
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethylated guanosine and uridine modifications in S. cerevisiae mRNAs modulate translation elongation†
Joshua D.
Jones
 a,
Monika K.
Franco
b,
Tyler J.
Smith
a,
Laura R.
Snyder
a,
Anna G.
Anders
a,
Brandon T.
Ruotolo
a,
Robert T.
Kennedy
ab and
Kristin S.
Koutmou
*ab
a,
Monika K.
Franco
b,
Tyler J.
Smith
a,
Laura R.
Snyder
a,
Anna G.
Anders
a,
Brandon T.
Ruotolo
a,
Robert T.
Kennedy
ab and
Kristin S.
Koutmou
*ab
aDepartment of Chemistry, University of Michigan, 930 N University, Ann Arbor, MI 48109, USA. E-mail: kkoutmou@umich.edu; Tel: +1-734-764-5650
bProgram in Chemical Biology, University of Michigan, 930 N University, Ann Arbor, MI 48109, USA
First published on 20th February 2023
Abstract
Chemical modifications to protein encoding messenger RNAs (mRNAs) influence their localization, translation, and stability within cells. Over 15 different types of mRNA modifications have been observed by sequencing and liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) approaches. While LC-MS/MS is arguably the most essential tool available for studying analogous protein post-translational modifications, the high-throughput discovery and quantitative characterization of mRNA modifications by LC-MS/MS has been hampered by the difficulty of obtaining sufficient quantities of pure mRNA and limited sensitivities for modified nucleosides. We have overcome these challenges by improving the mRNA purification and LC-MS/MS pipelines. The methodologies we developed result in no detectable non-coding RNA modifications signals in our purified mRNA samples, quantify 50 ribonucleosides in a single analysis, and provide the lowest limit of detection reported for ribonucleoside modification LC-MS/MS analyses. These advancements enabled the detection and quantification of 13 S. cerevisiae mRNA ribonucleoside modifications and reveal the presence of four new S. cerevisiae mRNA modifications at low to moderate levels (1-methyguanosine, N2-methylguanosine, N2,N2-dimethylguanosine, and 5-methyluridine). We identified four enzymes that incorporate these modifications into S. cerevisiae mRNAs (Trm10, Trm11, Trm1, and Trm2, respectively), though our results suggest that guanosine and uridine nucleobases are also non-enzymatically methylated at low levels. Regardless of whether they are incorporated in a programmed manner or as the result of RNA damage, we reasoned that the ribosome will encounter the modifications that we detect in cells. To evaluate this possibility, we used a reconstituted translation system to investigate the consequences of modifications on translation elongation. Our findings demonstrate that the introduction of 1-methyguanosine, N2-methylguanosine and 5-methyluridine into mRNA codons impedes amino acid addition in a position dependent manner. This work expands the repertoire of nucleoside modifications that the ribosome must decode in S. cerevisiae. Additionally, it highlights the challenge of predicting the effect of discrete modified mRNA sites on translation de novo because individual modifications influence translation differently depending on mRNA sequence context.
Introduction
Post-transcriptional modifications to RNA molecules influence their structure, localization, stability, and function.1,2 Over 150 different nucleoside modifications exist within non-coding RNAs (ncRNA) and many are important, or even essential, for cellular processes including protein synthesis.1,3 The biological significance of ncRNA modifications is underscored by decades of observations implicating the dysfunction and mis-regulation of ncRNA modifying enzymes in cancer and other diseases.4–9 Although a handful of chemical modifications [N7-methylguanosine (m7G), N6-methyladenosine (m6A), inosine(I), and 5-methylcytidine (m5C)] have long been detected in protein coding messenger RNAs (mRNAs), recently there has been an explosion in the discovery of additional mRNA modifications enabled by technological advances in RNA sequencing. There are now over 15 different types of modified nucleosides reported within mRNAs,1,10–17 and it is becoming rapidly apparent that, akin to their ncRNA counterparts, modifications likely modulate molecular function of mRNAs.While the impacts of ncRNA modifications on gene expression are extensively documented, the consequences of mRNA modifications are just beginning to be evaluated. One of the most abundant and well-studied mRNA modifications, m6A, is implicated in multiple facets of the mRNA lifecycle including nuclear export,18–20 mRNA stability,21–23 and translational efficiency.22,24–28 Given this wide range of potential roles it is unsurprising that sequencing based studies suggest that the mis-regulation of m6A is linked to a host of diseases including endometrial cancer29 and type 2 diabetes.30 Initial largely correlative studies of m6A distribution provide an example of the biological impact mRNA modifications might have, but the quantitative investigation of m6A and other mRNA modifications is required to determine the biological role (if any) of each of the thousands of individual modified mRNA sites. Furthermore, it is vital to continue exploring the chemical diversity of modifications in mRNAs, as the >10-fold larger variety of modifications found in ncRNA raises the possibility that the breadth of the chemical landscape in mRNA nucleosides might still remain to be revealed.
The development of further sensitive, quantitative techniques to detect mRNA modifications is essential to direct future investigations into the molecular level consequences of this emerging class of RNA modifications. Deep sequencing based technologies capable of mapping the location of mRNA modifications have enabled their widespread study. Nonetheless, like all methods, these approaches have limitations – they are computationally laborious, not generally quantitative, and typically detect a single type of modification at a time. Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) technologies12,31–34 have the potential to complement the knowledge obtained by sequencing based modification mapping approaches. The sensitivity and specificity of LC-MS/MS methodologies have long made it the method of choice for identifying and extensively characterizing protein35 and ncRNA modifications.36–44 Currently published LC-MS/MS methods can assay for up to 40 ribonucleosides in a single analysis and use calibration curves constructed with nucleoside standards to enable quantification,31 though they do not report on where individual modifications reside throughout the transcriptome.13 Despite the demonstrated utility of LC-MS/MS methodologies for studying chemical modifications to biomolecules, LC-MS/MS platforms are not widely used to examine mRNA modifications.
Here, we identify two factors that have impeded the application of LC-MS/MS to mRNA modification analysis: the quantity of mRNA required for current LC-MS/MS sensitivities, and the difficulty of obtaining highly pure mRNA. We integrated an improved chromatographic approach with enhanced mRNA purification and validation processes to overcome these limitations and develop a robust workflow for mRNA modification characterization. Our method is capable of quantifying 50 ribonucleoside variants in a single analysis. Using this method, we found that purified S. cerevisiae mRNA samples contain four previously undetected modifications, 1-methylguanosine (m1G), N2-methylguanosine (m2G), N2,N2-dimethylguanosine (m22G) and 5-methyluridine (m5U), that are likely incorporated into mRNAs both enzymatically (Trm10, Trm11, Trm1, and Trm2) and non-enzymatically. Investigations into the impact of these mRNA modifications on translation elongation in a fully purified in vitro translation system demonstrate that the inclusion of the methylated nucleosides into mRNA codons can slow amino acid addition by the ribosome. Collectively, our findings advance available chromatography and mRNA purification and validation methods to enhance the high-confidence and high-throughput detection of modified nucleosides by LC-MS/MS, and support a growing body of evidence that the inclusion of mRNA modifications alter the peptide elongation during protein synthesis.
Results and discussion
Development of highly sensitive LC-MS/MS method for simultaneously quantifying 50 ribonucleosides
Quantitative ribonucleoside LC-MS/MS methods typically rely on reversed phase chromatography to separate ribonucleosides prior to detection by multiple reaction monitoring (MRM) on a triple quadrupole mass spectrometer.31,36,38,45,46 These approaches have reported limits of detection (LODs) down to ∼60 attomole for select ribonucleosides using standard mixtures with canonical and modified nucleosides at equal concentration.36 However, the abundance of unmodified and modified nucleosides in RNAs are not equivalent in cells, with canonical bases existing in 20- to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold higher concentrations than RNA modifications (Fig. 1(A)). In currently available chromatography methods, modified nucleosides (e.g., m5U, m1G, m1Ψ, and s2U) commonly coelute with canonical nucleosides, reducing the detectability of some modified bases.36,38,39 Coelution limits the utility of available LC-MS/MS methods because it results in ion suppression of modified nucleoside signals, with abundant canonical nucleosides outcompeting modified nucleosides for electrospray droplet surface charge. Additionally, this phenomenon makes calibration curves non-linear and worsens the quantifiability of modifications at concentrations necessary for mRNA modification analyses. Recent efforts have been made to derivatize ribonucleosides prior to LC-MS/MS analysis to increase sensitivity and retention on reversed-phase chromatography.32,47–49 The analogous benzoyl chloride derivatization of neurochemicals has previously been an important separation strategy for many neurochemical monitoring applications.50,51 However, labeling strategies are unlikely to prove as useful for investigating mRNA modifications because derivatizing agents are typically nucleobase specific, limiting the ability of LC-MS/MS assays to be multiplexed.32,47,48 Furthermore, labeling increases the amount of mRNA sample required due to additional sample preparation steps following derivatization. This is an important consideration given that mRNAs represent only ∼1–2% of the total RNAs in a cell, and purifying sufficient quantities of mRNA for LC-MS/MS analysis is challenging.
000-fold higher concentrations than RNA modifications (Fig. 1(A)). In currently available chromatography methods, modified nucleosides (e.g., m5U, m1G, m1Ψ, and s2U) commonly coelute with canonical nucleosides, reducing the detectability of some modified bases.36,38,39 Coelution limits the utility of available LC-MS/MS methods because it results in ion suppression of modified nucleoside signals, with abundant canonical nucleosides outcompeting modified nucleosides for electrospray droplet surface charge. Additionally, this phenomenon makes calibration curves non-linear and worsens the quantifiability of modifications at concentrations necessary for mRNA modification analyses. Recent efforts have been made to derivatize ribonucleosides prior to LC-MS/MS analysis to increase sensitivity and retention on reversed-phase chromatography.32,47–49 The analogous benzoyl chloride derivatization of neurochemicals has previously been an important separation strategy for many neurochemical monitoring applications.50,51 However, labeling strategies are unlikely to prove as useful for investigating mRNA modifications because derivatizing agents are typically nucleobase specific, limiting the ability of LC-MS/MS assays to be multiplexed.32,47,48 Furthermore, labeling increases the amount of mRNA sample required due to additional sample preparation steps following derivatization. This is an important consideration given that mRNAs represent only ∼1–2% of the total RNAs in a cell, and purifying sufficient quantities of mRNA for LC-MS/MS analysis is challenging.
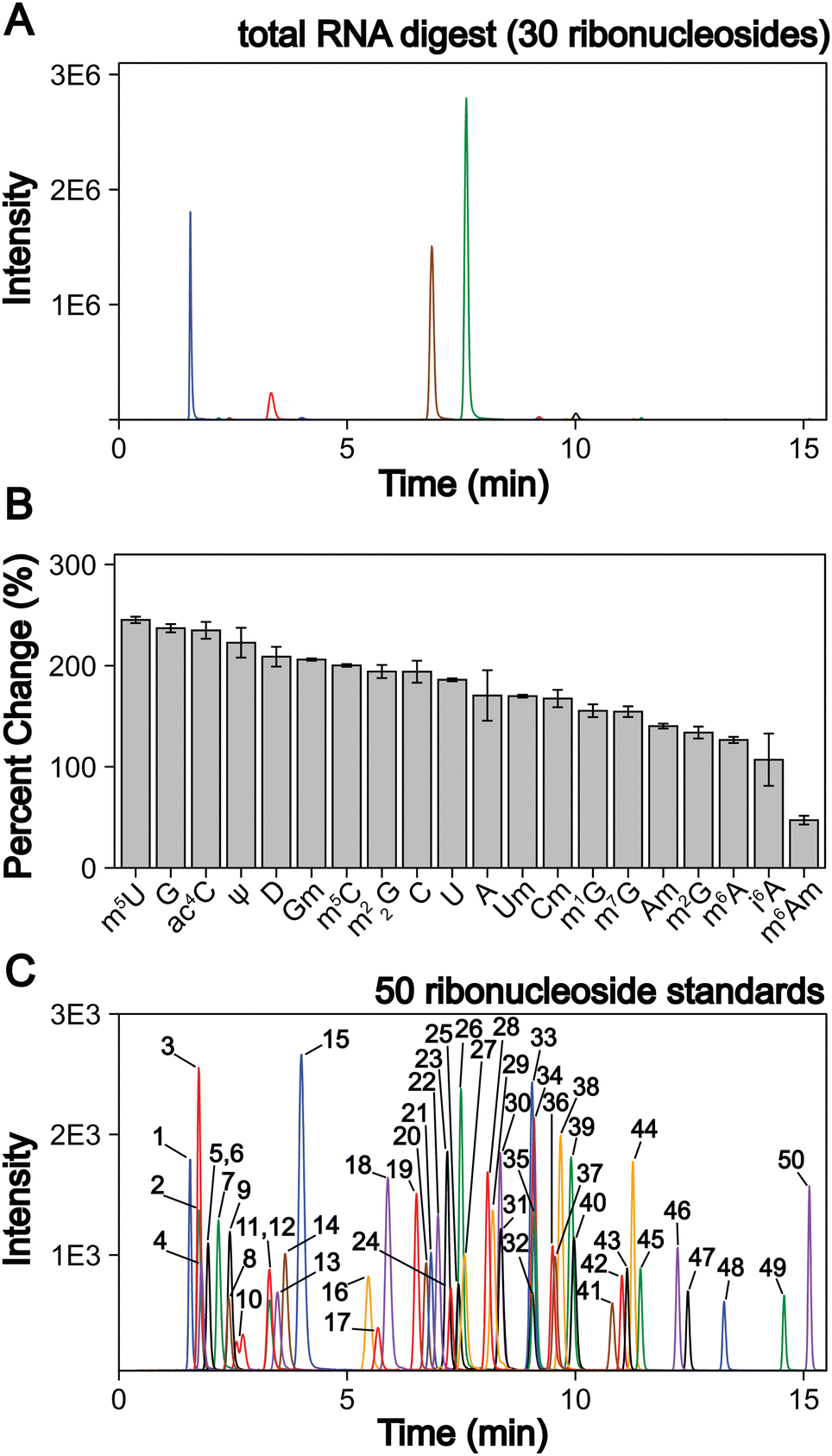 | ||
| Fig. 1 LC-MS/MS method development to quantify 50 ribonucleosides in a single analysis. (A) Extracted ion chromatogram for the 30 ribonucleosides (4 canonical bases and 26 naturally occurring modifications) detected in a S. cerevisiae total RNA digestion displaying that the canonical bases exist at much larger levels than the ribonucleoside modifications. (B) LC-MS/MS signal percent improvement using 1 mm chromatography at 100 μL min−1 compared to 2 mm chromatography at 400 μL min−1. (C) Extracted ion chromatogram for 50 ribonucleoside standards (4 canonical bases, 45 naturally occurring modifications, and 1 non-natural modifications). The concentrations of each ribonucleoside standards within the standard mix and their corresponding peak numbers are displayed in Table S2 (ESI†). For the chromatograms, each color peak represents a separate ribonucleoside in the method, and the colors are coordinated between panel (A) and (C). | ||
We addressed these limitations by first improving upon existing chromatography techniques. Current methods normally utilize 2 mm internal diameter (I.D.) columns that require higher flow rates (300 to 400 μL min−1), which worsens ionization efficiencies compared to smaller I.D. chromatography with lower flow rates. We utilized a 1 mm I.D. column with flow rates at 100 μL min−1 to lessen these effects. In principle, even smaller bore columns (i.e., “nano-LC”), which are commonly used in in proteomics,52 could be used. Indeed, some studies have shown their effectiveness for nucleosides.53,54 However, we selected a 1 mm I.D. column because smaller bore columns can suffer from robustness issues in some conditions and the low binding capacity of polar nucleosides results in poor peak shapes in nano-LC due to relatively large injection volumes. Furthermore, the stationary phases used in nano-LC present a limitation, as porous graphitic carbon columns yield poor chromatographic performance for some ribonucleosides (e.g., methylated guanosine modifications) and many C18 phases have low binding capacity for some ribonucleosides (e.g., cytidine and pseudouridine) making them difficult to retain. Therefore, we used a polar endcapped C18 column to provide more retention and good performance for all nucleosides. We also used mobile phase buffers previously shown to provide high ESI-MS sensitivity for modified ribonucleosides.36 These alterations combined improved the limit of detection (LOD) of the assay by 50 to 250% for all nucleosides tested compared to standard 2 mm I.D. chromatography at 400 μL min−1 (Fig. 1(B)) while maintaining adequate ribonucleoside binding capacity for early eluting ribonucleosides.
