
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetics of the nitrate-mediated photooxidation of monocarboxylic acids in the aqueous phase†
Yuting
Lyu
 ab,
Jany Ting Chun
Chow
a and
Theodora
Nah
*ab
ab,
Jany Ting Chun
Chow
a and
Theodora
Nah
*ab
aSchool of Energy and Environment, City University of Hong Kong, Hong Kong SAR, China. E-mail: theodora.nah@cityu.edu.hk; Tel: +852 3442 5578
bState Key Laboratory of Marine Pollution, City University of Hong Kong, Hong Kong SAR, China
First published on 26th January 2023
Abstract
The photooxidation of organic compounds by hydroxyl radicals (·OH) in atmospheric aqueous phases contributes to both the formation and aging of secondary organic aerosols (SOAs), which usually include carboxylic acids. Hydrogen peroxide (H2O2) and inorganic nitrate are two important ·OH photochemical sources in atmospheric aqueous phases. The aqueous phase pH is an important factor that not only controls the dissociation of carboxylic acids and consequently their ·OH reactivities, but also the production of ·OH and other reactive species from the photolysis of some ·OH photochemical precursors, particularly inorganic nitrate. While many studies have reported on the aqueous pH-dependent photodegradation rates of carboxylic acids with ·OH produced by H2O2 photolysis, the aqueous pH-dependent photodegradation rates of carboxylic acids with ·OH produced by inorganic nitrate photolysis have not been studied. In this work, we investigated the pH-dependent (pH 2 to 7) aqueous photooxidation of formic acid (FA), glycolic acid (GA), and pyruvic acid (PA) initiated by the photolysis of ammonium nitrate (NH4NO3). The observed reaction rates of the three carboxylic acids were controlled by the [NH4NO3]/[carboxylic acid] concentration ratio. Higher [NH4NO3]/[carboxylic acid] concentration ratios resulted in faster photodegradation rates, which could be attributed to the higher concentrations of ·OH produced from the photolysis of higher concentrations of NH4NO3. In addition, the observed photodegradation rates of the three carboxylic acids strongly depended on the pH. The highest photodegradation rate was observed at pH 4 for FA, whereas the highest photodegradation rates were observed at pH 2 for GA and PA. The observed pH-dependent FA and GA photodegradation rates were due to the combined effects of the pH-dependent ·OH formation from NH4NO3 photolysis and the differences in ·OH reactivities of dissociated vs. undissociated FA and GA. In contrast, the observed pH-dependent PA photodegradation rate was due primarily to the pH-dependent decarboxylation of PA initiated by light. These results highlight how the aqueous phase pH and inorganic nitrate photolysis can combine to influence the degradation rates of carboxylic acids, which can have significant implications for how the atmospheric fates of carboxylic acids are modeled for regions with substantial concentrations of inorganic nitrate in cloud water and aqueous aerosols.
Environmental significanceLow-molecular-weight carboxylic acids (LMWCA) and inorganic nitrate are ubiquitous components of cloud water and aqueous aerosols. The reactive fates of LMWCA in atmospheric aqueous phases are of interest to the scientific community since they participate in important atmospheric processes. Aqueous-phase inorganic nitrate photolysis produces reactive species that can react with LMWCA to influence their reactive fates. We investigated the role of pH on the aqueous degradation kinetics of formic acid, glycolic acid, and pyruvic acid during nitrate-mediated photooxidation. The pH and inorganic nitrate photolysis combined to influence the pH-dependent degradation rates of these LMWCA, which has significant implications for how the fates of LMWCA are modeled for regions with substantial concentrations of inorganic nitrate in cloud water and aqueous aerosols. |
1 Introduction
Atmospheric aerosols play important roles in air quality and climate change. Secondary organic aerosols (SOAs) typically contribute more than half of the mass fraction of organic aerosols in most areas.1,2 Atmospheric aqueous phases, such as cloud water and aqueous aerosols, are important media for the formation and aging of aqueous SOAs (aqSOAs).3–7 The fates of aqSOA components in atmospheric aqueous phases depend on the composition of the aqueous phase since some of the inorganic (e.g., inorganic nitrate and hydrogen peroxide) and organic (e.g., organic peroxides) compounds in the aqueous phase produce reactive oxidative species upon illumination,8–11 which can react with aqSOA components and form lower and higher molecular weight oxygenated organic compounds.3–5 Low molecular weight carboxylic acids are ubiquitous aqSOA components of cloud water and aqueous aerosols in urban, rural, and remote environments.12–17 They play important roles in many atmospheric processes such as contributing to aqueous phase acidity levels and participating in aqueous reactions.3–5,18 Low molecular weight carboxylic acids in cloud water usually originate from the aqueous processing of water-soluble organic compounds (WSOCs).3 However, in aqueous aerosols, they are attributed to both aqueous processing (in the case of dicarboxylic acids) and the formation of nonvolatile carboxylate salts during long-range transport (in the case of monocarboxylic acids).7,12,19The hydroxyl radical (·OH) is widely recognized to be an important oxidant in atmospheric aqueous phases.20 Many organic compounds have high reaction rates with ·OH in the aqueous phase, most of which will lead to lifetimes close to those in the gas phase.20,21 Although ·OH can be transferred from the gas phase to the aqueous phase, it can also be formed in situ by the aqueous photolysis of hydrogen peroxide, nitrate, nitrite, and light-absorbing organic matter containing photolabile molecules (e.g., organic peroxides and metal–organic complexes), as well as by dark Fenton and photo-Fenton processes.8,20,22–24 Hydrogen peroxide (H2O2) is believed to be the main photochemical source of ·OH in cloud water in most regions.6,23 For the typical range of pH values of atmospheric aqueous phases (pH 2 to 7),25 ·OH production from H2O2 photolysis does not depend significantly on the pH.26 However, the pH can impact the oxidation kinetics of acidic and basic organic compounds since it controls the fraction of the acidic/basic organic compound that is present in its neutral vs. deprotonated/protonated forms which have different ·OH reactivities.27–29 In the case of carboxylic acids, due to the electrophilic nature of ·OH, the reaction of ·OH with the deprotonated carboxylate group via electron transfer occurs at a faster rate than hydrogen (H) atom abstraction by ·OH.30–32 This results in carboxylic acids generally having lower ·OH reactivities at lower pH, though the ·OH reactivities of low molecular weight carboxylic acids are more sensitive to pH compared to the ·OH reactivities of high molecular weight carboxylic acids.27,32,33
Inorganic nitrate is a major component of cloud water and aqueous aerosols in many regions.6,34,35 The concentrations of inorganic nitrate in cloud water and aqueous aerosols are usually higher in densely populated urban areas where high levels of nitrogen oxides are present.6,34,35 In addition, inorganic nitrate is becoming increasingly important in areas with large curtailment in sulfur dioxide emissions and/or with high ammonia emissions, which result in nitric acid being neutralized by excessive ammonia.36–38 The importance of inorganic nitrate as a ·OH photochemical source in continental clouds is typically second only to H2O2 photolysis,6,11,39 though inorganic nitrate photolysis is reportedly the main source of ·OH in some areas such as California and Louisiana.24 The production of ·OH and other reactive species by inorganic nitrate photolysis is strongly influenced by the pH. This is because nitrous acid (HNO2) production from inorganic nitrate photolysis is favored over the production of nitrite ions (NO2−) at around pH ≤ 3.5.40 Since HNO2 has a higher quantum yield for ·OH formation than NO2− in the near-UV region, higher ·OH formation rates are expected at pH ≤ 3.5.40,41 Due to reductions in the emissions of acidic precursors into the atmosphere, the average pH of cloud water and aqueous aerosols in many parts of Europe, North America, and China has been steadily increasing.25 Hence, given the ubiquitous nature of carboxylic acids and inorganic nitrate in cloud water and aqueous aerosols, it is necessary to understand how the pH influences the nitrate-mediated photooxidation of carboxylic acids.
In this study, we investigated the effect of pH (2 to 7) on the aqueous degradation kinetics of three carboxylic acids during photooxidation initiated by inorganic nitrate photolysis. The three carboxylic acids used were formic acid, glycolic acid, and pyruvic acid (Fig. 1). Formic acid was chosen because it is one of the most abundant carboxylic acids in atmospheric aqueous phases.42 Glycolic acid and pyruvic acid were chosen since they are the simplest α-hydroxy and α-keto carboxylic acids, which are commonly detected in atmospheric aqueous phases.43–45 Sources of these three carboxylic acids include direct emissions from biogenic and anthropogenic sources, and production from atmospheric chemical processes.12,42,43,46–50 The three carboxylic acids have Henry's law constants larger than 8000 M per atom,51 so they are water soluble and can partition easily into atmospheric aqueous phases where they can undergo reactions with ·OH. Given that inorganic nitrate is formed from anthropogenic nitrogen oxides and these carboxylic acids have biogenic sources, the nitrate-mediated photooxidation of these carboxylic acids can be viewed as a mechanism by which anthropogenic emissions facilitate the aging of biogenic aqSOAs. Results from this study provide insights into how the pH and inorganic nitrate photolysis combine to influence the overall photodegradation rates of carboxylic acids in atmospheric aqueous phases, which will be useful for modeling the reactive fates of carboxylic acids in regions with inorganic nitrate-rich cloud water and/or aqueous aerosols. The reactive fates of carboxylic acids in atmospheric aqueous phases are of huge interest since they can participate in important aqueous reactions and are key contributors to aqueous phase and precipitation acidity,3–5,18 which have important implications for atmospheric chemistry and ecosystem health.
