
Evaluation of transfection efficacy, biodistribution, and toxicity of branched amphiphilic peptide capsules (BAPCs) associated with mRNA†
Nitish
Kunte
 a,
Matthew
Westerfield
a,
Erin
McGraw
a,
Jiyeong
Choi
b,
Tosin
Akinsipe
a,
Susan K.
Whitaker
c,
Andrew
Brannen
d,
Peter
Panizzi
e,
John M.
Tomich
b and
L. Adriana
Avila
*a
a,
Matthew
Westerfield
a,
Erin
McGraw
a,
Jiyeong
Choi
b,
Tosin
Akinsipe
a,
Susan K.
Whitaker
c,
Andrew
Brannen
d,
Peter
Panizzi
e,
John M.
Tomich
b and
L. Adriana
Avila
*a
aDepartment of Biological Sciences, Auburn University, Auburn, AL- 36849, USA. E-mail: adriana.avila@auburn.edu
bSchool of Integrative Plant Science, Cornell University, NY- 14853, USA
cDepartment of Biochemistry and Molecular Biophysics, 141 Chalmers Hall, Kansas State University, Manhattan, Kansas- 66506, USA
diThera-medical, Lexington, MA 02421, USA
eDepartment of Drug Discovery and Development, Harrison College of Pharmacy, Auburn University, Auburn, AL 36849, USA
First published on 13th October 2022
Abstract
Nanoparticles (NPs) have been shown to be a suitable mRNA delivery platform by conferring protection against ribonucleases and facilitating cellular uptake. Several NPs have succeeded in delivering mRNA intranasally, intratracheally, and intramuscularly in preclinical settings. However, intravenous mRNA delivery has been less explored. Only a few NPs have been tested for systemic delivery of mRNA, many of which are formulated with polyethylene glycol (PEG). The incorporation of PEG presents some tradeoffs that must be carefully considered when designing a systemic delivery model. For example, while the addition of PEG may prolong circulation time by preventing early clearance by the mononuclear phagocytic system (MPS), it has also been reported that treating patients with PEGylated drugs can result in hypersensitivity reactions due to anti-PEG antibodies. Thus, it is desirable to have alternative PEG-free delivery methods for mRNA to avoid these adverse effects while preserving the beneficial effects. Our research group developed BAPCs (branched amphiphilic peptide capsules), a peptide-based nanoparticle that resists disruption by chaotropes, proteases, and elevated temperature, thus displaying significant stability and shelf-life. In this study, we demonstrated that similarly to PEG, mRNA shields the BAPC cationic surface to avoid early clearance by the MPS. Multispectral optoacoustic tomography (MSOT) and fluorescence reflectance imaging were imaging techniques used to analyze biodistribution within major MPS organs. Analysis of pro-inflammatory cytokine expression showed that BAPC-mRNA complexes do not cause chronic inflammation. Additionally, BAPCs enhance intracellular delivery of mRNA with negligible cytotoxicity or oxidative stress. These results might pave the way for future therapeutic applications of BAPCs as a delivery platform for systemic mRNA delivery.
1. Introduction
Messenger RNA (mRNA) is emerging as a promising molecular tool to provide therapeutic proteins to specific tissues.1,2 Clear advantages of mRNA over DNA include regulated expression and lack of insertional mutagenesis. In addition, mRNA displays more sustainable and lasting effects compared to the direct delivery of therapeutic proteins.1,2 Intravenous mRNA delivery has been less explored than local delivery due to challenges associated with rapid mRNA degradation by serum nucleases and potential immune responses.3–5 Nanoparticles (NPs) have proven to be an efficient strategy to protect mRNA against ubiquitous endonucleases, reduce immune response, and also promote cellular internalization.3,6Nanomicelles, lipopolyplexes, and polymeric cationic NPs have been assessed for systemic delivery of mRNA showing some promising preliminary results. Nonetheless, all of these NPs contain polyethylene glycol (PEG) in their formulations.5–8 Incorporation of PEG is essential to improve the shelf-life stability and to avoid opsonization. Opsonized NPs are removed from the bloodstream within seconds via phagocytosis, thereby promoting sequestration by the mononuclear phagocytic system (MPS).9 Despite the versatility of PEG, recent studies have demonstrated that some individuals develop PEG-specific antibody response and hypersensitivity reactions, which translates to lower therapeutic responses to PEGylated NPs carrying mRNA therapeutics.10,11 Thus, it is desirable to explore alternative NPs-based delivery to overcome these shortcomings.
Our research team developed a peptide-based nanoparticle named BAPCs (branched amphipathic peptide capsules) that can be associated with nucleic acids such as mRNA to promote cellular internalization and prevent degradation by ribonucleases.12,13 BAPCs are formed through the assembly of two branched amphipathic peptides [bis(Ac-FLIVI)-K-KKKK-CO-NH2 and bis(Ac-FLIVIGSII)-K-KKKK-CO-NH2] mixed at an equimolar concentration in water. The BAPC-forming peptides contain a cationic C-terminus attached to branched hydrophobic segments, thus mimicking bilayer-forming phosphoglycerides in their overall architecture.13–17 BAPCs stand out in the crowded field of nanoparticle delivery systems owing to two crucial factors: (1) BAPCs do not require the use of surfactants such as PEG and (2) they resist disruption by chaotropes, anionic detergents, proteases, and elevated temperature (∼95 °C).18
Once formed, the BAPCs cationic surface interacts electrostatically with nucleic acids, acting as nucleation center around which nucleic acids wind.12,18 Published studies have demonstrated that the association of BAPCs with plasmid DNA led to the formation of BAPC-DNA complexes that enhanced cellular uptake of a green fluorescent protein (eGFP)-coding plasmid DNA in a variety of cell lines.15 Furthermore, BAPCs carrying a DNA vaccine against HPV-16 elicited immune response in tumor bearing mice after intramuscular administration. Animals vaccinated with the BAPC-DNA complexes showed tumor regression and improved survival rate.12
The main purpose of this article was to assess the biodistribution and toxicity of mRNA associated with BAPCs after intravenous injection in mice. In addition, we also evaluated the ability of BAPCs to deliver mRNA into HEK293T cells. The rationale behind evaluating these parameters is to evaluate the potential of BAPCs to serve as a systemic delivery platform for therapeutic mRNA. For in vitro delivery experiments, HEK293T cells were transfected with BAPC-Ova complexes formed by association of Ovalbumin mRNA (Ova-mRNA) with varying concentrations of BAPCs. Transfection efficiency was evaluated by assessment of Ova protein expression using western blot analysis. In addition, gene delivery efficiency was also evaluated using the reporter gene eGFP.19 According to our results, the association of mRNA on the BAPCs surface resulted in successful expression of both proteins. In vivo biodistribution and toxicity studies were performed in B6 albino mice. BAPC-mRNA complexes labeled with the infrared IRDye-800CW were administered intravenously and tracked 1 h, 3 h and 24 h following injection using Multispectral Optoacoustic Tomography (MSOT) or fluorescence reflectance imaging.20–22 The IRDye-800CW was selected due to the minimal absorption of near infrared light by biological tissues.23 The presence of pro-inflammatory cytokines was also evaluated in blood 1 h and 24 h after injection. Transmission electronic microscopy (TEM) and dynamic light scattering (DLS) indicated that, as previously observed for other nucleic acids, conjugation of mRNA on the BAPCs cationic surface led to the formation of ∼150 nm BAPC-mRNA complexes. Thus, instead of PEG, mRNA is adsorbed on the peptide nano-capsule surface, providing enhanced circulation time and longer shelf-life.
Vesicular structures composed entirely of pure peptides are relatively rare. Furthermore, only a few of them have been tested for nucleic acid delivery.24,25 According to our findings, association of mRNA to BAPCs surface serves several purposes: (1) to protect mRNA against enzyme degradation, (2) to avoid early sequestration by spleen and liver and (3) to enhance intracellular delivery of mRNA. Results from this article lay the foundation for future therapeutic applications in which systemic delivery of mRNA is required.
2. Materials and methods
2.1. Materials and reagents
2,2,2-Trifluoroethanol (TFE) (Tokyo Chemical Industry Ltd), IRDye-800CW (Li–Cor, Lincoln, NE, USA), 12-well plates (TPP tissue culture plates, Corning, Costar, NY, USA), DMEM (Corning Inc., NY, USA), HEK-293T cells (ATCC CRL-11268), Accutase (Innovative cell technologies, San Diego, CA, USA) pH 7.4 phosphate buffered saline (PBS) without Ca2+ and Mg2+ (VWR Radnor, PA, USA), ovalbumin (OVA) mRNA (TriLink BioTechnologies Inc., San Diego, CA, USA), enhanced Green Fluorescent Protein (eGFP) mRNA (TriLink BioTechnologies Inc., San Diego, CA, USA), ssRNA ladder (New England Biolabs, MA, USA), 10× MOPS buffer (Quality Biological, Gaithersburg, MD, USA), 37% formaldehyde (VWR Radnor, PA, USA), SYBR Green II Nucleic Acid Gel Stain (Rockville, MD, USA), agarose (JT Baker, Phillipsburg, NJ, USA), TEM carbon film copper grid (FCF300-Cu) (Electron Microscopy Sciences, Ft, Washington, PA, USA), Cell cytotoxicity assay kit (Abcam, Cambridge, MA, USA), EnzyChrom™ Aspartate and Alanine Transaminase assay kit (BioAssay Systems, Hayward, CA, USA), (BioAssay Systems, Hayward, CA, USA). ECL reagent (Promega, Madison, WI), Restore plus western blot stripping buffer (Thermo Scientific, Waltham, MA, USA), mouse monoclonal anti-ovalbumin antibody (MA5-15307, Invitrogen, Carlsbad, CA, USA), Mouse mono-clonal anti-β actin antibody (MA1-91399, Invitrogen, Carlsbad, CA, USA), HRP conjugated goat anti-mouse (405-306, Biolegend, San Diego, CA, USA). N-Methyl-2-pyrrolidinone (Advanced Chemtech, Louisville, KY), N,N-dimethylformamide (Fisher Scientific, Hampton, NH), trifluoroacetic acid (MilliporeSigma, St Louis, MO), triisopropylsilane (MilliporeSigma, St Louis, MO), dichloromethane (MilliporeSigma, St Louis, MO), 4-methyltrityl (MilliporeSigma, St Louis, MO), CellROX™ Deep Red Reagent (Thermo Scientific, Waltham, MA, USA), LEGENDplex™ cytokine assay kit (BioLegend, USA).2.2 Peptide synthesis
The peptides bis(Ac-FLIVI)-K-KKKK-CO-NH2 and bis(Ac-FLIVIGSII)-K-KKKK-CO-NH2, were synthesized and cleaved as previously described.18 The cleaved peptides were purified by reversed phase HPLC and characterized on a quadrupole orbitrap mass spectrometer (Orbitrap Explorus 120).13 Peptide isotope patterns were deconvoluted with the Xtract Deconvolution algorithm in FreeStyle 1.8 software (Thermo Fisher).2.3. Cell culture
HEK293T cells were maintained at 37 °C containing 5% CO2 and humidity. Cells were cultured in DMEM media supplemented with 10% (v/v) fetal bovine serum, 1× non-essential amino acids, and 1× penicillin and streptomycin (Complete media). Cell lines were passaged by treatment with Accutase every third to fourth day.2.4 BAPCs and BAPC-mRNA complexes preparation
The two branched peptides were dissolved separately in 2,2,2-trifluoroethanol (TFE) and peptide concentration was determined using the absorbance of phenylalanine at 257.5 nm. Upon determining concentrations, the peptides were mixed at equimolar ratios to achieve final concentration of 1000 μM, then dried using Centrivap Vacuum Concentrator (Labconco). BAPCs were formed by resuspending dried peptides in sterile dI H2O at 25 °C and allowed to stand for 10 minutes before solution was cooled to 4 °C for 1 h to control size. After the 4 °C incubation, 1 μg eGFP or ovalbumin mRNA was mixed with 40 μM, 80 μM and 120 μM of BAPCs to generate N/P ratios of 12.5, 25 and 37.5, respectively. The N/P ratio was calculated by diving the number positively charged amine groups in peptides by negatively charged phosphate groups on the mRNA backbone. Unless otherwise indicated, all subsequent experiments used OVA-mRNA complexed with BAPCs.2.5. IRDye-800CW labeled BAPCs
The IRDye-800CW was conjugated to the (ε)-amino of the C-terminal lysine of bis-(Ac-FLIVI)-K-KKKK-CO-NH2. Peptide synthesis was performed as previously described, with the exception of the (ε)-amino group of the C-terminal lysine containing a 4-methyltrityl (Mtt) protecting group, while the (ε)-amino of all other lysine residues was protected with tert-butyloxycarbonyl (Boc). After the peptide was fully synthesized, the resin was treated with 1% TFA in DCM with 5% TIS for 30 min. This low concentration of TFA selectively removes the Mtt protecting group, resulting in specific labeling. Then, the IRDye-800CW was dissolved in NMP and DMF and incubated with the peptide overnight. The IRDye-800CW adducted sequence was incorporated at the prescribed mole percentage along with the unlabeled bis(Ac-FLIVIGSII)-K-KKKK-CO-NH2.2.6 Dynamic light scattering (DLS) and zeta potential (ZP)
The BAPC-mRNA complexes with 12.5, 25, and 37.5 N/P ratios were prepared as previously described. Particle size and zeta potentials for all samples were determined on a Zetasizer Nano ZSP (Malvern Instruments Ltd, Westborough, MA). Samples were analyzed in dIH2O at 25 °C. Measurements were performed with n = 3 for ZP and n = 6 for DLS. To test the stability in serum containing media, DLS readings were performed in PBS containing 10% FBS.2.7 Transmission electron microscopy
BAPCs-mRNA complexes for TEM analysis were formed by mixing 80 μM BAPCs with 1 μg of Ova mRNA respectively (N/P = 25). Approximately 5 μL of the solution containing the BAPC-mRNA complexes was deposited on TEM grids for 10 min (FCF 300-Cu, Formvar® Electron Microscopy Sciences, Hatfield, PA, USA). To test the stability in serum containing media, complexes were formed, and then transferred into a solution containing 10% FBS before the deposition on the grid. Subsequently, the grid was quickly washed three times with dIH2O and negatively stained with a multi-isotope 2% uranyl acetate. Samples were dried for 2 h before analysis and images were acquired in TEM Tecnai Spirit T12 (Thermo-Fisher, formerly FEI).2.8 Gel retardation assay
The BAPC-mRNA complexes for electrophoresis analysis were formed at N/P ratio of 12.5, 18.75, 25, 37.5, 133. After 10 min of incubation at room temperature, all samples were mixed with RNA gel loading buffer containing SYBR green stain in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Then, samples were loaded on a 2% agarose gel containing 1× MOPS-formaldehyde buffer and electrophoresed at 100 V for 45 min. 1 μg of only mRNA and ssRNA ladder were included as controls. After electrophoresis, the mRNA bands were visualized using ImageQuant LAS 4000 (GE Healthcare, Pittsburgh, PA, USA).
:1 ratio. Then, samples were loaded on a 2% agarose gel containing 1× MOPS-formaldehyde buffer and electrophoresed at 100 V for 45 min. 1 μg of only mRNA and ssRNA ladder were included as controls. After electrophoresis, the mRNA bands were visualized using ImageQuant LAS 4000 (GE Healthcare, Pittsburgh, PA, USA).
2.9 In vitro toxicity and ROS quantitative assay
HEK293T cells were seeded in triplicate at 10000 cells per well in 96-well plate. The next day, media was replaced with fresh DMEM and treated with BAPC-mRNA complexes at 12.5, 25 and 37.5 N/P ratios along with their only BAPCs counterpart. According to the MTT cell cytotoxicity assay kit instructions (Abcam – ab112118), cells need to be incubated with Component A (cytotoxicity reagent) for 1–4 h after the treatment. However, in order to assess the short-term toxicity of BAPC complexes generated during the first 4 h treatment period, HEK293T cells were incubated with the BAPC formulations for 1 h, followed by the addition of Component A (without removing the treatments) and an additional 3 h incubation. This makes the total incubation time of cells with BAPCs-mRNA complexes 4 h. After this total incubation time, absorbance intensity was recorded at 570 nm and 605 nm, and percentage of cell viability was calculated according to the formula provided by the company: % cell viability = 100 × [(Abs (sample) − Abs (blank)/(Abs (control) − Abs (blank)]. For the ROS quantitative assay, HEK293T cells we treated as described above with BAPC-mRNA complexes. ROS production was measured using the cellROX Oxidative stress kit, following company instructions. Briefly, after treatment with the BAPC-mRNA formulations, cells were incubated with 5 μM cellROX reagent for 30 min. Then, the reagent was washed three times with PBS and fluorescence was measured using Cytation3 fluorometer (BioTek Instruments, Inc., VT, USA) at excitation/emission wavelength of 640/665 nm.
2.10 In vitro mRNA delivery
For transfection experiments, HEK293T cells were seeded at 200000 cells per mL in 12-wall-plate. Upon overnight incubation, media was replaced with fresh media containing BAPC-mRNA complexes of varying N/P ratios (12.5, 25, and 37.5). Then, cells were incubated at 37 °C for 4 h with the complexes before replacing with fresh DMEM media. After 24 h, Ova expression was analyzed using western blotting. For western blotting, transfected cells were detached and washed twice with 1 mL of PBS. Protein lysate was prepared by heating the cell pellet mixed with 2× laemmli buffer at 95 °C for 10 min and immediately placed on ice for 5 min. Cell debris was removed by centrifugation at 15000 rpm for 15 min. An equal amount of protein lysate was resolved using 10% (w/v) polyacrylamide Tris-glycine SDS gels. Proteins were transferred using wet transfer unit (Bio-Rad, Hercules, CA, USA) at 100 V for 1.5 h to PVDF membranes. Membrane was then blocked with blocking buffer (5% non-fat dry milk in Tris-buffered saline/0.1% Tween 20) for 1 h at room temperature and then incubated overnight at 4 °C with a mouse monoclonal anti-ovalbumin antibody diluted at 1:1000 in 4 mL of blocking buffer. The membrane was washed thrice with TBST and incubated with HRP conjugated goat anti-mouse antibody diluted 1:2000 in blocking buffer for 1 h at room temperature. The membrane was again washed three times with TBST, and the ovalbumin protein was detected using ECL reagent and ImageQuant LAS 4000 (GE Healthcare, Pittsburgh, PA, USA). The antibody labeling from the membrane was removed using Restore plus western blot stripping buffer and membrane was reprobed with a mouse mono-clonal anti-β actin antibody (1:2000) and HRP conjugated goat anti-mouse (1:2000) antibody as per above mentioned protocol.
Similarly, HEK293T cells were transfected with BAPC-(eGFP) mRNA complexes of same N/P ratios. Transfection efficiency of HEK293T cells treated with BAPC-eGFPmRNA complexes was monitored by fluorescence microscopy and quantified by flow cytometry (MACSQuant® Analyzer 10). Cell transfected with mRNA jetMessenger® (Polyplus, NY, USA) were used as a positive control. For microscopy analysis, cells were fixed using 4% paraformaldehyde, mounted on glass slides using anti-fade mounting media and imaged under FITC channel of fluorescence microscope (Olympus IX73 fluorescent microscope with a DP74 camera (Tokyo, Japan). For flow cytometry analysis, cells were harvested, washed, and stained with 7-aminoactinomycin D (7-AAD) to exclude dead cells and GFP expression was analyzed using FITC channel. Data was processed using FlowLogic™ software.
2.11 Mice
Female B6-albino mice at 6–8 weeks of age were supplied by Jackson Laboratories All procedures involving animal handling and treatment were approved by Auburn University's Institutional Animal Care and Use Committee (IACUC- 2020-3656). MSOT imaging and toxicity studies were performed on healthy female B6 Albino mice aged 8–10 weeks.2.12 Determination of in vivo biodistribution using MSOT imaging
Healthy B6-albino mice (2 mice for each formulation) were shaved to reduce background signal from keratin. Animals were anesthetized using isoflurane vaporizer set at 1–2% with continuous supply of room air at a flow rate of 1 L min−1. Following, BAPC-mRNA complexes of 25 N/P ratio (1600 μM of BAPCs mixed with 20 μg eGFP-mRNA) were injected using a PE10 tail vein catheter. After 1, 3 or 24 h post-injection, animals were placed into a small animal holder that was connected to isoflurane vaporizer. Generous amount of ultrasound gel was applied evenly on the skin and animal along with holder was placed inside the MSOT water bath with the ventral side of the mouse facing upwards and the dorsal side facing the center of the 270-degree transducer array. Breathing rate of all animals was monitored throughout the imaging process via an integrated webcam in the MSOT imaging chamber. Illumination wavelengths were set up at 700, 730, 760, 778, 800, 850, and 875 nm, with 10 averaged frames per wavelength. Pre-scan images were acquired and served as a control for direct comparison to MSOT images post-injection. MSOT images were reconstructed using a back-projection algorithm, followed by spectral unmixing using linear regression. Wavelengths acquired included those necessary to spectrally unmix oxyhemoglobin, deoxyhemoglobin, and IRDye® 800CW to determine the contribution of each component to the overall optoacoustic signal. Optoacoustic images were obtained using an MSOT inVision 256-TF small animal imaging system (iThera Medical GmbH, Munich, Germany). A tunable optical parametric oscillator pumped by an Nd:YAG laser provided excitation pulses (9 ns duration) at wavelengths ranging from 680 nm to 980 nm at a repetition rate of 10 Hz, a wavelength tuning speed of 10 ms and a peak pulse energy of 100 mJ at 730 nm. Ten arms of a fiber bundle provided even illumination of a ring-shaped light strip of approximately 8 mm width. For ultrasound detection, 256 toroidally focused ultrasound transducers, arranged in a concave array (270° angular coverage) with a 4 cm radius of curvature, and with a center frequency of 5 MHz (60% bandwidth), were used. Reconstruction, spectral unmixing, fluence correction, and quantification was performed using viewMSOT v4.0.3.2.2.13. Fluorescence reflectance imaging
After completion of MSOT imaging, all the mice were euthanized (1, 3 or 24 h post injection) via cardiac puncture and cervical dislocation while the animals were still under anesthesia. The heart, lungs spleen, liver and kidneys were collected and fluorescence intensity of IRDye-800CW was assessed using IVIS Lumina XRMS system (Caliper Life Sciences, Waltham, MA, USA). Images were acquired at excitation and emission wavelengths of 745 nm and 800 nm respectively with exposure time 1 s. Fluorescence levels of the organs was quantified using ImageJ software. Region of interest around each organ was manually selected upon background subtraction. This procedure was performed for each image slice where organ fluorescence was visible. The average intensity of each ROI was calculated by following formula: volume average intensity = integrated density − (area of selected region × mean fluorescence of background readings).2.14 Multiplex cytokine assay
Mice subjected to MSOT analysis were euthanized as described previously, and blood (∼500 μL) was collected 1 h post injection via cardiac puncture. Serum was separated from blood by centrifugation at 2000g for 10 min at 4 °C within 2 h after collection. Subsequently, serum samples were diluted 1:1 with PBS. Inflammatory cytokine response in mice was assessed by quantifying the levels of IL-1β, IFN-γ, TNF-α and IL6 in the serum samples using LEGENDplex™ multiplex cytokine assay kit (BioLegend, USA). Samples were processed using flow cytometry (MACSQuant® Analyzer 10) and data was analyzed with the FlowLogic™ software.
3. Results and discussion
3.1 Biophysical characterization of the BAPC-mRNA complexes
To assemble BAPCs, the two peptides, bis(Ac-FLIVIGSII)-K-KKKK-CO-NH2 and bis(Ac-FLIVI)-K-KKKK-CO-NH2, were mixed at equimolar concentration in 2,2,2, trifluoroethanol (TFE). In this solvent, both peptides are in a monomeric state, and do not aggregate. Once combined, the solvent is removed under vacuum, and water is added to induce nanocapsule formation. TEM was used to confirm the BAPCs vesicular structure after this assemble process. As shown in Fig. 1A, BAPCs form spherical capsules with a size ranging from 20 to 50 nm. To control the size, rehydrated peptide is incubated at 25 °C for 10 min until capsules reach a size of ∼50 nm, followed by incubation at 4 °C for 1 h, and then rewarmed to 25 °C.26 As previously reported, this temperature-shift treatment induces a locked-in conformation, thus preventing capsule fusion.16 Previous TEM analyses have confirmed a bilayer formation within this type of capsule structure, where the hydrophobic peptide branches reside within the bilayer while lysine residues remain exposed to solvent interface and provide a net positive surface charge which facilitates the interaction with mRNA.16Fig. 1B corroborates that the BAPCs surface appears to be uniformly coated by mRNA. This association yielded the formation of 50–200 nm clusters, suggesting that the interaction with mRNA is a multi-molecular process in which multiple BAPCs interact with more than one mRNA molecule (Fig. 1C). These interactions are the result of the lysyl groups present in the branched peptides and the phosphates in the mRNA backbone. Notably, the size and geometry of BAPCs and the BAPCs-mRNA complexes remains stable after exposure to serum containing media (Fig. S1†).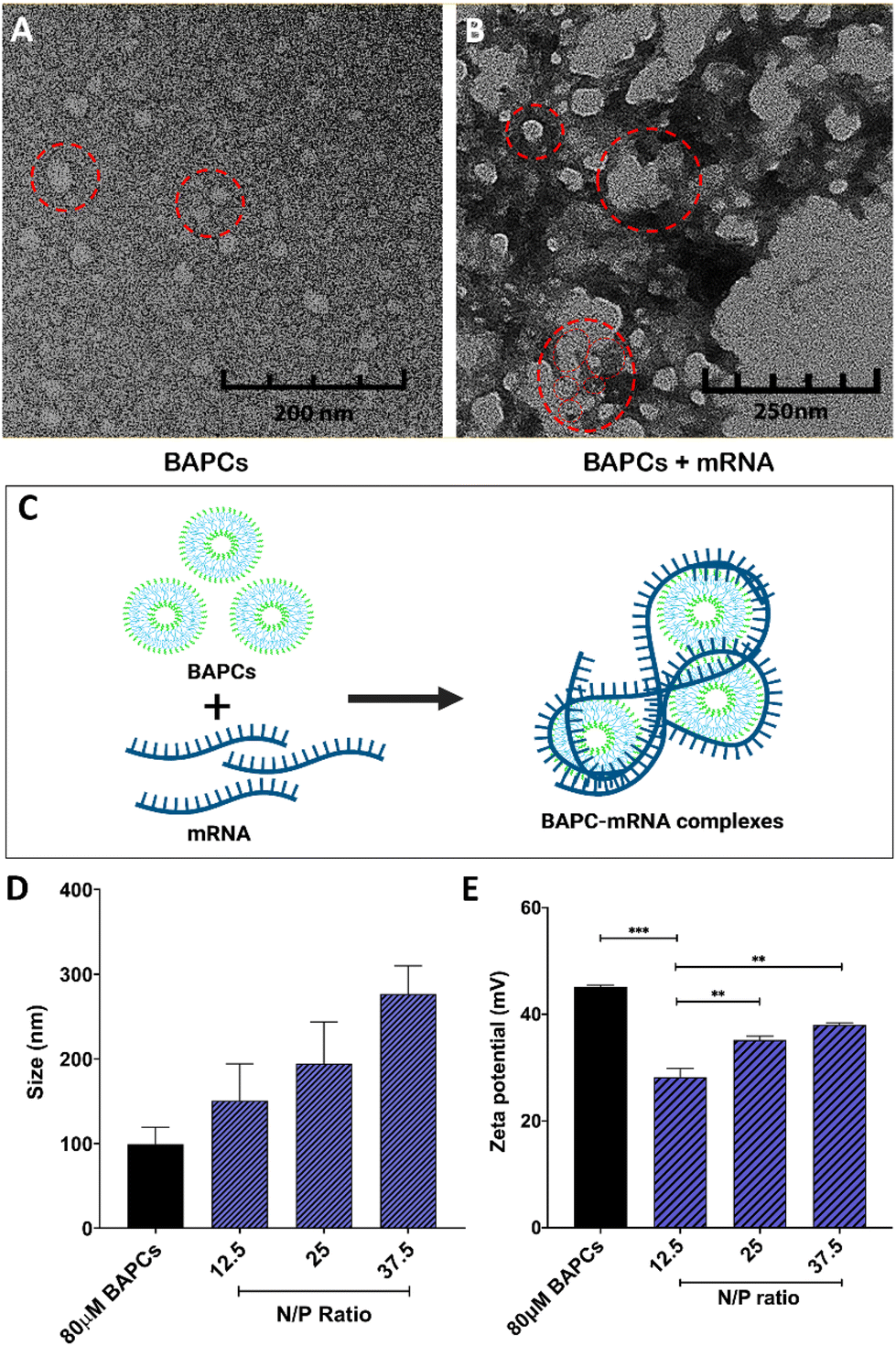 | ||
| Fig. 1 Biophysical characterization of BAPC-mRNA complexes: TEM analysis of (A) bare BAPCs and (B) BAPC-mRNA complexes at 12.5 N/P ratio. Red circles in A indicates single BAPC (monodispersed solution) and in B indicates cluster of BAPCs with mRNA wrapped around them leading to formation of complexes. (C) Schema of BAPC-mRNA cluster formation (D) analysis of size distribution by DLS of BAPC-mRNA complexes at different N/P ratios. (E) Zeta potential of BAPC-mRNA complexes at different N/P ratios. Differences between values were compared by ANOVA using Tuckey as post-test. Statistical significance: (***) p < 0.001; (****) p < 0.0001. Non-statistical significance (ns) was considered when p > 0.05. | ||
The biophysical properties of BAPCs and the BAPC-mRNA complexes were also evaluated by dynamic light scattering (DLS) and zeta potential (ZP). The BAPC-mRNA complexes were analyzed at different N/P ratios. As shown in Fig. 1D, bare BAPCs displayed a size ranging from 80 to 110 nm with a PDI ∼0.2, indicating that the sample contained monodispersed peptide vesicles. The BAPC-mRNA complexes showed a minor increase in size proportional to the incremental increase of N/P ratio (Fig. 1D). To estimate the surface charge of the complexes, ZP values were obtained at different N/P ratios. For bare BAPCs, ZP was ∼45 mV, and after mRNA association, the ZP value changed to ∼36 mV (Fig. 1E). A plausible explanation for the reduction of the ZP is the shielding of the lysine positive charges by the mRNA, confirming the interaction between BAPCs and mRNA (Fig. 1E). Literature suggests that nanoparticles with ZP < 40 mV showed improved blood circulation and low toxicity, suggesting that mRNA might serve as a suitable surface coating for BAPCs for in vivo delivery applications.12,27–29
In order to avoid excessive use of materials and undesirable effects produced by higher nanoparticle dosage or excessive mRNA use, it is crucial to determine the optimal mRNA binding capacity of BAPC-mRNA complexes. These parameters help to maximize treatment effectiveness and reduce therapeutic cost. To determine the mRNA loading capacity of BAPCs, we employed an electrophoretic retardation assay.30 The electrostatic interactions between mRNA and the BAPCs cationic surface led to a reduced migration into the agarose gel compared with unbound mRNA. We prepared BAPC-mRNA complexes by mixing varying amount of mRNA (1000 ng, 750 ng, 500 ng, 250 ng, and 100 ng) with constant amount of BAPCs (40 μM) to achieve BAPC-mRNA complexes of N/P ratio of 12.5, 18.75, 25, 37.5 and 133, respectively. The electrophoretic mobility of these complexes was assessed in a 2% RNA denaturing agarose gel (Fig. 2). BAPC-mRNA complexes with >25 N/P ratio did not show visible mRNA bands, suggesting that at that ratio there is a complete saturation of mRNA binding sites. These results also indicated that BAPC are capable of shielding mRNA, conferring with this protection against ribonucleases.31
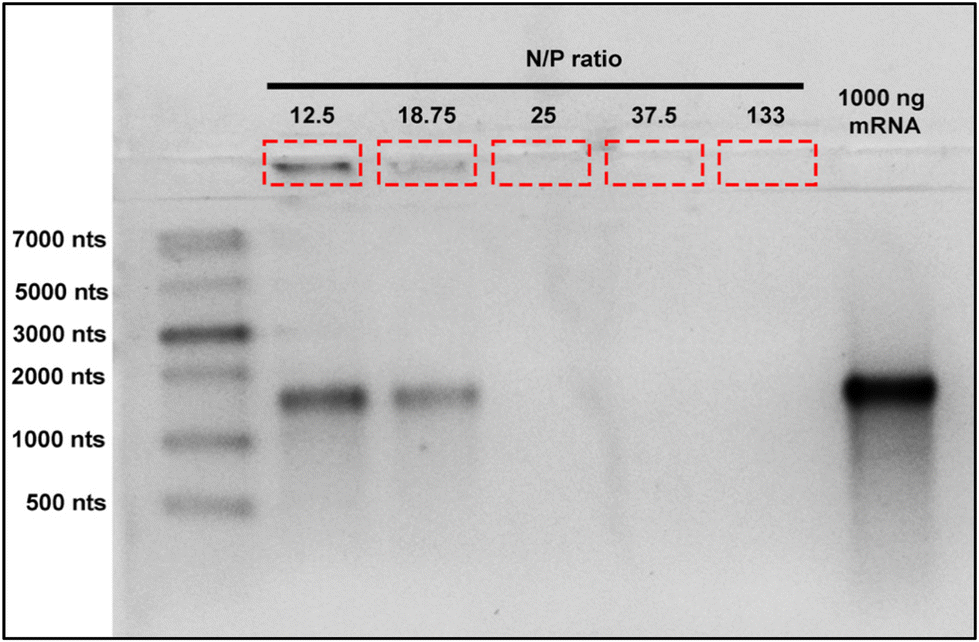 | ||
| Fig. 2 BAPC loading capacity: BAPCs binding capacity was assessed by the electrophoretic mobility shift assay. BAPC-mRNA complexes were formed by mixing fixed amount of BAPCs (40 μM) with varying mRNA concentrations (100 ng–1000 ng) and resolved on 2% RNA denaturing gel. mRNA bands indicated by red box shows BAPC-mRNA complexes retained in the wells, while bands around 1400 nts are unbound mRNA. | ||
3.2 In vitro transfection efficiency of BAPCs coupled with mRNA
To confirm BAPCs potential to enhance intracellular uptake of mRNA into mammalian cells, mRNA encoding for the chicken ovalbumin (Ova) protein was complexed with BAPCs and co-incubated with HEK293T cells. The Ova protein induces a T cell-dependent antigenic response and is commonly used as a model antigen for studying adaptive immune responses in several animal models.32,33 Cells were co-incubated with the complexes at the specific N/P ratios of 12.5, 25 and 37.5 for 4 h in reduced serum media. Optimal in vitro transfection parameters for BAPCs such as incubation times and transfection media have been previously optimized for different cell types.18,31 Western blotting was used to analyze expression of the Ova protein. The housekeeping protein β-actin was included to compare protein abundance across treatments groups.31 Cells treated with the complexes at the lowest N/P ratio (12.5) showed the highest Ova expression, equivalent to the commercial transfection reagent jetMessenger® (Polyplus) (Fig. 3A).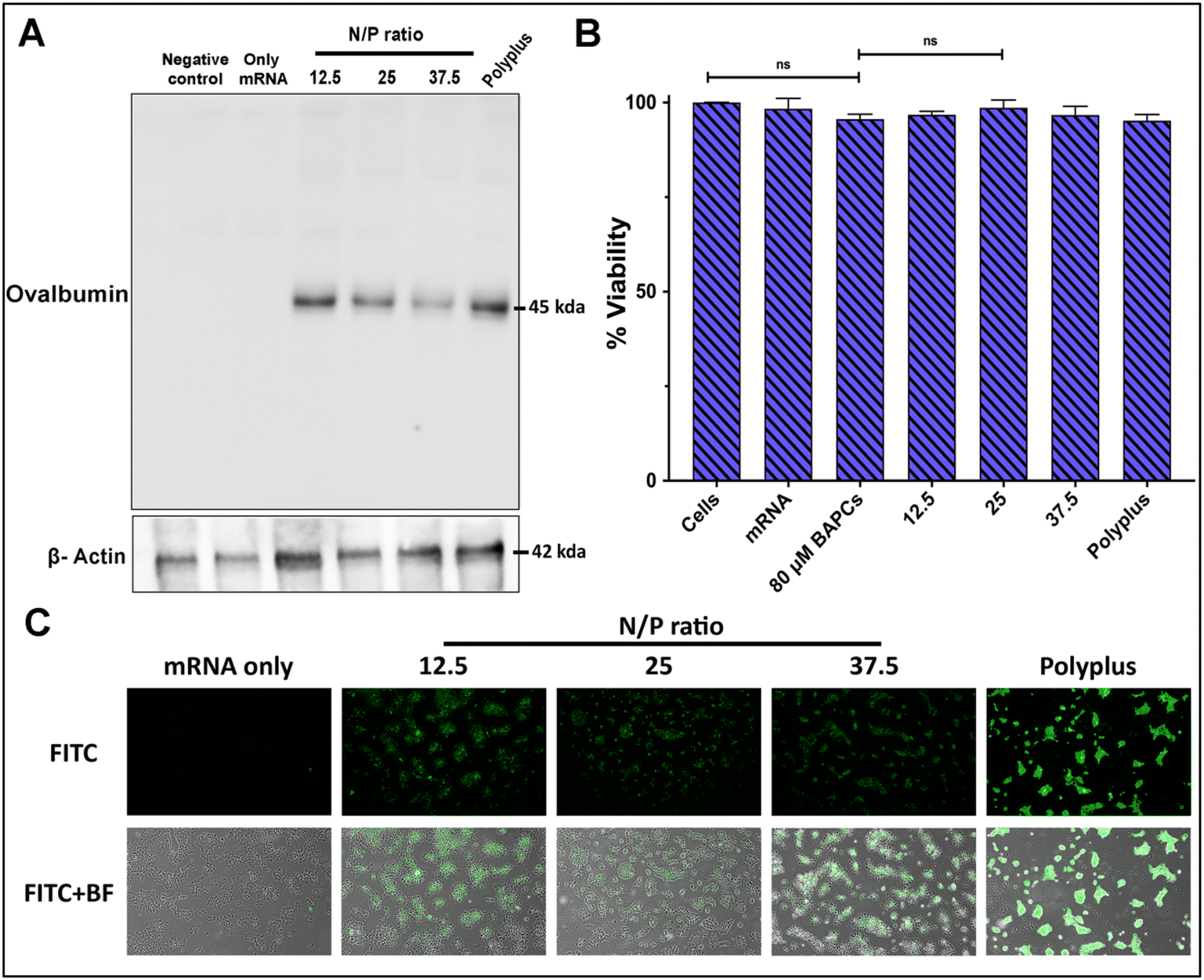 | ||
| Fig. 3 BAPCs-mediated in vitro mRNA delivery and cytotoxicity: comparison of transfection efficiency of BAPC-mRNA complexes with varying N/P ratio in HEK293T cells. (A) Western blot for ovalbumin protein along with β-actin as loading control. (B) Cell viability of each formulation during the initial 4 h treatment period determined by the MTT assay (C) fluorescence images of eGFP expressing HEK293T cells 24 h post-treatment (10× magnification). Differences between values were compared by one way ANOVA using Tukey post-test. Statistical significance: (*) p < 0.05. Non-statistical significance (ns) was considered when p > 0.05. n = 3. | ||
None of the BAPCs formulations displayed cellular toxicity according to a colorimetric MTT assay based on the reduction of a tetrazolium equivalent dye by dehydrogenases – enzymes associated with the endoplasmic reticulum and the mitochondria that reflect the cellular metabolic activity34 (Fig. 3B). Nonetheless, mechanisms of cellular toxicity can be diverse. We also analyzed the production of reactive oxygen species (ROS) induced by the BAPCs-mRNA complexes. ROS can disrupt mitochondrial activity, cause DNA damage, and lipid peroxidation. This leads to deterioration of the cell membrane, making it more vulnerable to further oxidation. A key element involved in nanoparticle-induced ROS is the presence of prooxidant functional groups on the reactive surface of nanoparticles.35 In the case of BAPCs, mRNA is the only element surrounding the nanoparticle, and no production of ROS species was observed compared with untreated cells (Fig. S2†). Cellular morphology appeared to be normal after treatment with the BAPC-mRNA formulations, whereas cells treated with jetMessenger® developed altered morphologies.
In addition to Ova expression analysis, we tested the ability of BAPCs to deliver reporter mRNA coding for eGFP in HEK293T cells (Fig. 3C). We applied the same transfection conditions used for the delivery of Ova-mRNA. Gene delivery efficiency was monitored qualitatively by fluorescence microscopy and quantified using flow cytometry (Fig. S3†). The dye 7-AAD was used to identify and then exclude dead cells from the analysis. Similar to Ova-mRNA results, expression profile was higher in cells treated with complexes at the lowest N/P ratio (12.5). A plausible explanation for obtaining higher in vitro transfection rates at lower ratios can be related with the dissociation rate of mRNA after cellular internalization. According with literature review, intracellular proteins participate in the release of the cargo from nanoparticles.36 We hypothesize that at higher ratios, multiple BAPCs interact with more than one mRNA, thus resulting in the formation of clusters with tightly bound mRNA. This causes unfavorable mRNA dissociation, potentially leading to lower in vitro transfection.
3.3 In vivo biodistribution
Nanoparticles used for systemic drug delivery applications must exhibit prolonged blood circulation in order to achieve targeted and controlled release of therapeutics. Unmodified cationic NPs readily adsorb opsonin proteins on their surface following intravenous administration. Opsonized NPs are removed from the bloodstream within seconds via phagocytosis, thereby promoting NP localization in major MPS organs such as the liver and spleen.37 Accumulation of NPs carrying mRNA is commonly observed in these organs, resulting in chronic toxicity and truncated therapeutic effects.1 Although the underlying mechanisms of NP opsonization are not fully understood, NP surface charge plays a crucial role in opsonin binding.38 It has been observed that NPs with low ZP have a much lower opsonization rate than more intensely charged particles. Hence, a broad range of approaches are being applied to reduce the opsonization of charged NPs.36 One of the most widely used approach is modification of the NP surface by adsorption or grafting of chemical groups such as PEG that can prevent molecular interactions between NPs and opsonin proteins.The sole purpose of this section was to elucidate if instead of PEG, the association of mRNA to BAPCs surface helps circumvent entrapments by liver and spleen in B6-albino mice. To achieve this goal, we employed MSOT imaging, in which irradiation by ultrashort laser pulses (1–100 nanoseconds) generates ultrasound waves, which are then used for image construction. Since ultrasound waves are scattered to a lesser degree in tissues than light, MSOT provides a significantly higher spatial resolution compared to fluorescence imaging.39,40 In addition, multiple wavelengths are used to allow simultaneous detection of multiple compounds, including natural chromophores such as oxy- and deoxy-hemoglobin and external probes (i.e., NPs).
To track the BAPC-mRNA complexes (N/P = 25) using MSOT, we conjugated the infrared (IRDye-800CW) dye to the (ε)-lysine in one of the forming BAPCs peptides. Despite obtaining the best in vitro expression with the N/P = 12.5, we selected the N/P = 25 for in vivo studies to avoid potential immunogenicity induced by free mRNA. According to the gel retardation, at N/P = 25 all mRNA is being associated with the BAPCs and no unbound mRNA was detected. The complexes were administered via tail vein, and biodistribution in the liver, spleen and three major blood vessels was monitored 1 and 24 h following the injection (Fig. 4A). MSOT images acquired from mice injected with PBS and only IRDye-800CW dye were included as controls (Fig. 4F & G). Biodistribution in liver, spleen and major blood vessels were monitored for BAPCs and BAPC-mRNA complexes (Fig. 4H). MSOT signal was corrected by subtracting the signal acquired prior to the treatments from the signal acquired post-treatment. Bare BAPCs showed a stronger MSOT signal in the spleen and liver compared to the BAPC-mRNA complexes, indicating that the surface association of mRNA prevents sequestration within these organs. Furthermore, detection of MSOT signal in the aorta and portal vein confirms the presence of BAPC-mRNA complexes in blood 1 h after delivery, displaying a longer half-life in blood circulation than bare BAPCs (Fig. 4I).
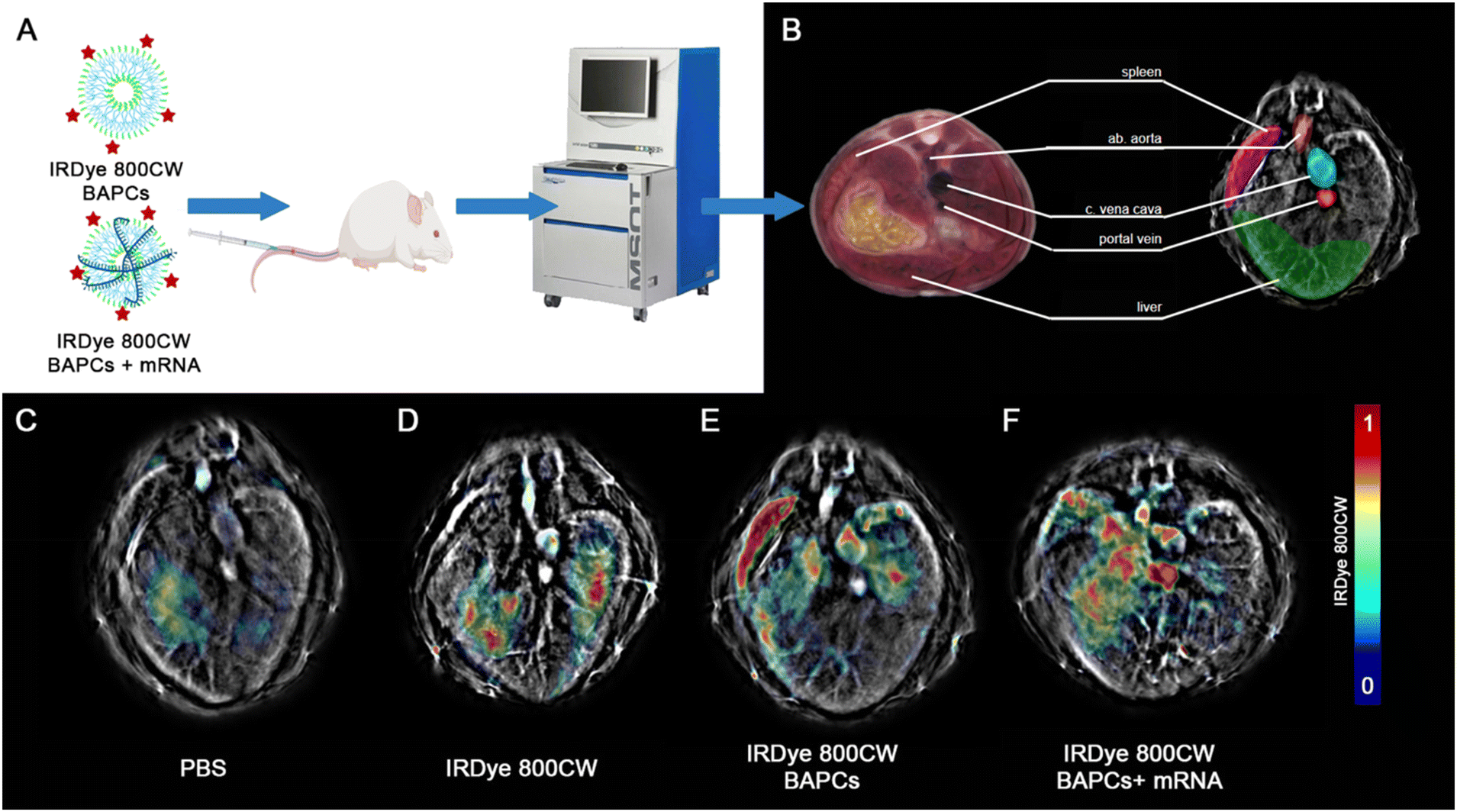 | ||
| Fig. 4 Multispectral optoacoustic tomography of BAPCs and BAPCs conjugated with mRNA at 1 h. Post injection: (A) brief experimental outline of in vivo imaging experiment using MSOT. Panel B highlights relevant anatomical features from a murine cryo-sliced atlas and corresponding features visible with MSOT. Panel C–F shows representative slices from each group, with spectrally unmixed signal from IRDye-800CW overlaid on each respective 875 nm single-wavelength images. Slices show the approximate transverse sections as those outlined in (B). Panels C–F represent mice injected with (C) PBS, (D) IRDye-800CW dye alone, (E) IRDye-800CW tagged BAPCs, and (F) IRDye-800CW-BAPCs conjugated with mRNA. | ||
Clearance of both formulations was observed 24 h post-injection. No noticeable MSOT signal was detected in the spleen, liver, or lungs (Fig. S4†). Clearance after this period is desirable since nanoparticle accumulation in liver can lead to damage of hepatic cells, which in turn can affect critical processes such as detoxification, glycogen storage, bile formation, protein synthesis, and general homeostasis.41 Buildup in the spleen could negatively impact the innate and adaptive immune response, which is involved in the elimination of other pathogenic microorganisms, immune response to vaccines, and immune surveillance.42
Fluorescence reflectance imaging analysis was also performed in mice 1, 3 and 24 h post injection. Animals were sacrificed, and heart, lungs, liver, spleen, and kidneys were harvested. The fluorescence images of all the organs were acquired using IVIS Lumina XRMS system to support MSOT analysis. Mice that received PBS and only IRDye-800cw did not show fluorescence signal in any organ (Fig. 5A & B). Mice injected with IRDye-800CW-BAPCs showed high fluorescence signal in liver and spleen compared to controls at 1 h and 3 h post-injection (Fig. 5C and Fig. S5†). In contrast, mice administered with IRDye-800CW-BAPCs complexed with mRNA showed significantly lower fluorescence intensity in spleen and liver, and increased signal in lung. The absence of fluorescence in the kidneys suggest that none of the formulations is excreted through renal filtration (Fig. 5D). A semi-quantitative measure of organ fluorescence using ImageJ software supported the observations indicated above (Fig. 5E). For bare BAPCs liver showed a higher fluorescence intensity compared to any other organs at both time points, which could be the result of the clearance process via biliary excretion, or a potential reabsorption from the gastro-intestinal tract.43 For 24 h biodistribution, fluorescence levels are significantly decreased in spleen, liver, and lungs.
 | ||
| Fig. 5 Fluorescence reflectance imaging of BAPCs and BAPCs conjugated with mRNA at 1, 3 and 24 h post injection. (A) PBS only (B) IRDye-800CW only (C) IRDye-800CW tagged BAPCs (D) IRDye-800CW tagged BAPCs complexed with mRNA. (E) Quantitative analysis of the organ biodistribution profile of BAPCs. Fluorescence value was obtained by individually selecting region of interest (ROI) for each organ and subtracting background signals from the PBS-treated mice. Differences between values were compared by ANOVA using Tuckey as post-test. Statistical significance: (*) p < 0.05; (**) p < 0.01. Non-statistical significance (ns) was considered when p > 0.05. | ||
Overall, MSOT and fluorescence reflectance imaging analysis support the notion that surface properties of NPs is major a deciding factor of NPs biodistribution. Functionalization of BAPCs with mRNA influence its biodistribution profile by avoiding accumulation in spleen and liver. These results suggest that BAPC-mRNA complexes will be a useful delivery platform for systemic delivery of mRNA in vivo.
3.4 Effect of BAPC-mRNA complexes on in vivo cytokine release profile
Cytokines are proteins that play a key role in the modulation of both innate and adaptive immune responses. Quantification of cytokine expression can help in prediction of the immunomodulatory effects of NPs and potential inflammatory response.44 To assess cytokine response induced by BAPCs and BAPC-mRNA complexes at 1 and 24 h following injection, we employed a multiplex cytokine assay. This assay uses the same basic principle as sandwich ELISA, in which the target cytokine is bound to specific antibodies conjugated to beads. Subsequently, streptavidin-phycoerythrin (SA-PE)-tagged detection antibodies are added to recognize cytokines captured by antibody-bead complexes. Fluorescence intensity of the detection antibody is proportional to the amount of bound analytes (Fig. S6†). The panels used in this assay allowed simultaneous quantification of multiple cytokines, including interferon (IFN-γ), interleukins (IL-1β, IL-6), and tumor necrotic factor (TNF)-α. IL-6 and TNF-α cytokines are expressed by almost all cell types. On the other hand, IFN-γ is produced only by T lymphocytes and natural killer cells, while IL-1β is expressed by dendritic cells, macrophages, endothelial and epithelial cells.IL-1β is a pro-inflammatory cytokine responsible for the recruitment of immune cells to the site of inflammation commonly triggered by infections. It is also involved in a variety of cellular activities such as cellular differentiation and apoptosis.45 Mice treated with BAPCs or BAPC-mRNA complexes did not show change in IL-1β expression at any timepoints, indicating that none of the BAPC formulations induced severe inflammation (Fig. 6A & B). On the other hand, IL-6 is both a pro- and anti-inflammatory cytokine. During tissue injury, it regulates inflammatory and repair processes by recruiting neutrophils and platelets to inflammation site as well as induces the synthesis of acute-phase proteins such as C-reactive protein, serum amyloid A.46,47 Mice injected with bare BAPCs showed elevated IL-6 expression level compared with PBS control group at 1 h and 24 h, but no change in IFN-γ and TNF-α cytokine levels observed (Fig. 6A). This increased expression could be the result of IL6 mediated repair of tissue damage induced by BAPCs upon accumulation into the MPS organs. Nonetheless, IL6 also play critical role in inflammation. During a viral infection or systemic administration of exogenous RNA, this cytokine stimulates inflammatory response by activation of Toll-like receptors (TLR) and RIG-I signaling pathways, followed by expression of pro-inflammatory cytokines such as IL-6, IFN-γ and TNF-α.48–50 Mice injected with BAPC-mRNA complexes showed moderate increase in IL6, IFN-γ and TNF-α levels only at 1 h. This elevated cytokine expression could be the result of mRNA recognition by immune cells immune cells, and stimulation of a transient inflammatory response. Notably, IL-6, IFN-γ and TNF-α returned to basal level 24 h post injection, which indicates that after that period of time, cytokine levels shift back toward a non-inflammatory environment (Fig. 6B).
 | ||
| Fig. 6 Cytokine expression assay: cytokine profile of (A) BAPCs and (B) BAPC-mRNA complexes post 1 and 24 h injections. All treatment groups are compared with cytokine levels of PBS and IRDye-800CW dye injected mice at 1 h timepoint. Differences between values were compared by ANOVA using Tuckey as post-test. Statistical significance: (*) p < 0.05; (**) p < 0.01. Non-statistical significance (ns) was considered when p > 0.05. | ||
4. Conclusions
In this study, we demonstrated that the association of BAPCs with mRNA resulted in the formation of 50–350 nm BAPC-mRNA complexes with ZP values ∼36 mV. This association is the consequence of a multi-molecular process driven by the electrostatic interaction of the lysine residues exposed on the surface of the BAPCs and the phosphates in the mRNA backbone. The formed complexes were capable of delivering Ova-mRNA and eGFP mRNA into HEK293T cells, without inducing oxidative stress or altering the cell metabolism. Despite having the mRNA exposed on the NP surface, observing GFP and OVA expression in transfected cells indicates that mRNA is not being fully degraded by enzymes. We hypothesized that some target sites for RNAses are no longer accessible to the catalytic core responsible for RNA degradation after the mRNA association to the BAPCs surface.51Currently there is significant literature available discussing NP-mediated delivery of mRNA, yet the majority of them address mostly local delivery methods. A key factor in systemic delivery of mRNA is the premature sequestration of nanomaterials by liver and spleen following administration.1 Nonetheless, the NP surface can be modified to avoid this undesired sequestration process that impairs therapeutic effects. Our results demonstrated that BAPCs with mRNA adducted on the surface displayed a reduced accumulation in the spleen and liver, contributing to increased circulation times. An increased accumulation of BAPC-mRNA complexes was observed in lungs 1 h and 3 h post injection, which suggest that the complexes might be a suitable candidate for lung-targeted therapies. Lung accumulation is likely enhanced by specific receptors or endocytic pathways present in pulmonary endothelial cells. It has been reported that endothelial cell heterogeneity plays a key role on the cellular uptake of nanoparticles.52 Imaging analysis after 24 h confirm that both BAPCs and BAPCs-mRNA complexes are cleared out most likely via biliary excretion, avoiding with this potential chronic toxicity. In addition, proinflammatory cytokine assessment showed BAPC-mRNA complexes are not highly immunogenic since they induced only a transient increase in IFN-γ, TNF-α and IL6 levels. Overall, the avoidance of liver and spleen combined with clearance after 24 h and low immunogenicity suggests that BAPCs can be an alternative delivery platform to enable systemic delivery of mRNA.
Conflicts of interest
Andrew Brannen is an employee of iThera Medical, the vendor for the MSOT inVision used in this study. The authors otherwise state that there are no conflicts of interest in connection with this article.References
- D. Loughrey and J. E. Dahlman, Non-liver mRNA Delivery, Acc. Chem. Res., 2021, 55, 13–23 CrossRef PubMed.
- Y. Xiao and J. Shi, Lipids and the Emerging RNA Medicines, Chem. Rev., 2021, 121, 12109–12111 CrossRef PubMed.
- A. Khurana, P. Allawadhi, I. Khurana, S. Allwadhi, R. Weiskirchen and A. K. Banothu, et al., Role of nanotechnology behind the success of mRNA vaccines for COVID-19, Nano Today, 2021, 38, 101142 CrossRef PubMed.
- S. Guan and J. Rosenecker, Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems, Gene Ther., 2017, 24, 133–143 CrossRef PubMed.
- S. Uchida, H. Kinoh, T. Ishii, A. Matsui, T. A. Tockary and K. M. Takeda, et al., Systemic delivery of messenger RNA for the treatment of pancreatic cancer using polyplex nanomicelles with a cholesterol moiety, Biomaterials, 2016, 82, 221–228 CrossRef PubMed.
- Y. Wang, H. H. Su, Y. Yang, Y. Hu, L. Zhang and P. Blancafort, et al., Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy, Mol. Ther., 2013, 21, 358–367 CrossRef PubMed.
- F. Perche, T. Benvegnu, M. Berchel, L. Lebegue, C. Pichon and P. A. Jaffrès, et al., Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA, Nanomedicine, 2011, 7, 445–453 CrossRef PubMed.
- P. S. Kowalski, U. Capasso Palmiero, Y. Huang, A. Rudra, R. Langer and D. G. Anderson, Ionizable amino-polyesters synthesized via ring opening polymerization of tertiary amino-alcohols for tissue selective mRNA delivery, Adv. Mater., 2018, 30, 1801151 CrossRef PubMed.
- C. Meng, Z. Chen, G. Li, T. Welte and H. Shen, Nanoplatforms for mRNA Therapeutics, Adv. Ther., 2021, 4, 2000099 CrossRef CAS.
- K. Shiraishi and M. Yokoyama, Toxicity and immunogenicity concerns related to PEGylated-micelle carrier systems: a review, Sci. Technol. Adv. Mater., 2019, 20, 324–336 CrossRef CAS PubMed.
- T. T. H. Thi, E. H. Pilkington, D. H. Nguyen, J. S. Lee, K. D. Park and N. P. Truong, The importance of Poly(ethylene glycol) alternatives for overcoming PEG immunogenicity in drug delivery and bioconjugation, Polymers, 2020, 12, 298 CrossRef CAS PubMed.
- L. A. Avila, L. R. M. M. Aps, N. Ploscariu, P. Sukthankar, R. Guo and K. E. Wilkinson, et al., Gene delivery and immunomodulatory effects of plasmid DNA associated with Branched Amphiphilic Peptide Capsules, J. Controlled Release, 2016, 241, 15–24 CrossRef CAS PubMed.
- P. Sukthankar, L. A. Avila, S. K. Whitaker, T. Iwamoto, A. Morgenstern and C. Apostolidis, et al., Branched amphiphilic peptide capsules: Cellular uptake and retention of encapsulated solutes, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2296–2305 CrossRef CAS PubMed.
- P. Sukthankar, S. Gudlur, L. A. Avila, S. K. Whitaker, B. B. Katz and Y. Hiromasa, et al., Branched oligopeptides form nanocapsules with lipid vesicle characteristics, Langmuir, 2013, 29, 14648–14654 CrossRef CAS PubMed.
- L. A. Avila, S. Y. Lee and J. M. Tomich, Synthetic In Vitro Delivery Systems for Plasmid DNA in Eukaryotes, J. Nanopharm. Drug Deliv., 2014, 2, 17–35 CrossRef.
- S. Gudlur, P. Sukthankar, J. Gao, L. A. Avila, Y. Hiromasa and J. Chen, et al., Peptide Nanovesicles Formed by the Self-Assembly of Branched Amphiphilic Peptides, PLoS One, 2012, 7, e45374 CrossRef CAS PubMed.
- S. M. Barros, S. K. Whitaker, P. Sukthankar, L. A. Avila, S. Gudlur and M. Warner, et al., A review of solute encapsulating nanoparticles used as delivery systems with emphasis on branched amphipathic peptide capsules, Arch. Biochem. Biophys., 2016, 596, 22–42 CrossRef CAS PubMed.
- S. D. M. Barros, L. A. Avila, S. K. Whitaker, K. E. Wilkinson, P. Sukthankar and E. I. C. Beltrão, et al., Branched Amphipathic Peptide Capsules: Different Ratios of the Two Constituent Peptides Direct Distinct Bilayer Structures, Sizes, and DNA Transfection Efficiency, Langmuir, 2017, 33, 7096–7104 CrossRef PubMed.
- A. Reiser, D. Woschée, N. Mehrotra, R. Krzysztoń, H. H. Strey and J. O. Rädler, Correlation of mRNA delivery timing and protein expression in lipid-based transfection, Integr. Biol., 2019, 11, 362–371 CrossRef PubMed.
- Y. P. Fang, C. H. Chuang, P. C. Wu, Y. b. Huang, C. C. Tzeng and Y. L. Chen, et al., Amsacrine analog-loaded solid lipid nanoparticle to resolve insolubility for injection delivery: Characterization and pharmacokinetics, Drug Des., Dev. Ther., 2016, 10, 1019 CrossRef PubMed.
- S. P. Egusquiaguirre, N. Beziere, J. L. Pedraz, R. M. Hernández, V. Ntziachristos and M. Igartua, Optoacoustic imaging enabled biodistribution study of cationic polymeric biodegradable nanoparticles, Contrast Media Mol. Imaging, 2015, 10, 421–427 CrossRef PubMed.
- M. K. Gurka, D. Pender, P. Chuong, B. L. Fouts, A. Sobelov and M. W. McNally, et al., Identification of pancreatic tumors in vivo with ligand-targeted, pH responsive mesoporous silica nanoparticles by multispectral optoacoustic tomography, J. Controlled Release, 2016, 231, 60–67 CrossRef PubMed.
- L. Zhang, C. D. Wallace, J. E. Erickson, C. M. Nelson, S. M. Gaudette and C. S. Pohl, et al., Near infrared readouts offer sensitive and rapid assessments of intestinal permeability and disease severity in inflammatory bowel disease models, Sci. Rep., 2020, 10, 1–12 CrossRef PubMed.
- N. Yi, B. Oh, H. A. Kim and M. Lee, Combined delivery of BCNU and VEGF siRNA using amphiphilic peptides for glioblastoma, J. Drug Targeting, 2014, 22, 156–164 CrossRef PubMed.
- S. Tarvirdipour, C. A. Schoenenberger, Y. Benenson and C. G. Palivan, A self-assembling amphiphilic peptide nanoparticle for the efficient entrapment of DNA cargoes up to 100 nucleotides in length, Soft Matter, 2020, 16, 1678–1691 RSC.
- P. Sukthankar, S. K. Whitaker, M. Garcia, A. Herrera, M. Boatwright and O. Prakash, et al., Thermally Induced Conformational Transitions in Nascent Branched Amphiphilic Peptide Capsules, Langmuir, 2015, 31, 2946–2955 CrossRef CAS PubMed.
- A. Vonarbourg, C. Passirani, P. Saulnier, P. Simard, J. C. Leroux and J. P. Benoit, Evaluation of pegylated lipid nanocapsules versus complement system activation and macrophage uptake, J. Biomed. Mater. Res., Part A, 2006, 78, 620–628 CrossRef CAS PubMed.
- T. Merdan, K. Kunath, H. Petersen, U. Bakowsky, K. H. Voigt and J. Kopecek, et al., PEGylation of poly(ethylene imine) affects stability of complexes with plasmid DNA under in vivo conditions in a dose-dependent manner after intravenous injection into mice, Bioconjugate Chem., 2005, 16, 785–792 CrossRef CAS PubMed.
- S. Bhattacharjee, DLS and zeta potential - What they are and what they are not?, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- H. Yasar, A. Biehl, C. de Rossi, M. Koch, X. Murgia and B. Loretz, et al., Kinetics of mRNA delivery and protein translation in dendritic cells using lipid-coated PLGA nanoparticles, J. Nanobiotechnol., 2018, 16, 1–19 CrossRef PubMed.
- L. A. Avila, L. R. M. M. Aps, P. Sukthankar, N. Ploscariu, S. Gudlur and L. Šimo, et al., Branched amphiphilic cationic oligopeptides form peptiplexes with DNA: A study of their biophysical properties and transfection efficiency, Mol. Pharm., 2015, 12, 706–715 CrossRef CAS PubMed.
- M. Pei, R. Xu, C. Zhang, X. Wang, C. Li and Y. Hu, Mannose-functionalized antigen nanoparticles for targeted dendritic cells, accelerated endosomal escape and enhanced MHC-I antigen presentation, Colloids Surf., B, 2021, 197, 111378 CrossRef PubMed.
- N. Habibi, S. Christau, L. J. Ochyl, Z. Fan, A. Hassani Najafabadi and M. Kuehnhammer, et al., Engineered Ovalbumin Nanoparticles for Cancer Immunotherapy, Adv. Ther., 2020, 3, 2000100 CrossRef.
- R. Scherließ, The MTT assay as tool to evaluate and compare excipient toxicity in vitro on respiratory epithelial cells, Int. J. Pharm., 2011, 411, 98–105 CrossRef PubMed.
- M. Pešić, A. Podolski-Renić, S. Stojković, B. Matović, D. Zmejkoski and V. Kojić, et al., Anti-cancer effects of cerium oxide nanoparticles and its intracellular redox activity, Chem.-Biol. Interact., 2015, 232, 85–93 CrossRef PubMed.
- C. van Bruggen, D. Punihaole, A. R. Keith, A. J. Schmitz, J. Tolar and R. R. Frontiera, et al., Quinine copolymer reporters promote efficient intracellular DNA delivery and illuminate a protein-induced unpackaging mechanism, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32919–32928 CrossRef PubMed.
- D. E. Owens and N. A. Peppas, Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- K. Xiao, Y. Li, J. Luo, J. S. Lee, W. Xiao and A. M. Gonik, et al., The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles, Biomaterials, 2011, 32, 3435–3446 CrossRef PubMed.
- T. Anani, A. Brannen, P. Panizzi, E. C. Duin and A. E. David, Quantitative, real-time in vivo tracking of magnetic nanoparticles using multispectral optoacoustic tomography (MSOT) imaging, J. Pharm. Biomed. Anal., 2020, 178, 112951 CrossRef CAS PubMed.
- N. Bhutiani, W. E. Grizzle, S. Galandiuk, D. Otali, G. W. Dryden and N. K. Egilmez, et al., Noninvasive imaging of colitis using multispectral optoacoustic tomography, J. Nucl. Med., 2017, 58, 1009–1012 CrossRef CAS PubMed.
- A. Boey and H. K. Ho, All Roads Lead to the Liver: Metal Nanoparticles and Their Implications for Liver Health, Small, 2020, 16, 2000153 CrossRef CAS PubMed.
- X. D. Zhang, D. Wu, X. Shen, P. X. Liu, F. Y. Fan and S. J. Fan, In vivo renal clearance, biodistribution, toxicity of gold nanoclusters, Biomaterials, 2012, 33, 4628–4638 CrossRef CAS PubMed.
- N. Waegeneers, A. Brasseur, E. van Doren, S. van der Heyden, P. J. Serreyn and L. Pussemier, et al., Short-term biodistribution and clearance of intravenously administered silica nanoparticles, Toxicol. Rep., 2018, 5, 632–638 CrossRef CAS PubMed.
- J. J. M. Lasola, A. L. Cottingham, B. L. Scotland, N. Truong, C. C. Hong and P. Shapiro, et al., Immunomodulatory nanoparticles mitigate macrophage inflammation via inhibition of pamp interactions and lactate-mediated functional reprogramming of nf-κb and p38 mapk, Pharmaceutics, 2021, 13, 1841 CrossRef CAS PubMed.
- M. Khatri, D. Bello, A. K. Pal, J. M. Cohen, S. Woskie and T. Gassert, et al., Evaluation of cytotoxic, genotoxic and inflammatory responses of nanoparticles from photocopiers in three human cell lines, Part. Fibre Toxicol., 2013, 10, 1–22 CrossRef PubMed.
- T. Tanaka, M. Narazaki and T. Kishimoto, Il-6 in inflammation, Immunity, And disease, Cold Spring Harbor Perspect. Biol., 2014, 6, a016295 CrossRef PubMed.
- C. A. Fielding, R. M. McLoughlin, L. McLeod, C. S. Colmont, M. Najdovska and D. Grail, et al., IL-6 Regulates Neutrophil Trafficking during Acute Inflammation via STAT3, J. Immunol., 2008, 181, 2189–2195 CrossRef CAS PubMed.
- C. Gabay, Interleukin-6 and chronic inflammation, Arthritis Res. Ther., 2006, 8, 1–6 CrossRef PubMed.
- J. Sancéau, T. Kaisho, T. Hirano and J. Wietzerbin, Triggering of the human interleukin-6 gene by interferon-γ and tumor necrosis factor-α in monocytic cells involves cooperation between interferon regulatory factor-1, NFeκB, and Sp1 transcription factors, J. Biol. Chem., 1995, 270, 27920–27931 CrossRef PubMed.
- W. Wu, K. K. Dietze, K. Gibbert, K. S. Lang, M. Trilling and H. Yan, et al., TLR ligand induced IL-6 counter-regulates the anti-viral CD8 + T cell response during an acute retrovirus infection, Sci. Rep., 2015, 5, 1–14 Search PubMed.
- H. K. Wayment-Steele, D. S. Kim, C. A. Choe, J. J. Nicol, R. Wellington-Oguri and A. M. Watkins, et al., Theoretical basis for stabilizing messenger RNA through secondary structure design, Nucleic Acids Res., 2021, 49, 10604–10617 CrossRef PubMed.
- A. Aliyandi, S. Satchell, R. E. Unger, B. Bartosch, R. Parent and I. S. Zuhorn, et al., Effect of endothelial cell heterogeneity on nanoparticle uptake, Int. J. Pharm., 2020, 587, 119699 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01314b |
| This journal is © The Royal Society of Chemistry 2022 |