
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Circular valorization of coffee silverskin through supercritical CO2 for the production of functional extracts†
Stefania
Marzorati
 *a,
Amparo
Jiménez-Quero
b,
Alessio
Massironi
a,
Rita
Nasti
a and
Luisella
Verotta
a
*a,
Amparo
Jiménez-Quero
b,
Alessio
Massironi
a,
Rita
Nasti
a and
Luisella
Verotta
a
aDepartment of Environmental Science and Policy, Università degli Studi di Milano, Via Celoria 2, 20133, Milano, Italy. E-mail: stefania.marzorati@unimi.it
bDepartment of Chemistry, Division of Glycoscience, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Alba Nova University Centre, Roslagstullsbacken 21, 114 21, Stockholm, Sweden
First published on 8th March 2023
Abstract
The development of sustainable procedures for the valorization of industrial biomass waste represents a major challenge for the scientific community. Among the residual biomasses, coffee silverskin could represent an attractive source of bioactive compounds. In spite of its promising applications, silverskin is still underutilized and nowadays is still discarded by roasters as solid waste in landfill. This work presents the application of a green unconventional extraction strategy, supercritical CO2, with a particular focus on the extractability of polar compounds from green coffee beans and their related wasted biomass, coffee silverskin, by means of the addition of ethanol as a co-solvent, allowing us to shift the window of extractables towards more polar molecules. The results point out the ability of the supercritical fluid to efficiently target chlorogenic acids, minimizing the excessive use of organic solvents and avoiding the presence of inorganic or organic acids. The use of a co-solvent, at the expense of reducing the selectivity towards target species, was demonstrated to efficiently co-extract other classes of compounds directly connected to the relevance of silverskin's phytochemical profile, dealing mainly with its antioxidant and prebiotic properties, pointing at interesting applications in functional food products.
Sustainability spotlightIn the context of circular economy, the agri-food chain represents the sector in which there is the highest production of wasted biomasses with a direct strong potential to be further investigated. This, joint with the growing interest in the nutraceutical industry that is continuously raising, pushes research to seek techniques to extract bioactive compounds starting from these wastes. Our research merges the valorization of a biomass residual, coffee silverskin, responsible for the production of 2400 tons of waste per year only in Italy, with the development of a green and sustainable technique, supercritical CO2, to target its polar fraction to be exploited in the nutraceutical field. This approach is perfectly aligned with the following UN sustainable development goals: responsible consumption and production (SDG 12), industry, innovation, and infrastructure (SDG 9) and climate action (SDG 13). |
Introduction
Biomass valorization constitutes a key aspect of the circular economy and an opportunity to unlock the full potential of the bioeconomy and enhance the possible synergies between different sectors. Within circular models, agriculture and in particular the agri-food chain represent the sector in which there is the highest production of waste biomass with a direct strong potential to be further investigated.1 This, together with a growing interest in the nutraceutical industry, has led to research to find new bioactive molecules and techniques to extract and isolate them from these wastes. Among the food-chain-related wastes, plant residuals could represent an attractive source of more sustainable bioactive compounds for the nutraceutical and/or functional foods sector.2The exploitation of byproducts towards the generation of added-value compounds should meet and merge the need to develop alternative and innovative extraction techniques that are able to guarantee the sustainability of the entire process, from biomass selection to the technological methods to final added-value generation. In this context, unconventional extraction techniques are promising approaches able to enhance compounds recovery in a greener way compared to conventional solvent-based technologies. Among them, subcritical water and supercritical carbon dioxide extraction are attracting more and more interest. These extraction techniques are efficient, economical, and promising routes for the recovery of bioactives from different matrices. When the target compounds could undergo thermal degradation, supercritical CO2 (sc-CO2) might be a preferable choice, in order to maintain all the antioxidant profiles in the extracts.3,4 The sc-CO2 technology, employing CO2 as extraction fluid is indeed characterized by low environmental impacts, the use of organic solvents can be avoided or minimized, ensuring safer and selective processes directly on the biomass without any pretreatment, with the possibility to recycle the employed CO2 in industrial plants.5 Following these global trends, where the interest in “green” products and technologies is growing, this work presents as a core strategy a supercritical CO2 extraction methodology with a particular focus on the extractability of polar compounds. Since sc-CO2 alone displays a solvating power similar to n-hexane, most of the literature is focused on the primary and direct use of the supercritical fluid to target the lipidic fraction and, more in general, non-polar compounds.6 On the other hand, the addition of a co-solvent in a relatively low ratio enables the modification of the supercritical fluid polarity, allowing the shifting of the window of extractables towards more polar molecules.7 This approach has already been discussed in the literature; however, it is still underdeveloped, probably due to the loss of certain selectivity attributable to the presence of a co-solvent.8,9 Nevertheless, if the loss of selectivity is evaluated together with an investigation of the co-extracted compounds and their potential added value, a sustainable platform might be designed, in a more complete biorefinery concept, where the effect of co-extraction has an interesting economical outcome from the process perspective. Other molecules, apart from the targeted ones, could be beneficial towards specific applications in particular in the nutraceutical field, where they could have great potential as prebiotics or as building blocks for functional materials.10–12
In this wide context, this work presents for the first time the valorization of the polar fraction from coffee silverskin by means of supercritical CO2 in the presence of a co-solvent. Silverskin, the thin layer directly in contact with the coffee bean removed during the roasting process, representing about 4.2% (w/w) of coffee beans and accounting for a production of 2400 tons of waste produced per year only in Italy, was selected as an interesting case study together with its related original biomass, green coffee beans, as the term of comparison.13 In spite of its promising applications, silverskin is still underutilized and nowadays is still discarded by the roasters as solid waste in landfill. To date, the exploitation of silverskin has been focused mainly on its lipidic content, explored also by means of environmentally-friendly techniques such as sc-CO2, but resulting in the loss of its high-value polar content14,15. This latter connects with the relevance of silverskin's phytochemical profile, dealing mainly with its antioxidant and prebiotic properties, mostly related to the presence of phenolics and, in particular, chlorogenic acids.16–18 To the best of our knowledge, no investigations are present in the literature related to the extraction of phenolics from coffee silverskin by means of a green extraction strategy such as supercritical CO2. In addition, the aim of the present work was to study the effect of co-solvent addition to the supercritical fluid and investigate the presence and nature of the co-extracted compounds.
Materials and methods
Reagents and starting biomasses
All the chemicals were used without further treatment. Acetonitrile and water, when used for liquid chromatography, were purchased from Sigma-Aldrich (Italy) as ultra-performance liquid chromatography grade. Formic acid, trifluoracetic acid, ethanol, n-hexane, caffeine (powder, ReagentPlus®), and 5-caffeoylquinic acid (≥95%) standards were purchased from Sigma-Aldrich (Italy). A carbon dioxide tube (CO2 purity 4.5) was purchased from Sapio (Italy). Chemicals, p-coumaric, caffeic, and ferulic acid standards were purchased from Sigma-Aldrich (SE).Green coffee beans (GCB) of Robusta species from Vietnam and micronized coffee silverskin (CS) were kindly donated by an Italian roasting company (Ideal Caffè snc, Verderio) and then pulverized using a knife blender at its maximum velocity for 3 min.
Delipidization of GCB and CS
The extraction yield of lipids was calculated as: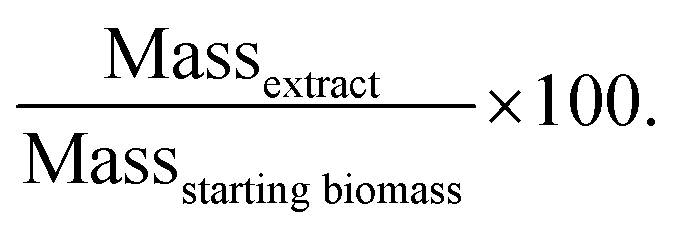
Three replicates were performed for each experiment.
Following the results from our previous work on coffee silverskin,19 the operative pressure was set at 300 bar in all experiments, while the temperature of the vessel and the temperature of the restrictor block were maintained at 60 °C and 75 °C, respectively, throughout the extraction period. For each extraction experiment, many cycles, each one comprising 30 min of maceration time in static conditions and 10 min of dynamic conditions, were performed until no evident extracted mass was further gained. Under dynamic conditions, the valves were opened and the extract was collected in a vial, keeping a CO2 gas constant flow rate of 8 SCFH (standard cubic feet per hour). In the case of the lab-scale supercritical fluid extractor, the duration of static and dynamic cycles (and hence the final number of cycles) was optimized on the basis of the need for a specific soaking time under static conditions before opening the valves to run the dynamic process to collect the extract. The extracts were then stored at −20 °C for subsequent analysis, whilst the defatted biomasses were collected from the vessel and subjected to further experiments.
Chlorogenic acid extraction
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol (1:1) mixture was injected inside the vessel, as a result of preliminary trials aimed to assess the best co-solvent mixture. The operative supercritical pressure was set at 300 bar, while the temperature of the vessel and the temperature of the restrictor block were maintained at 60 °C and 75 °C, respectively, throughout the extraction period. A static maceration 30 min period was maintained at the beginning, followed by a dynamic period of 1 hour maintaining an ethanol flow rate of 0.5 mL min−1 (corresponding to 0.25% v/v of the CO2 amount) with a CO2 gas flow rate constant at 8 SCFH (standard cubic feet per hour). After this first cycle, many cycles, comprising 15 min of maceration time in static conditions and 1 hour of dynamic conditions with co-solvent addition, were performed. The extracted solutions were collected and ethanol was evaporated using a rotary evaporator (Buchi) and then under a vacuum pump.
:ethanol = 7:3 (+1% formic acid). The suspension was magnetically stirred at room temperature for 4 hours, centrifuged at 6000 rpm for 5 min, and then filtered on a Buchner funnel. The solvent was evaporated using a rotary evaporator (Buchi) and then under a vacuum pump.
:ethanol (1:1) mixture was injected inside the vessel, as a result of preliminary trials aimed to assess the best co-solvent mixture. The operative supercritical pressure was set at 300 bar, while the temperature of the vessel and the temperature of the restrictor block were maintained at 60 °C and 75 °C, respectively, throughout the extraction period. A static maceration 30 min period was maintained at the beginning, followed by a dynamic period of 1 hour maintaining an ethanol flow rate of 0.5 mL min−1 (corresponding to 0.25% v/v of the CO2 amount) with a CO2 gas flow rate constant at 8 SCFH (standard cubic feet per hour). After this first cycle, many cycles, comprising 15 min of maceration time in static conditions and 1 hour of dynamic conditions with co-solvent addition, were performed. The extracted solutions were collected and ethanol was evaporated using a rotary evaporator (Buchi) and then under a vacuum pump.
:ethanol = 7:3 (+1% formic acid). The suspension was magnetically stirred at room temperature for 4 hours, centrifuged at 6000 rpm for 5 min, and then filtered on a Buchner funnel. The solvent was evaporated using a rotary evaporator (Buchi) and then under a vacuum pump.
Characterization of the extracts
Before sample injections, six dilutions of 5-caffeoylquinic acid (5-CQA) aqueous solution (515 μg mL−1) and six dilutions of caffeine aqueous solution (220 μg mL−1) were prepared in the range 5.15–515 μg mL−1 for 5-CQA and in the range 2.2–220 μg mL−1 for caffeine. Standard solutions were filtered (0.2 μm polypropylene filters) and injected three times in the UPLC system. 5-CQA and caffeine were eluted as sharp peaks. In the operative concentration range, the trend was linear, with no saturation effects that could bend the linearity.
The area under each peak was quantified using the instrumental software and plotted versus the concentration. The best fit of experimental data in the plot “peak area at 324 nm vs. [standard concentration]” was then used for 5-CQA and caffeine quantification in each extract.
The best fit of experimental data in the plot of “A324nmvs. [5-CQA]” was a straight-line represented by the following mathematical equation: y = 6.1 × 107x − 3 × 105, resulting in an R2 equal to 0.996. The slope m and intercept q of the regression line with their respective standard deviations were (6.1 × 107 ± 2 × 106) and (−3 × 105 ± 4 × 105), respectively. The calculated equation of the regression line was then employed to determine the 5-CQA concentration in each extract.
The best fit of the experimental data in the plot of “A324nmvs. [caffeine]” was a straight-line, represented by the following mathematical equation: y = 4.91 × 107x + 7 × 104, resulting in an R2 equal to 0.999. The slope m and intercept q of the regression line with their respective standard deviations were (4.91 × 107 ± 8 × 105) and (7 × 104 ± 9 × 104), respectively. The calculated equation of the regression line was then employed to determine the caffeine concentration in each extract.
When necessary, chromatographic separation was followed by mass spectrometry (LCQ Fleet Thermofisher) analysis. A negative electrospray mode was used for the ionization of molecules with a capillary voltage of −42 V, at a capillary temperature of 275 °C. The heater temperature was set at 150 °C, the gas flow rate was 20 (arb) and the spray voltage was 5 kV. The monitored mass range was from m/z 50 to 1000. Before the sample injection, mass spectrometry parameters were optimized using a commercial standard of 5-caffeoyquinic acid.
:1, v/v), filtered through Chromacol 0.2 μm filters (Scantec Nordic, Sweden), and analyzed on the HPLC using the ZORBAX StableBond C18 column (Agilent Technologies, USA) fitted with photodiode array detection at 200–400 nm (Waters 2695, MA, USA). Samples that were not saponified were diluted with methanol 2% (v/v) acetic acid (1:1, v/v), filtered through Chromacol 0.2 μm filters (Scantec Nordic, Sweden), and subjected to the HPLC for analysis. The phenolic acids were separated in accordance with a previous method using p-coumaric, caffeic, and ferulic acid as standards.26
Results and discussion
Biomasses milling
One of the most critical operating parameters affecting the extraction yield is particle size. The solid matrix must be blended to increase the mass transfer area. Enhancing the exposed surface area to the solvent makes the extractions more effective in terms of yield, facilitating the mass transfers. Solutes can be in fact located on the surface of the cells of the vegetable matrix or in intracellular spaces. As the particle size of the sample decreases, the extracted solute would travel shorter diffusional paths along the solid substrate, and mass transfer barriers, such as cell walls, would be destroyed. However, it was also reported by Coats et al. that some adverse hydrodynamic effects may occur when excessively small particles are treated in a packed bed.28,29 Particle size is hence considered a critical parameter, influencing the yield of extraction processes.30Coffee silverskin (CS) was provided as already micronized in the form of fine powder. Optical microscopy assessed an average particle size of about 100 μm. Green coffee beans (GCB) were subjected to blending before extraction. Due to the high content of lipids, the GCB fatty biomass could not be transformed into a fine powder as silverskin, not even by embrittlement by means of liquid nitrogen before blending. In this case, the minimum achievable particle size was in the range of 500–1000 μm, as detected by an optical microscope. GCB particle size was in any case in the range of recommendations found in the literature.30 Pictures of the pristine and blended biomasses are shown in the ESI in Fig. S1.†
Delipidization extraction yields and kinetics
The lipid content of CS was already reported in a recent publication.19 For the purpose of this work, a preliminary delipidization of both biomasses was performed in order to ease the successive extraction of the target polar compounds. The presence of excessive lipids would hinder the accessibility of the supercritical solvent (and the co-solvent) towards the generation of the necessary interface between the fluid and the solutes. The presence of lipids in the biomasses could be responsible for a certain hindrance in the targeted polar compounds extraction, rendering the biomass less prone to be penetrated by the extraction fluid. Moreover, lipids could contaminate the extracts later obtained in sc-CO2 in the presence of the co-solvent.The lipid content was determined by conventional solvent-based procedures by triplicate trials, corresponding to 5.0 ± 0.5% and 1.4 ± 0.1% (w/w on dry biomass) for GCB and CS samples, respectively.
When the extraction was performed in supercritical CO2, the optimization of process parameters followed the results from the previous work by Nasti et al., set at 60 °C and 300 bar. Cycle by cycle, with an alternation of static and dynamic periods, an incremental mass of extract was obtained from both GCB and CS, and the extraction process was maintained active until no evident mass gain was observed, thus corresponding to the maximum extractable yield. These values corresponded to 6.2 ± 0.1% and 1.4 ± 0.2% for GCB and CS, respectively. It was expectable that these yield values are comparable to the corresponding numbers obtained by conventional extraction. A slight difference was observed for GCB. The lipids yield obtained by means of sc-CO2 was 20% higher than the conventional extract. This can be attributed to the impossibility of finely micronizing GCB, hence making the biomass-hexane solid–liquid interface contact less effective, due to a lower exposed surface area. The penetration inside the porosity of the supercritical fluid, partially behaving like a gas in terms of permeability, was, on the other hand more effective, reaching pores inaccessible to hexane, thus enhancing the mass transfer and hence the yield.31
sc-CO2 delipidization extraction kinetics are displayed in Fig. 1. Both extractions follow the model explained by da Silva et al., primarily based on two mass transfer mechanisms: solubility and diffusion.32 The model assumes two district modes of extraction: initial constant rate extraction that is controlled by solubility, and falling rate extraction that is controlled by diffusivity. An initial period of extraction (the steepest, represented by the first four points in each dataset) corresponds to the depletion of lipids found on the particle surface, while the final period represents the extraction from inside the porosity, where the mass transfer is more hindered and the extraction rate slows down until it zeroes.32,33 For the purpose of this work, during the delipidization process, the extraction was interrupted after 15/16 cycles when a plateau was reached, meaning that no more lipids could be obtained in the process.
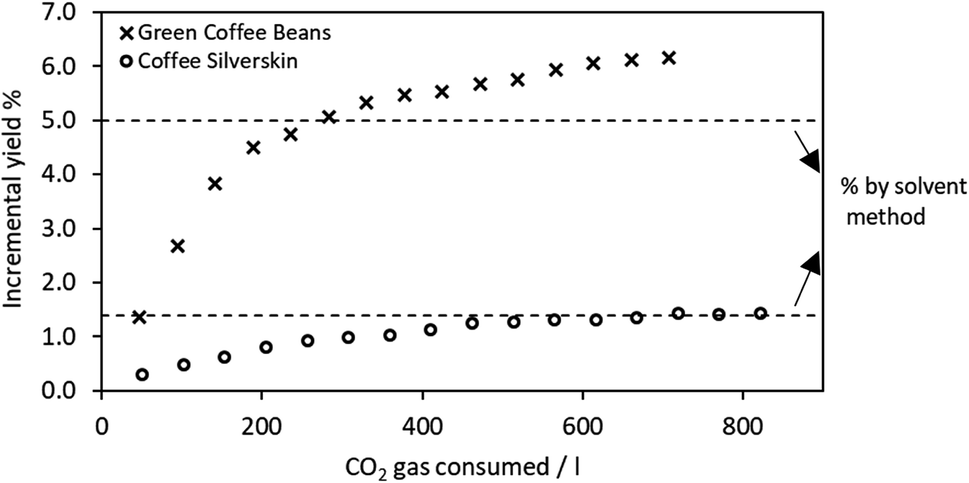 | ||
| Fig. 1 Extraction kinetics (incremental yield vs. CO2 utilization) during supercritical delipidization of GCB and CS. Dashed lines correspond to extraction yields obtained by the solvent method. | ||
All the extracts obtained in the delipidization process were subjected to UPLC analysis, and none of them displayed evidence of chlorogenic acid presence. The details on CS lipidic extract composition have been reported in a recent paper by Nasti et al.19
Chlorogenic acid extraction
Chlorogenic acids, due to their well-known antioxidant and interesting biological activities, were considered the main target compounds adopting supercritical fluid in the presence of a co-solvent.20,34 For comparison purposes, a solvent-based extraction was carried out employing a literature method with the aim of effectively extracting all the chlorogenic acid content from the delipidized biomasses. In the conventional extraction, a 1:10 solid-to-liquid ratio was employed since it represents a good compromise between not exceeding the amount of solvent and guaranteeing an exhaustive extraction of targeted compounds.
The first three sc-CO2 extracts obtained in the presence of co-solvent from GCB delipidized biomass and the first five extracts obtained in the presence of co-solvent from delipidized CS biomass were all similar to samples obtained during delipidization and almost completely insoluble in water. After the mentioned cycles, successive extracts displayed an almost complete water-solubility.
Fig. 2 displays the chromatograms recorded at 324 nm (maximum absorbance for chlorogenic acids) for the solvent-based extracts and a selection of representative samples from sc-CO2 extraction cycles.
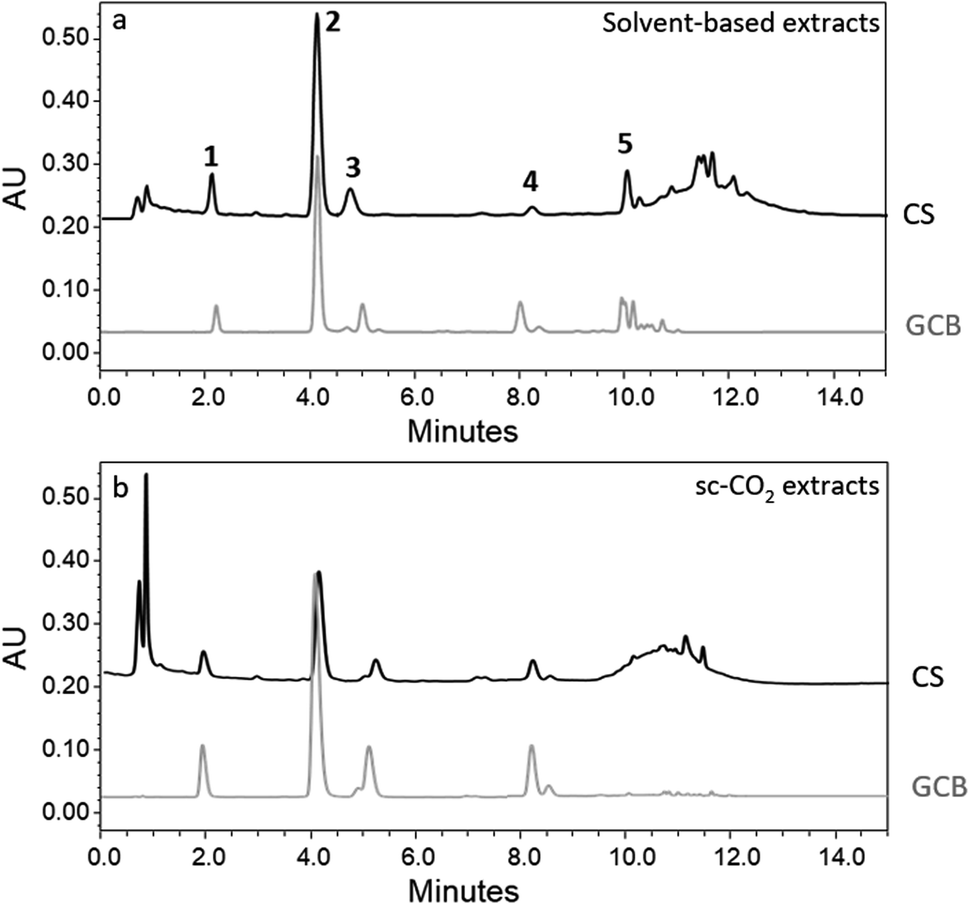 | ||
| Fig. 2 UPLC-UV chromatographic profile monitored at 324 nm of: (a) conventional solvent-based extracts, (b) sc-CO2 extracts. Peaks assignments: (1) 3-CQA, (2) 5-CQA, (3) 4-CQA, (4) FQA, (5) di-CQA. | ||
The UPLC data show an almost complete overlap of the chromatograms at 324 nm, corresponding to the maximum absorption of chlorogenic acids, relative to all four samples, irrespective of their extraction method. This leads to the conclusion that sc-CO2 in the presence of the co-solvent was effective in the extraction of chlorogenic acids to the same extent as the solvent-based method, from a qualitative point of view. Some slight differences are detected in the last part of the chromatogram, after 10 min as the retention time, where some small peaks are present in all chromatograms, differing from sample to sample. Due to the low area of the corresponding peaks, these differences were not further investigated, but tentative attributions to ubiquitary flavonoids such as quercetin and kaempferol could be carried out on the basis of the work by Nzekoue et al.35
The UPLC-UV chromatogram shows a main peak characterized by a retention time of 4.1 min (peak 2 in Fig. 2) attributable to 5-CQA, the most abundant chlorogenic acid in coffee, as confirmed by co-injection of the commercial standard and further mass spectrometry results.
The assignments of the other peaks in the chromatograms were confirmed by mass spectrometry. Peaks 1, 2, and 3 were attributed to 3-caffeoyquinic acid (3-CQA), 5-caffeoyquinic acid (5-CQA) and 4-caffeoyquinic acid (4-CQA), respectively, and all were characterized by the presence of the molecular ion signal at m/z = 353.5 corresponding to [M–H]−. Isomers were recognized on the basis of the literature data.36 Peak 4 was attributed to feruloylquinic acid (FQA), characterized by the presence of the molecular ion signal at m/z = 367.7 corresponding to [M–H]−. Peak 5 was assigned to dicaffeoylquinic acids (di-CQA), due to the presence of the molecular ion signal at m/z = 515.4, corresponding to [M–H]−, with no further investigation on the identity of the specific isomers.
Extraction results are consistent with the literature, where a previous work on the coffee silverskin extract obtained by acidified water (1% aqueous formic acid) at 70 °C for 1 h reported the presence of 3-CQA, 4-CQA, 5-CQA, and feruloylquinic acids.37
Supercritical CO2 has already been employed in the literature targeting phenolics from green coffee beans. De Azevedo employed ethanol and isopropyl alcohol as co-solvents in temperature ranges similar to this work, concluding that chlorogenic acids were extracted only from coffee beans when isopropyl alcohol was used. However, they evaluated chlorogenic acid extraction efficiency as poor, explained by the competition between these molecules and more polar components present in the coffee beans (e.g., sugars) and by the inability of the solvents to break down the caffeine–chlorogenic acid interactions.38 In this work, the presence of an enhanced possibility of hydrogen bonding due to the presence of an ethanol/water co-solvent mixture, could have resulted in an increased competition and hence in breaking the hydrogen bonds between caffeine and chlorogenic acid.
Some other coffee by-products were also investigated targeting the chlorogenic acid by sc-CO2, such as spent coffee grounds and coffee husks for valorization purposes. 15% of ethanol as co-solvent was the best choice, providing higher recovery of chlorogenic acids in supercritical conditions.39
Concerning silverskin, to the best of our knowledge, no investigations have been reported in the literature related to chlorogenic acid extraction from this biomass by means of supercritical CO2.
After the identification of the main phenolics, the most abundant chlorogenic acid in the samples, 5-CQA, was quantified.
As reported in Table 1, 5-CQA was quantified as 4.4% of green coffee beans and 0.29% of silverskin, when the extraction was run by means of a conventional solvent-based strategy. Results for green coffee beans are consistent with the literature, depending on the plant species, harvest time, and geographical origin of the plant. 2.7–6.5% of 5-CQA was reported by Ky et al. employing GCB of species C. liberica var. dewevrei from the Ivory Coast.40 Jeszka-Skowron et al. found chlorogenic acid content varying from 3.41% per dry mass in Arabica type from Laos or Rwanda to 8.16% in Robusta coffee from Indonesia.41
| Sample name | Starting biomass | Extraction method | 5-CGA (on dry biomass) (%) | 5-CGA (in extract) (%) |
|---|---|---|---|---|
| a The concentration of 5-CQA changes in each extract from the different successive sc-CO2 cycles. In the table are reported the highest values, obtained during the 11th sc-CO2 cycle for GCB and during the 6th sc-CO2 cycle for CS. | ||||
| GCB-solv | GCB | Solvent | 4.4 | 11.7 |
| CS-solv | CS | Solvent | 0.29 | 2.1 |
| GCB-scCO2 | GCB | sc-CO2 + co-solvent | 3.5 | 16.8a |
| CS-scCO2 | CS | sc-CO2 + co-solvent | 0.37 | 33.9a |
The content of chlorogenic acid quantified in coffee silverskin is also consistent with the literature. In particular, 5-CQA was extracted at 0.1–0.2% (w/w dry matter) from CS in the temperature range of 25–180 °C by Narita et al.42 Bresciani et al. reported similar content of chlorogenic acid, with an abundance of circa. 0.2% of 5-CQA in the dry biomass.43 Subcritical water was also employed by Ginting et al., targeting chlorogenic acids, reaching a percentage of 0.27% (w/w dry CS) at 147.8 °C.44
The solvent-based extraction was supposed to be exhaustive in chlorogenic acids' complete extraction from both the delipidized biomasses. After assessing for consistency the effectiveness of the extraction of chlorogenic acids from GCB and CS with solvents with the literature, data were assumed to represent the reference when studying the recovery of the same phenolics by means of supercritical fluids.
5-CQA was then quantified in all extracts deriving from supercritical extraction and results were compared to the solvent-based extraction. When the water solubility of the sc-CO2-samples was low (first extracts obtained during the initial cycles in sc-CO2), they were first dissolved in dichloromethane and then counter-extracted in water. Only the water phase was injected in UPLC.
In terms of 5-CQA recovery, the results are displayed in Fig. 3.
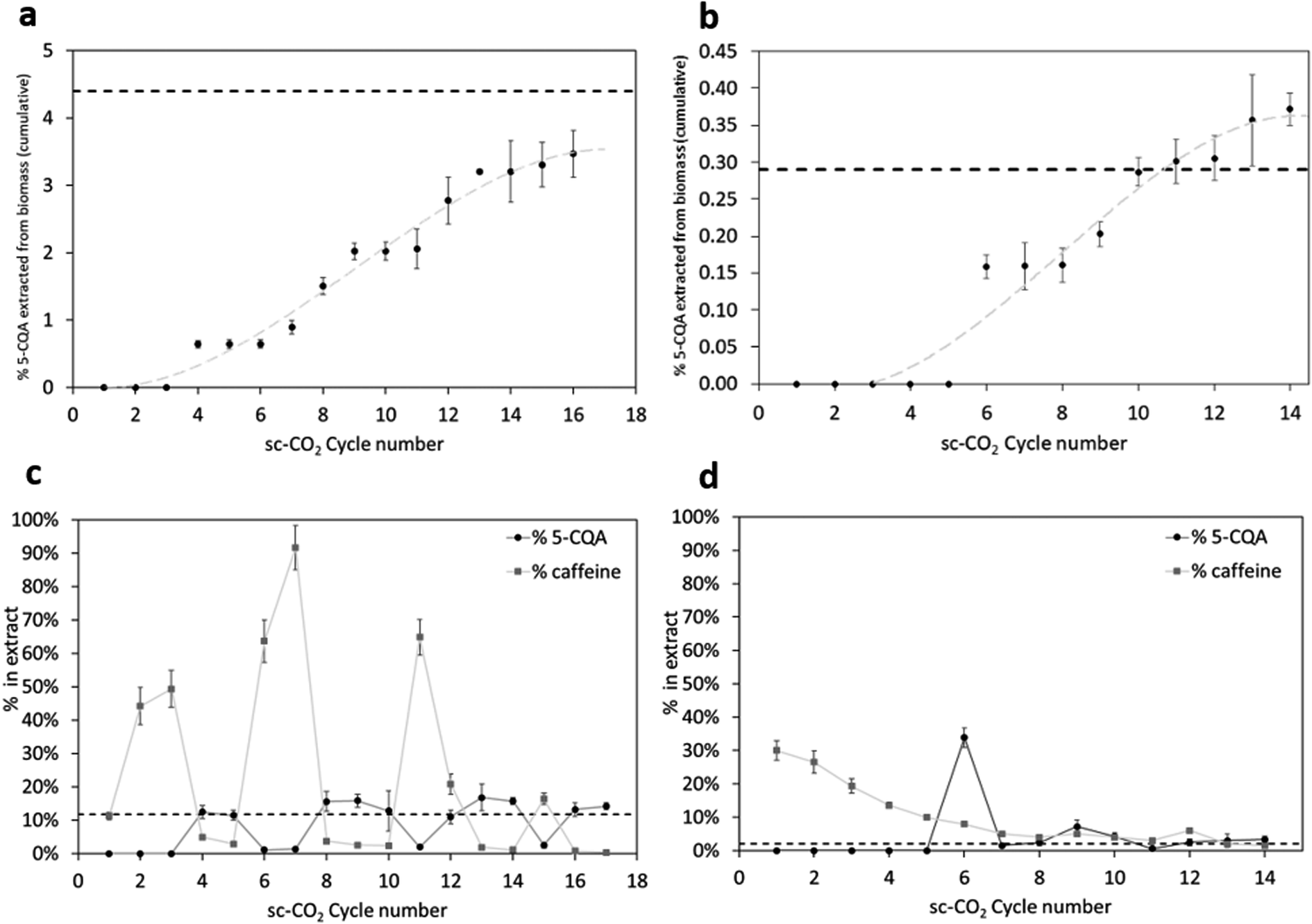 | ||
| Fig. 3 Extraction kinetics of 5-CQA (incremental yield on dry biomass vs. progressive extraction cycle) from (a) delipidized GCB and (b) delipidized CS. % of 5-CQA and % of caffeine in each extract from (c) delipidized GCB and (d) delipidized CS. Dashed lines represent the % of 5-CQA obtained by solvent-method. | ||
Fig. 3a and b show the % of the cumulative yield of 5-CQA (on dry biomass) extracted over time, for each cycle of sc-CO2 in the presence of the co-solvent. The dashed lines represent the % of 5-CQA (on dry biomass) extracted by means of the solvent method. The extractions of 5-CQA from both GCB and CS follow similar kinetics, with an initial absence of chlorogenic acids in the samples, then following the model again by da Silva et al. with a steep extraction rate followed by a lower extraction rate, where the mass transfer occurs mainly by diffusion inside the solid particles.32 The initial period where no 5-CQA is detected in the samples can be explained by the wetting of the biomass by the co-solvent being too low, rendering it unable to drag out (even with the aid of the gas flow) affine compounds, such as chlorogenic acids, in terms of polarity.
For GCB (Fig. 3a), the first three cycles were ineffective in 5-CQA extraction, whilst, from the 4th cycle onward, 5-CQA was positively found in the extracts. After the 16th cycle, 3.5% of 5-CQA (on dry biomass) was reached. This value is slightly lower, but comparable, to the % of 5-CQA obtained by solvent-based extraction, equal to 4.4%. The extraction was not continued further, due to the amount of co-solvent already used. Further use of the organic solvent, higher than 10 times the mass of the solid to be extracted, would not be reasonable when applying a “green technique” such as supercritical fluid extraction. Regarding chlorogenic acid recovery from CS extraction, as shown in Fig. 3b, the first five cycles provided extracts in which 5-CQA was absent. Its extraction began with the 6th cycle. From the 11th cycle onward, the amount of extracted 5-CQA was even higher than that obtained by means of a solvent strategy, reaching a maximum extraction yield of 0.37% (the content of 5-CQA in CS when the extraction was run in conventional conditions was 0.29%). The micronization of the sample, together with the enhanced penetrability of the fluid mixture in supercritical conditions, might have facilitated the mass transfer process of chlorogenic acids. For the same reason as above (not to exceed the logical amount of co-solvent employed), the extraction was stopped on the 14th cycle.
Besides the 5-CQA recovery, it was also interesting to quantify, cycle by cycle during the sc-CO2 extraction process, the 5-CQA content of each extract, as a necessary dataset to study how the selectivity towards chlorogenic acids varies over the time of sc-CO2 extraction. Explicative data are displayed in Table 1 in the last column, where it appears evident that supercritical extracts are enriched in chlorogenic acids more than the solvent-based samples, confirming the increased selectivity of the sc-CO2 technique. GCB solvent-based extract contains only 11.7% of 5-CQA, while one of the extracts obtained in sc-CO2 reaches 16.8%. Results are even more rewarding when considering coffee silverskin: solvent-based extract contains only 2.1% of 5-CQA, while an sc-CO2 extract reaches 33.9%.
Apart from these data, relative to the most rewarding sc-CO2 extracts (in terms of 5-CQA content), the % of 5-CQA in each extract changes during the extraction kinetics under supercritical conditions. Fig. 3c and d are explicative. The dashed lines represent the 5-CQA enrichment in the conventional extracts, while each point of the plot represents the sc-CO2 samples' enrichment in terms of 5-CQA content. For GCB, the trend is oscillating. This might be explained by the non-micronized biomass particles hindering the mass transfer processes and hence releasing phenolics in a non-linear way, being trapped inside different channels' sizes and morphologies. Anyhow, most of the extracted samples display enhanced 5-CQA content compared to the solvent-based extract, with most of the points above the dashed line. Interestingly, the caffeine content follows a complementary behavior: samples characterized by a high content of caffeine are composed of low content of 5-CQA and vice versa, as clearly visible in Fig. 3c and d.
Caffeine is characterized by solubilities in organic solvents that are completely different from the solubilities of chlorogenic acids, almost insoluble in most organic solvents.45,46
When the extraction was performed starting from GCB, caffeine was immediately extracted during the first extraction cycles (caffeine has a higher solubility in pure sc-CO2 than chlorogenic acids), which were those characterized by a lower wetting of the matrix by the co-solvent. Over time (and over cycles) increasing the wetting of the matrix corresponded to an easier extractability of polar compounds, and in fact, chlorogenic acids were extracted to a higher extent than caffeine, flushed out from the vessel together with the co-solvent. At the end of this process, almost complete co-solvent removal corresponded to a decrease in the wetting of the matrix, thus making caffeine more easily extractable in the subsequent cycles. This behavior can explain the alteration in the presence of more caffeine (or more chlorogenic acids) in the extracts, hence related to a higher/lower presence of the co-solvent in the vessel. In the case of CS, due to the lower percentages of caffeine and chlorogenic acids present in the biomasses, this phenomenon might have been hidden.
CS extracts behave in a completely different way. The first extract containing 5-CQA (6th cycle) is also the highest one in terms of 5-CQA content (33.9%), exceeding more than 15 times the content of the solvent-based extract (2.1%). About 50% by weight of the total 5-CQA present in the dry biomass was extracted during the 6th cycle, likely facilitated by the micronized particles that favor the contact between the fluid and the biomass, and hence the mass transfers. These results point out the possibility to interrupt the extraction at this cycle, to collect an extract that is selectively enriched in 5-CQA. On the other hand, caffeine follows a decreasing trend, being mostly extracted at the beginning of cycles, then decreasing over time and over extraction cycles.
Overall, aiming at extracting polar compounds such as phenolics in sc-CO2, the presence of a co-solvent (such as ethanol) as a polarity modifier is demonstrated to be indispensable, justified by the increase in the solubility of polar compounds in the mixture ethanol/sc-CO2, compared to the solubility in pure sc-CO2.41,47
Carbohydrates, phenolics, and conjugate profiles
As discussed in the previous section, not only does the solubility of the target compound increase with the use of co-solvent, but also the diversity of other compounds solubilized by the same solvent, which increases the yield as mentioned above, at the expense of reducing the process selectivity.In a biorefinery concept, the effect of co-extraction has an interesting economical outcome from a process perspective. The use of conventional solvent extraction methods could solubilize other molecules apart from the targeted phenolics due to them having similar physicochemical properties or because of molecular interactions of the phenolics with other macromolecules in the biomass such as carbohydrates or lignin. The effect of sc-CO2 extraction has been proven for the disruption of the plant cell wall as a pretreatment but also for the solubilization of carbohydrates.48–50 Based on that, considering the potential co-extraction of different biomass fractions from the same treatment, the characterization of co-extracted species was performed on both conventional extracts and on the 6th cycles of sc-CO2 extracts, to understand the profile of the obtained samples. In the case of conventional solvent extraction, the method is based on the capacity of the specific solvent to solubilize polar compounds from a matrix. However, the sc-CO2 also promotes a physical extraction process by pressurized conditions on the flow rate in the reactor, implying that other polar compounds from the biomass can be co-extracted. Since carbohydrates are the main components of GCB and CS biomasses, they have been analyzed to assess and quantify their co-extraction together with the polar phenolics. For this determination, the initial extracts were characterized by high-performance anion-exchange chromatography-pulsed amperometric detection analysis (HPAEC-PAD) in order to study the presence of free sugars. In addition, acidic hydrolysis of the extracts was performed with trifluoroacetic acid (TFA), aiming to check the presence of the complex carbohydrate molecules in the extracts derived from the biomass used in the study. The comparison allows an understanding of the complexity of the carbohydrates in monomeric, oligomeric, and polymeric forms co-extracted during the process. In the case of sc-CO2 conditions, the carbohydrate co-extraction is more important in the case of GCB, as displayed in Fig. 4a. After TFA hydrolysis, a clear increase in the complexity of monosaccharide profiles of the different samples was observed, as displayed in Fig. 4b. For instance, the presence of other sugars such as fucose, arabinose, rhamnose, xylose, and mannose was detected by HPAEC-PAD. The sugars confirmed the most probable presence of galactoglucomannans, glucans, arabinogalactans, and, in the case of CS, pectins were also deducted by the presence of rhamnose. This is in agreement with previous studies on CS for the extraction of pectins.51,52 The occurrence of those sugars was also followed by an increase of the solubilized glucose by the hydrolytic effect of the polymeric carbohydrate species. However, the galactose content seems to be affected by the TFA hydrolysis, more clearly in the GCB samples; in the case of GCB-sc-CO2, the galactose after TFA hydrolysis was 20% of the initial free amount (Fig. 4a). The partial degradation of free sugars has been presented previously and the susceptibility of degradation in the case of arabinose and galactose more than in the case of free glucose has been discussed.53,54 From the SEC analysis, it was highlighted that most of the extracts were of a lower molecular size until 200 Da, clearly represented in Fig. 4c, with most of the signal in the small range of the molecular size. The greater increase of monosaccharides after TFA hydrolysis from the CS samples is directly correlated with the presence of longer polymeric fractions, close to 100000 Da presented in the magnified image in Fig. 4d. The molecular distribution of the CS extract showed the presence of longer polymeric fractions that were co-extracted by both conventional solvent and sc-CO2. This also correlates with the lower amounts of free sugars shown in Fig. 4a. In the case of GCB, the molecular distribution of the extracts is in the range of oligomers and short polymers, with populations around 2000 Da for sc-CO2 extraction and 10000 Da for the conventional solvent extraction. The use of LC-ESI-MS before and after saponification of the samples allowed the identification of certain alternative phenolic molecules in monomeric forms such as ferulic acid at m/z = 195 [M + H]+ and m/z = 177, corresponding to its dehydrated adduct [M + H–H2O]+, caffeic acid at m/z = 181 [M + H]+ and m/z = 163 [M + H–H2O]+, as well as certain dimeric forms such as di-ferulic acid at m/z = 369 [M + H–H2O]+ and m/z = 351 [M + H–2H2O]+ as reported in Table S1.† These fractions, commonly present in other lignocellulosic materials such as cereal brans,55 were also identified by comparing them with available standards (reported in Fig. S2†). Moreover, it was possible to identify the presence of carbohydrate-phenolic conjugates at m/z = 509 (feruloyl hexo-pentose), m/z = 345 (caffeoyl hexose), and m/z = 329 (feruloyl pentose), which were distinctly present in the CS samples and were structures that had been presented previously in other types of biomasses such as tomatoes or cereals56,57. In the case of GCB, the possible presence of conjugates is reported only after sc-CO2 treatment, at m/z = 499 (caffeoyl hexo-pentose) and m/z = 367 (caffeoyl hexose), demonstrating the increased potential of co-extraction of this technique (Fig. S3†). The different elution times could correspond with different linkages in the sugar-phenolic conjugates. These results correlate with the previously mentioned co-extraction of phenolics with carbohydrates reported in Fig. 4. In the case of sc-CO2, it is the first time, to our knowledge, that these conjugates have been recognized after extraction. Other studies using subcritical water extraction have proven the protection of the link between carbohydrates and phenolic moieties present in natural cell wall extracts.27 These conjugates could have great potential as prebiotics or as building blocks for functional materials since the phenolics are able to provide their characteristic antioxidant properties to the carbohydrates fraction.58,59
 | ||
| Fig. 4 Monosaccharide profile and molecular weight distribution (Da) of the extracts from conventional solvent extraction and supercritical CO2 + EtOH. (a) Carbohydrate composition after direct solubilization; (b) carbohydrate composition after TFA hydrolysis; (c) complete molecular weight distribution; (d) oligomeric and polymeric molecular weight distribution of the extracts after direct solubilization (zooming of (c)). | ||
Conclusions
The results highlighted the ability of the supercritical fluid to efficiently target polar compounds, such as chlorogenic acids, in the presence of other beneficial co-extracted compounds, minimizing the excessive use of organic solvents and avoiding the presence of inorganic or organic acids conventionally employed when solvent-based methods are used. To the best of our knowledge, no other works are present in the literature targeting chlorogenic acids from coffee silverskin employing the sc-CO2 strategy. From the view of the application, the feasibility of the technique of supercritical CO2 in the presence of a co-solvent should meet a solvent usage threshold in order to be environmentally and economically convenient in comparison to conventional solvent-based methods. For example, less use of an organic co-solvent is advisable and should be the object of future studies and technological improvements, in order to further reduce the use of ethanol/water and to decrease the time and energy requirements for solvent removal.The scalability of sc-CO2 technology is not well-documented in the literature, and future studies are needed to better understand the practical application of this technology and related implications when co-solvents are employed.
The advantage of sc-CO2 connects also with the possibility to recover extracts in different batches over time since it requires alternation of static and dynamic periods. Extract separation over time allows a selection and isolation of the most promising as the ones characterized by high polyphenol content and a negligible amount of caffeine. This profile is desirable since caffeine itself has some biological properties that are not always compatible with the nutraceutical purposes of extracts containing chlorogenic acids.
Overall, the results further confirm that CS can be considered a good raw material for the production of chlorogenic acid-enriched extracts, with the advantage of being cheaper and more sustainable than green coffee beans. A very interesting application could be related to prebiotics in food products such as yogurt and derivatives since coffee chlorogenic acids have already been demonstrated to increase the growth of total bacteria to a similar magnitude to fructooligosaccharides, which are the positive prebiotic control.60 This assertion could also be supported by the fact that a considerable proportion of ingested chlorogenic acids reaches the large intestine, giving them the possibility of exerting beneficial effects in the large gut.61 Upcoming studies are desirable to assess the nutraceutical and antioxidant profiles of these extracts in order to confirm the potential commercial interest in this ambitious and virtuous “waste-to-products” valorization chain.
Author contributions
Data curation: S. M., A. M., R. N. and A. J. Q.; funding acquisition: L. V., S. M. and A. J. Q.; investigation: A. M., R. N., S. M. and A. J. Q.; project administration: L. V., S. M. and A. J. Q.; supervision: L. V., S. M. and A. J. Q.; conceptualization: L. V., S. M., R. N. and A. J. Q.; validation: A. M., R. N., S. M. and A. J. Q.; visualization: A. M., R. N., S. M. and A. J. Q.; writing – original draft: S. M. and A. J. Q.; writing – review and editing: A. M., R. N., L. V., S. M. and A. J. Q.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Cariplo Foundation and Innovhub (CirCO project, Grant No. 2017-0988) and by the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 101037796). The authors thank Dr Ermelinda Falletta and Dr Marco Schiavoni for chlorogenic acids LC-MS analyses and Ideal Caffè snc (Verderio) for providing the biomass.References
- I. Vural Gursel, B. Elbersen, K. P. H. Meesters and M. van Leeuwen, Sustain, 2022, 14, 1–21 CrossRef.
- C. Zhou and Y. Wang, Sci. Technol. Adv. Mater., 2020, 21, 787–804 CrossRef CAS PubMed.
- S. O. Essien, B. Young and S. Baroutian, Trends Food Sci. Technol., 2020, 97, 156–169 CrossRef CAS.
- A. Vandeponseele, M. Draye, C. Piot and G. Chatel, Green Chem., 2020, 22, 8544–8571 RSC.
- E. Ibáñez, J. A. Mendiola and M. Castro-Puyana, Encyclopedia of Food and Health, 2015, pp. 227–233 Search PubMed.
- R. M. Couto, J. Fernandes, M. D. R. G. da Silva and P. C. Simões, J. Supercrit. Fluids, 2009, 51, 159–166 CrossRef CAS.
- M. D. Luque de Castro and M. M. Jiménez-Carmona, TrAC, Trends Anal. Chem., 2000, 19, 223–228 CrossRef CAS.
- M. D. Luque de Castro and M. T. Tena, TrAC, Trends Anal. Chem., 1996, 15, 32–37 CrossRef CAS.
- M. W. Tham and K. C. Liew, Eur. J. Wood Wood Prod., 2014, 72, 67–72 CrossRef CAS.
- S. R. Falsafi, S. P. Bangar, V. Chaudhary, E. Hosseini, Z. Mokhtari, A. C. Karaca, M. K. Samota, D. Goswami, V. Krishnan, G. Askari and H. Rostamabadi, Carbohydr. Polym., 2022, 298, 120074 CrossRef CAS PubMed.
- S. Yilmaz-Turan, P. Lopez-Sanchez, A. Jiménez-Quero, T. S. Plivelic and F. Vilaplana, Food Hydrocoll., 2022, 28, 107575 CrossRef.
- V. Manasa, A. Padmanabhan and K. A. Anu Appaiah, Waste Manag., 2021, 120, 762–771 CrossRef CAS PubMed.
- N. Kumar, R. Weldon and J. G. Lynam, Biocatal. Agric. Biotechnol., 2021, 36, 102145 CrossRef CAS.
- A. Procentese, F. Raganati, G. Olivieri, M. E. Russo and A. Marzocchella, Appl. Microbiol. Biotechnol., 2019, 103, 1021 CrossRef CAS PubMed.
- R. Nasti, A. Galeazzi, S. Marzorati, F. Zaccheria, N. Ravasio, G. L. Bozzano, F. Manenti and L. Verotta, Waste and Biomass Valorization, 2021, 12, 6021–6033 CrossRef CAS.
- V. S. Ribeiro, A. E. Leitão, J. C. Ramalho and F. C. Lidon, Food Res. Int., 2014, 61, 39–47 CrossRef CAS.
- F. Rodrigues, A. Palmeira-de-Oliveira, J. Das Neves, B. Sarmento, M. H. Amaral and M. B. P. P. Oliveira, Pharm. Biol., 2015, 53, 386–394 CrossRef CAS PubMed.
- Y. Narita and K. Inouye, Food Res. Int., 2014, 61, 16–22 CrossRef CAS.
- R. Nasti, A. Galeazzi, S. Marzorati, F. Zaccheria, N. Ravasio, L. B. Giulia, F. Manenti and L. Verotta, Waste and Biomass Valorization, 2021, 12, 6021 CrossRef CAS.
- G. Navarra, M. Moschetti, V. Guarrasi, M. R. Mangione, V. Militello and M. Leone, J. Chem., 2017, 2017, 6435086 Search PubMed.
- M. L. Suárez-Quiroz, A. Alonso Campos, G. Valerio Alfaro, O. González-Ríos, P. Villeneuve and M. C. Figueroa-Espinoza, J. Food Compos. Anal., 2014, 33, 55–58 CrossRef.
- A. Rai, R. Shukla, S. Sawant, R. Shetye, D. Bopte and H. Gavandi, NCIFEH Conference Proceeding, 2018, p. 269 Search PubMed.
- L. S. McKee, H. Sunner, G. E. Anasontzis, G. Toriz, P. Gatenholm, V. Bulone, F. Vilaplana and L. Olsson, Biotechnol. Biofuels, 2016, 9, 1–13 CrossRef PubMed.
- P. Comino, H. Collins, J. Lahnstein, C. Beahan and M. J. Gidley, Food Hydrocoll., 2014, 41, 219–226 CrossRef CAS.
- C. Antoine, S. Peyron, V. Lullien-Pellerin, J. Abecassis and X. Rouau, J. Cereal. Sci., 2004, 39, 387–393 CrossRef CAS.
- C. Menzel, C. González-Martínez, A. Chiralt and F. Vilaplana, Carbohydr. Polym., 2019, 214, 142–151 CrossRef CAS PubMed.
- A. C. Ruthes, A. Martínez-Abad, H. T. Tan, V. Bulone and F. Vilaplana, Green Chem., 2017, 19, 1919–1931 RSC.
- J. M. Del Valle, M. Jiménez and J. C. De la Fuente, J. Supercrit. Fluids, 2003, 25, 33–44 CrossRef CAS.
- H. B. Coats and M. R. Wingard, J. Am. Oil Chem. Soc., 1950, 27, 93–96 CrossRef CAS.
- C. G. Pereira and M. A. A. Meireles, Food Bioprocess Technol., 2010, 3, 340–372 CrossRef CAS.
- P. A. Uwineza and A. Waśkiewicz, Molecules, 2020, 25, 3847 CrossRef CAS PubMed.
- R. P. F. F. da Silva, T. A. P. Rocha-Santos and A. C. Duarte, TrAC, Trends Anal. Chem., 2016, 76, 40–51 CrossRef CAS.
- R. N. Patel, S. Bandyopadhyay and A. Ganesh, Energy Convers. Manag., 2011, 52, 652–657 CrossRef CAS.
- S. I. M. L. F. Ballesteros and J. A. Teixeira, Food Bioprocess Technol., 2014, 7, 3493 CrossRef.
- F. K. Nzekoue, S. Angeloni, L. Navarini, C. Angeloni, M. Freschi, S. Hrelia, L. A. Vitali, G. Sagratini, S. Vittori and G. Caprioli, Food Res. Int., 2020, 133, 109128 CrossRef CAS PubMed.
- L. Regazzoni, F. Saligari, C. Marinello, G. Rossoni, G. Aldini, M. Carini and M. Orioli, J. Funct. Foods, 2016, 20, 472 CrossRef CAS.
- L. Bresciani, L. Calani, R. Bruni, F. Brighenti and D. Del Rio, Food Res. Int., 2014, 61, 196–201 CrossRef CAS.
- A. B. A. De Azevedo, P. Mazzafera, R. S. Mohamed, S. A. B. Vieira De Melo and T. G. Kieckbusch, Braz. J. Chem. Eng., 2008, 25, 543–552 CrossRef CAS.
- K. S. Andrade, R. T. Gonalvez, M. Maraschin, R. M. Ribeiro-Do-Valle, J. Martínez and S. R. S. Ferreira, Talanta, 2012, 88, 544–552 CrossRef CAS PubMed.
- C. L. Ky, M. Noirot and S. Hamon, J. Agric. Food Chem., 1997, 45, 786–790 CrossRef CAS.
- A. Daraee, S. M. Ghoreishi and A. Hedayati, J. Supercrit. Fluids, 2019, 144, 19–27 CrossRef CAS.
- Y. Narita and K. Inouye, Food Chem., 2012, 135, 943–949 CrossRef CAS PubMed.
- L. Bresciani, L. Calani, R. Bruni, F. Brighenti and D. Del Rio, Food Res. Int., 2014, 61, 196–201 CrossRef CAS.
- A. R. Ginting, T. Kit, W. Mingvanish and S. P. Thanasupsin, Sustainability, 2022, 14, 8435 CrossRef CAS.
- Q. V. Vuong and P. D. Roach, Separ. Purif. Rev., 2014, 43, 155–174 CrossRef CAS.
- K. Dibert, E. Cros and J. Andrieu, J. Food Eng., 1989, 10, 1–11 CrossRef.
- Y. Wu, B. Liu, Y. Chang and Q. Wang, J. Liq. Chromatogr. Relat. Technol., 2015, 38, 443–450 CrossRef CAS.
- L. Vaahtera, J. Schulz and T. Hamann, Nat. Plants, 2019, 5, 924–932 CrossRef PubMed.
- Y. Jiang, Y. Feng, B. Lei and H. Zhong, Int. J. Biol. Macromol., 2020, 161, 1506–1515 CrossRef CAS PubMed.
- T. Gu, M. A. Held and A. Faik, Environ. Technol., 2013, 34, 1735–1749 CrossRef CAS PubMed.
- L. Wen, Z. Zhang, M. Zhao, R. Senthamaraikannan, R. B. Padamati, D. W. Sun and B. K. Tiwari, Int. J. Food Sci. Technol., 2020, 55, 2242–2250 CrossRef CAS.
- V. Gottstein, M. Bernhardt, E. Dilger, J. Keller, C. M. Breitling-Utzmann, S. Schwarz, T. Kuballa, D. W. Lachenmeier and M. Bunzel, Foods, 2021, 10, 1–18 CrossRef PubMed.
- S. I. Mussatto, L. M. Carneiro, J. P. A. Silva, I. C. Roberto and J. A. Teixeira, Carbohydr. Polym., 2011, 83, 368–374 CrossRef CAS.
- Y. Mao, R. Lei, J. Ryan, F. Arrutia Rodriguez, B. Rastall, A. Chatzifragkou, C. Winkworth-Smith, S. E. Harding, R. Ibbett and E. Binner, Food Chem., 2019, 2, 100026 CAS.
- R. C. Rudjito, A. C. Ruthes, A. Jiménez-Quero and F. Vilaplana, ACS Sustain. Chem. Eng., 2019, 7, 13167–13177 CrossRef CAS.
- R. C. Rudjito, A. Jiménez-Quero, M. Hamzaoui, S. Kohnen and F. Vilaplana, Green Chem., 2020, 22, 8337–8352 RSC.
- A. Carrillo-López and E. Yahia, J. Food Sci., 2013, 78, 1839–1844 CrossRef PubMed.
- J. Ou and Z. Sun, J. Funct. Foods, 2014, 7, 90–100 CrossRef CAS.
- S. Yilmaz-Turan, A. Jiménez-Quero, C. Menzel, D. M. de Carvalho, M. E. Lindström, O. Sevastyanova, R. Moriana and F. Vilaplana, Carbohydr. Polym., 2020, 250, 116916 CrossRef CAS PubMed.
- L. Wang, X. Pan, L. Jiang, Y. Chu, S. Gao, X. Jiang, Y. Zhang, Y. Chen, S. Luo and C. Peng, Front. Nutr., 2022, 9, 1–22 Search PubMed.
- F. Tomas-Barberan, R. García-Villalba, A. Quartieri, S. Raimondi, A. Amaretti, A. Leonardi and M. Rossi, Mol. Nutr. Food Res., 2014, 58, 1122–1131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00037k |
| This journal is © The Royal Society of Chemistry 2023 |