
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revisiting the origin of green emission in Cs4PbBr6
Koushik
Biswas

Department of Chemistry and Physics, Arkansas State University, State University, Arkansas 72467, USA. E-mail: kbiswas@astate.edu
First published on 20th July 2022
Abstract
It is probably acceptable to state that the recent debate over the origin of green emission in Cs4PbBr6 and/or CsPbBr3 has been narrowed down to two major possibilities: (i) a defect-mediated event in Cs4PbBr6, or (ii) a radiative recombination of charge carriers at CsPbBr3 inclusions embedded within Cs4PbBr6. The first scenario is relatively broad in terms of identifying specific defects that could act as recombination centers, although Br-vacancy has been proposed as a potential candidate. In the case of the second possibility, the omnipresence of CsPbBr3 nanoinclusions even in pure phase Cs4PbBr6 is also a matter of contention. Resolving this “green emission question” is consequential to the further development of the Cs–Pb–X (X = halogen) system of perovskite-inspired optoelectronic materials. Here, we will briefly survey the current status of the literature and offer some perspectives with respect to the two possibilities noted above.
 Koushik Biswas | Koushik received his PhD in Physics from Texas Tech University (Lubbock, TX). After working as a postdoctoral associate at the National Renewable Energy Lab and Oak Ridge National Lab, he joined as a member of the faculty at Arkansas State University, Jonesboro. Here, he continues to work in the field of computational materials. His primary areas of interest include semiconductors and insulators in the field of optoelectronics. |
Perovskite halides, both hybrids (organic-inorganic) and all-inorganic, have been at the center of attention for over a decade due to their excellent optoelectronic properties. The surge in research activity among these ionic compounds has had at least two positive impacts. Continued progress in characterization and optimization has brought the leading perovskite candidates closer to commercialization, as is evident in the case of lead-halide perovskites reaching a power conversion efficiency of well over 20% for photovoltaic (PV) applications.1 The other benefit is the proliferation of studies around halide materials that are promising in a number of applications including solar cells, light emitting diodes (LEDs) and scintillators.2,3 In addition to ease of synthesis and comprising of abundant constituents, some of these perovskites seem to defy the common understanding regarding the propensity of deep defects that would be detrimental to device applications. In contrast, they often exhibit excellent charge transport and light absorbing properties. These remarkable developments are opening new possibilities in a broad class of perovskite-inspired halides.
Low-dimensional perovskites
Effective design principles are being implemented in the search for new perovskite-type compounds.4,5Fig. 1 shows such a concept where the 1-1-3 composition of the AIBIIX3 halide (AI = monovalent, BII = divalent, X = halogen) transmutes to 0-dimensional (0D) K2PtCl6-type (2-1-6), 2-dimensional (2D) layered Cs3Fe2Cl9-type or a 0D face sharing dimer phase (3-2-9), and 3-dimensional (3D) double perovskite structure (2-1-1-6) – all of which retain the [BX6] octahedra in the crystal framework. Here, the dimensionality (i.e., 0D, 2D, and 3D) refers to connectivity between the [BX6] octahedra. For example, the 2-1-6 compounds feature isolated octahedra with A-ions filling the intervening voids. The 3-2-9 perovskite derivatives may appear in the 0D dimer phase or the layered 2D phase, as shown in Fig. 1. In general, the 2-1-6 and 3-2-9 compounds owing to their lower structural dimensionality lead to larger gap and poorer charge transport as compared to the state-of-the-art 3D perovskite CH3NH3PbI3. This may be unfavorable for PV but does not preclude them as efficient light emitters, although multivalency of some constituent elements can induce harmful defect properties which may need to be addressed (e.g., Sn changing its oxidation state from 4+ to 2+ and producing deep centers).6 These examples demonstrate the versatility of crystal structures and chemical compositions available in the broad family of perovskites and perovskite derivatives, with tunable optoelectronic properties.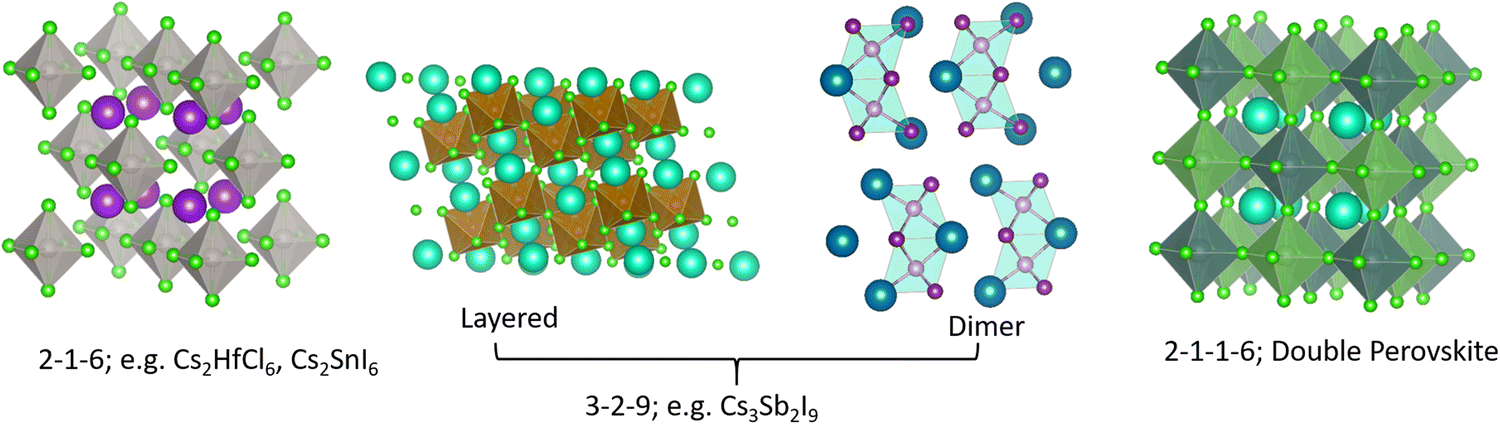 | ||
| Fig. 1 Different structure types having chemical compositions 2-1-6, 3-2-9, 2-1-1-6; they may be termed as descendants from the AIBIIX3 (1-1-3) perovskite family. | ||
The Cs–Pb–Br halides
The above discussion reminds us about the vast materials space offered by the perovskite-inspired halides that are waiting to be explored. Among these possibilities is the Cs–Pb–Br family comprising of CsPbBr3, CsPb2Br5 and Cs4PbBr6.7–11 Single crystals of CsPb2Br5 have been recently reported.12 It possesses a tetragonal lattice where Cs ions are sandwiched between Pb2Br5 polyhedral layers. There are earlier reports of similar compounds with formula-type AB2X5, where the A and B sites are occupied by alkali, alkaline-earth or ns2 ions, and X = Cl, Br, or I. Based on relative ionic size ratio, it has been found that the tetragonal structure is usually favored when an ns2 ion (e.g., Pb2+) occupies the ‘B’ site, as in the case of CsPb2Br5.13,14 Several of these AB2X5 compounds may be important optoelectronic materials, especially as scintillators.15 In the following text, however, we focus on the ternary CsPbBr3 and Cs4PbBr6 who are at the center of much debate on the subject of green emission.CsPbBr3 is already promising as a semiconductor radiation detector material.16 Its iodide counterpart, i.e., CsPbI3 has been reported as an all-inorganic candidate for efficient solar cells.17,18 Both, CsPbBr3 and Cs4PbBr6 feature the quintessential [PbBr6] octahedra, with Cs ions occupying interstitial positions or voids created between those octahedra. But there is an essential difference between the two with respect to octahedral connectivity. At room temperature, CsPbBr3 crystals adopt an orthorhombic structure displaying a continuous network of corner-sharing octahedra, and are popularly described as a 3D halide perovskite.19,20 On the other hand, the structure of Cs4PbBr6 comprises of isolated octahedra with little wavefunction overlap and therefore referred as a 0D halide.21,22 This structural difference has important consequences on their respective band structure and associated optoelectronic properties. Electronic structure calculations have shown the dispersive band edges of CsPbBr3 with a band gap ∼2.3 eV, whereas Cs4PbBr6 expectedly has flat bands and a larger fundamental gap of ∼3.9 eV.23,24 Experimental band gaps roughly agree with these values.25–27 Luminescence from CsPbBr3 single crystals is weak due to shallow excitons that are easily ionized to free carriers and energy transport is expected to be limited by shallow or deep traps. In Cs4PbBr6, however, the flat conduction and valence bands raise the opportunity for carriers to self-trap, and if they survive at room temperature, then charge transport and recombination may be dominated by polarons or excitons. Within this context we arrive at the dispute surrounding fast, green emission observed in Cs4PbBr6 and/or CsPbBr3 samples approximately in the range 520–540 nm. Hereinafter, we refer to it as ∼520 nm emission. Notwithstanding the apparent agreement between emission wavelength and band gap of CsPbBr3, the origin of luminescence could be a complex affair. Although it is a point of disagreement, recent reports seem to have narrowed it down to two main possibilities. Acknowledging the significant volume of the recent literature on this specific issue, we briefly explore a few around these two possibilities.
Embedded CsPbBr3 inclusions
Over the past several years, evidence has been gathering in favor of CsPbBr3 inclusions embedded in large-gap Cs4PbBr6 as the source of green emission. Cs4PbBr6 has a tendency to have impurity-phase CsPbBr3 due to incongruent melting, as noted initially by Nikl et al.25 After presenting evidence of phase coexistence, they attributed the observed ∼545 nm emission to traces of CsPbBr3 in melt-grown Cs4PbBr6 bulk crystals and thin films. Several recent reports have discussed the solution synthesis of emissive and non-emissive Cs4PbBr6 bulk crystals, powders, films and nanocrystals (NCs).26,28–34 Here, ‘emissive’ represents green luminescence. If we consider that luminescence is weak from CsPbBr3 single crystals at room temperature, but CsPbBr3 NCs have strong green photoluminescence (PL) quantum yield, then one could infer that emissive Cs4PbBr6 possibly contains CsPbBr3 impurities.25,35 Indeed, Akkermann et al. found that Cs4PbBr6 films doped with 2% CsPbBr3 exhibited green PL similar to CsPbBr3 NCs, while pure Cs4PbBr6 NCs and films did not.28 It is notable that the 2% ‘doped’ sample produced similar X-ray diffraction (XRD) to the pure Cs4PbBr6. There are several reports on successful growth of non-emissive and green-emitting Cs4PbBr6.26,36–40Fig. 2 shows an example of Cs4PbBr6 emissive and non-emissive nanocrystal suspension, while the larger crystals appear yellow under white light and bright green upon ultraviolet (UV) excitation.36Fig. 3a, as reported by Zhang et al.,26 shows solution processed, millimeter-size transparent Cs4PbBr6 crystals having no visible PL at room temperature and a broad UV peak around 370 nm. After vacuum annealing at 150 °C the crystals turned pale yellow and a bright green luminescence peak appeared around 525 nm as shown in Fig. 3b, similar to those shown in Fig. 2c and d. According to Zhang et al.,26 a small absorption peak (not shown here) appearing at ∼2.4 eV in the yellow samples points towards the presence of CsPbBr3.26 Chen et al. mentioned that the initial nonstoichiometry induces the formation of CsPbBr3 nanocrystals in centimeter-sized crystalline Cs4PbBr6 matrices.41 There are many more experimental reports that indicate or support CsPbBr3 inclusions in Cs4PbBr6.42–50 | ||
| Fig. 2 Nanocrystal suspensions of (a) emissive and (b) non-emissive Cs4PbBr6 under day light and UV exposure, respectively. Larger crystals of Cs4PbBr6 under (c) white and (d) UV light. Reprinted (adapted) with permission from ref. 36. Copyright 2019 American Chemical Society. | ||
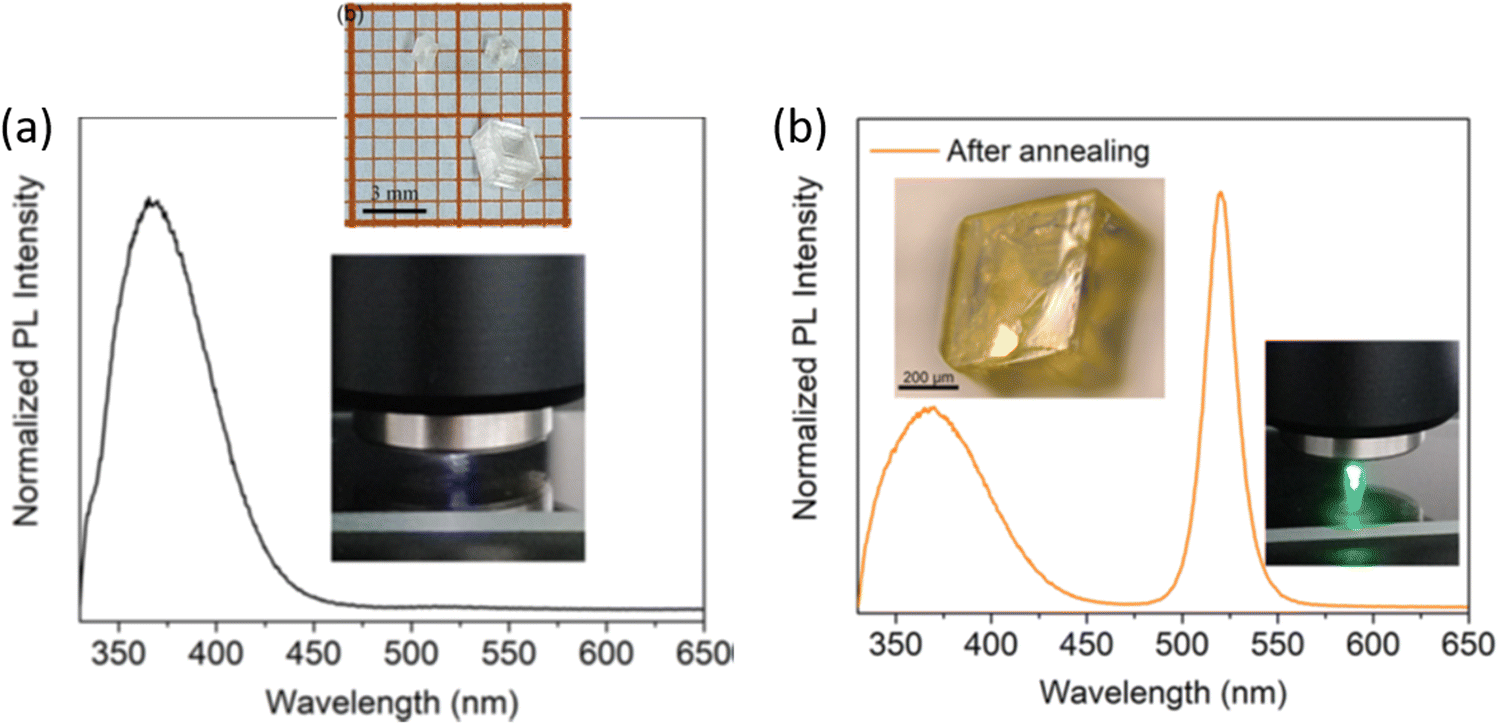 | ||
| Fig. 3 (a) PL spectrum of as-grown Cs4PbBr6 crystals showing no green luminescence (inset: photo of the transparent crystals and during PL using 325 nm excitation). (b) PL of the pale yellow samples obtained after annealing the crystals, with green luminescence around ∼520 nm. Reprinted (adapted) with permission from ref. 26. Copyright 2018 American Chemical Society. | ||
Although these reports suggest the CsPbBr3 phase within Cs4PbBr6, complicating factors, namely, XRD and transmission electron microscopy (TEM) do not find evidence of CsPbBr3 and results are nearly identical in emissive vs. non-emissive samples.28,36,51,52 This observation is not entirely unexpected due to the limitations of these techniques, failing to detect low concentrations of nanometer sized CsPbBr3 islands.36,50 While XRD and TEM do not reveal any signature of CsPbBr3, Raman spectroscopy data suggested otherwise as discussed by Qin et al.36 A Raman active mode at ∼29 cm−1 in emissive Cs4PbBr6 coincided with a doublet feature at 28–30 cm−1 observed in CsPbBr3. This study also reported on the pressure dependent evolution of Raman peaks and green PL intensity in emissive Cs4PbBr6. First, they established a 310 cm−1 Raman line as a spectral signature to identify CsPbBr3 in Cs4PbBr6. Under hydrostatic pressure, the green PL and the 310 cm−1 line follow a similar trend – both disappear at ∼2 GPa; thus offering some convincing data in favor of CsPbBr3 NCs as the source of ∼520 nm emission. Further evidence is provided by Riesen et al. who reported cathodoluminescence (CL) imaging with better spatial resolution of highly bright spots with wavelengths ranging between 500 and 540 nm that appeared in the Cs4PbBr6 aggregate.53 The similarity of the CL spectrum of the bright spots with the CsPbBr3 PL spectra corroborates their presence in the form of inclusions within Cs4PbBr6.
The work by Shin et al. also concluded that the origin of sub-bandgap luminescence in Cs4PbBr6 films was due to residual CsPbBr3.54 The 3.9 eV absorption edge in these films are attributed to excitons. Thereafter, the excitons are confined in the lower gap CsPbBr3, resulting in PL at 2.4 eV. The “emitter-in-matrix” design principle by Cao et al. provides additional impetus to the CsPbBr3 inclusion hypothesis.55 They adopted this concept to describe the scintillation properties of CsPbBr3 NCs embedded inside the Cs4PbBr6 host, demonstrating the superior performance of CsPbBr3@Cs4PbBr6 thin films for X-ray imaging applications.
Petralanda et al. implemented ab initio molecular dynamics, offering additional arguments in favor of the CsPbBr3 inclusion hypothesis.56 By monitoring the time evolution of the ground (singlet) and excited (triplet) states as a function of temperature they found clear signatures of exciton–phonon coupling with increasing temperature. Hence, according to them, a mechanism for thermal quenching of excitonic emission exists in 0D Cs4PbBr6. Incidentally, thermal quenching of Cs4PbBr6 emission has been reported in the experimental literature.25 Computational work by Kang et al. revealed possible excitonic structures in Cs4PbBr6 with emission in the UV range (∼387 nm), whereas similar triplet structures, meaning strongly bound excitons, could not be stabilized in the 3D CsPbBr3.23 Those calculations did not reveal any Cs4PbBr6 excitons that emit in the green. We should also note that transitions from purely triplet to singlet states are forbidden and therefore expected to be slow which goes against the observed fast green PL. According to calculations, possible type-I band alignment in concert with radiative or nonradiative energy transfer would favor the collection and concentration of carriers into the CsPbBr3 recombination islands, thus favoring the concept of luminescence at CsPbBr3 inclusions in the Cs4PbBr6 host.23,57
Aside from CsPbBr3 inclusions, other compositions have been proposed as a potential source of ∼520 nm emission. For instance, Ray et al. discussed the presence of Cs2PbBr4 inclusions alongside CsPbBr3.37 Interestingly, Sun et al. also proposed the emergence of a 2D phase, Csn+1PbnBr3n+1, as emitting centers during the crystallization stage of Cs4PbBr6.58 At n = 1, the 2D phase should be Cs2PbBr4, which is predicted to be direct gap, ∼2.29 eV, and therefore a credible source of green emission.37,59
Br-Vacancy or similar defects
Another strong hypothesis regarding green emission in pure phase Cs4PbBr6 suggests a defect-mediated mechanism.38,51,60–68 Bastiani et al. proffered the existence of intrinsic green luminescence centers, likely Br-vacancy (VBr).69 Their Cs4PbBr6 crystals had a pale green color under ambient light and appeared bright green when exposed to 365 nm UV radiation. Utilizing temperature dependent PL, they showed rapid quenching of PL intensity that was reversible up to about 180 °C and became irreversible above that temperature. This observation coupled with their temperature dependent XRD, which showed the appearance of CsPbBr3 peaks at elevated temperatures led them to conclude that formation of CsPbBr3 phase may actually oppose green emission instead of enabling it. The authors of ref. 69 also reported similar electron diffraction patterns of emissive and non-emissive Cs4PbBr6 with no reflection that could be ascribed to CsPbBr3 in either sample. We remind our readers that emissive in this case refers to green luminescence. Altogether, they found that green emission in Cs4PbBr6 is independent of CsPbBr3 which points to the possibility of an intrinsic defect center that produces gap states where radiative recombination can occur. Since halogen vacancies are expected to be abundant and often create sub-gap states in insulating halides, VBr in Cs4PbBr6 was proposed as a possible candidate.29,69 These reports were followed by those of Yin et al. who performed a comprehensive study of native defects in CsPbBr3, Cs2PbBr5, and Cs4PbBr6 using density functional theory (DFT).24 According to their calculations, VBr is shallow in the former two compounds, but it is a deep defect in Cs4PbBr6 with a +/0 transition level ∼2.3 eV above the valence band maximum (VBM). Several more native defects produced deep midgap states in Cs4PbBr6. Nevertheless, VBr appeared to be the most likely candidate due to its low formation energy under Br-poor conditions. It is important to note that this study,24 and previous works by the same group have reported on the phase purity of experimentally grown emissive samples of Cs4PbBr6 with no traces of CsPbBr3. It conflicts with other groups’ claim of phase coexistence in Cs4PbBr6.36,37,50,53,58Based on a detailed analysis of femtosecond transient absorption spectroscopy Liu et al. attributed green luminescence in Cs4PbBr6 single crystals to exciton migration to a defect level occurring within several hundred femtoseconds, followed by exciton relaxation and recombination in the picosecond and the subnanosecond or nanosecond time scale, respectively.70 Chen et al. inferred a similar mechanism where excitons are trapped potentially at halogen related defect states in Cs4PbBr6 which then recombine to generate the observed luminescence.64
Recently, Cha et al. investigated the magnetic properties of Cs4PbBr6 that provide new insights into this issue.40 Single crystals were found to have paramagnetic components in emissive Cs4PbBr6, while the non-emissive crystals exhibited exclusively diamagnetic behavior. This notable difference in magnetic response of the emissive crystals is due to VBr which serve as paramagnetic domains and green emission centers.
Studies reported by Yang et al. also support the defect mechanism, but offer yet another reason for green emission.71 In this case, Cs4PbBr6 micro-particles were synthesized involving ethanol as a polar solvent. Assisted by DFT calculations, they infer that the impact of the OH- group (presumably from ethanol) was to reduce the band gap of the Cs4PbBr6 host from 3.75 eV to 3.5 eV, also downshifting the Br-vacancy level from 2.75 eV to 2.44 eV, which approximately matches with the ∼520 nm green emission. In another report, Hu et al. found that interstitial hydroxyl induces a calculated sub-gap level at 2.6 eV and is therefore a possible reason for green PL.72 Similar effects of hydroxyl level inside Cs4PbBr6 have been reported.73
Using first-principles theory Jung et al. reported several uncharacteristic findings about the nature of point defects in Cs4PbBr6.74 The calculated equilibrium chemical potential diagram shown in Fig. 4 verifies the narrow (shaded) region of stability of Cs4PbBr6, bound by the competing binary and ternary phases. Three points marked A, B, and C in the stable region of Cs4PbBr6 perhaps correspond to Br-rich, stoichiometric (i.e., neither rich nor poor), and Pb-rich conditions, respectively. The defect formation energies of various native defects corresponding to points A, B and C are also shown in Fig. 4. According to this report,74 most native defects are deep with heavy compensation among the positive and negative charged defects and low equilibrium carrier concentrations of less than 109 cm−3. Their contention that the VBr concentration will remain low conflicts with the low formation energy (hence, high concentration) under Br-poor conditions reported by Yin et al.24 Based on an analysis of the configuration coordinate diagram of several prominent defects including VBr, Jung et al. ruled out the possibility of visible emission except for the curious case of the BrCs (Br at Cs site) defect. A closer inspection of this defect revealed that the antisite BrCs actually bonds with two neighboring Br ions forming a Br3 molecular species. Furthermore, they suggested a radiative mechanism involving BR−3 that could result in green emission.74
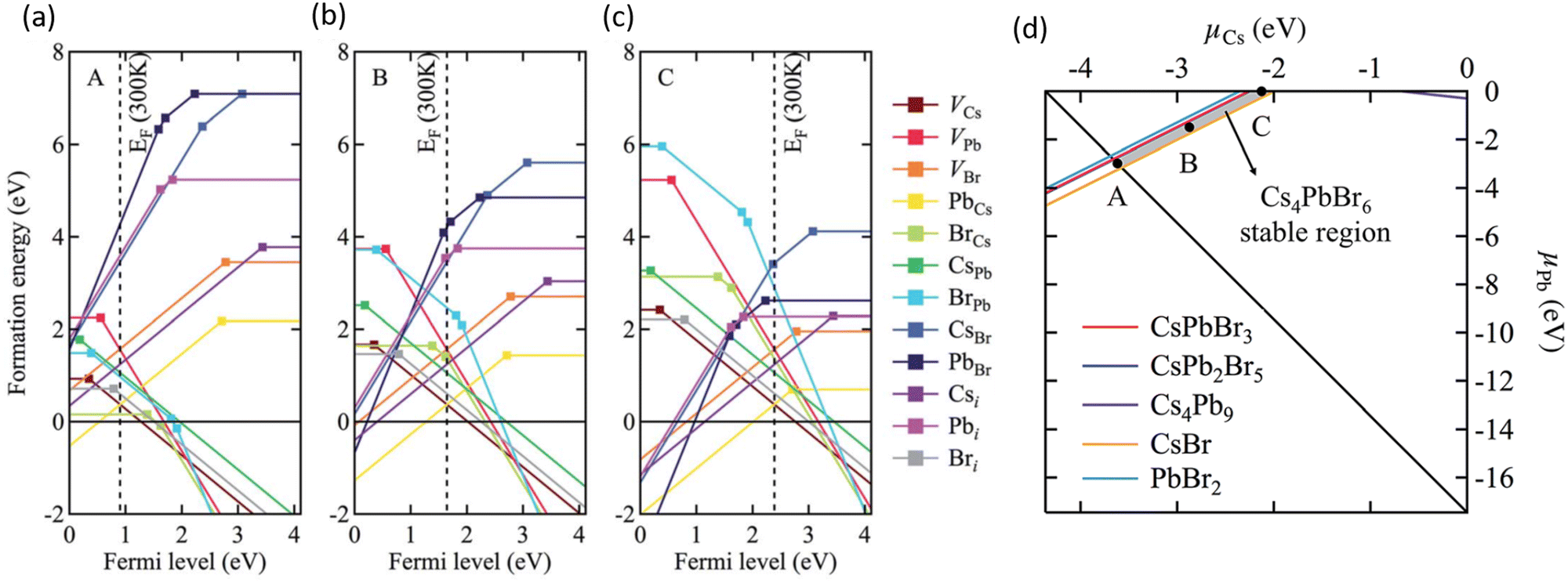 | ||
| Fig. 4 (a–c) Calculated formation energy of native defects as a function of Fermi energy inside the band gap of Cs4PbBr6. The zero of energy is set at the VBM. The three figures correspond to points A, B and C shown in (d) which depicts the calculated ranges of chemical potentials of stable Cs4PbBr6 under equilibrium. Reprinted (adapted) with permission from ref. 74. Copyright 2019 The Royal Society of Chemistry. | ||
Concluding remarks
Looking back at the volume of scientific research dedicated to green PL in the ternary Cs–Pb–Br system, it is clear that strong arguments are presented by both sides. The case of extrinsic causes such as an impurity phase (or inclusions) is shown in Fig. 5a. It presents a plausible scenario where CsPbBr3 inclusions appear within Cs4PbBr6, with type-I band edge alignment between the two phases. It facilitates the collection of excited charge carriers from Cs4PbBr6 to CsPbBr3 islands and subsequent recombination, producing an ∼520 nm emission. This scenario is muddled by the reports of green-emitting Cs4PbBr6 where XRD and high resolution TEM did not find any evidence of CsPbBr3. The PL properties of CsPbBr3 and Cs4PbBr6 are very similar, which only adds to the confusion. As already stated, there are also reports of emissive and non-emissive Cs4PbBr6 with identical XRD patterns. In fact, the successful growth of non-emissive Cs4PbBr6 may be strong evidence in favor of the inclusion hypothesis. Even if CsPbBr3 is present due to incongruent melting and the narrow region of stability of pure Cs4PbBr6 (Fig. 4d), uniform fabrication techniques (across research groups) that are able to reproduce both emissive and non-emissive samples will go a long way towards resolving the role of CsPbBr3@Cs4PbBr6 in the ∼520 nm emission. Similarly, on the computational side, reports of type-I or type-II band alignment between Cs4PbBr6 and CsPbBr3 need to be clarified too.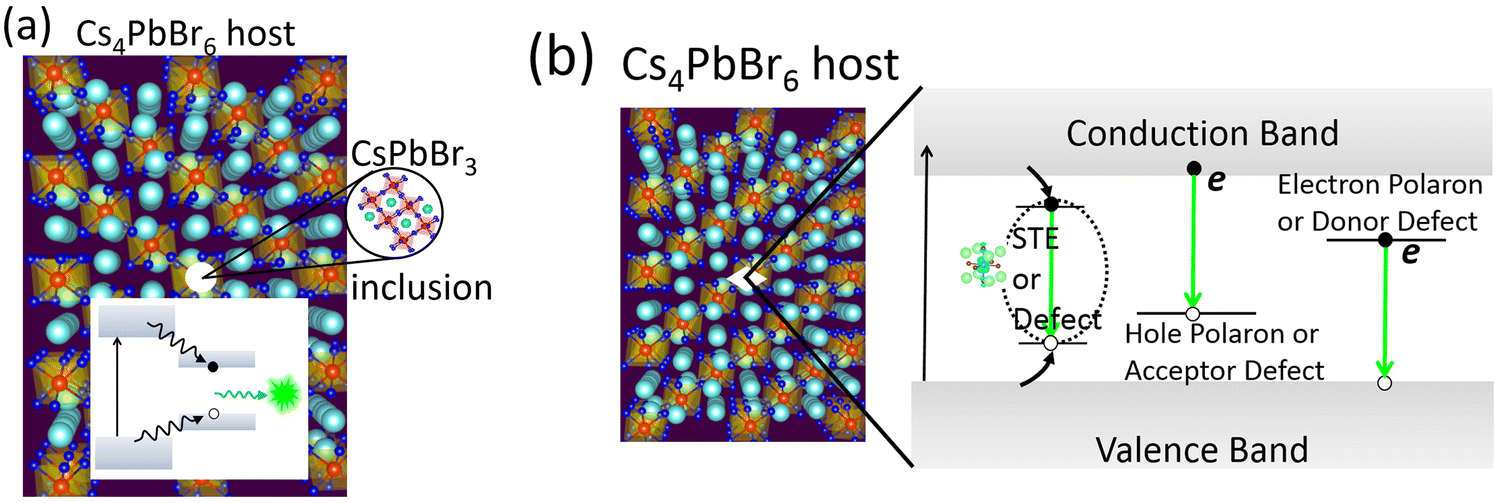 | ||
| Fig. 5 (a) CsPbBr3 emitters appearing as nano-inclusions within Cs4PbBr6. A type-I band alignment will favor the collection and subsequent recombination of charge carriers, producing green PL. (b) Possible optical transitions involving self-trapped excitons, (electron or hole) polarons, and defects inside the band gap of Cs4PbBr6 as the source of green PL. | ||
There are additional issues regarding the Cs4PbBr6/CsPbBr3 host–island interface that deserve careful consideration. For example, Ling et al. suggested shallow states created at the interface are responsible for the enhancement of green PL in the Cs4PbBr6/CsPbBr3 composite.48 Xu et al. proposed that the enhanced oscillator strength of imbedded CsPbBr3 nanocrystals (relative to its extended crystal form) may be due to the lower dielectric constant of the surrounding Cs4PbBr6 matrix, reminiscent of dielectric confinement.42 According to reports, the static dielectric constant of Cs4PbBr6 (∼6 to 8) is indeed lower than CsPbBr3 (∼20).23,74,75 It is important to determine whether the chemical correspondence between the host and island present a fortunate case where the aforementioned shallow states and dielectric confinement act in concert to aid stronger emission. Clarifying these aspects is essential to optimizing the PL properties by engineering the shape, size and distribution of emitting centers in a Cs4PbBr6 matrix.
As a final remark, a recent work on ‘dots-in-a-matrix’ by Ning et al. demonstrated the successful solution growth of epitaxially aligned PbS quantum dots passivated inside CH3NH3PbI3 films, which they termed poly-heterocrystalline solids.76 They found that the photogenerated carriers in the larger gap CH3NH3PbI3 matrix can be efficiently transferred to the smaller gap quantum dots for subsequent emission in the infrared region – thus leveraging the best features of both components. A similar situation is possible in the form of CsPbBr3 emitters in Cs4PbBr6, as schematically depicted in Fig. 5a. Indeed, the CsPbBr3@Cs4PbBr6 scintillators by Cao et al.55 is rooted in this principle. In the context of CsPbBr3@Cs4PbBr6 scintillators, Prof. Williams and colleagues cautioned about some pitfalls when it comes to applying them as energy resolving radiation detectors, where light yield from bulk crystals is important.77 Even if energy transfer is efficient from the Cs4PbBr6 matrix to CsPbBr3 nanoinclusions (as in quantum dots in a matrix), a small Stokes shift could lead to the problem of self-absorption of the emitted light by CsPbBr3. It may be severe when material thickness is required to be in the centimeter range to stop high energy ionizing radiation. This is why scintillators for spectroscopic γ-ray detection are either self-activated or doped with activator ions who provide the needed Stokes shift. The self-absorption problem is not a big limitation in the case of (nonspectroscopic) thin film applications for X-ray imaging or electroluminescent devices (LEDs, for example).
The defect-mediated green emission hypothesis in Cs4PbBr6 also has its own set of unanswered questions and accompanying opportunities. Fig. 5b shows a schematic representation of potential optical transitions that could lead to ∼520 nm emission. It includes the case a self-trapped exciton, although no such signature has been detected theoretically or experimentally, thus far. Among the potential defects, whether it is VBr, Br3 or some other point defect, fast emission dictates efficient energy transfer at room temperature to the defect site for radiative recombination. Upon excitation, self-trapped excitons (STEs) are expected to form via electron–phonon coupling. However, the 375 nm STE emission band of Cs4PbBr6 begins quenching at 100 K and becomes weak by 250 K.25,56 Du et al. linked the quenching at higher temperature to the significant electronic coupling between [PbBr6] octahedra and spectral overlap between STE absorption and emission (small Stokes shift). Accordingly, they suggest a mechanism of exciton migration via nonradiative resonant energy transfer between the [PbBr6] octahedra, and increased chance of energy loss at lattice defects.75,78
It has been suggested that the appearance of an Urbach tail in the absorption spectrum of Cs4PbBr6 is indicative of localized states in the gap. A rather large Urbach energy of about 1.7 eV has been estimated.51 For argument's sake, subtracting this value from the Cs4PbBr6 band gap of ∼3.9 eV results in an energy close to the green luminescence. On the contrary, Kajino et al. recently evaluated the Urbach energy of CsPbBr3/Cs4PbBr6 to be about 16.6 meV, dominated by phonon interactions.79 Irrespective of its value, the contribution to the Urbach energy may come from any physically valid mechanism including electron–phonon interactions and defect states. While defect-trapped excitons are a possible route for exciton relaxation (and subsequent emission), an in-depth study will be necessary to justify defects as the primary cause of broadened absorption edge of Cs4PbBr6.80
Besides the need for energy transfer to a radiative center, nonradiative processes may be in competition, whose rate depends on the carrier capture cross-section of a charged or neutral defect and its ionization energy with respect to the conduction or valence band edge. The mere abundance of a defect at a deep level is not sufficient for optical transition at a specific wavelength. For instance, let us pick VBr where there is agreement about its deep level inside the band gap of Cs4PbBr6. Its different charge states are positive where the defect level is empty (V+Br in semiconductor point defect nomenclature), neutral or singly occupied (V0Br), and a fully occupied negative charge state (V−Br). Note that the equilibrium defect levels corresponding to each of the charged states have different energetic positions because of differences in lattice relaxation and the energy cost of doing so. Therefore, defect induced single particle levels may or may not reside inside the band gap, although in this case it appears they are inside the gap. Regarding the question of photon emission involving these levels, it is possible either due to decay of an electron from the conduction band minimum (CBM) to an unoccupied state or due to recombination of an electron in the occupied defect state with a hole at the VBM (see Fig. 5b). The first transition communicates with electrons at the CBM while the latter with holes at the VBM. Theory and experimentation have not yet clarified which defect's initial and final states could result in such a transition producing ultrafast green PL. It is not to say that defects in Cs4PbBr6 is the unlikely cause, in fact, there are plenty of examples among scintillators where activators (dopants behaving like point defects) produce sub-band gap states for efficient and fast optical decay, as in LaBr3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ce.81 If true, defect-related emission from pure bulk crystals of Cs4PbBr6 will be beneficial in medical diagnostic applications like time-of-flight positron-emission tomography which rely more on time resolution along with light yield, and consequently, require fast scintillation decay, preferably in the sub-nanosecond timescale.
:Ce.81 If true, defect-related emission from pure bulk crystals of Cs4PbBr6 will be beneficial in medical diagnostic applications like time-of-flight positron-emission tomography which rely more on time resolution along with light yield, and consequently, require fast scintillation decay, preferably in the sub-nanosecond timescale.
In conclusion, there are two opposing sets of thought, where one believes CsPbBr3 nanocrystals and inclusions, and the other believes defects in Cs4PbBr6 to be the sole cause of green luminescence. Resolving this dichotomy is essential not just for scientific reasons, but is also necessary for successful practical applications which demand an answer to the question – are we growing CsPbBr3, Cs4PbBr6 or a composite like dots-in-a-matrix?
Epilogue
Koushik is grateful to have had many stimulating and enlightening conversations with Prof. Richard T. Williams on this topic, and generally in the field of scintillators and radiation detectors. In addition to his scientific contributions, his kind mentorship will be sorely missed.Conflicts of interest
The author declares no competing financial interests.Acknowledgements
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. We acknowledge Dr Mahua Choudhury and Anastasia Jerman for organizing references.References
- Best Research Cell-Efficiency Chart, (https://www.nrel.gov/pv/cell-efficiency.html).
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed.
- X. Wang, T. Li, B. Xing, M. Faizan, K. Biswas and L. Zhang, J. Phys. Chem. Lett., 2021, 12, 10532–10550 CrossRef CAS PubMed.
- J. R. Long, L. S. McCarty and R. H. Holm, J. Am. Chem. Soc., 1996, 118, 4603–4616 CrossRef CAS.
- E. G. Tulsky and J. R. Long, Chem. Mater., 2001, 13, 1149–1166 CrossRef CAS.
- K. Biswas, S. Lany and A. Zunger, Appl. Phys. Lett., 2010, 96, 201902 CrossRef.
- M. Cola, V. Massarotti, R. Riccardi and C. Sinistri, Z. Naturforsch., A: Phys. Sci., 1971, 26, 1328–1332 CAS.
- T. Udayabhaskararao, L. Houben, H. Cohen, M. Menahem, I. Pinkas, L. Avram, T. Wolf, A. Teitelboim, M. Leskes, O. Yaffe, D. Oron and M. Kazes, Chem. Mater., 2018, 30, 84–93 CrossRef CAS.
- M. Liu, J. Zhao, Z. Luo, Z. Sun, N. Pan, H. Ding and X. Wang, Chem. Mater., 2018, 30, 5846–5852 CrossRef CAS.
- I. Y. Kuznetsova, I. S. Kovaleva and V. A. Fedorov, Russ. J. Inorg. Chem., 2001, 46, 1730–1735 Search PubMed.
- M. Rodová, J. Brožek, K. Knížek and K. Nitsch, J. Therm. Anal. Calorim., 2003, 71, 667–673 CrossRef.
- I. Dursun, M. De Bastiani, B. Turedi, B. Alamer, A. Shkurenko, J. Yin, A. M. El-Zohry, I. Gereige, A. AlSaggaf, O. F. Mohammed, M. Eddaoudi and O. M. Bakr, ChemSusChem, 2017, 10, 3746–3749 CrossRef CAS PubMed.
- H. P. Beck, G. Clicqué and H. Nau, Z. Anorg. Allg. Chem., 1986, 536, 35–44 CrossRef CAS.
- D. Becker and H. P. Beck, Z. Anorg. Allg. Chem., 2004, 630, 1924–1932 CrossRef CAS.
- B. Kang, C. M. Fang and K. Biswas, J. Phys. D: Appl. Phys., 2016, 49, 395103 CrossRef.
- Y. He, M. Petryk, Z. Liu, D. G. Chica, I. Hadar, C. Leak, W. Ke, I. Spanopoulos, W. Lin, D. Y. Chung, B. W. Wessels, Z. He and M. G. Kanatzidis, Nat. Photonics, 2021, 15, 36–42 CrossRef CAS.
- G. Eperon, G. Paternò, R. Sutton, A. Zampetti, A. Haghighirad, F. Cacialli and H. Snaith, J. Mater. Chem. A, 2015, 3, 19688–19695 RSC.
- X. Zhang, M. E. Turiansky and C. G. Van de Walle, Cell Rep. Phys. Sci., 2021, 2, 100604 CrossRef CAS.
- M. Rodova, J. Brozek, K. Knizek and K. Nitsch, J. Therm. Anal. Calorim., 2003, 71, 667–673 CrossRef CAS.
- C. C. Stoumpos, C. D. Malliakas, J. A. Peters, Z. Liu, M. Sebastian, J. Im, T. C. Chasapis, A. C. Wibowo, D. Y. Chung, A. J. Freeman, B. W. Wessels and M. G. Kanatzidis, Cryst. Growth Des., 2013, 13, 2722–2727 CrossRef CAS.
- M. Velázquez, A. Ferrier, S. Péchev, P. Gravereau, J.-P. Chaminade, X. Portier and R. Moncorgé, J. Cryst. Growth, 2008, 310, 5458–5463 CrossRef.
- C. K. Moller, On the structure of caesium hexahalogeno-plumbates (ii), Mater.-Fys. Medd., Mater.-Fys. Medd. K. Dan. Vidensk. Selsk, Munksgaard, København, 1960, vol. 32, pp. 1–13 Search PubMed.
- B. Kang and K. Biswas, J. Phys. Chem. Lett., 2018, 9, 830–836 CrossRef CAS PubMed.
- J. Yin, H. Yang, K. Song, A. M. El-Zohry, Y. Han, O. M. Bakr, J.-L. Brédas and O. F. Mohammed, J. Phys. Chem. Lett., 2018, 9, 5490–5495 CrossRef CAS PubMed.
- M. Nikl, E. Mihokova, K. Nitsch, F. Somma, C. Giampaolo, G. P. Pazzi, P. Fabeni and S. Zazubovich, Chem. Phys. Lett., 1999, 306, 280–284 CrossRef CAS.
- Z. Zhang, Y. Zhu, W. Wang, W. Zheng, R. Lin, X. Li, H. Zhang, D. Zhong and F. Huang, Cryst. Growth Des., 2018, 18, 6393–6398 CrossRef CAS.
- M. Sebastian, J. A. Peters, C. C. Stoumpos, J. Im, S. S. Kostina, Z. Liu, M. G. Kanatzidis, A. J. Freeman and B. W. Wessels, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 235210 CrossRef.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- J. Yin, Y. Zhang, A. Bruno, C. Soci, O. M. Bakr, J.-L. Brédas and O. F. Mohammed, ACS Energy Lett., 2017, 2, 2805–2811 CrossRef CAS.
- G. Jin, D. Zhang, P. Pang, Z. Ye, T. Liu, G. Xing, J. Chen and D. Ma, J. Mater. Chem. C, 2021, 9, 916–924 RSC.
- H. Yang, Y. Zhang, J. Pan, J. Yin, O. M. Bakr and O. F. Mohammed, Chem. Mater., 2017, 29, 8978–8982 CrossRef CAS.
- X. Wei, J. Liu, H. Liu, X. Lei, H. Qian, H. Zeng, F. Meng and W. Deng, Inorg. Chem., 2019, 58, 10620–10624 CrossRef CAS PubMed.
- J. Jiang, D. Wang, M. Wu, P. Peng, F.-F. Li, F. Liu, R. Jing, X. Ma, Y. Chao, Z. Xiao and Q. Jiang, APL Mater., 2020, 8, 071115 CrossRef CAS.
- R. Bose, Y. Zheng, T. Guo, J. Yin, M. N. Hedhili, X. Zhou, J.-F. Veyan, I. Gereige, A. Al-Saggaf, Y. N. Gartstein, O. M. Bakr, O. F. Mohammed and A. V. Malko, ACS Appl. Mater. Interfaces, 2020, 12, 35598–35605 CrossRef CAS PubMed.
- L. N. Quan, R. Quintero-Bermudez, O. Voznyy, G. Walters, A. Jain, J. Z. Fan, X. Zheng, Z. Yang and E. H. Sargent, Adv. Mater., 2017, 29, 1605945 CrossRef PubMed.
- Z. Qin, S. Dai, V. G. Hadjiev, C. Wang, L. Xie, Y. Ni, C. Wu, G. Yang, S. Chen, L. Deng, Q. Yu, G. Feng, Z. M. Wang and J. Bao, Chem. Mater., 2019, 31, 9098–9104 CrossRef CAS.
- A. Ray, D. Maggioni, D. Baranov, Z. Dang, M. Prato, Q. A. Akkerman, L. Goldoni, E. Caneva, L. Manna and A. L. Abdelhady, Chem. Mater., 2019, 31, 7761–7769 CrossRef CAS PubMed.
- Y. Zhang, M. I. Saidaminov, I. Dursun, H. Yang, B. Murali, E. Alarousu, E. Yengel, B. A. Alshankiti, O. M. Bakr and O. F. Mohammed, J. Phys. Chem. Lett., 2017, 8, 961–965 CrossRef CAS PubMed.
- D. Chen, Z. Wan, X. Chen, Y. Yuan and J. Zhong, J. Mater. Chem. C, 2016, 4, 10646–10653 RSC.
- J.-H. Cha, H.-J. Lee, S. H. Kim, K. C. Ko, B. J. Suh, O. H. Han and D.-Y. Jung, ACS Energy Lett., 2020, 5, 2208–2216 CrossRef CAS.
- X. Chen, F. Zhang, Y. Ge, L. Shi, S. Huang, J. Tang, Z. Lv, L. Zhang, B. Zou and H. Zhong, Adv. Funct. Mater., 2018, 28, 1706567 CrossRef.
- J. Xu, W. Huang, P. Li, D. R. Onken, C. Dun, Y. Guo, K. B. Ucer, C. Lu, H. Wang, S. M. Geyer, R. T. Williams and D. L. Carroll, Adv. Mater., 2017, 29, 1703703 CrossRef PubMed.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS PubMed.
- F. Palazon, C. Urso, L. De Trizio, Q. Akkerman, S. Marras, F. Locardi, I. Nelli, M. Ferretti, M. Prato and L. Manna, ACS Energy Lett., 2017, 2, 2445–2448 CrossRef CAS PubMed.
- C. de Weerd, J. Lin, L. Gomez, Y. Fujiwara, K. Suenaga and T. Gregorkiewicz, J. Phys. Chem. C, 2017, 121, 19490–19496 CrossRef PubMed.
- L. Yang, D. Li, C. Wang, W. Yao, H. Wang and K. Huang, J. Nanopart. Res., 2017, 19, 258 CrossRef.
- F. Palazon, G. Almeida, Q. A. Akkerman, L. De Trizio, Z. Dang, M. Prato and L. Manna, Chem. Mater., 2017, 29, 4167–4171 CrossRef CAS PubMed.
- Y. Ling, L. Tan, X. Wang, Y. Zhou, Y. Xin, B. Ma, K. Hanson and H. Gao, J. Phys. Chem. Lett., 2017, 8, 3266–3271 CrossRef CAS PubMed.
- G. Hu, W. Qin, M. Liu, X. Ren, X. Wu, L. Yang and S. Yin, J. Mater. Chem. C, 2019, 7, 4733–4739 RSC.
- F. Cao, D. Yu, X. Xu, Z. Han and H. Zeng, J. Phys. Chem. C, 2021, 125, 3–19 CrossRef CAS.
- L. Wang, H. Liu, Y. Zhang and O. F. Mohammed, ACS Energy Lett., 2020, 5, 87–99 CrossRef CAS.
- Q. A. Akkerman, A. L. Abdelhady and L. Manna, J. Phys. Chem. Lett., 2018, 9, 2326–2337 CrossRef CAS PubMed.
- N. Riesen, M. Lockrey, K. Badek and H. Riesen, Nanoscale, 2019, 11, 3925–3932 RSC.
- M. Shin, S.-W. Nam, A. Sadhanala, R. Shivanna, M. Anaya, A. Jiménez-Solano, H. Yoon, S. Jeon, S. D. Stranks, R. L. Z. Hoye and B. Shin, ACS Appl. Energy Mater., 2020, 3, 192–199 CrossRef CAS.
- F. Cao, D. Yu, W. Ma, X. Xu, B. Cai, Y. M. Yang, S. Liu, L. He, Y. Ke, S. Lan, K.-L. Choy and H. Zeng, ACS Nano, 2020, 14, 5183–5193 CrossRef CAS PubMed.
- U. Petralanda, G. Biffi, S. C. Boehme, D. Baranov, R. Krahne, L. Manna and I. Infante, Nano Lett., 2021, 21, 8619–8626 CrossRef CAS PubMed.
- J. Yin, J.-L. Brédas, O. M. Bakr and O. F. Mohammed, Chem. Mater., 2020, 32, 5036–5043 CrossRef CAS.
- R. Sun, N. Liu, W. Zheng, J. Zhang, N. Li, H. Lian, H. Liu and Y. Zhang, Chem. Mater., 2021, 33, 3721–3728 CrossRef CAS.
- J.-H. Yang, Q. Yuan and B. I. Yakobson, J. Phys. Chem. C, 2016, 120, 24682–24687 CrossRef CAS.
- X. Liu, W. Xie, Y. Lu, X. Wang, S. Xu and J. Zhang, J. Mater. Chem. C, 2022, 10, 762–767 RSC.
- J. Zhang, A. Wang, L. Kong, L. Zhang and Z. Deng, J. Alloys Compd., 2019, 797, 1151–1156 CrossRef CAS.
- H. Zhang, Q. Liao, Y. Wu, J. Chen, Q. Gao and H. Fu, Phys. Chem. Chem. Phys., 2017, 19, 29092–29098 RSC.
- S. Zou, C. Liu, R. Li, F. Jiang, X. Chen, Y. Liu and M. Hong, Adv. Mater., 2019, 31, 1900606 CrossRef PubMed.
- X. Chen, D. Chen, J. Li, G. Fang, H. Sheng and J. Zhong, Dalton Trans., 2018, 47, 5670–5678 RSC.
- S. Seth and A. Samanta, J. Phys. Chem. Lett., 2017, 8, 4461–4467 CrossRef CAS PubMed.
- S. Seth and A. Samanta, J. Phys. Chem. Lett., 2018, 9, 176–183 CrossRef CAS PubMed.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- M. I. Saidaminov, J. Almutlaq, S. Sarmah, I. Dursun, A. A. Zhumekenov, R. Begum, J. Pan, N. Cho, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2016, 1, 840–845 CrossRef CAS.
- M. De Bastiani, I. Dursun, Y. Zhang, B. A. Alshankiti, X.-H. Miao, J. Yin, E. Yengel, E. Alarousu, B. Turedi, J. M. Almutlaq, M. I. Saidaminov, S. Mitra, I. Gereige, A. AlSaggaf, Y. Zhu, Y. Han, I. S. Roqan, J.-L. Bredas, O. F. Mohammed and O. M. Bakr, Chem. Mater., 2017, 29, 7108–7113 CrossRef CAS.
- R.-T. Liu, X.-P. Zhai, Z.-Y. Zhu, B. Sun, D.-W. Liu, B. Ma, Z.-Q. Zhang, C.-L. Sun, B.-L. Zhu, X.-D. Zhang, Q. Wang and H.-L. Zhang, J. Phys. Chem. Lett., 2019, 10, 6572–6577 CrossRef CAS PubMed.
- L. Yang, T. Wang, X. Yang, M. Zhang, C. Pi, J. Yu, D. Zhou, X. Yu, J. Qiu and X. Xu, Opt. Express, 2019, 27, 31207–31216 CrossRef CAS PubMed.
- M. Hu, C. Ge, J. Yu and J. Feng, J. Phys. Chem. C, 2017, 121, 27053–27058 CrossRef CAS.
- X. Wang, J. Yu, M. Hu, Y. Wu, L. Yang, W. Ye and X. Yu, J. Lumin., 2020, 221, 116986 CrossRef CAS.
- Y.-K. Jung, J. Calbo, J.-S. Park, L. D. Whalley, S. Kim and A. Walsh, J. Mater. Chem. A, 2019, 7, 20254–20261 RSC.
- D. Han, H. Shi, W. Ming, C. Zhou, B. Ma, B. Saparov, Y.-Z. Ma, S. Chen and M.-H. Du, J. Mater. Chem. C, 2018, 6, 6398–6405 RSC.
- Z. Ning, X. Gong, R. Comin, G. Walters, F. Fan, O. Voznyy, E. Yassitepe, A. Buin, S. Hoogland and E. H. Sargent, Nature, 2015, 523, 324–328 CrossRef CAS PubMed.
- R. T. Williams, W. W. Wolszczak, X. Yan and D. L. Carroll, ACS Nano, 2020, 14, 5161–5169 CrossRef CAS PubMed.
- M.-H. Du and F. A. Reboredo, Opt. Mater.: X, 2020, 8, 100066 CAS.
- Y. Kajino, S. Otake, T. Yamada, K. Kojima, T. Nakamura, A. Wakamiya, Y. Kanemitsu and Y. Yamada, Phys. Rev. Mater., 2022, 6, L043001 CrossRef CAS.
- K. S. Song and R. T. Williams, Self-trapped excitons, Springer, Berlin, New York, 1996 Search PubMed.
- E. V. D. van Loef, P. Dorenbos, C. W. E. van Eijk, K. Krämer and H. U. Güdel, Appl. Phys. Lett., 2001, 79, 1573–1575 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |