
Surface-modified nanotherapeutics targeting atherosclerosis
Wenpan
Li
a,
Karina Marie
Gonzalez
a,
Jinha
Chung
a,
Minhyeok
Kim
a and
Jianqin
Lu
 *abcd
*abcd
aSkaggs Pharmaceutical Sciences Center, Department of Pharmacology & Toxicology, R. Ken Coit College of Pharmacy, The University of Arizona, Tucson, Arizona 85721, USA. E-mail: lu6@pharmacy.arizona.edu; Fax: +520-626-2466; Tel: +520-626-1786
bNCI-designated University of Arizona Comprehensive Cancer Center, Tucson, Arizona 85721, USA
cBIO5 Institute, The University of Arizona, Tucson, Arizona 85721, USA
dSouthwest Environmental Health Sciences Center, The University of Arizona, Tucson, 85721, USA
First published on 15th August 2022
Abstract
Atherosclerosis is a chronic and metabolic-related disease that is a serious threat to human health. Currently available diagnostic and therapeutic measures for atherosclerosis lack adequate efficiency which requires promising alternative approaches. Nanotechnology-based nano-delivery systems allow for new perspectives for atherosclerosis therapy. Surface-modified nanoparticles could achieve highly effective therapeutic effects by binding to specific receptors that are abnormally overexpressed in atherosclerosis, with less adverse effects on non-target tissues. The main purpose of this review is to summarize the research progress and design ideas to target atherosclerosis using a variety of ligand-modified nanoparticle systems, discuss the shortcomings of current vector design, and look at future development directions. We hope that this review will provide novel research strategies for the design and development of nanotherapeutics targeting atherosclerosis.
1. Introduction
1.1 Pathological properties and immunological mechanisms in atherosclerosis
Atherosclerosis is a chronic inflammatory disease of the arterial wall caused by abnormal blood lipids. Hypertension and high blood cholesterol levels were thought to be the primary factors affecting the pathogenesis of atherosclerosis, but new research demonstrated that disease progression is also regulated by innate and adaptive immune responses.1–3 In addition, inflammatory processes also play an important role in all stages of disease progression.4,5Pathological studies show that atherosclerosis mainly occurs in the turbulent areas of blood flow in the arterial wall,6 while in laminar flow areas, the incidence of plaque is very low.7 Arterial endothelial dysfunction at the site of atherosclerosis allows low-density lipoprotein (LDL) particles to pass through the endothelial layer and accumulate in the intima. Oxidized LDL nanoparticles activate the CD36 receptor on macrophages and form complexes with TLR4 and TLR6 receptors, leading to NF-κB activation and chemokine expression. Chemokines stimulate immune cell infiltration into the intima. Then atherosclerotic plaque begins to develop and macrophages engulf more and more LDL particles to form foam cells,8 which are prone to apoptosis and eventually necrosis. Smooth muscle cells begin to synthesize extracellular matrix components (e.g., proteoglycans, collagen) that constitute the plaque scaffold and fibrous cap covering, increasing the overall plaque size. At the same time, co-occurring cholesterol crystals within macrophages induce interleukin-1-beta (IL-1β) synthesis, thereby enhancing the inflammatory state within the plaque. Interactions with other plaque-infiltrating immune cells—neutrophils, T and B lymphocytes, and dendritic cells—lead to plaque instability and rupture, clinically manifesting as acute coronary syndrome.9,10
Numerous innate immune cells, including macrophages, dendritic cells (DCs), and monocytes, etc., are associated with atherosclerosis progression.11 At vascular sites of atherosclerosis, lipoproteins and chemokines (CCR2) accumulate subendothelially and trigger the influx of monocytes into the vessel wall. Monocytes differentiate into macrophages and monocyte-derived DCs that can present antigens to T cells.12 Monocyte-derived macrophages initiate uptake and clearance of lipoproteins, resulting in the formation of lipid-rich foam cells.13 The production of foam cells further triggers a cascade of inflammatory cytokines and chemokines, leading to the infiltration and activation of pro-atherosclerotic immune cells. When inflammatory macrophages accumulate, they increase the production of molecules encoding MHC class II, Fcγ receptor I (also known as CD64), costimulatory molecules CD80 and CD86, and proinflammatory cytokines such as IL-6, tumor necrosis factor (TNF) and IL-1β. For dendritic cells, conventional DCs (cDCs) and plasmacytoid DCs (pDCs) have been detected in atherosclerotic lesions.14 cDCs take up antigens in the vessel wall and activate draining lymph nodes or spleen antigen-specific naive T cells in atherosclerotic lesions, and maturation of plaque DCs in the presence of Toll-like receptor (TLR) agonists, risk-related ligands, and proinflammatory cytokines may favor pro-atherosclerosis. Similarly, in lesions with a necrotic core, CLEC9A-dependent sensing of necrotic cells by CD8α+ DCs promotes atherosclerosis by inhibiting the production of anti-inflammatory IL-10.15
Adaptive immunity is a key regulator of atherosclerosis.16 Studies have demonstrated that the role of MHC class II-mediated activation of CD4+ T cells by various APCs and the role of CD4+ TH cell subsets in atherosclerosis.17 At the same time, CD8+ T cells and lipid antigen-specific natural killer T cells and γδ T cells also play an important role in atherosclerosis. Studies have shown that CD8+ T cells accumulate in the aorta of atherosclerotic mice. Interferon-gamma, perforin, and granzyme B produced by CD8+ T cells promote vascular inflammation and lesion growth through apoptotic cell lysis.18,19
1.2 Current status of atherosclerosis treatment
At present, the primary treatment for atherosclerosis in clinical practice is lifestyle changes, such as healthy diet and exercise, followed by medicine or surgery. Clinical treatment drugs mainly include hypolipidemic drugs (statins), cholesterol absorption inhibitors (ezetimibe), angiotensin-converting enzyme (ACE) inhibitors (Captopril), beta-blockers (Propranolol) which may lower blood pressure and antiplatelet drugs such as aspirin to prevent blood from clotting and clogging arteries. These drugs can delay the progression of atherosclerosis. In addition, some new therapeutic targets are also being investigated, such as activators of peroxisome proliferator-activated receptor (PPAR), and endoglin receptor, etc.20,21 However, for the atherosclerotic plaque area, these drugs cannot specifically accumulate on the plaque site, cannot specifically prevent plaque rupture and plaque elimination, and may also bring systemic side effects. Therapeutic drugs exhibit off-target effects and fail to achieve effective therapeutic concentrations at tolerable doses. For example, rosiglitazone (PPARγ agonist), has anti-inflammatory and anti-atherosclerotic effects, but has also shown to cause weight gain, edema and fluid retention, and increased risk of heart failure.22,23 The side effects of edema are mainly caused by the off-target effects of PPARγ, which leads to enhanced reabsorption of sodium and water in the kidney.24 Statins (HMG-CoA reductase inhibitors) inhibit the synthesis of cholesterol by the liver and at the same time reduce the level of LDL in the blood circulation, thereby producing cardio protection and reducing the mortality of coronary heart disease. However, statins cannot relieve the local oxidative damage and inflammation of atherosclerosis.25 Moreover, satins have narrow therapeutic indications with severe systemic side effects. Although anti-cytokine antibodies and cytokine secretion inhibitors can reduce the inflammatory response that leads to atherosclerosis, they lack tissue specificity that can cause toxic side effects in clinical use.26 The suboptimal efficacy of these conventional therapies further demonstrates the urgent need to develop alternative effective treatment strategies for atherosclerosis.1.3 Nanotechnology-based drug delivery
Nanotechnology has brought major advances in the diagnosis and treatment of diseases, and the attractiveness of these nanoparticles for medical purposes is based on their important and unique properties, such as their large surface-to-mass ratio, their quantum properties, and their ability to adsorb and carry substances such as drugs, probes, nucleic acids (e.g., mRNA, siRNA) and proteins. Nanoparticles can be composed of organic materials such as lipids,27–29 polymers,30–39 metallic or inorganic materials such as iron oxides,40,41 gold42 and silicon,43 or a combination of these materials. The fabrication of nanoparticles can be precisely controlled, which allows them to vary in size, and to control their shape, surface charge, stability, and various other properties, ultimately affecting their in vivo behavior.44 The mode of administration or disease biology,45 heterogeneity between lesion sites may also influence the delivery efficiency of nanoparticles.46 Despite the influence of these factors, their delivery efficiency can be increased through the modification and optimization of nanoparticles. Furthermore, nanoparticles exhibit high surface area-to-volume ratios, allowing the use of surface coatings for modification with various molecules. This has special implications for interactions with cells. Among different nanomedicine applications, nanoparticle-based drug delivery has been the most widely explored. Advantages include, but are not limited to, improved pharmacokinetics of drugs and enable therapeutic use of drugs with certain disadvantages, such as low water solubility, poor bioavailability, poor tissue specificity, rapid metabolism, or severe side effects.47In the field of cardiovascular therapy, the advent of nanotechnology-based drug delivery brings new hope for improving the treatment of atherosclerosis as nanoparticles can solve the solubility problems plaguing most developed drugs, can also protect the encapsulated payloads from being recognized and eliminated by opsonization, thereby significantly prolonging the circulation time of the delivered drugs, as well as can enhance the targeting efficiency to diseased tissues based on EPR effects.48,49 However, the plaque site of the peripheral atherosclerotic area is small and the blood flow in the blood vessel is large, making it difficult for the nanoparticles to stay in the lesion site in large quantities and therefore posing a dilemma for the clinical use of nanoparticles. Fortunately, the active nano-drug delivery system based on ligand modification can specifically target the lesion, improving the treatment efficiency of drugs and reducing toxic side effects. Nanoparticles even eliminate plaque at the cellular level by modulating the response of leukocytes or macrophages to plaque accumulation.
Therefore, the ligands that recognize pro-inflammatory cell surface proteins expressed in early stages of atherosclerosis can provide an excellent opportunity to construct targeted nanodrug delivery systems as inflammatory cells are recruited in this process.
2. Surface-modified multifunctional nanoparticles for atherosclerosis
2.1 Cell adhesion molecules (CAMs)
Cell adhesion molecules (CAMs) play an important role in inflammatory cell recruitment and mediate cam-based ligand internalization, thus having significant implications for the targeted delivery of nanoparticles. Intercellular cell adhesion molecule-1 (ICAM-1) is a member of CAM immunoglobulin superfamily glycoproteins. Icam-1 is usually expressed in the lumen of endothelial cells and is significantly upregulated approximately 20–50 times after local inflammation. Immunohistochemical analysis showed that ICAM-1 was also strongly expressed in atherosclerotic vascular cells, thus demonstrating its role in disease progression. Some peptides have been shown to specifically target ICAM-1.50,51 Based on this property, cyclo(1,12)PenITDGEATDSGC (cLABL) peptide was used to target nanoparticles to the monolayer of vascular endothelial cells (HUVEC), where intercellular adhesion molecule-1 (ICAM-1) expression was significantly upregulated.52 In this study, poly (DL-lactate-coglycolic acid) nanoparticles conjugated with polyethylene glycol and cLABL rapidly bound to HUVEC. Specific resistance experiments further demonstrated that the binding of nanoparticles is specifically mediated by the binding of surface peptides to ICAM-1 on HUVEC. Compared with untargeted PLGA-PEG nanoparticles, targeted nanoparticles are rapidly endocytosed and transported to lysosomes to a greater extent. This experiment demonstrates that cLABL peptide-mediated nanoparticles targeting ICAM-1 eventually lead to therapeutic agents targeting inflammatory sites where ICAM-1 expression is upregulated.53Similar to ICAM-1, the vascular cell adhesion molecule (VCAM-1) is also a candidate target for the design of targeted delivery of nanoparticles.54,55 VCAM-1 is a transmembrane protein, a member of the immunoglobulin super-family, expressed by endotheliocytes and other cell types under stimulation of cytokines like IL-4, IL-1β, and TNF-α. In particular, VCAM-1 has been investigated because it is scarcely expressed by healthy endothelium while it is rapidly upregulated in damaged and lesion-predisposed vascular regions.56–58
The researchers prepared targeted liposomes containing DSPE-PEG2000-maleimide59 and carried out a coupling reaction with the terminal cysteine-SH of the targeting peptide VHPKQHRGGSKGC. The authors confirmed the binding of targeted liposomes to TNF-α-stimulated endothelial cells with high expression of VCAM-1 through flow cytometry and fluorescence microscopy. They also proved that the binding of activated cells was higher than that of non-activated cells and that it can be completely blocked by pre-incubating the soluble recombinant VCAM-1 protein. This experiment further demonstrates the guiding effect of targeting peptides on nanoparticles. Similarly, Kheirolomoom et al. used this peptide to develop a new type of cationic lipid particle for the delivery of anti-miR-712 drugs.60 The targeting properties of the nanoparticles were tested on immortalized TNF-α-stimulated murine endothelial cells (iMAEC). It was observed via fluorescence microscopy that compared with non-targeted lipid particles and unstimulated cells; the amount of targeted lipid particles accumulated in the stimulated cells significantly increased. Most importantly, the targeting particles only induced a significant reduction in the expression of miRNA-712 in activated cells. The results of frontal confocal imaging of the carotid artery endothelium showed that the accumulation of nanoparticles in the intimal layer of the VCAM-1 area overexpressing in ApoE−/− mice was significantly increased.
In addition to being used to deliver therapeutic drugs, various strategies have been investigated using nanoparticles for imaging purposes based on high affinity peptides targeting VCAM-1. Through a modified phage display method, Several VCAM-1 binding peptides were identified by Kelly et al. and a peptide with the sequence VHSPNKK was discovered.61 Using peptides, the authors developed a targeted imaging system cyanine dye (CLIO-Cy5.5) composed of labeled cross-linked iron oxide NP. Through in vitro analysis, the specificity of targeted nanoparticles for endothelial cells was found to be higher than that for macrophages. In addition, there is evidence that CLIO-Cy5.5 can target inflamed tissues. This nano-system was also tested on ApoE−/− mice and was detected by magnetic resonance imaging (MRI) in vivo. These detected in vivo changes in MR signal proved the cumulative accumulation of nanoparticles in atherosclerotic lesions. Based on this work, the authors also showed the capability of detecting early atherosclerotic lesions in young mice and of targeting human VCAM-1 in ex vivo human carotid endarterectomy specimens.62
2.2 Integrin
Integrins, molecules also implicated in atherosclerosis, are heterodimeric glycoproteins constituted by the non-covalent association of an α- and a β-subunit, with molecular masses ranging from 100 to 160 kDa.63 Studies have shown that blood vessels around newly formed plaques also show increased expression of αvβ3 integrin. Because of this, integrin can provide nutrients to developing atherosclerotic plaques, promote intraplaque hemorrhage, and is therefore considered both a target for diagnostic probes and a potential therapeutic.64–66 Imaging techniques targeting αvβ3 integrins have been used in experimental studies of atherosclerosis. Synthesis of lipid-coated perfluorooctyl bromide-cored NPs loaded with large amounts of lipophilic Gd chelates and complexed with non-peptide αvβ3 covalently bound to PEG distearoylethanolamine derivatives body is functionalized and has targeting atherosclerosis ability. These NPs were shown to target the abdominal aorta of atherosclerotic rabbits in vivo.67 Furthermore, the researchers conducted studies to develop anti-angiogenic treatment regimens based on theragnostic ανβ3-targeted fumagillin nanoparticles. These nanoparticles were able to reduce the neovascularization signal by 50% to 75% at 1 week and maintain this effect for 3 weeks. However, atherosclerotic rabbits treated with statins alone had no anti-vascular effects at 8 weeks. This experiment further proves that integrin-modified nanoparticles can improve the therapeutic efficiency of drugs for atherosclerosis.68RGD is another excellent targeting moiety that activates endothelial cell integrin αvβ 3 and has proven successful for imaging angiogenesis in numerous preclinical settings.69 Based on the RGD peptide, the researchers developed a new radiotracer 99mTc-labeled RGD peptide (99mTc-IDA-D-[c(RGDfK)]2) and used it for single-photon detection of glioblastoma via emission computed tomography (SPECT) imaging.70 This formulation has a high specific integrin-binding affinity compared to other reported RGD monomer-based formulations.71,72 Similar to imaging tumor angiogenesis, this radiotracer is expected to efficiently image new blood vessel-rich atherosclerotic plaques and demonstrate better imaging performance. On this basis, Zhang's team also used cRGDfK peptide to prepare apoptotic body-mimetic liposomes (AP-Lipo) for the selective delivery of PPARγ agonist pioglitazone (PIO) into atherosclerotic macrophages, by upregulating anti-inflammatory M2 macrophages while minimizing the side effects of PPARγ agonists (Fig. 1). After intravenous injection, the cRGDfK peptide modified on AP-Lipo can specifically bind to the integrin αvβ3 receptor of activated vascular endothelial cells in atherosclerotic plaques, promoting its penetration into the plaque.73
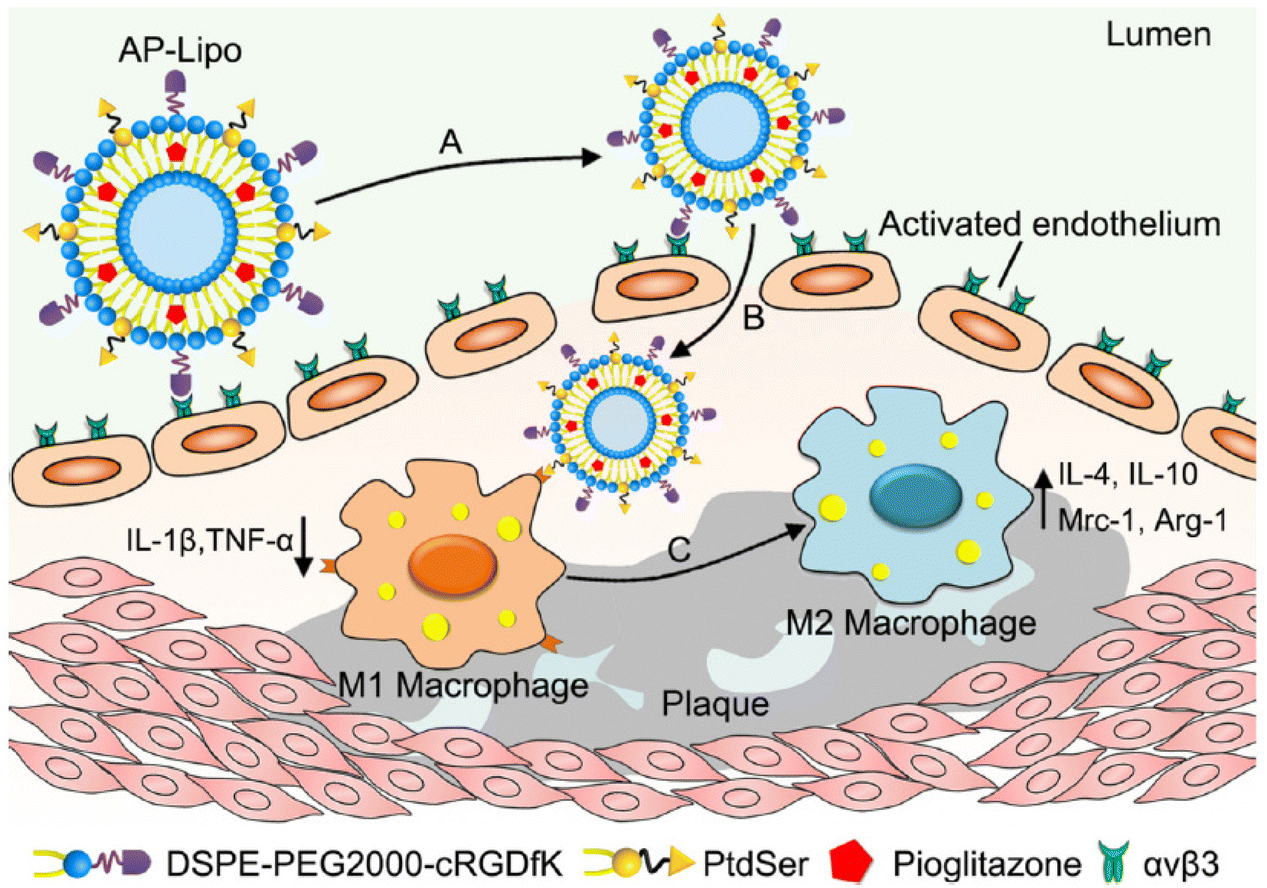 | ||
| Fig. 1 AP-Lipo in situ upregulates anti-inflammatory macrophages for atherosclerosis regression. (A) AP-Lipo selectively targeted to the activated vascular endothelial cells by the interaction between cRGDfK and ανβ3 integrin. (B) AP-Lipo entered atherosclerotic plaques through enhanced permeability and retention effect and would be recognized and uptake by M1 macrophages due to the “eat-me” signal of PtdSer. (C) AP-Lipo generated anti-inflammatory response by increasing M2 macrophages polarization or increasing the M2 macrophages number in atherosclerotic plaques. Adapted with permission from ref. 73. Copyright (2019) Elsevier. | ||
2.3 Collagen
Collagen in atherosclerotic lesions can also serve as a target for atherosclerotic plaques. Studies have shown that collagen type IV is abundant in blood vessels and is easily exposed when tissue permeability increases due to the formation of atherosclerotic plaques.74,75 More importantly, as atherosclerosis progresses, the amount of type IV collagen increases in atherosclerotic plaques.76 Studies have shown that collagen type IV targeting peptides are effective ligands for nanoparticles targeting atherosclerosis.77,78 Farokhzad's group synthesized Col IV peptide-conjugated targeting polymers by conjugating the targeting peptide KLWVLPK to PLGA-PEG-Mal using maleimide chemistry via the free thiol of the C-terminal GGGC linker (Fig. 2).77 They used the nanoparticles to target the delivery of the anti-inflammatory cytokine interleukin 10 (IL-10) to atherosclerotic plaques. In vitro and in vivo studies showed that the most potent nanoparticle, Col-IV IL-10 (NP22), significantly alleviated acute inflammation in a self-limited peritonitis model and was shown to be more effective than IL-10. Furthermore, Col-IV IL-10 nanoparticles prevented the formation of vulnerable plaques by increasing fibrous cap thickness and reducing advanced necrotic cores in a lesion model in high-fat-fed LDL−/− mice. These results demonstrate the efficacy and pro-resolution potential of this targeted modification of nanoparticles for controlled delivery of potent IL-10 cytokines for the treatment of atherosclerosis. They also used this targeted nanoparticle to deliver Ac2-26 peptides to treat inflammatory responses.79 In addition to type IV collagen, the change of type I collagen composition is also an important indicator of plaque progression, which is related to the thickness of the fibrous cap, plaque stability and atherosclerotic injury. In this study, Chen et al. developed high-density lipoprotein particles using type I collagen-binding peptide GKWH[CTTKFPHHYC] for magnetic resonance imaging (MRI) in mouse models.80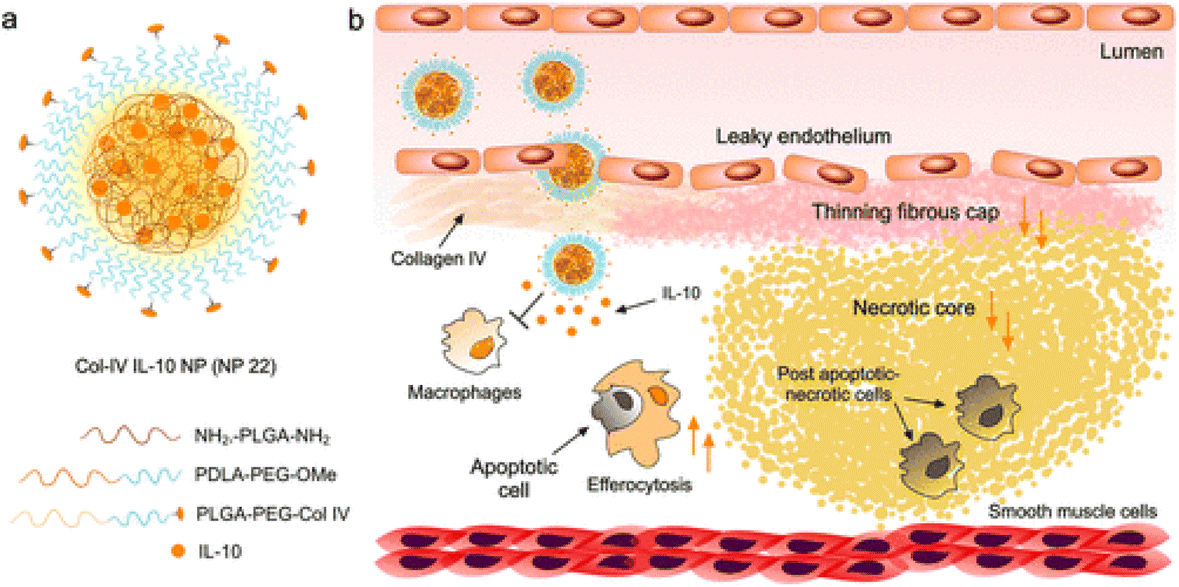 | ||
| Fig. 2 Targeted anti-inflammatory NP design and application to the resolution of inflammation in atherosclerotic plaques. (a) Col-IV IL-10 NP (NP22) identified from screening 24 formulations was fabricated by nanoprecipitation using a glass microfluidic rapid-mixing chip. The NPs were formed in a single self-assembly step and consisted of a blend of NH2-PLGA-NH2, PDLA-PEG-OMe, PLGA-PEG-Col IV, and IL-10 in addition to D-(+)-glucosamine hydrochloride as an IL-10-stabilizing additive and cryoprotectant. (b) The targeted NPs can enter plaques via leaky endothelial junctions and bind to exposed collagen IV (Col IV) and release their therapeutic inflammation-resolving IL-10 payload within the plaque over time, resulting in increased efferocytosis, an increase in cap size, and a decrease in necrotic core size (orange arrows). Adapted with permission from ref. 77. Copyright (2016) American Chemical Society. | ||
2.4 Fibrin
Fibrin is a major component of thrombi that form on the surface of atherosclerotic plaques.81 Fibrin accumulates within atherosclerotic lesions and contributes to the growth of thrombi and may also reflect the inflammatory state of atherosclerosis.82 Therefore, the presence of fibrin in mature plaques becomes a specific marker for the targeted delivery of imaging and anticoagulant agents to the site of atherosclerotic lesions. Holger's group uses peptide Ac-RWQPCPAESWT-Cha-CWDPGGGK-NH2, containing the fibrin binding motif RWQPCPAESWT-Cha-CWD prepared peptide modified Iron Oxide Nanoparticle-Micelles (FibPep-ION-Micelle) platform for in vivo, non-invasive imaging of fibrin in preclinical disease models of thrombus-related pathologies and atherosclerosis.83 Previous studies have shown that pentapeptides cysteine-arginine-glutamine-lysine-alanine (CREKA) screened by phage display can bind to plasma protein clotting plaques in blood vessels. The Erkki team then used the peptide to develop modular multifunctional micelles that targeted atherosclerotic plaques in ApoE−/− mice (Fig. 3).84 Experimental results showed that fluorescent-labeled micelles bind to the entire surface of the plaque, especially concentrated in the shoulder of the plaque, which is prone to rupture. The results also showed that targeted micelles could enrich anticoagulant leeches in plaques compared with untargeted micelles. These experimental results further demonstrate that fibrin-modified nanoparticles can effectively improve its targeting ability to arteriosclerosis.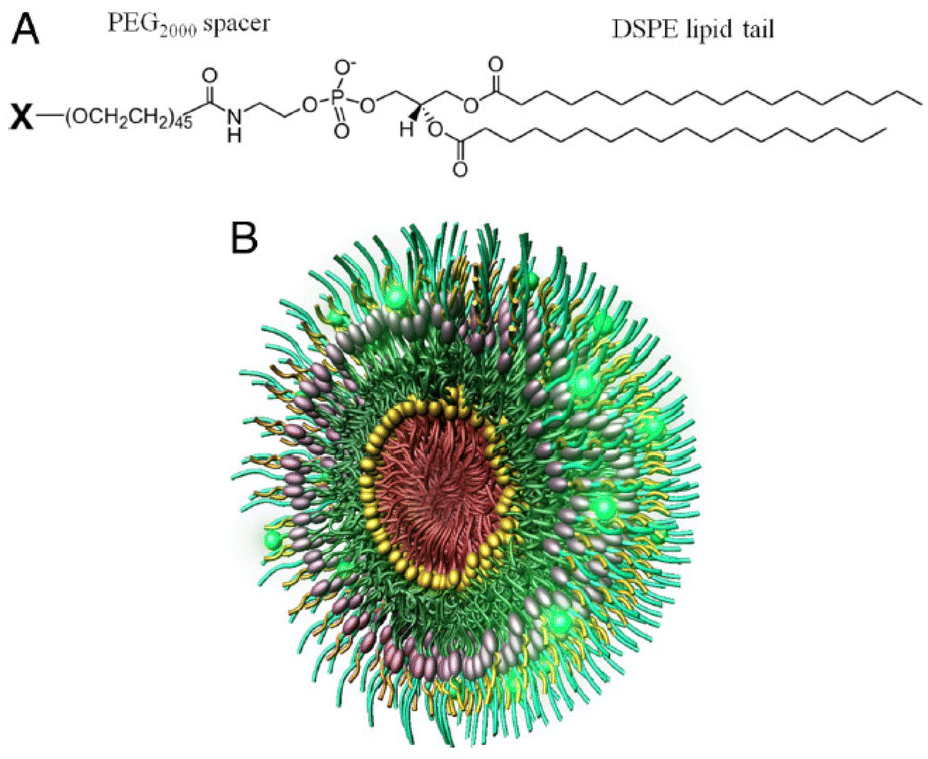 | ||
| Fig. 3 Construction of modular multifunctional micelles. (A) Individual lipopeptide monomers are made up of a DSPE tail, a poly (ethylene glycol) (PEG2000) spacer, and a variable polar head group (X) of CREKA, FAM-CREKA, FAM, N-acetylcysteine, Cy7, or hirulog. The monomers were combined to form various mixed micelles. (B) The 3D structure of FAM-CREKA/Cy7/hirulog mixed micelle. Adapted with permission from ref. 84. Copyright (2009) PNAS. | ||
To increase treatment efficiency, combination nanotherapies have also been explored. Wang et al., used CREKA peptide to prepare ROS-responsive self-assembling prodrug micelles for co-delivery of simvastatin, α-Tocopherol and ticagrelor. Simvastatin is conjugated to an α-tocopherol polyethylene glycol derivative via a ROS-responsive cleavable thioketal bond to form a simvastatin nanoprodrug (TPTS). TPTS has both hydrophilic and hydrophobic components, and it self-assembled into nanoparticles. A fibronectin targeting drug delivery system (TPTS/C/T) was further constructed using CREKA peptide to synergistically deliver ticagrelor to atherosclerotic plaque sites, which achieved efficient anti-inflammatory activity in an ApoE−/− mouse model with excellent atherosclerosis therapeutic efficacy and a promising biosafety profile.85
2.5 Chemokine receptor
Atherosclerosis is an inflammatory disease characterized by enhanced immune activity and the accumulation of lipid-rich plaques. The activation of endothelial cells within the arterial wall secretes monocyte chemoattractant protein 1 (MCP-1, also known as Called CC chemokine ligand 2) that binds to the CC chemokine receptor CCR2.86,87 Aggregated monocytes differentiate into macrophages, leading to further tissue damage by secreting more cytokines, chemokines, and toxic oxygen and nitrogen free radicals.88 Mononuclear cell accumulation in the arterial wall was observed in fatty streaks, one of the earliest visible lesion features in human and experimental atherosclerosis. In an ApoE-knockout mouse model of atherosclerosis, monocyte accumulation is continuous and proportional to disease progression. Therefore, CCR2-based monocyte targeting is also a strategy for the diagnosis and treatment of atherosclerosis.89,90 To construct monocyte-targeting peptide amphiphile micelle (PAM), the CCR2-binding motif (residues 13–35) of MCP-1(YNFTNRKISVQRLASYRRITSSK), was used to modify the PAM. The results demonstrate that MCP-1 PAMs can bind to monocytes in vitro and that MCP-1 PAMs can detect and differentiate between early and late atherosclerotic aortas (Fig. 4).91 Therefore, MCP-1 PAMs are promising and safe novel nanoparticles capable of monitoring the progression of atherosclerosis. We believe that the nanoparticles can also be further used for atherosclerosis treatment.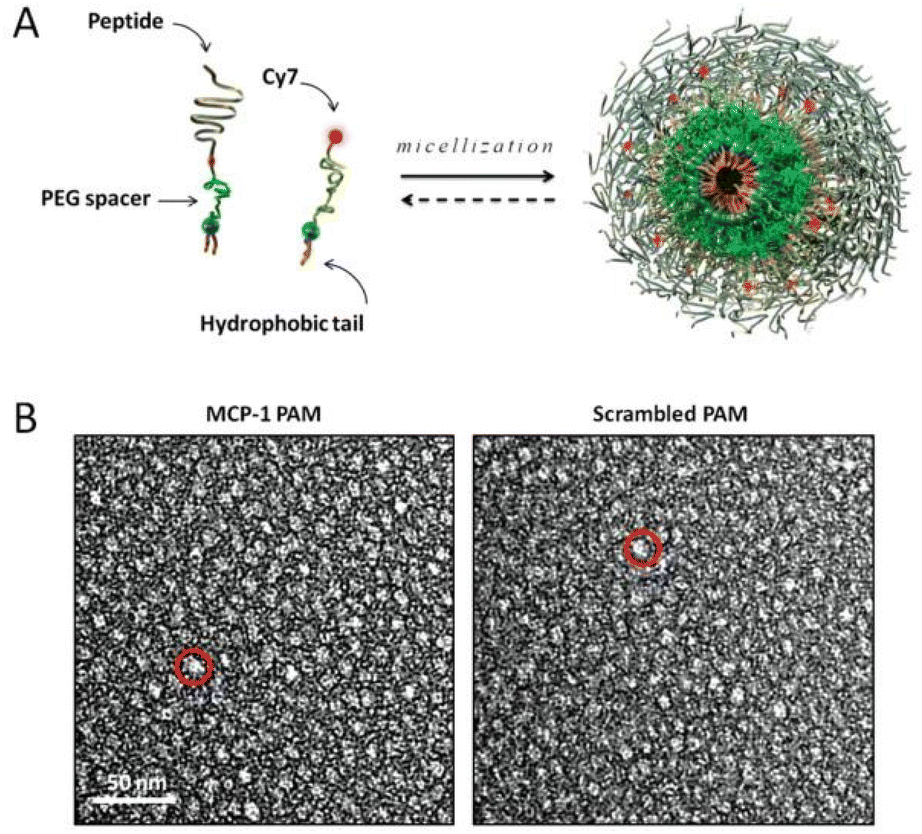 | ||
Fig. 4 Design and structure of MCP-1 PAMs. (A) Schematic depicting PAM self-assembly. PAs consist of a di-stearoyl hydrophobic tail (two 18-carbon chains) and a PEG spacer that was conjugated to the MCP-1 peptide that corresponds to the CCR2-binding motif (residues 13–35), scrambled peptide, or the Cy7 fluorophore. Fluorescently labeled, mixed micelles consisted of peptide-containing and Cy7-labeled amphiphiles in a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 molar ratio. (B) Representative TEM images of MCP-1 PAMs (L) and scrambled PAMs (R). Both micelles are spherical in shape with a diameter on the order of 10 nm. Adapted with permission from ref. 91. Copyright (2015) Wiley. :10 molar ratio. (B) Representative TEM images of MCP-1 PAMs (L) and scrambled PAMs (R). Both micelles are spherical in shape with a diameter on the order of 10 nm. Adapted with permission from ref. 91. Copyright (2015) Wiley. | ||
Besides CCR2, chemokine receptor 5 (CCR5) has been reported as an active participant in late atherosclerosis and as a prognostic biomarker of plaque stability.92–94 It is also widely studied as a potential receptor for targeting atherosclerosis.95–97 To develop nanoparticles targeting atherosclerosis, the researchers chose the D-Ala1-peptide T-amide (DAPTA) peptide as the targeting ligand for the CCR5 receptor. A biocompatible polymethylmethacrylate core/polyethylene glycol shell amphiphilic comb nanoparticles were prepared and labeled with 64Cu (64Cu-DOTA-DAPTA) for CCR5-targeted imaging in an ApoE−/− injury model. 64Cu-DOTA-DAPTA tracer showed specific PET imaging of CCR5 in ApoE−/− mice. Compared with the sham-operated model, the targeted nanoparticles had higher accumulation in the lesioned model. Competitive PET receptor blockade studies further confirmed the specific uptake of nanoparticles for the CCR5 receptor.97
2.6 Scavenger receptor B1
Class B1 scavenger receptors (SR-B1) are membrane glycoproteins.98,99 As cell surface receptors, they can recognize different ligands to participate in different biological functions100,101 including endocytosis, phagocytosis, adhesion, and signal transduction. SR-BI exhibits broad ligand specificity and also recognizes low-density lipoproteins (LDLs),102 oxidized LDLs,103 and very low-density lipoproteins.104,105 Studies have shown that SR-BI plays an important role in high-density lipoprotein (HDL) metabolism.106,107 SR-BI can bind to HDL and mediate selective lipid uptake.107,108 As an endogenous particle, HDL is mainly composed of phospholipids and amphiphilic α-helical apolipoprotein A-I (ApoA-I).109,110 The structure of HDL provides new opportunities for the transport of cholesteryl esters and lipophilic compounds including drugs.110,111 The limited sources of endogenous HDL and the complicated extraction process restrict its widespread use. Therefore, reconstituted HDL nanoparticles (rHDL NPs) become ideal candidates in vitro.112–114 In 2003, the Nissen et al. group validated the anti-atherosclerotic efficacy of rHDL NPs in acute coronary patients,115 laying a solid foundation for their use in the treatment of anti-arteriosclerosis.Inspired by this study, the researchers started the research about rHDL NPs as a drug delivery system and expected to further intensify the efficacy of atherosclerosis treatment by exploiting the synergistic effect of therapeutic drugs with rHDL NPs.116,117 For example, Mulder's group reported an injectable rHDL NPs that delivers statins to atherosclerotic plaques (Fig. 5).118 In this research, they identified the anti-inflammatory effect of statin loading rHDL in vitro and showed that this effect is mediated through inhibition of the mevalonate pathway. They also intravenously injected statin-rHDL nanoparticles in a mouse model with atherosclerosis (apolipoprotein E knockout) and demonstrated that they could accumulate in atherosclerotic lesions. Finally, they demonstrate that a three-month low-dose statin-rHDL regimen (15 mg kg−1 simvastatin, 10 mg kg−1 ApoA-1, 2 injections/week) effectively inhibits plaque inflammation progression, while a one-week high-dose regimen (60 mg kg−1 simvastatin, 40 mg kg−1 ApoA-1, 4 injections per week) significantly reduces inflammation in advanced atherosclerotic plaques. This experiment demonstrates that statin-rHDL represents a novel and potent atherosclerotic nanotherapy that directly targets plaque inflammation.
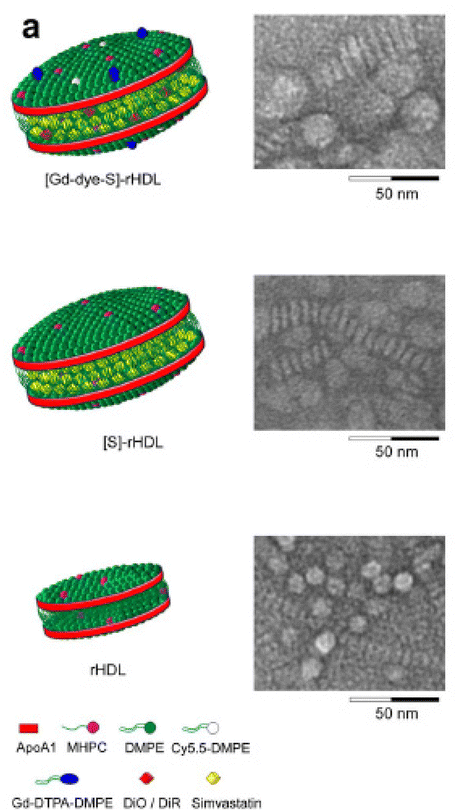 | ||
| Fig. 5 Schematic representations of the nanoparticle formulations and in vitro efficacy data (a) schematic representation of dual gadolinium and fluorescent dye (Cy5.5, DiO, DiR) labeled statin containing reconstituted high-density lipoprotein ([Gd-dye-S]-rHDL), statin containing rHDL ([S]-rHDL), and rHDL. Negative staining transmission electron microscopy (TEM) images of each of the aforementioned particles showed the typical disk-like morphology. The circular shapes are nanoparticles viewed enface, while the striped configurations are rouleaux of nanoparticles viewed from the side. Adapted with permission from ref. 118. Copyright (2014) Springer Nature. | ||
Studies have shown that drugs in rHDL are prone to leakage in the circulation because of lecithin cholesterol acyltransferase-induced remodeling (LCAT), a phenomenon that reduces the amount of drug delivered to the lesion. To deliver more drug into target cells for improved efficacy, some researchers employed a dual ligands-modification strategy to improve the targeting efficiency of nanoparticles. In addition to ApoA-1 ligand targeting, lovastatin (LT)-loaded d-rHDL was concurrently coated with arachidonic acid (AA), 20-carbon chain with four double bonds and negative charge.119 Studies have shown that with the increasing amount of AA modification, AA-LT-d-rHDL has a lower reactivity with LCAT, allowing it to significantly reduce the LCAT-induced drug leakage behavior and effectively inhibit the formation of macrophage-derived foam cells. The main reason why rHDL can bind to macrophage-derived foam cells is that foam cells overexpress SR-BI that can specifically recognize rHDL and take rHDL into foam cells through endocytosis.120,121 However, in vivo experiments in mice demonstrated that rHDL NPs may still accumulate in the liver because SR-BI is also overexpressed in hepatocytes.122–124 Therefore, in order to reduce the uptake of rHDL-encapsulated drugs by the liver and actively increase the accumulation of drugs in atherosclerotic lesions, researchers attempted to modify rHDL with hyaluronic acid (HA).125 Because HA is a ligand for CD44, the CD44 receptor is highly expressed in inflammatory sites such as atherosclerosis. Based on this, the researchers selected lovastatin (LT) as the model drug and further constructed HA-modified rHDL loaded with LT (HA-LT-rHDL) (Fig. 6).125 The nanoparticles would reduce liver recognition by shielding SR-BI with HA coating, thereby prolonging the circulation time in the blood, and then massively accumulating in atherosclerotic lesions through CD44-mediated targeting. Once across the endothelium, HA is degraded by the abundant hyaluronidase (HAase) within the plaque,126 which then exposes drug-loaded rHDL, and finally, foam cells endocytose rHDL into the cell via SR-BI on the surface.
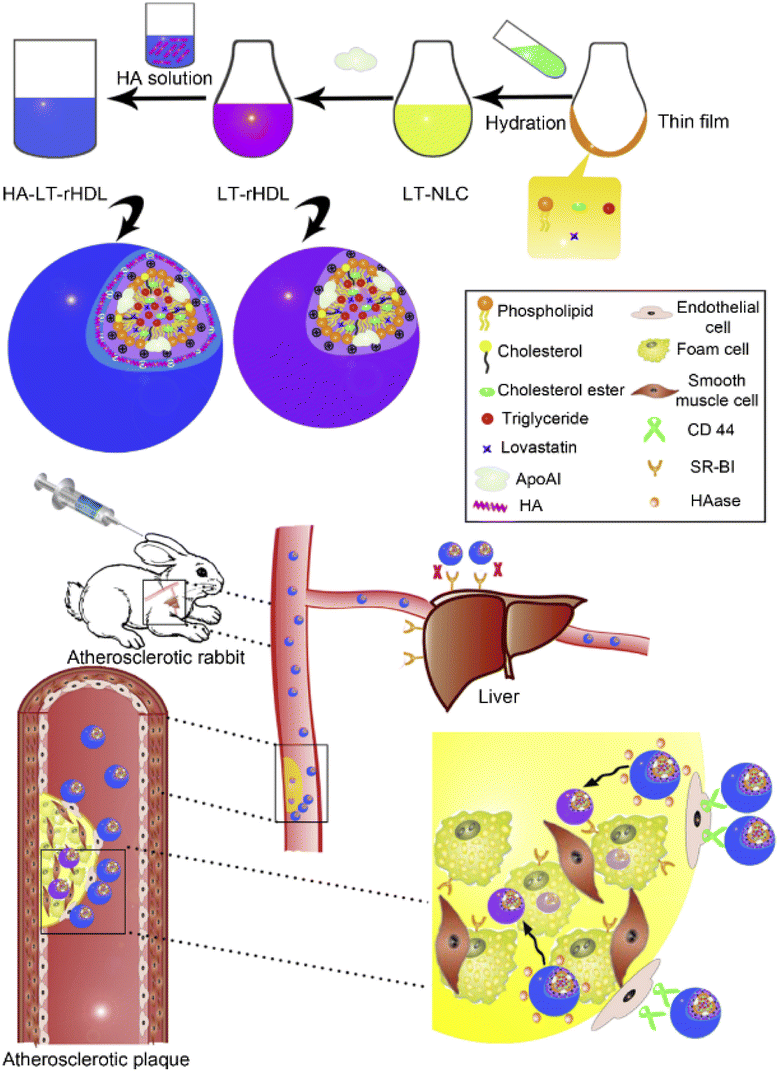 | ||
| Fig. 6 Schematic diagram on preparation methods and atherosclerotic lesion targeting property of HA-LT-rHDL. Adapted with permission from ref. 125. Copyright (2014) Elsevier. | ||
In addition to delivering chemical drugs, the researchers also designed dual-targeted nanoparticles for co-delivery of chemical drugs and gene drugs. On the basis of previous studies, they constructed PLGA as the core and coated it with a lipid bilayer by electrostatic adsorption as the inner layer and apolipoprotein A-I (apoA-I) as the macrophage-targeted middle layer, hyaluronic acid (HA)-DOPE as the outermost layer targeted by endothelial cells. The co-delivery of LOX-1 siRNA and Atorvastatin through the dual-targeted nanoparticles can synergistically act on plaque regression by preventing lipid deposition and accelerating intracellular lipid removal.127
3. Discussion and clinical translation challenges
In order to improve the targeting efficiency of nanoparticles in atherosclerotic sites, researchers designed various ligand-modified targeting nanoparticles by taking advantage of the physiological properties of highly expressed receptors in the lesion site. In this review, we summarize the design ligand modification strategies for targeting nanoparticles based on the current design of atherosclerosis-targeting nanoparticles (Table 1). We summarize the design ofligand modification strategies for atherosclerosis-targeting nanoparticles. We can clearly see that the currently used ligands are all concentrated in polypeptides, which boasts inherent biocompatibility and biodegradability, providing additional advantages for biomaterials. Unlike antibodies, peptides are small enough in molecular weight to fit into shallow or hydrophobic binding pockets without compromising specificity or affinity. The targeting ability is enhanced by attaching multiple polypeptide molecules to a single nanoparticle compared to antibodies with larger molecular weights.128 What's more, unlike proteins, peptides that do not exist in nature can be synthesized in the laboratory using liquid or solid-phase synthesis methods. The versatility of chemical design is achieved through changing amino acid sequence or primary structure. In addition, the development of random peptide libraries selected for interaction with specific epitopes by phage display technology is another research strategy.129| Receptor targeted | Ligand | Type of nanomedicine | Drug loaded | Ref. |
|---|---|---|---|---|
| Cell adhesion molecules (CAMs) | cyclo(1,12)PenITDGEATDSGC (cLABL) | Poly(DL-lactic-co-glycolic acid) nanoparticles (PLGA) | None | 53 |
| VHPKQHRGGSKGC | Cationic lipoparticles | Anti-miR-712 | 60 | |
| VHSPNKK | Iron oxide nanoparticles | Cy5.5 | 61 | |
| Integrin | Peptidomimetic vitronectin antagonist | Paramagnetic nanoparticles | Gd chelates | 67 |
| αvβ3-Integrin antagonist | Iaramagnetic nanoparticles | Fumagillin | 68 | |
| IDA-D-[c(RGDfK)]2 | 99mTc-IDA-D-[c(RGDfK)]2 | 99mTc | 70 | |
| cRGDfK | Apoptotic body biomimic liposome | Pioglitazone (PIO) | 73 | |
| Collagen | KLWVLPK | Col-IV IL-10 nanopartcles | Interleukin 10 | 77 |
| GKWH[CTTKFPHHYC] | High density lipoprotein | Gadolinium diethylenetriaminepentaacetate-bis(stearylamide) (Gd-DTPA-BSA) | 80 | |
| Fibrin | Ac-RWQPCPAESWT-Cha-CWDPGGGK-NH2 | Iron Oxide Nanoparticle-Micelles (FibPep-ION-Micelle) | Iron oxide | 83 |
| CREKA | Micelles | Carboxyfluorescein, hirulog peptide | 84 | |
| CREKA | Micelles | Simvastatin, α-tocopherol and ticagrelor | 85 | |
| Chemokine receptor | MCP-1(YNFTNRKISVQRLASYRRITSSK) | Peptide amphiphile micelle | Cy7 | 91 |
| D-Ala1-peptide T-amide (DAPTA) peptide | Polymethylmethacrylate core/polyethylene glycol shell amphiphilic comb nanoparticles | 64Cu | 97 | |
| Scavenger receptor B1 | Apolipoprotein A-I | Reconstituted HDL nanoparticles(rHDL) | Simvastatin | 118 |
| Apolipoprotein A-I and arachidonic acid (AA) | rHDL | Lovastatin | 119 | |
| Apolipoprotein A-I and hyaluronic acid (HA) | rHDL | Lovastatin | 125 | |
| Apolipoprotein A-I and hyaluronic acid (HA) | PLGA | LOX-1 siRNA and Atorvastatin | 127 |
Although these polypeptide-modified nanoparticles can effectively improve the targeting efficiency, there are differences among them. For example, studies have evaluated the targeting ability of both RGD peptides and collagen IV targeting peptides. In this experiment, cRGD or collagen IV targeting (Col IV-tg-) modified pluronic nanocarriers containing iron oxide nanoparticles (IONPs) were prepared. The ability to target atherosclerotic plaques was evaluated in an apolipoprotein E-deficient (Apo E−/−) mouse model. The results suggest that cRGD-based targeting is more effective than Col IV-tg-peptide in the early stages of atherosclerosis.130 In addition, compared to other targeting peptides, apolipoprotein A-I also has anti-inflammatory properties and promotes reverse cholesterol transport, which clears cholesterol from lipid-rich macrophage foam cells to the liver for elimination.131,132 This characteristic makes the rDHL nanoparticles composed of apolipoprotein A-I have great potential for clinical translation. The impact of different ligands on the delivery efficiency of nanoparticles needs further investigation, and the differences of current targeting approaches also provide more stringent requirements for further optimizing the design of nanoparticles.
Even with promising results in preclinical studies, many development hurdles arose for atherosclerosis-targeted nanomedicines, limiting their success in clinical translation. Among the various hurdles, the first is the large-scale production of targeted nanoparticles as it faces continual issues such as multi-step preparation which poses challenges to the reproducibility of its preparation.133–135 In the process of clinical translation, we also need to understand atherosclerosis is a long-term chronic disease. Long-term repeated intravenous administration is not ideal and is likely to result in poor compliance, yielding obstacles to impede the clinical translation. In addition, alternative effective delivery strategies including local or oral drug delivery platforms are needed to complement the intravenous injections.136 For instance, one study investigated the self-assembled oral nanoparticles of chitosan (CS) and aspartic acid (PAA) loaded with (−)-epigallocatechin gallate (EGCG), which reduced the gastrointestinal side effects of EGCG and improved the efficacy of oral EGCG for the treatment of atherosclerosis in rabbits.137
Second, although nanoparticles have been proven safe in preclinical studies, further research is needed on the specific metabolic pathways and metabolites of nanomaterials. For example, some inorganic materials such as silicon,138,139 gold,140,141 and iron142,143 cannot be metabolized in the body, so there is a risk of nephrotoxicity. Therefore, novel designed nanoparticles should be rigorously evaluated before conducting human studies.
Third, there is also the issue of the adaptability of targeting ligands and highly expressed receptors at the lesion site.144,145 When targeting vascular endothelial cells, it is difficult to avoid the aggregation of nanocarriers in the pulmonary vasculature and the hepatic reticuloendothelial system. Although nanoparticles can be modified with PEG or HA to reduce the uptake of nanoparticles by the liver,125 this strategy makes it difficult to avoid the uptake of nanoparticles in the lung, which has been reported to account for 30% of the endothelial surface in vivo and receives the entire cardiac output.146,147 Therefore, when designing atherosclerosis-targeting nanoparticles, differences in cell surface expression molecules in other organs need to be assessed in detail to avoid off-target effects. For this issue, the intracellular signaling pathways at the lesion site should also be further elucidated, which is beneficial for screening new molecular markers to better target the lesion site. For example, new ligands that effectively bind these targets can be rapidly discovered using advanced phage display technology.148,149 Thus, highly specific, non-immunogenic, and small-sized ligands, including peptides and peptidomimetics, are used in the transformation of them into nanocarriers, leading to the development of more selective and efficient nano-drug delivery systems.
Moreover, further elucidation of the underlying functional mechanisms of nanoparticles is required in future studies, such as strategies regarding the mechanisms of interaction between immune cells and nanoparticles and especially regarding the fate of nanoparticles and their components after deposition in atherosclerotic lesions and the impact on immune cells.150–153 Such research is expected to help construct nanoparticles for targeted delivery to obtain specific responses and future studies are needed to improve the clinical outcomes of targeted therapy.
4. Conclusion
In recent decades with the development of nanotechnology, nanoparticle-based drug delivery systems have emerged as a powerful platform for improving the treatment and diagnosis of atherosclerosis. Although nanoparticles can significantly improve the in vivo circulation time, tissue targeting, and reduce toxic and other side effects of drugs, atherosclerotic blood flow is large, the flow rate is fast, and the retention capacity of nanoparticles in the lesion site is poor, resulting in low drug delivery efficiency. We know that the surrounding environment for atherosclerotic lesions also undergoes significant biological changes, such as high expression of inflammatory factors and receptors, which are significantly different from surrounding healthy tissue, thus providing a basis for the design of targeted nanoparticles. This article summarizes the current specific receptors based on atherosclerosis and the targeted nanoparticles designed based on the biology of these receptors. These studies solved the bottleneck of nanoparticle-mediated drug entry into atherosclerosis to an extent, realized the targeted drug delivery at the lesion site, and effectively improved the efficacy of the treatment of atherosclerosis. Although these reported nanoparticles exhibit satisfactory preclinical findings, it will be challenging to translate the findings of these studies into possible work. Future research needs to focus on enhancing its safety, stability, and efficacy in vitro and in vivo. At the same time, we should also pay attention to atherosclerosis as a systemic disease and the treatment plan combining nanoparticles with gene therapy and monoclonal antibody to further improve the treatment efficiency. We believe that these strategies will open new horizons for the treatment of atherosclerosis.Conflicts of interest
The authors report no conflicts of interest in this work.Acknowledgements
This work was supported in part by a Startup Fund from the R. Ken Coit College of Pharmacy at The University of Arizona and a PhRMA Foundation Research Starter Grant in Drug Delivery, and by National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM147002.References
- P. Libby, Nature, 2021, 592, 524–533 CrossRef CAS PubMed.
- M. Rafieian-Kopaei, M. Setorki, M. Doudi, A. Baradaran and H. Nasri, Int. J. Prev. Med., 2014, 5, 927 Search PubMed.
- P. Roy, M. Orecchioni and K. Ley, Nat. Rev. Immunol., 2022, 22, 251–265 CrossRef CAS PubMed.
- P. Libby, Clin. Chem., 2021, 67, 131–142 CrossRef PubMed.
- O. Soehnlein and P. Libby, Nat. Rev. Drug Discovery, 2021, 20, 589–610 CrossRef CAS PubMed.
- C. Su, N. V. Menon, X. Xu, Y. R. Teo, H. Cao, R. Dalan, C. Y. Tay and H. W. Hou, Lab Chip, 2021, 21, 2359–2371 RSC.
- C. M. Warboys, N. Amini, A. de Luca and P. C. Evans, F1000 Med. Rep., 2011, 3, 1–8 Search PubMed.
- X.-H. Yu, Y.-C. Fu, D.-W. Zhang, K. Yin and C.-K. Tang, Clin. Chim. Acta, 2013, 424, 245–252 CrossRef CAS PubMed.
- C. Weber and H. Noels, Nat. Med., 2011, 17, 1410–1422 CrossRef CAS PubMed.
- M. Kowara and A. Cudnoch-Jedrzejewska, Int. J. Mol. Sci., 2021, 22, 3513 CrossRef CAS PubMed.
- P. Libby, A. H. Lichtman and G. K. Hansson, Immunity, 2013, 38, 1092–1104 CrossRef CAS PubMed.
- C. V. Jakubzick, G. J. Randolph and P. M. Henson, Nat. Rev. Immunol., 2017, 17, 349–362 CrossRef CAS PubMed.
- I. Tabas and A. H. Lichtman, Immunity, 2017, 47, 621–634 CrossRef CAS PubMed.
- A. Zernecke, Arterioscler., Thromb., Vasc. Biol., 2015, 35, 763–770 CrossRef CAS PubMed.
- Y. Haddad, C. Lahoute, M. Clément, L. Laurans, S. Metghalchi, L. Zeboudj, A. Giraud, X. Loyer, M. Vandestienne and J. Wain-Hobson, Circ. Res., 2017, 121, 234–243 CrossRef CAS PubMed.
- H. Winkels, E. Ehinger, M. Vassallo, K. Buscher, H. Q. Dinh, K. Kobiyama, A. A. J. Hamers, C. Cochain, E. Vafadarnejad and A.-E. Saliba, Circ. Res., 2018, 122, 1675–1688 CrossRef CAS PubMed.
- A. Zernecke, H. Winkels, C. Cochain, J. W. Williams, D. Wolf, O. Soehnlein, C. S. Robbins, C. Monaco, I. Park and C. A. McNamara, Circ. Res., 2020, 127, 402–426 CrossRef CAS PubMed.
- T. Kyaw, A. Winship, C. Tay, P. Kanellakis, H. Hosseini, A. Cao, P. Li, P. Tipping, A. Bobik and B.-H. Toh, Circulation, 2013, 127, 1028–1039 CrossRef CAS PubMed.
- T. T. P. Seijkens, K. Poels, S. Meiler, C. M. Van Tiel, P. J. H. Kusters, M. Reiche, D. Atzler, H. Winkels, M. Tjwa and H. Poelman, Eur. Heart J., 2019, 40, 372–382 CrossRef CAS PubMed.
- S. P. Mahamuni, R. D. Khose, F. Menaa and S. L. Badole, Biomedicines, 2012, 2, 137–146 Search PubMed.
- P. G. Jamkhande, P. G. Chandak, S. C. Dhawale, S. R. Barde, P. S. Tidke and R. S. Sakhare, Saudi Pharm. J., 2014, 22, 179–190 CrossRef PubMed.
- A. Rubenstrunk, R. Hanf, D. W. Hum, J.-C. Fruchart and B. Staels, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2007, 1771, 1065–1081 CrossRef CAS PubMed.
- A. Zinn, S. Felson, E. Fisher and A. Schwartzbard, Clin. Cardiol., 2008, 31, 397–403 CrossRef PubMed.
- B. Staels, Cell Metab., 2005, 2, 77–78 CrossRef CAS PubMed.
- A. M. Gotto, J. Am. Coll. Cardiol., 2003, 41, 1205–1210 CrossRef CAS PubMed.
- R. Klingenberg and G. K. Hansson, Eur. Heart J., 2009, 30, 2838–2844 CrossRef CAS PubMed.
- N. Filipczak, J. Pan, S. S. K. Yalamarty and V. P. Torchilin, Adv. Drug Delivery Rev., 2020, 156, 4–22 CrossRef CAS PubMed.
- Z. Wang, N. Little, J. Chen, K. T. Lambesis, K. T. Le, W. Han, A. J. Scott and J. Lu, Nat. Nanotechnol., 2021, 16, 1130–1140 CrossRef CAS PubMed.
- J. Lu, X. Liu, Y.-P. Liao, F. Salazar, B. Sun, W. Jiang, C. H. Chang, J. Jiang, X. Wang and A. M. Wu, Nat. Commun., 2017, 8, 1–14 CrossRef PubMed.
- E. A. Wyatt and M. E. Davis, Mol. Pharm., 2020, 17, 717–721 CAS.
- Y. Matsumoto, J. W. Nichols, K. Toh, T. Nomoto, H. Cabral, Y. Miura, R. J. Christie, N. Yamada, T. Ogura and M. R. Kano, Nat. Nanotechnol., 2016, 11, 533–538 CrossRef CAS PubMed.
- Y. Huang, J. Lu, X. Gao, J. Li, W. Zhao, M. Sun, D. B. Stolz, R. Venkataramanan, L. C. Rohan and S. Li, Bioconjugate Chem., 2012, 23, 1443–1451 CrossRef CAS PubMed.
- J. Lu, Y. Huang, W. Zhao, R. T. Marquez, X. Meng, J. Li, X. Gao, R. Venkataramanan, Z. Wang and S. Li, Biomaterials, 2013, 34, 1591–1600 CrossRef CAS PubMed.
- X. Zhang, J. Lu, Y. Huang, W. Zhao, Y. Chen, J. Li, X. Gao, R. Venkataramanan, M. Sun and D. B. Stolz, Bioconjugate Chem., 2013, 24, 464–472 CrossRef CAS PubMed.
- X. Zhang, Y. Huang, W. Zhao, Y. Chen, P. Zhang, J. Li, R. Venkataramanan and S. Li, Mol. Pharm., 2014, 11, 2807–2814 CrossRef CAS PubMed.
- X. Zhang, Y. Huang, W. Zhao, H. Liu, R. Marquez, J. Lu, P. Zhang, Y. Zhang, J. Li and X. Gao, Biomacromolecules, 2014, 15, 4326–4335 CrossRef CAS PubMed.
- J. Lu, W. Zhao, Y. Huang, H. Liu, R. Marquez, R. B. Gibbs, J. Li, R. Venkataramanan, L. Xu and S. Li, Mol. Pharm., 2014, 11, 4164–4178 CrossRef CAS PubMed.
- J. Lu, W. Zhao, H. Liu, R. Marquez, Y. Huang, Y. Zhang, J. Li, W. Xie, R. Venkataramanan and L. Xu, J. Controlled Release, 2014, 196, 272–286 CrossRef CAS PubMed.
- J. Lu, C. Liu, P. Wang, M. Ghazwani, J. Xu, Y. Huang, X. Ma, P. Zhang and S. Li, Biomaterials, 2015, 62, 176–187 CrossRef CAS PubMed.
- S. M. Dadfar, K. Roemhild, N. I. Drude, S. von Stillfried, R. Knüchel, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2019, 138, 302–325 CrossRef CAS PubMed.
- C. He, J. Lu and W. Lin, J. Controlled Release, 2015, 219, 224–236 CrossRef CAS PubMed.
- N. Elahi, M. Kamali and M. H. Baghersad, Talanta, 2018, 184, 537–556 CrossRef CAS PubMed.
- M. Li, F. Cheng, C. Xue, H. Wang, C. Chen, Q. Du, D. Ge and B. Sun, Langmuir, 2019, 35, 14688–14695 CrossRef PubMed.
- W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS PubMed.
- Y. Dölen, M. Valente, O. Tagit, E. Jäger, E. A. W. Van Dinther, N. K. van Riessen, M. Hruby, U. Gileadi, V. Cerundolo and C. G. Figdor, OncoImmunology, 2020, 9, 1738813 CrossRef PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- Y. Zi, K. Yang, J. He, Z. Wu, J. Liu and W. Zhang, Adv. Drug Delivery Rev., 2022, 114449 CrossRef CAS PubMed.
- J. Fang, W. Islam and H. Maeda, Adv. Drug Delivery Rev., 2020, 157, 142–160 CrossRef CAS PubMed.
- H. Yusuf-Makagiansar and T. J. Siahaan, Pharm. Res., 2001, 18, 329–335 CrossRef CAS PubMed.
- S. A. Tibbetts, D. S. S. Jois, T. J. Siahaan, S. H. Benedict and M. A. Chan, Peptides, 2000, 21, 1161–1167 CrossRef CAS.
- N. Zhang, C. Chittasupho, C. Duangrat, T. J. Siahaan and C. Berkland, Bioconjugate Chem., 2008, 19, 145–152 CrossRef CAS PubMed.
- N. Zhang, C. Chittasupho, C. Duangrat, T. J. Siahaan and C. Berkland, Bioconjugate Chem., 2008, 19, 145–152 CrossRef CAS PubMed.
- P. Dosta, I. Tamargo, V. Ramos, S. Kumar, D. W. Kang, S. Borrós and H. Jo, Adv. Healthcare Mater., 2021, 10, 2001894 CrossRef CAS PubMed.
- H. Yang, F. Zhao, Y. Li, M. Xu, L. Li, C. Wu, H. Miyoshi and Y. Liu, Int. J. Nanomed., 2013, 8, 1897 Search PubMed.
- M. I. Cybulsky, K. Iiyama, H. Li, S. Zhu, M. Chen, M. Iiyama, V. Davis, J.-C. Gutierrez-Ramos, P. W. Connelly and D. S. Milstone, J. Clin. Invest., 2001, 107, 1255–1262 CrossRef CAS PubMed.
- M. J. Davies, J. L. Gordon, A. J. H. Gearing, R. Pigott, N. Woolf, D. Katz and A. Kyriakopoulos, J. Pathol., 1993, 171, 223–229 CrossRef CAS PubMed.
- K. Ley and Y. Huo, J. Clin. Invest., 2001, 107, 1209–1210 CrossRef CAS PubMed.
- M. Calin, D. Stan, M. Schlesinger, V. Simion, M. Deleanu, C. A. Constantinescu, A.-M. Gan, M. M. Pirvulescu, E. Butoi and I. Manduteanu, Eur. J. Pharm. Biopharm., 2015, 89, 18–29 CrossRef CAS PubMed.
- A. Kheirolomoom, C. W. Kim, J. W. Seo, S. Kumar, D. J. Son, M. K. J. Gagnon, E. S. Ingham, K. W. Ferrara and H. Jo, ACS Nano, 2015, 9, 8885–8897 CrossRef CAS PubMed.
- K. A. Kelly, J. R. Allport, A. Tsourkas, V. R. Shinde-Patil, L. Josephson and R. Weissleder, Circ. Res., 2005, 96, 327–336 CrossRef CAS PubMed.
- M. Nahrendorf, F. A. Jaffer, K. A. Kelly, D. E. Sosnovik, E. Aikawa, P. Libby and R. Weissleder, Circulation, 2006, 114, 1504–1511 CrossRef CAS PubMed.
- Y. Takada, X. Ye and S. Simon, Genome Biol., 2007, 8, 1–9 CrossRef PubMed.
- S. A. Wickline, A. M. Neubauer, P. M. Winter, S. D. Caruthers and G. M. Lanza, J. Magn. Reson. Imaging, 2007, 25, 667–680 CrossRef PubMed.
- J. R. McCarthy, Adv. Drug Delivery Rev., 2010, 62, 1023–1030 CrossRef CAS PubMed.
- K. T. Fitzgerald, C. A. Holladay, C. McCarthy, K. A. Power, A. Pandit and W. M. Gallagher, Small, 2011, 7, 705–717 CrossRef CAS PubMed.
- P. M. Winter, A. M. Morawski, S. D. Caruthers, R. W. Fuhrhop, H. Zhang, T. A. Williams, J. S. Allen, E. K. Lacy, J. D. Robertson and G. M. Lanza, Circulation, 2003, 108, 2270–2274 CrossRef CAS PubMed.
- P. M. Winter, S. D. Caruthers, H. Zhang, T. A. Williams, S. A. Wickline and G. M. Lanza, JACC, 2008, 1, 624–634 Search PubMed.
- D. M. McDonald and P. L. Choyke, Nat. Med., 2003, 9, 713–725 CrossRef CAS PubMed.
- Y. S. Song, J. H. Kim, B. C. Lee, J. H. Jung, H. S. Park and S. E. Kim, Cancer Biother.Radiopharm., 2018, 33, 396–402 CrossRef CAS PubMed.
- K.-H. Lee, K.-H. Jung, S.-H. Song, D. H. Kim, B. C. Lee, H. J. Sung, Y.-M. Han, Y. S. Choe, D. Y. Chi and B.-T. Kim, J. Nucl. Med., 2005, 46, 472–478 CAS.
- R. Haubner, H.-J. Wester, U. Reuning, R. Senekowitsch-Schmidtke, B. Diefenbach, H. Kessler, G. Stöcklin and M. Schwaiger, J. Nucl. Med., 1999, 40, 1061–1071 CAS.
- Y. Wu, Y. Zhang, L. Dai, Q. Wang, L. Xue, Z. Su and C. Zhang, J. Controlled Release, 2019, 316, 236–249 CrossRef CAS PubMed.
- E. J. Chung, Exp. Biol. Med., 2016, 241, 891–898 CrossRef CAS PubMed.
- G. Fredman, N. Kamaly, S. Spolitu, J. Milton, D. Ghorpade, R. Chiasson, G. Kuriakose, M. Perretti, O. Farokhzad and I. Tabas, Sci. Transl. Med., 2015, 7, 275ra220–275ra220 Search PubMed.
- K. Murata, T. Motayama and C. Kotake, Atherosclerosis, 1986, 60, 251–262 CrossRef CAS PubMed.
- N. Kamaly, G. Fredman, J. J. R. Fojas, M. Subramanian, W. Choi II, K. Zepeda, C. Vilos, M. Yu, S. Gadde and J. Wu, ACS Nano, 2016, 10, 5280–5292 CrossRef CAS PubMed.
- M. Kim, A. Sahu, G. B. Kim, G. H. Nam, W. Um, S. J. Shin, Y. Y. Jeong, I.-S. Kim, K. Kim and I. C. Kwon, J. Controlled Release, 2018, 269, 337–346 CrossRef CAS PubMed.
- N. Kamaly, G. Fredman, M. Subramanian, S. Gadde, A. Pesic, L. Cheung, Z. A. Fayad, R. Langer, I. Tabas and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6506–6511 CrossRef CAS PubMed.
- Y. Chen, M. Molnár, L. Li, P. Friberg, L.-M. Gan, H. Brismar and Y. Fu, PLoS One, 2013, 8, e83805 CrossRef PubMed.
- M. Ząbczyk, J. Natorska and A. Undas, J. Clin. Med., 2021, 10, 2999 CrossRef PubMed.
- F. Guo, J. Liu, C. Wang, N. Liu and P. Lu, Biochem. Biophys. Res. Commun., 2009, 390, 942–946 CrossRef CAS PubMed.
- L. W. E. Starmans, D. Burdinski, N. P. M. Haex, R. P. M. Moonen, G. J. Strijkers, K. Nicolay and H. Grüll, PLoS One, 2013, 8, e57335 CrossRef CAS PubMed.
- D. Peters, M. Kastantin, V. R. Kotamraju, P. P. Karmali, K. Gujraty, M. Tirrell and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9815–9819 CrossRef CAS PubMed.
- R. Zhao, X. Ning, M. Wang, H. Wang, G. Xing, L. Wang, C. Lu, A. Yu and Y. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 25080–25092 CrossRef CAS PubMed.
- P. E. Szmitko, C.-H. Wang, R. D. Weisel, J. R. de Almeida, T. J. Anderson and S. Verma, Circulation, 2003, 108, 1917–1923 CrossRef PubMed.
- P. Libby, Circulation, 2001, 104, 365–372 CrossRef CAS PubMed.
- X. Zhang and D. M. Mosser, J. Pathol., 2008, 214, 161–178 CrossRef CAS PubMed.
- M. K. Georgakis, J. Bernhagen, L. H. Heitman, C. Weber and M. Dichgans, Eur. Heart J., 2022, 43, 1799–1808 CrossRef PubMed.
- Y. Yan, M. Thakur, E. P. C. van der Vorst, C. Weber and Y. Döring, Atherosclerosis, 2021, 330, 95–106 CrossRef CAS PubMed.
- E. J. Chung, L. B. Mlinar, K. Nord, M. J. Sugimoto, E. Wonder, F. J. Alenghat, Y. Fang and M. Tirrell, Adv. Healthcare Mater., 2015, 4, 367–376 CrossRef CAS PubMed.
- M. P. Quinones, H. G. Martinez, F. Jimenez, C. A. Estrada, M. Dudley, O. Willmon, H. Kulkarni, R. L. Reddick, G. Fernandes and W. A. Kuziel, Atherosclerosis, 2007, 195, e92–e103 CrossRef CAS PubMed.
- J. Rolin and A. A. Maghazachi, J. Leukocyte Biol., 2014, 95, 575–585 CrossRef PubMed.
- E. P. C. van der Vorst, Y. Döring and C. Weber, J. Mol. Med., 2015, 93, 963–971 CrossRef CAS PubMed.
- L. Detering, A. Abdilla, H. P. Luehmann, J. W. Williams, L.-H. Huang, D. Sultan, A. Elvington, G. S. Heo, P. K. Woodard and R. J. Gropler, Mol. Pharm., 2021, 18, 1386–1396 CrossRef CAS PubMed.
- H. P. Luehmann, L. Detering, B. P. Fors, E. D. Pressly, P. K. Woodard, G. J. Randolph, R. J. Gropler, C. J. Hawker and Y. Liu, J. Nucl. Med., 2016, 57, 1124–1129 CrossRef CAS PubMed.
- H. P. Luehmann, E. D. Pressly, L. Detering, C. Wang, R. Pierce, P. K. Woodard, R. J. Gropler, C. J. Hawker and Y. Liu, J. Nucl. Med., 2014, 55, 629–634 CrossRef CAS PubMed.
- G. Valacchi, C. Sticozzi, Y. Lim and A. Pecorelli, Ann. N. Y. Acad. Sci., 2011, 1229, E1–E7 CrossRef PubMed.
- X.-P. Yang, M. J. Amar, B. Vaisman, A. V. Bocharov, T. G. Vishnyakova, L. A. Freeman, R. J. Kurlander, A. P. Patterson, L. C. Becker and A. T. Remaley, J. Lipid Res., 2013, 54, 2450–2457 CrossRef CAS PubMed.
- M. Panchoo and A. Lacko, Biochem. Biophys. Res. Commun., 2018, 495, 614–620 CrossRef CAS PubMed.
- M. Z. Ashraf and N. Gupta, Int. J. Biochem. Cell Biol., 2011, 43, 697–700 CrossRef CAS PubMed.
- L. J. Goldstein and S. M. Brown, Annu. Rev. Biochem., 1977, 46, 897–930 CrossRef PubMed.
- S. Parthasarathy, A. Raghavamenon, M. O. Garelnabi and N. Santanam, Free Radic. Antioxid., 2010, 403–417 CAS.
- D. Gao, M. Z. Ashraf, N. S. Kar, D. Lin, L. M. Sayre and E. A. Podrez, J. Biol. Chem., 2010, 285, 4447–4454 CrossRef CAS PubMed.
- R. Pechlaner, S. Tsimikas, X. Yin, P. Willeit, F. Baig, P. Santer, F. Oberhollenzer, G. Egger, J. L. Witztum and V. J. Alexander, J. Am. Coll. Cardiol., 2017, 69, 789–800 CrossRef CAS PubMed.
- K. T. Trajkovska and S. Topuzovska, Anatol. J. Cardiol., 2017, 18, 149 CAS.
- A. Rigotti, H. E. Miettinen and M. Krieger, Endocr. Rev., 2003, 24, 357–387 CrossRef CAS PubMed.
- T. J. F. Nieland, M. Penman, L. Dori, M. Krieger and T. Kirchhausen, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15422–15427 CrossRef CAS PubMed.
- X. Mei and D. Atkinson, Arch. Med. Res., 2015, 46, 351–360 CrossRef CAS PubMed.
- M. C. Phillips, J. Lipid Res., 2013, 54, 2034–2048 CrossRef CAS PubMed.
- A. G. Lacko, N. A. Sabnis, B. Nagarajan and W. J. McConathy, Front. Pharmacol., 2015, 6, 247 Search PubMed.
- J. Chen, X. Zhang, R. Millican, J. E. Creutzmann, S. Martin and H.-W. Jun, Nano Convergence, 2020, 7, 1–14 CrossRef CAS PubMed.
- A. Gupta, R. Sharma, K. Kuche and S. Jain, Int. J. Pharm., 2021, 596, 120272 CrossRef CAS PubMed.
- S. Marrache and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9445–9450 CrossRef CAS PubMed.
- S. E. Nissen, T. Tsunoda, E. M. Tuzcu, P. Schoenhagen, C. J. Cooper, M. Yasin, G. M. Eaton, M. A. Lauer, W. S. Sheldon and C. L. Grines, J. Am. Med. Assoc., 2003, 290(17), 2292–2300 CrossRef CAS PubMed.
- R. Kuai, D. Li, Y. E. Chen, J. J. Moon and A. Schwendeman, ACS Nano, 2016, 10, 3015–3041 CrossRef CAS PubMed.
- C. M. Madsen, A. Varbo and B. G. Nordestgaard, Arterioscler., Thromb., Vasc. Biol., 2021, 41, 128–140 CAS.
- R. Duivenvoorden, J. Tang, D. P. Cormode, A. J. Mieszawska, D. Izquierdo-Garcia, C. Ozcan, M. J. Otten, N. Zaidi, M. E. Lobatto, S. M. Van Rijs and W. J. Mulder, Nat. Commun., 2014, 5(1), 1–12 Search PubMed.
- H. He, L. Liu, H. Bai, J. Wang, Y. Zhang, W. Zhang, M. Zhang, Z. Wu and J. Liu, Pharm. Res., 2014, 31, 1689–1709 CrossRef CAS PubMed.
- W. J. S. De Villiers and E. J. Smart, J. Leukocyte Biol., 1999, 66, 740–746 CrossRef CAS PubMed.
- D. A. Chistiakov, A. A. Melnichenko, V. A. Myasoedova, A. V. Grechko and A. N. Orekhov, J. Mol. Med., 2017, 95, 1153–1165 CrossRef CAS PubMed.
- C. D. Rodrigues, M. Hannus, M. Prudêncio, C. Martin, L. A. Gonçalves, S. Portugal, S. Epiphanio, A. Akinc, P. Hadwiger and K. Jahn-Hofmann, Cell Host Microbe, 2008, 4, 271–282 CrossRef CAS PubMed.
- L. Malerød, L. Juvet, T. Gjøen and T. Berg, Cell Tissue Res., 2002, 307, 173–180 CrossRef PubMed.
- Y. Zhang, J. R. Da Silva, M. Reilly, J. T. Billheimer, G. H. Rothblat and D. J. Rader, J. Clin. Invest., 2005, 115, 2870–2874 CrossRef CAS PubMed.
- L. Liu, H. He, M. Zhang, S. Zhang, W. Zhang and J. Liu, Biomaterials, 2014, 35, 8002–8014 CrossRef CAS PubMed.
- P. T. Bot, G. Pasterkamp, M. J. Goumans, C. Strijder, F. L. Moll, J. P. De Vries, S. T. Pals, D. P. De Kleijn, J. J. Piek and I. E. Hoefer, Eur. J. Clin. Invest., 2010, 40, 818–827 CrossRef CAS PubMed.
- Y. Zhao, H. Gao, J. He, C. Jiang, J. Lu, W. Zhang, H. Yang and J. Liu, J. Controlled Release, 2018, 283, 241–260 CrossRef CAS PubMed.
- S. Raha, T. Paunesku and G. Woloschak, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 269–281 CrossRef CAS.
- E. J. Chung, Exp. Biol. Med., 2016, 241, 891–898 CrossRef CAS PubMed.
- M. Kim, A. Sahu, G. B. Kim, G. H. Nam, W. Um, S. J. Shin, Y. Y. Jeong, I. S. Kim, K. Kim, I. C. Kwon and G. Tae, J. Controlled Release, 2018, 269, 337–346 CrossRef CAS PubMed.
- A. M. Chamberlain, A. R. Folsom, S. R. Heckbert, W. D. Rosamond and M. Cushman, Blood, 2008, 112, 2675–2680 CrossRef CAS PubMed.
- M. Navab, G. M. Anantharamaiah, S. T. Reddy and A. M. Fogelman, Nat. Clin. Pract. Cardiovasc. Med., 2006, 3, 540–547 CrossRef CAS PubMed.
- S. S. Imam, S. Alshehri, M. M. Ghoneim, A. Zafar, O. A. Alsaidan, N. K. Alruwaili, S. J. Gilani and M. Rizwanullah, Polymers, 2021, 13, 4036 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- M. Rizwanullah, M. Z. Ahmad, M. M. Ghoneim, S. Alshehri, S. S. Imam, S. Md, N. A. Alhakamy, K. Jain and J. Ahmad, Pharmaceutics, 2021, 13, 2039 CrossRef CAS PubMed.
- S. Shi, N. Kong, C. Feng, A. Shajii, C. Bejgrowicz, W. Tao and O. C. Farokhzad, Adv. Healthcare Mater., 2019, 8, 1801655 CrossRef PubMed.
- Z. Hong, Y. Xu, J.-F. Yin, J. Jin, Y. Jiang and Q. Du, J. Agric. Food Chem., 2014, 62, 12603–12609 CrossRef CAS PubMed.
- H. A. Santos, E. Mäkilä, A. J. Airaksinen, L. M. Bimbo and J. Hirvonen, Nanomedicine, 2014, 9, 535–554 CrossRef CAS PubMed.
- M. P. Donnorso, E. Miele, F. De Angelis, R. La Rocca, T. Limongi, F. C. Zanacchi, S. Marras, R. Brescia and E. Di Fabrizio, Microelectron. Eng., 2012, 98, 626–629 CrossRef.
- H. Daraee, A. Eatemadi, E. Abbasi, S. Fekri Aval, M. Kouhi and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 410–422 CrossRef CAS PubMed.
- A. Kumar, X. Zhang and X.-J. Liang, Biotechnol. Adv., 2013, 31, 593–606 CrossRef CAS PubMed.
- T. Vangijzegem, D. Stanicki and S. Laurent, Expert Opin. Drug Delivery, 2019, 16, 69–78 CrossRef CAS PubMed.
- J. Estelrich, E. Escribano, J. Queralt and M. A. Busquets, Int. J. Mol. Sci., 2015, 16, 8070–8101 CrossRef CAS PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- A. Narang, R.-K. Chang and M. A. Hussain, J. Pharm. Sci., 2013, 102, 3867–3882 CrossRef CAS PubMed.
- S. Muro, C. Garnacho, J. A. Champion, J. Leferovich, C. Gajewski, E. H. Schuchman, S. Mitragotri and V. R. Muzykantov, Mol. Ther., 2008, 16, 1450–1458 CrossRef CAS PubMed.
- D. R. Lewis, K. Kamisoglu, A. W. York and P. V. Moghe, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 400–420 CAS.
- W. Jaroszewicz, J. Morcinek-Orłowska, K. Pierzynowska, L. Gaffke and G. Węgrzyn, FEMS Microbiol. Rev., 2022, 46, fuab052 CrossRef PubMed.
- K. Nagano and Y. Tsutsumi, Viruses, 2021, 13, 178 CrossRef CAS PubMed.
- A. Mahdavi Gorabi, N. Kiaie, Ž. Reiner, F. Carbone, F. Montecucco and A. Sahebkar, Biomolecules, 2019, 9, 416 CrossRef PubMed.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delacher, K. Tron, G. U. Nienhaus, A. Musyanovych, V. Mailander and K. Landfester, ACS Nano, 2011, 5, 1657–1669 CrossRef CAS PubMed.
- A. Cervadoro, R. Palomba, G. Vergaro, R. Cecchi, L. Menichetti, P. Decuzzi, M. Emdin and S. Luin, Front. Bioeng. Biotechnol., 2018, 177, 1–10 Search PubMed.
- N. Oh and J.-H. Park, Int. J. Nanomed., 2014, 9, 51 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |