
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccessing three-branched high-affinity cereblon ligands for molecular glue and protein degrader design†
Robert
Kuchta
a,
Christopher
Heim‡
 bc,
Alexander
Herrmann‡
b,
Samuel
Maiwald‡
b,
Yuen Lam Dora
Ng
d,
Izidor
Sosič
e,
Tim
Keuler
a,
Jan
Krönke
d,
Michael
Gütschow
a,
Marcus D.
Hartmann
*bc and
Christian
Steinebach
*a
bc,
Alexander
Herrmann‡
b,
Samuel
Maiwald‡
b,
Yuen Lam Dora
Ng
d,
Izidor
Sosič
e,
Tim
Keuler
a,
Jan
Krönke
d,
Michael
Gütschow
a,
Marcus D.
Hartmann
*bc and
Christian
Steinebach
*a
aPharmaceutical Institute, Pharmaceutical & Medicinal Chemistry, University of Bonn, An der Immenburg 4, Bonn D-53121, Germany. E-mail: c.steinebach@uni-bonn.de
bMax Planck Institute for Biology Tübingen, Tübingen D-72076, Germany. E-mail: marcus.hartmann@tuebingen.mpg.de
cInterfaculty Institute of Biochemistry, University of Tübingen, Tübingen, 72076, Germany
dCharité, Department of Internal Medicine with Focus on Hematology, Oncology and Tumor Immunology, Berlin D-12203, Germany
eFaculty of Pharmacy, University of Ljubljana, Ljubljana SI-1000, Slovenia
First published on 3rd January 2023
Abstract
The Petasis borono-Mannich reaction was employed for an alternative entry towards three-branched cereblon ligands. Such compounds are capabable of making multiple interactions with the protein surface and possess a suitable linker exit vector. The high-affinity ligands were used to assemble prototypic new molecular glues and proteolysis targeting chimeras (PROTACs) targeting BRD4 for degradation. Our results highlight the importance of multicomponent reactions (MCRs) in drug discovery and add new insights into the rapidly growing field of protein degraders.
MCRs represent an attractive approach to synthesize biologically relevant molecules.1 Prominent examples of such complexity-generating transformations are the Biginelli, Strecker, Ugi, and Petasis reaction.2 Due to the rapid and catalyst-free access to structural diversity, these reactions were successfully applied in drug discovery campaigns.3 Among the reaction types mentioned above, the Strecker synthesis is a powerful tool to prepare biogenic and non-natural α-aminoacids.4 In the classical Strecker protocol,5 an amine (or ammonia) attacks the carbonyl carbon of an aldehyde to form an iminium ion, followed by the nucleophilic attack of cyanide, leading to a highly functionalized α-aminonitrile. Subsequent hydrolysis provides the desired amino acid. Although this reaction type generates tailor-made amino acids, two common limitations are safety concerns regarding the use of highly toxic cyanides and the need to hydrolyze the nitriles, which might limit the scope of the reaction.4 An alternative approach towards α-amino acids is the Petasis borono-Mannich reaction, which utilizes amines, aldehydes, and boronic acids as the nucleophile component.6 This boron-based MCR combines a diverse substrate scope with other merits such as mild reaction conditions and wide availability of starting materials.1
Our previous work has been devoted to mechanistic investigations of anti-myeloma agents such as the immunomodulatory drugs (IMiDs) pomalidomide and lenalidomide (Fig. 1),7–9 as well as medicinal chemistry in the field of targeted protein degradation with particular focus on PROTACs that hijack the E3 ligase cereblon (CRBN).10–15 Recently, a new class of CRBN ligands has been coined, i.e. CRBN E3 ligase modulators (CELMoDs), which are typically based on aminoglutarimide as the core binding moiety,17–21 and to which examples such as iberdomide (CC-220, Fig. 1) and mezigdomide (CC-92480) belong.16,22,23 The structural elongation in iberdomide compared to lenalidomide enabled additional interactions with CRBN or ligase substrates leading to a more pronounced degradation of the CRBN neosubstrates, namely the transcription factors Ikaros (IKZF1) and Aiolos (IKZF3). Recent medicinal chemistry projects also generated libraries of molecular glues with extended side chains, which induced pronounced degradation of G1 to S phase transition 1 protein (GSPT1)24–26 or Helios (IKZF2).27 The work reported in this manuscript aims at developing three-branched high-affinity CRBN ligands via MCRs. The expected high affinity of these ligands might be accompanied by increased cellular potency of CELMoDs or PROTACs that can be derived from such entities.
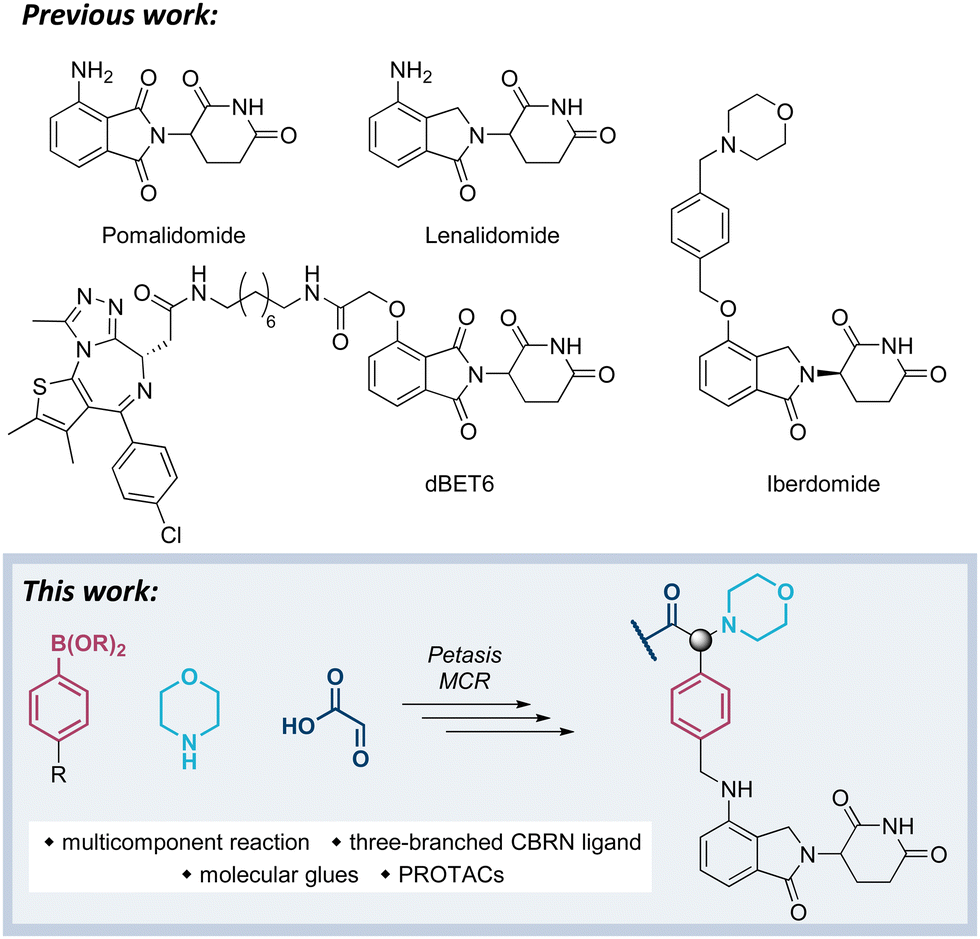 | ||
| Fig. 1 Compilation of IMiDs, CELMoDs, and PROTACs. | ||
Visual inspection of the co-crystal structure of CC-220 in complex with CRBN (Fig. 2A) revealed further interactions of the morpholine ring with the protein surface and suggested the benzylic carbon between the morpholine nitrogen and the aromatic side chain of CC-220 as a convenient anchor point for a third branch as an exit vector. To demonstrate the importance of making such additional interactions with the surface of CRBN, we designed a series of related aminoglutarimides whose structural expansion ranged from the acetyl derivative 1 to the CC-220 analogue 5 (Fig. 2B and Scheme S1, ESI†). Increased molecular size, as well as higher polarity, accompany the fragment growth. Sets of experimentally determined (Table 1) and calculated physico-chemical properties (Table S1, ESI†) are provided. The former set comprises distribution coefficients at pH 7.4 (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D) to rate the lipophilicity, the permeability surrogate CHI determined by immobilized artificial membrane (IAM) chromatography, and human serum albumin (HSA) binding values. Predicted ADME properties (Table S1, ESI†) encompass topological polar surface area (TPSA) values, solubility as well as permeability and hERG binding descriptors. Concerns regarding the latter should be validated experimentally.
D) to rate the lipophilicity, the permeability surrogate CHI determined by immobilized artificial membrane (IAM) chromatography, and human serum albumin (HSA) binding values. Predicted ADME properties (Table S1, ESI†) encompass topological polar surface area (TPSA) values, solubility as well as permeability and hERG binding descriptors. Concerns regarding the latter should be validated experimentally.
 | ||
| Fig. 2 (A) The crystal structure of CRBN in complex with CC-220 (PDB: 5V3O)16 reveals a possible exit vector for linker attachment. (B) Strategy towards three-branched CRBN recruiters: stepwise increase of the ligand size and incorporation of an exit vector by multicomponent reactions. | ||
| Cmpd | logD7.4a |
CHIIAMb | %HSA bindingc | IC50d (μM) | K i (μM) | % IKZF3e deg |
|---|---|---|---|---|---|---|
| a Distribution coefficients at pH 7.4 were estimated by an HPLC-based method. b Chromatographic hydrophobicity index values referring to IAM chromatography (CHIIAM values), an estimate for drug-membrane interactions and permeability.28 c Protein binding values were estimated by an HPLC-based method. d Affinity values determined in a competitive MST assay as described in the Method sections (see ESI). e Percentage of degraded IKZF3 after 24 h treatment of 106 OPM-2 cells per mL with 10 μM of each compound. f Not determined. g CRBN binding data taken from ref. 29. | ||||||
| 1 | n.d.f | n.d. | n.d. | 481 ± 340 | 247 ± 177 | 20 |
| 2 | –1.7 | –1.1 | 4.0 | 107 ± 85 | 52 ± 44 | 4 |
| 3 | –0.4 | 4.4 | 12 | 18.9 ± 1.5 | 6.40 ± 0.8 | 84 |
| 4 | 2.2 | 28.5 | 89 | 7.9 ± 1.7 | 0.7 ± 0.9 | 60 |
| 5 | 1.6 | 22.5 | 86 | 8.6 ± 1.1 | 1.1 ± 0.6 | 93 |
| 6 | 2.0 | 25.3 | 90 | 10.5 ± 1.6 | 2.0 ± 0.8 | 57 |
To determine relative binding affinities of these IMiD analogues, we used a custom thermophoresis-based competitive CRBN binding assay. Therein, the dose-dependent out-competition of a fluorescent cereblon ligand (the reporter BODIPY-uracil) is quantified, and affinity values (IC50 and Ki) are derived from binding curves (Table 1 and Fig. S1, ESI†).29 In this assay, acylated derivatives 1 and 2 exhibited significantly lower affinity than lenalidomide (3). In comparison, compounds 4 and 5, capable of forming additional interactions with CRBN via the aromatic side chain, showed the highest affinity. With an anilinic nitrogen present in 2–5, these compounds contain a possible degron needed to induce degradation of neosubstrates such as IKZF3. Their impact on causing the proteasomal destruction of IKZF3 at a concentration of 10 μM in OPM2 cells is reported in Table 1. As expected, most compounds showed significant degradation of the CRBN neosubstrate IKZF3 (Fig. S2, ESI†).
The first attempt to add a third branch to 5 was via an MCR yielding the racemic α-aminonitrile 6 (Scheme S1, ESI†). Its synthesis was achieved by reacting semi-protected terephthalaldehyde, morpholine, and potassium cyanide under typical Strecker conditions. Subsequently, this building block was coupled to lenalidomide via reductive amination. Importantly, robust CRBN affinity was maintained for this highly functionalized derivative 6 (Table 1). However, hydrolysis of the nitrile failed under basic and acidic conditions due to the decomposition of the three-branched moiety. Despite good CRBN affinity and potent activity on neosubstrates of 6, this structure lacked an opportunity to explore further structure-activity relationships (SARs) of three-branched CRBN ligands. By employing a linear synthetic strategy, we tried to attach a Boc-methylamino functionality at the outer phenylene carbon, which might be amenable for derivatization (Scheme S2, ESI†). However, this sequence was not only disadvantageous due to its complexity but also failed at later stages. Given the synthetic difficulties that we encountered, we considered the Petasis MCR to be highly suitable for synthesizing the desired three-headed tail. In a more recent modification of the borono-Mannich reaction, solvents of increased acidity were tested.30 The use of hexafluoroisopropanol (HFIP) significantly accelerated reaction conversions and extended the substrate scope of the Petasis reaction. We employed these conditions to react glycolic acid with morpholine and 4-(bromomethyl)benzeneboronic acid in HFIP at room temperature (Scheme 1). The in situ generated amino acid was esterified by adding EDC and a catalytic amount of 4-dimethylaminopyridine (DMAP) to give building block 7. Regioselective alkylation of lenalidomide at the anilinic nitrogen31 provided the activated ester 8, which represented an excellent starting point for further SAR investigations. For instance, 8 was reacted with excess n-propylamine under very mild conditions to give amide derivative 9 in good yield after a 4 h reaction time. Compound 9 exhibited the highest affinity measured in our array of compounds, and greater than the affinity we previously obtained for iberdomide,29 despite being synthesized as a racemic mixture via this sequence. Among all derivatives tested, 9 also showed the highest impact on cell viability decrease in multiple myeloma cell lines (Fig. 3) and other malignant hematopoietic cell lines tested (Fig. S3, ESI†). Global proteome analysis of the effects of the CRBN-directed molecular glue 9 was performed using diaPASEF-based mass spectrometry32 in MM.1S cells upon treatment with the compound at 0.1 mM for three hours. Of the total of 7170 unique proteins identified, IKZF1 and IKZF3 appeared as the most significantly downregulated proteins (Fig. 4), thus further explaining the impact on cell viability of MM.1S cells.
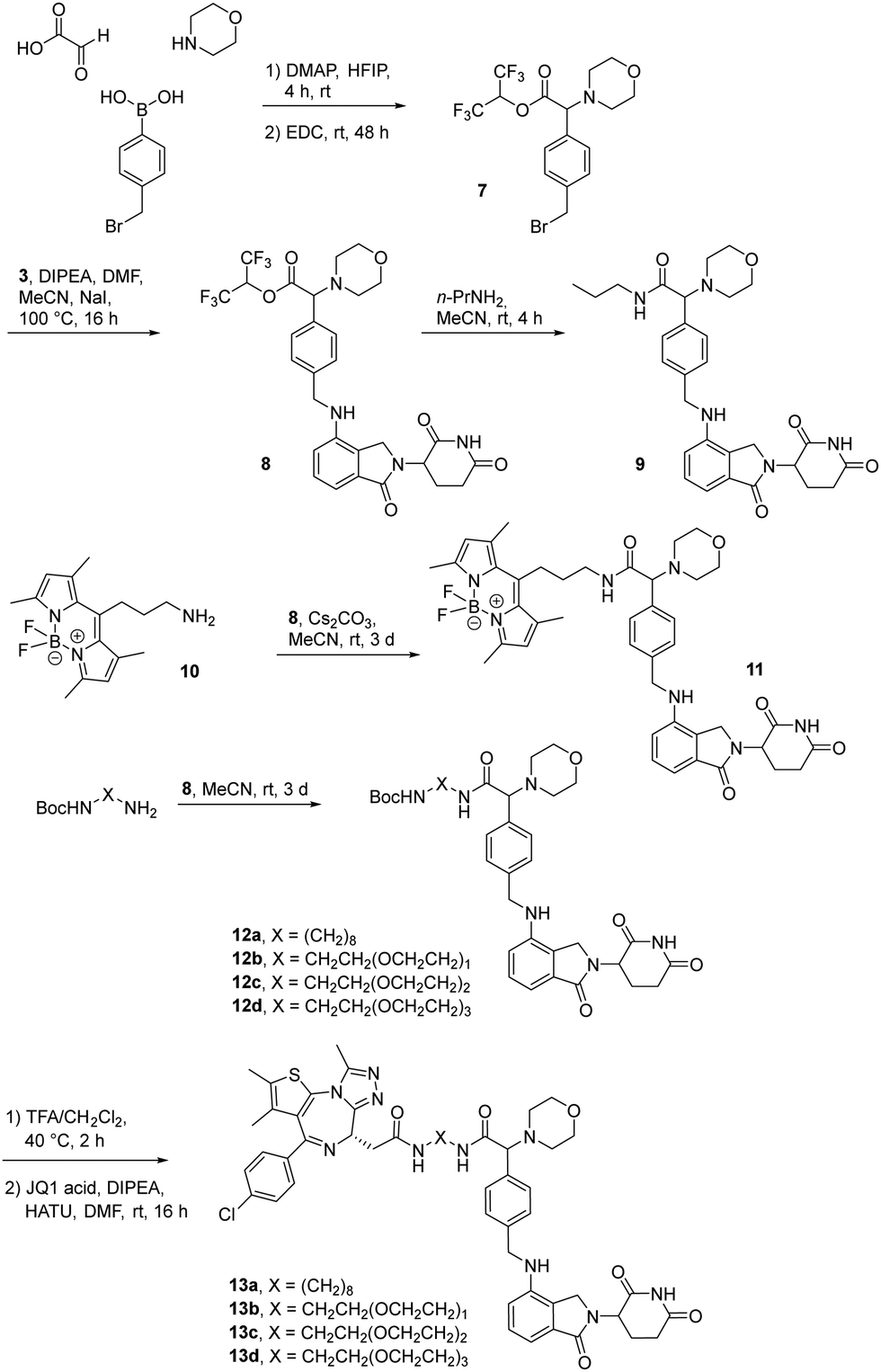 | ||
| Scheme 1 The Petasis borono-Mannich reaction enabled the synthesis of active ester 8, a versatile building block for the synthesis of tool compounds and PROTACs targeting the E3 ligase CRBN. | ||
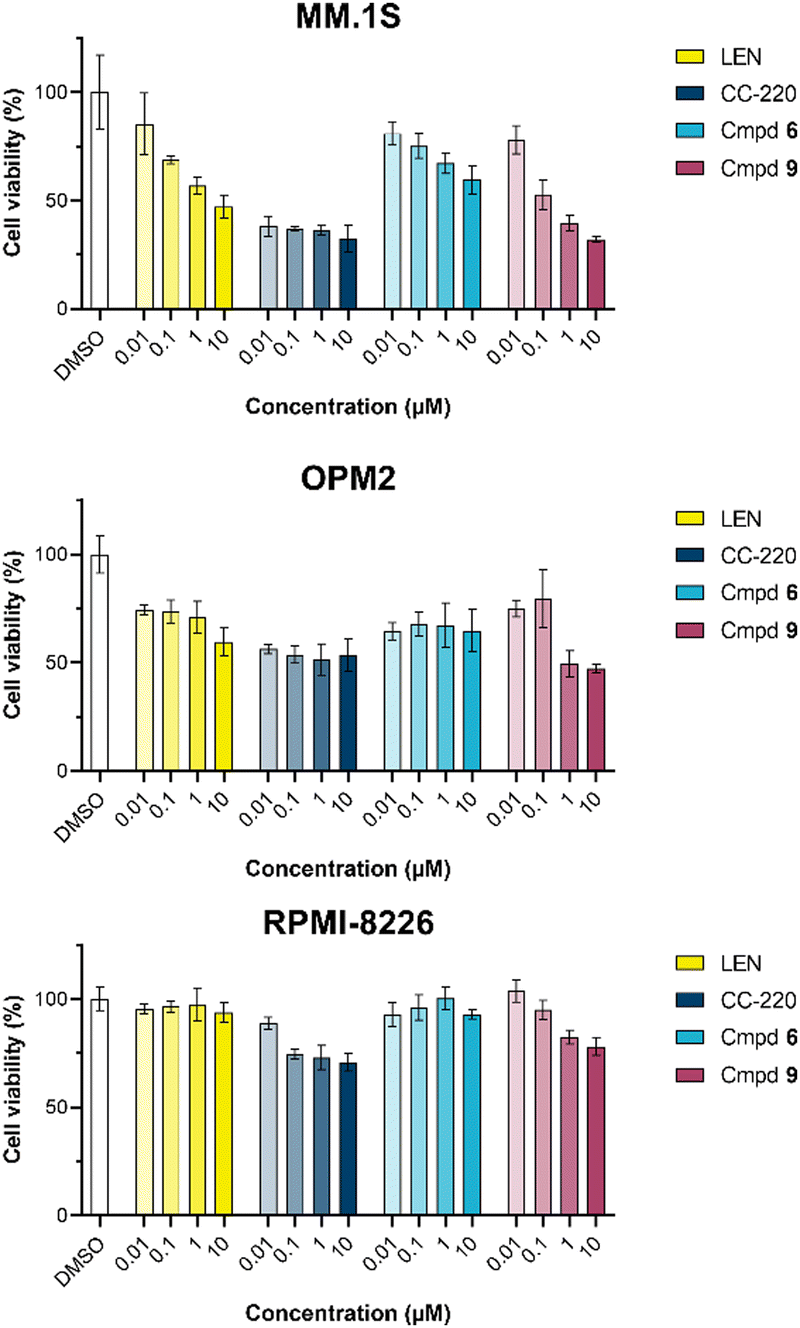 | ||
| Fig. 3 CellTiter-Glo luminescent cell viability assay upon a 96 h treatment with lenalidomide (LEN), CC-220, or three-branched compounds 6 and 9 in the respective multiple myeloma cell lines. Data are shown as mean ± s.d. (n = 3). | ||
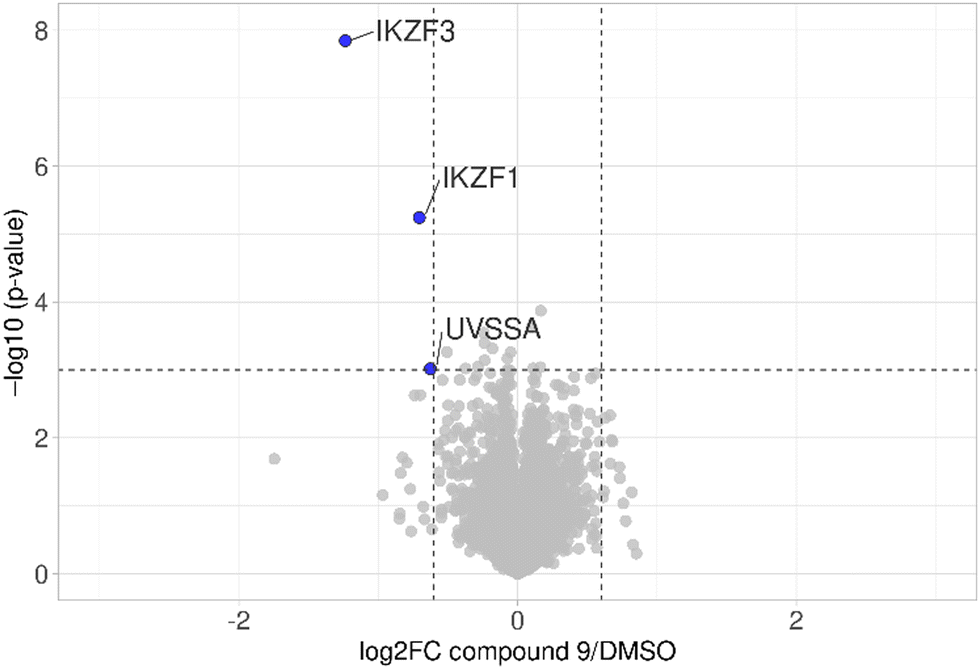 | ||
| Fig. 4 diaPASEF quantitative proteomics for molecular glue 9. MM.1S cells were treated with compound 9 at 0.1 μM for 3 h. Bioconductor's limma package was used to perform statistical analysis of degrader treatment compared to DMSO vehicle treatment. The identified proteins were plotted as log2 fold change (PROTAC/DMSO) versus −log10 of the p-value. Proteins with −log10 (p-value) > 3 (p-value < 0.001) and log2 fold change > 0.6 or < −0.6 (translating to 1.5-fold up- or down-regulation) were considered to have significantly changed in abundance. Data are the mean of biological duplicates. | ||
Motivated by the outstanding activities of 9, a fluorogenic reporter 11 based on this new scaffold was synthesized by utilizing the amino-functionalized BODIPY derivative 10.3311 showed a lower affinity than 9 (Table 2), but could be successfully utilized in the competitive MST assay (Fig. S4, ESI†). Delighted by the ease of the synthetic entry via the Petasis reaction and the high affinity of three-branched amide derivatives, we aimed at developing chimeric degrader molecules based on this ligand. In the case of VHL-based PROTAC molecules, specific modifications at the E3 recruiting unit resulted in entities with enhanced ligase affinity, which often translated to improved cellular target degradation.11,34 By combining 9 with different mono-protected bis-amines, the linker-connected derivatives 12a–12d were obtained in excellent yields. After cleavage of the Boc protecting group in acidic media and subsequent HATU-mediated coupling to JQ1, a well-studied bromodomain inhibitor,35 BRD4-targeting PROTACs 13a–13d containing different linker shapes were realized. Similar to a JQ1-derivative with two stereogenic centers,36 diastereomeric differentiability was not evident in the chromatographic and spectroscopic data. For this set of PROTACs, physicochemical properties, and BRD4 degradation were determined (Table 2). Increasing the molecular structure of the CRBN ligand significantly elevated lipophilicity and was accompanied by higher HSA binding values. Among the four new PROTACs tested, 13a–13d induced pronounced degradation of BRD4 (Fig. 5A), and appropriate control experiments demonstrated the involvement of the ubiquitin-proteasome system (Fig. 5B). Quantitative proteomics of the 6886 proteins detected confirmed an apparent down-regulation of JQ1-targeted proteins BRD2/3/4. In contrast, IMiD off-targets remained deregulated (Fig. S5, ESI†). However, iberdomide-type PROTACs 13 were less active compared to highly advanced thalidomide-based PROTACs such as ARV-825 and dBET6.37,38 We presume that an extended set of linkers may further improve the efficacy PROTACs derived from our new CRBN-binding chemotype.
| Cmpd | logD7.4a |
CHIIAMb | % HSA bindingc | K i/Kdd (μM) | DC50e (nM) | % BRD4f deg. |
|---|---|---|---|---|---|---|
| a Distribution coefficients at pH 7.4 were estimated by an HPLC-based method. b Chromatographic hydrophobicity index values referring to IAM chromatography (CHIIAM values), an estimate for drug-membrane interactions and permeability.28 c Protein binding values were estimated by an HPLC-based method. d Affinity values determined in a competitive MST assay as described in the Method sections (see ESI). e Concentration at which 50% of BRD4 has been degraded after 24 h treatment of Namalwa cells with each compound ranging from 0.1 to 1000 nM. f Percentage of degraded BRD4 after 24 h treatment of Namalwa cells with 0.1 μM of each compound. g Not determined. h Affinity was determined as a Kdvia initial fluorescence and is reported in place of the Ki. | ||||||
| 9 | 1.7 | 21.1 | 81 | 0.2 ± 0.7 | n.d.g | n.d. |
| 11 | 3.2 | n.d. | n.d. | 11.0 ± 13.5h | n.d. | n.d. |
| 13a | 3.1 | 34.5 | 94 | n.d. | 6.1 ± 2.4 | 95 |
| 13b | 2.5 | 29.6 | 91 | n.d. | 88 ± 53 | 38 |
| 13c | 2.5 | 29.2 | 90 | n.d. | 162 ± 41 | 22 |
| 13d | 2.5 | 28.7 | 90 | n.d. | 83 ± 80 | 52 |
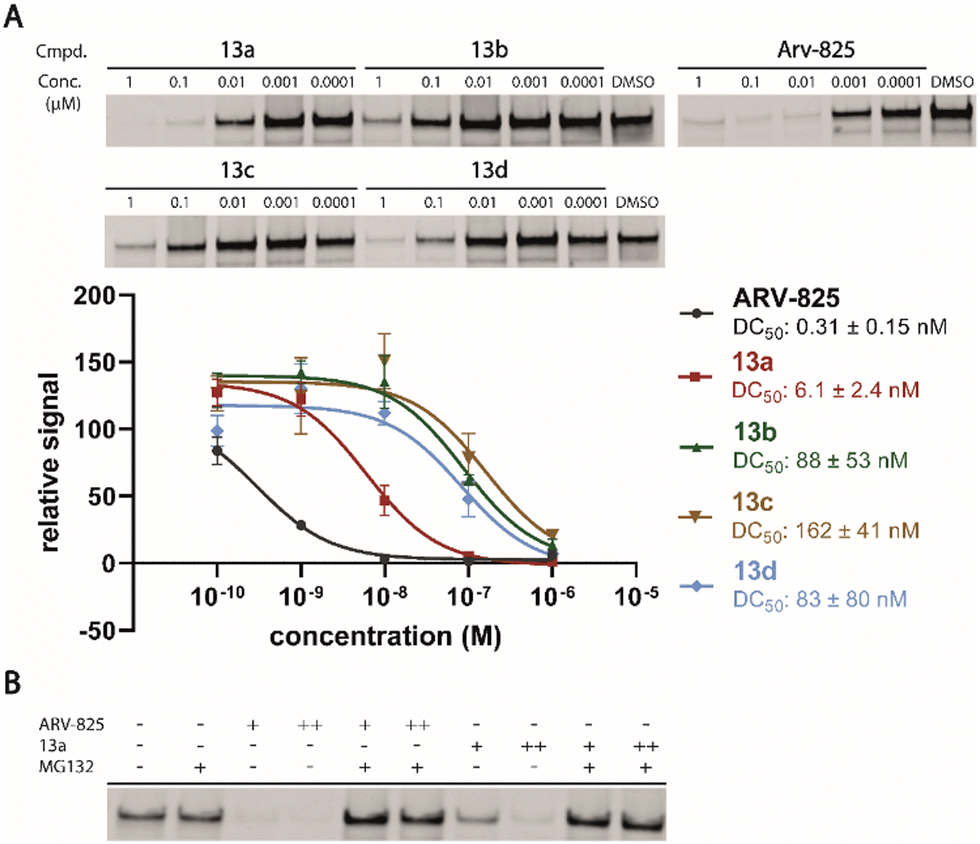 | ||
| Fig. 5 (A) PROTACs 13a–13d induced degradation of BRD4. Western blot analysis of BRD4 protein levels in Namalwa cells treated with compounds for 24 h. DC50 values were obtained by fitting the mean intensities to a dose-response model (Hill slope = 1). The signal was normalized with total protein stain and against DMSO. Data are shown as mean ± s.d. (n = 3). (B) Confirmation of proteasome-based mechanism of BRD4 degradation upon ARV-825 and 13a treatment. Namalwa cells were treated for 24 h with ARV-825 or 13a (+, 0.1 μM; ++, 1 μM) alone and with MG132 (5 μM). | ||
The work that we report herein impressively demonstrates the power of three-component reactions to provide tailor-made synthetic products. So far, MCRs have attracted little attention in the field of E3 ligase ligand discovery and optimization.39,40 With the aid of the Petasis reaction, easy access to three-winged substances with excellent CRBN binding affinities was realized. Their effects on neosubstrates, such as IKZF3, seem to be highly correlated with the affinity data. It was possible to show that chimeric degraders can be assembled via this novel exit vector. Active ester 8 is an excellent starting point for further research into structure-degradation relationships of three-branched CRBN modulators and PROTACs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support by the DFG (Kr-3886/2-1 and SFB-1074 to J. K.) and the ARRS (P1-0208 and J1-2485 to I. S.) and institutional funds of the Max Planck Society. T. K. was supported by a fellowship from the Jürgen Manchot Foundation. M. G. was supported by the Volkswagen Foundation. We thank Aleša Bricelj, Maja Frelih, Matic Proj, Marion Schneider, Rabea Voget, and Glencora Wolffhugel for their support. We thank Katherine A. Donovan, Eric Fischer, and the Fischer Lab Degradation Proteomics Initiative for the collection of the proteomics data supported by NIH CA214608 and CA218278. Open Access funding provided by the Max Planck Society.Notes and references
- P. Wu, M. Givskov and T. E. Nielsen, Chem. Rev., 2019, 119, 11245–11290 CrossRef CAS
.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef
- C. J. Gerry and S. L. Schreiber, Nat. Rev. Drug Discovery, 2018, 17, 333–352 CrossRef CAS PubMed
- W. Masamba, Molecules, 2021, 26, 1707 CrossRef CAS PubMed
- A. Strecker, Justus Liebigs Ann. Chem., 1854, 91, 349–351 CrossRef
- N. A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1993, 34, 583–586 CrossRef CAS
- J. Krönke, E. C. Fink, P. W. Hollenbach, K. J. MacBeth, S. N. Hurst, N. D. Udeshi, P. P. Chamberlain, D. R. Mani, H. W. Man, A. K. Gandhi, T. Svinkina, R. K. Schneider, M. McConkey, M. Järås, E. Griffiths, M. Wetzler, L. Bullinger, B. E. Cathers, S. A. Carr, R. Chopra and B. L. Ebert, Nature, 2015, 523, 183–188 CrossRef
- J. Krönke, N. D. Udeshi, A. Narla, P. Grauman, S. N. Hurst, M. McConkey, T. Svinkina, D. Heckl, E. Comer, X. Li, C. Ciarlo, E. Hartman, N. Munshi, M. Schenone, S. L. Schreiber, S. A. Carr and B. L. Ebert, Science, 2014, 343, 301–305 CrossRef PubMed
- C. Heim, S. Maiwald, C. Steinebach, M. K. Collins, J. Strope, C. H. Chau, W. D. Figg, M. Gütschow and M. D. Hartmann, Biochem. Biophys. Res. Commun., 2021, 534, 67–72 CrossRef CAS PubMed
- C. Steinebach, H. Kehm, S. Lindner, L. P. Vu, S. Köpff, Á. López Mármol, C. Weiler, K. G. Wagner, M. Reichenzeller, J. Krönke and M. Gütschow, Chem. Commun., 2019, 55, 1821–1824 RSC
- C. Steinebach, Y. L. D. Ng, I. Sosic, C.-S. Lee, S. Chen, S. Lindner, L. P. Vu, A. Bricelj, R. Haschemi, M. Monschke, E. Steinwarz, K. G. Wagner, G. Bendas, J. Luo, M. Gütschow and J. Krönke, Chem. Sci., 2020, 11, 3474–3486 RSC
- C. Steinebach, S. Lindner, N. D. Udeshi, D. C. Mani, H. Kehm, S. Köpff, S. A. Carr, M. Gütschow and J. Krönke, ACS Chem. Biol., 2018, 13, 2771–2782 CrossRef CAS PubMed
- A. Murgai, I. Sosič, M. Gobec, P. Lemnitzer, M. Proj, S. Wittenburg, R. Voget, M. Gütschow, J. Krönke and C. Steinebach, Chem. Commun., 2022, 58, 8858–8861 RSC
- I. Sosič, A. Bricelj and C. Steinebach, Chem. Soc. Rev., 2022, 51, 3487–3534 RSC
- T. Keuler, B. König, N. Bückreiß, F. B. Kraft, P. König, L. Schäker-Hübner, C. Steinebach, G. Bendas, M. Gütschow and F. K. Hansen, Chem. Commun., 2022, 58, 11087–11090 RSC
- M. E. Matyskiela, W. Zhang, H.-W. Man, G. Muller, G. Khambatta, F. Baculi, M. Hickman, L. LeBrun, B. Pagarigan, G. Carmel, C.-C. Lu, G. Lu, M. Riley, Y. Satoh, P. Schafer, T. O. Daniel, J. Carmichael, B. E. Cathers and P. P. Chamberlain, J. Med. Chem., 2018, 61, 535–542 CrossRef CAS PubMed
- I. Boichenko, K. Bär, S. Deiss, C. Heim, R. Albrecht, A. N. Lupas, B. Hernandez Alvarez and M. D. Hartmann, ACS Omega, 2018, 3, 11163–11171 CrossRef CAS PubMed
- C. Heim, D. Pliatsika, F. Mousavizadeh, K. Bär, B. Hernandez Alvarez, A. Giannis and M. D. Hartmann, J. Med. Chem., 2019, 62, 6615–6629 CrossRef CAS PubMed
- S. Ichikawa, H. A. Flaxman, W. Xu, N. Vallavoju, H. C. Lloyd, B. Wang, D. Shen, M. R. Pratt and C. M. Woo, Nature, 2022, 610, 775–782 CrossRef CAS PubMed
- C. Heim, A.-K. Spring, S. Kirchgäßner, D. Schwarzer and M. D. Hartmann, Biochem. Biophys. Res. Commun., 2022, 637, 66–72 CrossRef CAS PubMed
- M. Krasavin, M. Adamchik, A. Bubyrev, C. Heim, S. Maiwald, D. Zhukovsky, P. Zhmurov, A. Bunev and M. D. Hartmann, Eur. J. Med. Chem., 2022, 246, 114990 CrossRef PubMed
- A. K. Nooka and S. Lonial, Cancer J., 2019, 25, 19–31 CrossRef CAS PubMed
- C. Heim and M. D. Hartmann, Acta Crystallogr., Sect. D: Struct. Biol., 2022, 78, 290–298 CrossRef CAS PubMed
- A. D. Huber, Y. Li, W. Lin, A. N. Galbraith, A. Mishra, S. N. Porter, J. Wu, R. R. Florke Gee, W. Zhuang, S. M. Pruett-Miller, J. Peng and T. Chen, ACS Med. Chem. Lett., 2022, 13, 1311–1320 CrossRef CAS PubMed
- G. Nishiguchi, F. Keramatnia, J. Min, Y. Chang, B. Jonchere, S. Das, M. Actis, J. Price, D. Chepyala, B. Young, K. McGowan, P. J. Slavish, A. Mayasundari, J. A. Jarusiewicz, L. Yang, Y. Li, X. Fu, S. H. Garrett, J. B. Papizan, K. Kodali, J. Peng, S. M. Pruett Miller, M. F. Roussel, C. Mullighan, M. Fischer and Z. Rankovic, J. Med. Chem., 2021, 64, 7296–7311 CrossRef CAS
- C. E. Powell, G. Du, J. Che, Z. He, K. A. Donovan, H. Yue, E. S. Wang, R. P. Nowak, T. Zhang, E. S. Fischer and N. S. Gray, ACS Chem. Biol., 2020, 15, 2722–2730 CrossRef CAS PubMed
- E. S. Wang, A. L. Verano, R. P. Nowak, J. C. Yuan, K. A. Donovan, N. A. Eleuteri, H. Yue, K. H. Ngo, P. H. Lizotte, P. C. Gokhale, N. S. Gray and E. S. Fischer, Nat. Chem. Biol., 2021, 17, 711–717 CrossRef CAS PubMed
- K. Valko, C. M. Du, C. D. Bevan, D. P. Reynolds and M. H. Abraham, J. Pharm. Sci., 2000, 89, 1085–1096 CrossRef CAS
- S. Maiwald, C. Heim, B. Hernandez Alvarez and M. D. Hartmann, ACS Med. Chem. Lett., 2021, 12, 74–81 CrossRef CAS PubMed
- H. Jourdan, G. Gouhier, L. Van Hijfte, P. Angibaud and S. R. Piettre, Tetrahedron Lett., 2005, 46, 8027–8031 CrossRef CAS
- X. Qiu, N. Sun, Y. Kong, Y. Li, X. Yang and B. Jiang, Org. Lett., 2019, 21, 3838–3841 CrossRef CAS PubMed
- F. Meier, A.-D. Brunner, M. Frank, A. Ha, I. Bludau, E. Voytik, S. Kaspar-Schoenefeld, M. Lubeck, O. Raether, N. Bache, R. Aebersold, B. C. Collins, H. L. Röst and M. Mann, Nat. Methods, 2020, 17, 1229–1236 CrossRef CAS PubMed
- F. Heisig, S. Gollos, S. J. Freudenthal, A. El-Tayeb, J. Iqbal and C. E. Müller, J. Fluoresc., 2014, 24, 213–230 CrossRef CAS PubMed
- X. Han, C. Wang, C. Qin, W. Xiang, E. Fernandez-Salas, C.-Y. Yang, M. Wang, L. Zhao, T. Xu, K. Chinnaswamy, J. Delproposto, J. Stuckey and S. Wang, J. Med. Chem., 2019, 62, 941–964 CrossRef CAS PubMed
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed
- J. Devonport, L. Sully, A. K. Boudalis, S. Hassell-Hart, M. C. Leech, K. Lam, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, J. Spencer, A. Vargas and G. E. Kostakis, JACS Au, 2021, 1, 1937–1948 CrossRef CAS PubMed
- J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755–763 CrossRef CAS PubMed
- G. E. Winter, A. Mayer, D. L. Buckley, M. A. Erb, J. E. Roderick, S. Vittori, J. M. Reyes, J. di Iulio, A. Souza, C. J. Ott, J. M. Roberts, R. Zeid, T. G. Scott, J. Paulk, K. Lachance, C. M. Olson, S. Dastjerdi, S. Bauer, C. Y. Lin, N. S. Gray, M. A. Kelliher, L. S. Churchman and J. E. Bradner, Mol. Cell, 2017, 67, 5–18 CrossRef CAS PubMed
- B. Wang, J. Liu, I. Tandon, S. Wu, P. Teng, J. Liao and W. Tang, Eur. J. Med. Chem., 2021, 219, 113425 CrossRef CAS PubMed
- I. P. Bhela, A. Ranza, F. C. Balestrero, M. Serafini, S. Aprile, R. M. C. Di Martino, F. Condorelli and T. Pirali, J. Med. Chem., 2022, 65, 15282–15299 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Schemes S1 and S2, Fig. S1–S5; biological, chemical, and physicochemical methods; synthetic procedures; structures, 1H NMR, 13C NMR and MS data. See DOI: https://doi.org/10.1039/d2cb00223j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |