
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
4,7-Diarylbenzo[c][1,2,5]thiadiazoles as fluorophores and visible light organophotocatalysts†‡
Dominic
Taylor
 a,
Thomas
Malcomson
b,
Adilet
Zhakeyev
c,
Shengxian
Cheng
d,
Georgina M.
Rosair
a,
Jose
Marques-Hueso
c,
Zhengtao
Xu
e,
Martin J.
Paterson
a,
Scott J.
Dalgarno
*a and
Filipe
Vilela
*a
a,
Thomas
Malcomson
b,
Adilet
Zhakeyev
c,
Shengxian
Cheng
d,
Georgina M.
Rosair
a,
Jose
Marques-Hueso
c,
Zhengtao
Xu
e,
Martin J.
Paterson
a,
Scott J.
Dalgarno
*a and
Filipe
Vilela
*a
aInstitute of Chemical Sciences, School of Engineering and Physical Science, Heriot-Watt University, Riccarton, Edinburgh EH14 4AS, UK. E-mail: S.J.Dalgarno@hw.ac.uk; F.Vilela@hw.ac.uk
bDepartment of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK
cInstitute of Sensors, Signals and Systems, School of Engineering and Physical Science, Heriot-Watt University, Riccarton, Edinburgh EH14 4AS, UK
dDepartment of Chemistry, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong
eInstitute of Materials Research and Engineering (IMRE), Agency of Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis, Singapore 138634
First published on 19th August 2022
Abstract
Electron donor–acceptor (D–A) systems based on the benzo[c][1,2,5]thiadiazole (BTZ) motif have been extensively researched for use in photovoltaics or as fluorescent sensors. However, their use as potential visible-light organophotocatalysts has not received any in-depth study. Here we report the synthesis, characterisation, and application of a library of 26 D–A compounds based on the BTZ group. By varying the donor groups whilst keeping the BTZ acceptor group the same, we have been able to systematically modify the photocatalyst's optoelectronic and photophysical properties. These photocatalysts were then validated using a Minisci-type decarboxylative alkylation of electron deficient heteroarenes as a test reaction under both batch and continuous flow conditions.
Introduction
Organic π-conjugated electron donor–acceptor (D–A) systems have firmly established themselves as a powerful design motif for applications such as organic photovoltaics, fluorophores and photocatalysts.1–4 Among the endless combinations of donor and acceptor groups, the benzo[c][1,2,5]thiadiazole (BTZ) motif has received appreciable attention as a strongly electron accepting moiety, primarily for use in photovoltaic applications but also as fluorescent sensors or bioimaging probes for lipid droplets, mitochondria and plasma membranes.5–12 The BTZ group has also been researched for photocatalytic applications although this has mainly been limited to heterogeneous systems involving BTZ containing metal–organic frameworks (MOFs),13–15 covalent organic frameworks (COFs),16–18 and conjugated porous polymers (CPPs).19–23In recent years, anthropogenic climate change and globally depleting resources have stimulated discussion within the chemical community about methods to perform environmentally sustainable chemistry, including the use of light as a “traceless” reagent. Concordant with this has been the rise of photoredox catalysis as a powerful tool for enabling organic transformations, with current state of the art photoredox catalysts based on iridium and ruthenium polypyridyl complexes.24,25 However, the cost and scarcity of the precious metals they contain calls into question the long term environmental sustainability of photocatalysis and contradicts the green ideals that modern photochemistry strives towards.26 Extensive research has been conducted into the use of organophotocatalysts as alternatives but these are mainly limited to a few chemical families including carbazoyl cyanobenzenes,27 benzophenones, xanthenes, thiazines and acridiniums.25,28 Recent examples have highlighted that the properties of various families of organophotocatalysts can be tailored towards more favorable redox potentials and photophysical properties. For example, Miyake and coworkers have demonstrated that installation of electron donating groups to phenoxazine photocatalysts was more conducive towards high reduction potentials than electron withdrawing groups.29 Similarly, Zeitler and coworkers have demonstrated that carbazoyl cyanoarenes can be tuned with the goal of achieving both strongly oxidizing and reducing photocatalysts from a common scaffold.27
Inspired by these systematic approaches towards tailoring the properties of photocatalysts, we sought to perform a similar study of photocatalysts based on the 4,7-diarylBTZ motif (see Fig. 1).5 While there has been selected reports of homogeneous BTZ photocatalysis arising from intramolecular charge transfer between the donor and acceptor units, there has been no in-depth exploration of how their photophysical properties and excited state redox potentials can be altered by manipulating available structural levers.30–35 The reaction that was selected to test this library of BTZ photocatalysts was the decarboxylative C–H functionalisation of heteroarenes, previously explored by Glorius and coworkers.36 This Minisci-type reaction was noted to provide a direct method to alkylate a wide variety of substrates, including pharmaceutical analogues, using visible light and without the need for pre-functionalisation of substrates.
 | ||
| Fig. 1 Structure of the 4,7-diarylBTZ photocatalysts with the BTZ group highlighted in pink (solid line) and the aryl groups highlighted blue (dashed lines). | ||
While the research conducted into homogeneous BTZ photocatalysis is limited in scope, more in-depth research has been devoted towards their fluorescence properties.5,7 The focus of research into BTZ fluorescence includes methods to tune the wavelengths of emission (particularly towards lower energy red light) and spans applications such as sensors,37 bioimaging agents,38 thermally activated delayed fluorophores,39 and organic LED devices.40 With this in mind, we sought to examine the fluorescence properties of our library of BTZ molecules in parallel with assessing their photocatalytic performance.
Herein we describe the synthesis of a library of 26 organophotocatalysts based on the BTZ motif and examine the effect that different structural modifications had on their optoelectronic and photophysical properties, with specific reference to tuning their absorption, fluorescence, and redox properties. The BTZ photocatalyst library was then validated using the decarboxylative C–H functionalisation of heteroarenes as a test reaction.
Results and discussion
Design and synthesis of photocatalyst library
In the design of this BTZ photocatalyst library (Fig. 1), we aimed to investigate how different structural modifications to the parent compound 4,7-diphenylbenzo[c][1,2,5]thiadiazole (pH-BTZ) influenced the compounds photophysical properties. In this study we elected to keep the BTZ core (the acceptor) constant across all photocatalysts and focus solely on investigating the effects of changing the donor aryl groups. With this in mind, our general design considerations included targeting changes that we envisaged would allow access to a wide window of redox potentials and bathochromically shift the absorption of light from ultraviolet to visible wavelengths. This led us to investigate the effect that introducing heavy atoms or electron donating/electron withdrawing substituent groups, extending π-conjugation, restricting rotation of the aryl groups, and replacing the phenyl rings with heterocycles had on the photocatalyst. The structures of all of the photocatalysts investigated as part of this study are shown in Fig. 2 along with the abbreviations used throughout this study.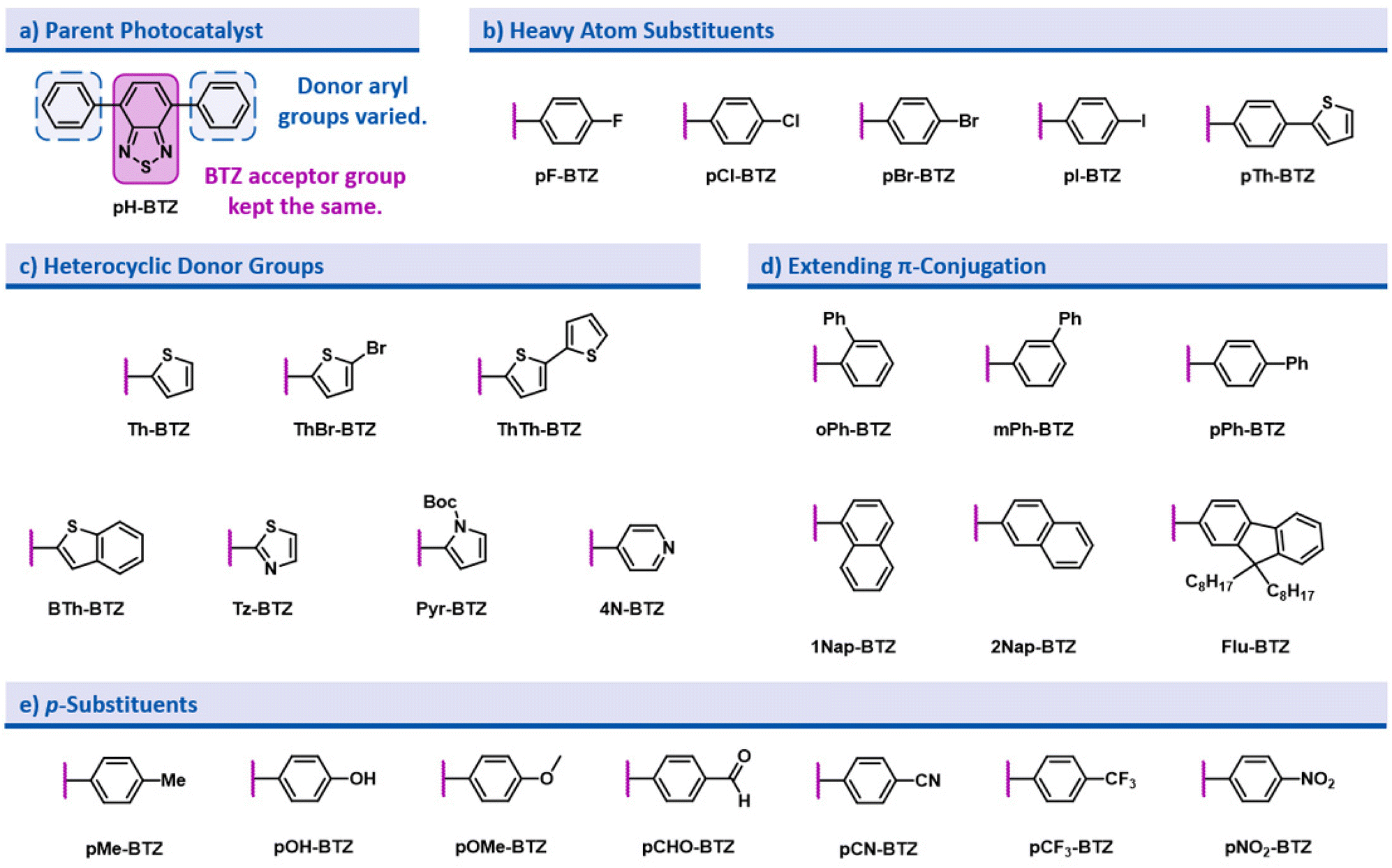 | ||
| Fig. 2 Summary of the different derivative structures of the parent photocatalyst pH-BTZ (a) that were investigated, and the abbreviations used. The different types of modifications made to pH-BTZ were (b) the addition of heavy atoms, (c) replacing the phenyl ring with heterocycles, (d) extension of π-conjugation and (e) the addition of p-substituents. | ||
The starting point for the synthesis of the photocatalyst library was 4,7-dibromobenzo[c][1,2,5]thiadiazole (Br2BTZ), as this can be cheaply synthesised through high yielding literature procedures on a large scale.41,42 With other common organophotocatalysts, such as acridiniums,28 BODIPYs,43 and phenothiazines,44 derivatisation can often be difficult or labourious: this can be a lengthy process as the syntheses often involve multiple steps. In comparison, by mainly employing Suzuki–Miyaura cross coupling we were able to take advantage of the diversity of aryl boronic acids that were commercially available and synthesise a large proportion of our library in a single step with moderate to excellent yields. For selected photocatalysts, additional reactions such as bromination, iodination or Miyaura borylation with a subsequent Suzuki–Miyaura cross couplings were also utilised (see section 2.2 of the ESI‡). Using these reactions, we were able to synthesise a total of 26 different prospective photocatalysts to investigate: in each case, UV-Vis. absorption/emission spectra and cyclic voltammograms were recorded and photostability assessed before moving onto testing their photocatalytic performance.
Absorption and emission properties
For each of the BTZ photocatalyst that was synthesised, their UV-Vis absorption and visible emission spectra were recorded in chloroform solution (see Table 1 and Fig. S30–S55 in the ESI‡). In the case pH-BTZ, which as the parent photocatalyst was used as a reference for all modifications, the maximum of light absorption (λabs) and emission (λem) were located at 380 and 482 nm respectively. The introduction of electron donating substituent groups in the para-position led to slight bathochromic shifts in both absorption and emission. The largest bathochromic shift in both absorbance and emission was observed with the introduction of a para-methoxy group (λabs of 409 and λem of 525 nm respectively). This shift in absorption and emission can be rationalised in terms of an increase in both the lowest unoccupied molecular orbital (LUMO) and, to a greater degree, the highest occupied molecular orbital (HOMO) energy levels of the photocatalyst, with the overall effect being a reduction in the HOMO–LUMO energy gap. Electron withdrawing groups produced the opposite result, leading to hypsochromic shifts in light absorption and emission. Of the electron withdrawing groups tested, the largest hypsochromic shift in absorption was produced by the introduction of trifluoromethyl group (λabs of 369 nm) while for emission this was produced by the nitro group (λem of 455 nm).| Entry | Compound |
λ
abs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) /nm
/nm |
λ
ema,b/nm |
Stokes shift/nm | ε M , /×103 M−1 cm−1 | PLQYa,d/% |
|---|---|---|---|---|---|---|
| a In CHCl3 solution. b Excitation wavelength was the wavelength of maximum absorption of the photocatalyst. c The molar attenuation coefficient (εM) was measured as described in section 4.2 of the ESI.‡ d Absolute PLQY. | ||||||
| 1 | pH-BTZ | 380 | 482 | 102 | 7.5 ± 0.1 | 86.4 |
| 2 | pF-BTZ | 381 | 495 | 114 | 7.5 ± 0.3 | 93.3 |
| 3 | pCl-BTZ | 383 | 490 | 107 | 10.4 ± 0.2 | 94.3 |
| 4 | pBr-BTZ | 381 | 492 | 111 | 9.8 ± 0.4 | 93.4 |
| 5 | pI-BTZ | 384 | 475 | 91 | 13.7 ± 0.2 | 91.9 |
| 6 | pTh-BTZ | 409 | 525 | 116 | 17.9 ± 0.6 | 86.9 |
| 7 | Th-BTZ | 446 | 552 | 106 | 12.1± 0.4 | 88.1 |
| 8 | ThBr-BTZ | 455 | 556 | 101 | 17.4 ± 0.5 | 91.4 |
| 9 | ThTh-BTZ | 505 | 611 | 106 | 22.8 ± 0.2 | 58.0 |
| 10 | BTh-BTZ | 450 | 548 | 98 | 16.6 ± 0.3 | 96.2 |
| 11 | Tz-BTZ | 428 | 514 | 86 | 17.3 ± 0.6 | 92.7 |
| 12 | Pyr-BTZ | 397 | 529 | 132 | 5.6 ± 0.1 | 73.4 |
| 13 | 4N-bTZ | 359 | 437 | 78 | 12.3 ± 0.3 | 66.1 |
| 14 | oPh-BTZ | 373 | 486 | 113 | 6.5 ± 0.2 | 100 |
| 15 | mPh-BTZ | 381 | 479 | 98 | 10.2 ± 0.4 | 92.3 |
| 16 | pPh-BTZ | 398 | 502 | 104 | 18.2 ± 0.3 | 97.3 |
| 17 | 1Nap-BTZ | 376 | 504 | 128 | 5.6 ± 0.1 | 23.6 |
| 18 | 2Nap-BTZ | 396 | 509 | 113 | 15.5 ± 0.3 | 66.2 |
| 19 | Flu-BTZ | 420 | 526 | 106 | 19.9 ± 0.1 | 96.8 |
| 20 | pMe-BTZ | 391 | 499 | 108 | 13.0 ± 0.4 | 92.7 |
| 21 | pOH-BTZ | 404 | 524 | 120 | 11.3 ± 0.1 | 89.1 |
| 22 | pOMe-BTZ | 409 | 525 | 116 | 9.8 ± 0.4 | 94.0 |
| 23 | pCHO-BTZ | 377 | 464 | 87 | 25.2 ± 0.2 | 85.2 |
| 24 | pCN-bTZ | 372 | 457 | 85 | 18.3 ± 0.3 | 93.9 |
| 25 | pCF3-BTZ | 369 | 459 | 90 | 9.4 ± 0.3 | 88.2 |
| 26 | pNO2-BTZ | 377 | 455 | 78 | 23.1 ± 1.0 | 4.87 |
The second type of modification that produced interesting shifts in absorption and emission was extending the π-conjugated structure of pH-BTZ. For example, replacing the 4,7-phenyl groups with 2-naphthyl groups in 2Nap-BTZ introduced a bathochromic shift in both light absorption and emission (λabs of 396 and λem of 509 nm). In comparison, the isomeric 1Nap-BTZ demonstrated a slight hypsochromic shift in absorption (376 nm), which could arise from a disruption of π-conjugation by introducing the sterically bulky 1-naphthyl groups. Some evidence of this was provided by computational modelling of the energy barrier to rotation of 1Nap-BTZ and pH-BTZ (see Fig. S88 and S90‡). As can be seen in these figures, the lowest energy conformation for pH-BTZ was determined to be when the phenyl groups sat at around 40° to the BTZ group. By comparison, the minimum energy for 1Nap-BTZ was around 70°, with an energy barrier to rotation of the 1-naphthyl groups of around 63 kJ mol−1 (compared to 8 kJ mol−1 for pH-BTZ). Similar trends were also observed by the introduction of phenyl groups in the ortho-, meta- and para-positions of pH-BTZ. In the case of pPh-BTZ, the phenyl group situated in the para-position extended π-conjugation and introduced a bathochromic shift in light absorption (with λabs of 398 nm). Conversely, the ortho-isomer oPh-BTZ featured a hypsochromic shift in absorption (λabs at 373 nm), which could also be explained by the steric bulk of the ortho-phenyl groups disrupting π-conjugation (see Fig. S91‡). With the meta-isomer, mPh-BTZ, the phenyl group was neither in a position to suitably extend π-conjugation in a meaningful way or twist the molecule, leading to no significant change in either absorption or emission.
The most dramatic changes in the absorption and emission spectra were observed when the phenyl rings were replaced with various heterocycles. For example, replacing the phenyl rings of pH-BTZ with 2-thienyl groups, to give Th-BTZ, resulted in a significant bathochromic shift in λabs to 446 nm and λem to 552 nm, again largely driven by an increase in the HOMO energy relative to the LUMO. The addition of a second 2-thienyl group to form ThTh-BTZ further shifted λabs to 505 nm, which represents a total bathochromic shift in absorption from pH-BTZ of 125 nm. ThTh-BTZ also featured the lowest energy emission of the entire series, with λem at 611 nm. This can be contrasted with 4N-BTZ, with the electron deficient 4-pyridyl group, which exhibited the highest energy absorption (λabs of 359 nm) and emission (λem of 437 nm) in the entire series.
The dependence of λabs and λem in various polar and non-polar solvents was also assessed for pH-BTZ (see Table S1‡). It was observed that, while λabs remained mainly unaffected by the solvent used λem varied by up to 31 nm, with more polar solvents leading to bathochromic shifts in emission (see Fig. S56 and S57‡). The dependence of the spectroscopic properties of pH-BTZ upon solvent can be visualised through a Lippert–Mataga plot of Stokes’ shift against the solvent polarity parameter (see Fig. S58‡), where a straight-line plot was obtained.45 The increased Stokes’ shift in more polar solvent suggests that the excited state of pH-BTZ has a higher dipole moment than its ground state, indicating a redistribution of charge following photoexcitation and the formation of an intramolecular charge transfer state.46–48 A linear relationship between the observed Stokes’ shift and solvent polarity was also attained when the Reichardt ET(30) parameter was used as an alternative measure of solvent polarity (see Fig. S59‡).49,50 Absolute photoluminescence quantum yields (PLQY) in chloroform solution were also measured for the entire library, with values in the range of 4.87–100% observed (see Table 1).
Photostability of BTZ photocatalysts
The photostability of each BTZ photocatalyst was assessed by irradiating a solution of the photocatalyst dissolved in oxygenated CDCl3 for 18 hours and comparing 1H NMR spectra recorded before and after. Most of the library exhibited good photostability, with little to no photobleaching observed during this time. Only Pyr-BTZ, 2Nap-BTZ and pCHO-BTZ exhibited significant photodecomposition (see Table S3‡).Electrochemical potentials
To assess the prospective performance of the library towards photoredox catalysis, cyclic voltammetry was performed on each photocatalyst to extract their ground state oxidation and reduction potentials. The S0 (v = 0) → S1 (v = 0) energy difference (E0,0) was estimated from the intercept between normalised absorption and emission spectra, then used to predict the excited state redox potentials as described by Romero and Nicewicz.25 Due to the low solubility of selected BTZ photocatalysts in acetonitrile (the typical solvent for cyclic voltammetry), DCM was used instead as this provided better dissolution of the photocatalysts whilst still allowing a wide range of potentials to be assessed (see Table S35 and Fig. S94–118‡).51In general, it was observed that the excited state reduction potentials 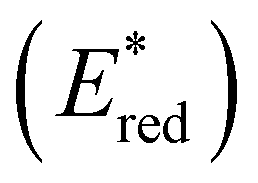 were far more sensitive towards changing the aryl donor groups than the excited state oxidation potentials
were far more sensitive towards changing the aryl donor groups than the excited state oxidation potentials 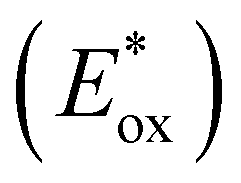 were. In comparison to pH-BTZ
were. In comparison to pH-BTZ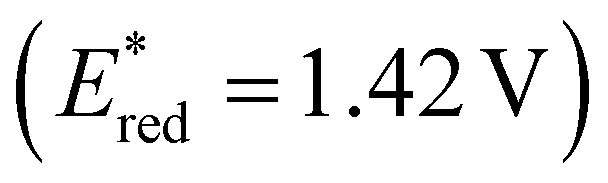 ,
,  was increased by the addition of electron withdrawing substituents onto the phenyl ring (p-CHO, p-CN, p-CF3 and p-NO2). The most extreme example of this was with the nitro-substituted pNO2-BTZ, which exhibited an
was increased by the addition of electron withdrawing substituents onto the phenyl ring (p-CHO, p-CN, p-CF3 and p-NO2). The most extreme example of this was with the nitro-substituted pNO2-BTZ, which exhibited an  of 1.91 V. Similarly, replacing the phenyl rings with electron deficient heterocycles such the 4-pyridyl groups of 4N-BTZ or the 2-thiazolyl groups of Tz-BTZ also increased
of 1.91 V. Similarly, replacing the phenyl rings with electron deficient heterocycles such the 4-pyridyl groups of 4N-BTZ or the 2-thiazolyl groups of Tz-BTZ also increased 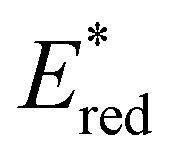 from 1.42 V to 1.88 V and 1.51 V respectively. Introducing electron donating groups (p-OH, p-OMe and p-Me) or electron rich heterocycles (such as pyrrole or thiophene rings) produced the opposite effect and led to a reduction in
from 1.42 V to 1.88 V and 1.51 V respectively. Introducing electron donating groups (p-OH, p-OMe and p-Me) or electron rich heterocycles (such as pyrrole or thiophene rings) produced the opposite effect and led to a reduction in  . Overall, the susceptibility of
. Overall, the susceptibility of 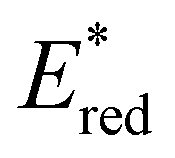 towards aryl group derivatisation allowed a wide window of potentials in the range of 0.95–1.91 V to be covered. The maximum
towards aryl group derivatisation allowed a wide window of potentials in the range of 0.95–1.91 V to be covered. The maximum 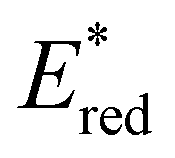 of 1.91 V attained for pNO2-BTZ is higher than some common organophotocatalysts, such as thiazines, BODIPYs and carbazoyl cyanobenzenes, and approaches the
of 1.91 V attained for pNO2-BTZ is higher than some common organophotocatalysts, such as thiazines, BODIPYs and carbazoyl cyanobenzenes, and approaches the 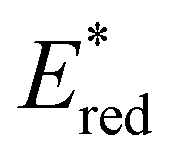 displayed by some widely utilised acridinium photocatalysts (for comparison the
displayed by some widely utilised acridinium photocatalysts (for comparison the 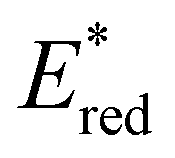 of Mes-Acr-Me+ is 2.08 V) (Fig. 3).27,28,52–54
of Mes-Acr-Me+ is 2.08 V) (Fig. 3).27,28,52–54
 | ||
Fig. 3 Comparison between the excited state reduction potentials 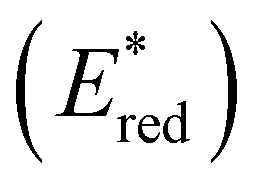 of selected BTZ photocatalysts (above axis) and common photocatalysts discussed in literature (below axis).27,28,52–54 of selected BTZ photocatalysts (above axis) and common photocatalysts discussed in literature (below axis).27,28,52–54 | ||
In comparison to the wide range of  , donor group variation produced a narrower window of
, donor group variation produced a narrower window of 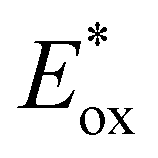 , with values in the range of −1.11 to −1.49 V, but no clear correlation between the type of modification made and the value of
, with values in the range of −1.11 to −1.49 V, but no clear correlation between the type of modification made and the value of 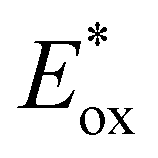 . In addition, several of the photocatalysts tested underwent chemical reactions at positive potentials during cyclic voltammetry.
. In addition, several of the photocatalysts tested underwent chemical reactions at positive potentials during cyclic voltammetry.
Computational results
In order to supplement the experimental characterisation, density functional theory (DFT) calculations were also carried out on all members of the photocatalyst library. Investigation of the rotational barriers around the donor–acceptor dihedral angles were undertaken via calculation of fully relaxed scans to determine global geometric minima (Fig. S88–92‡), revealing the distinct difference in the geometric behavior of phenyl and thiophene based donor groups. Photocatalysts with phenyl-based donor groups possessed a barrier of approximately 8–10 kJ mol−1 with geometric minima at 45 and 135°, showing a preference to avoid a planar geometry. In comparison the thiophene-based donor groups show a geometric preference for a planar geometry in which the sulphur atom is orientated at a 180° dihedral angle to the sulphur of the acceptor group with a rotational barrier of 15–20 kJ mol−1. A noted outlier in this trend was the 2-thiazolyl donor group (Tz-BTZ) which, while maintaining planarity, shows a preference to have the sulphurs orientated in the same direction (a dihedral of 0°) with a barrier of approx. 27 kJ mol−1; this is likely due to the less favorable interactions between in-plane lone pairs situated on the nitrogen atoms in both donor and acceptor groups when an angle of 180° was obtained. Alteration of the chalcogen atoms of the donor groups of Th-BTZ to either selenium or tellurium also resulted in a steady change in preference for a 0° dihedral angle with barriers of approximately 17 and 22 kJ mol−1 respectively (see Fig. S92‡).In addition to the experimental spectra presented, time dependent-DFT (TD-DFT) spectra were also obtained for each photocatalyst to further assess both their absorption and emission properties. A summary of the predicted absorption and emission maxima and a comparison to the experimental properties can be found in Table S4.‡ Computational absorption spectra were shown to be in strong agreement with those derived experimentally while the emission spectra obtained showed marginally less agreement (see Fig. S60–85 and Table S4‡). Computational analysis revealed that the lowest energy absorption band observed in each spectrum was characterised by a single state, S1, of the photocatalyst. In comparison, the second strong absorption band was characterised by states ranging from S2 to S10, depending on the photocatalyst involved.
Natural transition orbitals (NTOs) corresponding to the excitations to the bright states responsible for the first two peaks of the absorption spectra were calculated for all synthesised photocatalysts and can be found in Tables S5–S30.‡55 A general observation was that the hole orbital for the vast majority of bright transitions presented character on both the aryl donors and the benzo-portion of the BTZ acceptor group (see Fig. 4). In comparison, the particle orbital for the S0 → S1 transition shows movement of charge from the aryl groups, becoming localised over the entirety of the BTZ acceptor group (e.g., S0 → S1Th-BTZ). The particle orbital of the S0 → S3 transition of the Th-BTZ compound provides an example of the dominant transition in the second spectral peak in which less character is removed from the aryl groups and, instead of consolidating to the entire BTZ moiety, isolates to the 5-membered portion of the moiety. These results further reinforce the presence of an intramolecular charge transfer mechanism during light absorption, with electron density transferring from the aryl donor groups that constitute part of the hole orbital to the particle orbital localised on the BTZ group.
 | ||
| Fig. 4 NTO representations of the hole (left) and particle (right) orbitals of the bright states responsible for the two lower energy peaks observed in the absorption spectra. The S1 state of Th-BTZ (top) represents the common transition responsible for the lowest energy peak while the S2 transitions of Th-BTZ and pH-BTZ (middle and bottom, respectively) represent the two different transitions that may contribute to the second peak. | ||
Single crystal X-ray structures
For a number of the BTZ photocatalysts investigated as part of this library, we were able to grow single crystals suitable for X-ray diffraction and subsequently determine their crystal structure. The crystal structures of pF-BTZ, pCl-BTZ, pI-BTZ, ThTh-BTZ, BTh-BTZ, oPh-BTZ, mPh-BTZ, pPh-BTZ, 1Nap-BTZ, 2Nap-BTZ, pMe-BTZ, pOH-BTZ and pCF3-BTZ are all presented in the ESI (see Fig. S2–S29‡) and stored in the Cambridge Structural Database (CCDC 2179269–2179281‡). Unsurprisingly, most of the para-substituted BTZ photocatalysts possessed fairly similar structures. Due to the low solubility of pOH-BTZ in most solvents, the single crystal had to be grown from THF solution, which led to the formation of a solvate consisting of pOH-BTZ molecules engaged in hydrogen bonding with the THF solvent molecule.BTh-BTZ and ThTh-BTZ were both observed to be completely planar in the crystal state, which is consistent with the previously reported crystal structure of Th-BTZ.56 The planar structure of BTh-BTZ also echoes the preference to also be planar in solution as we determined using DFT calculations (see above). The structures of BTh-BTZ and ThTh-BTZ also exhibited some disorder regarding the relative orientation of the thiophene rings.
Photocatalytic application of BTZ library
To test our BTZ photocatalysts, we selected the alkylation of electron deficient heteroarenes via a Minisci-style C–H radical functionalisation as a test photoredox reaction.57 Nitrogen containing heteroarenes, such as quinolines, pyridines, quinazolines etc., are abundant structural motifs in nature that have a become commonplace features among various pharmaceutical compounds.58 As many of these heteroarenes feature alkyl regions, including examples such as Paperverine, Bendamustine and Dabigatran (Fig. 5a), several methods for selective alkylation have been explored by various groups (Fig. 5b).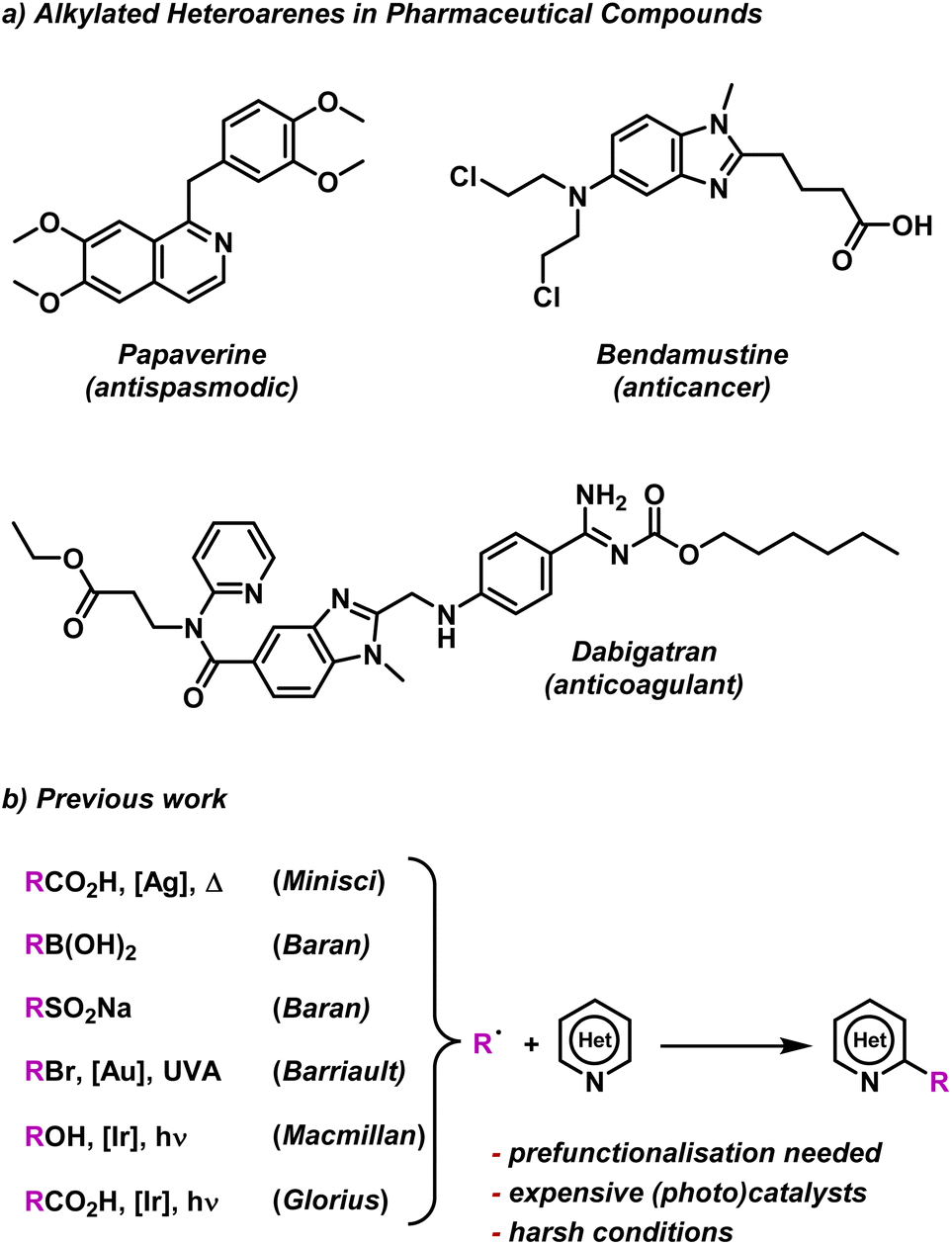 | ||
| Fig. 5 (a) Common pharmaceutical molecules containing alkylated heterocycles. (b) Examples of previous methods to alkylate electron deficient heterocycles.36,59–64 | ||
The classical Minisci reaction generates alkyl radicals (R˙) via a Ag(I) catalysed decarboxylation of carboxylic acids using S2O82− as the oxidant.59 The scope of possible R˙ sources has since been expanded to include a wide variety of functional groups, including aryl boronic acids and sulfinates.60–62 However, such methods often present harsh conditions, like the conventional Minisci coupling, or require pre-functionalisation of substrates. Barriault and co-workers have developed a method to alkylate heteroarenes using R˙ generated photocatalytically from widely available alkyl bromides.63 However, this method utilised high loadings of gold catalysts and UVA irradiation (315–400 nm). The high energy UV light could lower the functional group tolerance of this protocol, a problem that would be remedied by the switching to lower energy visible light. Methods for alkylation of heteroarenes via visible light photoredox catalysis have been reported by both Macmillan and coworkers and Glorius and coworkers, using alcohols and carboxylic acids as the R˙ sources respectively.36,64 While these methods both address the issues of harsh reaction conditions, low functional group tolerance and required pre-functionalisation associated with other protocols, expensive iridium-based photocatalysts were employed.
