
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radiolabelling an 18F biologic via facile IEDDA “click” chemistry on the GE FASTLab™ platform†
Louis
Allott‡
 abc,
Ala
Amgheib‡
ad,
Chris
Barnes
a,
Marta
Braga
a,
Diana
Brickute
a,
Ning
Wang
a,
Ruisi
Fu
a,
Sadaf
Ghaem-Maghami
ad and
Eric O.
Aboagye
*a
abc,
Ala
Amgheib‡
ad,
Chris
Barnes
a,
Marta
Braga
a,
Diana
Brickute
a,
Ning
Wang
a,
Ruisi
Fu
a,
Sadaf
Ghaem-Maghami
ad and
Eric O.
Aboagye
*a
aComprehensive Cancer Imaging Centre, Faculty of Medicine, Department of Surgery and Cancer, Imperial College London, Hammersmith Hospital, Du Cane Road, London, W12 0NN, UK. E-mail: eric.aboagye@imperial.ac.uk
bPositron Emission Tomography Research Centre, Faculty of Health Sciences, University of Hull, Cottingham Road, Kingston upon Hull, HU6 7RX, UK
cDepartment of Biomedical Sciences, Faculty of Health Sciences, University of Hull, Cottingham Road, Kingston upon Hull, HU6 7RX, UK
dFaculty of Medicine, Department of Surgery and Cancer, Imperial College London, Hammersmith Hospital, Du Cane Road, London, W12 0NN, UK
First published on 15th April 2021
Abstract
The use of biologics in positron emission tomography (PET) imaging is an important area of radiopharmaceutical development and new automated methods are required to facilitate their production. We report an automated radiosynthesis method to produce a radiolabelled biologic via facile inverse electron demand Diels–Alder (IEDDA) “click” chemistry on a single GE FASTLab™ cassette. We exemplified the method by producing a fluorine-18 radiolabelled interleukin-2 (IL2) radioconjugate from a trans-cyclooctene (TCO) modified IL2 precursor. The radioconjugate was produced using a fully automated radiosynthesis on a single FASTLab™ cassette in a decay-corrected radiochemical yield (RCY, d.c.) of 19.8 ± 2.6% in 110 min (from start of synthesis); the molar activity was 132.3 ± 14.6 GBq μmol−1. The in vitro uptake of [18F]TTCO-IL2 correlated with the differential receptor expression (CD25, CD122, CD132) in PC3, NK-92 and activated human PBMCs. The automated method may be adapted for the radiosynthesis of any TCO-modified protein via IEDDA chemistry.
Introduction
Positron emission tomography (PET) is a functional molecular imaging modality used in the clinic to inform the diagnosis and progression of diseases like cancer.1,2 Targeted radiopharmaceuticals are required to trace or quantify biological processes associated with disease states (i.e. energy metabolism, proliferation, receptor expression) and an active research and development community is working towards producing such molecules for clinical translation. The transition of radiopharmaceuticals from the laboratory bench into clinical evaluation requires automated radiosynthesis under good manufacturing processes (GMP) conditions, to produce consistent sterile patient doses and reduce radiation exposure to production staff.3,4 As we ask evermore complex clinical questions, the development of new radiopharmaceuticals with intricate and sensitive chemical structures often provide answers; however, their efficient radiolabelling and automated production can be challenging. The implementation of radiolabelled peptides and proteins as targeting vectors in nuclear medicine is increasing owed to their unparalelled targeted affinity, selectivity and specificity;5 however, few GMP compatible automated radiolabelling processes have been reported.6,7 We and others have developed various automated radiolabelling procedures for their production, including an automated procedure for copper catalysed azide alkyne cycloaddition (CuAAC) “click” chemistry for an 18F somatostatin analogue ([18F]FET-βAG-TOCA) which has progressed into phase III clinical trials;8,9 a generic automated radiolabelling procedure for the aluminium-[18F]fluoride method to produce [18F]NOTA-octreotide and [18F]NOTA-RGDfK radioconjugates and an automated procedure for solid-supported reductive amination radiochemistry.10–12Fluorine-18 prosthetic group (PG) strategies, where a small organic molecule is radiolabelled independently of a biomolecule and later conjugated under mild conditions, can be used to maintain the structural integrity of the protein (Fig. 1).13 Reported PG strategies and their automated procedures support the production of radioconjugates from micromolar (μmol) quantities of biomolecule precursors, typically peptides (ca. 1–4 kDa) which are inexpensive to synthesise in multi-milligram quantities.8,14 Targeted proteins (>5 kDa) are often made in small batches (μg to mg) as they are expensive to produce and are therefore radiolabelled at low total protein quantities (μg, nmol) to reduce cost, but also because of the challenge in separating the radioconjugate from the unlabelled biomolecule, which can negatively influence molar activity (Am). There are few examples where targeted proteins are radiolabelled with an 18F-PG using fully automated procedures, and to the best of our knowledge, no examples where an entire radiosynthesis has been accomplished on a single cassette-based automated platform.
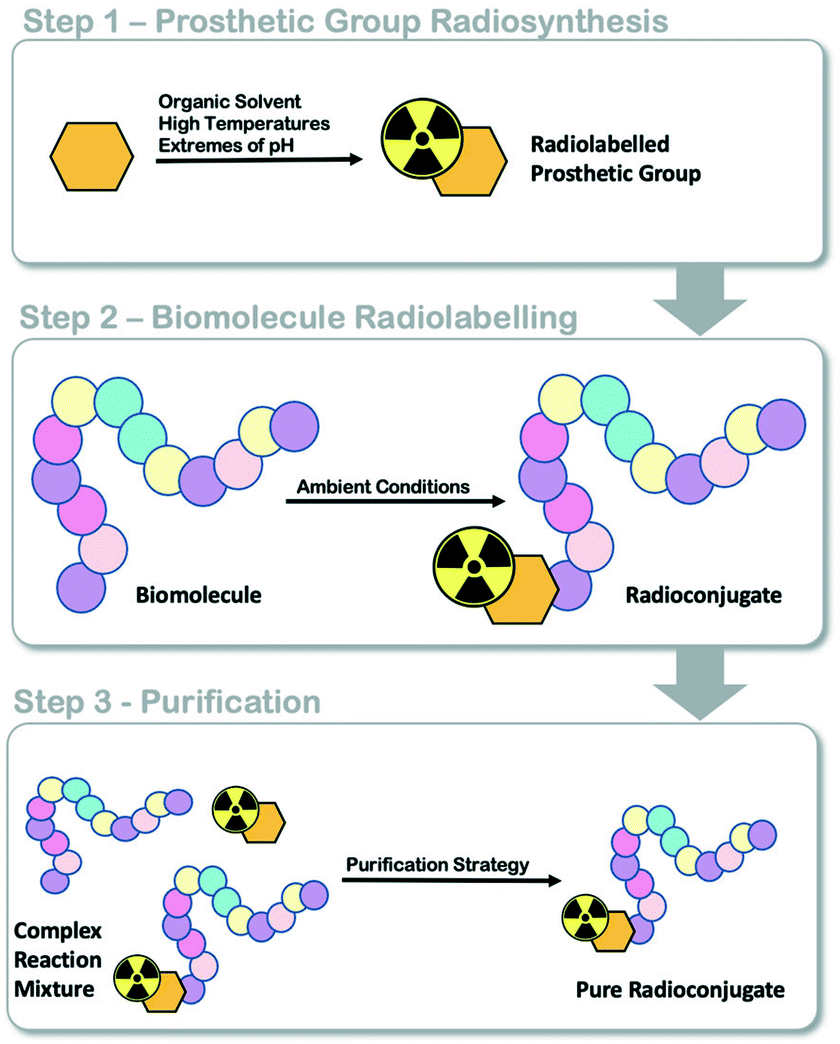 | ||
| Fig. 1 Schematic representation of fluorine-18 prosthetic group (PG) strategies for radiolabelling peptides and proteins. | ||
The biologic interleukin-2 (IL2) is of great interest as it targets the IL2-receptor (IL2R) expressed on activated T-cells. IL2R is comprised of IL2Rα (CD25), IL2Rβ (CD122) and IL2Rγ (CD132) subunits resulting in a variable affinity receptor.15 Recombinant IL2 is commercially available as Proleukin™ (Novartis) and is used in the treatment of metastatic renal cell carcinoma and melanoma; typically administered as intravenous infusion or subcutaneous injection.16,17 Targeting the high affinity CD25 using IL2-based radioconjugates could potentially enable the detection of T-cell activation in solid tumours and, thus monitoring of response to immunotherapy.3,18,19 Gialleonardo et al. (2012) reported a fluorine-18 IL2 radioconjugate ([18F]FB-IL2) which was synthesised using the N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB) prosthetic group and later translated the radiochemistry into a GMP compatible production to support a clinical trial.6,20 Unfortunately, the lengthy radiosynthesis of [18F]SFB and the complexity of radiolabelling proteins using automated platforms required the use of two hot-cells and two automated radiosynthesis platforms. This process is not attractive to GMP production facilities. DeVries et al. are addressing this concern and have developed metal-based radiolabelling strategies;21 but the question remains, if we can develop an 18F-PG strategy for the automated production of proteinous radioconjugates using a single cassette-based platform?
The use of inverse electron demand Diels–Alder (IEDDA) chemistry to radiolabel a protein modified with a trans-cyclooctene (TCO) moiety by ligating an 18F-tetrazine prosthetic group has been reported but an automated radiolabelling procedure has not been described.22 We report a fully automated 18F-PG strategy used to radiolabel TCO-modified IL2 using IEDDA “click” chemistry. We believe this to be the first fully automated radiosynthesis of an IL2 radioconjugate using an 18F-PG strategy on a single cassette-based automated platform. The reported method may be adapted for use in radiolabelling any TCO-modified protein and “noteworthy considerations” are suggested throughout for translating this work to other biomolecules of interest.
Experimental procedures
Detailed materials and methods appear in the ESI.† Radiochemical yields (RCY) are decay corrected (d.c.) to the start of radiosynthesis, in line with the “Consensus nomenclature rules for radiopharmaceutical chemistry – Setting the record straight”.23 Experimental procedures for the synthesis of compounds 1–5 and their characterisation by NMR and mass spectrometry appear in the ESI† (Fig. S1–S12). Experimental procedure for in vitro metabolite analysis, cell culture and flow cytometry are reported in the ESI† (Fig. S22–S24).Materials and methods
Anhydrous solvents and reagents were purchased from Sigma Aldrich (Gillingham, UK) and were used without additional purification. TCO-PEG4-NHS ester was purchased from Jena Bioscience (Jena, Germany). Proleukin™ was procured from Novartis Pharmaceuticals (London, UK). Flash column chromatography purification was performed on silica gel (Merck Kieselgel 60 F254 320–400 mesh). [18F]Fluoride was produced by a GE PETtrace cyclotron by 16 MeV irradiation of enriched [18O]H2O target, supplied by Alliance Medical Radiopharmacy Ltd (Warwick, UK). Automated radiosynthesis were performed using the GE FASTlab™ automated synthesis module (GE Healthcare Life Sciences, Amersham, UK). Solid phase extraction (SPE) cartridges were purchased from Waters (Elstree, Hertfordshire, UK) and used according to the manufacturers recommended guidelines. Semi-preparative RP-HPLC was performed using a Shimadzu LC20-AT pump attached to a custom-built system, equipped with an Agilent Eclipse XDB-C18, 5 μ (250 × 9.4 mm) column using an isocratic mobile phase of MeCN (44%), H2O (56%) and 0.1% H3PO4 (14.8 M) at a flow rate of 3 mL min−1. Reaction efficiency and radioactive product identity was determined by RP-HPLC using an Agilent 1200 series instrument connected to a flow-ram detector (Lablogic, Sheffield, UK). This study used a PC3 cell line, gifted from Prof. Charlotte Bevan, Imperial College London and a NK-92 cell line purchased from ATCC (Teddington, UK). Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood samples obtained from healthy volunteers under the approval of the West London Research Ethics Committee (Reference 10/H0707/7 and 12/WA/0196).Synthesis of TCO-PEG4-IL2
To each of three vials of Proleukin™ (18 × 106 IU, ∼1.3 mg) was added water (250 μL), which was combined into a single vial to give a Proleukin™ (3.96 mg, 255 nmol) in water (750 μL, 5.28 mg mL−1). A Zeba™ spin desalting column (7 kDa MWCO, 5 mL) was equilibrated with a pH 8 bioconjugation solution containing NaHCO3 (0.1 M) and SDS (0.05 % w/v), as per the manufacturers recommended equilibration protocol. The Proleukin™ solution was exchanged into the NaHCO3/SDS solution and protein recovery was determined to be quantitative by UV-vis (nanodrop). A fresh stock of TCO-PEG4-NHS ester was prepared in DMF (255 nmol μL−1) and an aliquot (10.6 μL) was added to the Proleukin™ solution in a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (TCO-PEG4-NHS ester:Proleukin™). The reaction was gently shaken for 2 h at ambient temperature. A Zeba™ spin desalting column (7 kDa MWCO, 5 mL) was equilibrated in a storage solution containing SDS (0.05% w/v) and PSB. The Proleukin™ reaction mixture was loaded onto the column and purified. The resulting solution contained TCO-PEG4-IL2, and the protein content was determined by UV-vis spectroscopy (nanodrop) and BCA assay; aliquots of 200 μg were prepared for radiolabelling and stored at −20 °C. The number of TCO moieties per molecule of IL2 was determined by nanodrop and the full experimental procedure described in the ESI.†
:1 ratio (TCO-PEG4-NHS ester:Proleukin™). The reaction was gently shaken for 2 h at ambient temperature. A Zeba™ spin desalting column (7 kDa MWCO, 5 mL) was equilibrated in a storage solution containing SDS (0.05% w/v) and PSB. The Proleukin™ reaction mixture was loaded onto the column and purified. The resulting solution contained TCO-PEG4-IL2, and the protein content was determined by UV-vis spectroscopy (nanodrop) and BCA assay; aliquots of 200 μg were prepared for radiolabelling and stored at −20 °C. The number of TCO moieties per molecule of IL2 was determined by nanodrop and the full experimental procedure described in the ESI.†
Automated radiosynthesis of [18F]FB-Tz
A previously reported radiosynthesis for automating reductive amination radiochemistry using solid-supported cyanoborohydride cartridges was adapted for the synthesis of [18F]FB-Tz.10 In summary, [18F]FBA was synthesised to which precursor 1 (35 mg) in MeCN (1.5 mL) and triethylamine (40 μL) was added. After warming and reduction using the BH3CN− cartridge following the published protocol, the reaction mixture was diluted in phosphate buffer (5 mL, pH 2.4) and purified by semi-preparative HPLC. The cut peak was diluted in phosphate buffer (45 mL, pH 2.4) and trapped on a tC18 plus SPE cartridge which was subsequently flushed with nitrogen. [18F]FB-Tz was eluted with EtOH.Automated radiosynthesis of [18F]FBoxTz
A detailed description of the automated radiosynthesis setup can be found in the ESI† (Fig. S14). This radiosynthesis required the water bag (position 15) to be modified to include H3PO4 (0.1% v/v) and SDS (0.05% w/v). A GE FASTLab™ system was programmed to trap aqueous [18F]fluoride from [18O]water on a QMA bicarbonate cartridge (position 4–5) which was eluted into the reactor using a solution containing KHCO3 (3.5 mg mL−1, H2O 200 μL) and Kryptofix-222 (6.0 mg mL−1, MeCN 800 μL). The [18F]fluoride was dried at 120 °C (9 min) and 70 °C (5 min) before the addition of 4-formyl-N,N,N-trimethylanilinium triflate (3 mg in 1.4 mL MeCN). The reaction was heated to 90 °C for 6.6 min to synthesise [18F]FBA. After cooling, a solution containing 3 (8 mg in 1 mL MeCN) and aniline hydrochloride (6 mg in 400 μL H2O) was added to the [18F]FBA in the reactor and warmed to 40 °C for 10 min to synthesise [18F]FBoxTz. The reaction mixture was diluted for semi-preparative HPLC purification (7.5 mL H2O + 0.1% H3PO4). The isolated [18F]FBoxTz (tR = 21–24 min) was cut into a vial containing a dilution mixture (35 mL H2O + 0.1% H3PO4) and returned to the FASTLab™ for trapping on a tC18 SPE. The trapped [18F]FBoxTz was washed with the water bag solution (2 × 5 mL) and dried under N2. [18F]FBoxTz was eluted from the SPE cartridge in ethanol. The sequence continued for the automated radiosynthesis of [18F]TTCO-IL2.Automated radiosynthesis of [18F]TTCO-IL2
[18F]FBoxTz was eluted into an off-board reactor with ethanol (500 μL) containing TCO-PEG4-IL2 (200 μg, 2–3 mg mL−1) and allowed to react for 15 min at ambient temperature. The reaction mixture containing [18F]TTCO-IL2 was diluted with the water bag solution (9 mL) and loaded onto a tC2 SPE cartridge. Unreacted [18F]FBoxTz was eluted to waste by washing with a 50% EtOH solution. The desired [18F]TTCO-IL2 was eluted from the cartridge using 100% EtOH. The minimum SA was determined by the following equation: [radioactivity in product vial (MBq)]/200 μg = specific activity, SA (MBq μg−1); or by integrating the area under the radioactive peak at the 210 nm wavelength and comparing area to a reference standard of TCO-PEG4-IL2. Radiochemical purity was determined by radio-HPLC (tR = 08:48 mm:ss) and radio-TLC (Rf = 0).Radiochemical stability of [18F]TTCO-IL2
A vial of [18F]TTCO-IL2 in the standard formulation was synthesised using the automated procedure starting from a high activity of [18F]fluoride (19.6 ± 2.4 GBq, n = 3). An aliquot of radioactivity (4.4 ± 0.9 MBq) was diluted in 0.1% TFA (500 μL) and analysed by HPLC for each 0, 0.5, 1, 2, 3, 4, 5 and 6 h time point. Radio-TLC was also used (iTLC silica sheet, mobile phase: 2:1 EtOAc:hexane) for 1, 2, 3, 4, 5 and 6 h time points. The experiment was repeated three times from separate productions.
In vitro radioactive uptake
To assess the in vitro uptake of [18F]TTCO-IL2, 72 h PHA-stimulated PBMCs (approximately 1 × 106 cells per tube) were incubated with 0.74 MBq of [18F]TTCO-IL2 at 37 °C and 5% CO2. PC3 cells were seeded in a 6-well plate at a seeding density of 0.3 × 106 cells per well 24 hours prior to administration of [18F]TTCO-IL2. Approximately 1 × 106 of NK-92 cells were incubated with 0.74 MBq of [18F]TTCO-IL2 at room temperature. After 1 hour, cells were washed twice with 500 μL of ice-cold PBS and lysed with 1 mL ice-cold RIPA buffer (ThermoFisher) for 10 minutes on ice. Cell-bound radioactivity was measured, and decay corrected using Packard Cobra II gamma counter (Perkin Elmer). The radiopharmaceutical uptake was normalised to total cellular protein. Uptake was expressed as % radioactivity per mg protein. To determine the effect of [18F]TTCO-IL2 on IL2R signalling, NK-92 cells were incubated with 0.74 MBq [18F]TTCO-IL2 or unlabelled recombinant IL2 (214 ng mL−1) for 1 hour at 37 °C and 5% CO2. Cells were washed as previously described and fixed with 4% paraformaldehyde for 20 minutes before proceeding with phosphorylated-STAT5 (Tyr694) analysis using flow cytometry. Data were summarised and analysed using Prism GraphPad 7 software. Statistical analysis was performed using an unpaired two-tailed t-test.Results and discussion
The use and automation of 18F-PG strategies to radiolabel proteins is challenging for several reasons:1. 18F-PG strategies can be challenging to synthesise. Depending on the PG, multistep radiosynthesis and purification are required to access the desired 18F-PG. The PG needs to be produced in high Am for radiolabelling biomolecules through second order reactions to avoid self-competition at reactive sites.
2. 18F-PGs can be unstable. N-Hydroxysuccinimide esters (e.g. [18F]SFB) rapidly hydrolyse in aqueous media at the pH required for efficient amide bond formation with lysine amino acids (8–9 pH). Competitive hydrolysis reduces the efficiency in radiolabelling the molecule of interest, impacting both radiochemical yield (RCY) and potentially, molar activity (Am).
3. Cassette-based automated platforms have a limited capacity. These platforms are primarily designed for small-molecule radiochemistry where SN2 18F-fluorination is followed by deprotection and a purification step; radiolabelling biomolecules through 18F-PG strategies is more complex in terms of i) number of synthetic steps, ii) types of purification strategy, iii) low reagent concentrations and thus iv) low volume reagent handling.
“Click” chemistry has been proved to be an effective strategy for radiolabelling complex molecules.8,9,14,22,24,25 We proposed that the IEDDA “click” reaction could address each of these limitations and produce a fully automated single-cassette based method for radiolabelling biomolecules.22 Given the challenges faced in producing an efficient radiosynthesis of [18F]FB-IL2, and its clinical relevance, we developed an automated IEDDA “click” method to radiolabel a novel TCO-modified IL2 conjugate (TCO-PEG4-IL2) that we produced for this project.
Developing a TCO-modified IL2 conjugate
The abundant availability of TCO reagents allowed for a simple route to accessing a TCO-containing IL2 precursor by conjugating commercially available TCO-PEG4-NHS ester to recombinant IL2 (Proleukin™). In brief, the lyophilised recombinant IL2 (1.3 mg vial) was reconstituted in water to a concentration of 5 mg mL−1 and exchanged into a sodium bicarbonate (0.1 M, pH 8) conjugation medium using Zeba™ spin desalting columns. The TCO-PEG4-NHS ester was added to the reaction mixture in a 12:1 molar ratio, similar to that used in the development of [99mTc]Tc-HYNIC-IL2.26 After gentle shaking for 2 hours at room temperature, the bioconjugate was purified by Zeba™ spin desalting column. The protein concentration was determined by bicinchoninic acid (BCA) assay and typical yields were ca. 60%. The average number of TCO moieties per molecule of IL2 was 0.74 ± 0.02, determined by UV-vis spectroscopy. The TCO-PEG4-IL2 bioconjugate was aliquoted into 1.5 mL microcentrifuge tubes for radiolabelling and stored at −20 °C. HPLC analysis of TCO-PEG4-IL2 resulted in a single peak at 08:18 mm:ss (ESI† Fig. S19).
It was considered that functionalising IL2 with TCO-PEG4 could change the biological properties of the protein. Prodrug strategies for IL2 have been developed by decorating the surface of the protein with large PEG chains to modulated the affinity, target selectivity and in vivo pharmacokinetics of the drug.27 Additionally, small modifications to the protein structure of IL2 have been reported to influence binding modes.28–30 To investigate the influence of our modification to IL2, the in vitro biological properties of [18F]TTCO-IL2 have been evaluated and are reported later.
Development of a convenient 18F-tetrazine prosthetic group
There are many examples of 18F-radiolabelled tetrazines produced by different radiosynthesis methods and in variable RCY, fuelled by the increased interest in pre-targeted immuno-PET;34–37 however, new 18F-tetrazines were developed specifically as PGs that could be accessed in a high RCY, Am and in few radiosynthetic steps. The direct 18F-fluorination of a tetrazine is notoriously difficult due to instability in the harsh reaction conditions required to promote nucleophilic chemistry. The modular assembly of two novel 18F-tetrazines was therefore favoured and reductive amination chemistry, as well as oxime chemistry, were evaluated (Scheme 1A). The radiosynthesis of N-(4-[18F]fluorobenzyl)-1-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)methanamine ([18F]FB-Tz) used simple reductive chemistry reaction between an amine-containing tetrazine (1) and 4-[18F]fluorobenzaldehyde ([18F]FBA). The automated radiochemistry was derived from our previously published work on the development solid-supported cyanoborohydride reducing agent cartridges.10 [18F]FB-Tz was successfully produced in a 8.7 ± 1.1% RCY (d.c.) using a fully automated procedure in 80 min. To compare the influence of reaction chemistry on the overall radiochemical properties of the PG, we developed E-2-(((4-[18F]fluorobenzylidene)amino)oxy)-N-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)acetamide ([18F]FBoxTz) to utilise oxime chemistry.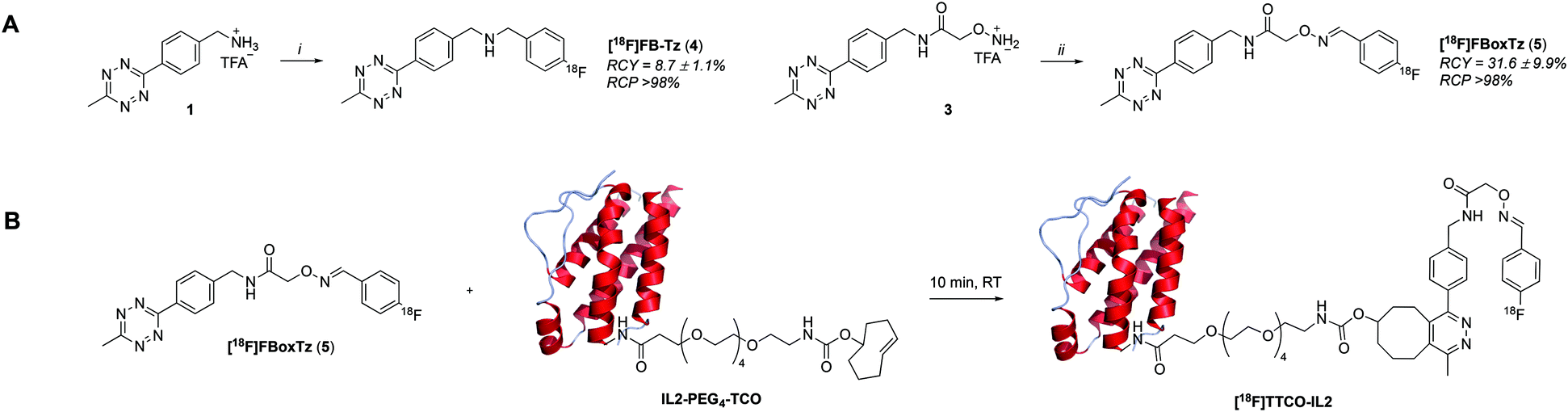 | ||
| Scheme 1 A) Two radiosynthetic routes to 18F-tetrazines: N-(4-[18F]fluorobenzyl)-1-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)methanamine ([18F]FB-Tz) and E-2-(((4-[18F]fluorobenzylidene)amino)oxy)-N-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)acetamide ([18F]FBoxTz). Reaction conditions: i) [18F]FBA, solid-supported cyanoborohydride cartridge, MeCN/H2O; ii) [18F]FBA, aniline hydrochloride, MeCN/H2O, 40 °C, 10 min. B) The reaction between [18F]FBoxTz and TCO-PEG4-IL2 to produce the radioconjugate [18F]TTCO-IL2. | ||
[18F]FBoxTz removed the need for an on-board reducing agent by exploiting oxime ligation between the aminoxy functional group and [18F]FBA. The radiochemistry was simple to automate and produced [18F]FBoxTz in a 31.6 ± 9.9% RCY (d.c.) in 85 min. In brief, [18F]fluoride was dried and [18F]FBA synthesised and precursor 3 added along with aniline hydrochloride as a catalyst. The resulting [18F]FBoxTz was purified by semi-preparative HPLC and the radioactive product reformulated using a tC18 solid phase extraction (SPE) cartridge, which was eluted with ethanol. The [18F]FBoxTz radiosynthesis not only gave a higher RCY (d.c.) compared to [18F]FB-Tz (ca. 32% vs. 9%), the purity profile was much improved and therefore fewer competing tetrazine impurities would be introduced into the IEDDA “click” step. The low RCY of [18F]FB-Tz was likely due to instability of the tetrazine under reducing conditions, converting into an unreactive dihydrotetrazine.38 Additionally, the radiosynthesis used fewer positions on the FASTLab™ cassette which would allow greater flexibility in implementing the entire radiolabelling method onto the single cassette (Fig. 2).
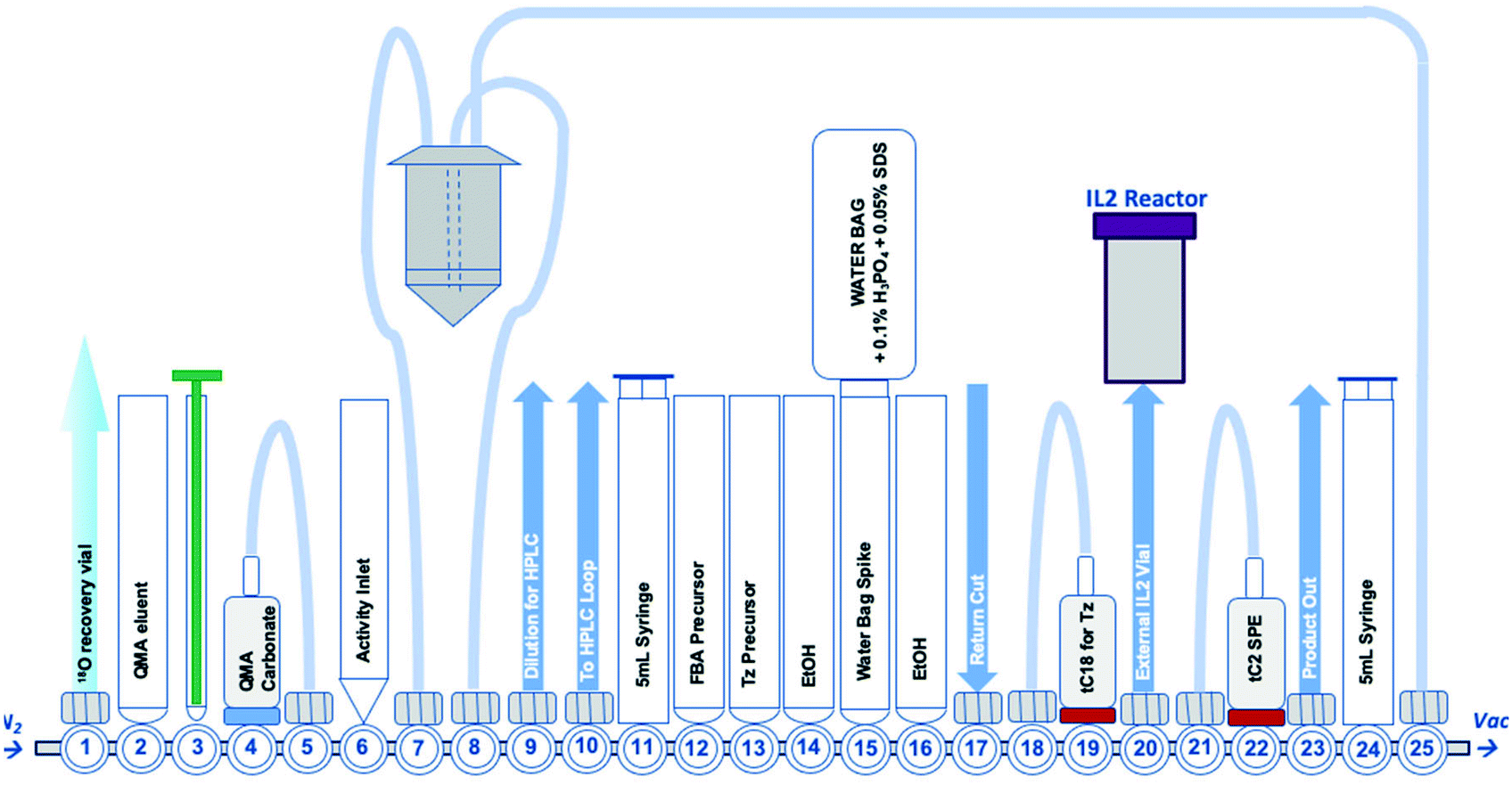 | ||
| Fig. 2 A schematic representation of the GE FASTLab™ cassette developed for the automated radiosynthesis of [18F]TTCO-IL2; a detailed description of the cassette setup appears in the ESI† (Fig. S14). | ||
Automating the IEDDA “click” reaction
With an efficient 18F-tetrazine radiosynthesis developed, the cassette and sequence were expanded to automate the IEDDA “click” chemistry (Fig. 2). This reaction occurs quickly at ambient temperature, at low concentration of reagents and in the absence of metal catalysts meaning there were few variables to optimise other than the quantity of TCO-PEG4-IL2 precursor. A key radiochemical parameter for IL2 radioconjugates is a high final Am; IL2 is an agonist and there are reports of effective low-dose IL2 therapy (100 μg per dose).41,42 There is a precedent for PET and SPECT IL2 radioconjugates to been administered at <50 μg of total protein per dose, therefore we aimed to develop a similar protocol for this IL2 radioconjugate.3,18,19,43 The automated procedure and cassette layout for [18F]FBoxTz was adapted to elute the 18F-PG in ethanol into an external Wheaton vial containing the TCO-PEG4-IL2 precursor which reacted for 15 min before purification; purification of the radioconjugate is described later in this manuscript. The efficiency of the IEDDA “click” reaction was monitored by HPLC and radio-TLC. The full radiosynthesis is described in the ESI† (Fig. S14).A maximum quantity of TCO-PEG4-IL2, 200 μg per radiolabelling reaction was investigated to produce clinically relevant Am of [18F]TTCO-IL2. The automated synthesis produced [18F]TTCO-IL2 in 19.8 ± 2.6% RCY (d.c.) (n = 14) within 110 min from the start of synthesis in >98% RCP. The radiochemical yield was not influenced by the starting activity of [18F]fluoride, however the molar activity increased linearly (R2 = 0.9744) with starting activities ranging from 1.0–21.8 GBq (ESI,† Fig. S15). Three productions using 19.6 ± 2.4 GBq of [18F]fluoride gave an Am of 132.3 ± 14.6 GBq μmol−1 (SA: 8.5 ± 0.8 MBq μg−1) which is sufficient for biological evaluation and clinical studies; in theory, a patient dose could be administered 2.5 h post radiosynthesis and still be within the 50 μg per dose IL2 limit. The recovery of the radioconjugate from the cassette was high, with negligible radiolabelled protein binding to plastic consumables.
It was assumed that reducing the quantity of TCO-PEG4-IL2 precursor from 200 μg to 100 μg would improve the Am if the IEDDA “click” efficiency remained the same. This hypothesis was tested by reducing the precursor quantity and reaction concentration by half; Am did not improve as the overall RCY was diminished by 50% (Table 1). A TCO-PEG4-IL2 precursor quantity of 200 μg was maintained for all further experiments.
| Entry 1 | Entry 2 | Entry 3 | |
|---|---|---|---|
| a n = 3. b n = 11 repeats. | |||
| TCO-PEG4-IL2 (μg) | 100a | 200b | 200a |
| Starting activity of [18F]fluoride (GBq) | 7.5 ± 1.5 | 6.9 ± 2.2 | 19.6 ± 2.4 |
| Specific activity (MBq μg−1) | 3.8 ± 1.5 | 3.5 ± 1.2 | 8.5 ± 0.8 |
| Molar activity (GBq μol−1) | 59.4 ± 22.7 | 54.3 ± 18.0 | 132.3 ± 14.6 |
| Radiochemical yield (% RCY, d.c.) | 5.1 ± 1.6 | 10.2 ± 1.2 | 8.7 ± 0.7 |
Purification of [18F]TTCO-IL2
Selecting an appropriate purification strategy for a radioconjugate is specific to the physicochemical properties of the protein of interest. IL2 radioconjugates have been effectively purified by tC2 SPE, a strategy made possible by the stability of the biomolecule to high concentrations of organic solvents.6,44 This is not the case for all proteins, which can denature under such conditions; however, some small protein fragments (e.g. affibody molecules) and DNA aptamers can tolerate high concentrations of organic solvent.22,45 [18F]TTCO-IL2 was purified by tC2 by adapting previously described procedures.6,46,47 This method removed unreacted [18F]FBoxTz and small-molecule impurities independently of [18F]TTCO-IL2, which was finally eluted into a formulation vial in >98% radiochemical purity (RCP).Summary of complete automated radiosynthesis
The automated radiosynthesis was simple to set up using commercially available components and reliably produced large batches of [18F]TTCO-IL2 (1.7 ± 0.2 GBq, n = 3). The radiosynthesis from [18F]fluoride to formulated [18F]TTCO-IL2 was complete in 110 min and the product was radiochemically stable (99.9 ± 0.1%) for the duration of testing (6 h) (ESI† Fig. S20 and S21). The identity of [18F]TTCO-IL2 was confirmed by radio-HPLC and a representative chromatogram is shown in Fig. 3. The RCY (d.c.) was 19.8 ± 2.6% (n = 14) and the molar activity was 132.3 ± 14.6 GBq μmol−1 from 19.6 ± 2.4 GBq of [18F]fluoride. Since completing this study, we have successfully produced GMP grade precursor 3 through a commercial research organisation to support future clinical translation of this strategy.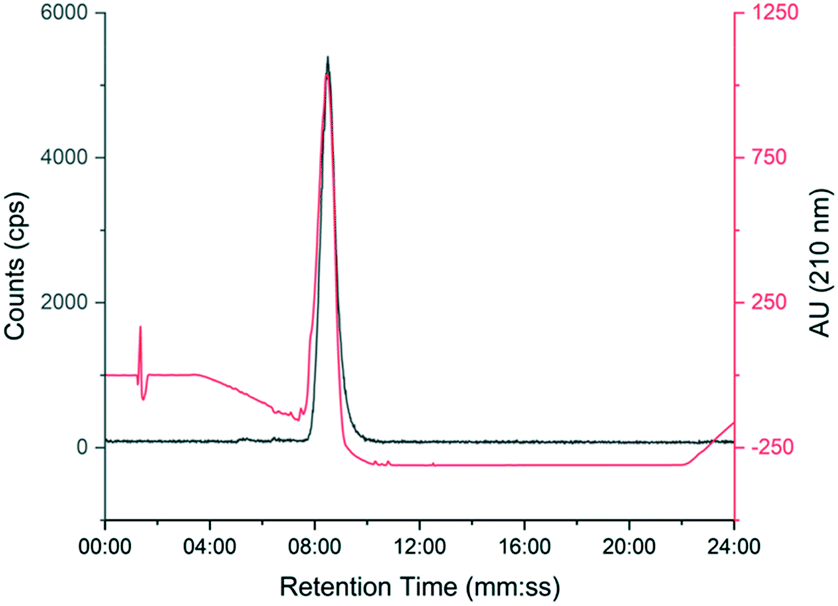 | ||
| Fig. 3 Representative HPLC chromatogram showing [18F]TTCO-IL2 (grey line) co-injected with non-reactive TCO-PEG4-IL2 (red line). | ||
In vitro evaluation of [18F]TTCO-IL2
[18F]TTCO-IL2 was evaluated in vitro against IL2R positive and negative cell lines, to confirm the retention of biological activity after radiolabelling. This was important to assess if any component of the conjugation or radiolabelling procedure had negatively influenced the integrity of IL2; additionally, this is the first time a TCO modified IL2 conjugate has been described and it was important to determine if TCO-PEG4 had perturbed its receptor recognition.The in vitro uptake of [18F]TTCO-IL2 in cell lines with differential IL2R expression was investigated (Fig. 4A and B). NK-92 cells and human PBMCs were stimulated to upregulate CD25 and consequently form the high affinity IL2R. PC3 prostate cancer cell line did not express CD25 and CD122, and therefore was used as a negative control. IL2R expression was confirmed by FACS analysis (Fig. 4C). The in vitro uptake of [18F]TTCO-IL2 is in agreement with the differential expression of CD25 in stimulated PBMCs, NK-92 and PC3 cells, and comparable to other IL2-based PET probes.21 Uptake was low in CD25 and CD122 negative PC3 cells indicating negligible non-specific binding. Analysis of phosphorylated STAT5 (Tyr694), critical for IL2 signalling, in NK-92 cells incubated with a dose of [18F]TTCO-IL2 (which also contains TCO-PEG4-IL2) demonstrated insignificant effect on STAT5 phosphorylation compared to recombinant IL2 at the same concentration (Fig. S26†). This suggests that [18F]TTCO-IL2 may not elicit an immune response when administered at a low concentration. The metabolic profile of [18F]TTCO-IL2 was determined in vitro using human liver microsomes (HLM) and mouse liver S9. The radioconjugate was stable (ca. 92% parent remaining in 60 min) for both species, although extraction efficiencies were low (19.5 ± 1.5% for HLM and 27.5 ± 3.3% for mouse liver S9), likely resulting from precipitation or aggregation of [18F]TTCO-IL2 caused by the extraction process from the biological milieu (Fig. S22†).
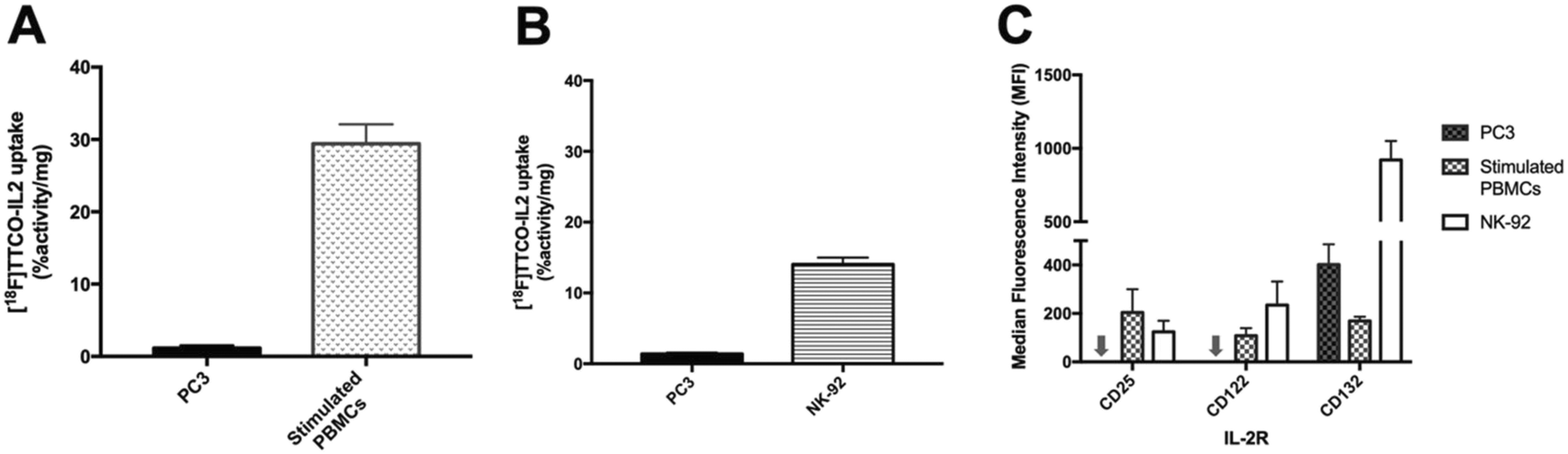 | ||
| Fig. 4 A) In vitro uptake of [18F]TTCO-IL2 (0.74 MBq, 85 ng–3.2 μg protein) in PC3 and PHA-stimulated PBMCs (72 h) at 37 °C. Two independent experiments were performed for PBMCs and PC3 (n = 6). B) In vitro uptake of [18F]TTCO-IL2 (0.74 MBq, 85–264 ng protein) in PC3 and NK-92 at room temperature (RT). Three independent experiments were performed from PC3 and NK-92 (n = 6). Data expressed as mean ± SEM. Significant difference between PC3 and NK-92/PHA-stimulated PBMCs is indicated by **** (p < 0.0001). Statistical difference is analysed using unpaired t-test. C) Surface expression of IL2R (CD25, CD122, CD132) in PC3 cells, NK-92 cells treated with 6.1 ng mL−1 (100 U mL−1) IL-2, and PHA-stimulated PBMCs (72 h) – arrow represents PC3 cell line with 0 MFI. Data are presented as median fluorescence intensity (MFI) from five donors for PBMCs and three independent experiments for PC3 and NK-92 cells. | ||
Conclusion
To exemplify the utility of adapting IEDDA “click” chemistry to radiolabel proteins in a fully automated, single-cassette based procedure, we describe a method for radiolabelling a TCO-modified IL2 protein using the GE FASTLab™ platform. [18F]TTCO-IL2, a biologically active radioconjugate towards the IL2R was produced in 110 min from a single-cassette radiosynthesis. The in vivo evaluation of [18F]TTCO-IL2 is underway, but early data suggests uptake in target enriched tissues such as the lymphatic system; given the complexity of biological models of immune response, these data will be reported elsewhere. It is hoped that that the work presented here will be useful to the PET radiochemistry community for automating the radiolabelling of biomolecules of interest.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Imperial College NIHR Biomedical Research Centre award (WSCC_P62585), the Medical Research Council Grant (MC-A652-5PY80) and Imperial College Experimental Cancer Medicines award C1312/A25149. We thank those who donated blood for this work. We would like to thank “The Friends of Hammersmith Hospital” (Registered Charity No: 209182) for the funding the purchase of our PosiRAM radio-metabolite detector used in this study.Notes and references
- A. Gallamini, C. Zwarthoed and A. Borra, Cancers, 2014, 6, 1821–1889 CrossRef PubMed.
- E. M. Rohren, T. G. Turkington and R. E. Coleman, Radiology, 2004, 231, 305–332 CrossRef PubMed.
- L. Allott and E. O. Aboagye, Mol. Pharmaceutics, 2020, 17(7), 2245–2259 CrossRef CAS PubMed.
- L. Bruton and P. J. H. Scott, in Handbook of Radiopharmaceuticals, 2020, pp. 437–456 Search PubMed.
- C. Rangger and R. Haubner, Pharmaceuticals, 2020, 13, 22 CrossRef CAS PubMed.
- E. L. van der Veen, I. F. Antunes, P. Maarsingh, J. Hessels-Scheper, R. Zijlma, H. H. Boersma, A. Jorritsma-Smit, G. A. P. Hospers, E. G. E. de Vries, M. N. Lub-de Hooge and E. F. J. de Vries, EJNMMI radiopharm. chem., 2019, 4, 1–15 CrossRef PubMed.
- P. B. Iveson, M. Glaser, B. Indrevoll, J. Shales, D. Mantzilas, L. Omtvedt, S. K. Luthra, D. Hiscock and J. Grigg, J. Labelled Compd. Radiopharm., 2019, 62, 925–932 CrossRef CAS PubMed.
- L. Allott, C. Barnes, D. Brickute and E. O. Aboagye, React. Chem. Eng., 2019, 4, 569–574 RSC.
- S. R. R. Dubash, N. Keat, P. Mapelli, F. Twyman, L. Carroll, K. Kozlowski, A. Al-Nahhas, A. Saleem, M. Huiban, R. Janisch, A. Frilling, R. Sharma, E. O. O. Aboagye, N. Keat, P. Mapelli, F. Twyman, L. Carroll, K. Kozlowski, A. Al-Nahhas, A. Saleem, M. Huiban, R. Janisch, A. Frilling, R. Sharma and E. O. O. Aboagye, J. Nucl. Med., 2016, 57, 1207–1213 CrossRef CAS PubMed.
- L. Allott, C. Barnes, D. Brickute, S. F. J. Leung and E. O. Aboagye, React. Chem. Eng., 2019, 4, 1748–1751 RSC.
- L. Allott, C. Da Pieve, D. R. Turton and G. Smith, React. Chem. Eng., 2017, 2, 68–74 RSC.
- T. Tshibangu, C. Cawthorne, K. Serdons, E. Pauwels, W. Gsell, G. Bormans, C. M. Deroose and F. Cleeren, EJNMMI radiopharm. chem., 2020, 5, 4 CrossRef PubMed.
- O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed.
- L. Allott, S. Dubash and E. O. Aboagye, Cancers, 2020, 12, E865 CrossRef PubMed.
- Y. Ohashi, T. Takeshita, K. Nagata, S. Mori and K. Sugamura, J. Immunol., 1989, 143, 3548–3555 CAS.
- S. Hotte, T. Waldron, C. Canil and E. Winquist, Can Urol Assoc J, 2007, 1, 27–38 Search PubMed.
- T. Petrella, I. Quirt, S. Verma, A. E. Haynes, M. Charette and K. Bak, Cancer Treat. Rev., 2007, 33, 484–496 CrossRef CAS PubMed.
- A. Annovazzi, L. Biancone, R. Caviglia, M. Chianelli, G. Capriotti, S. J. Mather, R. Caprilli, F. Pallone, F. Scopinaro and A. Signore, Eur. J. Nucl. Med. Mol. Imaging, 2003, 30, 374–382 CrossRef CAS PubMed.
- A. Signore, A. Annovazzi, R. Barone, E. Bonanno, C. D'Alessandria, M. Chianelli, S. J. Mather, U. Bottoni, C. Panetta, D. Innocenzi, F. Scopinaro and S. Calvieri, J. Nucl. Med., 2004, 45, 1647–1652 Search PubMed.
- V. Di Gialleonardo, A. Signore, A. W. J. M. Glaudemans, R. A. J. O. Dierckx and E. F. J. De Vries, J. Nucl. Med., 2012, 53, 679–686 CrossRef PubMed.
- E. L. van der Veen, F. V. Suurs, F. Cleeren, G. Bormans, P. H. Elsinga, G. A. P. Hospers, M. N. Lub-de Hooge, E. G. E. de Vries, E. F. J. de Vries and I. F. Antunes, J. Nucl. Med., 2020, 61(9), 1355–1360 CrossRef CAS PubMed.
- C. Da Pieve, L. Allott, C. D. Martins, A. Vardon, D. M. Ciobota, G. Kramer-Marek and G. Smith, Bioconjugate Chem., 2016, 27, 1839–1849 CrossRef CAS PubMed.
- H. H. Coenen, A. D. Gee, M. Adam, G. Antoni, C. S. Cutler, Y. Fujibayashi, J. M. Jeong, R. H. Mach, T. L. Mindt, V. W. Pike and A. D. Windhorst, Nucl. Med. Biol., 2017, 55, 5–11 CrossRef PubMed.
- L. Iddon, J. Leyton, B. Indrevoll, M. Glaser, E. G. Robins, A. J. T. George, A. Cuthbertson, S. K. Luthra and E. O. Aboagye, Bioorg. Med. Chem. Lett., 2011, 21, 3122–3127 CrossRef CAS PubMed.
- A. Challapalli, L. M. Kenny, W. A. Hallett, K. Kozlowski, G. Tomasi, M. Gudi, A. Al-Nahhas, R. C. Coombes and E. O. Aboagye, J. Nucl. Med., 2013, 54, 1551–1556 CrossRef CAS PubMed.
- U. Karczmarczyk, P. Garnuszek, M. Maurin, V. Di Gialleonardo, F. Galli, A. Signore and R. Mikołajczak, Nucl. Med. Biol., 2010, 37, 795–803 CrossRef CAS PubMed.
- D. Charych, S. Khalili, V. Dixit, P. Kirk, T. Chang, J. Langowski, W. Rubas, S. K. Doberstein, M. Eldon, U. Hoch and J. Zalevsky, PLoS One, 2017, 12, e0179431 CrossRef PubMed.
- V. S. De Paula, K. M. Jude, S. Nerli, C. R. Glassman, K. C. Garcia and N. G. Sgourakis, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7183–7192 CrossRef CAS PubMed.
- S. M. Zurawski and G. Zurawski, EMBO J., 1992, 11, 3905–3910 CrossRef CAS PubMed.
- D. V. Liu, L. M. Maier, D. A. Hafler and K. D. Wittrup, J. Immunother., 2009, 39, 887–894 CrossRef PubMed.
- Q. Zhou, Biomedicines, 2017, 5, 1–15 CrossRef PubMed.
- M. Morais and M. T. Ma, Drug Discovery Today: Technol., 2018, 30, 91–104 CrossRef PubMed.
- B. E. Cook, P. Adumeau, R. Membreno, K. E. Carnazza, C. Brand, T. Reiner, B. J. Agnew, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 1789–1795 CrossRef CAS PubMed.
- E. M. F. Billaud, S. Belderbos, F. Cleeren, W. Maes, M. Van De Wouwer, M. Koole, A. Verbruggen, U. Himmelreich, N. Geukens and G. Bormans, Bioconjugate Chem., 2017, 28, 2915–2920 CrossRef CAS PubMed.
- Z. Li, H. Cai, M. Hassink, M. L. Blackman, R. C. D. Brown, P. S. Conti and J. M. Fox, Chem. Commun., 2010, 46, 8043–8045 RSC.
- L. Qiu, W. Mao, H. Yin, H. Tan, D. Cheng and H. Shi, Contrast Media Mol. Imaging, 2019 DOI:10.1155/2019/9182476.
- J. P. Meyer, J. L. Houghton, P. Kozlowski, D. Abdel-Atti, T. Reiner, N. V. K. Pillarsetty, W. W. Scholz, B. M. Zeglis and J. S. Lewis, Bioconjugate Chem., 2016, 27, 298–301 CrossRef CAS PubMed.
- J. Sauer, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, Oxford, 1996, pp. 901–955 Search PubMed.
- A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Chem. Sci., 2014, 5, 4545–4553 RSC.
- Q. Zheng, H. Xu, H. Wang, W.-G. H. Du, N. Wang, H. Xiong, Y. Gu, L. Noodleman, K. B. Sharpless, G. Yang and P. Wu, J. Am. Chem. Soc., 2021, 143, 3753–3763 CrossRef CAS PubMed.
- M. Tahvildari and R. Dana, J. Immunol., 2019, 203, 2749–2755 CrossRef CAS PubMed.
- A. Kosmaczewska, Int. J. Mol. Sci., 2014, 15, 18574–18592 CrossRef CAS PubMed.
- Committee for Medicinal Products for Human Use. ICH guideline M3 (R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals (EMA/CPMP/ICH/286/1995), EMA, 2009.
- M. Chianelli, A. Signore, A. R. Fritzberg and S. J. Mather, Nucl. Med. Biol., 1997, 24, 579–586 CrossRef CAS PubMed.
- K. K. H. Stanlis and J. R. McIntosh, J. Histochem. Cytochem., 2003, 51, 797–808 CrossRef CAS PubMed.
- Y. Gao, Q. Luo, C. Luo, X. Ma, J. Shi and F. Wang, J. Nucl. Med., 2020, 61(supplement 1), 161 Search PubMed.
- A. Signore, A. Picarelli, A. Annovazzi, K. E. Britton, A. B. Grossman, E. Bonanno, B. Maras, D. Barra and P. Pozzilli, Nucl. Med. Commun., 2003, 24, 305–316 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, HPLC chromatograms. See DOI: 10.1039/d1re00117e |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |