
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crosslinked p(MMA) particles by RAFT emulsion polymerisation: tuning size and stability†
Catherine J.
Marsden
ab,
Colum
Breen
ab,
James D.
Tinkler
 a,
Thomas R.
Berki
ab,
Daniel W.
Lester
c,
Jonathan
Martinelli
d,
Lorenzo
Tei
d,
Stephen J.
Butler
*b and
Helen
Willcock
*a
a,
Thomas R.
Berki
ab,
Daniel W.
Lester
c,
Jonathan
Martinelli
d,
Lorenzo
Tei
d,
Stephen J.
Butler
*b and
Helen
Willcock
*a
aDepartment of Materials, Loughborough University, Leicestershire, LE11 3TU, UK. E-mail: h.willcock2@lboro.ac.uk
bDepartment of Chemistry, Loughborough University, Leicestershire, LE11 3TU, UK
cPolymer Characterisation Research Technology Platform, University of Warwick, Coventry, CV4 7AL, UK
dDipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale, Viale Teresa Michel 11, 15121, Alessandria, Italy
First published on 1st June 2022
Abstract
The controlled synthesis of amphiphilic di-block copolymers allows a large array of nanostructures to be created, including block copolymer particles, which have proved valuable for biomedical applications. Despite progress in targeting specific block copolymer architectures, control over the size and stability of spherical particles is less well explored. Here, we report the use of RAFT emulsion polymerisation to synthesise a library of p(MMA) particles, crosslinked with ethylene glycol dimethacrylate and stabilised by brush-like poly(ethylene glycol)-based polymers. We successfully synthesised a range of block copolymer particles, offering stability up to p(MMA)1000, with DLS reporting stable particle diameters of 33–176 nm and PDI < 0.2. DLS and AFM studies showed a general increase in particle diameter with increasing amounts of p(MMA). The use of a PEG methacrylate monomer with a methyl ether end group resulted in more well defined and stable particles than those with hydroxyl end groups. The copolymerisation of a suitably functionalized Gd(III) complex into the shell of the spherical p(MMA) particles resulted in Gd-loaded particles that were investigated in detail by 1H NMR relaxometry, demonstrating that the Gd complex was successfully incorporated into the particles. This study will inform the synthesis of core–shell particles with optimised stability and targeted sizes, and show a simple method to incorporate a molecular sensor, generating a macromolecular imaging agent.
Introduction
The synthesis of amphiphilic di-block copolymers gives access to a wide variety of architectures, such as worms, vesicles, and spherical particles. Vesicles and particles have been found particularly useful as drug delivery systems1–5 and diagnostic or theranostic agents.6–8 The size of these nanostructures can be tuned to influence their characteristics, allowing direct injection into the bloodstream.9 Furthermore, particle diameters below 200 nm may allow evasion of the immune system,9 whereas structures smaller than 100 nm can show enhanced antimicrobial effects due to increased interactions with microorganisms.10Reversible addition–fragmentation chain-transfer (RAFT)-emulsion polymerisation techniques offer an attractive approach to synthesising polymeric particles.11 RAFT polymerisation uses a chain transfer agent (CTA) to synthesise polymers with a predictable molecular weight, low molecular weight dispersity (Đ), and capacity for continued chain growth.12 A wide range of di-block copolymer particles have been studied for biomedical applications, many of which are based on the monomer, methyl methacrylate (MMA).2,13,14 MMA is chosen for its biocompatibility,15 inherent resistance to chemical hydrolysis,13 and hydrophobic nature, which allows the transportation of water-insoluble drugs around the body.2 However, several reports have detailed unexpected restrictions on the ability to stabilise p(MMA)-based particles synthesised through RAFT-emulsion polymerisation, requiring adjustments that either add additional steps to the synthesis or alter the particle's surface characteristics.16–18 These adjustments include the addition of hydrophobic character to the stabilising block and the use of a more anionic stabilising block such as methacrylic acid.17,18 For example, Charleux and co-workers combined the use of methacrylic acid and blocks of brush-like PEG chains to stabilise p(MMA) particles, however, they observed deviation from spherical particles to fibres and vesicles when the p(MMA) degree of polymerisation (DP) exceeded 400.16
The use of hydrophilic, brush-like polymers to stabilise hydrophobic nanostructures has received little attention. Notably, Perrier and co-workers compared brush-like PEG stabilising blocks to a linear polymer (α-D-mannopyran-1-oxyethyl acrylamide or ManAM) in the stabilisation of butyl acrylate.4 Particles stabilised with brush-like polymers were shown to be significantly smaller in size than those stabilised by linear hydrophilic blocks. The authors postulated that this decrease in size was due to the increased stability offered by the highly hydrophilic and bulky side chains of the PEG-stabilising system. Linear PEG chains are more commonly used on the surface of block copolymer particles,19,20 offering enhanced blood circulation times and a reduced immune response.21–23 Brush-like PEG polymers have also been shown to exhibit these properties, along with additional benefits. The bulkiness of brush-like systems may offer enhanced stabilisation, and the degree of polymerisation can be easily tuned through RAFT polymerisation techniques.23–25
We proposed that the under-used, brush-like PEG polymers could present a suitable method of stabilising p(MMA)-based particles, without the need for incorporation of additional stabilising groups, which may elongate the synthesis or alter the character of the shell.16–18 Here, we report the production of p(MMA) particles with excellent control over stability across a broad range of diameters, from 33 to 176 nm, utilising stabilising blocks based on either oligo(ethylene glycol) methacrylate (OEGMA) or oligo(ethylene glycol) methyl ether methacrylate (OEGMEM).
Results and discussion
Particle design
The particles were designed according to Fig. 1 and are based on the production of biocompatible particles with a hydrophobic p(MMA) core. The PEG-based monomers, OEGMA and OEGMEM, have higher hydrophilicity than MMA which promotes self-assembly into particle structures.9,26 To increase the stability of these particles, an ethylene glycol dimethacrylate (EGDMA) crosslinker was used. The formation of the hydrophilic polymer shell, p(OEGMA) or p(OEGMEM), was controlled using the RAFT agent 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid (CTP).27 This generated a macromolecular chain transfer agent (mCTA) which controlled the subsequent chain extension reaction with MMA and EGDMA to produce the core–shell particles (Fig. 1).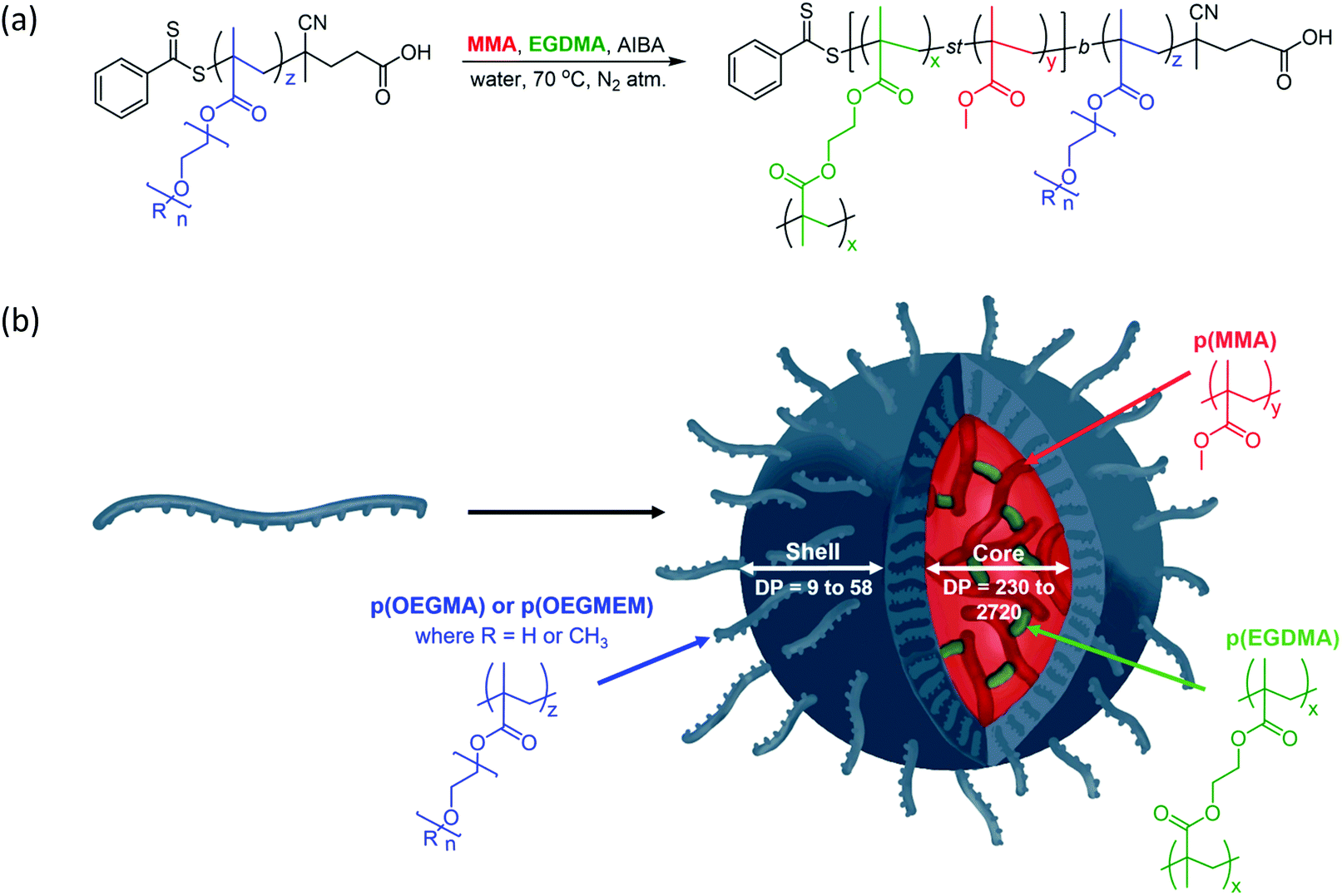 | ||
| Fig. 1 (a) Synthesis of block copolymer particles via chain extension of the mCTA p(OEGMEM) (R = CH3, n = 9), or p(OEGMA) (R = H, n = 6), with hydrophobic monomer MMA and crosslinker EGDMA. The DPs of the polymer blocks are varied between the following limits: x = 2 to 25, y = 230 to 2720, and z = 9 to 58. (b) Illustration of the particle synthesis, showing the self-assembly of the di-block copolymers into a spherical particle structure. | ||
Synthesis of p(OEGMA)z and p(OEGMEM)z
The first step of the synthesis was to polymerise the PEG monomers, OEGMEM and OEGMA, using RAFT agent CTP, based on the procedure by Roy et al.27 The use of lower molecular weight monomers (500 g mol−1 and 360 g mol−1) resulted in an optimised initial monomer concentration of 0.4 M, and a RAFT agent to initiator ratio, [CTP]/[I], of 5. The initiator was varied according to the solvent used, 4,4′-azobis(4-cyanopentanoic acid) (ACVA) when in dioxane and 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AIBA) in water/DMSO solutions. These conditions allowed for the completion of both small (5 mL) and large scale (100 mL) polymerisations in a reasonable time frame (4 to 24 hours). Whilst the polymerisation was shown to work in dioxane, aqueous solvent mixtures are more suited for future biological applications, and hence were used preferentially in this work. Purification of the polymers synthesised in 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 water/DMSO mixture was achieved by dialysis. The reactions were tracked by NMR spectroscopy using trioxane as an internal reference, with all reactions exceeding 80% conversion (Table S1 and eqn (S1)†).
