owing to the low extracellular protease activity within B cell follicles,57 FDCs can preserve antigens for extended periods of time, serving as important antigen depots to facilitate BCR affinity maturation.58 Some recent biomaterial strategies can target antigen delivery to these intermediate cells for B cell antigen acquisition. We will highlight several relevant studies in following sections.
Follicular B cells rely on follicular T helper (Tfh) cells for critical survival signals in order to further differentiate and mature. Tfh cells are a specialized subtype of CD4+ T cells licensed for B cell follicle entry by expressing the transcription factor Bcl-6 and the chemokine receptor CXCR5 (C–X–C chemokine receptor type 5), derived from naive CD4+ T cells that receive antigen presentation by dendritic cells in the LNs59 (Fig. 1B). They support B cell survival by providing T cell help via secreted cytokines, such as IL-4, and the surface expression of CD40L.
B cells activated by cognate antigens migrate to the T–B cell borders and establish cognate interactions with Tfh cells for T cell help59 (Fig. 1C). A subset of these B cells will spontaneously coalesce into tight clusters in close association with FDCs, giving rise to microanatomical structures within B cell follicles, namely germinal centers (GCs)61,62 (Fig. 1D). B cell entry into the GCs is significantly influenced by their germline affinity of BCRs to antigen60 (Fig. 1E). Non-specific B cells are largely excluded from GCs.63,64 B cells of high germline antigen affinity proliferate and differentiate extrafollicularly likely due to strong BCR stimulation,61 and develop into early memory B cells (MBCs) and plasmablasts that produce antibodies with little somatic mutations on BCRs.62–64 Only B cells with a relatively median germline antigen affinity are selected for GC entry. The density of the peptide/MHC II (pMHCII) complex expressed by B cells was also shown to regulate GC selections, where B cells with a low pMHCII density are largely excluded from GC entry.65
Within GCs, B cells undergo Darwinian-like selection and affinity maturation. B cells organize into two distinct zones, i.e. the light zone (LZ) and dark zone (DZ) (Fig. 1D and E). The LZs are composed of Tfh cells, FDCs, and LZ B cells or centrocytes, while the DZs contain predominantly DZ B cells or centroblasts and newly defined dark zone FDCs.66,67 LZ B cells and DZ B cells differ in several important aspects. DZ B cells rapidly proliferate and carry out activation-induced deaminase (AID) mediated somatic hypermutation (SHM) to foster the affinity maturation of immunoglobulin genes.68 AID introduces random mutations on immunoglobulin genes, leading to a range of modifications of BCR antigen affinity. Mutations that lead to increased antigen affinity will improve B cell competitive fitness in the LZ, while lower affinity or nonfunctional mutations will undergo apoptosis.69 AID enzymes also initiate immunoglobulin class-switching from IgM to downstream isotypes IgG, IgE, or IgA.70 While the primary goal of DZ B cells is to undergo proliferation, LZ B cells, in contrast, express more activation markers, such as CD83 and CD86, along with upregulating BCRs and CD40, to facilitate affinity selection.71,72
Tfh cells play a central regulatory role in B cell affinity selection, owing to their critical function of inducing B cell survival in the LZ.73 The often-limited Tfh cell population necessitates affinity-driven competition among LZ B cells for T cell help. High-affinity LZ B cells can attract and maintain cognate interactions with Tfh cells through the secretion of chemokines74 and expression of a high density of pMHC II complexes and ICOSL (inducible co-stimulatory molecule ligand).75 Conversely, low-affinity LZ B cells express a lower density of pMHC II complexes, and are thus unable to compete for Tfh cell help, leading to apoptosis.75 The survival advantage of high-affinity LZ B cells ultimately improves the average affinity of given germinal centers and leads to the production of high-affinity antibodies (Fig. 1E). There are two main mechanisms through which the low-affinity B cells undergo apoptosis. B cell apoptosis in the LZ due to the lack of T cell help, i.e. “death by neglect”, was initially thought to be the primary mechanism.75 However, recent literature has demonstrated that the strength of T cell help received by LZ B cells determines the B cell survival in the DZ, with low-affinity B cells undergoing apoptosis within the DZ.76–78 These two models of affinity selection potentially co-exist during GC reactions. Understanding this pathway can guide efforts to improve antigen affinity. Additionally, Tfh cells may also contribute to affinity selection by actively inducing B cell apoptosis within the LZ through the Fas ligand and granzyme B secretion.79 Due to the crucial functions of Tfh cells in regulating B cell immunity, a number of studies have designed strategies to modulate T cells in order to improve antibody responses. We will discuss some of these studies in the section below.
The longevity of the GC reaction is an important factor for antibody affinity maturation. The persistence of GCs allows for continuous replenishment by naive B cells, a feature that leads to the extensive mutation of B cells and broadens the breadth of antibody responses.80–82 Vaccine-induced long-lived germinal center reactions have also been observed in patients who received the SARS-CoV-2 mRNA vaccine, generating robust humoral immune responses.83,84 We will highlight a variety of biomaterial platforms that achieved extended GC reactions via sustained antigen delivery.
Ultimately, B cells survive and exit the GC as either long-lived plasma cells (LLPCs) or MBCs (Fig. 1E). While the molecular mechanisms regulating the post-GC B cells’ fate decisions remain elusive, antibody affinity has long been established as a crucial factor in determining the fate of GC B cells.85,86 High-affinity GC B cells are exported as LLPCs for the production of high-affinity antibodies to exert immune functions, such as neutralization and opsonization.8 GC B cells with relatively lower antigen affinity are more likely to develop into MBCs prior to the differentiation of LLPCs.63,64 One important cytokine that has been linked to MBC differentiation is Tfh cell-derived IL-9, but the mechanism is still unclear.87,88 The detailed mechanisms responsible for MBC recall are being actively investigated.89,90 MBC GC reentry appears to be rather infrequent and dependent on the context of secondary exposure.90–92 Once entered, MBCs participate in GC affinity selection processes and differentiate into effector B cells.91,93
The mechanism of GC termination is still poorly understood (Fig. 1E). But a slew of factors, including antigen availability, feedback by soluble antibodies, suppression by T follicular regulatory (Tfr) cells, and metabolic inhibition, have all been shown to play unique roles in regulating the duration of GC reactions.94 Recently, canonical Tfh cells are also shown to initiate GC contraction by expressing Foxp3 transcription factors.95 Future progress in this field will have significant implications for vaccination and autoimmunity. Other strategies aiming to suppress B cell functions through inhibitory co-receptor stimulation or depletion have shown early successes. Some of the biomaterial-aided design will be discussed in our review.
Other than the abovementioned molecular and cellular mechanisms, B cell responses are also significantly influenced by factors that are often outside of conventional biomaterial engineering control, such as the B cell precursor frequency and B cell immune history. These factors contribute to individual differences when responding to the same antigens and complicate B cell responses to complex antigens. We would like to highlight the impacts of these factors in the following two aspects:
2.1. Epitope immunodominance
B cells are regularly exposed to complex protein antigens during infection or vaccination. Among a broad range of potential epitopes in a protein antigen, often only a few epitopes will be focused on by B cells to generate antibodies against while many others are neglected.96,97 Similar observations have also been made during T cell responses.98,99 This phenomenon, referred to as immunodominance, is described by Crotty et al., as “immunodominance is the natural focusing of an immune response toward a specific number of B cell or T cell clones at the expense of expansion of other epitope-specific B or T cells”.100 While therapeutic immune responses can feature an immunodominant epitope bias, for rapidly mutating pathogens, such as HIV and influenza virus, immunodominance can be detrimental by distracting immune responses away from broadly neutralizing epitopes.38,101 Two main contributing factors for epitope immunodominance are (1) the germline epitope BCR affinity and (2) the precursor frequency of epitope-specific germline B cells.100,102 Firstly, BCR antigen affinity determines the fate of B cells during GC entry and GC selection, as described above (Fig. 1E). During the initial responses to complex antigens, GCs are primarily composed of antigen-specific B cells with a significant portion of B cells showing no detectable binding affinity.103 Over time, the average antigen affinity of GC B cells increases and B cells with non-detectable affinity will be gradually eliminated. This natural affinity selection process creates a considerable hurdle for low-affinity germline B cells to overcome in order to mature into LLPCs.66 Secondly, the frequency of germline B cell clones specific for a given antigen is largely undetermined, but its impact on the robustness of the B cell responses is crucial. HIV vaccine studies demonstrated that the average frequency of germline B cells for VRC01-class bnAbs in the naive repertoire is about only 1 in 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.100,104 This is considerably lower than the frequency of naive B cells specific for other common antigens. For example, the frequency of B cells against phycoerythrin (PE), a common fluorophore, is 1:5000.105 The frequency of B cells specific for the spike protein of SARS-CoV-2 virus is recently determined to be 1:43000 in human PBMC, still much higher than HIV bnAb precursor B cells.106 The paucity of HIV bnAb-precursor germline B cells dramatically limits the effectiveness of an HIV vaccine to recruit antigen-specific B cells into GCs for affinity maturation, thus diminishing the probability of eliciting bnAbs through vaccination. Collectively, antigen immunodominance and the individual B cell repertoire likely determine vaccine efficacy. The development of vaccine strategies targeting neutralizing epitopes that are subdominant requires considering this factor.
000.100,104 This is considerably lower than the frequency of naive B cells specific for other common antigens. For example, the frequency of B cells against phycoerythrin (PE), a common fluorophore, is 1:5000.105 The frequency of B cells specific for the spike protein of SARS-CoV-2 virus is recently determined to be 1:43000 in human PBMC, still much higher than HIV bnAb precursor B cells.106 The paucity of HIV bnAb-precursor germline B cells dramatically limits the effectiveness of an HIV vaccine to recruit antigen-specific B cells into GCs for affinity maturation, thus diminishing the probability of eliciting bnAbs through vaccination. Collectively, antigen immunodominance and the individual B cell repertoire likely determine vaccine efficacy. The development of vaccine strategies targeting neutralizing epitopes that are subdominant requires considering this factor.
2.2. Immunological imprinting
The immune history impacts the subsequent immune responses induced by vaccines or health conditions. This phenomenon is originally referred to as “original antigenic sin”, coined by Francis in the 1960s.107 Due to the negative connotation and relatively narrow utility of this terminology, this phenomenon is now generally referred to as “immunological imprinting”.108,109 The immunological imprinting effect is multifaceted. In some cases, the immune history will bias responses toward the original exposure, limiting the diversity of antibodies induced by subsequent vaccinations.89,109 In other cases, previous exposure accelerates the subsequent responses as the typical immune memory responses. These conflicting effects of prior exposure are the result of an interplay between immunological memory and cross-reactivity. Cross-reactive MBCs can promote MBCs to rapid expansion and plasma cell differentiation during subsequent exposure to different viral strains. It can also guide MBCs to enter GCs for the production of antibodies, often with broader reactivities and stronger responses against the original strain.110 However, the frequency of this occurrence is relatively low.91 Conversely, the pre-existing cross-reactive serum antibodies may inhibit B cell activation through FcγRIIB1 signaling, thus limiting antibody production against mutated antigens.111 Rapid antigen clearance by pre-existing antibodies is another mechanism by which the original immune responses may be counterproductive for subsequent antigen exposures. Collectively, the implications of immunological imprinting on vaccination or immunotherapy are rather complex and context dependent. Therefore, it is imperative to consider the immune history of the test subjects during therapy design and data interpretation.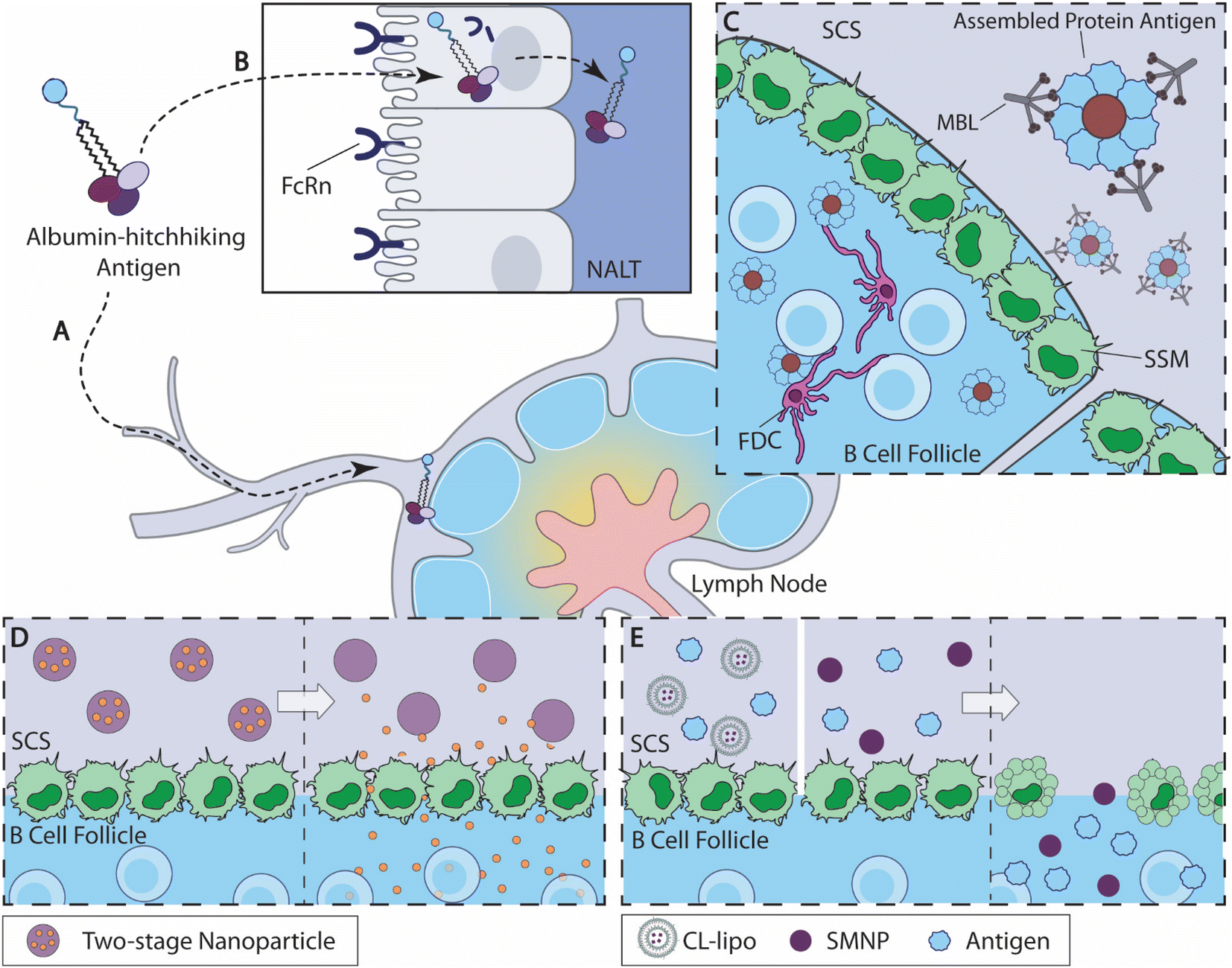 | ||
| Fig. 2 Overview of B cell-targeted antigen delivery strategies. (A) Albumin-binding amph-vaccine accumulates in dLN by albumin hitchhiking.115 (B) Intranasally delivered amph-vaccines are transported to the nasal associated lymphoid tissue (NALT) through a mucosal epithelial layer mediated by neonatal Fc receptor (FcRn).117 (C) Highly glycosylated self-assembled HIV nanoparticle antigens activate the mannose-binding lectin (MBL) complement pathway and are acquired by SSM cells from the subcapsular sinus (SCS) for antigen delivery to the B cell follicles.121 (D) Two-stage nanoparticles accumulate at the SCS (left), and release small-molecule drug to the B cell follicle as the linkers degrade at predetermined kinetics (right).134 (E) When antigens are administered with CL-lipo137 or SMNP138(left), the SSM barrier cells are depleted and antigens are accumulated in the B cell follicles (right). | ||
2.2.1.1 Albumin hitchhiking for antigen delivery. Inspired by rapid LN accumulation by compounds that bind to serum albumin, Liu et al. designed albumin-binding lipid-conjugated vaccines, namely amph-vaccines115 (Fig. 2A). This design exploited the natural trafficking of endogenous albumin within the lymphoid tissues, leading to a rapid accumulation of antigens and adjuvants within the dLN following subcutaneous administration. Lipid tail modification to a CpG oligonucleotide adjuvant increased LN accumulation by 12-fold over an unmodified control after subcutaneous immunization. Moreover, amph-vaccines show clear colocalization with LN B cell follicles through immunohistochemical imaging, indicating that the albumin-mediated antigen trafficking overcomes SSM barriers.115 This strategy is currently being evaluated for the LN targeting of a cancer vaccine in a phase 1 clinical trial (NCT04853017).116
Mucosal immunity is critical for preventing infection,9–11 but the efficacy of mucosal vaccination is often hampered by rapid antigen clearance at mucosal barriers.39 Leveraging the amph-vaccine design, Hartwell et al. developed amphiphile lipid-conjugated eOD-GT8 (germline-targeting engineered outer domain of HIV gp120 proteins) as an HIV mucosal vaccine (amph-eOD)117 (Fig. 2B). After intranasal administration, amph-eODs were transported to the lymphatic vessels and the nasal-associated lymphoid tissue (NALT) within 6 h and remained detectable 7 days after immunization, while unmodified antigens showed a minimal level of accumulation. The enhanced accumulation of amph-vaccines promoted B cell responses and improved the quantities of both GC B cells and eOD-binding GC B cells relative to the unmodified control. When combined with a STING agonist, cdGMP, or SMNP adjuvant (discussed later), amph-eOD nasal immunizations promoted high levels of IgA responses systemically or within the mucosal tissues, while eOD antigen alone mostly failed to elicit any IgA responses. Amph-eOD vaccine also showed superior immunogenicity in non-human primates (NHPs) relative to eOD control. The researchers identified that neonatal Fc receptor (FcRn) mediated albumin transportation facilitated the transit of amph-vaccine cross the mucosal barriers.117 These studies represent an exciting broadly applicable strategy to improve antigen accumulation in the dLNs.
2.2.1.2 Promote SSM-mediated antigen delivery. SSM cells preferentially acquire antigens that are opsonized or deposited with complement proteins from afferent lymph for antigen presentation to B cells.43 Therefore, increasing antigen retention by SSM cells has long been explored for antigen delivery, even before the role of SSM in antigen presentation was defined.118 Initial attempts have largely focused on the modification of antigens with antibodies against CD11c119 or CD169 (an SSM-specific receptor).120 Through purposeful design, recent versions of biomaterial-based vaccines showed improved efficacy in leveraging SSM-mediated antigen presentation to drive B cell responses.
Tokatlian et al. designed two kinds of self-assembled protein nanoparticle, eOD-60mer and MD39–8mer, and both achieved impressive levels of antigen accumulation in the follicle121 (Fig. 2C). The eOD-60mer, a ∼32 nm-diameter nanoparticle that carries eOD antigens, is assembled from eOD-lumazine synthase fusion proteins; and the MD39-8mer, a ∼40 nm-diameter nanoparticle that carries MD39 (modified HIV gp140 trimer proteins), is assembled from MD39 fused with archaeal ferritin. Both nanoparticles exhibited significant levels of colocalization between antigens and the FDC network on day 3 and day 7 following subcutaneous administration in BALB/c mice, whereas their corresponding monomers accumulated primarily in the subcapsular sinus and medullary areas. Immunization with these particles, compared with unassembled protein controls, elicited antigen-specific IgG with elevated titers and binding affinities and higher frequencies of GC B cells and Tfh cells. As the acquisition of antigens is largely mediated by SSM within LN,43,48 the authors speculated the observed FDC nanoparticle retention was likely mediated by SSM antigen relay following complement activation. Involvement of the complement system was experimentally confirmed by the fact that any disruption of the mannose-binding lectin (MBL) complement pathway or deglycosylation of the eOD-60mer nanoparticles could severely compromise the nanoparticle vaccine's follicle accumulation and immunogenicity. Thus, these self-assembled highly glycosylated protein nanoparticles stimulated MBL activation and were likely captured and handed off to FDCs by SSMs. This study highlights an engineering opportunity to improve B cell antigen delivery.
Martin et al. further explored the utility of complement pathways for antigen delivery into B cell follicles.122 Antibody-opsonized antigens can access B cell follicles by engaging SSM, which enhance the magnitude and the duration of antibody responses.123–125 The authors sought to activate complement pathways using immune complexes (IC) formed between soluble HIV Env trimer immunogen (BG505) and macaque-derived monoclonal antibodies (RM19R mAb) that bind to the base of BG505. In BALB/c mice, the administration of RM19R mAb showed a dose-dependent enhancement effect for BG505 accumulation within dLNs, suggesting that the formation of IC between BG505 and RM19R mAb facilitated B cell follicle access by the antigens. While the formation of IC did not show a statistically significant improvement on antigen follicle access in NHPs partly due to limited sample sizes, IC-mediated antigens appeared to be more focused within FDC networks than antigen alone. The authors also evaluated antigen follicle accumulation in NHPs, following the administration of self-assembled BG505 SOSIP-T33_dn2 nanoparticles that carry 4 copies of BG505 SOSIP HIV trimers. Unexpectedly, free BG505 trimer proteins exhibited greater antigen fluorescence intensity. However, both light sheet imaging of cleared lymph node tissue and immunohistology imaging showed higher levels of antigen presence in B cell follicles in the nanoparticle immunization group, as compared with free trimer immunizations. The authors suggested that such a discrepancy may be caused by several factors, including LN accumulation kinetic differences and accidental protein removal during the tissue clearing process. Taken together, these studies suggest that both the immune complex and self-assembled antigen-displaying particles can be effective in promoting antigen follicular localizations.
Aside from leveraging complement activation to promote SSM-mediated antigen acquisition, an SSM-specific receptor, CD169, has also been explored for antigen delivery through ligand design. Antigen-conjugated anti-CD169 antibodies elicited antibody responses with markedly improved quantity, affinity, and frequencies of GC B cells.120 James Paulson and co-workers screened a library of about 8400 substituted carboxylic acids that potentially bind to CD169 and identified a potent ligand of the CD169 receptor, namely TCCNeu5Ac.126 Liposomes displaying TCCNeu5Ac ligands (TCCNeu5Ac-liposomes) showed binding specificity for CD169+ macrophages both in vitro and in vivo.127,128 Due to SSM's key role in T cell antigen presentations, TCCNeu5Ac-liposomes successfully delivered antigens to SSM and amplified the immune responses of natural killer cells and CD8+ T cells.128–132 Gao et al. recently designed TCCNeu5Ac-displaying acetylated dextran nanoparticles that carried both protein antigens and imiquimod, a TLR7/8 agonist, as adjuvant, to demonstrate the enhancing effect of TCCNeu5Ac ligand-mediated SSM-targeted delivery on B cell responses.133 These nanoparticles showed a significant targeting effect toward CD169+ macrophages in vitro and elicited more potent antibody responses and CD8+ T cell responses as compared with the non-targeting control nanoparticles. Using SSM-targeting nanoparticles that carry SARS-CoV-2 RBD proteins showed a similar improvement in antibody responses in both mice and rabbits, as compared with either protein control or PEG nanoparticles. While these studies have not directly shown the impact of CD169-targeting strategies on antigen follicle accumulation or the dynamics of B cell responses, recent data demonstrated the feasibility and effectiveness of SSM-targeted antigen delivery for improving antibody responses, along with T cell responses.
2.2.1.3 Circumvent SSM barrier regulation. In addition to actively engaging SSM for antigen acquisition, several research teams engineered molecular strategies that evade SSM regulations and efficiently deliver the cargo into the B cell follicle.
As discussed previously, proteins with a molecular weight lower than 70 kDa or particles with diameters lower than 5 nm can directly diffuse into B cell follicles without being impeded by SSM barriers.43 To take advantage of this feature, Schudel et al. designed a two-stage release nanoparticle system that can rapidly reach dLN and avoided the barrier control by SSM via a timed cargo release at the subcapsular sinus134 (Fig. 2D). These nanoparticles were synthesized by crosslinking thiolated poly(propylene sulfide) (PSS)/Pluronic F127 micelles (OND-NP) with thiol-reactive oxanorbornadiene (OND) linkers. Depending on the substitution, these OND linkers exhibit different degradation kinetics, independent of pH and solvent conditions, with half-lives of fragmentation ranging from 17 min to 29 h at 37 °C. Using rhodamine dye as cargo, the researchers demonstrated that the nanoparticle-mediated cargo accumulates within the dLNs following i.d. administration, while neither free dye nor dye carried by rapidly degrading nanoparticles reached the dLN, indicating that both nanoparticle-mediated transport and properly timed release are necessary to achieve optimal follicle-targeted release. Using a linker with a half-life around 8 h, OND-NP showed a significant improvement in cargo acquisitions by both barrier cells and para(cortex) cells, relative to the non-degradable control. For example, OND-NP-mediated cargo delivery showed 100-fold more antigen acquisition by B cells, as compared with the non-degradable control or free cargo. The release kinetics of OND-NP influences the cargo's cellular distribution within the dLN, with the slower degradation rate skewing toward more cargo accumulation in the paracortex cells. The OND-NP-mediated delivery of CpG oligonucleotide showed an improved activation of B cells, cDCs (conventional dendritic cells) and pDCs (plasmacytoid dendritic cells), relative to free CpG and disulfide-linker controls, and ultimately promoted immune infiltration within tumor tissues. Hence, this two-stage release OND-NP system can improve cargo access to B cell follicles by timed release and shows promise for B cell-targeted delivery.
Another recent engineering strategy for follicular B cell antigen delivery is to selectively deplete SSM. Inspired by several previous studies where the depletion of SSM promoted antibody production,135,136 Chan and coworkers sought to evaluate the impact of SSM depletion on the immunogenicity of gold nanoparticle (AuNP) vaccines137 (Fig. 2E). The research team depleted SSM using clodronate liposomes (CL-lipo), and found that SSM depletion resulted in a rapid accumulation of AuNP and prolonged retention of AuNP in the follicles for over 2 weeks, while PBS-lipo-treated mice did not show a comparable level of antigen accumulation. The enhanced follicle antigen accumulation driven by CL-lipo-mediated depletion correlated with higher GC B cell frequencies and elevated antibody productions for both free antigens and antigen delivered by AuNPs. Interestingly, the authors demonstrated that inhibitions of macrophage uptake with gadolinium chloride, carrageenan, or dextran sulfate 500 all showed enhancement effects for antigen follicular accumulation, similar to CL-lipo, suggesting that SSM uptake negatively impacts nanoparticle vaccine efficacy. SSM depletion is a potentially versatile strategy to improve nanoparticle antigen accumulation within B cell follicles and the resulted immune responses.
Similar SSM depletion-led antigen accumulation also contributed to the strong immunogenicity of a novel nanoparticle adjuvant (SMNP)138 (Fig. 2E). Silva et al. formulated SMNP by incorporating the TLR4 agonist MPLA into saponin-based immune-stimulating complexes (ISCOM). During immunization, SMNP showed a superior ability to promote GC reactions and antibody productions over several other adjuvants tested. SMNP was shown to promote antigen uptake by B cells and enhance the activation and proliferation of antigen-specific B cells, in addition to improving the cytokine secretion by antigen-specific Tfh cells.138 The research team identified two mechanisms that contribute to the enhanced immunogenicity of SMNP. Immunofluorescence imaging of dLNs revealed that SMNP administration led to the reduction of SSM, similar to CL-lipo treatment. Moreover, SMNP accelerated antigen trafficking by enlarging the lymphatic vessel within the first hour of administration according to intravital imaging. The detailed mechanism of this enhanced lymphatic flow was further confirmed to be driven by mast cell activity induced by SMNP using mast cell-deficient mice, where an absence of mast cells reduced the immunogenicity of SMNP. SMNP also showed a potent adjuvant effect in NHPs when administered with the aforementioned HIV nanoparticle vaccine, MD39-8mers.121 Therefore, the SMNP adjuvant promotes strong immune responses by amplifying antigen accumulation in B cell follicles by disrupting SSM barriers and enlarging lymphatic vessels.
These corroborating studies collectively demonstrate that circumventing SSM regulations either by releasing small molecule cargos within subcapsular sinus cargos or by disrupting SSM cells can effectively deliver biologics to B cell follicles.
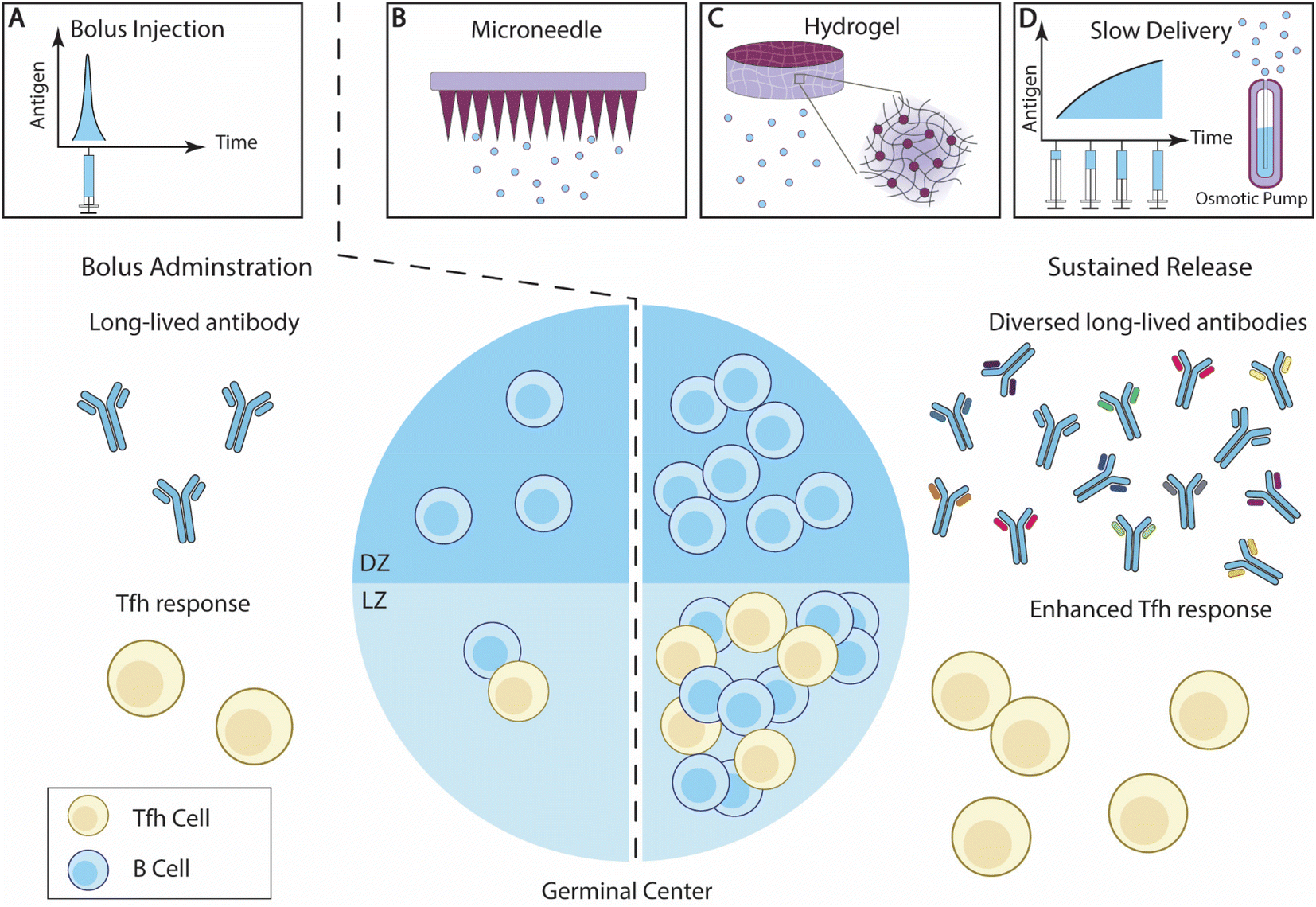 | ||
| Fig. 3 Overview of sustained antigen delivery for enhanced B cell responses. (A) Bolus administration of vaccine elicits antibody responses with limited diversity, and relatively low Tfh cell responses. (B–D) The sustained delivery of B cell antigens enhances GC reactions, and potentially increases the ratio of LZ B cells and DZ B cells. The breadth of the antibody responses and Tfh cell responses is also improved. Effective sustained antigen delivery strategies include (B) microneedle (MN) patches; (C) hydrogel; and (D) slow administration through exp-inc dosing regimen or an osmotic pump.140 | ||
Diverse biomaterial strategies have been developed to control the release of antigens or drugs. Some of these designs have been recently reviewed elsewhere.25,147 Here, we aim to highlight recent studies that demonstrated the impact of sustained antigen release on B cell immunity.
2.2.2.1. Microneedle patches. Microneedle (MN) patches, consisting of an array of micron-scale needles, can introduce antigens into the epidermis and dermis layers.148 Dermal immune cells, such as dermal DCs and Langerhans cells, are thus activated by the MN transcutaneous vaccinations and initiated immune responses.149 Extended antigen release can be achieved by developing novel vaccine-loading strategies or an MN architecture design (Fig. 3B).
DeMuth et al. engineered a series of MN strategies to control the delivery of antigens. Through the layer-by-layer (LbL) approach, the researchers designed a DNA vaccine by interweaving layers of antigen encoding plasmid DNA (pDNA) and the adjuvant polyinosinic:polycytidylic acid, poly(I:C), and biodegradable cationic poly(β-amino-ester) (PBAE) onto poly(L-lactide) (PLLA)-based MN patches.150 Nucleic acids delivered through the MN patches remained detectable over 20 days after administration, whereas pDNA introduced via intradermal (id) injections was cleared within 2 days. MN immunizations elicited antigen-specific antibody responses with a titer 3-fold higher relative to id immunizations.150 The research team also deposited layers of interbilayer-crosslinked multilamellar vesicles (ICMVs) using similar LbL strategies.151,152 MN-mediated OVA-loaded ICMV delivery generated OVA-specific antibody titers that were 10-fold higher than with the id administration of ICMV particles, and were about 100-fold higher than the id administration of OVA protein alone.
To further control MN-mediated antigen delivery kinetics, DeMuth et al. engineered composite MN patches composed of a dissolvable poly(acrylic acid) (PAA) base and protein-loaded PLGA bulk or microparticle-based MN tips.153 After application to the skin, the PAA-based bases and pedestals were rapidly dissolved by the interstitial fluid within a day, leaving the PLGA MN tips embedded at the injection sites and enabling a long-term delivery of antigens for over 20 days. The research team subsequently improved the composite MN design by loading the protein antigens within silk protein-based MN tips due to silk's advantage in high-density loading.154 Antigens delivered using silk-based MN remained detectable at the administration sites for over 16 days. The prolonged antigen delivery improved the frequency of CD8+ memory T cells and the antibody titer and avidity. Leveraging this composite MN patch design, Boopathy et al. encapsulated the aforementioned HIV antigen, MD39 trimers, together with adjuvants, pam3CSK4 and polyI:C, within silk protein MN tips.155 The MD39 proteins maintained their antigenicity after being released from the MN, and remained in the skin for over 14 days, in contrast to the rapid clearance by intradermal injection. MN-mediated sustained release also improved antigen access to FDCs in the dLNs, along with the development of GC B cells and Tfh cells, particularly trimer-specific Tfh cells. The improvement in cellular responses led to a significantly higher production of MD39-specific IgG antibodies.
Chen et al. designed fully-embedded chitosan-based MN tips for antigen sustained release.156,157 Chitosan was selected for its biocompatibility and biodegradability. Upon application, chitosan MN tips are separated from the MN base and are embedded within the skin to act as vaccine depots, and the polyvinyl alcohol/polyvinyl pyrrolidone (PVA/PVP) supporting patch can be dissolved. The PVA/PVP chitosan MN patches sustained OVA protein antigen release for about 21 days. Hemagglutinin (HA) protein-loaded PVA/PVP chitosan MN patches generated a 4-fold increase in virus-specific IgG compared with intramuscular (IM) immunizations, resulting in 100% survival and minimal weight loss after an influenza virus challenge.158
Prausnitz and coworkers designed MN patches that encapsulated lyophilized influenza vaccines within PVP MN tips through photopolymerization.159 This manufacturing process preserved vaccine antigenicity, and MN immunization and IM immunization induced comparable titers of antibodies. Moreover, MN immunization generated superior long-term viral protection and higher titers of antigen-specific IgA in bronchoalveolar lavage fluid (BALF), relative to IM administration.159 The research team later integrated active self-healing encapsulation (ASE) PLGA particles160 within polyvinyl alcohol and sucrose, to develop microparticle-based MN patches.161 This design leveraged ASE particle-mediated protein loading through Alhydrogel absorption to facilitate facile protein antigen long-term delivery.
In addition to improving the biomaterials used for MNs, several research groups have innovated the manufacturing process to modulate antigen release or to accelerate the manufacture process. Tran et al. designed MN patches with an array of core–shell microneedles composed of PLGA with varying degradability kinetics.162 The PLGA shell of embedded MN tips would degrade at various time points depending on the molecular weight and terminal groups of the PLGA polymers, thus releasing the loaded antigens at different stages. This multi-stage release of antigens achieved prime and boost immunization through a single administration, with the last dose of antigens released 40 days after MN immunization. Immunization with these core–shell MN patches encapsulating Prevnar-13, a clinical vaccine against S. pneumoniae infections, achieved strong antigen-specific antibody responses, and improved the protection against bacterial challenges relative to bolus s.c. immunizations.162 DeSimone and colleagues utilized continuous liquid interface production (CLIP) technology to rapidly fabricate MN with control over a variety of design parameters and achieved cargo loading and delivery.163,164 To improve antigen loading, the researchers designed faceted MN with significantly larger surface areas than their square pyramidal counterparts, and thus loaded 36% more antigen.165 CLIP-printed MNs prolonged the presence of both antigen and adjuvants at injection sites relative to bolus SC immunization. This design also achieved antigen dose sparing. At 10-fold lower antigen dosing, MN-induced antibody responses were nearly unaltered, while all other tested administration methods showed diminished responses.
Collectively, these studies demonstrate that MN patches can effectively control antigen release. Additionally, their diverse designs potentially offer avenues for future innovations to improve transcutaneous delivery.
2.2.2.2 Hydrogels. Hydrogels are composed of a network of hydrophilic polymer chains and a large amount of water content, thus resembling tissue physical properties, and exhibiting excellent biocompatibility once embedded. A hydrogel loaded with hydrophilic drugs can serve as a local drug depot at the administration site and control drug release through a number of approaches, including tuning mesh sizes, modulating drug/polymer interactions, and altering degradation rates.166 The versatility of hydrogel makes it an excellent choice to control the delivery of antigens and adjuvants25,167 (Fig. 3C).
Injectable hydrogels that can be introduced into local tissue through injection are particularly attractive platforms for vaccine delivery.168–170 A prominent example of hydrogel-mediated antigen sustained release is the polymer-nanoparticle (PNP) hydrogel system developed by Appel and colleagues.141,170 The PNP hydrogel is crosslinked through non-covalent reversible hydrophobic interactions between poly(ethylene glycol)-b-poly(lactic acid) (PEG-PLA) nanoparticles and hydroxypropyl methylcellulose derivatives (HPMC).170 Shear stress disrupts the crosslinking, enabling the hydrogels to flow through syringes. The hydrophobic interaction reestablishes rapidly once the shear stress is removed, allowing the hydrogels to recover. Roth et al. designed PNP hydrogels encapsulating OVA antigens and poly(I:C) adjuvants.141 By altering the concentration of nanoparticles and HPMC, the researchers produced PNP hydrogels with different yield stresses. Antigens delivered with stronger hydrogels persisted at the administration sites for over 28 days, with a retention half-life of 7.7 days. Relative to bolus immunizations, sustained antigen delivery enhanced GC B cell responses, especially leading to elevated ratios of LZ to DZ B cells, suggesting enhanced affinity selection activities. The improved GC response naturally leads to high levels of antibody responses induced by hydrogel vaccines.141,171 The PNP platform has also been used for the sustained delivery of other protein antigens, including influenza HA protein,172 and SARS-CoV-2 receptor-binding domain (RBD).173 Using imidazoquinolinone-conjugated PEG-PLA nanoparticles, the researchers achieved a sustained delivery of small-molecule TLR7/8a adjuvants, and enhanced antigen immunogenicity.187,188 A similar improvement was also achieved when delivering a self-assembled multivalent RBD-functional nanoparticle (RBD-NP) antigen using a PNP hydrogel.174 These findings underscore the effectiveness and versatility of PNP hydrogel-mediated sustained vaccine delivery in generating robust, enduring, and broad antibody responses.
Several other research teams have also explored different types of hydrogel to sustain antigen release. Mueller et al. designed PEG hydrogel nanoparticles using particle replication in nonwetting templates (PRINT) technology to control the delivery of the conjugated protein antigens.175,176 PRINT particle-mediated antigen delivery rapidly accumulated within dLNs in about 2 h, and persisted within dLNs and B cell follicles for over 15 days. The rapid and persistent antigen delivery promoted T cell proliferation and enhanced antigen-specific antibody titers to nearly 1000-fold higher relative to protein antigen alone. Other particle/hydrogel hybrid systems have also shown similar sustained antigen release and excellent antibody responses.177,178 Methacrylated (MA)-alginate cryogels form porous structures by covalently crosslinking around ice crystals, thus enabling the encapsulation of biologics. Shih et al. further improved the design by incorporating ionic crosslinkers to allow needle administration.179 The dual-crosslinking cryogels showed improved mechanical properties and prolonged protein release for over 15 days. Cryogel-mediated antigen delivery improves antibody responses and successfully prevented tumor growth in a prophylactic model. Peptide and DNA-based hydrogels have also been developed to achieve prolonged antigen delivery and showed enhanced antibody responses.180–182
Collectively, hydrogels show an excellent ability to sustain antigen release and are suitable for a wide range of antigens and adjuvants. The highly tunable chemical and physical properties allow for the modulation of cargo-release kinetics. With new designs of hydrogel materials on the rise,183,184 hydrogel-mediated antigen delivery shows significant potential for improving vaccine effectiveness.
2.2.2.3. Slow vaccine administration. Extending the vaccine administration schedule has recently been investigated for its effect on B cell and antibody responses (Fig. 3D). Tam et al. designed and compared several vaccination regimens for their effectiveness in generating antibody responses.139 They showed that extended vaccine administration prolonged antigen availability within follicles in mice, as compared with the bolus immunizations. Among the extended delivery regimens, exponentially increasing (exp-inc) dosing over 7 days showed a greater improvement of antibody titer and neutralization potential. Exp-inc administration also dramatically improved many aspects of the B cell responses, including the frequencies of GC B cells, plasma cells, and activated B cells. Extended antigen delivery using embedded osmotic pumps (OP) in mice showed a similar improvement to exp-inc, confirming the advantages of slow antigen delivery in boosting B cell immunity. Joyce et al. investigated extended-delivery strategies using MN patches and demonstrated that this strategy is broadly applicable for various antigens, including inactivated polio, tetanus toxoid, influenza, and measles.185
Slow vaccine administration was similarly effective in NHPs. Both OP-mediated slow antigen delivery and exp-inc administration can induce a higher frequency of HIV-specific B cells and GC Tfh cells, along with elevated antibody titers and broadened antibody diversity in NHP.140 It was also shown through BCR sequencing and single-cell sequencing that slow delivery shifted the immune responses away from distracting immunodominant antigens. Extending the duration of priming immunization alone has shown a significant improvement in B cell responses. Crotty and colleagues demonstrated that slow priming immunizations promoted a higher number of SHM mutations in GC B cells and diverged both memory B cells and antibody responses away from focusing on immunodominant epitopes.186 Thus, this strategy can generate a higher antibody titer and broader neutralization capability against viral targets. These studies show that extended administration holds great promise in inducing therapeutic antibody responses against challenging viral targets like HIV.
Sustained antigen delivery promotes the recruitment of antigen-specific B cells and drives GC Tfh cell development. These fundamental improvements on a cellular level significantly enhance the quantity, quality and breadth of antibodies elicited by immunization. Future innovations providing more precise control over antigen release kinetics using platforms that improve patient compliance will potentially translate these exciting preclinical discoveries into clinical success.
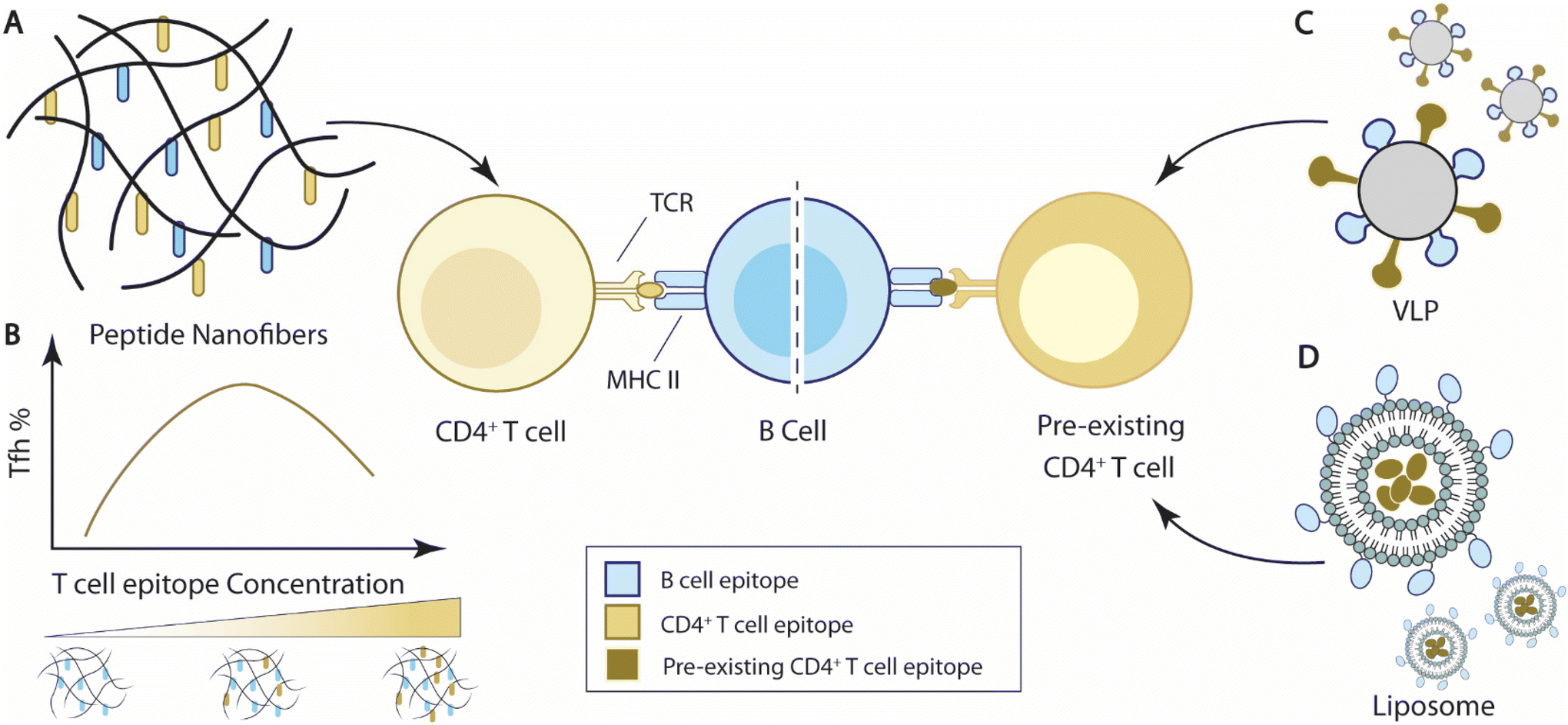 | ||
| Fig. 4 Overview of current strategies to integrate T cell help for B cell immunity. (A) Self-assembled peptide nanofibers with appended peptide epitopes for both T cells and B cells to elicit T cell help for B cell responses. (B) T cell epitope concentrations carried by nanofibers impact Tfh cell responses and B cell immune responses. In the representative bell curve, the media level of T cell concentration elicits the maximal frequency of Tfh cells.193 (C) Virus-like particles (VLPs) carrying epitopes for pre-existing T cells can elicit T cell help from pre-existing T cells for B cell responses.209 (D) Liposomal nanoparticles encapsulate antigens for pre-existing T cells and elicit T cell help for B cell responses against B cell antigens displayed on the nanoparticles.210,211 | ||
Collier and coworkers designed a self-assembled vaccine platform based on fibrillizing β-sheet peptides (acetyl-QQKFQFQFEQQ-amide), namely Q11189–191 (Fig. 4A). Driven by a mixture of electrostatic interaction, hydrogen bonding, and π–π stacking of aromatic groups, Q11 peptides spontaneously form a network of nanofibers in PBS solution. Modified with appendant peptide antigens, the self-assembled Q11 nanofibers alone can raise high levels of antigen-specific antibody and T cell responses similar to, if not better than, adjuvant emulsified antigens without inducing a significant inflammatory reaction.189–192 The self-assembling feature of Q11 peptides enabled the “mix-match” of B cell epitopes and T cell epitopes for vaccine design. Pompano et al. leveraged this modularity feature and designed nanofiber vaccines carrying B cell peptide epitopes against Staphylococcus aureus, E214 peptides, and synthetic non-natural T cell peptide epitopes, PADRE193 (Fig. 4B). The co-assembled E214-Q11/PADRE-Q11 nanofibers elicit E214-specific antibody responses, while separate injections of E214-Q11 nanofibers and PADRE-Q11 nanofibers failed to elicit such antibody responses, suggesting that the co-assembled nanofibers generated T cell help for B cell responses. Importantly, the dosing of the PADRE epitope on the Q11 nanofibers altered the effector phenotypes of the induced PADRE-specific CD4+ T cells. The total number of PADRE-specific CD4+ T cells, including Th1 (Tbet+) and Th2 (Gata3+) CD4+ T cells, showed a negative correlation with PADRE concentrations, with the highest cell numbers at the lowest tested PADRE concentration (0.005 mM). But the frequency of PADRE-specific Tfh cells peaked at a relatively higher formulation concentration, between 0.05–0.10 mM (Fig. 4B). The level of Tfh cell responses also influenced the antibody responses at the corresponding concentrations.193 This observation is corroborated by the fact that Tfh cell differentiation requires a higher strength of TCR signaling relative to Th1 cell differentiation.194 The self-assembling nature of the peptide nanofiber vaccines thus facilitated the optimization of T cell epitope dosing to improve B cell responses.
Guided by this design principle, Collier and colleagues designed peptide nanofiber vaccines against several inflammatory cytokines, including tumor necrosis factor (TNF) and IL-17, by maximizing T cell help through T cell epitope dosing titration.195,196 The anti-cytokine antibody responses showed therapeutic effects in both LPS-induced acute inflammation and an imiquimod-induced psoriasis murine model. The influence of T cell epitope dosing on B cells was also consistently demonstrated when nanofiber vaccines were administered through the mucosal route. Kelly et al. modified Q11 peptides with either PEG or proline–alanine–serine modification to enable nanofibers to persist at the sublingual immunization site and induce mucosal immune responses.197 Through an optimization of T cell epitope dosing, the authors maximized the antibody responses against three epitopes of ion receptor proteins of uropathogenic Escherichia coli (UPEC) after sublingual immunization.198 Combined with STING and TLR9 agonists, the nanofiber sublingual vaccination generated protective pathogen-specific IgG and IgA within the urinary tract and prevented transurethral UPEC infection without significant disruption of the microbiome.198 These examples illustrate the versatility and effectiveness of optimizing T cell epitope dosing to improve B cell responses.
In addition to Tfh cells, vaccines induce other effector T cells that can promote the therapeutic effectiveness of antibody-dependent vaccines. Utilizing the adjuvanting effect of the complement protein, C3dg, Hainline et al. designed an anti-TNF vaccine formed by Q11 nanofibers co-assembled with C3dg bearing a beta-tail handle (βtail-C3dg).199,200 C3dg co-assembled Q11 nanofibers were able to raise high-level therapeutic antibodies against TNF and showed a protective effect in imiquimod-induced psoriasis. Interestingly, this protection was found to be critically CD4+ T cell-dependent, as CD4+ T cell depletion after immunization abolished the survival benefit in the acute inflammation model, suggesting that vaccine-induced T cell responses play a significant role in the observed anti-inflammation protection.
Several other studies have also used similar co-assembling strategies to improve CD4+ T cell responses for therapeutic immunity. Files et al. designed self-adjuvanting a peptide nanofiber vaccine based on β-sheet KFE8 peptides (FKFEFKFE) carrying a Mycobacterium tuberculosis (Mtb)-specific CD4+ T cell epitope, Ag85B.201 Following BCG primary immunizations, booster immunizations with KFE8-Ag85B nanofibers and KFE8-Ag85B nanofiber-pulsed DCs both elicited improved Ag85B-specific lung tissue resident memory T (Trm) cell responses, relative to primary immunization alone. Antigen-specific cytokine secretions were also improved by booster immunizations. However, the elevated CD4+ Trm responses alone were unable to ameliorate the bacterial burden after Mtb aerosol challenges, highlighting the need for crosstalk between T cells and B cells for combating challenging infections.
Collier and colleagues also designed another fibrillizing alpha-helical peptide, Coil29 (QARILEADAEILR-AYARILEAHAEILRAQ), that can generate T cell help for antibody responses.202–205 The Coil29 nanofiber vaccine showed a self-adjuvanting effect similar to the β-sheet peptide nanofibers mentioned above.202 This strategy is notable for having an exceptional ability to elicit antibody responses compared with either Q11 nanofibers or other commercial adjuvants. This superior humoral immunogenicity later contributed to the induction of Coil29-specific Tfh cells by the Coil29 nanofiber vaccine.203 Leveraging the strong immunogenicity and self-assembling features of this platform, Collier and coworkers designed Coil29-based cancer vaccines that simultaneously induced strong tumor antigen-specific antibody and CD8+ T cell responses in an EGFRvIII-expressing murine melanoma model.204 In both prophylactic and therapeutic tumor models, the combination of cellular and humoral responses outperformed either response alone in tumor inhibition, highlighting that tumor-specific antibodies can be leveraged to synergize cytotoxic CD8+ T cells for tumor treatment. Incorporating T cell checkpoint (aPD-L1) and phagocytosis checkpoint (aCD47) blockade antibodies further improves the antitumor therapeutic effect of the nanofiber vaccine. Other applications of the peptide-based vaccine platform are outside the scope of this manuscript, and readers are referred to other comprehensive reviews on this topic of interest.206–208
Another tested strategy to elicit T cell help for antibody responses is to induce responses from pre-existing T cell populations for T cell help. Bachmann and colleagues designed virus-like particles (CMVTT) by replacing the first 12 N-terminal amino acids of the Cucumber Mosaic Virus (CMV) envelope proteins with tetanus toxin (TT) T cell epitopes as a vaccine delivery platform, in order to leverage pre-existing TT-specific T cells209 (Fig. 4C). Through chemical conjugation, CMVTT can be linked to antigens of interest and elicit TT-specific T cell help to improve antigen-specific antibody responses. The team utilized this platform and improved vaccine protection in tetanus-immunized mice against three different antigens, dimeric murine IL-17A, cat allergen, and β-amyloid antigen. The therapeutic effects were abolished if the antigens were delivered using unmodified CMV particles, suggesting CMVTT indeed elicited pre-existing T cell responses for improved antibody responses.209
Hills et al. engineered liposomal vaccine particles that displayed the malaria circumsporozoite (CSP) antigen and encapsulated either ovalbumin OVA323–339 or murine cytomegalovirus (MCMV) m09133–147 peptides to improve antibody protection against malaria210 (Fig. 4D). Vaccines with CSP nanoparticles incorporating OVA323–339 peptides stimulated significantly higher titers of CSP-specific antibodies in OVA-immunized mice than PBS control mice, suggesting that OVA-specific immunity improved antibody responses against CSP. A similar improvement was also demonstrated when MCMV chronically infected mice were immunized with CSP(m09133–147), further demonstrating the utility of pre-existing immunity in vaccine-induced antibody responses. Wallis et al. subsequently developed liposomal vaccine particles that displayed ErbB-2 and encapsulated OVA323–339 peptides to improve antibodies against the cancer surface protein ErbB-2, also known as HER-2.211
Leveraging pre-existing T cells can accelerate GC formation and amplify the magnitude of GC responses,212–215 but the expansion on antigen-specific B cells appears to be minimal in other studies.216 Additionally, while the enhancement effect is clearly T cell epitope dependent, the exact phenotype of the effector T cells involved in this process remains unclear. It is likely that circulating Tfh cells play a critical role in this process as they sustain a memory-like phenotype and maintain reprogramming plasticity.217–221 Memory T cells may also be involved in this process.222 Further clarification of the cellular mechanisms during secondary exposure will accelerate the development of biomaterial platforms to exploit specific pre-existing immune cell populations for improved immune responses.
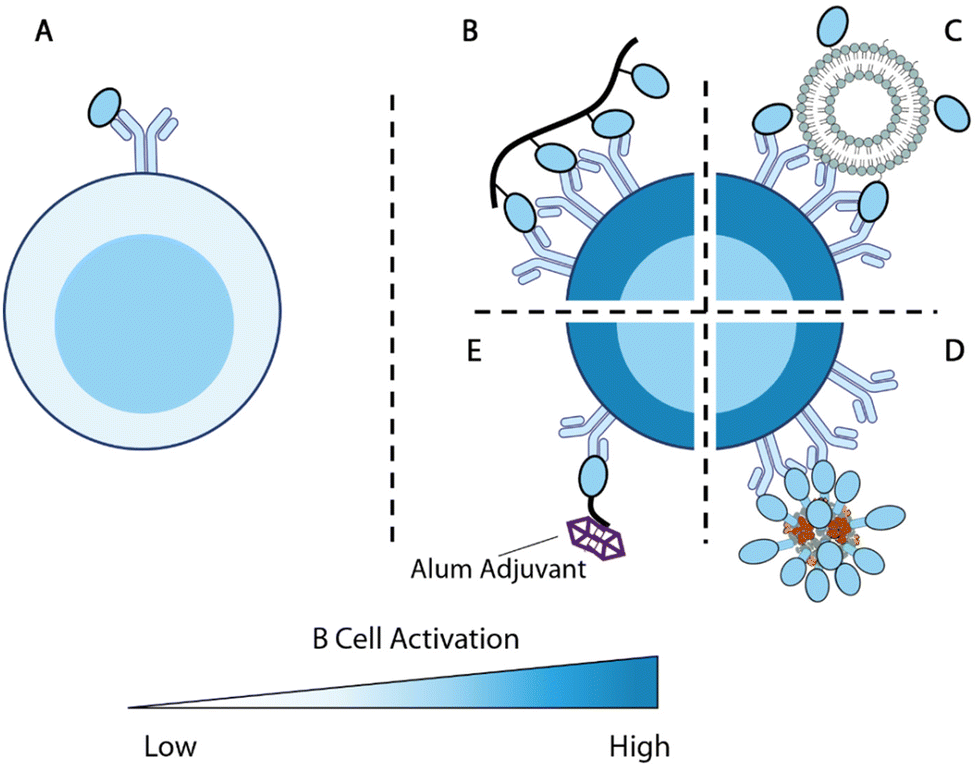 | ||
| Fig. 5 Overview of antigen engineering strategies for B cell responses. (A) Engagement of a single antigen induces limited B cell activation. (B–D) Biomaterial-mediated multivalent antigen presentations promote B cell activations. (B) Multivalent display of antigens along a polymer backbone.225 (C) Nanoparticle displays multiple B cell antigens on the particle surface.152 (D) Computationally designed self-assembled enanoparticles.228,229 (E) B cell antigen modified with alum-binding peptides carries alum adjuvant to antigen-specific B cells for enhanced immunity.231 | ||
Kiessling and colleagues set to explore this concept by designing polymer conjugates that display multiple copies of T cell or B cell antigens225,226 (Fig. 5B). Polymer backbones were prepared through ring-opening metathesis polymerization (ROMP) of a norbornene monomer with an N-hydroxy succinimidyl (NHS) ester. The obtained polymers were subsequently functionalized with 2,4-dinitrophenyl hapten (DNP) as B cell antigens and OVA323–329 T cell peptide antigens. High molecular weight antigen-carrying polymers showed a superior ability to stimulate T cell activation relative to low molecular weight polymers during in vitro DO-11.10 T cell stimulation. Multivalent polymer conjugate antigens are also more effective at promoting B cell activation as compared with protein alone.225 Moreover, both the degree of substitution of B cell antigens on polymers and the molecular weight of polymers positively impact the antigenicity in vivo. The 300-mer polymers with 12% B cell antigen substitutions (the tested highest substitution and molecular weight) showed the highest level of antigen-specific IgG responses in mice. B cells stimulated by this formulation were also the most potent in promoting T cell activation.226
Nanoparticles have also been used to display multiple copies of antigens to improve B cell responses. One of the pioneering examples is ICMV liposomal nanoparticle antigen114,152 (Fig. 5C). The ICMV particles kinetically entrap protein antigens within liposomes. The lipid layers were crosslinked through dithiol and cations to improve the nanoparticle stability. When administered with adjuvants, nanoparticle displayed and encapsulated antigens elicited nearly 10-fold higher antibody responses compared with encapsulated antigens.114 This suggests that surface antigens are important for B cell responses, potentially due to their BCR engagement functions.
King and coworkers developed Rosetta modeling software and computationally designed protein assemblies.227 A two component nanoparticle system assembled from trimers and pentamers was selected for vaccine delivery, with the F glycoprotein trimer (DS-Cav1) of Respiratory syncytial virus (RSV) linked to the trimers228 (Fig. 5D). These multivalent nanoparticle antigens showed strong immunogenicity and promoted higher quantities of GC B cells and Tfh cells relative to each component alone, confirming that the multivalent display of antigens boosts immune responses. Such a strategy has also been successfully deployed for SARS-CoV-2 vaccine design, with RBD replacing DS-Cav1 on the trimer component.229 Immunizations with these particles in NHP elicited antibodies with superior neutralizing capacity to antibodies in human convalescent sera.
The optimal spacing between B cell antigens to promote BCR crosslinking was recently determined using DNA origami nanoassemblies.230 Veneziano et al. treated Ramos B cells with rigid DNA rod scaffolds that carried two eOD antigens at various distances to determine the optimal antigen distance for B cell activation. As the distance between the two antigens increased from 1 nm, the calcium influx gradually improved, indicating rising levels of B cell activation. Such activation signals plateaued when the distance between the two antigens reached 28 nm. When the B cells were treated with DNA nanoparticles displaying five antigens with controlled distances, the calcium influx signals maximized at an antigen-space of 22 nm. This study exemplifies the precise control provided by biomaterial design and revealed an important parameter for multivalent antigen design.
Alum-binding antigens are another noteworthy strategy to enhance B cell activation. Moyer et al. modified protein antigens with a short peptide of repeating phosphoserine (pSer) and enabled antigen retention by alum adjuvant231(Fig. 5E). Using fluorescently labeled eOD antigens, the researchers demonstrated that the pSer modification improved antigen persistence at the injection site when administered with alum. Antigens carrying longer pSer peptides exhibited longer retention, stronger B cell activation, and higher antibody titers. Alum-binding antigen can efficiently accumulate within B cell follicles and, surprisingly, deliver alum crystals inside eOD-binding B cells, indicating that alum-binding eOD antigens are delivered in an alum-bound form in vivo. B cells that internalized alum salt upregulated genes associated with antigen processing and presentations. Modification of MD39 antigens with pSer4 at the stem focused the antibody responses against productive epitopes and significantly improved the elicitation of neutralizing antibodies.
Collectively, these studies demonstrated that biomaterials provide versatile control over antigen valency and antigen immunogenicity for enhanced B cell activation. These antigen modifications effectively promoted GC responses and ultimately improved the quality and magnitude of antibody responses. These design principles revealed by these studies can instruct future biomaterial design for the generation of potent B cell responses.
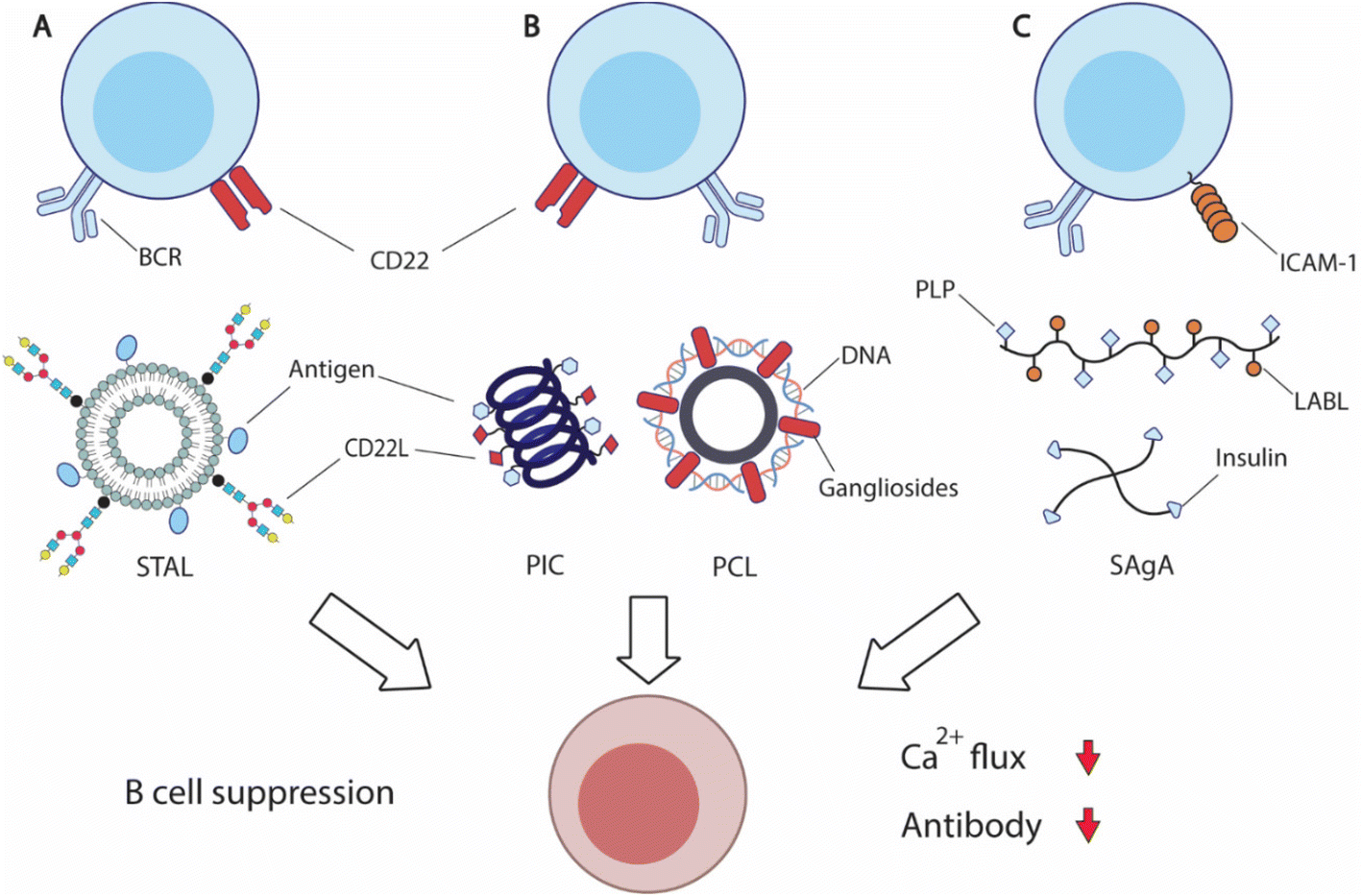 | ||
| Fig. 6 Overview of strategies to suppress antigen-specific B cells. (A) Siglec-engaging tolerance-inducing antigenic liposomes (STAL) display antigens and synthetic siglec CD22L suppresses antigen-specific B cells by engaging inhibitory CD22 receptor and B cell receptor (BCR). (B) Helical polyisocyanopeptide (PIC)251 and PEGylated cationic liposome (PCL)252 display both antigens and glycan or peptide CD22L respectively to inhibit B cell functions. (C) Soluble antigen array (SAgA) is designed based on hyaluronic acid (HA) polymers or 4-arm PEG polymers to deliver antigens, myelin sheath peptide antigen (PLP) or insulin, with inhibitory peptide (LABL) for intercellular adhesion molecule-1 (ICAM-1) to inhibit B cell activation.254,258 | ||
CD22 (Siglec-2) and Siglec-G are members of sialic-acid binding immunoglobulin-type lectin, serving as important B cell inhibitory co-receptors.237,238 CD22 specifically binds to ligands carrying α2–6-linked sialic acids, while siglec-G binds to α2–3-linked sialic acids. Co-ligation of BCRs and CD22 or Siglec-G downregulates BCRs, potentially leading to B cell apoptosis. Because CD22 is more commonly expressed on traditional B cells and conserved between mice and humans, it is an attractive therapeutic target for B cell inhibition.
Paulson and coworkers have carried out extensive research in an effort to translate the inhibitory function of CD22 into effective treatment. The researchers designed a library of synthetic siglec ligands with C-9 biphenyl substitution on the sialoside glycans.239 Through in vitro screening, the researchers identified that the best ligand for mouse CD22 (mCD22) was BPANeuGc-PAA and the best performing ligand for human CD22 (hCD22) was BPCNeuAc.239–241 By conjugating the synthetic CD22 ligands along with antigens of therapeutic interest, the team produced a series of siglec-engaging tolerance-inducing antigenic liposomes (STAL) capable of inducing antigen-specific tolerance in several therapeutic models242–247 (Fig. 6A). B cell inhibition by STAL was initially demonstrated in mice for both nitrophenol and hen egg lysozyme (HEL). STAL-treated mice produced 100-fold lower IgG responses when challenged with the corresponding antigens than untreated mice.242 These B cell suppression effects of STAL were absent in CD22-KO mice, confirming that the observed antibody reduction was CD22-dependent. Additionally, HEL-displaying STALs only induced tolerance toward HEL, but not OVA antigens, indicating the antigen-specificity of STAL-induced tolerance. The research team demonstrated the effectiveness of STAL technology in several preclinical models for allergy or autoimmunity. STAL effectively suppressed B cell responses against peanut allergens Ah2, and reduced IgE production and anaphylaxis in mice.244,247 STAL nanoparticles also induced B cell tolerance toward autoantigen cyclic citrullinated peptides (CCP), and ameliorated the disease burden of rheumatoid arthritis.245 STAL displaying glucose-6-phosphate isomerase (GPI) antigen also showed a therapeutic effect in a K/BxN mouse model that spontaneously develops joint inflammation due to the production of GPI-specific autoantibodies.246 To further improve the therapeutic efficacy, the team also designed rapamycin-encapsulating GPI-STAL nanoparticles to induce tolerogenic responses from both B and T cells.248,249 This therapeutic strategy also alleviated the arthritis symptoms in the K/BxN mouse model.249
The codelivery of antigens with CD22 ligands for B cell suppression has also been achieved by other biomaterial designs. Courtney et al. designed synthetic ROMP polymers carrying both antigens and amine-substituted trisaccharides (Neu5Ac α2,6 Gal β1,4 Glc) that functioned as ligands for CD22.250In vitro treatment with the obtained polymers on B cells induced CD22 phosphorylation and attenuated tyrosine phosphorylation, resulting in B cell inhibition. Kristyanto et al. delivered the same CD22 ligands with CCP antigens in polyisocyanopeptides (PICs) that form stable helical filaments251 (Fig. 6B). PIC-treatment inhibited auto-reactive B cells in an antigen-dependent fashion. Qelliny et al. designed PEGylated cationic liposomes (PCLs) that carried pDNA and gangliosides, another ligand for CD22, to inhibit antibody responses against DNA and PEG252 (Fig. 6B). Taken together, these studies demonstrate that targeting the B cell inhibitory coreceptor CD22 can achieve antigen-specific B cell suppression. It holds significant promise in treating allergy and autoimmune disorders, without compromising protective immunity.
Berkland and coworkers designed hyaluronic acid (HA) polymers that carry both myelin sheath peptide antigen (PLP) and intercellular adhesion molecule-1 (ICAM-1) inhibitory peptide (LABL), namely soluble antigen array (SAgA)253,254 (Fig. 6C). The co-delivery of antigens and inhibitory peptides suppressed the autoreactive immunity against myelin proteins and ameliorated the disease symptoms in murine experimental autoimmune encephalomyelitis. Mechanistically, SAgA treatment induced BCR clustering and tempered calcium flux, and non-hydrolysable linkers between polymers and cargos showed higher binding avidity.255,256 The research team also utilized the SAgA strategy to deliver insulin and LABL to induce insulin tolerance.257 SAgA treatment induced anergy for insulin-binding B cells from nonobese diabetic (NOD) mice, while insulin protein alone stimulated B cell activation. The research team also explored four-arm PEG polymers for soluble antigen delivery.258
Different from the pathogenic role that conventional B cells played in autoimmunity, B1a cells play a therapeutic role in autoimmunity by secreting regulatory cytokine IL-10 or by producing protective low-affinity natural antibodies against antigens, such as phosphocholine (PC).259 Curvino et al. designed Q11 nanofiber vaccines with a multivalent display of PC antigens (PCM-Q11) to stimulate B1a cells for the treatment of inflammation.282 Intraperitoneal immunizations of PCM-Q11 promoted significant antigen uptake by B1a cells and elicited PC-specific antibody responses. Strikingly, PCM-Q11 immunizations showed a protective effect in both acute and chronic DSS-induced colitis. Sera collected from PCM-Q11 immunized mice showed similar therapeutic benefits, indicating that the observed protection against inflammation in part is driven by PC-specific antibodies. This study demonstrated an effective approach to induce B1a cells for treatment.
Collectively, these studies highlight the therapeutic potential of B cell-targeted therapy in treating autoimmunity, allergy, and inflammation. Future strategies can potentially benefit from targeting alternative inhibitory B cell co-receptors. Regulatory T cells can also potentially synergize with B cell-targeted therapy for the treatment of autoimmunity.
3. Outlook and conclusion
This review has discussed a number of biomaterial engineering strategies that regulate B cell immune responses through controlling fundamental aspects of B cell immune responses, including antigen trafficking, antigen presentation, antigen retention and T–B cognate interaction. Yet, it remains challenging to fully realize the therapeutic potential of B cells for the treatment of various illnesses, including autoimmune conditions,3 and cancers,7 and infections by pathogens that are difficult to develop vaccines against, such as HIV,260 malaria,261 and tuberculosis.254 As new mechanistic insights into these pathologies emerge, future biomaterial engineers can accelerate the translation of these discoveries into novel therapeutics.B cells shape tumor microenvironments through cytokine secretions, tumor-associated antibody production, and antigen presentations to T cells.7,262 Tumor-infiltrated B cells can also spontaneously organize into tertiary lymphoid structures (TLSs) with T cells and positively contribute to immunotherapies.15,16,19,263–265 Yet, engineering strategies to harvest anti-tumor B cell immunity remain limited. Raising tumor-specific antibodies through vaccination have only showed limited efficacy, largely due to immunological escape and lack of T cell involvement.266,267 The aforementioned combinatory Coil29 vaccine, along with other combinatorial therapies, demonstrated that inducing synergistic anti-tumor B cell and CD8+ T cell responses is a promising approach to enhance anti-tumor B cell immunity and to prevent immunological escape by promoting antigen spreading.204,267 Inducing TLS formation within tumor tissue is another potential strategy to leverage B cells for tumor treatment, because of the strong association between the presence of TLS and favorable clinical outcomes.268 Hydrogel encapsulating a combination of chemokines has been shown to promote the neogenesis of lymphoid structures.269 Recent innovations in biomaterial-based sustained delivery platforms can potentially accelerate the development of TLS-related therapeutics and uncover the therapeutic mechanisms of TLS.
The holy grail of vaccine research is the elicitation of bnAbs against the conserved viral antigens on rapidly mutating pathogens such as HIV and influenza.260 However, this is inherently difficult for several reasons, including low frequencies of bnAb-precursor B cells, the immunodominance of non-neutralizing epitopes, and the extraordinarily high SHM rate required for bnAbs.102,270 Prolonged and enhanced germinal center reactions have an increased likelihood to recruit rare bnAb-precursor B cells for GC reactions and promote bnAb-driving mutations.140,142,186 To this end, sustained release using hydrogel delivery systems, MN patches, microparticles, and slow administration have all demonstrated various levels of effectiveness in promoting GC responses and improving antibody quality and diversity.139–141,162,186 Slow vaccine administration, in particular, achieved impressive bnAb-focusing B cell responses. Future biomaterial engineering strategies that achieve similarly extended antigen delivery kinetics using a single administration can improve bnAb elicitation while avoiding potential patient compliance issues. Individual immune and infection histories also complicate the responses to vaccination, and potentially negatively impact bnAb responses.108 Repeated nasal influenza immunizations in children focused antibody responses against conserved antigens and successfully induced bnAbs against influenza, suggesting immunization in young populations can potentially circumvent the impact of immunological imprinting.271 Therefore, biomaterial engineers should also take the potential patient populations into consideration when designing bnAb-inducing vaccines.
Vaccine development against pathogenic bacteria is another area that is receiving increased research attention. Different from antibiotics that deplete the gut microbiota and potentially drive antibiotic resistance, vaccination approaches can specifically eliminate pathogenic bacterial strains without negative impacts on the gut microbiome.283 Future anti-bacterial vaccine research can greatly benefit from the identification of pathogenic bacteria-specific antigen targets and strategies to eliminate intracellular bacterial pathogens.272,273
Not only can biomaterials engineering design translate immunological insights, but the spatial tempo control on both the molecular and cellular levels provided by biomaterials platforms can further deepen our fundamental understanding of B cell immunology.274 The 3D hydrogel coculture system for GC B cells helped untangle the influence of individual molecular factors on B cell responses and revealed epigenic factors involved in GC B cell responses.275–278 The ability to capture GC B cell dynamics within this hydrogel system also enabled the in vitro screening of glycoconjugate vaccines and significantly accelerated the antigenicity evaluations as compared with the animal immunization-based approach.279 A B-cell lymphoma in vitro model based on a similar strategy informed the design of a novel combination therapy to attenuate treatment resistance.280 Hydrogel has also been incorporated into microfluidic devices to assess B cell responses against vaccines.281 These studies collectively showcased the potential of biomaterials in facilitating B cell immunobiology and vaccine screening by serving as reductionist models for germinal centers or disease tissues.
In this review, we have provided an overview of the current understanding of B cell biology in the context of some recent advances in biomaterial engineering strategies to modulate B cell immunity from the past decade. New discoveries surrounding GC dynamics, neuroimmunology, the gut–brain axis, and other advances have sparked intense research interest in the field of B cell immunity. These newly unveiled cellular and molecular insights not only provide a deeper understanding of human biology, but also give rise to potential therapeutic opportunities. Leveraging biomaterial engineering strategies will undoubtedly continue to accelerate the translation of these new B cell biology insights and improve therapeutics for human health.
Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
This manuscript is supported by the Biomedical and Chemical Engineering Department of Syracuse University and NIH/NIGMS R35GM147560.References
- D. R. Burton, Nat. Rev. Immunol., 2002, 2, 706–713 CrossRef CAS PubMed.
- M. Aziz, N. E. Holodick, T. L. Rothstein and P. Wang, Immunol. Res., 2015, 63, 153–166 CrossRef CAS PubMed.
- H. U. Scherer, D. van der Woude and R. E. M. Toes, Nat. Rev. Rheumatol., 2022, 18, 371–383 CrossRef CAS PubMed.
- K. Yanaba, J. Bouaziz, T. Matsushita, C. M. Magro, E. W. St. Clair and T. F. Tedder, Immunol. Rev., 2008, 223, 284–299 CrossRef CAS PubMed.
- E. C. Rosser and C. Mauri, Immunity, 2015, 42, 607–612 CrossRef CAS PubMed.
- C. Sautès-Fridman, J. Verneau, C.-M. Sun, M. Moreira, T. W.-W. Chen, M. Meylan, F. Petitprez and W. H. Fridman, Semin. Immunol., 2020, 48, 101406 CrossRef PubMed.
- C. M. Laumont, A. C. Banville, M. Gilardi, D. P. Hollern and B. H. Nelson, Nat. Rev. Cancer, 2022, 22, 414–430 CrossRef CAS PubMed.
- L. L. Lu, T. J. Suscovich, S. M. Fortune and G. Alter, Nat. Rev. Immunol., 2018, 18, 46–61 CrossRef CAS PubMed.
- S. A. Wellford, A. P. Moseman, K. Dao, K. E. Wright, A. Chen, J. E. Plevin, T.-C. Liao, N. Mehta and E. A. Moseman, Immunity, 2022, 55, 2118–2134 CrossRef CAS PubMed.
- C. E. Nelson, S. Namasivayam, T. W. Foreman, K. D. Kauffman, S. Sakai, D. E. Dorosky, N. E. Lora, N. T. I. Program, K. Brooks, E. L. Potter, N. L. Garza, B. A. P. Lafont, R. F. Johnson, M. Roederer, A. Sher, D. Weiskopf, A. Sette, E. de Wit, H. D. Hickman, J. M. Brenchley, L. E. Via, D. L. Barber, A. Abdi, E. K. Dayao, J. D. Fleegle, F. Gomez, M. K. Piazza, K. M. Repoli, B. Y. Sloan, A. L. Butler, A. M. Walker, D. M. Weiner, M. J. Woodcock and A. Vatthauer, Sci. Immunol., 2022, 7, eabo0535 CrossRef CAS PubMed.
- J. M. Brady, M. Phelps, S. W. MacDonald, E. C. Lam, A. Nitido, D. Parsons, C. L. Boutros, C. E. Deal, W. F. Garcia-Beltran, S. Tanno, H. Natarajan, M. E. Ackerman, V. D. Vrbanac and A. B. Balazs, Sci. Transl. Med., 2022, 14, eabn9662 CrossRef CAS PubMed.
- K. Rawat, A. Tewari, M. J. Morrisson, T. D. Wager and C. V. Jakubzick, eLife, 2021, 10, e69713 CrossRef CAS PubMed.
- R. D. Mazor, N. Nathan, A. Gilboa, L. Stoler-Barak, L. Moss, I. Solomonov, A. Hanuna, Y. Divinsky, M. D. Shmueli, H. Hezroni, I. Zaretsky, M. Mor, O. Golani, G. Sabah, A. Jakobson-Setton, N. Yanichkin, M. Feinmesser, D. Tsoref, L. Salman, E. Yeoshoua, E. Peretz, I. Erlich, N. M. Cohen, J. M. Gershoni, N. Freund, Y. Merbl, G. Yaari, R. Eitan, I. Sagi and Z. Shulman, Cell, 2022, 185, 1208–1222 CrossRef CAS PubMed.
- A. M. Weis and J. L. Round, Cell Host Microbe, 2021, 29, 334–346 CrossRef CAS PubMed.
- F. Petitprez, A. de Reyniès, E. Z. Keung, T. W.-W. Chen, C.-M. Sun, J. Calderaro, Y.-M. Jeng, L.-P. Hsiao, L. Lacroix, A. Bougoüin, M. Moreira, G. Lacroix, I. Natario, J. Adam, C. Lucchesi, Y. Laizet, M. Toulmonde, M. A. Burgess, V. Bolejack, D. Reinke, K. M. Wani, W.-L. Wang, A. J. Lazar, C. L. Roland, J. A. Wargo, A. Italiano, C. Sautès-Fridman, H. A. Tawbi and W. H. Fridman, Nature, 2020, 577, 556–560 CrossRef CAS PubMed.
- B. A. Helmink, S. M. Reddy, J. Gao, S. Zhang, R. Basar, R. Thakur, K. Yizhak, M. Sade-Feldman, J. Blando, G. Han, V. Gopalakrishnan, Y. Xi, H. Zhao, R. N. Amaria, H. A. Tawbi, A. P. Cogdill, W. Liu, V. S. LeBleu, F. G. Kugeratski, S. Patel, M. A. Davies, P. Hwu, J. E. Lee, J. E. Gershenwald, A. Lucci, R. Arora, S. Woodman, E. Z. Keung, P.-O. Gaudreau, A. Reuben, C. N. Spencer, E. M. Burton, L. E. Haydu, A. J. Lazar, R. Zapassodi, C. W. Hudgens, D. A. Ledesma, S. Ong, M. Bailey, S. Warren, D. Rao, O. Krijgsman, E. A. Rozeman, D. Peeper, C. U. Blank, T. N. Schumacher, L. H. Butterfield, M. A. Zelazowska, K. M. McBride, R. Kalluri, J. Allison, F. Petitprez, W. H. Fridman, C. Sautès-Fridman, N. Hacohen, K. Rezvani, P. Sharma, M. T. Tetzlaff, L. Wang and J. A. Wargo, Nature, 2020, 577, 549–555 CrossRef CAS PubMed.
- S. Biswas, G. Mandal, K. K. Payne, C. M. Anadon, C. D. Gatenbee, R. A. Chaurio, T. L. Costich, C. Moran, C. M. Harro, K. E. Rigolizzo, J. A. Mine, J. Trillo-Tinoco, N. Sasamoto, K. L. Terry, D. Marchion, A. Buras, R. M. Wenham, X. Yu, M. K. Townsend, S. S. Tworoger, P. C. Rodriguez, A. R. Anderson and J. R. Conejo-Garcia, Nature, 2021, 591, 464–470 CrossRef CAS PubMed.
- L. Vanhersecke, M. Brunet, J.-P. Guégan, C. Rey, A. Bougouin, S. Cousin, S. L. Moulec, B. Besse, Y. Loriot, M. Larroquette, I. Soubeyran, M. Toulmonde, G. Roubaud, S. Pernot, M. Cabart, F. Chomy, C. Lefevre, K. Bourcier, M. Kind, I. Giglioli, C. Sautès-Fridman, V. Velasco, F. Courgeon, E. Oflazoglu, A. Savina, A. Marabelle, J.-C. Soria, C. Bellera, C. Sofeu, A. Bessede, W. H. Fridman, F. L. Loarer and A. Italiano, Nat. Cancer, 2021, 2, 794–802 CrossRef CAS PubMed.
- R. Cabrita, M. Lauss, A. Sanna, M. Donia, M. S. Larsen, S. Mitra, I. Johansson, B. Phung, K. Harbst, J. Vallon-Christersson, A. van Schoiack, K. Lövgren, S. Warren, K. Jirström, H. Olsson, K. Pietras, C. Ingvar, K. Isaksson, D. Schadendorf, H. Schmidt, L. Bastholt, A. Carneiro, J. A. Wargo, I. M. Svane and G. Jönsson, Nature, 2020, 577, 561–565 CrossRef CAS PubMed.
- F. E. Lund and T. D. Randall, Nat. Rev. Immunol., 2010, 10, 236–247 CrossRef CAS PubMed.
- N. Komatsu and H. Takayanagi, Nat. Rev. Rheumatol., 2022, 18, 415–429 CrossRef CAS PubMed.
- A. Schudel, D. M. Francis and S. N. Thomas, Nat. Rev. Mater., 2019, 4, 415–428 CrossRef PubMed.
- P. Yousefpour, K. Ni and D. J. Irvine, Nat. Rev. Bioeng., 2023, 1, 107–124 CrossRef PubMed.
- J. A. Finbloom, C. Huynh, X. Huang and T. A. Desai, Nat. Rev. Bioeng., 2023, 1, 139–152 CrossRef.
- B. S. Ou, O. M. Saouaf, J. Baillet and E. A. Appel, Adv. Drug Delivery Rev., 2022, 187, 114401 CrossRef CAS PubMed.
- G. A. Roth, V. C. T. M. Picece, B. S. Ou, W. Luo, B. Pulendran and E. A. Appel, Nat. Rev. Mater., 2022, 7, 174–195 CrossRef CAS PubMed.
- E. A. Gosselin, H. B. Eppler, J. S. Bromberg and C. M. Jewell, Nat. Mater., 2018, 17, 484–498 CrossRef CAS PubMed.
- M. O. Dellacherie, B. R. Seo and D. J. Mooney, Nat. Rev. Mater., 2019, 4, 379–397 CrossRef.
- N. J. Shah, A. S. Mao, T.-Y. Shih, M. D. Kerr, A. Sharda, T. M. Raimondo, J. C. Weaver, V. D. Vrbanac, M. Deruaz, A. M. Tager, D. J. Mooney and D. T. Scadden, Nat. Biotechnol., 2019, 37, 293–302 CrossRef CAS PubMed.
- N. J. Shah, A. J. Najibi, T.-Y. Shih, A. S. Mao, A. Sharda, D. T. Scadden and D. J. Mooney, Nat. Biomed. Eng., 2020, 4, 40–51 CrossRef CAS PubMed.
- A. H. Morris, K. R. Hughes, R. S. Oakes, M. M. Cai, S. D. Miller, D. N. Irani and L. D. Shea, Nat. Commun., 2020, 11, 3871 CrossRef CAS PubMed.
- K. Sadtler, A. Singh, M. T. Wolf, X. Wang, D. M. Pardoll and J. H. Elisseeff, Nat. Rev. Mater., 2016, 1, 16040 CrossRef CAS.
- L. Chung, D. R. Maestas, F. Housseau and J. H. Elisseeff, Adv. Drug Delivery Rev., 2017, 114, 184–192 CrossRef CAS PubMed.
- L. R. Nih, S. Gojgini, S. T. Carmichael and T. Segura, Nat. Mater., 2018, 17, 642–651 CrossRef CAS PubMed.
- C. L. Stabler, Y. Li, J. M. Stewart and B. G. Keselowsky, Nat. Rev. Mater., 2019, 4, 429–450 CrossRef CAS PubMed.
- K. S. Corbett, D. K. Edwards, S. R. Leist, O. M. Abiona, S. Boyoglu-Barnum, R. A. Gillespie, S. Himansu, A. Schäfer, C. T. Ziwawo, A. T. DiPiazza, K. H. Dinnon, S. M. Elbashir, C. A. Shaw, A. Woods, E. J. Fritch, D. R. Martinez, K. W. Bock, M. Minai, B. M. Nagata, G. B. Hutchinson, K. Wu, C. Henry, K. Bahl, D. Garcia-Dominguez, L. Ma, I. Renzi, W.-P. Kong, S. D. Schmidt, L. Wang, Y. Zhang, E. Phung, L. A. Chang, R. J. Loomis, N. E. Altaras, E. Narayanan, M. Metkar, V. Presnyak, C. Liu, M. K. Louder, W. Shi, K. Leung, E. S. Yang, A. West, K. L. Gully, L. J. Stevens, N. Wang, D. Wrapp, N. A. Doria-Rose, G. Stewart-Jones, H. Bennett, G. S. Alvarado, M. C. Nason, T. J. Ruckwardt, J. S. McLellan, M. R. Denison, J. D. Chappell, I. N. Moore, K. M. Morabito, J. R. Mascola, R. S. Baric, A. Carfi and B. S. Graham, Nature, 2020, 586, 567–571 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- K. E. Stephenson, K. Wagh, B. Korber and D. H. Barouch, Annu. Rev. Immunol., 2020, 38, 673–703 CrossRef CAS PubMed.
- H. F. Haddad, E. F. Roe and J. H. Collier, Biomater. Sci., 2023, 11, 1625–1647 RSC.
- Y. Shi, J. Controlled Release, 2021, 339, 156–163 CrossRef CAS PubMed.
- D. S. W. Lee, O. L. Rojas and J. L. Gommerman, Nat. Rev. Drug Discovery, 2021, 20, 179–199 CrossRef CAS PubMed.
- J. Wang, J. Yang and J. Kopeček, Acta Biomater., 2022, 137, 1–19 CrossRef CAS PubMed.
- F. D. Batista and N. E. Harwood, Nat. Rev. Immunol., 2009, 9, 15–27 CrossRef CAS PubMed.
- K. A. Pape, D. M. Catron, A. A. Itano and M. K. Jenkins, Immunity, 2007, 26, 491–502 CrossRef CAS PubMed.
- R. Roozendaal, T. R. Mempel, L. A. Pitcher, S. F. Gonzalez, A. Verschoor, R. E. Mebius, U. H. von Andrian and M. C. Carroll, Immunity, 2009, 30, 264–276 CrossRef CAS PubMed.
- Y.-N. Zhang, J. Lazarovits, W. Poon, B. Ouyang, L. N. M. Nguyen, B. R. Kingston and W. C. W. Chan, Nano Lett., 2019, 19, 7226–7235 CrossRef CAS PubMed.
- Y. R. Carrasco and F. D. Batista, Immunity, 2007, 27, 160–171 CrossRef CAS PubMed.
- T. G. Phan, I. Grigorova, T. Okada and J. G. Cyster, Nat. Immunol., 2007, 8, 992–1000 CrossRef CAS PubMed.
- T. Junt, E. A. Moseman, M. Iannacone, S. Massberg, P. A. Lang, M. Boes, K. Fink, S. E. Henrickson, D. M. Shayakhmetov, N. C. D. Paolo, N. van Rooijen, T. R. Mempel, S. P. Whelan and U. H. von Andrian, Nature, 2007, 450, 110–114 CrossRef CAS PubMed.
- T. G. Phan, J. A. Green, E. E. Gray, Y. Xu and J. G. Cyster, Nat. Immunol., 2009, 10, 786–793 CrossRef CAS PubMed.
- T. I. Arnon, R. M. Horton, I. L. Grigorova and J. G. Cyster, Nature, 2013, 493, 684–688 CrossRef CAS PubMed.
- L. C. Davies, S. J. Jenkins, J. E. Allen and P. R. Taylor, Nat. Immunol., 2013, 14, 986–995 CrossRef CAS PubMed.
- S. M. Lewis, A. Williams and S. C. Eisenbarth, Sci. Immunol., 2019, 4, eaau6085 CrossRef CAS PubMed.
- A. Reboldi and J. G. Cyster, Immunol. Rev., 2016, 271, 230–245 CrossRef CAS PubMed.
- E. A. Koppel, C. W. Wieland, V. C. M. van den Berg, M. Litjens, S. Florquin, Y. van Kooyk, T. van der Poll and T. B. H. Geijtenbeek, Eur. J. Immunol., 2005, 35, 2962–2969 CrossRef CAS PubMed.
- Y. Fang, C. Xu, Y.-X. Fu, V. M. Holers and H. Molina, J. Immunol., 1998, 160, 5273–5279 CrossRef CAS.
- A. Aung, A. Cui, L. Maiorino, A. P. Amini, J. R. Gregory, M. Bukenya, Y. Zhang, H. Lee, C. A. Cottrell, D. M. Morgan, M. Silva, H. Suh, J. D. Kirkpatrick, P. Amlashi, T. Remba, L. M. Froehle, S. Xiao, W. Abraham, J. Adams, J. C. Love, P. Huyett, D. S. Kwon, N. Hacohen, W. R. Schief, S. N. Bhatia and D. J. Irvine, Science, 2023, 379, eabn8934 CrossRef CAS PubMed.
- B. A. Heesters, R. C. Myers and M. C. Carroll, Nat. Rev. Immunol., 2014, 14, 495–504 CrossRef CAS PubMed.
- J. G. Cyster and C. D. C. Allen, Cell, 2019, 177, 524–540 CrossRef CAS PubMed.
- S. Crotty, Immunity, 2019, 50, 1132–1148 CrossRef CAS PubMed.
- T. D. Chan, D. Gatto, K. Wood, T. Camidge, A. Basten and R. Brink, J. Immunol., 2009, 183, 3139–3149 CrossRef CAS PubMed.
- X. Liu, B. Liu and H. Qi, Curr. Opin. Immunol., 2023, 82, 102308 CrossRef CAS PubMed.
- K. A. Pape, J. J. Taylor, R. W. Maul, P. J. Gearhart and M. K. Jenkins, Science, 2011, 331, 1203–1207 CrossRef CAS PubMed.
- F. J. Weisel, G. V. Zuccarino-Catania, M. Chikina and M. J. Shlomchik, Immunity, 2016, 44, 116–130 CrossRef CAS PubMed.
- C.-H. Yeh, T. Nojima, M. Kuraoka and G. Kelsoe, Nat. Commun., 2018, 9, 928 CrossRef PubMed.
- G. D. Victora and M. C. Nussenzweig, Annu. Rev. Immunol., 2022, 40, 1–30 CrossRef PubMed.
- N. B. Pikor, U. Mörbe, M. Lütge, C. Gil-Cruz, C. Perez-Shibayama, M. Novkovic, H.-W. Cheng, C. Nombela-Arrieta, T. Nagasawa, M. A. Linterman, L. Onder and B. Ludewig, Nat. Immunol., 2020, 21, 649–659 CrossRef CAS PubMed.
- J. M. D. Noia and M. S. Neuberger, Annu. Rev. Biochem., 2007, 76, 1–22 CrossRef PubMed.
- I. Stewart, D. Radtke, B. Phillips, S. J. McGowan and O. Bannard, Immunity, 2018, 49, 477–489 CrossRef CAS PubMed.
- J. A. Roco, L. Mesin, S. C. Binder, C. Nefzger, P. Gonzalez-Figueroa, P. F. Canete, J. Ellyard, Q. Shen, P. A. Robert, J. Cappello, H. Vohra, Y. Zhang, C. R. Nowosad, A. Schiepers, L. M. Corcoran, K.-M. Toellner, J. M. Polo, M. Meyer-Hermann, G. D. Victora and C. G. Vinuesa, Immunity, 2019, 51, 337–350 CrossRef CAS PubMed.
- G. D. Victora, T. A. Schwickert, D. R. Fooksman, A. O. Kamphorst, M. Meyer-Hermann, M. L. Dustin and M. C. Nussenzweig, Cell, 2010, 143, 592–605 CrossRef CAS PubMed.
- S. Finkin, H. Hartweger, T. Y. Oliveira, E. E. Kara and M. C. Nussenzweig, Immunity, 2019, 51, 324–336 CrossRef CAS PubMed.
- S. Crotty, Nat. Rev. Immunol., 2015, 15, 185–189 CrossRef CAS PubMed.
- B. Liu, Y. Lin, J. Yan, J. Yao, D. Liu, W. Ma, J. Wang, W. Liu, C. Wang, L. Zhang and H. Qi, Nature, 2021, 592, 133–137 CrossRef CAS PubMed.
- C. D. C. Allen, T. Okada and J. G. Cyster, Immunity, 2007, 27, 190–202 CrossRef CAS PubMed.
- A. D. Gitlin, C. T. Mayer, T. Y. Oliveira, Z. Shulman, M. J. K. Jones, A. Koren and M. C. Nussenzweig, Science, 2015, 349, 643–646 CrossRef CAS PubMed.
- J. Pae, J. Ersching, T. B. R. Castro, M. Schips, L. Mesin, S. J. Allon, J. Ordovas-Montanes, C. Mlynarczyk, A. Melnick, A. Efeyan, A. K. Shalek, M. Meyer-Hermann and G. D. Victora, J. Exp. Med., 2020, 218, e20201699 CrossRef PubMed.
- Z. Long, B. Phillips, D. Radtke, M. Meyer-Hermann and O. Bannard, Sci. Immunol., 2022, 7, eabm0775–eabm0775 CrossRef CAS PubMed.
- R. Razzaghi, S. Agarwal, N. Kotlov, O. Plotnikova, K. Nomie, D. W. Huang, G. W. Wright, G. A. Smith, M. Li, K. Takata, M. Yamadi, C. Yao, J. J. O'Shea, J. D. Phelan, S. Pittaluga, D. W. Scott and J. R. Muppidi, J. Exp. Med., 2020, 218, e20201173 CrossRef PubMed.
- J. S. Turner, W. Kim, E. Kalaidina, C. W. Goss, A. M. Rauseo, A. J. Schmitz, L. Hansen, A. Haile, M. K. Klebert, I. Pusic, J. A. O'Halloran, R. M. Presti and A. H. Ellebedy, Nature, 2021, 595, 421–425 CrossRef CAS PubMed.
- R. V. H. de Carvalho, J. Ersching, A. Barbulescu, A. Hobbs, T. B. R. Castro, L. Mesin, J. T. Jacobsen, B. K. Phillips, H.-H. Hoffmann, R. Parsa, M. C. C. Canesso, C. R. Nowosad, A. Feng, S. R. Leist, R. S. Baric, E. Yang, P. J. Utz and G. D. Victora, Cell, 2023, 186, 131–146 CrossRef CAS PubMed.
- T. Hägglöf, M. Cipolla, M. Loewe, S. T. Chen, L. Mesin, H. Hartweger, M. A. ElTanbouly, A. Cho, A. Gazumyan, V. Ramos, L. Stamatatos, T. Y. Oliveira, M. C. Nussenzweig and C. Viant, Cell, 2023, 186, 147–161 CrossRef PubMed.
- J. S. Turner, J. A. O'Halloran, E. Kalaidina, W. Kim, A. J. Schmitz, J. Q. Zhou, T. Lei, M. Thapa, R. E. Chen, J. B. Case, F. Amanat, A. M. Rauseo, A. Haile, X. Xie, M. K. Klebert, T. Suessen, W. D. Middleton, P.-Y. Shi, F. Krammer, S. A. Teefey, M. S. Diamond, R. M. Presti and A. H. Ellebedy, Nature, 2021, 596, 109–113 CrossRef CAS PubMed.
- W. Kim, J. Q. Zhou, S. C. Horvath, A. J. Schmitz, A. J. Sturtz, T. Lei, Z. Liu, E. Kalaidina, M. Thapa, W. B. Alsoussi, A. Haile, M. K. Klebert, T. Suessen, L. Parra-Rodriguez, P. A. Mudd, S. P. J. Whelan, W. D. Middleton, S. A. Teefey, I. Pusic, J. A. O'Halloran, R. M. Presti, J. S. Turner and A. H. Ellebedy, Nature, 2022, 604, 141–145 CrossRef CAS PubMed.
- C. Viant, G. H. J. Weymar, A. Escolano, S. Chen, H. Hartweger, M. Cipolla, A. Gazumyan and M. C. Nussenzweig, Cell, 2020, 183, 1298–1311 CrossRef CAS PubMed.
- T. Inoue and T. Kurosaki, Nat. Rev. Immunol., 2023, 1–13 CAS.
- Y. Wang, J. Shi, J. Yan, Z. Xiao, X. Hou, P. Lu, S. Hou, T. Mao, W. Liu, Y. Ma, L. Zhang, X. Yang and H. Qi, Nat. Immunol., 2017, 18, 921–930 CrossRef CAS PubMed.
- S. Takatsuka, H. Yamada, K. Haniuda, H. Saruwatari, M. Ichihashi, J.-C. Renauld and D. Kitamura, Nat. Immunol., 2018, 19, 1025–1034 CrossRef CAS PubMed.
- A. Schiepers, M. F. L. van ‘t Wout, A. J. Greaney, T. Zang, H. Muramatsu, P. J. C. Lin, Y. K. Tam, L. Mesin, T. N. Starr, P. D. Bieniasz, N. Pardi, J. D. Bloom and G. D. Victora, Nature, 2023, 615, 482–489 CrossRef CAS PubMed.
- J. S. Turner, J. Q. Zhou, J. Han, A. J. Schmitz, A. A. Rizk, W. B. Alsoussi, T. Lei, M. Amor, K. M. McIntire, P. Meade, S. Strohmeier, R. I. Brent, S. T. Richey, A. Haile, Y. R. Yang, M. K. Klebert, T. Suessen, S. Teefey, R. M. Presti, F. Krammer, S. H. Kleinstein, A. B. Ward and A. H. Ellebedy, Nature, 2020, 586, 127–132 CrossRef CAS PubMed.
- L. Mesin, A. Schiepers, J. Ersching, A. Barbulescu, C. B. Cavazzoni, A. Angelini, T. Okada, T. Kurosaki and G. D. Victora, Cell, 2020, 180, 92–106 CrossRef CAS PubMed.
- M. Kuraoka, C.-H. Yeh, G. Bajic, R. Kotaki, S. Song, I. Windsor, S. C. Harrison and G. Kelsoe, Sci. Immunol., 2022, 7, eabn5311 CrossRef CAS PubMed.
- K. J. Kenderes, R. C. Levack, A. M. Papillion, B. Cabrera-Martinez, L. M. Dishaw and G. M. Winslow, Cell Rep., 2018, 24, 824–837 CrossRef CAS PubMed.
- T. Arulraj, S. C. Binder, P. A. Robert and M. Meyer-Hermann, Front. Immunol., 2021, 12, 705240 CrossRef CAS PubMed.
- J. T. Jacobsen, W. Hu, T. B. R. Castro, S. Solem, A. Galante, Z. Lin, S. J. Allon, L. Mesin, A. M. Bilate, A. Schiepers, A. K. Shalek, A. Y. Rudensky and G. D. Victora, Science, 2021, 373, eabe5146 CrossRef CAS PubMed.
- C. Havenar-Daughton, D. G. Carnathan, A. T. de la Peña, M. Pauthner, B. Briney, S. M. Reiss, J. S. Wood, K. Kaushik, M. J. van Gils, S. L. Rosales, P. van der Woude, M. Locci, K. M. Le, S. W. de Taeye, D. Sok, A. U. R. Mohammed, J. Huang, S. Gumber, A. Garcia, S. P. Kasturi, B. Pulendran, J. P. Moore, R. Ahmed, G. Seumois, D. R. Burton, R. W. Sanders, G. Silvestri and S. Crotty, Cell Rep., 2016, 17, 2195–2209 CrossRef CAS PubMed.
- J. K. Hu, J. C. Crampton, A. Cupo, T. Ketas, M. J. van Gils, K. Sliepen, S. W. de Taeye, D. Sok, G. Ozorowski, I. Deresa, R. Stanfield, A. B. Ward, D. R. Burton, P. J. Klasse, R. W. Sanders, J. P. Moore and S. Crotty, J. Virol., 2015, 89, 10383–10398 CrossRef CAS PubMed.
- D. Wellington, Z. Yin, B. M. Kessler and T. Dong, Curr. Opin. Virol., 2021, 50, 183–191 CrossRef CAS PubMed.
- A. Tarke, J. Sidney, C. K. Kidd, J. M. Dan, S. I. Ramirez, E. D. Yu, J. Mateus, R. da S. Antunes, E. Moore, P. Rubiro, N. Methot, E. Phillips, S. Mallal, A. Frazier, S. A. Rawlings, J. A. Greenbaum, B. Peters, D. M. Smith, S. Crotty, D. Weiskopf, A. Grifoni and A. Sette, Cell Rep. Med., 2021, 2, 100204 CrossRef CAS PubMed.
- R. K. Abbott and S. Crotty, Immunol. Rev., 2020, 296, 120–131 CrossRef CAS PubMed.
- J. J. Guthmiller, J. Han, H. A. Utset, L. Li, L. Y.-L. Lan, C. Henry, C. T. Stamper, M. McMahon, G. O'Dell, M. L. Fernández-Quintero, A. W. Freyn, F. Amanat, O. Stovicek, L. Gentles, S. T. Richey, A. T. de la Peña, V. Rosado, H. L. Dugan, N.-Y. Zheng, M. E. Tepora, D. J. Bitar, S. Changrob, S. Strohmeier, M. Huang, A. García-Sastre, K. R. Liedl, J. D. Bloom, R. Nachbagauer, P. Palese, F. Krammer, L. Coughlan, A. B. Ward and P. C. Wilson, Nature, 2022, 602, 314–320 CrossRef CAS PubMed.
- R. K. Abbott, J. H. Lee, S. Menis, P. Skog, M. Rossi, T. Ota, D. W. Kulp, D. Bhullar, O. Kalyuzhniy, C. Havenar-Daughton, W. R. Schief, D. Nemazee and S. Crotty, Immunity, 2018, 48, 133–146 CrossRef CAS PubMed.
- M. Kuraoka, A. G. Schmidt, T. Nojima, F. Feng, A. Watanabe, D. Kitamura, S. C. Harrison, T. B. Kepler and G. Kelsoe, Immunity, 2016, 44, 542–552 CrossRef CAS PubMed.
- C. Havenar-Daughton, A. Sarkar, D. W. Kulp, L. Toy, X. Hu, I. Deresa, O. Kalyuzhniy, K. Kaushik, A. A. Upadhyay, S. Menis, E. Landais, L. Cao, J. K. Diedrich, S. Kumar, T. Schiffner, S. M. Reiss, G. Seumois, J. R. Yates, J. C. Paulson, S. E. Bosinger, I. A. Wilson, W. R. Schief and S. Crotty, Sci. Transl. Med., 2018, 10, eaat0381 CrossRef PubMed.
- C. Havenar-Daughton, R. K. Abbott, W. R. Schief and S. Crotty, Curr. Opin. Immunol., 2018, 53, 209–216 CrossRef CAS PubMed.
- J. Feldman, J. Bals, C. G. Altomare, K. St. Denis, E. C. Lam, B. M. Hauser, L. Ronsard, M. Sangesland, T. B. Moreno, V. Okonkwo, N. Hartojo, A. B. Balazs, G. Bajic, D. Lingwood and A. G. Schmidt, Sci. Immunol., 2021, 6, eabl5842 CrossRef CAS PubMed.
- T. Francis, Proc. Am. Philos. Soc., 1960, 104, 572–578 Search PubMed.
- M. Koutsakos and A. H. Ellebedy, Immunity, 2023, 56, 909–913 CrossRef CAS PubMed.
- K. Röltgen, S. C. A. Nielsen, O. Silva, S. F. Younes, M. Zaslavsky, C. Costales, F. Yang, O. F. Wirz, D. Solis, R. A. Hoh, A. Wang, P. S. Arunachalam, D. Colburg, S. Zhao, E. Haraguchi, A. S. Lee, M. M. Shah, M. Manohar, I. Chang, F. Gao, V. Mallajosyula, C. Li, J. Liu, M. J. Shoura, S. B. Sindher, E. Parsons, N. J. Dashdorj, N. D. Dashdorj, R. Monroe, G. E. Serrano, T. G. Beach, R. S. Chinthrajah, G. W. Charville, J. L. Wilbur, J. N. Wohlstadter, M. M. Davis, B. Pulendran, M. L. Troxell, G. B. Sigal, Y. Natkunam, B. A. Pinsky, K. C. Nadeau and S. D. Boyd, Cell, 2022, 185, 1025–1040 CrossRef PubMed.
- D. Schaefer-Babajew, Z. Wang, F. Muecksch, A. Cho, M. Loewe, M. Cipolla, R. Raspe, B. Johnson, M. Canis, J. DaSilva, V. Ramos, M. Turroja, K. G. Millard, F. Schmidt, L. Witte, J. Dizon, I. Shimeliovich, K.-H. Yao, T. Y. Oliveira, A. Gazumyan, C. Gaebler, P. D. Bieniasz, T. Hatziioannou, M. Caskey and M. C. Nussenzweig, Nature, 2023, 613, 735–742 CrossRef CAS PubMed.
- A. Roghanian, R. J. Stopforth, L. N. Dahal and M. S. Cragg, J. Leukocyte Biol., 2018, 103, 1077–1088 CrossRef CAS PubMed.
- D. J. Irvine, M. A. Swartz and G. L. Szeto, Nat. Mater., 2013, 12, 978–990 CrossRef CAS PubMed.
- S. Shukla, J. T. Myers, S. E. Woods, X. Gong, A. E. Czapar, U. Commandeur, A. Y. Huang, A. D. Levine and N. F. Steinmetz, Biomaterials, 2017, 121, 15–27 CrossRef CAS PubMed.
- J. J. Moon, H. Suh, A. V. Li, C. F. Ockenhouse, A. Yadava and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1080–1085 CrossRef CAS PubMed.
- H. Liu, K. D. Moynihan, Y. Zheng, G. L. Szeto, A. V. Li, B. Huang, D. S. V. Egeren, C. Park and D. J. Irvine, Nature, 2014, 507, 519–522 CrossRef CAS PubMed.
- S. Pant, M. Furqan, R. M. Abdul-Karim, V. Chung, C. E. Devoe, M. L. Johnson, A. D. Leal, H. Park, Z. A. Wainberg, E. Welkowsky, C. M. Haqq, E. M. O'Reilly and C. D. Weekes, J. Clin. Oncol., 2022, 40, TPS2701–TPS2701 CrossRef.
- B. L. Hartwell, M. B. Melo, P. Xiao, A. A. Lemnios, N. Li, J. Y. H. Chang, J. Yu, M. S. Gebre, A. Chang, L. Maiorino, C. Carter, T. J. Moyer, N. C. Dalvie, S. A. Rodriguez-Aponte, K. A. Rodrigues, M. Silva, H. Suh, J. Adams, J. Fontenot, J. C. Love, D. H. Barouch, F. Villinger, R. M. Ruprecht and D. J. Irvine, Sci. Transl. Med., 2022, 14, eabn1413 CrossRef CAS PubMed.
- P. R. Taylor, S. Zamze, R. J. Stillion, S. Y. C. Wong, S. Gordon and L. Martinez-Pomares, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1963–1968 CrossRef CAS PubMed.
- R. Kratzer, F.-X. Mauvais, A. Burgevin, É. Barilleau and P. van Endert, J. Immunol., 2010, 184, 6855–6864 CrossRef CAS PubMed.
- H. Veninga, E. G. F. Borg, K. Vreeman, P. R. Taylor, H. Kalay, Y. Kooyk, G. Kraal, L. Martinez-Pomares and J. M. M. den Haan, Eur. J. Immunol., 2015, 45, 747–757 CrossRef CAS PubMed.
- T. Tokatlian, B. J. Read, C. A. Jones, D. W. Kulp, S. Menis, J. Y. H. Chang, J. M. Steichen, S. Kumari, J. D. Allen, E. L. Dane, A. Liguori, M. Sangesland, D. Lingwood, M. Crispin, W. R. Schief and D. J. Irvine, Science, 2019, 363, 649–654 CrossRef CAS PubMed.
- J. T. Martin, C. A. Cottrell, A. Antanasijevic, D. G. Carnathan, B. J. Cossette, C. A. Enemuo, E. H. Gebru, Y. Choe, F. Viviano, S. Fischinger, T. Tokatlian, K. M. Cirelli, G. Ueda, J. Copps, T. Schiffner, S. Menis, G. Alter, W. R. Schief, S. Crotty, N. P. King, D. Baker, G. Silvestri, A. B. Ward and D. J. Irvine, npj Vaccines, 2020, 5, 72 CrossRef CAS PubMed.
- R. Kumar, M. Tuen, J. Liu, A. Nàdas, R. Pan, X. Kong and C. E. Hioe, Vaccine, 2013, 31, 5413–5421 CrossRef CAS PubMed.
- Y. Chen, R. Wilson, S. O'Dell, J. Guenaga, Y. Feng, K. Tran, C.-I. Chiang, H. E. Arendt, J. DeStefano, J. R. Mascola, R. T. Wyatt and Y. Li, J. Immunol., 2016, 197, 3982–3998 CrossRef CAS PubMed.
- C. E. Hioe, R. Kumar, C. Upadhyay, M. Jan, A. Fox, V. Itri, K. K. Peachman, M. Rao, L. Liu, N. C. Lo, M. Tuen, X. Jiang, X.-P. Kong and S. Zolla-Pazner, Front. Immunol., 2018, 9, 2441 CrossRef PubMed.
- C. M. Nycholat, C. Rademacher, N. Kawasaki and J. C. Paulson, J. Am. Chem. Soc., 2012, 134, 15696–15699 CrossRef CAS PubMed.
- N. Kawasaki, J. L. Vela, C. M. Nycholat, C. Rademacher, A. Khurana, N. van Rooijen, P. R. Crocker, M. Kronenberg and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7826–7831 CrossRef CAS PubMed.
- L. J. Edgar, N. Kawasaki, C. M. Nycholat and J. C. Paulson, Cell Chem. Biol., 2019, 26, 131–136 CrossRef CAS PubMed.
- W. C. Chen, N. Kawasaki, C. M. Nycholat, S. Han, J. Pilotte, P. R. Crocker and J. C. Paulson, PLoS One, 2012, 7, e39039 CrossRef CAS PubMed.
- M. Perdicchio, J. M. Ilarregui, M. I. Verstege, L. A. M. Cornelissen, S. T. T. Schetters, S. Engels, M. Ambrosini, H. Kalay, H. Veninga, J. M. M. den Haan, L. A. van Berkel, J. N. Samsom, P. R. Crocker, T. Sparwasser, L. Berod, J. J. Garcia-Vallejo, Y. van Kooyk and W. W. J. Unger, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3329–3334 CrossRef CAS PubMed.
- D. van Dinther, H. Veninga, S. Iborra, E. G. F. Borg, L. Hoogterp, K. Olesek, M. R. Beijer, S. T. T. Schetters, H. Kalay, J. J. Garcia-Vallejo, K. L. Franken, L. B. Cham, K. S. Lang, Y. van Kooyk, D. Sancho, P. R. Crocker and J. M. M. den Haan, Cell Rep., 2018, 22, 1484–1495 CrossRef CAS PubMed.
- A. J. Affandi, J. Grabowska, K. Olesek, M. L. Venegas, A. Barbaria, E. Rodríguez, P. P. G. Mulder, H. J. Pijffers, M. Ambrosini, H. Kalay, T. O'Toole, E. S. Zwart, G. Kazemier, K. Nazmi, F. J. Bikker, J. Stöckl, A. J. M. van den Eertwegh, T. D. de Gruijl, G. Storm, Y. van Kooyk and J. M. M. den Haan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 27528–27539 CrossRef CAS PubMed.
- Y. Gao, W. Wang, Y. Yang, Q. Zhao, C. Yang, X. Jia, Y. Liu, M. Zhou, W. Zeng, X. Huang, S. Chiu, T. Jin and X. Wu, Adv. Sci., 2023, 10, 2204598 CrossRef CAS PubMed.
- A. Schudel, A. P. Chapman, M.-K. Yau, C. J. Higginson, D. M. Francis, M. P. Manspeaker, A. R. C. Avecilla, N. A. Rohner, M. G. Finn and S. N. Thomas, Nat. Nanotechnol., 2020, 15, 491–499 CrossRef CAS PubMed.
- E. Tonti, N. Jiménez de Oya, G. Galliverti, E. A. Moseman, P. Di Lucia, A. Amabile, S. Sammicheli, M. De Giovanni, L. Sironi, N. Chevrier, G. Sitia, L. Gennari, L. G. Guidotti, U. H. von Andrian and M. Iannacone, Cell Rep., 2013, 5, 323–330 CrossRef CAS PubMed.
- F. Pucci, C. Garris, C. P. Lai, A. Newton, C. Pfirschke, C. Engblom, D. Alvarez, M. Sprachman, C. Evavold, A. Magnuson, U. H. von Andrian, K. Glatz, X. O. Breakefield, T. R. Mempel, R. Weissleder and M. J. Pittet, Science, 2016, 352, 242–246 CrossRef CAS PubMed.
- Y.-N. Zhang, W. Poon, E. Sefton and W. C. W. Chan, ACS Nano, 2020, 14, 9478–9490 CrossRef CAS PubMed.
- M. Silva, Y. Kato, M. B. Melo, I. Phung, B. L. Freeman, Z. Li, K. Roh, J. W. V. Wijnbergen, H. Watkins, C. A. Enemuo, B. L. Hartwell, J. Y. H. Chang, S. Xiao, K. A. Rodrigues, K. M. Cirelli, N. Li, S. Haupt, A. Aung, B. Cossette, W. Abraham, S. Kataria, R. Bastidas, J. Bhiman, C. Linde, N. I. Bloom, B. Groschel, E. Georgeson, N. Phelps, A. Thomas, J. Bals, D. G. Carnathan, D. Lingwood, D. R. Burton, G. Alter, T. P. Padera, A. M. Belcher, W. R. Schief, G. Silvestri, R. M. Ruprecht, S. Crotty and D. J. Irvine, Sci. Immunol., 2021, 6, eabf1152 CrossRef CAS PubMed.
- H. H. Tam, M. B. Melo, M. Kang, J. M. Pelet, V. M. Ruda, M. H. Foley, J. K. Hu, S. Kumari, J. Crampton, A. D. Baldeon, R. W. Sanders, J. P. Moore, S. Crotty, R. Langer, D. G. Anderson, A. K. Chakraborty and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6639–E6648 CrossRef CAS PubMed.
- K. M. Cirelli, D. G. Carnathan, B. Nogal, J. T. Martin, O. L. Rodriguez, A. A. Upadhyay, C. A. Enemuo, E. H. Gebru, Y. Choe, F. Viviano, C. Nakao, M. G. Pauthner, S. Reiss, C. A. Cottrell, M. L. Smith, R. Bastidas, W. Gibson, A. N. Wolabaugh, M. B. Melo, B. Cossette, V. Kumar, N. B. Patel, T. Tokatlian, S. Menis, D. W. Kulp, D. R. Burton, B. Murrell, W. R. Schief, S. E. Bosinger, A. B. Ward, C. T. Watson, G. Silvestri, D. J. Irvine and S. Crotty, Cell, 2019, 177, 1153–1171 CrossRef CAS PubMed.
- G. A. Roth, E. C. Gale, M. Alcántara-Hernández, W. Luo, E. Axpe, R. Verma, Q. Yin, A. C. Yu, H. L. Hernandez, C. L. Maikawa, A. A. A. Smith, M. M. Davis, B. Pulendran, J. Idoyaga and E. A. Appel, ACS Cent. Sci., 2020, 6, 1800–1812 CrossRef CAS PubMed.
- K. M. Cirelli and S. Crotty, Curr. Opin. Immunol., 2017, 47, 64–69 CrossRef CAS PubMed.
- M. Bajénoff, O. Wurtz and S. Guerder, J. Immunol., 2002, 168, 1723–1729 CrossRef PubMed.
- R. Obst, H.-M. van Santen, R. Melamed, A. O. Kamphorst, C. Benoist and D. Mathis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15460–15465 CrossRef CAS PubMed.
- J. Merkenschlager, S. Finkin, V. Ramos, J. Kraft, M. Cipolla, C. R. Nowosad, H. Hartweger, W. Zhang, P. D. B. Olinares, A. Gazumyan, T. Y. Oliveira, B. T. Chait and M. C. Nussenzweig, Nature, 2021, 591, 458–463 CrossRef CAS PubMed.
- C. Havenar-Daughton, J. H. Lee and S. Crotty, Immunol. Rev., 2017, 275, 49–61 CrossRef CAS PubMed.
- W. Li, J. Tang, D. Lee, T. R. Tice, S. P. Schwendeman and M. R. Prausnitz, Nat. Rev. Mater., 2022, 7, 406–420 CrossRef.
- T. Sheng, B. Luo, W. Zhang, X. Ge, J. Yu, Y. Zhang and Z. Gu, Adv. Drug Delivery Rev., 2021, 179, 113919 CrossRef CAS PubMed.
- B. Combadiere and C. Liard, Hum. Vaccines, 2011, 7, 811–827 CrossRef CAS PubMed.
- P. C. DeMuth, Y. Min, B. Huang, J. A. Kramer, A. D. Miller, D. H. Barouch, P. T. Hammond and D. J. Irvine, Nat. Mater., 2013, 12, 367–376 CrossRef CAS PubMed.
- P. C. DeMuth, J. J. Moon, H. Suh, P. T. Hammond and D. J. Irvine, ACS Nano, 2012, 6, 8041–8051 CrossRef CAS PubMed.
- J. J. Moon, H. Suh, A. Bershteyn, M. T. Stephan, H. Liu, B. Huang, M. Sohail, S. Luo, S. H. Um, H. Khant, J. T. Goodwin, J. Ramos, W. Chiu and D. J. Irvine, Nat. Mater., 2011, 10, 243–251 CrossRef CAS PubMed.
- P. C. DeMuth, W. F. Garcia-Beltran, M. L. Ai-Ling, P. T. Hammond and D. J. Irvine, Adv. Funct. Mater., 2013, 23, 161–172 CrossRef CAS PubMed.
- P. C. DeMuth, Y. Min, D. J. Irvine and P. T. Hammond, Adv. Healthcare Mater., 2014, 3, 47–58 CrossRef CAS PubMed.
- A. V. Boopathy, A. Mandal, D. W. Kulp, S. Menis, N. R. Bennett, H. C. Watkins, W. Wang, J. T. Martin, N. T. Thai, Y. He, W. R. Schief, P. T. Hammond and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16473–16478 CrossRef CAS PubMed.
- M.-C. Chen, S.-F. Huang, K.-Y. Lai and M.-H. Ling, Biomaterials, 2013, 34, 3077–3086 CrossRef CAS PubMed.
- M.-C. Chen, K.-Y. Lai, M.-H. Ling and C.-W. Lin, Acta Biomater., 2018, 65, 66–75 CrossRef CAS PubMed.
- Y.-H. Chen, K.-Y. Lai, Y.-H. Chiu, Y.-W. Wu, A.-L. Shiau and M.-C. Chen, Acta Biomater., 2019, 97, 230–238 CrossRef CAS PubMed.
- S. P. Sullivan, D. G. Koutsonanos, M. del P. Martin, J. W. Lee, V. Zarnitsyn, S.-O. Choi, N. Murthy, R. W. Compans, I. Skountzou and M. R. Prausnitz, Nat. Med., 2010, 16, 915–920 CrossRef CAS PubMed.
- K.-G. H. Desai and S. P. Schwendeman, J. Controlled Release, 2013, 165, 62–74 CrossRef CAS PubMed.
- J. M. Mazzara, L. J. Ochyl, J. K. Y. Hong, J. J. Moon, M. R. Prausnitz and S. P. Schwendeman, Bioeng. Transl. Med., 2019, 4, 116–128 CrossRef CAS PubMed.
- K. T. M. Tran, T. D. Gavitt, N. J. Farrell, E. J. Curry, A. B. Mara, A. Patel, L. Brown, S. Kilpatrick, R. Piotrowska, N. Mishra, S. M. Szczepanek and T. D. Nguyen, Nat. Biomed. Eng., 2021, 5, 998–1007 CrossRef CAS PubMed.
- J. R. Tumbleston, D. Shirvanyants, N. Ermoshkin, R. Janusziewicz, A. R. Johnson, D. Kelly, K. Chen, R. Pinschmidt, J. P. Rolland, A. Ermoshkin, E. T. Samulski and J. M. DeSimone, Science, 2015, 347, 1349–1352 CrossRef CAS PubMed.
- A. R. Johnson, C. L. Caudill, J. R. Tumbleston, C. J. Bloomquist, K. A. Moga, A. Ermoshkin, D. Shirvanyants, S. J. Mecham, J. C. Luft and J. M. DeSimone, PLoS One, 2016, 11, e0162518 CrossRef PubMed.
- C. Caudill, J. L. Perry, K. Iliadis, A. T. Tessema, B. J. Lee, B. S. Mecham, S. Tian and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2102595118 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- A. Singh and N. A. Peppas, Adv. Mater., 2014, 26, 6530–6541 CrossRef CAS PubMed.
- C. B. Rodell, A. L. Kaminski and J. A. Burdick, Biomacromolecules, 2013, 14, 4125–4134 CrossRef CAS PubMed.
- N. C. Wickremasinghe, V. A. Kumar and J. D. Hartgerink, Biomacromolecules, 2014, 15, 3587–3595 CrossRef CAS PubMed.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 6295 CrossRef CAS PubMed.
- G. A. Roth, O. M. Saouaf, A. A. A. Smith, E. C. Gale, M. A. Hernández, J. Idoyaga and E. A. Appel, ACS Biomater. Sci. Eng., 2021, 7, 1889–1899 CrossRef CAS PubMed.
- O. M. Saouaf, G. A. Roth, B. S. Ou, A. A. A. Smith, A. C. Yu, E. C. Gale, A. K. Grosskopf, V. C. T. M. Picece and E. A. Appel, J. Biomed. Mater. Res., Part A, 2021, 109, 2173–2186 CrossRef CAS PubMed.
- E. C. Gale, A. E. Powell, G. A. Roth, E. L. Meany, J. Yan, B. S. Ou, A. K. Grosskopf, J. Adamska, V. C. T. M. Picece, A. I. d'Aquino, B. Pulendran, P. S. Kim and E. A. Appel, Adv. Mater., 2021, 33, 2104362 CrossRef CAS PubMed.
- B. S. Ou, O. M. Saouaf, J. Yan, T. U. J. Bruun, J. Baillet, X. Zhou, N. P. King and E. A. Appel, Adv. Healthcare Mater., 2023, 12, e2301495 CrossRef PubMed.
- J. P. Rolland, B. W. Maynor, L. E. Euliss, A. E. Exner, G. M. Denison and J. M. DeSimone, J. Am. Chem. Soc., 2005, 127, 10096–10100 CrossRef CAS PubMed.
- S. N. Mueller, S. Tian and J. M. DeSimone, Mol. Pharm., 2015, 12, 1356–1365 CrossRef CAS PubMed.
- Y. Wang, J. Wu, Q. Fan, M. Zhou, Z. Yue, G. Ma and Z. Su, Adv. Healthcare Mater., 2014, 3, 670–681 CrossRef CAS PubMed.
- C. Korupalli, W.-Y. Pan, C.-Y. Yeh, P.-M. Chen, F.-L. Mi, H.-W. Tsai, Y. Chang, H.-J. Wei and H.-W. Sung, Biomaterials, 2019, 216, 119268 CrossRef CAS PubMed.
- T. Shih, S. O. Blacklow, A. W. Li, B. R. Freedman, S. Bencherif, S. T. Koshy, M. C. Darnell and D. J. Mooney, Adv. Healthcare Mater., 2018, 7, e1701469 CrossRef PubMed.
- Y. Umeki, M. Saito, Y. Takahashi, Y. Takakura and M. Nishikawa, Adv. Healthcare Mater., 2017, 6, 1700355 CrossRef PubMed.
- C. Yang, F. Shi, C. Li, Y. Wang, L. Wang and Z. Yang, ACS Biomater. Sci. Eng., 2018, 4, 2000–2006 CrossRef CAS PubMed.
- B. M. Friedrich, D. W. C. Beasley and J. S. Rudra, Vaccine, 2016, 34, 5479–5482 CrossRef CAS PubMed.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS PubMed.
- A. C. Daly, L. Riley, T. Segura and J. A. Burdick, Nat. Rev. Mater., 2020, 5, 20–43 CrossRef CAS PubMed.
- J. C. Joyce, H. E. Sella, H. Jost, M. J. Mistilis, E. S. Esser, P. Pradhan, R. Toy, M. L. Collins, P. A. Rota, K. Roy, I. Skountzou, R. W. Compans, M. S. Oberste, W. C. Weldon, J. J. Norman and M. R. Prausnitz, J. Controlled Release, 2019, 304, 135–145 CrossRef CAS PubMed.
- J. H. Lee, H. J. Sutton, C. A. Cottrell, I. Phung, G. Ozorowski, L. M. Sewall, R. Nedellec, C. Nakao, M. Silva, S. T. Richey, J. L. Torres, W.-H. Lee, E. Georgeson, M. Kubitz, S. Hodges, T.-M. Mullen, Y. Adachi, K. M. Cirelli, A. Kaur, C. Allers, M. Fahlberg, B. F. Grasperge, J. P. Dufour, F. Schiro, P. P. Aye, O. Kalyuzhniy, A. Liguori, D. G. Carnathan, G. Silvestri, X. Shen, D. C. Montefiori, R. S. Veazey, A. B. Ward, L. Hangartner, D. R. Burton, D. J. Irvine, W. R. Schief and S. Crotty, Nature, 2022, 609, 998–1004 CrossRef CAS PubMed.
- D. Yu, L. S. K. Walker, Z. Liu, M. A. Linterman and Z. Li, Nat. Immunol., 2022, 23, 1157–1168 CrossRef CAS PubMed.
- J. R. Harris and J. Markl, Micron, 1999, 30, 597–623 CrossRef CAS PubMed.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS PubMed.
- J. S. Rudra, S. Mishra, A. S. Chong, R. A. Mitchell, E. H. Nardin, V. Nussenzweig and J. H. Collier, Biomaterials, 2012, 33, 6476–6484 CrossRef CAS PubMed.
- J. S. Rudra, T. Sun, K. C. Bird, M. D. Daniels, J. Z. Gasiorowski, A. S. Chong and J. H. Collier, ACS Nano, 2012, 6, 1557–1564 CrossRef CAS PubMed.
- J. Chen, R. R. Pompano, F. W. Santiago, L. Maillat, R. Sciammas, T. Sun, H. Han, D. J. Topham, A. S. Chong and J. H. Collier, Biomaterials, 2013, 34, 8776–8785 CrossRef CAS PubMed.
- R. R. Pompano, J. Chen, E. A. Verbus, H. Han, A. Fridman, T. McNeely, J. H. Collier and A. S. Chong, Adv. Healthcare Mater., 2014, 3, 1898–1908 CrossRef CAS PubMed.
- N. J. Tubo, A. J. Pagán, J. J. Taylor, R. W. Nelson, J. L. Linehan, J. M. Ertelt, E. S. Huseby, S. S. Way and M. K. Jenkins, Cell, 2013, 153, 785–796 CrossRef CAS PubMed.
- C. Mora-Solano, Y. Wen, H. Han, J. Chen, A. S. Chong, M. L. Miller, R. R. Pompano and J. H. Collier, Biomaterials, 2017, 149, 1–11 CrossRef CAS PubMed.
- L. S. Shores, S. H. Kelly, K. M. Hainline, J. Suwanpradid, A. S. MacLeod and J. H. Collier, Front. Immunol., 2020, 11, 1855 CrossRef CAS PubMed.
- S. H. Kelly, Y. Wu, A. K. Varadhan, E. J. Curvino, A. S. Chong and J. H. Collier, Biomaterials, 2020, 241, 119903 CrossRef CAS PubMed.
- S. H. Kelly, N. L. Votaw, B. J. Cossette, Y. Wu, S. Shetty, L. S. Shores, L. A. Issah and J. H. Collier, Sci. Adv., 2022, 8, eabq4120 CrossRef CAS PubMed.
- K. M. Hainline, L. S. Shores, N. L. Votaw, Z. J. Bernstein, S. H. Kelly, C. N. Fries, M. S. Madhira, C. A. Gilroy, A. Chilkoti and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018627118 CrossRef CAS PubMed.
- G. A. Hudalla, T. Sun, J. Z. Gasiorowski, H. Han, Y. F. Tian, A. S. Chong and J. H. Collier, Nat. Mater., 2014, 13, 829–836 CrossRef CAS PubMed.
- M. A. Files, K. F. Naqvi, T. B. Saito, T. M. Clover, J. S. Rudra and J. J. Endsley, npj Vaccines, 2022, 7, 48 CrossRef CAS PubMed.
- Y. Wu, P. K. Norberg, E. A. Reap, K. L. Congdon, C. N. Fries, S. H. Kelly, J. H. Sampson, V. P. Conticello and J. H. Collier, ACS Biomater. Sci. Eng., 2017, 3, 3128–3132 CrossRef CAS PubMed.
- Y. Wu, S. H. Kelly, L. Sanchez-Perez, J. H. Sampson and J. H. Collier, Biomater. Sci., 2020, 8, 3522–3535 RSC.
- Y. Wu, H. Wen, Z. J. Bernstein, K. M. Hainline, T. S. Blakney, K. L. Congdon, D. J. Snyder, J. H. Sampson, L. Sanchez-Perez and J. H. Collier, Sci. Adv., 2022, 8, eabm7833 CrossRef CAS PubMed.
- C. N. Fries, Y. Wu, S. H. Kelly, M. Wolf, N. L. Votaw, S. Zauscher and J. H. Collier, Adv. Mater., 2020, 32, e2003310 CrossRef PubMed.
- A. N. Tsoras and J. A. Champion, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 337–359 CrossRef CAS PubMed.
- C. L. O'Neill, P. C. Shrimali, Z. P. Clapacs, M. A. Files and J. S. Rudra, Acta Biomater., 2021, 133, 153–167 CrossRef PubMed.
- I. W. Hamley, ACS Appl. Bio Mater., 2022, 5, 905–944 CrossRef CAS PubMed.
- A. Zeltins, J. West, F. Zabel, A. E. Turabi, I. Balke, S. Haas, M. Maudrich, F. Storni, P. Engeroff, G. T. Jennings, A. Kotecha, D. I. Stuart, J. Foerster and M. F. Bachmann, npj Vaccines, 2017, 2, 30 CrossRef PubMed.
- T. Hills, P. G. Jakeman, R. C. Carlisle, P. Klenerman, L. W. Seymour and R. Cawood, PLoS One, 2016, 11, e0166383 CrossRef PubMed.
- J. Wallis, P. Katti, A. M. Martin, T. Hills, L. W. Seymour, D. P. Shenton and R. C. Carlisle, Eur. J. Pharm. Sci., 2020, 152, 105456 CrossRef CAS PubMed.
- E. Gage, N. Hoeven, N. D. Cauwelaert, S. E. Larsen, J. Erasmus, M. T. Orr and R. N. Coler, Eur. J. Immunol., 2019, 49, 266–276 CrossRef CAS PubMed.
- A. T. DiPiazza, S. Fan, A. Rattan, M. L. DeDiego, F. Chaves, G. Neumann, Y. Kawaoka and A. J. Sant, J. Immunol., 2019, 203, 1502–1508 CrossRef CAS PubMed.
- T. W. Ng, A. S. Wirchnianski, A. Z. Wec, J. M. Fels, C. T. Johndrow, K. O. Saunders, H.-X. Liao, J. Chan, W. R. Jacobs, K. Chandran and S. A. Porcelli, J. Immunol., 2020, 205, 425–437 CrossRef CAS PubMed.
- L. Guo, H. Xie, Z. Zhang, Z. Wang, S. Peng, Y. Niu and Z. Shang, Vaccines, 2021, 9, 786 CrossRef CAS PubMed.
- D. F. Besavilla, L. Reusch, J. Enriquez, K. Schön and D. Angeletti, Front. Immunol., 2023, 14, 1243164 CrossRef CAS PubMed.
- P. T. Sage, D. Alvarez, J. Godec, U. H. von Andrian and A. H. Sharpe, J. Clin. Invest., 2014, 124, 5191–5204 CrossRef PubMed.
- R. Cubas, J. van Grevenynghe, S. Wills, L. Kardava, B. H. Santich, C. M. Buckner, R. Muir, V. Tardif, C. Nichols, F. Procopio, Z. He, T. Metcalf, K. Ghneim, M. Locci, P. Ancuta, J.-P. Routy, L. Trautmann, Y. Li, A. B. McDermott, R. A. Koup, C. Petrovas, S. A. Migueles, M. Connors, G. D. Tomaras, S. Moir, S. Crotty and E. K. Haddad, J. Immunol., 2015, 195, 5625–5636 CrossRef CAS PubMed.
- M. Koutsakos, A. K. Wheatley, L. Loh, E. B. Clemens, S. Sant, S. Nüssing, A. Fox, A. W. Chung, K. L. Laurie, A. C. Hurt, S. Rockman, M. Lappas, T. Loudovaris, S. I. Mannering, G. P. Westall, M. Elliot, S. G. Tangye, L. M. Wakim, S. J. Kent, T. H. O. Nguyen and K. Kedzierska, Sci. Transl. Med., 2018, 10, eaan8405 CrossRef PubMed.
- M. Künzli, D. Schreiner, T. C. Pereboom, N. Swarnalekha, L. C. Litzler, J. Lötscher, Y. I. Ertuna, J. Roux, F. Geier, R. P. Jakob, T. Maier, C. Hess, J. J. Taylor and C. G. King, Sci. Immunol., 2020, 5, eaay5552 CrossRef PubMed.
- A. M. Izmirly, A.-N. Pelletier, J. Connors, B. Taramangalam, S. O. Alturki, E. A. Gordon, S. O. Alturki, J. C. Mell, G. Swaminathan, V. Karthik, M. A. Kutzler, E. G. Kallas, R.-P. Sekaly and E. K. Haddad, PLoS Pathog., 2022, 18, e1009903 CrossRef CAS PubMed.
- S. Crotty, Cold Spring Harbor Perspect. Biol., 2018, 10, a032102 CrossRef PubMed.
- D. Allman, J. R. Wilmore and B. T. Gaudette, Immunol. Rev., 2019, 288, 128–135 CrossRef CAS PubMed.
- J. S. Turner, F. Ke and I. L. Grigorova, Cell Rep., 2018, 25, 1395–1403 CrossRef CAS PubMed.
- N. R. Bennett, D. B. Zwick, A. H. Courtney and L. L. Kiessling, ACS Chem. Biol., 2015, 10, 1817–1824 CrossRef CAS PubMed.
- N. R. Bennett, C. M. Jarvis, M. M. Alam, D. B. Zwick, J. M. Olson, H. V.-T. Nguyen, J. A. Johnson, M. E. Cook and L. L. Kiessling, Biomacromolecules, 2019, 20, 4370–4379 CrossRef CAS PubMed.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed.
- J. Marcandalli, B. Fiala, S. Ols, M. Perotti, W. de van der Schueren, J. Snijder, E. Hodge, M. Benhaim, R. Ravichandran, L. Carter, W. Sheffler, L. Brunner, M. Lawrenz, P. Dubois, A. Lanzavecchia, F. Sallusto, K. K. Lee, D. Veesler, C. E. Correnti, L. J. Stewart, D. Baker, K. Loré, L. Perez and N. P. King, Cell, 2019, 176, 1420–1431 CrossRef CAS PubMed.
- A. C. Walls, B. Fiala, A. Schäfer, S. Wrenn, M. N. Pham, M. Murphy, L. V. Tse, L. Shehata, M. A. O'Connor, C. Chen, M. J. Navarro, M. C. Miranda, D. Pettie, R. Ravichandran, J. C. Kraft, C. Ogohara, A. Palser, S. Chalk, E.-C. Lee, K. Guerriero, E. Kepl, C. M. Chow, C. Sydeman, E. A. Hodge, B. Brown, J. T. Fuller, K. H. Dinnon, L. E. Gralinski, S. R. Leist, K. L. Gully, T. B. Lewis, M. Guttman, H. Y. Chu, K. K. Lee, D. H. Fuller, R. S. Baric, P. Kellam, L. Carter, M. Pepper, T. P. Sheahan, D. Veesler and N. P. King, Cell, 2020, 183, 1367–1382 CrossRef CAS PubMed.
- R. Veneziano, T. J. Moyer, M. B. Stone, E.-C. Wamhoff, B. J. Read, S. Mukherjee, T. R. Shepherd, J. Das, W. R. Schief, D. J. Irvine and M. Bathe, Nat. Nanotechnol., 2020, 15, 716–723 CrossRef CAS PubMed.
- T. J. Moyer, Y. Kato, W. Abraham, J. Y. H. Chang, D. W. Kulp, N. Watson, H. L. Turner, S. Menis, R. K. Abbott, J. N. Bhiman, M. B. Melo, H. A. Simon, S. H.-D. la Mata, S. Liang, G. Seumois, Y. Agarwal, N. Li, D. R. Burton, A. B. Ward, W. R. Schief, S. Crotty and D. J. Irvine, Nat. Med., 2020, 26, 430–440 CrossRef CAS PubMed.
- R. W. Jain and V. W. Yong, Nat. Rev. Immunol., 2022, 22, 513–524 CrossRef CAS PubMed.
- C. J. Kushner, S. Wang, N. Tovanabutra, D. E. Tsai, V. P. Werth and A. S. Payne, JAMA Dermatol., 2019, 155, 1404–1409 CrossRef PubMed.
- C. Markmann and V. G. Bhoj, Immunol. Rev., 2021, 303, 154–167 CrossRef CAS PubMed.
- N. W. C. J. van de Donk and S. Z. Usmani, Front. Immunol., 2018, 9, 2134 CrossRef PubMed.
- Q. Cheng, A. Pelz, A. Taddeo, L. Khodadadi, J. Klotsche, B. F. Hoyer, T. Alexander, A. Thiel, G. Burmester, A. Radbruch and F. Hiepe, Eur. J. Immunol., 2020, 50, 284–291 CrossRef CAS PubMed.
- K. A. Brzezicka and J. C. Paulson, Mol. Aspects Med., 2023, 90, 101140 CrossRef CAS PubMed.
- J. C. Poe and T. F. Tedder, Trends Immunol., 2012, 33, 413–420 CrossRef CAS PubMed.
- B. E. Collins, O. Blixt, S. Han, B. Duong, H. Li, J. K. Nathan, N. Bovin and J. C. Paulson, J. Immunol., 2006, 177, 2994–3003 CrossRef CAS PubMed.
- P. R. Crocker, O. Blixt, B. E. Collins, I. M. van den Nieuwenhof and J. C. Paulson, J. Biol. Chem., 2003, 278, 31007–31019 CrossRef PubMed.
- O. Blixt, S. Han, L. Liao, Y. Zeng, J. Hoffmann, S. Futakawa and J. C. Paulson, J. Am. Chem. Soc., 2008, 130, 6680–6681 CrossRef CAS PubMed.
- M. S. Macauley, F. Pfrengle, C. Rademacher, C. M. Nycholat, A. J. Gale, A. von Drygalski and J. C. Paulson, J. Clin. Invest., 2013, 123, 3074–3083 CrossRef CAS PubMed.
- F. Pfrengle, M. S. Macauley, N. Kawasaki and J. C. Paulson, J. Immunol., 2013, 191, 1724–1731 CrossRef CAS PubMed.
- K. A. Orgel, S. Duan, B. L. Wright, S. J. Maleki, J. C. Wolf, B. P. Vickery, A. W. Burks, J. C. Paulson, M. D. Kulis and M. S. Macauley, J. Allergy Clin. Immunol., 2017, 139, 366–369 CrossRef CAS PubMed.
- K. J. Bednar, C. M. Nycholat, T. S. Rao, J. C. Paulson, W.-P. Fung-Leung and M. S. Macauley, ACS Chem. Biol., 2019, 14, 644–654 CrossRef CAS PubMed.
- A. Srivastava, B. M. Arlian, L. Pang, T. K. Kishimoto and J. C. Paulson, ACS Chem. Biol., 2021, 16, 1985–1993 CrossRef CAS PubMed.
- L. C. Hardy, J. M. Smeekens, D. Raghuwanshi, S. Sarkar, G. C. Daskhan, S. Rogers, C. Nycholat, S. Maleki, A. W. Burks, J. C. Paulson, M. S. Macauley and M. D. Kulis, J. Allergy Clin. Immunol., 2022, 150, 1476–1485 CrossRef CAS PubMed.
- L. Pang, M. S. Macauley, B. M. Arlian, C. M. Nycholat and J. C. Paulson, ChemBioChem, 2017, 18, 1226–1233 CrossRef CAS PubMed.
- K. A. Brzezicka, B. M. Arlian, S. Wang, M. Olmer, M. Lotz and J. C. Paulson, ACS Nano, 2022, 16, 20206–20221 CrossRef CAS PubMed.
- A. H. Courtney, E. B. Puffer, J. K. Pontrello, Z.-Q. Yang and L. L. Kiessling, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2500–2505 CrossRef CAS PubMed.
- H. Kristyanto, M. D. Holborough-Kerkvliet, L. Lelieveldt, Y. Bartels, R. Hammink, K. A. J. van Schie, R. E. M. Toes, K. M. Bonger and H. U. Scherer, ACS Biomater. Sci. Eng., 2022, 8, 1486–1493 CrossRef CAS PubMed.
- M. R. Qelliny, T. Shimizu, N. E. Elsadek, S. E. Emam, H. Takata, Z. M. A. Fathalla, A. K. Hussein, K. A. Khaled, H. Ando, Y. Ishima and T. Ishida, Mol. Pharm., 2021, 18, 2406–2415 CrossRef CAS PubMed.
- J. Sestak, M. Mullins, L. Northrup, S. Thati, M. L. Forrest, T. J. Siahaan and C. Berkland, J. Controlled Release, 2013, 168, 334–340 CrossRef CAS PubMed.
- B. L. Hartwell, A. S. Hall, D. Swafford, B. P. Sullivan, A. Garza, J. O. Sestak, L. Northrup and C. Berkland, Mol. Pharm., 2016, 13, 330–343 CrossRef CAS PubMed.
- B. L. Hartwell, C. J. Pickens, M. Leon and C. Berkland, Biomacromolecules, 2017, 18, 1893–1907 CrossRef CAS PubMed.
- B. L. Hartwell, F. J. Martinez-Becerra, J. Chen, H. Shinogle, M. Sarnowski, D. S. Moore and C. Berkland, Biomacromolecules, 2016, 17, 710–722 CrossRef CAS PubMed.
- M. A. Leon, S. M. Wemlinger, N. R. Larson, J. K. Ruffalo, J. O. Sestak, C. R. Middaugh, J. C. Cambier and C. Berkland, Mol. Pharm., 2019, 16, 1563–1572 CrossRef CAS PubMed.
- S. N. Johnson, J. D. Griffin, C. Hulbert, B. J. DeKosky, J. W. Thomas and C. J. Berkland, ACS Appl. Bio Mater., 2020, 3, 6319–6330 CrossRef CAS PubMed.
- B. Duan and L. Morel, Autoimmun. Rev., 2006, 5, 403–408 CrossRef CAS PubMed.
- B. F. Haynes, K. Wiehe, P. Borrow, K. O. Saunders, B. Korber, K. Wagh, A. J. McMichael, G. Kelsoe, B. H. Hahn, F. Alt and G. M. Shaw, Nat. Rev. Immunol., 2023, 23, 142–158 CrossRef CAS PubMed.
- I. A. Cockburn and R. A. Seder, Nat. Immunol., 2018, 19, 1199–1211 CrossRef CAS PubMed.
- G. V. Sharonov, E. O. Serebrovskaya, D. V. Yuzhakova, O. V. Britanova and D. M. Chudakov, Nat. Rev. Immunol., 2020, 20, 294–307 CrossRef CAS PubMed.
- M. Meylan, F. Petitprez, E. Becht, A. Bougoüin, G. Pupier, A. Calvez, I. Giglioli, V. Verkarre, G. Lacroix, J. Verneau, C.-M. Sun, P. Laurent-Puig, Y.-A. Vano, R. Elaïdi, A. Méjean, R. Sanchez-Salas, E. Barret, X. Cathelineau, S. Oudard, C.-A. Reynaud, A. de Reyniès, C. Sautès-Fridman and W. H. Fridman, Immunity, 2022, 55, 527–541 CrossRef CAS PubMed.
- A. T. Ruffin, A. R. Cillo, T. Tabib, A. Liu, S. Onkar, S. R. Kunning, C. Lampenfeld, H. I. Atiya, I. Abecassis, C. H. L. Kürten, Z. Qi, R. Soose, U. Duvvuri, S. Kim, S. Oesterrich, R. Lafyatis, L. G. Coffman, R. L. Ferris, D. A. A. Vignali and T. C. Bruno, Nat. Commun., 2021, 12, 3349 CrossRef CAS PubMed.
- C. Sautès-Fridman, F. Petitprez, J. Calderaro and W. H. Fridman, Nat. Rev. Cancer, 2019, 19, 307–325 CrossRef PubMed.
- J. H. Sampson, A. B. Heimberger, G. E. Archer, K. D. Aldape, A. H. Friedman, H. S. Friedman, M. R. Gilbert, J. E. H. II, R. E. McLendon, D. A. Mitchell, D. A. Reardon, R. Sawaya, R. J. Schmittling, W. Shi, J. J. Vredenburgh and D. D. Bigner, J. Clin. Oncol., 2010, 28, 4722–4729 CrossRef PubMed.
- K. D. Moynihan, C. F. Opel, G. L. Szeto, A. Tzeng, E. F. Zhu, J. M. Engreitz, R. T. Williams, K. Rakhra, M. H. Zhang, A. M. Rothschilds, S. Kumari, R. L. Kelly, B. H. Kwan, W. Abraham, K. Hu, N. K. Mehta, M. J. Kauke, H. Suh, J. R. Cochran, D. A. Lauffenburger, K. D. Wittrup and D. J. Irvine, Nat. Med., 2016, 22, 1402–1410 CrossRef CAS PubMed.
- S. Aoyama, R. Nakagawa, J. J. Mulé and A. W. Mailloux, Front. Immunol., 2021, 12, 675538 CrossRef CAS PubMed.
- Y. Kobayashi and T. Watanabe, Front. Immunol., 2016, 7, 316 Search PubMed.
- J. G. Jardine, D. W. Kulp, C. Havenar-Daughton, A. Sarkar, B. Briney, D. Sok, F. Sesterhenn, J. Ereño-Orbea, O. Kalyuzhniy, I. Deresa, X. Hu, S. Spencer, M. Jones, E. Georgeson, Y. Adachi, M. Kubitz, A. C. deCamp, J.-P. Julien, I. A. Wilson, D. R. Burton, S. Crotty and W. R. Schief, Science, 2016, 351, 1458–1463 CrossRef CAS PubMed.
- S. Yegorov, D. B. Celeste, K. B. Gomes, J. C. Ang, C. Vandenhof, J. Wang, K. Rybkina, V. Tsui, H. D. Stacey, M. Loeb and M. S. Miller, Cell Rep. Med., 2022, 3, 100509 CrossRef CAS PubMed.
- A. Osterloh, Vaccines, 2022, 10, 751 CrossRef CAS PubMed.
- J. C. Barrett, B. D. Ulery, A. Trent, S. Liang, N. A. David and M. V. Tirrell, ACS Biomater. Sci. Eng., 2017, 3, 144–152 CrossRef CAS PubMed.
- A. Dauner, P. Agrawal, J. Salvatico, T. Tapia, V. Dhir, S. F. Shaik, D. R. Drake and A. M. Byers, Vaccine, 2017, 35, 5487–5494 CrossRef CAS PubMed.
- A. Purwada, S. B. Shah, W. Beguelin, A. M. Melnick and A. Singh, ACS Biomater. Sci. Eng., 2017, 3, 214–225 CrossRef CAS PubMed.
- A. Purwada, S. B. Shah, W. Béguelin, A. August, A. M. Melnick and A. Singh, Biomaterials, 2019, 198, 27–36 CrossRef CAS PubMed.
- T. Nojima, K. Haniuda, T. Moutai, M. Matsudaira, S. Mizokawa, I. Shiratori, T. Azuma and D. Kitamura, Nat. Commun., 2011, 2, 465 CrossRef PubMed.
- W. Béguelin, M. A. Rivas, M. T. C. Fernández, M. Teater, A. Purwada, D. Redmond, H. Shen, M. F. Challman, O. Elemento, A. Singh and A. M. Melnick, Nat. Commun., 2017, 8, 877 CrossRef PubMed.
- T. D. Moeller, S. B. Shah, K. Lai, N. Lopez-Barbosa, P. Desai, W. Wang, Z. Zhong, D. Redmond, A. Singh and M. P. DeLisa, ACS Cent. Sci., 2023, 9, 787–804 CrossRef CAS PubMed.
- S. B. Shah, C. R. Carlson, K. Lai, Z. Zhong, G. Marsico, K. M. Lee, N. E. F. Vélez, E. B. Abeles, M. Allam, T. Hu, L. D. Walter, K. E. Martin, K. Gandhi, S. D. Butler, R. Puri, A. L. McCleary-Wheeler, W. Tam, O. Elemento, K. Takata, C. Steidl, D. W. Scott, L. Fontan, H. Ueno, B. D. Cosgrove, G. Inghirami, A. J. García, A. F. Coskun, J. L. Koff, A. Melnick and A. Singh, Nat. Mater., 2023, 22, 511–523 CrossRef CAS PubMed.
- G. Goyal, P. Prabhala, G. Mahajan, B. Bausk, T. Gilboa, L. Xie, Y. Zhai, R. Lazarovits, A. Mansour, M. S. Kim, A. Patil, D. Curran, J. M. Long, S. Sharma, A. Junaid, L. Cohen, T. C. Ferrante, O. Levy, R. Prantil-Baun, D. R. Walt and D. E. Ingber, Adv. Sci., 2022, 9, 2103241 CrossRef PubMed.
- E. J. Curvino, E. F. Roe, H. F. Haddad, A. R. Anderson, M. E. Woodruff, N. L. Votaw, T. Segura, L. P. Hale and J. H. Collier, Nat. Biomed. Eng., 2023 DOI:10.1038/s41551-023-01139-6 , 38012308.
- S. H. Kelly, N. L. Votaw, B. J. Cossette, Y. Wu, S. Shetty, L. S. Shores, L. A. Issah and J. H. Collier, Sci. Adv., 2022, 8(47), eabq4120, DOI:10.1126/sciadv.abq4120 , 36417519.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |