
Exploring the oxygen evolution electrocatalysis of an amine-based cobalt metal–organic framework†
Jade Nadine S.
Ang
 ,
Manjunath
Chatti
,
Khang N.
Dinh
,
Stuart R.
Batten
*,
Alexandr N.
Simonov
* and
David R.
Turner
*
,
Manjunath
Chatti
,
Khang N.
Dinh
,
Stuart R.
Batten
*,
Alexandr N.
Simonov
* and
David R.
Turner
*
School of Chemistry, Monash University, Clayton, VIC 3800, Australia. E-mail: david.turner@monash.edu
First published on 1st March 2023
Abstract
When immobilised on a suitable electrode surface, metal organic frameworks (MOFs) can effectively promote the oxygen evolution reaction (OER) – the anode process of water electrolysis for the green hydrogen synthesis – most commonly through the electrooxidative decomposition of the framework into thermodynamically stable (oxy)hydroxides/oxides. In this study, the potential advantageous electrocatalytic effect of the macrocyclic amine core coordinating to the cobalt ions in an interdigitating 2D sheet framework is investigated. Nickel foam electrodes modified with Co-TMBT-MOF at a low loading of 0.25 mg cm−2 (0.004 mmolCo cm−2) sustained the OER rate of 20 mA cm−2 (80 A gMOF−1) in 1 M KOH at ambient temperature at a stabilised overpotential of only 0.294 ± 0.005 V on a timescale of 20 h. Physical characterisation of the electrode after the OER tests confirmed transformation of the MOF into the catalytically active cobalt oxyhydroxide.
Design, System, ApplicationThe system reported in this manuscript incorporates the molecular design of a strongly chelating ligand used as part of the metal–organic framework. In doing so, the stability of the material is greatly enhanced, preventing leaching of the metal and decomposition of the framework. This stability enables the material to be used and processed as an aqueous suspension. In the current study we demonstrate that this allows effective deposition on nickel foam as a pre-catalyst for the OER. |
Introduction
As the demand for energy is rapidly exceeding the supply, the renewable energy research sector has been pulling out all the stops to find cleaner alternatives to fossil fuels. Electrochemical water splitting is one of the most widely studied routes to developing sustainable energy systems.1–3 Practical water electrolysis requires the use of either acidic or alkaline electrolytes. The latter presents an important advantage of the improved stability of the device components without the need for use of noble metals and other highly expensive components, and is the focus herein.While both cathode and anode half-reactions of the water electrolysis process require catalysts to achieve practical rates at economically feasible overpotentials, the oxygen evolution reaction (OER) is a major contributor of the two to the energy efficiency losses.4,5 Hence, a great variety of OER electrocatalysts continue to be developed using different combinations of transition metal (hydr)oxides/oxyhydroxides,6–8 chalcogenides,9–11 pnictides,12–14 and their composites.15–17
Among different strategies to the design of OER catalysts, the use of metal–organic frameworks (MOFs), or porous coordination polymers (PCPs), has recently attracted some attention due to the very high internal surface area, tailorable pore size, ability to embed different functionalities, and the variety of metal nodes pertinent to MOFs.18–22 Hypothetically, these characteristics give the potential for electrically conductive MOFs to present a very high concentration of catalytically active sites and rapid mass/charge transport rates as compared to the conventional bulk metal oxide derivatives.23 However, it is highly unlikely for any MOF to maintain its structural integrity during the OER, even during model ambient temperature tests, due to an obvious thermodynamic instability of these materials under the very harsh conditions of a water splitting anode.24 Indeed, it has been expectedly demonstrated that electrochemical testing of MOFs induces their transformation into corresponding transition metal (hydr)oxides, which present the genuine catalytic species for the OER. Hence, the ability to produce a high-performance MOF-derived electrocatalyst will strongly depend on the initial stability of the MOF and its ability to preserve the initial texture with very high accessibility of the metal sites upon transformation into the oxide-based catalyst. Formation of catalysts in this way can give enhanced surface areas or different nanostructures than other methods.
To date, numerous transition metal-based MOFs, especially cobalt-based, have been used to produce electrodes with highly promising catalytic activity for the OER.23,25–31 Some of the noteworthy results obtained at ambient temperature using a 1 M KOH electrolyte solution are surveyed here. Yu et al. used biphenyl-4,4′-dicarboxylic acid (H2BPDC) to form Co-MOF nanosheets directly on a nickel foam (NF) electrode substrate using a hydrothermal method.26 The 3D macroporous structure of the NF paired with the 2D Co-MOF nanosheets enabled the material to achieve the OER rate of 20 mA cm−2 (4.6 A gMOF−1) at an overpotential of ca. 0.29 V. Zhao et al. studied NiCo-MOF nanosheets using benzene-1,4-dicarboxylate as the organic ligand and suggested that the unsaturated metal surface atoms served as the dominant catalytic active centres of the OER.32 However, their X-ray absorption data suggest that the NiCo-MOF was completely transformed into transition metal oxyhydroxides, which effectively catalysed the OER with the reaction rate of 10 mA cm−2 (50 A gMOF−1) achieved at an overpotential of ca. 0.25 V. Zhuang et al. used FeCo-MOF nanosheets with 1,3,5-benzenetricarboxylate as the ligand and observed a similar synergistic effect between Co and Fe.33 Similarly, Tian et al. also formed a FeCo-MOF with nitrogen sites incorporated (as triazole) into the structure using 2,5-bis(1,2,4-triazol-1-yl)terephthalate (BTTA2−) as the organic component. Their MOF-derived electrocatalyst displayed an OER rate of 10 mA cm−2 (20 A gMOF−1) at a very low overpotential of ca. 0.23 V. Their study also demonstrated the importance of using a highly conductive substrate like NF and doping with other metals for enhanced activity.34
Further insight into the usage of the different ligand components in MOF precatalysts should be explored to effectively utilise these materials. In this study, a ligand containing an amine-based macrocyclic core with biphenyl-carboxylate arms was designed to explore the functional advantage of having a metal chelating site in potentially enhancing stability (Fig. 1, bottom). The new Co-TMBT-MOF material was explored as a precursor for a cobalt-based oxygen evolution catalyst operating in alkaline electrolyte solutions.
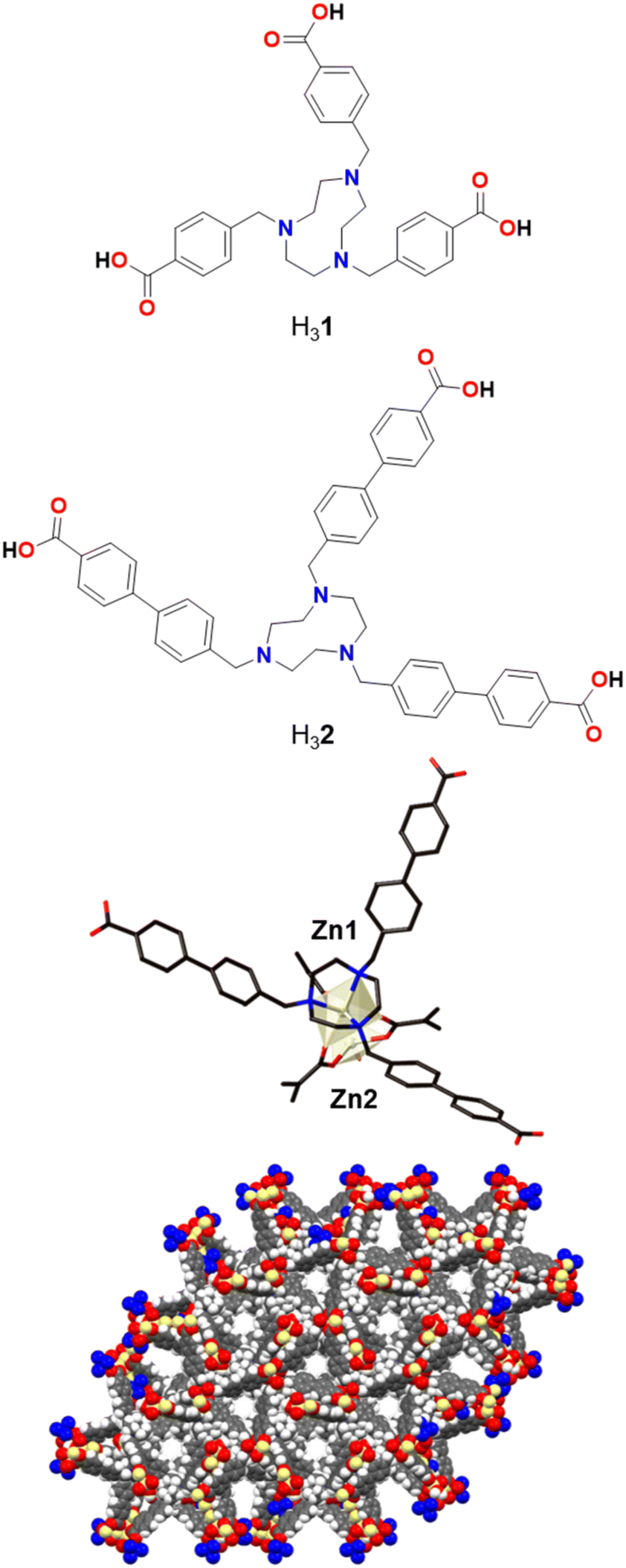 | ||
| Fig. 1 Designed TACN-based tricarboxylic acid H32 with extended, rigid arms compared to the previously reported H31 (top), magnified view of the dinuclear Zn cluster in poly-[Zn2(2)(OH2)]Cl (middle), and view of the pore channels along the crystallographic c-axis (bottom). | ||
Experimental
Materials
All starting materials were of ACS reagent grade and used as received from commercial suppliers (Alfa Aesar, Merck, Oakwood, and Sigma Aldrich) without further purification. 1,4,7-Triazacyclononane trihydrochloride (TACN·3HCl) was synthesised using a previously reported literature procedure.35 Ni foam (NF, >99.99%; thickness 1.6 mm; surface density 346 g m−2; porosity ≥95%, 80–110 pores per inch) was obtained from Marketech International. Pt wire was purchased from A&E Metals. All solutions were prepared using ultrapure deionised water with resistivity of 18.2 MΩ cm at 23 °C derived from Sartorius Arium Comfort I Ultrapure water system H2O-I-1-UV-T; the same quality water was used for all procedures requiring H2O.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (2:1 vol.) mixture at 70 °C for 24 hours under N2. The reaction mixture was filtered through Celite under vacuum to remove the Pd catalyst. The filtrate was concentrated and the organic product was extracted into ethyl acetate after washing with distilled H2O and brine. Purification was conducted using column chromatography with 10:90 to 20:80 vol. ethyl acetate in petroleum benzine (87% yield). 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 8.4 Hz, 2H, ArH), 7.65 (d, J = 8.4 Hz, 2H, ArH), 7.53 (d, J = 8.1 Hz, 2H, ArH), 7.28 (d, J = 7.6 Hz, 2H, ArH), 3.94 (s, 3H, RCO2–CH3), 2.41 (s, 3H, ArCH3).
:90 to 20:80 vol. ethyl acetate in petroleum benzine was performed to purify the product (95% yield). 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.7 Hz, 2H, ArH), 7.65 (d, J = 8.6 Hz, 2H, ArH), 7.60 (d, J = 8.3 Hz, 2H, ArH), 7.49 (d, J = 8.3 Hz, 2H, ArH), 4.55 (s, 2H, BrCH2), 3.94 (s, 3H, RCO2–CH3).
:H2O (60 mL, 1:2 vol.) for 48 hours at 100 °C and rendered acidic (pH 3) with concentrated HCl. The product precipitated out of the solution and was recovered by filtration (2.068 g, 65% yield). 1H NMR (600 MHz, DMSO-d6) δ 7.96 (m, 6H, ArH), 7.67 (m, 12H, ArH), 7.29 (d, J = 7.8 Hz, 6H, ArH), 3.92 (s, 6H, R2N–CH2), 2.89 (s, 12H, TACN–CH2). 13C NMR (151 MHz, DMSO-d6): δ 167.04, 143.48, 138.70, 130.56, 129.96, 127.09, 126.64, 118.93, 72.20, 57.03, 44.46, 39.94, 39.80, 39.66, 39.52, 39.38, 39.24, 39.10. m/z (ES+): 760.33 ([M + H]+) 53.0%, calculated for C48H46N3O6+, MW = 760.33. FTIR (υ/cm−1): 3405 m, 2954w, 2886w, 2814m, 2499w, 2113w, 1794w, 1702s, 1607s, 1396s, 1272s, 1107s, 1006m, 958m, 821m, 770s, 754s, 698s. Found: C, 63.60; H, 5.34; N, 4.68%; calculated for C48H45N3O6·3HCl·2H2O: C, 63.68; H, 5.79; N, 4.64%.
:H2O (2:1 vol.) mixture at 70 °C for 24 hours under N2. The reaction mixture was filtered through Celite under vacuum to remove the Pd catalyst. The filtrate was concentrated and the organic product was extracted into ethyl acetate after washing with distilled H2O and brine. Purification was conducted using column chromatography with 10:90 to 20:80 vol. ethyl acetate in petroleum benzine (87% yield). 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 8.4 Hz, 2H, ArH), 7.65 (d, J = 8.4 Hz, 2H, ArH), 7.53 (d, J = 8.1 Hz, 2H, ArH), 7.28 (d, J = 7.6 Hz, 2H, ArH), 3.94 (s, 3H, RCO2–CH3), 2.41 (s, 3H, ArCH3).
:90 to 20:80 vol. ethyl acetate in petroleum benzine was performed to purify the product (95% yield). 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.7 Hz, 2H, ArH), 7.65 (d, J = 8.6 Hz, 2H, ArH), 7.60 (d, J = 8.3 Hz, 2H, ArH), 7.49 (d, J = 8.3 Hz, 2H, ArH), 4.55 (s, 2H, BrCH2), 3.94 (s, 3H, RCO2–CH3).
:H2O (60 mL, 1:2 vol.) for 48 hours at 100 °C and rendered acidic (pH 3) with concentrated HCl. The product precipitated out of the solution and was recovered by filtration (2.068 g, 65% yield). 1H NMR (600 MHz, DMSO-d6) δ 7.96 (m, 6H, ArH), 7.67 (m, 12H, ArH), 7.29 (d, J = 7.8 Hz, 6H, ArH), 3.92 (s, 6H, R2N–CH2), 2.89 (s, 12H, TACN–CH2). 13C NMR (151 MHz, DMSO-d6): δ 167.04, 143.48, 138.70, 130.56, 129.96, 127.09, 126.64, 118.93, 72.20, 57.03, 44.46, 39.94, 39.80, 39.66, 39.52, 39.38, 39.24, 39.10. m/z (ES+): 760.33 ([M + H]+) 53.0%, calculated for C48H46N3O6+, MW = 760.33. FTIR (υ/cm−1): 3405 m, 2954w, 2886w, 2814m, 2499w, 2113w, 1794w, 1702s, 1607s, 1396s, 1272s, 1107s, 1006m, 958m, 821m, 770s, 754s, 698s. Found: C, 63.60; H, 5.34; N, 4.68%; calculated for C48H45N3O6·3HCl·2H2O: C, 63.68; H, 5.79; N, 4.64%.
Materials characterisation
Electrochemical measurements
Electrochemical analysis was undertaken in a three-electrode configuration using an H-cell with the working and auxiliary compartments separated by a low-porosity (P4) ceramic frit and a Bio-Logic VSP electrochemical workstation at ambient temperature (23 ± 2 °C). Aqueous 1 M KOH electrolyte solution was saturated with O2 in the working electrode compartment.Working electrodes were prepared using Ni foam substrates with an active geometric area of 0.5 cm × 0.5 cm, which were cleaned by sonicating in concentrated HCl (ca. 32 wt%), rinsed and sonicated again in water and then in ethanol for 10 min each media. The working electrodes were dried overnight in a desiccator prior to use. The MOFs were deposited by drop-casting a suspension onto the NF working electrode substrates. The suspension was prepared using 5 mg of Co-TMBT-MOF, 50 μL of 5 wt% Nafion (ethanol solution), 250 μL of isopropanol, and 700 μL of water. The mixture was uniformly dispersed by sonication (Elmasonic E-60H 6-Quart bath) for 30 min before it was drop-cast onto the substrates. Loading levels were varied from 0.10 to 1.00 mg cm−2 (Fig. S5†).
A high surface area Pt wire was used as an auxiliary electrode. The Hg|HgO|1 M KOH reference electrode was confined in a glass tube with a P4 ceramic frit. All the potentials reported were converted to the reversible hydrogen electrode (RHE) scale using the experimentally measured potential of the reference electrode against the custom-made RHE8 filled with 1 M KOH. Uncompensated resistance (Ru) was calculated from the electrochemical impedance spectra, which were collected at potentials where negligible faradaic processes were present, before and after experiments in a frequency range 200 kHz to 0.1 Hz at an amplitude 0.010 V. Typical Ru values were 0.3–0.4 Ω. Correction for the ohmic losses was done manually post-analysis by subtracting the IRu product. All current densities are reported normalised to the geometric surface area of the electrodes or the amount of the catalytically relevant material.
Results and discussion
MOF synthesis and structure
A previously reported tris-benzylcarboxylic acid TACN ligand, H31 (Fig. 1), forms a Zn-MOF, poly-[Zn2(1)(H2O)]Cl, which was used for CO2 adsorption due to its porosity and rigidity.43 Building on this design strategy, in which a central TACN pocket is able to tightly bind a transition metal and yet the structure still provides accessible pore space, a longer derivative using 4′-methylbiphenyl-4-carboxylic acid arms, H32, was synthesised in order to increase the pore space to provide easier access to the metal sites.Initially, we were able to obtain faint yellow crystals of poly-[Zn2(2)(OH2)]Cl·1.5DMF·6.5H2O, Zn-TMBT-MOF, directly from the heated reaction (see Experimental). The MOF is a 3D framework based on a dinuclear cluster (Fig. 1). Zn1 is coordinated to the three nitrogen atoms of the TACN core and to three carboxylate oxygen atoms. Zn2 is disordered over two sites and tetracoordinated to three carboxylate oxygen atoms as well as an aqua ligand. Each 2 ligand is coordinated to four Zn2 clusters (one via the TACN ring and three via the carboxylates), and in turn each Zn2 cluster is coordinated to four 2 ligands – three by carboxylate groups which generate a Zn2(μ2-CO2)3 motif, and one by the TACN group. This generates two interpenetrating 3D nets; if the nodes are defined as the 2 ligand and the Zn2 clusters then a uninodal 4-connected (65·8) net of unusual topology is generated. Alternatively, if the Zn2(CO2)3(TACN) moiety is considered as a node, then it can be described as a 6-connected 32·44·56·62·7 net.
The structure contains solvent-filled 1D channels equivalent to 48% of the crystal volume. A pore diameter of approximately 6 Å measured between the shortest C⋯C distance (including van der Waals radii) exists along the crystallographic c-axis (Fig. 1). The degree of solvation was assigned through chemical analysis, and the presence of the chloride counter-anion inferred from microanalysis and the analogous nature of the framework to those previously reported. Although this 3D Zn framework displays remarkable unrestricted pore channel accessibility, this study will focus on the Co-TMBT-MOF material, below, as cobalt-based compounds are well known to be good OER catalysts.
The 2D Co-TMBT-MOF adopts a very different structure to the zinc material, surprisingly considering the nature of the metals involved, with a purple crystalline material isolated directly from the reaction (see Experimental). SC-XRD shows the formation of poly-[Co(H2)(OH2)]·2DMF·4H2O as parallel interdigitated 2D sheets (Fig. 2). Each ligand coordinates to three metal ions, one through the TACN ring, and two others through the carboxylate arms. The third arm of each ligand remains protonated and uncoordinated; these arms project outwards on either side of the plane of the sheet. Each metal ion, which exists as a single ion node rather than the binuclear node in the Zn-MOF, coordinates to three ligands, one through the TACN ring, and two others through the carboxylate arms (there is also an additional water ligand). Thus, if one considers the metal and the ligand as 3-connecting nodes, the sheet has (6,3) topology (with both nodes topologically identical). However, it is perhaps better to consider a Co(H2)(OH2) moiety as the node, where the Co is bound to the TACN ring of the ligand, in which case each node is connected to four others and the topology is (4,4) (Fig. 2 middle). As expected from the design strategy, the secondary amines of the core group holding the metal in place providing extra stability for the framework, as evidenced by its stability in air after filtering and leaving exposed to the atmosphere for 24 h before determining its PXRD pattern (Fig. S4a†).
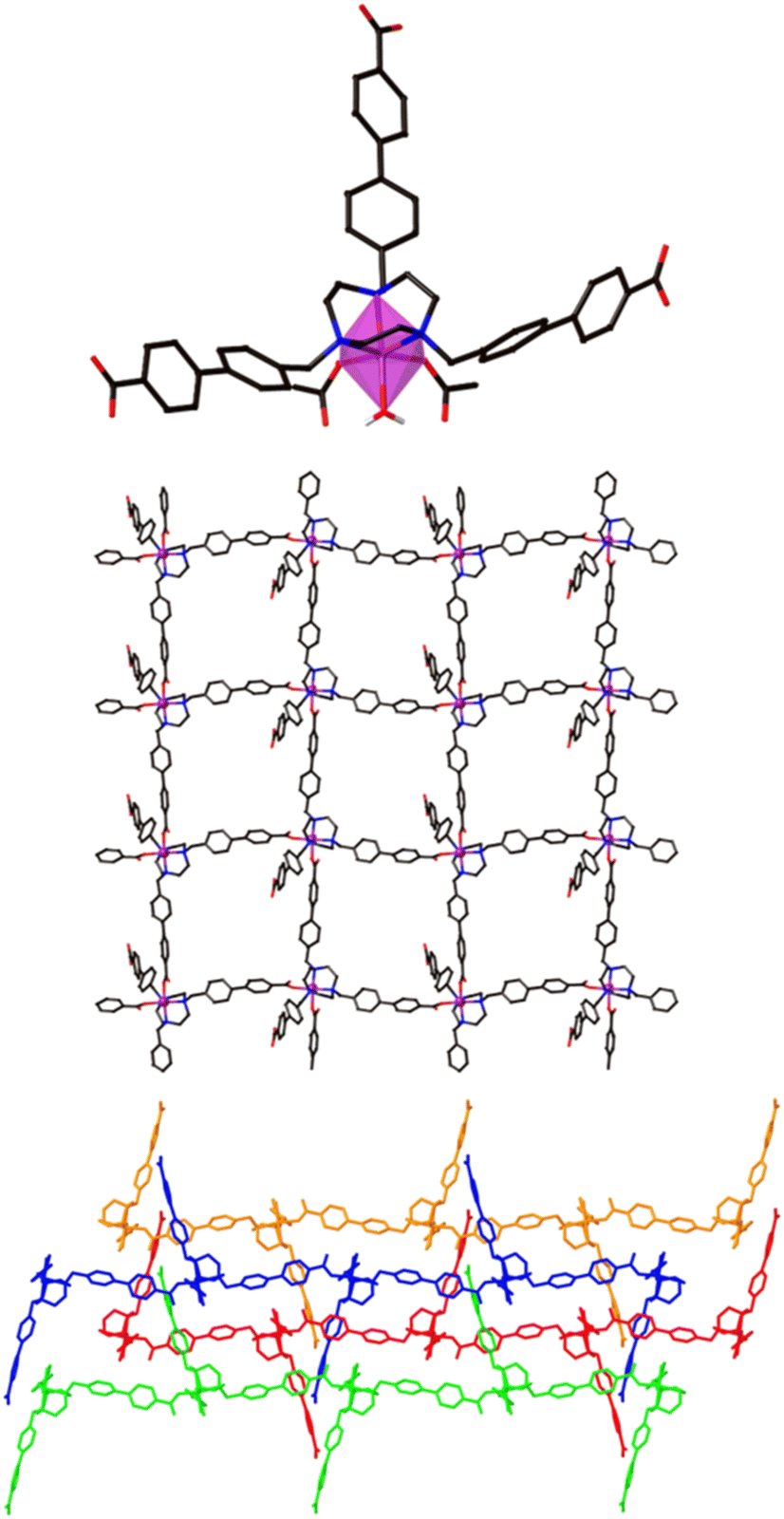 | ||
| Fig. 2 Magnified view of the hexacoordinated Co node in the structure of poly-[Co2(H2)(OH2)]·2DMF·4H2O (top), simplified view of the framework viewed along the crystallographic a-axis (middle), and an expanded view to highlight how the sheets stack with interdigitation in the x direction (bottom). | ||
The sheets stack in the x direction and are highly interdigitated (Fig. 2 bottom), with the uncoordinated carboxylic acid arm of each ligand penetrating a window in each of two adjoining sheets and forming a hydrogen bond with the non-coordinated atom of a monodentate carboxylate of the sheet that is three removed. Each window in a sheet is in turn penetrated by two of these uncoordinated ligand arms, one from each of two adjoining sheets which lie on the same side of the original sheet. The interdigitation is such that all the windows in one row are penetrated by sheets from one side, while all the windows in the next row are penetrated by sheets from the other side. Even with this interdigitation, 1D channels exist parallel to the crystallographic a axis in which solvent molecules reside which could not be resolved from the single crystal X-ray diffraction data.
Electrocatalytic activity
Electrocatalytic oxygen evolution activity of the nickel foam electrodes modified with as synthesised Co-TMBT-MOF was investigated in 1 M KOH electrolyte solution at ambient temperature. During the cyclic voltammetry measurements (Fig. S6a†), the performance of Co-TMBT-MOF/NF improved with cycling suggesting an in situ chemical and structural transformation until stabilisation after ca. 10 cycles at 0.005 V s−1. This transformation is most likely associated with the electrochemical oxidation of the metal organic species, which eventually leads to the formation of more catalytically active cobalt oxyhydroxide species. The highest performance was achieved at the initial MOF loading of 0.25 mg cm−2, suggesting that higher amounts of the material limit the charge transport. Importantly, the background performance of the Ni foam substrate was negligibly low as compared to the MOF-modified electrodes in the investigated potential range (Fig. 3a).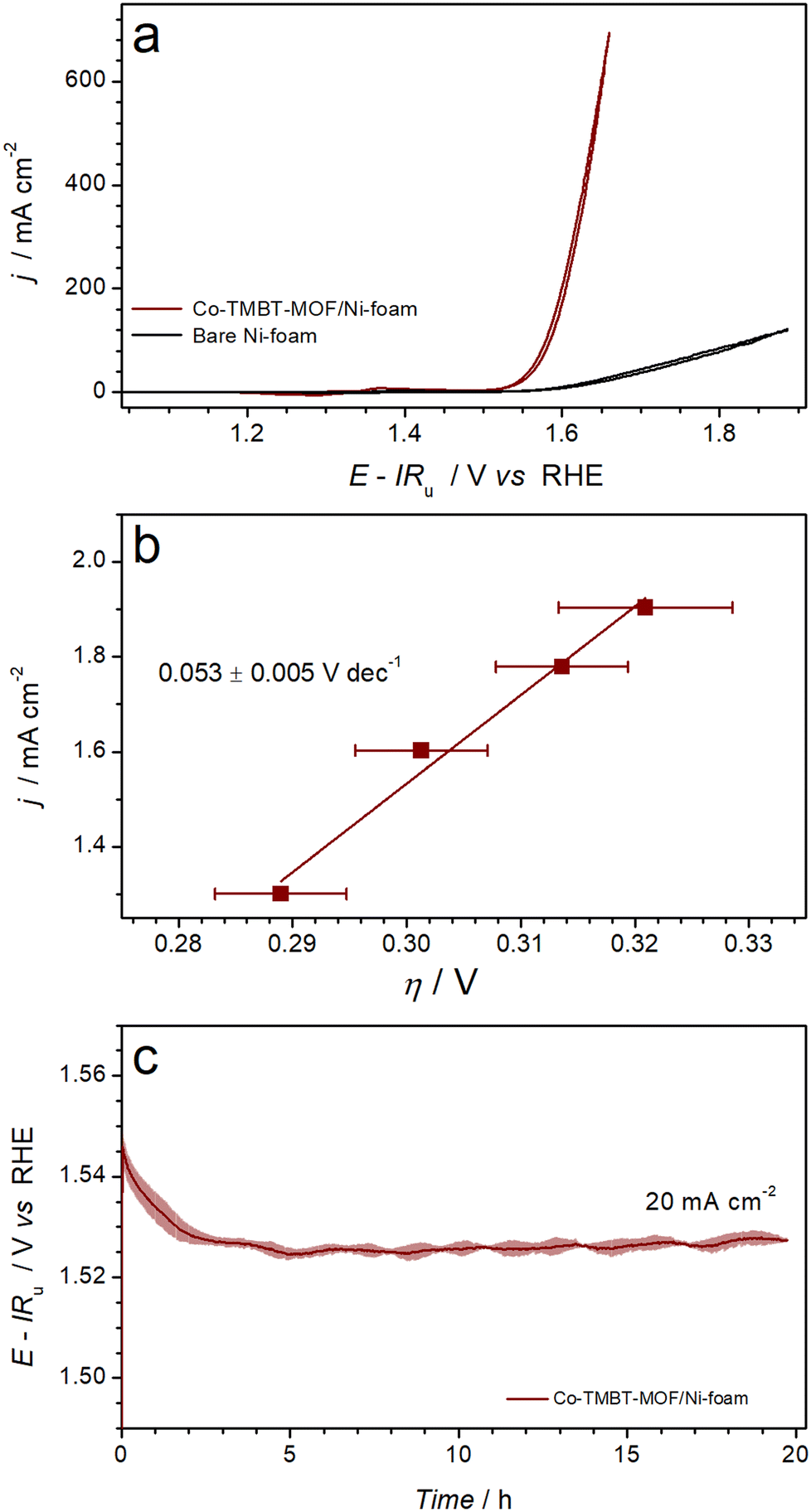 | ||
| Fig. 3 Electrocatalytic OER performance of Ni foam electrodes modified with Co-TMBT-MOF (0.25 mg cm−2) in 1 M KOH at 23 ± 2 °C: (a) quasi-stabilised cyclic voltammetry (scan rate 0.005 V s−1; 10th scans) with black curve showing background data for the unmodified Ni foam electrode, (b) OER polarisation plots derived from potentiometric measurements (Fig. S6b†), and (c) longer-term galvanostatic OER at 20 mA cm−2. Data in panels b and c are shown as mean (symbols and solid line) ± standard deviation (error bars and shading) derived from tests of three independent samples. | ||
Upon stabilisation of the voltammetric response, the electrocatalytic activity of the Co-TMBT-MOF/NF electrodes was investigated by short-term galvanostatic measurements (Fig. S6b†), which were used to construct an OER polarisation plot (Fig. 3b). The potential dependence of the current density was close to linear within the 20–80 mA cm−2 range indicating insignificant charge-transport limitations within the catalyst (Fig. S6b†). The corresponding Tafel slope was 0.053 ± 0.005 V dec−1.
When tested in galvanostatic mode on a longer timescale, the Co-TMBT-MOF/NF electrodes exhibited even further improvements in the catalytic performance (Fig. 3c). Eventually, the quasi-steady state was achieved after ca. 5 h of operation at 20 mA cm−2, which required only 0.294 ± 0.005 V overpotential, and the performance did not change further indicating that chemical transformations of Co-TMBT-MOF are complete at this stage. The electrocatalytic performance of these electrodes was highly reproducible, as confirmed through tests of three independent MOF samples (Fig. 3b and c and S6b and c†).
Comparisons of the OER performance reported herein to those of the recently reported Co-MOF-derived catalysts indicates that Co-TMBT-MOF/NF is among the top-performing electrodes of this class (Table S1†). The only material outperforming Co-TMBT-MOF/NF was derived from the FeCo-BTTA-MOF,23 confirming the well-known promoting effect of iron on the OER activity. It should be also noted though that no measures were taken herein to purify the electrolyte solutions from the Fe impurities, meaning that the Co-TMBT-MOF/NF electrodes were likely modified with very low amounts of iron-based species, although they could not be detected by XPS (vide infra). Overall, the Co-TMBT-MOF/NF electrode with the MOF loading of 0.25 mg cm−2 (0.004 mmolCo cm−2) maintained an OER rate of 20 mA cm−2 (80 A gMOF−1 or 4.5 A gCo−1) at a low overpotential of 0.294 ± 0.005 V. This activity is notably higher than that reported for CoOOH on NF, which required an overpotential of 0.37 ± 0.01 V at 10 mA cm−2 in 1 M KOH.44,45
Electrode characterisation
The Co-TMBT-MOF/NF electrodes were characterised before and after 20 h long OER tests at 20 mA cm−2 to ascertain the origin of the high electrocatalytic activity. The X-ray diffraction peaks for Co-TMBT-MOF and products of its transformations cannot be unambiguously detected from the assembled electrode due to the low mass loading (Fig. S4b†). SEM images before the electrochemical measurements showed deposition of well-defined Co-TMBT-MOF grains with approximately 10 μm length and 2 μm width onto the NF substrate (Fig. 4a). After the OER tests, these small grains were transformed into larger sheet-like microstructures with the size of ca. 20–30 μm (Fig. 4b), as well as smaller grain-like nanostructures with the dimensions of a few hundred nanometres (Fig. 4c). The EDS mapping of the electrodes after the OER tests confirmed the presence of Co, as well as, quite unexpectedly, of N (Fig. S6†). Apparently, the duration of the tests used herein was insufficient for a complete elimination of the ligand from the electrode surface. | ||
| Fig. 4 SEM images of Co-TMBT-MOF/NF before (top) and after the 20 h (centre and bottom) OER tests in 1 M KOH at 20 mA cm−2 and 23 ± 2 °C. | ||
XPS analysis was used to establish the chemical nature of the observed morphological changes. The C 1s and N 1s spectra suggest decomposition and possible dissolution of the organic ligand from the metal centre, as concluded from the significant decrease of the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.6 eV) C 1s peak (Fig. S9b†) and the disappearance of the C–N (402.5 eV) and M–N (399.6 eV) N 1s signals (Fig. S9c†) after the OER tests. The presence of aliphatic C–OH on the surface of the as-prepared Co-TMBT-MOF/NF electrode is corroborated by a broad peak at 532.7 eV in the O 1s spectra, whereas much narrower O 1s signals shifted towards the lower binding energies typical of metal hydroxides23,46,47 were recorded for the tested electrode. Notable changes were also induced to the Co 2p spectra. Notwithstanding the low signal-to-noise ratio associated with the low concentration of Co on the electrode surface, the change of the initial spectra, likely dominated by the Co2+ species,48,49 to a set of signals typical of cobalt oxyhydroxide was detected (Fig. 5).23,50,51 These have been referenced against existing literature of characterised standards of Co materials.50 FTIR spectra of the electrode after the OER also showed a significant decrease in the intensity of the characteristic peaks of Co-TMBT-MOF confirming its electrochemically induced decomposition (Fig. S9†).
O (288.6 eV) C 1s peak (Fig. S9b†) and the disappearance of the C–N (402.5 eV) and M–N (399.6 eV) N 1s signals (Fig. S9c†) after the OER tests. The presence of aliphatic C–OH on the surface of the as-prepared Co-TMBT-MOF/NF electrode is corroborated by a broad peak at 532.7 eV in the O 1s spectra, whereas much narrower O 1s signals shifted towards the lower binding energies typical of metal hydroxides23,46,47 were recorded for the tested electrode. Notable changes were also induced to the Co 2p spectra. Notwithstanding the low signal-to-noise ratio associated with the low concentration of Co on the electrode surface, the change of the initial spectra, likely dominated by the Co2+ species,48,49 to a set of signals typical of cobalt oxyhydroxide was detected (Fig. 5).23,50,51 These have been referenced against existing literature of characterised standards of Co materials.50 FTIR spectra of the electrode after the OER also showed a significant decrease in the intensity of the characteristic peaks of Co-TMBT-MOF confirming its electrochemically induced decomposition (Fig. S9†).
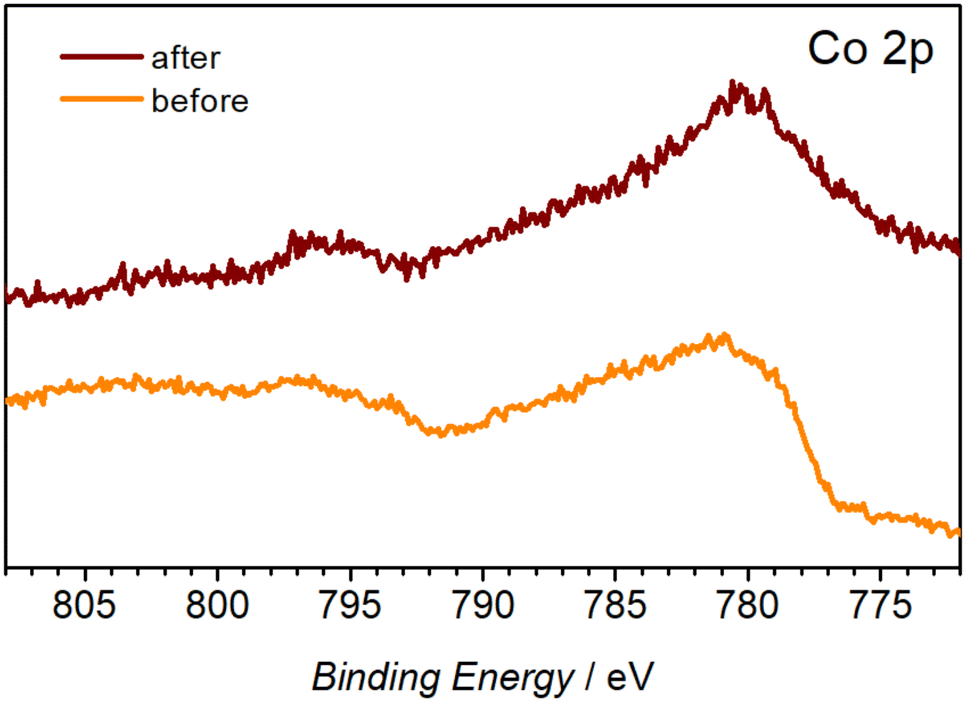 | ||
| Fig. 5 High-resolution Co 2p XP spectra of the Co-TMBT-MOF/NF electrodes before and after the 20 h OER tests in 1 M KOH at 20 mA cm−2 and 23 ± 2 °C. | ||
ICP-MS of the electrolyte solution after 20 h of the OER (20 mA cm−2) catalysed by Co-TMBT-MOF/NF electrodes detected the presence 0.01 μM of Co which is equivalent to 0.07 mol% of the amount of cobalt initially deposited onto the electrode in the form of the MOF. The low amount of Co ions released after the activation is indicative of the electrochemical stability of the MOF-derived catalyst.
Conclusions
Crystal engineering plays an important role in ensuring that MOFs are rationally designed towards their targeted applications. The synthesised two-fold 2D sheet framework of poly-[Co2(H2)(OH2)]·2DMF·4H2O, Co-TMBT-MOF, provides a strong chelating TACN core for Co. The assembled Co-TMBT-MOF/NF electrode demonstrated a lower OER overpotential of 0.294 ± 0.005 V at a lower mass loading (0.25 mg cm−2) than most of the reported Co-MOF derived catalysts. Durability tests show promising electrochemical stability with no decay for at least 20 h. Further investigation into the origin of the OER activity of the assembled electrode has shown that the chemical and structural transformation of the MOF into metal hydroxides and oxyhydroxides gives the primary active species for the OER.52 This study helps confirm the usability of Co-MOFs as pre-catalysts regardless of them undergoing certain transformations as the electrochemical stability proves to be far more important for this application.Author contributions
Conceptualisation, JNSA/MC/ANS/SRB/DRT; investigation, JNSA/MC/KND; writing – original draft, JNSA; writing – review & editing, JNSA/MC/KND/SRB/ANS/DRT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Parts of this research were conducted at the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the ACRF detector. The authors acknowledge the use of the facilities within the Monash Centre for Electron Microscopy and the Monash X-Ray Platform. JNSA acknowledges Monash University for the Monash Graduate Scholarship (MGS) and Monash International Tuition Scholarship (MITS), ANS acknowledges Australian Research Council for funding (FT200100317).Notes and references
- J. R. Rostrup-Nielsen, Catal. Rev.: Sci. Eng., 2004, 46, 247–270 CrossRef CAS.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- D. R. MacFarlane, J. Choi, B. H. R. Suryanto, R. Jalili, M. Chatti, L. M. Azofra and A. N. Simonov, Adv. Mater., 2020, 32, 1904804 CrossRef CAS PubMed.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57–69 CrossRef PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- P. W. Menezes, A. Indra, A. Bergmann, P. Chernev, C. Walter, H. Dau, P. Strasser and M. Driess, J. Mater. Chem. A, 2016, 4, 10014–10022 RSC.
- M. Chatti, A. M. Glushenkov, T. Gengenbach, G. P. Knowles, T. C. Mendes, A. V. Ellis, L. Spiccia, R. K. Hocking and A. N. Simonov, Sustainable Energy Fuels, 2018, 2, 1561–1573 RSC.
- D. Simondson, M. Chatti, S. A. Bonke, M. F. Tesch, R. Golnak, J. Xiao, D. A. Hoogeveen, P. V. Cherepanov, J. L. Gardiner, A. Tricoli, D. R. MacFarlane and A. N. Simonov, Angew. Chem., Int. Ed., 2021, 60, 15821–15826 CrossRef CAS PubMed.
- I. H. Kwak, H. S. Im, D. M. Jang, Y. W. Kim, K. Park, Y. R. Lim, E. H. Cha and J. Park, ACS Appl. Mater. Interfaces, 2016, 8, 5327–5334 CrossRef CAS PubMed.
- W. Chen, Y. Liu, Y. Li, J. Sun, Y. Qiu, C. Liu, G. Zhou and Y. Cui, Nano Lett., 2016, 16, 7588–7596 CrossRef CAS PubMed.
- J.-H. Kim, K. Kawashima, B. R. Wygant, O. Mabayoje, Y. Liu, J. H. Wang and C. B. Mullins, ACS Appl. Energy Mater., 2018, 1, 5145–5150 CAS.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- A. Dutta, A. K. Samantara, S. K. Dutta, B. K. Jena and N. Pradhan, ACS Energy Lett., 2016, 1, 169–174 CrossRef CAS.
- F. Yu, H. Zhou, Z. Zhu, J. Sun, R. He, J. Bao, S. Chen and Z. Ren, ACS Catal., 2017, 7, 2052–2057 CrossRef CAS.
- X. Zou, J. Su, R. Silva, A. Goswami, B. R. Sathe and T. Asefa, Chem. Commun., 2013, 49, 7522–7524 RSC.
- X. Yu, M. Zhang, W. Yuan and G. Shi, J. Mater. Chem. A, 2015, 3, 6921–6928 RSC.
- H. Chen, Y. Gao, L. Ye, Y. Yao, X. Chen, Y. Wei and L. Sun, Chem. Commun., 2018, 54, 4979–4982 RSC.
- G. R. Desiraju, J. J. Vittal and A. Ramanan, Crystal Engineering: A Text Book, World Scientific, 2011 Search PubMed.
- Y. Gong, H.-F. Shi, P.-G. Jiang, W. Hua and J.-H. Lin, Cryst. Growth Des., 2014, 14, 649–657 CrossRef CAS.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS PubMed.
- X. Zhao, B. Pattengale, D. Fan, Z. Zou, Y. Zhao, J. Du, J. Huang and C. Xu, ACS Energy Lett., 2018, 3, 2520–2526 CrossRef CAS.
- S. A. Patil, N. K. Shrestha, A. I. Inamdar, C. Bathula, J. Jung, S. Hussain, G. Nazir, M. Kaseem, H. Im and H. Kim, Nanomaterials, 2022, 12, 1916 CrossRef CAS PubMed.
- J. Tian, F. Jiang, D. Yuan, L. Zhang, Q. Chen and M. Hong, Angew. Chem., Int. Ed., 2020, 59, 13101–13108 CrossRef CAS PubMed.
- E. A. Flügel, V. W.-H. Lau, H. Schlomberg, R. Glaum and B. V. Lotsch, Chem. – Eur. J., 2016, 22, 3676–3680 CrossRef PubMed.
- K. Ge, S. Sun, Y. Zhao, K. Yang, S. Wang, Z. Zhang, J. Cao, Y. Yang, Y. Zhang, M. Pan and L. Zhu, Angew. Chem., Int. Ed., 2021, 60, 12097–12102 CrossRef CAS PubMed.
- J. Yu, Y. Cao, Q. Liu, Y. Luo, Y. Liu, X. Shi, A. M. Asiri, T. Li and X. Sun, ChemNanoMat, 2021, 7, 906–909 CrossRef CAS.
- R. K. Tripathy, A. K. Samantara and J. N. Behera, Dalton Trans., 2019, 48, 10557–10564 RSC.
- Q. Qian, Y. Li, Y. Liu, L. Yu and G. Zhang, Adv. Mater., 2019, 31, 1901139 CrossRef PubMed.
- S. Ibrahim, I. Majeed, E. Hussain, A. Badshah, Y. Qian, D. Zhao, D. R. Turner and M. A. Nadeem, Inorg. Chim. Acta, 2019, 486, 684–693 CrossRef CAS.
- S. Ibrahim, K. Shehzadi, B. Iqbal, S. Abbas, D. R. Turner and M. A. Nadeem, J. Colloid Interface Sci., 2019, 545, 269–275 CrossRef CAS PubMed.
- S. Li, Y. Gao, N. Li, L. Ge, X. Bu and P. Feng, Energy Environ. Sci., 2021, 14, 1897–1927 RSC.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- L. Zhuang, L. Ge, H. Liu, Z. Jiang, Y. Jia, Z. Li, D. Yang, R. K. Hocking, M. Li, L. Zhang, X. Wang, X. Yao and Z. Zhu, Angew. Chem., 2019, 131, 13699–13706 CrossRef.
- B. Zhang, Y. Zheng, T. Ma, C. Yang, Y. Peng, Z. Zhou, M. Zhou, S. Li, Y. Wang and C. Cheng, Adv. Mater., 2021, 33, 2006042 CrossRef CAS PubMed.
- S. H. Eitel, M. Bauer, D. Schweinfurth, N. Deibel, B. Sarkar, H. Kelm, H.-J. Krüger, W. Frey and R. Peters, J. Am. Chem. Soc., 2012, 134, 4683–4693 CrossRef CAS PubMed.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
- D. Aragao, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- G. Ortiz, S. Brandes, Y. Rousselin and R. Guilard, Chem. – Eur. J., 2011, 17, 6689–6695 CrossRef CAS PubMed.
- S. Zhang, T. Yu, H. Wen, Z. Ni, Y. He, R. Guo, J. You and X. Liu, Chem. Commun., 2020, 56, 15387–15405 RSC.
- K. N. Dinh, M. Chatti, T. D. Nguyen, D. A. Hoogeveen, D. R. MacFarlane and A. N. Simonov, ACS Energy Lett., 2022, 7, 3910–3916 CrossRef CAS.
- J. Xu, X. Zhu and X. Jia, ACS Sustainable Chem. Eng., 2019, 7, 16629–16639 CrossRef CAS.
- G. Beamson and D. Briggs, J. Chem. Educ., 1993, 70, A25 Search PubMed.
- C. V. Schenck, J. G. Dillard and J. W. Murray, J. Colloid Interface Sci., 1983, 95, 398–409 CrossRef CAS.
- N. R. Babij, E. O. McCusker, G. T. Whiteker, B. Canturk, N. Choy, L. C. Creemer, C. V. D. Amicis, N. M. Hewlett, P. L. Johnson, J. A. Knobelsdorf, F. Li, B. A. Lorsbach, B. M. Nugent, S. J. Ryan, M. R. Smith and Q. Yang, Org. Process Res. Dev., 2016, 20, 661–667 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- D. Wu, Y. Wei, X. Ren, X. Ji, Y. Liu, X. Guo, Z. Liu, A. M. Asiri, Q. Wei and X. Sun, Adv. Mater., 2018, 30, 1705366 CrossRef PubMed.
- W. Zheng and L. Y. S. Lee, ACS Energy Lett., 2021, 6, 2838–2843 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical and characterisation data. CCDC 2220213 and 2220214. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2me00259k |
| This journal is © The Royal Society of Chemistry 2023 |