
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Empowering clean water whilst safeguarding water distribution pipeline integrity: towards manganese- and iron-free lime hydrate for water treatment†
Dávid
Kocsis
*abc,
Rhys A.
Ward
bc,
Christopher R.
Meyer
*a,
Michael
Thompson
d,
Timothy J.
Prior
 d,
Stephen M.
Kelly
cd,
Nathan S.
Lawrence
bc and
Jay D.
Wadhawan
*bc
d,
Stephen M.
Kelly
cd,
Nathan S.
Lawrence
bc and
Jay D.
Wadhawan
*bc
aSingleton Birch, Ltd., Melton Ross Quarries, Barnetby DN38 6AE, North Lincolnshire, UK. E-mail: dkocsis@singletonbirch.co.uk; chrismeyer747@gmail.com; j.wadhawan@hull.ac.uk
bDepartment of Chemical Engineering, The University of Hull, Cottingham Road, Kingston-upon-Hull HU6 7RX, UK
cAura Innovation Centre, Bridgehead Business Park, Meadow Road, Hessle HU13 0GD, UK
dDepartment of Chemistry, The University of Hull, Cottingham Road, Kingston-upon-Hull HU6 7RX, UK
First published on 12th January 2023
Abstract
Hydrated limes are amongst the most economically valuable alkalis used by the water industry for the treatment of potable water. They are typically manufactured from the thermal decomposition of high purity limestones. However, the latter contain both manganese and iron impurities, which are transformed into the oxides Mn3O4 and Fe2O3 on burning in kilns (between 900–1100 °C) during the manufacture of lime, and are retained in the lime hydrate upon slaking. These impurities can be released through oxidation by conventional water disinfection chemicals (such as alkaline hypochlorite) during the use of lime hydrate as the alkaline pH modifier during conventional operations in water treatment works. This work investigates the redox mechanisms for manganese and iron removal from lime hydrate using alkaline hypochlorite: for manganese, interfacial electron transfer occurs first leading to dissolution as permanganate; in the case of iron impurities, solubility is encouraged in oxygenated solutions first through formation of solid ferrite, with oxidative dissolution of ferrite to ferrate. As expected for activation-controlled reactions, the oxidative dissolution is enhanced with increased temperatures; mapping the dissolution process with time allows for the unravelling of “rule-of-thumb” relationships for impurity removal of ∼1% min−1 for manganese and ∼3% min−1 for iron at 90 °C in alkaline hypochlorite.
Water impactReducing the amount of manganese and iron impurities present in drinking water treatment chemicals can improve both the aesthetic water quality and the integrity of its distribution pipelines, as it reduces the occurrence of discoloured water and pipescale. Herein, we develop treatments to remove these species from lime hydrate manufactured for the water industry, and provide “rules-of-thumb” for their scale-up. |
Introduction
Although access to safe drinking water is a mandated human right,1 clean water distribution has been plagued by pipeline integrity issues, even in high-income countries, for at least the last 90 years.2 In England, drinking water, supplied by water companies and authorities, is subjected to economic, environmental and public health regulation, by the Water Services Regulation Authority (Ofwat), the Environment Agency, and the Drinking Water Inspectorate (DWI), respectively.3 Drinking water quality is governed by compliance with legislative standards (such as the European Drinking Water Directive 98/83/EC), which are based on guidelines published by the World Health Organisation.4 These standards set upper limits on the presence of a wide range of ions and compounds, predominantly based on their toxicity; potable water is frequently contaminated by both natural effects (associated with groundwater/reservoir hydrology and biology),5 and anthropogenic causes.6 These impurities can be removed in water treatment works through the addition of chemical coagulants (such as alum and ferric sulphate) followed by clarification and filtration, with a subsequent disinfection step using, amongst others, chlorine or ozone, to remove micro-organisms.7 There is an additional consequence for drinking water treatment that results from the historical use of lead piping infrastructure in its distribution: the corrosion of this material is a public health hazard (most infamously demonstrated through the 2014–19 Flint water crisis);8 plumbosolvency control in the water mains relies on tiny solubility products, necessitating the addition of phosphates during water treatment, so as to maintain an extremely low concentration of soluble lead ions.Manganese and iron are two essential mineral elements which are, likewise, regulated as a result of both toxicity (as Mn2+), and their propensity (as black and red oxides) to cause pipe-scale4,9 – the lining of the water distribution pipelines, causing restricted flow, and ultimately requiring more energy to be expended so as to deliver water to consumers than in a de-scaled pipeline. Although iron in drinking water is generally present at sufficiently low concentrations so as not to present a health hazard (there is no toxicity-based limit on iron in drinking water), manganese in drinking water may have adverse effects on intellectual and cognitive development, causing it to have an upper guideline value of 400 ppb.4,9 Nevertheless, the guidelines recommend upper thresholds on manganese (50 ppb) and iron (200 ppb), to reduce “metallic taste” and prevent so-called “black water” and “red water” phenomena, respectively.4,9 In the UK, these two discoloured water types are often a source of customer contacts (complaints) with water companies and authorities: in 2020, 1.8 M consumers (up from 1.1 M in 2019) were affected by the supply of tap water with an unaesthetic appearance (colour, taste and odour), as catalogued in the Chief Inspector's Report from the DWI,10 with the largest number of complaints stemming from North-West England, the Midlands and Wales – areas where the water supply is fed by upland surface waters.10,11 Such contacts cause the water companies and authorities to be fined by Ofwat. These problems with drinking water distribution arise from a variety of factors, such as the increase in water use due to hot weather,10,11 the inadequate treatment of raw waters,11,12 or during the improvement of the energy efficiency of water distribution by dislodging manganese- and iron-based oxide debris and pipe-scale using increased hydraulic pressure.9,11,12 Since the solubility of manganese and iron oxides is both redox- and pH-sensitive, control of manganese and iron in the water supply is achieved through (1) understanding reservoir geology and water hydrology,11 (2) designing water treatment works to incorporate elaborate filtration operations,13 which may be gravel-based, or involve “greensand” (glauconite, (K, Na)(FeIII, Al, Mg)2(Si, Al)4O10(OH)2, coated with manganese dioxide), and (3) reducing the amount of manganese-oxidising bacterial biofilms,9,12a,14via efficient disinfection of the water supply.9,15,16
It is also important to control the impurity level present in the chemicals used to treat drinking water: the volume of chemicals used in water treatment means that even small reductions can provide benefit. Alkalis (pH modifiers) represent one of the largest group (by volume) of chemicals used by the UK water industry (in excess of 70 kTe y−1),17 with a slight favouring (53%) of slaked lime (portlandite, lime hydrate, Ca(OH)2) over caustic soda (NaOH).17a Lime is unusual in that it can be used both to remove manganese from reservoir water,2,18a,b and, as observed in KwaZulu-Natal, South Africa, brown lime (added during the treatment process) can be a source of manganese, even when the raw water has appreciable amounts of manganese.18c Lime softening of water through impurity removal is thought to occur via carbonation of portlandite,19a followed by heavy metal removal through sorption.19a,b Removal of impurities containing silicon and phosphorous can be achieved through their reactive addition to lime or hydrate.19c In the UK, water-grade alkalis currently conform to European Standard CEN TC 164/WG9/TG5, with purity criteria for both caustic (BS EN 896) and lime (BS EN 12518) regulating the concentrations of fluorides, radionuclides and heavy metals (arsenic, cadmium, chromium, mercury, nickel, lead, antimony and selenium), with further limits imposed on lime for SiO2, Al2O3, Fe2O3, MnO2 and CaCO3 impurities.17a This mandates the use of lime derived from industrial grade limestones only for water treatment. Such limestones are often referred to as pure carbonates (>95 wt% combined CaCO3 and MgCO3), which are readily available across the globe: Fig. 1a depicts the distribution of the total area (4.1 million km2) of pure carbonate outcrops in the world,20 with the distribution of the manufacture of 420 MTe y−1 of lime (the average global production rate in 2016–2020)21a presented in Fig. 1b. Using the data for the UK as an example, we estimate that ca. 2 wt% of all lime production is required for water treatment purposes,21b so that the global requirement of lime hydrate is ca. 11 MTe y−1 – a quantity that is not economically viable to manufacture without calcination of limestone.
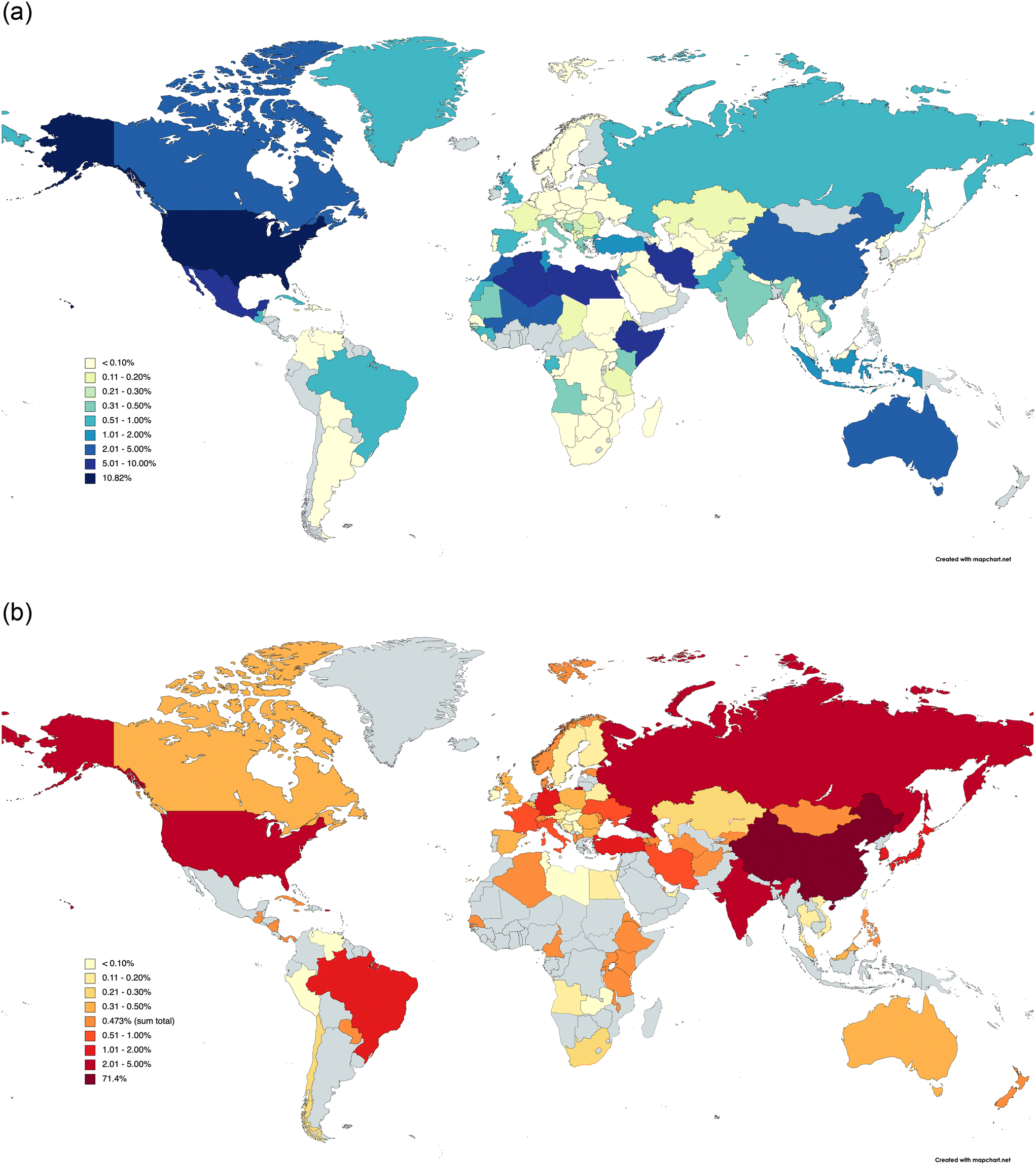 | ||
| Fig. 1 World distribution maps illustrating the country breakdown of (a) pure carbonate rock outcrops, and (b) lime production. In both cases, data are expressed as a percentage of the world totals: (a) 4.1 million km2 and (b) 420 MTe y−1 (average for 2016–2022), with the country with the greatest pure carbonate rock outcrop area (USA) or the highest lime production (Republic of China) indicated separately. Note that in (b), only the sum total of the individual countries is given with the colour corresponding to 0.473%, and that other nations (in grey) may have produced lime over the 2016–2020 period. Data taken from (a) ref. 20, and (b) ref. 21, with both maps being drawn using software available at MapChart.Net. | ||
The British Geological Survey have mapped the areas where pure calcium carbonate outcrops occur in England and Wales (see Table 1);22 current UK lime manufacturing23 uses either the high-, or very high-, purity limestones found in North Lincolnshire (Welton formation of Cretaceous chalk)24 or the Derbyshire Dome (Bee Woo formation of Carboniferous limestones).25 However, although these are all >97 wt% CaCO3, they are typically not “manganese-free”, and therefore may introduce manganese into the drinking water distribution network, unless first cleaned to remove the manganese impurities.