In order to the test the suitability of BTZ photocatalysts towards a Minisci-type reaction, we adapted a protocol developed by Glorius and coworkers.36 The reaction conditions were first optimised using pH-BTZ as the photocatalyst (Table 2, entry 1). Using lepidine as the substrate, cyclohexanecarboxylic acid (10 equivalents) as the alkyl radical source, ammonium persulfate (2 equivalents) as the oxidant and DMSO as the solvent, we were able to attain a conversion of 48% in 16 h using a 12 W LED module (410–420 nm). From these initial conditions, we then varied the choice of solvent and oxidant, although we were unable to improve from the initial conditions. Replacing the ammonium persulfate oxidant with potassium persulfate (K2S2O8, entry 2) still permitted the reaction to occur, although with significantly diminished conversion. Similarly, replacing DMSO with other solvents severely lowered the conversion (entries 3 and 4). A potential reason for this could be due to S2O82− reportedly decomposing more readily in DMSO compared to other solvents to generate the reactive species.65 Control experiments were also performed, showing that the reaction did not proceed in the absence of light, oxidant and photocatalyst (entries 5–7). Additionally, not degassing the reaction also drastically reduced the conversion attained (entry 8). We also observed that the conversion was drastically reduced by the addition of TEMPO or CeCl3, which would act as radical traps for R˙ and the sulfate radical anion respectively.66,67 This would suggest the involvement of both the cyclohexyl and sulfate radicals in the reaction, in agreement with the mechanism previously proposed by Glorius and coworkers.36
|
|
||||
|---|---|---|---|---|
| Entrya | Photocatalyst | Oxidant | Solvent | Conversionb (%) |
| a Reaction conditions: lepidine (0.3 mmol), oxidant (0.6 mmol), cyclohexane carboxylic acid (3 mmol), photocatalyst (0.015 mmol), solvent (3 mL), 12 W 410–420 nm LED, 16 h. b Conversion determined by 1H NMR. c Reaction performed in the dark. d Reaction was performed without degassing the reaction mixture with nitrogen. e Reaction performed for 40 h. f Isolated yield in brackets. N.R. = no reaction. | ||||
| 1 | pH-BTZ | (NH4)2S2O8 | DMSO | 48 |
| 2 | pH-BTZ | K2S2O8 | DMSO | 15 |
| 3 | pH-BTZ | (NH4)2S2O8 | DMF | N.R. |
| 4 | pH-BTZ | (NH4)2S2O8 | MeCN | 4 |
| 5 | pH-BTZ | — | DMSO | <1 |
| 6c | pH-BTZ | (NH4)2S2O8 | DMSO | N.R. |
| 7 | — | (NH4)2S2O8 | DMSO | N.R. |
| 8d | pH-BTZ | (NH4)2S2O8 | DMSO | 4 |
| 9 | oPh-BTZ | (NH4)2S2O8 | DMSO | 53 |
| 10 | mPh-BTZ | (NH4)2S2O8 | DMSO | 25 |
| 11 | pPh-BTZ | (NH4)2S2O8 | DMSO | N.R. |
| 12 | 1Nap-BTZ | (NH4)2S2O8 | DMSO | 57 |
| 13 | 2Nap-BTZ | (NH4)2S2O8 | DMSO | 16 |
| 14e,f | 1Nap-BTZ | (NH4)2S2O8 | DMSO | >99 (73) |
Using these initial conditions, we then proceeded to screen our photocatalyst library: the key results are summarised in Table 2, while all reaction conditions tested can be found in Table S36.‡ In most cases, derivatisation of pH-BTZ had a detrimental effect on the conversion of the reaction. Furthermore, no correlation could be observed between the conversion and the excited state oxidation or reduction potentials. This suggested that the reaction was not solely dependent on the optoelectronic properties of the photocatalyst but could also be influenced by other factors, such as the lifetime of the excited state. We observed that, of all of the modifications made, only limiting rotation of the aryl side group by introducing significant steric bulk resulted in any increase in conversion. Replacing the 4,7-phenyl groups of pH-BTZ with 1-naphthyl groups to form 1Nap-BTZ increased the conversion attained to 57%. For comparison, the 2-naphthyl analogue, 2Nap-BTZ, resulted in a conversion of 16%. These results were echoed by the installation of a phenyl ring in either the ortho-, meta- or para-positions. The highest conversion of 53% was produced by oPh-BTZ while its meta- and para-isomers, mPh-BTZ and pPh-BTZ, gave conversions of 25% and 0% respectively. To further increase the conversion obtained using 1Nap-BTZ, we subsequently increased the reaction time from 16 hours to 40 hours, leading to a conversion of >99%.
These initial optimisation results can be directly compared with the work performed by Glorius and coworkers to gauge how BTZ compared to established photoredox catalysts.36 Using Mes-Acr-Me+ and [Ru(bpy)3](PF6)2 (at 2 mol% loading), the authors reported attaining conversions of 36 and 28% in 16 hours, suggesting that 1Nap-BTZ, along with pH-BTZ and oPh-BTZ, are comparable to some well investigated organic and metal-based photoredox catalysts. However, the authors also reported that by employing [Ir(dF(CF3)ppy)2(dtbpy)][PF6] (at 0.5 mol% loading) as the photocatalyst, 94% conversion could be obtained within 90 minutes. This indicates that 1Nap-BTZ was not the optimum photocatalyst for the reaction shown in Table 2, however, this could be offset by the lower costs of using BTZ based organophotocatalysts compared to the more expensive [Ir(dF(CF3)ppy)2(dtbpy)][PF6].
Using these optimised reaction conditions and employing 1Nap-BTZ as our photocatalyst, we then proceeded to investigate the scope of the reaction with selected heteroarenes and carboxylic acids (Scheme 1). Under the reaction conditions, various electron deficient nitrogen-based heterocycles including quinolines, pyridines, quinazolines and benzimidazoles (1–6) were successfully alkylated (31–73% yield). Expanding the scope of carboxylic acids revealed that the reaction was suitable for installing primary, secondary and tertiary alkyl groups (1, 7 and 8) in good yields (47–73%). However, in the case of 8, additional oxidant was required to attain full conversion, possibly due to the lower stability of the primary radicals generated. N-Acetyl glycine was also employed as a carboxylic acid in this reaction, with a yield of 95% obtained (9), demonstrating that this reaction could also be extended to amino acids. To demonstrate the pharmaceutical relevance of this reaction, we also applied 1Nap-BTZ to the synthesis of analogue of the antispasmodic drug Papaverine using isoquinoline and homoveratric acid (10, 57% yield).
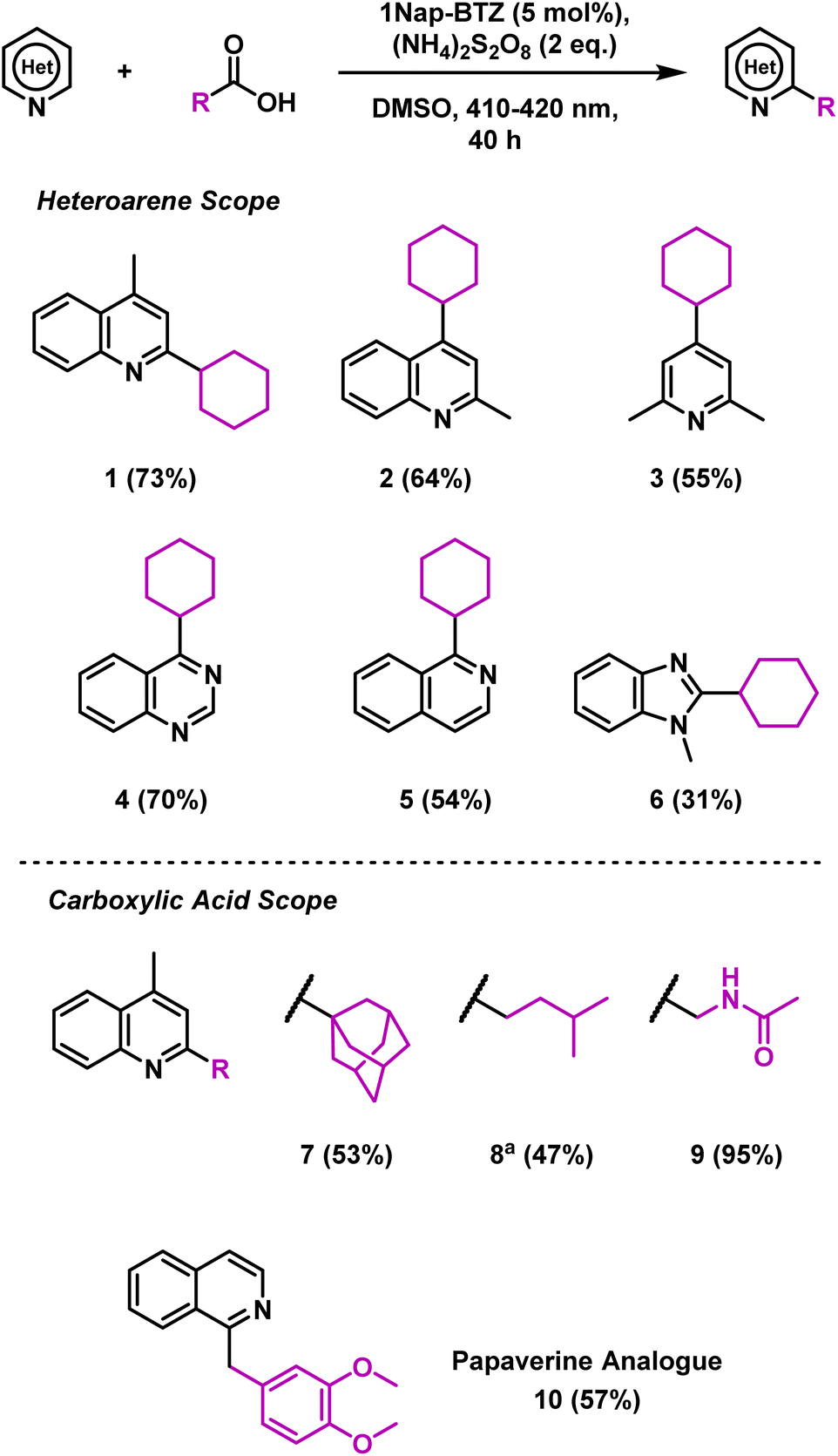 | ||
| Scheme 1 Scope of heteroarenes and carboxylic Acids. Reaction conditions: heteroarene (0.3 mmol), (NH4)2S2O8 (0.6 mmol), carboxylic acid (3 mmol), 1Nap-BTZ (0.015 mmol), DMSO (3 mL), 12 W 410–420 nm LED, 40 h. Isolated yields shown in brackets. a4 equivalents of (NH4)2S2O8 used. | ||
Under the optimised reaction conditions 1Nap-BTZ required 40 hours to achieve complete conversion. To lower this reaction time, we also investigated performing this reaction under continuous flow conditions. By flowing the reaction mixture through narrow transparent tubes, a smaller pathlength for the incident light was achieved in comparison to the performing the reaction in a vial. This resulted in increased and more uniform photon flux, which has been well reported to drastically accelerate photochemical reactions.68,69
Using a commercial flow reactor (Easy-Photochem E-Series, Vapourtec), we began by testing the alkylation of lepidine using cyclohexanecarboxylic acid under analogous conditions to the optimised batch conditions (Table 2, entry 12). The reaction mixture was continually recirculated through a 5 mL coil of tubing at a rate of 1 mL min−1 under irradiation from the same LED that was utilised in the batch reactions (Fig. 6). After 16 hours, a conversion of >99% conversion was achieved, which was a significant improvement compared to the 40 hours required under batch conditions. It should be noted that the external LED module could only irradiate the 5 mL coil from a single side, which is not the most effective means of delivering light. We were further able to reduce the time required by switching to the flow machines built in LED module (UV-150 reactor module equipped with a 420 nm LED, Fig. S121‡), which has a power of 60 W compared to 12 W for the in-house built LED array. This light source is designed to irradiate the tubing from the middle of the reactor coil, which presents a more efficient 360° method of supplying photons to the reaction. Employing this module reduced the time required to achieve >99% conversion to 100 minutes, which is an overall 24-fold decrease in the reaction time needed under the initial batch conditions.
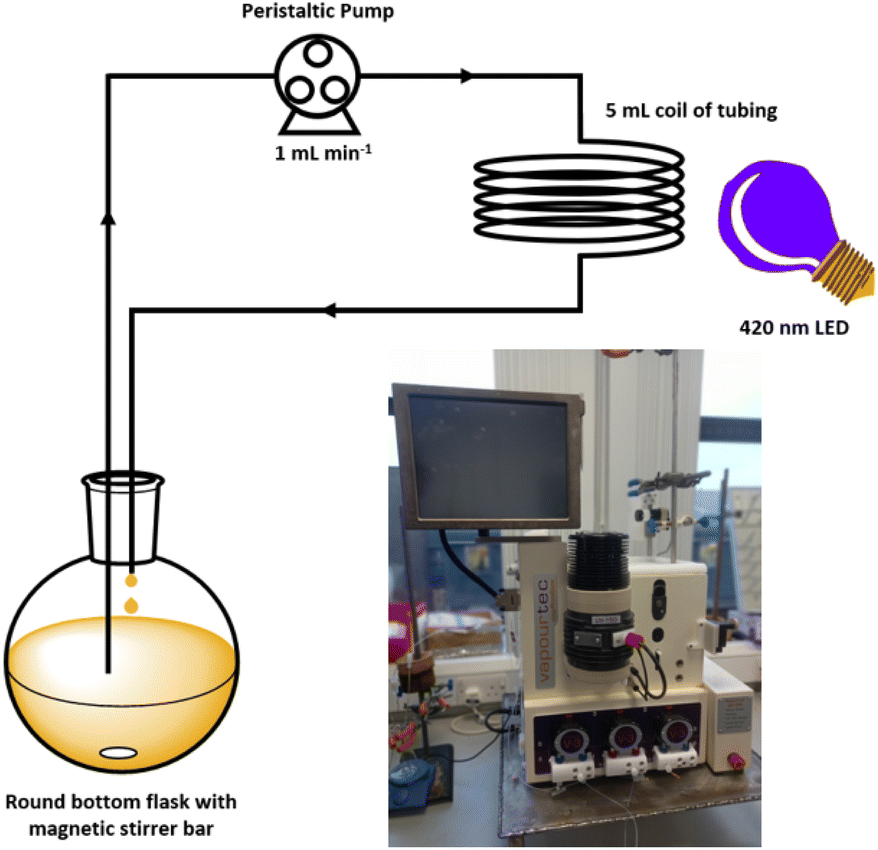 | ||
| Fig. 6 Schematic representation of the continuous flow set up used to reduce the time required for the Minisci-type reaction. Inset picture shows the Easy-Photochem flow system from Vapourtec equipped with the UV-150 photoreactor. | ||
Conclusions
In conclusion, a library of 26 electron donor–acceptor photocatalysts based on the BTZ group have been synthesised and studied, both experimentally and computationally. By varying the electron donor groups whilst keeping the BTZ constant, systematic variations in the photophysical and optoelectronic properties of the photocatalysts were obtained. This included tuning λabs to cover a range of 359–505 nm and λem to cover a range of 437–611 nm, with the introduction of various heteroarene groups producing the starkest variation in properties. Furthermore, was observed to be sensitive to the nature of the electron donor groups present on the photocatalyst: the presence of electron rich donor groups reduced the value of
was observed to be sensitive to the nature of the electron donor groups present on the photocatalyst: the presence of electron rich donor groups reduced the value of 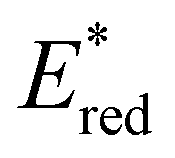 while electron deficient donor groups instead raised
while electron deficient donor groups instead raised 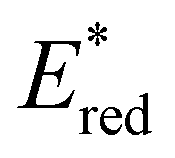 . This ultimately allowed
. This ultimately allowed 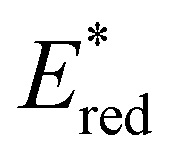 to cover a range of values from 0.95 V for ThTh-BTZ to 1.91 V for pNO2-BTZ. These photocatalysts were then implemented in a Minisci-type alkylation of heteroarenes via carboxylic acid decarboxylation, resulting in good to excellent yields. Although the reaction under batch conditions required a long time for completion (>99% in 40 hours using 1Nap-BTZ), the use of continuous flow conditions allowed this time to be reduced to less than two hours, which further highlights the benefits of combining photoredox catalysis with flow technologies. In this study, we selected to solely focus on investigating the effect that changing the electron donor aryl groups had on the properties of the photocatalyst. This leaves the door open for further exploration of the strength of the acceptor BTZ core as a means to tune the properties of the photocatalyst, including substitution of the 5,6-positions, replacement of the sulphur atom with other chalcogen elements and/or extending the BTZ core with fused aromatic ring(s).
to cover a range of values from 0.95 V for ThTh-BTZ to 1.91 V for pNO2-BTZ. These photocatalysts were then implemented in a Minisci-type alkylation of heteroarenes via carboxylic acid decarboxylation, resulting in good to excellent yields. Although the reaction under batch conditions required a long time for completion (>99% in 40 hours using 1Nap-BTZ), the use of continuous flow conditions allowed this time to be reduced to less than two hours, which further highlights the benefits of combining photoredox catalysis with flow technologies. In this study, we selected to solely focus on investigating the effect that changing the electron donor aryl groups had on the properties of the photocatalyst. This leaves the door open for further exploration of the strength of the acceptor BTZ core as a means to tune the properties of the photocatalyst, including substitution of the 5,6-positions, replacement of the sulphur atom with other chalcogen elements and/or extending the BTZ core with fused aromatic ring(s).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
FV and SJD thank the EPSRC funded (EP/L016419/1) CRITICAT centre for doctoral training for DT's PhD funding. We acknowledge Vapourtec for their invaluable technical support. MJP thanks the EPSRC for funding through grants EP/P001459, EP/T021675 and EP/V006746. JMH and AZ thank the EPSRC for support under Grant EP/T013680/1. The authors also thank Stuart Thomson and Anna Gakamsky from Edinburgh Instruments Ltd for their help with the optical characterisation.References
- X. Wan, C. Li, M. Zhang and Y. Chen, Acceptor–donor–acceptor type molecules for high performance organic photovoltaics – chemistry and mechanism, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- W. Che, Y. Xie and Z. Li, Structural Design of Blue–to–Red Thermally–Activated Delayed Fluorescence Molecules by Adjusting the Strength between Donor and Acceptor, Asian J. Org. Chem., 2020, 9, 1262–1276 CrossRef CAS.
- Q.-A. Wu, F. Chen, C.-C. Ren, X.-F. Liu, H. Chen, L.-X. Xu, X.-C. Yu and S.-P. Luo, Donor–acceptor fluorophores as efficient energy transfer photocatalysts for [2 + 2] photodimerization, Org. Biomol. Chem., 2020, 18, 3707–3716 RSC.
- H. Deol, G. Singh, M. Kumar and V. Bhalla, Phenazine-Based Donor Acceptor Systems as Organic Photocatalysts for “Metal-free” C–N/C–C Cross-Coupling, J. Org. Chem., 2020, 85, 11080–11093 CrossRef CAS PubMed.
- B. A. D. Neto, A. A. M. Lapis, E. N. Da Silva Júnior and J. Dupont, 2,1,3-Benzothiadiazole and derivatives: Synthesis, properties, reactions, and applications in light technology of small molecules, Eur. J. Org. Chem., 2013, 228–255 CrossRef CAS.
- J. Du, M. C. Biewer and M. C. Stefan, Benzothiadiazole building units in solution-processable small molecules for organic photovoltaics, J. Mater. Chem. A, 2016, 4, 15771–15787 RSC.
- B. A. D. Neto, P. H. P. R. Carvalho and J. R. Correa, Benzothiadiazole Derivatives as Fluorescence Imaging Probes: Beyond Classical Scaffolds, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS PubMed.
- Y. Yu and C. Dong, An efficient colorimetric and fluorescent probe for the detection of fluoride ions based on a benzothiadiazole derivative, Anal. Methods, 2015, 7, 9604–9608 RSC.
- S. Uchiyama, K. Kimura, C. Gota, K. Okabe, K. Kawamoto, N. Inada, T. Yoshihara and S. Tobita, Environment-sensitive fluorophores with benzothiadiazole and benzoselenadiazole structures as candidate components of a fluorescent polymeric thermometer, Chem. – Eur. J., 2012, 18, 9552–9563 CrossRef CAS PubMed.
- G. A. Medeiros, J. R. Correa, L. P. de Andrade, T. O. Lopes, H. C. B. de Oliveira, A. B. Diniz, G. B. Menezes, M. O. Rodrigues and B. A. D. Neto, A benzothiadiazole-quinoline hybrid sensor for specific bioimaging and surgery procedures in mice, Sens. Actuators, B, 2021, 328, 128998 CrossRef CAS.
- H. Appelqvist, K. Stranius, K. Börjesson, K. P. R. Nilsson and C. Dyrager, Specific Imaging of Intracellular Lipid Droplets Using a Benzothiadiazole Derivative with Solvatochromic Properties, Bioconjugate Chem., 2017, 28, 1363–1370 CrossRef CAS PubMed.
- A. A. R. Mota, P. H. P. R. Carvalho, B. C. Guido, H. C. B. de Oliveira, T. A. Soares, J. R. Corrêa and B. A. D. Neto, Bioimaging, cellular uptake and dynamics in living cells of a lipophilic fluorescent benzothiadiazole at low temperature (4 °C), Chem. Sci., 2014, 5, 3995 RSC.
- J.-K. Jin, K. Wu, X.-Y. Liu, G.-Q. Huang, Y.-L. Huang, D. Luo, M. Xie, Y. Zhao, W. Lu, X.-P. Zhou, J. He and D. Li, Building a Pyrazole–Benzothiadiazole–Pyrazole Photosensitizer into Metal–Organic Frameworks for Photocatalytic Aerobic Oxidation, J. Am. Chem. Soc., 2021, 143, 21340–21349 CrossRef CAS PubMed.
- W.-Q. Zhang, Q.-Y. Li, Q. Zhang, Y. Lu, H. Lu, W. Wang, X. Zhao and X.-J. Wang, Robust Metal–Organic Framework Containing Benzoselenadiazole for Highly Efficient Aerobic Cross-dehydrogenative Coupling Reactions under Visible Light, Inorg. Chem., 2016, 55, 1005–1007 CrossRef CAS PubMed.
- S. Goswami, C. E. Miller, J. L. Logsdon, C. T. Buru, Y.-L. Wu, D. N. Bowman, T. Islamoglu, A. M. Asiri, C. J. Cramer, M. R. Wasielewski, J. T. Hupp and O. K. Farha, Atomistic Approach toward Selective Photocatalytic Oxidation of a Mustard-Gas Simulant: A Case Study with Heavy-Chalcogen-Containing PCN-57 Analogues, ACS Appl. Mater. Interfaces, 2017, 9, 19535–19540 CrossRef CAS PubMed.
- S. Wang, Q. Sun, W. Chen, Y. Tang, B. Aguila, Y. Pan, A. Zheng, Z. Yang, L. Wojtas, S. Ma and F. S. Xiao, Programming Covalent Organic Frameworks for Photocatalysis: Investigation of Chemical and Structural Variations, Matter, 2020, 2, 416–427 CrossRef CAS.
- S. Li, L. Li, Y. Li, L. Dai, C. Liu, Y. Liu, J. Li, J. Lv, P. Li and B. Wang, Fully Conjugated Donor-Acceptor Covalent Organic Frameworks for Photocatalytic Oxidative Amine Coupling and Thioamide Cyclization, ACS Catal., 2020, 10, 8717–8726 CrossRef CAS.
- W. Chen, Z. Yang, Z. Xie, Y. Li, X. Yu, F. Lu and L. Chen, Benzothiadiazole functionalized D-A type covalent organic frameworks for effective photocatalytic reduction of aqueous chromium(VI), J. Mater. Chem. A, 2019, 7, 998–1004 RSC.
- D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Conjugated porous polymers: incredibly versatile materials with far-reaching applications, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC.
- D. Taylor, S. J. Dalgarno and F. Vilela, Structure–function Relationship in Conjugated Porous Polymers, in Concepts and Design of Materials Nanoarchitectonics, 2022, pp. 226–246 Search PubMed.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Surface Area Control and Photocatalytic Activity of Conjugated Microporous Poly(benzothiadiazole) Networks, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- H. Urakami, K. Zhang and F. Vilela, Modification of conjugated microporous poly-benzothiadiazole for photosensitized singlet oxygen generation in water, Chem. Commun., 2013, 49, 2353–2355 RSC.
- R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. I. Zhang, Poly(benzothiadiazoles) and Their Derivatives as Heterogeneous Photocatalysts for Visible-Light-Driven Chemical Transformations, ACS Catal., 2018, 8, 4735–4750 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- E. Speckmeier, T. G. Fischer and K. Zeitler, A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L. C. Campeau, D. Nicewicz and D. A. DiRocco, Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed.
- B. G. McCarthy, R. M. Pearson, C.-H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, Structure–Property Relationships for Tailoring Phenoxazines as Reducing Photoredox Catalysts, J. Am. Chem. Soc., 2018, 140, 5088–5101 CrossRef CAS PubMed.
- Y. Takaguchi, H. Miyake, T. Izawa, D. Miyamoto, R. Sagawa and T. Tajima, Molecular design of benzothiadiazole-based dyes for working with carbon nanotube photocatalysts, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 707–711 CrossRef CAS.
- R. Li, D. W. Gehrig, C. Ramanan, P. W. M. Blom, F. F. Kohl, M. Wagner, K. Landfester and K. A. I. Zhang, Visible Light-Mediated Conversion of Alcohols to Bromides by a Benzothiadiazole-Containing Organic Photocatalyst, Adv. Synth. Catal., 2019, 361, 3852–3859 CrossRef CAS.
- M. Bartolini, V. Gombac, A. Sinicropi, G. Reginato, A. Dessì, A. Mordini, J. Filippi, T. Montini, M. Calamante, P. Fornasiero and L. Zani, Tuning the Properties of Benzothiadiazole Dyes for Efficient Visible Light-Driven Photocatalytic H2 Production under Different Conditions, ACS Appl. Energy Mater., 2020, 3, 8912–8928 CrossRef CAS.
- E. Broumidis, C. M. S. Jones, M. Koyioni, A. Kourtellaris, G. O. Lloyd, J. Marques-Hueso, P. A. Koutentis and F. Vilela, 8,8′-(Benzo[c,][1,2,5]thiadiazole-4,7-diyl)bis(quinolin-4(1H)-one): a twisted photosensitizer with AIE properties, RSC Adv., 2021, 11, 29102–29107 RSC.
- Z. Wang, H. Li, Z. Wang, M. Suleman, Y. Wang and P. Lu, Photocatalytic Approach for Construction of 5,6-Dihydroimidazo[2,1-a]isoquinolines and Their Luminescent Properties, J. Org. Chem., 2021, 86, 8101–8111 CrossRef CAS PubMed.
- S. Bhattacharyya, S. R. Ali, M. Venkateswarulu, P. Howlader, E. Zangrando, M. De and P. S. Mukherjee, Self-Assembled Pd12 Coordination Cage as Photoregulated Oxidase-Like Nanozyme, J. Am. Chem. Soc., 2020, 142, 18981–18989 CrossRef CAS PubMed.
- R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, Visible Light-Mediated Direct Decarboxylative C-H Functionalization of Heteroarenes, ACS Catal., 2017, 7, 4057–4061 CrossRef CAS.
- M. Alfonso, A. Espinosa, A. Tárraga and P. Molina, Multifunctional Benzothiadiazole-Based Small Molecules Displaying Solvatochromism and Sensing Properties toward Nitroarenes, Anions, and Cations, ChemistryOpen, 2014, 3, 242–249 CrossRef CAS PubMed.
- J. Zhang, A. Konsmo, A. Sandberg, X. Wu, S. Nyström, U. Obermüller, B. M. Wegenast-Braun, P. Konradsson, M. Lindgren and P. Hammarström, Phenolic Bis-styrylbenzo[c,]-1,2,5-thiadiazoles as Probes for Fluorescence Microscopy Mapping of Aβ Plaque Heterogeneity, J. Med. Chem., 2019, 62, 2038–2048 CrossRef CAS PubMed.
- F. Ni, Z. Wu, Z. Zhu, T. Chen, K. Wu, C. Zhong, K. An, D. Wei, D. Ma and C. Yang, Teaching an old acceptor new tricks: rationally employing 2,1,3-benzothiadiazole as input to design a highly efficient red thermally activated delayed fluorescence emitter, J. Mater. Chem. C, 2017, 5, 1363–1368 RSC.
- Z. Wang, Z. Wang, P. Lu and Y. Wang, Preparation and Photoluminescent Properties of Three 5-Amino Benzothiadiazoles (5-amBTDs), Chem. – Asian J., 2020, 15, 3519–3526 CrossRef CAS PubMed.
- B. A. D. Neto, A. S. A. Lopes, G. Ebeling, R. S. Gonçalves, V. E. U. Costa, F. H. Quina and J. Dupont, Photophysical and electrochemical properties of π-extended molecular 2,1,3-benzothiadiazoles, Tetrahedron, 2005, 61, 10975–10982 CrossRef.
- J. P. Heiskanen, P. Vivo, N. M. Saari, T. I. Hukka, T. Kastinen, K. Kaunisto, H. J. Lemmetyinen and O. E. O. Hormi, Synthesis of Benzothiadiazole Derivatives by Applying C-C Cross-Couplings, J. Org. Chem., 2016, 81, 1535–1546 CrossRef CAS PubMed.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, BODIPY dyes in photodynamic therapy, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- M. Abdallah, T. T. Bui, F. Goubard, D. Theodosopoulou, F. Dumur, A. Hijazi, J. P. Fouassier and J. Lalevée, Phenothiazine derivatives as photoredox catalysts for cationic and radical photosensitive resins for 3D printing technology and photocomposite synthesis, Polym. Chem., 2019, 10, 6145–6156 RSC.
- E. Lippert, Dipolmoment und Elektronenstruktur von angeregten Molekülen, Z. Naturforsch., A: Astrophys., Phys. Phys. Chem., 1955, 10, 541–545 CrossRef.
- M. Chen, H. Nie, B. Song, L. Li, J. Z. Sun, A. Qin and B. Z. Tang, Triphenylamine-functionalized tetraphenylpyrazine: Facile preparation and multifaceted functionalities, J. Mater. Chem. C, 2016, 4, 2901–2908 RSC.
- S. Chen, X. Li and L. Song, A fluorescent photochromic diarylethene based on naphthalic anhydride with strong solvatochromism, RSC Adv., 2017, 7, 29854–29859 RSC.
- D. Jana and S. Jana, Donor-Pyrene-Acceptor Distance-Dependent Intramolecular Charge-Transfer Process: A State-Specific Solvation Preferred to the Linear-Response Approach, ACS Omega, 2020, 5, 9944–9956 CrossRef CAS PubMed.
- C. Reichardt, Empirical Parameters of the Polarity of Solvents, Angew. Chem., Int. Ed. Engl., 1965, 4, 29–40 CrossRef.
- C. Reichardt, Solvatochromic Dyes as Solvent Polarity Indicators, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- S. P. Pitre, C. D. McTiernan and J. C. Scaiano, Library of Cationic Organic Dyes for Visible-Light-Driven Photoredox Transformations, ACS Omega, 2016, 1, 66–76 CrossRef CAS PubMed.
- D. Wang, X. Guo, H. Wu, Q. Wu, H. Wang, X. Zhang, E. Hao and L. Jiao, Visible Light Excitation of BODIPYs Enables Dehydrogenative Enamination at Their α-Positions with Aliphatic Amines, J. Org. Chem., 2020, 85, 8360–8370 CrossRef CAS PubMed.
- D. M. Schultz, J. W. Sawicki and T. P. Yoon, An improved procedure for the preparation of Ru(bpz)3(PF6)2 via a high-yielding synthesis of 2,2′-bipyrazine, Beilstein J. Org. Chem., 2015, 11, 61–65 CrossRef PubMed.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- C. B. Nielsen, A. J. P. White and I. McCulloch, Effect of Fluorination of 2,1,3-Benzothiadiazole, J. Org. Chem., 2015, 80, 5045–5048 CrossRef CAS PubMed.
- R. S. J. Proctor and R. J. Phipps, Recent Advances in Minisci-Type Reactions, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Nucleophilic character of alkyl radicals—VI, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS.
- I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, Direct C−H Arylation of Electron-Deficient Heterocycles with Arylboronic Acids, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS PubMed.
- Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Practical and innate carbon hydrogen functionalization of heterocycles, Nature, 2012, 492, 95–99 CrossRef CAS PubMed.
- G. X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Photoredox-mediated Minisci C-H alkylation of N-heteroarenes using boronic acids and hypervalent iodine, Chem. Sci., 2016, 7, 6407–6412 RSC.
- T. McCallum and L. Barriault, Direct alkylation of heteroarenes with unactivated bromoalkanes using photoredox gold catalysis, Chem. Sci., 2016, 7, 4754–4758 RSC.
- J. Jin and D. W. C. MacMillan, Alcohols as alkylating agents in heteroarene C-H functionalization, Nature, 2015, 525, 87–90 CrossRef CAS PubMed.
- D. R. Sutherland, M. Veguillas, C. L. Oates and A. L. Lee, Metal-, Photocatalyst-, and Light-Free, Late-Stage C-H Alkylation of Heteroarenes and 1,4-Quinones Using Carboxylic Acids, Org. Lett., 2018, 20, 6863–6867 CrossRef CAS PubMed.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Mechanistic Studies in Photocatalysis, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- C. Wang, R. Chen, R. Zhang and N. Zhang, Simple spectrophotometric determination of sulfate free radicals, Anal. Methods, 2018, 10, 3470–3474 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
Footnotes |
| † Dedicated to the memory of Professor John S. Fossey. |
| ‡ Electronic supplementary information (ESI) available: Full synthesis and characterisation of photocatalyst library and substrate scope, details of computational calculations, single crystal X-ray structures. CCDC 2179269–2179281 and 2179284. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo01316a |
| This journal is © the Partner Organisations 2022 |