
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEducational series: turning monomers into crosslinked polymer networks
M. A. Sachini N.
Weerasinghe
a,
Obed J.
Dodo
 a,
Chamoni W. H.
Rajawasam
a,
Ibrahim O.
Raji
a,
Shiwanka V.
Wanasinghe
a,
Dominik
Konkolewicz
*a and
Nethmi
De Alwis Watuthanthrige
*b
a,
Chamoni W. H.
Rajawasam
a,
Ibrahim O.
Raji
a,
Shiwanka V.
Wanasinghe
a,
Dominik
Konkolewicz
*a and
Nethmi
De Alwis Watuthanthrige
*b
aDepartment of Chemistry and Biochemistry, Miami University, 651 E High St, Oxford, OH 45056, USA. E-mail: d.konkolewicz@miamiOH.edu
bLaboratory of Polymeric Materials, Department of Materials, ETH Zurich, Vladimir-Prelog-Weg 5, Zurich 8093, Switzerland. E-mail: nethmi.dealwis@mat.ethz.ch
First published on 20th September 2023
Abstract
Multifunctional monomers enable the synthesis of polymer networks by adapting the polymerization methods used for conventional linear polymer synthesis. Both step and chain polymerization methods can be combined with multifunctional monomers to generate networks with essentially infinite molecular weight. Traditional networks are held together through covalent crosslinks that derive from the multifunctional monomers. Additional approaches to network like materials involve polymer phase separation, forming non-covalent crosslink-points. This contribution provides an overview of the approaches and chemistries available for polymer network synthesis, and serves as a resource for researchers exploring polymer networks or attempting alternative approaches to network development.
Introduction
Polymer networks are used in many applications, ranging from high performance thermosets, elastomers, adhesives, and hydrogels which can have biological uses. Crosslinks are the key feature of polymer networks, which distinguishes them from other types of polymeric structures.1,2 These crosslinks bind the polymer chains together creating a percolated, or space occupying mesh.3 This generates a polymer where in principle the macroscopic scale material is a single molecule, with an incredibly high molecular weight that approaches infinity. In addition to generating a very high molecular weight polymer, the crosslinks between polymer chains alter the materials properties. Unlike conventional isolatable macromolecules, polymer networks are no longer soluble, but rather only capable of swelling in a good solvent.4 Networks also resist flow and maintain structural stability, even above any melting or glass transition temperatures. This makes polymer networks appealing for high performance applications, but the presence of crosslinks can make their synthesis substantially more challenging, and additionally, many of the characterization tools favored by chemists are no longer applicable.5The first synthesis of polymer networks coincides with the generally accepted first synthesis of Bakelite in 1909.6,7 Bakelite is synthesized by the condensation of phenol with formaldehyde, as shown in Scheme 1. Subsequently, various approaches have been developed for the synthesis of crosslinked macromolecules. The common building blocks used to synthesize polymer network are multifunctional monomers. These multifunctional monomers act as branching points, which eventually connect multiple polymer chains to form crosslinked networks. Networks can be synthesized by both chain and step polymerizations. In chain polymerization, also called addition or chain growth polymerization, there is an active end group, such as a radical, anion, cation or metal, that adds monomer and transforms the previously active monomer to an inert unit and converts the monomer to the same type of active end group. In step polymerizations, also called step growth polymerization, the monomers react with each other without requiring a specific active end group, and the generated oligomers typically react with one another to create even higher molecular weight polymers. Note that IUPAC terminology separates step polymerization into polycondensation or polyaddition, depending on whether there is a small molecule released upon polymer coupling.8,9
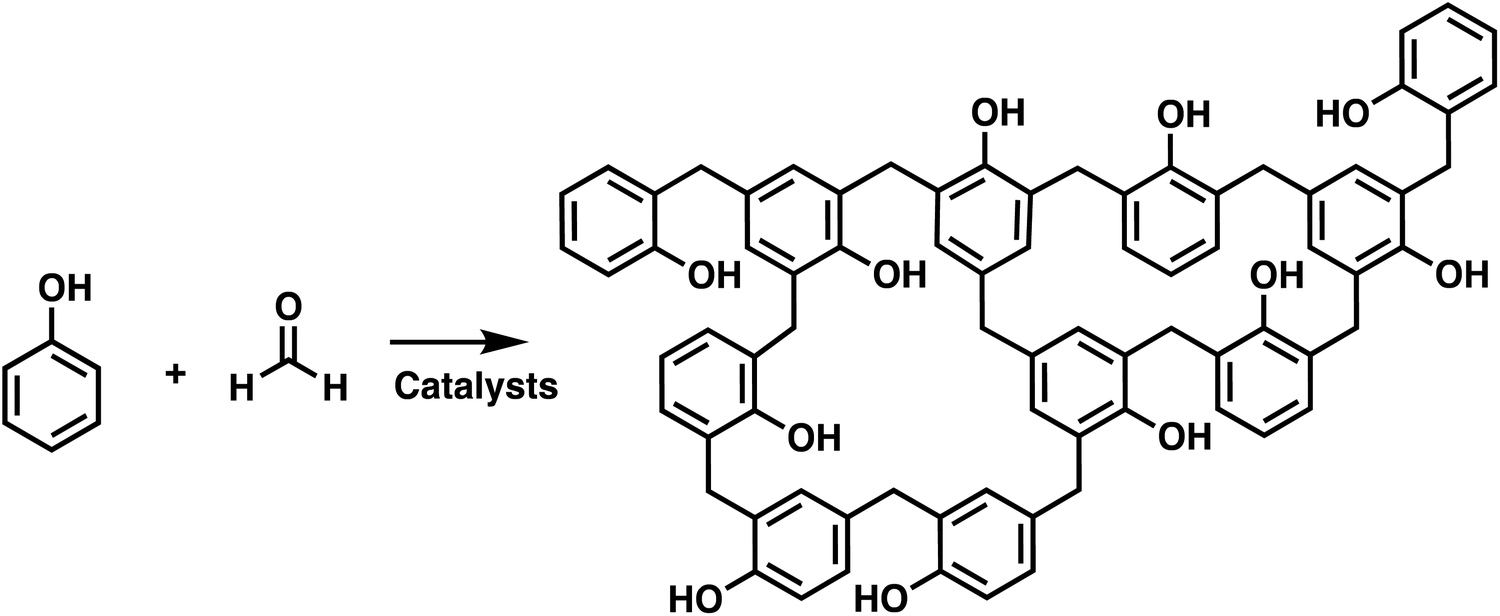 | ||
| Scheme 1 Synthesis of Bakelite by condensation of phenol and formaldehyde.6 | ||
Although both chain and step polymerization processes have been used to synthesize linear polymers, the addition of multifunctional monomers, transitions from the synthesis of linear chains towards branched and eventually crosslinked polymers. In this series of 2 articles, an overview of the synthesis and characterization of polymer networks is presented. The synthetic approaches to polymer networks through various methods and estimates of the transition from soluble (sol) to gelled (gel) state are given by various syntheses. Common chemistries used in network synthesis are highlighted. In the second contribution to this series, the methods used to characterize networks are presented.5 These two articles on polymer networks together are designed as a framework for researchers to design their own polymer networks and an overview on the methods available to evaluate their properties. These two articles can serve as a first entry point into polymer networks and complement many excellent reviews in the field,1,3,10 which can serve as sources for detailed characterization or synthesis beyond the focus of these shorter contributions.
Overview of synthesis of polymer networks
The synthesis of polymer networks has facilitated the design and instigation of multifunctionality in polymeric materials, making them highly appealing for a wide range of demanding emerging applications including coatings, self-healing materials, shape memory systems, optical indicators and sensors.11A crosslinked polymer network possesses a minimum of two pathways connecting most points within the polymers. The extensive crosslinking fosters the development of three-dimensional mesh-like structures, thereby introducing multifunctionality into polymer chains (Scheme 2a). This transition in topology entails a shift from linear chains to branched polymers and ultimately leads to the formation of networks with more intricate architectures such as single double and triple networks.12 Further, network structures can be varied in perfection, resulting in perfect, almost perfect to imperfect networks (Scheme 2b).2
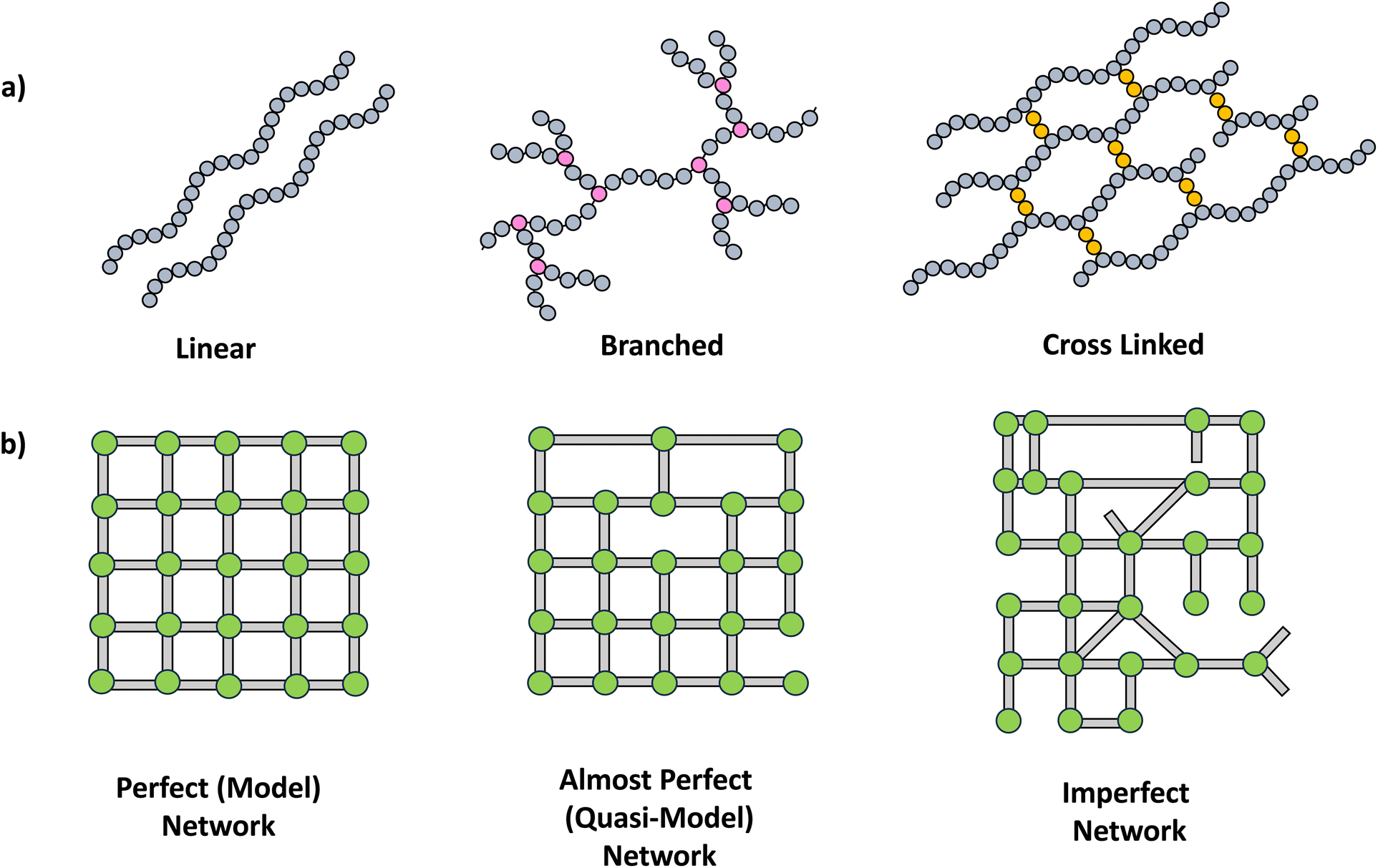 | ||
| Scheme 2 a) Schematic drawing of polymer structures, linear, branched and crosslinked. (b) Types of networks. | ||
The synthesis of polymer networks involves either polymerization with a multifunctional monomer or a pre-synthesized polymer featuring crosslinkable units in the backbone or at the chain ends. Crosslinks between these monomers or pre-polymers can be established through various bond types, including static covalent bonds, dynamic covalent bonds (disulfide linkages, Diels–Alder, transamination, transesterification, olefin metathesis), as well as physical interactions (hydrophobic interactions, van der Waals interaction, metal ligand coordination and coulombic interactions).13 These bond types can be utilized individually or in combinations to enhance the functionality of the polymer networks.14 The resulting properties of the networks, range from gels and elastomers to thermosets and thermoplastic elastomers, depending on the monomers and chemistries employed.3
Crosslinking can be facilitated through a diverse range of techniques such as light irradiation, thermal conditions, pH, addition of salts, electrochemistry, sonochemistry.1,2,15–18 However, the network properties fundamentally rely on the selection of appropriate chemistries and optimization of the crosslinking conditions, and control over the crosslink density.
This overview primarily focuses on four distinct synthetic routes for polymer network formation. The four routes discussed are: step polymerization of multifunctional monomers, chain polymerization in the presence of a multifunctional crosslinker, polymerization of functional monomers coupled with crosslinking through step growth mechanisms, and self-assembly processes exemplified by urethanes.
Step polymerization processes
In step polymerization, bifunctional or multifunctional monomers with mutually reactive groups react with each monomer in the absence of activating groups. Monomers first form dimers, trimers, oligomers and eventually form high molecular weight polymers.19,20 At the initial stages of polymerization, monomers are consumed rapidly with the initial low degree of polymerization due to formation of short fragments. These fragments grow continuously throughout coupling steps increasing the degree of polymerization. Step polymerization can occur through condensation with the elimination of small molecules or addition by rearrangement and without the elimination of small molecules.21 Polyesters, polyamides and polyethers are some of common examples for polymers made by condensation, while polyurethanes and epoxy resins are some important polymers made by addition. Step polymerization is capable of forming both linear polymers and polymer networks based on the functionality of the monomer/monomers. Linear step polymerization is extensively applied to synthesize commercially important polymers such as poly(ethylene terephthalate) and nylon.22,23 In linear step polymerization, as depicted in Scheme 3, difunctional monomers (such as AB or A2 + B2) undergo stepwise reactions, resulting in the formation of difunctional dimers, trimers, tetramers, or oligomers with reactive end groups at both ends. These entities then proceed to grow into linear polymer chains.19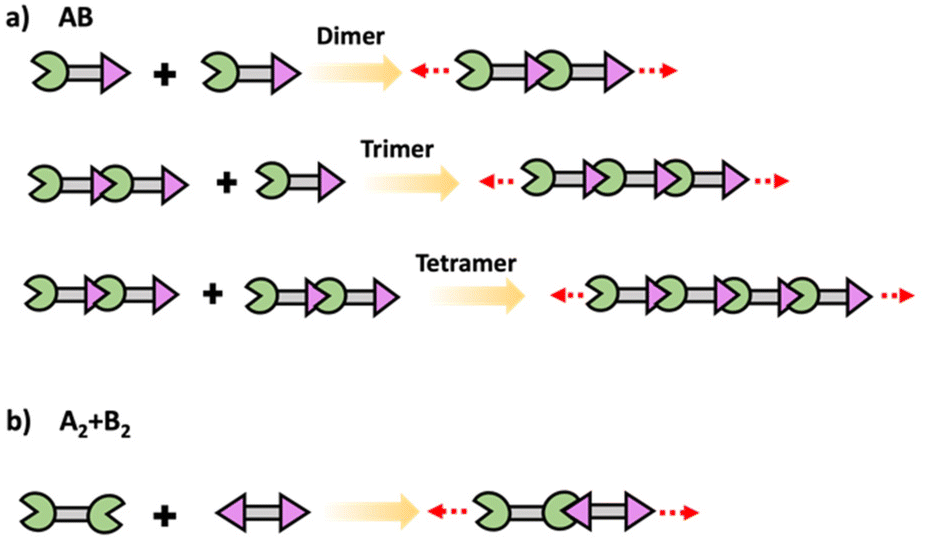 | ||
| Scheme 3 Schematic of conventional step polymerization to yield linear polymers using difunctional (a) AB and (b) A2 + B2 monomers. Formation of difunctional dimers, trimers and tetramers are shown in (a) Reactive functional groups are represented as A and B. | ||
Crosslinked network polymers can be synthesized using multifunctional monomers including A3 + B2, A3 + B3 or A4 + B2 in step polymerization and as schematically drawn in Scheme 4.22 Polymer chains grow similar to that in linear polymer formation. However, resulting oligomers can have the same number or even more numbers of reactive end groups than the original monomer, thus potentially accelerating the growth of that macromolecule compared to linear step polymerization. Each time a multifunctional monomer incorporates into the backbone and reacts with monomers a branch point is formed. These branch points act as crosslinks and ultimately result in a crosslinked polymer network as shown in Scheme 4.
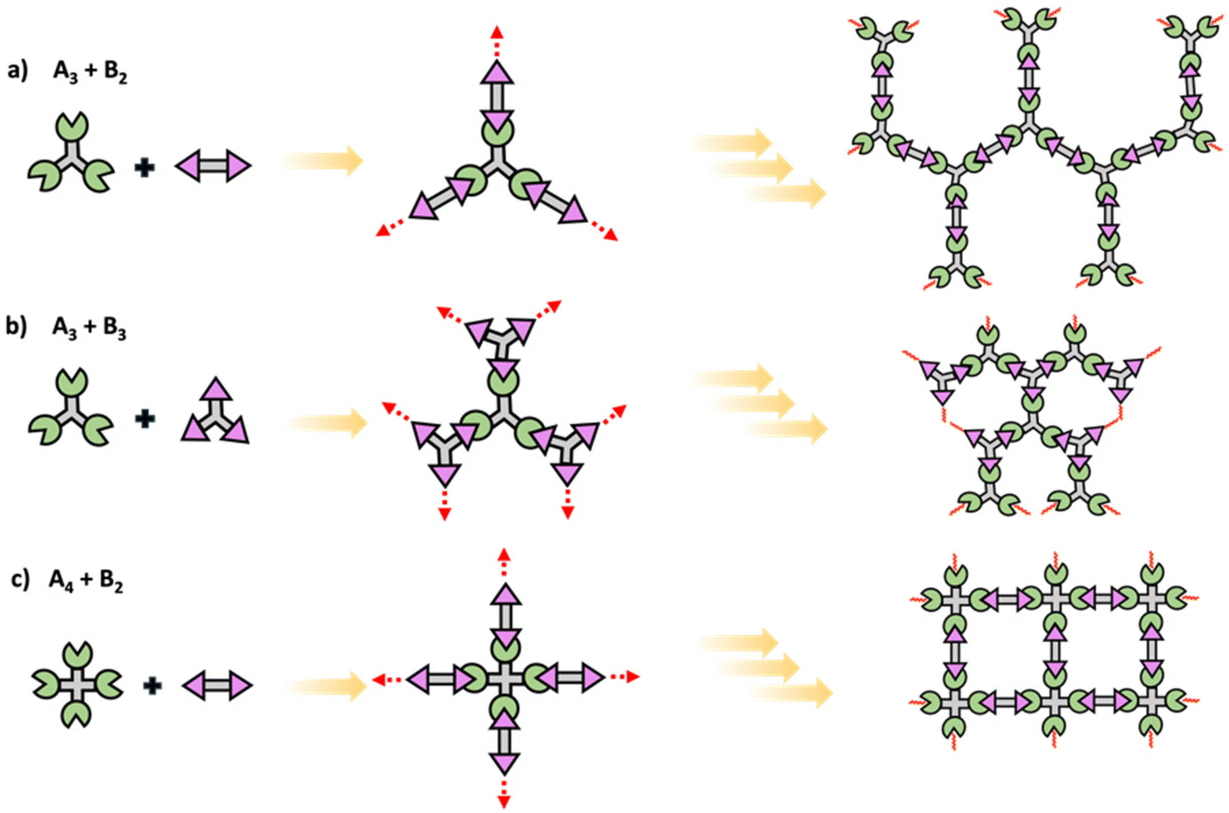 | ||
| Scheme 4 Schematic of conventional step polymerization to yield polymer networks using multifunctional (a) A3 + B2, (b) A3 + B3, and (c) A4 + B2 monomers. | ||
Further, as the reaction progresses the number average molecular weight (xn) increases and can be approximated under ideal conditions using Flory theory as shown below for step polymerization.22,24,25
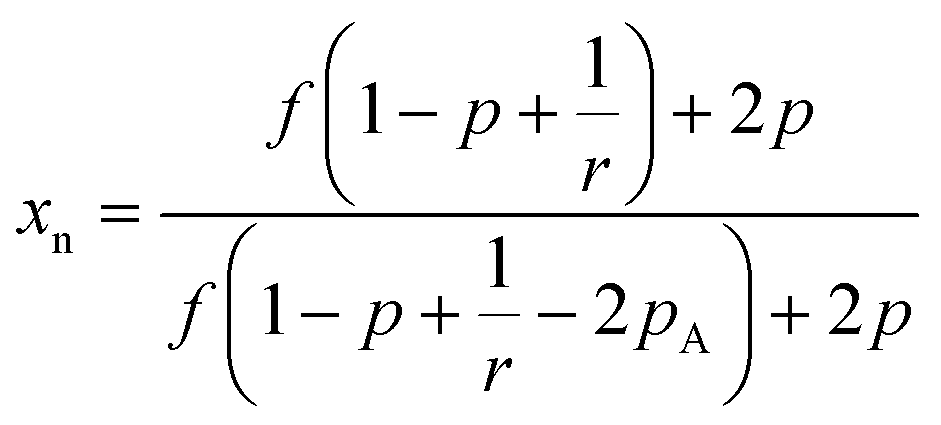 | (1) |
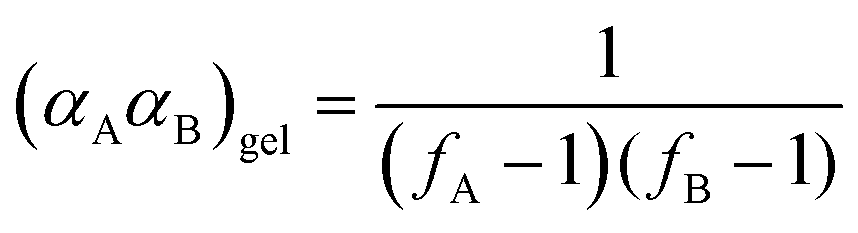 | (2) |
As indicated in the equation above at a critical conversion, xn approaches infinity and at this point gelation occurs and forms the first network. This point is known as the gel point, at this point a flowy liquid reaction mixture turns into a gel resulting in an immobilized polymer network.19 The conversion at the gel point can be determined by following the eqn (2). Further, the formation of loops can be expected in practical situations removing reactive end groups and in that case the conversion needed to reach gelation will be higher than that predicted by the equation above.
Many commercially used polymers such as epoxy–amine networks,26–28 poly(urethane) networks,29 and formaldehyde networks7 are synthesized using multifunctional step polymerization. Structures and synthesis of those polymer networks are illustrated in Scheme 5. Usually, difunctional monomers are reacted with monomers with functionality greater than 2 and mostly either one of them are prepolymers with two/multiple functional groups. Epoxy–amine networks are synthesized by first reacting epichlorohydrin and bisphenol A to prepare prepolymers with epoxide end groups followed by reacting the prepolymer with multifunctional amine monomers through addition reaction between epoxide and amine.26,27 Diisocyanates and multifunctional alcohols (polyols prepolymers) are utilized to prepare poly(urethane) networks which are important as elastomers and for their application in synthetic forms.28 Similar to epoxy–amine and poly(urethane) network synthesis, usually difunctional formaldehydes react with phenol, urea, or melamine with functionality higher than 2 to result phenol-formaldehyde networks, urea–formaldehyde networks, or melamine–formaldehyde networks respectively. Bakelite is a well-known thermosetting polymer which made from phenol and formaldehyde condensation.7 In addition to these traditional polymer networks made by step polymerization, various approaches such as thiol–ene or thiol–yne polymerization are developed recently to prepare network polymers by reacting multifunctional thiol monomers with -ene or -yne monomers as shown in Scheme 5.30 Further, it is noted that these new approaches are capable of forming a highly crosslinked polymer network with a delayed gel point and a homogeneous network through a radical mediated step growth mechanism.31–33
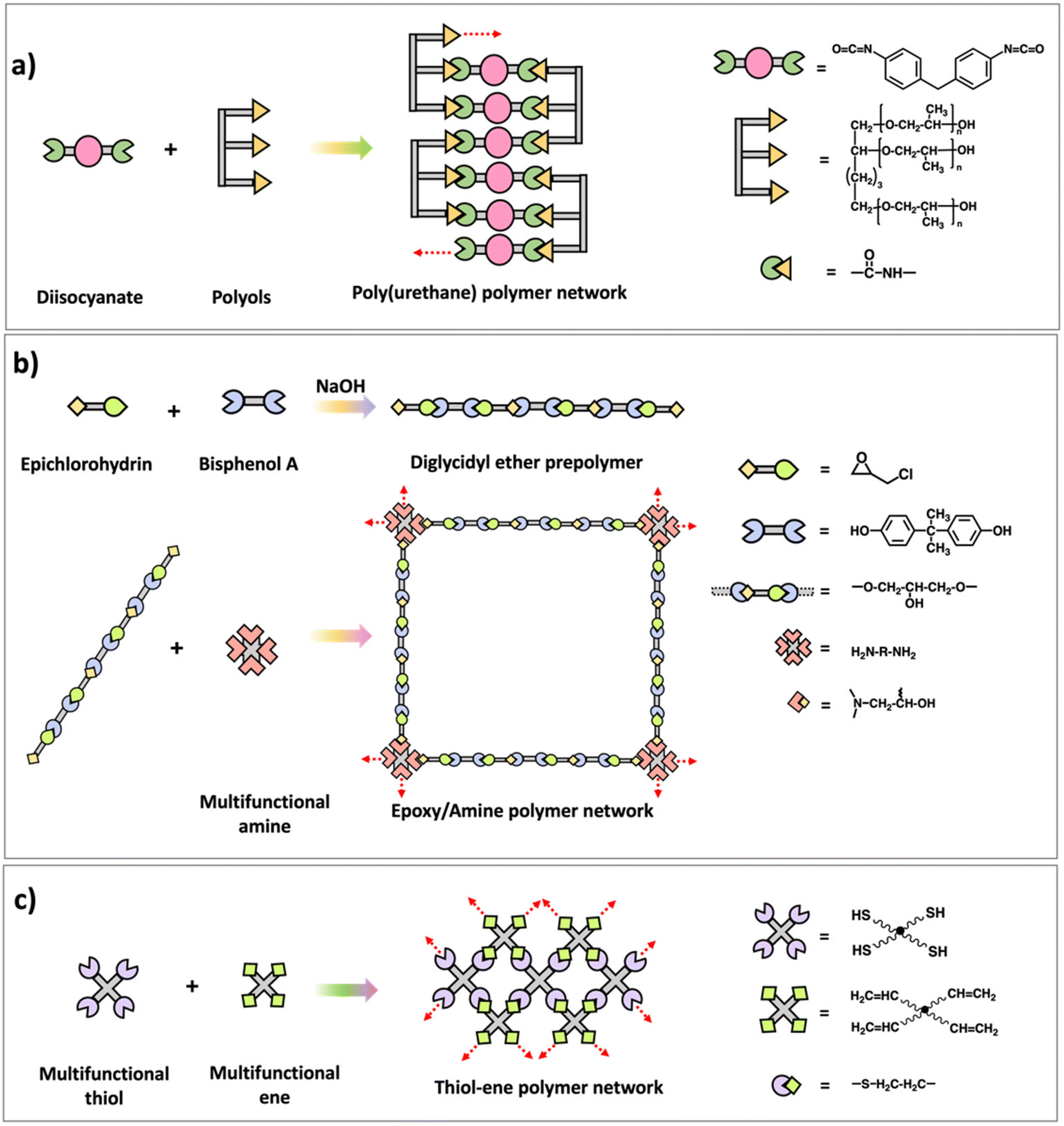 | ||
| Scheme 5 Synthesis scheme of (a) poly(urethane) polymer network, (b) epoxy–amine polymer network, and (c) thiol–ene polymer network. | ||
Chain polymerization processes
In chain polymerization, polymer growth occurs by sequential addition of unsaturated monomers to a terminal active center. Examples of chain polymerization processes include free radical, cationic, anionic, and metathesis. The reactive end group attacks the monomers resulting in a longer chain and a newly created active center. This is shown in Scheme 6a for free radical polymerization. When simple monofunctional monomers are employed, linear chains are obtained. However multifunctional monomers could lead to branched and even hyperbranched polymers which can eventually result in networks. In principle, even a difunctional monomer is sufficient for making polymer networks by chain polymerization.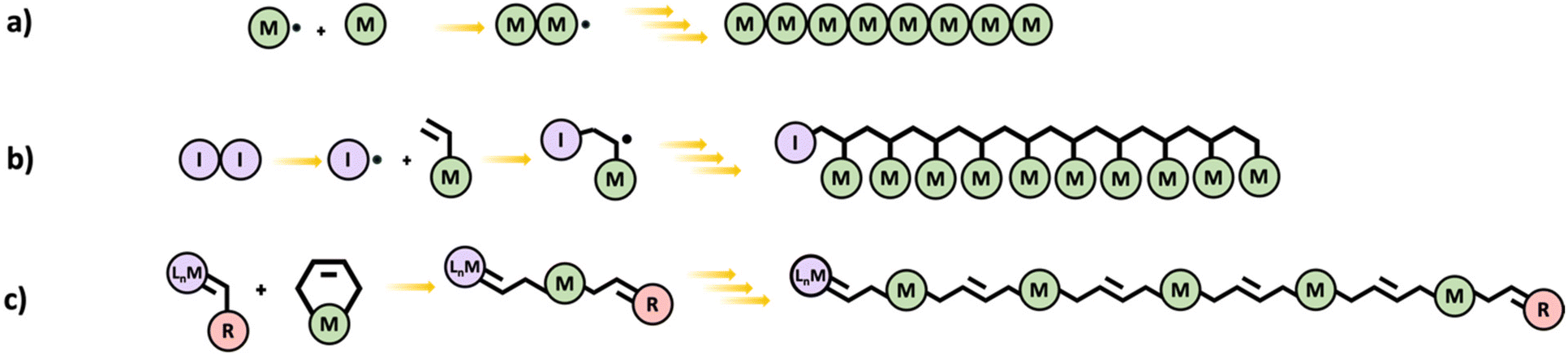 | ||
| Scheme 6 Schematic diagrams of (a) chain polymerization (generic mechanism), (b) vinyl polymerization, and (c) ROMP. | ||
Free radical polymerization (FRP) proceeds via consecutive addition of monomers (almost exclusively vinyl monomers) to an active free-radical center.34 The reaction can be initiated by thermal, chemical, or photoactive initiator molecules. FRP begins at the initiation stage which involves two steps. Initially, the initiator undergoes homolytic splitting into two free radicals and in the second step, one of the free radicals adds on to a monomer. Propagation is the next stage of FRP, and it involves a sequentially rapid addition of monomers to the active free-radical center of a polymer leading to growth of the polymer chain. In the termination stage, growth of a polymer chain is terminated by destruction of the active center either through a process called bimolecular reaction or disproportionation (Scheme 6b).
Cationic polymerization of vinyl monomers occurs by the creation of cationic active centers from the reaction of monomers with electrophiles at the initiation stage.35 Propagation proceeds predominantly via successive head-to-tail addition of vinyl monomers to a carbocation active center and forming a new carbocation in the process. This process continues until available monomers are consumed or until growing polymer chains terminates, which is the final stage. Unlike FRP where active centers bear no electronic charge, in cationic polymerization, termination can occur by either unimolecular rearrangement of ion pair or by chain transfer.
In anionic polymerization, initiation involves a nucleophilic attack on a monomer by an electron rich or anionic (negatively charged group) initiator resulting in the formation of a reactive carbanion. Hence initiation can be achieved by addition of either a negative (Nu:−) or neutral (Nu:) nucleophile to a monomer and a good example for this process is the addition of amide to styrene. Termination can be achieved by proton abstraction. In the case of a growing styrene chain, termination occurs by chain transfer to solvent, wherein proton is abstracted from ammonia leading to termination of chain growth.36
Olefin metathesis is catalyzed in the presence of a transition metal complex and Scheme 6c gives a generally representation of the reaction.37 Ring-opening metathesis polymerization (ROMP) however employs the use of metal carbenes in the presence of homogeneous catalysts to prepare uniform and stereoregular linear polymers and copolymer chains from cyclic unsaturated alkenes such as cyclobutene, cyclopentene, norbornene, etc.
Ring-opening polymerization (ROP) is another form of chain polymerization that can be used to prepare polymers that are not easily accessible by other methods.38 For this process, terminal end group of the polymer chain acts as the active center responsible for the addition of cyclic monomers by ring-opening reactions. Relief of steric repulsion and bond-angle strain are the main driving force for ring-opening in cyclic monomers. ROP typically requires an initiator and proceeds various mechanisms including anionic, cationic, free-radical, and metathesis polymerizations.
The functionality of a monomer indicates the number of chain links that can be obtained and does not necessarily depend on the number of functional groups present in the monomer. Monofunctional monomers lead to materials that chain extend and do not experience cross-linking or branching. Such materials often have lower viscosity, flexibility of formula, and better adhesion potential due to the linear structure of resulting polymer chains. Adding multifunctional monomers, typically difunctional, introduces branching points, which can expand to crosslinks as the network continues to form. Branching and crosslinking must occur by a sequence where first the difunctional monomer incorporates into the growing chain, forming a pendant group, followed by incorporation of the pendant group into a second growing polymer chain (Scheme 7). Although possible, difunctional monomers can typically be combined with monofunctional ones in these polymerizations depending on targeted material property.
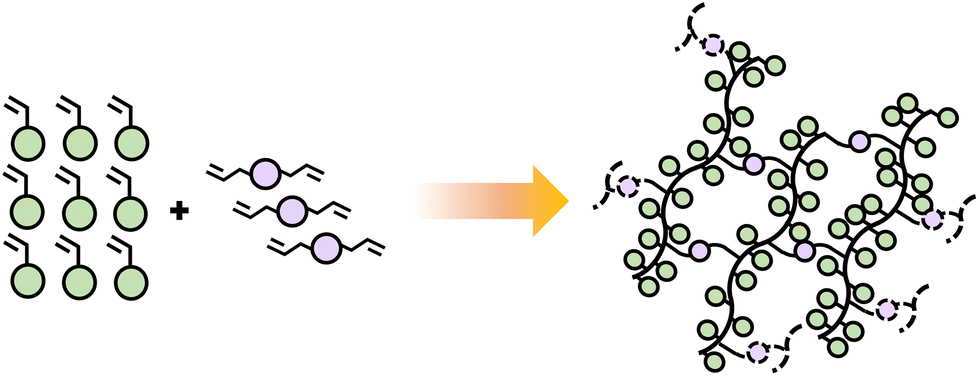 | ||
| Scheme 7 Schematic diagram illustrating the polymerization of difunctional monomers combined with monofunctional, forming linear chains and networks. | ||
It is important to note that not all chain growth mechanisms are equal since they produce polymers with varying backbone uniformity. FRP gives networks with relatively low uniformity, due to fast reaction rates, leading to pockets of high crosslink density and pockets of long chains connecting the densely crosslinked clusters. Reversible–deactivation radical polymerization (RDRP) can be used to achieve better control of chain growth leading to uniformity in chain length of polymer backbone.39 In RDRP, the active site of the polymer chain is retained upon deactivation; instead, it can resume propagation, leading to increased chain length when reacting with monomers. Typically, this process involves a simultaneous initiation of chain growth leading to a narrow molar mass distribution of polymer chains in contrast to the broader distributions observed in FRP. Examples of RDRP techniques include reversible-addition–fragmentation chain-transfer radical polymerization (RAFT) and atom-transfer radical polymerization (ATRP). Scheme 8a gives the distinction in polymer synthesis using the aforementioned RDRP methods in comparison to FRP. Up until now, several groups have published detailed reviews presenting in-depth discussion of RDRP methods, along with multiples examples from the literature.40–42
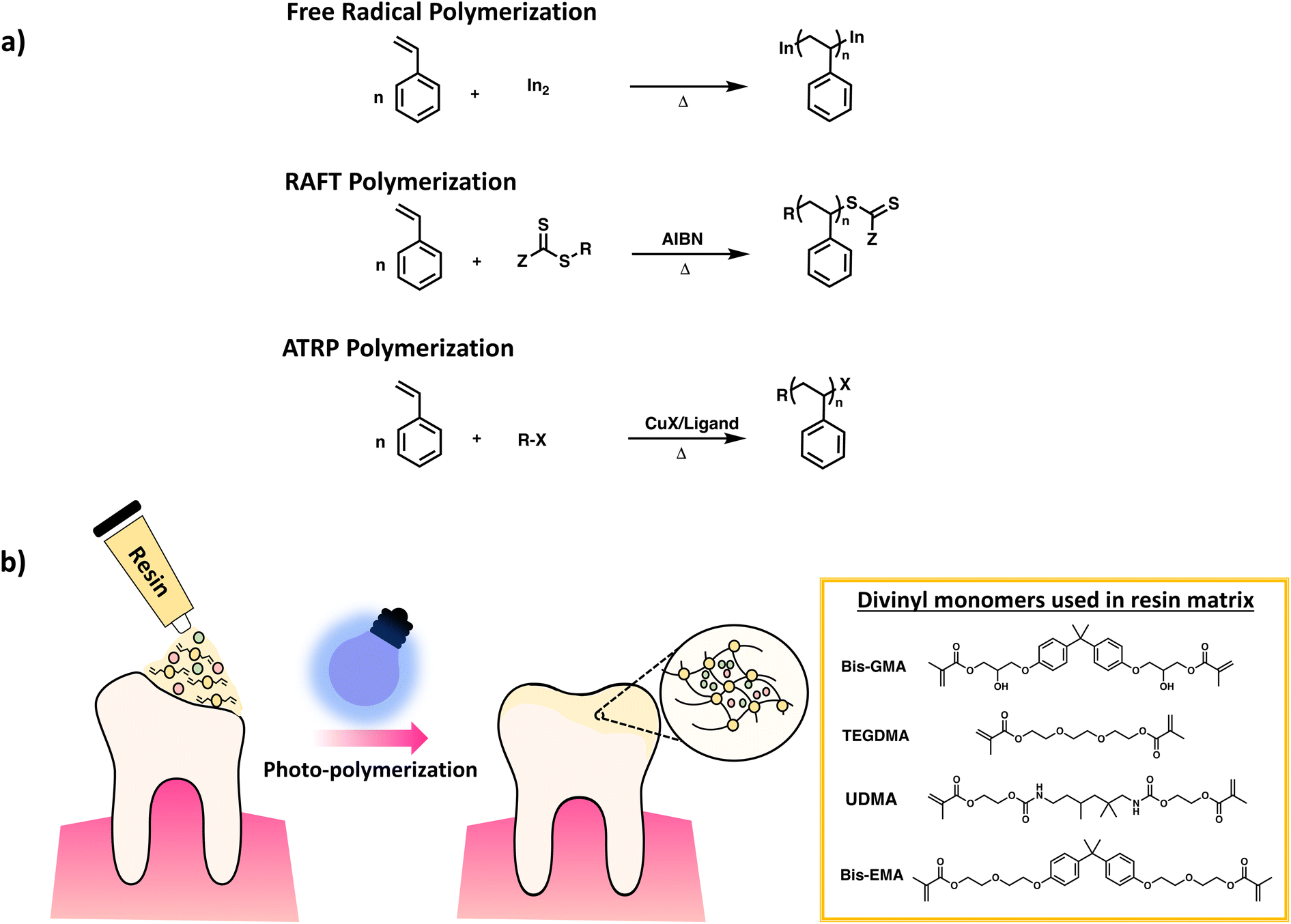 | ||
| Scheme 8 Schematic diagram of (a) synthesis of poly(styrene) via FRP, RAFT and ATRP. (b) Crosslinking of DVMs for dental resins reproduced with modifications from ref. 50 with permission from John Wiley and Sons, copyright 2021. | ||
Gelation or gel transition in non-linear polymers is a key factor to consider under chain polymerization. This usually occurs in branched or network polymer wherein continued linking leads to an immobilized single macroscopic molecule at a point now referred to as gel point.43 At this point, the gel loses fluidity and becomes highly viscous, resulting in a gel network that swells in contrast to dissolving in solvent.44 Gelation can be predicted by several approaches which include the Carothers theory of gelation and the Flory–Stockmayer (FS) theory of gelation now adapted as the statistical approach.45 In free radical polymerization involving a mixture mono vinyl monomers (MVM) and divinyl monomers (DVMs), the critical vinyl group conversion at the gel point (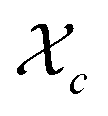 ) can be estimated using the eqn (3).46
) can be estimated using the eqn (3).46
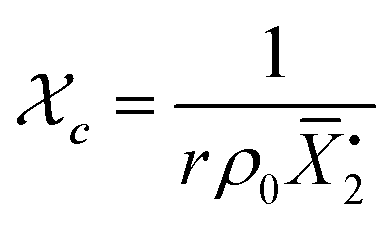 | (3) |
In this equation, ρ0 represents the initial molar fraction of the vinyl groups contributed by the DVMs, 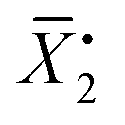 denotes the average chain length of the primary molecule and r signifies the unequal reactivity of the vinyl groups between MVM and DVM.
denotes the average chain length of the primary molecule and r signifies the unequal reactivity of the vinyl groups between MVM and DVM.
Through application of the FS theory, the theoretical gel point of the RDRP systems can be calculated by using the eqn (4).47,48
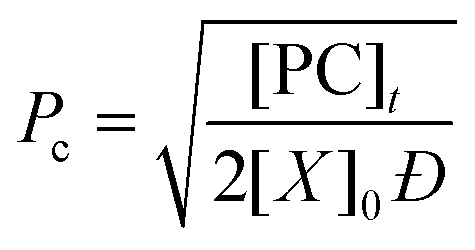 | (4) |
Examples of chain growth polymerization mostly focus on divinyl polymerization, often by radical polymerization due to simplicity of use. Industrially used examples of chain polymerization leading to crosslinked networks includes hydrogels such as contact lenses, polyacrylamide gels, ion exchange resins, dental resins (Scheme 8b), and GPC column packing materials3,49,50
Post polymerization crosslinking
Crosslinking of polymers is an essential process in the field of polymer science that transforms branched or linear polymer chains into polymeric network structures.51 Post-polymerization crosslinking is a highly effective processes of forming polymeric network structures after the primary chain is synthesized.52 Crosslinking after polymerization can be successfully accomplished on polymers containing pendant reactive groups (Scheme 9).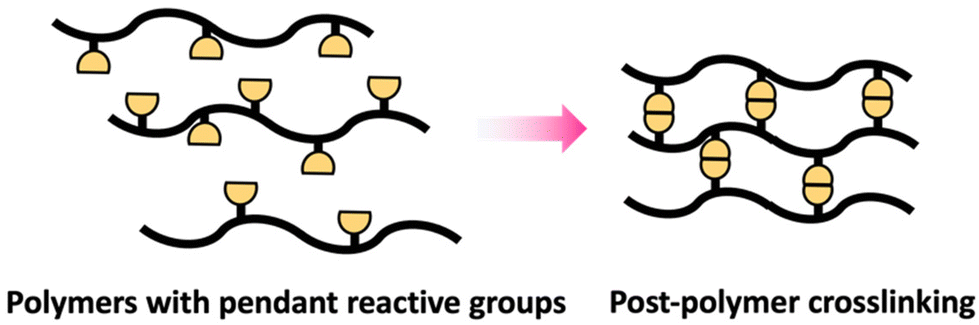 | ||
| Scheme 9 Schematic of polymers containing pendant reactive groups. | ||
These polymers are generally designed to include reactive pendant groups along the polymer chain's backbone.53 Epoxy groups are an example that can be used as pendant group in polymer chain due to their shelf stability and ability to efficiently react with suitable nucleophiles like amines and alcohols.54 The crosslinking degree can be modulated to attain desired qualities via controlling the crosslinking agent's stoichiometry and conditions of reaction.53 Additionally, post-polymerization can be achieved through polymer with reactive terminal groups as in telechelic polymer.55
Similar to polymers with pendant reactive groups, these terminal groups enable highly regulated crosslinking and post-polymerization crosslinking can be attained through a variety of methods. This could be accomplished through reacting the terminal group with a crosslinking agent to form a crosslinked network (Scheme 10b) or interaction between terminal groups on different polymer chains (Scheme 10a).55 The advantage of the telechelic polymer approach is that the molecular weight between crosslinks can be regulated by the primary polymer length. Selecting suitable functional groups that comprise the pendant or reactive group is an important feature of these approaches. They should not only be stable during and after the initial polymerization, but they should also have high reactivity for post-polymerization crosslinking.51–53 Generally, isocyanates,56 amines,57 and epoxides54 are some of the suitable functional groups that have been extensively explored for post-polymerization crosslinking resulting in polymer networks with unique properties for targeted applications.
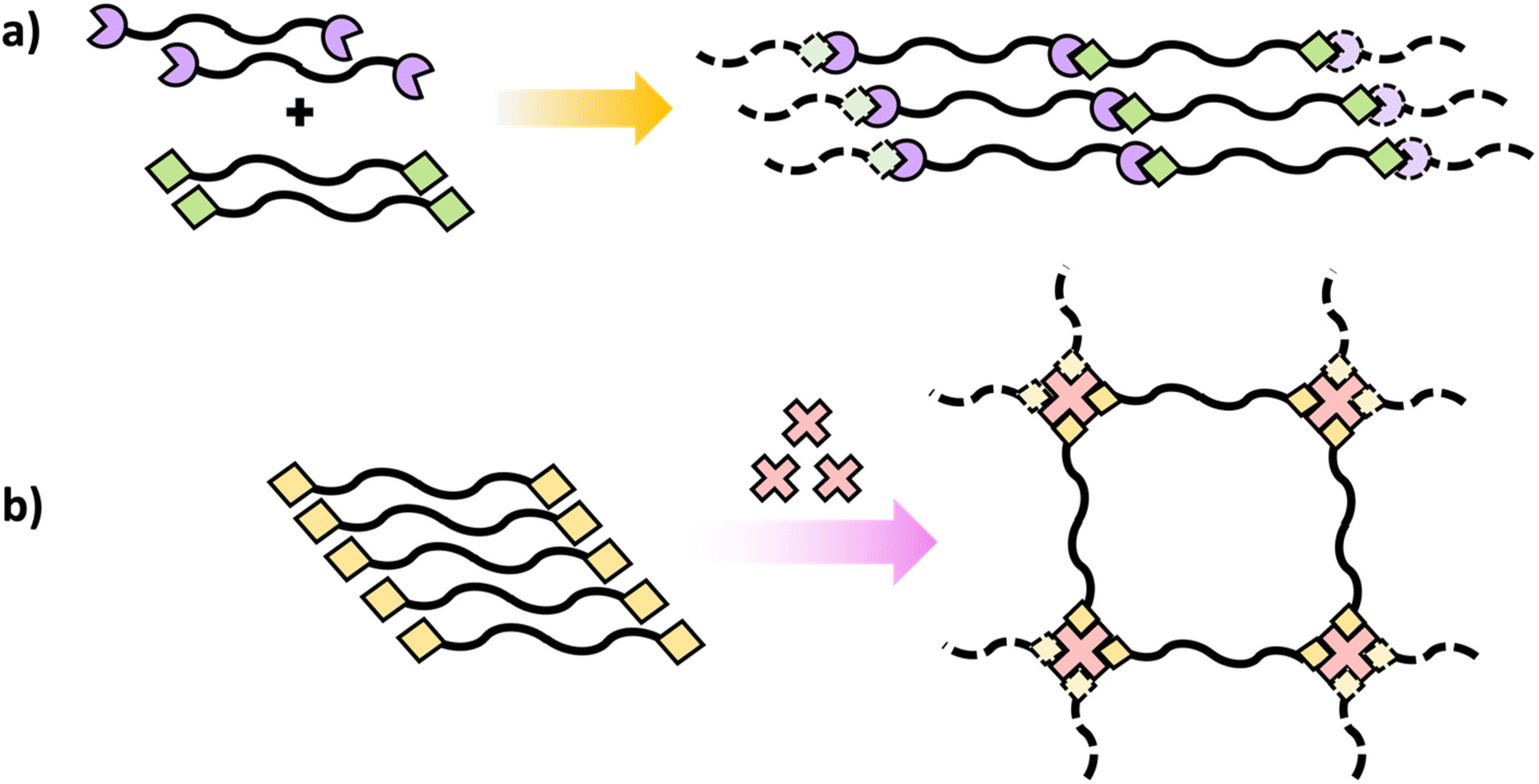 | ||
| Scheme 10 Schematic diagrams of polymers containing terminal reactive groups (a) with another polymer with terminal reactive groups and (b) with the addition of a multifunctional crosslinker. | ||
An industrially relevant example of post polymerization crosslinking is the vulcanization of rubber which involves crosslinking of polyisoprene (rubber) with sulfur to obtain a material with improved durability (Scheme 11).58
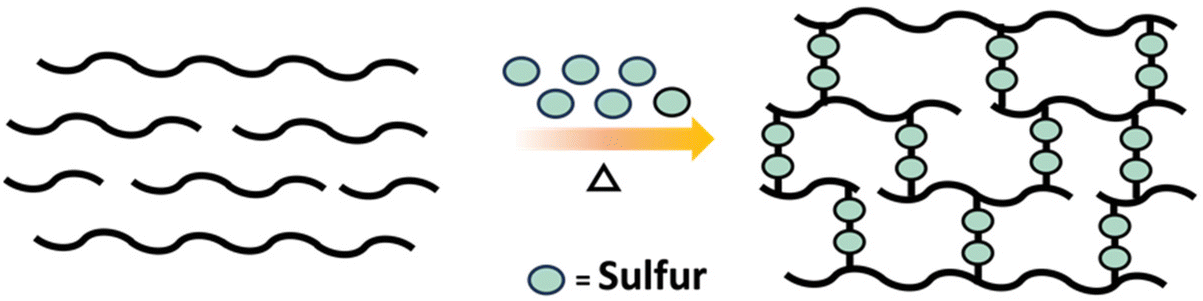 | ||
| Scheme 11 Schematic diagram of vulcanization of rubber using sulfur. | ||
Networks by phase separation
Polymer network like properties can be achieved in the absence of covalent crosslinks by leveraging phase separation and microaggregation of polymer chains through non-covalent interactions. A material consisting of immiscible or poorly miscible polymer components, leads to the formation of distinct regions or microaggregates. These microaggregates effectively connect the polymer chains through weak physical interactions such as van der Waals forces, ionic interactions, hydrogen bonding, and aromatic stacking. As a result, the material exhibits network-like behavior, including enhanced mechanical properties. Notable examples include polyurethanes, which intrinsically form hard and soft domains from their backbone units and multiblock thermoplastic elastomers based on hard–soft–hard triblock copolymers.Polyurethane structure, illustrated in Scheme 12a, consists of a hard segment and a soft segment. Phase separation in polyurethanes primarily relies on hydrogen bonding. The degree of phase separation is influenced by the lengths of the hard and soft segments as well as the hydrogen bonding interactions and temperature. Significantly, the strength the material stem from the hydrogen bonding between the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups within the hard domain.59,60 By modulating these factors, physical and mechanical properties of these polyurethane can be tailored, allowing for wide range of applications such as coatings, adhesives, textile and medical applications.61
O groups within the hard domain.59,60 By modulating these factors, physical and mechanical properties of these polyurethane can be tailored, allowing for wide range of applications such as coatings, adhesives, textile and medical applications.61
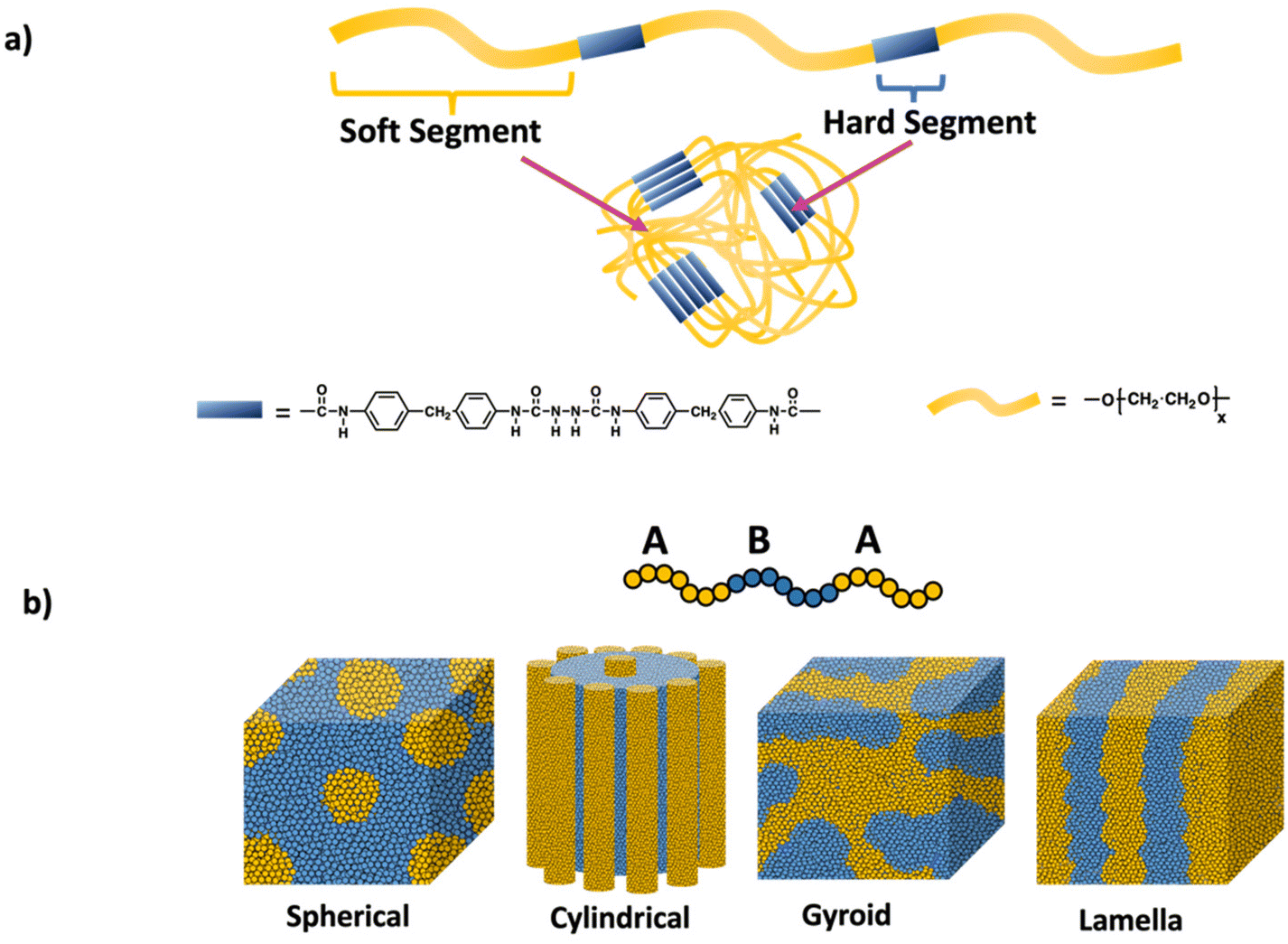 | ||
| Scheme 12 a) Polyurethane networks and (b) self-assembly of ABA triblock copolymers into different domains (spherical, cylindrical, gyroid, and lamella) through physical crosslinks. | ||
Another notable example of materials exhibiting network-like behavior is multiblock thermoplastic elastomers, especially ABA triblock copolymers. These elastomers consist of a soft midblock and two hard, glassy blocks at the ends (ABA structure). The monomer chosen for the hard blocks typically has glass transition temperature above room temperature, while the soft polymer has a glass transition temperature far below room temperature. As a result of the incompatibility between these polymer units, microphase separation occurs, leading to the formation of two distinct domains: one rich in the hard block and the other rich in the soft block as shown in Scheme 12b. Prominent examples of such triblock elastomers include polystyrene-poly(butadiene)-polystyrene (SBS)62 and polystyrene-poly(isoprene)-polystyrene (SIS).63,64 In these copolymers, the π–π stacking of the styrene blocks creates a physically crosslinked material, while the elastomeric behavior is provided by the soft midblock. Recent advancements have introduced new ABA thermoplastic elastomers with enhanced properties, such as self-healing65 and degradability.66 Adjusting the length and composition of the blocks allows the mechanical properties to be modulated for specific applications.67
Conclusions
There are many approaches to the synthesis of polymer networks, although they generally adapt from the traditional step and chain polymerization approaches. Multifunctionality is key to network formation, since the polymerized multifunctional monomers serve as crosslinks in the network. Once the polymers cross the gel-point the material has effectively infinite molecular weight, with there being a pathway of covalent bonds connecting any two parts of the polymer gel. Phase separation can enhance materials properties and in certain cases also lead to network-like properties from linear chains. Due to the insolubility caused by the effectively infinite molecular weight of polymer networks, their characterization is more complex than soluble polymers and will be highlighted in a subsequent contribution.Author contributions
D.K. and N.D.A.W were involved in conceptualization and project administration. S.V.W., M.A.S.N.W., O.J.D., I.O.R., C.W.H.R, N.D.A.W and D.K. were involved in visualization. M.A.S.N.W., O.J.D., I.O.R., C.W.H.R, N.D.A.W and D.K. were involved in writing the original draft. All authors were involved in editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr John Matson for discussion of chain vs. step mechanisms. M. A. S. N. W. and D. K. were supported by the National Science Foundation (CHE-2203727) for description of step polymerization processes and overview of network materials. I. O. R. was supported by the National Science Foundation (DMR-1749730) for descriptions of Post Polymerization Crosslinking. This work was partially supported by the United States Department of Energy, Office of Science, Basic Energy Sciences, under Award No. DE-SC0018645 (supporting C. W. H. R) for descriptions of phase separation in networks. N.D.A.W was supported by the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement no. 101030516 for descriptions of overall approaches to network synthesis.References
- Y. Gu, J. Zhao and J. A. Johnson, Polymer Networks: From Plastics and Gels to Porous Frameworks, Angew. Chem., Int. Ed., 2020, 59(13), 5022–5049 CrossRef CAS PubMed.
- C. S. Patrickios, Polymer Networks: Recent Developments, in Macromolecular symposia, Wiley Online Library, 2010, Vol. 291, pp 1–11 Search PubMed.
- C. K. Varnava and C. S. Patrickios, Polymer Networks One Hundred Years after the Macromolecular Hypothesis: A Tutorial Review, Polymer, 2021, 215, 123322 CrossRef CAS.
- T. Kaiser, Highly Crosslinked Polymers, Prog. Polym. Sci., 1989, 14(3), 373–450 CrossRef CAS.
- S. P. O. Danielsen, H. K. Beech, S. Wang, B. M. El-Zaatari, X. Wang, L. Sapir, T. Ouchi, Z. Wang, P. N. Johnson and Y. Hu, Molecular Characterization of Polymer Networks, Chem. Rev., 2021, 121(8), 5042–5092 CrossRef CAS PubMed.
- L. H. Baekeland , Method of Making Insoluble Products of Phenol and Formaldehyde. Google Patents December 7, 1909.
- L. H. Baekeland, The Synthesis, Constitution, and Uses of Bakelite, Ind. Eng. Chem., 1909, 1(3), 149–161 CrossRef CAS.
- C. K. Luscombe, G. Moad, R. C. Hiorns, R. G. Jones, D. J. Keddie, J. B. Matson, J. Merna, T. Nakano, G. T. Russell and P. D. Topham, A Brief Guide to Polymerization Terminology (IUPAC Technical Report), Pure Appl. Chem., 2022, 94(9), 1079–1084 CrossRef CAS.
- C. H. Chan, J.-T. Chen, W. S. Farrell, C. M. Fellows, D. J. Keddie, C. K. Luscombe, J. B. Matson, J. Merna, G. Moad and G. T. Russell, Reconsidering Terms for Mechanisms of Polymer Growth: The “Step-Growth” and “Chain-Growth” Dilemma, Polym. Chem., 2022, 13(16), 2262–2270 RSC.
- V. Zhang, B. Kang, J. V. Accardo and J. A. Kalow, Structure–Reactivity–Property Relationships in Covalent Adaptable Networks, J. Am. Chem. Soc., 2022, 144(49), 22358–22377 CrossRef CAS PubMed.
- R. F. T. Stepto, J. I. Cail and D. J. R. Taylor, Polymer Networks: Principles of Formation, Structure and Properties, Polimery, 2000, 45(7–8), 455–464 CAS.
- P. A. Panteli and C. S. Patrickios, Multiply Interpenetrating Polymer Networks: Preparation, Mechanical Properties, and Applications, Gels, 2019, 5(3), 36 CrossRef CAS PubMed.
- J. C. Hernández-Ortiz and E. Vivaldo-Lima, Crosslinking. Handb. Polym. Synth. Charact. Process, 2013, pp. 187–204 Search PubMed.
- B. Zhang, N. De Alwis Watuthanthrige, S. V. Wanasinghe, S. Averick and D. Konkolewicz, Complementary Dynamic Chemistries for Multifunctional Polymeric Materials, Adv. Funct. Mater., 2022, 32(8), 2108431 CrossRef CAS.
- A. Bagheri, C. M. Fellows and C. Boyer, Reversible Deactivation Radical Polymerization: From Polymer Network Synthesis to 3D Printing, Adv. Sci., 2021, 8(5), 2003701 CrossRef CAS PubMed.
- E. Blasco, M. Wegener and C. Barner-Kowollik, Photochemically Driven Polymeric Network Formation: Synthesis and Applications, Adv. Mater., 2017, 29(15), 1604005 CrossRef PubMed.
- A. Palma–Cando and U. Scherf, Electrochemically Generated Thin Films of Microporous Polymer Networks: Synthesis, Properties, and Applications, Macromol. Chem. Phys., 2016, 217(7), 827–841 CrossRef.
- A. R. S. S. Kumar, A. Padmakumar, U. Kalita, S. Samanta, A. Baral, N. K. Singha, M. Ashokkumar and G. Qiao, Ultrasonics in Polymer Science; Applications and Challenges, Prog. Mater. Sci., 2023, 101113 CrossRef.
- V. Schamboeck, P. D. Iedema and I. Kryven, Dynamic Networks That Drive the Process of Irreversible Step-Growth Polymerization, Sci. Rep., 2019, 9(1), 2276 CrossRef PubMed.
- K. Y. Choi and K. B. McAuley, Step–Growth Polymerization, Polym. React. Eng., 2007, 273–314 CAS.
- M. Ueda, Sequence Control in One-Step Condensation Polymerization, Prog. Polym. Sci., 1999, 24(5), 699–730 CrossRef CAS.
- L. De Keer, P. H. M. Van Steenberge, M.-F. Reyniers and D. R. D'hooge, Going beyond the Carothers, Flory and Stockmayer Equation by Including Cyclization Reactions and Mobility Constraints, Polymers, 2021, 13(15), 2410 CrossRef CAS PubMed.
- M. Shakiba, E. Rezvani Ghomi, F. Khosravi, S. Jouybar, A. Bigham, M. Zare, M. Abdouss, R. Moaref and S. Ramakrishna, Nylon—A Material Introduction and Overview for Biomedical Applications, Polym. Adv. Technol., 2021, 32(9), 3368–3383 CrossRef CAS.
- P. J. Flory, Molecular Size Distribution in Three Dimensional Polymers. I. Gelation1, J. Am. Chem. Soc., 1941, 63(11), 3083–3090 CrossRef CAS.
- C. Wen, R. Odle and S. Cheng, Molecular Weight Distribution of Branched Polymers: Comparison between Monte Carlo Simulation and Flory-Stockmayer Theory, Polymers, 2023, 15(7), 1791 CrossRef CAS PubMed.
- F.-L. Jin, X. Li and S.-J. Park, Synthesis and Application of Epoxy Resins: A Review, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- H. Q. Pham and M. J. Marks, Epoxy Resins, in Ullmann's Encycl. Ind. Chem, 2000 Search PubMed.
- M. C. C. Ferrer, D. Babb and A. J. Ryan, Characterisation of Polyurethane Networks Based on Vegetable Derived Polyol, Polymer, 2008, 49(15), 3279–3287 CrossRef CAS.
- S. S. Mahapatra, S. K. Yadav, H. J. Yoo, J. W. Cho and J.-S. Park, Highly Branched Polyurethane: Synthesis, Characterization and Effects of Branching on Dispersion of Carbon Nanotubes, Composites, Part B, 2013, 45(1), 165–171 CrossRef CAS.
- D. S. Kazybayeva, G. S. Irmukhametova and V. V. Khutoryanskiy, Thiol-Ene “Click Reactions” as a Promising Approach to Polymer Materials, Polym. Sci., Ser. B, 2022, 64(1), 1–16 CrossRef CAS.
- J. W. Chan, J. Shin, C. E. Hoyle, C. N. Bowman and A. B. Lowe, Synthesis, Thiol− Yne “Click” Photopolymerization, and Physical Properties of Networks Derived from Novel Multifunctional Alkynes, Macromolecules, 2010, 43(11), 4937–4942 CrossRef CAS.
- N. B. Cramer, J. P. Scott and C. N. Bowman, Photopolymerizations of Thiol− Ene Polymers without Photoinitiators, Macromolecules, 2002, 35(14), 5361–5365 CrossRef CAS.
- N. B. Cramer, S. K. Reddy, A. K. O'Brien and C. N. Bowman, Thiol− Ene Photopolymerization Mechanism and Rate Limiting Step Changes for Various Vinyl Functional Group Chemistries, Macromolecules, 2003, 36(21), 7964–7969 CrossRef CAS.
- A. B. Scranton, J. Klier and N. A. Peppas, Statistical Analysis of Free-Radical Copolymerization/Crosslinking Reactions Using Probability Generating Functions: Reaction Directionality and General Termination, Macromolecules, 1991, 24(6), 1412–1415 CrossRef CAS.
- P. H. Plesch, The Chemistry of Cationic Polymerization, Elsevier, 2016 Search PubMed.
- M. Morton, Anionic Polymerization: Principles and Practice, Elsevier, 2012 Search PubMed.
- A. Fürstner, Olefin Metathesis and Beyond, Angew. Chem., Int. Ed., 2000, 39(17), 3012–3043 CrossRef.
- E. J. Goethals, D. Van Meirvenne, G. Trossaert and R. Deveux, Polymer Networks via Cationic Ring–opening Polymerization, in Makromolekulare Chemie. Macromolecular Symposia, Wiley Online Library, 1990, vol. 32, pp 11–20 Search PubMed.
- C. W. A. Bainbridge, A. Wangsadijaya, N. Broderick and J. Jin, Living Polymer Networks Prepared by Controlled Radical Polymerization Techniques, Polym. Chem., 2022, 13(11), 1484–1494 RSC.
- C. L. Moad and G. Moad, Fundamentals of Reversible Addition–Fragmentation Chain Transfer (RAFT), Chem. Teach. Int., 2020, 3(2), 3–17 CrossRef.
- K. Matyjaszewski, Atom Transfer Radical Polymerization: From Mechanisms to Applications, Isr. J. Chem., 2012, 52(3–4), 206–220 CrossRef CAS.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, A Comparison of RAFT and ATRP Methods for Controlled Radical Polymerization, Nat. Rev. Chem., 2021, 5(12), 859–869 CrossRef CAS PubMed.
- J. L. Mann, R. L. Rossi, A. A. A. Smith and E. A. Appel, Universal Scaling Behavior during Network Formation in Controlled Radical Polymerizations, Macromolecules, 2019, 52(24), 9456–9465 CrossRef CAS PubMed.
- F.-Y. Lin, M. Yan and E. W. Cochran, Gelation Suppression in RAFT Polymerization, Macromolecules, 2019, 52(18), 7005–7015 CrossRef CAS.
- R. J. Young and P. A. Lovell, Introduction to Polymers, CRC press, 2011 Search PubMed.
- O. Okay, Kinetics of Gelation in Free Radical Crosslinking Copolymerization, Polymer, 1994, 35(12), 2613–2618 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Synthesis of Functional Polymers with Controlled Architecture by CRP of Monomers in the Presence of Cross-Linkers: From Stars to Gels, Prog. Polym. Sci., 2009, 34(4), 317–350 CrossRef CAS.
- K. Matyjaszewski, Controlled/Living Radical Polymerization: Progress in ATRP, NMP, and RAFT, ACS Publications, 2000 Search PubMed.
- R. H. Wiley, Crosslinked Styrene/Divinylbenzene Network Systems, Pure Appl. Chem., 1975, 43(12), 57–75 CrossRef CAS.
- L. Lopes-Rocha, L. Ribeiro-Gonçalves, B. Henriques, M. Özcan, M. E. Tiritan and J. C. M. Souza, An Integrative Review on the Toxicity of Bisphenol A (BPA) Released from Resin Composites Used in Dentistry, J. Biomed. Mater. Res., Part B, 2021, 109(11), 1942–1952 CrossRef CAS PubMed.
- W. Kuhn and G. Balmer, Crosslinking of Single Linear Macromolecules, J. Polym. Sci., 1962, 57(165), 311–319 CrossRef CAS.
- G. Odian, Principles of Polymerization, John Wiley & Sons, 2004 Search PubMed.
- H. R. Kricheldorf, Handbook of Polymer Synthesis, CRC press, 1991, vol. 24 Search PubMed.
- R. Bagheri, B. T. Marouf and R. A. Pearson, Rubber-Toughened Epoxies: A Critical Review, J. Macromol. Sci., Part C: Polym. Rev., 2009, 49(3), 201–225 CAS.
- M. A. Tasdelen, M. U. Kahveci and Y. Yagci, Telechelic Polymers by Living and Controlled/Living Polymerization Methods, Prog. Polym. Sci., 2011, 36(4), 455–567 CrossRef CAS.
- F. Biedermann, E. A. Appel, J. Del Barrio, T. Gruendling, C. Barner-Kowollik and O. A. Scherman, Postpolymerization Modification of Hydroxyl-Functionalized Polymers with Isocyanates, Macromolecules, 2011, 44(12), 4828–4835 CrossRef CAS.
- M. E. Elhalwagy, A. S. Elsherbiny and A. H. Gemeay, Amine-Rich Polymers for Water Purification Applications, Mater. Today Chem., 2023, 27, 101344 CrossRef CAS.
- D. Craig, The Vulcanization of Rubber with Sulfur, Rubber Chem. Technol., 1957, 30(5), 1291–1346 CrossRef CAS.
- F. M. de Souza, P. K. Kahol and R. K. Gupta, Introduction to Polyurethane Chemistry, in Polyurethane Chemistry: Renewable Polyols and Isocyanates, ACS Publications, 2021, pp 1–24 Search PubMed.
- H. S. Lee, Y. K. Wang and S. L. Hsu, Spectroscopic Analysis of Phase Separation Behavior of Model Polyurethanes, Macromolecules, 1987, 20(9), 2089–2095 CrossRef CAS.
- Q. Zhang, G. Zhang, J. Xu, C. Gao and Y. Wu, Recent advances on ligin-derived polyurethane polymers, Rev. Adv. Mater. Sci., 2015, 40(2), 146–154 CAS.
- H. T. Ban, T. Kase, M. Kawabe, A. Miyazawa, T. Ishihara, H. Hagihara, Y. Tsunogae, M. Murata and T. Shiono, A New Approach to Styrenic Thermoplastic Elastomers: Synthesis and Characterization of Crystalline Styrene− Butadiene− Styrene Triblock Copolymers, Macromolecules, 2006, 39(1), 171–176 CrossRef CAS.
- B. J. Dair, C. C. Honeker, D. B. Alward, A. Avgeropoulos, N. Hadjichristidis, L. J. Fetters, M. Capel and E. L. Thomas, Mechanical Properties and Deformation Behavior of the Double Gyroid Phase in Unoriented Thermoplastic Elastomers, Macromolecules, 1999, 32(24), 8145–8152 CrossRef CAS.
- Y. Matsumiya, H. Watanabe, A. Takano and Y. Takahashi, Uniaxial Extensional Behavior of (SIS) p-Type Multiblock Copolymer Systems: Structural Origin of High Extensibility, Macromolecules, 2013, 46(7), 2681–2695 CrossRef CAS.
- J.-X. Yang, Y.-Y. Long, L. Pan, Y.-F. Men and Y.-S. Li, Spontaneously Healable Thermoplastic Elastomers Achieved through One-Pot Living Ring-Opening Metathesis Copolymerization of Well-Designed Bulky Monomers, ACS Appl. Mater. Interfaces, 2016, 8(19), 12445–12455 CrossRef CAS PubMed.
- N. De Alwis Watuthanthrige, J. A. Reeves, M. T. Dolan, S. Valloppilly, M. B. Zanjani, Z. Ye and D. Konkolewicz, Wavelength-Controlled Synthesis and Degradation of Thermoplastic Elastomers Based on Intrinsically Photoresponsive Phenyl Vinyl Ketone, Macromolecules, 2020, 53(13), 5199–5207 CrossRef CAS.
- P. Maji and K. Naskar, Styrenic Block Copolymer–based Thermoplastic Elastomers in Smart Applications: Advances in Synthesis, Microstructure, and Structure–Property Relationships—A Review, J. Appl. Polym. Sci., 2022, 139(39), e52942 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |