
Multi-walled carbon nanotube supported manganese selenide as a highly active bifunctional OER and ORR electrocatalyst†
Harish
Singh
a,
McKenzie
Marley-Hines
ab,
Shatadru
Chakravarty
c and
Manashi
Nath
 *a
*a
aDepartment of Chemistry, Missouri University of Science and Technology, Rolla, MO 65409, USA. E-mail: nathm@mst.edu
bDepartment of Chemical and Biochemical Engineering, Missouri University of Science and Technology, Rolla, MO 65409, USA
cAdvanced Materials Characterization Laboratory/Materials Research Center, Missouri University of Science and Technology, Rolla, Missouri 65409, USA
First published on 8th February 2022
Abstract
Transition metal selenides have attracted intensive interest as cost-effective electrocatalysts for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) because of the continuous thrust in sustainable energy conversion. In this article a Mn-based bifunctional electrocatalyst, MnSe, has been identified which shows efficient OER and ORR activity in alkaline medium. The catalytic activity could be further enhanced by using multiwalled carbon nanotubes (MWCNTs) which increases the charge transfer and electronic conductivity of the catalyst composite. This MnSe@MWCNT catalyst composite exhibits a very low overpotential of 290 mV at 10 mA cm−2, which outperforms state-of-the-art RuO2 as well as other oxide based electrocatalysts. Furthermore, the composite's facile OER kinetics was evidenced by its small Tafel slope of 54.76 mV dec−1 and low charge transfer resistance, indicating quick transport of the reactant species at the electrode interface. The MnSe@MWCNT also exhibited efficient electrocatalytic activity for ORR with an Eonset of 0.94 V, which is among the best reported to date for chalcogenide based ORR electrocatalysts. More importantly, this MnSe-based ORR electrocatalyst exhibits high degree of methanol tolerance, showing no degradation of catalyst performance in the presence of copious quantities of methanol, thereby out-performing the state-of-the-art Pt electrocatalyst. The catalyst composite also exhibited exceptional functional and compositional stability for OER and ORR after a prolonged period of continuous operation in alkaline medium. The surface Raman analysis after OER revealed the retention of manganese selenide surface with evidence of oxo coordination, confirming the formation of an (oxy)selenide as the active surface for OER. Such efficient bifunctional OER and ORR activity makes this MnSe based catalyst attractive for overall electrolysis in regenerative as well as direct methanol fuel cells.
Introduction
The perils of global warming and environmental pollution will be critical in the near future, forcing researchers to find a potential solution for clean energy generation from sustainable resources. Among the various solutions being pursued, regenerative fuel cells have attracted considerable attention due to the escalating energy demand and environmental concerns. Renewable energy technologies such as regenerative fuel cells (RFCs), electrocatalytic water splitting (EWS), and metal–air batteries (MABs) include the fundamental electrochemical reactions from oxygen reduction (ORR) to oxygen evolution (OER). These alternative “clean” energy technologies are supported through the use of electrocatalysts that increase the efficiency of these fundamental energy conversion processes. Precious metal-based materials, such as Pt and RuO2, are widely regarded as state-of-the-art electrocatalysts for the ORR and OER, respectively.1–4 However, their large-scale application is limited by economic constraints and insufficient reserves for the precious elements Pt and Ru. Intensive research over the last few years, on the other hand, has resulted in identification of several non-precious metal-based systems as highly efficient OER and ORR electrocatalysts. In particular, transition metal oxides,5 chalcogenides,6–9 pnictides,9,10 and layered double hydroxides (LDHs) have shown promising OER electrocatalytic activity. On the other hand, heteroatom-doped carbon materials, Fe, Co, and Mn-based metal hydroxides, and chalcogenides have shown promise as potential electrode materials for the ORR in fuel cells.6,11–15The oxygen evolution reaction (OER) and the oxygen reduction reaction (ORR) take place at the same electrode in electrochemical devices (EWS, MABs, or RFCs), making it a bifunctional electrode, wherein the OER can be considered as charging while the ORR is the discharging process. However, due to a scarcity of bifunctional oxygen electrodes which can support both the OER and ORR, the electrocatalysts would also be required to be electrochemically stable over a wide range of potentials, and these renewable energy technologies are still facing limitations with regard to practical usage. Several bifunctional electrocatalysts have recently been investigated, including alloys of Pt and Ir/Ru catalysts, for ORR and OER.16–18 However, the high cost of precious metals precludes their widespread use in electrochemical energy conversion and storage devices. As far as general electrochemical activities are concerned, they also have intrinsic disadvantages, such as high activation barriers, low specific capacitance, unstable chemical structures, and limited cycle life. To overcome these concerns, research has focused on non-precious transition metal based compositions wherein several bifunctional catalysts have been developed for both ORR and OER including Co3O4/CNTs,19 MnxOy/NC,20 CoxOy/NC,20 NixCoyO4/Co–NG,21 and BaTiO3−x.22
Recently, catalysts based on transition metal chalcogenides (TMCs) have gained considerable attention compared to metal oxides and alloys because of their unprecedented high activity.23–26 This is especially relevant because the rich stoichiometry alterations possible in metal-rich chalcogenides make them highly efficient, inexpensive, and robust as well as a viable substitute for noble metals as electrocatalysts for the OER and ORR. TMCs also have promising prospects for use in sensors27,28 as well as in a plethora of emerging renewable energy technologies.29–33 Among nonprecious metal-based catalysts, Ni and Co-based compositions have been studied widely for electrocatalytic activities.8,21,30,34 One of the critical criteria necessary for improved electrochemical activity is that the catalytic centre should exhibit facile electrochemical tuning with a low energy barrier. This can be achieved by using transition metals that can shuttle between various stable oxidation states. In that regard, Mn-based compositions can be very attractive since Mn can achieve various oxidation states with ease. Moreover, such Mn-based compounds are also lucrative due to their rich electrochemical properties, high abundance, low cost, and less toxicity. However, there have been few reports of Mn-based compounds being used as OER catalysts,25,26 and as ORR catalysts (mainly oxides) in alkaline batteries.35,36 It has been observed that the valence state of Mn is critical for the ORR performance, as the Mn(III) ion in MnOx typically exhibits greater activity than Mn(II) or Mn(IV). In our current investigation, an attempt has been made to modify the oxidation state of Mn by synthesizing MnSe which further helped to develop a bifunctional OER/ORR catalyst.
A major drawback of TMCs in terms of their limited electrical conductivity has hindered their large-scale industrial application in oxygen electrocatalysis.37,38 Carbon materials, on the other hand, due to their high electrical conductivity and specific surface area, provide reliable and consistent support for the integration and assembly of electrocatalysts.39,40 Earth-abundant conductive carbon materials and their derivatives, such as carbon nanotubes (CNTs) and graphene, along with non-precious metal based electrocatalysts, thus have the capability to replace noble metal alloys in practical fuel cell applications.41 Multi-walled carbon nanotubes (MWCNTs) have unique thermal and mechanical properties that make them appealing for the development of new materials, in addition to the electrical properties that they inherit from graphene.42,43 Their hollow interiors can be filled with a variety of nanomaterials, isolating and shielding them from their surrounding environment – an extremely useful property for fuel cell application.44–46 The numerous surface defects on the carbon support result in abundant homogeneous nucleation sites, and in conjunction with strong coordination interactions, this provides numerous sites for nanoparticle stabilization, resulting in increased specific surface area that leads to enhanced catalytic activity. Furthermore, their enhanced electron transfer and mass diffusion properties can also be used to improve the OER and ORR current density.47–49 Recently, Lee and co-workers have also reported a series of studies on WS2-CNT composites which demonstrate surprisingly better performance in OER and ORR compared to support-free WS2.50 Nevertheless, carbon materials by themselves are not sufficiently active and stable relative to a bifunctional catalysis, especially as they are prone to serious corrosion under a high anodic potential as well as in a highly alkaline medium.51,52 The absence of carbon, on the other hand, would result in a low conductivity of the catalyst, reducing the efficiency of electrocatalysis. However, the controlled synthesis of nano-heterostructures integrating multiple nanostructures is challenging and usually uses more complex and expensive multi-step synthesis processes.53,54
In the present study, MnSe nanostructures have been investigated as bifunctional electrocatalysts for OER and ORR in alkaline medium. The MnSe nanostructures were also mixed with carbon nanostructures to form hybrid composites to see if they can compensate for each other's shortcomings to ameliorate the catalytic activities. Herein, we provide a protocol for the development of a MnSe@MWCNT bifunctional catalyst that can simultaneously perform the OER and ORR with high efficiency and superior stability and also exhibits methanol tolerance in an alkaline medium. In this study, MnSe@MWCNT hetero-nanostructures were produced by a facile one-pot synthesis strategy using an in situ solvothermal reaction of MWCNTs with manganese and selenium sources. The MnSe@MWCNT composite was synthesized through a one-step hydrothermal synthesis process at a moderately low temperature. The MnSe@MWCNT composite showed an overpotential of 290 mV at 10 mA cm−2 which is significantly better than those of state-of-the-art precious metal oxides and comparable to those of some of the best OER electrocatalysts reported so far.55,56 Although manganese selenide based compositions have recently been reported as OER electrocatalysts,25,26 it must be noted that in these previous reports, the MnSe composition was doped with other transition metals such as Fe, Co, etc. Such doping has been known to alter the composition of the catalytically active site, changing its intrinsic activity. Moreover, the pure MnSe and MnSe@MWCNT composite reported in this article show comparable activity to previously reported MnSe-based doped systems,25 even when grown on 2-dimensional OER-inactive substrates as opposed to Ni foam which leads to significantly enhanced activity due to the substrate effect. To the best of our knowledge, this is the first report of pure manganese selenide (MnSe) as a bifunctional OER/ORR electrocatalyst with high efficiency and high methanol tolerance. This current study not only paves the way for the development of new nonprecious metal-based OER/ORR electrocatalysts with superior stability while maintaining high activities, but it also paves the way for carbon–metal chalcogenide composites to be used as novel electrocatalysts in more energy conversion and storage processes. In addition to exhibiting excellent bifunctional OER/ORR activity this catalyst composition also exhibits superior methanol tolerance and stability over platinum@glassy carbon electrode (Pt/GCE). In this manuscript we have reported not only a bifunctional catalyst but have provided new knowledge about the evolution of the active surface composition and how to increase efficiency and functionality of the catalyst composite.
Experimental
Materials
All the reagents were of analytical grade and were used as received. Manganese chloride (MnCl2·4H2O) was obtained from Fisher Scientific. Sodium selenite (Na2SeO3) and hydrazine hydrate (N2H4·H2O) were purchased from Alfa Aesar and Acros Organics, respectively. MWCNTs (∼1–5 μm in length, 20–25 nm in outer diameter) were purchased from Strem Chemicals Inc. and treated in 3 M HNO3 solution at 80 °C for an hour before being used. Before usage, the Ni foam was soaked in dilute hydrochloric acid for 10 minutes and then cleaned several times with acetone, ethanol, and distilled water.Synthesis of MnSe
For the synthesis of manganese selenide, firstly, 0.2 M Na2SeO3 and 0.1 M MnCl2·4H2O were dissolved in 8 mL deionized water. After mixing for 20 min under vigorous stirring, hydrazine, N2H4·H2O (2 mL) was added to the mixture, and the mixture was stirred continuously for another 20 minutes. The final mixture was sealed in a 23 mL Teflon-lined stainless-steel autoclave and heated at 145 °C for 24 h. For direct growth of MnSe on nickel foam (MnSe/NF), pre-cleaned Ni foam was placed inside the Teflon-liner. The autoclave was then allowed to cool down naturally. The final black colored product was then centrifuged and washed several times with a mixture of ethanol and deionized water. Finally, the product obtained was dried at 60 °C in a vacuum oven. When Ni foam was used for direct growth, the as-obtained MnSe/NF was also rinsed several times with DI water and ethanol, and then dried at 60 °C in a vacuum oven.Synthesis of the MnSe@MWCNT composite
The MnSe@MWCNT composite was prepared as follows: 0.2 M Na2SeO3 and 0.1 M MnCl2·4H2O were dissolved in 8 mL deionized water and added to a 23 mL Teflon-lined stainless-steel autoclave. Then 2 mL of N2H4·H2O was added with vigorous stirring for 20 min. Finally, MWCNTs (2 mg) were added rapidly to the reaction mixture and stirred until being completely dissolved. For direct growth of MnSe@MWCNT on nickel foam (MnSe@MWCNT/NF), pre-cleaned Ni foam was also placed inside the Teflon-liner. The autoclave was sealed and maintained at 145 °C for 24 h. After cooling to room temperature naturally, the product was centrifuged, washed several times with a mixture of ethanol and deionized water, and dried under vacuum at 60 °C overnight. Similarly, when Ni foam was used, the as-obtained MnSe@MWCNT/NF was rinsed several times with DI water and ethanol, and then dried at 60 °C in a vacuum oven.Electrode preparation
To analyse the OER and ORR activity of hydrothermally synthesized MnSe and MnSe@MWCNT catalysts, a homogeneous catalyst ink was prepared by dispersing 4 mg of catalyst in 300 μL of ethanol mixed with Nafion (0.8 μL, 5 wt%). For OER studies, this mixture was ultrasonicated for about 1 h to prepare a homogeneous ink. 100 μL of the ink was drop-cast onto carbon cloth (geometric area of 0.283 cm−2). Before ORR studies, the glassy carbon (GC) electrode was polished with 1 and 0.05 μm alumina powder as well as sonicated first in isopropanol and then in distilled water. The catalyst ink (20 μL) was then drop-cast on the glassy carbon working electrode (geometric area of 0.196 cm−2) and dried under ambient conditions.Materials characterization
The crystallinity of the as-synthesized samples was analysed by powder X-ray diffraction (PXRD) using a Philips X'Pert X-ray diffractometer (PANalytical, Almelo, The Netherlands) with a CuKα radiation source (1.5418 Å). The morphology of the samples was examined using a scanning electron microscope (SEM, Hitachi S4700) with an accelerating voltage of 15 kV and a working distance of 10 mm. The as-synthesized samples were dispersed directly on the formvar-coated Cu TEM grid and analysed with a Tecnai F20 to collect high-resolution TEM (HRTEM) images. The chemical composition of the catalysts and the valence states of the elements were determined by X-ray photoelectron spectroscopy (XPS). The chemical composition was also analysed before and after the OER and ORR stability studies. All XPS analyses were conducted without sputtering on the pristine catalyst surface. The Raman spectra of all synthesized samples were collected using a HORIBA Jobin-Yvon LabRam ARAMIS Raman spectrometer equipped with a CCD detector. Thermo Scientific NEXSA surface analysis equipment was used to perform simultaneous XPS and surface Raman analyses. By combining both methodologies on the same platform, it is possible to examine the same positions concurrently on the surface to gain complete understanding of the material under optimal conditions in ultrahigh vacuum. The Nexsa system was equipped with a Thermo Scientific iXR Raman system (spectral dispersion of 2 cm−1 per CCD pixel element), with the Raman laser probe aligned with the X-ray spot. The surface Raman spectra were collected using a 532 nm laser with 2 mW output power and a 16 s acquisition duration. Calibration was performed using the 520 cm−1 Raman peak of a silicon reference standard. The Raman spectrometer features four aperture settings (25 μm and 50 μm pinholes, and 25 μm and 50 μm slits) that may be changed based on different sample features. The Raman spectrum signal was maximized using a 50 μm slit.Electrochemical characterization
The OER and ORR electrocatalytic performances of the as-prepared electrodes were studied by using an IviumStat potentiostat. The electrochemical measurements were performed in a three-electrode cell system with a graphite rod as the counter electrode, and Ag|AgCl (KCl-saturated) as the reference electrode. A 5 mm diameter rotating ring disk electrode (RRDE) was used as a working electrode for ORR studies while catalyst loaded carbon cloth was used for OER studies. For OER studies N2-saturated 1 M KOH was used and for ORR studies, the RRDE test was carried out in O2-saturated 1 M KOH at different rotation rates between 400 and 2000 rpm (10 mV s−1).The Koutecky–Levich (K–L) plots (J−1vs. ω−1/2) were obtained at different potentials between 0.2 V and 0.8 V and fitted to linear curves by using the following K–L equation (eqn (1)) to calculate the number of electrons transferred (n):57
| 1/J = 1/JL + 1/JK = 1/Bω1/2 + 1/JK | (1) |
| B = 0.62nFC0 (D0)2/3ν−1/6 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), n is the number of electrons transferred per molecule of oxygen, C0 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3), D0 is the coefficient of diffusion of O2 (1.9 × 10−5 cm2 s−1), and ν is the kinematic viscosity of the electrolyte (0.01 cm2 s−1).
485 C mol−1), n is the number of electrons transferred per molecule of oxygen, C0 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3), D0 is the coefficient of diffusion of O2 (1.9 × 10−5 cm2 s−1), and ν is the kinematic viscosity of the electrolyte (0.01 cm2 s−1).
Tafel plot
A key parameter to examine OER activity is the Tafel slope, which is calculated using the Tafel equation by fitting polarization data. As illustrated in eqn (3), the Tafel equation is the dependency of overpotential η on current density j.| η = a + 2.3RT/αnFlogj | (3) |
Electrochemically active surface area (ECSA)
The surface area of the catalyst has a significant impact on its catalytic activity. Thus, the electrochemically active surface area (ECSA) was determined using double-layer capacitance (CDL) in accordance with eqn (4):| ECSA = CDL/CS | (4) |
| iDL = CDL × ν | (5) |
Another important parameter is the Roughness Factor (RF) which defines surface roughness of the catalyst composite and can affect the observed catalytic properties. The ratio of ECSA to geometric electrode area (0.283 cm−2) was used to calculate RF.
Results and discussion
The as-synthesized catalysts were characterized using various techniques to determine their structural and morphological composition. PXRD was utilized to confirm the structural and phase purity of the hydrothermally synthesized samples. Fig. 1a shows the comparison of PXRD patterns of MnSe, MnSe@MWCNT, and MWCNT. The PXRD data confirmed the formation of pure MnSe corresponding to a cubic structure (lattice parameters a = b = c = 5.462 Å) in the Fm3m space group (225) [JCPDS no. 00-011-0683]. The presence of sharp diffraction peaks indicates the sample's purity and crystallinity. The as-synthesized MnSe@MWCNT hybrid material showed a similar PXRD pattern to that of the cubic MnSe phase indicating that the formation of a composite with MWCNT did not affect the crystallinity or phase purity of MnSe. MWCNT, on the other hand, did not show any diffraction peaks in the MnSe@MWCNT composite, perhaps due to the low carbon content and the relatively low diffraction intensity of the MWCNT peaks. Nevertheless, the presence of carbon nanotubes in the MnSe@MWCNT composite material was confirmed by Raman spectroscopy, XPS, and TEM.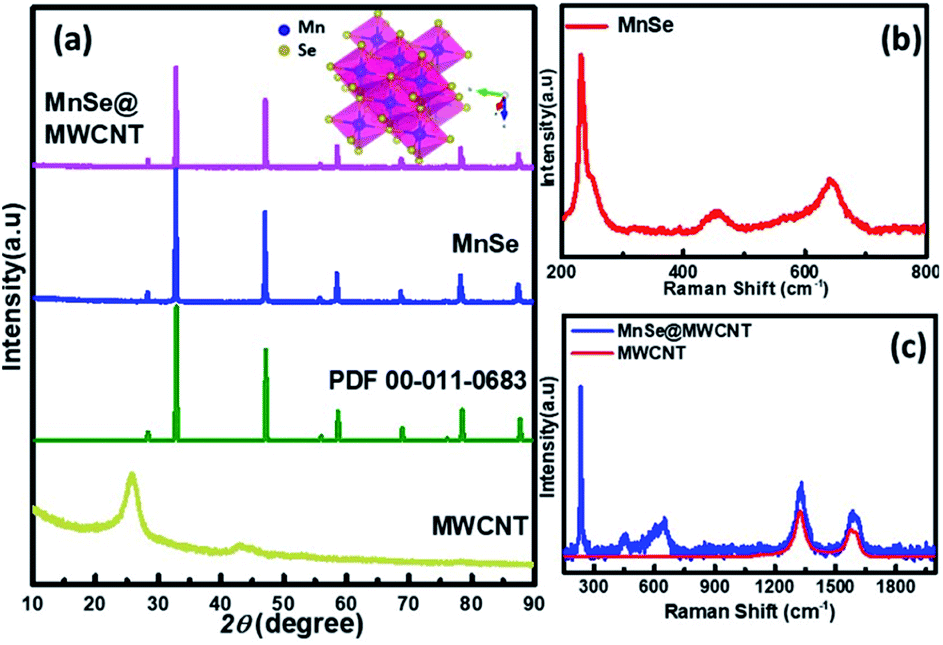 | ||
| Fig. 1 PXRD patterns of (a) MnSe@MWCNT, MnSe, and MWCNT compared with the MnSe standard pattern. Raman spectra of the as-synthesized (b) MnSe and (c) MnSe@MWCNT (blue curve) compared with that of MWCNT (red curve). | ||
Raman spectra of the MnSe sample were measured in the 200–800 cm−1 spectral range. Additionally, the peaks of the Se–Se stretching mode are observed at 254.44, 456.58 cm−1, and 646.28 cm−1 as shown in Fig. 1b, being close to the values previously reported.60,61 The Raman spectra of MnSe@MWCNT and pristine MWCNT showed the characteristic D band (1342 cm−1) and G band (1572 cm−1) as shown in Fig. 1c, which results from disorder and sp2 hybridized carbon atoms in MWCNT walls. The ID/IG ratio of the MnSe@MWCNT sample was found to be 1.42, which represents a large number of defects on the carbon nanotube surface.62 To investigate the surface elemental configuration and electronic states of the MnSe, X-ray photoelectron spectroscopy (XPS) was performed. Fig. 2 shows the XPS spectra where the peaks can be attributed to Mn 2p3/2, Mn 2p1/2, Se 3d5/2, and Se 3d3/2. From the deconvoluted spectrum of Mn, the peaks at 642.61 and 654.27 suggest the existence of Mn2+ and Mn3+. The presence of such mixed oxidation states in MnSe is consistent with the previously reported values.63 The Se 3d peak at 55.2 eV and the smaller peak at 56.0 eV are ascribed to Se 3d5/2 and Se 3d3/2, respectively. Interestingly, there was no evidence of the Mn-oxide phase, confirming that these samples were primarily of selenide composition. The C 1s spectra from the MnSe@MWCNT sample, on the other hand, showed peaks corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and carboxylate functionalities arising from the graphitic surface and functionalization of the MWCNT, respectively.
C and carboxylate functionalities arising from the graphitic surface and functionalization of the MWCNT, respectively.
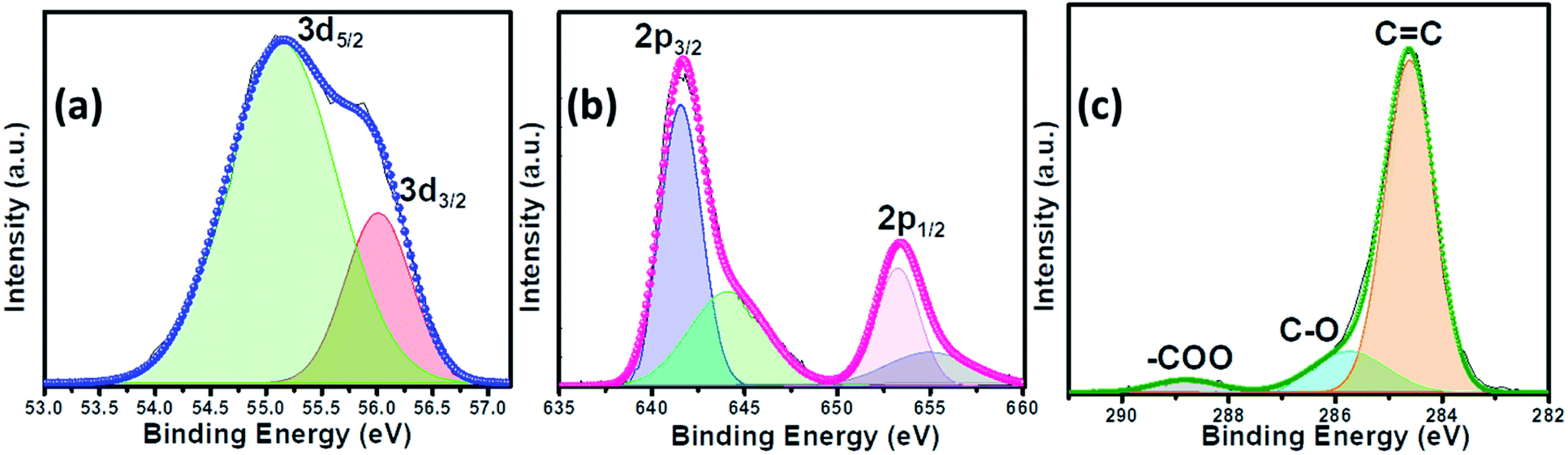 | ||
| Fig. 2 The deconvoluted XPS spectra of the MnSe@MWCNT composite electrode showing (a) Se 3d, (b) Mn 2p and (c) C 1s peaks. | ||
SEM and TEM analyses were performed to investigate the morphology of MnSe and the MnSe@MWCNT composite. The MnSe sample showed uniform dense structures with sizes in the range of 3–5 μm, as shown in the SEM image in Fig. 3a. The MnSe@MWCNT composite after hydrothermal reaction, on the other hand, showed MWCNT backbones decorated with numerous MnSe cubes or spheres as shown in Fig. 3b, where the light gray tubular agglomerations are the MWCNTs, while the cube-like particles are MnSe. A closer examination of the particle surface of both samples reveals that they have a multitude of small nanoparticles in the range of 30–60 nm. TEM images of MnSe (Fig. 3c) and MnSe@MWCNT (Fig. 3d) reveal the nanometer size particle distribution. In addition, Fig. 3e and f show the high-magnification TEM images showing lattice fringes with a clear interplanar spacing of 0.27 nm corresponding to the (200) lattice planes of MnSe confirm the crystallinity of these nanostructured grains.
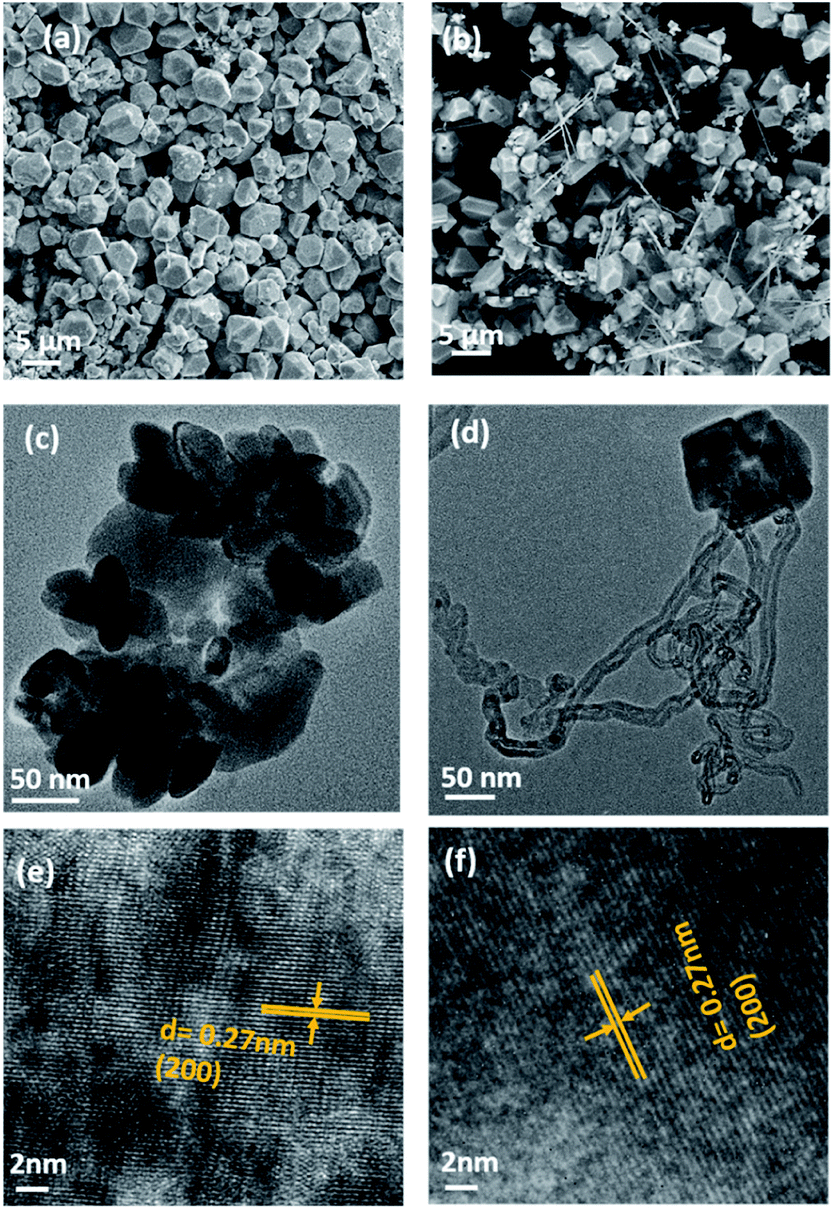 | ||
| Fig. 3 SEM images of (a) MnSe and (b) the MnSe@MWCNT composite. TEM images of (c) MnSe and (d) the MnSe@MWCNT composite. HRTEM images of (e) MnSe and (f) the MnSe@MWCNT composite showing the fringes corresponding to the (200) lattice planes of MnSe. | ||
Electrocatalytic performance for the OER
Electrocatalytic studies were performed using a standard 3-electrode set-up with graphite rod, Ag|AgCl, and MnSe/MnSe@MWCNT on carbon cloth as counter, reference, and working electrodes, respectively. The hydrothermally synthesized samples were drop-cast on carbon cloth as per the methodology described above to prepare the working electrodes. The electrochemical studies for OER were performed using linear sweep voltammetry (LSV) as well as one-step and multistep constant potential chronoamperometric techniques. The OER activity of the as-synthesized samples was determined using LSV at a scan rate of 10 mV s−1. The resistance of 1.0 M KOH was estimated to be 15.0 Ω and was used for iR correction of all the LSV plots reported in this work. The OER activity of MnSe and MnSe@MWCNT was compared with state-of-the-art catalyst RuO2 which was prepared using electrodeposition following a typical procedure outlined in the ESI.†Fig. 4a shows the OER activity of MnSe, MnSe@MWCNT, and RuO2 in N2-saturated 1.0 M KOH. The OER onset potentials of MnSe, MnSe@MWCNT and RuO2 were estimated to be 1.49, 1.47, and 1.57 V, respectively. The overpotentials at 10 mA cm−2 (η10) for MnSe, MnSe@MWCNT, and RuO2 were estimated to be 310, 290, and 380 mV, respectively. The LSV plots also showed that both MnSe and MnSe@MWCNT had better OER catalytic activity compared to RuO2. Interestingly, it was observed that the overpotential of the MnSe@MWCNT composite dropped significantly compared to that of MnSe signifying the positive effect of mixing with MWCNT. Apart from low overpotential, the MnSe@MWCNT composite also showed higher current density indicating that the highly conducting MWCNT facilitated charge transfer within the catalyst composite. The overpotential of MnSe@MWCNT is comparable to that of some of the best OER electrocatalysts reported to date, as shown in Table S1 (ESI).†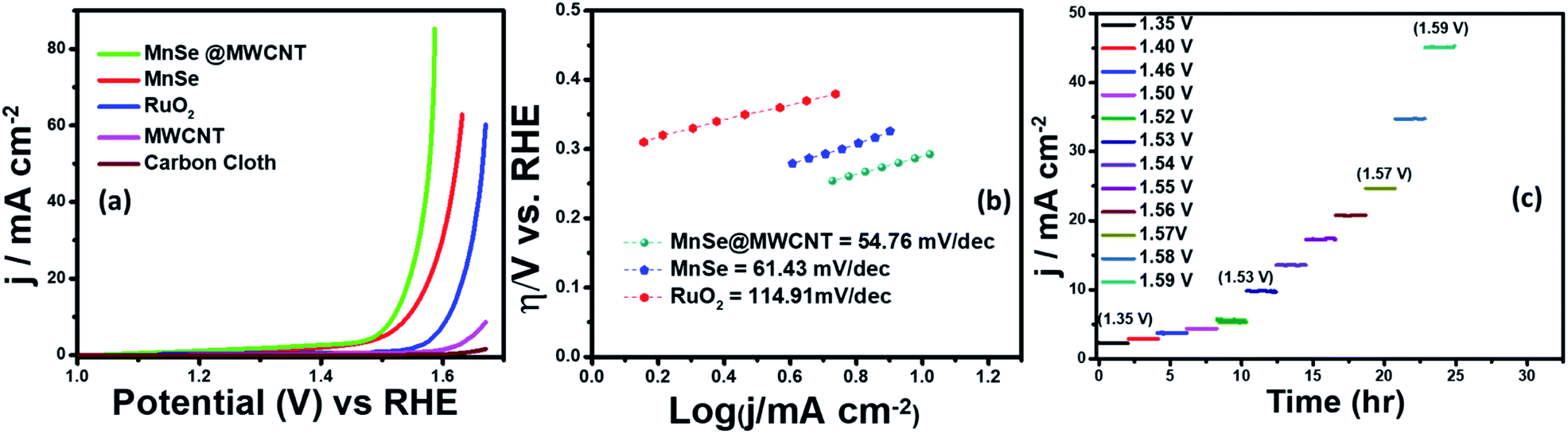 | ||
| Fig. 4 (a) LSV plots of the as-prepared MnSe, MnSe@MWCNT, MWCNT and carbon cloth electrodes compared with that of RuO2 in 1 M KOH solution measured at a scan rate of 10 mV s−1, confirming the enhanced OER activity of MnSe and the MnSe@MWCNT modified electrodes. (b) The Tafel plots of MnSe, MnSe@MWCNT and RuO2. (c) Multistep chronoamperometry (MCA) test of the MnSe@MWCNT composite electrode carried out at various constant applied potentials. | ||
Another important criterion for evaluating an electrocatalyst's OER performance is steady-state activity and durability under conditions of continuous operation. As shown in Fig. 4c, multistep chronoamperometry measurements for the OER were performed at various constant applied potentials to evaluate the electrochemical stability for the MnSe@MWCNT electrode. Specifically, in this experiment, the potential steps were varied from 1.35 V to 1.59 V vs. RHE where current density at each step was measured for ∼2 h as shown in Fig. 4c. It was observed that during long-term testing, there was no obvious deterioration of current density under different applied potentials, further confirming the excellent stability at low and high current densities, prominent mass transport, and mechanical robustness toward OER activity. To compare the OER activity of MnSe and the MnSe@MWCNT composite with other reported transition metal chalcogenides grown on metallic foam electrodes, and understand the effect of the substrate, the MnSe and MnSe@MWCNT catalyst composite were also assembled on Ni foam either through direct hydrothermal synthesis or drop-casting as described in the Experimental section. It was observed that the catalyst composite showed significantly enhanced current density at a lower overpotential (210 mV at 50 mA cm−2) when grown on Ni foam (Fig. S1(a))†, wherein the overpotentials were estimated from the reverse sweep of the CV plots as shown in Fig. S1(b).† The higher current density on Ni foam can be attributed to the 3-dimensional porous mesh-like architecture for Ni foam and its inherent OER activity. The overpotentials at higher current densities between MnSe-based composites and other reported systems are also compared in Table S2.† The ultimate usability of water electrolyzers can also be estimated by their ability to produce higher current density with less energy expense (i.e. lower overpotential). If a catalyst can produce higher current density, it signifies better potential for usage in practical devices. However, since we are interested in understanding the true intrinsic catalytic activity of these MnSe composites, further studies were conducted with carbon cloth as the substrate, which did not show any inherent activity.
Tafel slope and turnover frequency are critical parameters for determining the intrinsic catalytic activity of the as-synthesized nanostructures and for comprehending the OER mechanism. The Tafel plot is considered to be one of several methods for correlating the kinetic and thermodynamic properties of electrocatalytic systems and determining the rate at which oxidation current increases in response to the applied potential. The smaller the value of the Tafel slope, the faster the reaction kinetics is, which can increase current density and thus oxygen generation. In Fig. 4b, the Tafel slope values demonstrate that MnSe@MWCNT has the lowest Tafel slope of 54.76 mV dec−1 which even outperforms the RuO2 catalyst (114.91 mV dec−1). The low Tafel slope of MnSe@MWCNT suggests that it has comparable or better OER activity than most other recently reported Mn-based selenide OER catalysts (Table S1†). Another important factor for OER catalytic activity is the electrochemically active surface area (ECSA). An increase in ECSA typically results in enhanced exposure of the active sites which boosts catalytic performance.58,64 For direct comparison, the ECSA of each electrode was compared by estimating the double layer capacitance in the non-faradaic region obtained from cyclic voltammetry (CV) plots as shown in Fig. S2 (ESI).† The CV plot of each catalyst and corresponding capacitive current at 0.1 V versus Ag/AgCl were recorded at different scan rates in 1 M KOH to determine the ECSA which was estimated to be 10.48 and 19.3 cm2 for MnSe and MnSe@MWCNT, respectively. Increased ECSA results in increased surface roughness factor (RF), which can be considered as another factor that influences catalytic activity which is typically enhanced by the rough catalytic surface since more catalytic sites are exposed to the electrolyte. The RF value obtained for the MnSe@MWCNT electrode (68.2) is almost two times the value for the MnSe electrode (37.03), which is in good agreement with the improved catalytic activity observed for the MnSe@MWCNT nanocomposite. Table 1 presents the comparison of electrocatalytic parameters of all the catalysts for the OER reported in this article.
| OER | MnSe | MnSe@MWCNT | RuO2 |
|---|---|---|---|
| Onset potential/V | 1.49 | 1.47 | 1.57 |
| η @ 10 mA cm−2/mV | 310 | 290 | 380 |
| Tafel slope/mV dec−1 | 61.43 | 54.76 | 114.91 |
| ECSA/cm2 | 10.48 | 19.3 | — |
| RF | 37.03 | 68.2 | — |
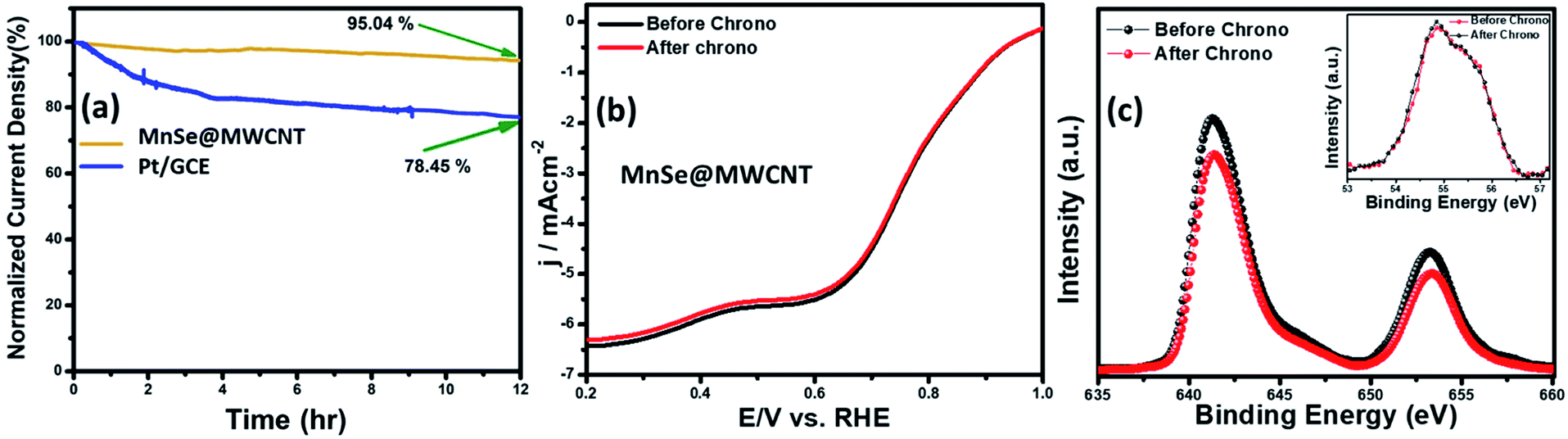
The long-term functional stability of the catalysts was also studied by chronoamperometry where a constant potential was applied, and the current density was measured for an extended period. Fig. S3† shows the 12 h stability studies for MnSe and the MnSe@MWCNT modified electrode at applied potentials of 1.55 and 1.52 V, respectively, to achieve 10 mA cm−2 current density. It was observed that both electrodes showed extensive stability for 12 h with minimal degradation of current density (9.8% and 4.7% for MnSe and MnSe@MWCNT, respectively). It must be noted here that most cutting-edge OER electrocatalysts are maintained as carbon-free owing to the degradation of carbon under the high anodic potential required for the OER. However, in this case, the presence of MWCNT in the MnSe@MWCNT composite doesn't show any instability in catalytic performance or loss of durability under long-term operation. The long-term stability of the composite MnSe@MWCNT electrode was also measured at 10 and 50 mA cm−2 current density as shown in Fig. 5a, which illustrated the extensive stability of these catalysts even at higher current density. As shown in Fig. 5b, there was no change in LSV plots even after long-term multistep chronoamperometry for 12 hours in the MnSe@MWCNT sample. These studies, specifically multistep, and long-term chronoamperometry results not only confirm the extended functional stability of these OER catalysts, but also demonstrate the positive effect of the carbon support on improving the surface stability of metal chalcogenide electrocatalysts which leads to more efficient OER activity. This also validated the superior mass transport properties, suggesting its superior operational stability in alkaline media.
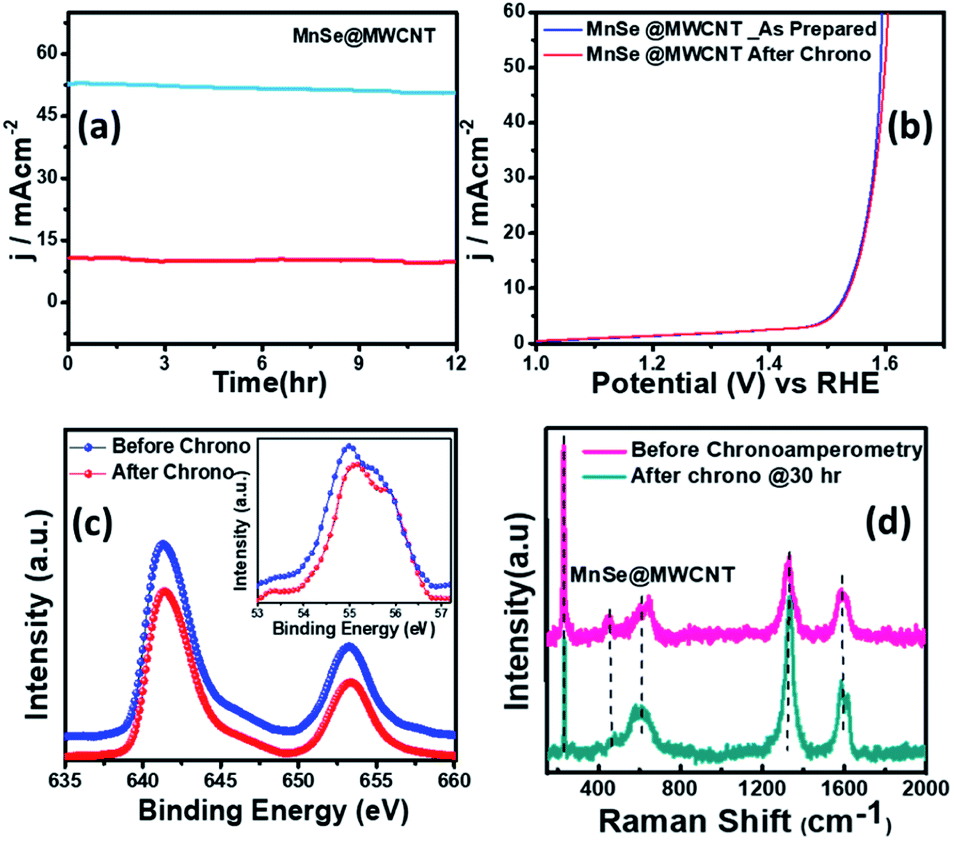 | ||
| Fig. 5 (a) Chronoamperometry plots illustrating the long-term stability of the MnSe@MWCNT modified electrode under constant overpotentials required to maintain current density at 10 and 50 mA cm−2. (b) Comparison of LSV plots for MnSe@MWCNT before and after chronoamperometry. (c) Comparison of the XPS peaks of Mn 2p and Se 3d (inset) before and after OER activity demonstrating the catalyst's robustness. (d) Raman spectra of MnSe@MWCNT before and after OER chronoamperometry. | ||
The compositional stability of these electrocatalysts after extended period OER studies was investigated with PXRD, XPS and Raman spectroscopy. Typically, PXRD, XPS, SEM and Raman were collected from the catalyst composite after a 12 h chronoamperometry study in 1 M KOH electrolyte. The XPS data of the MnSe@MWCNT composite revealed that there was no change of oxidation states or XPS peak positions of either Mn or Se as shown in Fig. 5c, confirming the compositional stability of the catalyst composite. Further information about the catalyst compositions before and after the OER is provided by Raman spectra. Interestingly, it was observed that the Raman spectra of the MnSe (Fig. S4†) and MnSe@MWCNT (Fig. 5d) catalysts showed no shift in peaks and no additional peaks after 30 h of OER, confirming that there were no structural changes in the catalytic composite. The PXRD patterns in Fig. S5† demonstrate that the crystallinity and phase purity of the MnSe@MWCNT phases remained unchanged after 12 h of OER in alkaline media. Similarly, a comparison of SEM images before and after OER activity revealed retention of similar morphology for hydrothermally synthesized MnSe and MnSe@MWCNT samples, as shown in Fig. S6.† Hence, from the results of PXRD, XPS, SEM and Raman spectra, it could be concluded that there was no degradation or bulk transformation of either the MnSe or MnSe@MWCNT catalyst after prolonged OER under an anodic potential.
However, it must be noted that in contrast to the bulk material, the surface of these electrocatalysts can exhibit varied physical and chemical properties post-activity owing to the presence of surface unsaturation and reactive intermediate adsorption. Because a surface atom is not surrounded by a uniform layer of atoms on all sides, these changes can have a major impact on the material's top atomic layer. This can result in different bonding potentials of the surface atoms which makes them more reactive than the bulk atoms. Hence it is important to characterize the surface of these catalyst composites to correctly understand evolution of the active catalyst interface. Identification of the active surface composition, especially for metal chalcogenides, has become increasingly important with the growing emphasis on these highly efficient catalytic systems. However, there is still no consensus on the actual surface composition, and researchers worldwide are still trying to get an answer by gathering evidence from various techniques. In this project, we have attempted to correctly identify the composition of the reactive surface through detailed surface analysis and electrochemical measurements. Hence, in addition to investigating bulk compositional stability, the change in surface composition of MnSe@MWCNT electrocatalysts after extended period OER studies was investigated by surface Raman spectroscopy integrated with XPS. Specifically, the experiment was performed by making sure that data were acquired from the same surface region by aligning the XPS analysis position precisely with the Raman analysis position. Fig. 6a demonstrates the working principle of surface Raman spectra acquisition for the MnSe@MWCNT composite. The region of investigation was focused on the central part of the functional electrode which was the most active area for the OER. Such surface specific analysis can be utilized to accurately study the evolution of the interface following active intermediate adsorption and conversion, thereby leading to the identification of the true composition of the catalyst surface. The Raman spectrum of the MnSe@MWCNT composite electrode after OER activity is shown in Fig. 6b. As can be seen from the Raman spectra, the characteristic D and G bands at 1342 and 1572 cm−1, respectively, corresponding to the MWCNT, were visible in both MnSe@MWCNT and pristine MWCNT surfaces. Additionally, absorptions at around 2680–2730 cm−1 were observed in both samples, which are believed to be the first overtone of the D-mode, commonly known as the G′ or 2D band. G′, the Raman double-resonance mode caused by disorders and/or defects in the MWCNT side walls, typically illustrates the degree of crystallinity in the nanotube walls. The ID/IG ratio, which describes the structural disorder of MWCNT or graphene, is proportional to D- and G-band intensities.65 After chronoamperometry stability studies under an anodic (OER) potential, the ID/IG ratio changed from 1.15 to 1.3, showing the possible formation of additional defects on the CNT surface which can be caused by the considerable influence of a prolonged period of OER activity. The peak for the Se–Se stretching mode in MnSe was observed at 254.44 cm−1.60,66 The sharp and broad peak at 636 cm−1 can be attributed to the Mn–Se stretching mode, which is consistent with an earlier report.61 After OER activity a small peak appeared at 738 cm−1 which can be ascribed to asymmetric stretching vibrations of SeO2 groups. However, the major Raman peaks at the surface were still consistent with the MnSe phase, suggesting that the composition and structure of the catalyst surface remained unchanged after chronoamperometry studies. It must be noted here that the surface Raman analysis after the OER failed to pick up any evidence for the formation of a Mn-oxide or hydroxide layer on the surface, confirming that the MnSe layer was indeed stable under continuous exposure at the low anodic potential required for the OER. Interestingly, an additional Raman peak at 746 cm−1 was observed after the OER (inset of Fig. 6b) which could be assigned to the formation of a local MnO cluster as has been proposed very recently,67 wherein the authors have identified the evolution of active surface species through the formation of this Mn–oxo linkage. In this article we propose that the OER on the MnSe surface occurs through the formation of similar Mn–O linkages (as evidenced by the Raman peak at 746 cm−1) on the selenide layer following the activation step initiated by –OH adsorption on the surface. This observation of formation of such an (oxy)selenide as the active surface composition for the MnSe-based catalyst composite further confirms our earlier observation that transition metal selenide catalysts evolve into a mixed anionic (hydroxy)chalcogenide surface during the OER.8 Accurate elucidation of the active surface composition will be transformative for the field and will provide critical knowledge for performing interface engineering and optimal catalyst surface design. Fig. S7† shows the high resolution XPS spectra of Mn, Se and C after 12 h of OER activity collected at the same spot as that where Raman spectral analysis was performed. The XPS peaks before and after catalytic activity did not demonstrate any shift in the Mn 2p and Se 3d peak locations. Both surface Raman and XPS indicate that there was no change in the surface composition of the MnSe@MWCNT.
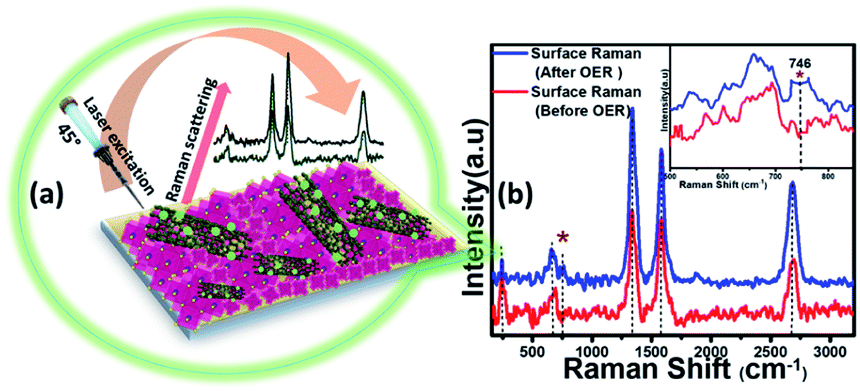 | ||
| Fig. 6 (a) Schematic representation for measuring surface Raman spectra. (b) Surface Raman spectra of the MnSe@MWCNT composite before and after OER chronoamperometry for 12 h. The inset shows magnified spectra from 500 to 850 cm−1. | ||
Electrocatalytic performance for the ORR
Apart from the intriguing OER performance, the as-synthesized MnSe nanocomposite-based electrodes also exhibit efficient ORR electrocatalytic activity, as illustrated in Fig. 7. The ORR activity was investigated through LSV measured in O2-saturated 1 M KOH with a rotating disk electrode (RDE) set-up at scanning frequencies of 10 mV s−1 and rotation rates of 400, 800, 1200, 1600 and 2000 rpm. LSV data for the MnSe@MWCNT, MnSe, and MWCNT are shown in Fig. 7 (a–d). The ORR activity of these Mn-based electrocatalysts was compared with that of electrodeposited Pt on a GCE, which shows standard ORR activity as previously reported (Fig. S8†).11 The ORR activity was also compared with that of a standard 20% Pt/C composite which showed an onset potential of 0.90 V vs. RHE (Fig. S9†) as has been reported earlier.44 It can be inferred from the polarization curves shown in Fig. 7 that the measured current density increased with increasing rotation speed due to enhanced diffusion of electrolyte on the electrode surface. The diffusion limiting current densities, onset potentials, and half-wave potentials have been determined for MnSe, MnSe@MWCNT, MWCNT and Pt/GCE from these plots. The onset potentials and half-wave potentials of MnSe@MWCNT were found to be 0.94 V and 0.86 V vs. RHE, respectively, along with a diffusion limiting current density of 6.49 mA cm−2 which was observed to be the highest among all electrocatalysts reported in this study. A bar plot of the limiting current density for different catalysts is shown in Fig. 8b. It shows that the MnSe@MWCNT electrode exhibits a current density of 6.02 mA cm−2 at a potential of 0.4 V (vs. RHE). However, MWCNT, Pt/GCE, and MnSe show significantly lower current densities of 3.06, 3.92 and 5.1 mA cm−2, respectively. The MnSe@MWCNT composite electrode's superior ORR activity compared to each of its constituents (i.e., MWCNT or MnSe) indicates a synergistic effect, most likely caused by the additional electron transfer pathways facilitated by the highly conductive MWCNT network.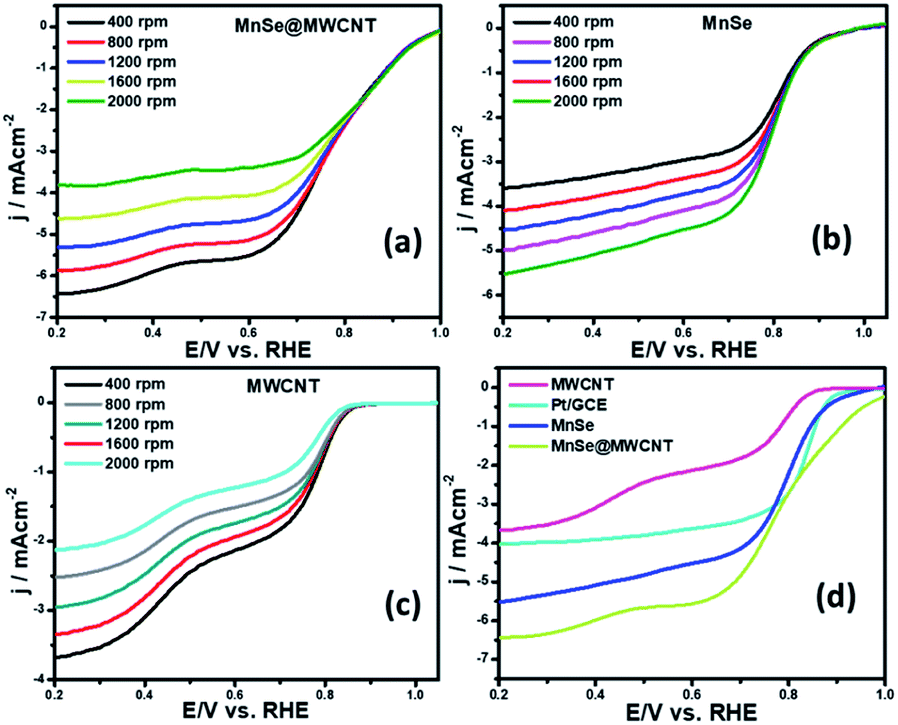 | ||
| Fig. 7 Linear sweep voltammograms of (a) the MnSe@MWCNT modified electrode, (b) MnSe, and (c) MWCNT collected using the RDE set-up. (d) Comparison of the ORR activity of these catalysts at a rotating speed of 2000 rpm. | ||
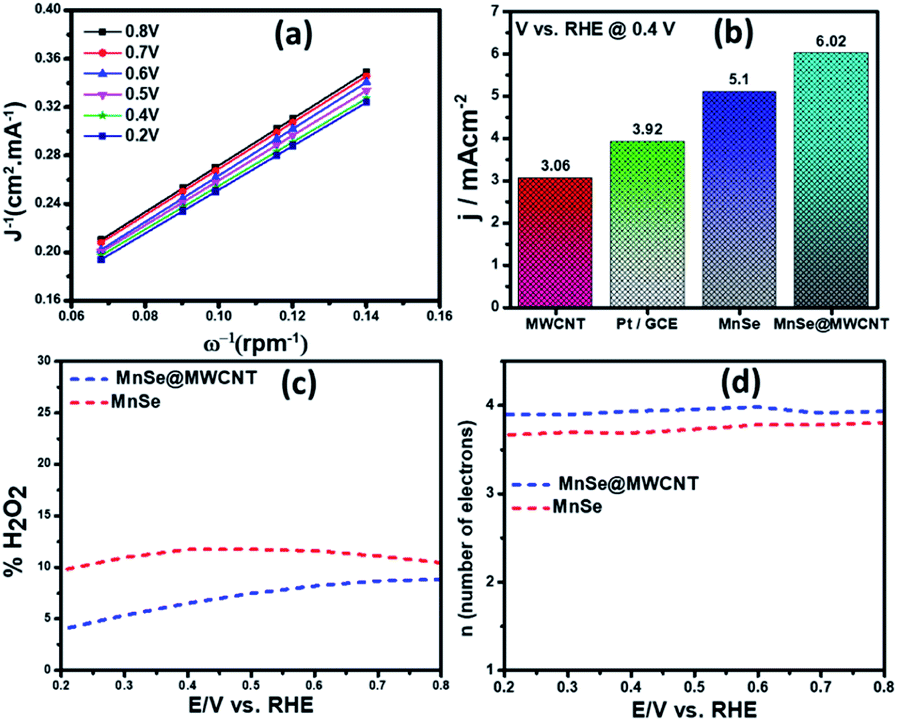 | ||
| Fig. 8 (a) K–L plots of the MnSe@MWCNT composite electrode in O2-saturated 1 M KOH. (b) Reduction current densities of MWCNT, Pt/GCE, MnSe, and MnSe@MWCNT. (c) H2O2 yield and (d) number of electrons for the MnSe@MWCNT modified electrode and MnSe at a disc rotation speed of 1200 rpm and scan rate of 10 mV s−1 in O2 saturated 1 M KOH. | ||
These results demonstrate that it has much better activity than the precious metal based (Pt) catalyst for ORR electrocatalysis in an alkaline medium. To identify the role of MWCNT as a carbon support in the electrocatalytic behaviour of the composite, the electrocatalytic properties of MWCNT were also determined and compared to those of the MnSe@MWCNT composite as shown in Fig. 7c and d. It was observed that while MWCNT was active for the ORR, the MnSe@MWCNT composite had much superior activity in terms of both onset potentials and limiting current density. This can be explained from the synergistic effect, wherein MnSe on MWCNT surfaces provides active sites for electrocatalytic activity, while MWCNTs provide conducting channels for efficient charge transfer and a large surface area, resulting in a highly active electrocatalyst for the ORR.44 To gain a more thorough understanding of the ORR electrocatalytic activity of these catalysts, further electrochemical analyses were carried out. Koutecky–Levich (K–L) curves were plotted at different potentials, as shown in Fig. 8a. The first-order reaction kinetics of dissolved oxygen with respect to dissolved oxygen concentration can be deduced from the linearity of the K–L graphs. The parallel behavior of the fitted lines also suggests similar electron transfer pathways for the ORR at different potentials. The disk current (ID) and the ring current (IR) obtained with the RRDE set-up were used to quantify the percentage of intermediate hydrogen peroxide, H2O2 production and the number of electrons (n) transferred. The H2O2 yield (%) and n were calculated using the following equations (eqn (6) and (7)), respectively.
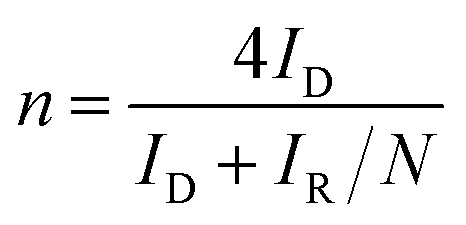 | (6) |
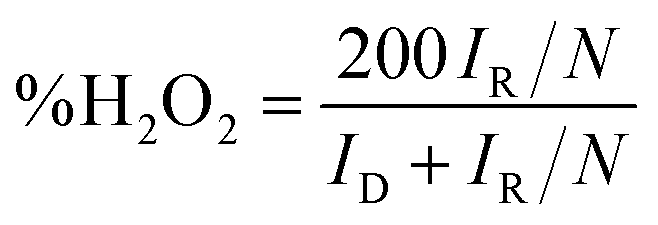 | (7) |
The number of electrons transferred (n) for MnSe@MWCNT was estimated to be 3.96 at a potential range of 0.8–0.2 V, respectively, from the slope of Koutecky–Levich plots, indicating a dominant four-electron reduction pathway in 1 M KOH solution forming water as the reduction product. MnSe by itself showed an n value of 3.84 indicative of a 4-electron pathway, while MWCNT showed an n value of 2.86, indicating a significant deviation from the 4-electron reduction pathway. Further proof for this reaction path has been obtained from electrochemical analysis using the rotating ring-disk electrode (RRDE) set-up to identify and quantify the reduction product.
The general reduction of oxygen occurs with two consecutive two-electron transfer processes that lead to the formation of a peroxide species (HO2−) which will thereafter yield hydrogen peroxide. The final form of hydroxide (OH−) is consequently reduced by two more electrons, while the direct four-electron method directly reduces O2 to OH. Consequently, the RRDE is considered a reliable and commonly used technique for studying the ORR catalytic pathway by detecting the formation of HO2−. Fig. 8c and d show the electron transfer numbers and HO2− yields from RRDE data for MnSe and MnSe@MWCNT. The estimated H2O2 yield, as can be seen, is less than 10% and 12% for MnSe@MWCNT and MnSe, respectively, within the potential range of 0.8–0.2 V vs. RHE. These observations indicate that the ORR pathway catalysed by MnSe and MnSe@MWCNT is primarily a 4-electron mechanism through the direct formation of hydroxyl species and water as final products, similar to commercially available Pt/GCE catalysts. It was also observed that the rate of H2O2 production remained constant for MnSe within the applied potential range, while it decreased slightly for the MnSe@MWCNT composite. The electron transfer rate in the MnSe@MWCNT composite can be expected to be higher than that in the MnSe based film due to the presence of MWCNT networks which facilitate charge transfer. Hence, at more cathodic potentials, as the rate of O2 reduction reaches a maximum, faster conversion to H2O occurs through 4 electron reduction pathways compared to that of H2O2 (2e− reduction) on the MnSe@MWCNT composite film. This faster conversion to H2O can be supported on the MnSe@MWCNT composite film due to its highly conducting nature, while for the MnSe film, limited conductivity leads to a steady conversion rate for H2O and H2O2. Hence there is a decrease in H2O2 yield for MnSe@MWCNT at more cathodic potentials.
The durability and stability of the catalysts are also essential to their functional applications, in addition to the superior catalytic performance. In Fig. 9a, the long-term stability of the composite catalysts MnSe@MWCNT and Pt/GCE for the ORR was checked through a constant potential chronoamperometry study at E1/2 potential and comparison of the LSV plots in O2-saturated 1 M KOH before and after 12 h of chronoamperometry studies. The LSV plots of the MnSe@MWCNT catalyst at a rotational speed of 2000 rpm before and after chronoamperometry are shown in Fig. 9b. It can be seen that after the stability test, E1/2 remains at the same potential. In comparison, following similar testing, the E1/2 of the Pt/GCE catalyst moves to a lower potential by more than ∼20 mV (Fig. S10†), suggesting the superior stability of the MnSe@MWCNT composite electrode for the ORR compared to the Pt/GCE. The robust connection between the MnSe nanoparticles and MWCNT network, which maintains structural integrity for efficient charge transfer, could be responsible for MnSe@MWCNT's superior stability for the ORR as well. Table 2 presents the comparison of electrocatalytic parameters of all the catalysts for the ORR reported in this article.
 | ||
| Fig. 9 (a) Durability test of the MnSe@MWCNT and Pt/GCE composite electrodes in O2-saturated 1 M KOH at a rotation speed of 1200 rpm. (b) LSV plots before and after chronoamperometry studies with MnSe@MWCNT. (c) Comparison of the XPS peaks of Mn 2p and Se 3d (inset) before and after the ORR chronoamperometry study. | ||
| Catalysts | ORR performance without methanol addition | ORR performance after methanol addition | |||||
|---|---|---|---|---|---|---|---|
| E onset vs. RHE (V) | E 1/2 | No. of transferred electrons | Diffusion limited current density (mA cm−2) | E onset vs. RHE (V) | E 1/2 | Diffusion limited current density (mA cm−2) | |
| MnSe@MWCNT | 0.94 | 0.86 | 3.96 | 6.02 | 0.94 | 0.86 | 5.91 |
| MnSe | 0.91 | 0.82 | 3.84 | 5.1 | — | — | — |
| MWCNT | 0.83 | 0.79 | 2.86 | 3.06 | — | — | — |
| Pt/GCE | 0.92 | 0.81 | 3.94 | 3.92 | 0.87 | 3.61 | |
The compositional stability of the composite electrode after prolonged ORR activity was confirmed through XPS. The XPS spectra of MnSe@MWCNT before and after the ORR stability test as shown in Fig. 9c confirmed that the composition of the catalyst was retained. The XPS spectrum obtained from the MnSe@MWCNT composite after prolonged ORR was superimposable on that of the as-prepared composite as shown in Fig. 9c, indicating no catalyst degradation.
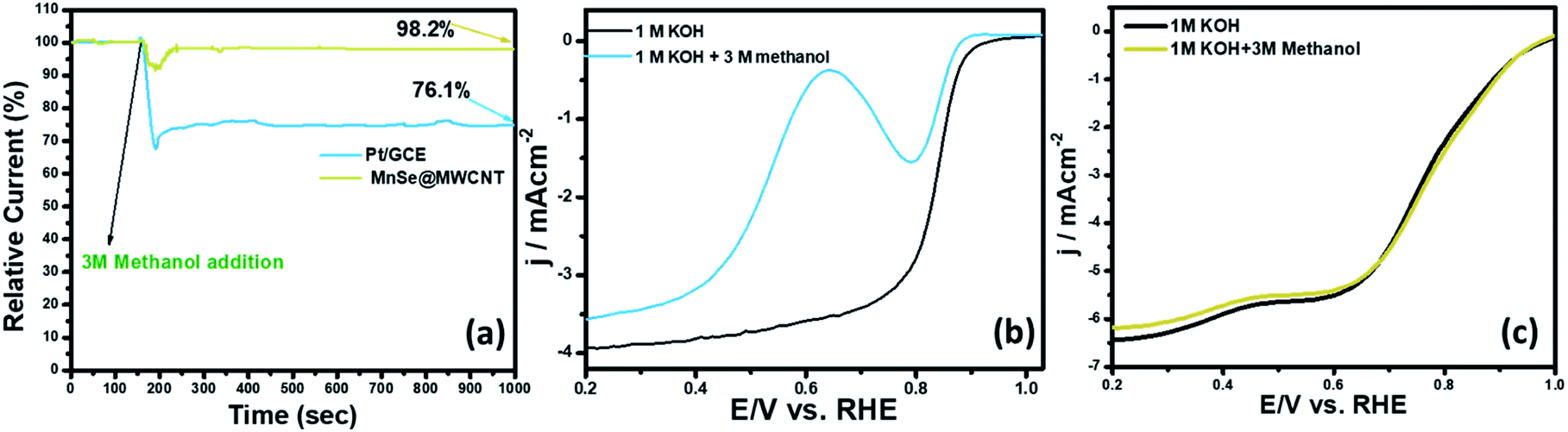
The methanol crossover effect and functional durability are some of the key criteria for the assessment of cathode stability in direct methanol fuel cells. In Fig. 10a, the catalyst-coated electrode's methanol crossover effect was determined by measuring the chronoamperometric response in O2-saturated 1 M KOH following the addition of 3 M methanol. The RDE LSV plots of MnSe@MWCNT and Pt/GCE in O2-saturated 3 M CH3OH + 1 M KOH solution are shown in Fig. 10b and c. In the presence of methanol, the ORR on Pt/GCE overlapped with methanol oxidation because Pt is active in the ORR potential zone for methanol oxidation leading to a reduction in ORR activities. However, the polarization curves for the MnSe@MWCNT composite electrode in the presence of methanol showed no loss of activity as can be seen in Fig. 10c, indicating a high degree of methanol tolerance. In contrast, a methanol oxidation peak for the Pt/GCE electrode appears at 0.59 V. Pt/GCE also exhibited a current loss of 24% following the addition of methanol after 1000 seconds as illustrated in the chronoamperometric response (Fig. 10a), while the current remained very stable for the MnSe@MWCNT catalyst composite, indicating that this electrode is highly selective to the ORR even in the presence of copious quantities of methanol. The i–t curve in Fig. 10a clearly demonstrates the excellent retention of the current density of around 98.2% for the MnSe@MWCNT catalyst in the presence of methanol, asserting its commercial viability in direct methanol fuel cells.
 | ||
| Fig. 10 (a) Comparison of the methanol tolerance for MnSe@MWCNT and Pt/GCE composite electrodes performed by adding methanol during the chronoamperometry study. LSV plots for (b) Pt/GCE and (c) MnSe@MWCNT electrodes in O2-saturated 1 M KOH and 1 M KOH + 3 M methanol. | ||
As discussed in the preceding sections, the synergistic effects between MnSe and MWCNT may explain the MnSe/MWCNT composite's remarkable methanol tolerance performance. Since each material (MnSe or MWCNT) alone has only moderate ORR and OER activity, the synergistic integration of MnSe and MWCNT provides excellent bifunctional electrocatalytic efficiency. Furthermore, MnSe particles dispersed within MWCNT networks not only provide the most active sites for catalysis, but can also effectively restrain catalyst particle aggregation, detachment, and Ostwald ripening, boosting electrocatalytic stability. The onset potential, along with other electrocatalytic parameters of MnSe@MWCNT, is comparable with those of some of the best-known ORR electrocatalysts reported to date as shown in Table S3 (ESI)†.
To gain better insight into the possible synergistic effect between the catalysts and carbon nanostructures, electrochemical impedance spectroscopy (EIS) of MnSe and MnSe@MWCNT catalysts was performed within a frequency range of 105–0.01 Hz and the results are shown in Fig. 11. The inset of Fig. 11 shows a simple equivalent circuit model that was used to fit the EIS data wherein Rf represents the resistance of the catalyst composite, RCT the charge transfer resistance at the electrode–electrolyte interface, ZW the Warburg impedance, and CPE the constant phase element. All these circuit elements were estimated from fitting of the equivalent circuit to the impedance plot and are presented in Table S4.† As shown in Fig. 11, the charge transfer resistance of the MnSe@MWCNT (RCT) was estimated to be 18.6 Ω and the value is substantially lower than that of MnSe (28.4 Ω). The resistance of the MnSe@MWCNT composite film was also lower than that of MnSe indicating enhanced charge transfer within the catalyst composite. This observation further confirmed that addition of MWCNT is beneficial for increasing charge transfer within the catalyst composite and electrode–electrolyte interface, resulting in enhancement of electrocatalytic performance for the MnSe@MWCNT composite electrode.
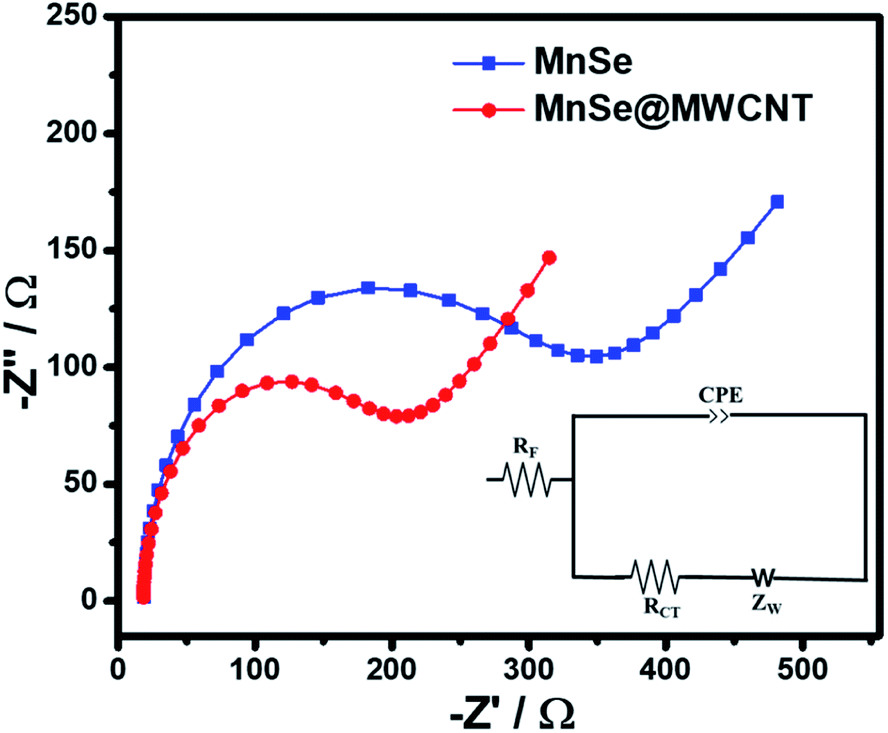 | ||
| Fig. 11 Nyquist plots of the MnSe and MnSe@MWCNT composite electrodes in the AC frequency range between 100 kHz and 0.1 Hz. The inset shows the fitted equivalent circuit. | ||
Conclusions
In summary, we have synthesized a manganese selenide based highly efficient bifunctional electrocatalyst active for OER and ORR in alkaline medium. Simple one-pot hydrothermal synthesis yielded these nanostructured catalysts. The OER activity of MnSe could be further improved by forming a composite with MWCNT, whereby the MnSe@MWCNT modified electrode shows a very low overpotential of 290 mV at 10 mA cm−2. Apart from low overpotential, MnSe and the MnSe@MWCNT modified electrode also exhibit exceptional functional and compositional stability after prolonged exposure to alkaline medium under an anodic potential. Detailed surface analysis revealed the formation of an oxychalcogenide as the active surface species explaining its high activity and stability. Furthermore, this composite electrode was also tested for methanol tolerance during the ORR. The MnSe@MWCNT electrode shows excellent methanol tolerance surpassing that of the state-of-the-art Pt/C electrocatalyst. Nanostructured MnSe on MWCNT surfaces provides active sites for electrocatalytic activity, while the MWCNT provides conducting channels and a large surface area. Hence, the high efficiency of electrocatalytic activity for the MnSe@MWCNT electrode can be attributed primarily to facile charge transfer within the catalyst composite and at the catalyst–electrode interface, its large surface area, abundance of active sites, and higher degree of surface roughness. The development of non-precious metal based nanostructured electrocatalyst composites will be expedited by this work leading to advancement in renewable energy conversion and storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge financial support from ACS PRF (54793-ND10), NSF (DMR 1710313) and Materials Research Centre (MRC), Missouri S&T for equipment usage.Notes and references
- L. Shi, T. Zhao, A. Xu and Z. Wei, ACS Catal., 2016, 6, 6285–6293 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 17, 552–556 CrossRef PubMed.
- W. Yang, W. Gong, Y. Shi, X. Wang, Y. Wang, J. Qiao, S. Zeng, J. Di and Q. Li, J. Mater. Chem. A, 2021, 9(29), 16187–16195 RSC.
- Y. F. Xu, Y. Chen, G. L. Xu, X. R. Zhang, Z. Chen, J. T. Li, L. Huang, K. Amine and S. G. Sun, Nano Energy, 2016, 28, 63–70 CrossRef CAS.
- H. Radinger, P. Connor, R. Stark, W. Jaegermann and B. Kaiser, ChemCatChem, 2021, 13, 1175–1185 CrossRef CAS.
- S. Umapathi, J. Masud, A. T. Swesi and M. Nath, Adv. Sustainable Syst., 2017, 1, 1–11 CAS.
- A. T. Swesi, J. Masud and M. Nath, Energy Environ. Sci., 2016, 9, 1771–1782 RSC.
- U. De Silva, J. See, W. P. R. Liyanage, J. Masud, J. Wu, W. Yang, W.-T. Chen, D. Prendergast and M. Nath, Energy Fuels, 2021, 35, 4403 Search PubMed.
- O. Mabayoje, A. Shoola, B. R. Wygant and C. B. Mullins, ACS Energy Lett., 2016, 1, 195–201 CrossRef CAS.
- Y. Lyu, J. Zheng, Z. Xiao, S. Zhao, S. P. Jiang and S. Wang, Small, 2020, 16, 1906867 CrossRef CAS PubMed.
- J. Masud and M. Nath, ACS Energy Lett., 2016, 1, 27–31 CrossRef CAS.
- Y. Cheng, S. He, S. Lu, J. Veder, B. Johannessen, L. Thomsen, M. Saunders, T. Becker, R. De Marco, Q. Li, S. Yang and S. P. Jiang, Adv. Sci., 2019, 6, 1802066 CrossRef PubMed.
- S. Guo, S. Zhang, L. Wu and S. Sun, Angew. Chem., Int. Ed., 2012, 51, 11770–11773 CrossRef CAS PubMed.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grützke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- M. Nath, U. De Silva, H. Singh, M. Perkins, W. P. R. Liyanage, S. Umapathi, S. Chakravarty and J. Masud, ACS Appl. Energy Mater., 2021, 4(8), 8158–8174 CrossRef CAS.
- B. S. Lee, H. Y. Park, M. K. Cho, J. W. Jung, H. J. Kim, D. Henkensmeier, S. J. Yoo, J. Y. Kim, S. Park, K. Y. Lee and J. H. Jang, Electrochem. Commun., 2016, 64, 14–17 CrossRef CAS.
- F. D. Kong, S. Zhang, G. P. Yin, N. Zhang, Z. B. Wang and C. Y. Du, Electrochem. Commun., 2012, 14, 63–66 CrossRef CAS.
- D. Mladenović, D. M. F. Santos, G. Bozkurt, G. S. P. Soylu, A. B. Yurtcan, Š. Miljanić and B. Šljukić, Electrochem. Commun., 2021, 124, 106963 CrossRef.
- M. Shamsuddin Ahmed, B. Choi and Y.-B. Kim, Sci. Rep., 2018, 8, 2543 CrossRef PubMed.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grützke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., 2014, 126, 8648–8652 CrossRef.
- Y. Hao, Y. Xu, J. Liu and X. Sun, J. Mater. Chem. A, 2017, 5, 5594–5600 RSC.
- C. F. Chen, G. King, R. M. Dickerson, P. A. Papin, S. Gupta, W. R. Kellogg and G. Wu, Nano Energy, 2015, 13, 423–432 CrossRef CAS.
- X. Cao, E. Johnson and M. Nath, J. Mater. Chem. A, 2019, 7, 9877–9889 RSC.
- W. Zhong, B. Xiao, Z. Lin, Z. Wang, L. Huang, S. Shen, Q. Zhang and L. Gu, Adv. Mater., 2021, 33, 2007894 CrossRef CAS PubMed.
- M. Sun, R.-T. Gao, X. Liu, R. Gao and L. Wang, J. Mater. Chem. A, 2020, 8, 25298–25305 RSC.
- G. Mei, H. Liang, B. Wei, H. Shi, F. Ming, X. Xu and Z. Wang, Electrochim. Acta, 2018, 290, 82–89 CrossRef CAS.
- S. Umapathi, H. Singh, J. Masud and M. Nath, Mater. Adv., 2021, 2(3), 927–932 RSC.
- H. Singh, J. Bernabe, J. Chern and M. Nath, J. Mater. Res., 2021, 36, 1413–1424 CrossRef CAS.
- M. Z. Xue and Z. W. Fu, Electrochem. Commun., 2006, 8, 1855–1862 CrossRef CAS.
- B. G. Amin, J. Masud and M. Nath, RSC Adv., 2019, 9, 37939–37946 RSC.
- P. D. Matthews, P. D. McNaughter, D. J. Lewis and P. O'Brien, Chem. Sci., 2017, 8, 4177–4187 RSC.
- J. He, A. Bhargav, H. Yaghoobnejad Asl, Y. Chen and A. Manthiram, Adv. Energy Mater., 2020, 10, 2001017 CrossRef CAS.
- A. Saxena, W. Liyanage, J. Masud, S. Kapila and M. Nath, J. Mater. Chem. A, 2021, 6, 4883–5230 Search PubMed.
- J. Masud, P.-C. Ioannou, N. Levesanos, P. Kyritsis and M. Nath, ChemSusChem, 2016, 9, 3128–3132 CrossRef CAS PubMed.
- F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, Chem. Mater., 2009, 22, 898–905 CrossRef.
- S. Sun, H. Miao, Y. Xue, Q. Wang, S. Li and Z. Liu, Electrochim. Acta, 2016, 214, 49–55 CrossRef CAS.
- Y. Zhong, X. H. Xia, F. Shi, J. Y. Zhan, J. P. Tu and H. J. Fan, Adv. Sci., 2016, 3(5), 1500286 CrossRef PubMed.
- C. Zhu, D. Gao, J. Ding, D. Chao and J. Wang, Chem. Soc. Rev., 2018, 47(12), 4332–4356 RSC.
- P. P. Patel, M. K. Datta, O. I. Velikokhatnyi, R. Kuruba, K. Damodaran, P. Jampani, B. Gattu, P. M. Shanthi, S. S. Damle and P. N. Kumta, Sci. Rep., 2016, 6, 1–14 CrossRef PubMed.
- N. Kazimova, K. Ping, M. Alam, M. Danilson, M. Merisalu, J. Aruväli, P. Paiste, M. Käärik, V. Mikli, J. Leis, K. Tammeveski, P. Starkov and N. Kongi, J. Catal., 2021, 395, 178–187 CrossRef CAS.
- D. M. Morales, M. A. Kazakova, S. Dieckhöfer, A. G. Selyutin, G. V. Golubtsov, W. Schuhmann and J. Masa, Adv. Funct. Mater., 2020, 30, 1905992 CrossRef CAS.
- S. H. Yetgin, J. Mater. Res. Technol., 2019, 8, 4725–4735 CrossRef CAS.
- M. N. Norizan, M. H. Moklis, S. Z. Ngah Demon, N. A. Halim, A. Samsuri, I. S. Mohamad, V. F. Knight and N. Abdullah, RSC Adv., 2020, 10, 43704–43732 RSC.
- M. S. Ahmed, B. Choi and Y.-B. Kim, Sci. Rep., 2018, 8, 2543 CrossRef PubMed.
- O. A. Oyetade and R. J. Kriek, Electrocatal, 2019, 11, 35–45 CrossRef.
- D. M. Morales, M. A. Kazakova, S. Dieckhöfer, A. G. Selyutin, G. V. Golubtsov, W. Schuhmann and J. Masa, Adv. Funct. Mater., 2020, 30, 1905992 CrossRef CAS.
- Y.-L. Zhang, K. Goh, L. Zhao, X.-L. Sui, X.-F. Gong, J.-J. Cai, Q.-Y. Zhou, H.-D. Zhang, L. Li, F.-R. Kong, D.-M. Gu and Z.-B. Wang, Nanoscale, 2020, 12, 21534–21559 RSC.
- M. Tavakkoli, E. Flahaut, P. Peljo, J. Sainio, F. Davodi, E. V. Lobiak, K. Mustonen and E. I. Kauppinen, ACS Catal., 2020, 10, 4647–4658 CrossRef CAS.
- D. M. Morales, M. A. Kazakova, S. Dieckhöfer, A. G. Selyutin, G. V. Golubtsov, W. Schuhmann and J. Masa, Adv. Funct. Mater., 2020, 30, 1905992 CrossRef CAS.
- A. P. Tiwari, D. Kim, Y. Kim and H. Lee, Adv. Energy Mater., 2017, 7, 1602217 CrossRef.
- M. Sakthivel and J. F. Drillet, Appl. Catal., B, 2018, 231, 62–72 CrossRef CAS.
- H. Singh, S. Zhuang, B. Ingis, B. B. Nunna and E. S. Lee, Carbon, 2019, 151, 160–174 CrossRef CAS.
- Y. Min, G. D. Moon, C. E. Kim, J. H. Lee, H. Yang, A. Soon and U. Jeong, J. Mater. Chem. C, 2014, 2, 6222–6248 RSC.
- S. Mishra, K. Song, K. C. Ghosh and M. Nath, ACS Nano, 2014, 8, 2077–2086 CrossRef CAS PubMed.
- J. Su, G. Xia, R. Li, Y. Yang, J. Chen, R. Shi, P. Jiang and Q. Chen, J. Mater. Chem. A, 2016, 4, 9204–9212 RSC.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 RSC.
- G. K. H. Wiberg, K. J. J. Mayrhofer and M. Arenz, Fuel Cells, 2010, 10, 575–581 CrossRef CAS.
- Q. Zhao, D. Zhong, L. Liu, D. Li, G. Hao and J. Li, J. Mater. Chem. A, 2017, 5, 14639–14645 RSC.
- A. T. Swesi, J. Masud and M. Nath, J. Mater. Res., 2016, 31, 2888–2896 CrossRef CAS.
- A. Milutinovi, Z. V Popovi, N. Tomi and S. Devi, Raman Spectroscopy of Polycrystalline-MnSe, 2004 Search PubMed.
- S. Sahoo, P. Pazhamalai, K. Krishnamoorthy and S. J. Kim, Electrochim. Acta, 2018, 268, 403–410 CrossRef CAS.
- L. Thi Mai Hoa, Diamond Relat. Mater., 2018, 89, 43–51 CrossRef CAS.
- H. Tang, X. Lu, H. Zhu, Y. Tian, R. Khatoon, Z. Zhu, Y.-J. Zeng, Q. Zhang and J. Lu, Ionics, 2020, 26, 43–49 CrossRef CAS.
- S. Rana, K. K. Yadav, S. K. Guchhait, S. T. Nishanthi, S. K. Mehta and M. Jha, J. Mater. Sci., 2021, 56, 8383–8395 CrossRef CAS.
- L. G. Cançado, A. Jorio, E. H. M. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
- J. Kucharek, R. Bożek and W. Pacuski, Acta Phys. Pol., A, 2019, 136(4), 598–602 CrossRef CAS.
- K. H. Cho, S. Park, H. Seo, S. Choi, M. Y. Lee, C. Ko and K. T. Nam, Angew. Chem., 2021, 133, 4723–4731 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: ECSA, chronoamperometry study, Raman spectra, XRD patterns, SEM images, XPS spectra after chronoamperometry studies, and LSV plots before and after chronoamperometry studies of Pt/GCE. See DOI: 10.1039/d1ta09864k |
| This journal is © The Royal Society of Chemistry 2022 |