| Location | Formation | CaO/wt% | Fe2O3/wt% | MnO/wt% |
|---|---|---|---|---|
| a Adapted from data provided as Tables 3 and 12 in ref. 22c. b Very high purity limestone is >98.5 wt% CaCO3, corresponding to >55.2 wt% CaO; high purity limestone has CaCO3 in the range 97–98.5 wt%, corresponding to CaO in the range 54.3–55.2 wt%, using conventional assessment criteria – see Table 5 of ref. 26a. Note that the Flamborough chalk formation is technically of medium purity. | ||||
| Carboniferous limestone | ||||
| Peak District | Bee low | 55.41 | 0.07 | 0.02 |
| Mendips | Burrington oolite | 55.24 | 0.01 | 0.00 |
| South Wales | Oxwich head | 55.80 | 0.02 | 0.01 |
| North Wales | Llandulas | 55.38 | 0.10 | 0.15 |
| North Pennines | Cove | 55.73 | 0.01 | 0.01 |
| Lake District | Park | 55.52 | 0.03 | 0.05 |
| Average (Carboniferous limestone) | 55.51 | 0.04 | 0.04 | |
| Standard deviation | 0.21 | 0.04 | 0.06 | |
| Cretaceous chalk | ||||
| Humberside | Welton | 54.81 | 0.04 | 0.05 |
| Humberside | Flamborough | 53.60 | 0.10 | 0.06 |
| Wiltshire | Upper chalk | 54.87 | 0.06 | 0.15 |
| Wiltshire | Upper chalk | 54.93 | 0.03 | 0.03 |
| Kent | Upper chalk | 55.45 | 0.04 | 0.04 |
| Suffolk | Upper chalk | 54.94 | 0.08 | 0.06 |
| Average (Cretaceous chalk) | 54.77 | 0.06 | 0.07 | |
| Standard deviation | 0.62 | 0.03 | 0.04 | |
In this paper, we illustrate redox chemical processes to treat water-grade lime derived from both high-purity and very high-purity limestones, so as to reduce the extent of manganese and iron impurities that they contain. We use insights into the physicochemical dynamics underpinning the impurity removal process to develop important design relationships for suitable process operations that could be integrated within existing lime manufacturing plants. We first provide an overview of the distribution of these impurities in both Carboniferous limestone and Cretaceous chalk (see ESI†), and indicate how these change during the lime cycle for the production of slaked lime.
Manganese and iron impurities in high purity limestones
The British Geological Survey describes limestones as being high purity, or very high purity depending on whether the mass fraction of CaCO3 present therein is between 97.0% and 98.5%, or above 98.5%, respectively.26 These are the only two categories of limestone considered to be suitable as industrial grade for use in the water treatment, iron and steel, chemicals, glass making, filters and pigments, paper and flue gas desulphurization industries.22,27Table 1 illustrates the typical compositional variation of both manganese (as MnO) and iron (as Fe2O3) impurities within high and very high-purity limestone (limestone and chalk) strata in England and Wales.22c It is clear that, with the exception of the Burlington oolite, all high-purity (Cretaceous) and very high-purity (Carboniferous) limestones in Table 1 are contaminated by both iron and manganese, with the slightly lower purity chalk being more uniform in its impurity distribution – the relative standard deviations are high for Carboniferous limestones (92% (Fe) and 141% (Mn)), but much lower for the Cretaceous chalk (47% (Fe) and 67% (Mn)), across all locations. This general picture is slightly misleading, since different parts of a limestone quarry can expose different depths of the limestone stratigraphy – for example, the Hindlow Quarry (Carboniferous limestone from the Peak District) is reported to have manganese levels between 80–160 ppm, and iron levels between 200–500 ppm;25 in contrast the Tunstead Quarry (Carboniferous limestone from the Peak District) has manganese levels in the range 80–960 ppm, with iron levels ranging between 200–3000 ppm.25 It appears that small variations in the rock lithology can lead to a wide variation in the level of the iron and manganese contaminants;22,26 such local variations can downgrade the purity of limestones,22,26 and, since limestones (including hard Northern Province chalk – see ESI†) are typically blasted from a rock face, it is not always possible to separate out these impurities.27Manganese and iron impurities in limestones derive from a number of sources, such as the iron sulphides (pyrite and marcasite) present in clay bands (wayboards), through dolomitization (minerals such as ankerite and kutnohorite), or through “accessory” minerals including tourmaline, garnet, magnetite, rhodochrosite and siderite.22,24,25,28 Since limestones are permeable, it is thought that incongruent reactions at the particle surfaces release Mn2+ and Fe2+ ions into either recharge or connate water, resulting in a purer calcite.28 Thus, under oxidising conditions (Eh > ca. 300 mV, see ESI†), manganese and iron can be deposited as dendrites at shallow fracture surfaces as the oxides (such as pyrolusite, MnO2), sesquioxides (braunite, Mn2O3, bixbyite, and haematite, Fe2O3), oxohydroxides (for example, manganite, MnOOH, and goethite, α-FeOOH), hydroxides and hydrated iron oxides (including limonite).29–32 Typically iron and manganese oxides in limestones occur in separate deposits. However, where they occur together, the iron oxides typically overlay the older manganese oxide deposits,29 indicating that the manganese oxides enable the precipitation of iron oxides.33 Under reducing conditions, the divalent impurities can be incorporated as interstitial solid solutions in the calcite (or, in the case of Limestone, calcite and aragonite) particles.29,34 Such solution/re-deposition chemical processes (see Fig. 2) are characteristic origins of manganese ores,29 including the famous deposits at Nikopol (Ukraine) and Chiaturi (in the Caucasus).35
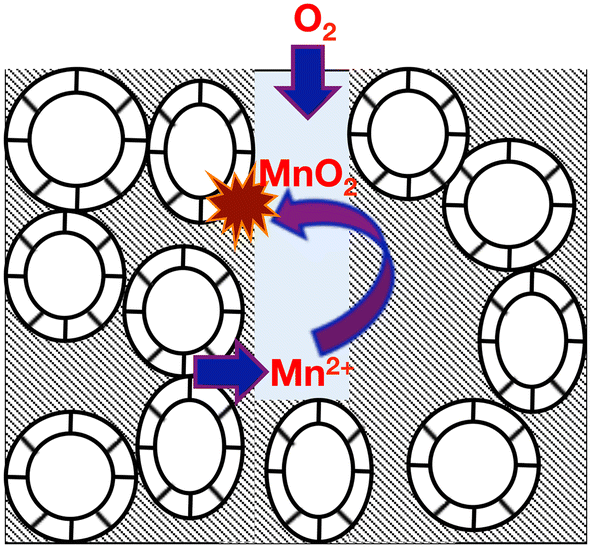 | ||
| Fig. 2 Schematic illustration of dissolution/re-precipitation reactions causing the mineralisation of manganese (as radial dendritic growths) through incongruent reactions at fracture surfaces in the presence of interstitial or connate or recharge water. Adapted from ref. 28. | ||
Manganese and iron impurities in slaked limes
The manufacture of lime hydrate first involves the comminution of limestones, followed by the stages of calcination and then reaction with water (see ESI†).27 During the calcination process to form quicklime (CaO, molar mass 56 g mol−1), carbon dioxide is lost from the calcite (CaCO3, molar mass 100 g mol−1), so that although there is a small degree of shrinkage of the rock,27 the concentration of solid impurities increases. Singleton Birch Ltd.'s high-calcium quicklime, Burnt Lime 40,36 although hard to quantify exactly owing to its high reactivity with atmospheric moisture and carbon dioxide, typically comprises >90 wt% CaO, with small degrees of impurities derived from both the limestone rock and the sulphur from the natural gas used in the calcination. Digestive analysis (see ESI†) of quicklime indicates a doubling of the manganese and iron concentration in lime compared with chalk (Fig. 3). No differences were found between the size fractions investigated (∼50 mm for Burnt Lime 40, or 3.35 mm finer screenings). The slight difference between the ratios for manganese and iron in lime versus the quarried rock, is indicative of the fact that both impurities derive from different oxidation forms in the chalk: the mass loss is consistent with Fe2O3 (FeIII) and Mn3O4 (hausmannite, where Jahn–Teller distorted MnIII occupies all of the octahedral sites, with MnII distributed in the tetrahedral sites in a cubic oxide lattice) being the impurities in quicklime, and Mn2O3 and either FeCO3 or FeS2 as the impurities in the chalk. This interpretation is consistent with the reported thermodynamic stability of manganese and iron oxides, carbonates and disulphides at kiln temperatures (900–1100 °C).37,38 Note that whilst the kiln temperature is controlled to avoid sintering (dead-burned lime), the maximum upper temperature is close to that for the tetragonal-to-cubic phase change of the hausmannite spinel (1170 °C).39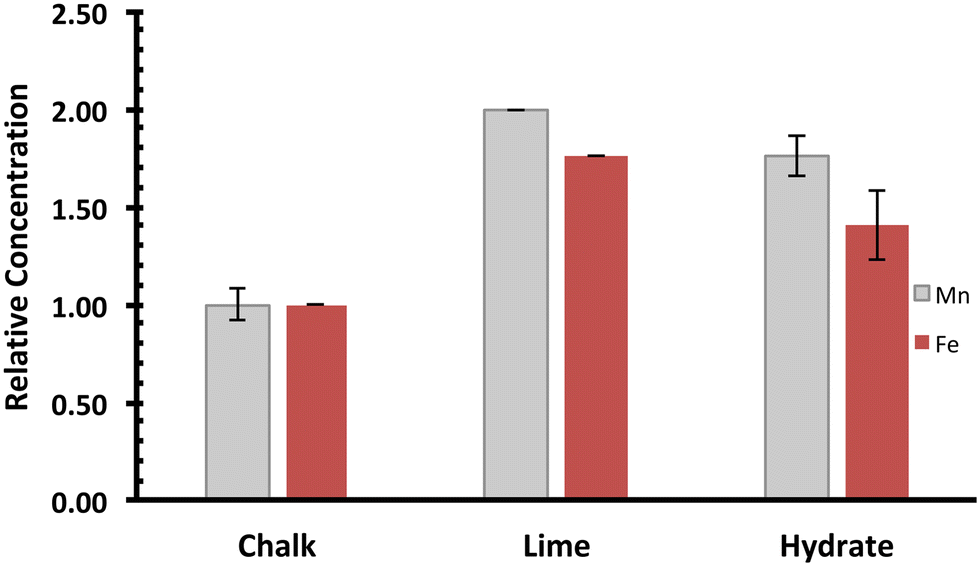 | ||
| Fig. 3 Manganese and iron impurities within the lime cycle (chalk is CaCO3; lime refers to CaO; hydrate refers to Ca(OH)2). Data obtained through HF digestion of the sample, followed by ICP-OES analysis (see ESI†). Note that in these plots, manganese (grey) and iron (red) levels, recorded as ppm (mg kg−1), have been normalised to the average manganese or iron level present in quarried chalk. Error bars indicate one standard deviation over at least three samples. | ||
Slaked lime is manufactured through reacting quicklime with a slight excess of the stoichiometric amount of water, to afford lime hydrate (Ca(OH)2). The exothermicity of this reaction (temperatures can reach up to 150 °C) causes the excess water to vaporise, so that the impurities will remain in the solid state. The lime hydrate is a fine white powder, with an average particle diameter of 5 μm, and is a porous material of extremely high specific surface area (>20 m2 g−1).36 As indicated in Fig. 3, on slaking the lime, the mass increase in going from CaO to Ca(OH)2 (molar mass 74 g mol−1) causes the impurity concentration to decrease by similar amounts for manganese and iron impurities. This suggests that the chemical nature of the manganese impurity is essentially unaffected by slaking, even if the iron contaminants form hydrated oxides. As indicated in ESI,† slaked lime manufactured by Singleton Birch Ltd. is typically ca. 97 wt% portlandite (Ca(OH)2). Accordingly, we may envisage that the particles of lime hydrate can be considered as existing with both “hard” core of CaCO3 surrounded by a “softer”, porous shell of Ca(OH)2. This visualisation is a little primitive, since re-carbonation of the surface of the intermediate lime particles can take place. Nevertheless, in terms of impurity distribution and dispersion, we consider the following: Fe2O3 (core/shell), FeS2/FeCO3, (core), Mn2O3 (core) and Mn3O4 (core/shell).
The manganese and iron impurities in lime hydrate cannot be removed by water alone, as will be discussed later. It follows that a solution-based chemical etching process is necessary to remove iron and manganese impurities from lime hydrate. However, as the hydrate buffers the solution at very high pH (ca. 12.3), an etching process involving reduction of the impurities to the soluble divalent ions is flawed, as this will cause the impurities to be precipitated as the (coloured) hydroxides onto the white hydrate.40 Accordingly, we consider the oxidative etching of the impurities.
Oxidative removal of manganese and iron impurities from slaked lime
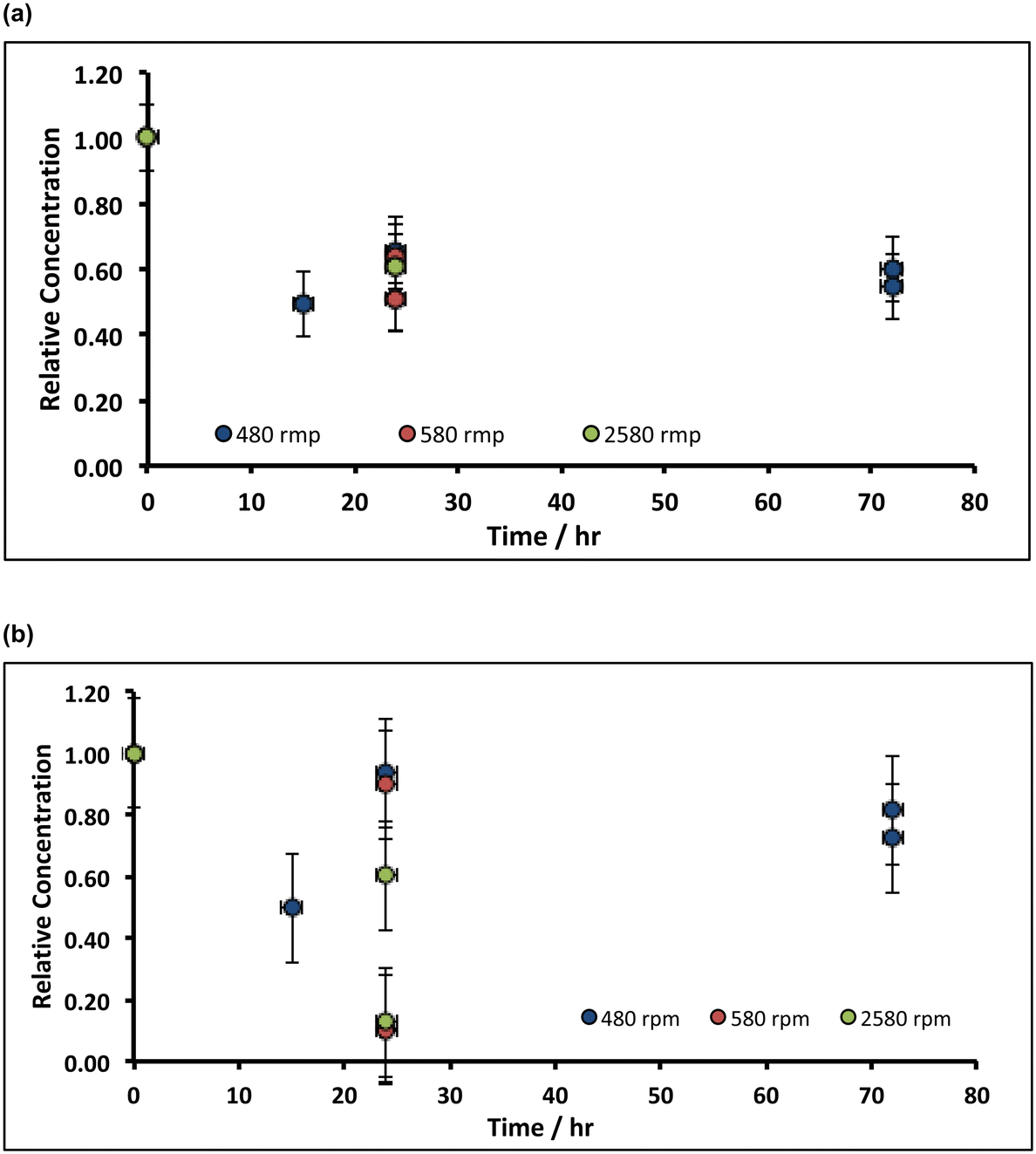
In preliminary experiments, 1 g of slaked lime were incubated overnight with 100 g (80 mL) of alkaline hypochlorite solution (∼8%, density ∼1.25 g mL−1) and left, without stirring, in an open beaker at ambient temperature, after which a pale pink supernatant was observed. Spectroscopic analysis (see ESI†) afforded both a split-peak at ca. 520 nm, corresponding to the permanganate (MnO4−) anion, and a peak at 505 nm, which matches-up with that expected from ferrate (FeO42−).41 Indeed, when the inverse spinel magnetite, Fe3O4, was treated under similar conditions, a purple supernatant, corresponding to ferrate was observed. These experiments were repeated using slaked lime manufactured from rocks derived from throughout the Welton formation of the Cretaceous chalk at the Melton Ross Quarry (North Lincolnshire, UK), in addition to slaked lime derived from Bee low Carboniferous limestone (Derbyshire, UK), both in the presence and absence of oxygen. In these experiments, after soaking the slaked lime in alkaline hypochlorite for a variable, fixed time periods, the pink supernatant was removed through vacuum filtration, and owing to the reduction of the supernatant by the filter paper (see ESI†),42 only the upper part of the dried filter cake was analysed via ICP-OES. Fig. 4 illustrates the temporal variation of both the manganese and the iron in the filter cake, where it is clear that after ca. 72 h of incubation, the manganese level drops by ca. 60%, and the iron content decreases by ca. 70%. These results are general, and hold irrespective of the origin of the slaked lime feedstock – Northern Province Cretaceous chalk (derived from a wide range of lithostratigraphies within the Welton formation) or Carboniferous limestone (Bee low formation). However, there is a subtle difference in the trends for manganese and iron: the filter cake becomes progressively depleted from manganese impurities during the treatment (irrespective of whether oxygen is present), at an initial rate of ∼1% h−1; in contrast, the removal of iron impurities is affected by the presence of oxygen in the solution. Under aerobic conditions, the iron content of the filter cake appears first to increase, and then decrease; however, there is no apparent loss of iron impurity under anaerobic conditions.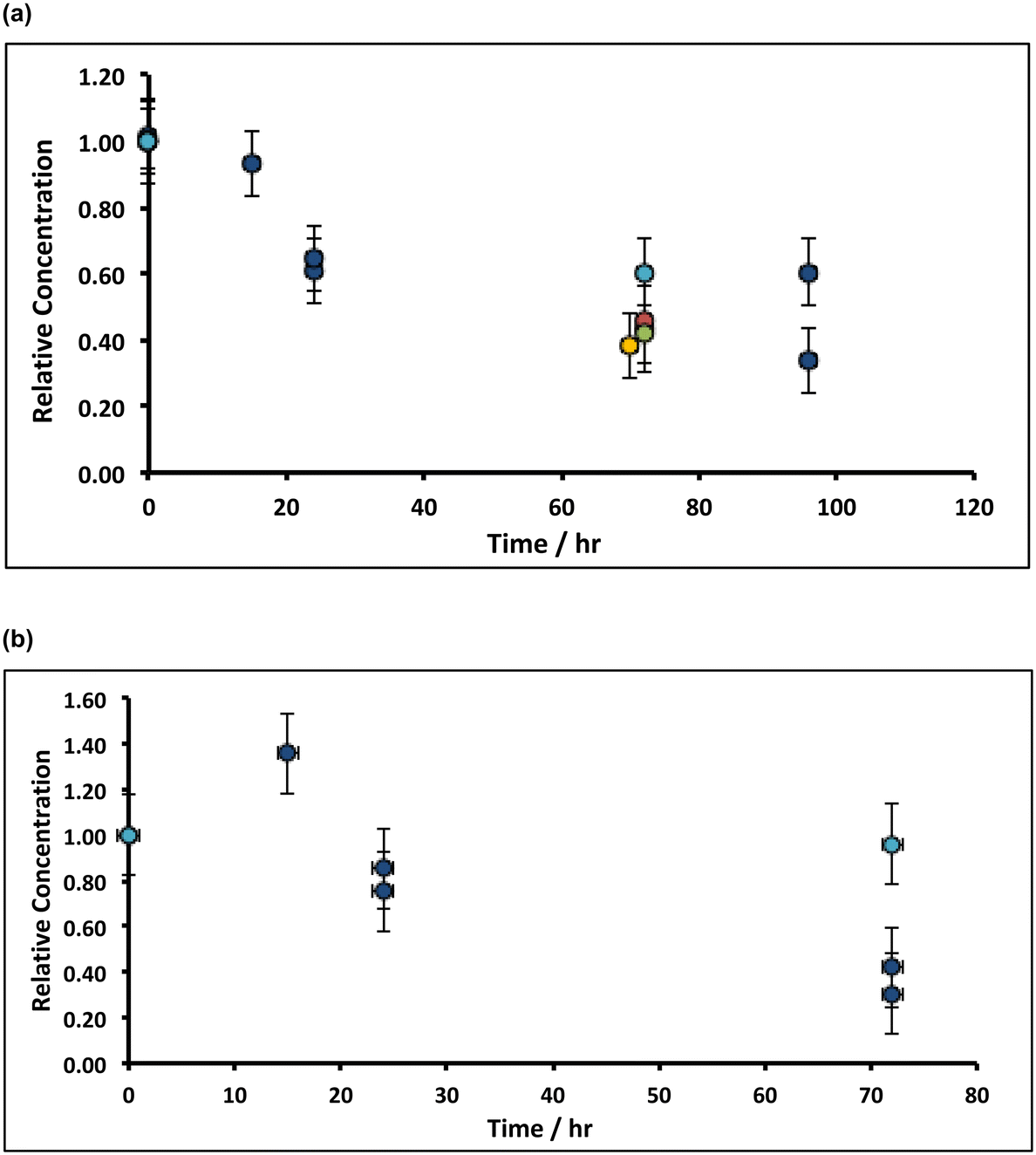 | ||
| Fig. 4 Variation of (a) Mn and (b) Fe under quiescent conditions during incubation with 8% alkaline hypochlorite solution. Note that the data have been normalised to the average manganese or iron level present in untreated sample (when time = 0 h, and where the relative concentration is unity). Unless stated, experiments were undertaken without oxygen purged-solutions. Key: Dark blue circles correspond to lime hydrate derived from Welton formation chalk derived from screenings obtained from the middle stratitographic section (between the Melton Ross Marl and the Barton Marl 1); red circles correspond to slaked lime derived from Bee Woo limestone; green and gold circles correspond to lime hydrate derived from chalk or chalk screenings, respectively, taken from the bottom bed (between the Grasby Marl to just below the Chalk Hill Marl, but above the black band); light blue circles correspond to conditions identical to the dark blue circles, except under anaerobic (oxygen-purged) conditions. Error bars indicate one standard deviation over at least three samples. | ||
These results indicate that the removal of both the manganese and the iron impurities occurs in slightly different ways, likely linked to the difference in the oxidation states of the two different transition metal ion impurities, and reaction mechanism. The manganese is present in Mn3O4 as both MnII and MnIII, so that oxidation to MnVII can be achieved through reaction with alkaline hypochlorite, without requiring the presence of oxygen.
2Mn3O4(s) + 13ClO−(aq) + 6OH−(aq) → 6MnO4−(aq) + 13Cl−(aq) + 3H2O(![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) ) ) | (A) |
| Mn3O4(s) + ClO−(aq) → Mn2O3(s) + MnO2(s) + Cl−(aq) Mn2O3(s) + ClO−(aq) → 2MnO2(s) + Cl−(aq) MnO2(s) + ClO−(aq) + 2OH−(aq) → MnO42−(aq) + Cl−(aq) + H2O() 2MnO42−(aq) + ClO−(aq) + H2O() → 2MnO4−(aq) + Cl−(aq) + 2OH−(aq) | (B) |
In contrast, since the iron impurities are present as FeIII, the oxidation to afford FeVI in the presence of concentrated sodium hydroxide (which stabilises the hypochlorite solution) must occur first through the solubilisation of the FeIII species,45
| Fe2O3(s) + 2OH−(aq) → 2FeO2−(aq) + H2O() | (C) |
In the absence of oxygen, there is little loss of iron from the filter-cake (Fig. 5) in the presence of alkaline hypochlorite, suggesting no oxidation takes place. However, in the presence of oxygen, the concentration of iron in the filter-cake first increases (by 40%), and then reduces to close to 60% of the original content. This is suggestive of an oxygen-catalysed FeIII dissolution process: oxygen is not sufficiently strong an oxidant in basic media to oxidise ferrite (FeO2−) to ferrate,
| FeO2−(aq) + 4OH−(aq) ⇄ FeO42−(aq) + 2H2O() + 3e− | (D) |
 | ||
| Fig. 5 Variation of (a) Mn and (b) Fe under stirred conditions (indicated by the rotation speed of the magnetic flea used) during incubation with 8% alkaline hypochlorite solution. Note that the data have been normalised to the average manganese or iron level present in untreated (and unstirred) sample (when time = 0 h, and where the relative concentration is unity). All experiments were undertaken under aerobic conditions with lime hydrate derived from Welton formation chalk derived from screenings obtained from the middle stratitographic section (between the Melton Ross Marl and the Barton Marl 1). Key: dark blue circles correspond to rotation speeds of 480 rpm; red circles correspond to rotation speeds of 580 rpm; green circles correspond to rotation speeds of 2580 rpm. Error bars indicate one standard deviation over at least three samples. | ||
In contrast, experiments using an ironstone (∼82% calcite, ∼17% goethite and <1% siderite, see ESI†) under reflux at 90 °C with alkaline hypochlorite did not give rise to a purple solution, indicating the need for ferrite formation to occur through dissolution prior to oxidation.
This confirms the nature of the oxidative etching process of iron-based impurities in lime, which can be written as the overall reaction:
| Fe2O3(s) + 3ClO−(aq) + 4OH−(aq) → 2FeO42−(aq) + 3Cl−(aq) + 2H2O() | (E) |
From a chemical process perspective, it is insightful to identify whether the chemical etching process is limited by slow transport of hypochlorite to the slaked lime particle surface, or by slow surface kinetics: in the former case, greater impurity removal will occur with increased agitation of the slaked lime/alkaline hypochlorite slurry; in the latter, greater removal necessitates longer incubation times. The comparison of Fig. 5 with Fig. 4 indicates that increasing the agitation of the hydrate/hypochlorite solution slurry does not translate into a greater loss of manganese from the resulting filter cake, implying that the etching is limited by the surface kinetics; this is less evident for the case of iron removal, as expected from the inferred solubilisation prior to oxidation (reaction (E)).
Owing to the control due to slow surface kinetics (ca. 50% of impurity removal through chemical etching using alkaline hypochlorite requires between 10–20 h), reaction variables (such as temperature and hypochlorite concentration) were next varied, so as both to unravel the mechanism further, and to optimise the impurity-removal chemistry, in order to develop a more useful and practical process operation.
Optimisation of dissolutive process chemistry
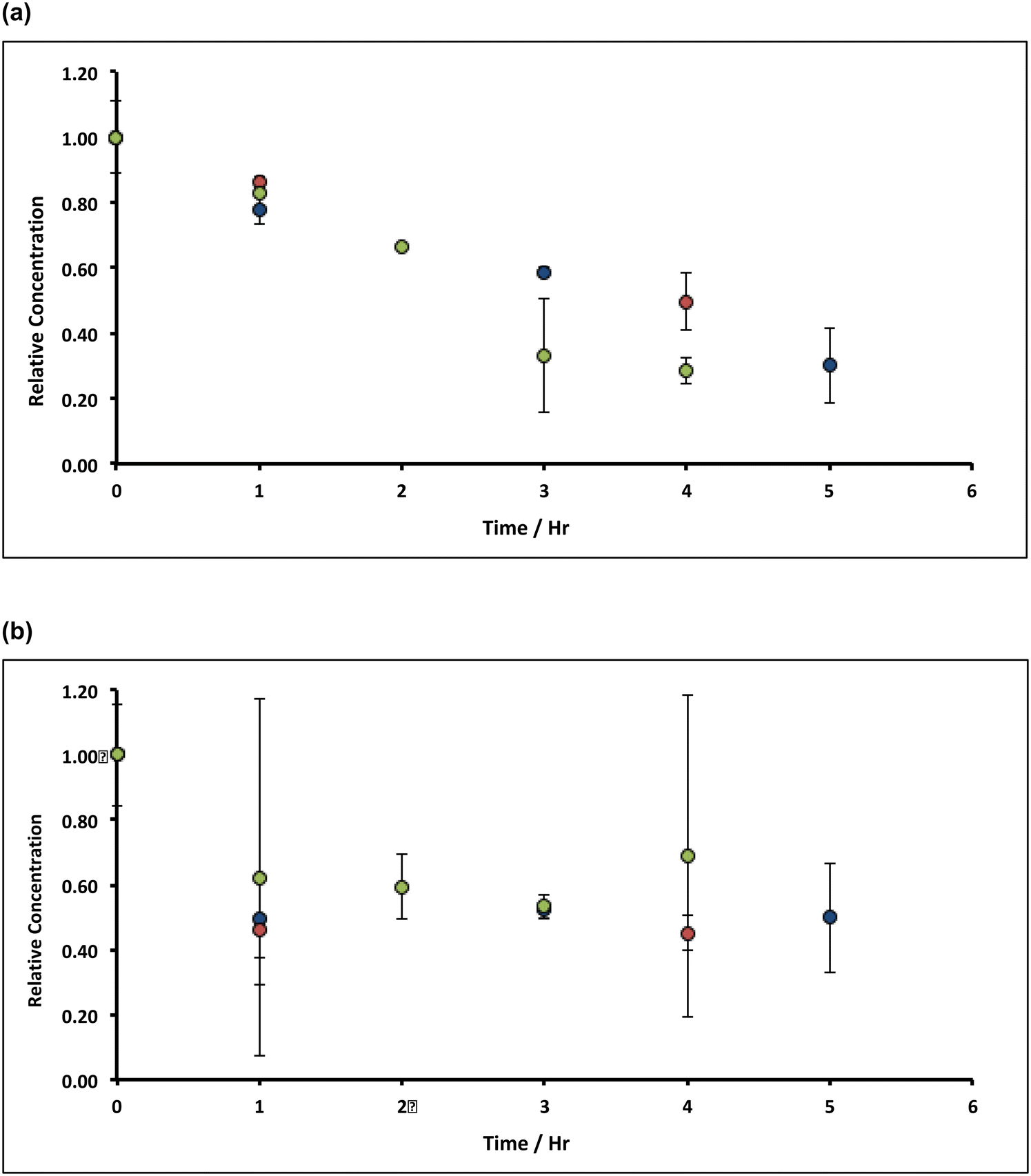
Under aerobic conditions, increasing the temperature from ambient (20 °C) to 90 °C in ∼8% alkaline hypochlorite solution, causes the initial rate of removal of both manganese and iron impurities in the filter cake to increase to ca. 1% min−1 (Mn) and ca. 3% min−1 (Fe), Fig. 6. This increase does not results from an apparent increase in solubility: the retrograde solubility of Ca(OH)2 with temperature,47 in addition to the common-ion effect, serves to reduce hydrate dissolution to negligible values (∼0.003 wt%, see ESI†). Indeed, control experiments using deionised water (1.0 g of lime hydrate equilibrated (20 min at 90 °C) in 80 mL of deionised water), reveal a 20% increase in the concentration of manganese in the solids, but ca. 60% decrease in iron (Fig. 6). Inasmuch as an increase in the manganese content is expected: ∼5 wt% of hydrate will dissolve (see ESI†), enriching the impurity concentration, the decrease in iron content of the resulting filter cake is consistent with the formation (and thermally enhanced solubility) of ferrite.45b Again, the effect of increasing the mass transport of hypochlorite (higher than that due to thermal convection) has only very limited effect (Fig. 6).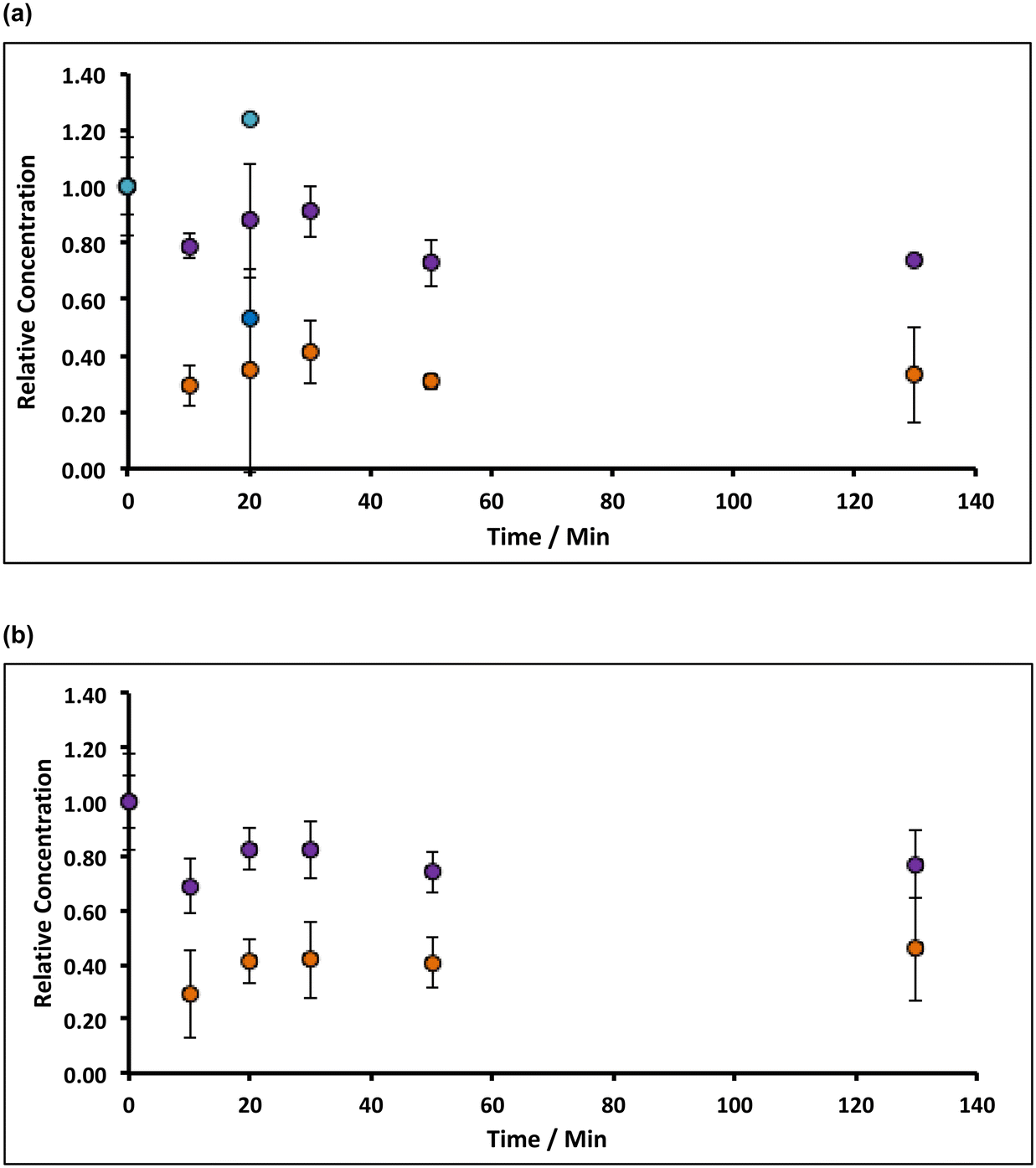 | ||
| Fig. 6 Variation of Mn and Fe concentrations in lime hydrate filter cake during incubation with 8% alkaline hypochlorite solution or deionised water at 90 °C, under (a) non-stirred but thermally convective, and (b) stirred at an arbitrary rotation speed of the magnetic flea used. Note that the data have been normalised to the average manganese or iron level present in untreated (and unstirred) sample (when time = 0 h, and where the relative concentration is unity). All experiments were undertaken under aerobic conditions with lime hydrate derived from Welton formation chalk derived from screenings obtained from the middle stratitographic section (between the Melton Ross Marl and the Barton Marl 1). Key: purple circles correspond to Mn concentrations using alkaline hypochlorite solution; sky blue circles correspond to Mn concentration using deionised water; orange circles correspond to Fe concentrations using alkaline hypochlorite solution; blue circles correspond to Fe concentration using deionised water. Error bars indicate one standard deviation over at least three samples. | ||
The plots in Fig. 6 indicate that prolonged incubation at 90 °C has little effect on the etching. This likely results from the thermal decomposition of hypochlorite, which can be catalysed by ferric oxide, and is thought48 to proceed via a slow initial disproportionation reaction of hypochlorite to chlorite, followed by faster oxidation to chlorate:
| 2ClO−(aq) → ClO2−(aq) + Cl−(aq) ClO−(aq) + ClO2−(aq) → ClO3−(aq) + Cl−(aq) | (F) |
 | ||
| Fig. 7 Variation of (a) Mn and (b) Fe concentrations in lime hydrate filter cake during variable time incubation intervals (1, 2 and 3 h) with 8% alkaline hypochlorite solution or deionised water at 90 °C whilst stirred at an arbitrary rotation speed of the magnetic flea used. Note that the data have been normalised to the average manganese or iron level present in untreated (and unstirred) sample (when time = 0 h, and where the relative concentration is unity). All experiments were undertaken under aerobic conditions with lime hydrate derived from Welton formation chalk derived from screenings obtained from the middle stratitographic section (between the Melton Ross Marl and the Barton Marl 1). Key: green, blue and red circles correspond to incubation times of 1, 2 and 3 h, respectively. Error bars indicate one standard deviation over at least three samples. | ||
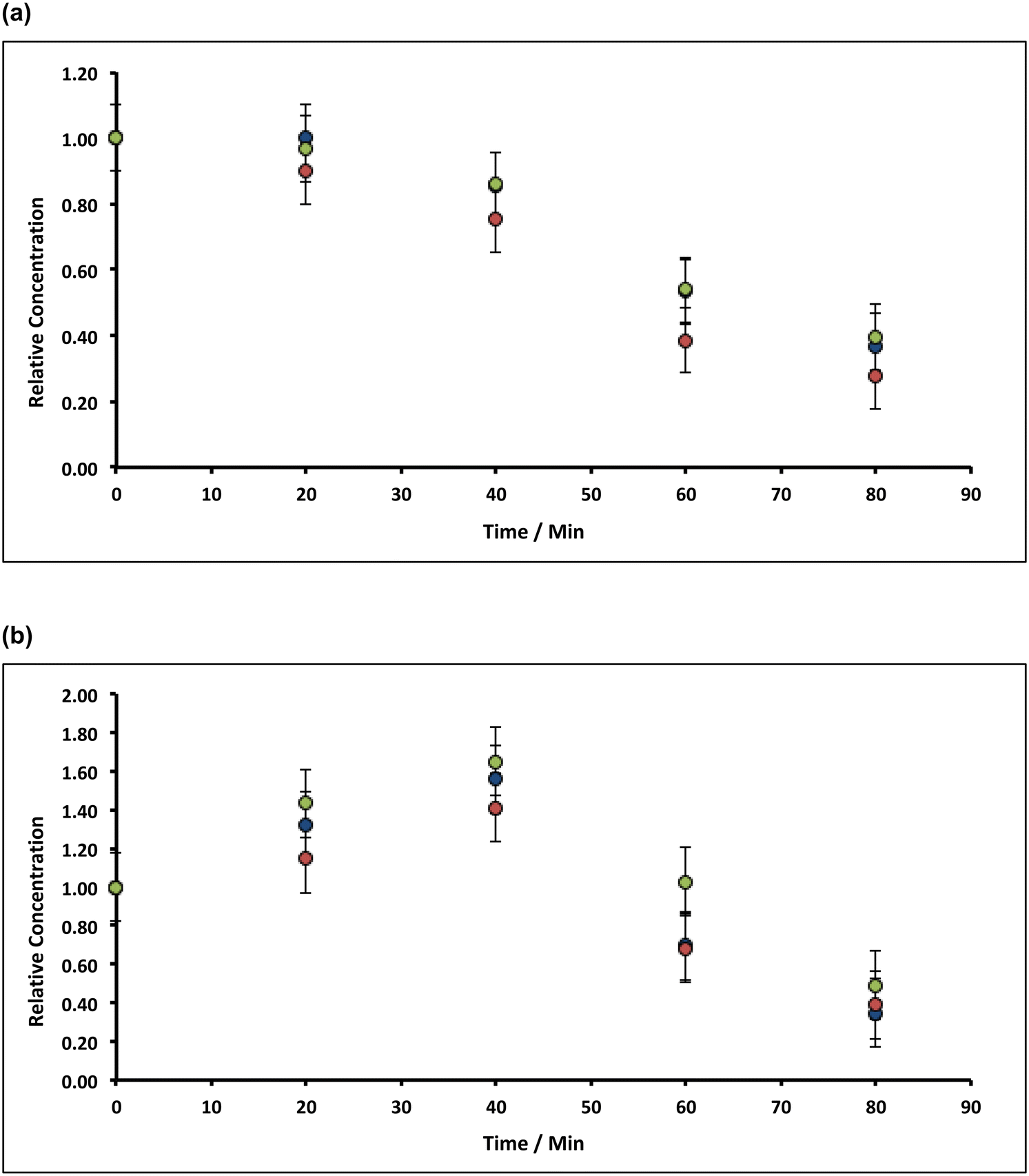 | ||
| Fig. 8 Variation of (a) Mn and (b) Fe concentrations in lime hydrate filter cake during fixed 20 min incubation cascades with 8% or 14% alkaline hypochlorite solution or deionised water at 90 °C whilst stirred at an arbitrary rotation speed of the magnetic flea used. Note that the data have been normalised to the average manganese or iron level present in untreated (and unstirred) sample (when time = 0 h, and where the relative concentration is unity). All experiments were undertaken under aerobic conditions with lime hydrate derived from Welton formation chalk derived from screenings obtained from the middle stratitographic section (between the Melton Ross Marl and the Barton Marl 1). Key: blue, red and green circles correspond to hypochlorite chloride levels of 8% (batch 1), 8% (batch 2) and 14%, respectively. Error bars indicate one standard deviation over at least three samples. | ||
Under these conditions of temperature (90 °C) and cascade interval (20 min), increasing the hypochlorite concentration from ∼8% to ∼14% (a concentration factor of 1.75), has little effect on the removal of manganese or iron (Fig. 8). This indicates that removal of both manganese and iron follows zeroth-order kinetics in hypochlorite in both cases. Focussing on the mechanistically less complicated manganese dissolution, the manganese-removal rate,
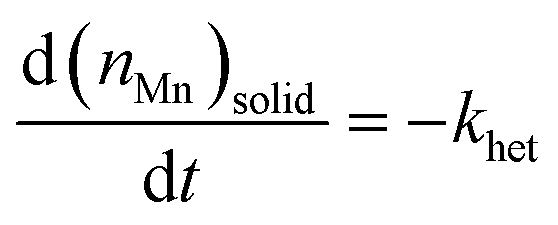 | (1) |
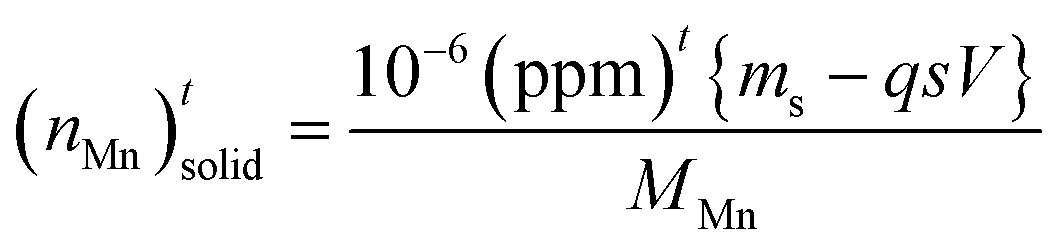 , in which (ppm)t is the manganese concentration in the filter-cake, ms is the mass of the virgin lime hydrate used at the start of the experimental cascade (in g), s is the solubility of the lime hydrate in the alkaline solution at the experimental temperature (in g L−1), V is volume of the alkaline hypochlorite solution used (in L), which is kept at a constant value throughout the cascade, q is the cascade number, with the first batch taking q = 1, and MMn is the molar mass of manganese (in g mol−1). This enables eqn (1) to be re-cast in terms of the solid manganese concentration, and integrated using the boundary condition t = 0, (ppm)t = (ppm)0, to yield eqn (2):
, in which (ppm)t is the manganese concentration in the filter-cake, ms is the mass of the virgin lime hydrate used at the start of the experimental cascade (in g), s is the solubility of the lime hydrate in the alkaline solution at the experimental temperature (in g L−1), V is volume of the alkaline hypochlorite solution used (in L), which is kept at a constant value throughout the cascade, q is the cascade number, with the first batch taking q = 1, and MMn is the molar mass of manganese (in g mol−1). This enables eqn (1) to be re-cast in terms of the solid manganese concentration, and integrated using the boundary condition t = 0, (ppm)t = (ppm)0, to yield eqn (2):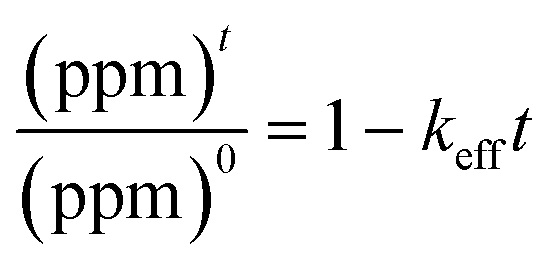 | (2) |
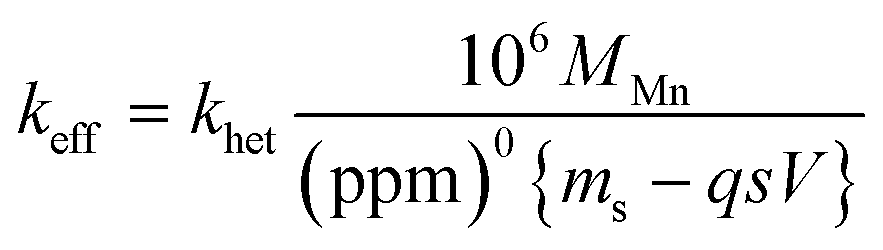 . For the extremely low lime hydrate solubilities (for further discussion, see ESI†), the corrected mass is essentially constant, so that plots of relative manganese concentration against time are linear, as observed in Fig. 9 for small cascade numbers. These afford values of khet = 6.1 ± 1.5 × 10−8 mol min−1 at 90 °C for the removal of manganese impurities in lime hydrate, which are independent of both the derived limestone type (Cretaceous chalk or Carboniferous limestone), and the mass of the material treated, suggesting all samples are approximately monodisperse and with similar particle sizes.
. For the extremely low lime hydrate solubilities (for further discussion, see ESI†), the corrected mass is essentially constant, so that plots of relative manganese concentration against time are linear, as observed in Fig. 9 for small cascade numbers. These afford values of khet = 6.1 ± 1.5 × 10−8 mol min−1 at 90 °C for the removal of manganese impurities in lime hydrate, which are independent of both the derived limestone type (Cretaceous chalk or Carboniferous limestone), and the mass of the material treated, suggesting all samples are approximately monodisperse and with similar particle sizes.
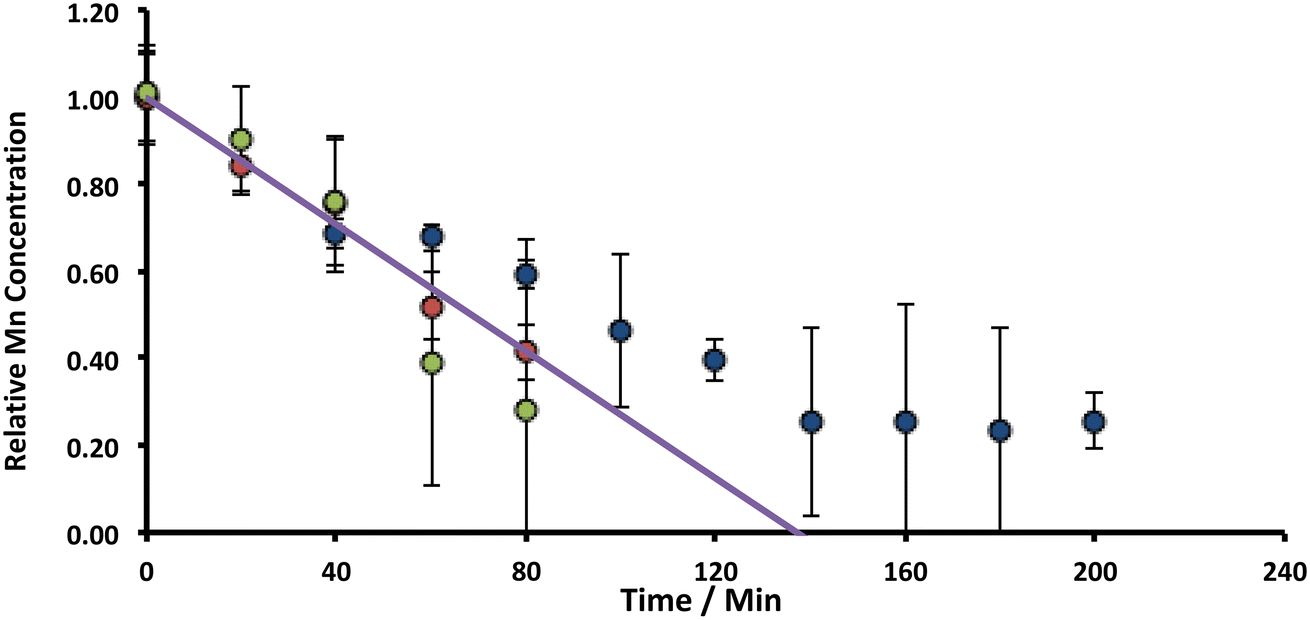 | ||
| Fig. 9 Variation of Mn concentrations in lime hydrate filter cake during fixed 20 min incubation cascades with 14% alkaline hypochlorite solution or deionised water at 90 °C whilst stirred at an arbitrary rotation speed of the magnetic flea used. Note that the data have been normalised to the average manganese or iron level present in untreated (and unstirred) sample (when time = 0 h, and where the relative concentration is unity). All experiments were undertaken under aerobic conditions. Blue and green circles correspond to lime hydrate derived from Welton formation chalk derived from screenings obtained from the middle stratitographic section (between the Melton Ross Marl and the Barton Marl 1), with initial hydrate mass of 2.0 and 1.0 g, respectively; red circles correspond to lime hydrate derived from Carboniferous limestone, with initial hydrate mass of 1.0 g. Error bars indicate one standard deviation over at least three samples. The purple line corresponds to eqn (2), with the average effective rate constant, keff = 7.3 ± 1.9 × 10−3 min−1. | ||
For manganese removal, these are pure kinetics – they have no contribution from mass transfer: under the approximation that the Sherwood number (Sh) is 2, viz.,49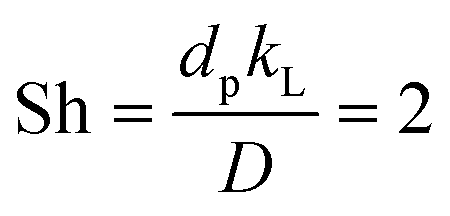 , an equation relevant for mass transfer to small particles, where dp is the slaked lime particle diameter (5 μm),36D is the diffusion coefficient of hypochlorite in aqueous alkaline solution (∼10−5 cm2 s−1), the mass transfer coefficient (kL) is estimated as being 0.04 cm s−1. In the experiments in Fig. 9, the etching rate constant at 90 °C is ∼10−9 mol s−1 from a surface area of36 ∼20 m2 g−1 using an hypochlorite concentration of ∼5 M, affording a mass transfer coefficient of ∼10−12 cm s−1, which is many orders of magnitude less than the transport-limited rate, confirming no contribution from mass transfer.50
, an equation relevant for mass transfer to small particles, where dp is the slaked lime particle diameter (5 μm),36D is the diffusion coefficient of hypochlorite in aqueous alkaline solution (∼10−5 cm2 s−1), the mass transfer coefficient (kL) is estimated as being 0.04 cm s−1. In the experiments in Fig. 9, the etching rate constant at 90 °C is ∼10−9 mol s−1 from a surface area of36 ∼20 m2 g−1 using an hypochlorite concentration of ∼5 M, affording a mass transfer coefficient of ∼10−12 cm s−1, which is many orders of magnitude less than the transport-limited rate, confirming no contribution from mass transfer.50
The low temperature data illustrated in Fig. 4 can also be analysed through eqn (2), to afford khet = 8.0 ± 3.4 × 10−10 mol min−1 at 20 °C for manganese removal from lime hydrate. Again, this is independent of the derived limestone type, and whether the etching is undertaken in the presence, or absence of oxygen. These low and high temperature rate constants enable the estimation of the activation barrier for the etching process to be 0.58 ± 0.07 eV, using an Arrhenius-type relationship. This activation energy is similar to literature estimates for the DC conductivity (via electron hopping between MnII and MnIII sites) within nano-crystalline (∼10 nm) Mn3O4 (0.45–0.69 eV),51 and suggests that the slow step for manganese etching is an interfacial electron transfer event:
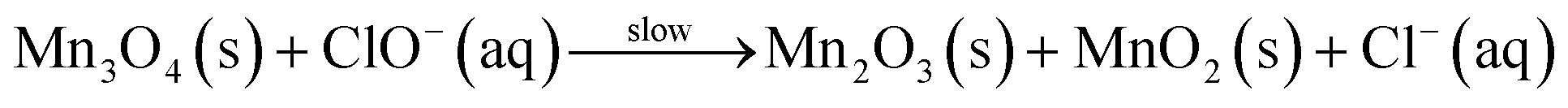 | (G) |
In order to confirm the dissolution rate as being controlled by an interfacial electron transfer process, ambient temperature experiments were undertaken in which the chemical oxidant was replaced by distilled water saturated with both ozone and lime hydrate (pH 12.3). In this case (data not shown), the value of khet was observed to be 2.2 ± 0.1 × 10−8 mol min−1 (when corrected for the greater solubility of lime hydrate in water than in sodium hydroxide solution). This value is significantly larger than that observed at ambient temperature for alkaline hypochlorite oxidants, even after accounting for the concentration differences53 between ozone (at saturation, ∼5 mM) and the hypochlorite solution used (∼2.5 M). Nevertheless, these results are consistent with the expected linear free energy nature of the activation/driving force within the normal Marcus region;54 the standard potentials at45a pH 14, E0B = +1.24 V vs. SHE (for O3/O2), with E0B = +0.89 V vs. SHE (for ClO−/Cl−) confirm the greater oxidising strength of ozone compared with hypochlorite, and, thence, the faster dissolution rate in the presence of ozone.
The observation of zeroth-order kinetics for manganese loss from slaked lime gives rise to a “rule-of-thumb” for the scale-up of this process chemistry. However, it is important to realise that after removal of ca. 75% of the original manganese impurity, a constant amount remains, as determined via ICP-OES (Fig. 9). This is “inactive” manganese – it is inaccessible and cannot be removed through a chemical etch with hypochlorite – either because it is too deep within the core of the hydrate particles (within the 3% calcite), or because it is in a different “shell” of the hydrate, or even in a different oxidation form, compared with the “active manganese”, and thus has a slower etching rate constant.
We next consider how the insights into this chemical etching chemistry can adapted to existing processes where lime is used for the drinking water distribution network.
Adaptation for end-use
Current methods the removal of soluble iron and manganese in drinking water typically uses elaborate filtration beds, each with a unit treatment cost of €0.12 m−3 (for a mid-sized water treatment plant).9 The removal of iron or manganese in drinking water treatment chemicals is not a standard practice, so that implementation of corrective actions, such as those indicated herein, necessarily has to be of low cost. Given this, considering typical water treatment plants employ pH modifiers such as lime, and oxidative disinfection chemicals such as chlorine or hypochlorite at a number of process stages, “manganese-free” lime is only needed if there is no filtration bed after the last combination of lime and oxidant. This reduces the demand for “manganese-free” lime.The chemical etching pathway demonstrated herein suffers from slow, zeroth-order heterogeneous kinetics, and this requires process heat to raise the temperature to 90 °C. However, these high temperatures facilitate the decomposition of the alkaline oxidant required, so that a cascade approach is required, if hypochlorite is used as the oxidant, which adds to the overall operational expenditure required: this work has demonstrated capability only using at a solid hydrate to liquid solution oxidant ratio of between 0.0125–0.025 g mL−1. Whilst further work may be able to demonstrate viability through increasing this ratio, an alternative is to employ the use of a different oxidant, such as the continual bubbling of chlorine gas in an aqueous lime slurry (“aqualime”). Although lime manufacturers do distribute aqualime, they tend not to have chlorine gas on-site. However, water treatment plants typically have opportunities to make aqualime on-plant, and sometimes may have the process safety infrastructure needed to use chlorine on-plant, so that water companies may use this chemistry. An additional advantage of performing this chemistry on a water treatment plant is that the permanganate and ferrate generated can be used to remove the odour associated with reduced gases (such as hydrogen sulphide and ammonia) that are typically present on such sites.
Conclusions
In this work, we have developed high pH redox chemistry that demonstrates how both manganese and iron impurities can be removed from lime hydrate derived from quarried limestones. The mechanism for the oxidative removal is different for both iron and manganese: in the former, oxygenated solutions are required to transform the surface ferric oxide into ferrite, which can be solubilised through oxidation with hypochlorite to yield soluble ferrate; in contrast, hausmannite is oxidised stepwise by hypochlorite (or ozone), ultimately to furnish soluble permanganate.These reactions are under activation control, with the kinetics of oxidation increasing with temperature. For manganese, these dissolution kinetics are zeroth-order that appear to be limited by the rate of electron transfer, enabling design rules for the up-scale of this process to be determined of ∼1% min−1 for manganese and ∼3% min−1 for iron, at 90 °C. However, it is important to realise that this chemistry cannot create a completely “manganese-free” or “iron-free” product: around 25% of the total manganese (“inactive” manganese) remains, irrespective of the feedstock (Carboniferous limestone or Cretaceous chalk).
Although further work is needed to examine the economic viability of this process chemistry at scale, it nevertheless provides an opportunity for manufacturers to develop suitable lime hydrate products that hold very low levels of manganese and iron impurities. Alternatively, given the simplicity of the operations, and that the treatment chemicals are similar to those already deployed for disinfection in the water industry, the impurity-removal process could potentially be integrated on the site of existing water treatment works.
Last, we note that control of manganese in lime products is important for processes other than in those employed by the water industry.55–57
Author contributions
Conceptualisation: DK, CRM, NSL, JDW. Methodology: DK, CRM, NSL, JDW. Investigation: DK. Formal analysis: DK, CRM, NSL, JDW. Software: DK, JDW. Validation: DK, RAW. ICP-OES: MT. X-Ray diffraction and analysis: TJP. Resources: DK, CRM, SMK, NSL, JDW. Data curation: DK, CRM, JDW. Writing – original draft: DK, CRM, NSL, JDW. Writing – review and editing: DK, RAW, CRM, MT, TJP, SMK, NSL, JDW. Visualisation: DK, CRM, NSL, JDW. Supervision: DK, CRM, NSL, JDW. Project administration: CRM, NSL, JDW. Funding acquisition: CRM, SMK, NSL, JDW.Conflicts of interest
DK and CRM are both employed by Singleton Birch Ltd.Acknowledgements
This work was funded by Singleton Birch Ltd. (50%), together with InnovateUK (25%) and the Engineering and Physical Sciences Research Council (25%), through a Knowledge Transfer Partnership (Programme Number 0110770). DK thanks The University of Hull for funding his MSc research studentship. RAW thanks Singleton Birch for funding. We thank Haydn Ward (ParagonID) for the generous donation of both magnetite and maghaemite used in this work, Pauline Smedley (British Geological Survey) for arranging access to ref. S25,† and Bernadette Ryan (Severn Trent, Plc) and Anna Taliana (Dŵr Cymru Welsh Water) for useful discussions. DK, NSL and JDW thank the Johnston Quarry Group for generously supplying a sample of Great Tew Ironstone. We are grateful to two anonymous reviewers who have improved the flow of this manuscript.Notes and references
- See, for example, United Nations General Assembly Resolution 64/292, The Human Right to Water and Sanitation, United Nations, adopted July 28, 2010 Search PubMed.
- E. C. Craig, E. L. Bean and R. W. Sawyer, Iron and lime in removal of manganese, J. – Am. Water Works Assoc., 1932, 24, 1762 CrossRef CAS.
- The complexity of the water distribution and regulatory framework in the United Kingdom is elegantly captured in a recent speech by Sir James Bevan, Chief Executive of the Environment Agency, Escaping the Jaws of Death: Ensuring Enough Water in 2050, Waterwise Conference, March 19, 2019, full transcript available at the following URL, https://www.gov.uk/government/speeches/escaping-the-jaws-of-death-ensuring-enough-water-in-2050, (Accessed December 28, 2021) Search PubMed.
- World Heath Organisation, Guidelines for Drinking Water Quality, World Heath Organisation, Geneva, 4th edn, (incl. 1st add.), 2017 Search PubMed.
- See, for example, D. K. Todd and L. W. Mays, Groundwater Hydrology, Wiley, New Delhi, 3rd edn, 2005 Search PubMed.
- (a) For example, the chalk aquifer of the East Riding of Yorkshire is contaminated by both atrazine (a pesticide) and dichloromethane (a non-aqueous phase liquid) at concentrations above European guideline values, q.v., I. N. Gale and H. K. Rutter, The Chalk Aquifer of Yorkshire, British Geological Survey Research Report RR/06/04, British Geological Survey, Keyworth, 2006 Search PubMed; (b) Likewise, “forever chemicals” such as perfluorinated hydrotopes have been found in soils, surface waters and groundwaters throughout England: see, for example, E. Pemberton, Poly- and Perfluoroalkyl Substances (PFAS): Sources, Pathways and Environmental Data, Chief Scientist's Report, Environment Agency, Bristol, 2021 Search PubMed.
- (a) J. Fawell and M. J. Nieuwenhuijsen, Contaminants in drinking water, Br. Med. Bull., 2003, 68, 199 CrossRef CAS PubMed; (b) S. Sharma and A. Bhattacharya, Drinking water contamination and treatment techniques, Appl. Water Sci., 2017, 7, 1043 CrossRef CAS.
- (a) S. Zahran, D. Mushinski, S. P. McElmurry and C. Keyes, Water lead exposure risk in Flint, Michigan after switchback in water source: implications for lead service line replacement policy, Environ. Res., 2020, 181, 108928 CrossRef CAS PubMed; (b) K. J. Pieper, M. Tang and M. A. Edwards, Flint water crisis caused by interrupted corrosion control: investigating “ground zero” home, Environ. Sci. Technol., 2017, 51, 2007 CrossRef CAS PubMed.
- Best Practice Guide on the Control of Iron and Manganese in Water Supply, ed. A. Postawa and C. Hayes, IWA Publishing, London, 2013 Search PubMed.
- Drinking Water Inspectorate, Drinking Water 2020: The Chief Inspector's Report for Drinking Water in England, Drinking Water Inspectorate, London, 2021 Search PubMed.
- (a) V. A. Pattinson, D. P. Butcher, J. C. Labadz and J. Shacklock, Water discolouration and the ôô of the reservoir, Phys. Chem. Earth, 1995, 20, 175 CrossRef; (b) S. Buss, Review of Spatial Factors Controlling Water Discolouration in England and Wales, Appendix 10, Technical Note 64116BN TN1D1, ESI, Ltd., 2018 Search PubMed.
- (a) L. I. Sly, M. C. Hodgkinson and V. Arunpairojana, Deposition of manganese in a drinking water distribution system, Appl. Environ. Microbiol., 1990, 56, 628 CrossRef CAS PubMed; (b) J. M. Cerrato, L. P. Reyes, C. N. Alvarado and A. M. Dietrich, Effect of PVC and iron materials on Mn(II) deposition in drinking water distribution systems, Water Res., 2006, 40, 2720 CrossRef CAS PubMed; (c) J. H. H. Vreeburg and J. B. Boxall, Discolouration in potable water distribution systems: a review, Water Res., 2007, 41, 519 CrossRef CAS PubMed.
- (a) For a thorough discussion of process improvements suggested by Severn Trent to improve their distribution of soft water, derived from the Elan Valley catchment in Wales, to the City of Birmingham (maximum flow of 450 ml per day), so as to reduce the iron, manganese, colour, turbidity and natural aluminium levels, see T. Schofield, R. Perkins and J. S. Simms, Frankley Water Treatment Works redevelopment: pilot-scale studies, Water Environ. J., 1991, 5, 370 CrossRef CAS; (b) A. E. Griffin, Significance and removal of manganese in water supplies, J. – Am. Water Works Assoc., 1960, 52, 1326 CrossRef.
- S.-W. Lee, M. Jones, C. Romano and B. M. Tebo, Formation of manganese oxide minerals by bacteria, in Redox-Reactive Minerals: Properties, Reactions and Applications in Natural Systems and Clean Technologies, ed. I. A. M. Ahmed and K. A. Hudson-Edwards, EMU Notes in Mineralogy, The Mineralogical Society of Great Britain and Ireland, London, 2017, ch. 7, vol. 17, p. 173 Search PubMed.
- D. Dixon, N. Anderson, B. Bolto, B. Chiswell, L. Sly, T. Hurse, K. Craig, G. Hamilton, B. Hutchinson, C. West and R. Woolley, The Removal of Manganese from Drinking Waters, Research Report No. 26, The Co-operative Research Centre for Water Quality and Treatment, Salisbury, Australia, 2006 Search PubMed.
- (a) We note that recent laboratory-scale research16b has illustrated how Mn2+ (at concentrations of 2 ppm in 0.1 M aqueous phosphate buffer) can be removed as Mn2O3 (lowering the aqueous concentration to ca. 900 ppb) via advanced electrochemical oxidation, although this technology has yet to be assessed for technical, environmental and economic feasibility; (b) S. T. McBeath, D. P. Wilkinson and N. J. D. Graham, Advanced electrochemical oxidation for the simultaneous removal of manganese and generation of permanganate oxidant, Environ. Sci.: Water Res. Technol., 2020, 6, 2405 RSC.
- (a) G. Dillon, P. Jackson, A. Ewence and J. Jonsson, Potential Contaminants in Drinking Water Treatment Chemicals: Final Report to the Department for the Environment, Food and Rural Affairs, Report No. Defra9033.03, WRc, plc, Swindon, 2003 Search PubMed; (b) Note that in the USA, similar standards are in operation, see NSF/ANSI 60-2016, Drinking Water Treatment Chemicals – Health Effects, NSF International, Michigan, 2016 Search PubMed; (c) P. Rumsby, H. Clegg, J. Jonsson, V. Benson, M. Harman, T. Doyle, L. Rushton, P. Warwick and D. Wilkinson, Speciation of Manganese in Drinking Water: Final Report for the Drinking Water Inspectorate, Report No. DWI70-2-276, WRc, plc, Swindon, 2014 Search PubMed.
- (a) S. R. Jenkins, L. Benefield, M. J. Keal and R. S. Peacock, Effective manganese removal using lime as an additive, J. – Am. Water Works Assoc., 1984, 76, 82 CrossRef CAS; (b) A. M. Silva, F. L. S. Cruz, R. M. F. Lima, M. C. Teixeira and V. A. Leão, Manganese and limestone interactions during minewater treatment, J. Hazard. Mater., 2010, 181, 514 CrossRef CAS PubMed; (c) D. L. Trollip, J. C. Hughes and L. W. Titshall, Sources of manganese in the residue from a water treatment plant, Water SA, 2013, 39, 265 CAS.
- (a) A. Hamdouni, G. Montes-Hernandez, M. Tlili, N. Findling, F. Renard and C. V. Putnis, Removal of Fe(II) from groundwater via aqueous portlandite carbonation and calcite-solution interactions, Chem. Eng. J., 2016, 283, 404 CrossRef CAS; (b) A. Sdiri, T. Higashi, T. Hatta, F. Jamoussi and N. Tase, Removal of heavy metals from aqueous solution by limestone, Int. J. Glob. Environ. Issues, 2012, 12, 171 CrossRef; (c) B. I. Whittington, The chemistry of CaO and Ca(OH)2 relating to the Bayer process, Hydrometallurgy, 1996, 43, 13 CrossRef CAS.
- (a) S. Caserini, N. Storni and M. Grosso, The availability of limestone and other raw materials for ocean alkalinity enhancement, Global Biogeochem. Cycles, 2022, 36, e2021GB007246 CrossRef CAS; (b) J. Hartmann and N. Moosdorf, The new global lithological map database GLiM: a representation of rock properties at the Earth surface, Geochem., Geophys., Geosyst., 2012, 13, 1 CrossRef.
- (a) Data taken from (i) USGS Mineral Commodity Summaries 2022, U.S. Geological Survey, Reston, Virginia, 2022, and (ii) the associated data release for the 2020 tables, available to download at https://www.usgs.gov/centers/national-minerals-information-center/lime-statistics-and-information, accessed January 6, 2023 Search PubMed; (b) This is in agreement with the proportion of end-use of ground calcium carbonate (9%) used for environmental protection (flue gas, drinking water and sewage treatment) reported in European Commission, Report on Critical Raw Materials for the EU: Non-Critical Raw Materials Profiles, 2014, Reference: Ares(2014)3690319-06/11/2014 Search PubMed.
- (a) Department of the Environment, Appraisal of High-Purity Limestones in England and Wales: A Study of Resources, Needs, Uses and Demands: Summary Report, Department of the Environment, London, 1990 Search PubMed; (b) D. J. Harrison, J. M. Hudson and B. Cannell, Appraisal of High-Purity Limestones in England and Wales: A Study of Resources, Needs, Uses and Demands: Part 1: Resources, British Geological Survey Mineral Resources Series WF/90/10, British Geological Survey, Keyworth, 1991 Search PubMed; (c) D. J. Harrison, High-purity limestones in England and Wales, Q. J. Eng. Geol., 1993, 26, 293 CrossRef; (d) D. J. Harrison, D. Highley, A. Bloodworth, R. Bate, D. Cameron, P. Lusty and D. Rayner, Limestone, Mineral Planning Factsheet prepared for the Office of the Deputy Prime Minister, British Geological Survey, Keyworth, 2006, available to download from https://www.mineralsUK.com, (accessed December 28, 2021) Search PubMed; (e) C. Mitchell, High-purity limestone quest, Ind. Miner., 2011, 48 Search PubMed.
- The British Lime Association (one of the constituent bodies of the Mineral Products Association) is the trade organisation for the companies that are responsible for producing at least 75% (ca. 1.2 MTe y−1) of the industrial lime sold in the UK: https://www.britishlime.org (accessed December 28, 2021). The British Calcium Carbonate Federation is the trade body representing the interests of UK producers and suppliers of chalk, limestone, marble, dolomite and precipitated calcium carbonate, whose members produce >3.5 MTe y−1 of calcium carbonate products: https://www.calciumcarbonate.org.uk, (accessed December 28, 2021).
- (a) G. D. Gaunt, T. P. Fletcher and C. J. Wood, Geology of the Country Around Kingston-upon-Hull and Brigg: Memoir for 1:50000 geological sheets 80 and 89 (England and Wales), British Geological Survey, Her Majesty's Stationary Office, London, 1992 Search PubMed; (b) F. Whitham, The geology of the Melton Ross chalk quarries, Humber Geologist, No. 12, Hull Geological Society, Kingston-upon-Hull, 1999, available online: https://www.hullgeolsoc.co.uk/meltonrs.htm, (accessed December 28, 2021) Search PubMed.
- (a) D. J. Harrison, The Limestone and Dolomite Resources of the Country Around Buxton, Derbyshire: Description of 1:25000 sheet SK 07 and parts of SK 06 and 08, Mineral Assessment Report 77 for the Institute of Geological Sciences, Her Majesty's Stationary Office, London, 1981 Search PubMed; (b) D. J. Harrison and K. A. Mcl. Adlam, Limestones of the Peak: A Guide to the Limestone and Dolomite Resources of the Peak District of Derbyshire and Staffordshire: Description of parts of 1:50000 geological sheets 99, 111, 112, 124 and 125, Mineral Assessment Report 144 for the British Geological Survey, Her Majesty's Stationary Office, London, 1985 Search PubMed.
- (a) F. C. Cox, D. McC. Bridge and J. H. Hull, Procedure for the Assessment of Limestone Resources, Mineral Assessment Report for the Institute of Geological Sciences, No. 30, Her Majesty's Stationary Office, London, 1977 Search PubMed; (b) D. J. Harrison, Industrial Minerals Laboratory Manual: Limestone, Mineralogy and Petrology Series Technical Report WG/92/29, British Geological Survey, Keyworth, 1993 Search PubMed; (c) D. J. Harrison, S. D. J. Inglethorpe, C. J. Mitchell, S. J. Kemp, P. Chaodumrong and M. Charusribandhu, Procedures for the Rapid Assessment of Limestone Resources, Overseas Geology Series Technical Report WC/98/1, British Geological Survey, Keyworth, 1998 Search PubMed.
- R. S. Boynton, Chemistry and Technology of Lime and Limestone, John Wiley and Sons, New York, 2nd edn, 1980 Search PubMed.
- (a) W. M. Edmunds, W. G. Darling, D. G. Kinniburgh, L. Dever and P. Vachier, Chalk Groundwater in England and France: Hydrogeochemistry and Water Quality, British Geological Survey Research Report SD/92/2, Keyworth, 1992 Search PubMed; (b) Whilst limestone rocks are generally known to give rise to karstland that characterised geomorphological features including caves, sink holes and dolines, the non-karstic nature of chalk landscapes has been challenged recently – see L. Maurice, A. R. Farrant, E. Mathewson and T. Atkinson, Karst hydrogeology of the Chalk and implications for groundwater protection, in The Chalk Aquifers of Northern Europe, ed. R. P. Farrell, N. Massei, A. E. Foley, P. R. Howlett and J. J. West, Geological Society Special Publications, Geological Society, London, 2023, vol. 517, in press: DOI:10.1144/SP517-2020-267.
- P. Shand and J. Bloomfield, Mineralisation of Shallow Fracture Surfaces in the Chalk and Implications for Contaminant Attenuation, British Geological Survey Hydrology Series Technical Report WD/95/15, British Geological Survey, Keyworth, 1995 Search PubMed.
- I. Casas, D. Casabona, L. Duro and J. de Pablo, The influence of haematite on the sorption of uranium(VI) onto granite filling fractures, Chem. Geol., 1994, 113, 319 CrossRef CAS.
- O. Landström and E. L. Tulborg, The Influence of Fracture Mineral/Groundwater Interaction on the Mobility of U, Th, REE and Other Trace Elements, SKB (Swed. Nucl. Fuel & Waste Manage. Co.), Technical Report 90-37, Stockholm, 1990 Search PubMed.
- K. A. Hudson-Edwards and D. Kossoff, Rôle of redox-reactive minerals in the re-use and remediation of mine wastes, in Redox-Reactive Minerals: Properties, Reactions and Applications in Natural Systems and Clean Technologies, ed. I. A. M. Ahmed and K. A. Hudson-Edwards, EMU Notes in Mineralogy, The Mineralogical Society of Great Britain and Ireland, London, 2017, ch. 12, vol. 17, p. 357 Search PubMed.
- (a) F. C. Cox and D. Mac. Bridge, The Limestone and Dolomite Resources of the Country Around Monyash, Derbyshire: Description of 1:25000 resource sheet SK 16, Mineral Assessment Report 26 for the Institute of Geological Sciences, Her Majesty's Stationary Office, London, 1977 Search PubMed; (b) P. Shand, W. M. Edmunds, A. R. Lawrence, P. L. Smedley and S. Burke, The Natural (Baseline) Quality of Groundwater in England and Wales, Environment Agency Technical Report NC/99/74/24, Groundwater Programme Research Report RR/07/06, British Geological Survey, Keyworth, 2007 Search PubMed.
- W. M. Edmunds, J. M. Cook, W. G. Darling, D. G. Kinniburgh, D. L. Miles, A. H. Bath, M. Morgan-Jones and J. N. Andrews, Baseline geochemical conditions in the Chalk aquifer, Berkshire, U.K.: a basis for groundwater quality management, Appl. Geochem., 1987, 2, 251 CrossRef CAS.
- W. R. Jones, Minerals in Industry, Penguin, London, 3rd edn, 1955 Search PubMed.
- A full range of chalk-derived lime products quarried and manufactured by Singleton Birch, Ltd., together with their physico-chemical characteristics, is available on their website: https://www.singletonbirch.co.uk, (accessed December 28, 2021).
- Thermal oxidation of rhodochrosite at kiln temperatures affords hausmannite: (a) L. Biernacki and S. Pokrzywnicki, The thermal decomposition of manganese carbonate: thermogravimetry and exoemission of electrons, J. Therm. Anal. Calorim., 1999, 5, 227 CrossRef ; both bixbyite and pyrolusite likewise decompose to hausmannite under thermal kiln conditions: ; (b) E. Alonso, C. Hutter, M. Romero, A. Steinfeld and J. Gonzalez-Aguilar, Kinetics of Mn2O3-Mn3O4 and Mn3O4-MnO redox reactions performed under concentrated thermal radiative flux, Energy Fuels, 2013, 27, 4884 CrossRef CAS; (c) N. Birkner and A. Navrotsky, Thermodynamics of manganese oxides: effects of particle size and hydration on oxidation-reduction equilibria among hausmannite, bixbyite and pyrolusite, Am. Mineral., 2015, 97, 1291 CrossRef ; the whole picture is further complicated by phase transformations: ; (d) A. N. Grundy, B. Hallstedt and L. J. Gauckler, Assessment of the Mn-O system, J. Phase Equilib., 2003, 24, 21 CAS.
- Oxidation of iron pyrite at kiln temperatures affords the sesquioxide: (a) G.-M. Schwab and J. Philinis, Reactions of iron pyrite: its thermal decomposition, reduction by hydrogen and air oxidation, J. Am. Chem. Soc., 1947, 69, 2588 CrossRef CAS; (b) T. Kennedy and B. T. Sturman, The oxidation of iron(II) sulphide, J. Therm. Anal., 1975, 8, 329 CrossRef CAS; (c) Y. Zhang, Q. Li, X. Liu, B. Xu, Y. Yang and T. Jiang, A thermodynamic analysis on the roasting of pyrite, Minerals, 2019, 9, 220 CrossRef CAS ; likewise, oxidation of siderite proceeds to magnetite: ; (d) B. M. French and P. E. Rosenberg, Siderite (FeCO3): thermal decomposition in equilibrium with graphite, Science, 1965, 147, 1283 CrossRef CAS PubMed ; and then maghemite: ; (e) M. Aliahmad and N. N. Moghaddam, Synthesis of maghemite (γ-Fe2O3) nanoparticles by thermal decomposition of magnetite (Fe3O4) nanoparticles, Mater. Sci.-Pol., 2013, 31, 264 CrossRef CAS; (f) Note that the kiln conditions are controlled to ensure sufficient fugacity of oxygen, in a manner similar to those in the Earth: see, for example, V. Stagno and S. Aulbach, Redox processes before, during, and after Earth's accretion affecting the deep carbon cycle, in Magma Redox Geochemistry, ed. R. Moretti and D. R. Neuville, Wiley, Hoboken, NJ, 2022, p. 21 Search PubMed.
- (a) D. S. McClure, The distribution of transition metal cations in spinels, J. Phys. Chem. Solids, 1957, 3, 311 CrossRef CAS; (b) J. D. Dunitz and L. E. Orgel, Electronic properties of transition metal oxides II: cation distribution amongst octahedral and tetrahedral sites, J. Phys. Chem. Solids, 1957, 3, 318 CrossRef CAS; (c) A. H. Eschenfelder, Ionic valences in manganese-iron spinels, J. Appl. Phys., 1958, 29, 378 CrossRef CAS; (d) S. K. Sahu, B. Huang, K. Lilova, B. F. Woodfield and A. Navrotsky, Thermodynamics of Fe3O4-Co3O4 and Fe3O4-Mn3O4 spinel solid solutions at the bulk and nanoscale, Phys. Chem. Chem. Phys., 2015, 17, 22286 RSC.
- (a) A. T. Stone and J. J. Morgan, Reduction and dissolution of manganese(III) and manganese(IV) oxides by organics: 1: reaction with hydroquinone, Environ. Sci. Technol., 1984, 18, 450 CrossRef CAS PubMed; (b) A. T. Stone, Reductive dissolution of manganese(III/IV) oxides by substituted phenols, Environ. Sci. Technol., 1987, 21, 979 CrossRef CAS PubMed; (c) K. L. Johnson, C. M. McCann and C. E. Clarke, Breakdown of organic contaminants in soil by manganese oxides: a short review, in Redox-Reactive Minerals: Properties, Reactions and Applications in Natural Systems and Clean Technologies, ed. I. A. M. Ahmed and K. A. Hudson-Edwards, EMU Notes in Mineralogy, The Mineralogical Society of Great Britain and Ireland, London, 2017, ch. 11, vol. 17, p. 313 Search PubMed; (d) B. Liu, Y. Zhang, M. Lu, Z. Su, G. Li and T. Jiang, Extraction and separation of manganese and iron from ferruginous manganese ores: a review, Miner. Eng., 2019, 131, 286 CrossRef CAS; (e) V. Singh, S. Pati and K. Kumar, Development of a process to produce manganese nanomaterials from low grade ferruginous manganese ores, Miner. Process. Extr. Metall., 2021, 130, 324 CAS.
- (a) S. Licht, V. Naschitz, L. Halperin, L. Lin, J. Chen, S. Ghosh and B. Liu, Analysis of ferrate(VI) compounds and super-iron Fe(VI) battery cathodes: FTIR, ICP, titimetric, XRD, UV/VIS, and electrochemical characterisation, J. Power Sources, 2001, 101, 167 CrossRef CAS; (b) Y. L. Wang, S. H. Ye, J. K. Bo, Y. Y. Wang and F. Wu, Electrochemical reduction mechanism of Fe(VI) at a porus Pt black electrode, J. Electrochem. Soc., 2009, 156, A572 CrossRef CAS; (c) S. Barışçı, F. Ulu, H. Särkkä, A. Dimoglo and M. Sillanpää, Electrosynthesis of ferrate(VI) ion using high purity iron electrodes: optimisation of influencing parameters on the process and investigating its stability, Int. J. Electrochem. Sci., 2014, 9, 3099 Search PubMed; (d) Y.-L. Wei, Y.-S. Wang and C.-H. Liu, Preparation of potassium ferrate from spent steel pickling liquid, Metals, 2015, 5, 1770 CrossRef; (e) J.-Q. Jiang, C. Stanford and M. Petri, Practical application of ferrate(VI) for water and wastewater treatment – site study approach, Water-Energy Nexus, 2018, 1, 42 CrossRef; (f) B. Shao, H. Dong and X. Guan, Rôle of ferrate(IV) and ferrate(V) in activating ferrate(VI) by calcium sulphite for enhanced oxidation of organic contaminants, Environ. Sci. Technol., 2019, 53, 894 CrossRef CAS PubMed; (g) P. C. W. Cheung, D. R. Williams, J. Barrett, J. Barker and D. W. Kirk, On the origins of some spectroscopic properties of “purple iron” (the tetroxoferrate(VI) ion) and its Pourbaix safe-space, Molecules, 2021, 26, 5266 CrossRef CAS PubMed; (h) Note that calcium ions can catalyse the decomposition of ferrate: L. Ma, W. W. Y. Lam, P.-K. Lo, K.-C. Lau and T.-C. Lau, Angew. Chem., Int. Ed., 2016, 55, 3012 CrossRef CAS PubMed.
- See, for example, S. G. Simpson, The effect of the presence of filter paper on permanganate-oxalate titrations, J. Ind. Eng. Chem., 1921, 13, 1152 CrossRef CAS.
- (a) B. E. Dixon and J. L. White, The reaction between manganese salts and sodium hypochlorite in the presence of certain other salts, J. Chem. Soc., 1927, 1469 RSC; (b) M. Valensi, Nouvelle méthode d'enseignement de la chimie minérale, Bull. Union Physiciens, 1950,(394), 61 Search PubMed; (c) Atlas of Electrochemical Equlibria in Aqueous Solutions, ed. M. Pourbaix, National Association of Corrosion Engineers, Houston, Texas, 2nd edn, 1974 Search PubMed.
- K. M. Love and A. Woronow, Chemical changes induced in aragonite using treatments for the destruction of organic material, Chem. Geol., 1991, 93, 291 CrossRef CAS.
- (a) W. M. Latimer, The Oxidation States of the Elements and their Potentials in Aqueous Solutions, Prentice-Hall, New York, 2nd edn, 1952 Search PubMed; (b) K. Ishikawa, T. Yoshioka, T. Sato and A. Okuwaki, Solubility of haematite in LiOH, NaOH and KOH solutions, Hydrometallurgy, 1997, 45, 129 CrossRef CAS; (c) S. Uchida, T. Sato and A. Okuwaki, Formation of NaxFeO2 by the oxidation of iron powder in concentrated sodium hydroxide solutions at elevated temperatures, J. Mater. Sci., 1995, 14, 633 CAS.
- (a) See, for example, B. M. Voelker and D. L. Sedlak, Iron reduction by photo-produced superoxide in seawater, Mar. Chem., 1995, 50, 93 CrossRef CAS; (b) S. Garg, G. Xing and T. D. Waite, Influence of pH on the kinetics and mechanism of photo-reductive dissolution of amorphous iron oxyhydroxide in the presence of natural organic matter: implications to iron bioavailability in surface waters, Environ. Sci. Technol., 2020, 54, 6771 CrossRef CAS PubMed.
- See, for example, (a) H. P. Hopkins Jr. and C. A. Wulff, The solution thermochemistry of polyvalent electrolytes: I: calcium hydroxide, J. Phys. Chem., 1965, 69, 6 CrossRef; (b) W. B. Euler, L. J. Kirschenbaum and B. Ruekberg, Determination of Ksp, ΔG0, ΔH0 and ΔS0, J. Chem. Educ., 2000, 77, 1039 CrossRef CAS.
- (a) J. R. Lewis, The catalytic decomposition of sodium hypochlorite solutions I: mechanism of the reaction, J. Phys. Chem., 1928, 22, 243 CrossRef; (b) M. W. Lister, Decomposition of sodium hypochlorite: the uncatalysed reaction, Can. J. Chem., 1956, 34, 465 CrossRef CAS.
- J. H. Atherton, Mechanism in two-phase reaction systems: coupled mass transfer and chemical reaction, in Research in Chemical Kinetics, ed. R. G. Compton and G. Hancock, Elsevier, Amsterdam, 1994, vol. 2, p. 193 Search PubMed.
- As an aside, we note that whilst calcite dissolution is typically controlled by surface kinetics – see R. G. Compton and P. R. Unwin, The dissolution of calcite in aqueous solution at pH < 4: kinetics and mechanism, Philos. Trans. R. Soc., A, 1990, 330, 1 Search PubMed and R. G. Compton and K. L. Pritchard, The dissolution of calcite at pH > 7: kinetics and mechanism, Philos. Trans. R. Soc., A, 1990, 330, 47 Search PubMed acidic dissolution of calcium carbonate from Southern Province Chalk has been shown to be mass transport controlled – see R. G. Compton, C. T. Walker, P. R. Unwin and W. A. House, Dissolution kinetics of Carrera Marble, Portland Stone and several limestones in acidic waters, J. Chem. Soc., Faraday Trans., 1990, 86, 849 RSC and that different species of coccolith hold different masses of calcium carbonate q.v., M. Yang, C. Batchelor-McAuley, S. Barton, R. E. M. Rickaby, H. A. Bouman and R. G. Compton, Opto-electrochemical dissolution reveals coccolith calcium carbonate content, Angew. Chem., Int. Ed., 2021, 60, 20999 CrossRef CAS PubMed.
- (a) E. M. Logothetis and K. Park, The electrical conductivity of Mn3O4, Solid State Commun., 1975, 16, 909 CrossRef CAS; (b) R. Metselaar, R. E. van Tol and P. Piercy, The electrical conductivity and thermoelectric power of Mn3O4 at high temperatures, J. Solid State Chem., 1981, 38, 335 CrossRef CAS; (c) S. E. Dorris and T. O. Mason, Electrical properties and cation valencies in Mn3O4, J. Am. Ceram. Soc., 1988, 71, 379 CrossRef CAS; (d) H. Dhaouadi, A. Madani and F. Touati, Mater. Lett., 2010, 64, 2395 CrossRef CAS; (e) V. C. Bose, K. Maniammal, G. Madhu, C. L. Veenas, A. S. A. Raj and V. Biju, IOP Conf. Ser.: Mater. Sci. Eng., 2015, 73, 012084 Search PubMed.
- J.-K. Wu, Kinetics of the reduction of hypochlorite ion, J. Electrochem. Soc., 1987, 134, 1462 CrossRef CAS.
- (a) A. Wilczak, W. R. Knocke, R. E. Hubel and M. E. Aieta, Manganese control during ozonation of water containing organic compounds, J. – Am. Water Works Assoc., 1993, 85, 98 CrossRef; (b) W. Li and S. T. Oyama, Mechanism of ozone decomposition on a manganese oxide catalyst: 2: steady-state and transient kinetic studies, J. Am. Chem. Soc., 1998, 120, 9047 CrossRef CAS; (c) J. A. Roth and D. E. Sullivan, Solubility of ozone in water, Ind. Eng. Chem. Fundam., 1981, 20, 137 CrossRef CAS.
- (a) N. S. Lawrence, B. Schöllhorn and J. D. Wadhawan, Asymmetric and anharmonic electrode kinetics: evaluation of a model for electron transfer with concerted rupture of weak, inner-shell interactions, ChemistrySelect, 2021, 6, 13331 CrossRef CAS; (b) B. Gilbert and G. A. Waychunas, The timescale of mineral redox reactions, in Redox-Reactive Minerals: Properties, Reactions and Applications in Natural Systems and Clean Technologies, ed. I. A. M. Ahmed and K. A. Hudson-Edwards, EMU Notes in Mineralogy, The Mineralogical Society of Great Britain and Ireland, London, 2017, ch. 4, vol. 17, p. 55 Search PubMed; (c) M. Sander, T. B. Hofstetter and C. A. Gorski, Electrochemical analyses of redox-active iron minerals: a review of non-mediated and mediated approaches, Environ. Sci. Technol., 2015, 49, 5862 CrossRef CAS PubMed.
- A. Lu, Y. Li, F. Liu, Y. Liu, H. Ye, Z. Zhuang, Y. Li, H. Ding and C. Wang, The photo-geochemical cycle of Mn oxides on the Earth's surface, Mineral. Mag., 2021, 85, 22 CrossRef CAS.
- S. Fraley, Manganese levels in limestone: operational issues in wet limestone flue gas scrubbing technologies, in Lime: Building on the 100-Year Legacy of the ASTM Committee C07, ed. M. L. Thomson and J. H. Brisch, Specialist Technical Publication, American Society for Testing and Materials, San Diego, 2012, vol. 1557, p. 167 Search PubMed.
- L. Lv, J. Yang, Z. Shen, Y. Zhou and J. Lu, Optimising the characteristics of calcium sulfate dehydrate in the flue gas desulfurisation process: investigation of the impurities in slurry: Cl−, Fe3+ and Mn2+, Chem. Ind. Chem. Eng. Q., 2017, 23, 293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ew00109h |
| This journal is © The Royal Society of Chemistry 2023 |