
Planar Blatter radicals through Bu3SnH- and TMS3SiH-assisted cyclization of aryl iodides: azaphilic radical addition†
Paulina
Bartos
 *a,
Małgorzata
Celeda
a,
Anna
Pietrzak
b and
Piotr
Kaszyński
*acd
*a,
Małgorzata
Celeda
a,
Anna
Pietrzak
b and
Piotr
Kaszyński
*acd
aFaculty of Chemistry, University of Łódź, 91-403 Łódź, Poland. E-mail: piotr.kaszynski@chemia.uni.lodz.pl
bFaculty of Chemistry, Łódź University of Technology, 90-924 Łódź, Poland
cCentre of Molecular and Macromolecular Studies, Polish Academy of Sciences, 90-363 Łódź, Poland
dDepartment of Chemistry, Middle Tennessee State University, Murfreesboro, Tennessee 37132, USA
First published on 10th December 2021
Abstract
Bu3SnH- and TMS3SiH-assisted cyclizations of iodoarene derivatives of benzo[e][1,2,4]triazine lead to the formation of planar Blatter radicals (2-phenyl-3H-[1,2,4]triazino[5,6,1-kl]phenoxazin-3-yls and 2-phenyl-3H-[1,2,4]triazino[5,6,1-kl]phenothiazin-3-yls) in yields of up to 96%. The cyclization step involves the thermodynamically favored (DFT: ΔG‡298 = 3.1 kcal mol−1 and ΔH = –55.3 kcal mol−1) unprecedented attack of a C-centered radical on the heterocyclic N atom. This method opens up access to functionalized sulfur-containing planar Blatter radicals for the first time. New radicals were characterized using spectroscopic (UV–vis, EPR), electrochemical, and single-crystal X-ray diffraction methods.
Introduction
Increasing interest in stable radicals as structural elements for advanced materials1–8 has resulted in increasing demand for their functional derivatives with tailored properties. Of particular interest is the benzo[e][1,2,4]triazinyl radical (Blatter radical)9 and its derivatives,10,11 which are exceptionally stable,12,13 and exhibit π-spin delocalization,14 a narrow electrochemical window,15,16 and low excitation energies.17 For these reasons, there is rapidly increasing interest in the exploration of this radical as a structural element for advanced materials, such as sensors,18 photodetectors19 and liquid crystalline photoconductors,20,21 and also for applications in spintronics22–24 and radical polymerization.25,26Recent advances10,11 in the chemistry of the benzo[e][1,2,4]triazinyl led to the discovery of planar Blatter radicals.27 The two parent radicals, 1O-a (2-Ph-3H-[1,2,4]triazino[5,6,1-kl]phenoxazin-3-yl) containing a phenoxazine ring and 1S-a based on phenothiazine (2-phenyl-3H-[1,2,4]triazino[5,6,1-kl]phenothiazin-3-yl), were obtained in low yields (20–25%) via the intramolecular azaphilic addition of ArLi, which was generated in situ from the appropriate derivative 2-a (Method A, Fig. 1).27 A much improved yield of 1O-a and access to its functional derivatives were demonstrated with the aza-Pschorr cyclization reaction (Method B).28–31 Further progress in the synthesis of ring-fused derivatives of 1O-a involved photocyclization (Mallory-type cyclization) of C(8)-substituted benzo[e][1,2,4]triazines (Method C, Fig. 1).32 Unfortunately, neither of the latter two methods is suitable for the preparation of sulfur-containing radicals 1S: strongly oxidative conditions are incompatible with the divalent sulfur in Method B, while in Method C the heavy atom effect causes a rapid decay of the S1 state.
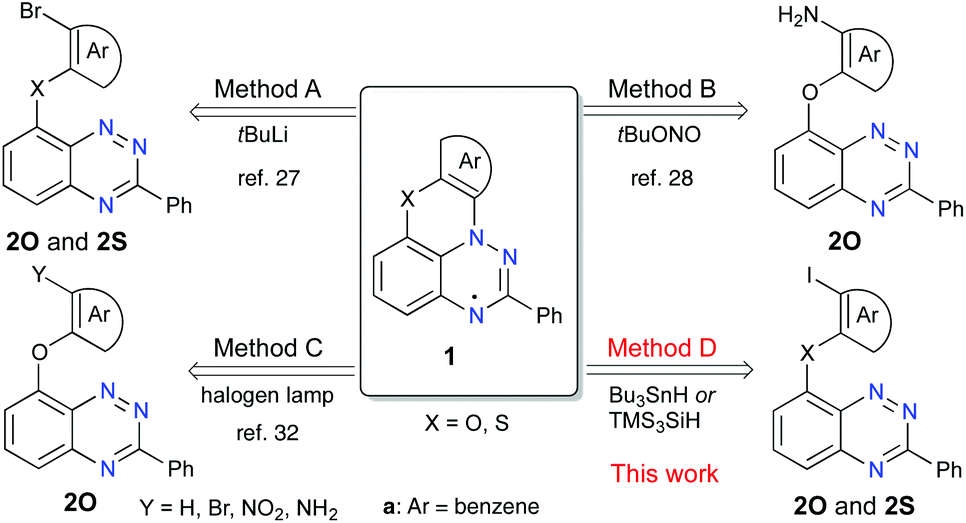 | ||
| Fig. 1 Three known (A–C) methods and one new (D) method for the preparation of planar benzo[e][1,2,4]triazin-4-yl radicals 1O and 1S. | ||
Successful Pschorr cyclization (Method B) implies an azaphilic addition of the transient aryl radical to the [1,2,4]triazine and suggests that other processes involving aryl radicals,33e.g. transformations of aryl halides in the presence of R3SnH34,35 or R3SiH,36,37 could also lead to N-cyclization and formation of the planar radical.
Radical chain cyclization reactions38,39 of aryl halides in the presence of R3SnH34,35 or R3SiH36,37 are well-established and have been used for the formation of carbocyclic derivatives (e.g. phenanthrenes40–42 and helicenes43,44) and for a variety of ring-fused heterocycles.38,39,45–48 In all these reactions, the key step is the attack of the transient C-centered radical onto the ring C-atom, which is preferred over the N-attack, if the two paths are available.49,50 Consequently, N-attack of an aryl radical has never been observed in tin- or silicon-hydride-assisted radical cyclization processes.
Herein, we present an efficient Bu3SnH- and TMS3SiH-assisted radical chain cyclization of aryl iodides 2 on the [1,2,4]triazine N-atom and the formation of planar Blatter radicals 1O and 1S (Method D, Fig. 1). The new method is tested on four previously reported radicals and applied to the preparation of the first functionalized sulfur-containing planar Blatter radical, ester 1S-b, as well as 1O-c. The two newly obtained radicals, 1O-c and 1S-b, are characterized by spectroscopic (UV-vis and EPR) and electrochemical methods, and the structure of the former radical is determined using a single crystal XRD method.
Results and discussion
Synthesis
Initial investigation focused on the cyclization reaction of the benzo[e][1,2,4]triazine 2O-a with Bu3SnH in toluene (Fig. 2). Following a literature procedure for similar cyclization reactions,39,44 a solution of the initiator AIBN was added to a solution of 2O-a and Bu3SnH in toluene at 80 °C over a 4 h period via a syringe pump (Method D-Sn). The initially formed leuco form, 1O-a-leuco, underwent slow oxidation to 1O-a during workup, and more rapidly during deposition onto the passivated SiO2. The crude radical 1O-a, which was isolated by filtration through passivated silica gel, contained significant amounts of organotin impurities. Therefore, following a modified analogous procedure24 crude 1O-a was oxidized with AgOTf to [1O-a]+, which was purified using chromatography. The pure salt was subsequently reduced with Zn/AcOH and the resulting 1O-a-leuco was exposed to air, giving pure radical 1O-a in 48–52% yield (Scheme 1). Using the same method, radicals 1O-b, 1O-c and 1S-a were obtained in comparable yields of 42–69% (Fig. 2). The yield of radical 1O-d was significantly lower than those of the others in the series (13% yield), mainly due to its lower stability on silica gel and alumina during the purification process.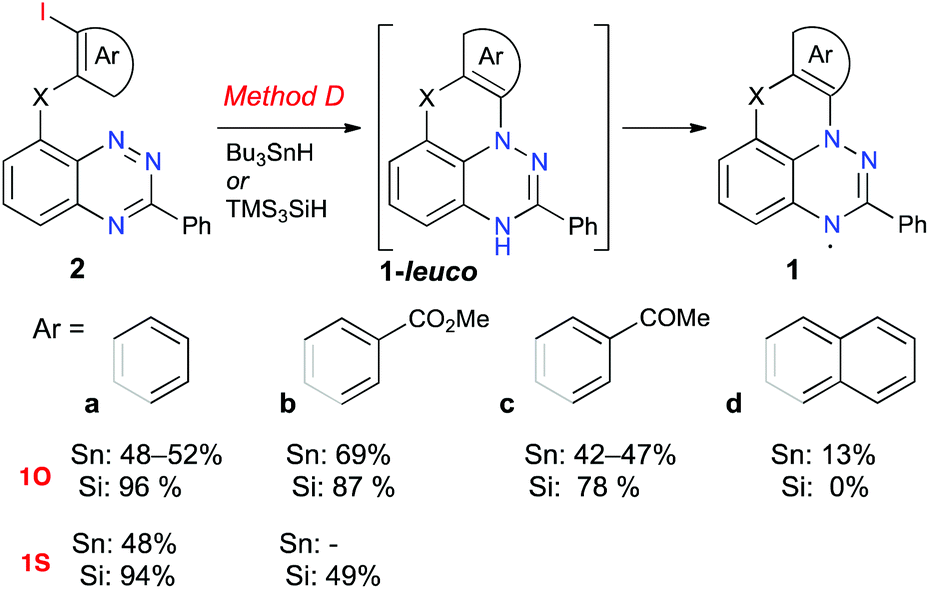 | ||
| Fig. 2 Cyclization of benzo[e][1,2,4]triazines 2 to radicals 1. Reagents and conditions, Method D: 1. Bu3SnH or TMS3SiH, AIBN, toluene, 80 °C, 4 h 2. Air. | ||
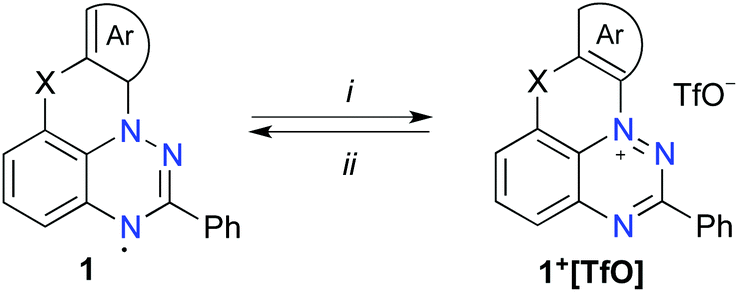 | ||
Scheme 1 Interconversion of cations 1+ and radicals 1. Reagents and conditions: (i) AgOTf, CH2Cl2/CH3CN (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 50 °C, 5–10 min (ii) 1. Zn powder, AcOH, CH2Cl2/CH3CN (9:1), 50 °C, 5 min; 2. Air. :1), 50 °C, 5–10 min (ii) 1. Zn powder, AcOH, CH2Cl2/CH3CN (9:1), 50 °C, 5 min; 2. Air. | ||
The difficulties with the removal of organotin byproducts prompted the investigation of an alternative method using TMS3SiH instead of Bu3SnH. Thus, treatment of benzo[e][1,2,4]triazine 2O-a with TMS3SiH in toluene in the presence of AIBN gave the desired radical 1O-a in an excellent yield of 96% (Method D-Si) without the need of going through the cation [1O-a]+ for purification purposes. This method allowed radicals 1O-a–1O-c and the parent sulfur-containing radical 1S-a to be obtained in higher yields of 48–94%. This method appears to be superior to Method D-Sn, and therefore was used to obtain radical 1S-b, the first functional derivative of 1S-a. The same Method D-Si did not work unfortunately for the extended radical 1O-d. The starting 2O-d was fully consumed, and only a complex mixture of polar products was formed.
The parent radicals 1O-a and 1S-a show no signs of decomposition after 6 years of storage in the solid-state under ambient conditions. Derivatives of 1O-a also exhibit long-term stability in the solid-state.
Precursors 2 were tested for photostability and photochemical formation of the corresponding radicals 1. The results demonstrated that all precursors 2 were stable under ambient conditions, while photocyclization of 2O-a–2O-c in CH2Cl2 solutions gave complex reaction mixtures from which 1O-a and 1O-c were isolated in 7% and 5% yield, respectively. As expected, attempts at the photocyclization of 8-(2-nitrophenylsulfanyl)-3-phenylbenzo[e][1,2,4]triazine (3S) did not yield the desired radical 1S-a and most of the substrate was recovered. This is in sharp contrast to a moderately efficient photocyclization of the analogous oxygen derivative 3O to form 1O-a (31% yield) under the same conditions.32
The requisite precursors 2O and 2S were obtained from 8-fluoro-3-phenylbenzo[e][1,2,4]triazine27 (4) by nucleophilic aromatic substitution reactions with the appropriate ortho-iodo phenols 5O (X = O) or thiols 5S (X = S) in the presence of NaH (Scheme 2).
 | ||
| Scheme 2 Synthesis of derivatives 2O and 2S. Reagents and conditions: (i) iodoarene 5, 60% NaH, DMSO, 80 °C, 6 h. | ||
The iodoarenes 5O and 5S-a were obtained using modified literature procedures by iodination of the appropriate phenols and diazotization of 2-iodoaniline, respectively.51 Iodomercaptan 5S-b was obtained from methyl 4-amino-3-iodobenzoate as shown in Scheme 3. Thus, treatment of 4-aminobenzoate 6 with pyridine iodine monochloride in MeOH gave methyl 4-amino-2-iodobenzoate (7) in 85% yield.52 To avoid the double iodination of 6 and to obtain high yields of 7, the reaction mixture was quenched with Na2S2O3 before the workup. The ester 7 was diazotized and then treated with EtOCSSK, subsequently with KOH/EtOH and finally with iodine. The resulting 4,4′-disulfanediylbis(3-iodobenzoic acid) (8) was partially purified and transformed to dimethyl ester 9 with diazomethane in THF. Finally, the disulfide 9 was reduced with NaBH4 in THF/MeOH according to a general literature procedure,53 giving methyl 3-iodo-4-mercaptobenzoate (5S-b).
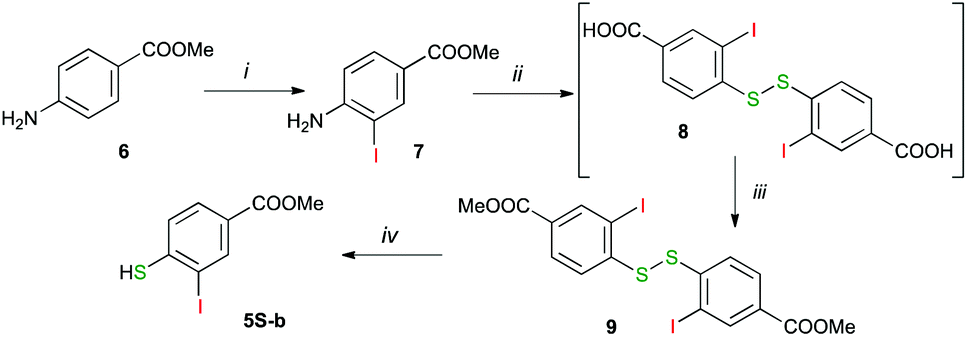 | ||
| Scheme 3 Synthesis of mercaptan 5S-b. Reagents and conditions: (i) 1. pyridine iodine monochloride, MeOH, rt, 2 h; 2. Na2S2O3; 85% yield; (ii) 1. NaNO2, aq HCl, ice bath; 2. EtOCSSK; 3. KOH, EtOH, reflux, 10 h; 4. I2, H2O; 40–56% yield, crude; (iii) CH2N2, THF/Et2O, 0 °C, 10 min; 70–72% yield; (iv) NaBH4, THF/MeOH, quant. | ||
Mechanistic considerations
The observed successful formation of 1-leuco in a radical chain process indicates that (a) the addition of the transient radical 10 to the N(1) atom is favorable and fast, and (b) the resulting radical 1 efficiently transfers an H atom from the donor R3E–H (E = Sn or Si, Fig. 3). DFT calculations at the B3LYP/6-31G(2d,p) level of theory performed in benzene dielectric medium indeed show a low barrier to the N(1) addition of radical 10O-a (ΔG‡298 = 3.1 kcal mol−1) and high exotherm for the formation of 1O-a (ΔH = –55.3 kcal mol−1, Fig. 3). More importantly, the H transfer takes place in spite of the relatively low N–H bond dissociation energy (BDE) in 1-leuco, calculated to be about 69 kcal mol−1,30 which compares to the experimental BDE of 73.4 ± 2 kcal mol−1 for Bu3SnH in benzene.54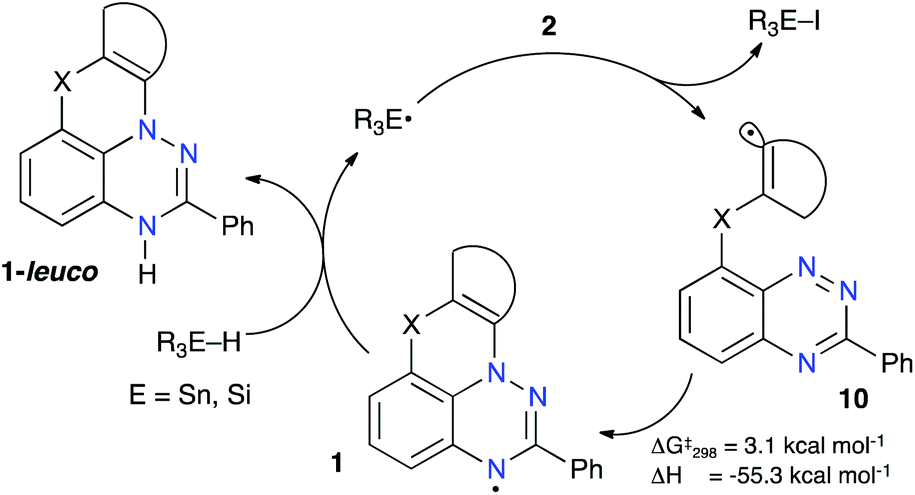 | ||
| Fig. 3 Proposed mechanism for the formation of 1-leuco in a radical chain process and DFT-derived thermochemical parameters for the N-addition step in 10O-a. | ||
Crystal and molecular structures
The structure of 10-acetyl derivative 1O-c was confirmed with a single-crystal XRD analysis (Fig. 4 and the ESI†). The analysis revealed that 1O-c crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit of 1O-c contains two molecules, for which the dimensions of the heterocyclic skeleton are consistent with those found in similar systems.27,29,30 The data showed that the heterocyclic core is nearly ideally planar. The C(2)–Ph ring is twisted from the coplanar orientation with the heterocycle by 4.2° and 5.3° in the two symmetry-independent molecules. The supramolecular system of 1O-c consists of two types of slipped stacks extending along the [1 0 0] direction. Each stack is formed by symmetry-equivalent molecules with an interplanar separation of 3.183 Å (for stack A) and 3.246 Å (for stack B) and slippage angles of 27.0° (for stack A) and 62.2° (for stack B). Stacks A are stabilized by C(2)⋯C(7a) close contacts of 3.228 Å.
. The asymmetric unit of 1O-c contains two molecules, for which the dimensions of the heterocyclic skeleton are consistent with those found in similar systems.27,29,30 The data showed that the heterocyclic core is nearly ideally planar. The C(2)–Ph ring is twisted from the coplanar orientation with the heterocycle by 4.2° and 5.3° in the two symmetry-independent molecules. The supramolecular system of 1O-c consists of two types of slipped stacks extending along the [1 0 0] direction. Each stack is formed by symmetry-equivalent molecules with an interplanar separation of 3.183 Å (for stack A) and 3.246 Å (for stack B) and slippage angles of 27.0° (for stack A) and 62.2° (for stack B). Stacks A are stabilized by C(2)⋯C(7a) close contacts of 3.228 Å.
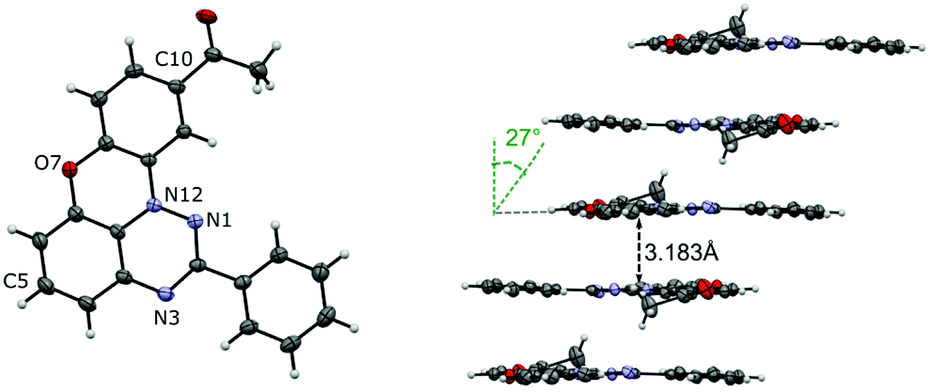 | ||
| Fig. 4 A displacement ellipsoid diagram for molecule A of 1O-c and slipped stack A indicating the distances between the average planes and the slippage angle. Ellipsoids are set at the 50% probability level and labelled according to the chemical structure. Selected intramolecular dimensions: N(1)–N(12), 1.360(1) Å; N(12)–C(11a), 1.399(1) Å; C(10)–Ac, 1.496(2) Å. For molecule B and other details, see the ESI.† | ||
Electronic spectroscopy and electrochemistry
The electronic properties of the new radicals 1O-c and 1S-b were analyzed using spectroscopic (UV–vis and EPR) and electrochemical methods. Data revealed that both radicals exhibit typical strong absorption in the UV region and broad, low-intensity absorption bands in the entire visible range. The absorption bands of sulfur analogues 1S-a and 1S-b are bathochromically shifted relative to those of the oxygen analogues and exhibit absorption maxima at about 680 nm and 755 (sh) nm (Fig. 5). Analysis of the spectra demonstrated that the optical band gaps, as determined from the absorption edge, are typically about 1.7 eV for radicals 1O-a–1O-d, while those of the sulfur analogs 1S-a and 1S-b are markedly lower at about 1.5 eV (Table 1).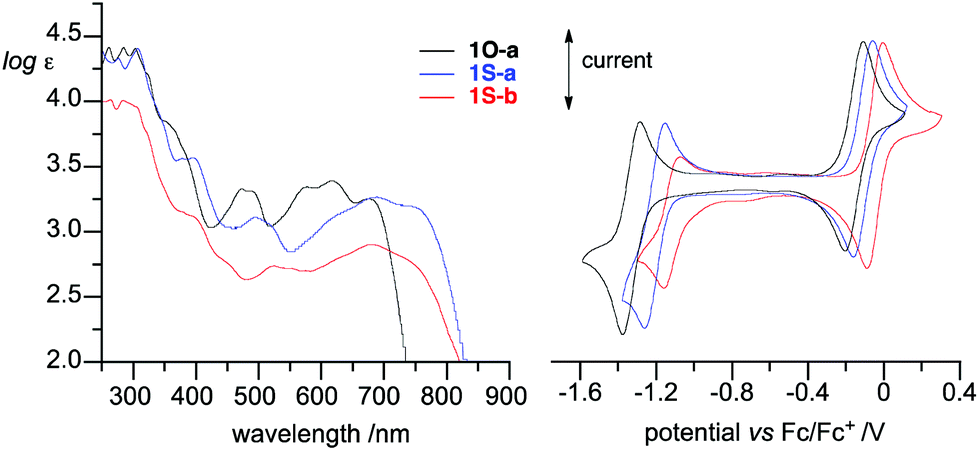 | ||
| Fig. 5 Left: UV/vis spectra in CH2Cl2 and right: Cyclic voltammograms (0.5 mM in CH2Cl2 [n-Bu4N]+PF6− (50 mM), at ∼20 °C, 50 mV s−1, glassy carbon working electrode) for radicals 1O-a (black), 1S-a (blue), and 1S-b (red). | ||
| Radical R |
E
1/2
−1/0
/V |
E
1/2
0/+1
/V |
E
cellb/V |
E
gc/eV |
|
|---|---|---|---|---|---|
|
a Potentials vs. Fc/Fc+.
b
E
cell = E1/20/+1 − E1/2−1/0.
c Optical band gap from the absorption edge.
d Ref. 28.
e Ref. 32. For details, see the ESI.†
|
|||||
| O-a | Hd | −1.317 | −0.154 | 1.163 | 1.66 |
| O-b | COOMed | −1.258 | −0.089 | 1.169 | 1.72 |
| O-c | Ac | −1.222 | −0.075 | 1.147 | 1.68 |
| O-d | Naphthe | −1.354 | −0.212 | 1.142 | 1.67 |
| S-a | H | −1.202 | −0.112 | 1.090 | 1.48 |
| S-b | COOMe | −1.103 | −0.047 | 1.056 | 1.51 |
Cyclic voltammetry revealed that the oxidation and reduction processes are quasi-reversible for all derivatives (Fig. 5, Table 1). The oxidation potentials E1/20/+1 in the “oxo” series increase slightly from −0.15 V for the parent 1O-a to −0.09 V for 1O-b and to −0.075 V for the 10-acetyl derivative 1O-c. Similarly, the reduction potentials E1/2−1/0 increase for C(10)–substituted derivatives 1O-b and 1O-c. This effect was recently correlated with the σm substituent parameter for a larger pool of 1O derivatives.30 Analysis of the pair of 1S derivatives reveals the same trend: there is an anodic shift of redox potentials upon substitution of the parent with the COOMe group (Table 1). Incidentally, the magnitude of this shift in E1/20/+1 is exactly 0.065 eV in both pairs of radicals, while the reduction potential is affected more strongly in the sulfur pair (ΔE1/2−1/0 = 0.059 V for the 1O pair and 0.099 V for the 1S pair).
EPR spectroscopy
Electron paramagnetic spectroscopy (EPR) revealed that the experimental hyperfine coupling constants (hfcc) aN for 1O-c and 1S-b (Fig. 6) are consistent with those for other planar benzo[e][1,2,4]triazinyl radicals.30 Analysis of the results indicates that the aN(12) value slightly decreases (from 7.42 G for 1S-a to 7.08 G for 1S-b), while the values for aN(1) and aN(3) increase, e.g. for aN(1), from 4.38 G for 1S-a to 4.52 G for 1S-b. This substituent effect is consistent with that observed in the oxygen analogues 1O.30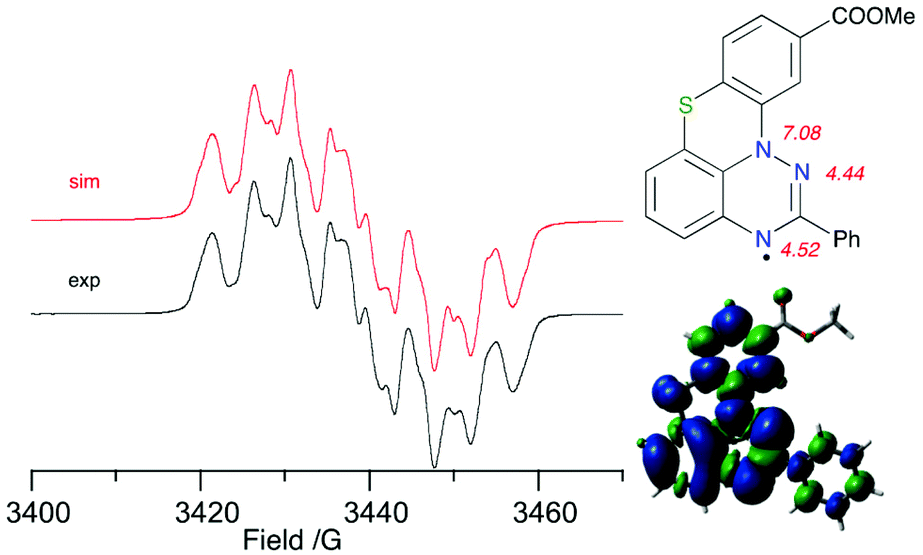 | ||
| Fig. 6 Left: experimental (black) and simulated (red) EPR spectra for 1S-b recorded in benzene at ca. 20 °C. Right: assignment of the resulting hfcc and total spin density in 1S-b. Contour values are plotted at ±0.02 (e per bohr3)1/2. | ||
Conclusions
We have demonstrated a new and efficient method for the formation of planar Blatter radical derivatives through Bu3SnH- and TMS3SiH-assisted cyclization of aryl iodides. Unlike in previous such reactions, the cyclization takes place at the heterocyclic N atom. The new method complements the existing methods A–C for the preparation of planar Blatter radicals with important advantages. Similarly to the Pschorr-type cyclization (Method B), it tolerates functional groups, such as COOMe and COMe, and allows for obtaining the desired products with significantly higher yields (Table 2). Most importantly, for the first time, it permits the preparation of functionalized derivatives of the [1,2,4]triazino[5,6,1-kl]phenothiazinyl (radical 1S-b), which was not possible through other methods. This discovery opens up possibilities for further exploration of functionalized [1,2,4]triazino[5,6,1-kl]phenothiazinyl radicals, especially of their magnetic properties in the solid state. This work is underway in our laboratory.| Radical | Method | |||||
|---|---|---|---|---|---|---|
| 1 | R | A | B | C | D-Sn | D-Si |
| a Not compatible. b Not attempted. For references see the text. c Naphtho-fused radical. d Low stability on isolation. | ||||||
| O-a | H | 20–25% | 57–64% | 7–64% | 48–52% | 96% |
| O-b | COOMe | a,b |
48–55% |
b |
69% | 87% |
| O-c | Ac | a,b |
b |
5% | 42–47% | 78% |
| O-d | c |
0% | 0% | 14–55% | (13%)d | 0% |
| S-a | H | 22–30% | a,b |
a |
48% | 94% |
| S-b | COOMe | a,b |
a,b |
0% | b |
49% |
The radical cyclization of aryl iodides method also gives the ring-extended derivatives, such as the naphtho-derivative 1O-d, but in this case, the photocyclization Method C appears to be more efficient. The newly discovered Method D for obtaining radicals 1 makes a significant contribution to the development of new materials for molecular electronics, spintronics, and functional self-organizing materials.
Computational details
Quantum-mechanical calculations were carried out using the Gaussian 09 suite of programs.55 Geometry optimizations were undertaken at the UB3LYP/6-31G(2d,p) level of theory using tight convergence limits and appropriate symmetry constraints. The transition state for the cyclization of 10O-a was located with the QST3 keyword. Calculations involving the cyclization of radical 10O-a to form 1O-a used SCF energies for reaction components in benzene dielectric medium requested with the SCRF(Solvent = Benzene) keyword (PCM model;56 single point calculations at the UB3LYP/6-311++G(2d,p)//UB3LYP/6-31G(2d,p) level of theory) and thermodynamic corrections obtained with the UB3LYP/6-31G(2d,p) method.Isotropic Fermi contact coupling constants for radical 1S-b were calculated using the UCAM-B3LYP/gen //UB3LYP/6-31G(2d,p) method in benzene dielectric medium requested with the SCRF(Solvent = Benzene) keyword (PCM model56). The basis set was requested with the GEN keyword: EPRIII basis for all light elements and 6-311 + G(2df) for the S atom.
Experimental section
General
Reagents and solvents were obtained commercially. Heat for reactions requiring elevated temperatures was supplied using oil baths. NMR spectra were obtained at 600 MHz (1H) and 151 MHz (13C) in CDCl3 and referenced to the solvent (δ = 7.26 ppm for 1H and δ = 77.16 ppm for 13C) or in DMSO-d6 and referenced to the solvent (δ = 2.50 ppm for 1H and δ = 39.52 ppm for 13C).57 IR spectra were recorded using a Nexus FT-IR Thermo Nikolet IR spectrometer in KBr tablets. UV spectra were measured in CH2Cl2 using a PerkinElmer Lambda 45 spectrophotometer. Melting points were determined using a Stuart SMP30 Advanced Digital Melting Point Apparatus and are uncorrected. High-resolution mass spectrometry (HRMS) measurements were performed using SYNAPT G2-Si High Definition Mass Spectrometry equipped with an ESI or APCI source and Quantitative Time-of-Flight (QuanTof) mass analyzer. Other mass spectrometry measurements were performed using a Varian 500-MS LC Ion Trap Spectrometer. In all cases little or no fragmentation was observed, and the M, [M + H] or [M − H] peaks were the most intense signals.The activity and effectiveness of the solid supports varied between suppliers and even between batches, and the exact conditions for isolation of the radicals required adjustments for each new container.
General procedure for Method D-Sn. Bu3SnH-assisted cyclization of aryl iodides 2
To a solution of aryl iodide 2 (0.25 mmol) in dry toluene (4 mL), Bu3SnH (0.50 mmol) was added and stirred at 80 °C under an atmosphere of Ar. Then, AIBN (0.15 mmol) dissolved in dry toluene (2 mL) was added to the reaction mixture over a period of 4 h using a syringe pump. After 4 h the reaction mixture was cooled and the solvent was evaporated to dryness. The residue was re-dissolved in ethyl acetate (20 mL), saturated aqueous potassium fluoride (20 mL) was added, and the mixture was stirred vigorously for 15 min. The organic fraction was separated, washed with brine (2 × 30 mL), dried (Na2SO4), and concentrated in vacuo. The crude product was separated using a short silica gel column passivated with Et3N (vide supra). To remove organotin impurities, the radical was oxidized to form the triflate salt 1+, purified, and subsequently reduced back to radical 1.:1) and AgOTf (0.30 mmol, 1.2 eq.) was added in one portion. The reaction mixture was stirred for 5–10 minutes at 50 °C until the substrate was no longer detectable by TLC (pet. ether/AcOEt, 9:1). The crude salt was purified using a short silica gel column with CH2Cl2/MeOH 9:1 as the eluent.
:1 CH2Cl2/CH3CN mixture (25 mL), and 100 μL of AcOH and 150 mg of zinc powder were added. The reaction mixture was stirred at 50 °C for 5 min until the starting salt 1+[OTf] was no longer detectable by TLC analysis (pet. ether/AcOEt, 9:1). The reaction mixture was cooled, filtered through Celite, washed with H2O (2 × 30 mL), dried (Na2SO4), and concentrated in vacuo. The resulting product was recrystallized by slow evaporation of a CH2Cl2/cyclohexane solution.
General procedure for Method D-Si. TMS3SiH-assisted cyclization of aryl iodides 2
To a solution of aryl iodide 2 (0.25 mmol) in dry toluene (4 mL), TMS3SiH (0.50 mmol) was added, and the solution was stirred at 80 °C under an atmosphere of Ar. Then, AIBN (0.15 mmol) dissolved in dry toluene (2 mL) was added to the reaction mixture over a period of 4 h using a syringe pump. After 4 h, the reaction mixture was cooled, and the solvent was evaporated to dryness. The crude product was absorbed on passivated SiO2 (vide supra), and radical 1 was isolated using a short silica gel column with passivated SiO2. The product was washed with warm cyclohexane (3×) and recrystallized from CH3CN.:5; blue-gray crystals. Analytical data identical to those previously reported.27 HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H13N3O: 299.1059; found: 299.1055.
:5; blue-gray crystals. Analytical data identical to the previously reported.28 HRMS (ESI-TOF) m/z [M + H]+ calcd for C21H15N3O3: 357.1113; found: 357.1120.
:1; blue-green crystals: mp 187–188 °C (MeCN); IR (KBr) v 1680, 1567, 1383, 1348, 1266, 1210, 1162, 1031, 791, 696 cm−1; UV (CH2Cl2) λmax (logε) 280 (3.58), 315 (4.24), 377 (3.64), 431 (3.29), 514.5 (3.19), 570 (3.18), 618 (3.24), 673 (3.13) nm; HRMS (ESI-TOF) [M + H]+m/z calcd for C21H15N3O2: 341.1164; found: 341.1166. Anal. Calcd for (C21H14N3O2)3·H2O: C, 72.82; H, 4.27; N, 12.13. Found: C, 72.99; H, 4.23; N, 12.42.
:5; blue-gray crystals. Analytical data identical to those previously reported.32 HRMS (ESI-TOF) [M + H]+m/z calcd for C23H15N3O: 349.1215; found: 349.1222.
:1 blue-green crystals. Analytical data identical to those previously reported.27 HRMS (ESI-TOF) [M + H]+m/z calcd for C19H13N3S: 315.0830; found: 315.0836.
:1; blue-green crystals: mp 185–186 °C (MeCN); IR (KBr) v 2956, 2925, 2854, 1728, 1470, 1436, 1404, 1249, 1080, 1013, 844, 754, 688 cm−1; UV (CH2Cl2) λmax (logε) 263 (4.02), 284 (4.02), 399 (3.12 sh), 524 (2.74), 679 (2.90), 746 (2.75 sh) nm; HRMS (ESI-TOF) [M + H]+m/z calcd for C21H15N3O2S: 373.0898; found: 373.0892. Anal. Calcd for C21H14N3O2S: C, 67.73; H, 3.79; N, 11.28. Found: C, 67.68; H, 3.93; N, 11.25.
Synthesis of iodoarenes 2. General procedure
To a stirred solution of phenol 5O or thiophenol 5S (1.10 mmol) dissolved in DMSO (4 mL), 60% NaH (45 mg, 1.10 mmol) was added in one portion. After 15 min of stirring, 8-fluoro-3-phenylbenzo[e][1,2,4]triazine27 (4, 225.2 mg, 1.00 mmol) was added, and the reaction was stirred under Ar at 80 °C for 6 h. After cooling, CH2Cl2 (30 mL) was added, and the organic layer was washed well with water (3 × 25 mL) and brine (25 mL). The combined organic extracts were dried (Na2SO4), and the solvent was evaporated. The resulting solid residue was adsorbed onto passivated silica and purified by column chromatography (passivated silica). The solvent was evaporated, and the product was recrystallized from MeCN.Synthesis of methyl-3-iodo-4-mercaptobenzoate (5S-b)
:1) giving 111–156 mg (40–56% yield) of disulfide 8 as a pale solid: mp > 230 °C (MeOH/H2O, 9:1); 1H NMR (600 MHz, DMSO-d6) δ 13.27 (s, 1H), 8.33 (s, 1H), 7.96 (dd, J1 = 8.3 Hz, J2 = 1.7 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H); IR (KBr) v 1690, 1580, 1542, 1412, 1369, 1296, 1244, 759 cm−1; HRMS (ESI-TOF) [M − H]−m/z calcd for C14H14I2O4S2: 556.7875; found: 556.7883. Anal. Calcd for C14H8I2O4S2: C, 30.13; H, 1.44. Found: C, 30.32; H, 1.57.
The acid was used in the next step without further purification.
:1) giving 103–106 mg (70–72% yield) dimethyl ester 9 as white crystals: mp 180–181 °C (AcOEt); 1H NMR (600 MHz, CDCl3) δ 8.45 (d, J = 1.7 Hz, 1H), 7.95 (dd, J1 = 8.3 Hz, J2 = 1.8 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 3.90 (s, 3H); 13C{1H} NMR (151 MHz, CDCl3) δ 165.2, 144.9, 140.5, 130.1, 130.0, 125.6, 94.2, 52.6; IR (KBr) v 1721, 1578, 1544, 1429, 1366, 1286, 1235, 1114, 1014, 832, 758 cm−1; HRMS (ESI-TOF) [M + H]+m/z calcd for C16H13I2O4S2: 586.8345; found: 586.8344. Anal. Calcd for C16H12I2O4S2: C, 32.78; H, 2.06; S, 10.94. Found: C, 32.79; H, 2.18; S, 11.16.
Preparation of other intermediates is described in the ESI.†
Author contributions
The manuscript was written through contributions of all authors and all authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Support for this work was provided by the National Science Center (2019/03/X/ST4/02006 and 2017/25/B/ST5/02851). The authors thank Dr. Paweł Tokarz for helpful discussions.Notes and references
- T. J. J. Muller and U. H. F. Bunz, Functional Organic Materials, Wiley, 2006 Search PubMed.
- I. Ratera and J. Veciana, Playing with organic radicals as building blocks for functional molecular materials, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- D. A. Wilcox, V. Agarkar, S. Mukherjee and B. W. Boudouris, Stable radical materials for energy applications, Annu. Rev. Chem. Biomol. Eng., 2018, 9, 83–103 CrossRef PubMed.
- Z. X. Chen, Y. Li and F. Huang, Persistent and stable organic radicals: Design, synthesis, and applications, Chem., 2021, 7, 288–332 CAS.
- S. Mukherjee and B. W. Boudouris, Organic Radical Polymers, Springer International Publishing, 2017 Search PubMed.
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Air-stable organic radicals: New-generation materials for flexible electronics?, Adv. Mater., 2020, 32, 1908015 CrossRef CAS PubMed.
- J. M. Hudson, T. J. H. Hele and E. W. Evans, Efficient light-emitting diodes from organic radicals with doublet emission, J. Appl. Phys., 2021, 129, 180901 CrossRef CAS.
- M. Deumal, V. Vela, M. Fumanal, J. Ribas-Arino and J. J. Novoa, Insights into the magnetism and phase transitions of organic radical-based materials, J. Mater. Chem. C, 2021, 9, 10624–10646 RSC.
- H. M. Blatter and H. Lukaszewski, A new stable free radical, Tetrahedron Lett., 1968, 9, 2701–2705 CrossRef.
- F. J. M. Rogers, P. L. Norcott and M. L. Coote, Recent advances in the chemistry of benzo[e][1,2,4]triazinyl radicals, Org. Biomol. Chem., 2020, 18, 8255–8277 RSC.
- Y. Ji, L. Long and Y. Zheng, Recent advances of stable Blatter radicals: Synthesis, properties and applications, Mater. Chem. Front., 2020, 4, 3433–3443 RSC.
- F. A. Neugebauer and I. Umminger, Über 1,4-dihydro-1,2,4-benzotriazinyl-radikale, Chem. Ber., 1980, 113, 1205–1225 CrossRef CAS.
- C. P. Constantinides, P. A. Koutentis, H. Krassos, J. M. Rawson and A. J. Tasiopoulos, Characterization and magnetic properties of a “super stable” radical 1,3-diphenyl-7-trifluoromethyl-1,4-dihydro-1,2,4-benzotriazin-4-yl, J. Org. Chem., 2011, 76, 2798–2806 CrossRef CAS PubMed.
- F. A. Neugebauer and G. Rimmler, ENDOR and triple resonance studies of 1,4-dihydro-1,2,4-benzotriazinyl radicals and 1,4-dihydro-1,2,4-benzotriazine radical cations, Magn. Reson. Chem., 1988, 26, 595–600 CrossRef CAS.
- A. A. Berezin, C. P. Constantinides, S. I. Mirallai, M. Manoli, L. L. Cao, J. M. Rawson and P. A. Koutentis, Synthesis and properties of imidazolo-fused benzotriazinyl radicals, Org. Biomol. Chem., 2013, 11, 6780–6795 RSC.
- A. A. Berezin, G. Zissimou, C. P. Constantinides, Y. Beldjoudi, J. M. Rawson and P. A. Koutentis, Route to benzo- and pyrido-fused 1,2,4-triazinyl radicals via N′-(het)aryl-N′-[2-nitro(het)aryl]hydrazides, J. Org. Chem., 2014, 79, 314–327 CrossRef CAS PubMed.
- G. Karecla, P. Papagiorgis, N. Panagi, G. A. Zissimou, C. P. Constantinides, P. A. Koutentis, G. Itskos and S. C. Hayes, Emission from the stable Blatter radical, New J. Chem., 2017, 41, 8604–8613 RSC.
- Y. Zheng, M.-s. Miao, M. C. Kemei, R. Seshadri and F. Wudl, The pyreno-triazinyl radical – magnetic and sensor properties, Isr. J. Chem., 2014, 54, 774–778 CrossRef CAS.
- Y. Zheng, M.-s. Miao, G. Dantelle, N. D. Eisenmenger, G. Wu, I. Yavuz, M. L. Chabinyc, K. N. Houk and F. Wudl, A solid-state effect responsible for an organic quintet state at room temperature and ambient pressure, Adv. Mater., 2015, 27, 1718–1723 CrossRef CAS PubMed.
- M. Jasiński, J. Szczytko, D. Pociecha, H. Monobe and P. Kaszyński, Substituent-dependent magnetic behavior of discotic benzo[e][1,2,4]triazinyls, J. Am. Chem. Soc., 2016, 138, 9421–9424 CrossRef PubMed.
- M. Jasiński, K. Szymańska, A. Gardias, D. Pociecha, H. Monobe, J. Szczytko and P. Kaszyński, Tuning the magnetic properties of columnar benzo[e][1,2,4]triazin-4-yls with the molecular shape, ChemPhysChem, 2019, 20, 636–644 CrossRef PubMed.
- F. Ciccullo, A. Calzolari, K. Bader, P. Neugebauer, N. M. Gallagher, A. Rajca, J. van Slageren and M. B. Casu, Interfacing a potential purely organic molecular quantum bit with a real-life surface, ACS Appl. Mater. Interfaces, 2019, 11, 1571–1578 CrossRef CAS PubMed.
- F. Ciccullo, N. M. Gallagher, O. Geladari, T. Chassé, A. Rajca and M. B. Casu, A derivative of the Blatter radical as a potential metal-free magnet for stable thin films and interfaces, ACS Appl. Mater. Interfaces, 2016, 8, 1805–1812 CrossRef CAS PubMed.
- J. Z. Low, G. Kladnik, L. L. Patera, S. Sokolov, G. Lovat, E. Kumarasamy, J. Repp, L. M. Campos, D. Cvetko, A. Morgante and L. Venkataraman, The environment-dependent behavior of the Blatter radical at the metal–molecule interface, Nano Lett., 2019, 19, 2543–2548 CrossRef CAS PubMed.
- M. Demetriou, A. A. Berezin, P. A. Koutentis and T. Krasia-Christoforou, Benzotriazinyl-mediated controlled radical polymerization of styrene, Polym. Int., 2014, 63, 674–679 CrossRef CAS.
- J. Areephong, K. M. Mattson, N. J. Treat, S. O. Poelma, J. W. Kramer, H. A. Sprafke, A. A. Latimer, J. Read de Alaniz and C. J. Hawker, Triazine-mediated controlled radical polymerization: new unimolecular initiators, Polym. Chem., 2016, 7, 370–374 RSC.
- P. Kaszyński, C. P. Constantinides and V. G. Young Jr., The planar Blatter radical: Structural chemistry of 1,4-dihydrobenzo[e][1,2,4]triazin-4-yls, Angew. Chem., Int. Ed., 2016, 55, 11149–11152 CrossRef PubMed.
- P. Bartos, B. Anand, A. Pietrzak and P. Kaszyński, Functional planar Blatter radical through Pschorr-type cyclization, Org. Lett., 2020, 22, 180–184 CrossRef CAS PubMed.
- A. A. Hande, C. Darrigan, P. Bartos, P. Baylère, A. Pietrzak, P. Kaszyński and A. Chrostowska, UV-photoelectron spectroscopy of stable radicals: the electronic structure of planar Blatter radicals as materials for organic electronics, Phys. Chem. Chem. Phys., 2020, 22, 23637–23644 RSC.
- P. Bartos, A. A. Hande, A. Pietrzak, A. Chrostowska and P. Kaszyński, Substituent effects on the electronic structure of the flat Blatter radical: Correlation analysis of experimental and computational data, New J. Chem., 2021, 45, 22876–22887 RSC.
- K. I. Shivakumar, D. Pociecha, J. Szczytko, S. Kapuściński, H. Monobe and P. Kaszyński, Photoconductive bent-core liquid crystalline radicals with a paramagnetic polar switchable phase, J. Mater. Chem. C, 2020, 8, 1083–1088 RSC.
- P. Bartos, V. G. Young and P. Kaszyński, Ring-fused 1,4-dihydro[1,2,4]triazin-4-yls through photocyclization, Org. Lett., 2020, 22, 3835–3840 CrossRef CAS PubMed.
- M. Gurry and F. Aldabbagh, A new era for homolytic aromatic substitution: Replacing Bu3SnH with efficient light-induced chain reactions, Org. Biomol. Chem., 2016, 14, 3849–3862 RSC.
- A. G. Davies, Tin organometallics, Compr. Organomet. Chem. III, 2007, 3, 809–883 CAS.
- A. J. Clark, Compounds of group 14 (Ge, Sn, Pb), in Science of Synthesis, ed. E. J. Thomas, Thieme, Stuttgart, 2003, vol. 5, pp. 205–274 Search PubMed.
- C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Thirty years of (TMS)3SiH: A milestone in radical-based synthetic chemistry, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed.
- C. Chatgilialoglu and J. Lalevée, Recent applications of the (TMS)3SiH radical-based reagent, Molecules, 2012, 17, 527–555 CrossRef CAS PubMed.
- W. R. Bowman and J. M. D. Storey, Synthesis using aromatic homolytic substitution – recent advances, Chem. Soc. Rev., 2007, 36, 1803–1822 RSC.
- W. R. Bowman, M. O. Cloonan and S. L. Krintel, Synthesis of heterocycles by radical cyclisation, J. Chem. Soc., Perkin Trans. 1, 2001, 2885–2902 RSC.
- R. K. Kawade, C. Hu, N. R. Dos Santos, N. Watson, X. Lin, K. Hanson and I. V. Alabugin, Phenalenannulations: Three-point double annulation reactions that convert benzenes into pyrenes, Angew. Chem., Int. Ed., 2020, 59, 14352–14357 CrossRef CAS PubMed.
- J. C. Estevez, M. C. Villaverde, R. J. Estevez and L. Castedo, Tributyltin(IV) hydride mediated free-radical syntheses of dehydrodibenzochromanones, dehydrodibenzocoumaranones and aristolactams, Tetrahedron, 1995, 51, 4075–4082 CrossRef CAS.
- H. Takiguchi, K. Ohmori and K. Suzuki, Synthesis and determination of the absolute configuration of cavicularin by symmetrization/asymmetrization approach, Angew. Chem., Int. Ed., 2013, 52, 10472–10476 CrossRef CAS PubMed.
- D. C. Harrowven, M. I. T. Nunn and D. R. Fenwick, [5]Helicenes by tandem radical cyclisation, Tetrahedron Lett., 2002, 43, 7345–7347 CrossRef CAS.
- D. C. Harrowven, I. L. Guy and L. Nanson, Efficient phenanthrene, helicene, and azahelicene syntheses, Angew. Chem., Int. Ed., 2006, 45, 2242–2245 CrossRef CAS PubMed.
- D. P. Curran and A. I. Keller, Radical additions of aryl iodides to arenes are facilitated by oxidative rearomatization with dioxygen, J. Am. Chem. Soc., 2006, 128, 13706–13707 CrossRef CAS PubMed.
- R. R. Castillo, C. Burgos, J. J. Vaquero and J. Alvarez-Builla, Radical intramolecular arylation of pyridinium salts: A straightforward entry to 7-hydroxypyrido[2,1-a]isoquinolinylium salts, Eur. J. Org. Chem., 2011, 619–628 CrossRef CAS.
- W. R. Bowman, A. J. Fletcher and G. B. S. Potts, Synthesis of heterocycles by radical cyclisation, J. Chem. Soc., Perkin Trans. 1, 2002, 2747–2762 RSC.
- S. Saito, K. Matsuo and S. Yamaguchi, Polycyclic π-electron system with boron at its center, J. Am. Chem. Soc., 2012, 134, 9130–9133 CrossRef CAS PubMed.
- S. M. Allin, W. R. S. Barton, W. R. Bowman and T. McInally, Radical cyclisation onto pyrazoles: Synthesis of withasomnine, Tetrahedron Lett., 2002, 43, 4191–4193 CrossRef CAS.
- F. Filace, P. A. Sańchez-Murcia, D. Sucunza, A. Peŕez-Redondo, J. Álvarez-Buílla, F. Gago and C. Burgos, Silyl assistance in the intramolecular addition of pyridyl radicals onto pyridines and quinolines, Eur. J. Org. Chem., 2016, 1891–1896 CrossRef CAS.
- For details see the ESI.†.
- J. Lalut, G. Santoni, D. Karila, C. Lecoutey, A. Davis, F. Nachon, I. Silman, J. Sussman, M. Weik, T. Maurice, P. Dallemagne and C. Rochais, Novel multitarget-directed ligands targeting acetylcholinesterase and σ1 receptors as lead compounds for treatment of Alzheimer's disease: Synthesis, evaluation, and structural characterization of their complexes with acetylcholinesterase, Eur. J. Med. Chem., 2019, 162, 234–248 CrossRef CAS PubMed.
- A. Ookawa, S. Yokoyama and K. Soai, Chemoselective reduction of diaryl bisulfides to thiols with sodium borohydride in mixed solvent containing methanol, Synth. Commun., 1986, 16, 819–825 CrossRef CAS.
- T. J. Burkey, M. Majewski and D. Griller, Heats of formation of radicals and molecules by a photoacoustic technique, J. Am. Chem. Soc., 1986, 108, 2218–2221 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS , and references therein.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- L. Almanqur, I. Vitorica-yrezabal, G. Whitehead, D. J. Lewis and P. O’Brien, Synthesis of nanostructured powders and thin films of iron sulfide from molecular precursors, RSC Adv., 2018, 8, 29096–29103 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic details, NMR spectra, details of XRD, spectroscopic (UV-vis and EPR) and electrochemical analyses and archives of DFT computational results. CCDC 2107124. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo01742j |
| This journal is © the Partner Organisations 2022 |