
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceObservationally constrained representation of brown carbon emissions from wildfires in a chemical transport model†
Soroush E.
Neyestani
 and
Rawad
Saleh
*
and
Rawad
Saleh
*
Air Quality and Climate Research Laboratory, School of Environmental, Civil, Agricultural, and Mechanical Engineering, University of Georgia, Athens, Georgia 30602, USA. E-mail: rawad@uga.edu
First published on 7th January 2022
Abstract
The month of August 2015 featured extensive wildfires in the Northwestern U.S. and no significant fires in Alaska and Canada. With the majority of carbonaceous aerosols (CA), including black carbon (BC) and brown carbon (BrC), over the U.S. dominated by emissions from Northwestern wildfires, this month presented a unique opportunity for testing wildfire BrC representation in the Weather Research and Forecasting model with chemistry (WRF-Chem). We performed parallel simulations that (1) did not account for BrC absorption, (2) accounted for BrC absorption, and (3) accounted for BrC absorption as well as its decay due to photobleaching. We used a comprehensive set of extensive and pseudo-intensive optical properties, namely the aerosol optical depth (AOD), aerosol absorption optical depth (AAOD), absorption Ångström exponent (AAE), and single scattering albedo (SSA) to constrain the model output against observations from the Aerosol Robotic Network (AERONET). We found that accounting for BrC absorption and photobleaching resulted in the best agreement with observations in terms of aerosol absorption (AAOD and AAE). However, the model severely underestimated AOD and SSA compared to observations. We attributed this discrepancy to missing scattering due to missing secondary organic aerosol (SOA) formation from wildfire emissions in the model. To test this hypothesis, we applied a zeroth-order representation of wildfire SOA, which significantly improved the AOD and SSA model-observation comparison. Our findings indicate that BrC absorption, the decay of its absorption due to photobleaching, as well as SOA formation should be accounted for in chemical transport models in order to accurately represent CA emissions from wildfires.
Environmental significanceWith the continued reduction in anthropogenic emissions driven by environmental regulation and the projected increase in the intensity of wildfires driven by increase in global temperatures and drought episodes, wildfires are expected to be the dominant source of carbonaceous aerosol (CA) in the U.S. in the near future. Therefore, predicting the air-quality and climate impacts of aerosols requires accurate representation of CA emitted by wildfires in chemical transport and climate models. Here, we utilize a comprehensive set of remote-sensing observations to constrain the representation of wildfire CA in models. Our analysis demonstrates the importance of accounting for light absorption by brown carbon and secondary organic aerosol formation in order to reconcile model output with observations. These findings improve the model ability to predict the effect of wildfire CA on the atmospheric energy budget and the CA inhalation exposure levels in regions impacted by wildfires. |
Introduction
Emissions from wildland fires are associated with significant impacts on public health1–3 and the climate.4,5 On the other hand, they play an essential ecological role that benefits natural resources and promotes ecosystem health and resilience.6,7 This tradeoff renders planning for the management and mitigation of wildland fires a major challenge,7 especially due to the chaotic nature of open fires which makes model predictions of their public-health and climate impacts highly uncertain.1,4,8 Wildland fires encompass wildfires, which are usually ignited unintentionally, and prescribed fires.7 On average in the U.S., prescribed fires (mostly in the Southeastern U.S.) and wildfires (mostly in the Western U.S.) cover similar burned areas annually of ∼3 million ha each, though Western wildfires exhibit significant year-to-year variability1 and have been increasing in intensity due to increase in global temperatures and drought episodes.9 With the continued decrease in anthropogenic emissions driven by emission standards and regulation,10 the fraction of air pollutants attributed to wildland fires has been steadily increasing and is projected to continue to do so in the future.11Wildland fires are major emitters of carbonaceous aerosol (CA), including organic aerosol (OA) and black carbon (BC). Numerous laboratory studies12–14 and field measurements15–17 have shown that OA from wildland fires is light-absorbing, and is categorized as brown carbon (BrC).18–20 BC is a strong absorber of solar radiation with a positive radiative forcing that is globally second only to carbon dioxide.21,22 BrC is comprised of various species with a wide range of light-absorption properties, usually quantified using the imaginary part of the refractive index (k). On average, BrC emitted from biomass burning (including wildland fires) exhibits mid-visible k values that are one to two orders of magnitude smaller than k of BC.20 However, BrC emissions are usually one to two orders of magnitude larger than BC,23 thus atmospheric BrC absorption is potentially equally important to BC. Furthermore, BrC exhibits absorption spectra that are largely skewed toward shorter visible and UV wavelengths, which can have important implications for photochemistry.24,25
Representing BrC absorption in chemical transport models is challenging. Reported estimates of the global direct radiative effect (DRE) of BrC absorption range between +0.03 W m−2 and +0.57 W m−2.24,26 This large range partly reflects the aforementioned large variation in BrC light-absorption properties, where different modeling studies have used different kBrC values. To account for variability in kBrC, experimental studies have developed parametrizations that correlated kBrC in biomass-burning emissions with the BC-to-BrC ratio (BC/BrC) in the emissions.12,27,28 Specifically, as BC/BrC increases, BrC becomes more absorbing (i.e. kBrC increases). The underlying reasoning is that a fraction of BrC is formed through the same route as BC, and as the combustion conditions become more conducive for BC formation, the light-absorption properties of the emitted BrC converge to those of BC.29
Further complicating the representation of BrC in chemical transport models is that BrC absorption decays upon aging in the atmosphere. This process, referred to as photobleaching, involves the destruction of BrC chromophores due to either direct photolysis or photochemically induced reactions with OH.15,30–33 Atmospheric observations of the evolution of wildfire plumes have shown that BrC absorption decays with e-folding timescales on the order of 1 day.18,34 Wang et al.35 implemented a photobleaching scheme in a chemical transport model and found that the global BrC absorption DRE dropped from +0.1 W m−2 to 0.048 W m−2 when they incorporated the effect of photobleaching. Similarly, Brown et al.36 reported a global BrC absorption DRE of +0.13 W m−2 and +0.06 W m−2 with and without photobleaching, respectively.
While the importance of light absorption by biomass-burning BrC has been established in experimental studies and atmospheric observations, the extent to which accounting for BrC absorption improves model performance is less clear. On one hand, several investigations have reported that accounting for BrC absorption resulted in better agreement between models and observations. Hammer et al.24 compared ultraviolet aerosol index (UVAI) values retrieved from Ozone Monitoring Instrument (OMI) data and those simulated by a global climate model and found that ignoring BrC absorption in the model caused a negative bias in UVAI over biomass-burning regions.24 Wang et al.35 used aircraft measurements over the U.S. to constrain model-simulated BrC absorption. They found that best model-measurement agreement was achieved by accounting for both BrC absorption and photobleaching. Similarly, June et al.37 found that ignoring photobleaching in a global chemical transport model led to overestimating aerosol absorption compared to Interagency Monitoring of Protected Visual Environments (IMPROVE) observations. On the other hand, Brown et al.36 found that even though implementing BrC absorption in a global chemical transport model led to better agreement between model absorption Ångström exponent (AAE) values over biomass-burning regions and those retrieved from Aerosol Robotic Network (AERONET) observations, the model underestimated single scattering albedo (SSA) values over these regions compared to AERONET. This finding was confirmed by a more comprehensive follow-up study which reported that all of 9 global models underestimated SSA over biomass-burning regions compared to measurements from 12 observational data sets.8 Indeed, that study reported that accounting for BrC absorption exacerbated the underestimation of SSA.
Here, we present a detailed investigation of the extent to which representing absorption by BrC from wildfire emissions in a chemical transport model improves the comparison with remote-sensing observations. Using the Weather Research and Forecasting model with chemistry (WRF-Chem),38 we performed simulations for the month of August 2015 over the U.S. where we treated OA emissions from wildfires as (1) non-absorbing, (2) absorbing (i.e. BrC), and (3) BrC with evolving light-absorption properties due to photobleaching. We then applied a comprehensive set of constraints retrieved from AERONET observations to assess the model performance for each treatment. The constraints included both extensive (AOD and AAOD) and pseudo-intensive (AAE and SSA) optical properties.
Experimental design
The goal of this study is to assess the importance of accounting for the absorption of BrC in wildfire CA emissions for accurately representing their interaction with solar radiation. To that end, we performed a series of WRF-Chem simulations over the U.S. for the month of August 2015. We chose this month because it featured extensive wildfire events in the Northwestern U.S., while no major fires were recorded in Alaska and Canada. Therefore, this simulation period presents a unique test case with high levels of wildfire CA emissions within the simulation domain and minimal transport from outside the domain boundaries. Over the month of August 2015, approximately 93% of the CA emissions in the U.S. were from biomass burning (7% were from anthropogenic sources), 92% of which were from the Northwestern wildfires. The impact of the CA emissions from the high wildfire activity is depicted in Fig. 1a, where the modeled monthly average wildfire CA column burden exceeded 50 mg m−2 over the Northwestern U.S. Fig. 1b shows the fraction of CA column burden over the U.S. attributed to the wildfire emissions, clearly reflecting that CA from wildfire emissions dominated the CA column burden over the majority of the U.S. regions. The contribution of the Northwestern wildfires to CA is the lowest in the Eastern U.S. due to both the long transport distance as well as the relatively high levels of anthropogenic CA and biogenic secondary organic aerosol (SOA) over that region.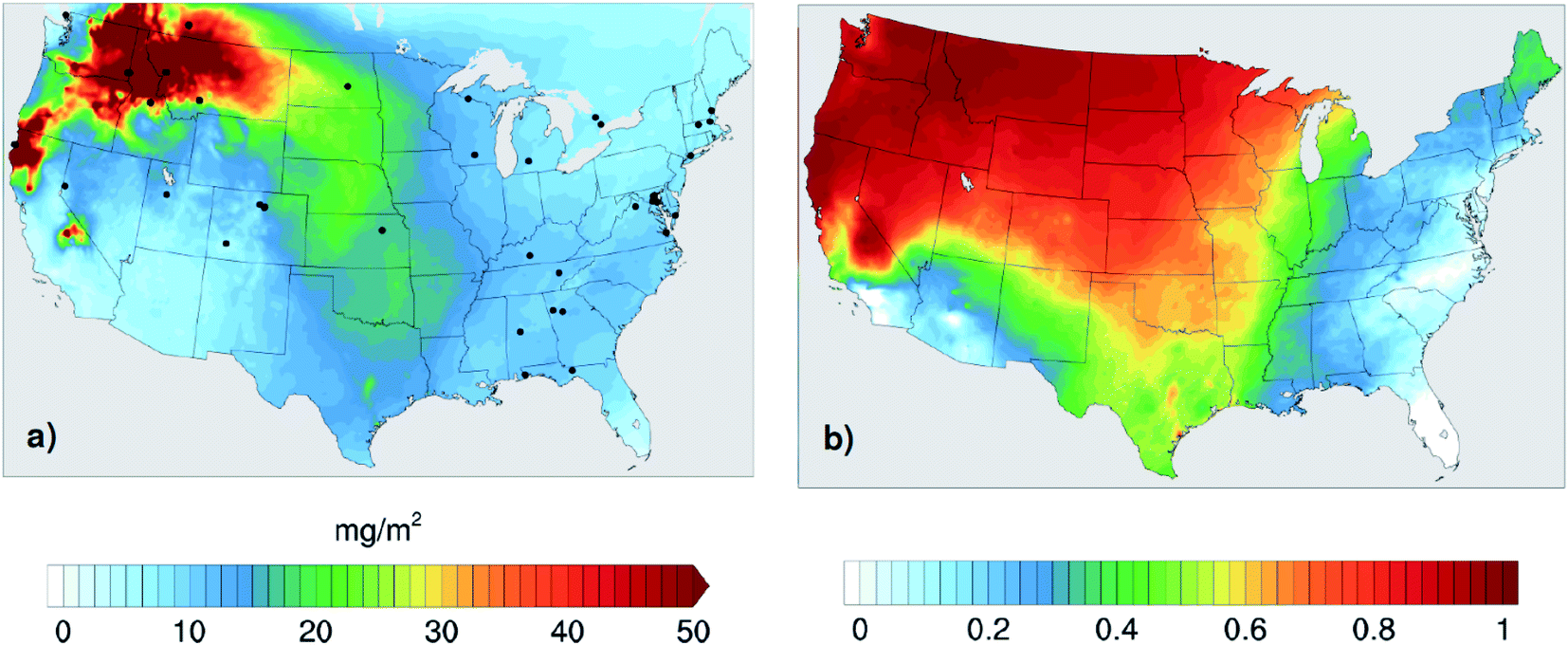 | ||
| Fig. 1 (a) Column burden of carbonaceous aerosol attributed to biomass-burning emissions averaged over the month of August 2015. As evident in the spatial distribution, emissions from Northwestern wildfires constitute the majority of biomass-burning emissions in the simulation period. Black dots show the locations of AERONET stations used in the model-observation comparisons. (b) The fraction of carbonaceous-aerosol column burden attributed to biomass-burning (wildfire) emissions. | ||
In order to assess the impact of representing BrC in wildfire emissions on CA optical properties, we performed 4 parallel simulations:
(1) Base: we treated all OA emissions, including biomass-burning OA, as non-absorbing. This is the default setting in WRF-Chem.
(2) BrC: we treated biomass-burning OA emissions as BrC based on the parameterization of Saleh et al.,28 as further elaborated below. More than 90% of biomass-burning OA emissions were attributed to Northwestern wildfires in the simulation period with little contribution from other biomass-burning sources. Therefore, biomass-burning BrC in this simulation is effectively wildfire BrC. Anthropogenic OA emissions as well as SOA from all precursors (biogenic, anthropogenic, and biomass burning) were treated as non-absorbing.
(3) BrC + bleaching: we accounted for the decay in BrC absorption due to photobleaching based on the parameterization of Wang et al.,35 as further elaborated below.
(4) BrC + bleaching + SOA: we incorporated a zeroth-order representation of SOA formation from the oxidation of vapors in wildfire emissions.
Model description
We employed WRF-Chem38 version 3.8. We set a single domain encompassing the contiguous U.S. with a horizontal resolution of 12 km (396 × 246 grid cells) and 30 pressure-based vertical layers extending up to 50 hPa. The model time step was 60 seconds and the output was saved as 3 hour averages. A 10 day spin-up time was added at the start of the simulation period. The initial and boundary conditions were processed for meteorology calculations using the National Center for Environmental Prediction (NCEP) final reanalysis data with a horizontal resolution of 1° × 1° and temporal resolution of 6 hours.39 We also implemented the reanalysis data to apply Four-Dimensional Data Assimilation (FDDA) nudging40 to the wind and temperature components in the simulations.We obtained both anthropogenic and biomass-burning emissions data from the EPA National Emission Inventory (NEI).41 In addition to satellite fire detection, wildland-fire emissions in NEI are obtained from fire information databases reported by national, regional, state, local, and tribal agencies.42 We preprocessed the NEI emissions for use in WRF-Chem using the Sparse Matrix Operator Kernel Emission (SMOKE)43 model version 3.6.5 and converted the SMOKE hourly emission output into WRF-Chem compatible input files using EPA_ANTHRO_EMIS code developed by NCAR.44 For computational efficiency, a simplified plume rise method45 was applied to the fire emission sources before using as input in WRF-Chem.
Gas-phase chemistry was processed using the Model for Ozone and Related Chemical Tracers (MOZART).46 Model of Emissions of Gases and Aerosols from Nature (MEGAN) version 2 (ref. 47) was used to calculate biogenic emissions online with meteorology. The Model for Simulating Aerosol Interactions and Chemistry (MOSAIC),48 which includes major aerosol species such as BC, organic carbon (OC), sulfate, nitrate, and ammonium, was selected for simulating aerosol microphysics. Within MOSAIC, gas–particle partitioning of semi-volatile organic species is simulated based on the volatility basis set framework49 and inorganic aerosol thermodynamics is simulated using the multi-component equilibrium solver over aerosols.50
We compared the monthly average BC and OA surface concentrations predicted by the model with those obtained from the Chemical Speciation Network (CSN) and the Interagency Monitoring of Protected Visual Environment (IMPROVE) network.51 The locations of the CSN and IMPROVE stations are given in ESI Fig. S1,† and the comparisons for BC and OA are given in Fig. S2a and b,† respectively. As shown in Fig. S2c,† the model achieves the performance goals defined by Boylan and Russell52 based on the mean fraction error (MFE) for the CSN comparison for both BC and OA. For the IMPROVE comparison, the model achieves the performance goals for BC and the less stringent performance criteria for the OA comparison. The reason for the larger MFE for the model-IMPROVE OA comparison relative to the model-CSN comparison is further discussed in ‘The missing scattering’ subsection.
WRF-Chem employs Mie theory to perform aerosol optical calculations (scattering coefficients and absorption coefficients) using MOSAIC size distributions and the complex refractive indices associated with each MOSAIC chemical constituent.53 We used the model default complex refractive indices except for wildfire BrC, which was parameterized as described in the next section. We employed an external mixing assumption for BC, and all the other aerosol components were treated as well-mixed within each size bin. We note that WRF-Chem does not provide this mixing state as an option. Therefore, we defined new (duplicate) size bins for BC particles which were only employed for optical calculations in the optical module and thus did not affect the aerosol microphysical and chemical processes in the chemical transport module.
Brown carbon parameterization
In its default configuration, WRF-Chem treats OA from all sources, including wildfires, as non-absorbing by assigning an OA imaginary part of a refractive index (kOA) of zero. In simulations 2, 3, and 4 (described above), we accounted for absorption by biomass-burning (mostly wildfire) OA, i.e. we treated wildfire OA as BrC. We applied the parameterization of Saleh et al.,28 which calculates kBrC of biomass-burning emissions as a function of the BC-to-BrC ratio (BC/BrC) in the emissions:| kBrC,550 = 0.016 × log(BC/BrC) + 0.03925 | (1) |
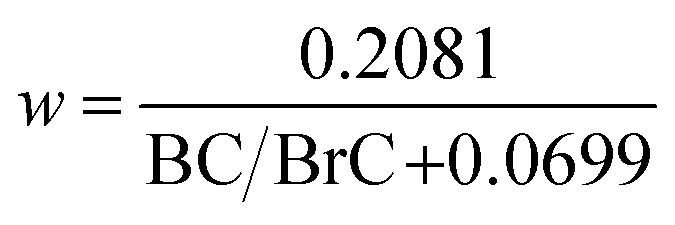 | (2) |
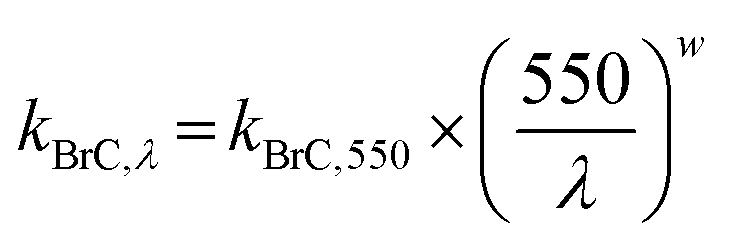 | (3) |
According to this parameterization, kBrC,550 increases and w decreases with increasing BC/BrC, signifying that the BrC absorption becomes stronger but has a flatter wavelength dependence. The inverse relationship between kBrC,550 and w has been observed in several studies.12,27,54 It is noteworthy that BC has w = 0 in the visible spectrum.
We note that in eqn (1) and (2), BC refers to biomass-burning BC and BrC refers to biomass-burning OA. Therefore, in order to implement the parameterization (eqn (1) and (2)) in WRF-Chem, we defined new species to separate the OA emissions into anthropogenic OA and biomass-burning OA (BrC) and the BC emissions into anthropogenic BC and biomass-burning BC.
In simulations 3 and 4 (described above), we accounted for the decay in BrC absorption by photobleaching15,30–33 upon aging in the atmosphere based on the parameterization of Wang et al.35 That study assumed that BrC absorption decreased following a first-order dynamic response with a time constant (i.e. lifetime) of approximately 1 day18,34 at an OH concentration of 5 × 105 molecules per cm3. Therefore, kBrC at each time step can be calculated as:
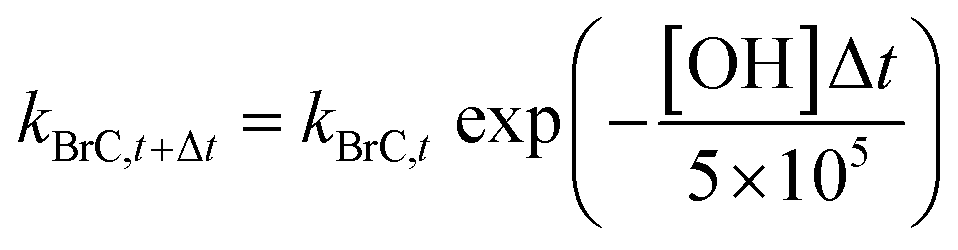 | (4) |
Following Wang et al.,35 we did not allow kBrC to drop below 1/4 of the original value (at t = 0), which is consistent with atmospheric observational studies showing that the decay in absorption associated with photobleaching plateaus at a certain threshold.18,34
Fig. S3a in the ESI† shows the spatial distribution of [OH] predicted by WRF-Chem for the month of August 2015, averaged over the first 8 layers of the model (where BrC effect is important). The corresponding BrC absorption half-lives, estimated from eqn (4), are shown in Fig. S3b.† The domain-average BrC absorption half-life was 0.52 days, which is consistent with the global-average half-life of 0.59 days reported by Brown et al.36
Observational constraints
In order to assess the optical treatments of BrC, we compared the model output with observations from the Aerosol Robotic Network (AERONET) observations.55Fig. 1 shows the locations of AERONET stations (black dots) used in this study.The model-observation comparisons are monthly averages (August 2015). They include both extensive optical properties, namely the aerosol absorption optical depth (AAOD, Fig. 2) and aerosol optical depth (AOD, Fig. 3), as well as pseudo-intensive optical properties, namely the absorption Ångström exponent (AAE, Fig. 4) and the single scattering albedo (SSA, Fig. 5). We use ‘pseudo’ to indicate that even though AAE and SSA do not depend on aerosol concentration, they are not true intensive optical properties (as for example, the complex refractive index) because they depend on particle size and mixing state of the aerosol.
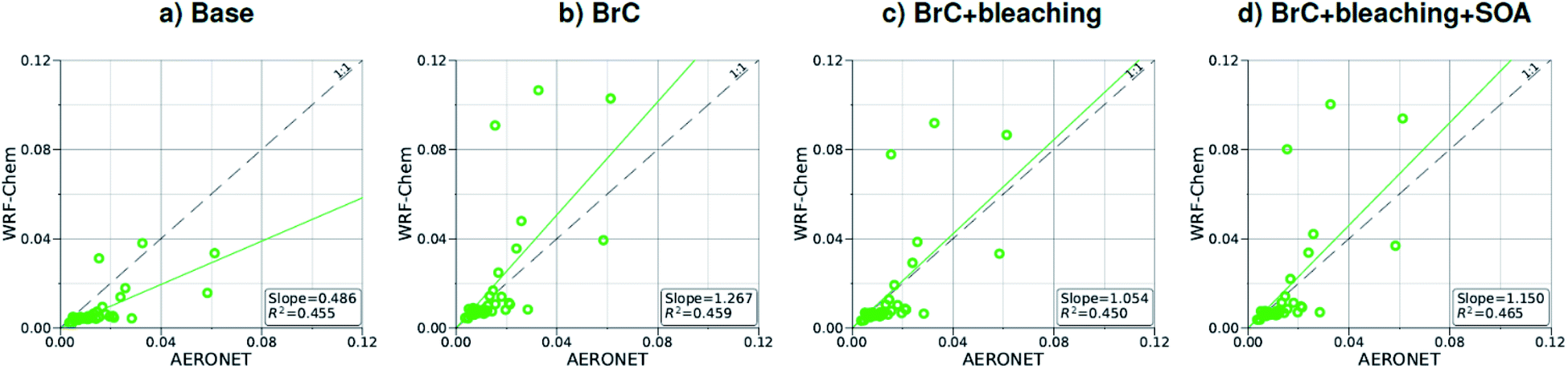 | ||
| Fig. 2 Comparison between monthly average AAOD at 440 nm for August 2015 obtained from AERONET observations and WRF-Chem output with four different model treatments of wildfire carbonaceous aerosol emissions. | ||
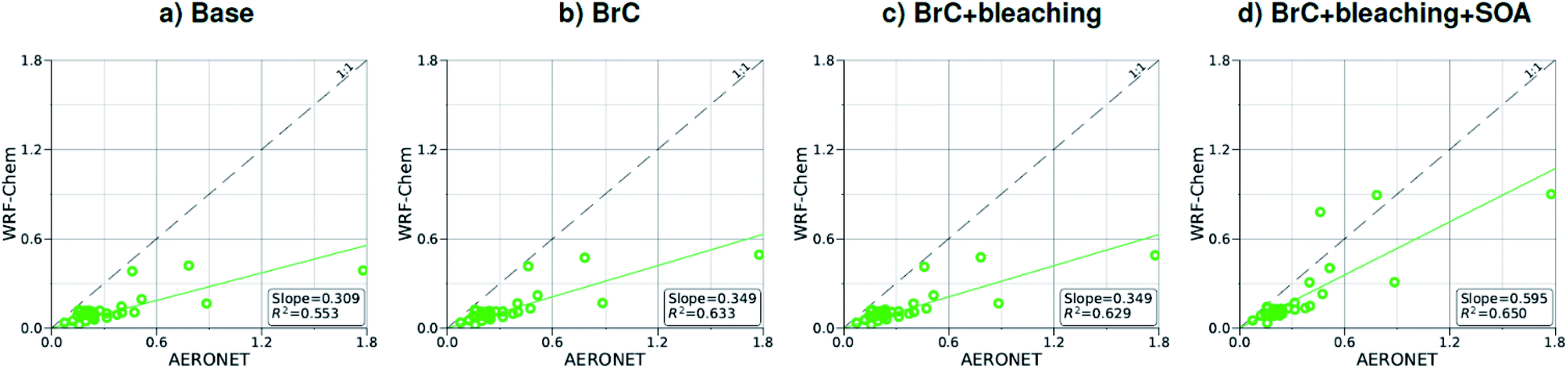 | ||
| Fig. 3 Comparison between monthly average AOD at 440 nm for August 2015 obtained from AERONET observations and WRF-Chem output with four different model treatments of wildfire carbonaceous aerosol emissions. | ||
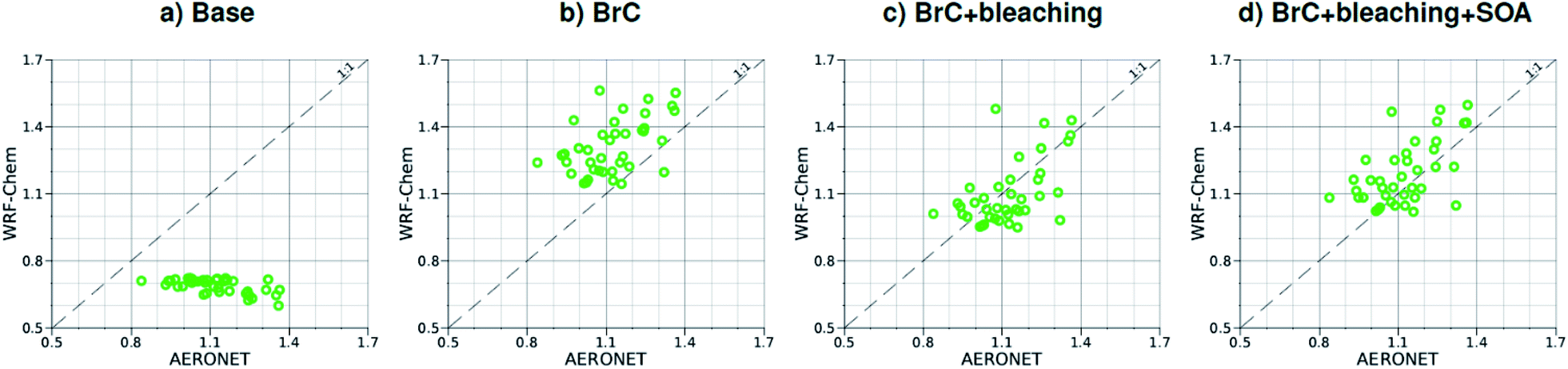 | ||
| Fig. 4 Comparison between monthly average AAE for August 2015 obtained from AERONET observations and WRF-Chem output with four different model treatments of wildfire carbonaceous aerosol emissions. | ||
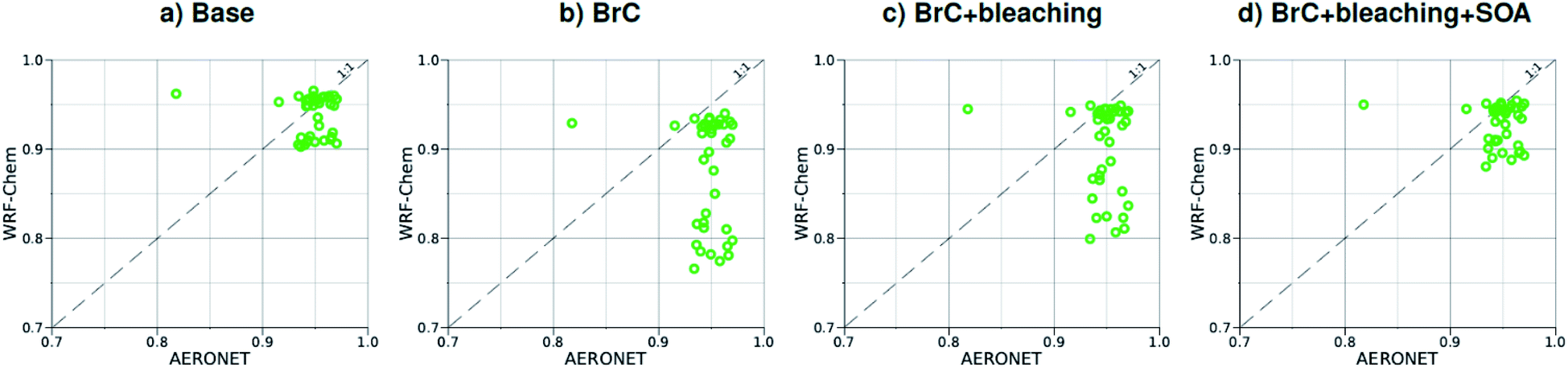 | ||
| Fig. 5 Comparison between monthly average SSA at 440 nm for August 2015 obtained from AERONET observations and WRF-Chem output with four different model treatments of wildfire carbonaceous aerosol emissions. | ||
AERONET inversion products use an inversion algorithm described in Dubovik & King56 where radiative transfer forward modeling is coupled with statistical estimation and constraints to optimize the inversion method. AERONET inversion products have two quality assurance levels.57 The first level (1.5) includes thresholds on azimuth and scattering angles while the second level (2.0) applies an extra set of criteria (e.g. omitting data points with AODs smaller than 0.4). For this study, we used level 1.5 AERONET inversion data to retain a large number of data points for comparison with the model results.
AERONET observations are reported at 440 nm, 675 nm, 870 nm, and 1020 nm. For the AAOD, AOD, and SSA comparisons, we focus on the observations at 440 nm, where BrC absorption is the strongest. Comparisons at 675 nm are shown in Fig. S4–S6 in the ESI.†
We calculated AAE from AERONET AAOD at 440 nm and 675 nm as:
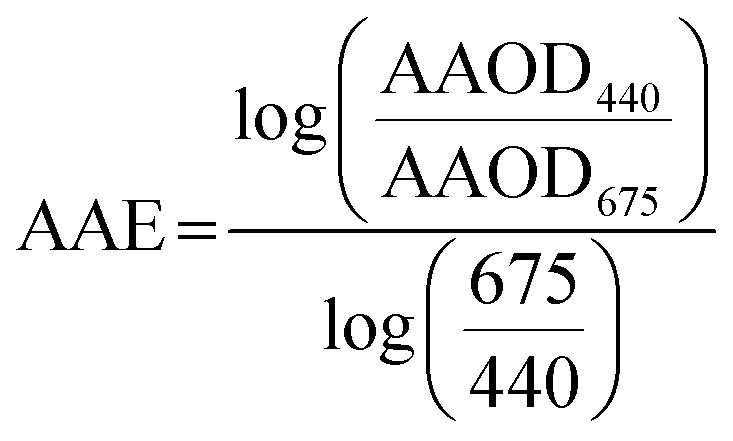 | (5) |
WRF-Chem optical properties are calculated at 300 nm, 400 nm, 600 nm, and 999 nm. Therefore, we converted WRF-Chem AAOD, AOD, and SSA values to 440 nm for comparison with AERONET observations. First, we calculated AAE and the extinction Ångström exponent (EAE) from WRF-Chem AAOD and AOD at 400 nm and 600 nm as:
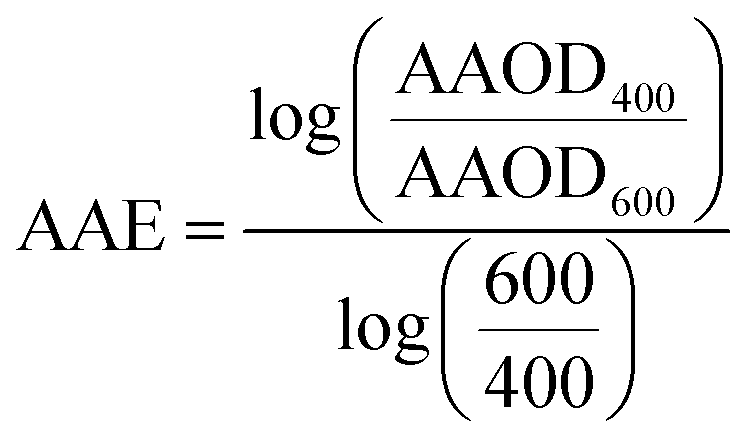 | (6) |
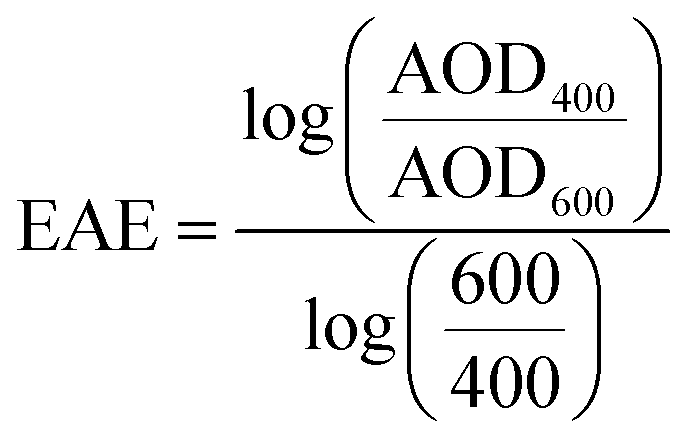 | (7) |
Then, we calculated AAOD and AOD at 440 nm as:
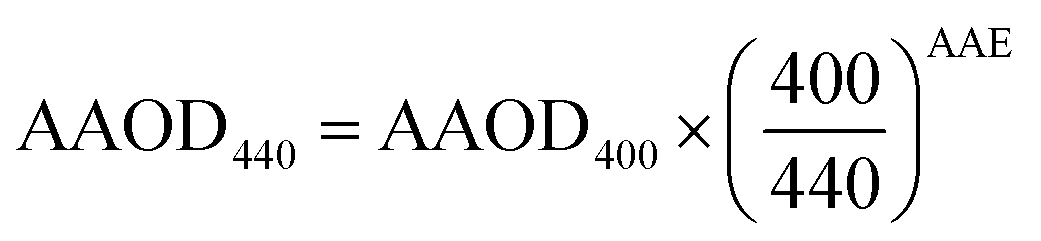 | (8) |
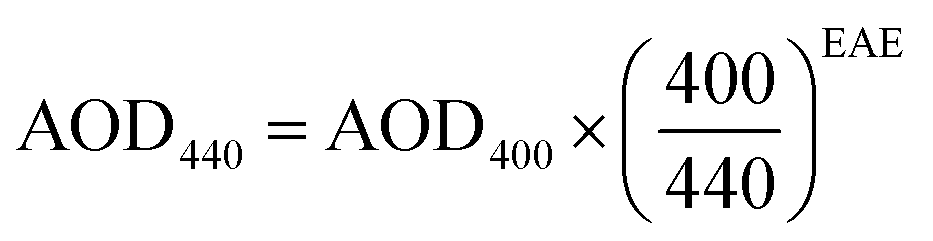 | (9) |
Model-observation comparison
The Base simulation
As shown in Fig. 2a, the slope of the linear fit to model AAOD versus AERONET AAOD at 440 nm is 0.49 for the Base simulation. The underestimation of aerosol absorption at 440 nm is larger than at 675 nm (slope = 0.62; Fig. S4a†), which is an indication of missing BrC absorption in the Base simulation. The effect of missing BrC absorption in the model is more evident in the AAE comparison (Fig. 4a). AERONET AAE values range between 0.8 and 1.4, which is consistent with variable contributions of BC and BrC to absorption, where smaller AAE values indicate BC-dominated absorption and larger AAE values indicate an increased contribution to absorption by BrC. On the other hand, the model AAE values exhibit a narrow range between 0.6 and 0.7 and no correlation with AERONET AAE because absorption in the Base simulation is solely dictated by BC. The AAE comparison in Fig. 4a indicates that missing BC absorption in the model cannot explain the underestimation of AAOD in the Base simulation because the Base simulation not only underestimates AAOD, but also its wavelength dependence. For instance, if one is to add BC to the model to reconcile the model and AERONET AAOD at 440 nm, that would lead to overestimating AAOD at 675 nm.The slope of model AOD versus AERONET AOD is 0.31 (Fig. 3a), which is smaller than the slope of the AAOD comparison. This indicates that in addition to the underestimation in aerosol absorption in the model, there is a more significant underestimation in scattering. As expected, because scattering is more underestimated on average than absorption in the model, a substantial fraction of model SSA values are smaller than AERONET SSA (Fig. 5a). The model SSA values are generally clustered in two groups, one in good agreement with AERONET and one lower than AERONET. This clustering can be understood by examining the spatial distribution of model and AERONET SSA in Fig. 6. The model SSA values are smallest in the Northwest, where wildfire CA is dominant, and increase toward the South and East, with the largest values along the Eastern region. The AERONET observations are mostly clustered in the Western region (where model SSA is smallest) and the Eastern region (where model SSA is the largest and closest to AERONET SSA), thus creating the two clusters in Fig. 5a. More importantly, comparing the spatial distributions of SSA (Fig. 6) and wildfire CA column burden (Fig. 1b) clearly indicates that the underestimation in model SSA (i.e. underestimation in aerosol scattering) is associated with the treatment of wildfire CA in the model. The missing wildfire CA scattering is further explored later in this section.
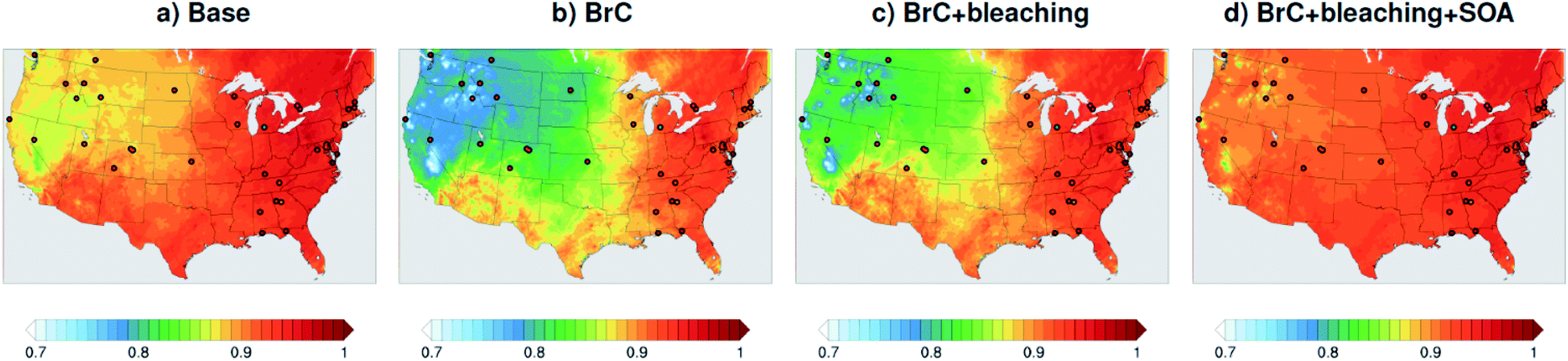 | ||
| Fig. 6 SSA at 440 nm averaged over the month of August 2015 from WRF-Chem output with four different model treatments of wildfire carbonaceous aerosol emissions. The colored dots show the locations and SSA values of AERONET stations used in the model-observation comparisons. | ||
Effect of incorporating brown carbon absorption
Model AAOD values in the BrC simulation are significantly larger than in the Base simulation. As shown in Fig. 2b, the slope of model AAOD versus AERONET AAOD for the BrC simulation is 1.23. The BrC simulation exhibits an improved model-observation AAOD comparison compared to the Base simulation (slope = 0.49; Fig. 2a), though incorporating BrC absorption leads to overestimating AAOD, on average. This is further evidenced in the AAE comparison. As shown in Fig. 4b, model AAE in the BrC simulation is better correlated with AERONET AAE compared to the Base simulation. However, almost all model AAE values are larger than AERONET AAE values. Overall, even though representing BrC based on the parameterization of Saleh et al.28 leads to overestimating aerosol absorption, it presents an improvement in the prediction of aerosol absorption compared to the Base simulation, where BrC is neglected.BrC absorption has a negligible effect on aerosol scattering and only affects the absorption component of aerosol extinction. Consequently, incorporating BrC absorption has only a small effect on the AOD model-observation comparison, as evident in comparing Fig. 3a and b. Because BrC absorption causes a substantial increase in absorption and negligible effect on scattering, this is reflected in a substantial decrease in SSA in the BrC simulation compared to the Base simulation. As shown in Fig. 5b, incorporating BrC absorption widens the gap between the two clusters described above and substantially worsens the SSA model-observation comparison.
As expected, accounting for the decay in BrC absorption due to photobleaching (BrC + bleaching simulation) leads to a reduction in both AAOD and AAE compared to the BrC simulation. As shown in Fig. 2c and 4c, the AAOD and AAE in the BrC + bleaching simulation exhibit very good agreement with AERONET observations. However, similar to the Base and BrC simulations, the BrC + bleaching simulation severely underestimates aerosol scattering as evident in the model-observation AOD comparison (Fig. 3c). Because incorporating photobleaching reduces BrC absorption, the SSA values in the BrC + bleaching simulation (Fig. 5c) are larger than in the BrC simulation (Fig. 5b) resulting in a slight improvement in the SSA model-observation comparison. However, SSA model-observation comparison in the BrC + bleaching simulation is still worse than the Base simulation (Fig. 5a).
To recap, neglecting BrC absorption (Base simulation) results in a significant underestimation of aerosol absorption as evidenced in the model-observation AAOD and AAE comparisons. Accounting for BrC absorption (BrC simulation) leads to overestimating AAOD and AAE, but results in a better comparison with observation. The best agreement in AAOD and AAE with observations is achieved when accounting for both BrC absorption and photobleaching (BrC + bleaching simulation). However, all simulations severely underestimate scattering compared to observations, as evident in the AOD comparisons. We note that the better model-observation agreement of SSA in the Base simulation compared to the BrC and BrC + bleaching simulations is rather serendipitous; it is due to the fact that the Base simulation underestimates both absorption and scattering.
The missing scattering
The model-observation comparisons employed in this study (Fig. 2, 3, 4, and 5) include both absorption and scattering, as well as extensive and pseudo-intensive properties. This comprehensive set of constraints enables dissecting the missing scattering problem described in the previous sub-section. For instance, the underestimation of AOD cannot be explained by an underestimation in wildfire CA emissions. Reconciling model and AERONET AODs would require increasing CA emissions by a factor of ∼3 (Fig. 3), which would lead to overestimating AAOD (Fig. 2). The underestimation of AOD cannot be explained by wrong BC/BrC or BrC optical properties either. Increasing the amount of BrC (or making BrC more absorbing and/or scattering) to reconcile the model and AERONET AOD would lead to either overestimating AAE, or AAOD, or both.A plausible explanation that satisfies the observational constraints is that the model largely underestimates SOA formation from wildfire emissions. Laboratory experiments58–61 and field measurements62–64 have reported SOA formation in wildfire plumes from the oxidation of vapor precursors as well as evaporation and subsequent oxidation of semi-volatile OA components,65 though to highly variable extents. We performed a simulation (BrC + bleaching + SOA) that involved a zeroth-order representation of wildfire SOA in addition to representing BrC and photobleaching. We treated SOA formation as direct emissions from wildfires alongside BrC (i.e. primary organic aerosol; POA) and BC. We assumed equal amounts of SOA and POA, which is within the range of values reported in the literature.61 For simplicity, we treated wildfire SOA as non-absorbing. Fig. 7a shows monthly average SOA column burdens over the model domain from the Base simulation. The relatively high levels of SOA over the Eastern U.S. are due to biogenic volatile organic compounds (VOCs) emissions, which are efficient SOA precursors. On the other hand, SOA formation from VOCs and other precursors (intermediate volatility and semi-volatile organic compounds)66 in wildfire emissions is severely underestimated in the model as evidenced by the low SOA levels in the Northwest incorporating BrC leads to a slight decrease in SOA concentrations (Fig. 6b and c) because BrC light absorption reduces the surface temperature slightly, which leads to a reduction in emission rates of biogenic VOCs.47 Representing wildfire SOA in the model (BrC + bleaching + SOA simulation) increases SOA column burden by about two orders of magnitudes over the wildfire regions in the Northwest (Fig. 7d).
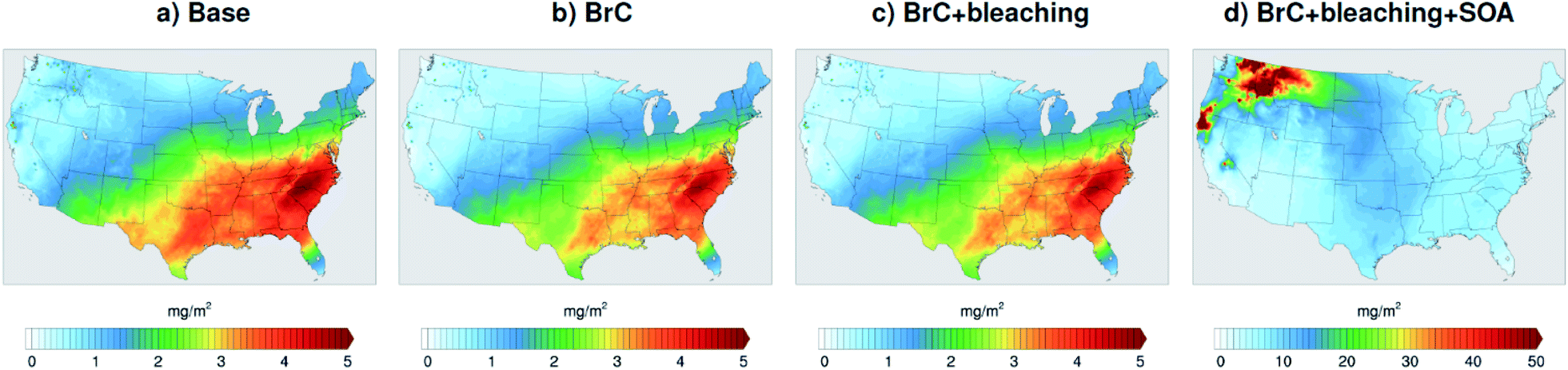 | ||
| Fig. 7 Column burden of SOA averaged over the month of August 2015 from WRF-Chem output with four different model treatments of wildfire carbonaceous aerosol emissions. | ||
Relative to the BrC + bleaching simulation, accounting for SOA formation in the BrC + bleaching + SOA simulation has a small effect on AAOD as shown in Fig. 2c and d (slope of AAOD comparison increases from 1.05 to 1.15) and AAE (Fig. 4c and d). However, accounting for SOA formation has a significant effect on AOD as shown in Fig. 3c and d (slope of AOD comparison increases from 0.35 to 0.60), thus largely reconciling the model SSA and AERONET SSA (Fig. 5d). Brown et al.8 performed a comprehensive SSA comparison between 9 global chemical transport/Earth system models and 12 observational data sets over globally distributed regions dominated by biomass-burning emissions. The study found that all models underestimated SSA compared to observations. Since those models did not include SOA formation from biomass-burning emissions,8 it is plausible that the underestimated scattering due to the missing SOA at least partly explains the inconsistency between the models and the observations.
The results obtained from the model-observation comparison performed in this study indicate that SOA formation from precursors in wildfire emissions is important and should be accounted for in chemical transport models.65,67 This SOA has implications not only to the radiative balance in the atmosphere, but also to air quality (public health). Including wildfire SOA in the model improved the comparison between surface-level OA concentrations predicted by the model and those obtained from IMPROVE, as evidenced by lower MFE of the BrC + bleaching + SOA simulation (ESI Fig. S2f†) compared to the Base simulation (ESI Fig. S2c†). We note that MFE of the model-CSN OA comparison was only slightly impacted by including wildfire SOA in the model. The reason is that IMPROVE stations (46 out of 118) are substantially more represented than CSN (14 out of 119) in the regions most impacted by wildfire emissions (ESI Fig. S1†). These results indicate that neglecting wildfire SOA can lead to underestimating aerosol surface concentrations, thus inhalation exposure, in regions impacted by wildfire emissions.
Conclusions
We employed a comprehensive set of optical properties retrieved from AERONET observations as complementary constraints for testing the representation of wildfire BrC in WRF-Chem. Specifically, the combination of AAOD (an extensive aerosol light-absorption property) and AAE (a pseudo-intensive light-absorption property that describes the wavelength dependence of absorption) comparisons showed that BrC absorption should be accounted for in order to reconcile model and observed absorption. One could match model and observed AAOD at a certain wavelength by scaling wildfire BC emissions, but that would lead to disagreement at other wavelengths if BrC absorption was ignored. Overall, the best model-observation agreement in terms of aerosol absorption was achieved by representing wildfire BrC absorption using the parameterization of Saleh et al.28 and its decay by photobleaching using the parameterization of Wang et al.35 However, in order to also reconcile model and observed AOD and SSA, we found that the model should account for relatively high levels of wildfire SOA (similar levels to wildfire POA). Neglecting wildfire SOA results in severely underestimating aerosol scattering. This finding indicates that modeling frameworks that do not account for wildfire SOA underestimate aerosol concentrations in regions impacted by wildfire emissions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Hunter Brown for the informative discussion on incorporating the brown carbon photobleaching scheme in WRF-Chem. We also thank the NASA's Aerosol Robotic Network (AERONET) for establishing and maintaining the sites used in this investigation. Numerical simulations were performed using the Georgia Advanced Computing Resource Center (GACRC) at the University of Georgia. Financial support was provided by the National Science Foundation, Division of Atmospheric and Geospace Sciences (AGS-1748080).References
- D. A. Jaffe, S. M. O’Neill, N. K. Larkin, A. L. Holder, D. L. Peterson, J. E. Halofsky and A. G. Rappold, J. Air Waste Manage. Assoc., 2020, 70(6), 583–615 CrossRef CAS PubMed.
- N. Fann, B. Alman, R. A. Broome, G. G. Morgan, F. H. Johnston, G. Pouliot and A. G. Rappold, Sci. Total Environ., 2018, 610–611, 802–809, DOI:10.1016/j.scitotenv.2017.08.024.
- A. E. Larsen, B. J. Reich, M. Ruminski and A. G. Rappold, J. Exposure Sci. Environ. Epidemiol., 2018, 28, 319–327 CrossRef CAS PubMed.
- I. N. Sokolik, A. J. Soja, P. J. DeMott and D. Winker, J. Geophys. Res.: Atmos., 2019, 124(23), 13005–13025, DOI:10.1029/2018JD029878.
- Core Writing Team, R. K. Pachauri and L. Meyer, Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Geneva, Switzerland, 2014 Search PubMed.
- J. J. O’Brien, J. K. Hiers, J. M. Varner, C. M. Hoffman, M. B. Dickinson, S. T. Michaletz, E. L. Loudermilk and B. W. Butler, Curr. For. Rep., 2018, 4(4), 161–177 Search PubMed.
- J. K. Hiers, J. J. O’Brien, J. M. Varner, B. W. Butler, M. Dickinson, J. Furman, M. Gallagher, D. Godwin, S. L. Goodrick, S. M. Hood, A. Hudak, L. N. Kobziar, R. Linn, E. L. Loudermilk, S. McCaffrey, K. Robertson, E. M. Rowell, N. Skowronski, A. C. Watts and K. M. Yedinak, Fire Ecol., 2020, 16, 11, DOI:10.1186/s42408-020-0070-8.
- H. Brown, X. Liu, R. Pokhrel, S. Murphy, Z. Lu, R. Saleh, T. Mielonen, H. Kokkola, T. Bergman, G. Myhre, R. B. Skeie, D. Watson-Paris, P. Stier, B. Johnson, N. Bellouin, M. Schulz, V. Vakkari, J. P. Beukes, P. G. van Zyl, S. Liu and D. Chand, Nat. Commun., 2021, 12, 277, DOI:10.1038/s41467-020-20482-9.
- M. D. Flannigan, M. A. Krawchuk, W. J. De Groot, B. M. Wotton and L. M. Gowman, Int. J. Wildland Fire, 2009, 18(5), 483–507 CrossRef.
- U.S. Environmental Protection Agency (EPA), Renewable Fuel Standard Program (RFS2) Regulatory Impact Analysis, 2010 Search PubMed.
- W. Knorr, F. Dentener, J. F. Lamarque, L. Jiang and A. Arneth, Atmos. Chem. Phys., 2017, 17(14), 9223–9236, DOI:10.5194/acp-17-9223-2017.
- C. D. McClure, C. Y. Lim, D. H. Hagan, J. H. Kroll and C. D. Cappa, Atmos. Chem. Phys., 2020, 20(3), 1531–1547, DOI:10.5194/acp-20-1531-2020.
- G. Adler, N. L. Wagner, K. D. Lamb, K. M. Manfred, J. P. Schwarz, A. Franchin, A. M. Middlebrook, R. A. Washenfelder, C. C. Womack, R. J. Yokelson and D. M. Murphy, Aerosol Sci. Technol., 2019, 53(9), 976–989, DOI:10.1080/02786826.2019.1617832.
- R. P. Pokhrel, N. L. Wagner, J. M. Langridge, D. A. Lack, T. Jayarathne, E. A. Stone, C. E. Stockwell, R. J. Yokelson and S. M. Murphy, Atmos. Chem. Phys., 2016, 16, 9549–9561 CrossRef CAS.
- H. Forrister, J. Liu, E. Scheuer, J. Dibb, L. Ziemba, K. L. Thornhill, B. Anderson, G. Diskin, A. E. Perring, J. P. Schwarz, P. Campuzano-Jost, D. A. Day, B. B. Palm, J. L. Jimenez, A. Nenes and R. J. Weber, Geophys. Res. Lett., 2015, 42(11), 4623–4630, DOI:10.1002/2015GL063897.
- Y. Zhang, H. Forrister, J. Liu, J. DIbb, B. Anderson, J. P. Schwarz, A. E. Perring, J. L. Jimenez, P. Campuzano-Jost, Y. Wang, A. Nenes and R. J. Weber, Nat. Geosci., 2017, 10, 486–489, DOI:10.1038/ngeo2960.
- J. Liu, E. Scheuer, J. Dibb, G. S. Diskin, L. D. Ziemba, K. L. Thornhill, B. E. Anderson, A. Wisthaler, T. Mikoviny, J. J. Devi, M. Bergin, A. E. Perring, M. Z. Markovic, J. P. Schwarz, P. Campuzano-Jost, D. A. Day, J. L. Jimenez and R. J. Weber, Atmos. Chem. Phys., 2015, 15(14), 7841–7858, DOI:10.5194/acp-15-7841-2015.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chem. Rev., 2015, 115(10), 4335–4382 CrossRef CAS PubMed.
- M. O. Andreae and A. Gelencsér, Atmos. Chem. Phys., 2006, 6(10), 3131–3148, DOI:10.5194/acp-6-3131-2006.
- R. Saleh, Curr. Pollut. Rep., 2020, 6, 90–104 CrossRef CAS.
- T. C. Bond, S. J. Doherty, D. W. Fahey, P. M. Forster, T. Berntsen, B. J. Deangelo, M. G. Flanner, S. Ghan, B. Kärcher, D. Koch, S. Kinne, Y. Kondo, P. K. Quinn, M. C. Sarofim, M. G. Schultz, M. Schulz, C. Venkataraman, H. Zhang, S. Zhang, N. Bellouin, S. K. Guttikunda, P. K. Hopke, M. Z. Jacobson, J. W. Kaiser, Z. Klimont, U. Lohmann, J. P. Schwarz, D. Shindell, T. Storelvmo, S. G. Warren and C. S. Zender, J. Geophys. Res.: Atmos., 2013, 118, 5380–5552 CAS.
- M. Z. Jacobson, Nature, 2001, 409, 695–697 CrossRef CAS PubMed.
- S. K. Akagi, R. J. Yokelson, C. Wiedinmyer, M. J. Alvarado, J. S. Reid, T. Karl, J. D. Crounse and P. O. Wennberg, Atmos. Chem. Phys., 2011, 11(9), 4039–4072, DOI:10.5194/acp-11-4039-2011.
- M. S. Hammer, R. V. Martin, A. Van Donkelaar, V. Buchard, O. Torres, D. A. Ridley and R. J. D. Spurr, Atmos. Chem. Phys., 2016, 16, 2507–2523 CrossRef CAS.
- D. S. Jo, R. J. Park, S. Lee, S. W. Kim and X. Zhang, Atmos. Chem. Phys., 2016, 16(5), 3413–3432, DOI:10.5194/acp-16-3413-2016.
- G. Lin, J. E. Penner, M. G. Flanner, S. Sillman, L. Xu and C. Zhou, J. Geophys. Res., 2014, 119, 7453–7476 Search PubMed.
- Z. Lu, D. G. Streets, E. Winijkul, F. Yan, Y. Chen, T. C. Bond, Y. Feng, M. K. Dubey, S. Liu, J. P. Pinto and G. R. Carmichael, Environ. Sci. Technol., 2015, 49, 4868–4877 CrossRef CAS PubMed.
- R. Saleh, E. S. Robinson, D. S. Tkacik, A. T. Ahern, S. Liu, A. C. Aiken, R. C. Sullivan, A. A. Presto, M. K. Dubey, R. J. Yokelson, N. M. Donahue and A. L. Robinson, Nat. Geosci., 2014, 7, 647–650 CrossRef CAS.
- R. Saleh, Z. Cheng and K. Atwi, Environ. Sci. Technol. Lett., 2018, 5(8), 508–513, DOI:10.1021/acs.estlett.8b00305.
- J. P. S. Wong, A. Nenes and R. J. Weber, Environ. Sci. Technol., 2017, 51(15), 8414–8421, DOI:10.1021/acs.est.7b01739.
- M. Zhong and M. Jang, Atmos. Chem. Phys., 2014, 14(3), 1517–1525, DOI:10.5194/acp-14-1517-2014.
- B. J. Sumlin, A. Pandey, M. J. Walker, R. S. Pattison, B. J. Williams and R. K. Chakrabarty, Environ. Sci. Technol. Lett., 2017, 4(12), 540–545, DOI:10.1021/acs.estlett.7b00393.
- R. Stevens and A. Dastoor, Atmosphere, 2019, 10(4), 168 CrossRef CAS.
- Y. Wang, M. Hu, P. Lin, Q. Guo, Z. Wu, M. Li, L. Zeng, Y. Song, L. Zeng, Y. Wu, S. Guo, X. Huang and L. He, Environ. Sci. Technol., 2017, 51(11), 5951–5961, DOI:10.1021/acs.est.7b00248.
- X. Wang, C. L. Heald, J. Liu, R. J. Weber, P. Campuzano-Jost, J. L. Jimenez, J. P. Schwarz and A. E. Perring, Atmos. Chem. Phys., 2018, 18(2), 635–653, DOI:10.5194/acp-18-635-2018.
- H. Brown, X. Liu, Y. Feng, Y. Jiang, M. Wu, Z. Lu, C. Wu, S. Murphy and R. Pokhrel, Atmos. Chem. Phys., 2018, 18(24), 17745–17768, DOI:10.5194/acp-18-17745-2018.
- N. A. June, X. Wang, L. W. A. Chen, J. C. Chow, J. G. Watson, X. Wang, B. H. Henderson, Y. Zheng and J. Mao, Geophys. Res. Lett., 2020, 47(23) DOI:10.1029/2020GL090332.
- G. A. Grell, S. E. Peckham, R. Schmitz, S. A. McKeen, G. Frost, W. C. Skamarock and B. Eder, Atmos. Environ., 2005, 39, 6957–6975 CrossRef CAS.
- National Centers for Environmental Prediction/National Weather Service/NOAA/U.S. Department of Commerce, NCEP FNL Operational Model Global Tropospheric Analyses, Continuing from July 1999. Research Data Archive at the National Center for Atmospheric Research, Computational and Information Systems Laboratory, DOI:10.5065/D6M043C6, accessed 5 June 2019.
- D. R. Stauffer, N. L. Seaman and F. S. Binkowski, Mon. Weather Rev., 2002, 119, 734–754 CrossRef.
- Technical Support Document (TSD) Preparation of Emissions Inventories for the Version 6.2, 2011 Emissions Modeling Platform, US Environmental Protection Agency, 2015 Search PubMed.
- N. K. Larkin, S. M. Raffuse, S. M. Huang, N. Pavlovic, P. Lahm and V. Rao, J. Air Waste Manage. Assoc., 2020, 70(11), 1165–1185, DOI:10.1080/10962247.2020.1802365.
- M. R. Houyoux and J. M. Vukovich, The Emission Inventory: Regional Strategies for the Future, 1999, pp. 1–11 Search PubMed.
- National Center for Atmospheric Research (NCAR), EPA_ANTHRO_EMIS tool, https://www.acom.ucar.edu/wrf-chem/download.shtml, accessed 6 June 2019 Search PubMed.
- J. Wang, C. Ge, Z. Yang, E. J. Hyer, J. S. Reid, B. N. Chew, M. Mahmud, Y. Zhang and M. Zhang, Atmos. Res., 2013, 122, 486–503, DOI:10.1016/j.atmosres.2012.05.009.
- L. K. Emmons, S. Walters, P. G. Hess, J. F. Lamarque, G. G. Pfister, D. Fillmore, C. Granier, A. Guenther, D. Kinnison, T. Laepple, J. Orlando, X. Tie, G. Tyndall, C. Wiedinmyer, S. L. Baughcum and S. Kloster, Geosci. Model Dev., 2010, 3, 43–67 CrossRef.
- A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer and C. Geron, Atmos. Chem. Phys., 2006, 6, 3181–3210 CrossRef CAS.
- R. A. Zaveri, R. C. Easter, J. D. Fast and L. K. Peters, J. Geophys. Res.: Atmos., 2008, 113(D13) DOI:10.1029/2007JD008782.
- N. M. Donahue, A. L. Robinson, C. O. Stanier and S. N. Pandis, Environ. Sci. Technol., 2006, 40, 2635–2643 CrossRef CAS PubMed.
- R. A. Zaveri, R. C. Easter and L. K. Peters, J. Geophys. Res.: Atmos., 2005, 110(D24) DOI:10.1029/2004JD005618.
- US Environmental Protection Agency, Air Quality System Data Mart [internet database], https://www.epa.gov/airdata, accessed 1 October 2019 Search PubMed.
- J. W. Boylan and A. G. Russell, Atmos. Environ., 2006, 40, 4946–4959 CrossRef CAS.
- J. C. Barnard, J. D. Fast, G. Paredes-Miranda, W. P. Arnott and A. Laskin, Atmos. Chem. Phys., 2010, 10, 7325–7340 CrossRef CAS.
- Z. Cheng, K. Atwi, O. El Hajj, I. Ijeli, D. Al Fischer, G. Smith and R. Saleh, Aerosol Sci. Technol., 2021, 55(1), 92–103, DOI:10.1080/02786826.2020.1820940.
- B. N. Holben, T. F. Eck, I. Slutsker, D. Tanré, J. P. Buis, A. Setzer, E. Vermote, J. A. Reagan, Y. J. Kaufman, T. Nakajima, F. Lavenu, I. Jankowiak and A. Smirnov, Remote Sens. Environ., 1998, 66(1), 1–16, DOI:10.1016/S0034-4257(98)00031-5.
- O. Dubovik and M. D. King, J. Geophys. Res.: Atmos., 2000, 105(D16), 20673–20696, DOI:10.1029/2000JD900282.
- B. N. Holben, T. F. Eck, I. Slutsker, A. Smirnov, A. Sinyuk, J. Schafer, D. Giles and O. Dubovik, in Remote Sensing of the Atmosphere and Clouds, 2006, vol. 6408 Search PubMed.
- A. P. Grieshop, N. M. Donahue and A. L. Robinson, Atmos. Chem. Phys., 2009, 9(6), 2227–2240, DOI:10.5194/acp-9-2227-2009.
- C. J. Hennigan, M. A. Miracolo, G. J. Engelhart, A. A. May, A. A. Presto, T. Lee, A. P. Sullivan, G. R. McMeeking, H. Coe, C. E. Wold, W. M. Hao, J. B. Gilman, W. C. Kuster, J. De Gouw, B. A. Schichtel, J. L. Collett, S. M. Kreidenweis and A. L. Robinson, Atmos. Chem. Phys., 2011, 11(15), 7669–7686, DOI:10.5194/acp-11-7669-2011.
- A. M. Ortega, D. A. Day, M. J. Cubison, W. H. Brune, D. Bon, J. A. De Gouw and J. L. Jimenez, Atmos. Chem. Phys., 2013, 13(22), 11551–11571, DOI:10.5194/acp-13-11551-2013.
- D. S. Tkacik, E. S. Robinson, A. Ahern, R. Saleh, C. Stockwell, P. Veres, I. J. Simpson, S. Meinardi, D. R. Blake, R. J. Yokelson, A. A. Presto, R. C. Sullivan, N. M. Donahue and A. L. Robinson, J. Geophys. Res., 2017, 122(11), 6043–6058, DOI:10.1002/2016JD025784.
- P. F. Decarlo, I. M. Ulbrich, J. Crounse, B. De Foy, E. J. Dunlea, A. C. Aiken, D. Knapp, A. J. Weinheimer, T. Campos, P. O. Wennberg and J. L. Jimenez, Atmos. Chem. Phys., 2010, 10(12), 5257–5280, DOI:10.5194/acp-10-5257-2010.
- M. J. Cubison, A. M. Ortega, P. L. Hayes, D. K. Farmer, D. Day, M. J. Lechner, W. H. Brune, E. Apel, G. S. Diskin, J. A. Fisher, H. E. Fuelberg, A. Hecobian, D. J. Knapp, T. Mikoviny, D. Riemer, G. W. Sachse, W. Sessions, R. J. Weber, A. J. Weinheimer, A. Wisthaler and J. L. Jimenez, Atmos. Chem. Phys., 2011, 11(23), 12049–12064, DOI:10.5194/acp-11-12049-2011.
- R. J. Yokelson, J. D. Crounse, P. F. DeCarlo, T. Karl, S. Urbanski, E. Atlas, T. Campos, Y. Shinozuka, V. Kapustin, A. D. Clarke, A. Weinheimer, D. J. Knapp, D. D. Montzka, J. Holloway, P. Weibring, F. Flocke, W. Zheng, D. Toohey, P. O. Wennberg, C. Wiedinmyer, L. Mauldin, A. Fried, D. Richter, J. Walega, J. L. Jimenez, K. Adachi, P. R. Buseck, S. R. Hall and R. Shetter, Atmos. Chem. Phys., 2009, 9(15), 5785–5812, DOI:10.5194/acp-9-5785-2009.
- B. B. Palm, Q. Peng, C. D. Fredrickson, B. H. Lee, L. A. Garofalo, M. A. Pothier, S. M. Kreidenweis, D. K. Farmer, R. P. Pokhrel, Y. Shen, S. M. Murphy, W. Permar, L. Hu, T. L. Campos, S. R. Hall, K. Ullmann, X. Zhang, F. Flocke, E. V. Fischer and J. A. Thornton, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(47), 29469–29477, DOI:10.1073/pnas.2012218117.
- A. P. Tsimpidi, V. A. Karydis, S. N. Pandis and J. Lelieveld, Atmos. Chem. Phys., 2016, 16(14), 8939–8962, DOI:10.5194/acp-16-8939-2016.
- M. Majdi, K. Sartelet, G. Maria Lanzafame, F. Couvidat, Y. Kim, M. Chrit and S. Turquety, Atmos. Chem. Phys., 2019, 19(8), 5543–5569, DOI:10.5194/acp-19-5543-2019.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ea00059d |
| This journal is © The Royal Society of Chemistry 2022 |