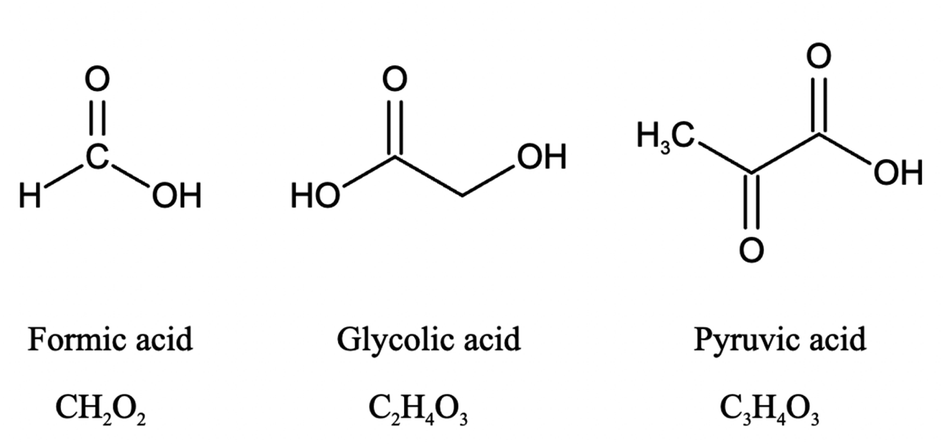 | ||
| Fig. 1 Chemical structures of the three carboxylic acids used in the study. | ||
2 Experimental methods
The chemicals used in this study were formic acid (FA, LC-MS grade) and ammonium nitrate (AN, ≥99.5%) which were purchased from Fisher Chemicals, glycolic acid (GA, 70 wt% in H2O) and pyruvic acid (PA, 98%) which were purchased from Sigma-Aldrich, H2O2 (35 wt% in H2O) which was purchased from Acros Organics, benzoic acid (BA, 99.5%) and p-hydroxybenzoic acid (PHBA, 99%) which were purchased from J&K Scientific, and sulfuric acid (H2SO4, 95%) and aqueous ammonia (NH4OH, 25%, v/v) which were purchased from VWR Chemicals. All the chemicals were used without further purification. Ultrapure water from a Milli-Q system (18.2 MΩ cm) was used to prepare all solutions. The concentrations of the carboxylic acids in the solutions used in the experiments were kept at 10 μM. The solutions also contained either 250 μM or 1250 μM AN. The concentrations of carboxylic acids and NO3− used in this study fall within the range of concentrations measured previously in cloud water.6,16,17 The pH of the solutions of FA, GA, and PA (with and without AN) was around 4. H2SO4 was added to the solutions to achieve pH 2 and 3, while NH4OH was added to solutions to achieve pH 7. The solution pH was measured using a pH meter (Mettler Toledo SevenMulti). The solution pH and carboxylic acid's dissociation constant (pKa) were used to determine the fraction of the carboxylic acid present in neutral (HA) vs. deprotonated (A−) forms (Fig. S1†). The fraction of the carboxylic acid present in the A− form (α) was calculated using the following equation: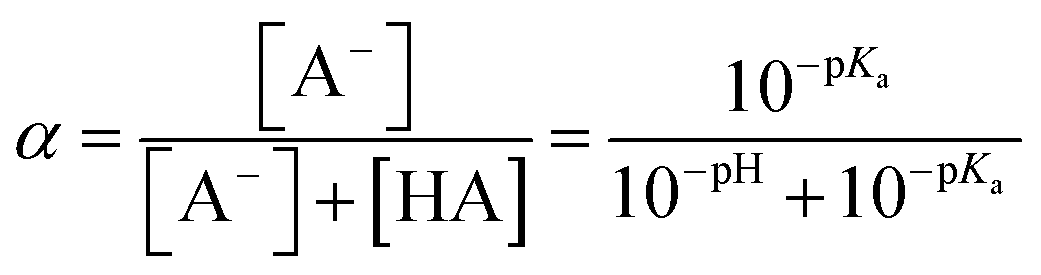 | (1) |
The pKas used to calculate α for FA, GA, and PA were 3.75,33 3.82,52 and 2.50,53 respectively. The addition of AN and H2SO4 or NH4OH to the solution will increase the solution's ionic strength. However, work by Yang et al. (2008) suggested that the solution ionic strength will not affect the kinetics of the reaction of ·OH with carboxylic acids, even at high ionic strengths of 0.05 to 0.16 M.54 Thus, we assumed that the ionic strengths had negligible effects on the photodegradation kinetics in our study since the highest solution ionic strength was 0.012 M (Table S1†).
All the experiments were conducted using a photoreactor (RPR-200, Rayonet, Southern New England UV Co.) equipped with sixteen UVB lamps (RPR-3000 Å, Southern New England UV Co.) which were located symmetrically around the photoreactor's interior walls. The UVB lamps had outputs centered at around 310 nm (Fig. S2†). The photoreactor's interior temperature was maintained at around 30 °C during the experiments by using a cooling fan positioned at the bottom of the photoreactor. During each experiment, the solutions were loaded into quartz tubes, which were placed on a rotating rack placed inside and in the middle of the photoreactor. Aliquots of the solutions were removed periodically for analysis of the carboxylic acid by using an anion ion chromatography (IC) system (Dionex ICS-1100, Thermo Fisher Scientific) using an isocratic elution method. Separation was achieved using a Dionex IonPac AS18 (4 × 250 mm) anion exchange column equipped with a Dionex IonPac AG18 guard column (4 × 50 mm). 23 mM potassium hydroxide was used as the eluent, and it was delivered at a flow rate of 1.0 mL min−1. IC analyses were performed on the same day of the experiment. All the experiments were repeated three times. The concentrations of the three carboxylic acids decreased when AN was present in the irradiated solutions. The control experiments showed that the three carboxylic acids did not react with AN in the dark. FA and GA concentrations did not decrease when AN was absent from the irradiated solutions. In contrast, the PA concentration decreased when AN was absent from the irradiated solutions.
Separate photooxidation experiments were performed using 10 μM benzoic acid (BA) as the probe molecule to estimate steady-state ·OH concentrations ([·OH]ss) in photooxidation experiments initiated by 250 μM AN photolysis.55 We used the same methodology as Yang et al. (2021, 2022) to estimate [·OH]ss.56,57 Briefly, PHBA was assumed to be formed solely from reactions of BA with ·OH at a yield of 0.17,22 which enabled us to use the measured time-dependent formation of PHBA to determine the time-dependent concentrations of BA that were consumed by ·OH. [·OH]ss was subsequently determined using the reaction rate constant of BA with ·OH (kBA+OH = 5.9 × 109 M−1 s−1).9 BA and PHBA were measured using an ultra-high performance liquid chromatography system (1290, Agilent) coupled to a high-resolution quadrupole-time-of-flight mass spectrometer (X500R QTOF MS/MS, Sciex) (UPLC-MS) equipped with an electrospray ionization (ESI) source operated in the negative mode. Prior to UPLC-MS analysis, solid phase extraction (SPE) was performed to remove inorganic salts in the samples. Descriptions of the SPE protocol and UPLC-MS analyses can be found in Section S1.† Due to difficulties in removing NO3− salts completely by SPE from the solutions used in photooxidation experiments initiated by 1250 μM AN photolysis prior to UPLC-MS analysis, we did not determine the [·OH]ss in these photooxidation experiments. This in turn prevented us from determining second-order rate constants for the three carboxylic acids from the measured pseudo-first order rate constants in the photooxidation experiments initiated by 1250 μM AN photolysis (Section 3.1). Hence, most of our discussion will be based on the results obtained from the photooxidation experiments initiated by 250 μM AN photolysis. Nevertheless, we do not expect our inability to determine the [·OH]ss in photooxidation experiments initiated by 1250 μM AN photolysis to impact the main conclusions of our study. This is because the three carboxylic acids demonstrated similar trends in their reactivities regardless of whether the photooxidation was initiated by 250 μM or 1250 μM AN photolysis.
3 Results and discussion
FA and GA only photodegraded during irradiation in the presence of AN, thus they only have one possible sink: oxidation by reactive species produced from AN photolysis. AN photolysis produces a variety of radicals such as ·OH, nitroso (NO˙) and nitrite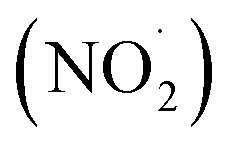 radicals (Table S2†),8,20,40,55,58 which can react with organic compounds. A comparison of results from previous studies indicates that the rates of reactions of saturated carboxylic acids with NO˙ and
radicals (Table S2†),8,20,40,55,58 which can react with organic compounds. A comparison of results from previous studies indicates that the rates of reactions of saturated carboxylic acids with NO˙ and 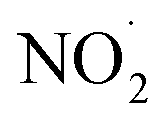 are likely 2 to 5 orders of magnitude slower than that of their reactions with ·OH.27,59–61 Since PA photodegraded during irradiation in the presence and absence of AN, it has two possible sinks: unimolecular photodissociation which will occur during irradiation in both the presence and absence of AN, and oxidation by reactive species during irradiation in the presence of AN. All the observed photodegradations of FA, GA, and PA followed apparent first order kinetics reasonably well (Fig. 2). Thus, we fitted the photodegradations of the three carboxylic acids using the following equation:
are likely 2 to 5 orders of magnitude slower than that of their reactions with ·OH.27,59–61 Since PA photodegraded during irradiation in the presence and absence of AN, it has two possible sinks: unimolecular photodissociation which will occur during irradiation in both the presence and absence of AN, and oxidation by reactive species during irradiation in the presence of AN. All the observed photodegradations of FA, GA, and PA followed apparent first order kinetics reasonably well (Fig. 2). Thus, we fitted the photodegradations of the three carboxylic acids using the following equation: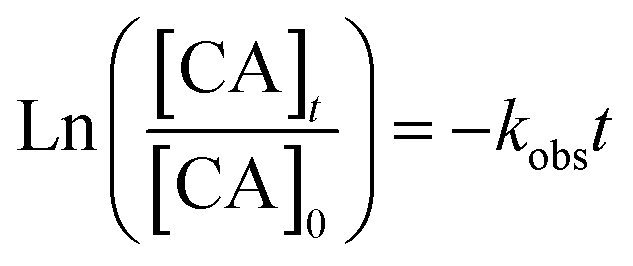 | (2) |
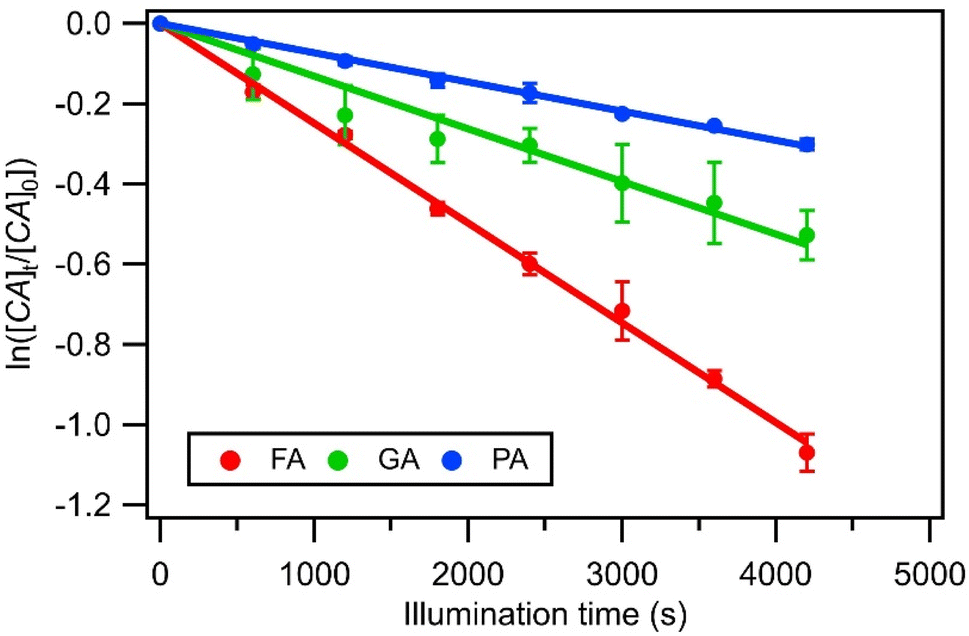 | ||
| Fig. 2 Photodegradations of FA, GA, and PA in nitrate-mediated photooxidation experiments using 250 μM AN at pH 4. Error bars represent standard deviations of repeated measurements. Lines show the exponential fit to the photodegradation data. | ||
3.1. Photooxidation of FA and GA
The kobs values for FA and GA were all in the order of 10−4 s−1. Fig. 3 shows how kobs for FA and GA depended on the pH during photooxidation initiated by the photolysis of 250 μM or 1250 μM AN. For both FA and GA, larger kobs values were obtained with 1250 μM AN compared to with 250 μM AN. This indicated that the kobs values were controlled partly by the [AN]/[CA] concentration ratio. Higher concentrations of ·OH and other reactive species were likely produced at higher [AN]/[CA] concentration ratios, which resulted in larger kobs values at higher [AN]/[CA] concentration ratios. The kobs values for FA and GA exhibited obvious pH dependence regardless of the [AN]/[CA] concentration ratio. The results from ordinary one-way ANOVA statistics tests indicated that the kobs values were significantly different (p < 0.01) between the four pH conditions for both FA and GA. Similar pH-dependent kobs trends were observed at different [AN]/[CA] concentration ratios. However, differences in the kobs values under different pH conditions were more prominent at higher [AN]/[CA] concentration ratios. In addition, the pH-dependent kobs trends for FA and GA were markedly different from one another. For FA, kobs(pH 4) > kobs(pH 7) > kobs(pH 3) > kobs(pH 2), whereas for GA, kobs(pH 2) > kobs(pH 3) > kobs(pH 4) > kobs(pH 7).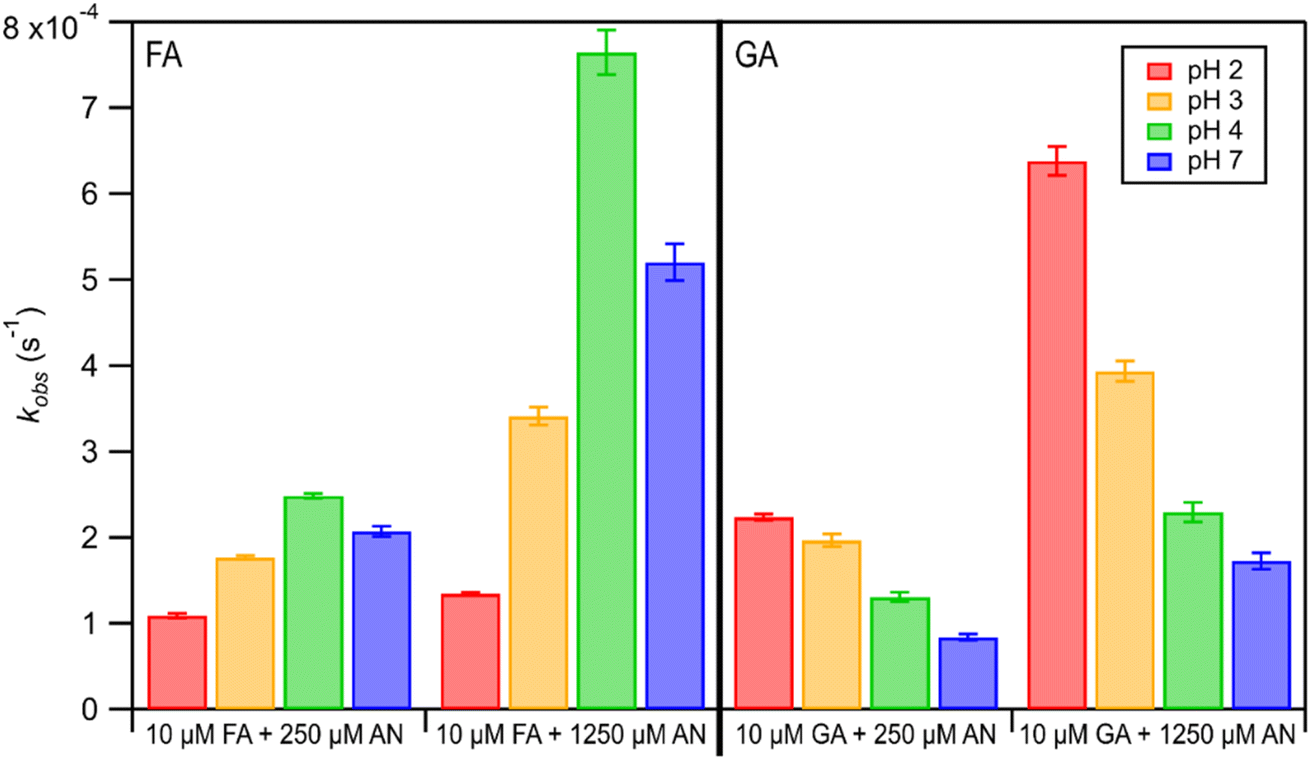 | ||
| Fig. 3 k obs of FA and GA at pH 2, 3, 4, and 7. The concentrations of FA and GA were fixed at 10 μM, while either 250 μM or 1250 μM AN were used in these experiments. Error bars indicate standard deviation of multiple experiments. | ||
We hypothesized that the pH-dependent kobs trends for FA and GA were a result of the pH controlling both the fraction of the carboxylic acid present in HA and A− forms (i.e., α) and ·OH production from AN photolysis. To test our hypothesis, we first determined the fractions of the FA and GA present in HA and A− forms at pH 2, 3, 4, and 7 using eqn (1). Our calculations showed that around 2% and 98% of the carboxylic acids were present as A− at pH 2 and 7, respectively (Table S3†). Due to the electrophilic nature of ·OH, A− has higher ·OH reactivities than HA, though the extent of this difference will depend on the carboxylic acid.27,30–32 For GA, the ·OH reactivity for its A− form is about 3.2 times higher than the ·OH reactivity for its HA form.62,63 In contrast, the ·OH reactivity for the A− form for FA is about 29 times higher than the ·OH reactivity for its HA form.27,33 Next, we estimated the ·OH concentrations in nitrate-mediated photooxidation experiments at pH 2, 3, 4, and 7. Fig. 4 shows the estimated [·OH]ss at different pH values determined from separate nitrate-mediated photooxidation experiments using 250 μM AN and BA as the probe molecule. The estimated [·OH]ss values were in the order of 10−13 M under the four pH conditions, which is within the range of ·OH concentrations in atmospheric aqueous phases (10−16 to 10−12 M).9 Differences in the estimated [·OH]ss values were statistically significant (p < 0.01). In general, higher [·OH]ss was observed at lower pH, which is consistent with past reports of higher ·OH formation rates at lower pH. Arakaki et al. (1999) previously reported that the ·OH formation rate constants at pH 1.9 are about 10-fold higher than those at pH 6.2.41 This is because HNO2 production from inorganic nitrate photolysis is favored over NO2− production at around pH ≤ 3.5.40 Since HNO2 has a higher quantum yield for ·OH formation than NO2− in the near-UV region, ·OH formation rates will be higher at pH ≤ 3.5.40,41 Even though the [·OH]ss in photooxidation experiments initiated by 1250 μM AN photolysis was not estimated, we expect a similar pH-dependent trend, albeit at higher [·OH]ss values, to the one shown in Fig. 4. In the subsequent discussion in this subsection, we use the photodegradation rates of FA and GA from photooxidation experiments initiated by 250 μM AN photolysis. Given the similar pH-dependent kobs trends (Fig. 3) observed regardless of the concentration of AN (i.e., 250 μM or 1250 μM) used to initiate photooxidation, we expect our general conclusions regarding the relationships between kobs and pH (Fig. 5) and the relationships between the calculated second-order rate constants and pH (Fig. S3†) to also apply to results from the photooxidation experiments initiated by 1250 μM AN photolysis.
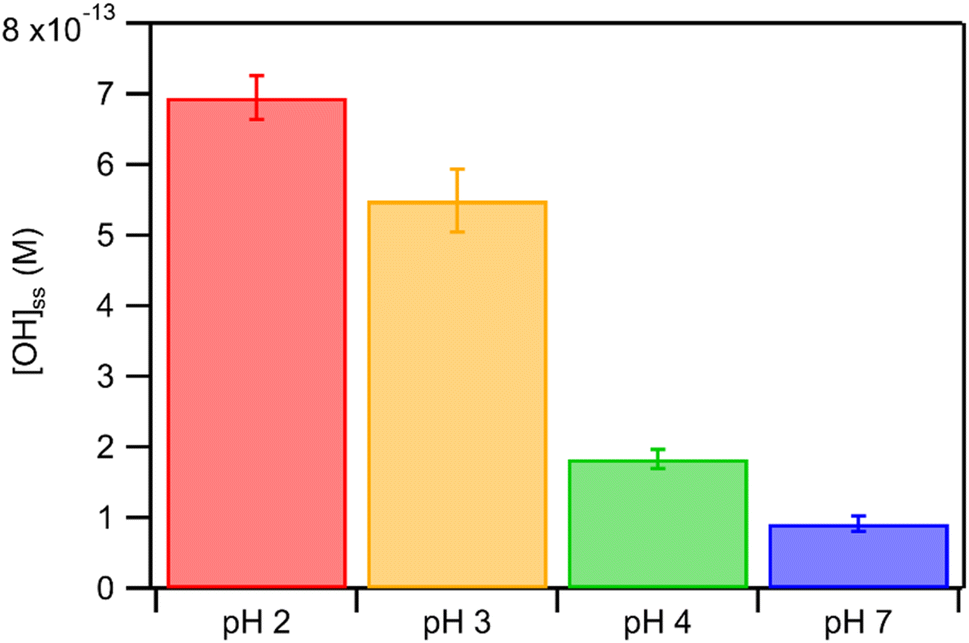 | ||
| Fig. 4 Estimated [·OH]ss in nitrate-mediated photooxidation experiments using 250 μM AN at pH 2, 3, 4, and 7. Error bars represent standard deviations of multiple experiments. | ||
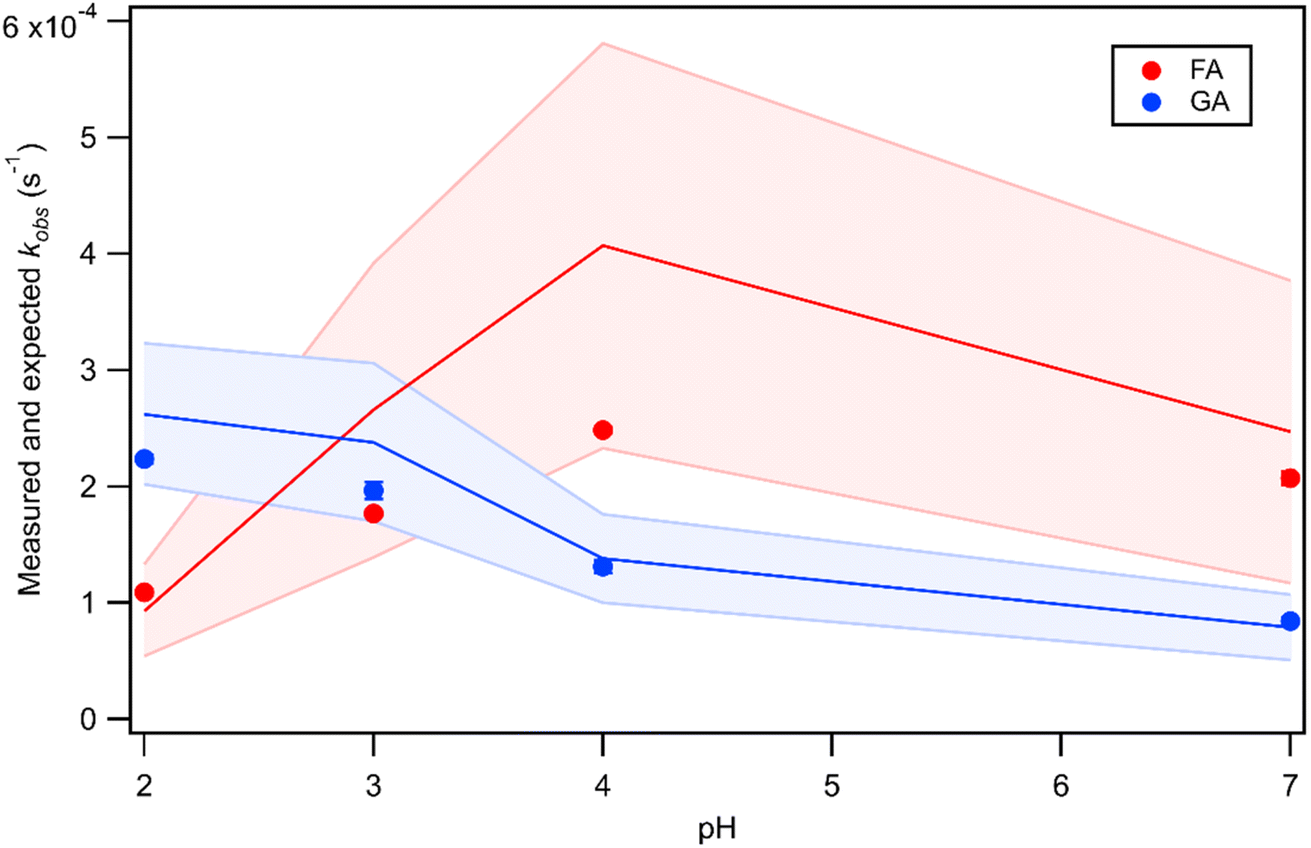 | ||
Fig. 5 Actual (symbols) vs. simulated (lines) relationships between kobs and pH for FA and GA. Simulated kobs values (i.e., kOH) were calculated using FA and GA dissociation fractions (Table S3†), estimated [OH]ss values (Fig. 4), and kHA+OHrxn and 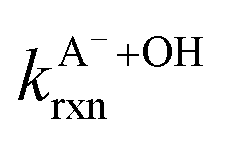 values for HA and A− (Table S4†). The bold lines denote simulated kobs values that were calculated using the average of kHA+OHrxn and values for HA and A− (Table S4†). The bold lines denote simulated kobs values that were calculated using the average of kHA+OHrxn and 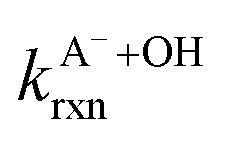 values obtained from the literature, while the shaded regions denote the 95% confidence intervals for simulated kobs values arising from standard deviations in the estimated [OH]ss values and literature kHA+OHrxn and values obtained from the literature, while the shaded regions denote the 95% confidence intervals for simulated kobs values arising from standard deviations in the estimated [OH]ss values and literature kHA+OHrxn and 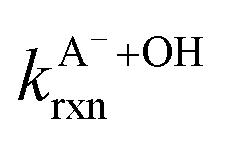 values. values. | ||
To test our hypothesis that the pH-dependent kobs trends for FA and GA (Fig. 3) were a result of the pH controlling both the fraction of the carboxylic acid present in HA and A− forms and ·OH production from AN photolysis, we had to make two simplifying assumptions. First, we assumed that the photodegradations of FA and GA during nitrate-mediated photooxidation were due primarily to their reactions with ·OH. The validity of this simplifying assumption relies on ·OH having higher reactivities compared to NO˙ and 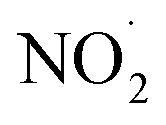 or larger concentrations of ·OH produced by AN photolysis compared to the concentrations of NO˙ and
or larger concentrations of ·OH produced by AN photolysis compared to the concentrations of NO˙ and 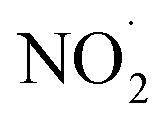 . Although previous studies have reported that approximately equal quantities of ·OH and
. Although previous studies have reported that approximately equal quantities of ·OH and 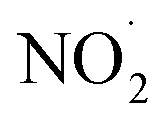 are produced by inorganic nitrate photolysis,59,60 the ·OH reactivities of saturated carboxylic acids are estimated to be 2 to 5 orders of magnitude higher than their NO˙ and
are produced by inorganic nitrate photolysis,59,60 the ·OH reactivities of saturated carboxylic acids are estimated to be 2 to 5 orders of magnitude higher than their NO˙ and 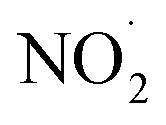 reactivities.27,59–61 While inorganic nitrate photolysis will also produce other reactive species such as hydroperoxide radicals
reactivities.27,59–61 While inorganic nitrate photolysis will also produce other reactive species such as hydroperoxide radicals 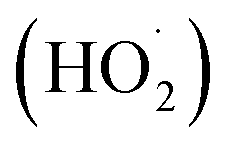 and superoxide ions (O2−˙) (Table S2†), results from previous studies suggest that their formation rates and reactivities with saturated carboxylic acids are likely at least 2 orders of magnitude lower than those of ·OH.55,64,65 Thus, our assumption appears to be reasonable. Second, we assumed that the ·OH production rate was constant throughout the experiment under each pH condition, so the ·OH concentration did not vary significantly with time and equaled the estimated [·OH]ss value at that pH. The simulated pseudo-first order rate constants for FA and GA (kOH) were subsequently calculated using the following equation:
and superoxide ions (O2−˙) (Table S2†), results from previous studies suggest that their formation rates and reactivities with saturated carboxylic acids are likely at least 2 orders of magnitude lower than those of ·OH.55,64,65 Thus, our assumption appears to be reasonable. Second, we assumed that the ·OH production rate was constant throughout the experiment under each pH condition, so the ·OH concentration did not vary significantly with time and equaled the estimated [·OH]ss value at that pH. The simulated pseudo-first order rate constants for FA and GA (kOH) were subsequently calculated using the following equation:
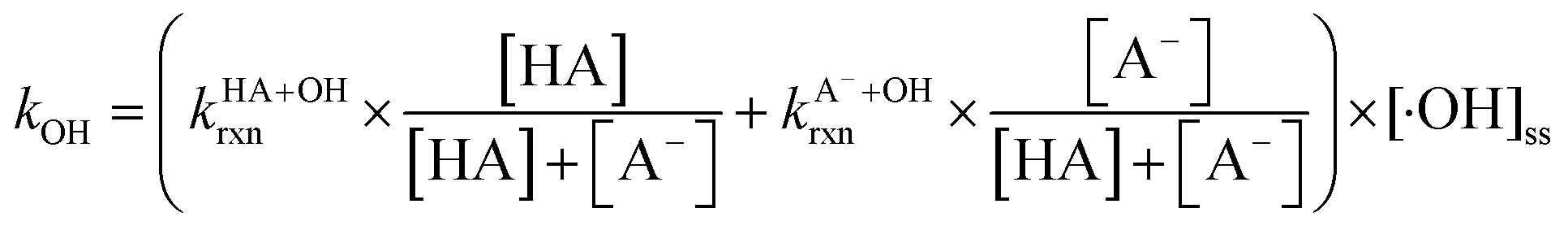 | (3) |
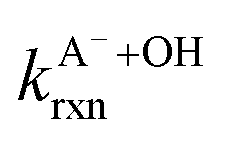 are the second-order rate constants for the reaction of ·OH with HA and A−, respectively, and
are the second-order rate constants for the reaction of ·OH with HA and A−, respectively, and 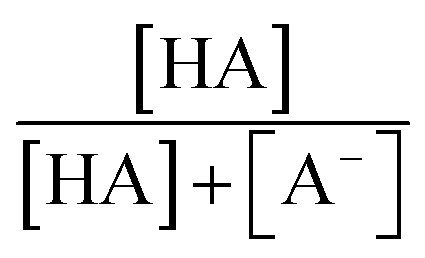 and
and 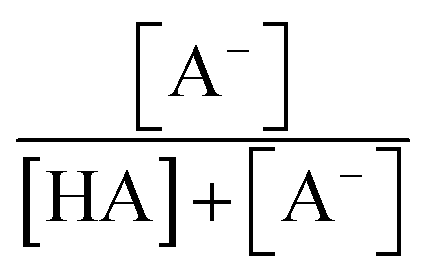 are the fractions of HA and A−, respectively. kHA+OHrxn and
are the fractions of HA and A−, respectively. kHA+OHrxn and 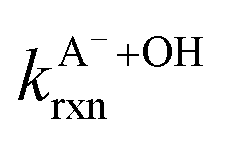 were obtained from the literature (Table S4†),27,33,62,63 while
were obtained from the literature (Table S4†),27,33,62,63 while 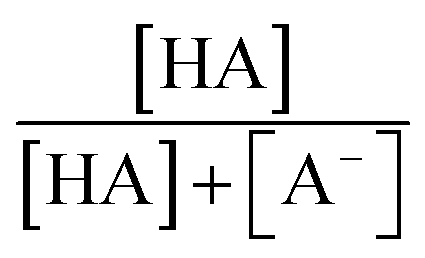 and
and 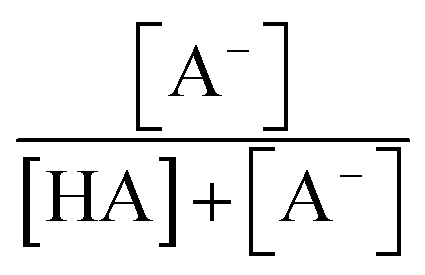 were calculated using eqn (1) and the pKas for the carboxylic acid (3.75 for FA33 and 3.82 for GA52) (Table S3†). Fig. 5 shows lines representing the simulated trends of kobs for FA and GA obtained from calculations of kOH values at different pH values using eqn (3), and then plotting the calculated kOH values as a function of pH. The measured kobs values for FA and GA generally agreed well with the simulated curves. More importantly, the simulated curves were able to reproduce the pH-dependent kobs trends for FA (i.e., kobs(pH 4) > kobs(pH 7) > kobs(pH 3) > kobs(pH 2)) and GA (i.e., kobs(pH 2) > kobs(pH 3) > kobs(pH 4) > kobs(pH 7)). Thus, these results supported our hypothesis that the pH-dependent kobs trends for FA and GA were a result of pH controlling both the fraction of the carboxylic acid present in HA and A− forms and ·OH production from AN photolysis. The results also suggested that our simplifying assumption that the photodegradations of FA and GA during nitrate-mediated photooxidation were due primarily to their reactions with ·OH was likely valid.
were calculated using eqn (1) and the pKas for the carboxylic acid (3.75 for FA33 and 3.82 for GA52) (Table S3†). Fig. 5 shows lines representing the simulated trends of kobs for FA and GA obtained from calculations of kOH values at different pH values using eqn (3), and then plotting the calculated kOH values as a function of pH. The measured kobs values for FA and GA generally agreed well with the simulated curves. More importantly, the simulated curves were able to reproduce the pH-dependent kobs trends for FA (i.e., kobs(pH 4) > kobs(pH 7) > kobs(pH 3) > kobs(pH 2)) and GA (i.e., kobs(pH 2) > kobs(pH 3) > kobs(pH 4) > kobs(pH 7)). Thus, these results supported our hypothesis that the pH-dependent kobs trends for FA and GA were a result of pH controlling both the fraction of the carboxylic acid present in HA and A− forms and ·OH production from AN photolysis. The results also suggested that our simplifying assumption that the photodegradations of FA and GA during nitrate-mediated photooxidation were due primarily to their reactions with ·OH was likely valid.
Previous studies have reported that the ·OH reactivities of saturated monocarboxylic acids will increase with pH as a result of A− having higher ·OH reactivities than HA.27,30–32 To determine whether our measured pH-dependent kobs trends for FA and GA (Fig. 3) were in line with previously reported pH-dependent trends of the ·OH reactivities of saturated carboxylic acids, we assumed that each kobs value was the product of the second-order rate constant (kII) and the estimated [·OH]ss at that pH. This simplifying assumption allowed us to calculate kII values for pH 2, 3, 4, and 7. Fig. S3† shows the calculated kII values at different pH values for FA and GA. Our calculated kII values for both FA and GA increased with the pH, which is consistent with previously reported pH-dependent trends of the ·OH reactivities of saturated carboxylic acids. The pH dependence of the calculated kII values for FA (C1) was significantly larger than that of GA (C2). Previous studies have reported larger pH dependence for the ·OH reactivities of smaller carboxylic acids compared to that of larger carboxylic acids.27,30–32 Amorim et al. (2020) explained that the large increase in ·OH reactivities with pH for FA was due to the electron transfer reaction of the deprotonated carboxylate group combined with the large carboxylate group effect on the FA molecule.27 This caused the C–H bond dissociation energy to decrease, which resulted in FA having substantially faster reactions with ·OH under higher pH conditions. In contrast, GA has multiple C–H sites from which ·OH can abstract an H atom, thus reducing the relative importance of the electron transfer reaction of its deprotonated carboxylate group.
3.2. Photolysis and photooxidation of PA
Unlike FA and GA, PA photodegraded during irradiation in the presence and absence of AN. Fig. 6 shows how kobs for PA depended on pH during direct photolysis (no AN) and photooxidation initiated by the photolysis of 250 μM or 1250 μM AN. All the kobs values for PA were in the order of 10−4 s−1. The kobs values for PA direct photolysis had the following trend: kobs(pH 2) > kobs(pH 3) > kobs(pH 4) ≈ kobs(pH 7). The results from ordinary one-way ANOVA statistics tests indicated that the kobs values were significantly different (p < 0.01) between pH 2, 3, and 4, but not between pH 4 and 7. The pH-dependent trend of kobs could be explained by the reaction mechanism of aqueous PA direct photolysis. PA is an α-keto carboxylic acid that is reversibly hydrated to form a gem-diol in an aqueous solution.66,67 Only the keto form of PA can be excited to undergo direct photolysis, which will proceed mainly through a photodecarboxylation reaction and lead to the formation of carbon dioxide and various aqueous-phase oxidation products.43,48 Since the presence of an acidic H atom is crucial for the photodecarboxylation reaction to occur, the HA form (which is more abundant at lower pH) is more prone to photodecarboxylation reactions compared to the A− form (which is more abundant at higher pH).43 Fig. S1† shows that almost all of the PA was present as A− at pH 4 and 7, whereas about 76% and 24% of PA were present as HA at pH 2 and 3, respectively. This explained why substantially higher kobs values were measured at pH 2 and 3, while lower and similar kobs values were measured at pH 4 and 7.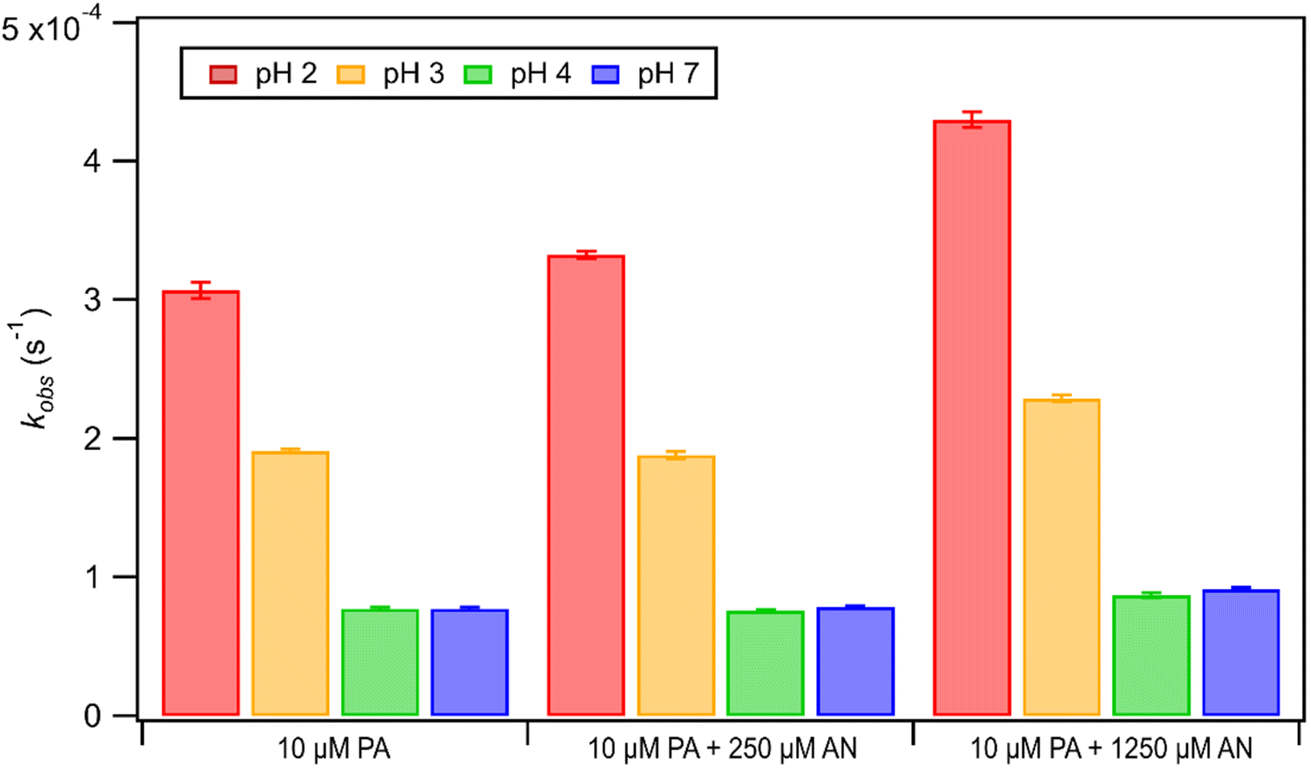 | ||
| Fig. 6 k obs of PA at pH 2, 3, 4, and 7. The PA concentration was fixed at 10 μM, while either 0 μM or 250 μM or 1250 μM AN were used in these experiments. Error bars indicate the standard deviation of multiple experiments. | ||
The kobs values for PA photooxidation initiated by the photolysis of 250 μM or 1250 μM AN showed the same pH-dependent trends as the kobs values for PA direct photolysis (i.e., kobs(pH 2) > kobs(pH 3) > kobs(pH 4) ≈ kobs(pH 7)). Larger kobs values were obtained for PA at higher [AN]/[CA] concentration ratios, which was likely due to the higher concentrations of ·OH and other reactive species produced at higher [AN]/[CA] concentration ratios. Comparisons of the kobs values from direct photolysis vs. photooxidation initiated by the photolysis of 250 μM indicated that only the difference in kobs values at pH 2 was statistically significant (p < 0.01). However, differences in the kobs values from direct photolysis vs. photooxidation initiated by the photolysis of 1250 μM were statistically significant (p < 0.01) for all four pH conditions. Nevertheless, comparisons of kobs values for PA direct photolysis vs. nitrate-mediated photooxidation indicated that PA direct photolysis was the main sink of PA during nitrate-mediated photooxidation at pH 2 to 7.
3.3. Atmospheric lifetimes of FA, GA, and PA
Using the pH-dependent photodegradation rates of these carboxylic acids calculated in this study, we examined how the partitioning of these carboxylic acids to the aqueous phase will affect their atmospheric lifetimes against reaction with ·OH (for FA and GA) and direct photolysis (for PA). Since these carboxylic acids can react both in the gas phase and aqueous phase, we used a simulation methodology similar to the one employed by Yang et al. (2021).28 Three scenarios were simulated: (1) gas phase only, (2) gas + aerosol, and (3) gas + cloud water. For FA and GA, we used kII values derived from nitrate-mediated (250 μM AN) photooxidation experiments (Fig. S3†). Similar to Yang et al. (2021),28 the ·OH concentrations were set to 1 × 106 molecules cm−3, 5 × 10−13 M, and 5 × 10−14 M for the gas phase, aerosols, and clouds, respectively, in our simulations. However, it should be noted that the ·OH concentrations in aqueous aerosols and cloud droplets depend on the environment (e.g., urban vs. rural vs. maritime) and span 4 orders of magnitude (10−16 M to 10−12 M).9 For PA, we used kobs values from direct photolysis experiments since direct photolysis is the dominant sink of PA. It should be noted that the photon flux in the photoreactor (Fig. S2†) is higher than the ambient photon flux. For instance, the photon flux in the photoreactor at 310 nm is about 10 times higher than the ambient flux at 310 nm for a summer day in Hong Kong at noon.68 Hence, the calculated first-order rate constants of PA in aqueous aerosols and clouds likely represent an upper limit. Details of the calculations performed and the parameters used for these simulations can be found in Section S2 and Table S5,† respectively. Our simulations do not account for dynamic equilibrium that may occur as the photodegradation of the carboxylic acid progresses, nor do they restrict the reactions of FA and GA with ·OH produced solely from inorganic nitrate photolysis. Fig. 7 shows that the photodegradation of FA and GA by reaction with ·OH in aqueous aerosols was estimated to be much faster than their photodegradation in the gas phase. This suggested that partitioning to aqueous aerosols will significantly reduce the lifetimes of FA and GA. While FA photodegradation in cloud water was estimated to be competitive with photodegradation in the gas phase, GA photodegradation was likely not important in cloud water. PA photodegradation via direct photolysis in aqueous aerosols was estimated to be competitive with its photodegradation in the gas phase, but its photodegradation was likely not important in cloud water.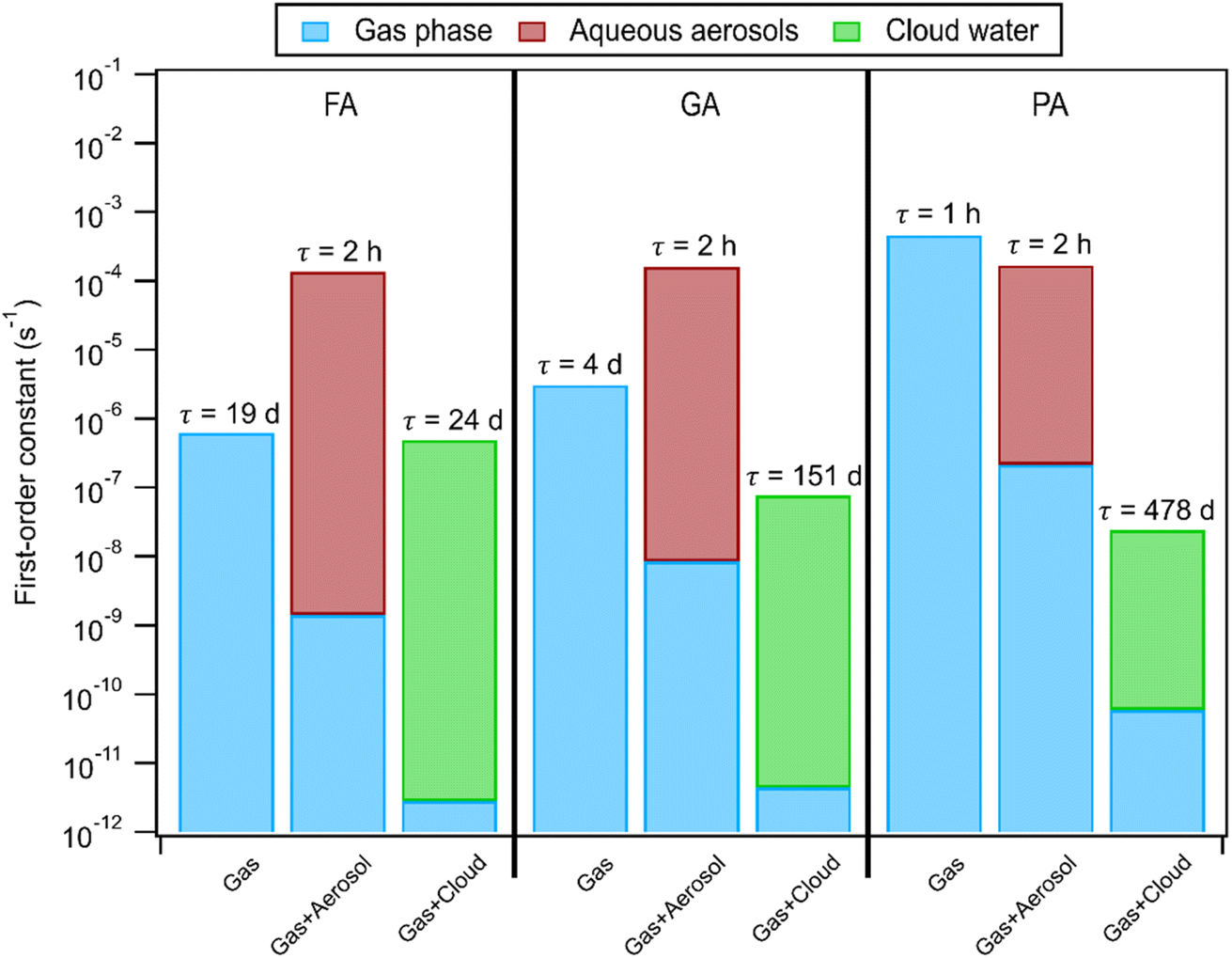 | ||
| Fig. 7 Calculated first-order rate constants and corresponding lifetimes of FA, GA, and PA against ·OH (FA and GA) and direct photolysis (PA) in the atmosphere. The blue portions in the gas + aerosol conditions and gas + cloud conditions represent the contributions from gas phase reactions. The y axis is presented in a logarithm scale to accommodate the drastically different first-order constants. | ||
4 Conclusion
In this study, we investigated the aqueous pH-dependent (pH 2 to 7) photooxidation of FA, GA, and PA initiated by AN photolysis. The photodegradation rates of the three carboxylic acids were controlled by the [AN]/[CA] concentration ratio. In general, higher [AN]/[CA] concentration ratios resulted in faster photodegradation rates, which could be attributed to the higher concentrations of ·OH produced from the photolysis of higher concentrations of AN. The photodegradation rates of the three carboxylic acids strongly depended on the pH. The highest photodegradation rate was observed at pH 4 for FA, whereas the highest photodegradation rates were observed at pH 2 for GA and PA. Our simulations revealed that the observed pH-dependent photodegradation rates for FA and GA were a result of pH controlling both the fraction of the carboxylic acid present in HA and A− forms and ·OH production from AN photolysis. In contrast, the pH-dependent PA photodegradation rates were due primarily to the pH-dependent decarboxylation of PA initiated by light. Nitrate-mediated photooxidation played a minor role in PA photodegradation, and direct photolysis was PA's main sink during nitrate-mediated photooxidation. To predict the fates of these carboxylic acids in the atmosphere, we examined how partitioning to aqueous aerosols and cloud water will affect their atmospheric lifetimes. Our simulations demonstrated that partitioning to aqueous aerosols will reduce the lifetimes of FA and GA against reactions with ·OH. However, GA photodegradation will likely not be important in cloud water, whereas FA photodegradation in cloud water will likely be competitive with gas-phase photodegradation. For PA, photodegradation in aqueous aerosols will likely be competitive with its photodegradation in the gas phase, but its photodegradation will likely not be important in cloud water.Overall, our results highlight how the pH and inorganic nitrate photolysis can combine to influence the overall photodegradation rates of carboxylic acids in atmospheric aqueous phases, which will be useful for modeling the reactive fates of carboxylic acids in regions with inorganic nitrate-rich cloud water and/or aqueous aerosols. This suggests that nitrate-mediated photooxidation needs to be considered during investigations of the atmospheric lifetimes of small carboxylic acids in regions with high levels of particulate inorganic nitrate. Further studies could be directed to elucidating the aqueous pH-dependent photodegradation rates of other short chain (e.g., dicarboxylic acids such as oxalic acid) and long chain carboxylic acids during nitrate-mediated photooxidation. In addition, while this study builds on our understanding of how aqueous nitrate-mediated photooxidation can contribute to the formation and aging of different aqSOA components, one caveat should be noted. In our study, we used solutions containing AN as the sole ·OH photochemical source because our study's focus was solely on nitrate-mediated photooxidation. However, H2O2 is believed to be the main photochemical source of ·OH in cloud water in most regions,6,23 and thus, inorganic nitrate and H2O2 are likely co-present in cloud water in many regions. Our preliminary work suggested that the overall ·OH production will depend on the concentrations of co-present inorganic nitrate vs. H2O2, which could lead to pH-dependent photodegradation rate trends different from those observed for nitrate-mediated photooxidation occurring in the absence of H2O2. For instance, while GA photodegraded at faster rates at pH 2 than at pH 7 during photooxidation initiated solely by AN photolysis (Fig. 3), the converse was observed during photooxidation initiated by co-present AN and H2O2 (Fig. S4†). In addition, the pH dependence of GA photodegradation rates decreased noticeably at higher [AN]/[H2O2] concentration ratios. Given the ubiquitous nature of inorganic nitrate and H2O2 as ·OH photochemical sources in cloud water, future investigations could focus on elucidating pH-dependent mechanisms that drive the photodegradation of carboxylic acids in aqueous phases containing both inorganic nitrate and H2O2. Understanding such mechanisms will enhance our knowledge of how pH influences the quantitative impact of inorganic nitrate on the photodegradation of carboxylic acids during cloud processing.
Data availability
Data can be accessed by request (E-mail: theodora.nah@cityu.edu.hk).Author contributions
Yuting Lyu: investigation, writing – original draft. Jany Ting Chun Chow: investigation. Theodora Nah: conceptualization, writing – review & editing, supervision.Conflicts of interest
There is no conflict to declare.Acknowledgements
This work was supported by the Hong Kong Research Grants Council (project numbers 21304919 and 11303321).References
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- B. Ervens, B. J. Turpin and R. J. Weber, Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 2011, 11, 11069–11102 CrossRef CAS.
- V. F. McNeill, Aqueous Organic Chemistry in the Atmosphere: Sources and Chemical Processing of Organic Aerosols, Environ. Sci. Technol., 2015, 49, 1237–1244 CrossRef CAS PubMed.
- Y. Tan, Y. B. Lim, K. E. Altieri, S. P. Seitzinger and B. J. Turpin, Mechanisms leading to oligomers and SOA through aqueous photooxidation: insights from OH radical oxidation of acetic acid and methylglyoxal, Atmos. Chem. Phys., 2012, 12, 801–813 CrossRef CAS.
- A. Bianco, M. Passananti, M. Brigante and G. Mailhot, Photochemistry of the Cloud Aqueous Phase: A Review, Molecules, 2020, 25, 423 CrossRef CAS PubMed.
- Y. B. Lim, Y. Tan, M. J. Perri, S. P. Seitzinger and B. J. Turpin, Aqueous chemistry and its role in secondary organic aerosol (SOA) formation, Atmos. Chem. Phys., 2010, 10, 10521–10539 CrossRef CAS.
- H. Herrmann, On the photolysis of simple anions and neutral molecules as sources of O−/OH, SOx− and Cl in aqueous solution, Phys. Chem. Chem. Phys., 2007, 9, 3935–3964 RSC.
- H. Herrmann, D. Hoffmann, T. Schaefer, P. Braeuer and A. Tilgner, Tropospheric Aqueous-Phase Free-Radical Chemistry: Radical Sources, Spectra, Reaction Kinetics and Prediction Tools, ChemPhysChem, 2010, 11, 3796–3822 CrossRef CAS PubMed.
- H. Herrmann, T. Schaefer, A. Tilgner, S. A. Styler, C. Weller, M. Teich and T. Otto, Tropospheric Aqueous-Phase Chemistry: Kinetics, Mechanisms, and Its Coupling to a Changing Gas Phase, Chem. Rev., 2015, 115, 4259–4334 CrossRef CAS PubMed.
- D. Vione, V. Maurino, C. Minero, E. Pelizzetti, M. A. J. Harrison, R. I. Olariu and C. Arsene, Photochemical reactions in the tropospheric aqueous phase and on particulate matter, Chem. Soc. Rev., 2006, 35, 441–453 CAS.
- Y. Chen, H. Guo, T. Nah, D. J. Tanner, A. P. Sullivan, M. Takeuchi, Z. Gao, P. Vasilakos, A. G. Russell, K. Baumann, L. G. Huey, R. J. Weber and N. L. Ng, Low-Molecular-Weight Carboxylic Acids in the Southeastern U.S.: Formation, Partitioning, and Implications for Organic Aerosol Aging, Environ. Sci. Technol., 2021, 55, 6688–6699 CrossRef CAS PubMed.
- T. Nah, H. Guo, A. P. Sullivan, Y. Chen, D. J. Tanner, A. Nenes, A. Russell, N. L. Ng, L. G. Huey and R. J. Weber, Characterization of aerosol composition, aerosol acidity, and organic acid partitioning at an agriculturally intensive rural southeastern US site, Atmos. Chem. Phys., 2018, 18, 11471–11491 CrossRef CAS.
- E. D. Baboukas, M. Kanakidou and N. Mihalopoulos, Carboxylic acids in gas and particulate phase above the Atlantic Ocean, J. Geophys. Res.: Atmos., 2000, 105, 14459–14471 CrossRef CAS.
- K. Kawamura, S. Steinberg, L. Ng and I. R. Kaplan, Wet deposition of low molecular weight mono- and di-carboxylic acids, aldehydes and inorganic species in Los Angeles, Atmos. Environ., 2001, 35, 3917–3926 CrossRef CAS.
- T. Li, Z. Wang, Y. Wang, C. Wu, Y. Liang, M. Xia, C. Yu, H. Yun, W. Wang, Y. Wang, J. Guo, H. Herrmann and T. Wang, Chemical characteristics of cloud water and the impacts on aerosol properties at a subtropical mountain site in Hong Kong SAR, Atmos. Chem. Phys., 2020, 20, 391–407 CrossRef CAS.
- M. Loflund, A. Kasper-Giebl, B. Schuster, H. Giebl, R. Hitzenberger and H. Puxbaum, Formic, acetic, oxalic, malonic and succinic acid concentrations and their contribution to organic carbon in cloud water, Atmos. Environ., 2002, 36, 1553–1558 CrossRef CAS.
- V. Shah, D. J. Jacob, J. M. Moch, X. Wang and S. Zhai, Global modeling of cloud water acidity, precipitation acidity, and acid inputs to ecosystems, Atmos. Chem. Phys., 2020, 20, 12223–12245 CrossRef CAS.
- Z. C. Yu, M. Jang and A. Madhu, Prediction of Phase State of Secondary Organic Aerosol Internally Mixed with Aqueous Inorganic Salts, J. Phys. Chem. A, 2021, 125, 10198–10206 CrossRef CAS PubMed.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Environmental Implications of Hydroxyl Radicals (center dot OH), Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- H. Herrmann, B. Ervens, H. W. Jacobi, R. Wolke, P. Nowacki and R. Zellner, CAPRAM2.3: A Chemical Aqueous Phase Radical Mechanism for Tropospheric Chemistry, J. Atmos. Chem., 2000, 36, 231–284 CrossRef CAS.
- C. Anastasio and K. G. McGregor, Chemistry of fog waters in California's Central Valley: 1. In situ photoformation of hydroxyl radical and singlet molecular oxygen, Atmos. Environ., 2001, 35, 1079–1089 CrossRef CAS.
- A. Bianco, M. Passananti, H. Perroux, G. Voyard, C. Mouchel-Vallon, N. Chaumerliac, G. Mailhot, L. Deguillaume and M. Brigante, A better understanding of hydroxyl radical photochemical sources in cloud waters collected at the puy de Dome station – experimental versus modelled formation rates, Atmos. Chem. Phys., 2015, 15, 9191–9202 CrossRef CAS.
- R. Kaur and C. Anastasio, Light absorption and the photoformation of hydroxyl radical and singlet oxygen in fog waters, Atmos. Environ., 2017, 164, 387–397 CrossRef CAS.
- H. O. T. Pye, A. Nenes, B. Alexander, A. P. Ault, M. C. Barth, S. L. Clegg, J. L. Collett Jr, K. M. Fahey, C. J. Hennigan, H. Herrmann, M. Kanakidou, J. T. Kelly, I. T. Ku, V. F. McNeill, N. Riemer, T. Schaefer, G. Shi, A. Tilgner, J. T. Walker, T. Wang, R. Weber, J. Xing, R. A. Zaveri and A. Zuend, The Acidity of Atmospheric Particles and Clouds, Atmos. Chem. Phys., 2020, 20, 4809–4888 CrossRef CAS PubMed.
- N. Daneshvar, M. A. Behnajady, M. K. A. Mohammadi and M. S. S. Dorraji, UV/H2O2 treatment of Rhodamine B in aqueous solution: influence of operational parameters and kinetic modeling, Desalination, 2008, 230, 16–26 CrossRef CAS.
- J. V. Amorim, S. Wu, K. Klimchuk, C. Lau, F. J. Williams, Y. Huang and R. Zhao, pH Dependence of the OH Reactivity of Organic Acids in the Aqueous Phase, Environ. Sci. Technol., 2020, 54(19), 12484–12492 CrossRef CAS PubMed.
- X. Yang, Y. Tao and J. G. Murphy, Kinetics of the oxidation of ammonia and amines with hydroxyl radicals in the aqueous phase, Environ. Sci.: Processes Impacts, 2021, 23, 1906–1913 RSC.
- A. Tilgner, T. Schaefer, B. Alexander, M. Barth, J. L. Collett, K. M. Fahey, A. Nenes, H. O. T. Pye, H. Herrmann and V. F. McNeill, Acidity and the multiphase chemistry of atmospheric aqueous particles and clouds, Atmos. Chem. Phys., 2021, 21, 13483–13536 CrossRef CAS PubMed.
- N. K. V. Leitner and M. Dore, Hydroxyl radical induced decomposition of aliphatic acids in oxygenated and deoxygenated aqueous solutions, J. Photochem. Photobiol., A, 1996, 99, 137–143 CrossRef.
- T. Schaefer, L. Wen, A. Estelmann, J. Maak and H. Herrmann, pH- and Temperature-Dependent Kinetics of the Oxidation Reactions of OH with Succinic and Pimelic Acid in Aqueous Solution, Atmosphere, 2020, 11, 320 CrossRef CAS.
- R. Zhao, A. K. Y. Lee, C. Wang, F. Wania, J. P. S. Wong, S. Zhou and J. P. D. Abbatt, The role of water in organic aerosol multiphase chemistry: Focus on partitioning and reactivity, in Advances in Atmospheric Chemistry, ed. J. R. Barker, A. L. Steiner and T. J. Wallington, World Scientific, 2016, vol. 1, pp. 95–184 Search PubMed.
- B. Ervens, S. Gligorovski and H. Herrmann, Temperature-dependent rate constants for hydroxyl radical reactions with organic compounds in aqueous solutions, Phys. Chem. Chem. Phys., 2003, 5, 1811–1824 RSC.
- Q. Zhang, J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang and D. R. Worsnop, Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 2007, 34, 13801 Search PubMed.
- Y. J. Li, Y. Sun, Q. Zhang, X. Li, M. Li, Z. Zhou and C. K. Chan, Real-time chemical characterization of atmospheric particulate matter in China: a review, Atmos. Environ., 2017, 158, 270–304 CrossRef CAS.
- J. J. West, A. S. Ansari and S. N. Pandis, Marginal PM(25): Nonlinear Aerosol Mass Response to Sulfate Reductions in the Eastern United States, J. Air Waste Manage. Assoc., 1999, 49, 1415–1424 CrossRef CAS PubMed.
- C. L. Heald, J. L. Collett Jr, T. Lee, K. B. Benedict, F. M. Schwandner, Y. Li, L. Clarisse, D. R. Hurtmans, M. Van Damme, C. Clerbaux, P. F. Coheur, S. Philip, R. V. Martin and H. O. T. Pye, Atmospheric ammonia and particulate inorganic nitrogen over the United States, Atmos. Chem. Phys., 2012, 12, 10295–10312 CrossRef CAS.
- M. Schaap, M. van Loon, H. M. ten Brink, F. J. Dentener and P. J. H. Builtjes, Secondary inorganic aerosol simulations for Europe with special attention to nitrate, Atmos. Chem. Phys., 2004, 4, 857–874 CrossRef CAS.
- P. Warneck, The relative importance of various pathways for the oxidation of sulfur dioxide and nitrogen dioxide in sunlit continental fair weather clouds, Phys. Chem. Chem. Phys., 1999, 1, 5471–5483 RSC.
- G. Marussi and D. Vione, Secondary Formation of Aromatic Nitroderivatives of Environmental Concern: Photonitration Processes Triggered by the Photolysis of Nitrate and Nitrite Ions in Aqueous Solution, Molecules, 2021, 26, 2550 CrossRef CAS PubMed.
- T. Arakaki, T. Miyake, T. Hirakawa and H. Sakugawa, pH dependent photoformation of hydroxyl radical and absorbance of aqueous-phase N(III) (HNO2 and NO2-), Environ. Sci. Technol., 1999, 33, 2561–2565 CrossRef CAS.
- P. Khare, N. Kumar, K. M. Kumari and S. S. Srivastava, Atmospheric formic and acetic acids: an overview, Rev. Geophys., 1999, 37, 227–248 CrossRef CAS.
- P. A. Leermakers and G. F. Vesley, The Photochemistry of α-Keto Acids and α-Keto Esters. I. Photolysis of Pyruvic Acid and Benzoylformic Acid, J. Am. Chem. Soc., 1963, 85, 3776–3779 CrossRef CAS.
- M. O. Andreae, R. W. Talbot and S.-M. Li, Atmospheric measurements of pyruvic and formic acid, J. Geophys. Res.: Atmos., 1987, 92, 6635–6641 CrossRef CAS.
- J. Liao, K. D. Froyd, D. M. Murphy, F. N. Keutsch, G. Yu, P. O. Wennberg, J. M. St Clair, J. D. Crounse, A. Wisthaler, T. Mikoviny, J. L. Jimenez, P. Campuzano-Jost, D. A. Day, W. Hu, T. B. Ryerson, I. B. Pollack, J. Peischl, B. E. Anderson, L. D. Ziemba, D. R. Blake, S. Meinardi and G. Diskin, Airborne measurements of organosulfates over the continental U.S, J. Geophys. Res.: Atmos., 2015, 120, 2990–3005 CrossRef CAS PubMed.
- T. Nah, Y. Ji, D. J. Tanner, H. Guo, A. P. Sullivan, N. L. Ng, R. J. Weber and L. G. Huey, Real-time measurements of gas-phase organic acids using SF6− chemical ionization mass spectrometry, Atmos. Meas. Tech., 2018, 11, 5087–5104 CrossRef CAS.
- Z. Gao, P. Vasilakos, T. Nah, M. Takeuchi, H. Chen, D. J. Tanner, N. L. Ng, J. Kaiser, L. G. Huey, R. J. Weber and A. G. Russell, Emissions, chemistry or bidirectional surface transfer? Gas phase formic acid dynamics in the atmosphere, Atmos. Environ., 2022, 274, 118995 CrossRef CAS.
- E. C. Griffith, B. K. Carpenter, R. K. Shoemaker and V. Vaida, Photochemistry of aqueous pyruvic acid, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11714–11719 CrossRef CAS PubMed.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. de Gouw, M. Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 2015, 15, 6283–6304 CrossRef CAS.
- F. Paulot, D. Wunch, J. D. Crounse, G. C. Toon, D. B. Millet, P. F. DeCarlo, C. Vigouroux, N. M. Deutscher, G. G. Abad, J. Notholt, T. Warneke, J. W. Hannigan, C. Warneke, J. A. de Gouw, E. J. Dunlea, M. De Maziere, D. W. T. Griffith, P. Bernath, J. L. Jimenez and P. O. Wennberg, Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 2011, 11, 1989–2013 CrossRef CAS PubMed.
- R. Sander, Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
- A. Williams, pKa Data Compiled by R. Williams, https://organicchemistrydata.org/hansreich/resources/pka/pka_data/pka-compilation-williams.pdf, updated 4/7/2022.
- C. J. Clarke, J. A. Gibbard, L. Hutton, J. R. R. Verlet and B. F. E. Curchod, Photochemistry of the pyruvate anion produces CO2, CO, CH3-, CH3, and a low energy electron, Nat. Commun., 2022, 13, 937 CrossRef CAS PubMed.
- L. Yang, M. B. Ray and L. E. Yu, Photooxidation of dicarboxylic acids—Part I: effects of inorganic ions on degradation of azelaic acid, Atmos. Environ., 2008, 42, 856–867 CrossRef CAS.
- J. Mack and J. R. Bolton, Photochemistry of nitrite and nitrate in aqueous solution: a review, J. Photochem. Photobiol., A, 1999, 128, 1–13 CrossRef CAS.
- J. W. Yang, W. C. Au, H. Law, C. H. Lam and T. Nah, Formation and evolution of brown carbon during aqueous-phase nitrate-mediated photooxidation of guaiacol and 5-nitroguaiacol, Atmos. Environ., 2021, 254, 118401 CrossRef CAS.
- J. Yang, W. C. Au, H. Law, C. H. Leung, C. H. Lam and T. Nah, pH affects the aqueous-phase nitrate-mediated photooxidation of phenolic compounds: implications for brown carbon formation and evolution, Environ. Sci.: Processes Impacts, 2022 10.1039/D2EM00004K.
- N. K. Scharko, A. E. Berke and J. D. Raff, Release of nitrous acid and nitrogen dioxide from nitrate photolysis in acidic aqueous solutions, Environ. Sci. Technol., 2014, 48, 11991–12001 CrossRef CAS PubMed.
- G. D. Chen, S. Hanukovich, M. Chebeir, P. Christopher and H. Z. Liu, Nitrate Removal via a Formate Radical-Induced Photochemical Process, Environ. Sci. Technol., 2019, 53, 316–324 CrossRef CAS PubMed.
- R. F. Zhang, M. S. Gen, T. M. Fu and C. K. Chan, Production of Formate via Oxidation of Glyoxal Promoted by Particulate Nitrate Photolysis, Environ. Sci. Technol., 2021, 55, 5711–5720 CrossRef CAS PubMed.
- E. Ford, M. N. Hughes and P. Wardman, Kinetics of the reactions of nitrogen dioxide with glutathione, cysteine, and uric acid at physiological pH, Free Radical Biol. Med., 2002, 32, 1314–1323 CrossRef CAS PubMed.
- J. A. Bell, E. Grunwald and E. Hayon, Kinetics of deprotonation of organic free radicals in water. Reaction of glycolate (HOCHCO2-), (HOCHCONH2), and (HOCCH3CONH2) with various bases, J. Am. Chem. Soc., 1975, 97, 2995–3000 CrossRef CAS.
- G. Scholes and R. L. Willson, γ-Radiolysis of aqueous thymine solutions. Determination of relative reaction rates of OH radicals, Trans. Faraday Soc., 1967, 63, 2983–2993 RSC.
- B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, Reactivity of Ho2/O-2 Radicals in Aqueous-Solution, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 CrossRef CAS.
- L. Bai, Y. Jiang, D. M. Xia, Z. S. Wei, R. Spinney, D. D. Dionysiou, D. Minakata, R. Y. Xiao, H. B. Xie and L. Y. Chai, Mechanistic Understanding of Superoxide Radical-Mediated Degradation of Perfluorocarboxylic Acids, Environ. Sci. Technol., 2022, 56, 624–633 CrossRef CAS PubMed.
- M. K. Maroń, K. Takahashi, R. K. Shoemaker and V. Vaida, Hydration of pyruvic acid to its geminal-diol, 2,2-dihydroxypropanoic acid, in a water-restricted environment, Chem. Phys. Lett., 2011, 513, 184–190 CrossRef.
- Y. Pocker, J. E. Meany, B. J. Nist and C. Zadorojny, Reversible hydration of pyruvic acid. I. Equilibrium studies, J. Phys. Chem., 1969, 73, 2879–2882 CrossRef CAS.
- Y. Li, Y. He, C. H. Lam and T. Nah, Environmental photochemistry of organic UV filter butyl methoxydibenzoylmethane: implications for photochemical fate in surface waters, Sci. Total Environ., 2022, 839, 156145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2em00458e |
| This journal is © The Royal Society of Chemistry 2023 |