
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic machinery in motion: controlling catalysis via speed†
Emad
Elramadi‡
 ,
Amit
Ghosh‡
,
Isa
Valiyev
,
Pronay Kumar
Biswas
,
Thomas
Paululat
and
Michael
Schmittel
*
,
Amit
Ghosh‡
,
Isa
Valiyev
,
Pronay Kumar
Biswas
,
Thomas
Paululat
and
Michael
Schmittel
*
Center of Micro and Nanochemistry and (Bio)Technology, Organische Chemie I & II, Universität Siegen, Adolf-Reichwein-Str. 2, D-57068, Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de; Tel: +49(0) 2717404356
First published on 21st June 2022
Abstract
Three 3-component copper(I)-based slider-on-deck systems served as catalysts for a click reaction showing a higher catalytic activity with increasing sliding speed. Upon addition of brake stones, the motion of the resulting 4-component machinery was slowed and eventually stopped (on the NMR time scale) with the effect that catalysis was reduced or obstructed.
One of the prerequisites of life is adaptive regulation in living organisms, e.g., the up or down modulation of enzymatic activity inside the cell by multiple control variables.1 Contrastingly, the activity in manmade catalytic machinery2 so far is mostly regulated by binary (photo)chemical inputs leading to ON–OFF3 or UP–DOWN4,5 regulation. To reach deeper cybernetic control,6 regulation by more than one input is desirable.7
Previously, we have presented catalytic rotors and sliders where the catalytic activity was correlated with the motional speed.8,9 The faster the motion, the higher was the catalytic activity, however, this could only be shown by comparing different machinery. Herein, this correlation will be confirmed using single catalytic machinery with a reversibly changeable speed controlling the catalytic activity. For multistep toggling of the speed an external input will be applied capitalizing on coordination and constitutional dynamic chemistry (CDC).
In detail, the slider-on-deck systems [Cu3(1)(2)]3+ were prepared through self-sorting10 from the tris-shielded phenanthroline deck 1 and one of three bipeds, the bis-lutidine 2a, the bis-picoline 2b10 or the bis-pyridine biped 2c11 in presence of copper(I) ions (Fig. 1). The temporarily free Cu+ center in the slider-on-deck was expected to catalyse a 1,3-dipolar cycloaddition via click chemistry.12 Based on the architecture of the slider-on-deck, it was conjectured that added 2-pyridine carboxaldehyde (3) would bind at the free Cu+-loaded phenanthroline13 affording [Cu3(1)(2)(3)]3+. Further addition of 8-aminoquinoline (4) was supposed to lead to a reaction with 3 affording the terpyridine-analogue 5,14 the latter being expected to form a strong HETTAP15-type complex. Due to the different binding affinity of 3 and 5, the dynamics of the biped in [Cu3(1)(2a–c)(3 or 5)]3+ should be modulated affecting catalysis.
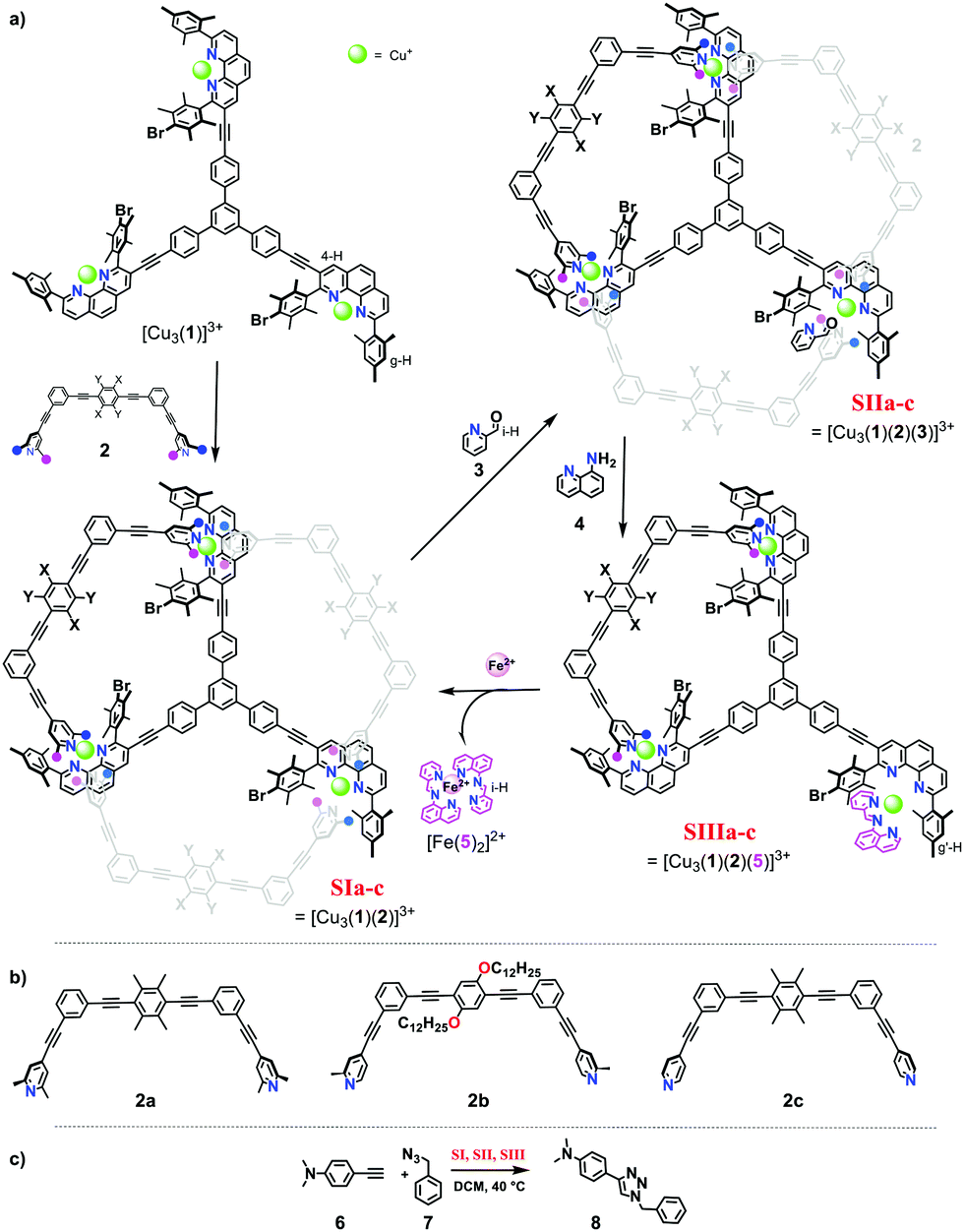 | ||
| Fig. 1 (a) A three-component slider-on-deck that is modulated upon addition of external stimuli (grey: biped in motion). (b) Molecular structure of biped 2a–c. (c) Model reaction catalysed by slider-on-deck systems. | ||
Firstly, both deck 1 and ligands 2a, b and c were synthesised by following analogous procedures. 2-Pyridine carboxaldehyde (3) and 8-amino quinoline (4) were commercially available.
When 1, 2, and copper(I) ions were mixed at rt in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 using CD2Cl2 as solvent, the slider-on-deck [Cu3(1)(2)]3+ formed both immediately and quantitatively. It was fully characterized by 1H NMR, 1H–1H COSY NMR, DOSY and mass spectroscopy. As anticipated, it showed a single set of signals for deck 1 and an upfield shift for the proton g-H signal of [Cu3(1)]3+ (Fig. 2) from 7.02 to 6.86, 6.87 and 7.03 ppm in SIa, SIb and SIc, respectively. In contrast, the proton signal 4-H showed a downfield shift from 8.84 to 8.87, 8.87 and 8.89 ppm in SIa, SIb and SIc, respectively. Upon binding of the lutidine units of 2a to the copper(I) phenanthroline stations the proton signals of b′-H shifted upfield by Δδ = 0.07 ppm. In case of bis-picoline biped 2b, the proton signals of d′, b′ and c′-H showed equally upfield shifts from 7.20, 7.27 and 8.50 ppm to 7.10, 7.17 and 7.54, respectively. Similarly, the signal groups of b′ and a′-H of bis-pyridine biped 2c shifted strongly upfield from 7.41 & 8.60 to 7.14 & 6.55 ppm, as shown in Fig. 2.
:1:3 using CD2Cl2 as solvent, the slider-on-deck [Cu3(1)(2)]3+ formed both immediately and quantitatively. It was fully characterized by 1H NMR, 1H–1H COSY NMR, DOSY and mass spectroscopy. As anticipated, it showed a single set of signals for deck 1 and an upfield shift for the proton g-H signal of [Cu3(1)]3+ (Fig. 2) from 7.02 to 6.86, 6.87 and 7.03 ppm in SIa, SIb and SIc, respectively. In contrast, the proton signal 4-H showed a downfield shift from 8.84 to 8.87, 8.87 and 8.89 ppm in SIa, SIb and SIc, respectively. Upon binding of the lutidine units of 2a to the copper(I) phenanthroline stations the proton signals of b′-H shifted upfield by Δδ = 0.07 ppm. In case of bis-picoline biped 2b, the proton signals of d′, b′ and c′-H showed equally upfield shifts from 7.20, 7.27 and 8.50 ppm to 7.10, 7.17 and 7.54, respectively. Similarly, the signal groups of b′ and a′-H of bis-pyridine biped 2c shifted strongly upfield from 7.41 & 8.60 to 7.14 & 6.55 ppm, as shown in Fig. 2.
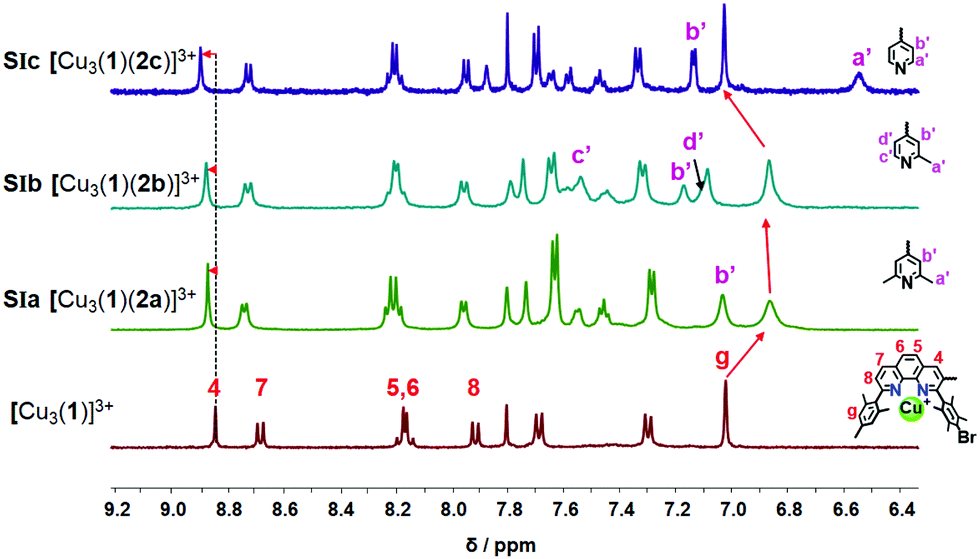 | ||
| Fig. 2 Comparison of partial 1H NMR spectra (CD2Cl2, 500 MHz, 298 K) of copper loaded deck [Cu3(1)]3+, SIa: [Cu3(1)(2a)]3+, SIb: [Cu3(1)(2b)]3+, SIc: [Cu3(1)(2c)]3+. For assignment, partial structures of 1 & 2a–c are given. | ||
To determine the sliding speed, the slider-on-deck assemblies [Cu3(1)(2)]3+ were studied by variable temperature (VT) 1H NMR spectroscopy (see ESI,† Fig. S54–S63). In case of SIa = [Cu3(1)(2a)]3+, the g-H proton peak of the deck showed coalescence at ca. −20 °C, whereas at −40 °C the signal split into two peaks at 6.78 and 6.73 ppm (ratio 2:1). The signal at 6.78 ppm was assigned to the lutidine-coordinated copper(I) phenanthroline, whereas the one at 6.73 ppm was attributed to the free copper-loaded phenanthroline. The exchange frequency in [Cu3(1)(2a)]3+ was determined as k298 = 2.4 kHz and the corresponding free activation energy as 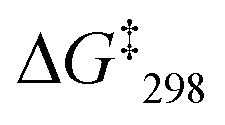 = 53.7 kJ mol−1. Similarly, the exchange frequency of [Cu3(1)(2b)]3+ was determined to k298 = 20 kHz and the corresponding free activation energy as
= 53.7 kJ mol−1. Similarly, the exchange frequency of [Cu3(1)(2b)]3+ was determined to k298 = 20 kHz and the corresponding free activation energy as 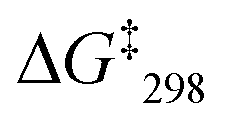 = 48.2 kJ mol−1.10 Finally, the VT 1H NMR spectrum of [Cu3(1)(2c)]3+ showed a singlet for the signal of g-H, which at −70 °C split into two signals at a ratio 2:1 at 6.95 and 6.98 ppm. The exchange frequency was determined as k298 = 42 kHz and the corresponding free activation energy
= 48.2 kJ mol−1.10 Finally, the VT 1H NMR spectrum of [Cu3(1)(2c)]3+ showed a singlet for the signal of g-H, which at −70 °C split into two signals at a ratio 2:1 at 6.95 and 6.98 ppm. The exchange frequency was determined as k298 = 42 kHz and the corresponding free activation energy 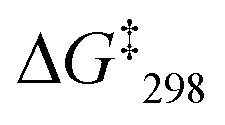 = 46.6 kJ mol−1.
= 46.6 kJ mol−1.
The binding of the pyridine head groups in bipeds 2a–c to deck 1 should vary as reflected by the association constants of pyridine, picoline and lutidine to [Cu(phenAr2)]+ that are logKpy = 3.20,16 logKpic = 3.43,17 and logKlu = 4.50.18 As expected, the sliding frequency declined with increasing binding affinity.
Upon addition of one equivalent of 2-pyridine carboxaldehyde (3) to a solution of [Cu3(1)(2)]3+ at rt, the four-component assembly [Cu3(1)(2)(3)]3+ formed instantly. A colour change from light yellow to deep red was noticed being characteristic for the complex motif [Cu(PhenAr2)(3)]+. Furthermore, in the 1H NMR, it showed only one set of signals for deck 1. The g-H proton peak in [Cu3(1)(2a)(3)]3+, [Cu3(1)(2b)(3)]3+ and [Cu3(1)(2c)(3)]3+ was broadened and shifted upfield to 6.80, 6.74, and 6.84 ppm, respectively, alike the aldehyde proton i-H signal that was shifted from 10.04 to 9.76, 9.65, and 9.61 ppm, respectively, as shown in Fig. 3.
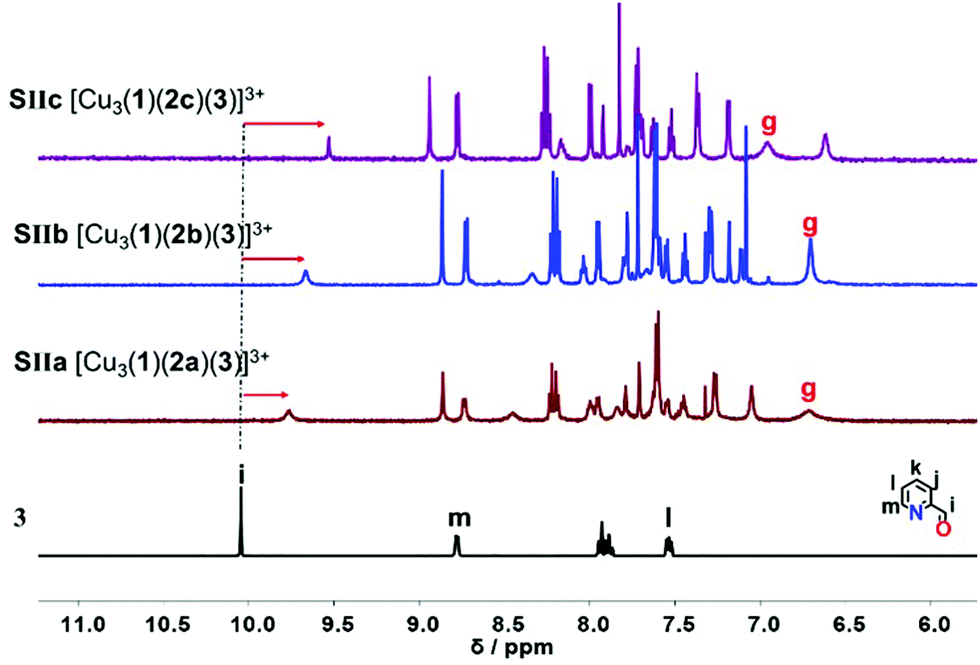 | ||
| Fig. 3 Comparison of partial 1HNMR spectra (CD2Cl2, 600 MHz, 298 K) of ligand 3, SIIa = [Cu3(1)(2a)(3)]3+, SIIb = [Cu3(1)(2b)(3)]3+, SIIc = [Cu3(1)(2c)(3)]3+. | ||
In the VT 1H NMR of [Cu3(1)(2a)(3)]3+, the signal of proton g-H coalesced at 10 °C and as the temperature reached −10 to −20 °C it split into two distinct signals at 6.78 and 6.54 ppm (ratio 2:1). The first signal was assigned to the lutidine-coordinated copper(I) phenanthroline while the signal at 6.54 ppm was attributed to the 2-pyridine carboxaldehyde-coordinated copper(I) phenanthroline unit. The exchange frequency and the free activation energy were determined to k298 = 1.6 kHz and of 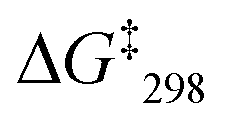 = 55.2 kJ mol−1. Thus, it shows slower sliding than [Cu3(1)(2a)]3+. The VT 1H NMR of [Cu3(1)(2b)(3)]3+ revealed splitting of the signal of proton 4-H at −25 °C into two distinct signals at 8.88 and 8.84 ppm (ratio 2:1) at −35 °C. The exchange frequency and the free activation energy were determined to k298 = 11 kHz and
= 55.2 kJ mol−1. Thus, it shows slower sliding than [Cu3(1)(2a)]3+. The VT 1H NMR of [Cu3(1)(2b)(3)]3+ revealed splitting of the signal of proton 4-H at −25 °C into two distinct signals at 8.88 and 8.84 ppm (ratio 2:1) at −35 °C. The exchange frequency and the free activation energy were determined to k298 = 11 kHz and 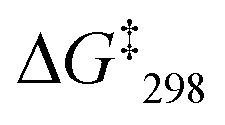 = 49.8 kJ mol−1. In the VT 1H NMR of [Cu3(1)(2c)(3)]3+ the signal of proton 4-H coalesced at −35 °C and was split into two distinct signals at 8.89 and 8.84 ppm (ratio 2:1) at −50 °C. The analysis provided an exchange frequency k298 = 26 kHz and
= 49.8 kJ mol−1. In the VT 1H NMR of [Cu3(1)(2c)(3)]3+ the signal of proton 4-H coalesced at −35 °C and was split into two distinct signals at 8.89 and 8.84 ppm (ratio 2:1) at −50 °C. The analysis provided an exchange frequency k298 = 26 kHz and 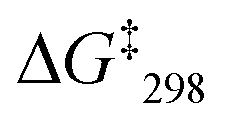 = 47.8 kJ mol−1 (see ESI,† Fig. S54–S63).
= 47.8 kJ mol−1 (see ESI,† Fig. S54–S63).
To investigate the catalytic activity of the three nanodevices [Cu3(1)(2a–c)]3+ in a click reaction, the reactants 6 and 7 (1:30:30) (the full mixture is denoted as States SIa–c) were mixed in CD2Cl2. After 10 h at 40 °C, the 1H NMR indicated a yield of 33%, 70% and 75% of 8, respectively. After addition of consumed amounts of 6 and 7 as well as of 2-pyridine carboxaldehyde (3) to form [Cu3(1)(2a–c)(3)]3+, setting up SIIa–c, the solution was heated again for 10 h at 40 °C. The yield of 8 increased by 13%, 8% and 5%. No yield was found in SIIIa–c (Fig. 4 and Table 1).
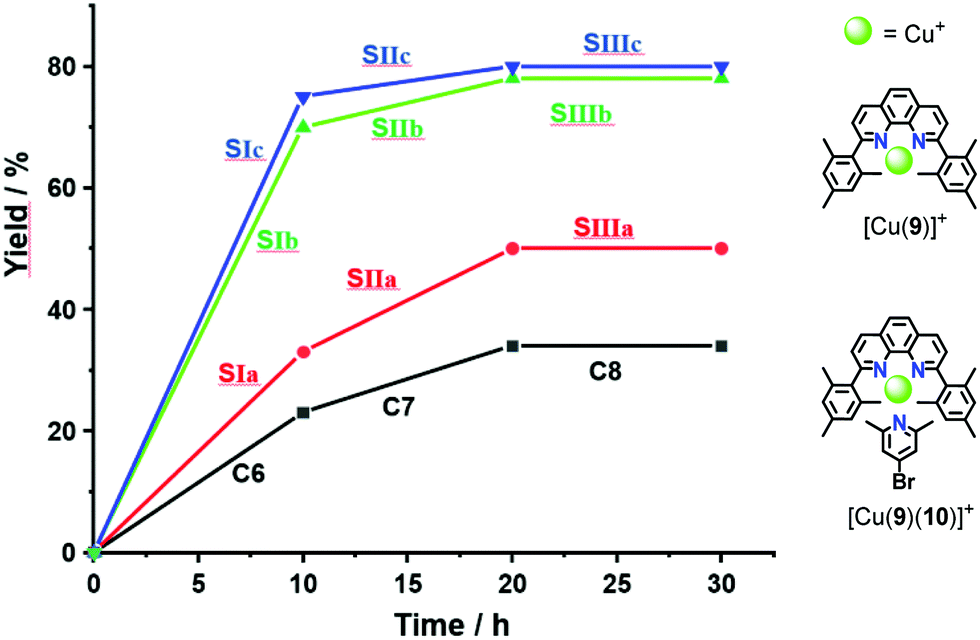 | ||
| Fig. 4 Yield of the click product 8 (from 6 and 7, each c = 1.20 × 10−2 M) in states SIa–c, SIIa–c (each c = 40.0 × 10−6 M) and with model complexes C6 = [Cu(9)]+, C7 = [Cu(9)(10)]+ (c = 1.20 × 10−3 M) after 10 h at 40 °C for each state. | ||
| State | Slider-on-deck | k 298 (kHz) |
|
Yield of 8 (%) |
|---|---|---|---|---|
| SIa | [Cu3(1)(2a)]3+ | 2.4 | 53.7 | 33 |
| SIb | [Cu3(1)(2b)]3+ | 20 | 48.2 | 70 |
| SIc | [Cu3(1)(2c)]3+ | 42 | 46.6 | 75 |
| SIIa | [Cu3(1)(2a)(3)]3+ | 1.6 | 55.2 | 13 |
| SIIb | [Cu3(1)(2b)(3)]3+ | 11 | 49.8 | 8 |
| SIIc | [Cu3(1)(2c)(3)]3+ | 26 | 47.8 | 5 |
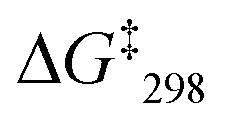
For deeper insight, the catalytic activity of the slider-on-deck needed to be assessed relative to that of model complexes. For instance, the yield of SIa (33% of 8) may be compared with that of C6 = [Cu(9)]+ + 2 × [Cu(9)(10)]+ (23% of 8) representing all binding sites in SIa. Analogously, the yield of SIIa (13% of 8) may be compared with that of C7 = 2 × [Cu(9)(10)]+ + [Cu(3)(9)]+ (11% of 8) this mixture embodying all binding sites in SIIa. In both cases, the catalytic activity of the slider-on-deck is higher. These examples suggest that both (a) thermodynamic and (b) kinetic aspects influence the catalytic activity: (a) dissociation of the complexes frees some of the copper(I) centres for catalysis. (b) On top, there are dynamic effects of motion in any slider-on-deck that liberate copper(I) centres by moving the biped foot to another location on the deck. The sliding motion may not only kick out the added ligand 3 in SIIa–c,7 but also bound product 8 in both SIa–c and SIIa–c.
As the sliding frequency increases in SIa–c, the click yield is higher. For instance, SIc furnished 75% of 8, while SIb afforded 70% and SIa only 33%. Clearly, the faster the sliding, the higher is the copper(I) availability due to kicking out the product and the higher is the catalytic activity. Yet, the binding strength of the biped (Npic > Npy) also plays a role in freeing 8 as otherwise the yield difference would be larger for SIb vs SIc.
On the other hand, considering the sliding speed, the situation looks opposite for SIIa–c. The slowest slider-on-deck in SIIa generated 13% of 8, while the faster ones in SIIb and SIIc afforded less, i.e., 8% and 5%, respectively. Using 1H NMR, we determined how much of 3 was liberated into solution in each of the slider-on-deck systems [Cu3(1)(2a)(3)]3+ (Fig. S74–75, Table S3 and S4, ESI,†). Accordingly, aldehyde 3 is being kicked out to a higher extent by the lutidine feet in [Cu3(1)(2a)(3)]3+ (47% of free 3) than in [Cu3(1)(2b)(3)]3+ (26% of free 3) and in [Cu3(1)(2c)(3)]3+ (19% of free 3). Here, the liberation seems to follow a thermodynamic motif: the stronger the binding of the biped the more of the brake may be liberated which is equivalent to temporarily freeing a copper(I) site for catalysis (Fig. 5).
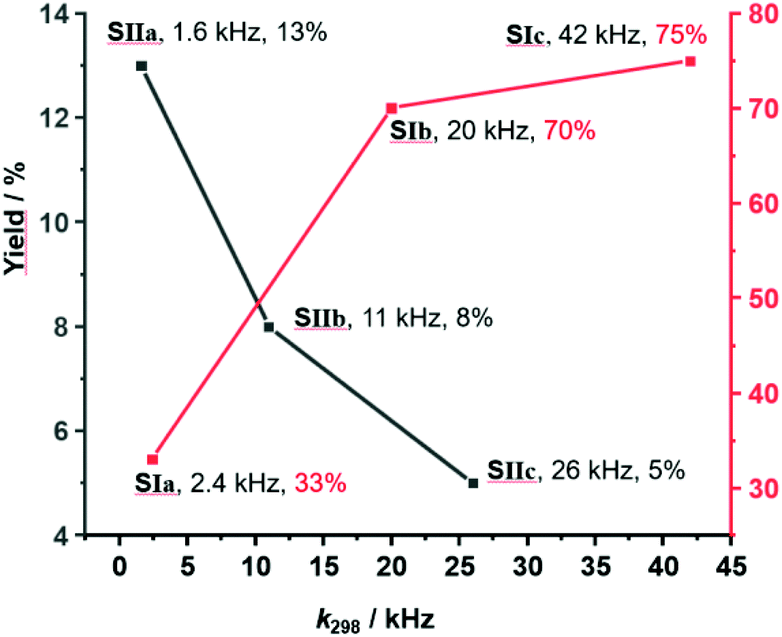 | ||
| Fig. 5 Representation of yield % vs. exchange frequency of SIa–c and SIIa–c showing an inverse relation. | ||
Finally, we chose catalyst [Cu3(1)(2a)(3)]3+ to evaluate its behaviour upon addition of one equiv. of 8-aminoquinoline (4), which caused the in situ formation of [Cu3(1)(2a)(5)]3+via imine bond formation. In the 1H NMR, the signal of mesityl protons g:g′:g′′-H showed three distinctive signals (ratio 4:1:1) (ESI,† Fig. S36). The larger peak at 6.80 ppm was attributed to the lutidine-coordinated copper phenanthroline moiety and the smaller signals at 6.33 and 6.18 ppm were assigned to the mesityl group of the copper phenanthroline bound to imine 5. The finding of two different mesityl g-H proton signals in one of the phenanthroline sites already indicated that the complex was not dynamic on the NMR timescale, a conclusion additionally supported by the EXSY analysis, because there was no cross peaks between g, g′-H proton signals (ESI,† Fig. S64).
Addition of 0.5 equiv. of iron(II) ions with respect to deck 1 enticed the imine away from the copper(I) phenanthroline into formation of the highly stable hexa-coordinated complex [Fe(5)2]2+. As a result, [Cu3(1)(2a)]3+ was regained and its dynamic motion reset.
Catalysis along SIa → SIIa → SIIIa → SIa (10 h at 40 °C) was first evaluated with deck 1, biped 2a and copper(I) ions (1:1:3) in presence of 6, 7 (10 equiv. each with respect to Cu+) via1H NMR analysis. Then, step-by-step, single inputs of 3, 4, and Fe2+ were added to furnish SIIa, SIIIa and SIa. The whole cycle was performed twice (Table 2). The starting state, SIa generated 33% of 8 in first cycle and 30% in the second cycle. Likewise, SIIa furnished 13% and 8%. The decreased yield in the 2nd cycle may be attributed to increased product inhibition. In contrast, in SIIIa, no catalytic activity was observed. As imine 5 blocks one of the copper(I) phenanthroline units, the biped 2a is unable to depart from the other two sites. By adding iron(II), SIa was regained and catalytic activity reignited (Table 2). Thus, a catalytic machinery is presented that changes its catalytic activity through “control and adaptability”.
In conclusion, by feeding the catalytic machinery with molecular brakestones, one can control both motional speed and catalytic activity in a stepwise and reversible manner from fully ON to OFF.
| State | k 298 (kHz) | Yield 1stcycle (%) | Yield 2ndcycle (%) |
|---|---|---|---|
| SIa = [Cu3(1)(2a)]3+ | 2.4 | 33 | 30 |
| SIIa = [Cu3(1)(2a)(3)]3+ | 1.6 | 13 | 10 |
| SIIIa = [Cu3(1)(2a)(5)]3+ | <10−4 | 0 | 0 |
We thank the DFG (Schm 647/20-2 and Schm 647/22-1, No 491092614) and the Univ. of Siegen for continued support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) E. Whitehead, The regulation of enzyme activity and allosteric transition, Prog. Biophys. Mol. Biol., 1970, 21, 321–397 CrossRef CAS PubMed; (b) G. G. Hammes, Multiple conformational changes in enzyme catalysis, Biochemistry, 2002, 41, 8221–8228 CrossRef CAS PubMed; (c) I. Zoi, J. Suarez, D. Antoniou, S. A. Cameron, V. L. Schramm and S. D. Schwartz, Modulating Enzyme Catalysis through Mutations Designed to Alter Rapid Protein Dynamics, J. Am. Chem. Soc., 2016, 138, 3403–3409 CrossRef CAS PubMed.
- (a) D. A. Leigh, V. Marcos and M. R. Wilson, Rotaxane Catalysts, ACS Catal., 2014, 4, 4490–4497 CrossRef CAS; (b) M. Schmittel, From self-sorted coordination libraries to networking nanoswitches for catalysis, Chem. Commun., 2015, 51, 14956–14968 RSC; (c) J. Choudhury, Recent developments on artificial switchable catalysis, Tetrahedron Lett., 2018, 59, 487–495 CrossRef CAS; (d) L. van Dijk, M. J. Tilby, R. Szpera, O. A. Smith, H. A. P. Bunce and S. P. Fletcher, Molecular machines for catalysis, Nat. Rev. Chem., 2018, 2, 117 CrossRef CAS.
- (a) M. Schmittel, S. De and S. Pramanik, Reversible ON/OFF nanoswitch for organocatalysis: mimicking the locking and unlocking operation of CaMKII, Angew. Chem., Int. Ed., 2012, 51, 3832–3836 CrossRef CAS PubMed; (b) V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, A Rotaxane-Based Switchable Organocatalyst, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed; (c) V. Blanco, D. A. Leigh, V. Marcos, J. A. Morales-Serna and A. L. Nussbaumer, A Switchable [2]Rotaxane Asymmetric Organocatalyst That Utilizes an Acyclic Chiral Secondary Amine, J. Am. Chem. Soc., 2014, 136, 4905–4908 CrossRef CAS PubMed; (d) S. Gaikwad, A. Goswami, S. De and M. Schmittel, A Metalloregulated Four-State Nanoswitch Controls Two-Step Sequential Catalysis in an Eleven-Component System, Angew. Chem., Int. Ed., 2016, 55, 10512–10517 CrossRef CAS PubMed.
- Some early key papers: (a) F. Würthner and J. Rebek, Light-Switchable Catalysis in Synthetic Receptors, Angew. Chem., Int. Ed. Engl., 1995, 34, 446–448 CrossRef; (b) R. Cacciapaglia, S. Di Stefano and L. Mandolini, The Bis-Barium Complex of a Butterfly Crown Ether as a Phototunable Supramolecular Catalyst, J. Am. Chem. Soc., 2003, 125, 2224–2227 CrossRef CAS PubMed; (c) J. Wang and B. L. Feringa, Dynamic control of chiral space in a catalytic asymmetric reaction using a molecular motor, Science, 2011, 331, 1429–1432 CrossRef CAS PubMed.
- (a) Some recent papers: P. Bora, S. Jakkampudi, R. Parella, N. Sakkani, Q. Dai, M. Bihani, H. D. Arman and J. C.-G. Zhao, Diastereodivergent synthesis of 4-oxocyclohexanecarbaldehydes by using the modularly designed organocatalysts upon switching on their iminium catalysis, Chem. Commun., 2021, 57, 5334–5337 RSC; (b) K. Nakamura, M. Kondo, C. G. Krishnan, S. Takizawa and H. Sasai, Azopyridine-based chiral oxazolines with rare-earth metals for photoswitchable catalysis, Chem. Commun., 2021, 57, 7414–7417 RSC.
- M. Schmittel and P. Howlader, Toward Molecular Cybernetics - the Art of Communicating Chemical Systems, Chem. Rec., 2021, 21, 523–543 CrossRef CAS PubMed.
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Systems chemistry, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- P. K. Biswas, S. Saha, T. Paululat and M. Schmittel, Rotating Catalysts Are Superior: Suppressing Product Inhibition by Anchimeric Assistance in Four-Component Catalytic Machinery, J. Am. Chem. Soc., 2018, 140, 9038–9041 CrossRef CAS PubMed.
- I. Paul, A. Goswami, N. Mittal and M. Schmittel, Catalytic Three-Component Machinery: Control of Catalytic Activity by Machine Speed, Angew. Chem., Int. Ed., 2018, 57, 354–358 CrossRef CAS PubMed.
- (a) Z. He, W. Jiang and C. A. Schalley, Integrative self-sorting: a versatile strategy for the construction of complex supramolecular architecture, Chem. Soc. Rev., 2015, 44, 779–789 RSC; (b) M. L. Saha and M. Schmittel, Degree of molecular self-sorting in multicomponent systems, Org. Biomol. Chem., 2012, 10, 4651–4684 RSC; (c) W. Jiang and C. A. Schalley, Integrative self-sorting is a programming language for high level self-assembly, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425–10429 CrossRef CAS PubMed.
- S. Saha, P. K. Biswas, I. Paul and M. Schmittel, Selective and reversible interconversion of nanosliders commanded by remote control via metal-ion signaling, Chem. Commun., 2019, 55, 14733–14736 RSC.
- A. Ghosh, I. Paul and M. Schmittel, Cooperative Effects in Switchable Catalysis, Angew. Chem., Int. Ed., 2021, 60, 20558–20562 CrossRef CAS PubMed.
- N. Mittal, I. Paul, S. Pramanik and M. Schmittel, Remote control of the reversible assembly/disassembly of supramolecular aggregates, Supramol. Chem., 2020, 32, 133–138 CrossRef CAS.
- S. Pramanik and I. Aprahamian, Hydrazone Switch-Based Negative Feedback Loop, J. Am. Chem. Soc., 2016, 138, 15142–15145 CrossRef CAS PubMed.
- M. L. Saha, S. Neogi and M. Schmittel, Dynamic heteroleptic metal-phenanthroline complexes: from structure to function, Dalton Trans., 2014, 43, 3815–3834 RSC.
- S. K. Samanta and M. Schmittel, Four-Component Supramolecular Nanorotors, J. Am. Chem. Soc., 2013, 135, 18794–18797 CrossRef CAS PubMed.
- N. Mittal, M. S. Özer and M. Schmittel, Four-Component Catalytic Machinery: Reversible Three-State Control of Organocatalysis by Walking Back and Forth on Track, Inorg. Chem., 2018, 57, 3579–3586 CrossRef CAS PubMed.
- S. Gaikwad, M. L. Saha, D. Samanta and M. Schmittel, Five-component trigonal nanoprism with six dynamic corners, Chem. Commun., 2017, 53, 8034–8037 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation, VT-NMR, spectral data. See DOI: https://doi.org/10.1039/d2cc02555h |
| ‡ Emad Elramadi and Amit Ghosh contributed equally |
| This journal is © The Royal Society of Chemistry 2022 |