
Tissue-derived carbon microbelt paper: a high-initial-coulombic-efficiency and low-discharge-platform K+-storage anode for 4.5 V hybrid capacitors†
Taoqiu
Zhang
,
Zhiefei
Mao
,
Xiaojun
Shi
,
Jun
Jin
,
Beibei
He
 ,
Rui
Wang
,
Yansheng
Gong
and
Huanwen
Wang
*
,
Rui
Wang
,
Yansheng
Gong
and
Huanwen
Wang
*
Engineering Research Center of Nano-Geomaterials of Ministry of Education, Faculty of Material and Chemistry, China University of Geosciences, Wuhan, 430074, China. E-mail: wanghw@cug.edu.cn
First published on 25th November 2021
Abstract
Hard carbon (HC) is a promising anode material for K+-storage due to its randomly oriented turbostratic structure. However, most reported HC anodes exhibit low initial coulombic efficiency (ICE) and no obvious discharge platform during K+-intercalation/deintercalation, thus restricting their practical application. Herein, cheap and renewable sanitary tissue is utilized as the precursor to construct a flexible self-supporting hard carbon microbelt paper (HCMB). As a binder-free anode, the HCMB can achieve a high ICE value of 88% with a high charge capacity below 1 V (204 mA h g−1 at 100 mA g−1), excellent rate capability (151 mA h g−1 at 1000 mA g−1) and superior cycling stability in a conventional KPF6-based electrolyte. More importantly, the HCMB-based anodes exhibit a rather low discharge platform, which is close to a graphite anode (0.25 V vs. K/K+). To demonstrate its practical use, a novel 4.5 V potassium ion capacitor (PIC) device is successfully constructed based on the HCMB anode and an activated carbon cathode together with a gel polymer electrolyte. The energy density of this hybrid system is up to 152 W h kg−1, and is still maintained as high as 112 W h kg−1 at a high power density of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 W kg−1. In addition, the effect of the carbonization temperature on the K+-storage behavior of HCMB and its comparison with carbon counterparts (graphite and soft carbon) are systematically investigated.
500 W kg−1. In addition, the effect of the carbonization temperature on the K+-storage behavior of HCMB and its comparison with carbon counterparts (graphite and soft carbon) are systematically investigated.
Broader contextPotassium-based energy storage systems are more affordable than their lithium counterparts due to the abundant potassium resources in the earth's crust and the similar redox potential between K+/K (−2.93 V) and Li/Li+ (−3.04 V). Potassium-ion capacitors (PICs) have attracted much attention because they integrate both the advantages of potassium-ion batteries (PIBs) and supercapacitors. However, the development of high-performance PICs faces a huge challenge because the current research into PIC anodes is directed towards high K+-storage capacity and rate capability, whereas the initial coulombic efficiency (ICE) and the charge specific capacity below 1.00 V are rarely studied. In this study, we report a self-supporting hard carbon microbelt (HCMB) derived from tissue paper and investigate its K-storage performance in comparison with graphite and soft carbon. HCMB exhibits a high ICE (88%), good rate performance/cycle stability, and most prominently, the HCMB has a discharge platform similar to that of graphite. These unique advantages of the HCMB anode endow the assembled PIC full device with an operating voltage of up to 4.5 V and a high energy density of 112 W h kg−1 at a fast charge rate of 23 s. We believe that this study provides insight into the energy-storage mechanisms of other carbon material families. |
Introduction
In the 21st century, the rapid development of science and technology greatly changed people's lifestyles and increased their dependence on 3C products (3C: computer, communication, and consumer electronics).1,2 To the best of our knowledge, lithium-ion batteries (LIBs) as a mainstream product for energy storage possess advantages of high energy density and continuous and stable energy output, almost dominating the 3C field. Unfortunately, severe shortage and uneven distribution of lithium resources in the earth's crust will definitely become a key factor in curbing the large-scale development of LIBs.3–5 Potassium-based energy storage technology is attracting more and more attention due to its unique characteristics: (1) potassium resources are abundant (2.09 wt% of the earth's crust, ≈1400 times the abundance of lithium);6 (2) the redox potential of K+/K (−2.93 V vs. SHE) is very close to that of Li/Li+ (−3.04 V vs. SHE) but lower than that of Na/Na+ (−2.71 V vs. SHE);7–9 (3) cheap and lightweight aluminium foil can be used as a current collector instead of copper foil, which will reduce the manufacturing cost of full cells;10 and (4) K+ ions have a fast ion diffusion rate in electrolytes due to their lower Stokes radii than those of Li+ and Na+ and weaker coulombic interaction with solvent molecules. These advantages are beneficial to constructing high-voltage/high-power potassium-ion batteries (PIBs) and potassium-ion capacitors (PICs).11–14Unfortunately, potassium has a larger ionic radius (1.38 Å) than lithium (0.76 Å) and sodium (1.02 Å),15 which usually leads to a huge volume expansion in potassium graphite intercalation compounds, corresponding to a theoretical capacity of 279 mA h g−1 in the full-potassiation state (KC8).16–18 The volume expansion (61% for K+) of graphite leads to the degradation and destruction of the graphite interlayer structure in commercial KPF6-based electrolytes, thus making the cyclability of graphite anodes unsatisfactory.19,20 Although potassium bis(fluoro-sulfonyl)imide (KFSI)-based concentrated electrolytes were reported to stabilize graphite-potassium anodes, the significantly higher cost of KFSI than KPF6 as well as FSI−-induced corrosion on Al foils at high potentials,21–23 will create a huge challenge when going from laboratory research to practical applications. The development of alternative K+-storing materials should not only guarantee high performance in the low-cost KPF6-based electrolyte, but also be suitable for large-scale production to fulfill their potential for practical applications.24
Among various anode materials, hard carbon (HC) is considered to be one of the most promising anode candidates due to its good microstructural tunability, high electrical conductivity, stable physicochemical properties, abundant raw material sources, and low environmental pollution during preparation and recycling.25–28 Because of their highly disordered microcrystalline structure, HC materials are able to take up more K+ ions, and are also able to achieve excellent rate capability and cycling stability.29,30 For example, bacterial cellulose was converted into a compressible carbon nanofiber foam (CNFF),31 which could maintain a stable K+-storage capacity of 158 mA h g−1 after 2000 cycles at 1000 mA g−1 and 122 mA h g−1 at 5000 mA g−1 for an additional 1000 cycles. Guo et al.32 reported an N-doped necklace-like hollow carbon fiber (NHC), which could deliver a K+-storage capacity of 293.5 mA h g−1 at 100 mA g−1, excellent rate capability (204.8 mA h g−1 at 2000 mA g−1) and cycling stability (161.3 mA h g−1 at 1000 mA g−1 after 1600 cycles). Alshareef et al.33 synthesized an edge-nitrogen-doping carbon 3D framework (3D-NTC) and a high K+-storage capacity of 473 mA h g−1 can be achieved. In addition, several S-doped HC materials have been designed for high-capacity potassium ion storage, including a sulfur-grafted hollow carbon sphere (SHCS,34 581 mA h g−1 at 25 mA g−1), sulfur-rich graphene nanobox (SGN,35 581 mA h g−1 at 50 mA g−1), and S/N codoped carbon nanofiber aerogel (S/N-CNFAs,36 356 mA h g−1 at 100 mA g−1).
Despite their the extraordinary K+-storage capacity, the above HC materials have two fatal drawbacks. The first one is their low initial coulombic efficiency (ICE), such as CNFF (ICE = ∼18%),31 NHC (ICE = ∼18%),32 3D-NTC (ICE = 61%),33 SHCS (ICE = 51%),34 SGN (ICE = 15.5%),35 and S/N-CNFAs (ICE = 53%),36 which is mainly caused by the large surface area and structural defects in HC.37,38 Another drawback is that the charge–discharge behavior of previously reported HC usually exhibits a sloping curve with a rather low charge capacity at <1 V vs. K/K+.39–41 During practical application, only the charge capacity at <1 V can be utilized in the discharge process of a full cell. Taking the above SHCS34 as an example, its discharge specific capacity is as high as 572 mA h g−1 in the potential range of 0.01–3 V, but the corresponding charge capacity from 0.01 to 1 V is only around 75 mA h g−1. Therefore, it is a major challenge to develop a low-cost HC material that has comprehensive K-storage properties in terms of high ICE, excellent rate capability, long cycling stability, low discharge platform and high charge capacity at <1 V.42
Herein, a flexible self-supporting hard carbon microbelt (HCMB) film has been successfully designed through simple carbonization of a very cheap precursor-sanitary tissue that is commonly used in our daily life. Sanitary tissue paper is usually made from pulp fibers, showing high flexibility.43 The tissue-derived HC inherits the primary morphology, namely “a closely interconnected microbelt framework”. Moreover, HCMB has high electronic conductivity and low surface area. In terms of K+-storage performance, this HCMB electrode exhibits a high ICE of 88%, as well as a significantly low charge–discharge plateau, which is comparable to graphite anodes (major discharge plateau at ∼0.25 V) and excellent rate/cycling performances. These unique merits of the HCMB anode endow the as-assembled PIC full device with an operation voltage up to 4.5 V and a high energy density of 112 W h kg−1 at a fast 23 s charge rate, which outperform previously reported PICs in the literature. Furthermore, the performance of HCMB is also better than that of graphite and pitch-derived soft carbon, demonstrating its powerful application in practical energy storage.
Results and discussion
Synthesis and characterization
The detailed fabrication process of flexible HCMB paper is shown in the experimental section. First, the tissue precursor with a three-dimensional cross-linked network structure (Fig. S1, ESI†) is pre-oxidized in air at 250 °C. During this process, the geometric area of the paper is reduced by approximately 5% due to the loss of tar in the precursor. In addition, the pre-oxidation can guarantee mechanical flexibility of HCMB (Fig. S2, ESI†). Second, the pre-oxidized paper was then treated at different temperatures to synthesize hard carbon samples, which maintain a similar morphology to the tissue (Fig. S3, ESI†). Typically, the sample obtained at 1300 °C under an argon atmosphere, is denoted as HCMB-1300. The size of the HCMB paper can be easily enlarged for large-scale applications. As shown in Fig. 1a, the HCMB-1300 paper is mechanically flexible and can be bent into any shape without breaking (including bending, rolling and recovering to its original shape). | ||
| Fig. 1 (a) Photographs of the tissue precursor and the HCMB paper at different bending states. (b–d) SEM images of HCMB-1300. (e) The TEM image of HCMB-1300. (f) Schematic of the HCMB-1300 lattice. | ||
The structural feature of the synthesized HCMB was first investigated by SEM and TEM. The low-magnification SEM image (Fig. 1b) indicates that many micron fibers are interwoven into a highly flexible and cross-linked network structure, which is beneficial for the high mechanical properties of the HCMB-1300 paper. The diameter of these fibers ranges from 10 to 15 μm, and the length generally varies with the paper size (Fig. 1c). The thickness of the HCMB paper is about 50 μm (Fig. S4, ESI†). The high-magnification SEM image (Fig. 1d) shows the rough surface of HCMB-1300, which is formed by the adhesion of fine pulp fibers on the surface during the papermaking process. In the TEM image of HCMB-1300, lattice fringes of the randomly and closely stacked carbon layers are clearly observed, corresponding to interlayer spacing as large as 0.44 nm (Fig. 1e). This typical hard carbon structure of HCMB-1300 would facilitate large-sized K+ intercalation and deintercalation (Fig. 1f).
The XRD pattern of HCMB-1300 exhibits two broad and weak diffraction peaks at 2θ = ∼23 and 43°, representing the (002) and (100) planes, respectively. This low crystallinity indicates a randomly oriented turbostratic structure of the carbon layer in HCMB-1300 (Fig. 2a). In contrast, XRD patterns of graphite show a sharp (002) peak, corresponding to a high crystallinity with the long-range-ordered lattice (Fig. S5a in the ESI†). In contrast, the graphitization degree of pitch-derived soft carbon at 1300 °C in Ar (Fig. S6a in the ESI†) is obviously improved as compared to HCMB-1300, but is slightly lower than that of graphite. The intensity and position of the diffraction peaks of the (002) crystal plane remain basically the same for the HCMB samples carbonized at different temperatures, which is mainly caused by their low crystallinity. The microstructure of the HCMB-700, HCMB-900, HCMB-1100 and HCMB-1300 samples was further investigated by Raman spectroscopy, and the strong D peak representing stacking disorder and structural defects, can be clearly observed in four samples at around 1351 cm−1 (Fig. 2b), which is almost not found in the Raman spectrum of graphite. Moreover, the graphite shows a strong 2D signal (perfect sp2 hybridization) (Fig. S5b in the ESI†), which is not observed in HCMB, demonstrating the existence of disordered species. The calculated peak area ratio (ID/IG) increases from 0.91 for HCMB-700, to 1.02 for HCMB-900, to 1.04 for HCMB-1100 and to 1.07 for HCMB-1100 (Tables S1 and S2, ESI†), indicating that small in-plane domains of graphitic structures can be increased at high carbonization temperatures.44
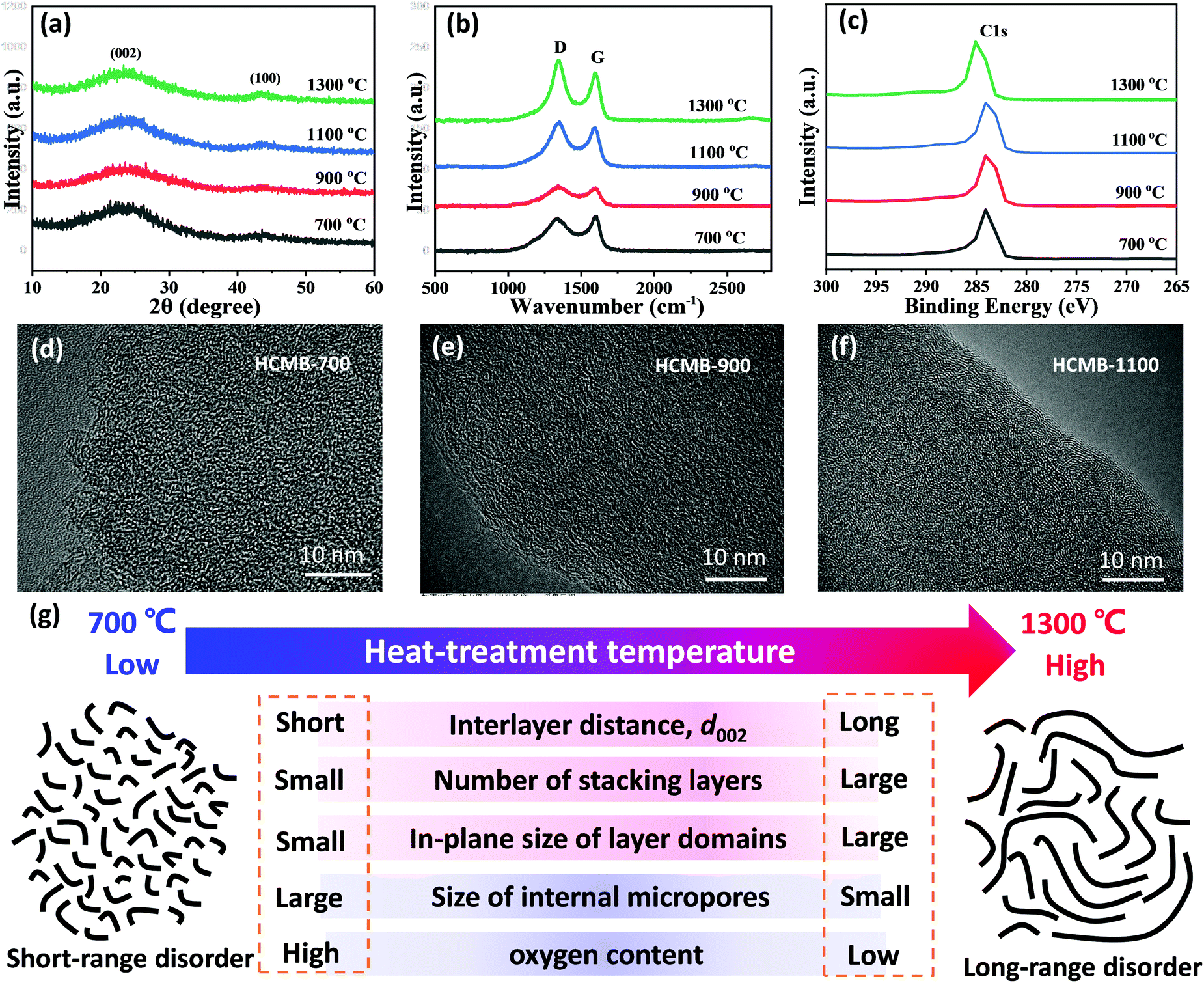 | ||
| Fig. 2 (a) XRD patterns, (b) Raman spectra, (c) XPS spectra, (d–f) TEM images and (g) schematic illustrations of the HCMB structure prepared at different temperatures and the structural parameters. | ||
To further understand the distribution of elements and the nature of the corresponding chemical bonds, XPS data of samples with different carbonation temperatures were collected. The most prominent peaks at ∼284.6 eV are designated as C 1s. This C peaks can be deconvoluted into the C–C, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (Fig. 2c and Fig. S7 in the ESI†). As compared to the other three samples, HCMB-1300 shows a higher binding energy (285.2 eV) and lower oxygen content (2.18%), demonstrating its large size of sp2 domains and less defects. TEM images (Fig. 2d) indicate that HCMB-700 shows a completely disordered structure and well-defined lattice fringes of the stacked carbon layers are not observed. This structure usually generates plenty of open pores.45,46 With an increase in the carbonization temperature (Fig. 2e and f), the carbon layers become gradually clear and bent, which can provide internal micropores as demonstrated in Jenkins–Kawamura and Ban's models.47,48 These pores are gradually closed during thermal treatments at high temperatures up to 1300 °C, which is in agreement with the results reported by Komaba et al.45 N2 adsorption/desorption isotherms further demonstrate this result (Fig. S8, ESI†). The surface area of HCMB is continuously decreased with elevated temperatures from 700 to 1300 °C. HCMB-700 has a higher surface area (216 m2 g−1) than HCMB-900 (68 m2 g−1), HCMB-1100 (4.5 m2 g−1) and HCMB-1300 (0.48 m2 g−1), suggesting that micropores on the surface of HCMB-700 are open for N2 molecules, and completely closed at 1300 °C. The closed pores in HCMB-1300 will decrease the irreversible capacity, thus resulting in a high ICE. All the above results indicate that the structural parameters of HCMB have strong dependence on carbonization temperature, as summarized in Fig. 2g. With increasing temperature, the carbon microstructure of HCMB transforms from open short-range disorder into closed long-range disorder.45,46
O bonds (Fig. 2c and Fig. S7 in the ESI†). As compared to the other three samples, HCMB-1300 shows a higher binding energy (285.2 eV) and lower oxygen content (2.18%), demonstrating its large size of sp2 domains and less defects. TEM images (Fig. 2d) indicate that HCMB-700 shows a completely disordered structure and well-defined lattice fringes of the stacked carbon layers are not observed. This structure usually generates plenty of open pores.45,46 With an increase in the carbonization temperature (Fig. 2e and f), the carbon layers become gradually clear and bent, which can provide internal micropores as demonstrated in Jenkins–Kawamura and Ban's models.47,48 These pores are gradually closed during thermal treatments at high temperatures up to 1300 °C, which is in agreement with the results reported by Komaba et al.45 N2 adsorption/desorption isotherms further demonstrate this result (Fig. S8, ESI†). The surface area of HCMB is continuously decreased with elevated temperatures from 700 to 1300 °C. HCMB-700 has a higher surface area (216 m2 g−1) than HCMB-900 (68 m2 g−1), HCMB-1100 (4.5 m2 g−1) and HCMB-1300 (0.48 m2 g−1), suggesting that micropores on the surface of HCMB-700 are open for N2 molecules, and completely closed at 1300 °C. The closed pores in HCMB-1300 will decrease the irreversible capacity, thus resulting in a high ICE. All the above results indicate that the structural parameters of HCMB have strong dependence on carbonization temperature, as summarized in Fig. 2g. With increasing temperature, the carbon microstructure of HCMB transforms from open short-range disorder into closed long-range disorder.45,46
Potassium-storage properties of HCMB in half-cells
The electrochemical characteristics of HCMBs for potassium storage were evaluated in a traditional ester electrolyte of 0.8 M KPF6/EC/DEC without any additives. It is noted that the ester/KPF6 electrolyte system is suitable for practical applications due to its low cost. Most previously reported hard carbon can achieve high performance in the ether electrolyte, but is hardly available in the ester electrolyte.23,49Fig. 3a–d shows the first three discharge/charge curves at 100 mA g−1 of HCMB with different carbonization temperatures. An obvious difference is observed in the K-storage behaviors of four representative samples, among which HCMB-1300 demonstrates its unique advantage. First, the sample with low carbonization temperatures exhibits a large irreversible capacity, thus leading to a low initial coulombic efficiency (ICE). For example, the reversible charge capacity and the ICE value for HCMB-700 is only 40 mA h g−1 and 22%, respectively. With the increase in carbonization temperature, the ICE value is gradually increased to 66% for HCMB-900, 80% for HCMB-1100 and 88% for HCMB-1300 (Fig. 3e). Moreover, the ICE of HCMB-1300 can be completely retained over four parallel measurements (Fig. 3f). To the best of our knowledge, the high ICE of 88% is unexpected for a typical hard carbon in the ester electrolyte, which is never reported for HC-based potassium storage (see Table 1). Second, the charge capacity below 1.00 V as an extremely important parameter in practical use, is gradually increased from 40 mA h g−1 (HCMB-700), 149 mA h g−1 (HCMB-900), 192 mA h g−1 (HCMB-1100), to 204 mA h g−1 (HCMB-1300). Another notable feature is the decrease in the discharge plateau with the increase of carbonization temperature. The HCMB-700 electrode shows no significant discharge plateau with a typical slope shape. When the temperature is increased to 900 °C, the slope capacity and the plateau capacity each account for 50% at the corresponding cut-off potential of 0.25 V. When the temperature further rises to 1100 °C and 1300 °C, the ratio of plateau capacity is determined to be 63% and 70%, respectively.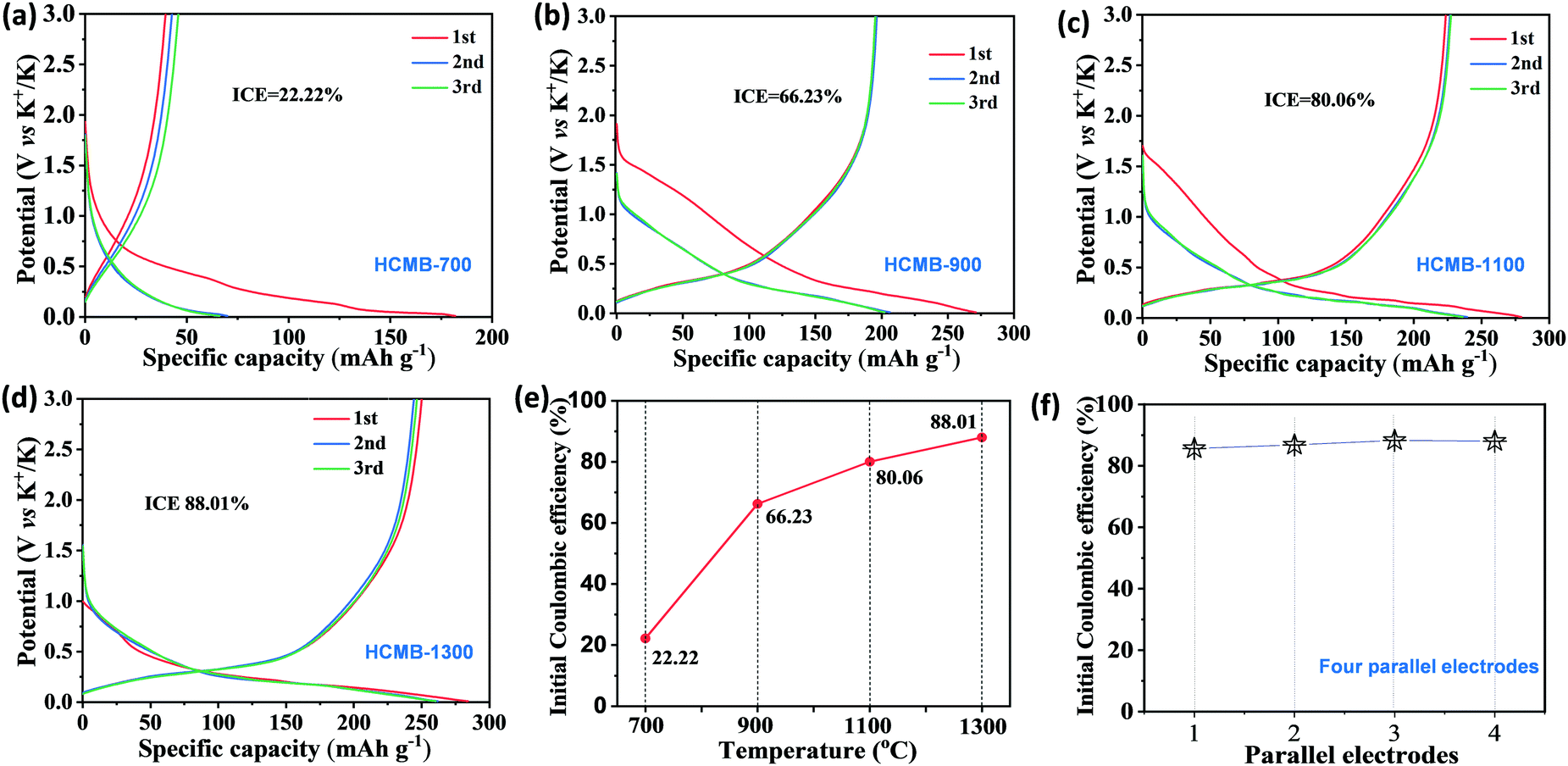 | ||
| Fig. 3 Galvanostatic charge–discharge (GCD) curves of HCMB electrodes with the KPF6/ester electrolyte. (a) HCMB-700, (b) HCMB-900, (c) HCMB-1100, (d) HCMB-1300, (e) initial coulombic efficiency (ICE), and (f) ICE values of HCMB-1300 from four parallel electrodes. | ||
| Anode | Potential range (V) | ICE % | Rate performance mA h g−1/(mA g−1) | Charge capacity below 1 V mA h g−1/(mA g−1) | Ref. |
|---|---|---|---|---|---|
| HCMB-1300 | 0.01–3 | 88 | 254 (100) 155 (1000) | 204 (100) | This work |
| 3DNFC | 0.01–2 | 24.3 | 309 (100) 111 (10000) |
∼120 (100) | 12 |
| GNC | 0.01–2 | 15.5 | 152 (1000) 122 (2000) | ∼50 (200) | 17 |
| NSG | 0.01–3 | ∼17.8 | 192 (100) 91.4 (20000) |
∼120 (100) | 18 |
| CNFF | 0.01–2.8 | ∼19.5 | 240 (50) 164 (1000) | ∼75 (50) | 31 |
| NHC | 0.01–2.6 | ∼14.7 | 277.8 (50) 204.8 (2000) | ∼100 (100) | 32 |
| SHCS | 0.01–3 | 51.4 | 400 (100) 110 (5000) | ∼100 (25) | 34 |
| S/N-CNFAs | 0.01–2.5 | 53 | 356 (100) 168 (2000) | ∼100 (100) | 36 |
| N/O–C@GF | 0.01–3 | 37.4 | 340 (100) 123 (5000) | ∼180 (100) | 57 |
| NOHPHC | 0.01–3 | 25 | 365 (25) 118 (5000) | ∼100 (50) | 58 |
| S/N-PCNs | 0.01–3 | 58.2 | 449 (1000) 107 (20000) |
∼100 (200) | 59 |
| MDPC | 0.01–3 | ∼13.5 | 266.4 (5) 57.6 (5000) | ∼120 (100) | 60 |
| 3D-Ti3C2 | 0.01–3 | ∼18.5 | 209 (50) 88 (2000) | ∼60 (50) | 54 |
| SnS2@C | 0.01–3 | 59.5 | 457.4 (50) 264.3 (2000) | ∼75 (100) | 61 |
| MoSe2/C | 0.01–3 | 63.4 | 382 (200) 244 (2000) | ∼60 (200) | 62 |
| FeVO4 | 0.01–3 | 48.2 | 300 (100) 180.2 (2000) | ∼30 (100) | 63 |
| Zn3V3O8 | 0.01–3 | 55.1 | 286 (100) 17.6 (2500) | ∼100 (100) | 64 |
| C-WS2@CNFs | 0.01–3 | 68.6 | 319 (50) 168 (10000) |
∼80 (50) | 9 |
| FeSe2/N–C | 0.01–3 | 37.8 | 277 (100) 155 (2000) | ∼5 (100) | 11 |
The GCD curves of HCMB-1300 at different current densities are measured to demonstrate the high rate capability (Fig. 4a). It is worth noting that the discharge plateau can be well maintained when the current density is increased from 100 to 2000 mA g−1. The specific capacities of four electrodes from their charge–discharge curves are calculated in Fig. 4b and Fig. S9–S11 (ESI†). At 100 mA g−1, HCMB-1300 can provide a high revisable specific capacity of 265 mA h g−1, significantly higher than that of HCMB-1100 (220 mA h g−1), HCMB-900 (184 mA h g−1) and HCMB-700 (110 mA h g−1). With the increase of 40-times higher current density to 2000 mA g−1, a still high capacity of 112 mA h g−1 can be achieved for HCMB-1300, also outperforming the other three samples. Fig. 4c and d show the cycle performance of HCMB-1300 at a small current density of 100 mA g−1. After 400 cycles, the reversible capacity of HCMB-1300 is 246 mA h g−1, corresponding to the capacity retention of 95% (relative to the 2nd cycle). A high-rate cycling test for HCMB-1300 was performed at 1000 mA g−1. As shown in Fig. 4e, the HCMB-1300 provides a high specific capacity of 174 mA h g−1 and maintains a capacity retention of nearly 90% over long-term cycles of up to 750 cycles.
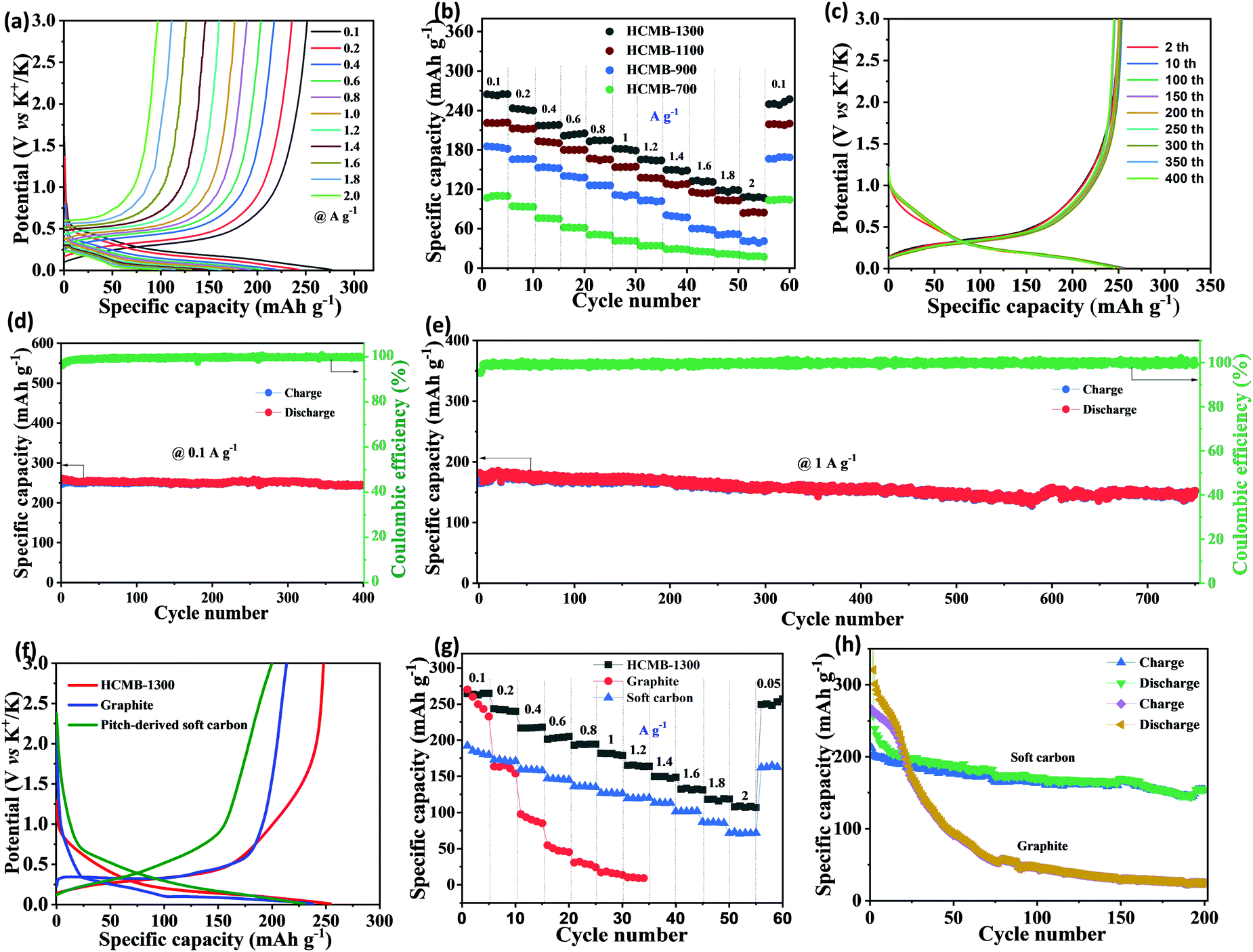 | ||
| Fig. 4 Potassium-storage properties in half-cells. (a) GCD curves of HCMB-1300 at different current densities, (b) rate capability of HCMB-700, HCMB-900, HCMB-1100 and HCMB-1300, (c) GCD curves of HCMB-1300 at 100 mA g−1, (d and e) cycling stability at 100 and 1000 mA g−1, (f) the 10th GCD curves of HCMB-1300 with commercial graphite and pitch-derived soft carbon at 100 mA g−1, (g) rate capability of HCMB-1300, graphite and soft carbon, and (h) cycling stability of graphite and soft carbon at 100 mA g−1. | ||
In order to demonstrate the advantages of HCMB-1300, we further investigated the potassium storage performance of two other types of carbon (graphite and soft carbon-derived from pitch) for comparison. The detailed results are presented in Fig. S12 and S13 (ESI†). According to the 10th charge–discharge curves (Fig. 4f), we can see that the graphite's discharge plateau is very close to that of HCMB-1300 (about 0.25 V), but the charge plateau of graphite is slightly higher that of HCMB-1300, indicating higher K-storage reversibility for the HCMB-1300 electrode. In addition, the slope capacity of HCMB-1300 is more obvious than that of graphite, which is a typical energy-storage characteristic of hard carbon. As compared to HCMB-1300, soft carbon does not exhibit a similar low discharge plateau, which is due to its flexible low-range lattice ordering structure in comparison with graphite. The charge capacity (below 1 V) is also higher for HCMB-1300 (204 mA h g−1) than for graphite (191 mA h g−1) and soft carbon (156 mA h g−1). The ICE value of graphite and soft carbon is determined to be about only 46% and 35%, respectively. The lower ICE reveals the higher side reactions of both electrodes during the first charge/discharge process and the excessive electrolyte consumption during the formation of SEI as well as the additional K+ loss from polymer binders.50–52 Previous studies have demonstrated that the insertion of K+ will induce a 60% volumetric expansion in graphite,16,22 thus seriously leading to the structural failure and poor cycling stability and rate capability. As expected, the K+-storage capacity of the graphite electrode is rapidly decreased to only 14 mA h g−1 at 1000 mA g−1. After 200 cycles at 100 mA g−1, the large capacity loss is clearly observed from the initial 267 mA h g−1 to 25 mA h g−1 at the 200th cycle. The rate and cycling performance of soft carbon is obviously better than that of graphite, but is still inferior to HCMB-1300. At a high current density of 1000 mA g−1, the soft carbon electrode exhibits a specific capacity of 126 mA h g−1, which is lower than that of HCMB-1300 (184 mA h g−1 at 1000 mA g−1). After 200 cycles, the capacity retention for soft carbon is around 72%.
The above results indicate that HCMB-1300 offers the best performance as compared to HCMB-700, HCMB-900, HCMB-1100, graphite and soft carbon. Moreover, based on the comprehensive K-storage performance in terms of the charge specific capacity below 1.00 V, the ICE value as well as the cycling stability, HCMB-1300 is also superior to the previously reported hard carbon materials (see the details in Table 1), commercial hard carbon (Fig. S14, ESI†), HCMB-1300 without pre-oxidation (Fig. S15, ESI†) and HCMB-1500 (Fig. S16, ESI†). Furthermore, the potassium storage performance of HCMB-1300 with the ether electrolyte of 1 M KTFSI in diethylene glycol dimethyl ether was also tested (Fig. S17, ESI†), and the ICE value is 83.2% and the revisable specific capacity is 214 mA h g−1 at 100 mA g−1, which are lower than those in the ester electrolyte of 0.8 M KPF6/EC/DEC. Such high ICE values and low discharge-platform as well as high charge capacity of HCMB-1300 arise from its structural characteristics: (1) the high carbonation temperature is beneficial to forming a closed and randomly oriented turbostratic structure, thus providing more graphitic microcrystal stripes with less defects.45,53 These curved carbon layers and the closed nanovoids may offer more active K storage sites to generate a high plateau capacity; (2) the freestanding configuration excludes the use of traditional current collectors (such as Cu or Al foil), polymer binders (such as PVDF) and conducting additive (such as super P), which will decrease the initial irreversible capacity during K-storage. In order to confirm this point, we test the K-storage performance of HCMB-1300 using the traditional slurry-coating technique under the same mass loading. As presented in Fig. S18 (ESI†), the ICE value of HCMB-1300 is reduced to about 68%, demonstrating the importance of the binder-free electrode in eliminating irreversible capacity; (3) the one-dimensional carbon microbelts allow for the formation of strong solid electrolyte inter films compared with using ordinary powder-type carbon samples.54–56 To investigate the microbelt change of HCMB-1300 after long term cycling, the electrode was disassembled after 750 cycles at 1000 mA g−1. As illustrated in Fig. S19 (ESI†), the microbelt morphology is well maintained after repeated K+ insertion/de-insertion, demonstrating high structural integrity.
To reveal the K+ storage mechanism of HCMB-1300, the CV curves at different scan rates are fitted to distinguish the diffusion-limited current and capacitive current. Fig. 5a shows the CV curves for HCMBs film-1300 for an initial three cycles at 0.1 mV s−1. The first CV curve shows a small irreversible cathodic peak at 1.55 V, which is related with the SEI film. In the second and third cycles, the CV curves are highly overlapped and the CV shape can still be retained when the scan rate is increased to 2 mV s−1 (Fig. S20a, ESI†), indicating its excellent electrochemical reversibility. According to the relationship between the peak current (i) with the scan rate (v) (i = avb),65 the b value of 0.5 is indicative of a diffusion-controlled process, while b = 1 reveals a fast capacitive process. Fig. 5b exhibits the log(i) vs. log(v) plot for two pairs of O1/R1 and O2/R2 peaks. It is calculated that the b value for O1 and O2 is 0.52 and 0.91, respectively. This result indicates that the low-potential plateau region (below 0.5 V) is associated with the slow intercalation of potassium ions into the graphitic nanodomain to form the compound KC8, as well as the extraction of potassium ions from the carbon structure. The high-potential slope region (above 0.5 V) belongs to the adsorption and desorption of K+ ions on the carbon surface including edges, defects, pores, and functional groups, which is important to achieve fast-charging performance and high-energy at high rates.27,44 This “intercalation–adsorption” mechanism is illustrated in Fig. 5c. The capacitive contribution at 1 mV s−1 is as low as 23.1% (Fig. S20b, ESI†), indicating that the potassium-storage kinetics is mainly diffusion-controlled.
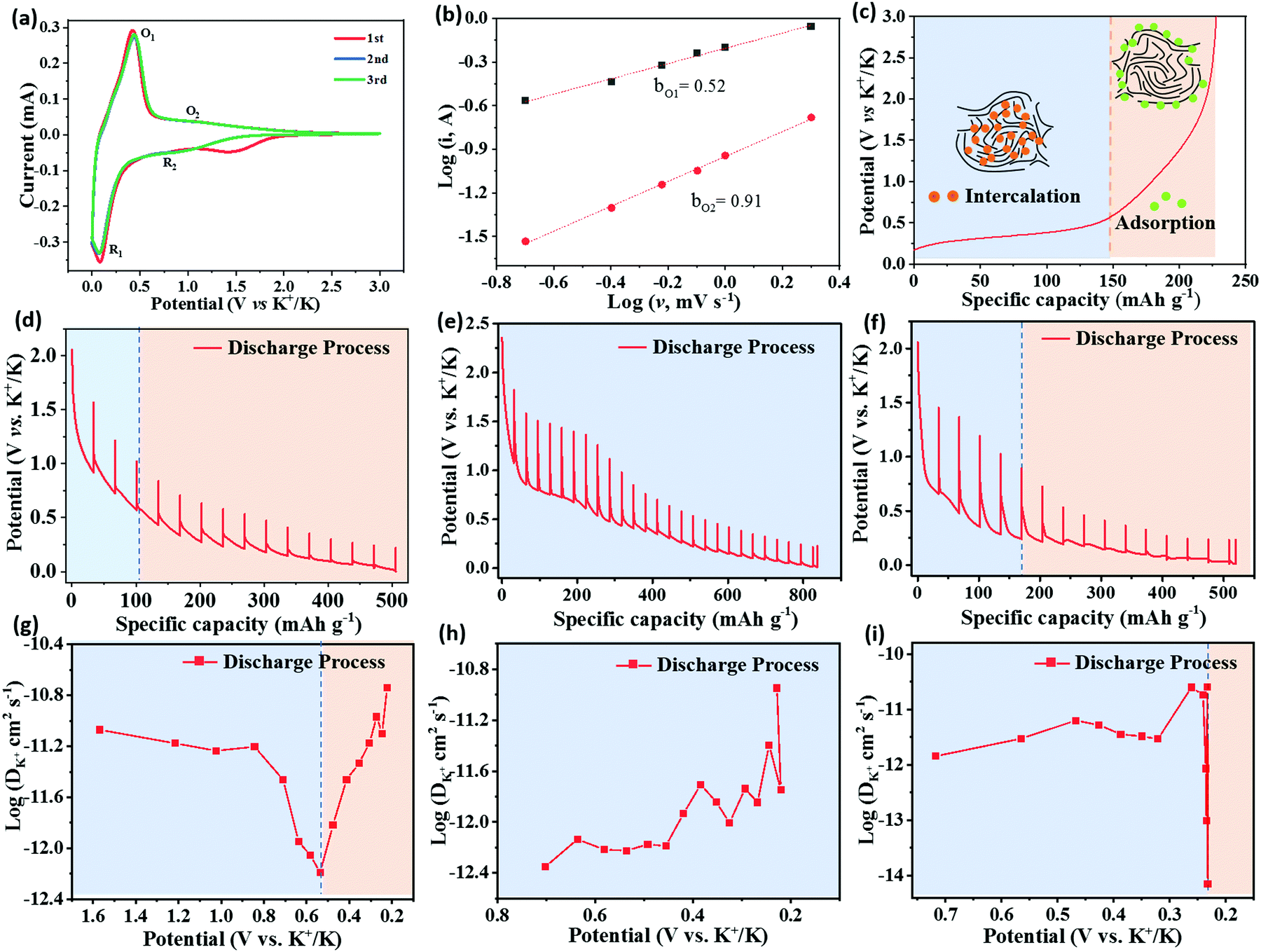 | ||
| Fig. 5 Potassium-storage mechanism. (a) CV curves of HCMB-1300 at 0.1 mV s−1, (b) plots of log(i) vs. log(v), (c) schematic illustration of the potassium storage mechanism, (d–f) galvanostatic intermittent titration technique (GITT) curves and (g–i) K+ apparent diffusion coefficients calculated from the GITT for (d and g) HCMB-1300, (e and h) soft carbon and (f and i) graphite. | ||
To further investigate the potassium storage behavior in different potential ranges, the galvanostatic intermittent titration technique (GITT) was employed to calculate the K diffusion coefficient of HCMB-1300, graphite and soft carbon in the discharging process. The detailed calculation process is shown in Fig. S21 (ESI†). Potassium storage of HCMB-1300 can be assigned to two regions consisting of a high-potential slope and a low-potential plateau (Fig. 5d), which demonstrates that their individual diffusion coefficients of discharge show a variation at 0.5 V. Furthermore, the slope region has a higher average diffusion coefficient than that of the plateau region (Fig. 5g), indicating that the diffusion-controlled plateau region is the rate determining step. In contrast, the soft carbon electrode shows only the plateau region and its diffusion coefficient value is slightly lower than that of HCMB-1300 in the high-potential range, which is due to its lower defect number (Fig. 5e and h). The diffusion coefficient of the graphite electrode shows an abrupt drop at a low potential of 0.25 V and its diffusion value is two orders of magnitude lower than that of HCMB-1300 in the same potential region (Fig. 5f and i). In particular, the buffering region between the plateau region and the slope region can be clearly found in the diffusion coefficient of the HCMB-1300 electrode, but is not found in graphite. We also use ex situ XRD to analyse the structural change during the K+ insertion/deinsertion process. From Fig. S22 (ESI†), we can see that the position of the (002) peak is almost not changed even after full discharge or full charge due to its low crystallinity, which is different from the stage-phase transition observed on graphite.
Electrochemical performance of potassium ion capacitor
Based on the excellent potassium storage performance demonstrated in the half-cell tests, a PIC full cell was assembled to demonstrate the feasibility for practical application (Fig. 6a). The HCMB-1300 membrane is used directly as an anode and coupled with an AC cathode (details in Fig. S23, ESI†). The sepiolite/PVDF–(HFP) membrane was employed as the matrix of the gel electrolyte (Fig. S24, ESI†). In order to take full advantage of the low discharge platform of HCMB-1300, the operating voltage window of the full cell in this work was set between 2.5 and 4.5 V, to provide a higher energy density. During charging, K+ in the electrolyte is inserted into the HCMB, while the PF6− anion is absorbed on the AC surface. During the discharge process, K+ and PF6− ions return into the electrolyte, and correspondingly the electrical supply is stopped. Before assembling the PIC, the pre-potassiation process for the HCMB anode is necessary to compensate for the irreversible K+ loss. The CV curves show a quasi-rectangular shape with no obvious redox peaks, indicating near ideal capacitance behavior (Fig. 6b). Not only that, the quasi-rectangular shape of the CV curve shows no significant distortion as the scan rate increases from 1 to 10 mV s−1, indicating high reversibility and good rate capability. The HCMB//AC PIC device also shows a very linear slope in the charge/discharge curves at different current densities (Fig. 6c and d), in agreement with the CV results. The specific capacitance of the PIC device is calculated to be 78 F g−1 at a current density of 0.1 A g−1. At a 50-times higher current density (5 A g−1), the capacitance retention is as high as 74%, showing excellent rate capability. Moreover, an acceptable cycling stability is achieved with 90% capacitance retention after 1000 cycles at 20 mV s−1 (Fig. 6e). In contrast to half cells that have superior cycling stability with no obvious decay, the PIC device exhibits capacity decay. The possible reasons for this may be attributed to an increase in the interfacial resistance and loss of active potassium ions with cycling. The energy and power densities of the HCMB//AC PIC device are obtained based on the total mass of the anode and the cathode and the results are shown in the Ragone plot from Fig. 6f. Remarkably, a maximum energy density of 152 W h kg−1 could be available at a power density of 350 W kg−1. With increasing power density to 17500 W kg−1 (corresponding to a fast 23 s charge rate), the PIC can still achieve a high energy density of 112 W h kg−1. These energy/power values are superior to other previously reported PICs, such as C-WS2@CNFs//ACNFs (42 W h kg−1 at 12600 W kg−1),9 3D-Ti3C2//HPAC (18.7 W h kg−1 at 7015.7 W kg−1),54 APN-HPCNF//PN-HPCNF (86 W h kg−1 at 7560 W kg−1),66 N-GQD@ASC-500//PC (50 W h kg−1 at 20000 W kg−1),26 MDPC//PDPC (21.6 W h kg−1 at 26000 W kg−1),60 and NHCS//ANHCS (19.1 W h kg−1 at 8203 W kg−1).67 Meanwhile, the energy storage performance of the HCMB//AC PIC is comparable to or even exceeds that of representative SICs and LICs. Furthermore, the as-fabricated PIC pouch cell has good mechanical flexibility (Fig. 6g). As illustrated in Fig. 6h, our PIC device can maintain 92% capacity even after folding to 180°, which arises from the robust flexibility of the interconnected carbon microbelt network.
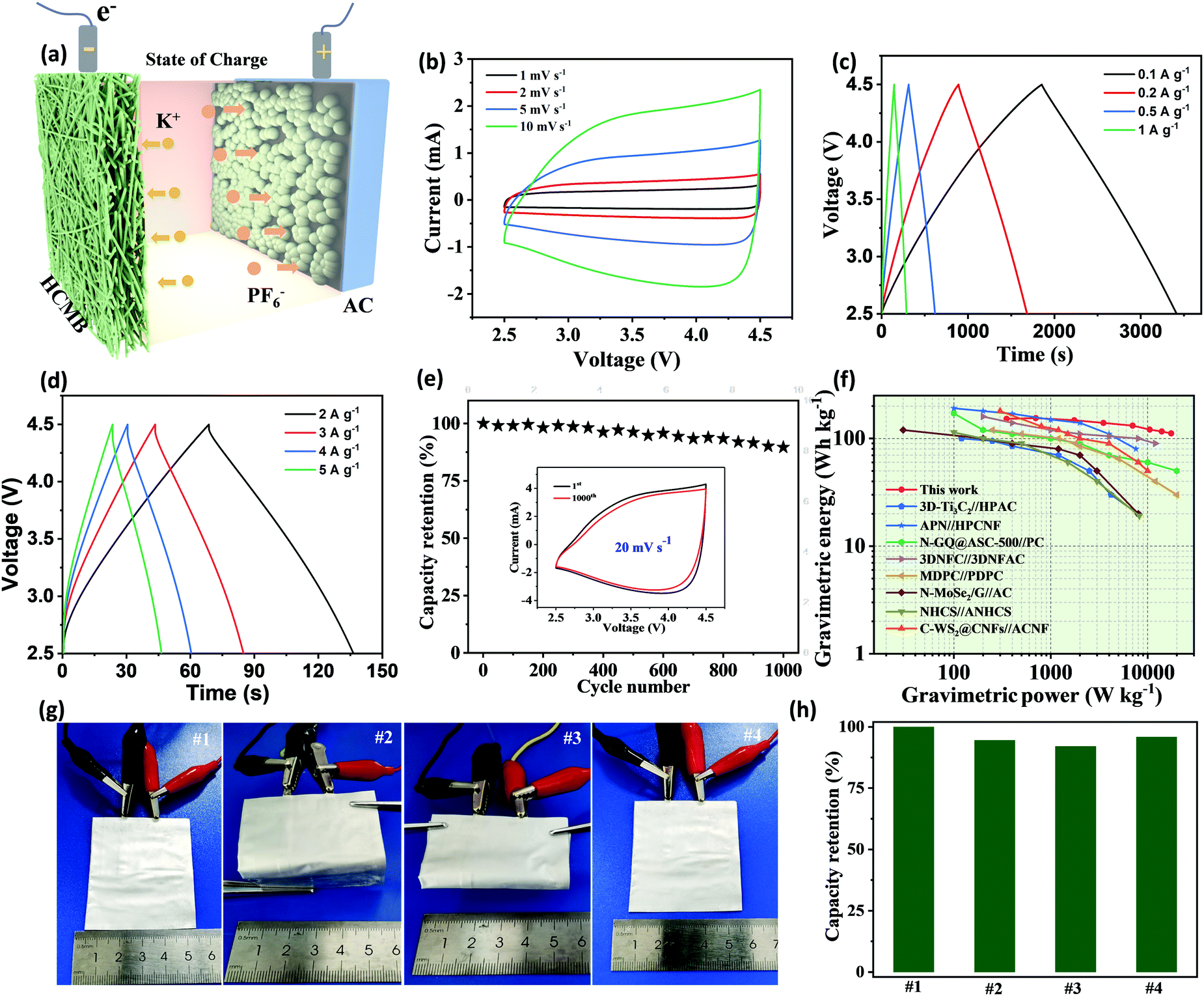 | ||
| Fig. 6 The HCMB//AC PIC full device. (a) Schematic of the PIC and their dual ion storage mechanism (during charge), (b) CV curves of the PIC at different scan rates ranging from 1 to 10 mV s−1, (c and d) GCD curves of the PIC at various current densities ranging from 0.1 to 5 A g−1, (e) cycling performance at 20 mV s−1, (f) Ragone plot, (g) photographs of the as-assembled PIC device, and (h) capacity retentions of flexible PIC measured at 0.1 A g−1 under various bent conditions. | ||
Conclusions
In summary, a flexible self-supporting hard carbon microbelt membrane was developed through pyrolysis of cheap and renewable sanitary tissue. Along with the increase of carbonation temperature, a closed and randomly oriented turbostratic structure with more graphitic microcrystal stripes is gradually formed, which may provide more active K storage sites to generate high plateau capacity. Compared to previously reported hard carbon, our designed HCMB shows a larger charge specific capacity below 1.00 V (204 mA h g−1 at 100 mA g−1) and higher average ICE (88%) as well as satisfactory rate/cycling performance in a conventional ester electrolyte. Moreover, the discharge plateau of HCMB is comparable to graphite and over 75% of the discharge plateau of the anode is concentrated below 0.25 V, thus enabling a high operation working voltage up to 4.5 V and a large energy density (152 W h kg−1) of the PIC full cell. More importantly, the PIC full cell based on the binder-free HCMB anode exhibits good mechanical flexibility and a high power density (17500 W kg−1 at a corresponding energy density of 112 W h kg−1). Considering the environmental friendliness of the raw materials as well as the wide abundancy of potassium resources, this work offers a feasible strategy for promoting the large-scale practical application of flexible K+-storage systems.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the National Natural Science Foundation of China (Grant No. 21975229), the Science and Technology Commission of Shanghai Municipality (19DZ2271500) and the CAS Key Laboratory of Carbon Materials (KLCMKFJJ2009).Notes and references
- H. Wang, C. Zhu, D. Chao, Q. Yan and H. J. Fan, Adv. Mater., 2017, 29, 1702093–1702111 CrossRef PubMed.
- Y. Wu, H. B. Huang, Y. Feng, Z. S. Wu and Y. Yu, Adv. Mater., 2019, 31, 1901414–1901429 CrossRef CAS PubMed.
- W. Zhang, J. Mao, S. Li, Z. Chen and Z. Guo, J. Am. Chem. Soc., 2017, 139, 3316–3319 CrossRef CAS PubMed.
- C. Yang, J. Chen, X. Ji, T. P. Pollard, X. Lu, C. J. Sun, S. Hou, Q. Liu, C. Liu, T. Qing, Y. Wang, O. Borodin, Y. Ren, K. Xu and C. Wang, Nature, 2019, 569, 245–250 CrossRef CAS PubMed.
- J. M. Lee, G. Singh, W. Cha, S. Kim, J. Yi, S. J. Hwang and A. Vinu, ACS Energy Lett., 2020, 5, 1939–1966 CrossRef CAS.
- H. D. Pham, N. R. Chodankar, S. D. Jadhav, K. Jayaramulu, A. K. Nanjundan and D. P. Dubal, Energy Storage Mater., 2021, 34, 475–482 CrossRef.
- M. Arnaiz, D. Shanmukaraj, D. Carriazo, D. Bhattacharjya, A. Villaverde, M. Armand and J. Ajuria, Energy Environ. Sci., 2020, 13, 2441–2449 RSC.
- J. Dong, Y. He, Y. Jiang, S. Tan, Q. Wei, F. Xiong, Z. Chu, Q. An and L. Mai, Nano Energy, 2020, 73, 104838–104846 CrossRef CAS.
- S. Geng, T. Zhou, M. Jia, X. Shen, P. Gao, S. Tian, P. Zhou, B. Liu, J. Zhou, S. Zhuo and F. Li, Energy Environ. Sci., 2021, 14, 3184–3193 RSC.
- Z. Y. Gu, J. Z. Guo, X. X. Zhao, X. T. Wang, D. Xie, Z. H. Sun, C. D. Zhao, H. J. Liang, W. H. Li and X. L. Wu, InfoMat, 2021, 3, 694–704 CrossRef CAS.
- J. Ge, B. Wang, J. Wang, Q. Zhang and B. Lu, Adv. Energy Mater., 2019, 10, 1903277–19032786 CrossRef.
- B. Yang, J. Chen, L. Liu, P. Ma, B. Liu, J. Lang, Y. Tang and X. Yan, Energy Storage Mater., 2019, 23, 522–529 CrossRef.
- M. Chen, L. Wang, X. Sheng, T. Wang, J. Zhou, S. Li, X. Shen, M. Zhang, Q. Zhang, X. Yu, J. Zhu and B. Lu, Adv. Funct. Mater., 2020, 30, 2004247–2004256 CrossRef CAS.
- J. Chen, B. Yang, H. Hou, H. Li, L. Liu, L. Zhang and X. Yan, Adv. Energy Mater., 2019, 9, 1803894–1803903 CrossRef.
- X. Shi, Y. Gan, Q. Zhang, C. Wang, Y. Zhao, L. Guan and W. Huang, Adv. Mater., 2021, 33, 2100837–2100849 CrossRef CAS PubMed.
- L. Fan, R. Ma, Q. Zhang, X. Jia and B. Lu, Angew. Chem., Int. Ed., 2019, 58, 10500–10505 CrossRef CAS PubMed.
- W. Zhang, J. Ming, W. Zhao, X. Dong, M. N. Hedhili, P. M. F. J. Costa and H. N. Alshareef, Adv. Funct. Mater., 2019, 29, 1903641–1903651 CrossRef.
- C. Lu, Z. Sun, L. Yu, X. Lian, Y. Yi, J. Li, Z. Liu, S. Dou and J. Sun, Adv. Energy Mater., 2020, 10, 2001161–2001171 CrossRef CAS.
- R. A. Adams, A. Varma and V. G. Pol, J. Power Sources, 2019, 410, 124–131 CrossRef.
- M. M. Rahman, C. Hou, S. Mateti, K. Tanwar, I. Sultana, A. M. Glushenkov and Y. Chen, J. Power Sources, 2020, 476, 228733–228742 CrossRef CAS.
- S. Liu, J. Mao, L. Zhang, W. K. Pang, A. Du and Z. Guo, Adv. Mater., 2021, 33, 2006313–2006322 CrossRef CAS PubMed.
- M. Gu, L. Fan, J. Zhou, A. M. Rao and B. Lu, ACS Nano, 2021, 15, 9167–9175 CrossRef CAS PubMed.
- L. Fan, S. Chen, R. Ma, J. Wang, L. Wang, Q. Zhang, E. Zhang, Z. Liu and B. Lu, Small, 2018, 14, 1801806–1801814 CrossRef PubMed.
- Y. Zhao, X. Shi, S. J. H. Ong, Q. Yao, B. Chen, K. Hou, C. Liu, Z. J. Xu and L. Guan, ACS Nano, 2020, 14, 4463–4474 CrossRef CAS PubMed.
- J. Cao, H. Xu, J. Zhong, X. Li, S. Li, Y. Wang, M. Zhang, H. Deng, Y. Wang, C. Cui, M. Hossain, Y. Cheng, L. Fan, L. Wang, T. Wang, J. Zhu and B. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 8497–8506 CrossRef CAS PubMed.
- C. Zhang, X. Liu, Z. Li, C. Zhang, Z. Chen, D. Pan and M. Wu, Adv. Funct. Mater., 2021, 31, 2101470–2101480 CrossRef CAS.
- H. J. Kang, Y. S. Huh, W. B. Im and Y. S. Jun, ACS Nano, 2019, 13, 11935–11946 CrossRef CAS PubMed.
- C.-D. Zhao, J.-Z. Guo, Z.-Y. Gu, X.-T. Wang, X.-X. Zhao, W.-H. Li, H.-Y. Yu and X.-L. Wu, Nano Res., 2021 DOI:10.1007/s12274-021-3577-7.
- S. Huang, Y. Lv, W. Wen, T. Xue, P. Jia, J. Wang, J. Zhang and Y. Zhao, Mater. Today Energy, 2021, 20, 100673–100683 CrossRef CAS.
- J. Zhang, L. Lai, H. Wang, M. Chen and Z. X. Shen, Mater. Today Energy, 2021, 21, 100747–100766 CrossRef CAS.
- H. Li, Z. Cheng, Q. Zhang, A. Natan, Y. Yang, D. Cao and H. Zhu, Nano Lett., 2018, 18, 7407–7413 CrossRef CAS PubMed.
- W. Yang, J. Zhou, S. Wang, W. Zhang, Z. Wang, F. Lv, K. Wang, Q. Sun and S. Guo, Energy Environ. Sci., 2019, 12, 1605–1612 RSC.
- W. Zhang, J. Yin, M. Sun, W. Wang, C. Chen, M. Altunkaya, A. H. Emwas, Y. Han, U. Schwingenschlogl and H. N. Alshareef, Adv. Mater., 2020, 32, 2000732–2000741 CrossRef CAS PubMed.
- J. Ding, H. Zhang, H. Zhou, J. Feng, X. Zheng, C. Zhong, E. Paek, W. Hu and D. Mitlin, Adv. Mater., 2019, 31, 1900429–1900438 CrossRef PubMed.
- Y. Sun, H. Wang, W. Wei, Y. Zheng, L. Tao, Y. Wang, M. Huang, J. Shi, Z. C. Shi and D. Mitlin, ACS Nano, 2021, 15, 1652–1665 CrossRef CAS PubMed.
- C. Lv, W. Xu, H. Liu, L. Zhang, S. Chen, X. Yang, X. Xu and D. Yang, Small, 2019, 15, 1900816–1900825 CrossRef PubMed.
- Z. Xu, M. Wu, Z. Chen, C. Chen, J. Yang, T. Feng, E. Paek and D. Mitlin, Adv. Sci., 2019, 6, 1802272–1802285 CrossRef PubMed.
- Y. Liu, H. Dai, Y. An, L. Fu, Q. An and Y. Wu, J. Mater. Chem. A, 2020, 8, 14993–15001 RSC.
- Z. Jian, Z. Xing, C. Bommier, Z. Li and X. Ji, Adv. Energy Mater., 2016, 6, 1501874–1501879 CrossRef.
- Z. Jian, S. Hwang, Z. Li, A. S. Hernandez, X. Wang, Z. Xing, D. Su and X. Ji, Adv. Funct. Mater., 2017, 27, 1700324–17003300 CrossRef.
- H. Ding, J. Zhou, A. M. Rao and B. Lu, Natl. Sci. Rev., 2021, 8, 279–285 CrossRef PubMed.
- X. Shi, Y. Zhang, G. Xu, S. Guo, A. Pan, J. Zhou and S. Liang, Sci. Bull., 2020, 65, 2014–2021 CrossRef CAS.
- Y. Li, Y. A. Samad, T. Taha, G. Cai, S. Y. Fu and K. Liao, ACS Sustainable Chem. Eng., 2016, 4, 4288–4295 CrossRef CAS.
- J. L. Xia, D. Yan, L. P. Guo, X. L. Dong, W. C. Li and A. H. Lu, Adv. Mater., 2020, 32, 2000447–2000455 CrossRef CAS PubMed.
- K. Kubota, S. Shimadzu, N. Yabuuchi, S. Tominaka, S. Shiraishi, M. Abreu-Sepulveda, A. Manivannan, K. Gotoh, M. Fukunishi, M. Dahbi and S. Komaba, Chem. Mater., 2020, 32, 2961–2977 CrossRef CAS.
- Y. Li, Y. Lu, Q. Meng, A. C. S. Jensen, Q. Zhang, Q. Zhang, Y. Tong, Y. Qi, L. Gu, M. M. Titirici and Y. S. Hu, Adv. Energy Mater., 2019, 9, 1902852–1902861 CrossRef CAS.
- G. M. Jenkins and K. Kawamura, Nature, 1971, 231, 175–176 CrossRef CAS PubMed.
- J. Yamanaka, E. Yasuda, H. Migita and Y. Tanabe, Carbon, 2021, 42, 1874–1877 Search PubMed.
- L. Fan, K. Lin, J. Wang, R. Ma and B. Lu, Adv. Mater., 2018, 30, 1800804–1800811 CrossRef PubMed.
- B. Wang, Z. Zhang, F. Yuan, D. Zhang, Q. Wang, W. Li, Z. Li, Y. A. Wu and W. Wang, Chem. Eng. J., 2022, 428, 131093–131112 CrossRef CAS.
- X. Wu, Y. Chen, Z. Xing, C. W. K. Lam, S. S. Pang, W. Zhang and Z. Ju, Adv. Energy Mater., 2019, 9, 1900343–1900389 CrossRef.
- Q. Zhang, B. Han, Y. Zou, S. Shen, M. Li, X. Lu, M. Wang, Z. Guo, J. Yao, Z. Chang and M. Gu, Adv. Mater., 2021, 33, 2102666–2102674 CrossRef CAS PubMed.
- Y. Liu, Y. X. Lu, Y. S. Xu, Q. S. Meng, J. C. Gao, Y. G. Sun, Y. S. Hu, B. B. Chang, C. T. Liu and A. M. Cao, Adv. Mater., 2020, 32, 2000505–2000513 CrossRef CAS PubMed.
- Y. Z. Fang, R. Hu, K. Zhu, K. Ye, J. Yan, G. Wang and D. Cao, Adv. Funct. Mater., 2020, 30, 2005663–2005673 CrossRef CAS.
- L. Zhou, Z. Cao, J. Zhang, H. Cheng, G. Liu, G. T. Park, L. Cavallo, L. Wang, H. N. Alshareef, Y. K. Sun and J. Ming, Adv. Mater., 2021, 33, 2005993–2006003 CrossRef CAS PubMed.
- Q. Zhang, X. Cheng, C. Wang, A. M. Rao and B. Lu, Energy Environ. Sci., 2021, 14, 965–974 RSC.
- S. Zeng, X. Chen, R. Xu, X. Wu, Y. Feng, H. Zhang, S. Peng and Y. Yu, Nano Energy, 2020, 73, 104807–104816 CrossRef CAS.
- J. Yang, Z. Ju, Y. Jiang, Z. Xing, B. Xi, J. Feng and S. Xiong, Adv. Mater., 2018, 30, 1700104–1700115 CrossRef PubMed.
- X. Hu, Y. Liu, J. Chen, L. Yi, H. Zhan and Z. Wen, Adv. Energy Mater., 2019, 9, 1901533–1901543 CrossRef CAS.
- M. Shao, C. Li, T. Li, H. Zhao, W. Yu, R. Wang, J. Zhang and L. Yin, Adv. Funct. Mater., 2020, 30, 2006561–2006571 CrossRef CAS.
- D. Li, L. Dai, X. Ren, F. Ji, Q. Sun, Y. Zhang and L. Ci, Energy Environ. Sci., 2021, 14, 424–436 RSC.
- W. Wang, B. Jiang, C. Qian, F. Lv, J. Feng, J. Zhou, K. Wang, C. Yang, Y. Yang and S. Guo, Adv. Mater., 2018, 30, 1801812–1801819 CrossRef PubMed.
- X. Niu, Y. Zhang, L. Tan, Z. Yang, J. Yang, T. Liu, L. Zeng, Y. Zhu and L. Guo, Energy Storage Mater., 2019, 22, 160–167 CrossRef.
- Z. Shi, Q. Ru, S. Cheng, X. Hou, F. Chen and F. Chi-Chung Ling, Energy Technol., 2020, 8, 1900754–1900765 Search PubMed.
- Y. Zhu and C. Wang, J. Phys. Chem. C, 2010, 114, 2830–2841 CrossRef CAS.
- X. Hu, G. Zhong, J. Li, Y. Liu, J. Yuan, J. Chen, H. Zhan and Z. Wen, Energy Environ. Sci., 2020, 13, 2431–2440 RSC.
- D. Qiu, J. Guan, M. Li, C. Kang, J. Wei, Y. Li, Z. Xie, F. Wang and R. Yang, Adv. Funct. Mater., 2019, 29, 1903496–1903504 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee03214c |
| This journal is © The Royal Society of Chemistry 2022 |