
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCritical assessment of advanced oxidation processes and bio-electrochemical integrated systems for removing emerging contaminants from wastewater
Yasser
Bashir
a,
Rishabh
Raj
 b,
M. M.
Ghangrekar
cb,
Arvind K.
Nema
a and
Sovik
Das
*a
b,
M. M.
Ghangrekar
cb,
Arvind K.
Nema
a and
Sovik
Das
*a
aDepartment of Civil Engineering, Indian Institute of Technology Delhi, New Delhi, 110016, India. E-mail: daassovik@iitd.ac.in
bSchool of Environmental Science and Engineering, Indian Institute of Technology Kharagpur, Kharagpur, 721302, India
cDepartment of Civil Engineering, Indian Institute of Technology Kharagpur, Kharagpur, 721302, India
First published on 19th September 2023
Abstract
The remediation of emerging contaminants (ECs) of concern, such as personal care products, antibiotics, endocrine-disrupting chemicals (EDCs), surfactants, pesticides, etc., is the need of the hour. Conventional wastewater treatment technologies such as the activated sludge process, trickling filter, constructed wetlands, coagulation and flocculation, adsorption, etc. are not designed to remove trace recalcitrant contaminants. This necessitates the need to devise novel technologies specifically to target bio-refractory microcontaminants present in wastewater. In this context, the present review article focuses on the remediation of ECs through advanced oxidation processes (AOPs) and integrated bio-electrochemical systems (BESs). In this critical assessment, the detailed mechanism, degradation efficiency, comparison, techno-economic and life cycle analysis, relative merits and demerits, and challenges and future prospects of electrochemical technologies (ETs) and integrated BESs are presented. The integrated BESs and hybrid AOPs have shown enormous potential for the degradation of ECs because of their low operational cost and environmental compatibility. Even though individual ETs are also promising, higher operational cost hinders their real-life applications. Therefore, more scaled-up investigations and efforts to overcome these challenges are required to accelerate the commercialization of these technologies.
Sustainability spotlightEmerging contaminants (ECs) are refractory pollutants which are toxic for human health as well as to aquatic lifeforms. Among forthcoming technologies, advanced oxidation processes (AOPs) and integrated bioelectrochemical systems (BESs) are highly competent for eliminating these contaminants of concern. In this context, the present article highlights the remediation of ECs via AOPs and integrated BESs by shedding light on the detailed mechanism, degradation efficiency, techno-economic and life cycle assessment, and relative pros and cons of both the technologies. Hence, the present review article is in line with the sustainable development goals (SDG) focussing on good health and well-being (SDG3), life below water (SDG4), clean water and sanitation (SDG6), and affordable and clean energy (SDG7). |
1. Introduction
Fresh water is probably the most precious natural resource that has nurtured the cradle of human civilization. Ironically, the development of society entails the hefty price of aquatic and terrestrial pollution. The ever-increasing population has elevated the stress on freshwater bodies, which is further compounded by the pollution of hydric sources. As per an estimate, if necessary measures are not taken, the water supply–demand gap can become as high as 40% by 2030.1Thus, in the current scenario, recycling wastewater for flushing, landscaping, and industrial usage, and even for potable use is the need of the hour. However, the detection of potentially toxic emerging contaminants (ECs) in treated effluents has cast a shadow over plausible wastewater recycling, especially for human consumption or reuse (Table 1). Among existing tertiary and quaternary treatment technologies, advanced oxidation processes (AOPs) have been advocated to abate ECs from the aquatic environment.2 Notably, photochemical oxidation and electrochemical advanced oxidation processes (EAOPs), such as electrochemical oxidation (EO), electrocoagulation (EC), electro-Fenton (EF) process, and photo-Fenton/Fenton type oxidation process, are touted as more efficient and sustainable alternatives for eliminating recalcitrant pollutants.3
| EC | Category | Concentration (μg L−1) | Location | References |
|---|---|---|---|---|
| a EC = emerging contaminant; EDC = endocrine disrupting compound; PFAS = per- and polyfluoroalkyl substances; WWTP = wastewater treatment plant; STP = sewage treatment plant. | ||||
| Methylparaben | EDC | 540 | Surface water, Nigeria | 29 |
| Dimethyl phthalate | EDC | 1405 | Landfill leachate, Poland | 30 |
| Tetracycline | Pharmaceutical | 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
Pharmaceutical industry effluent, China | 31 |
| Enrofloxacin | Pharmaceutical | 8.77 | Animal wastewater, China | 32 |
| Ciprofloxacin | Pharmaceutical | 246.1 | WWTP effluent, India | 33 |
| Diclofenac | Pharmaceutical | 203 | Pharmaceutical manufacturer's wastewater, South Korea | 34 |
| Ciprofloxacin | Pharmaceutical | ≥501 | STP, Durban, South Africa | 35 |
| Carbamazepine | Pharmaceutical | 6.3 | WWTP effluents, Europe, USA, Canada, Japan and South Korea | 36 |
| Perfluoro octane sulfonic acid | PFAS | 0.626 | Agricultural water samples, South Korea | 37 |
| Perfluorooctanoic acid | PFAS | 1.6302 | Surface water, Osaka, Japan | 38 |
| Metoprolol | Beta-blockers | 950 | Industrial effluent, India | 39 |
| Propranolol | Beta-blockers | 0.094 | Hospital wastewater, Italy | 40 |
The cost of fabricating and operating an (E)AOP reactor is exorbitant. To illustrate this, Stirling et al. (2020)4 estimated the annual cost of operating an approximately 6.5 L day−1 EAOP setup to be US$ 407. Meanwhile, the capital investment required to treat 1 m3 day−1 of Eriochrome Black T containing wastewater through EAOPs was approximated to be US$ 115397,5 which needs to be reduced considerably. Similarly, Dannys et al. (2016)6 indicated that the cost of assembling a 1 m3 dual chamber microbial fuel cell (MFC) was about US$ 289000. Further, when the reactor size is increased, the price increases even more. As an example, it was estimated that it would cost a staggering 4.2 million dollars to install a 113.14 m3 dual chamber MFC for a hydraulic retention time (HRT) of 1.5 h.7 Additionally, life cycle assessment (LCA) of chemical and electricity-driven advanced oxidation techniques such as UV heterogeneous photocatalysis (UV/TiO2), wet air oxidation (WAO), and EO employing boron doped diamond (BDD) electrodes has revealed the deteriorating impacts of these processes on human health, ecology, and climate.8 Therefore, it is necessary to economise the operation and offset the environmental impacts of these technologies for a sustainable wastewater treatment. To add further, a comparative LCA of different technologies for treating laundry wastewater highlighted the high environmental impacts of EF and EC processes owing to excessive electricity consumption and use of a non-sustainable electrode material.9 In this respect, the use of renewable energy can sustainably offset the environmental load of AOP treatment. In the past, AOPs have been fueled using solar energy for cost-effective tertiary treatment of wastewater.10 Even though solar energy is eco-friendly, it is unreliable due to intermittence, high installation cost and moderate efficiency.11,12 For instance, in 2009 the cost for installing 10 km solar lighting was estimated to be US$ 3090282, which was 1.64 times the installation price of a mercury lamp.13 This lacuna can be addressed using bio-electrochemical systems (BESs) that promise to provide a continuous renewable energy supply.14 To demonstrate this, Tian et al. (2017)15 were able to reduce 80.5% of thallium (initial concentration of 5 mg L−1) from groundwater using an MFC-powered electrochemical oxidation process. In another investigation, Sathe et al. (2021)16 demonstrated Fenton oxidation of sodium dodecyl sulphate (initial concentration of 20 mg L−1) in the cathodic chamber of an MFC, yielding almost 85% removal in a BEF system. Notably, the MFC is a one-of-a-kind technology that can harvest electricity from wastewater by utilizing electroactive microbes as bio-catalyst.17 The exoelectrogens break down the organics in the anodic chamber of the MFC producing H+ and e−. The H+ and e− are subsequently transferred to the cathodic chamber for reducing O2via the proton exchange membrane and external circuit, respectively.18 Further, the O2 reduction at the cathode is accompanied by the production of H2O and electricity.19
Apparently, the electrical power produced in an MFC is insubstantial (≤1.16 V) and it is not sufficient even for miniscule usages like charging cellular phones and powering light emitting diodes that requires a voltage level of 3 V or more.20 Nevertheless, this potential is sufficient to instigate H2O2 electrogeneration (E = −0.6 V), which further dissociates into ˙OH in the presence of a suitable catalyst. To demonstrate this, self-driven Fenton oxidation and photolysis can be stimulated in the MFC by the so-called bio-electro-Fenton (BEF) system and photocatalytic-microbial fuel cell (P-MFC), respectively. A detailed review of the application of these integrated systems in wastewater treatment has been carried out by Sathe et al. (2022)21 and Xu et al. (2021).22 There are several other configurations of MFC-driven AOP systems, such as three cathode reactors presented by Wang et al. (2022),23 which had dual cathodes for the production of H2O2 and Fe2+ simultaneously. Similarly, a microbial desalination cell merged Fenton oxidation system that produced power, desalinated brackish water, and performed the Fenton oxidation of anodic effluent in parallel was developed by Lan et al. (2019).24 Further, a bio-electrochemically powered EO process that solely operates on power produced from a single chambered MFC was demonstrated by Zhang et al. (2015)25 for decolourising methyl orange. These BES-driven systems dramatically reduce the cost of propelling AOPs and mitigate the environmental damage caused due to the consumption of chemicals and electricity during the operation of traditional AOPs.16 Since BES-driven AOPs do not require any external power, electricity consumption cost is also reduced. In this regard, the LCA performed by Sathe et al. (2021),16 demonstrated that bio-electro-Fenton was found to be much more environmentally suitable due to low electricity and resource requirements. Further, an estimated 1.5 to 10 times lower energy is required in hybrid-BES technologies compared to a conventional activated sludge process for wastewater treatment.26 However, a detailed comparison between AOPs and BES-driven AOPs in terms of operating cost is not available, and hence, it becomes difficult to gauge the scale of monetary benefits in BES–AOP systems. Moreover, integrating BESs can raise the technology readiness level (TRL) of photo/electro-chemical oxidation processes, thereby boosting the commercialisation prospects. At present, the TRL of the electro-Fenton process is at level 4 to 5,27 while photocatalytic oxidation is at an even lower TRL of 2 to 3.28
Hence, this review aims to inspect the cumulative development made in conjoining different BESs and AOPs either as integrated single units or as hybrid systems for eliminating ECs from wastewater. Though review articles on specific hybrid technology are available in the literature, to the best of our knowledge no attempt has been made to assess bio-electrochemically driven AOPs collectively. This article critically analyses the key parameters that govern the performance of BES–AOP hybrids and tactics for optimising treatment operations. Furthermore, a comparative techno-economic assessment supplemented with life cycle aspects has been included to track the economic viability and sustainability of BES–AOP coupled treatment schemes. Further, the explorations made in scaling-up bio-electrochemically powered hybrid reactors are incorporated along with the strategies required for addressing the critical issues limiting the field-scale implementation of these progressive technologies. Hence, the current review will assist seminal research that can catapult the transition of BES-driven AOPs to real-world applications.
2. Application of electrochemical and photochemical advanced oxidations for the remediation of emerging contaminants and their major drawbacks
The AOPs have shown the potential for the rapid degradation of harmful pollutants in water and wastewater, alleviating the problem of contamination of water bodies. Naturally, AOPs have attracted substantial attention due to their potency to convert aqueous contaminants into relatively benevolent species by exploiting reactive oxygen species (ROS).41 The AOPs are primarily utilised for biologically toxic or non-degradable substances found in wastewater, including volatile organic compounds, insecticides, aromatics, and petroleum components and are characterized based on the different working mechanisms.42 Electrochemical and photochemical oxidations are among those remediation techniques which are used for the remediation of ECs present in water bodies and both of these technologies are discussed in the subsequent sections.2.1 Electrochemical methods
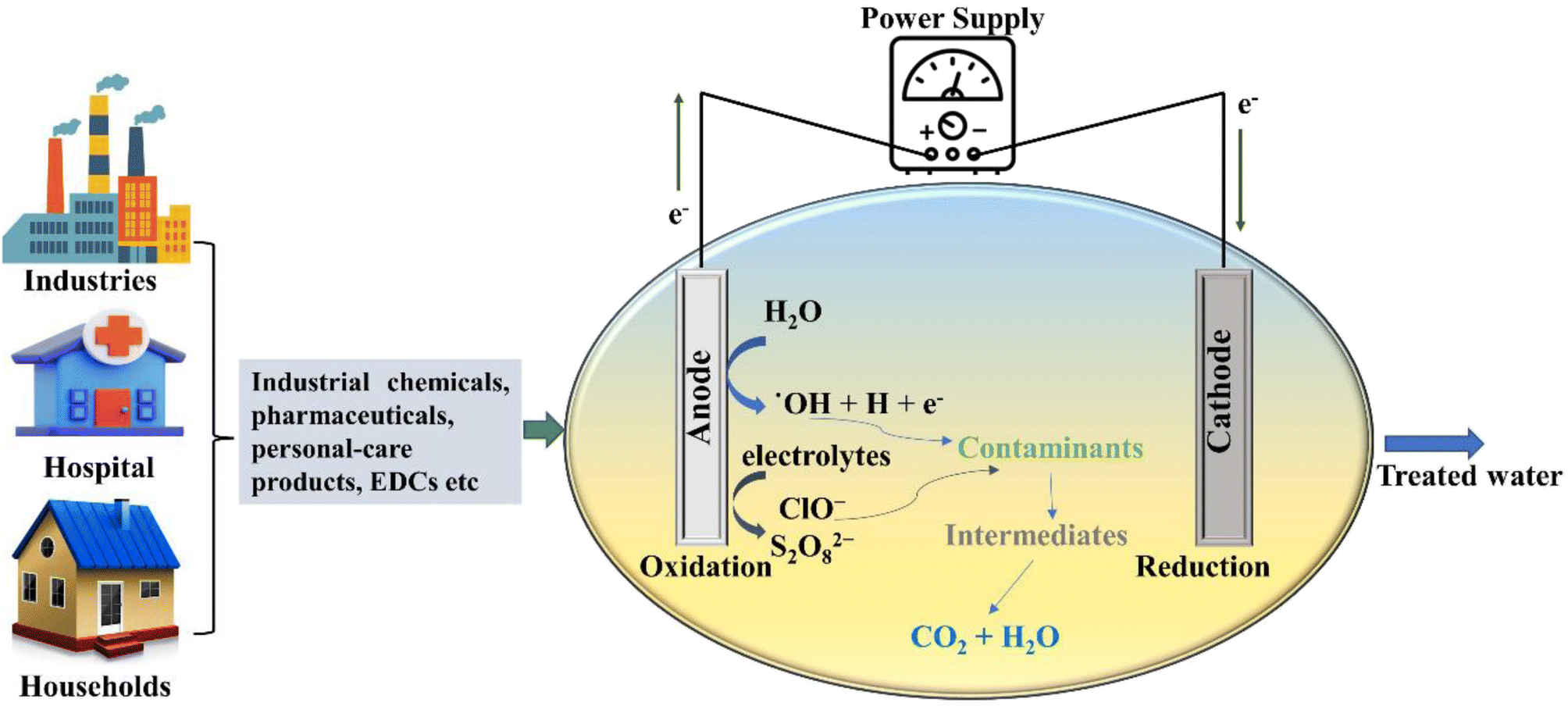 | ||
| Fig. 1 Schematic representation of the electrochemical oxidation process. | ||
During the EO process, oxidation of the organic pollutants can occur via two different mechanisms, namely (i) direct EO and (ii) indirect EO. In the direct EO process, oxidation of the target compound takes place at the surface of the anode via direct electron transfer without the involvement of any other reactive species. The direct EO only involves the mediation of the electrons having the ability to oxidize complex organic contaminants at a distinct electrode potential and results in the conversion of the contaminants into simple oxidation by-products, which are further disintegrated to innocuous inorganic compounds such as CO2 and H2O (eqn (1) and (2)).43
| M + H2O → M(˙OH) + H+ + e− | (1) |
| M(˙OH) + R → M + mCO2 + nH2O + H+ + e− | (2) |
In direct EO, prior adsorption of the pollutants over the anode surface is required, which is the rate limiting process and affects the overall degradation rate of the organic pollutants.45 Furthermore, the continuous oxidation process can lead to surface corrosion and poisoning of the anode, which is one the main constraints of the direct EO process. The efficacy of direct EO is highly dependent on the anode material employed. Depending on the potential for oxygen evolution anodes can be further subdivided into two categories that are active anode and inactive anode. Active anode materials like Pt, IrO2, and RuO2 have a lower oxygen evolution potential that promotes the formation of chemisorbed ˙OH. These chemically adsorbed radicals are relatively less potent, which results in partial and selective oxidation of non-biodegradable organic pollutants into biodegradable ones.46 On the other hand, inactive anodes such as SnO2, PbO2, and BDD have a higher oxygen evolution potential that allows the formation of physically-adsorbed ˙OH radicals. The physisorbed ˙OH can directly interact with pollutants, leading to their mineralisation.47,48 Among inactive anodes, BDD electrodes are extensively used for the removal of ECs due to their higher oxygen evolution potential, long life, stability, and superior catalytic activity.49 However, the high cost of the BDD anode prevents process scaling and application in the wastewater treatment industry for the removal of ECs.
In the indirect EO process, the oxidation of contaminants is exhibited with the assistance of electroactive species produced at the surface of the anode, which consists of the mediators employed for the electron transfer between organic compounds and the electrode.43 The EO process as a whole generates various kinds of oxidant species such as ROS, SO4˙−, and chlorine active species, which are widely used for treating wastewater (eqn (3)–(5)). Among different oxidising species, active chlorine is the most common and extensively used for treating wastewater due to its prevalence and low price.50 The active chlorine in the form of gaseous chlorine, hypochlorous or hypochlorite ions is produced by chlorides, which are present naturally in the waste stream or added artificially.
| 2Cl− → Cl2 + 2e− | (3) |
| Cl2 + H2O → HOCl + H+ + Cl− | (4) |
| HOCl → H+ + OCl− | (5) |
Electrochemically produced reactive chlorine species are generated irrespective of the electrode materials used, although dimensionally stable anodes (DSAs) have shown better catalytic activity towards these reactive species.45,51 In case of BDD and PbO2, oxidation of Cl− to Cl2 takes place efficiently, thus limiting the production of active species.51 Further, the Pt anode was found to assist the Cl2/H2O system by enhancing the formation of active chlorine species, which is dependent on the pH and Cl− concentration in the effluent.45
The idea of degrading organic contaminants via electrogenerated chlorine active species has attracted great interest at the industrial level because of the pervasive presence of chlorine in industrial effluents and other liquid waste streams. In fact, in some cases, the rate of chlorine-mediated oxidation of organic contaminants was considerably faster than that achieved by ˙OH radicals.41 Howbeit, chlorine oxidation can form organo-chlorinated by-products in the treated effluent, which are not amenable to mineralisation and have adverse health effects.45 Therefore, the threshold concentration of the chlorine concentration needs to be defined, which would allow the efficient degradation of organic contaminants, however, with less accumulation of undesired noxious products. It is important to take into account that the efficacy of both direct and indirect oxidation processes, which is dependent on various factors, namely, pH, temperature, reaction time, initial contaminant concentration, etc. However, direct oxidation occurs in the vicinity of the anode, and eventually it leads to surface passivation or corrosion of the anode surface. Hence, to safeguard the anode surface, indirect oxidation is often induced in tandem to direct oxidation by adding radical generating salts like NaCl and Na2SO4. In addition, reactive species are produced homogeneously in the bulk solution, which is more effective in oxidising the target compounds.
In the EF process, the formation of H2O2 takes place by a cathodic two electron oxygen reduction reaction (ORR) in which the gaseous oxygen gets dissolved in the electrolyte or bulk solution and from there onwards, it is transferred from the bulk solution to the diffusion layer and/or double layer. Next, the oxygen migrates from the diffusion/double layer to the active sites at the cathode, thus facilitating the ORR.52 Basically, the EF process has been developed to conquer the limitations of the traditional Fenton oxidation via in situ synthesis of H2O2 in the solution targeted for treatment. Moreover, in the same solution, the Fenton reaction is catalysed via electrochemical regeneration of Fe2+ under controlled conditions. The Fenton process is purely based on the oxidation of ˙OH radicals, the most powerful oxidizing agent. The production of ˙OH occurs by homogeneous Fenton reaction (eqn (6)) and works as the initiating reaction (eqn (7)–(10)).
| H2O2 + Fe2+ → Fe3+ + ˙OH + OH− | (6) |
| ˙OH + H2O2 → ˙HO2 + H2O | (7) |
| Fe3+ + ˙HO2 → Fe2+ + H+ + O2 | (8) |
| Fe2+ + ˙HO2 → Fe3+ + HO2− | (9) |
| Fe2+ + ˙OH → Fe3+ + OH− | (10) |
From eqn (6)–(8), the release of O2 is evident in the cycle, while following eqn (9) and (10), the termination reaction, leads to the end product with the reaction between two free radicals taking place. In this process, the ˙OH radical act as an intermediate species, which is a strong oxidizing agent that degrades the organic fraction into non-harmful compounds with the aid of hydroxylation or dehydrogenation.54
Moreover, based on the formation and accumulation of reagents, the EF process is characterized under different categories. When Fe2+ ions are introduced to the system externally with the simultaneous generation of H2O2 at the cathode via two electron reduction of O2, it is known as a cathodic EF process. In this process, the transportation, storage, and handling risks get reduced. In the second case, both the Fe2+ and H2O2 ions are externally introduced to the system and the regeneration of Fe2+ is accomplished by reducing the Fe3+ ions on the surface of the cathode, which is known as the Fe3+ cycling EF process. Similarly, the in situ generation of H2O2 is facilitated via two electron reduction of O2 and Fe2+ ions, which are regenerated by the continuous reduction of Fe3+ ions to the Fe2+ at the surface of the cathode and this process is known as cathode cycling. The final process deals with a sacrificial anode, in which H2O2 is induced externally, while the generation of Fe2+ is facilitated from the sacrificial anode (Fig. 2).55 Lately, in the so-called heterogeneous process application of solid Fenton catalysts is being explored that can attain comparable performance even at near neutral pH. Other benefits of heterogeneous EF include improved catalyst recycling, less operational cost, and reduced environmental impacts.
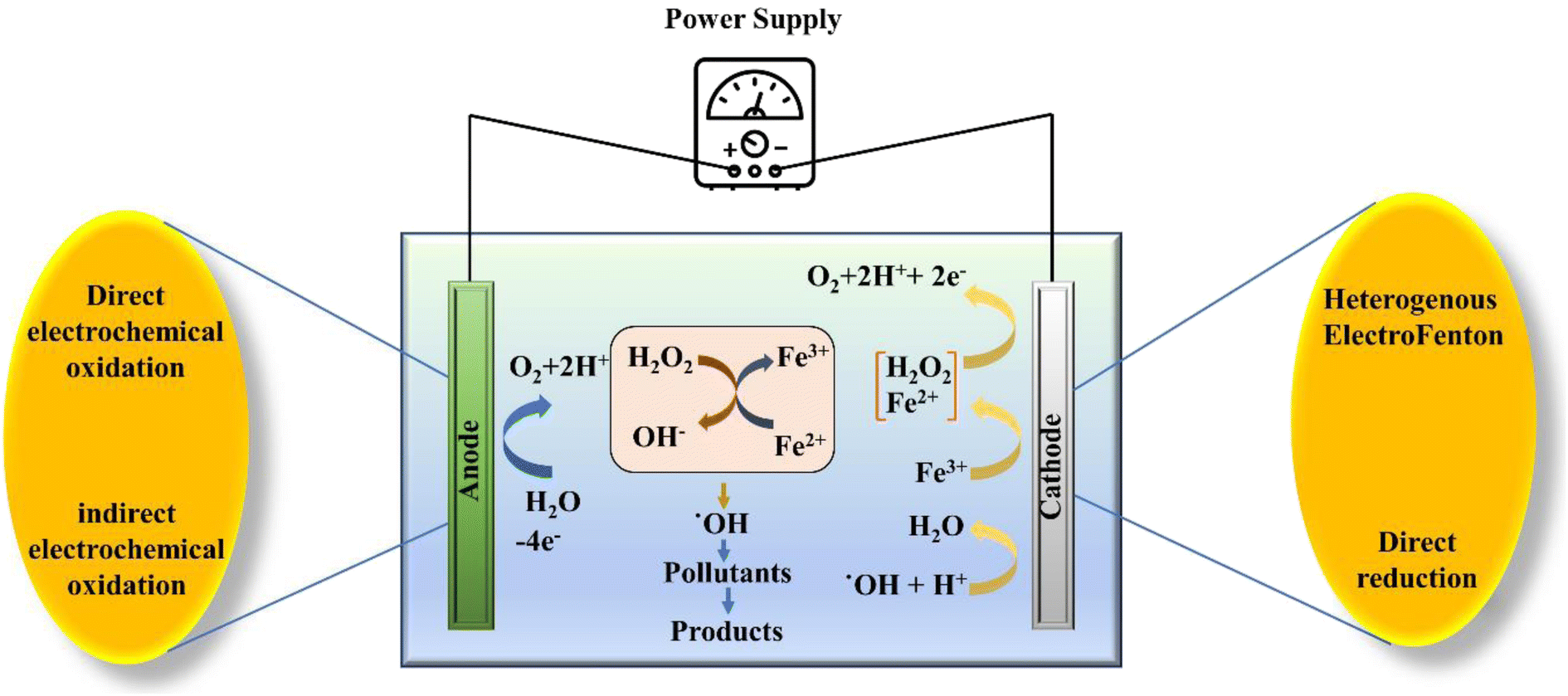 | ||
| Fig. 2 Schematic illustration of the electro-Fenton process. | ||
Furthermore, the efficiency and continuity of the H2O2 supply to the system are among the concerning factors, which affect the efficiency of the treatment obtained through the process of electrochemically mediated Fenton oxidation.56 Therefore, in the EF process, continuous production of H2O2 with the help of two electron cathodic reduction of O2 is required for the sustainable treatment of wastewater and the removal ECs.41 In order to warrant uninterrupted in situ H2O2 production highly porous carbon-based cathodes such as gas diffusion electrodes are used. These carbonaceous materials expedite the ORR by aiding the transfer of O2 on to the surface of the electrode. Nevertheless, despite the superior contaminant removal efficiency, various challenges such as high power consumption, poor stability and reusability of the recycled catalyst, and formation of toxic intermediates prevent the wider application and require further research to address these shortcomings.
2.2 Photocatalytic methods
The photocatalytic technology is a greener technology which emerged as a sustainable alternative for the removal and degradation of various organic contaminants.57 In the receiving water bodies, the concentration of ECs, such as dyes, phenolic compounds, surfactants, organohalides, hydrocarbons, pharmaceuticals and pesticides, has significantly increased in the last two decades, and these contaminants can be removed by adopting photocatalytic methods.58–60 Further, these pollutants are chemically stable, toxic and even carcinogenic and resistant towards decomposition in water leading to environmental contamination. To eliminate these contaminants from wastewater, various technologies such as membrane filtration, adsorption, and electrochemical reduction have been employed.61 However, the higher energy consumption of these conventional treatment technologies and low degradation efficiency due to the complex structure of these ECs make them persistent in the environment and limit the application of conventional technologies; thus a better alternative in this regard needs to be found.62 To fill this lacuna, photocatalytic methods have gained considerable attention and have emerged as an alternative for the degradation of these complexes.During the photocatalytic process, in the initial step, the excitation of electrons (e−) takes place, which results in the transfer of electrons from the valence band of the semiconductor to the conduction band, and leaves behind the holes (h+) in the valence band (eqn (11) and (12)). In the second step, the separation of photogenerated electrons and holes takes place. However, the bulk charges experience recombination along with the generation of heat, which results in the reduction of the excited charge carrier.57 Moreover, photo-electrocatalytic oxidation combines the electrochemical AOPs with photocatalysis, which exploits photoactive electrode materials, and they function as either electrodes or as photocatalysts.57,63 Furthermore, the electron transfer from the valence band to the conduction band of the catalyst generates an electron–hole pair that triggers a reaction at the surface of the catalyst and generates the highly oxidative ˙OH radicals and other reactive species (eqn (11) to (15)).
| Photoanode + hv → h+ + eCB− | (11) |
| h+ + H2O/OH− → ˙OH + H+ | (12) |
| eCB− + O2 → O2˙− | (13) |
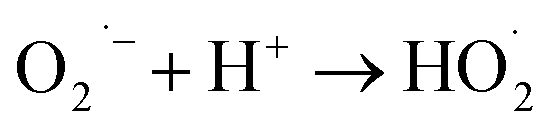 | (14) |
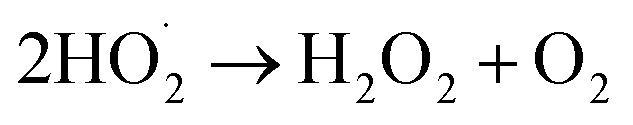 | (15) |
2.3 Drawbacks of electrochemical and photochemical treatment methods
There are several techno-economical drawbacks of the electrochemical and photochemical treatment systems that have withheld transition of these technologies from lab to field-scale. For example, in EO the formation of halogenated by-products such as ClO3−, ClO4−, and BrO3− results in detrimental effects like corrosion of the electrode's surface,64,65 while in EF the moderate H2O2 production and poor Fe2+/Fe3+ redox cycle suppresses the ROS production rate. Additionally, formation of inorganic by-products, halogenated organic compound, and ˙OH scavengers, and fouling of electrodes are also frequently encountered. Nevertheless, the main limitation of ETs is the forbidding electricity consumption that makes the treatment operation taxing.Similarly, in the case of photochemical treatment methods, the wide band gap energies, less absorption capability towards light, and fast recombination rate of photo-induced e− and h+ restrict the photo-oxidation efficiency of these systems. The contaminant removal rate is further retarded by the fast recombination rate of photoinduced electrons that results in lowering the quantity of photo-generated ROS, which are responsible for executing photodegradation.66,67 Another major issue with the photochemical oxidation process is the poor response of catalysts to visible light, which necessitates the use of artificial lighting sources, thereby increasing the treatment cost. To circumnavigate the aforementioned roadblocks, impetus is being laid on the development of cost-effective and greener materials that can subdue the construction and treatment cost of these advanced oxidation technologies. However, a feasible way to economise the operation of photo/electro-induced advanced oxidation is by integrating it with other treatment technologies, notably biological systems. The integrated processes can potentially enhance the treatment efficiency and can simultaneously achieve elimination of toxic pollutants such as pharmaceuticals and personal care products. Many technoeconomic assessments (TEA) and LCA investigations have also upheld the superiority of combined/hybrid systems over the standalone units. Hence, merging biotic-treatment with advanced oxidation can pave the way for sustainable wastewater treatment in the coming days.