:20 water/DMSO mixture was achieved by dialysis. The reactions were tracked by NMR spectroscopy using trioxane as an internal reference, with all reactions exceeding 80% conversion (Table S1 and eqn (S1)†).
Three DPs were assessed for each of the stabilising blocks p(OEGMA): DP 9, 31, and 46, and p(OEGMEM): DP 12, 26, and 58. DPs were calculated by comparison to aromatic end groups (Fig. S2†). Size Exclusion Chromatography (SEC) revealed high Đ for p(OEGMA) polymers (1.6 ≤ Đ ≤ 1.8), with high molecular weight shoulders seen for p(OEGMA)31 and p(OEGMA)46 (Fig. 2a).28 In contrast, the p(OEGMEM) polymers demonstrated narrow Đ in the range 1.1 ≤ Đ ≤ 1.2 (Fig. 2b). High Đ values are often a result of increased terminations, promoted by high levels of conversion.29,30 However, in the current work p(OEGMA)46 only reached 76% conversion and still demonstrated high Đ and a high molecular weight shoulder.
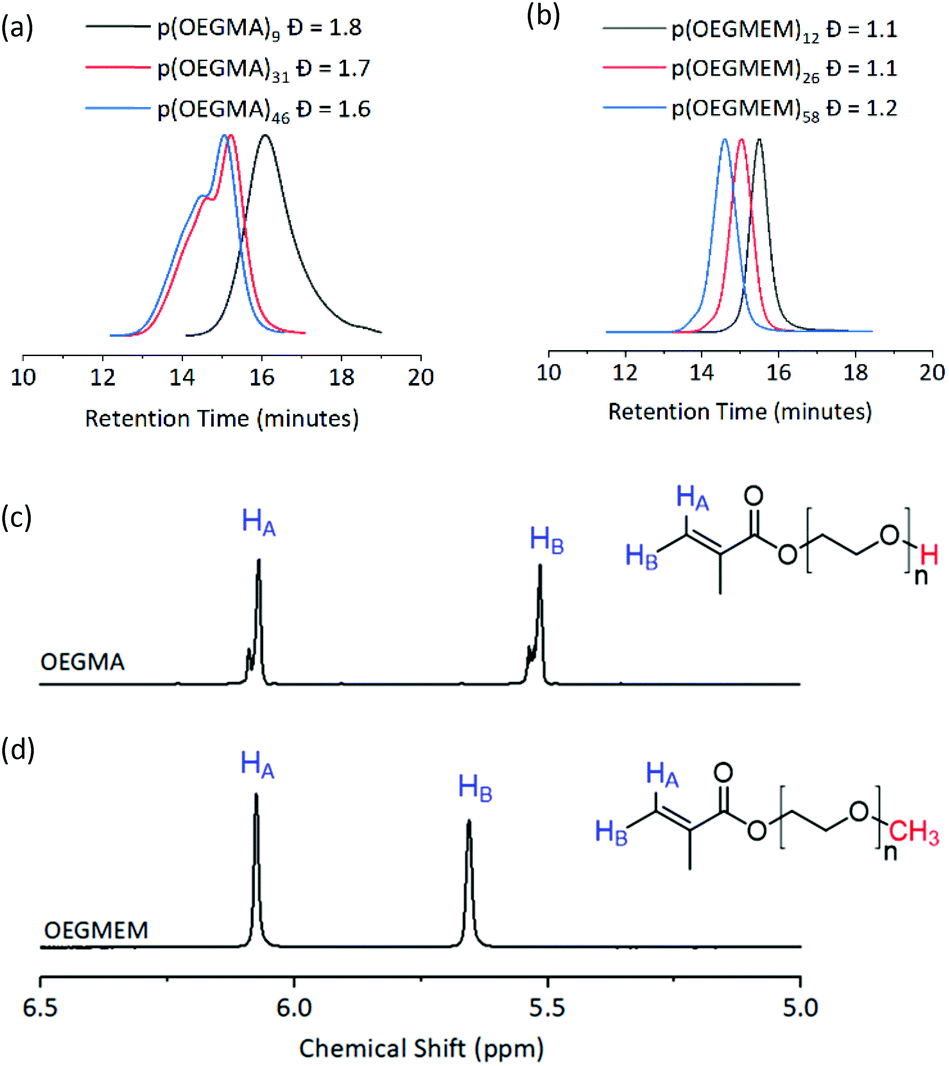 | ||
| Fig. 2 Top: SEC traces of the mCTA synthesised. (a) p(OEGMA) polymers with broad dispersity (1.6 ≤ Đ ≤ 1.8). (b) p(OEGMEM) polymers gave a narrow dispersity (1.1 ≤ Đ ≤ 1.2). mCTA synthesis summary, including SEC details, is given in Table S1.† Bottom: 1H NMR spectra of (c) OEGMA and (d) OEGMEM monomers, vinyl hydrogens in blue. Red atoms highlight the different end groups of the monomers. OEGMA possesses approximately 20 to 30% impurity by NMR integration. Full NMR spectra of the monomers is shown in Fig. S5.† | ||
We attempted to reduce the Đ of the synthesised p(OEGMA) by varying reaction temperature, monomer concentration, initiator concentration, solvent, degree of conversion and monomer batch. Despite these attempts, the high Đ and high molecular weight shoulder remained (Fig. S3†). For this sample it was not possible to conduct GPC with UV-detection to determine the presence of RAFT end groups due to limited solubility. However, the livingness of the chain was instead demonstrated by conducting a chain extension polymerisation on p(OEGMA)46 with MMA. SEC analysis showed only a small overlap of the p(OEGMA)46 and p(OEGMA)46-b-p(MMA)2150 traces, Fig. S4.† This suggests that the majority of the p(OEGMA)46 sample, including most of the high molecular weight shoulder, possessed RAFT end groups. Hence, it is unlikely that the large dispersity and high molecular weight shoulder was caused by radical coupling.
Analysis of the OEGMA starting material by NMR spectroscopy revealed the presence of more than one environment for each of the alkenyl hydrogens, indicating a 20–30% impurity based on the NMR peak integrations (Fig. 2c). We hypothesise that this is a dimethacrylate impurity present in the OEGMA monomer supply, similar to that seen in the methacrylate monomers used by Ratcliffe et al. and Blanazs et al.31,32 Both reports demonstrated access to lower Đ polymers by purification of their methacrylate monomers prior to polymerisation, using silica column chromatography. However, the higher molecular weight and broader dispersity of our OEGMA monomer led to streaking on thin layer chromatography and meant that purification in this manner was not possible in this case.
To confirm that a crosslinker impurity was causing the high molecular weight shoulder seen in the p(OEGMA)31 and p(OEGMA)46 SEC traces, the pure OEGMEM monomer was polymerised with varying amounts of EGDMA crosslinker. EGDMA concentrations of 0.1 mol%, 0.5 mol%, and 25 mol% were used, based on previous studies31,32 and OEGMA NMR integrations. The SEC traces of the two lower concentrations (0.1 and 0.5 mol%) showed a small high molecular weight shoulder, although much less pronounced than that of the p(OEGMA) samples (Fig. S6†). However, when using 25 mol% of crosslinker in the p(OEGMEM) polymerisation, gelation of the reaction mixture occurred. This supports the presence of a crosslinker impurity in the OEGMA, but at a significantly higher concentration than in other monomer samples reported in the literature, and lower than estimated from our NMR analysis of the OEGMA monomer.
Despite the high dispersity of the p(OEGMA) samples, we opted to proceed towards the synthesis of block copolymer particles. We compared the influence of using p(OEGMA) and p(OEGMEM) stabilising blocks, which differ in dispersity and end groups, as well as lengths of the PEG side chains. Monomers with more comparable molecular weights (OEGMA360 and OEGMEM300) were originally used, however p(OEGMEM300) had a lower critical solution temperature (LCST) similar to that of the emulsion polymerisation temperature and so OEGMEM500 was chosen to resolve this issue.33
Production of p(OEGMA)z-b-[p(MMA)y-st-p(EGDMA)x] and p(OEGMEM)z-b-[p(MMA)y-st-p(EGDMA)x] block copolymer particles
RAFT-emulsion polymerisation was utilised to extend the p(OEGMA) and p(OEGMEM) brush-like chains with MMA, producing block copolymers that possess varying degrees of hydrophilicity. These contrasting affinities for water generate entropic and enthalpic effects either avoiding or promoting interactions with surrounding water molecules, resulting in self-assembly.34–38 The RAFT-emulsion process is performed similarly to a standard RAFT polymerisation.11 The p(OEGMA) and p(OEGMEM) chains were used as an mCTA, allowing extension with MMA and EGDMA, as shown in Fig. 1. The initial monomer concentration of MMA used was maintained at 0.5 M, and variation in DP was achieved by altering the concentration of mCTA. The molar fraction of EGDMA to MMA was kept at EGDMAmol/MMAmol = 0.009. Once the mCTA, trioxane, and AIBA were dissolved in a water/DMSO mixture, the pH of the solution was adjusted to ca. pH 6.5, through the addition of sodium hydroxide (2 M). MMA was then added to the quickly stirring solution in a dropwise manner to create suspended monomer droplets. These monomer droplets act as monomer reservoirs in emulsion polymerisation, allowing the monomer to diffuse across the aqueous phase to the growing polymer particles.11,39,40 The solution was heated to 65 °C under inert conditions and EGDMA was added after 30 minutes (when the solution turned cloudy, indicating the presence of polymer particles). After another 1.5 hours, the reaction mixture was cooled before undergoing purification by dialysis against water (6 × 4 hours, 3500 MWCO, 4 L). Emulsion polymerisations were tracked in the same manner as the RAFT polymerisation, detailed in the ESI.† The majority of the emulsion polymerisations conducted exceeded 85% conversion.However, two of the particles targeting larger DPs, p(OEGMEM)26-b-[p(MMA)1730-st-p(EGDMA)25] and p(OEGMEM)58-b-[p(MMA)1500-st-p(EGDMA)25], only reached 69% and 60% conversion, respectively. It is possible that, despite purification by dialysis, a small amount of unreacted monomer may be present in these samples, which may have contributed towards particle instabilities.41,42
After purification, particle samples were stored in solution and only dried when required for atomic force microscopy (AFM). A brief study analysed dried and re-dispersed particles made up to the same concentration as the initial particle solution (Fig. S7†). The sample was sonicated in an attempt to break up particle aggregates. However, AFM standard error increased from 1.6 to 2.1 nm, and dynamic light scattering (DLS) showed an increase in Đ from 0.10 to 0.13. The average particle size in the redispersed sample was also shown to be higher than that of the initial particle sample, especially in the DLS analysis (97 nm initial sample, 131 nm redispersed sample). These changes could be due to increased aggregation of the particles upon redispersion.
Particle analysis
The use of AFM and DLS allowed analysis of the particles in both dry and hydrated states. The average particle size and PDI are summarised in Table 1. There are several trends identified from the data, discussed below.| Stabilising blocka | p(MMA) DPb | DLS | AFMe | |
|---|---|---|---|---|
| d , nm | PDId | |||
| a Average DP across several batches, calculated by 1H NMR end group analysis. b p(MMA) DP calculated using MMA equivalents added and NMR conversion. Calculation of conversion is described in the ESI.† c Z average diameter by DLS. d Average PDI of DLS measurement over three runs. e The average diameter of 50 particles measured in an AFM image, and the standard deviation corresponding to this. f More than one population of particles present by either DLS distribution, correlograms, or AFM. Apparent sizes stated above may be unrepresentative for these samples. | ||||
| p(OEGMEM)26 | 230 | 33 | 0.10 | 19 ± 1 |
| 460 | 97 | 0.10 | 68 ± 12 | |
| 740 | 176 | 0.10 | 113 ± 16 | |
| 990f | 114 | 0.21 | 203 ± 210 | |
| 1730f | 1133 | 0.68 | 128 ± 31 | |
| p(OEGMEM)58 | 250 | 63 | 0.03 | 36 ± 5 |
| 430 | 105 | 0.03 | 67 ± 12 | |
| 620 | 137 | 0.15 | 63 ± 9 | |
| 810 | 173 | 0.15 | 71 ± 8 | |
| 1500f | 363 | 0.43 | 203 ± 102 | |
| p(OEGMA)9 | 290 | 83 | 0.17 | 28 ± 3 |
| 500 | 228 | 0.73 | 28 ± 6 | |
| 730f | 1950 | 0.90 | 25 ± 4 | |
| 1080f | 241 | 0.35 | 30 ± 4.3 | |
| 2340f | 3020 | 0.86 | 63 ± 15 | |
| p(OEGMA)46 | 300f | 22 | 0.21 | 19 ± 4 |
| 600 | 32 | 0.27 | 15 ± 4 | |
| 790f | 57 | 0.20 | 24 ± 4 | |
| 1190f | 78 | 0.22 | 27 ± 5 | |
| 2720f | 414 | 0.48 | 41 ± 7 | |
Use of p(OEGMEM) stabilising block results in more stable particles
Table 1 allows a comparison between the ability of the two stabilising blocks, p(OEGMEM) and p(OEGMA), to stabilise p(MMA) particles. DLS data provides information on the particles in their hydrated state. DLS analysis demonstrates a much narrower size distribution of p(OEGMEM) particles in comparison to p(OEGMA) stabilised particles (Fig. 3). Eight of the ten samples p(OEGMEM)-stabilised samples shown in Table 1 gave PDI values in the range 0.03–0.21. In contrast, Table 1 reveals the broad PDI of many of the p(OEGMA) stabilised particles. When considering the DLS data, Fig. S10 and S11,† six of the ten p(OEGMA) particles analysed possessed more than one population of particle sizes, potentially due to crosslinking in the p(OEGMA) synthesis. The presence of crosslinked chains in the p(OEGMA) stabilising block may have restricted stabilisation and promoted crosslinking between particles. The different end groups of the stabilising blocks (OEGMA: alcohol, OEGMEM: methyl ether) could also be influencing particle stabilisation.43,44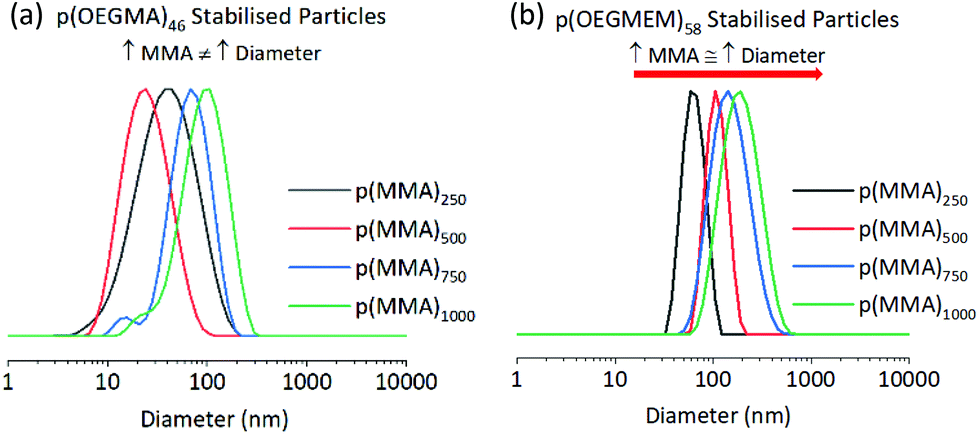 | ||
| Fig. 3 Comparison of (a) p(OEGMA)46-b-[p(MMA)y-st-p(EGDMA)x], and (b) p(OEGMEM)58-b-[p(MMA)y-st-p(EGDMA)x] particle DLS traces, showing intensity weighted data. The traces highlight the narrower dispersity of the p(OEGMEM)58 stabilised particles (b) in comparison to the p(OEGMA)46 stabilised particles (a), of which the Z-average diameters and PDIs are shown in Table 1. | ||
Typically, dry-state analysis of particles includes the use of scanning electron microscopy (SEM); however, the small size of most of these particles made SEM analysis challenging. Fortunately, AFM was a very useful technique, providing sufficiently high resolution for diameter measurements (e.g.Fig. 4a and b).45 When comparing p(OEGMEM) and p(OEGMA) stabilised samples, all AFM images showed particle-like architectures (Fig. S14–17†). However, several p(OEGMA)-stabilised samples appeared to contain fused particles (e.g.Fig. 4c and S16e†) supporting the presence of multiple populations seen in the DLS trace. Alternatively, it could be considered that the lower molecular weight of the OEGMA monomer units (and therefore shorter hydrophilic PEG chains) may reduce the ability of this stabilising block to stabilise spherical particle architectures. A smaller stabilising block increases the critical packing parameter,35,36,46 influencing the proportions of the space occupied by each block copolymer strand. This could result in different self-assembled structures such as cylindrical micelles, bilayer vesicles, or lamellar phases.47 Unfortunately, use of a more comparable monomer, OEGMEM300, gave mCTA with a LCST similar to that of the emulsion polymerisation temperature and so made chain extension difficult. Hence, OEGMEM500 was chosen to resolve this issue.33 However, despite differing DPs, the molecular weight of the p(OEGMA)46 stabilising block is theoretically very similar to that of p(OEGMEM)28 (16839 g mol−1 and 14279 g mol−1, respectively) and was measured to be similar to p(OEGMEM)58 (p(OEGMA)46 = 20600 g mol−1 and p(OEGMEM)58 = 20500 g mol−1), likely due to the high dispersity of p(OEGMA)46 seen by SEC (Fig. 2a and Table S1†). Therefore, these similarities offer an opportunity for comparison between the OEGMA-based and OEGMEM-based stabilising blocks.
 | ||
| Fig. 4 AFM images: (a) p(OEGMEM)58-b-[p(MMA)430-p(EGDMA)5], (b) p(OEGMEM)58-b-[p(MMA)620-p(EGDMA)8], (c) p(OEGMA)9-b-[p(MMA)730-p(EGDMA)8], (d) p(OEGMEM)58-b-[p(MMA)1500-p(EGDMA)25]. | ||
Overall, p(OEGMEM) stabilising blocks appeared to result in particles with a higher level of stability than p(OEGMA) stabilising blocks as measured by both DLS (Fig. 3) and AFM analysis. This result could be due to the difference in Đ of the stabilising blocks, as it has been shown previously that Đ of the stabilising block can have a significant influence on the size and stability of higher order structures in macromolecular systems.48–50
Larger stabilising blocks stabilise larger particles
The highest dispersities seen in the particle analysis corresponded to the largest p(MMA) cores; Table 1 reports PDI values of 0.68, 0.43, 0.86 and 0.48 for the particles with MMA DPs of around 2000. It is likely that in the presence of such a large amount of hydrophobic p(MMA), the stabilising block cannot stabilise the particles sufficiently. The presence of more than one particle size in the DLS distribution (Fig. S8d and e†) suggests the instability resulted in particle aggregation. Whilst the p(OEGMEM)58-b-[p(MMA)1500-st-p(EGDMA)25] DLS trace gave a narrower size distribution, both the DLS correlogram and AFM images indicate the presence of more than one population of particles (Fig. S9e† and Fig. 4d).Table 1 implies that larger stabilising blocks more efficiently stabilised p(MMA) particles at high DPs. The larger PEG-based blocks resulted in a narrower PDI, with samples p(OEGMEM)58-b-[p(MMA)1500-st-p(EGDMA)25] and p(OEGMA)46-b-[p(MMA)2720-st-p(EGDMA)25] showing PDIs less than 0.48 (Table 1). In contrast, the use of shorter hydrophilic blocks in the samples p(OEGMEM)26-b-[p(MMA)1730-st-p(EGDMA)25] and p(OEGMA)9-b-[p(MMA)2340-st-p(EGDMA)25], yielded higher PDI values of 0.68 and 0.86, respectively. This suggests that the shorter stabilising blocks, p(OEGMEM)26 and p(OEGMEM)9, are not bulky enough to sufficiently stabilise the large amount of hydrophobic p(MMA). The hydrophilic polymer p(OEGMEM)26 stabilised p(MMA) up to a DP of 750, after which the DLS trace demonstrated a broad range of particle sizes (Fig. S8d and e†) and a rise in PDI from 0.1 at p(MMA)750, to 0.21 at p(MMA)1000 (Table 1). The lack of stability afforded by the shorter p(OEGMEM)26 stabilising block may have induced flocculation of the particles, as observed by Armes and co-workers.18 On the other hand, the longer p(OEGMEM)58 stabilising block afforded particles with narrower size distributions, up to p(MMA)1000 (Fig. S9†), after which, sample p(OEGMEM)58-b-[p(MMA)1500-st-p(EGDMA)25] showed an increased range of particle sizes in the DLS correlogram and AFM (Fig. 4d).
Particles synthesised with smaller p(MMA)y = 230–790 blocks displayed similar PDI values for all particle samples, regardless of the p(OEGMEM) DP, as stabilisation with p(OEGMEM)26 and p(OEGMEM)58 gave similar PDIAVG values of 0.10 and 0.07 respectively. This implies that the shorter p(OEGMEM)26 possesses enough hydrophilic bulk to offer sufficient stabilisation of these smaller particles, and hence only limits stabilisation of particles with larger amounts of MMA.47
Increasing p(MMA) DP increases particle diameter
The DP of the p(MMA) block was varied to investigate the limits of particle size achievable. For each stabilising block used in Table 1, five DPs of p(MMA) were synthesised, with MMA equivalents of 250, 500, 750, 1000 and 2500 used in the emulsion polymerisation. Charleux and co-workers previously demonstrated progression from spheres to fibres and vesicles across a smaller range of p(MMA) DPs (200 to 700), when using a p([methacrylic acid]15-r-[OEGMEM950]15) copolymer as a stabilising block.16 However, in our work spherical particles were consistently produced, and a general increase in diameter was seen with an increase in p(MMA) DP, which is more comparable with that observed by Armes and co-workers. Armes demonstrated a library of spherical particles from 29 to 99 nm in diameter, with MMA DPs 50–2000 using a poly(methacrylic acid)56 stabilising block.18 Armes suggests that this increased stability was offered by the anionic nature of poly(methacrylic acid). Perhaps the large amount of hydrophilic PEG chains in this work gave a high level of stability, allowing the spherical architecture to be maintained at high MMA DPs.The larger PEG-based stabilising blocks shown in this work, p(OEGMEM)58 and p(OEGMA)46, gave serially increasing diameters by DLS. However, p(OEGMEM)26 and p(OEGMA)9 stabilised particles demonstrated fluctuating diameters between consecutive samples (Fig. 3). DLS distributions and correlograms for these samples indicate a range of particle populations, and thus some average diameters are likely unreliable (e.g., Fig. S8d†).
Brush-like stabilising blocks decrease the particle diameter
The use of PEG-based brush-like stabilising units resulted in particles smaller than expected. For example, the smallest stabilising block p(OEGMA)9 (3500 g mol−1), polymerised with a large amount of MMA (DP = 2340), resulted in particle diameters of 52 nm by AFM. This is much smaller than comparable particles in the literature, such as Tan et al.'s particles stabilised by linear OEGMEM (Mw = 2000 and 4000 g mol−1) with MMA DPs of ca. 750 and 1800 respectively, which resulted in particles with diameters of 1080 nm and 820 nm by SEM.19 The smaller average particle diameter seen here, despite similar molecular weights of both the stabilising block and the particle core, demonstrates the influence of the steric bulk of brush-like stabilising blocks in constricting the size of the block copolymer particles. This is in agreement with the work of Perrier and co-workers, who found that when comparing particles of the same core monomer DP (100) the use of brush-like stabilising blocks gave significantly smaller particle diameters (50 nm) than their linear counterparts (90 nm).4The influence of crosslinkers on particle diameters and stability
The addition of crosslinkers to block copolymer nano-assemblies is used to enhance structural stability against the influence of solvent, temperature, and other external stimuli.51,52 For future biological applications, the use of crosslinkers may prevent uncontrolled swelling or disintegration of the particles in the blood, ensuring the properties of the system are unchanged in a physiological environment.We postulated whether the addition of crosslinker before completion of the RAFT-mediated emulsion polymerisation had restricted particle growth. To test this, three particle samples with no crosslinker were synthesised: p(OEGMEM)24-b-p(MMA)220, 460, 860. Comparison with similar crosslinked samples (Table 2) demonstrated larger diameters for the un-crosslinked particles across both DLS and AFM studies. In some cases, the increase in diameter was almost double: p(OEGMEM)26-b-[p(MMA)230-st-p(EGDMA)2] at 33 nm compared to p(OEGMEM)24-b-p(MMA)220 at 61 nm, whereas other comparable samples had very similar diameters by DLS: p(OEGMEM)26-b-[p(MMA)460-st-p(EGDMA)5] at 97 nm compared to p(OEGMEM)24-b-p(MMA)460 at 104 nm. The PDI of the DLS data illustrates a negligible difference in the dispersity of the crosslinked and un-crosslinked samples, PDIAVG = 0.10 and 0.11 respectively. However, the AFM standard deviation of the largest un-crosslinked particles shows a much lower dispersity than that of the crosslinked particles (96 ± 13 vs. 203 ± 210). Given the larger diameters seen upon comparison, it may be beneficial to add crosslinker later in the particle synthesis in future studies, to avoid potential constriction of particle diameter.
| p(MMA) DPa | Crosslinked: p(OEGMEM)26-b-[p(MMA)y-st-p(EGDMA)x] | p(MMA) DPa | Un-Crosslinked: p(OEGMEM)24-b-p(MMA)y | ||||
|---|---|---|---|---|---|---|---|
| DLS db, nm | PDIc | AFM dd, nm | DLS db, nm | PDIc | AFM dd, nm | ||
| a Target DP of p(MMA). b DLS Z-average diameter of the particles across three runs. c DLS dispersity of the particles across three runs. d AFM average diameter of 50 measured particles. | |||||||
| 230 | 33 | 0.10 | 19 ± 1 | 220 | 61 | 0.10 | 44 ± 6 |
| 460 | 97 | 0.10 | 68 ± 12 | 460 | 104 | 0.06 | 91 ± 13 |
| 990 | 114 | 0.21 | 203 ± 210 | 860 | 129 | 0.16 | 96 ± 13 |
The synthesis of un-crosslinked samples allowed SEC analysis of the di-block copolymer particles, which showed a large PDI, 1.87–2.78 (Table S2†), and high molecular weight shoulder when using the refractive index (RI) detector. However, the high molecular weight shoulder was not visible in the corresponding UV trace (425 nm), demonstrating the absence of RAFT functionality (Fig. S20†). It has recently been shown that the use of hydrophilic initiators in emulsion polymerisation can reduce radical penetration of polymer particles, particularly when the particles possess glass transition temperatures higher than the temperature of the reaction (pMMA Tg ≈ 100 °C,53TR = 70 °C).54 Previous studies have noted that whilst the plasticising effect of unreacted monomer may lower the Tg inside the particles, this effect is not considered significant enough to have a large influence on radical penetration.54,55 Reduced radical penetration may reduce access of radicals to RAFT functionality contained within the particles, and therefore decreases the ability of RAFT agents to control the polymerisation. This can result in polymer particles with higher levels of dispersity and may be the reason for the high molecular weight shoulder and broad dispersity seen for these samples. The un-crosslinked samples may also contain some un-reacted p(OEGMEM)24, as the RI SEC traces show a peak at 16 minutes, which is similar to the retention time seen in the mCTA SEC analysis (Fig. S20†).
In order to confirm successful crosslinking of the EGDMA-containing particles, brief studies compared the stability of crosslinked and un-crosslinked particles in chloroform (Fig. S24 and S25†). Crosslinked samples were found to have decreased solubility in chloroform, which may be the reason for the variable Z-average values. However, DLS correlograms generated for the un-crosslinked particles closely resembled that of the p(OEGMEM) correlograms, Fig. S12,† suggesting dis-assembly of the particles. On the other hand, the correlograms of the crosslinked particles were more similar to that of the particles in water, which confirms that the EGDMA crosslinker added to these samples was effective in increasing the stability of the particle architectures.56
Copolymerisation with MRI-active monomer: Gd·L1
Having optimised the synthesis of PEG-stabilised p(MMA)-based particles and identified trends in DP influencing particle diameters and stability, we began introducing additional functionality into the particles. Previously, we demonstrated that decreasing the tumbling rate of MRI-active gadolinium(III) complexes, through incorporation into polymers, significantly increases the relaxivity of contrast agents, potentially decreasing the required dose in MRI imaging.57 The current work details copolymerisation of one of our previously reported monomeric Gd(III) complexes, Gd·L1, into the shell of spherical p(MMA) particles (Fig. 5).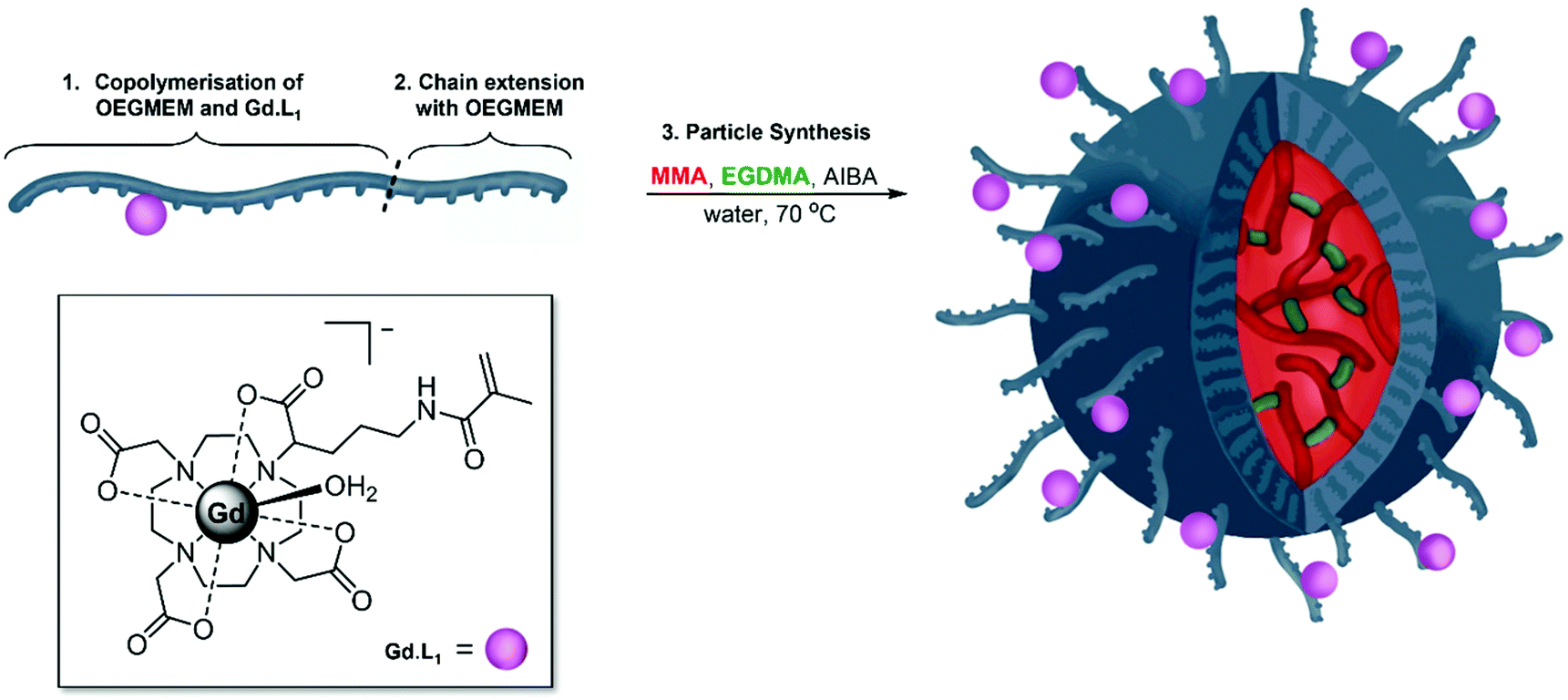 | ||
| Fig. 5 Illustration of the synthesis of Gd·L1 containing particles, synthesised in 3 steps. First (1) RAFT copolymerisation of OEGMEM500 and the previously synthesised Gd·L1 to give p(OEGMEM)50-st-p(GdL1)0.5; (2) chain extension of the random di-block copolymer with OEGMEM500, yielding [p(OEGMEM)50-st-p(GdL1)0.5]-b-p(OEGMEM)25. (3) Emulsion polymerisation of the GdL1-containing OEGMEM-based mCTA with MMA, to give GdL1-containing particles, [p(OEGMEM)50-st-p(GdL1)0.5]-b-p(OEGMEM)25-b-[p(MMA)1000-st-p(EGDMA)10]. | ||
The two-step particle synthesis allowed localisation of Gd·L1 into the particle shell, through addition of the complex to the p(OEGMEM) reaction mixture. The polymerisation was performed under the same conditions as the p(OEGMEM) synthesis detailed above (Fig. 1). To encourage polymerisation of the Gd·L1 complex, the OEGMEM monomer was added gradually, yielding the mCTA: p(OEGMEM)50-st-p(Gd·L1)0.5, if assuming 100% conversion of both monomers. This polymer was then purified, and chain extended with more OEGMEM to ensure that Gd·L1 was contained within the hydrophilic portion, yielding [p(OEGMEM)50-st-p(Gd·L1)0.5]-b-p(OEGMEM)25. The purified Gd·L1-containing shell was then used to form particles in the same manner as without Gd·L1, resulting in [p(OEGMEM)50-st-p(Gd·L1)0.5]-b-p(OEGMEM)25-b-[p(MMA)1000-st-p(EGDMA)10] particles, which will be referred to as: (Gd·L1)-P. The (Gd·L1)-P demonstrated an average diameter of 127 nm and PDI of 0.11 by DLS (Fig. S27†) and small changes in size with changes in pH, ca. 10 nm across pH 4–10, perhaps due to the terminal carboxylic acid group (Fig. S28†).44
The 1H NMR relaxometric properties of (Gd·L1)-P suspended in aqueous solutions were investigated to evaluate their efficacy as a diagnostic probe. The relaxivity values (r1) of these particles at 30 MHz and 298 and 310 K were 6.7 and 5.3 mM−1 s−1, respectively. These values are slightly higher than the monomeric Gd·L1 complex (6.2 and 5.0 mM−1 s−1, respectively), but much lower than those reported for other Gd-loaded polymeric or hybrid particles.58–60 Moreover, the same Gd-complex incorporated in linear 4-acryloylmorpholine (NAM) copolymers, p(NAM-r-Gd·L1), at different molar proportions of Gd·L1 (1 to 17 mol%) showed higher relaxivities, ranging from 17 to 18.4 mM−1 s−1 (at 30 MHz and 298 K).57 A similar r1 value was observed in the case of a GdDOTA-monoamide-functionalized mesoporous silicas SBA-15 (6.1 mM−1 s−1 at 30 MHz and 310 K);61 in that case the authors highlighted the presence of a large number (ca. 80%) of Gd-complex inside the pores of the hybrid material through which the water diffusion was reduced, lowering thus the overall relaxivity.
To get more insight into the relaxometric properties of (Gd·L1)-Ps, the nuclear magnetic relaxation dispersion (NMRD) profile of the (Gd·L1)-Ps was measured at 298 and 310 K over the frequency range 0.1–128 MHz which correspond to magnetic field strengths varying between 2.34 × 10−3 and 3 T (Fig. 6). The NMRD profiles are characterized by a r1 decrease in the 0.1–5 MHz frequency region, followed by a rather broad peak with a relatively modest amplitude at 30 MHz and a further decrease at higher fields. Such behaviour resembles the characteristics of slowly tumbling Gd-based nanosystems (long τR values). The data can be analysed according to the Solomon–Bloembergen–Morgan (SBM) equations of paramagnetic relaxation,58,62 modified by the incorporation of the Lipari–Szabo model-free approach,63,64 to assess some key molecular parameters that control the relaxivity of paramagnetic systems.‡
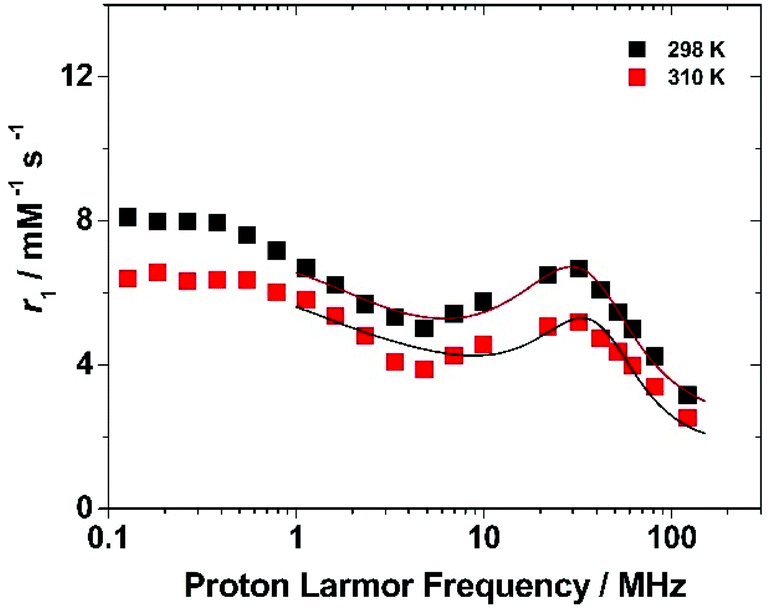 | ||
| Fig. 6 Nuclear magnetic relaxation dispersion (NMRD) profiles of Gd·L1-P particles (0.89 mM) at 298 (black squares) and 310 K (red squares). The solid lines represent the best results of the fitting to the experimental points. | ||
The high-field regions of the NMRD data were nicely fitted with the parameters reported in Table 3. In the best-fit procedure, some of the parameters were fixed at known or reasonable values. For example, τM was fixed to 330 ns, the value obtained for the linear p(NAM-r-Gd·L1) copolymers, and the global correlation time was fixed at 10 ns, to account for the slow tumbling motion of the particle (the results of the NMRD fits are not sensitive to the τRG value in the range of ca. 10 ns−1 ms).58 A full description of the fitting procedure is given in the ESI,† however, it should be noted that in accordance with the procedure used in case of the Gd-loaded SBA-15 mesoporous silicas, the coordinated water molecule (q) was left to vary in order to account for an “effective” concentration of Gd(III) that contributes to the observed relaxivity. The best fit for the NMRD profile was obtained with a τRL value of 0.41 ns which agrees with the degree of local rotational freedom of the complex observed in the case of the linear p(NAM-r-Gd·L1) polymer. On the other hand, considering the much higher τRL value, the S2 of 0.60 ± 0.05 implicates a higher coupling between the local and global motion of the system. Letting q vary, we obtained a value of 0.15 ± 0.02 which implies that a large number of Gd-complexes are hindered from contributing to the overall relaxivity. We can hypothesize several reasons for this result, among which the slow water diffusion inside the layer in which the complexes are located maybe the most reasonable.
| Parameter | (Gd·L1)-P | P(NAM-r-Gd·L1) |
|---|---|---|
| The Gd–Hw distance rGd–H was set to 3.0 Å, the distance of the closest approach of the outer-sphere solvent molecules to the Gd3+ ions, a, was fixed to 3.8 Å, and the water–solute relative diffusion coefficient, D, was fixed at 2.25 × 10−5 and 3.1 × 10−5 cm2 s−1 for 298 and 310 K, respectively.a Fixed to the value determined for the same Gd·L1 complex incorporated in the linear p(NAM-r-Gd·L1) copolymer.b Fixed to the value reported for Gd-loaded nanoparticles of similar size. | ||
| Δ 2/1019 s−2 | 1.9 ± 0.2 | 4.37 ± 0.06 |
| τ V/ps | 23 ± 2 | 42.1 ± 0.6 |
| τ M/ns | 330a | 330 |
| τ RL/ns | 0.41 ± 0.05 | 0.34 ± 0.01 |
| τ RG/ns | 10b | 2.7 |
| S 2 | 0.60 ± 0.05 | 0.175 |
| q | 0.15 ± 0.02 | 1 |
Conclusions
We have identified parameters within the synthesis of p(MMA) block copolymer particles that have controlled particle size and stability. A library of particles was generated encompassing a broad range of sizes, from tens to hundreds of nanometres. Our systematic investigation of the polymerisation of poly(ethylene glycol)-based monomers in the formation of the stabilising block found that OEGMEM gave much narrower Đ in the synthesis of p(OEGMEM) compared to p(OEGMA). Use of p(OEGMEM) to stabilise p(MMA) gave particles with a narrower size distribution, up to p(MMA)1000, whereas six of the ten p(OEGMA) samples showed multiple populations by DLS (Table 1).Importantly, the use of brush-like blocks offered a high level of stability without requiring additional anionic or more hydrophobic stabilising units and resulted in smaller systems than their linear counterparts. The use of p(OEGMEM) blocks offers opportunities for tuning the molecular weight, and the incorporation of molecular sensors and imaging agents onto the surface of the particle. This general concept was demonstrated by the introduction of an MRI-active Gd(III)-based contrast agent into the shell of the particles, promoting access to surrounding water molecules and showing a small increase in relaxivity compared to the free Gd·L1 monomer. Work is currently ongoing to reduce aggregation and increase water access to the contrast agents in Gd·L1-loaded particles, which should further improve the relaxivity values obtained.
Experimental
Below are representative synthesises for the production of the PEG mCTAs and block copolymer particles. Variation in DP is achieved through alterations to the CTA:Monomer equivalents, where the initial monomer concentration is kept constant. In the case of p(OEGMA) stabilised particles, initially, additional equivalents of MMA were added with the idea that the polymerisation would be stopped before 100%. However, the reaction progressed much faster than expected and reactions came close to 100%. Therefore, in the synthesis of all the p(OEGMEM)-stabilised particles, equivalents of MMA monomer added were that of the target DP.
Representative synthesis of p(OEGMEM)12: macro chain transfer agent
To a stirred 80:20 water/DMSO mixture (3.85 mL) was added OEGMEM500 (926 μL, 2.00 mmol, 10 equiv.), trioxane (14.0 mg, 0.16 mmol, 0.8 equiv.), CTP (56 mg, 0.2 mmol, 1 equiv.) and AIBA (220 μL stock solution, 0.04 mmol, 0.2 equiv.). The solution was purged with nitrogen for 5 minutes, before being stirred at 70 °C for 4 hours, at which point the reaction reached full conversion (>99%). The product was purified by dialysis (6 × 4 h, 3500 MWCO, 4 L deionised water) and lyophilized to yield a viscous pink oil. IR (vmax/cm−1, neat): 2868, 1728, 1452, 1349, 1247, 1094, 1034, 945, 852. Example p(OEGMEM) 1H NMR (400 MHz, D2O) δ 8.02–7.61 (5H, Aromatic H), 4.21 (2H, COOCH2 side chain), 3.81–3.64 (34H CH2OCH2 side chain), 3.41 (–OCH3 side chain), 1.93–0.92 (5H, CH2, CH3 backbone). Example p(OEGMEM) 13C NMR (101 MHz, D2O) δ 178.9, 178.0 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 71.1 (COOCH2 side chain), 69.9–69.6, 68.2, 64.9–64.7 (CH2OCH2 side chain), 58.2 (–OCH3 side chain), 49.0 (CQ backbone), 45.1 (CH2 backbone), 44.7 (CH3 backbone).
O), 71.1 (COOCH2 side chain), 69.9–69.6, 68.2, 64.9–64.7 (CH2OCH2 side chain), 58.2 (–OCH3 side chain), 49.0 (CQ backbone), 45.1 (CH2 backbone), 44.7 (CH3 backbone).
Representative synthesis of p(OEGMA)46: macro chain transfer agent
A 50 mg mL−1 stock solution of AIBA was produced. To a stirred mixture of 80:20 water/DMSO (4.3 mL) was added OEGMA360 (652 μL, 2.00 mmol, 50 equiv.), trioxane (14.0 mg, 0.16 mmol, 4 equiv.), CTP (11 mg, 0.04 mmol, 1 equiv.), and AIBA (40 μL stock solution, 0.008 mmol, 0.2 equiv.). The solution was purged with nitrogen for 5 minutes, then stirred at 70 °C for 4 hours, at which point the reaction reached 82% conversion. The reaction was cooled to room temperature and purified by dialysis (6 × 4 hours, 3500 MWCO, 4 L deionised water). The solvent was removed by lyophilization, to yield the product as a viscous pink oil. IR (vmax/cm−1, neat): 3453, 2870, 1724, 1450, 1351, 1247, 1096, 945, 861, 747, 691. Example p(OEGMA) 1H NMR (400 MHz, D2O) δ 8.05–7.63 (5H, Aromatic H), 4.22 (2H, COOCH2), 3.82–3.68 (22H, CH2OCH2 side chain), 2.92–0.94 (5H, CH2, CH3 backbone). Example p(OEGMA) 13C NMR (101 MHz, D2O) δ 179.1, 178.2 (CO), 71.9 (COOCH2 side chain), 69.7, 68.2, 64.9 (CH2OCH2 side chain), 60.5 (CH2OH), 49.0 (CQ backbone), 45.1 (CH2 backbone), 44.7 (CH3 backbone).
Representative synthesis of p(OEGMEM)26-b-p(MMA)230 particles
Prepare a 50 mg mL−1 stock solution of AIBA in water. In this exact order, poly(OEGMEM)26 (700 mg, 0.05 mmol, 1 equiv.), trioxane (90 mg, 1 mmol, 20 equiv.), AIBA (54 μL of stock solution, 0.01 mmol, 0.2 equiv.) were added to water (23 mL). The solution was adjusted to pH 6.5–7 using NaOH (2 M). Whilst stirring, MMA (1.3 mL, 12.5 mmol, 250 equiv.) was added dropwise. The solution was purged with nitrogen for 15 minutes, then heated at 70 °C for 30 minutes, or until the solution turned cloudy, at which point EGDMA (22.6 μL, 0.12 mmol, 2 equivalents) was added. The solution was heated for a further 1.5 h. The solution was purified by dialysis against water (6 × 4 h, 3500 MWCO, 4 L deionised water) to give the product, a cloudy pink solution. In general, the higher the DP of p(MMA) targeted, the more opaque the resulting solution. IR (vmax/cm−1, neat): 2993, 2950, 1724, 1448, 1269, 1239, 1191, 1142, 988, 984, 842, 749. Example un-crosslinked p(OEGMEM)24-b-p(MMA)2501H NMR (400 MHz, CDCl3) δ (ppm): 4.08 (2H, COOCH2), 3.64 (3H, MMA OCH3), 3.59 (34H, –CH2OCH2 PEG side chains), 3.37 (3H, –OCH3), 2.02–0.84 (CH3, CH2, MMA and PEG backbone). 13C NMR (101 MHz, CDCl3) δ (ppm): 72.2 (COOCH2), 70.8 (CH2OCH2 PEG), 59.3 (OCH3 PEG), 52.0 (OCH3 MMA), 45.1–44.8 (CH3, CH2, MMA and PEG backbone).Representative synthesis of p(OEGMA)46-b-p(MMA)2720 particles
Prepare a 50 mg mL−1 stock solution of AIBA in water. In this exact order, poly(OEGMA)46 (79 mg, 0.004 mmol, 1 equiv.), trioxane (90 mg, 1 mmol, 227 equiv.), AIBA (4.78 μL stock solution, 0.0009 mmol, 0.2 equiv.) were dissolved in water (23 mL). The solution was adjusted to pH 6.5–7 using NaOH (2 M), before the dropwise addition of MMA (1.3 mL, 12.2 mmol, 2750 equiv.). The solution was purged with nitrogen for 15 minutes and then heated at 70 °C for 30 minutes before the addition of EGDMA (20.7 μL, 0.12 mmol, 25 equiv.). The solution was heated for a further 3.5 hours. The resulting solution was purified by dialysis (6 × 4 hours, 3500 MWCO, 4 L deionised water) to yield a cloudy pink solution. In general, the higher the DP of p(MMA) targeted, the more opaque the resulting solution. IR (vmax/cm−1, neat): 2993, 2950, 1720, 1476, 1437, 1389, 1239, 1191, 1142, 841, 749. 1H NMR (400 MHz, CDCl3) δ (ppm): 4.06 (2H, COOCH2), 3.64 (3H, MMA OCH3), 3.60–3.30 (22H, –CH2OCH2 PEG side chains), 2.16–0.83 (CH3, CH2, MMA and PEG backbone). 13C NMR (101 MHz, CDCl3) δ (ppm): 72.1 (COOCH2), 70.7 (CH2OCH2 PEG), 51.9 (OCH3 MMA), 45.1–44.7 (CH3, CH2, MMA and PEG backbone).Synthesis of p(OEGMEM)50-r-p(Gd·L1)0.5-b-p(OEGMEM)25
:20 water/DMSO mixture (1.2 mL) in a 10 mL flask was added OEGMEM500 (278 μL, 0.6 mmol, 20 equiv.), trioxane (4.5 mg, 0.05 mmol, 2 equiv.), CTP (8.4 mg, 0.03 mmol, 1 equiv.), AIBA (32 μL stock solution, 0.006 mmol, 0.2 equiv.) and Gd·L1 (10 mg, 0.015 mmol, 0.5 equiv.). The solution was purged with nitrogen for 5 minutes, before being stirred at 70 °C for 2 hours (91% monomer conversion). A purged sample of OEGMEM500 (139 μL, 0.3 mmol, 10 equiv.) was then added and the reaction heated for a further 1.5 h. Two more aliquots of OEGMEM500 (139 μL, 0.3 mmol, 10 equiv.) were added once each batch of monomer was >70% consumed. In total, 50 equivalents of OEGMEM500 were used. The last two batches only required 1 h of reaction time. The product was purified by dialysis (6 × 4 h, 3500 MWCO, 4 L deionised water) and lyophilized to yield a viscous pink oil.
:20 water/DMSO mixture (1 mL) was added OEGMEM500 (218 μL, 0.5 mmol, 25 equiv.), trioxane (3.4 mg, 0.04 mmol, 2 equiv.), AIBA (20 μL stock solution, 0.004 mmol, 0.2 equiv.). The solution was purged with nitrogen for 5 minutes, before being stirred at 70 °C for 4 hours (77% conversion by NMR). The product was purified by dialysis (6 × 4 h, 3500 MWCO, 4 L deionised water) and lyophilized to yield a viscous pink oil. p(OEGMEM)50-r-p(Gd·L1)0.5-b-p(OEGMEM)251H NMR (400 MHz, D2O): δ 8.00–7.60 (Aromatic H), 4.20 (COOCH2 side chain), 3.80–3.65 (CH2OCH2 side chain), 3.41 (–OCH3 side chain), 1.96–0.96 (CH2, CH3 backbone). The 1H NMR spectrum was almost identical to that of p(OEGMEM), but with slightly broader signals due to the presence of Gd·L1.
Author contributions
CJM: Method optimisation, synthesis of homopolymers and block copolymer particles, SEC analysis of homopolymers, DLS analysis, writing, data processing. CB: Method optimisation, synthesis of homopolymers. JT: AFM analysis. TRB: Synthesis of the Gd·L1 complex. JM: NMRD analysis of the Gd-loaded particles. LT: Writing, analysis of Gd-loaded particles, design of Gd-loaded particles. DWL: Universal SEC analysis of block copolymers. SJB: Project design, writing, funding acquisition, supervision. HW: Project design, writing, funding acquisition, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Loughborough University PhD Studentship (T. R. B.) and the Engineering and Physical Sciences Research Council (EP/R513088/1) (C. J. M.). Authors are grateful for the Strategic Equipment Grant (EP/T006412/1) provided by the EPSRC, which funded AFM work. We are also grateful for the universally calibrated SEC analysis from the Polymer Characterisation Research Technology Platform (EP/V007688/1), at the University of Warwick.Notes and references
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- L. Dai and C. L. Si, Mater. Lett., 2017, 207, 213–216 CrossRef CAS.
- Q. Chen, S. Li, Z. Feng, M. Wang, C. Cai, J. Wang and L. Zhang, Int. J. Nanomed., 2017, 12, 6857–6870 CrossRef CAS PubMed.
- P. Gurnani, A. M. Lunn and S. Perrier, Polymer, 2016, 106, 229–237 CrossRef CAS.
- A. K. Pearce and R. K. O'Reilly, Bioconjugate Chem., 2019, 30, 2300–2311 CrossRef CAS PubMed.
- S. Indoria, V. Singh and M. F. Hsieh, Int. J. Pharm., 2020, 582, 119314 CrossRef CAS PubMed.
- M. Elsabahy, G. S. Heo, S. M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- I. Ali, M. Alsehli, L. Scotti, M. T. Scotti, S. T. Tsai, R. S. Yu, M. F. Hsieh and J. C. Chen, Polymers, 2020, 12, 598–630 CrossRef CAS PubMed.
- T. Smart, H. Lomas, M. Massignani, M. V. Flores-merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38–46 CrossRef CAS.
- G. R. Rudramurthy, M. K. Swamy, U. R. Sinniah and A. Ghasemzadeh, Molecules, 2016, 21, 1–30 CrossRef PubMed.
- P. A. Lovell and F. J. Schork, Biomacromolecules, 2020, 21, 4396–4441 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- F. A. Khan, S. Akhtar, D. Almohazey, M. Alomari, S. A. Almofty, I. Badr and A. Elaissari, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 1533–1542 CrossRef CAS PubMed.
- A. Bonfá, R. S. N. Saito, R. F. O. França, B. A. L. Fonseca and D. F. S. Petri, Mater. Sci. Eng., C, 2011, 31, 562–566 CrossRef.
- M. C. Hacker, J. Krieghoff and A. G. Mikos, in Principles of Regenerative Medicine, Elsevier, 3rd edn, 2019, p. 563 Search PubMed.
- W. Zhang, F. D'Agosto, P. Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS.
- M. Ramezanpour and A. R. Shirin-Abadi, J. Polym. Res., 2021, 28, 1189–1198 Search PubMed.
- D. H. H. Chan, A. A. Cockram, R. R. Gibson, E. L. Kynaston, C. Lindsay, P. Taylor and S. P. Armes, Polym. Chem., 2021, 12, 5760–5769 RSC.
- J. Tan, G. Zhao, Y. Lu, Z. Zeng and M. A. Winnik, Macromolecules, 2014, 47, 6856–6866 CrossRef CAS.
- L. Griveau, J. Delorme, J. Engström, P. Y. Dugas, A. Carlmark, E. Malmström, F. D'Agosto and M. Lansalot, Biomacromolecules, 2020, 21, 4479–4491 CrossRef CAS PubMed.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- J. Trmcic-Cvitas, E. Hasan, M. Ramstedt, X. Li, M. A. Cooper, C. Abell, W. T. S. Huck and J. E. Gautrot, Biomacromolecules, 2009, 10, 2885–2894 CrossRef CAS PubMed.
- Y. Zhang, Q. Yu, H. Huang, F. Zhou, Z. Wu, L. Yuan, D. Li and H. Chen, Soft Matter, 2010, 2616–2618 RSC.
- C. Boyer, M. R. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917–6926 CrossRef CAS.
- P. Alexandridis and B. Lindman, Amphiphilic Block Copolymers: Self Assembly and Applications, Elsevier, 1st edn, 2000 Search PubMed.
- D. Roy, G. Y. Berguig, B. Ghosn, D. Lane, S. Braswell, P. S. Stayton and A. J. Convertine, J. Invest. Dermatol., 2014, 5, 1791–1799 CAS.
- Agilent Technologies Inc., Primer, 2015, 1–32 Search PubMed.
- M. N. Antonopoulou, R. Whitfield, N. P. Truong, D. Wyers, S. Harrisson, T. Junkers and A. Anastasaki, Nat. Chem., 2021, 14, 304–312 CrossRef PubMed.
- J. Jennings, M. Beija, J. T. Kennon, H. Willcock, R. K. O'Reilly, S. Rimmer and S. M. Howdle, Macromolecules, 2013, 46, 6843–6851 CrossRef CAS.
- L. P. D. Ratcliffe, A. Blanazs, C. N. Williams, S. L. Brown and S. P. Armes, Polym. Chem., 2014, 5, 3643–3655 RSC.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- J. N. Israelachvili, Thermodynamic and geometric aspects of amphiphile aggregation into micelles, vesicles and bilayers, and the interactions between them, 1985 Search PubMed.
- J. N. Israelachvili, Intermolecular and surface forces, Elsevier Science Imprint, 9th edn, 2002 Search PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, Biochim. Biophys. Acta, 1977, 470, 185–201 CrossRef CAS.
- L. R. Pratt, Annu. Rev. Phys. Chem., 1985, 36, 433–449 CrossRef CAS.
- W. D. Harkins, J. Am. Chem. Soc., 1947, 69, 1428–1444 CrossRef CAS PubMed.
- W. D. Harkins, J. Chem. Phys., 1945, 13, 381–382 CrossRef CAS.
- M. Rolland, N. P. Truong, K. Parkatzidis, E. H. Pilkington, A. L. Torzynski, R. W. Style, E. R. Dufresne and A. Anastasaki, JACS Au, 2021, 1, 1975–1986 CrossRef CAS PubMed.
- K. Parkatzidis, N. P. Truong, M. Rolland, V. Lutz-Bueno, E. H. Pilkington, R. Mezzenga and A. Anastasaki, Angew. Chem., 2022, 61, e202113424 CAS.
- R. N. Carmean, C. A. Figg, G. M. Scheutz, T. Kubo and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 185–189 CrossRef CAS PubMed.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2015, 54, 1279–1283 CrossRef CAS PubMed.
- C. M. Hoo, N. Starostin, P. West and M. L. Mecartney, J. Nanopart. Res., 2008, 10, 89–96 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- D. Lombardo, M. A. Kiselev, S. Magazù and P. Calandra, Adv. Condens. Matter Phys., 2015, 11, 1–22 Search PubMed.
- O. Terreau, L. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601–5607 CrossRef CAS.
- S. George, R. Champagne-Hartley, G. Deeter, D. Campbell, B. Reck, D. Urban and M. Cunningham, Macromolecules, 2015, 48, 8913–8920 CrossRef CAS.
- R. Whitfield, N. P. Truong, D. Messmer, K. Parkatzidis, M. Rolland and A. Anastasaki, Chem. Sci., 2019, 10, 8724–8734 RSC.
- S. Li, G. Han and W. Zhang, Polym. Chem., 2020, 11, 4681–4692 RSC.
- J. R. Lovett, L. P. D. Ratcliffe, N. J. Warren, S. P. Armes, M. J. Smallridge, R. B. Cracknell and B. R. Saunders, Macromolecules, 2016, 49, 2928–2941 CrossRef CAS PubMed.
- S. W. Kuo, H. C. Kao and F. C. Chang, Polymer, 2003, 44, 6873–6882 CrossRef CAS.
- G. K. K. Clothier, T. R. Guimarães, G. Moad and P. B. Zetterlund, Macromolecules, 2022, 55(6), 1981–1991 CrossRef CAS.
- G. K. K. Clothier, T. R. Guimarães, G. Moad and P. B. Zetterlund, Macromolecules, 2021, 54, 3647–3658 CrossRef CAS.
- Malvern and M. Instruments, Tech. Note MRK656-01, 2011, 1–8 Search PubMed.
- T. R. Berki, J. Martinelli, L. Tei, H. Willcock and S. J. Butler, Chem. Sci., 2021, 12, 3999–4013 RSC.
- M. Botta and L. Tei, Eur. J. Inorg. Chem., 2012, 1945–1960 CrossRef CAS.
- M. A. Bruckman, X. Yu and N. F. Steinmetz, Nanotechnology, 2013, 24, 462001 CrossRef PubMed.
- F. Carniato, L. Tei and M. Botta, Eur. J. Inorg. Chem., 2018, 2018, 4936–4954 CrossRef CAS.
- F. Carniato, L. Tei, W. Dastrù, L. Marchese and M. Botta, Chem. Commun., 2009, 15, 1246–1248 RSC.
- A. E. Merbach, L. Helm and E. Toth, The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, John Wiley & Sons, New York, 2nd edn, 2013 Search PubMed.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4546–4559 CrossRef CAS.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4559–4570 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00337f |
| ‡ The Lipari–Szabo approach describes the rotational dynamics in terms of local (τRL) and global (τRG) rotational correlation times related by an order parameter, S2, that reflects the degree of correlation between the two types of motions and assumes the value of zero (S2 = 0) when these are completely independent, or the value of one (S2 = 1) when the complex is immobilized on the particle. |
| This journal is © The Royal Society of Chemistry 2022 |