In addition to switching columns, we also altered the chromatographic conditions, increasing the temperature (35 °C vs. 25 °C) and modifying mobile phase gradients to prevent coelution of the highly abundant canonical nucleosides with the modified nucleosides. Notably, in contrast to most available methods, m5U, m1G, m1Ψ do not coelute with unmodified nucleosides in our method (Fig. 1(C)). This greatly improves separation, resulting in the reduced ionization suppression of these nucleosides. Together, these advancements result in our method having a wider linear dynamic range than previous reports with over four orders of magnitude for most modifications and LODs down to 3 amol (0.6 pM) using a single internal standard and no derivatization steps. This method represents at least a 10-fold improvement over previous ultrahigh-performance LC (UHPLC) and nano-LC analyses for most modifications analyzed (Table S1 and Fig. S1–S4, ESI†). With these advances, the LC-MS/MS technique described here provides a linear dynamic range and LODs capable of analyzing both highly (i.e., ncRNA) and modestly (i.e., mRNA) modified RNAs without large sample requirements. In this method in-depth RNA modification analysis requires approximately 50 to 200 ng of total RNA or mRNA per replicate, which is achievable using standard eukaryotic and bacterial cell culture techniques. Overall, this assay can simultaneously quantify the 4 canonical nucleosides, 45 naturally occurring modified nucleosides, and 1 non-natural modified nucleoside (internal control) (Fig. 1(C), and Table S2, ESI†). This approach ameliorates current quantitative ribonucleoside LC-MS/MS methodologies by improving chromatographic conditions and characterizing quantifiability at nucleoside concentrations representative of typical RNA digest samples to enable higher confidence total RNA and mRNA modification analyses.
Three-stage mRNA purification and validation pipeline provides highly pure S. cerevisiae mRNA
The total cellular RNA pool is mainly comprised of the highly modified non-coding transfer and ribosomal RNAs (tRNAs, rRNA), with mRNA representing only a small percentage of RNA species in the cell. Unlike RNA-seq, LC-MS/MS nucleoside assays do not distinguish between modifications arising from ncRNA or mRNA. In total RNA digestions, mRNA modifications typically exist at concentrations 100-fold (or more) lower than ncRNA modifications.31 Therefore, even low-level contamination of tRNA and rRNA in purified mRNA samples can lead to inaccurate quantifications and false mRNA modification discovery. Most of the published mRNA purification pipelines use a combination of poly(A) enrichment and rRNA depletion steps to obtain mRNA.10,12,31,34,55–57 However, this is insufficient to remove all detectable signal from contaminating ncRNA modifications during LC-MS/MS analyses, especially those arising from tRNA.31,58 The inability to confidently obtain highly pure mRNA samples has limited the utility of nucleoside LC-MS/MS for studying these molecules. Recently, small RNA depletion steps have begun to be incorporated into mRNA purification pipelines to remove residual tRNA contamination;59 however, the highest efficiency purifications typically require expensive instrumentation and materials (liquid chromatograph and size exclusion column)33 or expertise in RNA gel purification.32 Despite these improvements, most reports do not provide the extensive mRNA purity quality controls necessary to confidently confirm the removal of ncRNA from mRNA samples. To apply the sensitive LC-MS/MS method we developed to studying mRNAs, we developed and implemented a three-stage purification pipeline comprised of a small RNA depletion step, two consecutive poly(A) enrichment steps, and ribosomal RNA depletion to selectively deplete the small ncRNA (e.g., tRNA and 5S rRNA) in addition to the 18S rRNA and 28S rRNA using fully commercial kits (Fig. 2). Additionally, we performed extensive quality control on our mRNA samples prior to LC-MS/MS analysis – assessing the purity of our mRNA following the three-stage purification pipeline using chip electrophoresis (Bioanalyzer), RNA-seq, qRT-PCR, and LC-MS/MS. The highly purified mRNA contained no detectable tRNA and rRNA peaks based on our Bioanalyzer electropherograms (Fig. 3(A) and Calculation S1, ESI†). Similarly, RNA-seq indicated the mRNA is enriched from 4.1% in our total RNA to >99.8% in our purified mRNA samples (Fig. 3(B) and Table S3, ESI†). Additionally, we observed a >3000-fold depletion of 25S and 18S rRNAs and an >9-fold enrichment of actin mRNA based on qRT-PCR (Fig. S5, ESI†). Similar purities by RNA-seq have been achieved without a small RNA depletion step,31,57 but we previously found that this protocol was insufficient to remove all contaminating ncRNA signals by LC-MS/MS levels, as low levels of tRNA- and rRNA-specific modified nucleosides are detected in the mRNA samples isolated solely by poly(A) pull-down.31 This observation is in line with the fact that RNA-seq does not accurately report on tRNA levels,60 making additional quality control analyses necessary to judge the extent of tRNA contamination. While the incorporation of tRNA compatible reverse transcriptases into the RNA-seq pipeline could be used to assay for tRNA contamination,60 we elected to apply a multiplexed LC-MS/MS assay for this purpose instead. Our LC-MS/MSprovides a direct, quantitative, and highly sensitive method to obtain multiple measurements for assessing if ncRNA contaminants are present above the LOD for our assays.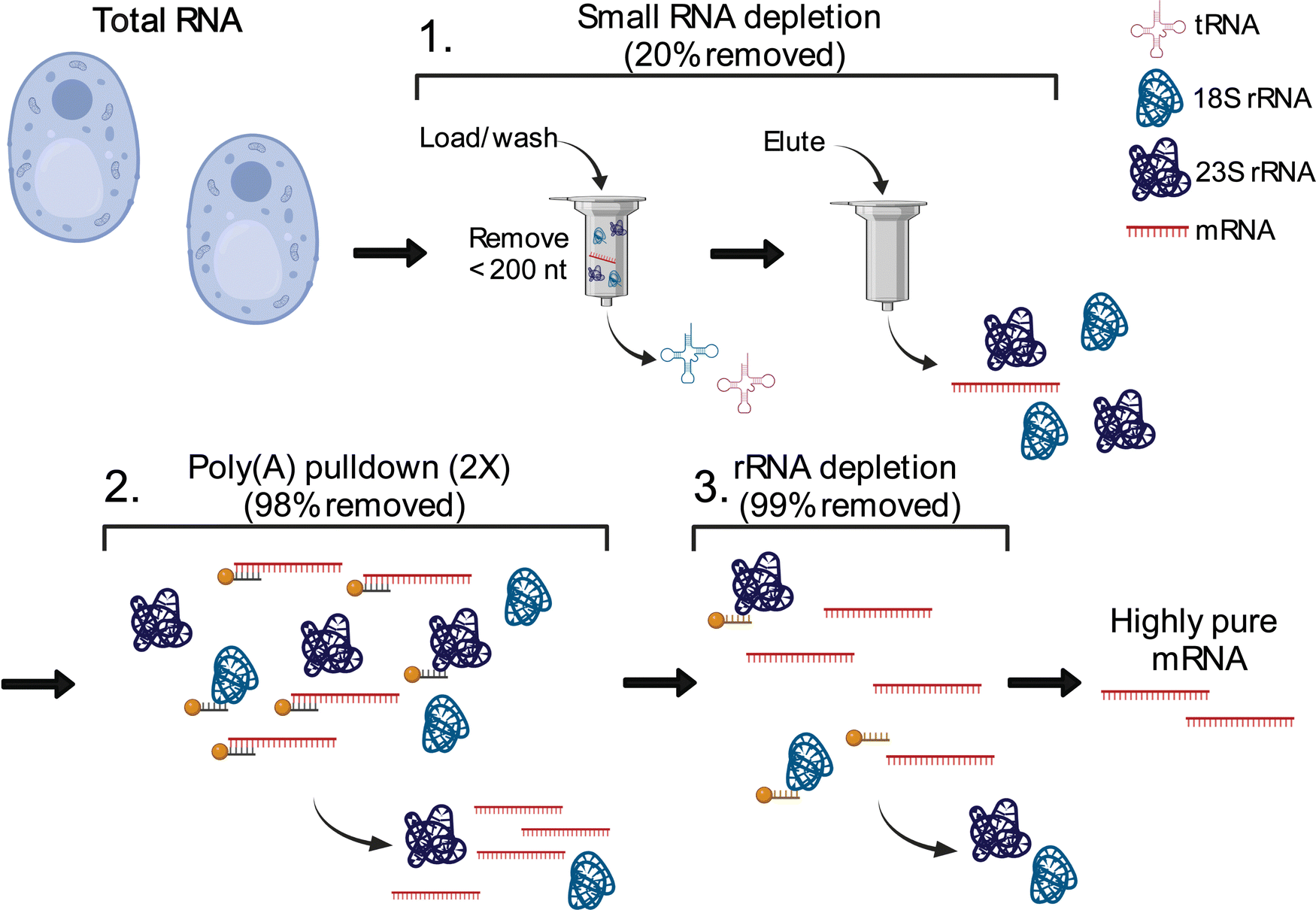 | ||
| Fig. 2 Three-stage mRNA purification pipeline. Total RNA from S. cerevisiae is purified to mRNA using a three-stage purification pipeline: (1) small RNA (e.g., tRNA and 5S rRNA) is depleted; (2) mRNA is enriched from the small RNA depleted fraction through two consecutive poly(A) enrichment steps; (3) remaining rRNA is depleted to result in highly purified mRNA. The displayed percent removed is the additive percent of total RNA removed throughout the three-stage purification pipeline. | ||
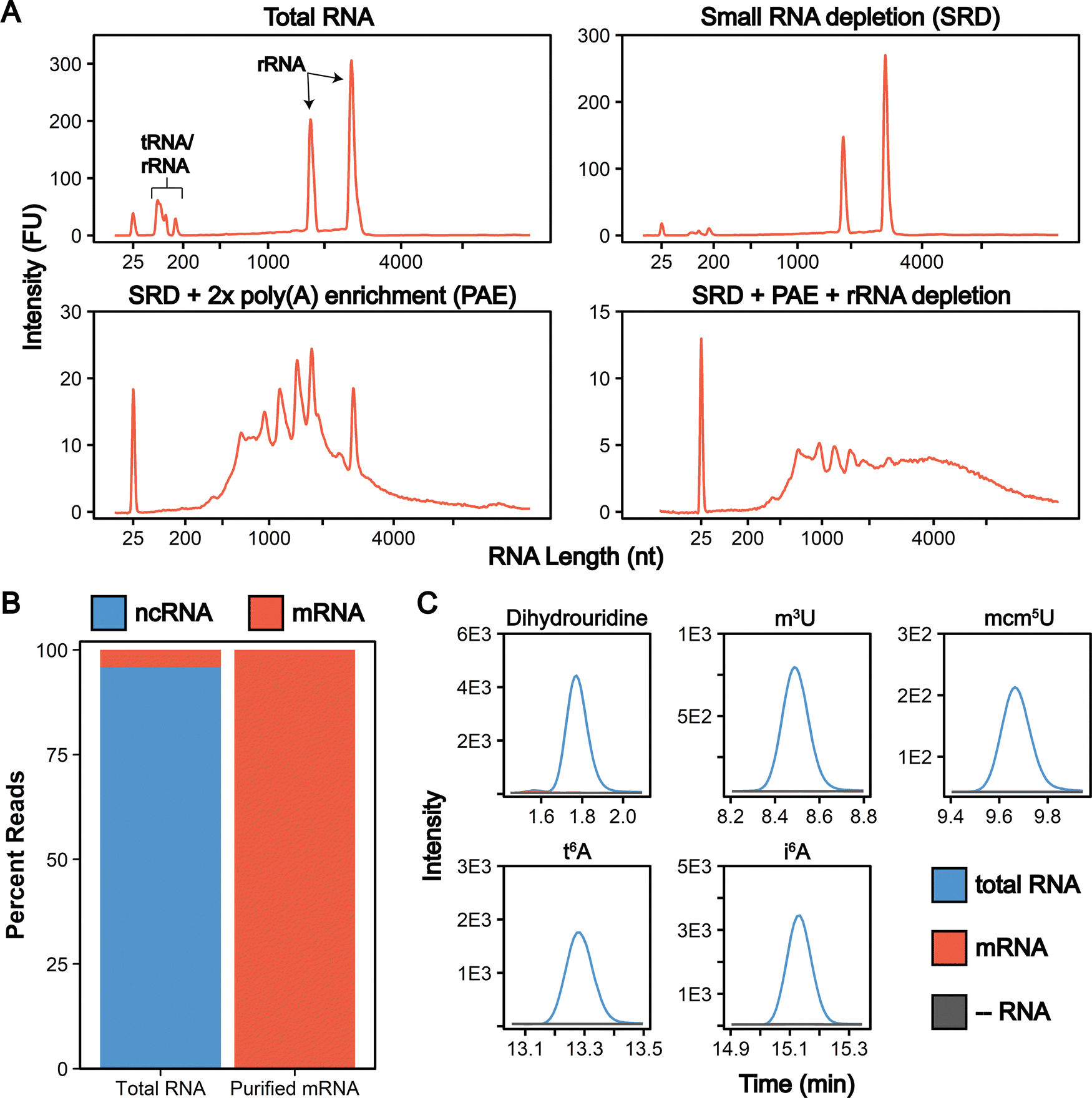 | ||
| Fig. 3 mRNA purity following three-stage purification pipeline. (A) Bioanalyzer electropherograms displaying the RNA distribution following each stage of our purification pipeline. (B) Average percentage of reads mapping to ncRNA (rRNA, tRNA, snRNA, etc.) and mRNA determined by RNA-seq of two biological replicate total RNA and purified mRNA samples. (C) Representative overlaid extraction ion chromatograms for five RNA modifications that exist solely in ncRNA. These five modifications, in addition to eight additional ncRNA modifications, were detected in our total RNA samples (blue) while not detected in our mRNA samples (red) above our control digestions without RNA added (grey). The LODs for DHU, m3U, mcm5U, t6A, and i6A are 530 amol, 45 amol, 29 amol, 21 amol, and 44 amol, respectively. | ||
To quantitatively evaluate the purity of our mRNA from the three-stage purification pipeline by LC-MS/MS we measured the levels of modifications present in our total RNA and purified mRNA (Fig. 2 and 4(A)). The level of the modifications in each sample was normalized to their corresponding canonical nucleosides (e.g., m6A/A) to account for any variations in RNA quantities digested. In our total RNA samples, we detected 26 out of 30 known S. cerevisiae ribonucleoside modifications that we assayed for; f5C, s2U, m2,7G, and m3G were not detected (Fig. 4(B) and Table S4, ESI†). This was expected because these modifications likely exist at levels below our LOD in our total RNA samples as they either arise from oxidative damage of m5C (f5C),61,62 are present at very low levels on S. cerevisiae tRNA (s2U),63–65 or are only found in low abundance snRNA and snoRNA (m2,7G and m3G).66–68 Additionally, we did not detect the 16 ribonucleoside modifications in our assay that have never been reported in S. cerevisiae (1 non-natural and 15 naturally occurring modifications that exist in other organisms) (Fig. 4(B) and Table S4, ESI†). Our purified mRNA samples contained markedly fewer modifications than total RNA, as anticipated. In addition to the 16 non-S. cerevisiae modifications, we did not detect 13 of the S. cerevisiae non-coding RNA modifications present in the total RNA samples (Fig. 3(C) and Table S4, ESI†).
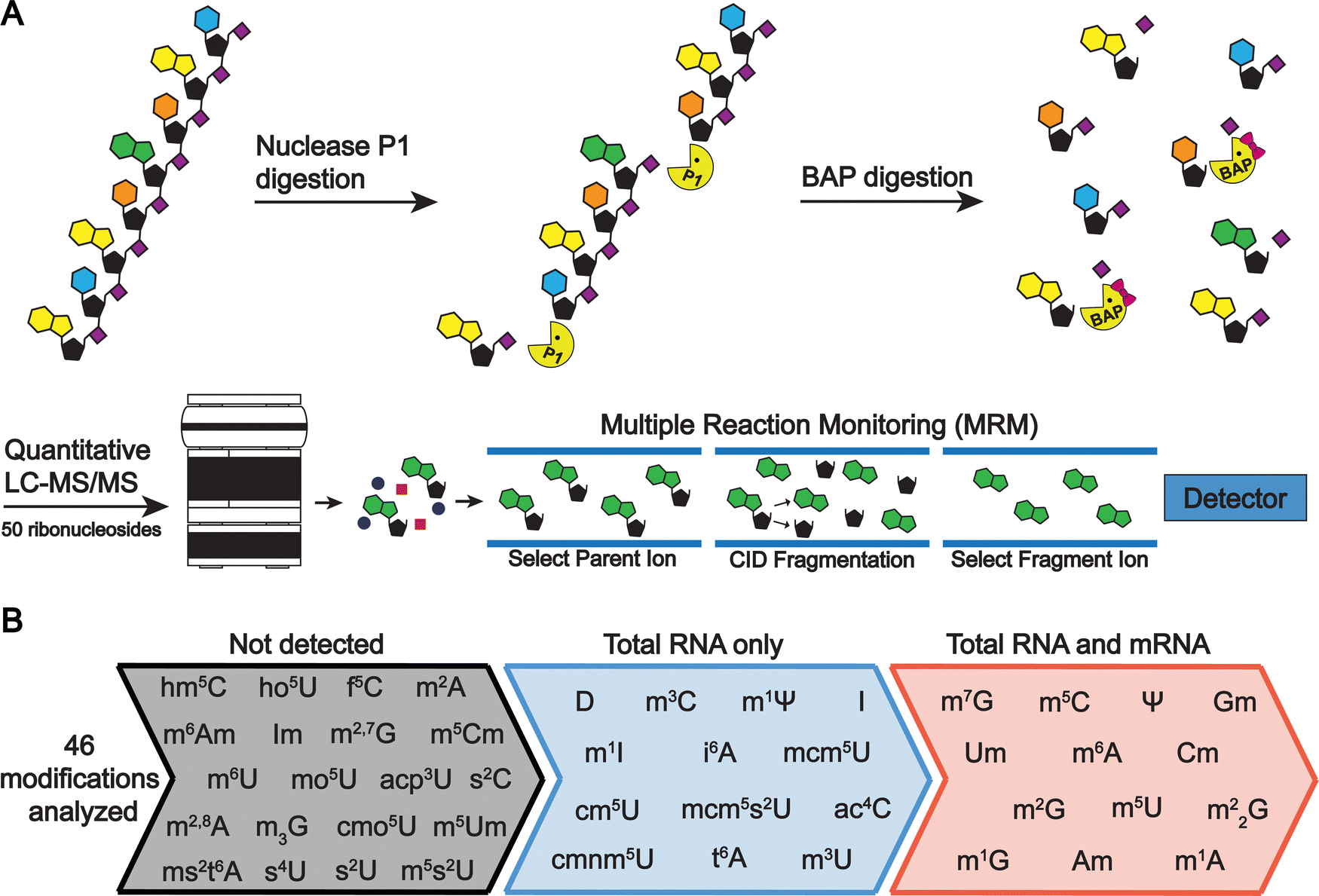 | ||
| Fig. 4 Enzymatic digestion and LC-MS/MS analysis of S. cerevisiae total RNA and mRNA. (A) RNA is enzymatic digested to ribonucleosides through a two-stage process. RNA is first digested to nucleotide monophosphates by nuclease P1 and then dephosphorylated to ribonucleosides by bacterial alkaline phosphatase. The resulting ribonucleosides are separated using reverse phase chromatography and then quantified using MRM on a triple quadrupole mass spectrometer. (B) S. cerevisiae total RNA and mRNA were analyzed using the LC-MS/MS method developed to quantify 46 modifications in a single analysis. In total RNA, 26 modifications were detected while 13 ribonucleosides were detected in the highly purified mRNA. | ||
All modifications not detected in the purified mRNA are reported to be exclusively located in S. cerevisiae tRNAs or rRNAs (e.g., i6A, m3C),3 result from oxidative damage (f5C),69 or were only previously detected in S. cerevisiae mRNAs purified from cells in grown under H2O2 stress (ac4C).31 The highly abundant dihydrouridine (DHU) modification provides a key example of such a common ncRNA modification that is not detected in our purified samples. DHU is located at multiple sites on every S. cerevisiae tRNA and is present at high levels (1.9 DHU/U%) in our total RNA samples (Tables S5 and S6, ESI†). However, we do not detect DHU above our LOD in our purified mRNA (Fig. 3(C)). Using our LOD data, we calculated the maximum tRNA contamination in our purified mRNA to be 0.002% since DHU is not present in S. cerevisiae rRNAs (Calculation S2, ESI†).
Previous studies have used a similar, but more limited, approach for judging mRNA purity by only measuring the levels of a select few ncRNA target modifications. While useful, these analyses provide a limited picture of the RNA modification landscape (and therefore possible levels of contamination) present in a sample. Because the LC-MS/MS assay described here quantifies up to 46 ribonucleoside modifications in a single analysis, we are able to use this method to thoroughly characterize our mRNA purity. These analyses ensure that rRNA and tRNA specific modifications are not present at detectable levels in our highly purified mRNA, corroborating our Bioanalyzer, qRT-PCR and RNA-seq findings (Fig. 3(C)). Our quality controls for mRNA purity give us confidence in downstream LC-MS/MS analyses. Multifaceted quality control data are not yet standard in mRNA modification LC-MS/MS studies, and we believe that the inclusion of controls (i.e., protocols presented here, or the combined application of LC-MS/MS and sequencing methods capable of detecting tRNAs) will increase the utilization of data generated from mRNA studies by LC-MS/MS analysis going forward.
Trm1, Trm2, Trm10 and Trm11 are involved in the formation of methylated guanosine and uridine modifications in S. cerevisiae mRNA
We detected 13 ribonucleoside modifications in purified mRNAs with abundances ranging from that of pseudouridine (0.023 Ψ/U%) to 1-methyladenosine (0.00014 m1A/A%) (Fig. 4(B) and Fig. S6, Tables S5, S6, ESI†). These abundances are lower than other previous mRNA modification LC-MS/MS analyses, including a previous S. cerevisiae study.31 We attribute this to the higher purity of our mRNAs than in previous LC-MS/MS studies, which leads to lower observed modification abundances since our samples lack detectable modifications arising from contaminating ncRNA species. While most of the modifications we observe in our samples were previously reported in in S. cerevisiae mRNA, we detected four modifications for the first time in S. cerevisiae (m1G, m2G, m22G, and m5U) (Fig. 5(A) and Fig. S7, ESI†). The detection of m1G and m5U in S. cerevisiae is in line with recent reports of these modifications in Arabidopsis thaliana and multiple mammalian cell lines at similar levels.32,34,70,71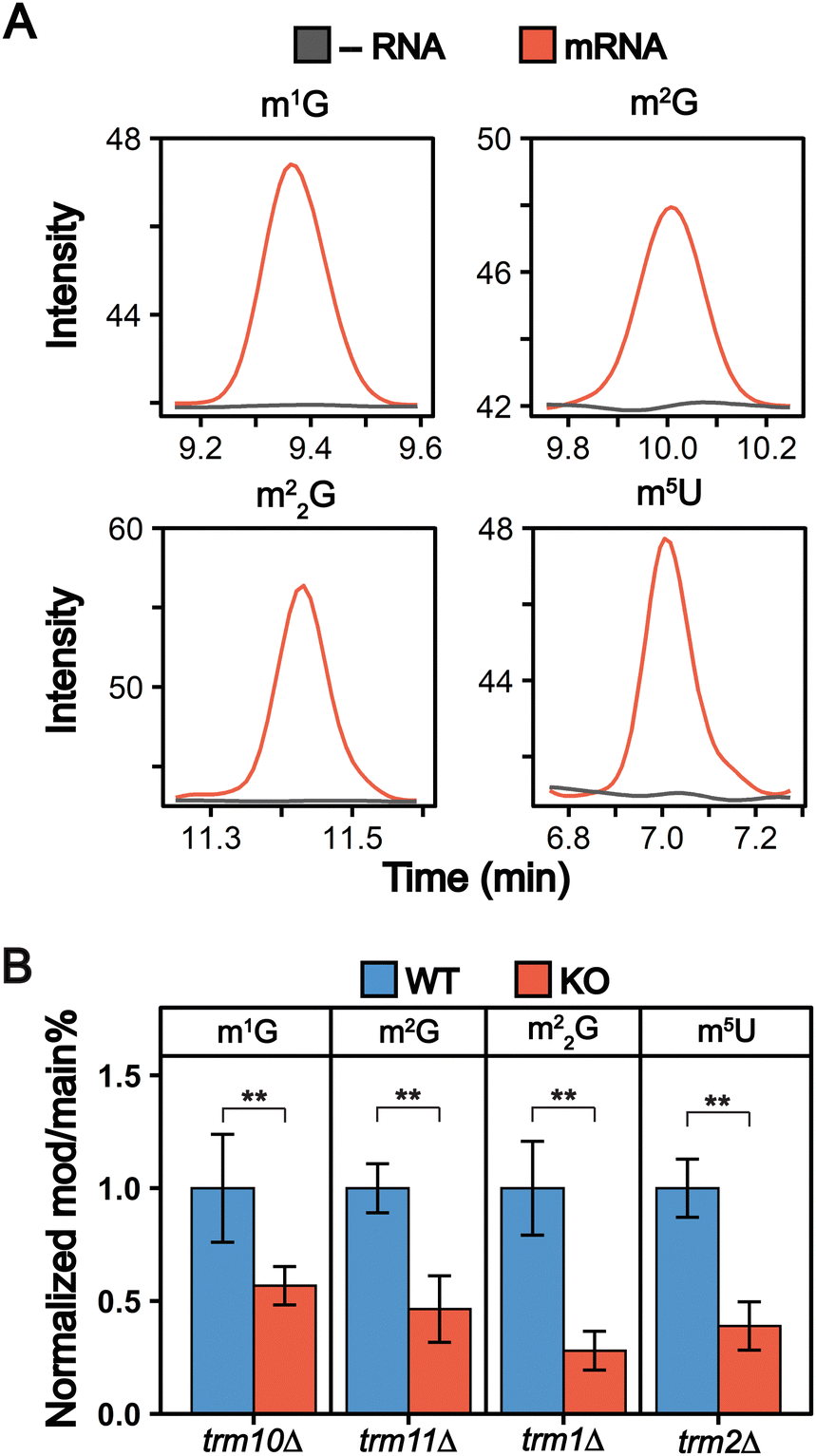 | ||
| Fig. 5 m1G, m2G, m22G, and m5U are present in S. cerevisiae mRNA. (A) Overlaid extracted ion chromatograms displaying m1G, m2G, m22G, and m5U are detected in our mRNA samples (red) above our digestion control samples without RNA added (grey). (B) m1G, m2G, m22G, and m5U are incorporated into S. cerevisiae mRNA by their corresponding tRNA modifying enzymes (Trm10, trm11, Trm1, and Trm2 respectively). The modification/main base% (e.g., m1G/G%) were normalized to their levels in the average WT mRNA levels. A significant decrease (**p < 0.01) was detected for all cases. The error bars are the standard deviation of the normalized mod/main base%. | ||
Many of mRNA modifications, such as pseudouridine, are incorporated by the same enzymes that catalyze their addition into tRNAs and rRNAs.3 We investigated if enzymes responsible for inserting m1G, m2G, m22G, and m5U into S. cerevisiae tRNAs (Trm10, Trm11, Trm1 and Trm2, respectively) also insert them into S. cerevisiae mRNAs. We compared the levels of m1G, m2G, m22G, and m5U in mRNA purified from wild-type and mutant (trm10Δ, trm11Δ, trm1Δ, and trm2Δ) S. cerevisiae. The abundance of all four modifications decreased significantly in mRNA purified from the knockout cell lines (Fig. 5(B) and Table S6, ESI†). While this indicates that these tRNA modifying enzymes can methylate S. cerevisiae mRNA nucleosides, it is notable that low levels of m1G, m2G, m22G, and m5U modifications are still detected in the mRNA purified from knockout cell lines (Fig. 5(B)). Several explanations could account for this. A second enzyme, Trm5, also catalyzes m1G addition into tRNAs and could possibly explain the remaining mRNA m1G signals. However, given that m1G and m2G were previously found as minor products of methylation damage in DNA and RNA,72–79 it is perhaps more likely that the remaining low-level signals that we detect arise from methylation associated RNA damage or minor off target methylation by other enzymes. Regardless of how they are incorporated, when present, these modifications have the potential to impact the function of mRNAs on which they exist.
m1G, m2G and m5U containing mRNA codons slow amino acid addition by the ribosome in a position dependent manner
The frequency of mRNA modification is analogous to that of N-linked and O-linked protein glycosylation, which occur at rates less than approximately 1% and 0.04% per target amino acid, respectively.80 Despite their low abundance, glycosylation influences protein localization and function81,82 and mis-regulation is linked to multiple diseases.83 Similarly, even though they are incorporated into less than 1% of all mRNA nucleosides, evidence is mounting that chemically modified nucleosides impact the mRNA lifecycle.13 mRNAs serve as substrates for the ribosome, and post-transcriptional modifications can alter ribosome decoding speed and accuracy by altering the hydrogen bonding patterns between the mRNA codons and incoming aminoacyl-tRNAs.24,25,84–95 Such perturbations to protein synthesis have significant consequences even when modifications are incorporated into mRNA transcripts at very low levels, as exemplified by the biological consequences of oxidatively damaged mRNAs, which exist at levels similar to that of m1G, m2G, m22G and m5U.79,96 We therefore sought to establish how the insertion of m5U, m1G, and m2G into mRNA codons impacts translation using a well-established reconstituted bacterial translation system24 (Fig. 6(A)). This system has long been used to conduct high-resolution kinetic studies investigating how the ribosome decodes mRNAs. Translation elongation is well conserved between bacteria and eukaryotes,97 and prior studies demonstrate that mRNA modifications (e.g. pseudouridine, N6-methyladenosine and 8-oxo-G) that slow elongation and/or change mRNA decoding elongation in the reconstituted E. coli system24,25,84,98 also do so in eukaryotes.24,99–101 m22G was not selected for study because the phosphoramidite required for mRNA oligonucleotide synthesis is not commercially available.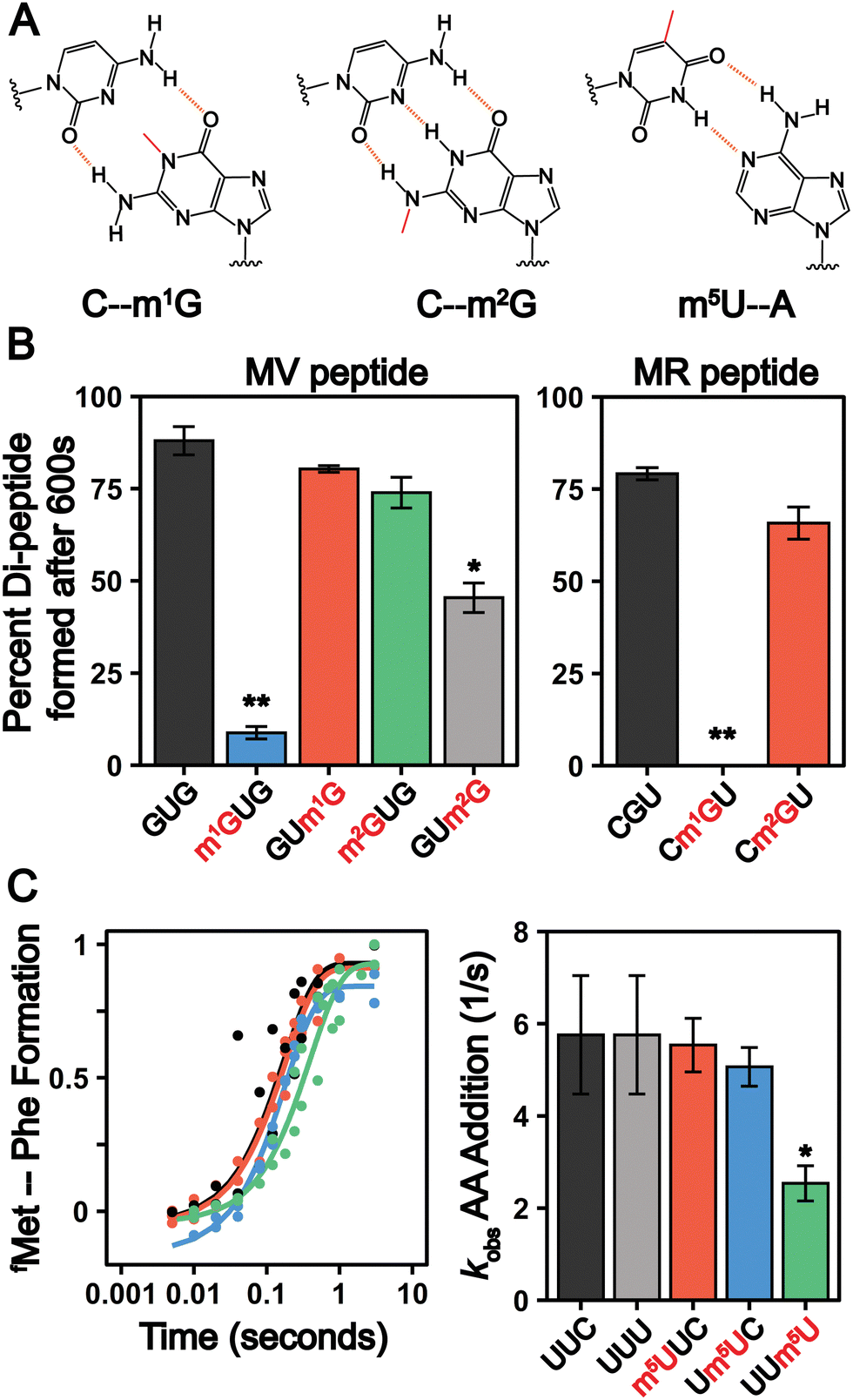 | ||
| Fig. 6 Methylated guanosine and uridine modifications alter amino acid addition. (A) Watson–Crick base pairing of m1G, m2G and m5U. The added methylation is displayed in red and the hydrogen bond interactions displayed as a dashed orange line. (B) Total peptide formation of translation reactions after 600 seconds using transcribed or single-nucleotide modified mRNAs encoding for either (left panel) Met-Val (GUG) or (right panel) Met-Arg (CGU) dipeptide. Error bars are the standard deviation. (C) Time courses displaying the formation of fMet-Phe dipeptide on an unmodified and singly modified UUC or UUU codons (left panel). Observed rate constants (right panel) were determined from the fit data. The error bars are the standard deviation of the fitted value of kobs. | ||
In our assays, 70S E. coli ribosome initiation complexes (ICs) with 35S-fMet-tRNAfMet programmed in the A site are formed on transcripts encoding Met-Phe, Met-Arg, or Met-Val dipeptides. Ternary complexes comprised of aminoacyl-tRNA:EF-Tu:GTP are added to the ICs to begin translation. Reactions are quenched at desired timepoints by KOH, and the unreacted 35S-fMet-tRNAfMet and dipeptide translation products are visualized by electrophoretic TLC (eTLC) (Fig. S8–S11, ESI†). We evaluated the extent of total dipeptide synthesis and/or the rate constants (kobs) for amino acid incorporation on unmodified (CGU, GUG, UUC, UUU) and modified (Cm1GU, Cm2GU, m1GUG, m2GUG, GUm1G, GUm2G, m5UUC, Um5UC, UUm5U) codons. The presence of modifications in the codons were verified by direct infusion ESI-MS or nanoelectrospray ionization (nESI)-MS (Fig. S12–S14, ESI†). Our assays reveal that the extent of amino acid addition is drastically reduced when m1G is present at the first or second position in a codon, but restored to normal levels when m1G is at the third nucleotide (Fig. 6(B) and Fig. S8–S10, ESI†). Codons containing m2G display more modest defects in dipeptide production, only reducing the extent of dipeptide synthesis by 1.9 ± 0.2-fold when m2G is in the third position of a codon (Fig. 6(B) and Fig. S8–S10, ESI†). These findings are consistent with a previous report indicating that insertion of a single m1G and m2G modification into a reporter mRNA codon lowers the overall protein production and translation fidelity in a position and codon dependent manner.89
m1G and m2G should both disrupt Watson–Crick base pairing between mRNAs and tRNAs (Fig. 6(A)) and might be expected to alter amino acid addition in similar ways. However, our results reveal that the insertion of m1G has a much larger consequence than m2G on peptide production. This can be partially rationalized by the fact that m1G would impede canonical Watson–Crick base-pairing by eliminating a central H-bond interaction, while m2G disrupts peripheral interactions (Fig. 6(A)). The N1-methylation of adenosine (m1A), structurally analogous to m1G, abolishes the ability of the ribosome to add amino acids,102 suggesting that the conserved N1 position on purine nucleobases is particularly crucial to tRNA decoding. The hydrogen bonding patterns possible between m2G and other nucleosides would be expected to closely resemble those of another well studied modification, inosine. Inosine also has a moderate (if any) impact on the rates of protein synthesis, though it can promote amino acid mis-incorporation.103,104 The limited consequence of both inosine and m2G on overall peptide production indicates that purine peripheral amines on the Watson–Crick face are less important than the N1 position for ensuring the rapid addition of amino acids by the ribosome.
In contrast to the guanosine modifications that we investigated, we did not observe changes in the overall amount of dipeptide product generated from mRNAs with m5U inserted into Phe codons (Fig. 6(C)). However, we did find that the inclusion of m5U modestly decreased the rate constants for amino acid addition (kobs) on Phe codons in a position dependent manner, similar to what we previously observed for Ψ-modified Phe codons.24 The rate constants (kobs) for Phe incorporation on unmodified and modified codons at the 1st and 2nd position were comparable, (∼5 s−1) (Fig. 6(C) and Fig. S11, ESI†). However, when m5U is in the 3rd position, the kobs value decreases by 2-fold (kobs,UUm5U = 2.5 s−1) (Fig. 6(C) and Fig. S11, ESI†). It is not clear how m5U and other modifications that do not change the Watson–Crick face of nucleobases (e.g., Ψ and 8-oxoG) alter amino acid addition by the ribosome.105 It is possible that such modifications alter nucleobase ring electronics to perturb the strength of the hydrogen bond donors and acceptors involved in mRNA:tRNA base pairing.
While the levels of the m1G, m2G, m22G and m5U modifications we measured in S. cerevisiae are lower than that of the best characterized modifications (m6A and Ψ), our findings suggest that they still have potential to impact biology. Although these data do not report on the ability of the modifications that we uncovered to control gene expression or identify the number of mRNAs that they are in, they do suggest that there will be consequences for translation if (and when) they are encountered by the ribosome. Additionally, given that the levels and distributions of mRNA modifications, both enzymatic and damage products, can change significantly in response to different environmental conditions, we anticipate that the modest levels of m1G, m2G, m22G and m5U modification that we observed in healthy, rapidly growing S. cerevisiae have the potential to increase under stress.16,31,96,106
The three modifications we investigated alter translation differently depending on their location within a codon. Such a context dependence has been observed for every mRNA modification investigated to date.105 Modifications have the capacity to change intra-molecular interactions within an mRNA, and interactions between mRNAs and proteins or rRNA. There is growing evidence that such factors, and not only tRNA anticodon:mRNA codon interactions, have a larger contribution to translation elongation than previously recognized. For example, ribosome stalling induced by the rare 8-oxo-guanosine damage modification has the potential to perturb ribosome homeostasis, and the small pauses in elongation induced by mRNA pseudouridine modifications can impact levels of protein expression in a gene specific manner.79,101 We posit that in addition to influencing the level of protein production, transient ribosome pauses might offer the cells an avenue for enhancing co-translational protein folding or provide sufficient time for RNA binding proteins to interact with a transcript.107,108 Future systematic biochemical and computational studies are needed to uncover the causes of the context dependent effects of mRNA modifications on translation that we and others have observed. This information will be broadly useful as researchers seek to identify which of the thousands of reported modified mRNA codons in the transcriptome are the most likely to have biological consequences when encountered by the ribosome.
Conclusions
Mass spectrometry based approaches are widely used to study protein post-translational modifications, but the application of similar techniques to investigate mRNA post-transcriptional modifications has not been widely adopted. We present mRNA purification, validation, and LC-MS/MS pipelines that enable the sensitive and highly multiplexed analysis of mRNA and ncRNA modifications. These developments enable us to confidently identify four previously unreported mRNA modifications in S. cerevisiae (m1G, m2G, m22G and m5U), demonstrating the utility of applying LC-MS/MS to discover and quantify mRNA modifications. Going forward, integrating quantitative LC-MS/MS approaches, like the one presented here, with sequencing based methodologies for transcriptome-wide modification mapping can facilitate the development of rigorous platforms to study mRNA modifications.69,109–118 In addition to revealing the enzymes that incorporate these modifications, we also demonstrate that the presence of m1G, m2G, and m5U in mRNA as consequences on protein synthesis. However, the impacts of modifications on amino acid addition are not uniform, with the position and identity of each modification resulting in a different outcome on dipeptide production. Our work is consistent with a growing body of evidence suggesting that the ribosome regularly encounters a variety of modified codons in the cell and that, depending on the identity and position of the modification, these interactions can alter the elongation step in protein synthesis.Experimental
S. cerevisiae cell growth and mRNA purification
Wild-type, Δtrm1, Δtrm2, Δtrm10 and Δtrm11 BY4741 S. cerevisiae (Horizon Discovery) were grown in YPD media as previously described.31 Knockout cells lines were grown on media including 200 μg mL−1 Geneticin (G418 Sulfate). Briefly, 100 mL of YPD media was inoculated with a single colony selected from a plate and allowed to grow overnight at 30 °C and 250 rpm. The cells were diluted to an OD600 of 0.1 with 300 mL of YPD and were grown to an OD600 of 0.6–0.8 at 30 °C and 250 rpm. The cell suspension was pelleted at 3220 × g at 4 °C and used for the RNA extraction.
S. cerevisiae cells were lysed as previously described with minor alterations.31,119 The cell pellet was resuspended in 12 mL of lysis buffer (60 mM sodium acetate pH 5.5, 8.4 mM EDTA) and 1.2 mL of 10% SDS. One volume (13.2 mL) of acid phenol:chloroform:isoamyl alcohol (125:24:1; Sigma-Aldrich, USA; P1944) was added and vigorously vortexed. The mixture was incubated in a water bath at 65 °C for five minutes and subsequently vigorously vortexed. The incubation at 65 °C and vortexing was repeated once. Then, the mixture was rapidly chilled in an ethanol/dry ice bath until the lysate became partially frozen. The lysate was then allowed to thaw and centrifuged for 15 min at 15000 × g. The resulting upper layer containing the total RNA was then washed three times with 13.2 mL phenol, and the phenol removed using two chloroform extractions of the same volume. The layer containing RNA was ethanol precipitated in the presence of 1/10th volume of 3 M sodium acetate and 2.5 volumes of ethanol. The precipitate was resuspended in water and ethanol precipitated a second time in the presence of 1/2 volume of 7.5 M ammonium acetate and 2.5 volumes of ethanol. The precipitated total RNA was pelleted at 12000 × g for 30 min, resuspended in 400 μL of water, and treated with 140 U RNase-free DNase I (Roche, 10 U μL−1) in the supplied digestion buffer at 37 °C for 30 min. The DNase I was removed by acid phenol-chloroform extraction. was ethanol precipitated in the presence of 1/10th volume of 3 M sodium acetate and 2.5 volumes of ethanol. The precipitate was resuspended in water and ethanol precipitated a second time in the presence of 1/2 volume of 7.5 M ammonium acetate and 2.5 volumes of ethanol. The precipitated RNA was pelleted and resuspended in water. The resulting total RNA was used for our LC-MS/MS, bioanalyzer, and RNA-seq analyses, and as a starting point for mRNA purification. For a 300 mL culture grown to an OD600 of 0.6, we obtain between 500 μg and 2 mg of total RNA.
mRNA was purified from total RNA through a three-stage purification pipeline. First, small RNA (tRNA and 5S rRNA) was depleted from 240 μg of total RNA using a Zymo RNA Clean and Concentrator-100 kit to purify RNAs >200 nt in length. Two consecutive poly(A) enrichment steps were applied to 125 μg of the resultant small RNA diminished samples using Dynabeads oligo-dT magnetic beads (Invitrogen, USA). The resulting poly(A) RNA was ethanol precipitated using 1/10th volume of 3 M sodium acetate pH 5.2 and 2.5 volumes of ethanol and resuspended in 14 μL of water. Then, we removed the residual 5S, 5.8S, 18S, and 28S rRNA using the commercial riboPOOL rRNA depletion kit (siTOOLs Biotech). The Bioanalyzer RNA 6000 Pico Kit (Agilent) was used to evaluate the purity of the mRNA prior to LC-MS/MS analysis.
qRT-PCR
DNase I treated total RNA and three-stage purified mRNA (200 ng) were reverse transcribed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) using the random hexamer primer. The resulting cDNA was diluted 5000-fold and 1 μL of the resulting mixture was analyzed using the Luminaris Color HiGreen qPCR Master Mix (Thermo Scientific) with gene-specific primers (Table S8, ESI†). RNA level fold changes were calculated using the Ct values for Act1, 18S rRNA, and 25S rRNA using the following equation: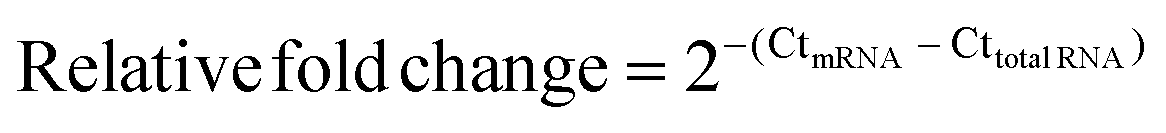
RNA-seq
The WT S. cerevisiae mRNA was analyzed by RNA-seq as previously described with minimal alterations.31 Briefly, 50 ng of DNase I treated total RNA and three-stage purified mRNA from the two biological replicates were fragmented using the TruSeq RNA Library Prep Kit v2 fragmentation buffer (Illumina). First-strand cDNA synthesis was performed using the random hexamer primer, and the second strand was synthesized using the Second Strand Master Mix. The resulting cDNA was purified with AMPure XP beads (Beckman Coulter), the ends were repaired, and the 3′ end was adenylated. Lastly, indexed adapters were ligated to the DNA fragments and amplified using 15 PCR cycles. Paired-end sequencing was performed for the cDNA libraries using 2.5% of an Illumina NovaSeq (S4) 300 cycle sequencing platform flow cell (0.625% of flow cell for each sample). All sequence data are paired-end 150 bp reads.FastQC (v0.11.9)120 was used to evaluate the quality of the raw and trimmed reads. Then, cutadapt (v1.18)121 was used to trim to paired-end 50 bp reads and obtain high quality clean reads with the arguments -u 10 -U 10 -l 50 -m 15 -q 10. Following, Bowtie2 (v2.2.5)122 was used to align the forward strand reads to S. cerevisiae reference genome (R64-1-1) with the default parameters. Following alignment, Rmmquant tool R package (v1.6.0)123 and the gene_biotype feature in the S. cerevisiae GTF file was used to count the number of mapped reads for each transcript and classify the RNA species, respectively. Only reads solely associated with an mRNA transcript are considered mRNA reads, while all others are considered ncRNA reads.
RNA digestions and LC-MS/MS analysis
RNA (200 ng) was hydrolyzed to composite mononucleosides using a two-step enzymatic digestion. The RNA was first hydrolyzed overnight to nucleotide monophosphates using 300 U μg−1 Nuclease P1 (NEB, 100000 U mL−1) at 37 °C in 100 mM ammonium acetate (pH 5.5) and 100 μM ZnSO4. Following, the nucleotides were dephosphorylated using 50 U μg−1 bacterial alkaline phosphatase (BAP, Invitrogen, 150 U μL−1) for 5 h at 37 °C in 100 mM ammonium bicarbonate (pH 8.1) and 100 μM ZnSO4. Prior to each reaction, the enzymes were buffer exchanged into their respective reaction buffers above using a Micro Bio-Spin 6 size exclusion spin column (Biorad) to remove glycerol and other ion suppressing constituents. After the reactions, the samples were lyophilized and resuspended in 9 μL of water and 1 μL of 400 nM 15N4-inosine internal standard.
The resulting ribonucleosides were separated using a Waters Acquity HSS T3 column (1 × 100 mm, 1.8 μm, 100 Å) with a guard column at 100 μL min−1 on a Agilent 1290 Infinity II liquid chromatograph interfaced to a Agilent 6410 triple quadrupole mass spectrometer. Mobile phase A was 0.01% (v/v) formic acid in water and mobile phase B was 0.01% (v/v) formic acid in acetonitrile. The gradient is displayed in Table S9 (ESI†). The autosampler was held at 4 °C, and 5 μL was injected for each sample. The eluting ribonucleosides were quantified using MRM and ionized using electrospray ionization in positive mode at 4 kV (Table S10, ESI†). The electrospray ionization conditions were optimized by infusing 500 nM uridine at 100 μL min−1 at 5% mobile phase B. The gas temperature was 350 °C, the gas flow rate was 10 L min−1, and the nebulizer gas pressure was 25 psi. After each RNA digestion sample, a wash gradient injection was performed to eliminate any column carryover of late eluting nucleosides (e.g., i6A) (Table S9, ESI†).
To compare the sensitivity between the 1 mm and 2 mm I.D. column chromatographies, a 2.1 mM Waters Acquity HSS T3 column (2.1 × 100 mm, 1.8 μm, 100 A) with a guard column was used at 400 μL min−1 using the same gradient and mobile phases described above. The source conditions for the 2.1 mm I.D. column were optimized by infusing 500 nM uridine at 400 μL min−1 at 5% mobile phase B. The gas temperature was 350 °C, the gas flow rate was 10 L min−1, and the nebulizer gas pressure was 55 psi. For both analyses, 5 μL of ribonucleoside standard mixes containing 1.4 μM canonical nucleosides and 72 nM modifications was injected.
To quantify RNA nucleosides calibration curves were created for the four main bases, 45 natural modified nucleosides, and 1 non-natural modified nucleoside using seven calibration points ranging over four orders of magnitude. 15N4-inosine (40 nM) was used as the internal standard for all ribonucleosides. The concentrations of ribonculeoside in the calibration curves standards can be found in Table 11 (ESI†). Suppliers for ribonucleoside standards can be found in Table 12 (ESI†). Automated peak integration was performed using the Agilent MassHunter Workstation Quantitative Analysis Software. All peaks were visually inspected to ensure proper integration. The calibration curves were plotted as the log10(response ratio) versus the log10(concentration (pM)) and the RNA sample nucleoside levels were quantified using the resulting linear regression. The limits of detection were calculated using:
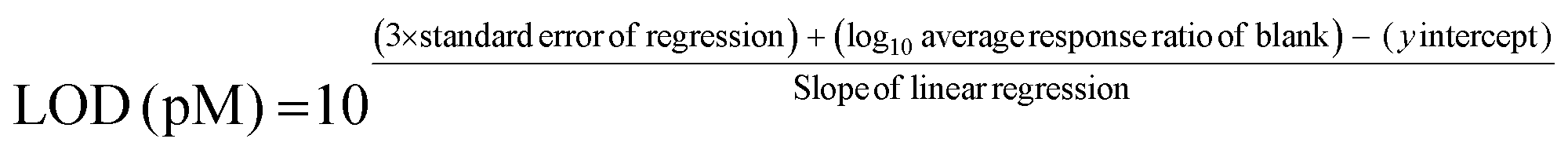
tRNA and mRNA for in vitro translation assay
Unmodified transcripts were prepared using run-off T7 transcription of Ultramer DNA templates that were purchased from Integrated DNA Technologies (Table S13, ESI†). HPLC purified modified mRNA transcripts containing 5-methyluridine, 1-methylguanosine, and N2-methylguanosine were purchased from Dharmacon (Table S14, ESI†). The homogeneity and accurate mass for most of the purchased modified oligonucleotides were confirmed by direct infusion ESI-MS prior to use by Dharmacon (Fig. S11–S13, ESI†). For the remaining purchased oligonucleotides lacking Dharmacon spectra, they were analyzed on a ThermoFisher Q-Exactive UHMR Hybrid Quadrupole-Orbitrap Mass Spectrometer in a negative ionization polarity. Samples were buffer exchanged into 100 mM ammonium acetate (AmOAc) using Micro Bio-Spin P-6 gel columns and directly infused via nanoelectrospray ionization (nESI). nESI was performed using borosilicate needles pulled and coated in-house with a Sutter p-97 Needle Puller and a Quorum SCX7620 mini sputter coater, respectively. The acquired native mass spectra were deconvoluted using UniDec124 in negative polarity (Fig. S11, ESI†).Native tRNA was purified as previously described with minor alterations.125 Bulk E. coli tRNA was either bought in bulk from Sigma-Aldrich or purified from a HB101 E. coli strain containing pUC57-tRNA that we obtained from Prof. Yury Polikanov (University of Illinois, Chicago). Two liters of media containing Terrific Broth (TB) media (TB, 4 mL glycerol per L, 50 mM NH4Cl, 2 mM MgSO4, 0.1 mM FeCl3, 0.05% glucose and 0.2% lactose (if autoinduction media was used)) were inoculated with 1:400 dilution of a saturated overnight culture and incubated with shaking at 37 °C overnight with 400 mg ml−1 of ampicillin. Cells were harvested the next morning by 30 min centrifugation at 5000 rpm and then stored at −80 °C. Extraction of tRNA was done by first resuspending the cell pellet in 200 mL of resuspension buffer (20 mM tris-Cl, 20 mM Mg(OAc)2 pH 7). The resuspended cells were then placed in Teflon centrifuge tubes with ETFE o-rings containing 100 mL acid phenol/chloroform/isoamyl alcohol mixture. The tubes were placed in a 4 °C incubator and left to shake for 1 h. After incubation, the lysate was centrifuged for 60 min at 3220 × g at 4 °C. The supernatant was transferred to another container and the first organic phase was then back-extracted with 100 mL resuspension buffer and centrifuged down for 60 min at 3220 × g at 4 °C. Aqueous solutions were then combined and a 1/10 volume of 3 M sodium acetate pH 5.2 was added and mixed well. Isopropanol was added to 20% and after proper mixing was centrifuged to remove DNA at 13700 × g for 60 min at 4 °C. The supernatant was collected, and isopropanol was added to 60% and was left to precipitate at −20 °C overnight. The precipitated RNA was pelleted at 13700 × g for 60 min at 4 °C and resuspended with approximately 10 mL 200 mM tris-acetate, pH 8.0. The RNA was incubated at 37 °C for at least 30 min to deacylate the tRNA. After incubation 1/10th volume of 3 M sodium acetate pH 5.2 and 2.5 volumes of ethanol was added to precipitate the RNA. Then, the mixture was centrifuged at 16000 × g for 60 min at 4 °C. The pellet was washed with 70% ethanol, resuspended in water, and desalted using an Amicon 10 kDa MWCO centrifugal filter prior to purification (Millipore-Sigma, USA).
Next, the tRNA was isolated using a Cytiva Resource Q column (6 mL) on a AKTA Pure 25M FPLC. Mobile phase A was 50 mM NH4OAc, 300 mM NaCl, and 10 mM MgCl2. Mobile phase B was 50 mM NH4OAc, 800 mM NaCl, 10 mM MgCl2. The resuspended RNA was filtered, loaded on the Resource Q column, and eluted with a linear gradient from 0–100% mobile phase B over 18 column volumes. Fractions were pulled and ethanol precipitated overnight at −20 °C.
The precipitated RNA was resuspended in water and filtered prior to purification on a Waters XBridge BEH C18 OBD Prep wide pore column (10 × 250 mm, 5 μm). Mobile phase A was 20 mM NH4OAc, 10 mM MgCl2, and 400 mM NaCl at pH 5 in 100% water. Mobile phase B was 20 mM NH4OAc, 10 mM MgCl2, and 400 mM NaCl at pH 5 in 60% methanol. The injection volume was 400 μL. A linear gradient of mobile phase B from 0–35% was done over 35 min. After 35 min, the gradient was increased to 100% mobile phase B over 5 min and held at 100% for 10 min, column was then equilibrated for 10 column volumes before next injection with mobile phase A. TCA precipitations were performed on the fractions to identify fractions containing the phenylalanine tRNA as well as measuring the A260 and amino acid acceptor activity.
Formation of E. coli ribosome initiation complexes
Ribosomes were purified from E. coli MRE600 as previously described.24 All constructs for translation factors were provided by Dr Rachel Green's lab unless specifically stated otherwise. The expression and purification of translation initiation and elongation factors were carried out as previously described in detail.24 Initiation complexes (ICs) were formed in 1 × 219-tris buffer (50 mM tris pH 7.5, 70 mM NH4Cl, 30 mM KCl, 7 mM MgCl2, 5 mM β-ME) with 1 mM GTP as previously described.125 70S ribosomes were incubated with 1 μM mRNA (with or without modification), initiation factors (1, 2, and 3) all at 2 μM final, and 2 μM of radiolabeled 35S-fMet-tRNAfMet for 30 min at 37 °C. After incubation, MgCl2 was added to a final concentration of 12 mM. The ribosome mixture was then layered onto 1 mL cold buffer D (20 mM tris-Cl, 1.1 M sucrose, 500 mM NH4Cl, 10 mM MgCl2, 0.5 mM disodium EDTA, pH 7.5) and centrifuged at 69000 rpm for 2 h at 4 °C. After pelleting, the supernatant was discarded into radioactive waste, and the pellet was resuspended in 1 × 219-tris buffer and stored at −80 °C.
In vitro amino acid addition assays
IC complexes were diluted to 140 nM with 1 × 219-tris buffer. Ternary complexes (TCs) were formed by first incubating the EF-Tu pre-loaded with GTP (1 × 219-tris buffer, 10 mM GTP, 60 μM EFTu, 1 μM EFTs) at 37 °C for 10 min. The EF-Tu mixture was incubated with the tRNA mixture (1 × 219-tris buffer, Phe-tRNAPhe (1–10 μM), 1 mM GTP) for another 15 min at 37 °C. After TC formation was complete, equal volumes of IC complexes (70 nM) and ternary complex (1 μM) were mixed either by hand or using a KinTek quench-flow apparatus. Discrete time-points (0–600 seconds) were taken as to obtain observed rate constants on m5U-containing mRNAs. Each time point was quenched with 500 mM KOH (final concentration). Time points were then separated by electrophoretic TLC and visualized using phosphorescence as previously described.24,125 Images were quantified with ImageQuant. The data were fit using (eqn 1):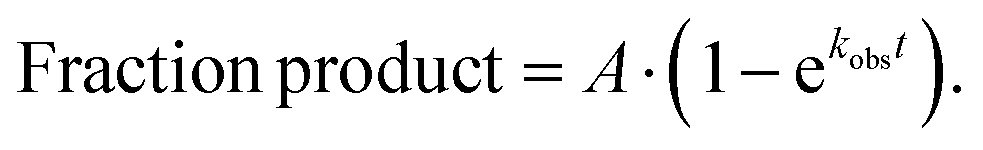 | (1) |
Author contributions
Joshua D. Jones: conceptualization, methodology, investigation, formal analysis, writing – original draft, writing – review & editing, visualization. Monika K. Franco: conceptualization, investigation, formal analysis, writing – original draft, writing – review & editing. Tyler J. Smith: conceptualization, investigation, formal analysis, writing – original draft, writing – review & editing. Laura R. Snyder: investigation, writing – review & editing. Anna G. Anders: investigation, writing – review & editing. Brandon T. Ruotolo: writing – review & editing. Robert T. Kennedy: writing – review & editing. Kristin S. Koutmou: conceptualization, writing – review & editing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We would like to acknowledge Dr B. Shay and Dr M. Sorenson for their thoughtful discussions regarding LC-MS/MS method development and sample preparation. We thank the following funding sources for their support: National Institutes of Health (NIGMS R35 GM128836 to K. S. K.), National Science Foundation (NSF CAREER 2045562 to K. S. K., GFRP to J. D. J., and NSF CHE 1904146 to R. T. K.), and Research Corporation for Science Advancement (Cottrell Scholar Award to K. S. K.).References
- P. J. McCown, A. Ruszkowska, C. N. Kunkler, K. Breger, J. P. Hulewicz, M. C. Wang, N. A. Springer and J. A. Brown, Naturally occurring modified ribonucleosides, Wiley Interdiscip. Rev.: RNA, 2020, 11, e1595 CAS.
- R. J. Ontiveros, J. Stoute and K. F. Liu, The chemical diversity of RNA modifications, Biochem. J., 2019, 476, 1227–1245 CrossRef CAS PubMed.
- P. Boccaletto, M. A. Machnicka, E. Purta, P. Piątkowski, B. Bagiński, T. K. Wirecki, V. de Crécy-Lagard, R. Ross, P. A. Limbach, A. Kotter, M. Helm and J. M. Bujnicki, MODOMICS: a database of RNA modification pathways. 2017 update, Nucleic Acids Res., 2018, 46, D303–D307 CrossRef CAS PubMed.
- N. Jonkhout, J. Tran, M. A. Smith, N. Schonrock, J. S. Mattick and E. M. Novoa, The RNA modification landscape in human disease, RNA, 2017, 23, 1754–1769 CrossRef CAS PubMed.
- X. Zhang, F. Trebak, L. A. C. Souza, J. Shi, T. Zhou, P. G. Kehoe, Q. Chen and Y. Feng Earley, Small RNA modifications in Alzheimer's disease, Neurobiol. Dis., 2020, 145, 105058 CrossRef CAS PubMed.
- M. Pereira, S. Francisco, A. S. Varanda, M. Santos, M. A. S. Santos and A. R. Soares, Impact of tRNA Modifications and tRNA-Modifying Enzymes on Proteostasis and Human Disease, Int. J. Mol. Sci., 2018, 19, 3738 CrossRef PubMed.
- J. Ramos, M. Proven, J. Halvardson, F. Hagelskamp, E. Kuchinskaya, B. Phelan, R. Bell, S. M. Kellner, L. Feuk, A.-C. Thuresson and D. Fu, Identification and rescue of a tRNA wobble inosine deficiency causing intellectual disability disorder, RNA, 2020, 26, 1654–1666 CrossRef CAS PubMed.
- J. T. Lant, M. D. Berg, I. U. Heinemann, C. J. Brandl and P. O’Donoghue, Pathways to disease from natural variations in human cytoplasmic tRNAs, J. Biol. Chem., 2019, 294, 5294–5308 CrossRef CAS PubMed.
- H. Lin, K. Miyauchi, T. Harada, R. Okita, E. Takeshita, H. Komaki, K. Fujioka, H. Yagasaki, Y. Goto, K. Yanaka, S. Nakagawa, Y. Sakaguchi and T. Suzuki, CO2-sensitive tRNA modification associated with human mitochondrial disease, Nat. Commun., 2018, 9, 1875 CrossRef PubMed.
- L. S. Zhang, C. Liu, H. Ma, Q. Dai, H. L. Sun, G. Luo, Z. Zhang, L. Zhang, L. Hu, X. Dong and C. He, Transcriptome-wide Mapping of Internal N7-Methylguanosine Methylome in Mammalian mRNA, Mol. Cell, 2019, 74, 1304–1316 CrossRef CAS PubMed.
- T. M. Carlile, M. F. Rojas-Duran, B. Zinshteyn, H. Shin, K. M. Bartoli and W. V. Gilbert, Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells, Nature, 2014, 515, 143–146 CrossRef CAS PubMed.
- J.-M. Chu, T.-T. Ye, C.-J. Ma, M.-D. Lan, T. Liu, B.-F. Yuan and Y.-Q. Feng, Existence of Internal N7-Methylguanosine Modification in mRNA Determined by Differential Enzyme Treatment Coupled with Mass Spectrometry Analysis, ACS Chem. Biol., 2018, 13, 3243–3250 CrossRef CAS PubMed.
- J. D. Jones, J. Monroe and K. S. Koutmou, A molecular-level perspective on the frequency, distribution, and consequences of messenger RNA modifications, Wiley Interdiscip. Rev.: RNA, 2020, e1586 CAS.
- D. P. Morse and B. L. Bass, Detection of Inosine in Messenger RNA by Inosine-Specific Cleavage, Biochemistry, 1997, 36, 8429–8434 CrossRef CAS PubMed.
- R. Desrosiers, K. Friderici and F. Rottman, Identification of Methylated Nucleosides in Messenger RNA from Novikoff Hepatoma Cells, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3971–3975 CrossRef CAS PubMed.
- S. Schwartz, D. A. Bernstein, M. R. Mumbach, M. Jovanovic, R. H. Herbst, B. X. León-Ricardo, J. M. Engreitz, M. Guttman, R. Satija, E. S. Lander, G. Fink and A. Regev, Transcriptome-wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA, Cell, 2014, 159, 148–162 CrossRef CAS PubMed.
- Y.-J. Feng, X.-J. You, J.-H. Ding, Y.-F. Zhang, B.-F. Yuan and Y.-Q. Feng, Identification of Inosine and 2′-O-Methylinosine Modifications in Yeast Messenger RNA by Liquid Chromatography–Tandem Mass Spectrometry Analysis, Anal. Chem., 2022, 94, 4747–4755 CrossRef CAS PubMed.
- I. A. Roundtree, G.-Z. Luo, Z. Zhang, X. Wang, T. Zhou, Y. Cui, J. Sha, X. Huang, L. Guerrero, P. Xie, E. He, B. Shen and C. He, YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs, eLife, 2017, 6, e31311 CrossRef PubMed.
- X. Yang, Y. Yang, B.-F. Sun, Y.-S. Chen, J.-W. Xu, W.-Y. Lai, A. Li, X. Wang, D. P. Bhattarai, W. Xiao, H.-Y. Sun, Q. Zhu, H.-L. Ma, S. Adhikari, M. Sun, Y.-J. Hao, B. Zhang, C.-M. Huang, N. Huang, G.-B. Jiang, Y.-L. Zhao, H.-L. Wang, Y.-P. Sun and Y.-G. Yang, 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m5C reader, Cell Res., 2017, 27, 606–625 CrossRef CAS PubMed.
- G. Zheng, J. A. Dahl, Y. Niu, P. Fedorcsak, C.-M. Huang, C. J. Li, C. B. Vågbø, Y. Shi, W.-L. Wang, S.-H. Song, Z. Lu, R. P. G. Bosmans, Q. Dai, Y.-J. Hao, X. Yang, W.-M. Zhao, W.-M. Tong, X.-J. Wang, F. Bogdan, K. Furu, Y. Fu, G. Jia, X. Zhao, J. Liu, H. E. Krokan, A. Klungland, Y.-G. Yang and C. He, ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility, Mol. Cell, 2013, 49, 18–29 CrossRef CAS PubMed.
- D. Dominissini and G. Rechavi, N4-acetylation of Cytidine in mRNA by NAT10 Regulates Stability and Translation, Cell, 2018, 175, 1725–1727 CrossRef CAS PubMed.
- X. Wang, Z. Lu, A. Gomez, G. C. Hon, Y. Yue, D. Han, Y. Fu, M. Parisien, Q. Dai, G. Jia, B. Ren, T. Pan and C. He, N6-methyladenosine-dependent regulation of messenger RNA stability, Nature, 2014, 505, 117–120 CrossRef PubMed.
- Y. Wang, Y. Li, J. I. Toth, M. D. Petroski, Z. Zhang and J. C. Zhao, N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells, Nat. Cell Biol., 2014, 16, 191–198 CrossRef CAS PubMed.
- D. E. Eyler, M. K. Franco, Z. Batool, M. Z. Wu, M. L. Dubuke, M. Dobosz-Bartoszek, J. D. Jones, Y. S. Polikanov, B. Roy and K. S. Koutmou, Pseudouridinylation of mRNA coding sequences alters translation, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23068–23074 CrossRef CAS PubMed.
- T. P. Hoernes, N. Clementi, K. Faserl, H. Glasner, K. Breuker, H. Lindner, A. Hüttenhofer and M. D. Erlacher, Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code, Nucleic Acids Res., 2016, 44, 852–862 CrossRef CAS PubMed.
- K. D. Meyer, D. P. Patil, J. Zhou, A. Zinoviev, M. A. Skabkin, O. Elemento, T. V. Pestova, S.-B. Qian and S. R. Jaffrey, 5′ UTR m6A Promotes Cap-Independent Translation, Cell, 2015, 163, 999–1010 CrossRef CAS PubMed.
- J. Zhou, J. Wan, X. Gao, X. Zhang, S. R. Jaffrey and S.-B. Qian, Dynamic m6A mRNA methylation directs translational control of heat shock response, Nature, 2015, 526, 591–594 CrossRef CAS PubMed.
- D. Arango, D. Sturgill, N. Alhusaini, A. A. Dillman, T. J. Sweet, G. Hanson, M. Hosogane, W. R. Sinclair, K. K. Nanan, M. D. Mandler, S. D. Fox, T. T. Zengeya, T. Andresson, J. L. Meier, J. Coller and S. Oberdoerffer, Acetylation of Cytidine in mRNA Promotes Translation Efficiency, Cell, 2018, 175, 1872–1886.e24 CrossRef CAS PubMed.
- X. Lin, G. Chai, Y. Wu, J. Li, F. Chen, J. Liu, G. Luo, J. Tauler, J. Du, S. Lin, C. He and H. Wang, RNA m6A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail, Nat. Commun., 2019, 10, 2065 CrossRef PubMed.
- D. F. De Jesus, Z. Zhang, S. Kahraman, N. K. Brown, M. Chen, J. Hu, M. K. Gupta, C. He and R. N. Kulkarni, m 6 A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes, Nat. Metab., 2019, 1, 765–774 CrossRef CAS PubMed.
- M. Tardu, J. D. Jones, R. T. Kennedy, Q. Lin and K. S. Koutmou, Identification and quantification of modified nucleosides in Saccharomyces cerevisiae mRNAs, ACS Chem. Biol., 2019, 14, 1403–1409 CrossRef CAS PubMed.
- Q.-Y. Cheng, J. Xiong, C.-J. Ma, Y. Dai, J.-H. Ding, F.-L. Liu, B.-F. Yuan and Y.-Q. Feng, Chemical tagging for sensitive determination of uridine modifications in RNA, Chem. Sci., 2020, 11, 1878–1891 RSC.
- L. Xu, X. Liu, N. Sheng, K. S. Oo, J. Liang, Y. H. Chionh, J. Xu, F. Ye, Y.-G. Gao, P. C. Dedon and X.-Y. Fu, Three distinct 3-methylcytidine (m3C) methyltransferases modify tRNA and mRNA in mice and humans, J. Biol. Chem., 2017, 292, 14695–14703 CrossRef CAS PubMed.
- H.-C. Duan, L.-H. Wei, C. Zhang, Y. Wang, L. Chen, Z. Lu, P. R. Chen, C. He and G. Jia, ALKBH10B Is an RNA N6-Methyladenosine Demethylase Affecting Arabidopsis Floral Transition, Plant Cell, 2017, 29, 2995–3011 CrossRef CAS PubMed.
- Y. Zhang, B. R. Fonslow, B. Shan, M.-C. Baek and J. R. Yates, Protein Analysis by Shotgun/Bottom-up Proteomics, Chem. Rev., 2013, 113, 2343–2394 CrossRef CAS PubMed.
- M. Basanta-Sanchez, S. Temple, S. A. Ansari, A. D’Amico and P. F. Agris, Attomole quantification and global profile of RNA modifications: Epitranscriptome of human neural stem cells, Nucleic Acids Res., 2016, 44, e26 CrossRef PubMed.
- C. T. Y. Chan, M. Dyavaiah, M. S. DeMott, K. Taghizadeh, P. C. Dedon and T. J. Begley, A Quantitative Systems Approach Reveals Dynamic Control of tRNA Modifications during Cellular Stress, PLoS Genet., 2010, 6, e1001247 CrossRef CAS PubMed.
- D. Su, C. T. Y. Chan, C. Gu, K. S. Lim, Y. H. Chionh, M. E. McBee, B. S. Russell, I. R. Babu, T. J. Begley and P. C. Dedon, Quantitative analysis of tRNA modifications by HPLC-coupled mass spectrometry, Nat. Protoc., 2014, 9, 828–841 CrossRef CAS PubMed.
- M. Heiss, F. Hagelskamp, V. Marchand, Y. Motorin and S. Kellner, Cell culture NAIL-MS allows insight into human tRNA and rRNA modification dynamics in vivo, Nat. Commun., 2021, 12, 389 CrossRef CAS PubMed.
- V. F. Reichle, S. Kaiser, M. Heiss, F. Hagelskamp, K. Borland and S. Kellner, Surpassing limits of static RNA modification analysis with dynamic NAIL-MS, Methods, 2019, 156, 91–101 CrossRef CAS PubMed.
- K. D. Clark, C. Lee, R. Gillette and J. V. Sweedler, Characterization of Neuronal RNA Modifications during Non-associative Learning in Aplysia Reveals Key Roles for tRNAs in Behavioral Sensitization, ACS Cent. Sci., 2021, 7, 1183–1190 CrossRef CAS PubMed.
- K. D. Clark, S. S. Rubakhin and J. V. Sweedler, Single-Neuron RNA Modification Analysis by Mass Spectrometry: Characterizing RNA Modification Patterns and Dynamics with Single-Cell Resolution, Anal. Chem., 2021, 93, 14537–14544 CrossRef CAS PubMed.
- M.-Y. Chen, Z. Gui, K.-K. Chen, J.-H. Ding, J.-G. He, J. Xiong, J.-L. Li, J. Wang, B.-F. Yuan and Y.-Q. Feng, Adolescent alcohol exposure alters DNA and RNA modifications in peripheral blood by liquid chromatography-tandem mass spectrometry analysis, Chin. Chem. Lett., 2022, 33, 2086–2090 CrossRef CAS.
- M.-Y. Chen, C.-B. Qi, X.-M. Tang, J.-H. Ding, B.-F. Yuan and Y.-Q. Feng, Comprehensive profiling and evaluation of the alteration of RNA modifications in thyroid carcinoma by liquid chromatography-tandem mass spectrometry, Chin. Chem. Lett., 2022, 33, 3772–3776 CrossRef CAS.
- S. Kellner, J. Neumann, D. Rosenkranz, S. Lebedeva, R. F. Ketting, H. Zischler, D. Schneider and M. Helm, Profiling of RNA modifications by multiplexed stable isotope labelling, Chem. Commun., 2014, 50, 3516–3518 RSC.
- S. P. Russell and P. A. Limbach, Evaluating the reproducibility of quantifying modified nucleosides from ribonucleic acids by LC–UV–MS, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 923–924, 74–82 CrossRef CAS PubMed.
- Y. Feng, C.-J. Ma, J.-H. Ding, C.-B. Qi, X.-J. Xu, B.-F. Yuan and Y.-Q. Feng, Chemical labeling – Assisted mass spectrometry analysis for sensitive detection of cytidine dual modifications in RNA of mammals, Anal. Chim. Acta, 2020, 1098, 56–65 CrossRef CAS PubMed.
- Y. Dai, C.-B. Qi, Y. Feng, Q.-Y. Cheng, F.-L. Liu, M.-Y. Cheng, B.-F. Yuan and Y.-Q. Feng, Sensitive and Simultaneous Determination of Uridine Thiolation and Hydroxylation Modifications in Eukaryotic RNA by Derivatization Coupled with Mass Spectrometry Analysis, Anal. Chem., 2021, 93, 6938–6946 CrossRef CAS PubMed.
- Y. Xie, K. A. Janssen, A. Scacchetti, E. G. Porter, Z. Lin, R. Bonasio and B. A. Garcia, Permethylation of Ribonucleosides Provides Enhanced Mass Spectrometry Quantification of Post-Transcriptional Modifications, Anal. Chem., 2022, 94, 7246–7254 CrossRef CAS PubMed.
- P. Song, O. S. Mabrouk, N. D. Hershey and R. T. Kennedy, In Vivo Neurochemical Monitoring Using Benzoyl Chloride Derivatization and Liquid Chromatography–Mass Spectrometry, Anal. Chem., 2012, 84, 412–419 CrossRef CAS PubMed.
- J.-M. T. Wong, P. A. Malec, O. S. Mabrouk, J. Ro, M. Dus and R. T. Kennedy, Benzoyl chloride derivatization with liquid chromatography–mass spectrometry for targeted metabolomics of neurochemicals in biological samples, J. Chromatogr. A, 2016, 1446, 78–90 CrossRef CAS PubMed.
- S. R. Wilson, T. Vehus, H. S. Berg and E. Lundanes, Nano-LC in proteomics: recent advances and approaches, Bioanalysis, 2015, 7, 1799–1815 CrossRef CAS PubMed.
- L. P. Sarin, S. D. Kienast, J. Leufken, R. L. Ross, A. Dziergowska, K. Debiec, E. Sochacka, P. A. Limbach, C. Fufezan, H. C. A. Drexler and S. A. Leidel, Nano LC-MS using capillary columns enables accurate quantification of modified ribonucleosides at low femtomol levels, RNA, 2018, 24, 1403–1417 CrossRef CAS PubMed.
- L. Fu, N. J. Amato, P. Wang, S. J. McGowan, L. J. Niedernhofer and Y. Wang, Simultaneous Quantification of Methylated Cytidine and Adenosine in Cellular and Tissue RNA by Nano-Flow Liquid Chromatography–Tandem Mass Spectrometry Coupled with the Stable Isotope-Dilution Method, Anal. Chem., 2015, 87, 7653–7659 CrossRef CAS PubMed.
- D. Dominissini, S. Nachtergaele, S. Moshitch-Moshkovitz, E. Peer, N. Kol, M. S. Ben-Haim, Q. Dai, A. Di Segni, M. Salmon-Divon, W. C. Clark, G. Zheng, T. Pan, O. Solomon, E. Eyal, V. Hershkovitz, D. Han, L. C. Doré, N. Amariglio, G. Rechavi and C. He, The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA, Nature, 2016, 530, 441–446 CrossRef CAS PubMed.
- D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek and G. Rechavi, Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq, Nature, 2012, 485, 201–206 CrossRef CAS PubMed.
- X. Li, P. Zhu, S. Ma, J. Song, J. Bai, F. Sun and C. Yi, Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome, Nat. Chem. Biol., 2015, 11, 592–597 CrossRef CAS PubMed.
- C. Legrand, F. Tuorto, M. Hartmann, R. Liebers, D. Jacob, M. Helm and F. Lyko, Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs, Genome Res., 2017, 27, 1589–1596 CrossRef CAS PubMed.
- M.-Y. Cheng, W.-B. Tao, B.-F. Yuan and Y.-Q. Feng, Methods for isolation of messenger RNA from biological samples, Anal. Methods, 2021, 13, 289–298 RSC.
- A. Behrens, G. Rodschinka and D. D. Nedialkova, High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq, Mol. Cell, 2021, 81, 1802–1815.e7 CrossRef CAS PubMed.
- B. Chen, B.-F. Yuan and Y.-Q. Feng, Analytical Methods for Deciphering RNA Modifications, Anal. Chem., 2019, 91, 743–756 CrossRef CAS PubMed.
- S. M. Huber, P. van Delft, L. Mendil, M. Bachman, K. Smollett, F. Werner, E. A. Miska and S. Balasubramanian, Formation and Abundance of 5-Hydroxymethylcytosine in RNA, ChemBioChem, 2015, 16, 752–755 CrossRef CAS PubMed.
- S. Laxman, B. M. Sutter, X. Wu, S. Kumar, X. Guo, D. C. Trudgian, H. Mirzaei and B. P. Tu, Sulfur Amino Acids Regulate Translational Capacity and Metabolic Homeostasis through Modulation of tRNA Thiolation, Cell, 2013, 154, 416–429 CrossRef CAS PubMed.
- C. Chen, B. Huang, M. Eliasson, P. Rydén and A. S. Byström, Elongator Complex Influences Telomeric Gene Silencing and DNA Damage Response by Its Role in Wobble Uridine tRNA Modification, PLoS Genet., 2011, 7, e1002258 CrossRef CAS PubMed.
- N. Shigi, Biosynthesis and functions of sulfur modifications in tRNA, Front. Genet., 2014, 5, 67 Search PubMed.
- J. Mouaikel, C. Verheggen, E. Bertrand, J. Tazi and R. Bordonné, Hypermethylation of the Cap Structure of Both Yeast snRNAs and snoRNAs Requires a Conserved Methyltransferase that Is Localized to the Nucleolus, Mol. Cell, 2002, 9, 891–901 CrossRef CAS PubMed.
- J. Mouaikel, J. M. Bujnicki, J. Tazi and R. Bordonné, Sequence–structure–function relationships of Tgs1, the yeast snRNA/snoRNA cap hypermethylase, Nucleic Acids Res., 2003, 31, 4899–4909 CrossRef CAS PubMed.
- S. Hausmann and S. Shuman, Specificity and Mechanism of RNA Cap Guanine-N2 Methyltransferase (Tgs1), J. Biol. Chem., 2005, 280, 4021–4024 CrossRef CAS PubMed.
- Y. Wang, Z. Chen, X. Zhang, X. Weng, J. Deng, W. Yang, F. Wu, S. Han, C. Xia, Y. Zhou, Y. Chen and X. Zhou, Single-Base Resolution Mapping Reveals Distinct 5-Formylcytidine in Saccharomyces cerevisiae mRNAs, ACS Chem. Biol., 2021, 17, 77–84 CrossRef PubMed.
- W. Dai, A. Li, N. J. Yu, T. Nguyen, R. W. Leach, M. Wühr and R. E. Kleiner, Activity-based RNA-modifying enzyme probing reveals DUS3L-mediated dihydrouridylation, Nat. Chem. Biol., 2021, 17, 1178–1187 CrossRef CAS PubMed.
- J.-M. Carter, W. Emmett, I. R. Mozos, A. Kotter, M. Helm, J. Ule and S. Hussain, FICC-Seq: a method for enzyme-specified profiling of methyl-5-uridine in cellular RNA, Nucleic Acids Res., 2019, 47, e113 CrossRef CAS PubMed.
- D. J. Ashworth, W. M. Baird, C.-J. Chang, J. D. Ciupek, K. L. Busch and R. G. Cooks, Chemical modification of nucleic acids. Methylation of calf thymus DNA investigated by mass spectrometry and liquid chromatography, Biomed. Mass Spectrom., 1985, 12, 309–318 CrossRef CAS PubMed.
- C.-J. Chang and C.-G. Lee, Chemical modification of ribonucleic acid. A direct study by carbon-13 nuclear magnetic resonance spectroscopy, Biochemistry, 1981, 20, 2657–2661 CrossRef CAS PubMed.
- C. J. Chang, J. D. Gomes and S. R. Byrn, Chemical modification of deoxyribonucleic acids: a direct study by carbon-13 nuclear magnetic resonance spectroscopy, J. Org. Chem., 1983, 48, 5151–5160 CrossRef CAS.
- J. C. Delaney and J. M. Essigmann, Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14051–14056 CrossRef CAS PubMed.
- P. Ø. Falnes, Repair of 3-methylthymine and 1-methylguanine lesions by bacterial and human AlkB proteins, Nucleic Acids Res., 2004, 32, 6260–6267 CrossRef CAS PubMed.
- P. J. Holland and T. Hollis, Structural and Mutational Analysis of Escherichia coli AlkB Provides Insight into Substrate Specificity and DNA Damage Searching, PLoS One, 2010, 5, e8680 CrossRef PubMed.
- J. O. Kang, Methylated Purine Bases in Hepatic and Colonic RNA of Rats Treated with 1,2-Dimethylhydrazine, Biochem. Med. Metab. Biol., 1994, 53, 52–57 CrossRef CAS PubMed.
- L. L. Yan and H. S. Zaher, How do cells cope with RNA damage and its consequences?, J. Biol. Chem., 2019, 294, 15158–15171 CrossRef CAS PubMed.
- G. A. Khoury, R. C. Baliban and C. A. Floudas, Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database, Sci. Rep., 2011, 1, 90 CrossRef CAS PubMed.
- E. Bieberich, in Glycobiology of the Nervous System, ed. R. K. Yu and C.-L. Schengrund, Springer, New York, NY, 2014, pp. 47–70 Search PubMed.
- H. H. Wandall, M. A. I. Nielsen, S. King-Smith, N. de Haan and I. Bagdonaite, Global functions of O-glycosylation: promises and challenges in O-glycobiology, FEBS J., 2021, 288, 7183–7212 CrossRef CAS PubMed.
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Glycosylation in health and disease, Nat. Rev. Nephrol., 2019, 15, 346–366 CrossRef PubMed.
- J. Choi, K.-W. Ieong, H. Demirci, J. Chen, A. Petrov, A. Prabhakar, S. E. O’Leary, D. Dominissini, G. Rechavi, S. M. Soltis, M. Ehrenberg and J. D. Puglisi, N(6)-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics, Nat. Struct. Mol. Biol., 2016, 23, 110–115 CrossRef CAS PubMed.
- J. Choi, G. Indrisiunaite, H. DeMirci, K.-W. Ieong, J. Wang, A. Petrov, A. Prabhakar, G. Rechavi, D. Dominissini, C. He, M. Ehrenberg and J. D. Puglisi, 2′- O -methylation in mRNA disrupts tRNA decoding during translation elongation, Nat. Struct. Mol. Biol., 2018, 25, 208–216 CrossRef CAS PubMed.
- T. P. Hoernes, N. Clementi, M. A. Juen, X. Shi, K. Faserl, J. Willi, C. Gasser, C. Kreutz, S. Joseph, H. Lindner, A. Hüttenhofer and M. D. Erlacher, Atomic mutagenesis of stop codon nucleotides reveals the chemical prerequisites for release factor-mediated peptide release, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E382–E389 CrossRef CAS PubMed.
- T. P. Hoernes, D. Heimdörfer, D. Köstner, K. Faserl, F. Nußbaumer, R. Plangger, C. Kreutz, H. Lindner and M. D. Erlacher, Eukaryotic Translation Elongation is Modulated by Single Natural Nucleotide Derivatives in the Coding Sequences of mRNAs, Genes, 2019, 10, 84 CrossRef CAS PubMed.
- B. H. Hudson and H. S. Zaher, O6-Methylguanosine leads to position-dependent effects on ribosome speed and fidelity, RNA, 2015, 21, 1648–1659 CrossRef PubMed.
- C. You, X. Dai and Y. Wang, Position-dependent effects of regioisomeric methylated adenine and guanine ribonucleosides on translation, Nucleic Acids Res., 2017, 45, 9059–9067 CrossRef CAS PubMed.
- Y. Fan, C. R. Evans, K. W. Barber, K. Banerjee, K. J. Weiss, W. Margolin, O. A. Igoshin, J. Rinehart and J. Ling, Heterogeneity of Stop Codon Readthrough in Single Bacterial Cells and Implications for Population Fitness, Mol. Cell, 2017, 67, 826–836.e5 CrossRef CAS PubMed.
- Y. Fan, J. Wu, M. H. Ung, N. De Lay, C. Cheng and J. Ling, Protein mistranslation protects bacteria against oxidative stress, Nucleic Acids Res., 2015, 43, 1740–1748 CrossRef CAS PubMed.
- T. Pan, Modifications and functional genomics of human transfer RNA, Cell Res., 2018, 28, 395–404 CrossRef CAS PubMed.
- E. M. Phizicky and A. K. Hopper, tRNA processing, modification, and subcellular dynamics: past, present, and future, RNA, 2015, 21, 483–485 CrossRef CAS PubMed.
- N. Ranjan and M. V. Rodnina, Thio-Modification of tRNA at the Wobble Position as Regulator of the Kinetics of Decoding and Translocation on the Ribosome, J. Am. Chem. Soc., 2017, 139, 5857–5864 CrossRef CAS PubMed.
- M. H. Schwartz and T. Pan, Temperature dependent mistranslation in a hyperthermophile adapts proteins to lower temperatures, Nucleic Acids Res., 2016, 44, 294–303 CrossRef CAS PubMed.
- Z. Shen, W. Wu and S. L. Hazen, Activated Leukocytes Oxidatively Damage DNA, RNA, and the Nucleotide Pool through Halide-Dependent Formation of Hydroxyl Radical, Biochemistry, 2000, 39, 5474–5482 CrossRef CAS PubMed.
- T. E. Dever, J. D. Dinman and R. Green, Translation Elongation and Recoding in Eukaryotes, Cold Spring Harbor Perspect. Biol., 2018, 10, a032649 CrossRef PubMed.
- E. N. Thomas, C. L. Simms, H. E. Keedy and H. S. Zaher, Insights into the base-pairing preferences of 8-oxoguanosine on the ribosome, Nucleic Acids Res., 2019, 47, 9857–9870 CrossRef CAS PubMed.
- B. Slobodin, R. Han, V. Calderone, J. A. F. O. Vrielink, F. Loayza-Puch, R. Elkon and R. Agami, Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-adenosine Methylation, Cell, 2017, 169, 326–337.e12 CrossRef CAS PubMed.
- L. L. Yan, C. L. Simms, F. McLoughlin, R. D. Vierstra and H. S. Zaher, Oxidation and alkylation stresses activate ribosome-quality control, Nat. Commun., 2019, 10, 5611 CrossRef CAS PubMed.
- O. Levi and Y. S. Arava, Pseudouridine-mediated translation control of mRNA by methionine aminoacyl tRNA synthetase, Nucleic Acids Res., 2021, 49, 432–443 CrossRef CAS PubMed.
- E. N. Thomas, K. Q. Kim, E. P. McHugh, T. Marcinkiewicz and H. S. Zaher, Alkylative damage of mRNA leads to ribosome stalling and rescue by trans translation in bacteria, eLife, 2020, 9, e61984 CrossRef CAS PubMed.
- T. P. Hoernes, K. Faserl, M. A. Juen, J. Kremser, C. Gasser, E. Fuchs, X. Shi, A. Siewert, H. Lindner, C. Kreutz, R. Micura, S. Joseph, C. Höbartner, E. Westhof, A. Hüttenhofer and M. D. Erlacher, Translation of non-standard codon nucleotides reveals minimal requirements for codon-anticodon interactions, Nat. Commun., 2018, 9, 4865 CrossRef PubMed.
- K. Licht, M. Hartl, F. Amman, D. Anrather, M. P. Janisiw and M. F. Jantsch, Inosine induces context-dependent recoding and translational stalling, Nucleic Acids Res., 2019, 47, 3–14 CrossRef CAS PubMed.
- M. K. Franco and K. S. Koutmou, Chemical modifications to mRNA nucleobases impact translation elongation and termination, Biophys. Chem., 2022, 285, 106780 CrossRef CAS PubMed.
- M. K. Purchal, D. E. Eyler, M. Tardu, M. K. Franco, M. M. Korn, T. Khan, R. McNassor, R. Giles, K. Lev, H. Sharma, J. Monroe, L. Mallik, M. Koutmos and K. S. Koutmou, Pseudouridine synthase 7 is an opportunistic enzyme that binds and modifies substrates with diverse sequences and structures, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2109708119 CrossRef PubMed.
- R. Rauscher and Z. Ignatova, Timing during translation matters: synonymous mutations in human pathologies influence protein folding and function, Biochem. Soc. Trans., 2018, 46, 937–944 CrossRef CAS PubMed.
- A. Re, T. Joshi, E. Kulberkyte, Q. Morris and C. T. Workman, in RNA Sequence, Structure, and Function: Computational and Bioinformatic Methods, ed. J. Gorodkin and W. L. Ruzzo, Humana Press, Totowa, NJ, 2014, pp. 491–521 Search PubMed.
- W. V. Gilbert, T. A. Bell and C. Schaening, Messenger RNA modifications: Form, distribution, and function, Science, 2016, 352, 1408–1412 CrossRef CAS PubMed.
- M. Helm and Y. Motorin, Detecting RNA modifications in the epitranscriptome: predict and validate, Nat. Rev. Genet., 2017, 18, 275–291 CrossRef CAS PubMed.
- Y. Motorin and M. Helm, Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies, Genes, 2019, 10, 35 CrossRef PubMed.
- S. Zaccara, R. J. Ries and S. R. Jaffrey, Reading, writing and erasing mRNA methylation, Nat. Rev. Mol. Cell Biol., 2019, 20, 608–624 CrossRef CAS PubMed.
- S. Schwartz and Y. Motorin, Next-generation sequencing technologies for detection of modified nucleotides in RNAs, RNA Biol., 2016, 14, 1124–1137 CrossRef PubMed.
- S. Thalalla Gamage, A. Sas-Chen, S. Schwartz and J. L. Meier, Quantitative nucleotide resolution profiling of RNA cytidine acetylation by ac4C-seq, Nat. Protoc., 2021, 16, 2286–2307 CrossRef CAS PubMed.
- A. V. Grozhik and S. R. Jaffrey, Distinguishing RNA modifications from noise in epitranscriptome maps, Nat. Chem. Biol., 2018, 14, 215–225 CrossRef CAS PubMed.
- J. Cui, Q. Liu, E. Sendinc, Y. Shi and R. I. Gregory, Nucleotide resolution profiling of m3C RNA modification by HAC-seq, Nucleic Acids Res., 2021, 49, e27 CrossRef CAS PubMed.
- V. Khoddami, A. Yerra, T. L. Mosbruger, A. M. Fleming, C. J. Burrows and B. R. Cairns, Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6784–6789 CrossRef CAS PubMed.
- M. Jain, R. Abu-Shumays, H. E. Olsen and M. Akeson, Advances in nanopore direct RNA sequencing, Nat. Methods, 2022, 19, 1160–1164 CrossRef CAS PubMed.
- M. E. Schmitt, T. A. Brown and B. L. Trumpower, A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae, Nucleic Acids Res., 1990, 18, 3091–3092 CrossRef CAS PubMed.
- S. Andrews, FastQC: a quality control tool for high throughput sequence data, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, (accessed 27 May 2021).
- M. Martin, Cutadapt removes adapter sequences from high-throughput sequencing reads, EMBnet. J., 2011, 17, 10–12 CrossRef.
- B. Langmead and S. L. Salzberg, Fast gapped-read alignment with Bowtie 2, Nat. Methods, 2012, 9, 357–359 CrossRef CAS PubMed.
- M. Zytnicki, mmquant: how to count multi-mapping reads?, BMC Bioinf., 2017, 18, 411 CrossRef PubMed.
- M. T. Marty, A. J. Baldwin, E. G. Marklund, G. K. A. Hochberg, J. L. P. Benesch and C. V. Robinson, Bayesian Deconvolution of Mass and Ion Mobility Spectra: From Binary Interactions to Polydisperse Ensembles, Anal. Chem., 2015, 87, 4370–4376 CrossRef CAS PubMed.
- J. G. Monroe, T. J. Smith and K. S. Koutmou, in Methods in Enzymology, ed. J. E. Jackman, Academic Press, 2021, vol. 658, pp. 379–406 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00229a |
| This journal is © The Royal Society of Chemistry 2023 |