
Enhanced emission by stacking of crown ether side chains in a 2D covalent organic framework†‡
Liu
Yuan§
ab,
Jun
Zhu§
a,
Shaofei
Wu
a and
Chunyan
Chi
 *a
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore, Singapore. E-mail: chmcc@nus.edu.sg
bState Key Laboratory of Electronic Thin Films and Integrated Devices, School of Optoelectronic Science and Engineering, University of Electronic Science and Technology of China, Chengdu 610054, China
First published on 20th December 2021
Abstract
Hydrazone compounds could be highly emissive in the solid state. However, most hydrazone-linking COFs are poorly luminescent. Here we report the enhancement of fluorescence in a 2D hydrazone-linking COF by side chain engineering. Stacking of the bulky and semi-rigid crown ether side chains restricts intramolecular rotation of the backbone around the hydrazone linkers, reducing the thermal dissipation and achieving 20-fold improved emission.
Photoluminescence (PL) covalent organic frameworks (COFs) are emerging porous crystalline materials showing specific sensing and detection abilities towards heavy metals,1–3 toxic gases,4 explosives,5,6 radicals,7 humidity,8 pH,9 bio-substances,10etc. Besides general applications found for porous materials,11–14 the light emitting property also endows them with potential applications in imaging, lasing and optoelectronics.15 The straightforward way to design highly photoluminescent COF materials is to incorporate strong emissive monomers. Despite occasional successes, this strategy fails in many cases mainly for two reasons. One is due to aggregation caused quenching induced by π–π stacking of monomers. The other one is available thermal dissipation pathways due to rotational labile linking. In the course of developing strong emissive COFs, methods like applying aggregation-induced emission (AIE) monomers,4 3D topology,16 avoiding eclipsed stacking,17 rigidifying linkers,18 “pinpoint surgery”19 and rotational restriction by hydrogen bonding20 have been used or discovered. These methods lead to synthesis of highly emissive, even white emissive COFs. Despite this profound progress, new strategies for improving the PL intensity of COF materials are still valuable.
Hydrazone-linking COFs, first reported by Uribe-Romo et al., could be highly crystalline and display excellent chemical and thermal stability.21 Due to these advantages, they are widely studied as a platform for a variety of applications.22–31 Hydrazone compounds are rotational-free and generally non-emissive in solution, but could be highly emissive in the solid state. This phenomenon is shown in Fig. S1 (ESI‡) for model compound MC2OMe. The emission in the aggregation state is due to restriction of intramolecular rotation (RIR) around single bonds. However, as a crystalline aggregation of hydrazone compounds by covalent linkages, most hydrazone COFs are weakly or non-emissive due to inefficient restriction of the bond rotation around hydrazone linkers. Besides, photo-induced electron transfer between the hydrazone linker and backbone moieties could also cause emission weakening.19 Li et al. developed highly emissive hydrazone COFs (PLQY up to 16.3%) by restriction of intramolecular bond rotation via intralayer and interlayer hydrogen bonds among highly organized layers.20 Jiang's group discovered that the emission of a hydrazone COF could be turned on by N–H bond deprotonation which eliminated the photo-induced electron transfer pathway.19
Here, we aim to enhance the PL intensity of hydrazone-linked COFs by side chain engineering. Four hydrazone COFs with the same skeleton but different side chains (R), as well as their model compounds, were synthesized (Scheme 1 and ESI‡). COF2OMe can represent the structural expansion and stacking of model compound MC2OMe. But unlike MC2OMe crystalline powder, this COF material is nearly non-emissive. When the two methoxyl groups were changed to bigger semi-rigid32 crown ether chains, the absolute PL quantum yield (QY) of the COFs improved, with larger crown ether chains leading to higher PLQY. A decent PLQY of 10.1% was found for COF21C7, close to its model compound MC21C7 (11.4%) in the crystalline state.
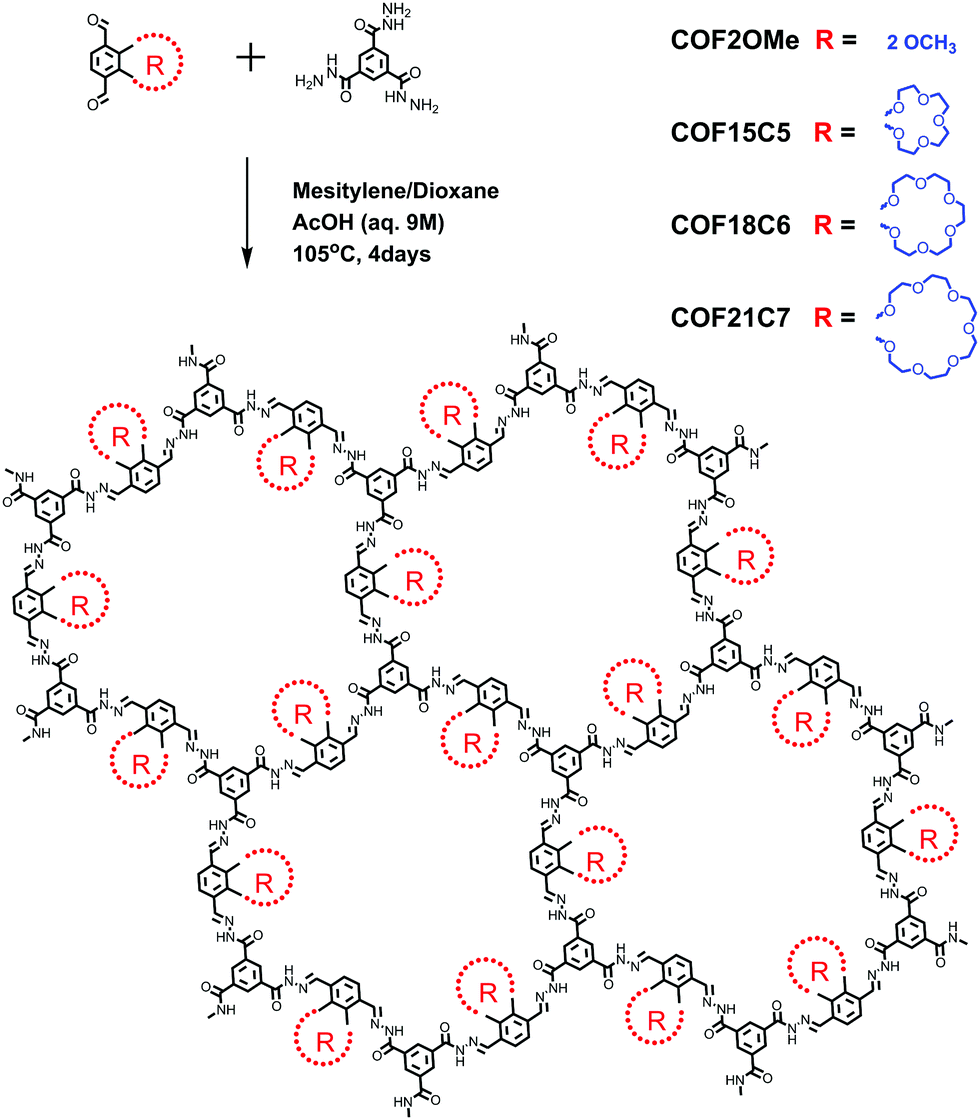 | ||
| Scheme 1 Synthesis and structure of model compounds and COF materials. | ||
All four COF materials have been successfully synthesized by imine condensation between benzene-1,3,5-tricarbohydrazide and the corresponding di-aldehyde under solvothermal conditions (see the ESI‡). Fourier transform infrared (FT-IR) spectra of the COFs, monomers and model compounds were obtained to confirm the formation of the COFs (Fig. S2–S5, ESI‡). For all of the COFs, their FT-IR spectra showed broad stretching vibration band of the imine bond (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) at 1650–1685 cm−1, which merged with the carbonyl (CO) stretch at 1652 cm−1 to form broad bands. Similar situation was also found for FT-IR spectra of other hydrazone COFs.22,27,28 This was further confirmed by their good agreement with the FT-IR spectra of the corresponding model compounds, which did not show the more commonly found (CN) stretch at ∼1610–1625 cm−1 either. Furthermore, the disappearance of the strong (N–H) stretching peak at 3300 cm−1 and aldehyde (CHO) stretching at ∼1700 cm−1, and the retention of (CON–H) stretching at 3205–3210 clearly demonstrates the consuming of monomers and the formation of the –CO–NH–NC– linking structure in the COFs. Bands at 2879–2950 cm−1 were assigned to the (OC–H) stretch of the side chains. And their relative intensity compared to low frequency bands increased as the COF's side chains grow bigger, in agreement with more –CH2– groups for larger crown ethers. To further confirm the chemical structure of these COFs, cross polarization magic angle spinning (CP-MAS) 13C NMR spectra of the solid samples were measured (Fig. S6, ESI‡). The peak at ∼157 ppm corresponding to CN linkages was observed for all COFs. Clear peaks were observed for COF2OMe, where the two signals at 164 ppm and 149 ppm were assigned to CO and side chain R-substituted benzene carbons, respectively, and the four peaks between 105 and 131 ppm were for the other aromatic carbons. For the crown ether COFs, peaks of CO carbon broadened and all peaks of crown ether dwelling benzene carbons shifted to lower fields. This might be caused by multiple conformations presented in the crown ether chains and backbone conformation “freezing” by tight stacking of these large semi-rigid cyclic groups. Elemental analysis reveals that the contents of carbon, hydrogen, and nitrogen of the COFs are close to the theoretically calculated values (ESI‡).
N–) at 1650–1685 cm−1, which merged with the carbonyl (CO) stretch at 1652 cm−1 to form broad bands. Similar situation was also found for FT-IR spectra of other hydrazone COFs.22,27,28 This was further confirmed by their good agreement with the FT-IR spectra of the corresponding model compounds, which did not show the more commonly found (CN) stretch at ∼1610–1625 cm−1 either. Furthermore, the disappearance of the strong (N–H) stretching peak at 3300 cm−1 and aldehyde (CHO) stretching at ∼1700 cm−1, and the retention of (CON–H) stretching at 3205–3210 clearly demonstrates the consuming of monomers and the formation of the –CO–NH–NC– linking structure in the COFs. Bands at 2879–2950 cm−1 were assigned to the (OC–H) stretch of the side chains. And their relative intensity compared to low frequency bands increased as the COF's side chains grow bigger, in agreement with more –CH2– groups for larger crown ethers. To further confirm the chemical structure of these COFs, cross polarization magic angle spinning (CP-MAS) 13C NMR spectra of the solid samples were measured (Fig. S6, ESI‡). The peak at ∼157 ppm corresponding to CN linkages was observed for all COFs. Clear peaks were observed for COF2OMe, where the two signals at 164 ppm and 149 ppm were assigned to CO and side chain R-substituted benzene carbons, respectively, and the four peaks between 105 and 131 ppm were for the other aromatic carbons. For the crown ether COFs, peaks of CO carbon broadened and all peaks of crown ether dwelling benzene carbons shifted to lower fields. This might be caused by multiple conformations presented in the crown ether chains and backbone conformation “freezing” by tight stacking of these large semi-rigid cyclic groups. Elemental analysis reveals that the contents of carbon, hydrogen, and nitrogen of the COFs are close to the theoretically calculated values (ESI‡).
Powder X-ray diffraction (PXRD) analyses were performed to determine the crystallinity of the COFs (Fig. 1). The experimental PXRD pattern of COF2OMe showed peaks at 3.60°, 6.15°, 7.14°, 9.34° and 26.3°, which could be assigned to the (100), (110), (200), (210) and (001) planes, respectively (Fig. 1a). The corresponding interlayer distance is 3.5 Å, calculated from the (001) diffraction. Very similar PXRD patterns were found for the other three COFs (Fig. 1c, e and g), but one more peak at around 10.5° was found in these materials, which was assigned to the (300) diffraction. All the (001) peaks were broad and located at very close positions, indicating similar interlayer packing distances around 3.5 Å. The sharp (100) diffraction peaks did show a decrease of angles from 3.60 to 3.53, 3.48 and 3.46 for COF2OMe, COF15C5, COF18C6 and COF21C7, respectively. Thus, the increase of the side chain size might have caused slight expansion of the backbone frames. To determine the crystal structure of the COFs, theoretical simulations were carried out to compare with the experimental ones. For all the COFs, the simulated PXRD patterns of the eclipsed structures (AA stacking) were in good agreement with the experimental PXRD patterns, either in peak positions or in relative intensities, while simulated staggered packing (AB stacking) showed deviations in relative intensities (Fig. 1a, c, e and g). Moreover, the Pawley refined PXRD profiles for AA stacking matched the experimental ones very well as evidenced by their negligible difference, with Rwp of 7.92%, 4.04%, 7.34%, 5.46% and Rp of 5.29%, 3.08%, 5.54%, 4.32% for COF2OMe, COF15C5, COF18C6 and COF21C7, respectively. It is worth noting that the AA stacking structure is generally found in hydrazone COFs. This eclipsed packing mode lays the foundation for restriction of backbone rotation by close staking.
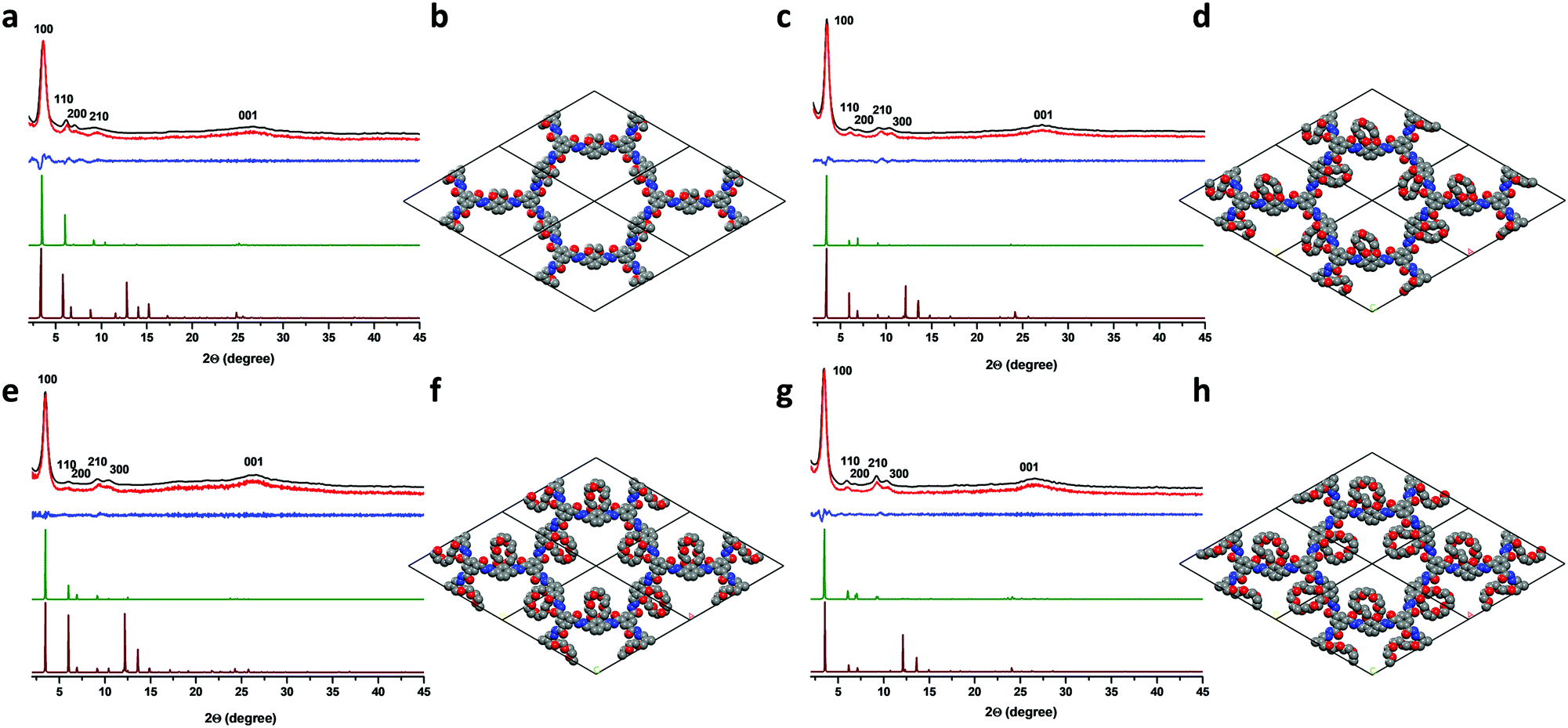 | ||
| Fig. 1 Powder XRD patterns of experimental (black), Pawley-refined (red), their difference (blue), simulated eclipsed (green) and staggered (brown) stacking for (a) COF2OMe, (c) COF15C5, (e) COF18C6 and (g) COF21C7. Simulated crystal structures of (b) COF2OMe, (d) COF15C5, (f) COF18C6 and (h) COF21C7. | ||
Though multiple diffraction peaks were observed for these COFs, their crystallinity was only at a moderate-to-low level. Full loading of finely-ground COF powders only gave a diffraction intensity of less than 4000 counts for all the (100) peaks, measured on a Bruker D8 Focus Powder X-ray diffractometer at 0.15° min−1 using Cu Kα radiation (40 kV, 40 mA). Besides, the crystallinity was also reflected by the low BET surface area for the COFs. Nitrogen adsorption–desorption measurements were carried out to investigate the porosity of these COF materials at 77 K, after activation by degassing the COF sample at 120 °C for 8 h. The sorption isotherms (Fig. S7, ESI‡) were all classified as type I nitrogen sorption isotherms according to the IUPAC classification. Though quite weak, a steep uptake of nitrogen gas at low pressure was observed for all COFs, indicating that the materials were microporous. Utilizing the Brunauer–Emmett–Teller (BET) model, low specific surface areas of 82, 18, 21 and 14 m2 g−1 were calculated for COF2OMe, COF15C5, COF18C6 and COF21C7, respectively. A nonlocal density functional theory (NLDFT) model was fitted to the isotherms of the COFs to estimate their pore size distributions. Only COF2OMe displayed a distinguishable pore size distribution profile centered at 2.63 nm, in good agreement with the expected pore sizes observed in the crystal simulations (Fig. S8, ESI‡). No centered pore size distributions were determined by NLDFT for the crown ether COFs. Despite oligomers that might reside inside the pores due to hydrogen-bonding interactions,26,33 bulk crown ethers occupied a large space in the pores, resulting in low BET surface area.
The morphology of the COFs was revealed by scanning electron microscopy (SEM) (Fig. S9, ESI‡). Nanoparticles and rods with non-uniform morphologies were observed for the COF2OMe. Nanoparticle (50–200 nm) aggregates were generally found for the other three COFs. The lack of regular and uniform shapes for these nanoparticles also indicates that these materials are not highly crystalline.
The luminescence property of these COF materials was investigated by solid-state fluorescence spectroscopy. When excited by 365 nm ultraviolet lamps, cyan to green luminescence with very similar PL bands centred at 503 nm was observed for all the COF powders (Fig. 2a). The luminescence brightness increased drastically as shown in the images (Fig. 2c inset). Thus, we measured the absolute PLQY of them in the solid state and the results were shown in Fig. 2c. COF2OMe exhibited a very low PLQY of 0.5%, which was slightly higher than the value of 0.4% for a hydrazone COF with very similar structure.20 Compared with COF2OMe, the latter three COFs exhibited successive increase of PLQY from 3.8% for COF15C5, to 5.5% for COF18C6, and finally to 10.1% for COF21C7, with a 20.2-fold improvement of PLQY from COF2OMe to COF21C7. In addition, a trend of band narrowing of the PL spectra from COF2OMe to the other three COFs was also observed (Fig. 2a), possibly due to more rotational freedom that leads to more energy relaxation pathways of the former, as depicted in Fig. 2d. The three crown ether COFs were tested for metal ion sensing, but no significant PL quenching was found for any metal ion (Fig. S14, ESI‡).
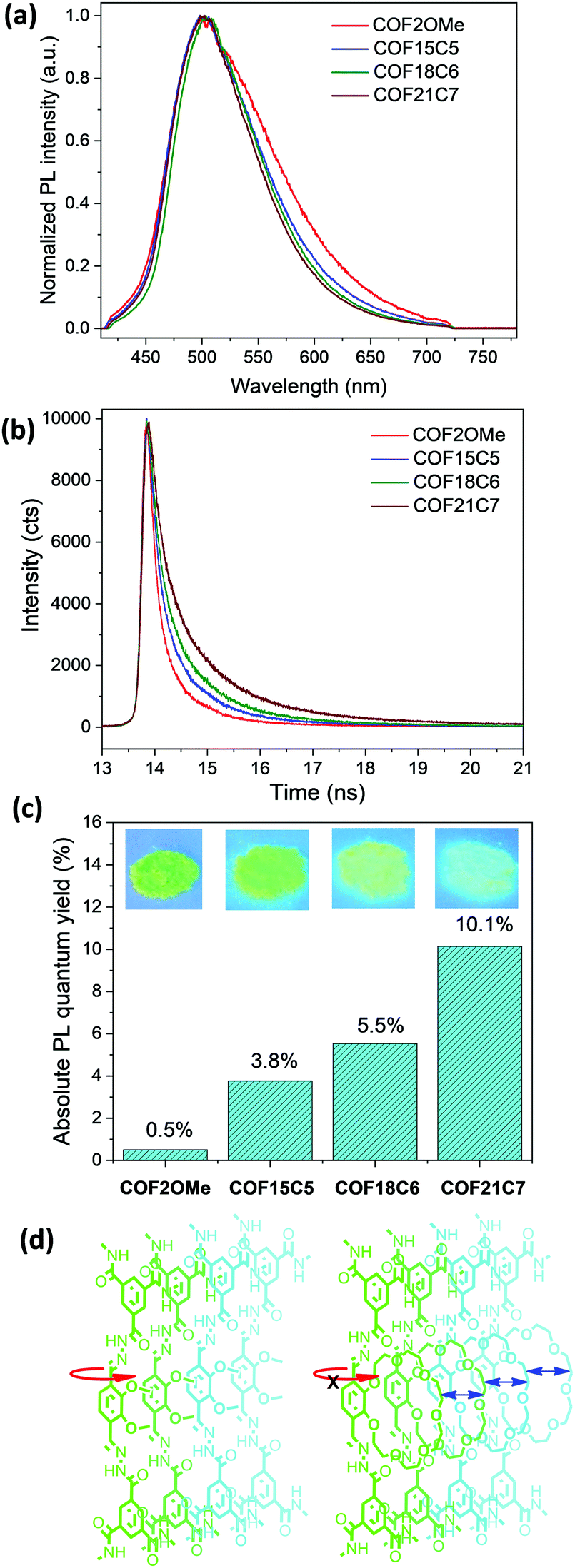 | ||
| Fig. 2 (a) Solid state photoluminescence spectra, (b) fluorescence decay curves, (c) absolute fluorescence quantum yield with inset fluorescence images of COF2OMe, COF15C5, COF18C6 and COF21C7 powders. (d) Schematic presentation of backbone rotation restriction by stacking of semi-rigid crown ether chains (red arrow indicates possible rotation mode, blue arrows represent repulsion force). | ||
Time-resolved fluorescence spectroscopy was performed to reveal the influence of chemical composition on the decay kinetics (Fig. 2b). Exponential fitting of the red curve indicated that COF2OMe had a PL lifetime of 0.33 ns. Upon substitution of the double methoxyl side chains by cyclic 15-crown-5, 18-crown-6, and 21-crown-7, the fluorescence lifetime of the COF materials increased to 0.44, 0.53, and 0.72 ns, respectively. This increment of PL lifetime proves the suppression of the intramolecular rotational energy relaxation pathway, due to reduced conformational freedom of hydrazone bridges by the stacking of bulky and semi-rigid crown ether chains.
In conclusion, we have explored side chain engineering as a strategy to improve the PL intensity of 2D hydrazone linked COFs. By decorating the COF pore walls with cyclic crown ether chains, the PLQY of the COF material is drastically increased by 20-fold from 0.5% to 10.1%. We ascribe the PL enhancement to the eclipsed stacking crystal structure that enables the close stacking of crown ether chains. The bulky and semi-rigid characteristics of the crown ether chains result in restriction of the backbone from easy rotation or vibration around the hydrazone linkers when closely packed. The strategy here may be extended to synthesizing fluorescent COF materials with desired functions by using specific bulky or rigid side groups.
We acknowledge the NRF-CRP grant (NRF-CRP16-2015-02), funded by the National Research Foundation, Prime Minister’s Office, Singapore. This work is also supported by MOE Tier 1 grant (R-143-000-B62-114) and Tier 2 grants (MOE2018-T2-1-152 and MOE-MOET2EP10120-0006).
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031 CrossRef CAS PubMed.
- Z. P. Li, Y. W. Zhang, H. Xia, Y. Mu and X. M. Liu, Chem. Commun., 2016, 52, 6613 RSC.
- D. M. Li, S. Y. Zhang, J. Y. Wan, W. Q. Zhang, Y. L. Yan, X. H. Tang, S. R. Zheng, S. L. Cai and W. G. Zhang, CrystEngComm, 2021, 23, 3594 RSC.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797 CrossRef CAS PubMed.
- W. Zhang, L. G. Qiu, Y. P. Yuan, A. J. Xie, Y. H. Shen and J. F. Zhu, J. Hazard. Mater., 2012, 221–222, 147 CrossRef CAS PubMed.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310 CrossRef CAS PubMed.
- W. Liu, Y. P. Cao, W. Z. Wang, D. Y. Gong, T. Cao, J. Qian, K. Iqbal, W. W. Qin and H. C. Guo, Chem. Commun., 2019, 55, 167 RSC.
- H. L. Qian, C. Dai, C. X. Yang and X. P. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 24999 CrossRef CAS PubMed.
- Y. Zhang, X. Shen, X. Feng, H. Xia, Y. Mu and X. Liu, Chem. Commun., 2016, 52, 11088 RSC.
- Y. Peng, Y. Huang, Y. Zhu, B. Chen, L. Wang, Z. Lai, Z. Zhang, M. Zhao, C. Tan, N. Yang, F. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698 CrossRef CAS PubMed.
- M. G. Mohamed, E. C. Atayde, B. M. Matsagar, J. Na, Y. Yamauchig, K. C. W. Wu and S. W. Kuo, J. Taiwan Inst. Chem. Eng., 2020, 112, 180 CrossRef CAS.
- L. Y. Zhang, J. H. Li, H. X. Zhang, Y. Liu, Y. M. Cui, F. C. Jin, K. Wang, G. Y. Liu, Y. L. Zhao and Y. F. Zeng, Chem. Commun., 2021, 57, 5558 RSC.
- M. G. Mohamed, M. Y. Tsai, C. F. Wang, C. F. Huang, M. Danko, L. Dai, T. Chen and S. W. Kuo, Polymers, 2021, 13, 221 CrossRef CAS PubMed.
- M. G. Mohamed, N. Y. Liu, A. F. M. El-Mahdy and S. W. Kuo, Microporous Mesoporous Mater., 2021, 311, 110695 CrossRef CAS.
- M. G. Mohamed, C. C. Lee, A. F. M. El-Mahdy, J. Luder, M. H. Yu, Z. Li, Z. Zhu, C. C. Chueh and S. W. Kuo, J. Mater. Chem. A, 2020, 8, 11448 RSC.
- G. Q. Lin, H. M. Ding, D. Q. Yuan, B. S. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302 CrossRef CAS PubMed.
- P. Albacete, J. I. Martínez, X. Li, A. Lo1E55ez-Moreno, S. Mena-Hernando, A. E. Platero-Prats, C. Montoro, K. P. Loh, E. M. Pérez and F. Zamora, J. Am. Chem. Soc., 2018, 140, 12922 CrossRef CAS PubMed.
- E. Q. Jin, J. Li, K. Y. Geng, Q. H. Jiang, H. Xu, Q. Xu and D. L. Jiang, Nat. Commun., 2018, 9, 4143 CrossRef PubMed.
- Z. P. Li, N. Huang, K. H. Lee, Y. Feng, S. S. Tao, Q. H. Jiang, Y. Nagao, S. Irle and D. L. Jiang, J. Am. Chem. Soc., 2018, 140, 12374 CrossRef CAS PubMed.
- X. Li, Q. Gao, J. Wang, Y. Chen, Z. H. Chen, H. S. Xu, W. Tang, K. Leng, G. H. Ning, J. Wu, Q. H. Xu, S. Y. Quek, Y. Lu and K. P. Loh, Nat. Commun., 2018, 9, 2335 CrossRef PubMed.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478 CrossRef CAS PubMed.
- G. Zhang, Y.-l. Hong, Y. Nishiyama, S. Bai, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2019, 141, 1227 CrossRef CAS PubMed.
- K. Gottschling, L. Stegbauer, G. Savasci, N. A. Prisco, Z. J. Berkson, C. Ochsenfeld, B. F. Chmelka and B. V. Lotsch, Chem. Mater., 2019, 31, 1946 CrossRef CAS PubMed.
- Z. Kang, Y. Peng, Y. Qian, D. Yuan, M. A. Addicoat, T. Heine, Z. Hu, L. Tee, Z. Guo and D. Zhao, Chem. Mater., 2016, 28, 1277 CrossRef CAS.
- W. J. Zhang, P. P. Jiang, Y. Wang, J. Zhang, Y. X. Gao and P. B. Zhang, RSC Adv., 2014, 4, 51544 RSC.
- G. Chen, H. H. Lan, S. L. Cai, B. Sun, X. L. Li, Z. H. He, S. R. Zheng, J. Fan, Y. Liu and W. G. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12830 CrossRef CAS PubMed.
- Y. Li, C. Wang, S. J. Ma, H. Y. Zhang, J. J. Ou, Y. M. Wei and M. L. Ye, ACS Appl. Mater. Interfaces, 2019, 11, 11706 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789 RSC.
- S. Mitra, H. S. Sasmall, T. Kundu, S. Kandambeth, K. Illath, D. D. Díaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513 CrossRef CAS PubMed.
- Y. Li, C. Wang, S. Ma, H. Zhang, J. Ou, Y. Wei and M. Ye, ACS Appl. Mater. Interfaces, 2019, 11, 11706 CrossRef CAS PubMed.
- W. J. Zhao, C. Y. Yu, F. Q. Chen,X. Y. Guan,H. Li, B. Tang, V. Valtchev, Y. S. Yan, S. L. Qiu and Q. R. Fang DOI:10.26434/chemrxiv.14157038.v1.
- M. G. Mohamed and S. W. Kuo, Macromolecules, 2020, 53, 2420 CrossRef CAS.
- X. R. Wang, X. Han, J. Zhang, X. W. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2016, 138, 12332 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Seth Marder on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures and structural characterization data; additional physical characterization data; calculation details. See DOI: 10.1039/d1cc03409j [ξ] Dedicated to Prof. Seth Marder on the occasion of his 60th birthday. |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |