
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical biology fluorescent tools for in vitro investigation of the multidrug resistant P-glycoprotein (P-gp) expression in tumor cells†
Pierre Daumar *a,
Antoine Goisnarda,
Clémence Duboisb,
Manon Rouxb,
Marie Depresleb,
Frédérique Penault-Llorcacd,
Mahchid Bamdada and
Emmanuelle Mounetoua
*a,
Antoine Goisnarda,
Clémence Duboisb,
Manon Rouxb,
Marie Depresleb,
Frédérique Penault-Llorcacd,
Mahchid Bamdada and
Emmanuelle Mounetoua
aUniversité Clermont Auvergne, Institut Universitaire de Technologie, UMR INSERM-UCA, U1240, Imagerie Moléculaire et Stratégies Théranostiques (IMoST), F-63000 Clermont-Ferrand, France. E-mail: pierre.daumar@uca.fr
bBIOMARQUEURS Company, 5 avenue Blaise Pascal, 63178 Aubière, France
cJean Perrin Comprehensive Cancer Center, F-63011 Clermont-Ferrand, France
dUniversité Clermont Auvergne, Faculté de Médecine, UMR INSERM-UCA, U1240, Imagerie Moléculaire et Stratégies Théranostiques (IMoST), F-63000 Clermont-Ferrand, France
First published on 8th September 2023
Abstract
Selective P-glycoprotein (P-gp)-targeted fluorescent conjugates are desirable tools to investigate the role of P-gp, a protein strongly implicated in mediating multidrug resistance and a major cause of chemotherapy failure. Herein, we report the development of 25 novel fluorescent small-molecule conjugates with varying physicochemical and optical properties, and their biological evaluation in a cell model as P-gp targeted constructs. This investigation revealed relationships between molecular structure and cell behavior and uncovered the capacity of conjugates with varying fluorophores to selectively target P-gp. Sulfocyanine 3 labeled conjugates (5, 10, 24, 29, 34) showed a particular intracellular staining pattern. Other conjugates bearing a boron dipyrromethene (BODIPY) core (3, 8, 13, 22, 27 (BODIPY FL), 12 (BODIPY 564/570) and 4, 9 (BODIPY 650/665)) or a 7-nitrobenz-2-oxa-1,3-diazole (NBD) core (11, 30) showed potential for global P-gp direct detection and quantification. These fluorescent conjugates holds key advantages over existing methods for drug resistance evaluation with regards to P-gp expression and could be used as innovative tools in preclinical assays and clinical diagnosis.
Introduction
The P-glycoprotein (P-gp, also known as ABCB1 or MDR1) belongs to a large class of membrane transporters known as the ATP-binding cassette (ABC) superfamily. Expressed ubiquitously in a wide range of cells, P-gp and other ABC transporters use energy derived from ATP hydrolysis to recognize and export substrates such as xenobiotics and toxins from cells, thus playing a pivotal role in physiological detoxification. Discovered more than forty years ago, P-gp has been deeply investigated, especially with regards to its implication in mediating multidrug resistance, a major cause of chemotherapy failure. Indeed, the most striking feature of this protein is perhaps its polyspecificity, or ability to transport a huge variety of structurally different substrates, including a number of hydrophobic drugs used for treatments of various pathologies, most notably cancers.1,2 By transporting drugs out of their target cells, it has been clearly evidenced that P-gp can strongly contribute to confer resistance to chemotherapy, especially when overexpressed.1,2 As such, P-gp has been consistently considered as an evident therapeutic target and its expression, a potential predictive marker of response to treatments. Consequently, several strategies have been explored to overcome P-gp-mediated drug resistance and improve treatment outcomes.3,4 Increasing intracellular drug accumulation through drug efflux inhibition has been the most thoroughly investigated approach, and a plethora of small molecules acting as P-gp inhibitors have been developed over the years to that end.5 Unfortunately, despite encouraging early results and an ever growing body of evidence suggesting a major role for P-gp in clinical multidrug resistance (along with other ABC transporters such as BCRP or MRP1), clinical translation has remained unsuccessful. As an explanation, experts acknowledge important methodological limitations when early P-gp targeting trials were designed, especially with respect to patient selection and the challenging task of reliably and systematically measuring P-gp expression in cells and tissues of interest such as tumors, the gastrointestinal tract or the blood–brain barrier.1,6,7 Consequently, it has appeared recently that innovative methods to detect and measure P-gp expression in cells or tissues would be highly beneficial to the field.To date, a lot of standard cellular and molecular biology approaches have been used to investigate P-gp levels in various samples. The list includes western blot, immunohistochemistry, immunofluorescence staining, reverse transcription quantitative real-time PCR (RT-qPCR) and more recently mass spectrometry-based proteomics techniques.8,9 Despite clear advantages, each approach also comes with its own set of drawbacks and limitations. Western blot, for example, is a multistep low throughput technique which provides only relative quantification. Mass spectrometry is a promising approach in terms of sensitivity and absolute quantification possibilities, but the development of methods remains quite challenging. Moreover, these two techniques, as macroscopic approaches, can only be applied on cell lysates and are thus unable to provide qualitative or quantitative data at the cell level. RT-qPCR offers many advantages such as the possibility of multiplex analyses, but again do not reflect cell heterogeneity in complex samples such as biopsies and, more importantly, mARN levels do not necessarily correlate the expression of the corresponding protein. Finally, unlike previous techniques, immunostaining approaches provide an interesting way to assess the location of a protein location in addition to its expression level in biological samples. Immunohistochemistry has been largely used to generate P-gp related data, especially from clinical samples, but has sometimes led to contradictory results and sensitivity issues have been reported.7,10 As for quantitative immunofluorescence, it has become a common method to characterize protein expression with high sensitivity in modalities such as flow cytometry or fluorescence microscopy. Nevertheless, despite high specificity, the use of costly fluorescent antibodies is also associated to heavy and time-consuming protocols, and their intrinsic biological and structural complexity can preclude the direct staining and analysis of certain types of samples.
The use of fluorescent small molecules has the potential to overcome limitations inherent to the use of antibodies while still allowing protein location and expression level studies in biological samples, with subcellular resolution and high sensitivity. As a consequence, over the last decade, along with the permanent optimization of optical imaging systems and image processing tools, there has been a remarkable growth in the development and use of fluorescent small molecules in biological sciences.11–14 Small-molecule fluorescent conjugates typically consist of a target specific ligand conjugated to an organic fluorophore for detection in the sample. Although the design of such constructs can be quite challenging – the ligand affinity for the target must be minimally altered after conjugation to the fluorophore – specifically designing P-gp targeted fluorescent conjugates would be highly beneficial to the field and complement the various fluorescent substrates reported to date for studying P-gp in in vitro functional assays.15–18
In this report, building on our expertise in studying P-gp in preclinical in vitro conditions, especially on cancer models,19–22 we describe the design and detail the synthesis of a series of P-gp targeted conjugates, for which a small peptidic core displaying fluorophores with varying optical properties was derivatized with aliphatic and aromatic groups. Based on flow cytometry and imaging-based experiments, we also contrast their in vitro performance and demonstrate their potential for P-gp detection and quantification in a cell model. Ultimately, these results could foster the clinical use of P-gp either as a biomarker or as a therapeutic target, as our approach based on new P-gp targeted fluorescent conjugates definitely meets the need for a simpler and innovative method for studying P-gp in biological samples.
Experimental section
Chemistry general information
All reagents and solvents were purchased from commercial sources and were used as received without further purification. Specifically, Lumiprobe NHS ester labeling dyes were purchased from Interchim (Montluçon, France) and NBD chloride was purchased from Fisher Scientific (Illkirch, France). Molecular biology grade DMSO (>99.9%) for biological experiments was purchased from Sigma-Aldrich (ref. D8418). TLC was performed on Merck (Darmstadt, Germany) silica gel 60 F-254 (ref. 1.05554.0001) or 60 RP-18 F-254S (ref. 1.05559.0001) aluminum-backed plates with visualization under UV (254 nm or 365 nm) or after staining with ninhydrin and heating. Preparative TLC were performed on Merck silica gel 60 F254 nm (20 × 20 cm) glass plates 0.5 mm (ref. 1.05744.0001). Column chromatography was performed on ultra pure silica gel 60 (40–60 μm) from Acros organics (ref. 360050050). All instruments were calibrated and maintained in accordance with standard quality-control procedures. 1H NMR spectra were recorded on a Bruker™ Avance apparatus (400 MHz) in deuterated solvents. Chemical shifts (δ) are presented in parts per million relative to TMS or to the residual solvent peak for 1H. Coupling constants (J) are given in hertz (Hz) and spectral splitting patterns are designated as singlet (s), doublet (d), triplet (t), quadruplet (q), multiplet or overlapped (m), doublet of doublets (dd), and broad (br). HRMS spectra were acquired on a Thermo Scientific™ ESI source apparatus with a “Q-Exactive” Orbitrap analyzer. Analytical and semi-preparative HPLC experiments were performed on a Shimadzu Prominence LC system equipped with a degasser (DGU-20A5R), quaternary pumps (LC-20AR), a photodiode array detector (DAD SPD-M20A), an autosampler (SIL-20AHT) and a fraction collector (FRC-10A). Shimadzu LabSolutions software (Shimadzu, France) was used for acquisition of chromatograms, spectra and integration data. Unless otherwise stated, the following gradient elution was applied using water and acetonitrile as solvents: 0–13 min, 5 to 95% acetonitrile; 13–17 min, 95% acetonitrile; 17–19 min, 95 to 5% acetonitrile; 19–23 min, 5% acetonitrile. For analytical experiments, the gradient was applied at a flow rate of 1 mL min−1 through a reversed-phase Zorbax® Eclipse Plus 3.5 μm C18 column (150 × 4.6 mm; Agilent Technologies, France) with an additional guard column. Sample volumes of 10 μL were injected. For semi-preparative experiments, the elution was applied at a flow rate of 6 mL min−1 through a Shim-pack GIST 5 μm C18 column (150 × 10 mm, Shimadzu 227-30020-03) with an additional Shim-pack GIST (G) C18 5 μm guard column (50 × 10 mm, 227-30036-01). Sample volumes of 500 μL were injected. DAD was set to record spectra between 190 and 750 nm and the monitoring wavelength was 254 nm unless otherwise stated. Compound purities of final compounds were calculated as the percentage HPLC peak area of the analyzed compound at the specified monitoring wavelength. All compounds employed were >95% pure. Reported chemical yields are unoptimized.Note for the 13C NMR spectra of the final compounds: most compounds were isolated in small quantities and long acquisition times did not result in satisfying 13C NMR spectra with sufficient resolution. This was attributed to the complexity and the floppy nature of the molecules, and also the presence of rotamers (observed in 1H NMR). Temperature variation for acquisition did not lead to significant changes in the spectra. For these reasons, 13C NMR spectra were not recorded. Identity and purity of all compounds however are supported by other means of characterization such as 1H NMR, HPLC-UV and HRMS.
Chemistry general procedures
Synthetic procedures for original compounds 2–19, 21–34, 36–39, 41, 44, 49
Biological evaluation
Results and discussion
Design of P-gp targeted fluorescent conjugates
A number of structurally unrelated compounds have been described to interact with the highly polyspecific P-gp, resulting in a wealth of structures from which fluorescence labeling can be investigated. However, the goal of our study was to obtain fluorescent conjugates that would bind to P-gp without being transported, allowing us to monitor a fluorescent signal representative of the protein expression at the cell level and not its activity. As a consequence, only non competitive P-gp inhibitors were considered for conjugation while reviewing the literature and the vast majority of P-gp interacting compounds described as substrates were ignored. We also paid attention to any data regarding the selectivity for P-gp over other MDR proteins, especially BCRP, which shares an extensive overlap of substrates with our target protein. This allowed us to further refine our search and identify two short peptidic derivatives, P1 and P2, as attractive candidates for the development of selective P-gp targeted fluorescent constructs (Fig. 1A). The formers were described recently and resulted from a reversin structure optimization for selective and potent P-gp binding.23 Their affinity to the protein was ascribed to both the hydrophobic nature of their side chains protected with bulky aromatic or alkyl groups, and the high level of constraint brought by the proline residue. Most importantly, the authors reported that the most potent compound P2 behaved as a non competitive inhibitor with low cytotoxicity. Additionally, from a practical point of view, the peptide scaffold lends itself to facile analog generation by employing the nitrogen atoms as conjugation handles (Fig. 1A), either after removal of a protecting group via a standard procedure or even directly in the case of P2. Finally, previous reports mention a large binding pocket for P-gp, likely to offer some flexibility in accommodating large ligands.23 Taking all these points into consideration, we reasoned that the peptidic scaffold was versatile enough and a good starting point to generate a panel of P-gp targeted fluorescent conjugates with varying degrees of hydrophobic and aromatic character, and also various emitting wavelengths.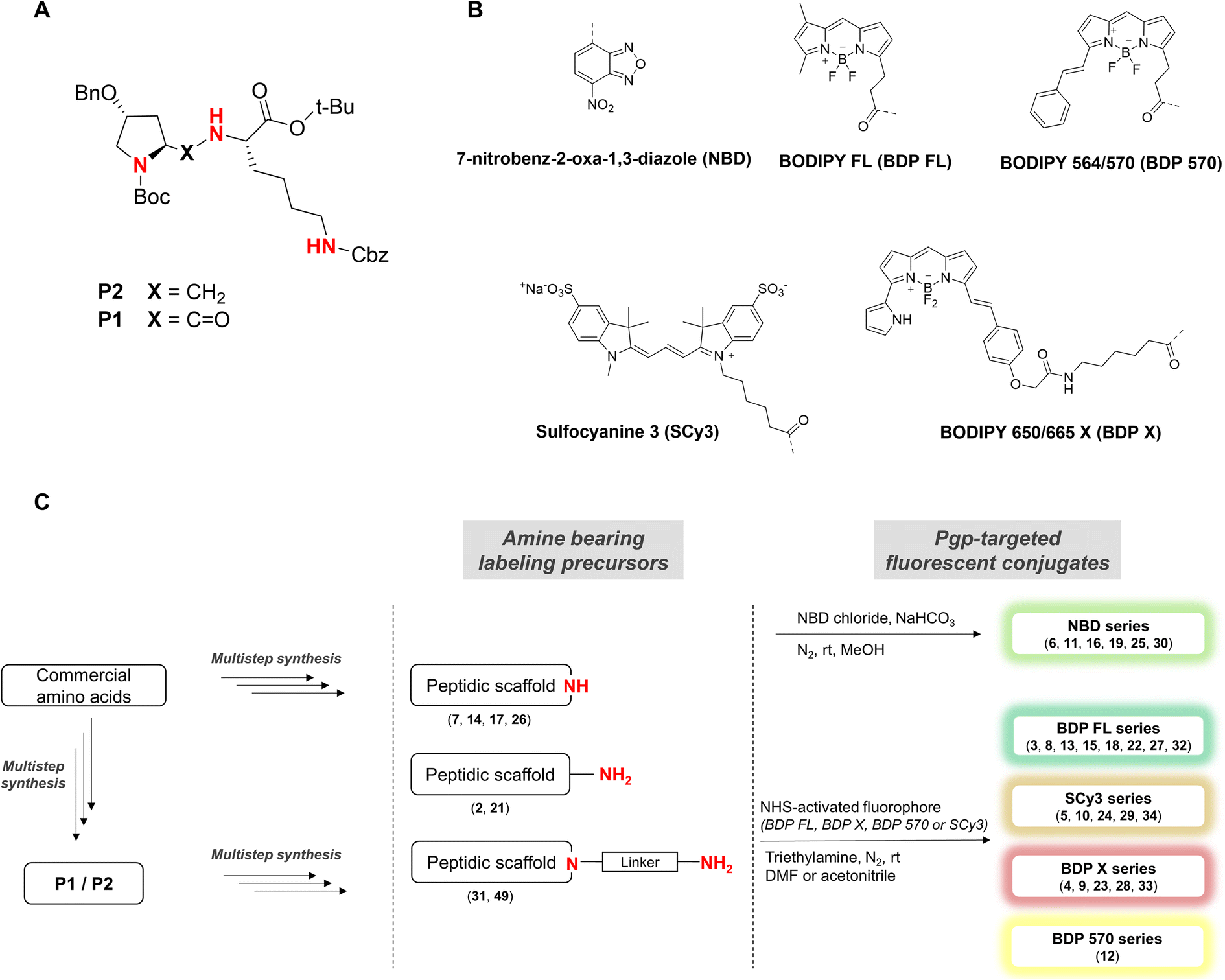 | ||
| Fig. 1 (A) Structures of previously described potent and selective P-gp inhibitors P1 and P2. Nitrogen atoms used as conjugation handles to fluorophores are shown in red; (B) structures of the five fluorophores used in the study. (C) Schematic illustration of the synthetic approach for fluorescent conjugates from commercial amino acids. Fluorophores were conjugated to the peptidic scaffold in the last step of the synthesis, either directly or via an aliphatic linker. | ||
Five well-characterized fluorophores were employed in our study to derivatize the peptidic scaffold, based either on a cyanine, a boron dipyrromethene (BODIPY), or the 7-nitrobenz-2-oxa-1,3-diazole (NBD) core. Specifically, the sulfocyanine 3 (SCy3), the BODIPY FL (BDP FL), the BODIPY 650/665 X (BDP X), the BODIPY 564/570 (BDP 570) and NBD were chosen (Fig. 1B). This selection allowed us to vary the optical properties of our compounds as well as determining characteristics likely to influence their affinity for the P-gp target and/or their cell distribution (e.g. steric hindrance, overall charge, cell permeability) as highlighted by many reports.11,14,24 Fluorophores were most often conjugated directly to the peptidic moiety, but aliphatic linkers were also used to produce some derivatives and potentially generate structure–activity relationships (SAR) (Fig. 1C). As a whole, following our design criteria, we synthesized a series of 25 peptides for their evaluation as P-gp targeted fluorescent conjugates. All the detailed structures and related physicochemical properties are summarized in Table 1.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Y | R1 | R2 | R3 | R4 | R5 | clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Pa Pa |
λEx/Emb [nm] |
| a Calculated with ChemDraw 20.1.b Maximum excitation and emission wavelengths evaluated in ethanol. | ||||||||
| 3 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
H | OBn | Boc | OtBu | BDP FL | 7.1 | 503/510 |
| 4 | CO |
H | OBn | Boc | OtBu | BDP X | 9.1 | 650/660 |
| 5 | CO |
H | OBn | Boc | OtBu | SCy3 | 2.7 | 559/569 |
| 6 | CO |
H | OBn | Boc | OtBu | NBD | 6.9 | 463/540 |
| 8 | CO |
H | OBn | BDP FL | OtBu | Cbz | 8.6 | 505/510 |
| 9 | CO |
H | OBn | BDP X | OtBu | Cbz | 9.8 | 650/662 |
| 10 | CO |
H | OBn | SCy3 | OtBu | Cbz | 3.4 | 556/572 |
| 11 | CO |
H | OBn | NBD | OtBu | Cbz | 6.6 | 463/540 |
| 12 | CO |
H | OBn | BDP570 | OtBu | Cbz | 9.7 | 565/575 |
| 13 | CO |
H | OBn | CO(CH2)5NH-BDP FL | OtBu | Cbz | 8.2 | 500/510 |
| 15 | CO |
H | H | BDP FL | OtBu | Cbz | 7.0 | 505/528 |
| 16 | CO |
H | H | NBD | OtBu | Cbz | 5.4 | 463/537 |
| 18 | CO |
H | OBn | BDP FL | OH | Cbz | 7.3 | 505/522 |
| 19 | CO |
H | OBn | NBD | OH | Cbz | 5.2 | 468/537 |
| 22 | CH2 | H | OBn | Boc | OtBu | BDP FL | 7.5 | 502/525 |
| 23 | CH2 | H | OBn | Boc | OtBu | BDP X | 9.5 | 638/681 |
| 24 | CH2 | H | OBn | Boc | OtBu | SCy3 | 0.8 | 558/578 |
| 25 | CH2 | H | OBn | Boc | OtBu | NBD | 7.4 | 462/540 |
| 27 | CH2 | H | OBn | BDP FL | OtBu | Cbz | 8.9 | 505/522 |
| 28 | CH2 | H | OBn | BDP X | OtBu | Cbz | 10.1 | 638/678 |
| 29 | CH2 | H | OBn | SCy3 | OtBu | Cbz | 1.4 | 560/581 |
| 30 | CH2 | H | OBn | NBD | OtBu | Cbz | 7.1 | 467/543 |
| 32 | CH2 | COCH2NH-BDP FL | OBn | Boc | OtBu | Cbz | 10.1 | 505/522 |
| 33 | CH2 | COCH2NH-BDP X | OBn | Boc | OtBu | Cbz | 12.1 | 646/676 |
| 34 | CH2 | COCH2NH-SCy3 | OBn | Boc | OtBu | Cbz | 5.7 | 556/578 |
Exploration of synthetic routes
To access final fluorescent conjugates, our general synthetic strategy relied on two main points: (i) using one of the nitrogen atoms of the peptidic scaffold as a conjugation handle, while maintaining other functional groups responsible for P-gp affinity; (ii) performing the conjugation in the last step of our synthetic routes with reaction to commercial fluorophores activated by an N-hydroxysuccinimide ester group (NHS) or to NBD chloride (Fig. 1C). Of note, the use of NBD chloride represents the easiest way to introduce this fluorophore but in our case it also provided an alternative non amide bond mode of conjugation, with potential to enlarge SAR.25,26 General procedures for conjugation are detailed in the experimental section (i.e., procedures C and D). Importantly, yields were unoptimized. As a consequence, yields were highly variable, often moderate and sometimes low, independently of the conjugation site. It is noteworthy that in the case of NDB coupling through nucleophilic substitution, yields were consistently excellent.In our strategy, P1 and P2 were key intermediates as they should allow access to amine labeling precursor after standard deprotection procedures. Therefore, those compounds were synthesized as previously reported.23 It is obvious from P2 structure that a nitrogen atom is directly available for conjugation (Fig. 1A). Unfortunately, coupling attempts with BDP FL NHS ester systematically failed in our hands, probably because of high steric hindrance. As a consequence, a glycinil spacer was introduced beforehand. Fmoc-Glycinyl chloride 46 was thus synthesized in two steps according to previous reports and then reacted with P2 to afford Fmoc protected intermediate 44 (Scheme 1).27,28 Subsequent deprotection in standard conditions gave amine bearing fluorescent labeling precursor 31, which was further reacted in a final step with NHS ester activated fluorophores, leading to novel compounds 32, 33, 34 in variable yields (Scheme 1).
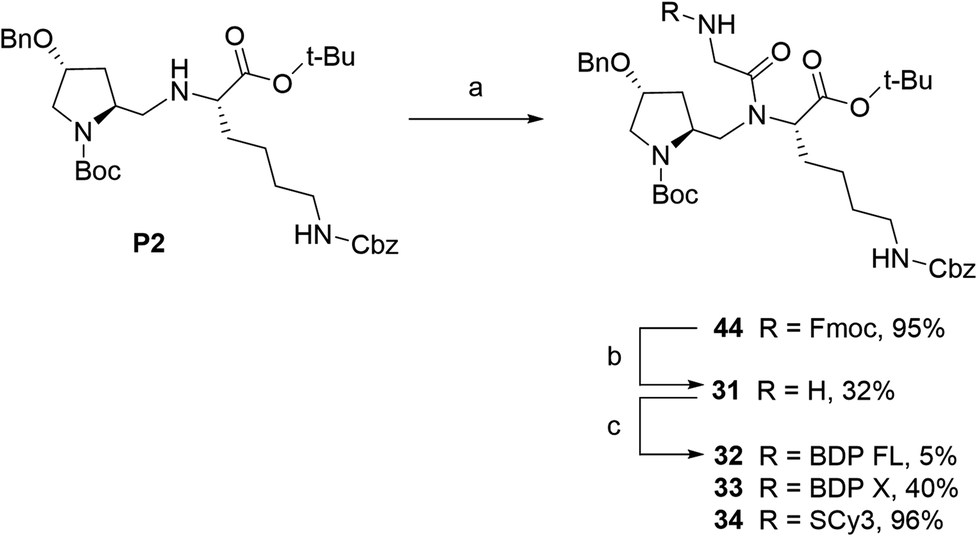 | ||
| Scheme 1 Synthesis of fluorescent conjugates 32–34. reagents and conditions: (a) dry DCM, N2, Fmoc-glycinyl chloride 46, 0 °C to rt, 30 min; (b) dry DMF, N2, diethylamine, rt, 1 h; (c) dry DMF (32)/acetonitrile (33, 34), N2, triethylamine, NHS-activated fluorophore, rt, 2 days (32)/2 h (33, 34). | ||
We next envisioned that a selective deprotection of the Boc-proline residue would lead to another interesting platform (secondary amine), either for direct coupling or for introduction of a spacer arm (Scheme 2). The challenge here was to use classical acidic conditions for Boc deprotection while leaving intact the tert-butylic ester protecting the C end of the peptidic scaffold, which could be detrimental for P-gp binding.29 Therefore, a classical procedure was adapted by performing acidic deprotection using chlorhydric acid in 1,4-dioxane over very short reaction times and at low temperature. This procedure allowed us to isolate labeling precursors 26 and 7, from P2 and P1 respectively (Scheme 2). From intermediate 26, direct conjugation led to another series of fluorescent conjugates (compounds 27–30, Scheme 2A). Despite the presence of two secondary amines, no regioselectivity issue was encountered, as the proline is much more accessible than the hindered aminomethylene group of the P2 scaffold, as previously experimented. Variable yields were observed, from very low for SCy3 NHS ester coupling to excellent for substitution reactions with NBD chloride. From key intermediate 7, final conjugation proceeded in moderate to excellent yields to generate another series of final fluorescent peptides (compounds 8–12, Scheme 2B). Compound 7 was also used to access amine 49, which presents a 6-aminohexanoic linker (Scheme 2B). Briefly, NHS ester 48 was obtained in two steps from commercial 6-amino hexanoic acid via a Fmoc protected intermediate according to well described procedures.30,31 Subsequent reaction with intermediate 7 in basic conditions led to unprotected amine 49, which was further coupled to NHS ester activated BDP FL to yield fluorescent peptide 13 (Scheme 2B).
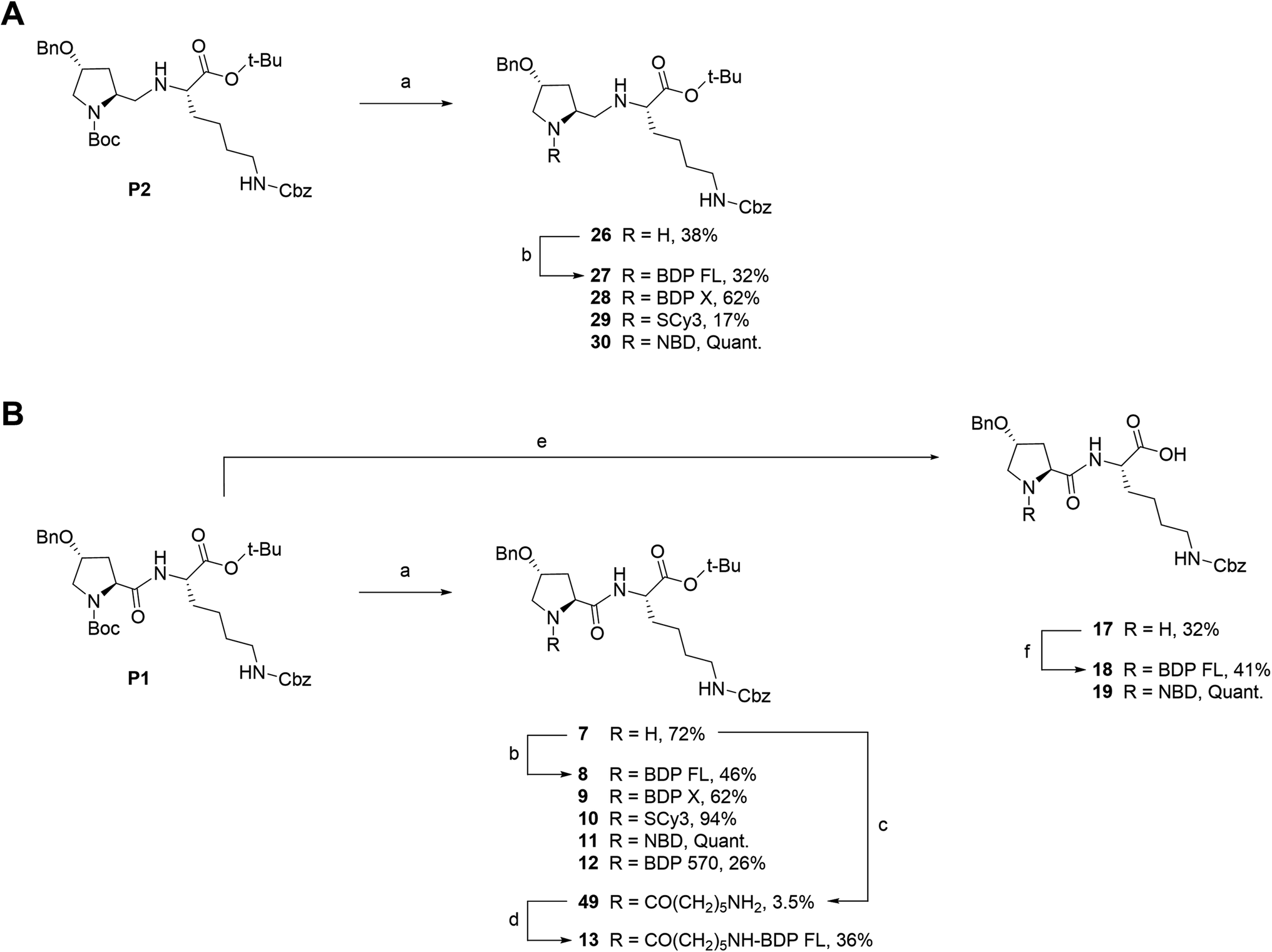 | ||
| Scheme 2 Synthesis of fluorescent conjugates 8–13, 18–19, 27–30. Reagents and conditions: (a) 4 N HCL in 1,4-dioxane, N2, 0 °C , rt, 30–45 min; (b) dry DMF (8, 9, 10, 12, 13, 27) or acetonitrile (28, 29), N2, triethylamine, NHS-activated fluorophore, rt, 45 min (28, 29)/12 h (27)/1 day (12)/3 days (8, 9)/6 days (10) or dry MeOH, N2, NaHCO3, NBD chloride, rt, overnight (11, 30); (c) dry DMF, N2, triethylamine, 6-(Fmoc-amino)caproic acid NHS ester 48, rt, 4 days; (d) dry DMF, N2, triethylamine, NHS-BDP FL, rt, 7 h; (e) 4 N HCL in 1,4-dioxane, N2, 0 °C , rt, overnight; (f) dry DMF, N2, triethylamine, NHS-BDP FL, rt, 5 days (18) or MeOH, N2, NaHCO3, NBD chloride, rt, overnight (19). | ||
Fluorescent P1 analogues 15, 16, 18 and 19 also present a fluorophore on the proline residue but supplementary structural modifications likely to impact their P-gp binding were brought to the peptidic scaffold in order to enlarge SAR (Table 1). Precisely, compounds 15 and 16 miss the benzyloxy group (OBn) on position 4 of the proline ring and compounds 18 and 19 present a polar and hydrophilic carboxylic acid functional group. For the synthesis, harsh acidic conditions applied to P1 allowed both Boc deprotection and conversion of the tert-butylic ester to acid to furnish intermediate 17 (Scheme 2B). This compound was subsequently used to access analogues 18 and 19 in moderate to quantitative yields after coupling to BDP FL and NBD fluorophores, respectively. Conjugates 15 and 16 were prepared in three steps from a commercial lysine and Boc protected proline (Scheme 3). To that end, very similarly to P1, peptidic intermediate 41 was obtained in high yield in a first step by the mixed anhydride method in presence of ethyl chloroformate. Subsequent selective Boc deprotection in conditions described earlier led to labeling precursor 14, and final coupling offered the desired compounds in excellent yields. Noteworthy, conjugation on this site to BDP FL via NHS ester coupling proceeded in much higher yield (i.e. 97%) than for analogues substituted on position 4 of the proline ring such as 8 (46%), 18 (41%) or 27 (32%). This result suggests that the OBn group on this position lessen the nucleophilicity of the nitrogen atom and/or contributes to steric hindrance.
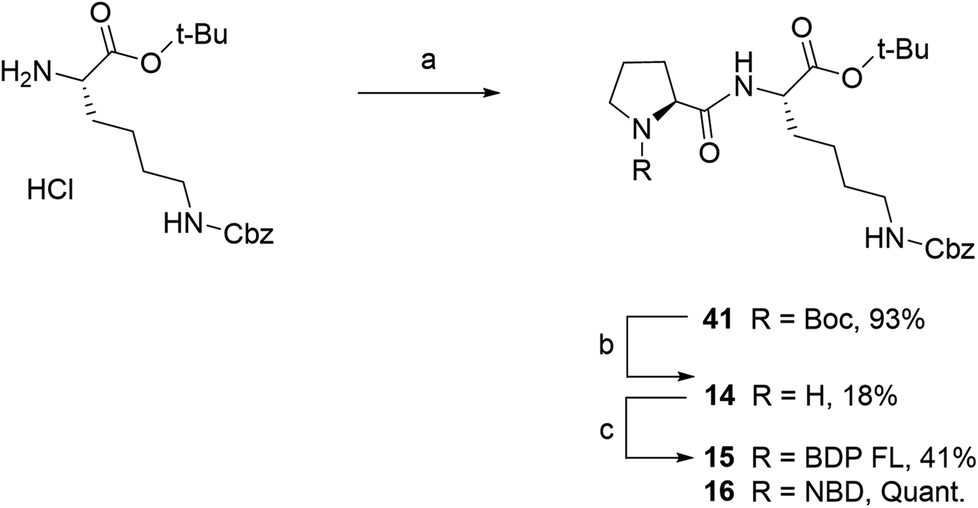 | ||
| Scheme 3 Synthesis of fluorescent conjugates 15–16. Reagents and conditions: (a) dry DCM, N2, ethyl chloroformate, triethylamine, N-(tert-butoxycarbonyl)-L-proline, 0 °C for 2 h , rt, overnight; (b) 4 N HCL in 1,4-dioxane, N2, 0 °C , rt, 40 min; (c) dry acetonitrile, N2, triethylamine, NHS-BDP FL, rt, 2 days or MeOH, N2, NaHCO3, NBD chloride, rt, 2 days. | ||
Finally, regarding the use of the lysine lateral chain as a conjugation site, a similar well known and adapted deprotection strategy from P1 and P2 would lead ideally to the free primary amine before the final coupling step (Fig. 1A/C). Therefore, it was first attempted to determine conditions leading to rapid and selective deprotection of the Cbz group of the lateral chain. However, experimental conditions would systematically lead to concomitant Boc deprotection and/or benzyl group removal on the proline moiety. It appeared that accessing a primary amine on this site imposed to elaborate a whole different synthetic scheme. The chosen strategy relied again on Fmoc chemistry, which can provide selective deprotection conditions over tert-butylic esters, Boc and Cbz groups (Scheme 4). Key intermediate lysine 37 was prepared in a 74% yield over two steps from commercial Boc protected lysine tert-butylic ester: the primary amine was Fmoc protected in the first step yielding intermediate 36, which afforded lysine 37 upon reaction in acidic conditions. From lysine 37, the Fmoc protected P1 analogue 38 was prepared in excellent yield by the mixed anhydride method. Subsequent selective Fmoc deprotection required to test several conditions, but finally led in a 58% yield to primary amine 2, which was then conjugated to the selected fluorophores BDP FL, BDP X, Scy3 and NBD to give new fluorescent peptides 3–6 (yields 24–100%, Scheme 4). Lysine 37 also allowed access to Fmoc protected P2 analogue 39 via a reductive amination step involving sodium cyanoborohydride as the reducing agent. Subsequent deprotection and coupling in conditions described earlier furnished the series of final fluorescent peptides 22–25 (yields 21–100%, Scheme 4).
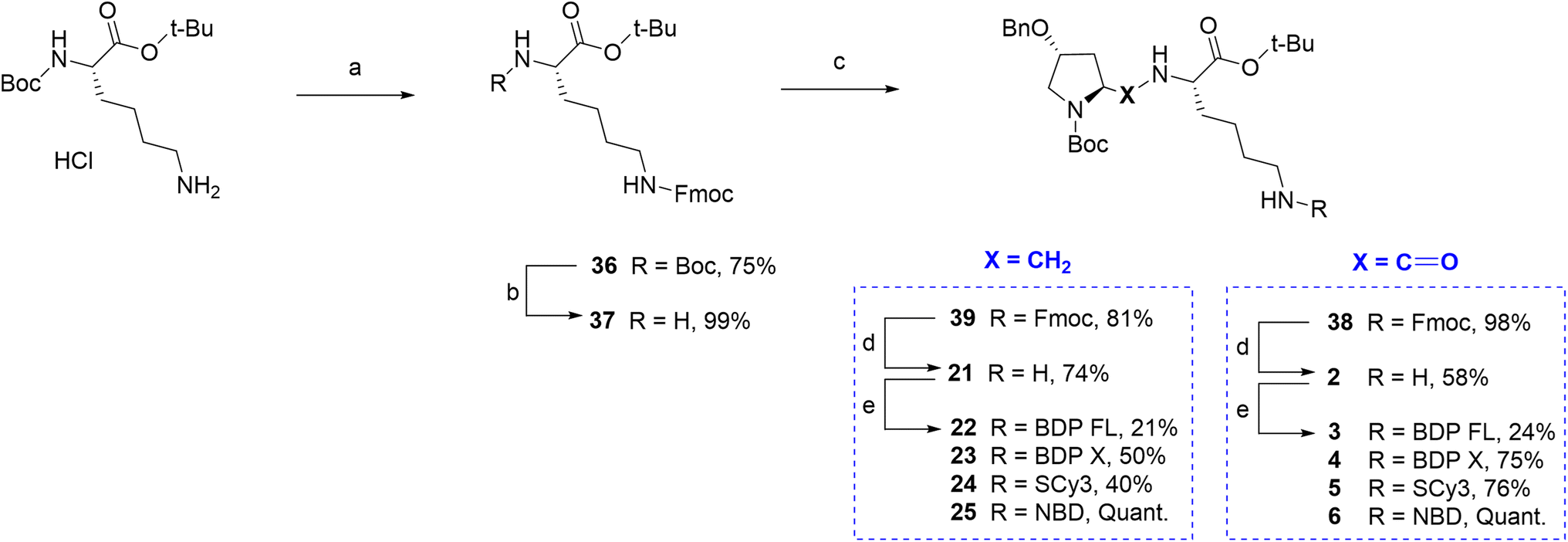 | ||
| Scheme 4 Synthesis of fluorescent conjugates 3–6, 22–25. Reagents and conditions: (a) 10% NaHCO3 aqueous solution, THF, Fmoc-NHS, 0 °C , rt, overnight; (b) 4 N HCL in 1,4-dioxane, N2, 0 °C , rt, 75 min; (c) MeOH, (4R)-benzyloxy-(2S)-formyl-pyrrolidine-1-carboxylic acid tert-butyl ester, NaBH3CN, CH3COOH, rt, 3 h; (d) dry DMF, N2, diethylamine, rt, 60 min; (e) dry DMF (3, 4, 5, 22)/acetonitrile (23, 24), N2, triethylamine, NHS-activated fluorophore, rt, 2 h (3)/18 h (4)/20 h (5)/3 days (22)/1 h (23)/45 min (24) or MeOH, N2, NaHCO3, NBD chloride, rt, overnight (6)/2 days (25). | ||
All intermediates and final fluorescent compounds were purified via column chromatography or reversed-phase HPLC, identified and characterized by classical analytical methods (1H NMR, UV-vis spectroscopy and ESI-HRMS (ESI Data†)). All final fluorescent peptides (i.e. 3–6, 8–13, 15, 16, 18, 19, 22–25, 27–30, 32–34) were obtained in >95% purity. Before biological evaluation, fluorescence spectra were recorded for all final conjugated compounds and also for commercial NHS activated fluorophores. With regards to excitation and emission wavelengths, which span over a large part of the visible spectrum, results showed no important differences between compounds bearing the same fluorophore, demonstrating that peptide conjugation did not affect the fluorescence characteristics of the fluorophore (Table 1, Fig. 2). With a library of 25 new P-gp targeted fluorescent peptides displaying different lipophilicity and optical properties at hands, the next step was to contrast their in vitro performance and interrogate their potential as efficient fluorescent conjugates for selective P-gp detection and quantification in a series of cell-based experiments.
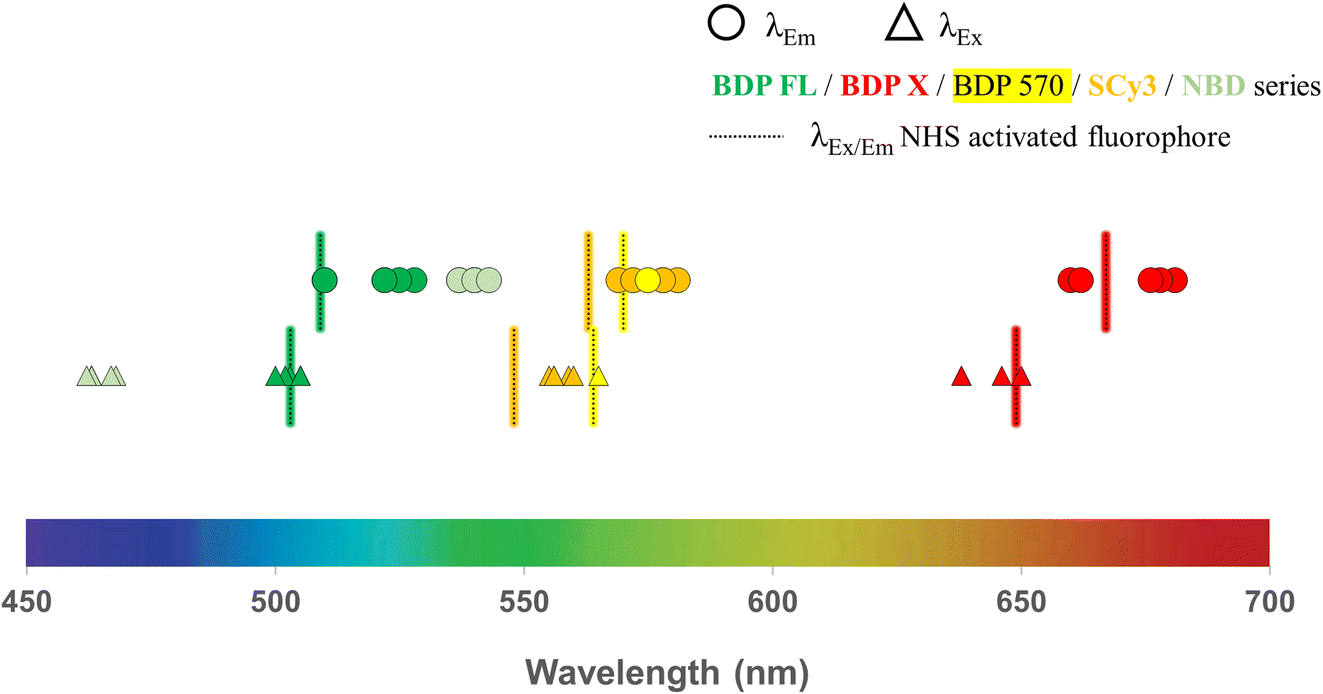 | ||
| Fig. 2 Fluorescence characteristics (excitation and emission wavelengths) determined from spectra recorded in ethanol solutions for all new P-gp targeted fluorescent conjugates and commercial NHS-activated fluorophores (BDP FL, BDP X, BDP 570 and SCy3 series). | ||
Biological evaluation
Flow cytometry experiments were carried out by incubating a suspension of DU4475 cells in cell culture media at 37 °C with the following compounds (1 μM): 3, 8, 13 (BDP FL series), 4 (BDP X series), 5, 10 (SCy3 series), 12 (BDP 570 series) and 30 (NBD series). This compound selection was considered as representative of our panel as they present all the fluorophores chosen for our study, different conjugation sites and a linker in the case of 13. Cell uptake kinetics was evaluated by comparing the mean fluorescence intensity of cells (MFI) to the control (unstained cells) at different time points over 2 hours (Fig. 3).
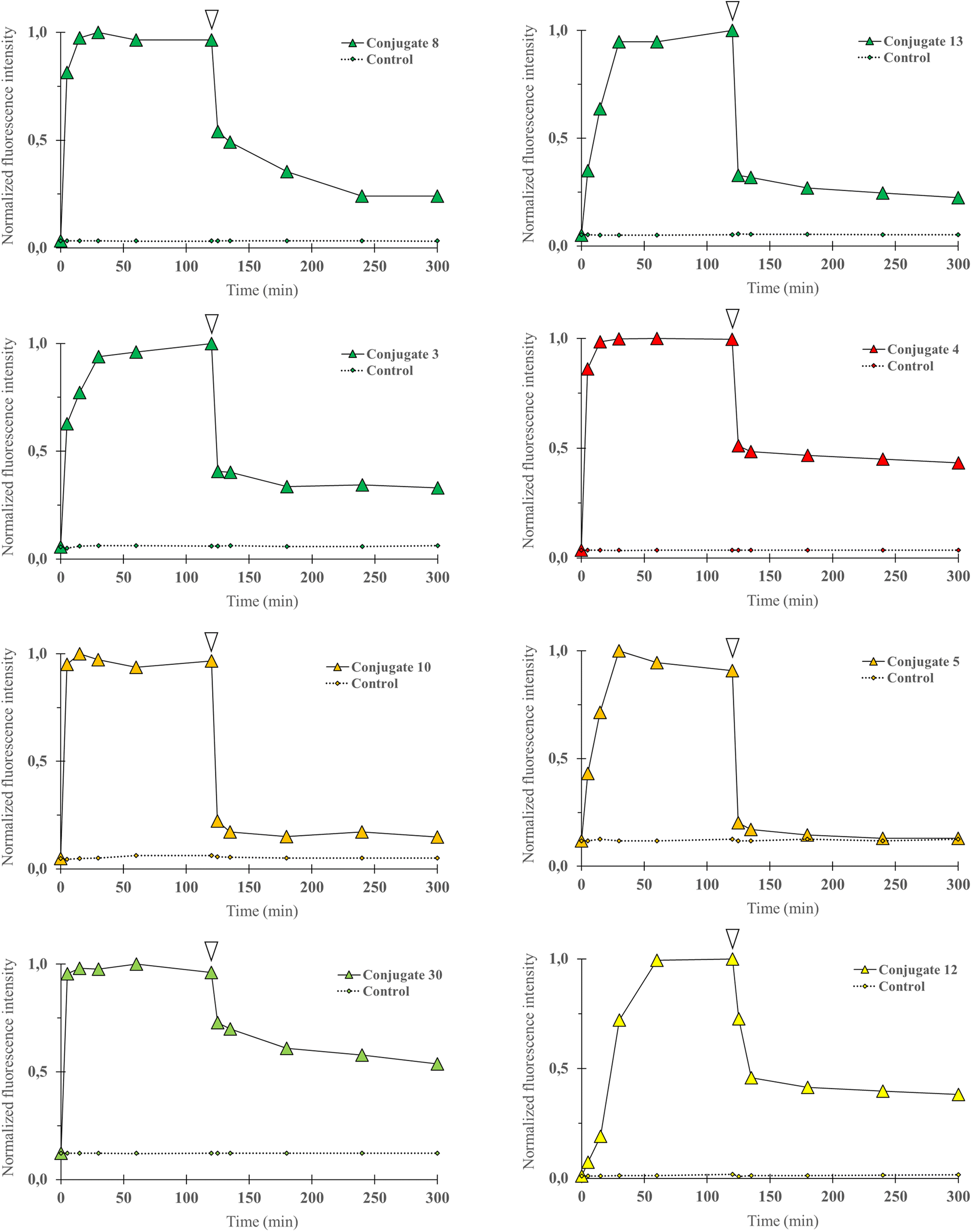 | ||
| Fig. 3 Cell uptake kinetics analyses were performed by flow cytometry. P-gp expressing DU4475 cells were incubated with a fluorescent conjugate (3–5, 8, 10, 12, 13 or 30) at 1 1 μM in cell culture media for 2 h at 37 °C and then rinsed in cold PBS (4 °C) for 3 h. Diagram shows normalized cells MFI at different time points upon incubation with a selected compound (0 to 120 min) and PBS rinse (120 to 300 min), and for control (unstained) cells. White arrows indicate the last data point before rinsing. Data were normalized to maximal MFI for each condition. For all compounds, MFI increased rapidly and then decreased during PBS rinse but remained higher than for control cells at all time points. | ||
Fluorescence signal was detectable for all compounds from the earlier time point (5 min), probing a very rapid cell uptake irrespective of the nature of the fluorophore and of the conjugate structure in general. As soon as 15 minutes and no longer than 60 minutes after incubation started, a plateau was observed in each experiment, suggesting that the cell–medium equilibrium was reached. After two hours, cells were removed from cell culture medium and rinsed with cold PBS for 3 hours, during which the MFI was regularly measured. A strong decrease was observed and a lower plateau was reached rapidly for all compounds. We noted that normalized values at the plateau seemed to be dependent on the type of fluorophore as the lowest values were observed for the SCy3 derivatives, intermediate values for the BODIPY derivatives, and the higher value for the NBD derivative 30. Beyond that, it is noteworthy that for all compounds the MFI remained systematically higher – although very close for compound 5 – than for its corresponding control (unstained cells). This could be explained by a novel cell–medium equilibrium, but could also result, at least partially, from a high affinity intracellular binding of our compounds. Overall, these results showed that the structural modifications that were brought to the peptidic scaffold were never detrimental for cell uptake.
We next aimed to demonstrate in an imaging-based experiment that our compounds could provide a fluorescent signal intensity closely dependent on the P-gp expression level of a given cell line. Indeed, when a selective binding is achieved for a fluorescent molecule, the expression level of the targeted protein determines the fluorescent signal intensity in imaging experiments. Therefore, DU4475 and SUM1315 breast cancer cell lines, which differentially express P-gp as determined by immunofluorescence intensity measurements (Fig. 4A, top row), were used in this assay.19,32 For each compound testing, cells were first fixed, incubated with a 1 μM PBS solution for 1 h, washed three times to remove unbound compound and imaged with a Cytation™3 MV (BioTek®, USA) fluorescence microscopy module. Working on fixed cells eliminated the potential bias related to cytotoxicity, although this was not observed in our previously reported study.20 Quantitative studies were finally performed with optimized imaging parameters for each compound by measuring the mean fluorescence intensity in cells. First of all, a measurable fluorescent signal was detected in each experimental condition thus supporting the previous conclusion in flow cytometry. Fig. 4A (bottom row) shows this result in the case of representative BDP FL conjugate 8, and also reveals a higher affinity (superior MFI) for the highest expressing DU4475 cells compared to SUM1315 cells, suggesting a selective binding to P-gp. This was confirmed in P-gp knockdown SUM1315 cells previously exposed to specific siRNAs (see Experimental section), for which both immunolabeling and compound 8 fluorescent signals were extinguished (Fig. 4A). Importantly, we actually observed that all compounds showed a higher affinity for DU4475 cells compared to SUM1315 cells. Indeed, when calculating the same signal intensity ratio as in our preliminary immunofluorescence microscopy experiment, values were systematically superior to 1.0 (Fig. 4B). Given the high number of tested compounds, this provided quite compelling evidence that, regardless of structural considerations, P-gp interaction plays a determining role in the cell binding mode of our peptidic compounds. As a matter of fact, Fig. 4B also reveals that for a rather large number of compounds, ratio values differ from the 2.4 reference value obtained in the control immunofluorescence experiment. For example, in the series of BDP FL conjugated peptides, compounds 8 and 13 gave close ratio values of 2.3 and 2.6 respectively, while compounds 22 and 27 gave much lower values of 1.3 and 1.7, respectively. As a whole, while establishing an evident link between the P-gp expression level and cell binding, our results also suggested that in our experimental conditions some compounds staining might not be limited to the plasma membrane as in the immunolabeling experiment. Consequently, we pursued our investigation with intracellular distribution studies.
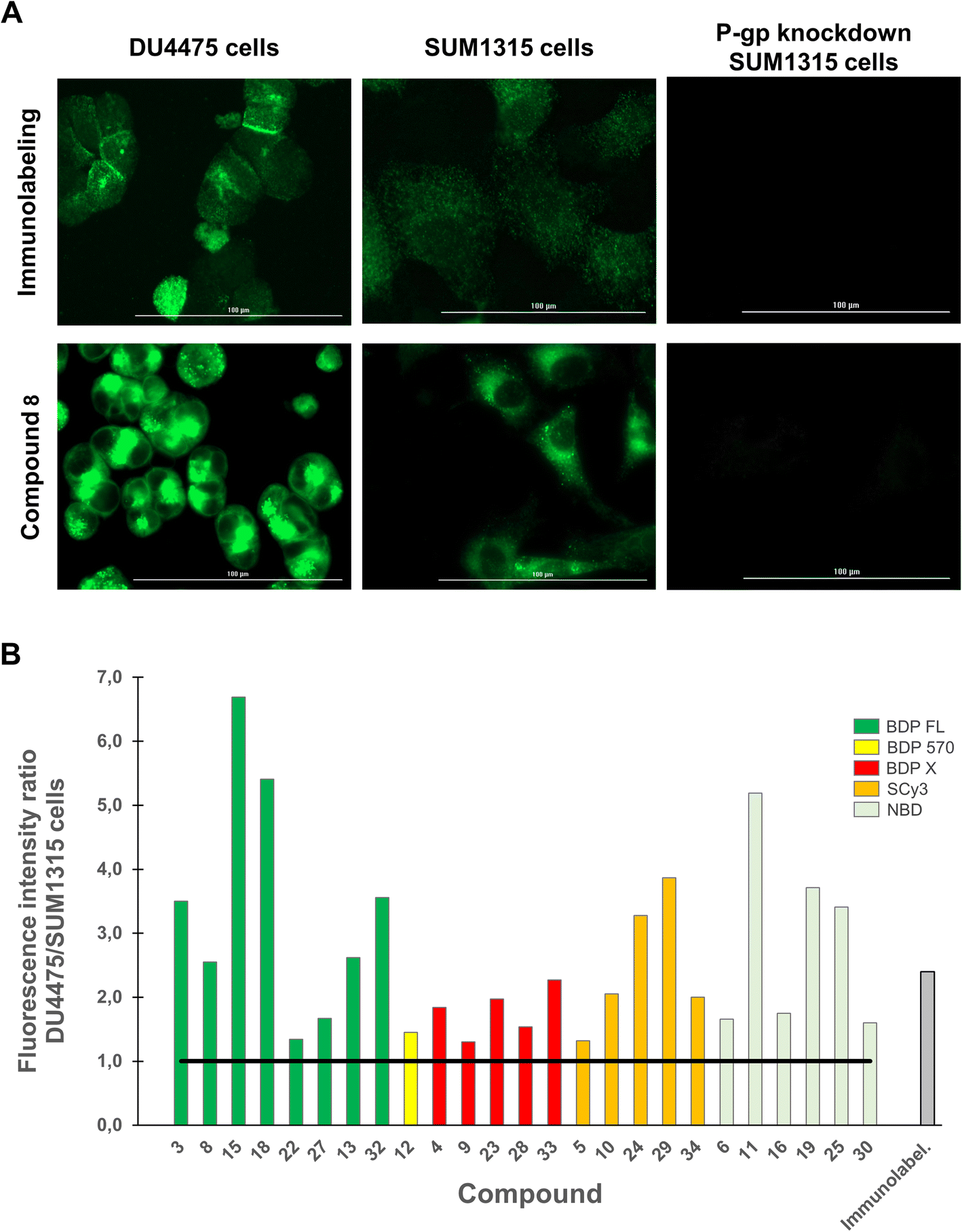 | ||
| Fig. 4 Quantitative fluorescence imaging studies were performed on DU4475 and SUM1315 cells, which present a differential basal P-gp expression. Cells were fixed, incubated with a fluorescent conjugate at 1 μM for 1 h, rinsed with PBS (3×) and imaged on a Cytation™3 MV (BioTek®, USA). For each condition, cells MFI was measured from 10 different imaging fields with optimized imaging parameters, which were kept constant for each fluorophore. (A) Images shows the P-gp immunolabeling (green signal, top row) and the distribution of conjugate 8 (BDP FL series, green signal, bottom row) in DU4475 cells, SUM1315 cells and P-gp knockdown SUM1315 cells. MFI is higher in DU4475 cells (higher P-gp expression level) as compared to MFI in SUM1315 cells (lower P-gp expression level), and no detectable signal was observed in control P-gp knockdown SUM1315 cells. (B) Histogram shows DU4475/SUM1315 cells MFI ratios measured for each P-gp-targeted fluorescent conjugates and for immunolabeling. Ratios are all superior to 1 (black solid line). | ||
Immunostaining showed an ubiquitous cell repartition for P-gp with a punctate fluorescence pattern possibly resulting from P-gp proteins clustering at the cell membrane (Fig. 5A). Most strikingly, several BDP FL, BDP 570 and NBD fluorophore bearing compounds (3, 6, 8, 11, 12, 13, 15, 22, 27, 30) led also to this characteristic staining pattern, suggesting a good colocalization, mostly driven by the P-gp targeting moiety (Fig. 5B/C/6A–C). Noteworthy, this was confirmed by the very different and highly diffuse staining patterns that were obtained in control experiments with BDP FL NHS ester and BDP FL 570 ester (Fig. S1†). The BDP 570 conjugate 12 and the BDP FL compounds in general gave bright images and high Pearson coefficients (0.66 to 0.84), except for compounds 15, 18 and 32, for which values dropped below 0.50. This showed that both the OBn group on position 4 of the proline residue and the tert-butylic ester of the lysine residue were important structural features for selective P-gp binding, and it also suggested that functionalizing the free secondary amine of P2 was highly detrimental. Beyond that, conjugation on other sites (compounds 3, 8, 13, 22, 27) and introduction of a spacer arm (compound 13) appeared as acceptable structural modifications for retaining P-gp binding (Fig. 6A/B). In the NBD series, similar results were observed for compounds 16 and 19, which miss the OBn group and the tert-butylic ester respectively, supporting the established SAR. Interestingly, we found that compounds 11 and 30 bearing the fluorophore on the proline residue gave much better results than their analogues 6 and 25, for which the NBD motif was introduced on the lateral chain of the lysine. Indeed, Pearson coefficients were 0.73 and 0.76 in the case of compounds 11 and 30, respectively, but values fell to around 0.4 for analogues 6 and 25 (Fig. 5B).
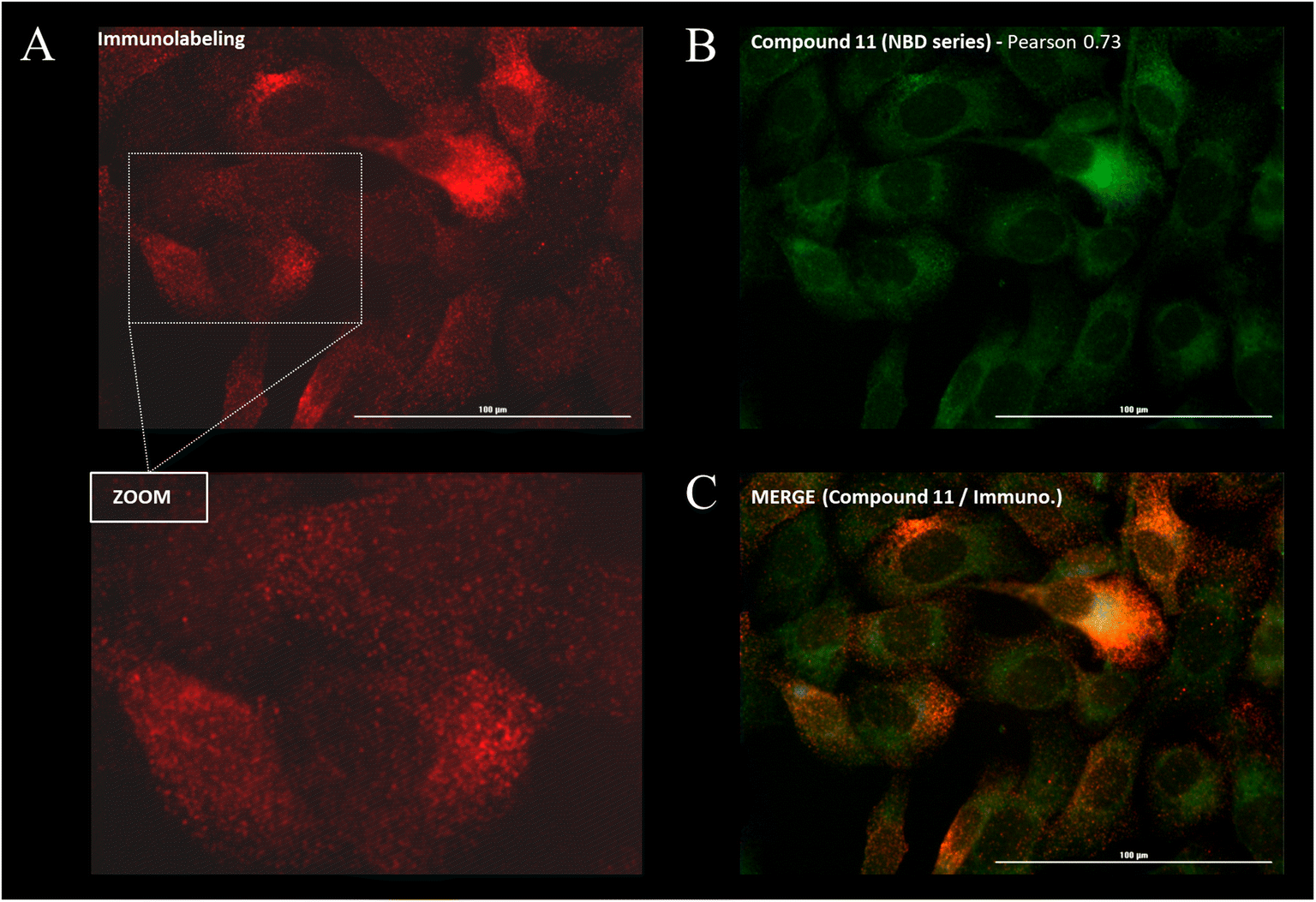 | ||
| Fig. 5 Intracellular distribution and immunofluorescence co-localization studies were performed by fluorescence imaging on SUM1315 cells for the 25 new P-gp-targeted conjugates. Cells were fixed, incubated with a fluorescent conjugate at 1 μM for 1 h, rinsed with PBS (3×) and then co-stained with a fluorescent anti-P-gp antibody before imaging on a Cytation™3 MV (BioTek®, USA). Images show characteristic staining patterns. (A) Immunostaining fluorescence pattern (red). The bottom panel (zoom) reveals the punctate pattern. (B) Compound 11 distribution (green). (C) Co-localization of compound 11 and immunostaining (orange). | ||
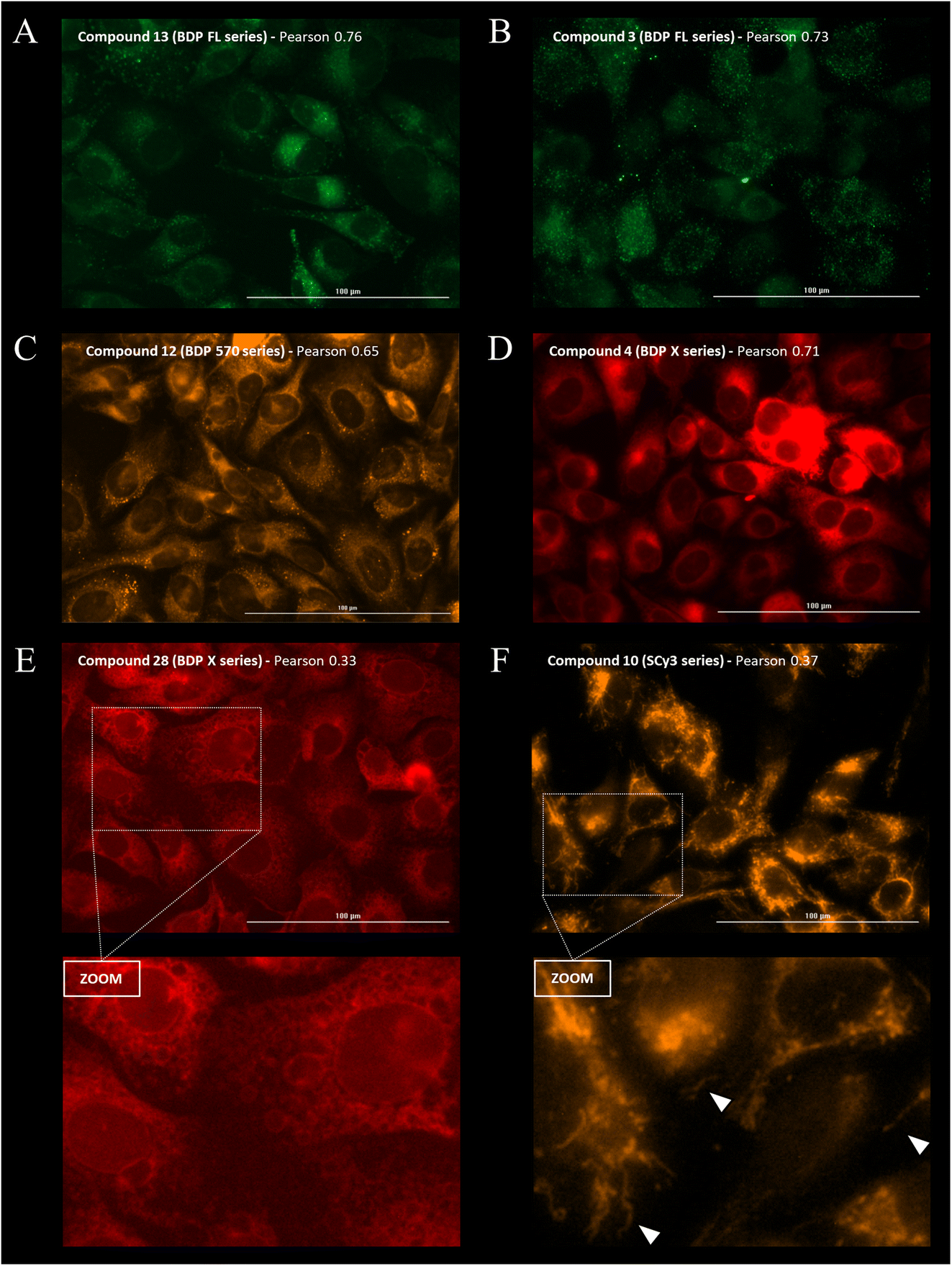 | ||
| Fig. 6 Intracellular distribution study was performed by fluorescence imaging for the 25 new P-gp-targeted conjugates. Cells were fixed, incubated with a fluorescent conjugate at 1 μM for 1 h, rinsed with PBS (3×) before imaging on a Cytation™3 MV (BioTek®, USA). Images show characteristic staining patterns for fluorescent conjugates of different series. (A) Compound 13 (green) distribution. (B) Compound 3 (green) distribution. (C) Compound 12 (orange) distribution. (D) Compound 4 (red) distribution. (E) Compound 28 distribution (red). The bottom panel (zoom) reveals staining of intracellular vesicle-like structures. (F) Compound 10 distribution (orange). The bottom panel (zoom) reveals staining of filamentous structures in the cytoplasmic region (white arrows). | ||
Compounds conjugated to the BDP X fluorophore gave a more diffuse signal throughout the cells with intense fluorescence from the perinuclear region, possibly in the endoplasmic reticulum (Fig. 6D/E). Interestingly, this was also observed upon incubation with BDP X NHS ester, indicating that this cell distribution might be partially driven by the fluorophore itself (Fig. S1†). Another striking feature that we noted is the localization of the fluorescent compounds at the membrane of intracellular vesicle-like structures, particularly visible for P2 derivatives 23, 28 and 33, for which Pearson coefficients were lower than for P1 derivatives 4 and 9 (Fig. 6D/E). Actually, it was previously reported that the use of the large and lipophilic BDP X fluorophore for small molecule labeling could strongly influence the pharmacokinetics and pharmacodynamics of the resulting compound, thus requiring optimized staining protocols with longer wash steps.33 In our study, we chose to compare the intracellular distribution of our conjugates following the same staining procedure regardless of the fluorophore. So, it appeared that while further investigations would be necessary in the case of some BDP X conjugated compounds in order to obtain high contrast images, conjugates 4 and 9 led to Pearson coefficients among the higher values, all series considered (Fig. 7).
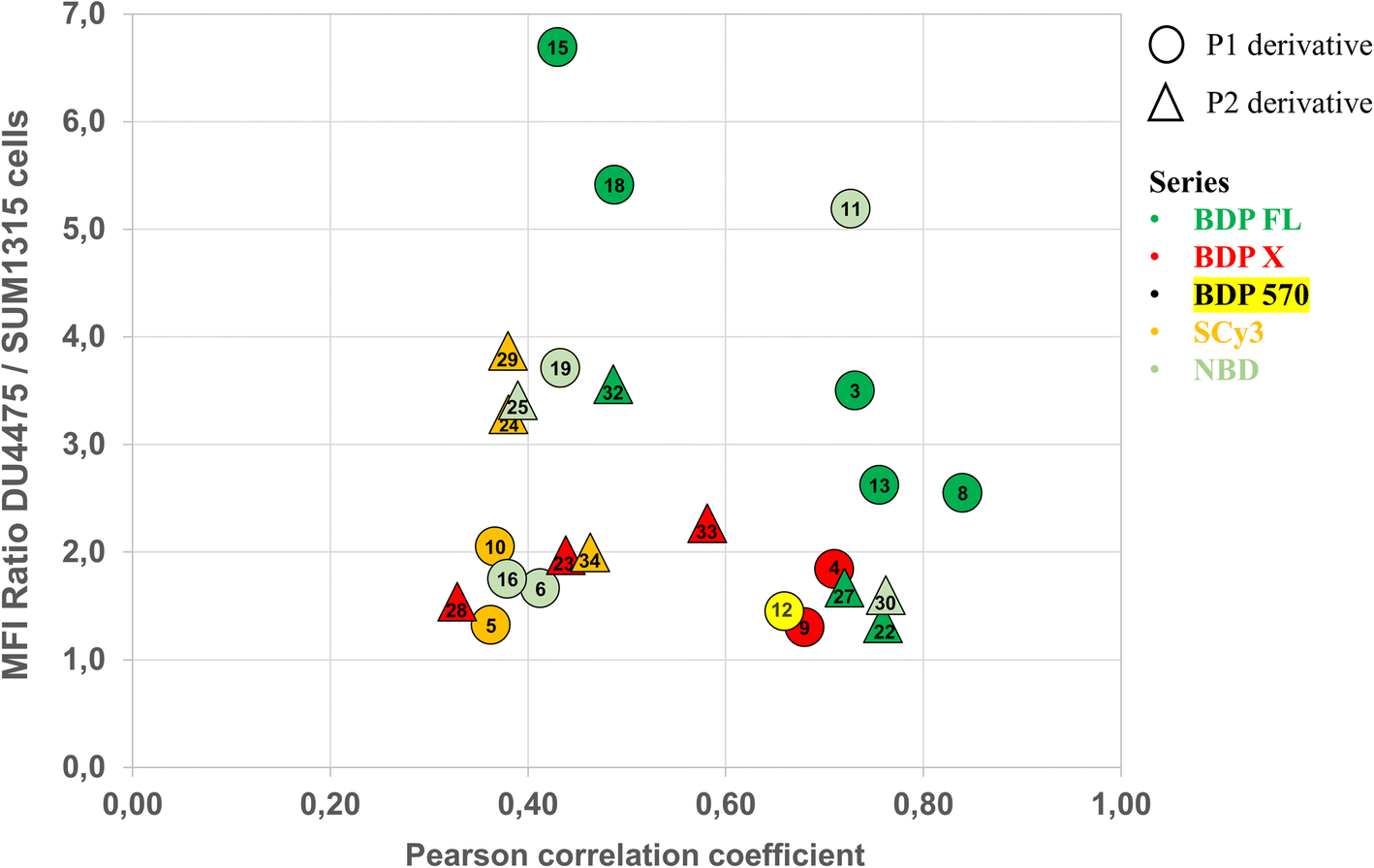 | ||
| Fig. 7 Diagram summarizing how structural features of the 25 new fluorescent conjugates (P1/P2 derivative and fluorophore structure) influence P-gp binding as determined by quantitative cell binding assay (cells MFI ratio) and intracellular distribution and immuno-fluorescence colocalization studies (Pearson correlation coefficient). | ||
Finally, staining with SCy3 labeled compounds led to a very different but characteristic pattern, which was observed for all compounds (5, 10, 24, 29 and 34), irrespective of structural considerations like the conjugation site for example (Fig. 6F). For this series of compounds, Pearson coefficients were low, varying from 0.36 to 0.46 with a mean value of 0.39, which indicates intracellular distribution and poor colocalization with the P-gp at the cell membrane, its main site of expression. Notably, the distribution of fluorescence was by no mean comparable to the nuclear staining observed in the control experiment with SCy3 NHS ester (Fig. S1†). This clearly indicated that distribution and cell binding were influenced by the peptidic targeting moiety. Precisely, the fluorescence distribution was not uniform but revealed instead filamentous structures in the cytoplasmic region, strongly reminiscent of mitochondria. Interestingly, it was previously reported that physiochemical properties for mitochondrial localization were closely related to those for P-gp inhibition, hence binding.34 Therefore, this series of SCy3 labeled compounds might be of interest to deepen this investigation and possibly clarify the link between distribution to mitochondria and P-gp expression.
Fig. 7 summarizes for all compounds the Pearson coefficient and the MFI ratio measured in the quantitative cell binding study. Taken together, cell binding and colocalization data revealed important SAR and uncovered the high capacity of several fluorescent conjugates – i.e. 3, 8, 13, 22, 27 in the BDP FL series, 12 in the BDP 570 series, 4, 9 in the BDP X series, and 11, 30 in the NBD series – to selectively target P-gp at the plasma membrane. Yet, it is important to acknowledge that in the field of fluorescent small molecules development, successful colocalization studies can be associated with higher Pearson values than those in the present report. Most certainly, it mainly stems from the ability of our compounds to permeate cells and target intracellular P-gp, unlike anti-P-gp antibodies, which are highly specific but inherently limited to targeting P-gp at the cell membrane. As mentioned earlier, it is undisputed that P-gp is mainly expressed at the plasma membrane where it plays its defensive role, but it is also well established that P-gp is localized in cellular organelles.1,35 Those intracellular localizations can be linked to synthesis and cell trafficking but can also mediate drug resistance, as evidenced by recent reports.35 Therefore, if our hypothesis is confirmed – further studies are underway – this will not only designate our compounds as innovative tools to study the role of P-gp in a large variety of samples via simple protocols, but also facilitate a deeper investigation of its implication in drug resistance with regards to its expression in intracellular organelles.
Conclusions
Following a rational design, we have conceived original multi-step synthetic routes to generate a library of 25 fluorescent compounds sharing a common peptidic structure derivatized with different aliphatic and aromatic groups, and displaying varying fluorophores. All compounds have been evaluated as P-gp targeted fluorescent conjugates in cell cultures. Most importantly, results of this investigation have clearly validated the P-gp targeting peptidic moiety as a versatile scaffold to build efficient conjugates, while confirming important SAR and generating new ones. In our methodology, with a relevant tumoral cell model and control immunostaining experiments, we have successfully identified compounds and experimental conditions allowing (i) the sensitive determination of a differential expression level between cell lines, and (ii) the selective detection of P-gp at the cell membrane, its main site of expression. This represents an original and innovative example of a small molecule-based approach for selective direct detection and quantification of P-gp in cells. We contend that this approach holds key advantages over the principal existing methods for protein detection such as immunofluorescence or immunoblotting, especially as it eschews the use of costly antibodies and the associated heavy, time-consuming protocols. Moreover, the varying lipophilicity of our new cell-permeant fluorescent conjugates revealed different staining patterns, which can be exploited to study intracellular P-gp in addition to its role at the plasma membrane. As a proof-of-concept reported herein, we have applied our methodology on simple models of cultured cells, but further studies will allow us to leverage the selectivity of our fluorescent conjugates for detecting and quantifying P-gp in a variety of more complex biological samples such as spheroids or clinical tumor samples. Ultimately, we are confident that the fluorescent conjugates reported herein will find innovative applications through simple flow cytometry or imaging based methodologies for detecting and quantifying P-gp. As an alternative to antibody-based methods, this approach will facilitate the preclinical and clinical evaluation of this relevant protein as a biomarker of resistance, especially in cancer treatments.Conflicts of interest
The authors declare the following competing financial interest(s): P. D., A. G., C. D., F. P. L., M. B. and E. M. are co-inventors on a patent application covering the method of P-gp detection, localization and quantification reported herein and as such may be entitled to a portion of any licensing fees and royalties generated by this technology. The authors declare no other potential conflict of interest relevant to this article.Acknowledgements
The authors thank the French government IDEX-ISITE initiative 16-IDEX-0001 (CAP 20-25, PRHI-3.2-CRI201216) and Région Auvergne-Rhône-Alpes (“Pack Ambition Recherche” 21.019016.01, ResLight Project) for partial funding. The authors are grateful to Martin Leremboure (UCA Partner) for technical assistance with HRMS experiments.Notes and references
- B. Sarkadi, L. Homolya, G. Szakács and A. Váradi, Physiol. Rev., 2006, 86, 1179–1236 CrossRef CAS PubMed.
- S. Lusvarghi, R. W. Robey, M. M. Gottesman and S. V. Ambudkar, F1000Research, 2020, 9, 1–12 Search PubMed.
- G. Szakács, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef PubMed.
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 48–58 CrossRef CAS PubMed.
- A. Palmeira, E. Sousa, M. H. Vasconcelos and M. M. Pinto, Curr. Med. Chem., 2012, 19, 1946–2025 CrossRef CAS PubMed.
- J. R. Vargas, E. G. Stanzl, N. N. H. Teng and P. A. Wender, Mol. Pharm., 2014, 11, 2553–2565 CrossRef CAS PubMed.
- R. W. Robey, K. M. Pluchino, M. D. Hall, A. T. Fojo, S. E. Bates and M. M. Gottesman, Nat. Rev. Cancer, 2018, 18, 452–464 CrossRef CAS PubMed.
- Z. Hamrang, Y. Arthanari, D. Clarke and A. Pluen, Microsc. Microanal., 2014, 20, 1329–1339 CrossRef CAS PubMed.
- Y. Mai, L. Dou, Z. Yao, C. M. Madla, F. K. H. Gavins, F. Taherali, H. Yin, M. Orlu, S. Murdan and A. W. Basit, Mol. Pharm., 2021, 18, 1895–1904 CrossRef CAS PubMed.
- M. Nedeljković, N. Tanić, M. Prvanović, Z. Milovanović and N. Tanić, Breast Cancer, 2021, 28, 727–736 CrossRef PubMed.
- M. Garland, J. J. Yim and M. Bogyo, Cell Chem. Biol., 2016, 23, 122–136 CrossRef CAS PubMed.
- T. Ueno and T. Nagano, Nat. Methods, 2011, 8, 642–645 CrossRef CAS PubMed.
- Y. Fu and N. S. Finney, RSC Adv., 2018, 8, 29051–29061 RSC.
- J. V. Jun, D. M. Chenoweth and E. J. Petersson, Org. Biomol. Chem., 2020, 18, 5747–5763 RSC.
- A. Rosati, L. Candussio, E. Crivellato, F. B. Klugmann, T. Giraldi, D. Damiani, A. Michelutti and G. Decorti, Cell. Oncol., 2004, 26, 3–11 CAS.
- E. Crivellato, L. Candussio, A. M. Rosati, F. Bartoli-Klugmann, F. Mallardi and G. Decorti, J. Histochem. Cytochem., 2002, 50, 731–734 CrossRef CAS PubMed.
- A. Sajid, N. Raju, S. Lusvarghi, S. Vahedi, R. E. Swenson and S. V. Ambudkar, Drug Metab. Dispos., 2019, 47, 1013–1023 CrossRef CAS PubMed.
- R. W. Robey, A. N. Robinson, F. Ali-Rahmani, L. M. Huff, S. Lusvarghi, S. Vahedi, J. M. Hotz, A. C. Warner, D. Butcher, J. Matta, E. F. Edmondson, T. D. Lee, J. S. Roth, O. W. Lee, M. Shen, K. Tanner, M. D. Hall, S. V. Ambudkar and M. M. Gottesman, Sci. Rep., 2021, 11, 1–15 CrossRef PubMed.
- R. Dufour, P. Daumar, E. Mounetou, C. Aubel, F. Kwiatkowski, C. Abrial, C. Vatoux, F. Penault-Llorca and M. Bamdad, Sci. Rep., 2015, 5, 12670 CrossRef CAS PubMed.
- A. Goisnard, P. Daumar, C. Dubois, C. Aubel, M. Roux, M. Depresle, J. Gauthier, B. Vidalinc, F. Penault-Llorca, E. Mounetou and M. Bamdad, Cancers, 2021, 13, 1–19 Search PubMed.
- E. Valton, I. Wawrzyniak, C. Amblard, B. Combourieu, M. L. Bayle, F. Desmolles, F. Kwiatkowski, F. Penault-Llorca and M. Bamdad, Biomarkers, 2017, 22, 566–574 Search PubMed.
- A. Goisnard, P. Daumar, C. Dubois, J. Gauthier, B. Vidalinc, F. Penault-Llorca, E. Mounetou and M. Bamdad, Fr. Pat., FR3101709 (A1), 2019, Eur. Pat. WO2021064191 (A1).
- O. Arnaud, A. Koubeissi, L. Ettouati, R. Terreux, G. Alamé, C. Grenot, C. Dumontet, A. Di Pietro, J. Paris and P. Falson, J. Med. Chem., 2010, 53, 6720–6729 CrossRef CAS PubMed.
- Z. Y. Kok, L. A. Stoddart, S. J. Mistry, T. A. M. Mocking, H. F. Vischer, R. Leurs, S. J. Hill, S. N. Mistry and B. Kellam, J. Med. Chem., 2022, 65, 8258–8288 CrossRef CAS PubMed.
- C. Milite, E. Barresi, E. Da Pozzo, B. Costa, M. Viviano, A. Porta, A. Messere, G. Sbardella, F. Da Settimo, E. Novellino, S. Cosconati, S. Castellano, S. Taliani and C. Martini, J. Med. Chem., 2017, 60, 7897–7909 CrossRef CAS PubMed.
- Z. L. Wang, F. F. Li, R. Quach, A. Ferrarese, A. Forgiarini, M. Ferrari, C. D'Amore, S. Bova, G. Orso, F. Fusi, S. Saponara, B. Hopkins, M. A. Brimble and D. Rennison, Bioorg. Med. Chem., 2022, 59, 116670 CrossRef CAS PubMed.
- L. J. Cruz, N. G. Beteta, A. Ewenson and F. Albericio, Org. Process Res. Dev., 2004, 8, 920–924 CrossRef CAS.
- G. Chaume, J. Simon, N. Lensen, J. Pytkowicz, T. Brigaud and E. Miclet, J. Org. Chem., 2017, 82, 13602–13608 CrossRef CAS PubMed.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, 2006, 1053–1082 Search PubMed.
- A. J. Ellison and R. T. Raines, Org. Biomol. Chem., 2018, 16, 7139–7142 RSC.
- A. Zumbuehl, D. Jeannerat, S. E. Martin, M. Sohrmann, P. Stano, T. Vigassy, D. D. Clark, S. L. Hussey, M. Peter, B. R. Peterson, E. Pretsch, P. Walde and E. M. Carreira, Angew. Chem., Int. Ed., 2004, 43, 5181–5185 CrossRef CAS PubMed.
- T. L. Bush, M. Payton, S. Heller, G. Chung, K. Hanestad, J. B. Rottman, R. Loberg, G. Friberg, R. L. Kendall, D. Saffran and R. Radinsky, Mol. Cancer Ther., 2013, 12, 2356–2366 CrossRef CAS PubMed.
- G. M. Thurber, T. Reiner, K. S. Yang, R. H. Kohler and R. Weissleder, Mol. Cancer Ther., 2014, 13, 986–995 CrossRef CAS PubMed.
- S. B. Fonseca and S. O. Kelley, ACS Med. Chem. Lett., 2011, 2, 419–423 CrossRef CAS PubMed.
- I. W. Gericke, G. Brandes and W. Löscher, Cells, 2022, 11(9), 1556, DOI:10.3390/cells11091556.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05093a |
| This journal is © The Royal Society of Chemistry 2023 |