3. Integrated bio-electrochemical technologies and hybrid advanced oxidation systems for emerging contaminants degradation
3.1 Need for integrating advanced oxidation processes and bio-electrochemical systems
The AOPs are incredibly effective in mineralising persistent organic pollutants (POPs) in wastewater, though the energy consumed in the treatment process is quite taxing. This situation can be salvaged by BESs that can fulfil the energy required for operating AOPs. In particular, integrating EAOPs with MFCs seems congruous as the degradation kinetics in both technologies is fundamentally governed by analogous electrochemistry. Further, such integrated systems can reap the advantages of biotic and abiotic processes, offering a wholesome treatment to wastewater. One such integrated technology is the so-called BEF, in which the well-known Fenton reaction is simulated in the cathodic chamber of an MFC for oxidising dyes, pharmaceuticals and other POPs, thus exemplifying their removal.21 Similarly, a synergy can be created by coupling photocatalysis and bioelectrochemistry for an improved abatement of refractory contaminants. The fundamental working principle of an in situ MFC-AOP unit can be visualised through Fig. 3. In addition to saving energy, these integrated systems have lower effluent toxicity, minimal sludge production, a wide range of working pH and high contaminant removal efficiency. Thus, it is apparent that combining an AOP and BES creates a synergy that suppresses the demerits of individual technologies, thus enhancing the acceptability and scalability of the integrated wastewater treatment system.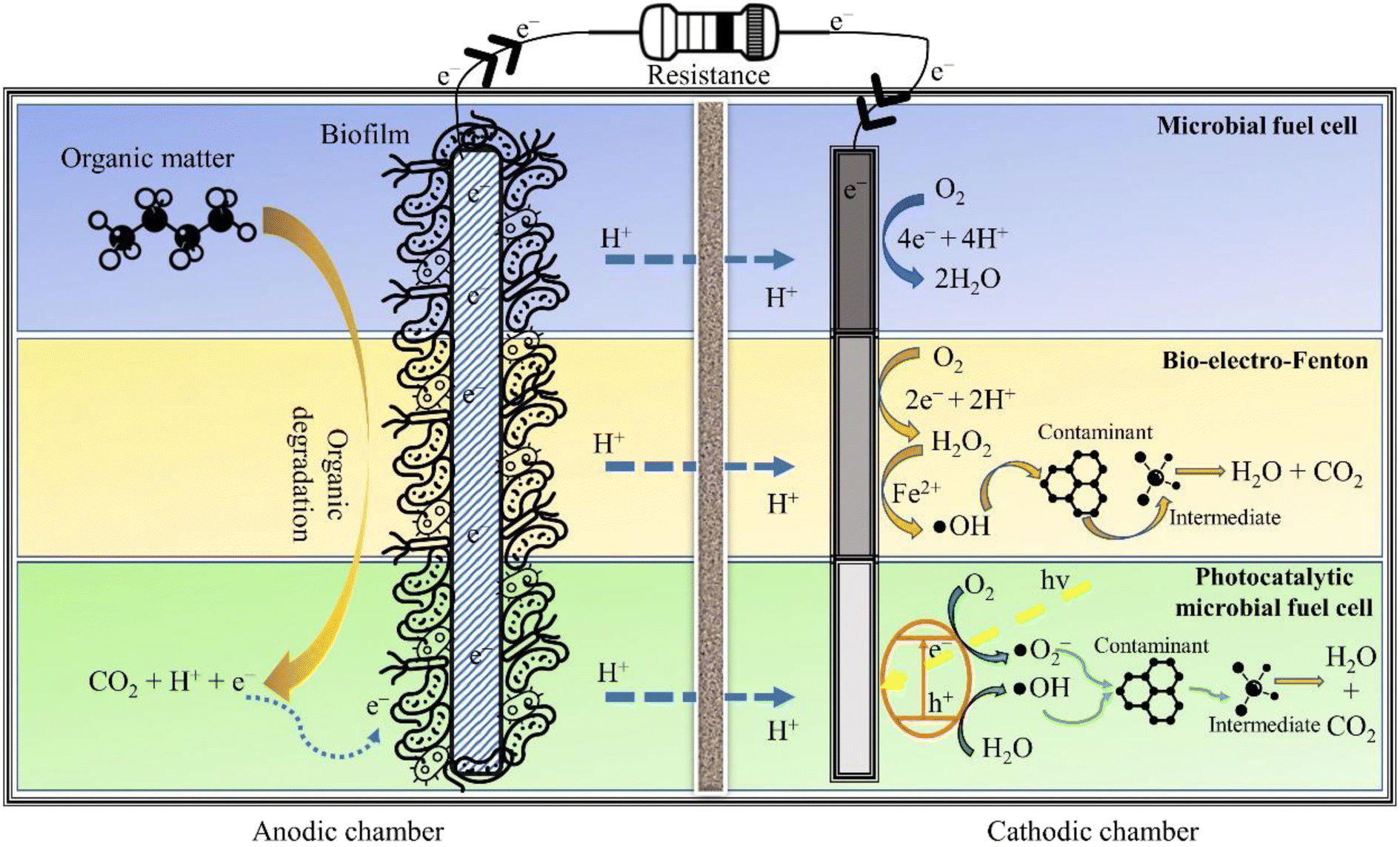 | ||
| Fig. 3 Fundamental working of the in situ integrated MFC-AOP system. | ||
3.2 The bio-electro-Fenton system
The in situ MFC-driven BEF can be stimulated in a single chamber MFC (Fig. 4) and dual chamber MFC (Fig. 3). The BEF process relies on the in situ production of H2O2, which is then catalysed by Fe2+ for producing reactive oxygen species, especially ˙OH. Further, the electrogeneration of H2O2 is achieved by 2e− reduction of O2 over a carbonaceous cathode. On the other hand, the biotic degradation of organic matter in the anodic chamber supplies the e− and H+ required to complete the reduction reaction. The Fe2+ then reacts with H2O2 to produce ˙OH and Fe3+. The Fe3+ is further reduced to Fe2+ at the cathode, ensuring the transient presence of Fe2+ in the wastewater. Since Fe2+ is electrochemically regenerated, it is required only in catalytic amounts. The entire operation is energy-free, barring the aeration necessary for sustaining the O2 level in the catholyte. Zhu and Ni (2009)68 demonstrated the first application of BEF by degrading p-nitrophenol in the cathodic chamber of the MFC using scrap iron as the Fenton catalyst. The Fenton catalyst used in BEF can be conventional homogeneous Fe (salt of iron dissolved in the catholyte) or a heterogeneous solid catalyst smeared over the cathode surface. The homogeneous Fenton process has relatively high degradation efficiency and requires Fe2+ in catalytic amounts. However, homogeneous reactions are only effective in a narrow acidic pH range (2.5 to 3.5), and the catalyst once used can't be recovered. On the other hand, heterogeneous catalysts are easily recoverable, perform reasonably well in a circumneutral pH environment and attain higher power output by boosting the ORR activity of the cathode. In this regard, several heterogeneous Fenton catalysts have been used in BEF systems, like Fe2O3/activated carbon, carbon nanotube/γ-FeOOH, and FeVO4.16,69,70 All these catalysts attained contaminant degradation efficiency greater than 80% though the time required was much higher than that of chemical oxidation methods. In fact, Li et al. (2020)71 reported a reaction time of 48 h for degrading 90% of erythromycin with the initial concentration of 50 μg L−1. The slower degradation rate in MFC-driven BEF systems can be attributed to the sluggish H2O2 production, which in turn leads to lower ˙OH generation. To ameliorate the formation of ˙OH and other reactive species, sometimes the BEF system is operated in microbial electrolysis cell (MEC) mode, i.e., under an external voltage supply. This method of increasing the cathodic potential accelerates the rate of two electron ORR, yielding higher H2O2 and ˙OH.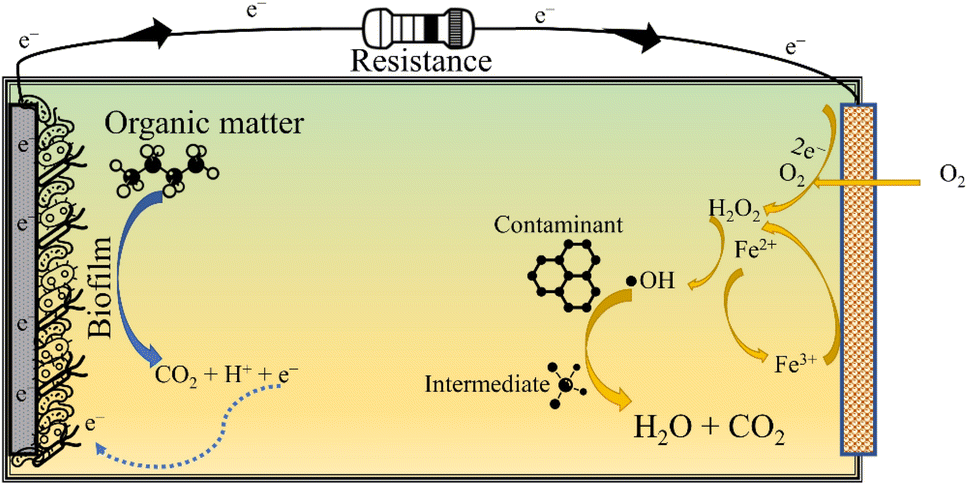 | ||
| Fig. 4 A typical single chamber MFC-driven in situ BEF system. | ||
Nevertheless, it is crucial to optimise the cell potential in MEC-driven BEF systems keeping in mind the tolerance level of anodic microbes, which is usually in the range of 0.2 V to 0.8 V.72 The optimised cathodic potential for maximum H2O2 production depends on the cathode material. For instance, Li et al. (2017)73 achieved the maximum degradation of aniline at an applied cathode potential of 0.45 V vs. a saturated calomel electrode (SCE) using a graphite cathode.73 At the same time, the carbon nanotube-coated graphite plate attained a maximum H2O2 yield and degradation efficiency at an applied cell potential of 0.6 V.74 A dip in treatment efficiency of BEF systems beyond the optimum applied potential can be explained by the electrochemical reduction of H2O2 to H2O at higher voltage.75 Also, the higher applied anodic potential in the MEC can severely damage the biofilm, choking the supply of H+ and e− required for the electrogeneration of H2O2 at the cathode.76 Hence, the applied potential in MEC-driven BEF systems should be meticulously optimised for an efficient and economical operation of MEC-BEF reactors.
3.3 Photocatalytic microbial fuel cell
A P-MFC is a modified MFC configuration fitted with either a photocathode (Fig. 3) or photoanode (Fig. 5) to execute photocatalytic oxidation of wastewater. Electrons and holes are generated when light (natural or ultraviolet, UV) is irradiated over the surface of the photocatalyst, which then reacts with water and O2 to form ˙OH and superoxide radicals (˙O2−). These reactive radicals oxidise the pollutant present in the wastewater to small and readily bio-metabolisable molecules such as carboxylic acids.77 In the case of the photoanode, the pollutants are effectively removed by the synergistic action of the photocatalyst and microorganisms. Moreover, electrons can be directly transported to the cathode to produce electricity, thus preventing electron–hole recombination. Additional electrons generated at the anode result in enhanced current production, improving power generation. Coating semiconductor photocatalysts such as α-Fe2O3 over a bioanode can also accelerate biofilm growth due to augmented extracellular electron transfer resulting from electron–hole photogeneration,78 while the pollutant degradation is achieved through the synergistic action of the photocatalyst and anodic microorganisms. To elucidate, Wang et al. (2022)79 connected a TiO2 photoanode in series with a conventional bioanode in a single chamber P-MFC, which removed 50% of o-chlorophenol with the initial concentration of 5 mg L−1 and attained a maximum power density of 301 mW m−2. Undoubtedly, anodic photo-oxidation improves the degradation efficiency of the coupled (bio)electrochemical systems; however, it can also damage the biofilm if the AOP is not controlled meticulously.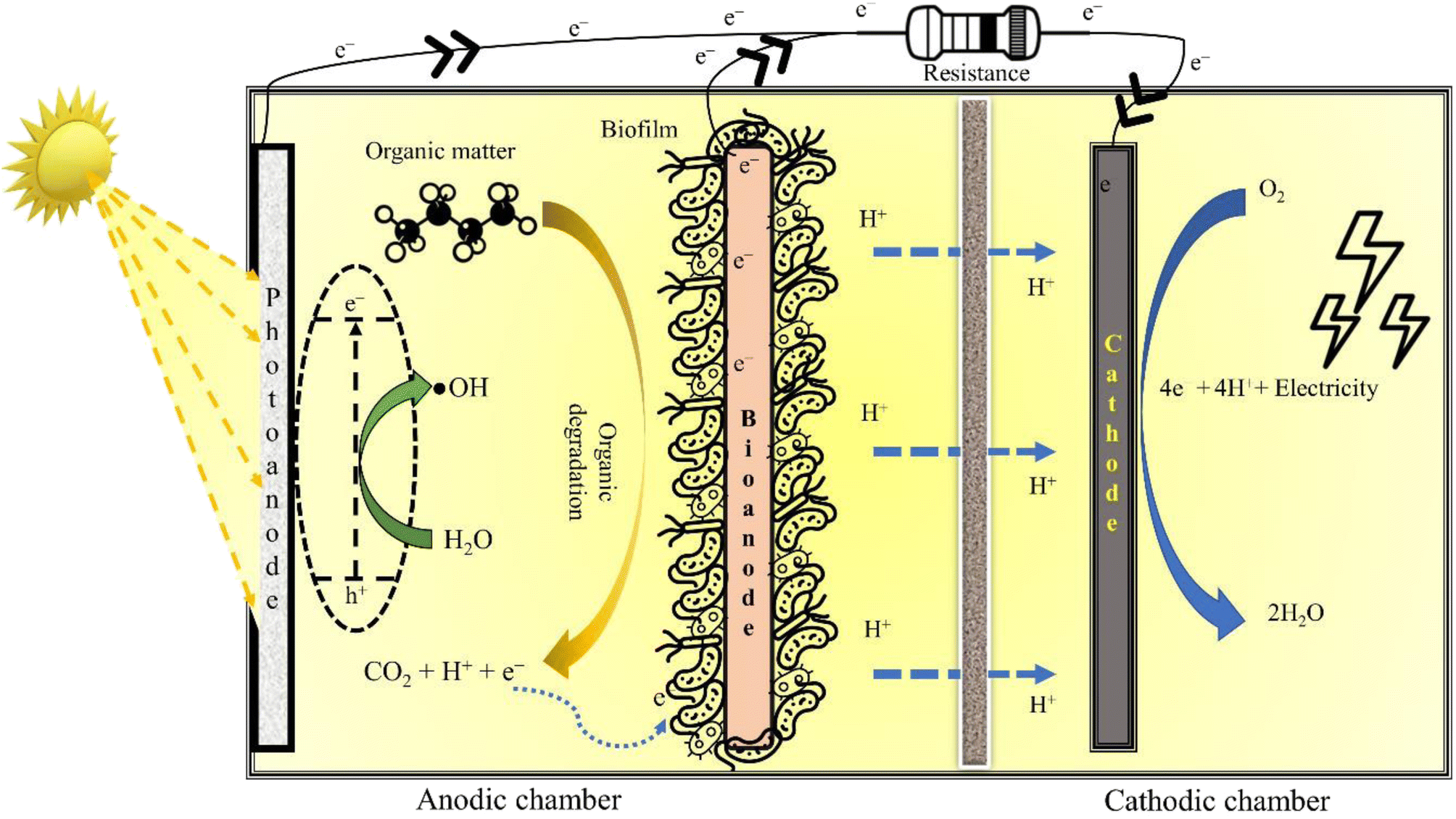 | ||
| Fig. 5 A schematic representation of a photo-anode P-MFC. | ||
To avoid disruption of biofilm, photocatalysis can be implemented in the abiotic cathodic chamber of the P-MFC. In this configuration of P-MFC, a photocathode is employed instead of a photoanode to facilitate the cathodic degradation of pollutants. It is well known that the infamous sluggish ORR of the cathode deprives MFCs of any significant up-scaled application. Hence, photocatalysts or photo-electrocatalysts can expedite the rate of this sluggish ORR by harvesting the energy of sunlight to activate the cathodic reaction. In this configuration, connecting the bioanode to the photocathode creates an internal bias in the MFC system. Electrons produced by the electrogens travel from the anode through the external circuit to the cathode and interact with photoinduced holes, releasing electrons, degrading organic matter, and reducing water.80 Many MFCs with the cathode smeared with photocatalytic materials, like natural rutile and lithium niobate, have displayed exemplary power performance.81,82 However, the salient feature of a photocathode-fitted P-MFC is its ability to photo-catalyse refractory pollutants. In this regard, photoactive cathode catalysts, like TiO2 and Pd-modified silicon nanowires, were reported to degrade more than 80% of sodium dodecyl sulphate and methyl orange.83,84
Nonetheless, most semiconductor materials respond only to UV light irradiation and thus are not economical for practical scenarios. Hence, the impetus has been on developing photocatalysts that can produce reliable performance under natural illumination. In this context, Hu and co-researchers utilized a P-MFC with a Ni decorated titanium carbide photocathode for treating chloramphenicol-contaminated water with an initial concentration of 30 mg L−1 under natural irradiation. The investigation identified ˙OH and ˙O2− produced from the photoactivation of water as the chief reactive radicals degrading 82% of chloramphenicol in a 36 h reaction period.85 Clearly, the slow degradation kinetics of natural light-driven P-MFCs makes them impractical and necessitates a suitable reactor design and modifications to meet the onsite demands.
3.4 Retrofitted and hybrid systems
Considering the complexities of a single-unit hybrid biochemical reactor, sometimes separate sequential treatment units seem more practical and workable in an actual setup. In such ex situ hybrid configurations, the power demand of the (E)AOP reactor is met by in parallel operating an MFC assembly (Fig. 6). Also, such treatment systems may provide more control over the operational parameters, which is vital for maintaining consistent effluent quality. For instance, Zhang et al. (2019)86 installed a separate photoelectrocatalysis reactor in parallel to an MFC for degrading phenol and aniline, and both the contaminants were photodegraded into readily biodegradable intermediates like hydroquinones, benzoquinones, and organic acids. Further, the effluent from the photoelectrocatalysis reactor was subjected to biological oxidation in the biotic chamber of the MFC, achieving 96% and 70% reduction in chemical oxygen demand (COD) from phenol (COD of 700 mg L−1) and aniline (COD of 165 mg L−1), respectively.86 Thus, these investigations emphasize on the fact that photochemical pre-treatment has positive effects on the treatment efficacy of the MFC.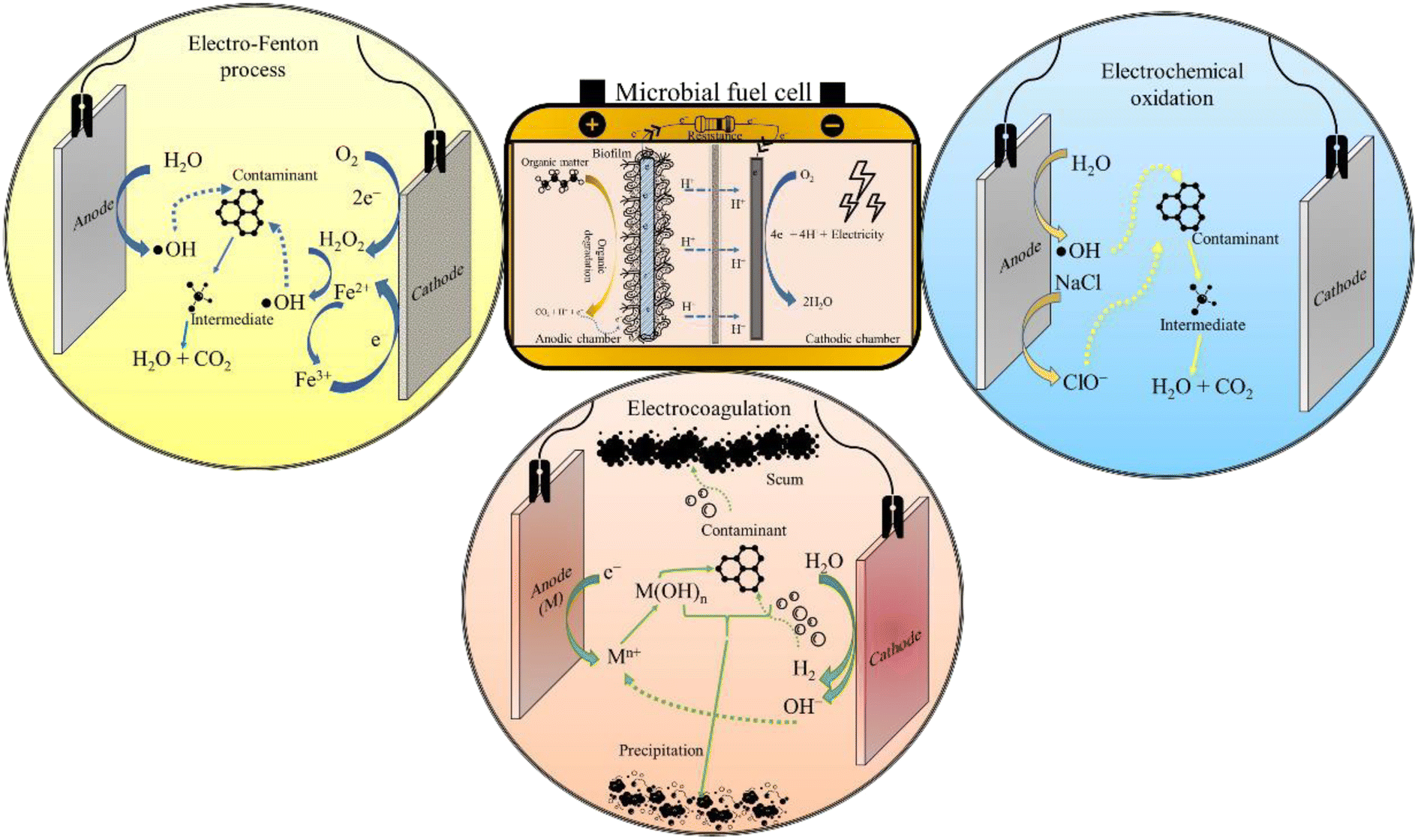 | ||
| Fig. 6 The ex situ MFC-AOP integrated systems. | ||
Further, in one investigation, the cathode chamber of the MFC was extended to accommodate an electrode arrangement for implementing EF oxidation.23 The reactor employed a triple cathode system yielding a maximum H2O2 concentration of 176 mg L−1, while 90% of Fe3+ (initial concentration of 28 mg L−1) was reduced to Fe2+ in 30 min, thus ensuring the adequate presence of Fe2+ in the solution for continuous catalysis of H2O2 into ˙OH.23 Further, in an attempt to reduce the dependency of wastewater treatment on fossil-derived electricity, the MFC has been used to power electrochemical systems, like EC, EF, and EO processes. In this regard, Mei and co-researchers operated an MFC driven EC reactor for treating oily bilge water with an initial COD of 2029 mg L−1.87 The coupled system eliminated 93% of organics, which was on par with conventionally operated EC.87 Similarly, a 100 mL benchtop EF reactor was driven using a single chamber MFC for degrading phenol.88 However, MFC-powered EF and EO processes are seldom reported in the literature. This may be because of the meagre voltage generation in the MFC (less than 2 V for stacked MFCs), which is far less than the oxygen evolution reaction potential of active electrodes (Pt, graphite and dimensionally stable anodes) as well as inactive electrodes (TiO2).89 Therefore, to meet the power demand of operating electrolysis reactors, multiple MFC reactors can be assembled in series. For instance, power generated from two single-chamber MFCs was used as a renewable energy source for operating the EO reactor for degrading pyridine and methyl orange.90 Further, Sun et al. (2020)91 reduced energy consumption by 58.2% by combining an MFC with the plasma oxidation process for mineralising methylene blue. This is just additional evidence of MFC's potential in propelling energy-efficient wastewater treatment. Past research has elucidated the self-powered wastewater treatment through integrated MFC systems, which could be especially handy for remote areas. According to Dziegielowski and co-workers, stacking 16 soil-MFCs resulted in 30-fold amplification of power (12.2 mW). Additionally, the soil-MFC stack, treating up to 2.8 L of wastewater per day in the field was able to charge a 600 mA h battery.92 However, the prospect of powering a full-size electrochemical reactor through MFCs is still questionable. This is due to the inconsistencies in power production of MFCs. Being a biological system, MFCs are susceptible to environmental and operational fluctuations, while the advanced treatment technologies are energy-consuming and need a consistent power supply for the entire operating period. Fluctuations or a drop in power could adversely impact the efficiency of treatment and may even damage the electromechanical components of the reactor. Hence, MFC assemblies that can ensure stable energy production should be developed for manifesting a reliable supply of energy, which could also power other remediation systems.
4. Performance comparison of conventional AOPs and integrated BESs
Both AOPs and BESs are innovative approaches for the treatment of wastewater containing refractory and emerging pollutants; however, BESs have an added advantage of concomitant bio-energy recovery (Table 2). The AOPs are competent technologies for the production of reactive species such as ˙OH,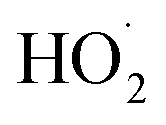 , SO4˙− and O2˙− that can effectively mineralise refractory organic pollutants.93 Though the exorbitant treatment cost of AOPs is an impending issue. Thus, these major challenges render this technology unaffordable at the commercial scale; however, the same can be overcome by promoting the use of low cost and efficient materials, use of renewable energy sources and adoption of integration strategies.93 However, the simultaneous generation of bioelectricity, wastewater treatment, and co-product recovery offered by BESs has the potential to reduce greenhouse gas emissions and produce renewable energy, and sequestering CO2 makes BESs advantageous over other contemporary technologies. Furthermore, researchers have employed novel BESs for the removal of various emerging pollutants present in wastewater.41 Besides, the presence of emerging pollutants in wastewater vastly affects the efficiency of BESs because of the intractable nature of the ECs and due to the inhibition of exoelectrogenic microbes residing in the BES resulting from inherent toxicity of these compounds.94
, SO4˙− and O2˙− that can effectively mineralise refractory organic pollutants.93 Though the exorbitant treatment cost of AOPs is an impending issue. Thus, these major challenges render this technology unaffordable at the commercial scale; however, the same can be overcome by promoting the use of low cost and efficient materials, use of renewable energy sources and adoption of integration strategies.93 However, the simultaneous generation of bioelectricity, wastewater treatment, and co-product recovery offered by BESs has the potential to reduce greenhouse gas emissions and produce renewable energy, and sequestering CO2 makes BESs advantageous over other contemporary technologies. Furthermore, researchers have employed novel BESs for the removal of various emerging pollutants present in wastewater.41 Besides, the presence of emerging pollutants in wastewater vastly affects the efficiency of BESs because of the intractable nature of the ECs and due to the inhibition of exoelectrogenic microbes residing in the BES resulting from inherent toxicity of these compounds.94
| Reactor details | Anodic volume (L) | Type of waste | Power density (mW m−2) | References |
|---|---|---|---|---|
| a NR: not reported. | ||||
| Dual chamber MFC | 0.06 | Synthetic wastewater | 457 | 100 |
| Dual chamber MFC | 0.35 | NR | 267.77 | 101 |
| Dual chamber MFC | 25 | Septic tank slurry | 0.32 | 102 |
| Dual chamber MFC | 120 | Human excreta | 7.29 ± 0.7 | 103 |
| Dual chamber MFC | 40 | Municipal wastewater | 2.8 | 104 |
| Dual chamber MFC | 120 | Human excreta | 6.82 ± 0.8 | 103 |
| Cubic dual chamber MFC | 1.8 | Pre-treatment of human faeces | 251 | 105 |
| Bioelectric toilet | 1500 | Human waste | 7.29 ± 0.7 | 106 |
Moreover, BESs have been proven to be greener and competent for the eradication of EC, for example pharmaceuticals, dyes, surfactants, and phenol from wastewater.95,96 However, in the real field various kinds of ECs are simultaneously present in wastewater, which are difficult to treat by the standalone technologies.97 In typical BESs, such as MFCs, MECs, MDCs and MCCs, the fouling of membrane, meagre power production, high fabrication and catalyst cost and in case of MECs, the external power requirements for the remediation of various contaminants, are the major obstructions in the way towards the broader application of these technologies.98 To overcome these hindrances, researchers have experimented with integrated BESs and hybridized BESs with other technologies such as constructed wetlands (CW), EF, adsorption, membrane bioreactors and also MECs integrated with MFCs, which were operated with the power drawn from the MFCs, and might prove to be a sustainable and economical alternative for remediating these ECs.96,97
The AOPs can be used as a pre-treatment or post-treatment based on the wastewater's characteristics. For instance, if the wastewater has contaminants toxic to microorganisms, then AOPs can be used as pre-treatment to enhance biodegradability and make it amenable for biological treatment. However, the cost of wastewater treatment, including capital and operation and maintenance costs via AOPs is very high. Corroborative results were outlined in research by Buthiyappan et al. (2015),99 where the phenol removal by O3 (2 mg L−1) was as low as $2 per 1000 gallons (3785.41 litres) compared to the ultrasound (US) method, which costed around $15536.59 per 1000 gallons (3785.41 litres). On the other hand, when the contaminant was changed to azo dye with the initial concentration of 12.4 mg L−1, the cost of removal by O3 was increased to $4.08 per 1000 gallons (3785.41 L), with the highest cost at $14203.7 per 1000 gallons (3785.41 L).107 Further, in comparison to AOPs, the capital expenditure for treating 100000 L day−1 of wastewater with 2000 mg L−1 BOD via MFCs was estimated to be $380528.108 Consequently, the cost for the wastewater treatment based on MFC technology comes out to be $3.946 per 1000 gallons (3785.41 L), which is lower than the minimum cost incurred using O3−-based technology ($4.08 per 1000 gallons (3785.41 L)).108 This estimate excludes the operational cost including maintenance, manpower and chemical cost for the MFC, which if included might increase the overall cost. Therefore, for a realistic comparison, operational tariffs must be included in the TEA along with other derived costs. Nevertheless, the integrated system has shown better performance in the degradation of various contaminants of concern. In this context, an investigation revealed that by adopting a light/peroxymonosulfate (PMS) AOP system, approximately 100% degradation of sulfamerazine with an initial concentration of 2 × 10−4 mg L−1 can be achieved.109 Meanwhile, an integrated MFC with CW has also exhibited a promising result of nearly 100% removal of sulfamethoxazole (SMX) antibiotic for a hydraulic retention time of 24 h and initial SMX concentration of 100 μg L−1.110 Furthermore, in comparison to the standalone CW system (76.3% removal of SMX with a hydraulic retention time (HRT) of 10 days), the CW-MFC exhibited a significant improvement in SMX removal, which was mainly due to the integration of both the technologies.111,112 From these investigations it is clear that both AOPs and integrated BESs have shown similar results; however, integrated systems have shown far better performance compared to standalone CW technology. Likewise, an investigation demonstrated that persulfate (PS)-based AOPs are also efficient in degrading emerging pollutants from wastewater.113 The same research indicated that the PS-based Fenton system achieved 96% degradation efficiency for 50 mg L−1 sulfamerazine within a time period of 30 min.113,114 Meanwhile, when compared with the hybrid system, an investigation by Yang and co-workers revealed that BEF systems have shown the degradation of metoprolol, a beta-blocker, to be nearly 95% within an HRT of 6 h from the initial pollutant concentration of 10 μg L−1.115 Furthermore, in comparison to the standalone CW system (76.3% removal of SMX with a hydraulic retention time (HRT) of 10 days), the CW-MFC exhibited a significant improvement in SMX removal, which was mainly due to the integration of both the technologies.112 Contrarily, MFCs have shown the degradation of SMX with approximately 85% efficacy within an operational time of 60 h.116 Thus, it is apparent that the PS-based AOP demonstrated higher removal efficiency for sulfamerazine, which is similar to the removal efficiency of the beta-blocker observed in BEF; however, the HRT for the degradation of the sulfamerazine by the hybrid system was quite lower than that of the standalone CW or MFC technology. Correspondingly, BESs are also used for hybridising with the AOPs, in the form of BEF, which is an emerging use of BESs. However, for better comparison, separate removal efficiency and associated energy consumption for metoprolol degradation in both BES and EF should be investigated, which will provide more clarity on the removal efficacy of these technologies. Thus, it can be concluded that both the integrated BES and AOPs are efficient and capable of achieving remarkable treatment efficiencies and they are highly efficient in the degradation of ECs. Further, both the technologies are cost intensive; however, integrated BESs have shown higher removal efficiency compared to standalone technologies such as MFC, MEC, and CW (Table 3). Besides, BESs are also capable of recovering value-added products and reducing the carbon footprints associated with the wastewater treatment process. Nevertheless, in-depth comparative explorations are required to optimise the functioning and reactor configuration of these integrated systems for on-site applications.
| Technology | Cathode material | Contaminants (concentration in mg L−1) | Valuable recovered | Removal efficiency (%) | Ref. | |
|---|---|---|---|---|---|---|
| a MCD = maximum current density; MCO = maximum current output; PD = power density, NR = not reported, CW-MFC = constructed wetland coupled microbial fuel cell, PMS = peroxymonosulfate. | ||||||
| Standalone technologies | MFC | Pt modified | Penicillin (50) | PD = 1277 mW m−2 | 98 | 117 |
| Carbon rods | New coccine (25) | MCD = 5500 mA m−2 | 83 | 118 | ||
| Graphite plate | Triclosan (10) | PD = 120 mW m2 | 94 | 119 | ||
| Carbon cloth coated with Pt/C | Sulfamethoxazole (10) | PD = 1790 mW cm−2 | 96 | 116 | ||
| MEC | Polished graphite plate | Nitrobenzene (50) | MCO = 2.58 mA | 98 | 120 | |
| NR | Quinoline (250) | MCO = 5 mA | 90.4 | 121 | ||
| Biogenic palladium modified graphite | Diclofenac (1) | NR | 92 ± 10 | 122 | ||
| Biogenic palladium modified graphite rod | Diatrizoate (2) | NR | 100 | 123 | ||
| Integrated technologies | Bio-electro Fenton system | Cubic graphite plate | Beta-blocker (metoprolol) (0.01) | NR | 95 | 115 |
| Carbon felt | Tetracycline (10) | PD = 93.13 mW m−2 | ∼100 | 124 | ||
| Waste derived iron catalyzed carbon felt | Surfactant (20) | PD = 117.5 mW m−2 | 85 | 125 | ||
| CW-MFC | NR | Sulfamethoxazole (0.1) | PD = 124.89 mW m−3 | 100 | 110 | |
| Light/PMS AOP | NR | Sulfamerazine (0.0002) | NR | 100 | 109 | |
5. Drawbacks of integrated bio-electrochemical and hybrid advanced oxidation systems and latest developments
Over the past few years, integrated bio-electrochemical and hybrid oxidation systems have been investigated as an effective platform for the degradation and mineralization of various contaminants present in wastewater.126 The main concern associated with the conventional wastewater treatment technologies such as activated sludge processes, trickling filters, sequencing batch reactor, traditional coagulation or flocculation processes, etc. is that they are energy intensive. This is due to the fact that aerobic treatment technologies require additional energy for aeration while chemical technologies require the addition of external chemicals, which make them costly. In addition, these technologies are not capable of efficiently removing recalcitrant micropollutants, which is evidenced by the presence of different types of emerging contaminants in the sludge and treated effluents of conventional wastewater treatment facilities across the globe.127 Moreover, the slow rate of treatment, large land footprint of processes based on ecological principles and high capital cost along with the upscaling issues of BESs have led to the development of integrated BESs.26 In this regard, the integrated BESs have proven to be more environment friendly compared to ETs, which is apparent because of the higher consumption of energy of the ETs. In this regard, an investigation revealed that EO has more negative impacts on the environment with approximately 55.03% contribution to the emission, which was nearly 22.8% more than that of the MFC and 42.2% higher than that of BEF.16 However, there are a few significant hindrances, which limits the large-scale application of the BESs, that need to be overcome. Basically, the bio-electrochemical integrated technologies harvest electricity from the microbial degradation; however, the energy recovery is very low and the fabrication cost is very high, which makes these technologies unaffordable.128 Furthermore, the degradation of the organic complexes is very slow and the cost of the electrodes is very high for these integrated technologies; thus, researchers are pondering to discover convenient alternatives in order to control the higher fabrication cost or to replace the costly electrodes with low-cost materials.The overall performance and efficiency of the integrated BESs and hybrid technology is highly contingent on the material of the electrode because of the electron transfer, adhesion of microbes and electrochemical activity, which are purely dependent on the type of electrode used in the system.110 To make the BESs economically affordable, researchers have investigated on the application of low-cost carbon-based electrodes such as carbon cloth, carbon felt, graphite fibre brush, graphite granules and graphite activated carbon as anode materials to enhance the performance of BESs. Moreover, in electrochemical systems, BDD, DSA, MMO, TiO2, PbO2, and SnO2 have been used to overcome the problem of corrosion and availability.129 The major drawback of these electrodes is their high cost; however, researchers are devoted to discovering superior alternatives by modifying the properties of the electrode materials to enhance the electron transfer efficacy with simultaneous cost reduction.
Nevertheless, the integration of BESs can make these technologies more versatile and pave the way for commercialization of the BESs for the treatment of wastewater and concomitant energy production.130 However, the practical application of BES is still challenging because of various critical issues such as poor degradability of real wastewater, inferior yield of valuables, high fabrication cost and membrane fouling, which needs to be overcome. Furthermore, the scaling-up of BES is a significant challenge to demonstrate the viability of these technology for practical applications and it is strongly dependent on the configuration, reactor design and mutual benefits of the technologies.131 However, in the scaling-up of the integrated technologies, the existing efforts in the direction of up-scaling of BESs might prove advantageous and pave the way in this direction.
In a similar way to BESs, the hybrid systems have also shown high efficacy in the treatment of wastewater and removal of various kinds of ECs such as beta blockers, EDCs, pesticides, antibiotics, and other pharmaceutical complexes.97 However, there are many challenges in up-scaling of the BESs for example fouling of membranes, higher energy demand, and higher operational and fabrication costs. Despite that, these technologies have shown tremendous potential for the removal of antibiotics, analgesics, lipid regulators, beta blockers, pesticides, and some miscellaneous pharmaceuticals, but still exhibited lower removal of some of the ECs.132 Nevertheless, the higher retention time and sludge management related problems limit the benefits of these systems. Likewise, the CW integrated system has also shown high effectiveness in the degradation of emerging pollutants; however, the requirement of large area and very high retention time have prevented its application on a larger scale.26
The advancement in the area of integrated and hybrid systems has shifted the focus on the minimization of resource consumption simultaneously achieving low cost, eco-friendliness, energy efficiency, and highly efficacious treatment to overcome the challenges associated with conventional technologies.26 Moreover, hybrid BES-based technology can function as a commercial technology for wastewater treatment with the concomitant recovery of value-added products such as bioenergy, biohydrogen, biodiesel, and biofuel, which will assist in achieving circularity and accomplishment of the sustainable development goals set by the United Nations (Fig. 7). In this context, mathematical models developed on the basis of the fundamental Butler–Volmer equation, Monod kinetics, and Nernst–Plank equation can be employed to rationalise and optimise the biotic and abiotic responses of a BES reactor.133,134 Models like the Taguchi method, Buckingham's pi theorem, black box models and many other electrochemical models have already been tested on MFCs.135 Moreover, artificial intelligence and machine learning are also being explored to optimise the performance of MFCs.136 However, these models are devised for very specific operating environments, which may not fit broader applications. Hence, for scaling-up of systems, the development of appropriate and reliable models will play an important role. To understand the role of various reactor configurations and operational parameters, these models might prove to be a powerful tool to predict the performance of a system. Therefore, further development of the overall integrated and hybrid systems is required for advancement of these technologies to realistic applications.137
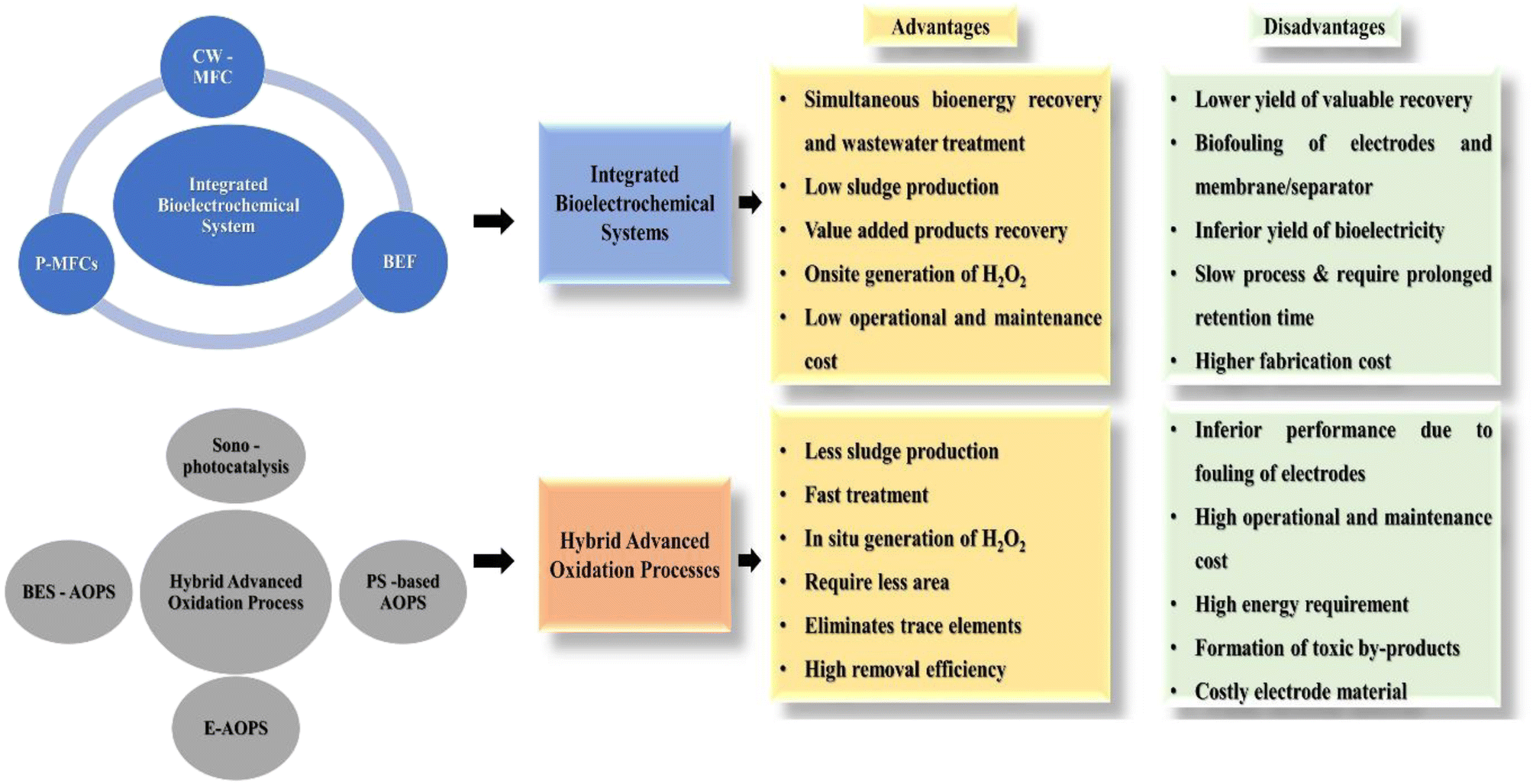 | ||
| Fig. 7 Advantages and disadvantages of integrated bioelectrochemical and hybrid advanced oxidation technologies. | ||
6. Life cycle and techno-economic evaluation of standalone advanced oxidation processes and bio-electrochemical integrated advanced oxidation systems
No matter how efficient a technology is, their scalability and acceptability is principally determined by the associated costs and is one of the major limiting factors in the commercialization process. For instance, in the context of up-scale applications, the performance of MFCs chiefly depends on design parameters, electrode material, wastewater characteristics, substrate conditions, and operational parameters. In the past, 720 L and 1500 L ceramic membrane based-MFC reactors have been operated for treating human excreta.103 Even though high COD removal (about 85%) and simultaneous disinfection was achieved by these up-scaled reactors, the maximum power and current output were just 75 mW and 239 mA, respectively. The electrical performance of field-scale MFCs is marred by increased internal resistance and overpotential losses due to larger rector components. Bio-fouling of the membrane and cathode are other operational nuisance often encountered at site settings. The hindrances in pragmatic application are further compounded by catalyst and assemblage cost, which increases proportionally with the reactor size.138 However, the AOPs usually involve applying a large amount of virgin chemicals, which is obviously unsustainable, when the system has to be scaled-up. This issue is addressed in electrochemically driven AOPs, where electrochemistry is tweaked to produce chemicals in situ in the system. Then again, in EAOPs, the electrical energy expense has to be accounted for while calculating the operational charges. Many estimates suggest that the cost of operating EAOPs is much lower than other advanced chemical oxidation processes. For example, the cost of treating real pharmaceutical wastewater was 10 times cheaper for the EF process (2.54 $ per m3) compared to the integrated coagulation and photocatalysis treatment.139 However, this cost is approximately nine times higher than for the conventional treatment of wastewater (0.28 $ per m3) and is thus not economically viable for more extensive applications.140 Recently, Lu et al. (2023)141 reported a treatment cost of 13.4 $ per m3 for treating landfill leachate by sequential electrochemical peroxidation and EF process, which is way too high to be commercially competitive. One plausible way of implementing advanced oxidation treatment economically is through a hybrid BES–AOP model. As per the estimate of Yang et al., the energy cost for treating 1 m3 of leachate in a BEF system operated in MEC mode (applied cell potential of 0.6 V) was 0.66 $ per m3, while the electrical consumption was estimated to be 4.8 kW h m−3.74 This value is about five fold lower than the electrical consumption reported for the EF treatment of leachate (19 kW h m−3).142 Hence, the cheaper operating cost of BES-driven AOPs can be ascribed to the reduced power consumption in these hybrid systems.Similarly, rhodamine B-contaminated wastewater was treated in a continuous flow zig-zag photocatalytic reactor at a cost of 9 $ per m3.143 In order to economise the photocatalysis treatment, Pham et al. (2022)144 integrated a microfiltration unit with the photocatalytic oxidation process for treating dye wastewater containing methylene blue. Installation of a microfiltration unit enabled the recovery and reuse of photocatalysts, thus bringing down the cost to 8.21 $ per m3.144 The price can be further marginalised in P-MFCs, which conjoins photocatalytic oxidation with MFCs. Evidently, BES-coupled AOP treatments are definitely more sustainable and commercially lucrative. Nevertheless, the slower degradation kinetics of these coupled reactors must not be ignored while gauging the cost in the field. To counter the sluggish reaction kinetics, the retention time can be increased. However, a higher retention time means a higher land footprint and larger reactor components will be required for treating a certain quantity of wastewater. Therefore, the surplus cost levied due to the slower treatment must be taken into account while performing the cost analysis of BES-driven advanced oxidation systems.
In addition to economic feasibility, the environmental viability of any technology is also of paramount importance. A technology can't be considered sustainable if it has very high environmental impacts. Therefore, the techno-economic evaluation of a system must also be supplemented with an LCA to ensure its sustainable implementation. As far as AOPs are concerned, the sustainability of the process is determined by the amount of energy and/or chemicals consumed during wastewater treatment. Naturally, a more energy-intensive treatment will have a low sustainability score. For instance, Chatzisymeon et al. (2013)8 performed LCA of three AOPs, namely, photocatalytic oxidation (UV/TiO2), wet air oxidation, and EO for treating the effluent from olive mills. As per the LCA of these three technologies, EO had the least environmental impact in terms of human health, fossil resource, and eco-system followed by wet air oxidation and UV/TiO2 treatment.8 As expected, electrochemical processes are more sustainable among different AOPs, probably due to the in situ production of reactive species. However, the significant environmental impact of EAOPs results from their excessive electricity consumption. This was also highlighted in a comparative LCA investigation, wherein the severity of the EF process on the environment was estimated to be approximately 30 times more than that of the photo-electro-Fenton process for degrading phenol.145 To make the EF process more sustainable, Zhang et al. (2022)146 suggested reusing iron sludge from conventional coagulation as the cathode, which can reduce fossil resource consumption and carbon emission by 73.7% and 97.1%, respectively. Nevertheless, further cost-effective measures such as using waste-derived electrodes and sustainable customisation like merging with biological systems should be implemented to enhance environmental sustainability of the EF process and other EAOPs.
In the past, coupling AOPs with BESs has been adopted as an effective way to reduce operating costs. Likewise, BES hybrids can be instrumental in offsetting the environmental loads of AOPs. The ability of BESs to recover resources in the form of energy or chemicals from wastewater reduces the environmental burden. Among different BESs, MFCs seem to be more propitious in mitigating environmental damage because they produce electricity instead of consuming it.147 In this regard, an MFC-powered EF oxidation was relatively more eco-friendly for eliminating surfactants, when compared to the treatment offered by a single MFC and EO.16 According to this estimate, the global warming potential was highest for the EO process with an estimated emission of 2236 kg CO2 eq., while the speculated emission of the MFC and BEF was 765 kg CO2 eq. and 663 kg CO2 eq., respectively.16 Likewise, solar-driven photocatalysis has emerged as an effective alternative owing to the lower ecological impacts. In an LCA of solar-based treatment for the abatement of micropollutants, solar/H2O2 was more sustainable than TiO2 photolysis and photo-Fenton process. Even though TiO2 and photo-Fenton treatment have greater environmental impacts, the mineralisation efficiency of these technologies is also higher, which means the effluent has a lower probability of containing harmful by-products. Henceforth, contaminant mineralisation and derived environmental benefits should be incorporated while deciding on the most suitable technology. Apparently, the MFC coupled with photocatalytic oxidation may further offset the environmental impacts by reducing the energy consumption. Howbeit, no effort has been made to quantify the repercussions of P-MFC on human and ecological health. Hence, additional investigation is required to dissect the impact of these novel hybrid systems based on different environmental factors. Therefore, the LCA outcomes will consequently guide the sustainable development of BES–AOP integrated systems.
7. Field-scale applications and future perspectives
The pilot-scale and on-site assessment is an acute stage in developing novel wastewater treatment technologies. Moreover, the performance of an upscaled prototype serves as a crucial parameter in assessing the TRL, an indicator of the technical maturity of a system. In this regard, many investigations on wastewater treatment in larger prototypes have been demonstrated (Table 2), notably for EAOPs. A 30 L h−1 EO reactor consisting of a 150 dia cell unit assembly (total anode area of 1.05 m2) was employed by Anglada et al. (2010)156 to treat landfill leachate. Meanwhile, Wang et al. (2014)157 developed an EO-CW combined unit to mitigate polluted surface water. Similarly, high COD (42%) and colour (85%) removal was attained in a 0.2 m3 EC unit treating olive mill wastewater with an initial COD concentration of 1000 mg L−1.158 Some researchers have even appraised the electrode performance and behaviour for up to two years in pilot-scale EC, which shows the extent of progress in the up-scaling of EAOPs.159 Moreover, among all EAOPs, the EF-based technologies are more competent in achieving rapid mineralisation of biorefractory contaminants. Accordingly, many pilot investigations have exhibited supporting results backing this claim. For example, Steter et al. (2018)160 designed a 2.5 L capacity (recirculation flow rate of 180 L h−1) solar photo-EF reactor fitted with a BDD anode and air diffusion cathode to treat paraben-spiked real secondary wastewater. This hybrid system achieved 66% mineralisation (initial total organic carbon of 110.8 mg L−1) at a relatively lower energy consumption rate of 84 kW h kg−1 of total organic carbon removed, which is encouraging for industrial implementation.160 In fact, in the review by Casado (2019),161 several commercialised EF-based reactors are enlisted, which exemplifies how far ETs have come in the field of wastewater treatment since their inception. In contrast, the BES–AOP hybrid reactors are still in the incipient stage, and explorations on the coupled up-scaled reactors are infrequent and only a few in number. To elaborate, a 110 L pilot MEC reactor (anode volume of 100 L, cathode volume of 10 L) was assessed by Sim et al. (2018)162 for the in situ synthesis of H2O2 using a gas diffusion cathode. The maximum accumulated H2O2 concentration in the catholyte was measured to be 98 mg L−1 at an applied cathode potential of −1.25 V vs. the saturated calomel electrode.162 Recovering H2O2 at such a low concentration from an upscaled setup is not economically viable. However, the gradual production of H2O2 can be exploited for facilitating Fenton oxidation. Subsequently, Zou et al. (2020)155 investigated on the application of a 20 L BEF reactor for degrading methylene blue for the first time. The pilot-BEF reactor almost entirely decolourised a 20 mg L−1 methylene blue solution, while attaining a mineralisation efficiency of 74%.155 The same research group also illustrated the complete elimination of six pharmaceutical compounds from real wastewater i.e., clofibric acid, diclofenac, carbamazepine, naproxen, ibuprofen, and ketoprofen (initial concentration of 500 μg L−1 each) in 26 h by imposing a cell potential of 0.1 V.154 Unquestionably, these investigations exemplify the up-scaling potential of BEF systems for treating complex wastewater (Table 4).| Technology | Operating conditions | Reactor volume/(flow rate) | Removal efficiency in % (parameter) | Ref. |
|---|---|---|---|---|
| a * = value directly calculated from the data provided in the article; # = six model pharmaceutical compounds used in the experiment (clofibric acid, diclofenac, carbamazepine, naproxen, ibuprofen, and ketoprofen); AOP = advanced oxidation process; BDD = boron doped diamond; BES = bio-electrochemical system; COD = chemical oxygen demand; TOC = total organic carbon, NA = not available; VUV/UV = vacuum ultraviolet/ultraviolet; UVC = ultraviolet-C. | ||||
| Electro-Fenton process | Anode: BDD; cathode: BDD; pH: 3; Fe2+: 16.8 mg L−1; contaminant: Black NT2 (250 mg L−1) | 4 L/(12 L min−1) | 100 (TOC) | 148 |
| Anode: Ti/IrO2; cathode: gas diffusion electrode; pH: 3; Fe2+: 5.6 mg L−1; contaminant: lamivudine (10 mg L−1) | 2 L/(NA) | 100 (lamivudine); 78.1 (TOC) | 149 | |
| Electrochemical oxidation | Anode: BDD; cathode: BDD; landfill leachate: 860 mg L−1 (COD) | NA/(11 L min−1) | 81 (COD) | 150 |
| Anode: BDD; cathode: carbon felt; landfill leachate: 860 mg L−1 (COD); pH: 3; contaminant: hydrochlorothiazide (29.8 mg L−1) | 1.67 L/(20.4 mL min−1) | 97 (TOC) | 151 | |
| Photochemical oxidation | UVC (95 W); H2O2: 400 mg L−1; gemifloxacin: 100 μg L | 120 L/(125 L min−1*) | 92 (gatifloxacin) | 152 |
| VUV/UV (120 W); aldicarb: 10 μg L−1 | 1.2 L/(20 L min−1*) | >98 (aldicarb) | 153 | |
| BES–AOP hybrid | Anode: carbon brush; cathode: graphite plate; Fe2+: 11.2 mg L−1; methylene blue = 20 mg L−1 | 20 L/(NA) | 74 (TOC) | 154 |
| Anode: carbon brush; cathode: graphite plate; Fe2+ = 0.16.8; applied cell voltage = 0.1 V; pharmaceutical compounds# = 500 μg L−1 | 20 L/(NA) | 100 (pharmaceutical compounds)# | 155 | |
Nonetheless, more pilot-scale explorations should be undertaken to test different configurations of BES–AOP hybrid systems to figure out the best alternatives. Further, one of the limiting drawbacks of EAOPs is the prohibitive cost of reactor components and operational charges. This is why, despite so many successful pilot trials, the TRL of BDD-driven EO systems has been stagnant at levels 4 to 5. Hence, coupling (E)AOPs with BES can be a plausible solution for raising the sustainability index of these hybrid technologies. Regarding EF systems, in addition to high power consumption, concern over cathode surface degradation has been raised in larger reactors. A detailed autopsy of the cathode by Salmerón et al. (2021)163 suggested that carbon layer gas diffusion cathodes erode in the acidic environment formed in the EF process, while in neutral pH, pore blocking occurs due to Fe precipitation. These nuisances can be alleviated by conjugating EF with an MFC in the BEF process. The BEF system operates under a much lower cathodic current density that minimises damage to cathodes. Also, the BEF process, in most cases, is sustained on self-generated electricity that can elevate the TRL of EF-based systems, which is currently at the intermediate level.164 Hence, to make BEF systems ready for application in the near future it is necessary to fabricate a low-cost cathode/cathode catalyst (heterogeneous) that demonstrates reliable performance in a circumneutral environment. Also, developing reactor assemblies that subdue aeration requirements can immensely boost the applicability of BEF systems.
Furthermore, the P-MFC is another emerging self-sustaining hybrid technology that is remarkably effective in eliminating recalcitrant contaminants. In spite of this, no pilot-scale investigation on P-MFCs has yet been reported in the literature. There are numerous challenges associated with P-MFCs, which hinders their application at a pragmatic scale. Primarily, in P-MFCs the ORR is intrinsically slow, which requires a very high overpotential to enhance the ORR kinetics. Consequently, it reduces the overall efficacy of the system because the energy of photogenerated electrons is limited due to the inherent nature of the photoanode. To overcome this issue, researchers have applied catalysts, such as platinum based catalysts, to enhance the ORR kinetics. However, these are prone to degradation over time, that leads to the decreased performance and stability of the systems. Furthermore, P-MFCs are reliant on photocatalysis and fuel cell principles to convert solar energy into electricity, which also make the system complex. Moreover, the system is highly dependent on the light source and the solar light is not a steady source as it can be affected by weather, restraining its application at the commercial scale. Together with the various technical challenges, long term stability and higher cost, which includes the fabrication and equipment maintenance, are also an additional burden that influences the overall cost of the system.165 Hence, future endeavor on enlarging P-MFC prototypes is necessary for bringing forward photo-driven BESs for contaminant remediation. Furthermore, retrofitting AOPs in a BES can have an induced effect on the biotic components of the coupled reactor. Hence, morphological changes and metagenomic variations in the microbial consortia should be looked into for the holistic optimisation of these integrated reactors. Such information will also help to decipher the electron transfer mechanism by a cellular or secretory mode that can assist in refining the designs of BES–AOP systems.
8. Conclusions
The critical assessment of AOPs and integrated BESs was carried out and application of these technologies for the remediation of ECs from wastewater was elucidated. Considering the recent advancements, both AOPs and integrated BESs have shown encouraging results for the removal of ECs of concern. In the analysed investigations, the integrated CW-MFC has shown a promising removal of EC compared to standalone systems. Furthermore, BEF has also exhibited a high EC removal of nearly 96% and a light/PMS AOP system has also shown nearly 100% degradation of ECs. Correspondingly, other integrated BESs and hybrid AOPs have also shown competence towards the removal of ECs from wastewater. Compared to conventional wastewater treatment technologies, which are not as efficient in the removal of trace contaminants of concern such as pharmaceuticals, personal care products, microplastics etc., traditional treatment technologies are not efficacious in the degradation and mineralization of trace contaminants while hybrid ETs and integrated BESs have shown promising results; however, the majority of research on these technologies is limited to lab scale due to various challenges, such as the requirement of costly electrodes and the non-selective reaction of ROS, which need to be addressed for commercialization of these technologies. Therefore, pilot-scale investigations are warranted, which might prove beneficial for understanding the technical feasibility as well as economic aspects of the integrated systems. Moreover, LCA and TEA can prove to be an effective tool in identifying the feasibility and sustainability of the AOPs and integrated BESs employed for the treatment of real wastewater containing ECs.Author contributions
Yasser Bashir: conceptualization, formal analysis, investigation, methodology, writing—original draft, writing—review and editing; Rishabh Raj: validation, writing—original draft, data curation, writing—review and editing; M. M. Ghangrekar: supervision, validation, writing—review and editing; Arvind K. Nema: validation, project administration, writing—review and editing; Sovik Das: project administration, resources, supervision, validation, writing—review and editing.Conflicts of interest
The authors declare that they do not have any financial interests or personal relationships that could be considered as potential competing interests.References
- M. An, L. Fan, J. Huang, W. Yang, H. Wu, X. Wang and R. Khanal, PLoS One, 2021, 16, e0247604 CrossRef CAS PubMed.
- M. K. Shahid, A. Kashif, A. Fuwad and Y. Choi, Coord. Chem. Rev., 2021, 442, 213993 CrossRef CAS.
- R. Raj, A. Tripathi, S. Das and M. Ghangrekar, Case Stud. Chem. Environ. Eng., 2021, 4, 100129 CrossRef CAS.
- R. Stirling, W. S. Walker, P. Westerhoff and S. Garcia-Segura, Electrochim. Acta, 2020, 338, 135874 CrossRef CAS.
- P. Canizares, R. Paz, C. Sáez and M. A. Rodrigo, J. Environ. Manage., 2009, 90, 410–420 CrossRef CAS PubMed.
- E. Dannys, T. Green, A. Wettlaufer, C. Madhurnathakam and A. Elkamel, J. Bioprocess. Biotech., 2016, 6, 2 Search PubMed.
- C. Abourached, M. J. English and H. Liu, J. Cleaner Prod., 2016, 137, 144–149 CrossRef CAS.
- E. Chatzisymeon, S. Foteinis, D. Mantzavinos and T. Tsoutsos, J. Cleaner Prod., 2013, 54, 229–234 CrossRef CAS.
- D. E. Santiago, M. J. H. Rodriguez and E. Pulido-Melian, J. Environ. Eng., 2021, 147, 03121001 CrossRef CAS.
- G. Maniakova, K. Kowalska, S. Murgolo, G. Mascolo, G. Libralato, G. Lofrano, O. Sacco, M. Guida and L. Rizzo, Sep. Purif. Technol., 2020, 236, 116249 CrossRef CAS.
- G. Vijayakumar, M. Kummert, S. A. Klein and W. A. Beckman, Sol. Energy, 2005, 79, 495–504 CrossRef.
- G. F. Nemet, E. O’Shaughnessy, R. Wiser, N. R. Darghouth, G. Barbose, K. Gillingham and V. Rai, Renewable Energy, 2017, 114, 1333–1339 CrossRef.
- M. Wu, H. Huang, B. Huang, C. Tang and C. Cheng, Renewable Energy, 2009, 34, 1934–1938 CrossRef.
- T. J. Arana and V. G. Gude, Int. Biodeterior. Biodegrad., 2018, 130, 91–97 CrossRef CAS.
- C. Tian, B. Zhang, A. G. Borthwick, Y. Li and W. Liu, Int. J. Hydrogen Energy, 2017, 42, 29454–29462 CrossRef CAS.
- S. Sathe, I. Chakraborty, V. S. Cheela, S. Chowdhury, B. Dubey and M. Ghangrekar, Bioresour. Technol., 2021, 341, 125850 CrossRef CAS PubMed.
- S. Das, R. Raj, S. Das and M. M. Ghangrekar, Environ. Sci. Pollut. Res., 2022, 1–18 Search PubMed.
- I. Chakraborty, G. D. Bhowmick, D. Nath, C. N. Khuman, B. K. Dubey and M. M. Ghangrekar, Int. Biodeterior. Biodegrad., 2021, 156, 105108 CrossRef CAS.
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schroder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5192 CrossRef CAS PubMed.
- J. Prasad and R. K. Tripathi, Energy Rep., 2022, 8, 10418–10433 CrossRef.
- S. Sathe, I. Chakraborty, B. Dubey and M. Ghangrekar, Environ. Res., 2022, 204, 112135 CrossRef CAS PubMed.
- Z. C. Xu, S. Y. Chen, S. Y. Guo, D. Wan, H. Xu, W. Yan, X. L. Jin and J. T. Feng, J. Power Sources, 2021, 501, 230000 CrossRef CAS.
- D. Wang, Y. Li, S. Hu, J. Hu, H. Hou, B. Liu, H. Zheng, X. Luo and H. Li, Sep. Purif. Technol., 2022, 299, 121704 CrossRef CAS.
- J. Lan, Y. X. Ren, Y. B. Lu, G. L. Liu, H. P. Luo and R. D. Zhang, Chem. Eng. J., 2019, 359, 1139–1149 CrossRef CAS.
- B. Zhang, Z. Wang, X. Zhou, C. Shi, H. Guo and C. Feng, Bioresour. Technol., 2015, 181, 360–362 CrossRef CAS PubMed.
- R. K. Yadav, S. Das and S. A. Patil, Trends Biotechnol., 2022, 41, 484–496 CrossRef PubMed.
- M. A. Montiel, I. F. Mena, J. Lobato, C. Saez and M. A. Rodrigo, Curr. Opin. Electrochem., 2022, 33, 100928 CrossRef CAS.
- A. Galushchinskiy, R. González-Gómez, K. McCarthy, P. Farràs and A. Savateev, Energy Fuels, 2022, 36, 4625–4639 CrossRef CAS PubMed.
- N. B. Bolujoko, O. O. Ogunlaja, M. O. Alfred, D. M. Okewole, A. Ogunlaja, O. D. Olukanni, T. A. Msagati and E. I. Unuabonah, Sci. Total Environ., 2022, 814, 152448 CrossRef CAS PubMed.
- P. Wowkonowicz and M. Kijeńska, PLoS One, 2017, 12, e0174986 CrossRef PubMed.
- J. Hou, C. Wang, D. Mao and Y. Luo, Environ. Sci. Pollut. Res., 2016, 23, 1722–1731 CrossRef CAS PubMed.
- R. Wei, F. Ge, M. Chen and R. Wang, J. Environ. Qual., 2012, 41, 1481–1486 CrossRef CAS PubMed.
- S. Mohapatra, C.-H. Huang, S. Mukherji and L. P. Padhye, Chemosphere, 2016, 159, 526–535 CrossRef CAS PubMed.
- W.-J. Sim, J.-W. Lee, E.-S. Lee, S.-K. Shin, S.-R. Hwang and J.-E. Oh, Chemosphere, 2011, 82, 179–186 CrossRef CAS PubMed.
- A. Faleye, A. Adegoke, K. Ramluckan, J. Fick, F. Bux and T. Stenström, Sci. Total Environ., 2019, 678, 10–20 CrossRef CAS PubMed.
- Y. Zhang, S.-U. Geißen and C. Gal, Chemosphere, 2008, 73, 1151–1161 CrossRef CAS PubMed.
- G.-H. Choi, D.-Y. Lee, D.-K. Jeong, S. Kuppusamy, Y. B. Lee, B.-J. Park and J.-H. Kim, J. Integr. Agric., 2017, 16, 1841–1851 CrossRef CAS.
- J. Campo, M. Lorenzo, F. Pérez, Y. Picó, M. la Farré and D. Barceló, Environ. Res., 2016, 147, 503–512 CrossRef CAS PubMed.
- P. K. Mutiyar and A. K. Mittal, Environ. Sci. Pollut. Res., 2014, 21, 7723–7736 CrossRef CAS PubMed.
- P. Verlicchi, M. Al Aukidy, A. Galletti, M. Petrovic and D. Barceló, Sci. Total Environ., 2012, 430, 109–118 CrossRef CAS PubMed.
- M. Priyadarshini, A. Ahmad, S. Das and M. M. Ghangrekar, J. Environ. Chem. Eng., 2022, 108230 CrossRef CAS.
- Y. Deng and R. Zhao, Curr. Pollut. Rep., 2015, 1, 167–176 CrossRef CAS.
- C. A. Martinez-Huitle and S. Ferro, Chem. Soc. Rev., 2006, 35, 1324–1340 RSC.
- S. O. Ganiyu, C. A. Martínez-Huitle and M. A. Oturan, Curr. Opin. Electrochem., 2021, 27, 100678 CrossRef CAS.
- S. Garcia-Segura, J. D. Ocon and M. N. Chong, Process Saf. Environ. Prot., 2018, 113, 48–67 CrossRef CAS.
- A. Li, J. Weng, X. Yan, H. Li, H. Shi and X. Wu, J. Electroanal. Chem., 2021, 898, 115622 CrossRef CAS.
- C. A. Martínez-Huitle and M. Panizza, Curr. Opin. Electrochem., 2018, 11, 62–71 CrossRef.
- S. Rathod, Thesis, University of Waterloo, 2020 Search PubMed.
- M. Zhou, H. Särkkä and M. Sillanpää, Sep. Purif. Technol., 2011, 78, 290–297 CrossRef CAS.
- M. Priyadarshini, I. Das, M. M. Ghangrekar and L. Blaney, J. Environ. Manage., 2022, 316, 115295 CrossRef CAS PubMed.
- I. Sánchez-Montes, J. F. Pérez, C. Sáez, M. A. Rodrigo, P. Cañizares and J. M. Aquino, Chemosphere, 2020, 238, 124575 CrossRef PubMed.
- P. Nidheesh and R. Gandhimathi, Desalination, 2012, 299, 1–15 CrossRef CAS.
- H. He and Z. Zhou, Crit. Rev. Environ. Sci. Technol., 2017, 47, 2100–2131 CrossRef CAS.
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- A. Babuponnusami and K. Muthukumar, J. Environ. Chem. Eng., 2014, 2, 557–572 CrossRef CAS.
- F. Deng, J. Jiang and I. Sirés, Carbon Lett., 2022, 1–18 Search PubMed.
- W. S. Koe, J. W. Lee, W. C. Chong, Y. L. Pang and L. C. Sim, Environ. Sci. Pollut. Res., 2020, 27, 2522–2565 CrossRef CAS PubMed.
- K. Noguera-Oviedo and D. S. Aga, J. Hazard. Mater., 2016, 316, 242–251 CrossRef CAS PubMed.
- A. Tripathi, R. Raj, S. Sathe, R. Y. Surampalli and M. Ghangrekar, Clean Technol. Environ. Policy, 2023, 1–18 Search PubMed.
- Y. Chen, J. Yang, L. Zeng and M. Zhu, Crit. Rev. Environ. Sci. Technol., 2022, 52, 1401–1448 CrossRef CAS.
- M. Mehrjouei, S. Müller and D. Möller, Chem. Eng. J., 2015, 263, 209–219 CrossRef CAS.
- C. Regmi, B. Joshi, S. K. Ray, G. Gyawali and R. P. Pandey, Front. Chem., 2018, 6, 33 CrossRef PubMed.
- B. Srikanth, R. Goutham, R. B. Narayan, A. Ramprasath, K. Gopinath and A. Sankaranarayanan, J. Environ. Manage., 2017, 200, 60–78 CrossRef CAS PubMed.
- B. P. Chaplin, in Electrochemical Water and Wastewater Treatment, Elsevier, 2018, pp. 451–494 Search PubMed.
- Á. Anglada, A. Urtiaga, I. Ortiz, D. Mantzavinos and E. Diamadopoulos, Water Res., 2011, 45, 828–838 CrossRef PubMed.
- G. Divyapriya, S. Singh, C. A. Martínez-Huitle, J. Scaria, A. V. Karim and P. Nidheesh, Chemosphere, 2021, 276, 130188 CrossRef CAS PubMed.
- X. T. Mai, D. N. Bui, V. Pham, T. H. Pham, T. T. L. Nguyen, H. D. Chau and T. K. N. Tran, Inorganics, 2022, 10, 211 CrossRef CAS.
- X. Zhu and J. Ni, Electrochem. Commun., 2009, 11, 274–277 CrossRef CAS.
- S. Li, T. Hua, C.-S. Yuan, B. Li, X. Zhu and F. Li, Bioresour. Technol., 2020, 298, 122501 CrossRef CAS PubMed.
- P. Xu, H. Xu and Z. Shi, Sep. Purif. Technol., 2018, 194, 457–461 CrossRef CAS.
- S. Li, Y. Liu, R. Ge, S. Yang, Y. Zhai, T. Hua, B. S. Ondon, Q. Zhou and F. Li, Sci. Total Environ., 2020, 699, 134160 CrossRef CAS PubMed.
- S. S. Lim, J.-M. Fontmorin, P. Izadi, W. R. W. Daud, K. Scott and E. H. Yu, Int. J. Hydrogen Energy, 2020, 45, 2557–2568 CrossRef CAS.
- X. Li, X. Jin, N. Zhao, I. Angelidaki and Y. Zhang, Water Res., 2017, 119, 67–72 CrossRef CAS PubMed.
- Z. Yang, S. Wu, H. Sun, S. G. Arhin, V. G. Papadakis, M. A. Goula, G. Liu, Y. Zhang, L. Zhou and W. Wang, J. Environ. Manage., 2022, 319, 115719 CrossRef CAS PubMed.
- H. Nadais, X. H. Li, N. Alves, C. Couras, H. R. Andersen, I. Angelidaki and Y. F. Zhang, Chem. Eng. J., 2018, 338, 401–410 CrossRef CAS.
- G. Baek, L. Shi, R. Rossi and B. E. Logan, Chem. Eng. J., 2021, 405, 126742 CrossRef CAS.
- X. C. Meng, Z. H. Zhang and X. G. Li, J. Photochem. Photobiol., C, 2015, 24, 83–101 CrossRef CAS.
- H. Feng, Y. Liang, K. Guo, N. Li, D. Shen, Y. Cong, Y. Zhou, Y. Wang, M. Wang and Y. Long, Water Res., 2016, 102, 428–435 CrossRef CAS PubMed.
- C. Wang, G. Wu, X. Zhu, Y. Xing, X. Yuan and J. Qu, Chemosphere, 2022, 293, 133517 CrossRef CAS PubMed.
- Q. P. Chen, J. H. Li, X. J. Li, K. Huang, B. X. Zhou, W. M. Cai and W. F. Shangguan, Environ. Sci. Technol., 2012, 46, 11451–11458 CrossRef CAS PubMed.
- Y. Li, A. H. Lu, H. R. Ding, S. Jin, Y. H. Yan, C. Q. Wang, C. P. Zen and X. Wang, Electrochem. Commun., 2009, 11, 1496–1499 CrossRef CAS.
- N. Touach, V. M. Ortiz-Martinez, M. J. Salar-Garcia, A. Benzaouak, F. Hernandez-Fernandez, A. P. de Rios, M. El Mahi and E. M. Lotfi, Particuology, 2017, 34, 147–155 CrossRef CAS.
- S. M. Sathe, G. D. Bhowmick, B. K. Dubey and M. M. Ghangrekar, Bioprocess Biosyst. Eng., 2020, 43, 2075–2084 CrossRef CAS PubMed.
- H. X. Han, C. Shi, L. Yuan and G. P. Sheng, Appl. Energy, 2017, 204, 382–389 CrossRef CAS.
- X. Hu, J. Qin, Y. Wang, J. Wang, A. Yang, Y. F. Tsang and B. Liu, J. Colloid Interface Sci., 2022, 628, 327–337 CrossRef CAS PubMed.
- M. Zhang, Y. Wang, P. Liang, X. Zhao, M. Liang and B. Zhou, Chemosphere, 2019, 214, 669–678 CrossRef CAS PubMed.
- X. X. Mei, H. M. Wang, D. X. Hou, F. L. Lobo, D. F. Xing and Z. J. Ren, Front. Environ. Sci. Eng., 2019, 13, 1–7 CrossRef CAS.
- X. P. Zhu and B. E. Logan, J. Hazard. Mater., 2013, 252, 198–203 CrossRef PubMed.
- P. V. Nidheesh, S. O. Ganiyu, C. Kuppam, E. Mousset, N. Samsudeen, H. Olvera-Vargas and G. Kumar, J. Water Process. Eng., 2022, 50, 103232 CrossRef.
- Z. J. Wang, B. G. Zhang, A. G. L. Borthwick, C. P. Feng and J. R. Ni, Chem. Eng. J., 2015, 280, 99–105 CrossRef CAS.
- Y. Sun, S. A. Cheng, Z. F. Lin, J. W. Yang, C. C. Li and R. N. Gu, J. Hazard. Mater., 2020, 384, 121307 CrossRef CAS PubMed.
- J. Dziegielowski, B. Metcalfe, P. Villegas-Guzman, C. A. Martínez-Huitle, A. Gorayeb, J. Wenk and M. D. Lorenzo, Appl. Energy, 2020, 278, 115680 CrossRef CAS.
- A. Stasinakis, Global NEST J., 2008, 10, 376–385 Search PubMed.
- H. Yuan and Z. He, Bioresour. Technol., 2015, 195, 202–209 CrossRef CAS PubMed.
- M.-H. Cui, W.-Z. Liu, Z.-E. Tang and D. Cui, Water Res., 2021, 203, 117512 CrossRef CAS PubMed.
- S. Wang, A. Adekunle and V. Raghavan, J. Cleaner Prod., 2022, 366, 132855 CrossRef CAS.
- A. Ahmad, M. Priyadarshani, S. Das and M. M. Ghangrekar, J. Basic Microbiol., 2022, 62, 201–222 CrossRef CAS PubMed.
- S. Okabe, Chemosphere, 2021, 274, 129715 CrossRef PubMed.
- A. Buthiyappan, A. R. A. Aziz and W. M. A. W. Daud, Rev. Chem. Eng., 2015, 31, 263–302 CAS.
- M. T. Noori, G. D. Bhowmick, B. R. Tiwari, M. Ghangrekar and C. Mukhrejee, MRS Adv., 2018, 3, 663–668 CrossRef CAS.
- Y. Wang, K. Zhong, H. Li, Y. Dai, H. Zhang, J. Zuo, J. Yan, T. Xiao, X. Liu and Y. Lu, J. Power Sources, 2021, 485, 229273 CrossRef CAS.
- I. Das, S. Das and M. Ghangrekar, Chem. Phys. Lett., 2020, 751, 137536 CrossRef CAS.
- I. Das, M. Ghangrekar, R. Satyakam, P. Srivastava, S. Khan and H. Pandey, J. Hazard., Toxic Radioact. Waste, 2020, 24, 04020025 CrossRef CAS.
- K. Tota-Maharaj and P. Paul, Int. J. Energy Environ. Eng., 2015, 6, 213–220 CrossRef CAS.
- I. Gajda, O. Obata, M. J. Salar-Garcia, J. Greenman and I. A. Ieropoulos, Bioelectrochemistry, 2020, 133, 107459 CrossRef CAS PubMed.
- D. A. Jadhav, I. Das, M. M. Ghangrekar and D. Pant, J. Water Process Eng., 2020, 38, 101566 CrossRef.
- N. N. Mahamuni and Y. G. Adewuyi, Ultrason. Chem., 2010, 17, 990–1003 CAS.
- J. J. Fornero, M. Rosenbaum and L. T. Angenent, Electroanalysis, 2010, 22, 832–843 CrossRef CAS.
- C. Cui, L. Jin, Q. Han, K. Lin, S. Lu, D. Zhang and G. Cao, Sci. Total Environ., 2016, 572, 244–251 CrossRef CAS PubMed.
- H. Wen, H. Zhu, B. Yan, Y. Xu and B. Shutes, Chemosphere, 2020, 250, 126252 CrossRef CAS PubMed.
- G. David, M. Rana, S. Saxena, S. Sharma, D. Pant and S. Prajapati, Int. J. Environ. Sci. Technol., 2022, 1–22 Search PubMed.
- Y. Liang, H. Zhu, G. Bañuelos, B. Shutes, B. Yan and X. Cheng, Chem. Eng. J., 2018, 341, 462–470 CrossRef CAS.
- D. Wu, K. Xia, C. Fang, X. Chen and Y. Ye, Catalysts, 2020, 10, 138 CrossRef CAS.
- J. X. Wu, B. Wang, G. Cagnetta, J. Huang, Y. J. Wang, S. B. Deng and G. Yu, Sep. Purif. Technol., 2020, 239, 116534 CrossRef CAS.
- X. Yang, R. Zou, K. Tang, H. R. Andersen, I. Angelidaki and Y. Zhang, Sci. Total Environ., 2021, 771, 145385 CrossRef CAS PubMed.
- W. Xue, F. Li and Q. Zhou, Bioresour. Technol., 2019, 289, 121632 CrossRef CAS PubMed.
- Q. Wen, F. Kong, H. Zheng, D. Cao, Y. Ren and J. Yin, Chem. Eng. J., 2011, 168, 572–576 CrossRef CAS.
- Y.-S. Oon, S.-A. Ong, L.-N. Ho, Y.-S. Wong, Y.-L. Oon, H. K. Lehl, W.-E. Thung and N. Nordin, Chem. Eng. J., 2018, 344, 236–245 CrossRef CAS.
- W. Xu, B. Jin, S. Zhou, Y. Su and Y. Zhang, Energies, 2020, 13, 761 CrossRef CAS.
- H. Luo, J. Hu, L. Qu, G. Liu, R. Zhang, Y. Lu, J. Qi, J. Hu and C. Zeng, Sci. Total Environ., 2019, 674, 336–343 CrossRef CAS PubMed.
- Y. Gao, X. Kong, A. Zhou, X. Yue, Y. Luo and Z. Defemur, Bioresour. Technol., 2020, 306, 123077 CrossRef CAS PubMed.
- B. D. Gusseme, M. Soetaert, T. Hennebel, L. Vanhaecke, N. Boon and W. Verstraete, Microb. Biotechnol., 2012, 5, 396–402 CrossRef PubMed.
- B. De Gusseme, T. Hennebel, L. Vanhaecke, M. Soetaert, J. Desloover, K. Wille, K. Verbeken, W. Verstraete and N. Boon, Environ. Sci. Technol., 2011, 45, 5737–5745 CrossRef CAS PubMed.
- F. Soltani, N. Navidjouy, H. Khorsandi, M. Rahimnejad and S. Alizadeh, RSC Adv., 2021, 11, 27160–27173 RSC.
- S. M. Sathe, I. Chakraborty, M. M. Doki, B. K. Dubey and M. M. Ghangrekar, Environ. Res., 2022, 212(113141) CAS.
- R. Gautam, J. K. Nayak, K. N. Talapatra and U. K. Ghosh, Mater. Today: Proc., 2021, 80, 2225–2259 Search PubMed.
- R. Raj, S. Das, S. Das and M. M. Ghangrekar, Groundw. Sustain. Dev., 2023, 100905 CrossRef.
- M. G. Waller and T. A. Trabold, Am. Soc. Mech. Eng., 2013, 5522 Search PubMed.
- A. Fernandes, M. Pacheco, L. Ciríaco and A. Lopes, Appl. Catal., B, 2015, 176, 183–200 CrossRef.
- B. E. Logan, Nat. Rev. Microbiol., 2009, 7, 375–381 CrossRef CAS PubMed.
- C. Santoro, C. Arbizzani, B. Erable and I. Ieropoulos, J. Power Sources, 2017, 356, 225–244 CrossRef CAS PubMed.
- W. Yan, Y. Xiao, W. Yan, R. Ding, S. Wang and F. Zhao, Chem. Eng. J., 2019, 358, 1421–1437 CrossRef CAS.
- D. Deb, R. Patel and V. E. Balas, Processes, 2020, 8, 583 CrossRef CAS.
- K. Rabaey, L. Angenent, U. Schroder and J. Keller, Bioelectrochemical Systems, IWA publishing, 2009 Search PubMed.
- G. Hernández-Flores, H. Poggi-Varaldo, O. Solorza-Feria, M. P. Noyola, T. Romero-Castañón and N. Rinderknecht-Seijas, Int. J. Hydrogen Energy, 2015, 40, 17421–17432 CrossRef.
- S. Gadkari, S. Gu and J. Sadhukhan, Chem. Eng. J., 2018, 343, 303–316 CrossRef CAS.
- S. Varjani, Sci. Total Environ., 2022, 156691 CrossRef CAS PubMed.
- D. A. Jadhav, I. Das, M. M. Ghangrekar and D. Pant, J. Water Process. Eng., 2020, 38, 101566 CrossRef.
- H. Olvera-Vargas, N. Gore-Datar, O. Garcia-Rodriguez, S. Mutnuri and O. Lefebvre, Chem. Eng. J., 2021, 404, 126524 CrossRef CAS.
- M. Molinos-Senante, F. Hernández-Sancho and R. Sala-Garrido, Sci. Total Environ., 2010, 408, 4396–4402 CrossRef CAS PubMed.
- W. Lu, S. H. Lei, N. Chen and C. P. Feng, Chem. Eng. J., 2023, 451, 138746 CrossRef CAS.
- M. Hassan, H. Olvera-Vargas, X. P. Zhu, B. Zhang and Y. L. He, J. Power Sources, 2019, 424, 220–244 CrossRef CAS.
- M. Bagal, G. Kumbhar, S. Shukla, A. Tiwari, D. Gajbhiye and A. Mohod, Chem. Eng. Res. Des., 2022, 188, 315–329 CrossRef CAS.
- D. C. Pham, T. M. D. Cao, M. C. Nguyen, T. D. Nguyen, V. H. Nguyen, V. H. Bui and T. T. T. Nguyen, Chem. Eng. Technol., 2022, 45, 1748–1758 CrossRef CAS.
- M. Magdy, M. G. Alalm and H. K. El-Etriby, J. Cleaner Prod., 2021, 291, 125923 CrossRef CAS.
- D. Zhang, S. W. Hu, Z. Q. Cao, H. B. Cao, Y. H. Zhao and H. Zhao, Resour., Conserv. Recycl., 2022, 185, 106475 CrossRef CAS.
- M. Y. Chin, Z. X. Phuang, K. S. Woon, M. M. Hanafiah, Z. Zhang and X. Liu, J. Environ. Manage., 2022, 320, 115778 CrossRef CAS PubMed.
- D. Villaseñor-Basulto, A. Picos-Benítez, N. Bravo-Yumi, T. Perez-Segura, E. R. Bandala and J. M. Peralta-Hernández, J. Electroanal. Chem., 2021, 895, 115492 CrossRef.
- J. An, Y. Feng, N. Wang, Q. Zhao, X. Wang and N. Li, J. Hazard. Mater., 2022, 428, 128185 CrossRef CAS PubMed.
- A. Anglada, D. Ortiz, A. M. Urtiaga and I. Ortiz, Water Sci. Technol., 2010, 61, 2211–2217 CrossRef CAS PubMed.
- H. Monteil, Y. Pechaud, N. Oturan, C. Trellu and M. A. Oturan, Chem. Eng. J., 2021, 404, 127048 CrossRef CAS.
- J. C. Espindola, M. Caianelo, N. Scaccia, C. Rodrigues-Silva, J. R. Guimaraes and V. J. P. Vilar, J. Environ. Chem. Eng., 2021, 9, 105060 CrossRef CAS.
- L. X. Yang, M. K. Li, W. T. Li, Y. J. Jiang and Z. M. Qiang, Chem. Eng. J., 2018, 342, 155–162 CrossRef CAS.
- R. Zou, I. Angelidaki, B. Jin and Y. Zhang, Environ. Int., 2020, 134, 105352 CrossRef CAS PubMed.
- R. Zou, I. Angelidaki, X. Yang, K. Tang, H. R. Andersen and Y. Zhang, Sci. Total Environ., 2020, 727, 138684 CrossRef CAS PubMed.
- A. Anglada, A. M. Urtiaga and I. Ortiz, J. Hazard. Mater., 2010, 181, 729–735 CrossRef CAS PubMed.
- C. R. Wang, M. R. Zhang, M. Ye, J. Wang and G. W. Li, J. Chem. Technol. Biotechnol., 2014, 89, 1599–1606 CrossRef CAS.
- A. K. Benekos, C. Zampeta, R. Argyriou, C. N. Economou, I. E. Triantaphyllidou, T. I. Tatoulis, A. G. Tekerlekopoulou and D. V. Vayenas, Process Saf. Environ., 2019, 131, 38–47 CrossRef CAS.
- S. R. S. Bandaru, A. Roy, A. J. Gadgil and C. M. van Genuchten, Water Res., 2020, 175, 115668 CrossRef CAS PubMed.
- J. R. Steter, E. Brillas and I. Sires, Appl Catal B-Environ, 2018, 224, 410–418 CrossRef CAS.
- J. Casado, J. Environ. Chem. Eng., 2019, 7, 102823 CrossRef CAS.
- J. Sim, R. Reid, A. Hussain, J. An and H. S. Lee, Biotechnol. Rep., 2018, 19, e00276 CrossRef PubMed.
- I. Salmerón, I. Oller, K. Plakas and S. Malato, Chemosphere, 2021, 275, 129962 CrossRef PubMed.
- V. Poza-Nogueiras, A. Moratalla, M. Pazos, A. Sanroman, C. Saez and M. A. Rodrigo, J. Electroanal. Chem., 2021, 895, 115475 CrossRef CAS.
- Y. Tong, J. Wei, R. Mo, H. Ma and F. Ai, Front. Chem., 2022, 10, 953434 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |