
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDDQ as a versatile and easily recyclable oxidant: a systematic review
Meshari A. Alsharifa,
Qandeel Alam Rajab,
Nida Abdul Majeedb,
Rabab. S. Jassasc,
Abdulrahman A. Alsimareed,
Amina Sadiqe,
Nafeesa Naeem b,
Ehsan Ullah Mughal
*b,
Reem I. Alsantalif,
Ziad Moussag and
Saleh A. Ahmed
*ahi
b,
Ehsan Ullah Mughal
*b,
Reem I. Alsantalif,
Ziad Moussag and
Saleh A. Ahmed
*ahi
aDepartment of Chemistry, Faculty of Applied Science, Umm Al-Qura University, 21955 Makkah, Saudi Arabia. E-mail: saahmed@uqu.edu.sa
bDepartment of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsantbtq@gmail.com
cDepartment of Chemistry, Jamoum University College, Umm Al-Qura University, 21955 Makkah, Saudi Arabia
dDepartment of Basic Science (Chemistry), College of Science and Humanities, Shaqra University, Afif, Saudi Arabia
eDepartment of Chemistry, Govt. College Women University, Sialkot-51300, Pakistan
fDepartment of Pharmaceutical Chemistry, Pharmacy College, Taif University, 888-Taif, Saudi Arabia
gDepartment of Chemistry, College of Science, United Arab Emirates University, P.O. Box 15551, Al Ain, Abu Dhabi, United Arab Emirates
hResearch laboratories unit, Faculty of Applied Science, Umm Al-Qura University, 21955 Makkah, Saudi Arabia
iDepartment of Chemistry, Faculty of Science, Assiut University, 71516 Assiut, Egypt
First published on 8th September 2021
Abstract
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) is the most widely used quinone with a high reduction potential, and it commonly mediates hydride transfer reactions and shows three accessible oxidation states: quinone (oxidized), semiquinone (one-electron-reduced), and hydroquinone (two-electron-reduced). DDQ has found broad utility as a stoichiometric oxidant in the functionalization of activated C–H bonds and the dehydrogenation of saturated C–C, C–O, and C–N bonds. The cost and toxicity of DDQ triggered recent efforts to develop methods that employ catalytic quantities of DDQ in combination with alternative stoichiometric oxidants. The aerobic catalytic approach was established for the selective oxidation of non-sterically hindered electron-rich benzyl methyl ethers and benzylic alcohols, and effectively extended to the oxidative deprotection of p-methoxybenzyl ethers to generate the alcohols in high selectivity. A combination of DDQ and protic acid is known to oxidize several aromatic donors to the corresponding cation radicals. The excited-state DDQ converts benzyls, heteroarenes, fluoroarenes, benzene, and olefins into their radical cation forms as well as chloride and other anions into their respective radicals. These reactive intermediates have been employed for the generation of C–C and C–X (N, O, or Cl) bonds in the synthesis of valuable natural products and organic compounds. To the best of our knowledge, however, there is still no review article exclusively describing the applications of DDQ in organic synthesis. Therefore, in the present review, we provide an overview of DDQ-induced organic transformations with their scope, limitations and the proposed reaction mechanisms.
 Ehsan Ullah Mughal | Ehsan Ullah Mughal was born in Wazirabad, Pakistan in 1978 and obtained his master's degree from Quaid-e-Azam University, Islamabad, Pakistan in organic chemistry. He obtained his PhD in 2013 from Bielefeld University, Germany under the supervision of Prof. Dr Dietmar Kuck. For postdoctoral studies, he joined the group of Prof. Dr Xinliang Feng, Max-Planck-Institute for Polymer Research, Mainz, Germany. In 2015, he started his independent research group at the Department of Chemistry in University of Gujrat, Gujrat, Pakistan. His current research interests focus on the design and synthesis of bioactive heterocycles, synthetic flavonoids, transition metals-based terpyridine complexes and their fabrication in dye-sensitized solar cells and nanographenes syntheses for applications in organic electronics. |
 Saleh A. Ahmed | Saleh Abdel-Mgeed Ahmed was born in Assiut, Egypt in 1968 and received his bachelor's and master's degrees from Assiut University, Egypt, and PhD in photochemistry (photochromism) under the supervision of Prof. Dr Heinz Dürr in Saarland University, Saarbrücken, Germany as DAAD fellow. During his last 27 years of stay in Europe, Japan, and USA, he had successfully published more than 195 publications and has more than 15 registered and filled patents in US-patent office. His current research interests include synthesis and photophysical properties of novel organic compounds such as material-based gels, electronic devices, solar energy conversion and bioactive materials. |
1. Introduction
Oxidation reactions play a vital role in organic synthesis and offer access to various important organic compounds and are key to the interconversion of functional groups from one to the other.1 Though frequent stoichiometric oxidants of inorganic nature have been conventionally used in many oxidation reactions, however, they are associated with some serious drawbacks. These drawbacks include low stability, high cost, and the formation of toxic or hazardous pollutants.2 In order to circumvent these problems, a versatile, mild and eco-friendly oxidant such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) has been employed in various organic reactions for the oxidation of chemical compounds. Besides serving as an oxidizing agent, it has proved useful as a multipurpose reactant for numerous organic chemical conversions.3–5DDQ has found a multitude of applications in the oxidation of various organic compounds such as ketones,6 alcohols,7 phenols,8 aromatic compounds,9 and heterocyclic structures, etc.10 Apart from that, due to the presence of two chlorine atoms along with two nitrile groups on the benzoquinone ring, it may also behave as a potential chlorinating agent. Consequently, DDQ could behave as the chlorinating agent as well as the oxidant simultaneously.11 It can also be used to remove the protective functional groups during deprotections of different chemical entities.12
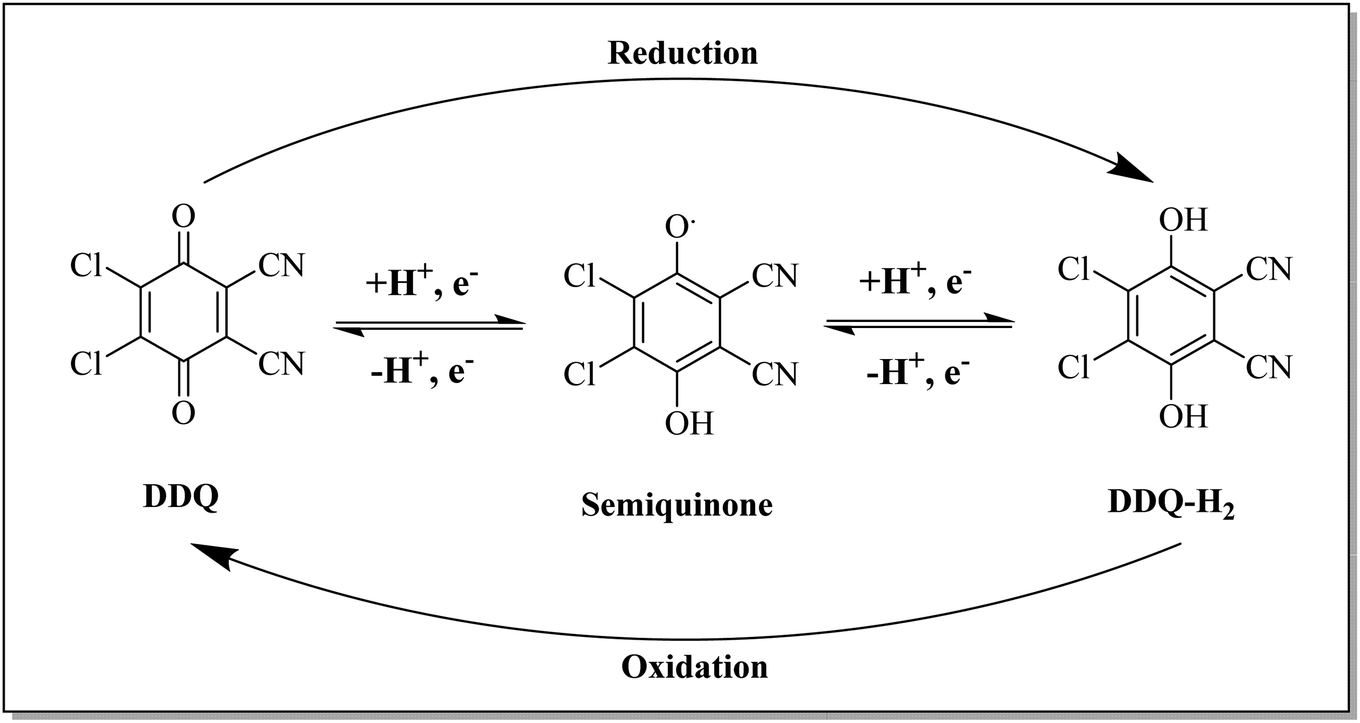
For dehydrogenation by DDQ, the hydride transfer mechanism has been proposed as shown in Scheme 1. The mechanism includes the transfer of hydride to the quinone oxygen followed by the transfer of a proton to the phenolate ion.13 DDQ is a strong oxidizing quinone that is indefinitely stable in dry conditions. It is widely employed for the dehydrogenation of organic molecules to form aromatic and α,β-unsaturated carbonyls, and the oxidation of activated methylene and hydroxy groups to carbonyl compounds.14 With the ongoing increase in demand for the development of selective organic reactions, certain reagents have witnessed substantial growth and expansion in their applications and now offer access to new synthesis opportunities.
 | ||
| Scheme 1 DDQ as an oxidant. | ||
Although several uses of DDQ have been reported in the literature from the pharmaceutical and specialty chemical industries, we shall restrict this article to an overview of organic reactions where DDQ has a significant contribution. Herein, we review the applications of DDQ that have been explored by several research groups and described in the literature. The applications are categorized according to different reaction types, with a special focus on the role of DDQ. In several instances, DDQ is coupled with some other oxidant for improved stability, decreased toxicity, and enhanced yield.15 This mini-review provides an overview of such DDQ-initiated organic transformations with their scope, limitations and discusses the proposed reaction mechanisms.
2. Synthesis of DDQ
This reagent was first synthesized by Thiele and Gunther in 1906 and is commercially available from several chemical vendors.4b In the laboratory, it can directly be synthesized by acid-catalyzed chlorination of 2,3-dicyanohydroquinone followed by treatment with an oxidant such as PbO2 under reflux conditions as depicted in Scheme 2. Furthermore, charcoal can also be used in the second step.16,17 DDQ is a yellow solid with a melting point of 213–215 °C. It can be stored in a dry environment, however, it is prone to decomposition in the presence of moisture. Notably, the rate of decomposition can be prevented by the presence of a strong acid as DDQ is very stable in aqueous mineral acid. It can be purified by crystallization from methylene chloride.18 The availability, excellent yields, high chemo- and regioselectivity, durability, and catalytic character are noteworthy features of DDQ. These features render it as a remarkable stoichiometric and catalytic oxidant for reactions in organic synthesis.19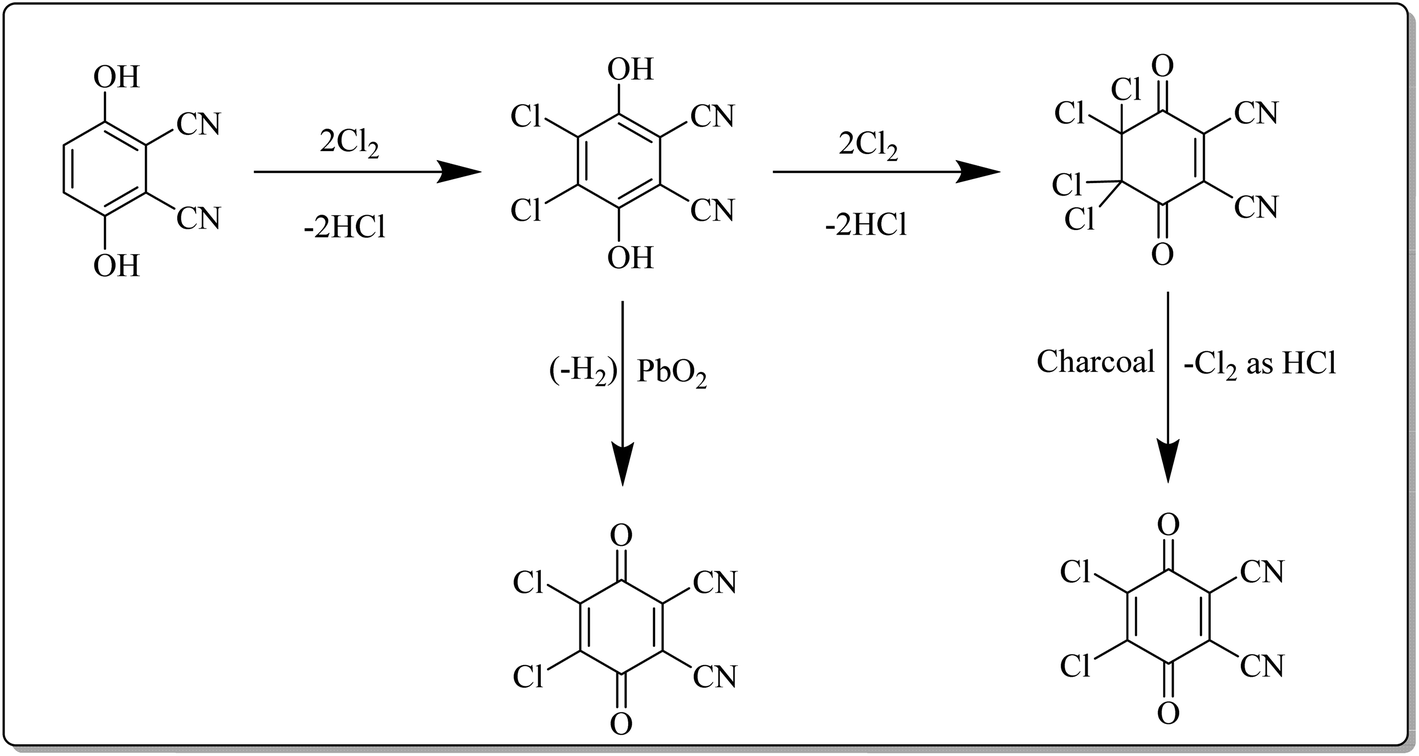 | ||
| Scheme 2 Synthesis of DDQ. | ||
3. General reactivity
DDQ is generally reactive against many classes of compounds like alcohols, ethers,20 ketones,21 α,β-unsaturated carbonyl compounds, arenes22 and imines23 as described above. Besides, it is used in C–H bond oxidation,24 and C–C bond formation reactions (Scholl reaction),25 protection and deprotection reactions, cross-coupling reactions,26a and in several other organic transformations such as DDQ-initiated visible-light reactions.26b We shall discuss these classes one by one in the following paragraphs.3.1. Oxidation of alcohols
In 1983, Harvey and co-workers reported that the oxidation of benzylic alcohols like compound 1 can be performed by treating with DDQ (Scheme 3). This outcome can be defined by the relative potential of solvent used to improve the charge-transfer complexation in the initial stage of the reaction between the alcohol and DDQ.27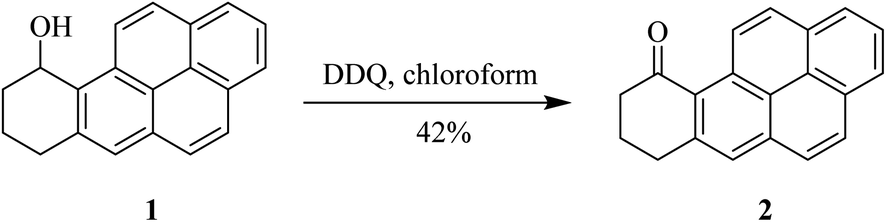 | ||
| Scheme 3 Benzylic alcohol oxidation by DDQ. | ||
Later on, Kasturi and co-workers (1993) described a procedure for the bis(2-hydroxyl-naphthyl)methane 3 oxidation by using DDQ, and the resulting products were spironaphthalenone (4a), dispironaphthalenoncs (4b), and 1,2-naphthoquinone-I-methide dimer (4c) (Scheme 4). Their studies towards the synthesis of novel organic compounds had scrutinized oxidative processes for the production of isomeric and dimeric products from bis(2-hydroxyl-naphthyl)methane.28
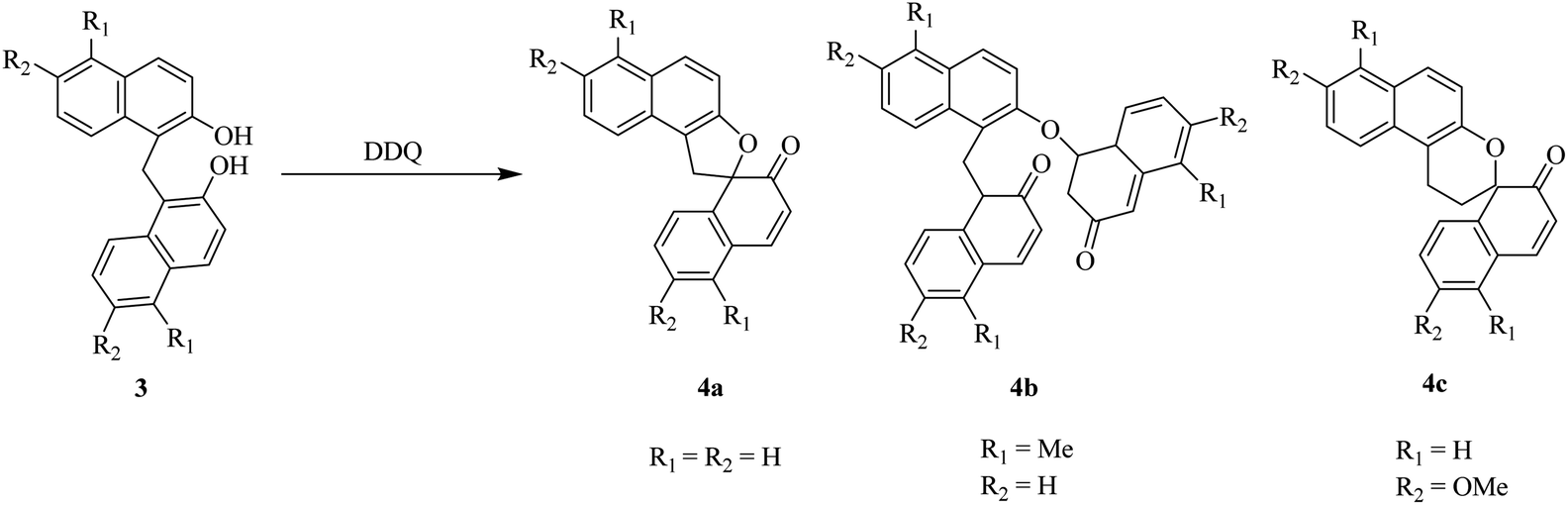 | ||
| Scheme 4 DDQ-induced oxidation of naphthol like compound 3. | ||
Thereafter, Pan and co-workers (2005) developed a mild and comprehensive method for the regioselective oxidation of chiral secondary 1,2-diols 5. The protocol highlights the synthetic utility of DDQ to affect the oxidation of sec-1,2-diols selectively at their benzylic or allylic hydroxyl group under mild ultrasound irradiation conditions. During this process, the arrangement of the adjoining chiral center did not change (Scheme 5).29
 | ||
| Scheme 5 Oxidation of 1,2-diols by DDQ. | ||
Progressively, Helquist and co-workers (2011) reported a protocol that describes the chemo-selective oxidation of alcohols in which a catalytic amount of DDQ served as the main oxidant, while a small quantity of co-oxidant Mn(OAc)3 was also used (Scheme 6; eqn (i)–(iii)). The oxidation of electron-rich chiral benzylic alcohols 7, 9, and 11 were carried out successfully to afford carbonyls, whereas benzylic alcohols that are not sufficiently electron-rich remained unreacted. The procedure was comparatively easy, cheap, less time-consuming, and showed noteworthy chemo-selectivity that favored allylic alcohols as compared to benzylic alcohols in as demonstrated in intramolecular competition studies.30
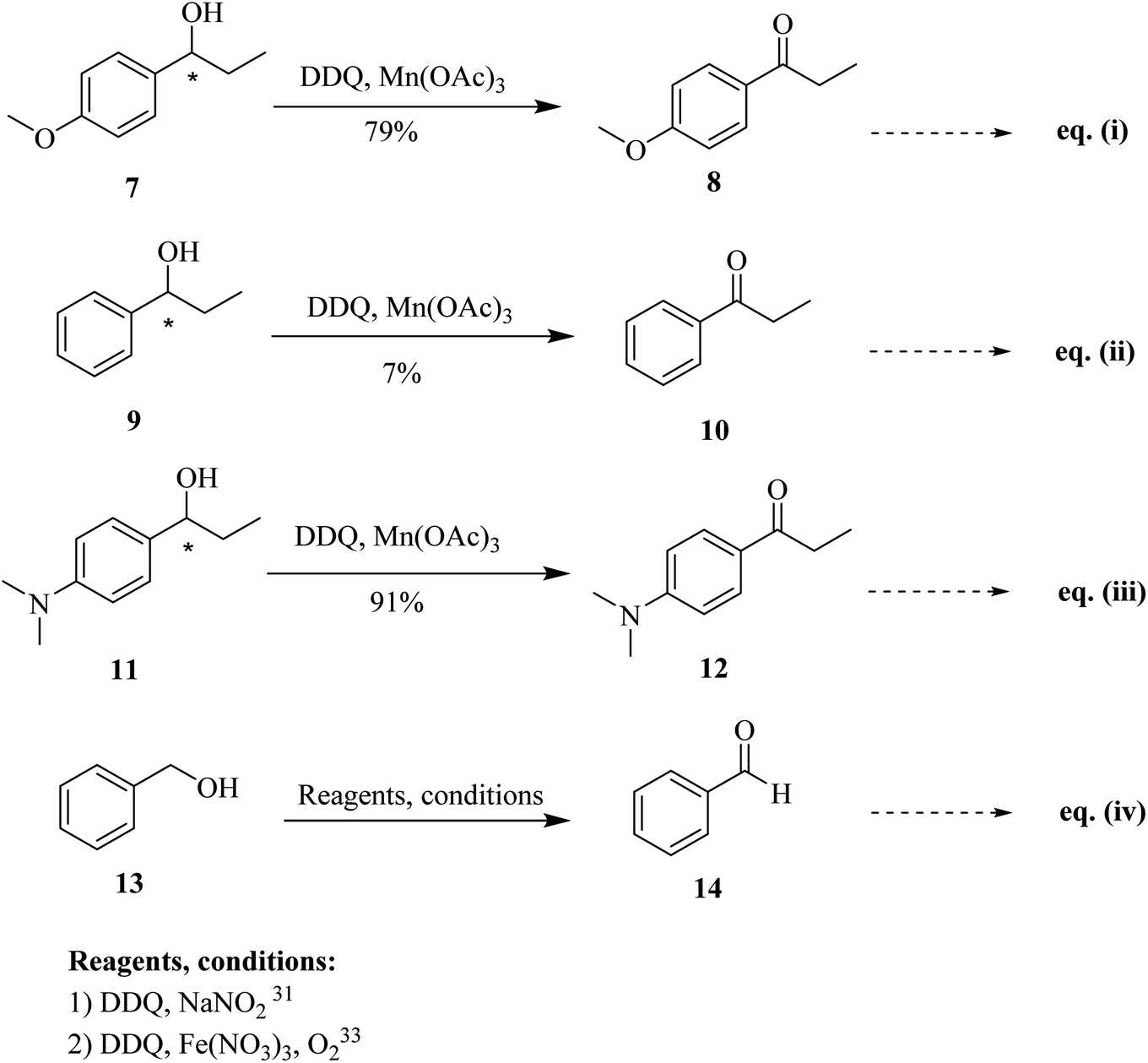 | ||
| Scheme 6 DDQ initiated oxidation of alcohols. | ||
In 2012, Gao and co-workers developed another simple procedure for the oxidation of benzylic alcohols such as 13 to formylbenzene 14 by taking a small quantity of DDQ as a catalyst. The co-catalyst used during this process was sodium nitrite (NaNO2), and oxygen (O2) was used as a finishing oxidant (Scheme 6; eqn (iv)). Nitric oxide was generated by NaNO2 in the presence of acetic acid. This step was crucial for the success of the catalytic cycle at 25 °C. This catalytic process is practically useful for the selective oxidation of unsaturated alcohols and is scalable as established in the large gram-scale conversion of cinnamyl alcohol to the corresponding aldehyde.31
In 2015, Westwood and co-workers used DDQ as a benzylic oxidant for lignin, because it exhibited excellent selectivity in oxidizing benzylic and allylic alcohols like 15. It was used in combination with oxygen and an appropriate co-oxidant. The substrate 15 was treated with the appropriate amount of DDQ at ambient temperature to produce ketone 16 in a high yield. The secondary benzylic alcohol can be selectively oxidized by controlling the amounts of DDQ and co-oxidant (Scheme 7).32
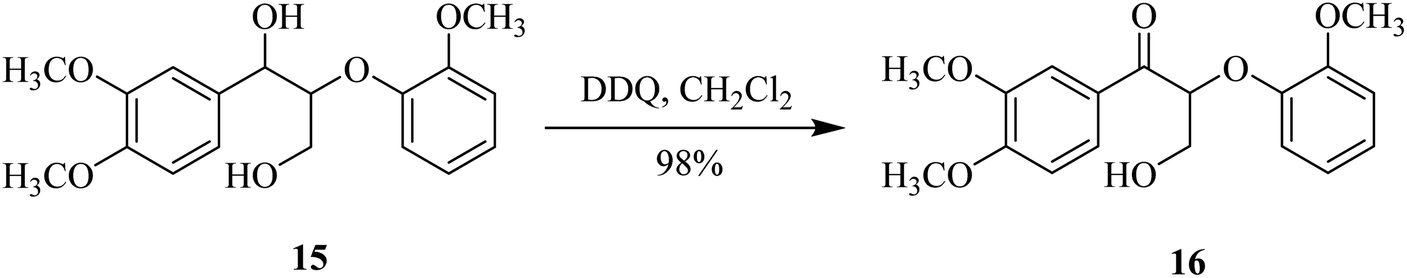 | ||
| Scheme 7 Selective benzylic oxidation of β-O-4 model compound 15. | ||
Moreover, the DDQ/tBuONO/O2 catalytic set-up showed significant selectivity and reactivity when used with 2-methoxyethanol for the oxidation of benzylic alcohols like 17, 19, and 21. The expected ketones 18, 20, and 22 thus obtained were isolated in excellent yields (Scheme 8).32
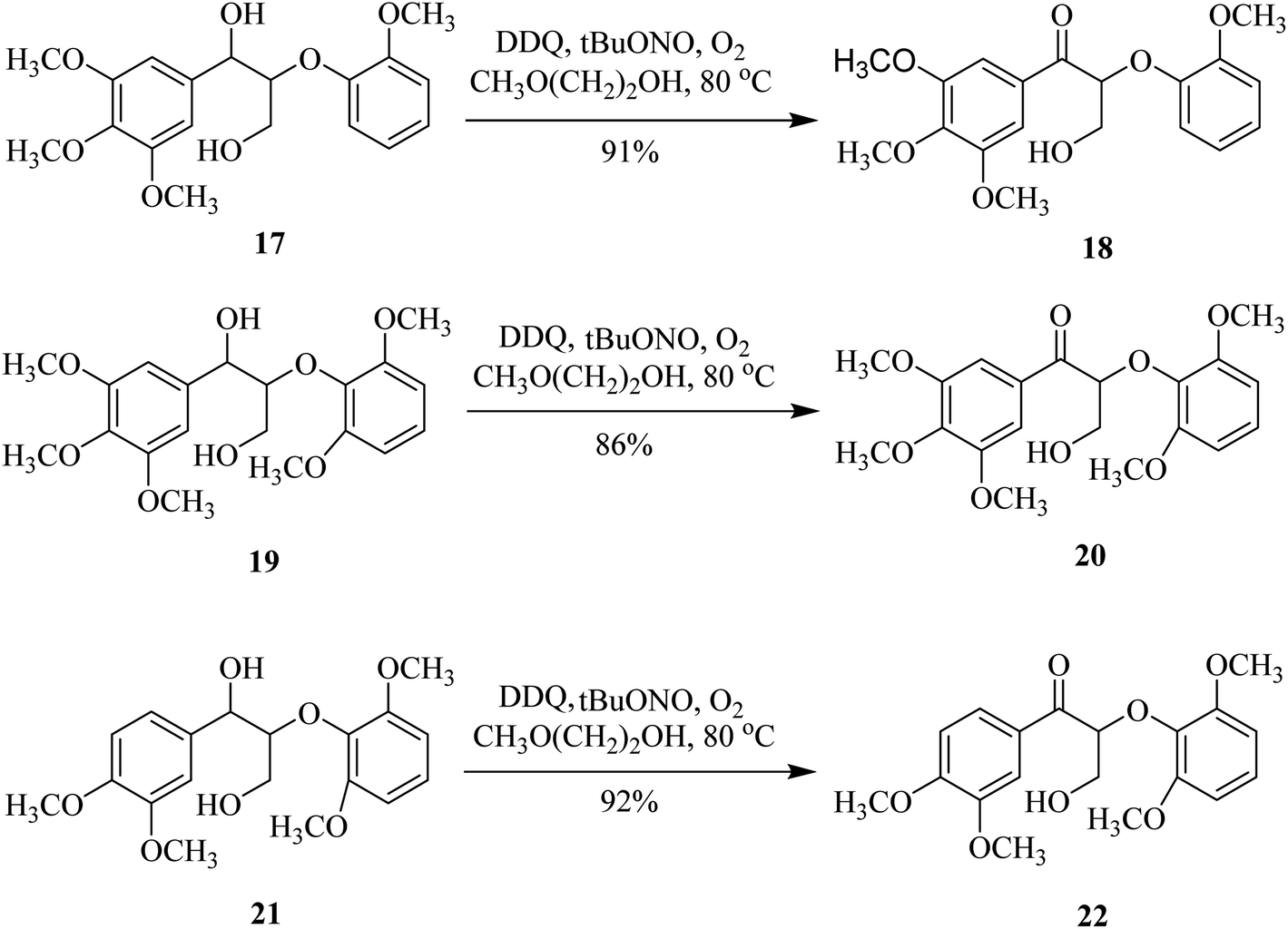 | ||
| Scheme 8 Selective benzylic alcohol oxidation by DDQ. | ||
Li and co-workers (2018) described an improved eco-friendly oxidative mixture comprising Fe(NO3)3 and DDQ to perform the same type of oxidation reported by Gao31 with O2 as the ultimate oxidant as mentioned above in Scheme 6, eqn (iv). This catalytic system displayed outstanding functional group tolerance and diversity and proved to oxidize several functionalities such as allylic, heterocyclic, propargylic, and benzylic alcohols to the corresponding carbonyl compounds in moderate to excellent yields. Furthermore, a reasonable reaction mechanism was also proposed in this report.33
More recently (2020), Jean and co-workers introduced a practical, scalable and selective method for the oxidation of diphenylmethanol 23 to benzophenone 24 by using a catalytic amount of the mixture DDQ and nitric acid (HNO3). However, that procedure was selectively used for the oxidation of allylic, benzylic, and propargylic alcohols (Scheme 9).34
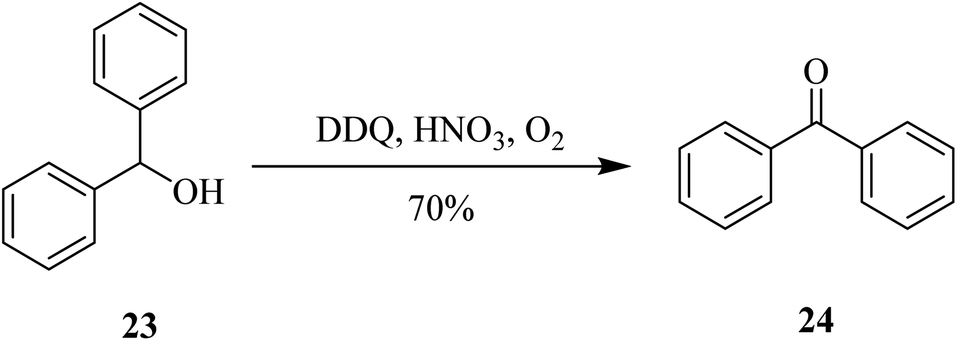 | ||
| Scheme 9 Oxidation of benzylic alcohols. | ||
The selective oxidation of alcohols has always been a thriving topic of discussion in organic synthesis. Mostly metallic oxidants, such as manganese oxides, hypervalent I2 and chromium oxides, are used for this purpose. Unfortunately, metallic oxidants produce toxic by-products and contribute to environmental pollution. Thus, to eliminate all these problematic reagents, the green oxidant DDQ serves as an eco-friendly alternative. The reagent has been found, as discussed above, to oxidize benzylic, phenolic, allylic, propargylic alcohols and chiral secondary 1,2-diols in a highly chemoselective and regioselective manner without causing any racemization to chiral substrates. Even more remarkable, DDQ may be used in catalytic amounts along with other co-oxidants to promote oxidations of alcohols. Interestingly, in many cases DDQ tolerates other green oxidants like molecular oxygen, which is inert because of the high energy barrier between alcohols and O2, and yet couples well with the cheap and less toxic oxidant DDQ to selectively oxidize alcohols. Hence, DDQ is being used preferably in such type of oxidations owing to its practical relevance.
3.2. Oxidation of ethers
Bhattacharya and co-workers (1989) reported that the reaction of silyl imidates of the 4-aza-3-keto steroids with DDQ produces lactams via the intermediacy of a unique adduct formation between the substrate and quinone oxidant followed by an electrocyclic reaction to form the double bond during this process to establish unsaturation. Similarly, ketones were found to react as silyl enol ethers with DDQ to furnish enones, as illustrated in Scheme 10 for the conversion of 25 to 26. This conversion takes place through the intermediacy of quinone-silyl enol ether adduct as shown below.35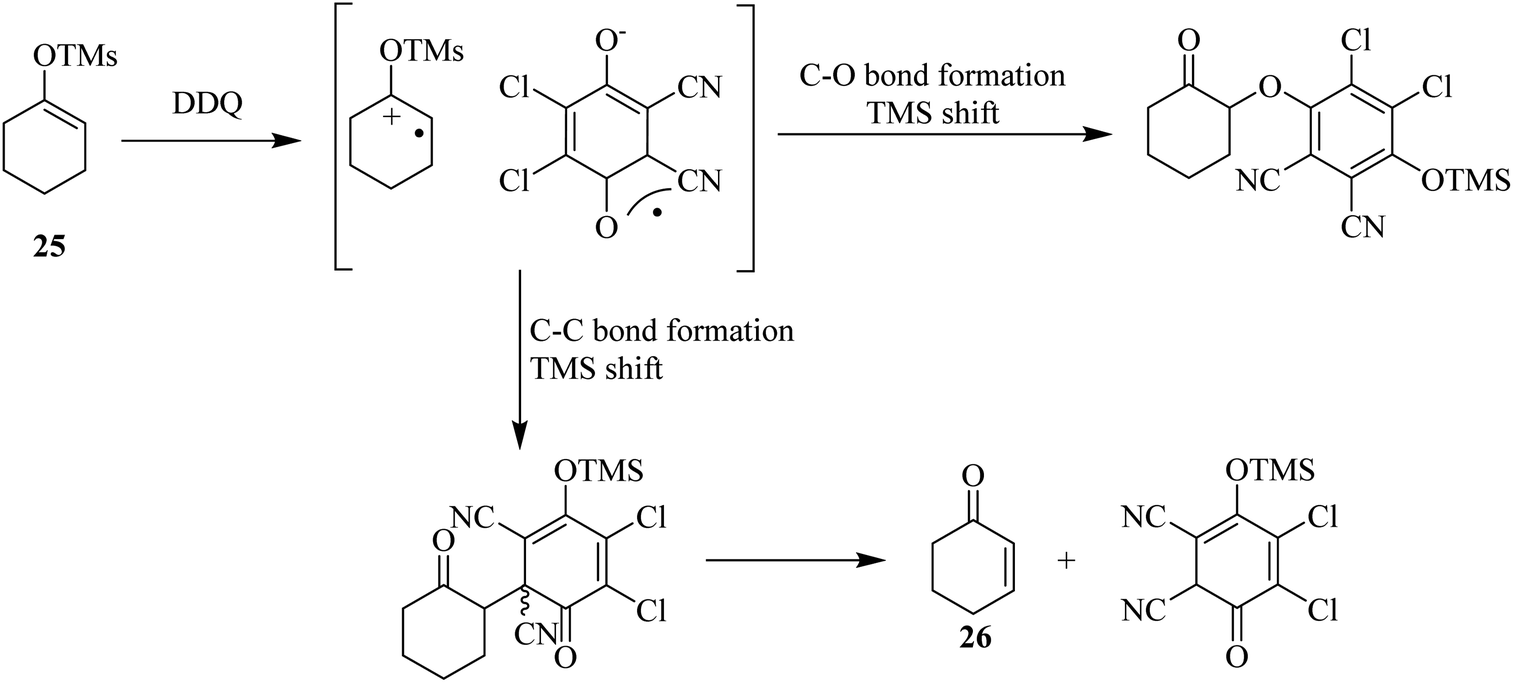 | ||
| Scheme 10 Oxidation of ethers to enones by DDQ. | ||
In the same year, Ruff and co-workers proposed that allylic and benzylic ethers like 27 undergo oxidation by DDQ to yield the substitution product 28 via an allylic cation intermediate (Scheme 11). Moreover, primary ethers can be oxidized with DDQ in high yield and give the corresponding esters. The oxidation may continue from 28 by a mechanism that includes abstraction of initial hydride to produce a stable benzylic or allylic cation, then the loss of a proton can occur followed by the formation of reduced DDQ–hydroquinone form, which can precipitate out of the reaction mixture usually in nonpolar solvents.36
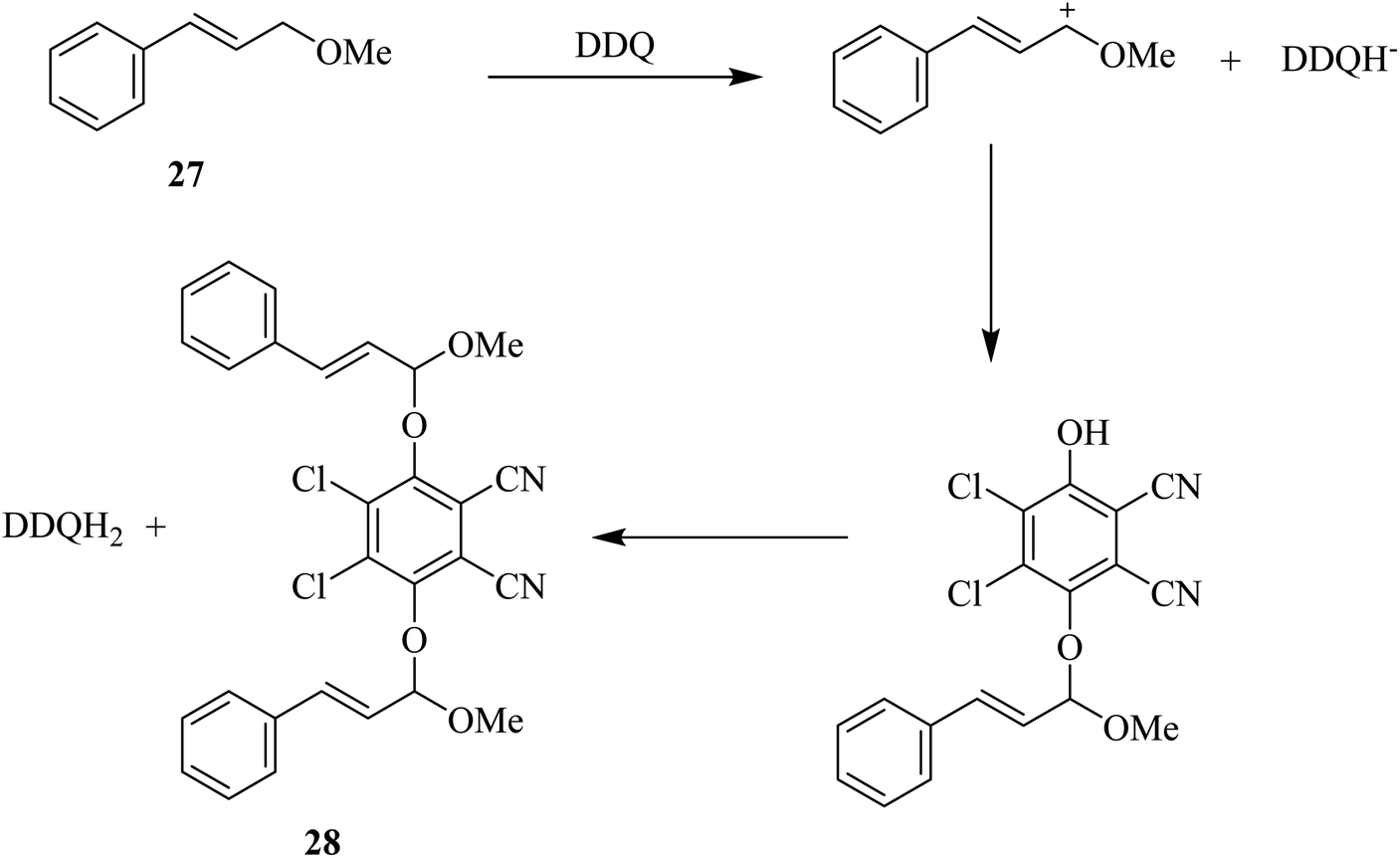 | ||
| Scheme 11 Oxidation of allylic ethers by DDQ. | ||
Later on, Wang and co-workers (1997) proposed the use of DDQ as an efficient oxidant for the oxidation of the bisbenzyl ethers into the corresponding monoaldehyde products as in the conversion of ether 29 to aldehyde 30 (Scheme 12). They also analogized this DDQ mediated oxidation of ethers with other oxidations of benzylic systems such as bisbenzyl alcohols and benzyl bromides. In this case, though, DDQ oxidation was comparatively advantageous and mild, generating monoaldehydes selectively. The authors also discussed the factors that impact reaction course and demonstrated the utility of the method by synthesizing several cyclic aldehydes intermediates.37
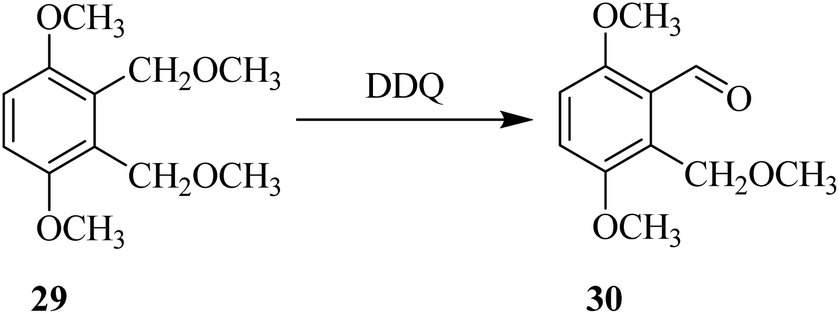 | ||
| Scheme 12 DDQ mediated selective benzylic oxidation. | ||
In 2017, West and co-workers explored the use of DDQ in the Nazarov cyclization reaction. This involves the oxidative cyclization of pentadienyl ethers like compound 31 to cyclopentenone derivative 32 (Scheme 13). Regioselective hydrogen abstraction can cause termination that retains both stereocenters formed during the electrocyclization process, which can yield chiral cyclopentanones like 32 containing an exocyclic double bond. They also demonstrated the use of DDQ as a catalyst with MnO2 as a terminal oxidant.38
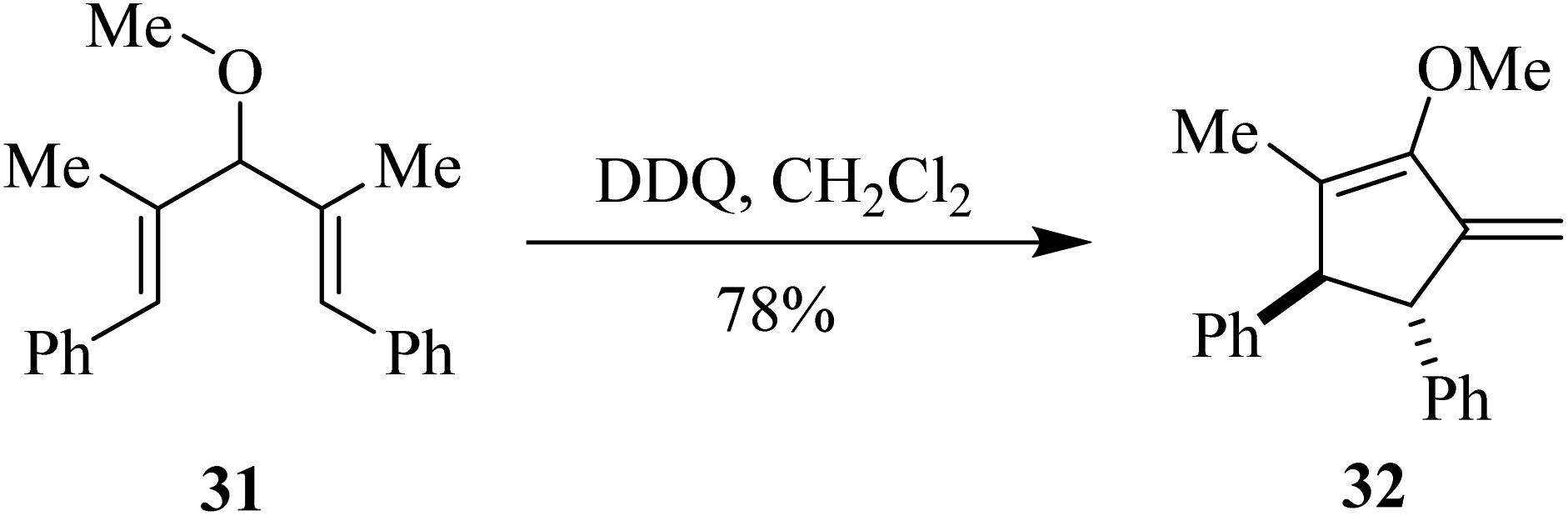 | ||
| Scheme 13 Oxidation-initiated cyclization of pentadienyl ethers by DDQ. | ||
Thereafter, Min and co-workers (2018) proposed a brief synthesis of 8-azabicyclo[3.2.1]-octanes via inter-and intramolecular Mannich-type reactions of N-aryl pyrrolidines with silyl enol ether. The main step in their strategy was an oxidative-Mannich coupling reaction between pyrrolidine derivatives like 33 and silyl enol ether to afford aminoketones like 34 (Scheme 14). Eventually, 34 would undergo intramolecular cyclization to produce the final target bicycle. The key oxidant utilized for these reactions was DDQ.39
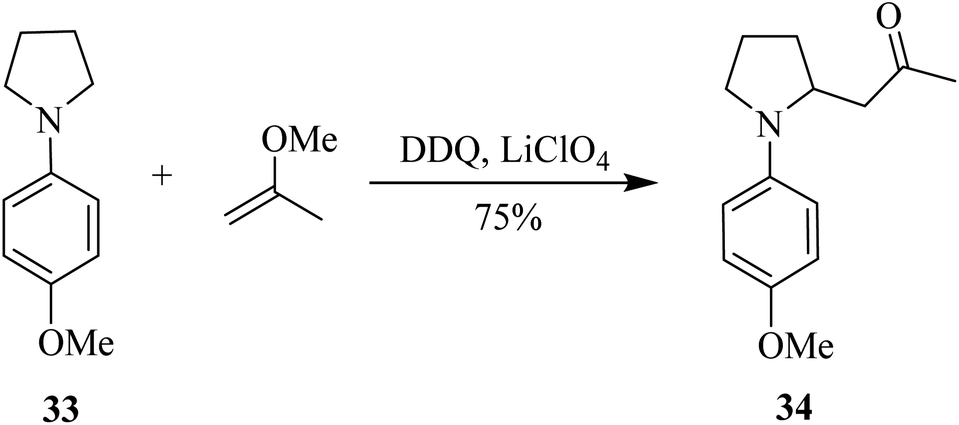 | ||
| Scheme 14 Construction of 8-azacyclooctanes by DDQ mediated reactions. | ||
The ether moiety is well known of its inertness, justifying the use of tetrahydrofuran and diethyl ether, among others, as solvents in organic synthesis. Thus, the selective oxidation of ethers is challenging in synthetic chemistry, often resulting in low yield and selectivity. Although many oxidants have been used in the past to promote ether oxidation reactions, most have failed to afford selective oxidation. Even though some reagents have succeeded in oxidizing ethers selectively, yields were often very low. Only DDQ proved as a suitable oxidant and succeeded in achieving both, selectivity, and high yield without any competing degradative oxidation due to its mild oxidation conditions. With DDQ, silyl enol ethers furnished enones, allylic and benzylic ethers gave substituted products, and bisbenzyl ethers produced monoaldehyde products, proving advantageous in generating monoaldehydes selectively compared to oxidations of benzylic systems such as bisbenzyl alcohols and benzyl bromides. Moreover, DDQ promoted the Nazarov cyclization reaction through the oxidative cyclization of pentadienyl ethers. Also, the synthesis of 8-azabicyclo[3.2.1]-octanes via inter-and intramolecular Mannich-type reactions of N-aryl pyrrolidines with silyl enol ether was efficiently carried out with DDQ. In addition, it is also used as deprotecting agent for the benzyl ethers function, a very difficult group to cleave.
3.3. C–C bond formation
In 2011, Todd and co-workers investigated the mechanism of DDQ controlled oxidative C–C bond-forming reactions. They employed DDQ in nitromethane (MeNO2) as solvent and reagent to perform cross-dehydrogenative couplings. This reaction generated β-nitroamine 36 from N-phenylisoquinoline 35 under oxidative conditions (Scheme 15). It is supposed that a reactive iminium ion might be generated as an intermediate by DDQ oxidation to couple the benzylic methylene group adjacent to the heteroatom. Here though, DDQ did not just play the role of an oxidant, rather a concerted type of nucleophilic reaction was thought to occur, and a hydride was transferred to DDQ to synthesize the nitroamines.40 The group was able to trap and characterized the putative iminium ion intermediate resulting from the oxidation of DDQ and react it further with other nucleophiles.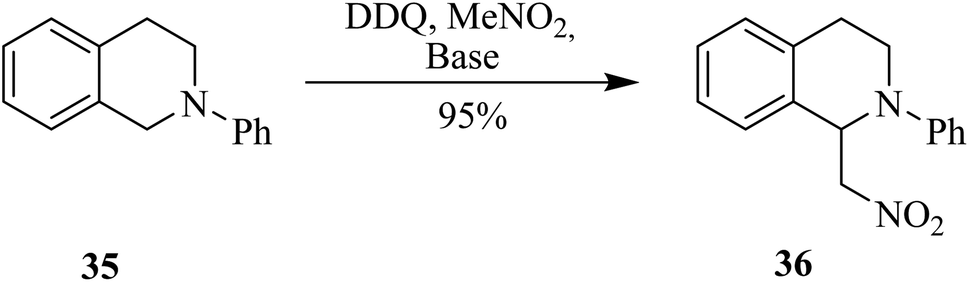 | ||
| Scheme 15 A carbon–carbon bond formation by DDQ. | ||
Likewise, Dehaen and co-workers (2013) developed an approach to synthesize multiple functionalized naphthodithiophene 38 structures by adding substitutions to dithienylbenzene 37 via DDQ/BF3-initiated oxidative cyclization reaction (Scheme 16). During this process, a C–C bond was established between two arene units. This newly reported DDQ-Lewis acid prompted method proved more efficient and high yielding in comparison to the already known FeCl3 catalyzed processes for performing such oxidative cyclization reactions (i.e., Scholl reaction). It is feasible to block other side-polymerization reactions by protecting the reactant 37. The extremely reactive α-positions of thiophenes can permit the addition of further functionalities.41
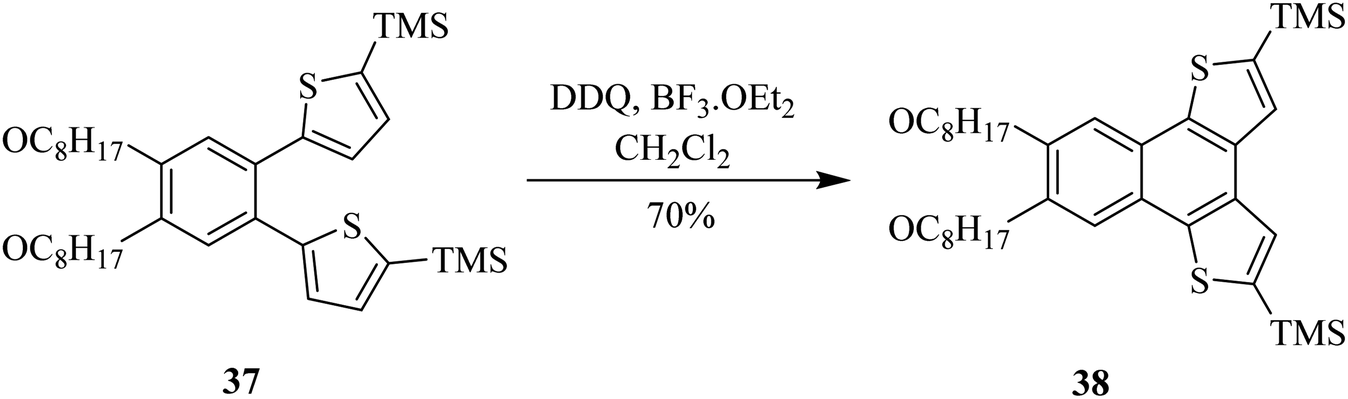 | ||
| Scheme 16 DDQ-mediated oxidative cyclization via Scholl reaction. | ||
In continuation, Tobe and co-workers (2017) proposed a procedure, under Scholl's reaction conditions, in which a spiral-polycyclic aromatic hydrocarbon 39a was treated with DDQ along with Sc(OTf)3 in chlorobenzene at a different range of temperatures. Surprisingly, instead of cyclodehydrogenation, an unexpectedly skeletal rearrangement was observed. The eight-membered ring in compound 39a was converted into the seven-membered ring structure 39b which was further converted into a six-membered ring analog 40 on further treatment with DDQ. They reported different yields at different temperatures as depicted in Scheme 17.42
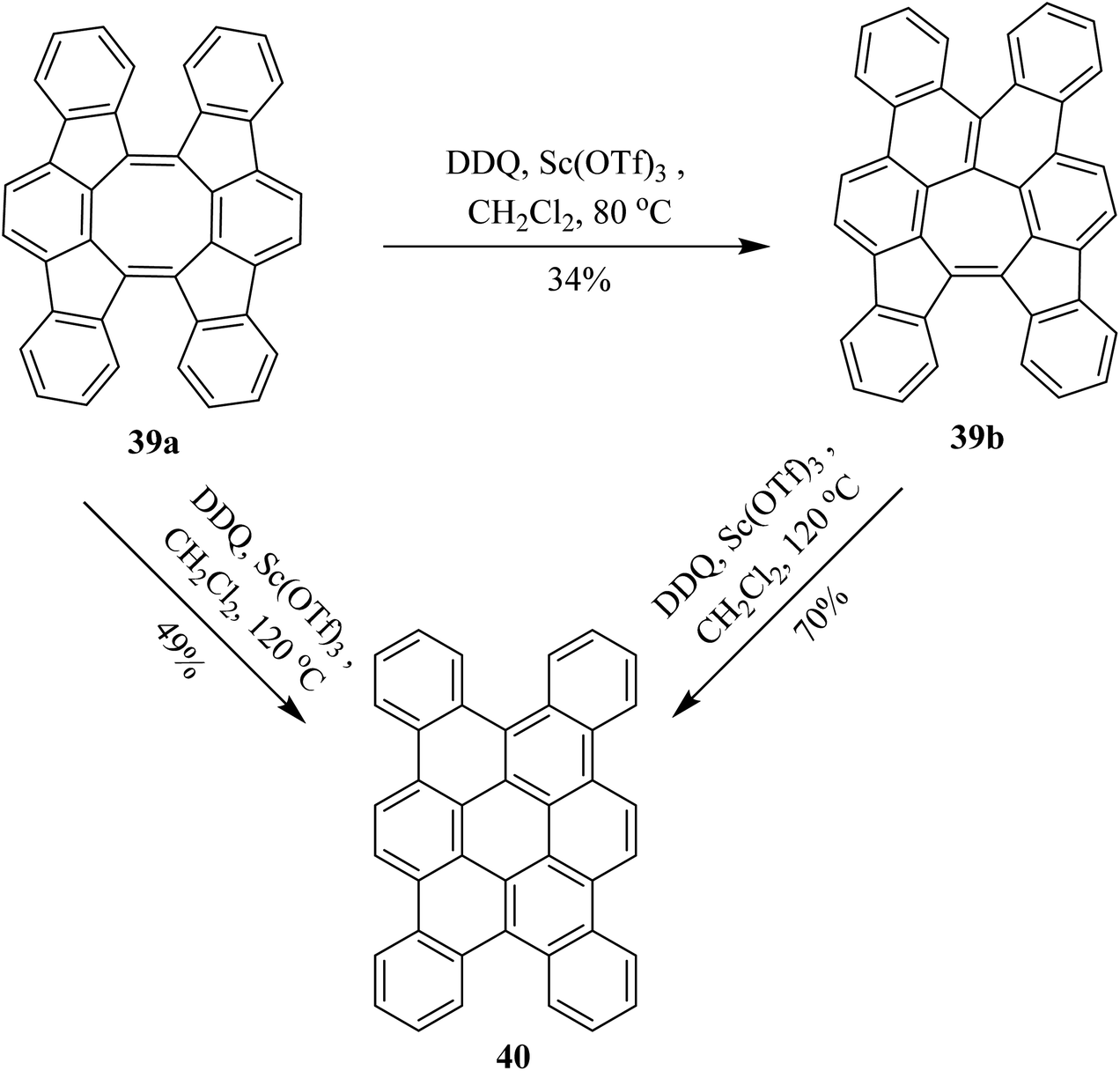 | ||
| Scheme 17 Skeletal rearrangement under Scholl's reaction conditions by DDQ. | ||
In 2018, Toyota and co-workers reported a C–C bond-forming procedure in which 9,10-diarylanthracene analog 41 was treated with DDQ in the presence of trifluoromethanesulfonic acid to furnish rubicene derivative 42 containing two five-membered rings in moderate to excellent yields (Scheme 18). This methodology could be beneficial for the preparation of a variety of functionalized rubicene derivatives, which may find applications as building blocks for the construction of curved or planner polycyclic aromatic hydrocarbons (PAH) as functional organic materials.43
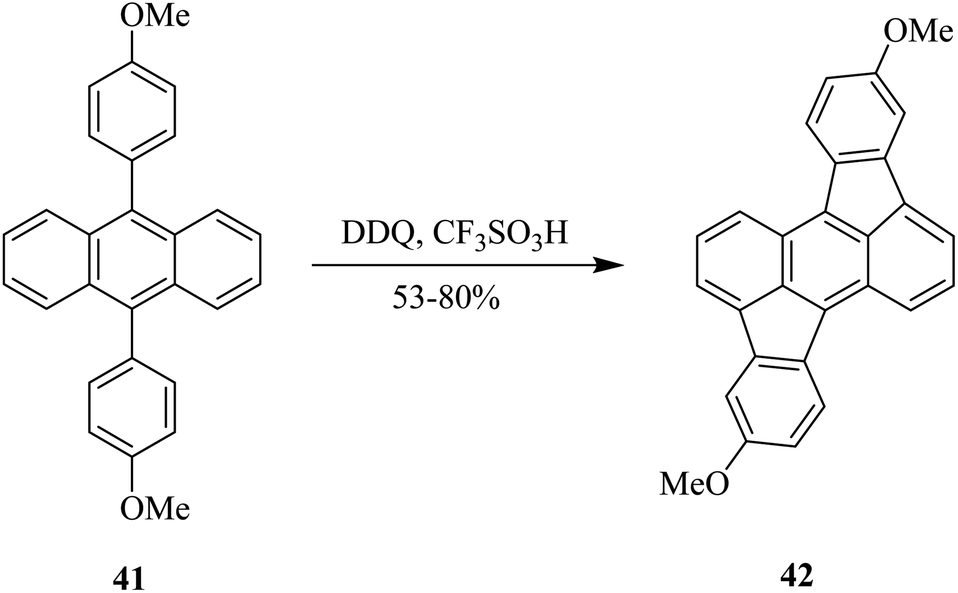 | ||
| Scheme 18 Synthesis of rubicene derivatives under Scholl's reaction conditions. | ||
In the current year (2021), Jana and co-workers used DDQ as a key mild oxidant in the chemical transformation of indole derivative 43 into indole-fluorene hybrid 44 in high yield. This approach involves electrophilic attack onto nearby arene position resulting in the formation of C–C bond. Noteworthy, dramatic improvement in product yield was observed in the presence of additives such as FeCl3 or molecular sieves (4 Å). This pathway offers high functional group tolerance as shown in Scheme 19.44
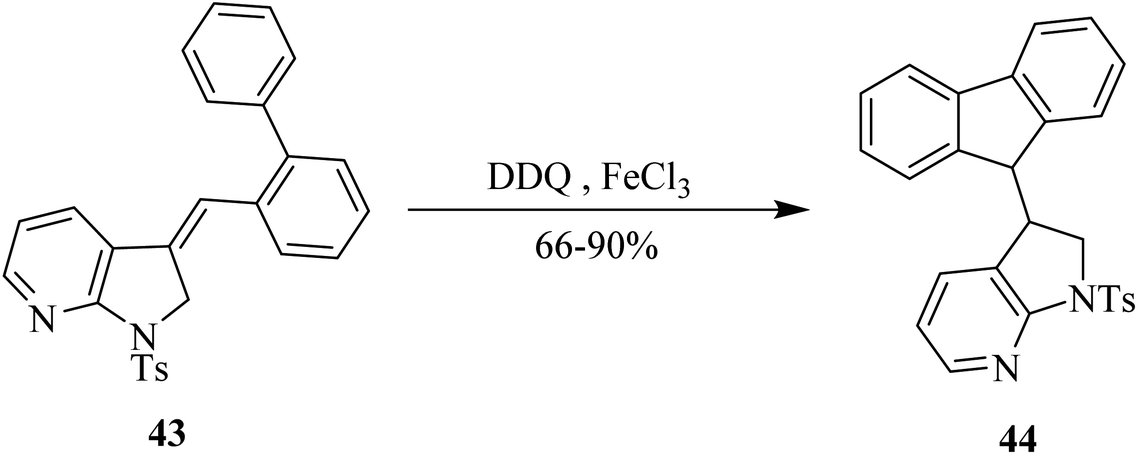 | ||
| Scheme 19 DDQ/FeCl3-controlled sequential oxidative C–C bond formation. | ||
During oxidative C–C bond-forming reactions, the control of cyclization and polymer formation is quite tricky. DDQ has provided solution to these challenging problems by controlling the cyclization during C–C bond formation and suppressing the process of polymerization. DDQ, owing to the mild oxidation conditions it offers, selectively constructs the C–C bonds to avoid polymerization. Furthermore, it is suitable oxidant to obtain the cyclization at the desired position, providing regioselective control. Thus, it provides facile synthetic route to construct planar and curved PAH systems.
3.4. Protection and deprotection by DDQ
DDQ also plays a vital role in promoting organic chemical conversions aimed toward the protection and deprotection of some functional groups. For instance, encouraged by previous reports, Oikawa and co-workers (1982) disclosed a facile and efficient method to remove the methoxybenzyl (PMB) protecting group of alcohols like 45 with DDQ in a dichloromethane-water mixture at 25 °C. They described that this type of deprotection via DDQ was selective, as all other typical protecting groups except methoxybenzyl remained unchanged due to this deprotection (Scheme 20).45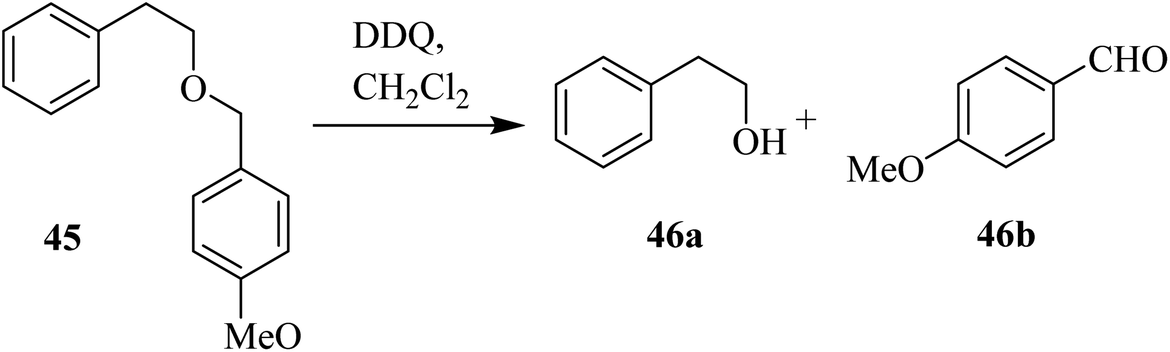 | ||
| Scheme 20 Removal of p-methoxybenzyl protection by DDQ. | ||
Using the same protocol, they also described the use of DDQ oxidant in the deprotection of the primary hydroxyl group in compound 47 under anhydrous conditions to generate an acid-sensitive methoxybenzylidene acetal 48 by intramolecular oxidative reaction (Scheme 21).46
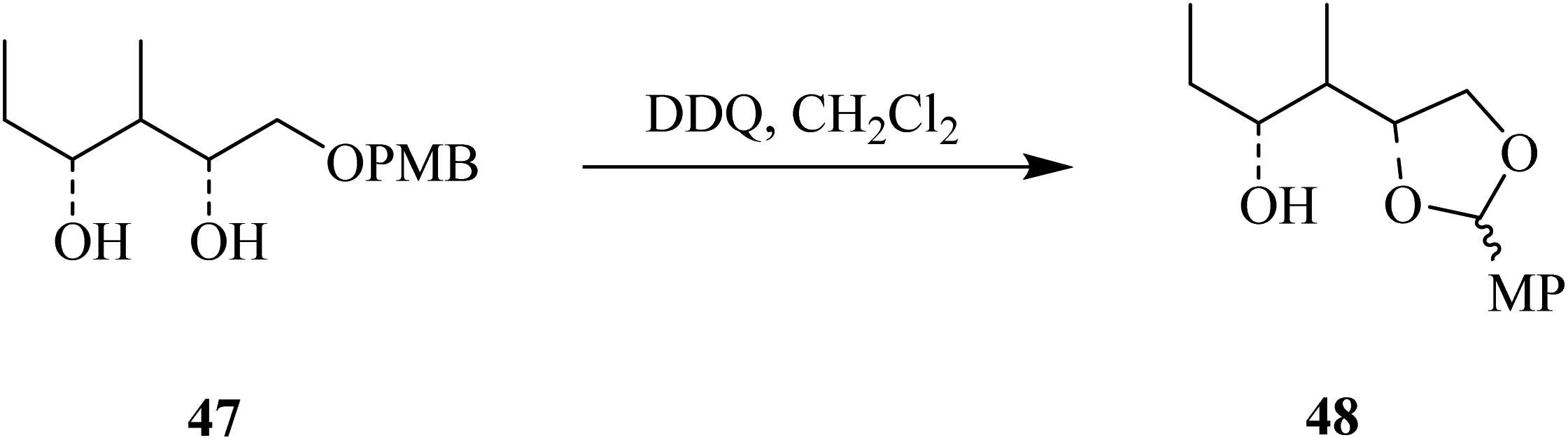 | ||
| Scheme 21 Deprotection of PMB-protected primary hydroxyl group by DDQ. | ||
Later on, the same research group (1984) proposed another DDQ-promoted deprotection method (Scheme 22). In this protocol, they demonstrated that the 3,4-dimethoxybenzyl protecting group 49 for the hydroxy functionality may easily be cleaved by DDQ oxidation under an inert atmosphere. This methodology was then applied to the preparation of macrolide and polyether antibiotics.47
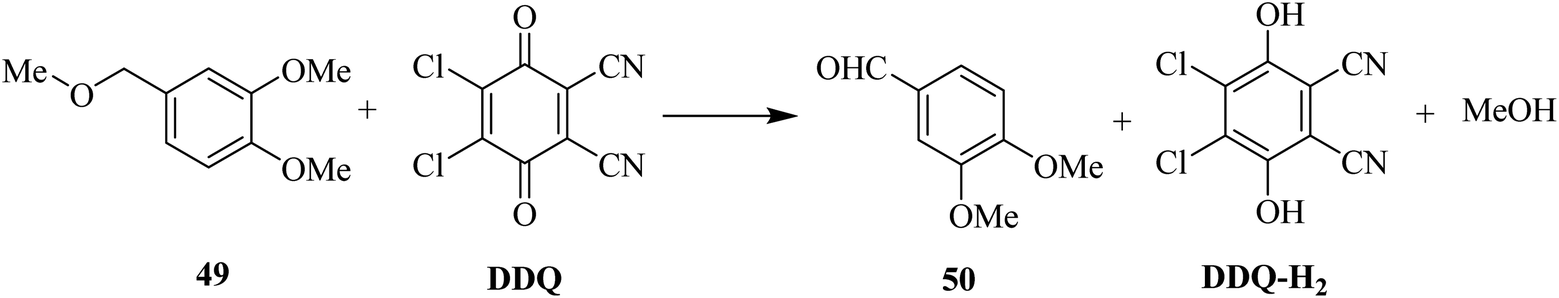 | ||
| Scheme 22 Cleavage of DMPM protecting group for hydroxy functionality by DDQ oxidation. | ||
In 1986, Yonemitsu and co-workers developed a method to remove the 4-methoxybenzyl (PMB) protection 51 with DDQ in wet dichloromethane (DCM) for alcohols at 25 °C (Scheme 23). These conditions tolerate a wide range of functional groups. Along with DDQ, 3,4-dimethoxybenzyl (DMPM) protection is a better reactive group as compared to PMB protection. They also presented deprotection of PMB, Bn, and DMPM groups selectively.48
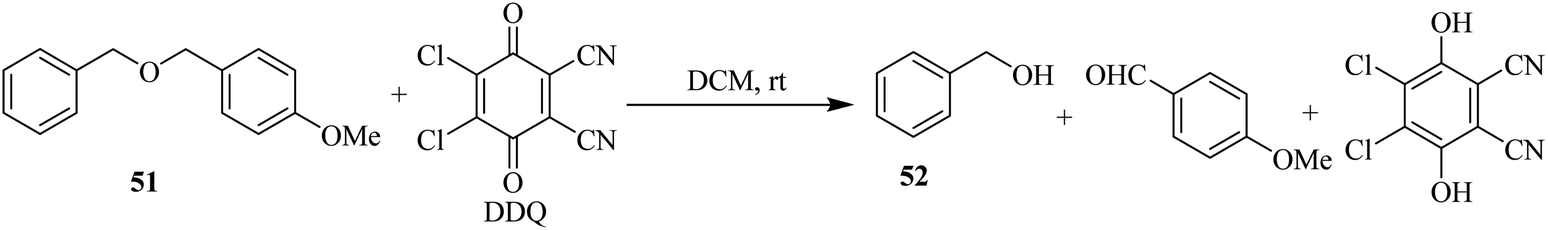 | ||
| Scheme 23 Deprotection of PMB for hydroxy functionality by DDQ. | ||
In 1994, McDonald and co-workers described the use of DDQ in deprotecting acetal 53 in high yield. In spite of offering mild oxidative conditions, this reagent exhibited low chemoselectivity in selectively removing the acetal function in the presence of other protecting groups such as benzyl (Bn) (Scheme 24).49
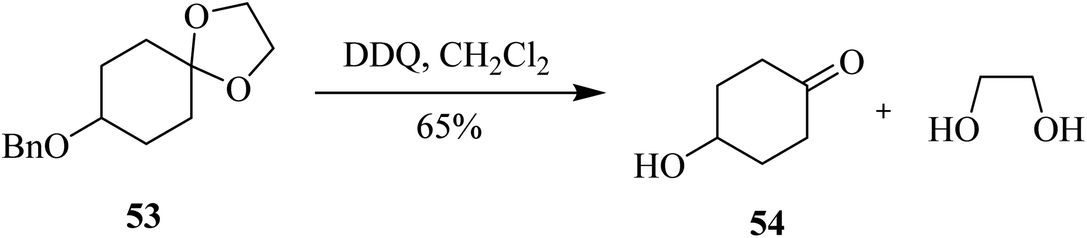 | ||
| Scheme 24 DDQ-mediated oxidative removal of acetal. | ||
Furthermore, Sampson and Honek (1999) devised a process to remove the protecting group diphenylmethyl amine 55 efficiently by converting the amine to an imine 56a via oxidation by DDQ (Scheme 25). The resulting imine can be simply hydrolyzed under mildly acidic conditions to give the salt 56b. This method is predominantly applied for the synthesis of α/β-amino phosphonates.50
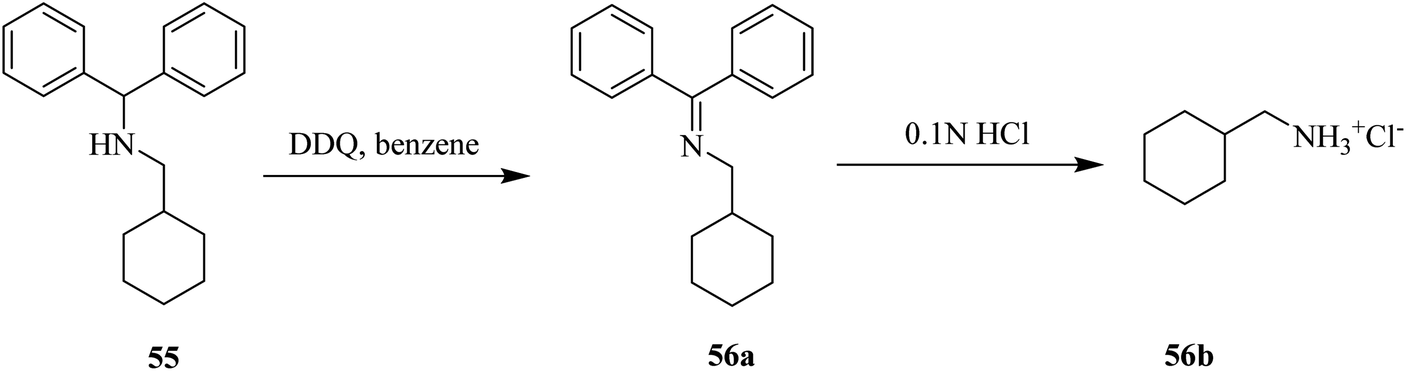 | ||
| Scheme 25 Oxidative deprotection of diphenyl methylamines by DDQ. | ||
Progressively, Sharma (2001) introduced a new process for the deprotection of the protecting group para-phenylbenzyl (PPB) as novel ‘PMB’ like protecting group 57 by DDQ (Scheme 26). Meanwhile, considering the cost associated with using DDQ in stoichiometric amounts and the formation of undesired quinol/hydroquinone by-products during this process, they treated the substrates with a mixture of DDQ and Mn(OAc)3 as coupled oxidative system to avoid these problems.51
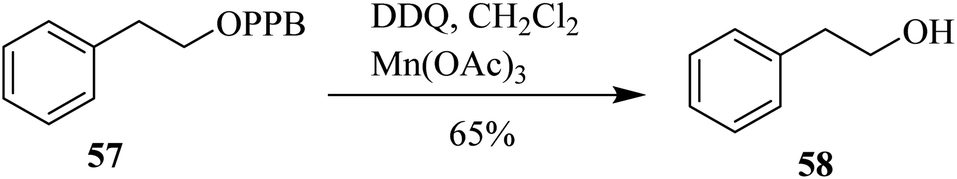 | ||
| Scheme 26 Deprotection of benzyl phenyl by DDQ. | ||
Afterward, Pan and co-workers (2002) synthesized 2-arylbenzoxazoles 60a, b starting from the condensation of o-aminophenols 59a, b with aromatic aldehydes followed by DDQ-initiated oxidative cyclization (Scheme 27). This one-pot strategy was highly significant for making separate aryl benzoxazole compounds in moderate to excellent yields. The results obtained from those experiments confirmed that DDQ is an effective and essential oxidative agent for the preparation of benzoxazole-containing biaryl structures.52
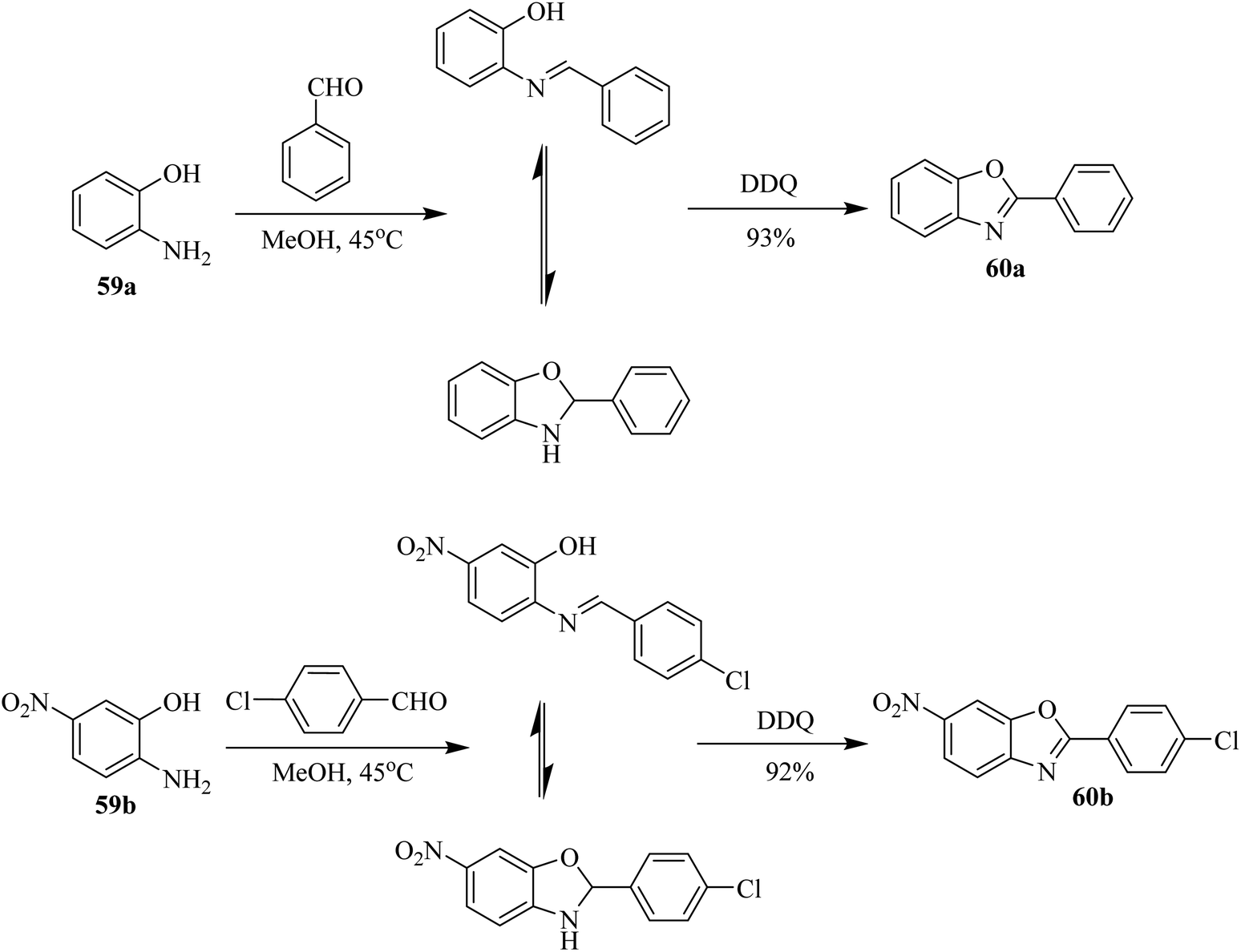 | ||
| Scheme 27 DDQ-promoted synthesis of 2-arylbenzoxazoles. | ||
In 2005, Toshima and co-workers designed a procedure for benzyl ethers deprotection 61 by employing DDQ under continuous irradiation of low energy radiations (Scheme 28). During their work, they found that these conditions are crucial for some compounds. Though the exact mechanism was unclear at that stage as well as the procedure also had some associated drawbacks, this new photo-induced deprotection procedure of benzyl ethers was a useful substitute for other applications in organic synthesis.53
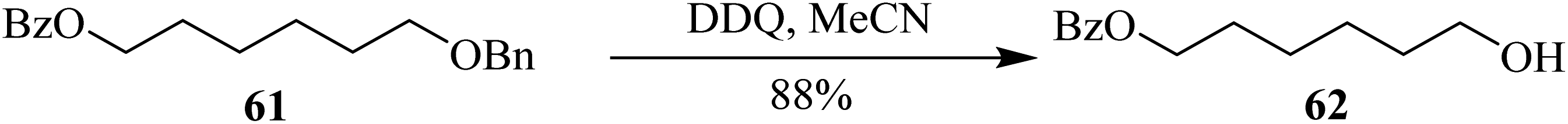 | ||
| Scheme 28 Deprotection of benzyl ethers using DDQ. | ||
Marra and co-workers (2010) described the oxidation of some hydroxyisochromans 63, 65, and 67 by using DDQ (Scheme 29). 1H NMR spectroscopy was used to check the reaction progress and several side-products were separated and further characterized. They observed the formation of some isochroman-6,7-diols during the reaction of dihydroxyphenyl ethanol with various substituted aromatic aldehydes. In this Scheme 29, the hemiacetal compounds 63, 65 and 67 are deprotected by DDQ and transformed into their corresponding/respective keto–enol form 64, 66, and 68. The signal of the respective ortho benzoquinone was observed in all cases after the addition of DDQ. In this process, the orthoquinone moiety was converted into the main oxidation product, a hydroxybenzophenone analog originating from C1-dibenzylic carbon oxidation to a carbonyl group such as in structures 64, 66, and 68.54
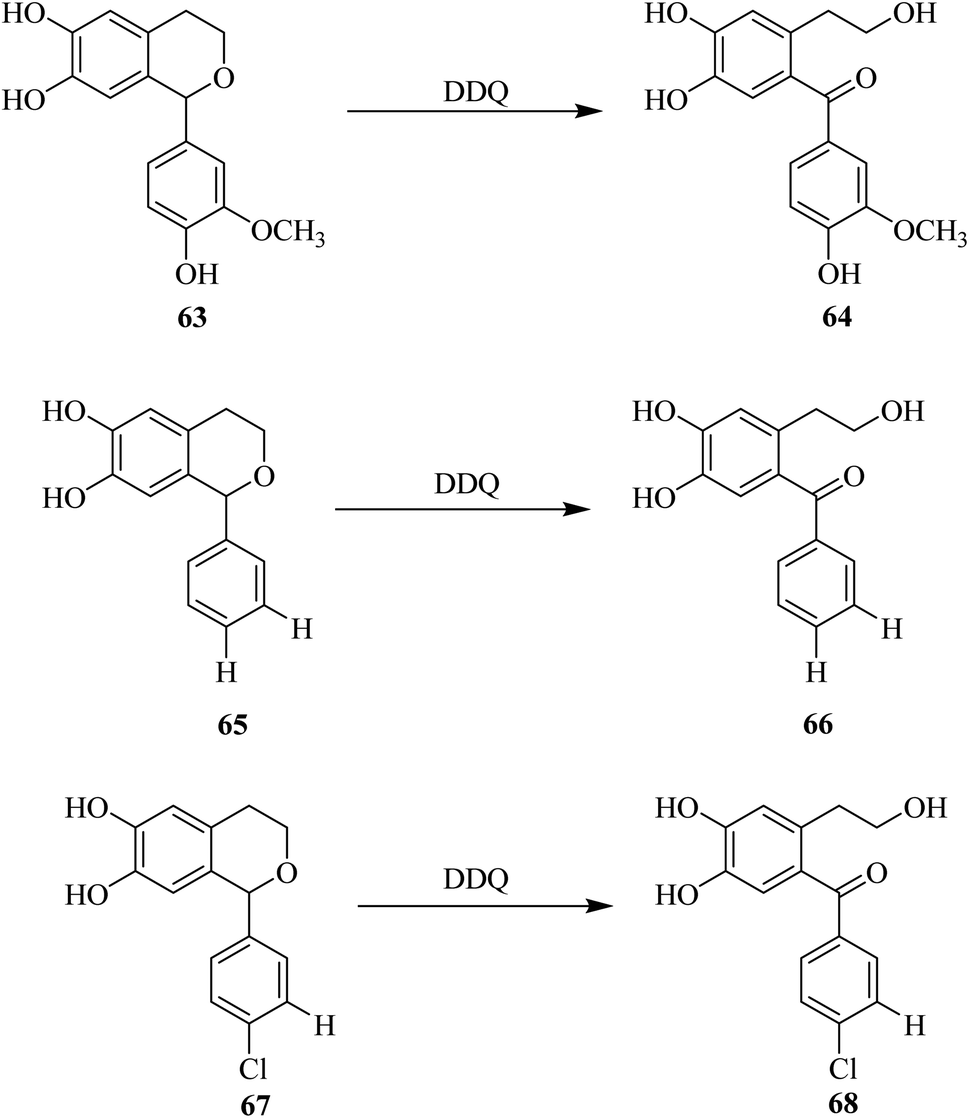 | ||
| Scheme 29 Deprotection of hydroxyisochromans by DDQ. | ||
Not long ago, Hu and co-workers (2013) disclosed a facile and promising protocol for the deprotection of benzyl ether 69 by DDQ-catalysed oxidation (Scheme 30). The reaction was carried out in an open atmosphere along with tert-butyl nitrite (TBN). The diverse groups of p-phenylbenzyl (PPB), benzyl (Bn), and p-methoxybenzyl ethers (PMB) were successfully deprotected under optimal conditions to their respective alcohols in high yields with high selectivity.55
 | ||
| Scheme 30 Deprotection of benzyl-type ethers by DDQ/tert-butyl nitrite mixture. | ||
Then Kumar and co-workers (2014) developed a chemoselective and efficient procedure for the deprotection of N-allylic amine 71 via DDQ assisted oxidation (Scheme 31). The utilization of DDQ offers a smooth and effective single-step removal of the allyl group with a wide range of tertiary amine derivatives that are protected orthogonally.56
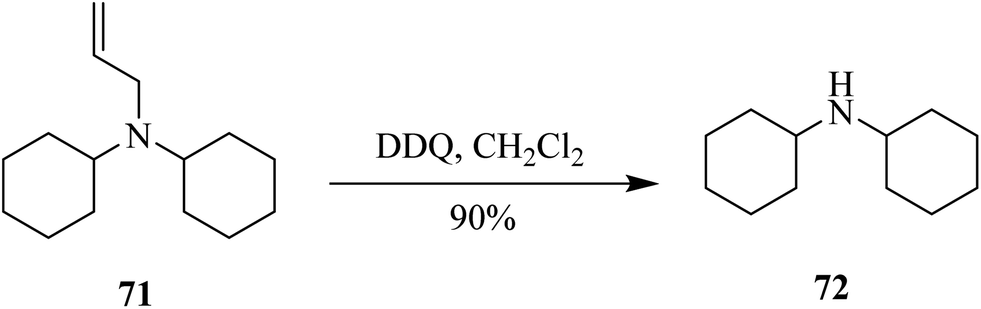 | ||
| Scheme 31 N-Allylic amine deprotection using DDQ. | ||
During the same year, Moody and co-workers demonstrated a process for p-methoxybenzyl ether 73 deprotection with high selectivity by employing a catalytic amount of DDQ along with sodium nitrite (NaNO2) under atmospheric oxygen that was taken as terminal oxidant (Scheme 32). Excellent yields were obtained for structurally diverse protected alcohols. These reaction conditions were reported as highly chemoselective to deprotect PMB type of protecting groups in the presence of other alcohol protecting groups like acetate, ester, benzyl ether, EOM, and silyl ethers, among others. This extensively used oxidant methodology is being utilized in the synthesis of numerous natural products.57
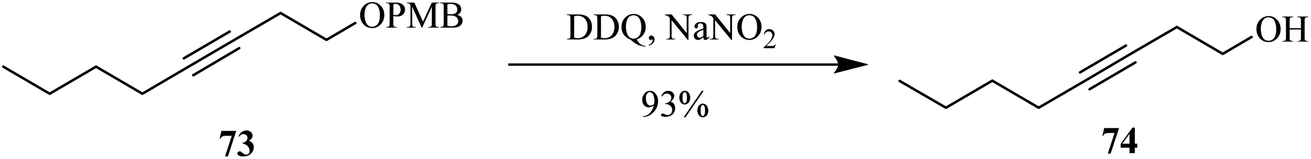 | ||
| Scheme 32 p-Methoxybenzyl ether deprotection by DDQ. | ||
Recently, Penta and co-workers (2017) devised a facile and effective protocol for the regioselective protection of primary alcohol 75 mostly present in nucleosides with a combination of DDQ and 4-methoxy-benzyl-2,7-dimethylpixylether (MBDPE) in high yield (Scheme 33). Interestingly, the same oxidative system can be used to quickly deprotect the same group in 76 in methanol. Each protocol was effectively applied to the synthesis of altered nucleosides at a gram-scale level. This procedure offered neutral conditions for the protection and deprotection of the dimethylpixyl group (DMPx) with high selectivity and their further deprotection in the presence of other typical protecting groups such as benzyl, benzoyl, and acetonide, among others.58
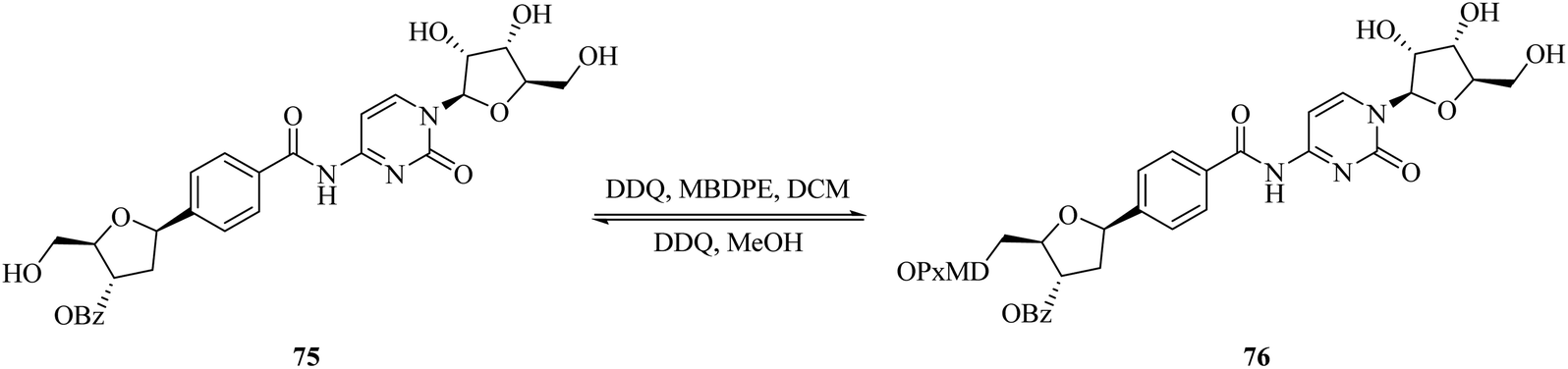 | ||
| Scheme 33 DDQ controlled regiospecific protection and deprotection of primary alcohol. | ||
The regio- and chemo-selective protection and deprotection of functional groups is one of the most essential tasks in modern organic synthesis and has been key to the successful execution of total synthesis of many small and large molecules. Since robust acid-resistant protecting groups are often required to withstand the harsh conditions of multi-step synthesis, the subsequent removal of such groups presents extreme challenges. DDQ has proven as a mild, and selective acid-resistant oxidant and is the reagent of choice which can be used in stoichiometric or catalytic amounts to deprotect alcohols, diols, allylic amines, and cleave hydroxyisochromans. The reagent cleaves the PPB, Bn, and PMB, which are otherwise very difficult to remove, very easily.
Being able to use DDQ in catalytic amount along other terminal oxidants like atmospheric oxygen or continuous irradiation is notable, reducing cost and avoiding the formation of undesired quinol/hydroquinone by-products. Although there are less applications of DDQ in protection reactions, the regioselective protection of primary alcohol groups in nucleosides with a combination of DDQ and 4-methoxy-benzyl-2,7-dimethylpixylether (MBDPE) is an important reaction. Further, as a benzylic oxidant, DDQ can selectively oxidize secondary benzylic alcohols, leaving primary alcohols intact for further manipulation. Also, it is used for electron-donating groups selectively and constructs charge transfer complexes with them to provide high yield products.
3.5. Oxidation of phenolic compounds
In 1965, Becker used DDQ against phenols as an effective oxidizing agent, and thus applied this reagent to oxidatively introduce substitution onto the phenolic ring (Scheme 34). This reaction was performed in methanolic solution at ambient temperature to give the substituted products in high yields. This reaction proceeds through oxidative dimerization by either C–O or C–C coupling. DDQ reacted promptly with the substrate 77a in the presence of CH3OH at ordinary temperature to produce colored intermediates with different stabilities. The resulting intermediate 77b was added to 4-hydroxydiphenyl thioether 77c to form the substitution product 78 in excellent yield. In the end, DDQ was obtained in reduced form (DQQ–H2).59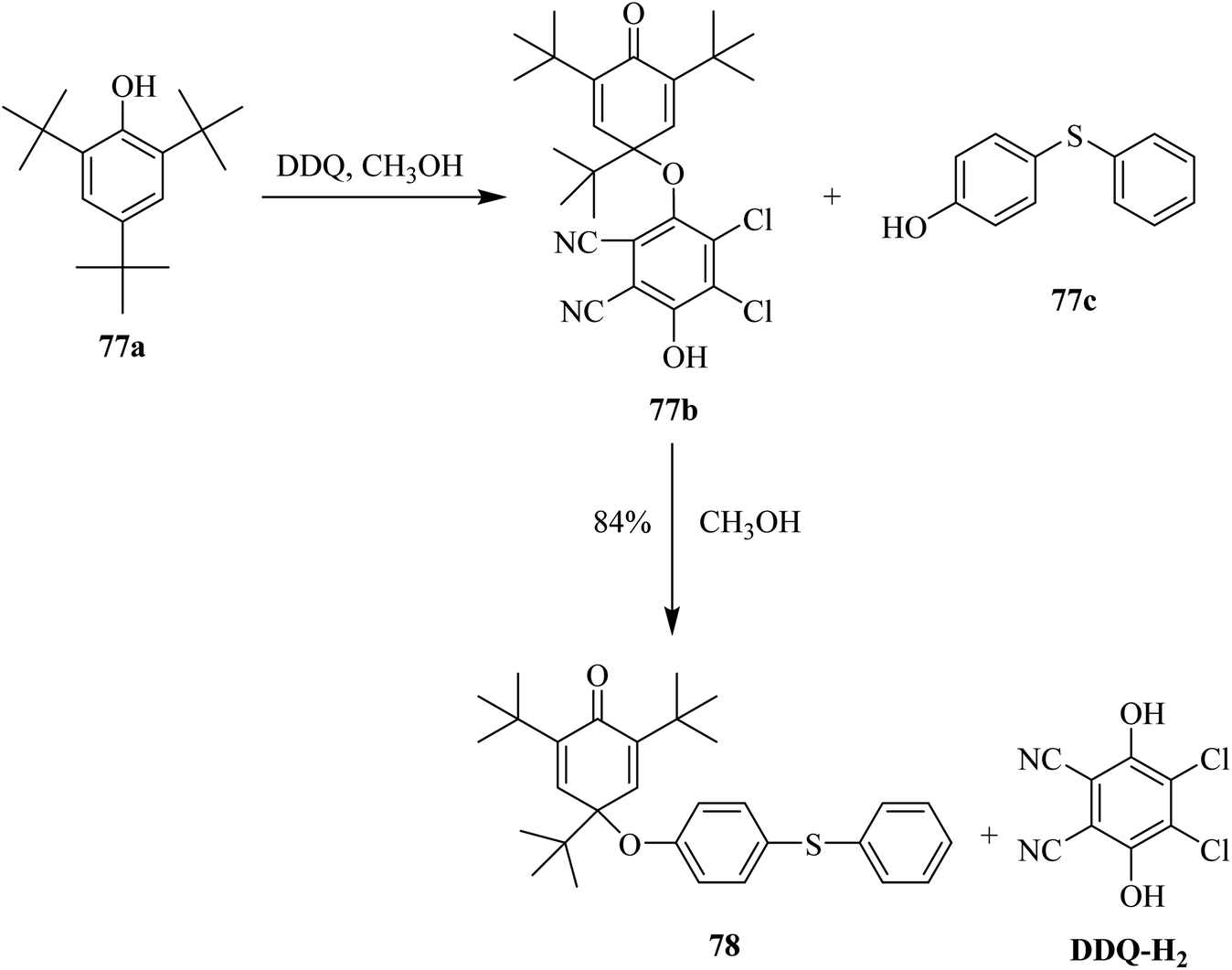 | ||
| Scheme 34 Oxidation of sterically hindered phenols by DDQ. | ||
Later on (1969), researchers explored the use of DDQ for the dehydrogenation of phenolic compounds. For example, the derivatives of p-cresol were treated with DDQ in the presence of methanol to yield the corresponding quinones. However, it was found that quinone methides type of unstable intermediates were formed. This problem was resolved by introducing bulky-alkyl groups onto the phenolic moiety (Scheme 35). As such, this strategy produced the desired quinone 80 from the phenol 79 via DDQ-assisted dehydrogenation in higher yield.60
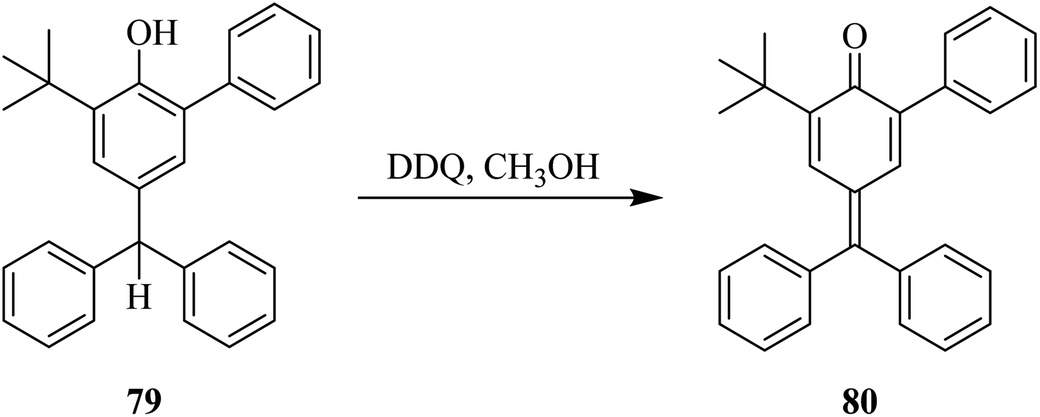 | ||
| Scheme 35 Oxidation of 4-hydroxytriphenylethane. | ||
Schofield and co-workers (1971) explored the use of DDQ in stoichiometric amounts (1.0 eq.) to perform oxidative cyclization of the phenol 81 to produce a mixture of 2-(4-hydroxyphenyl)benzo[b]furan 82a and 2,3-dihydro-2-(4-hydroxyphenyl)benzo[b]-furan 82b (Scheme 36). The latter compound was further subjected to dehydrogenation with DDQ (1.0 eq.) in benzene to furnish the target compound 82a in good yield.61
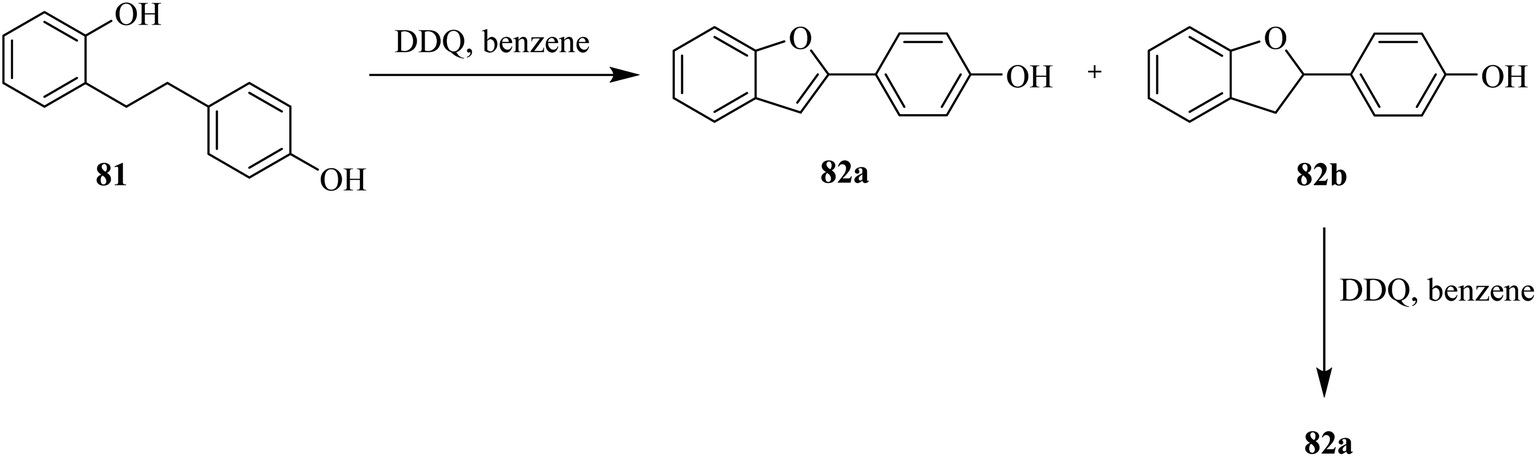 | ||
| Scheme 36 Oxidative cyclization of a substituted phenol by DDQ. | ||
Many typical oxidants have been used for the oxidation of phenols previously. DDQ is a novel oxidant for phenols that is capable of generating phenoxonium ion intermediate. Consequently, it is the only oxidant that can change the course of phenol oxidation. It oxidizes sterically hindered phenols quite easily in short time at room temperature. High yield products are often obtained by this most effective and cheap oxidant. Remarkably, DDQ oxidizes phenols 5500 times faster than chloranil and can be removed easily from reaction mixture via ion exchange resins.
3.6. Oxidation of steroids
In this section, we will shed light on the applications of DDQ in the reactions of steroids. In this regard, Pradhan and Ringold (1964) described the oxidation of a steroid 83 with DDQ to give dehydrogenated product 85 under anhydrous conditions. The sequence of reaction steps includes the abstraction of hydride at C-7, elimination of a proton at C-2 from the intermediate 84, and hydride elimination from C-1. However, in the presence of water, the reaction proceeds quickly through hydrolysis of the intermediate 84 rather than proceeding through a second path whereby it loses a proton to generate 86 in good yield (Scheme 37).62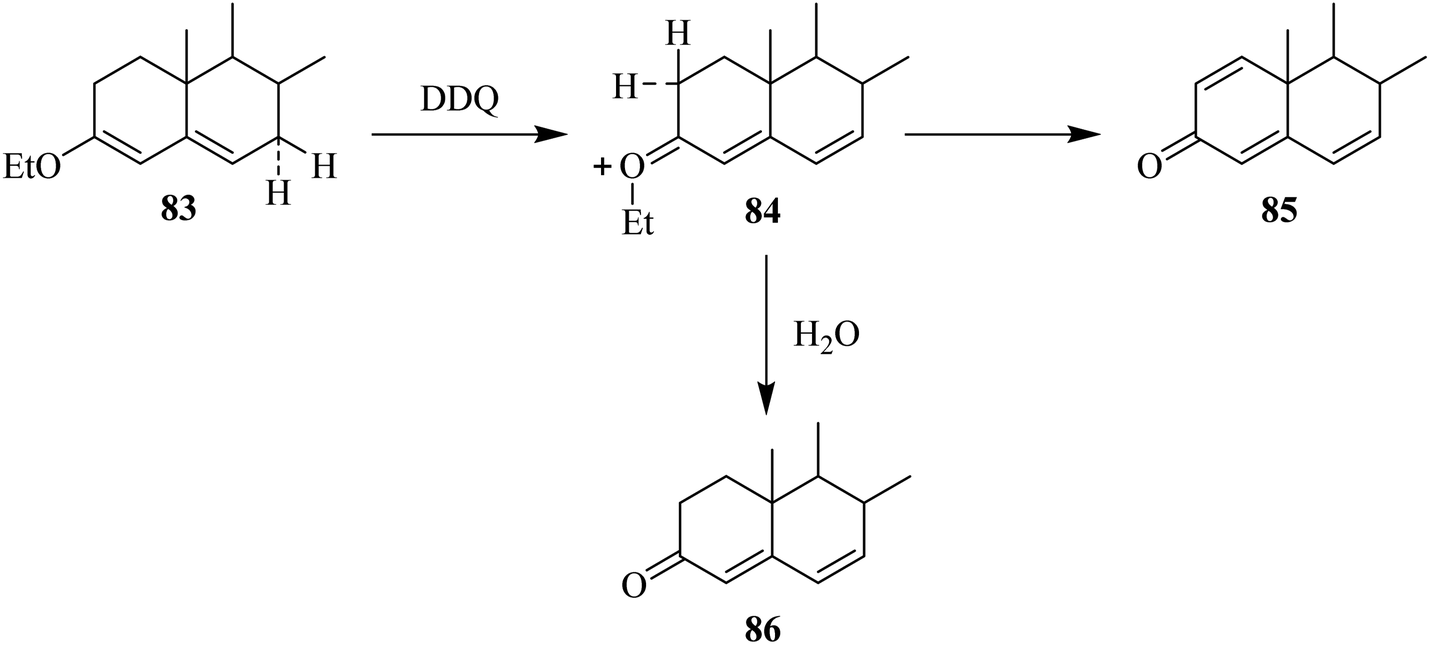 | ||
| Scheme 37 Oxidation of alkyl-enol ethers by DDQ. | ||
In 1994, Böhme and Kempfle proposed a mechanism for the formation of steroids-based fluorescent probes with an aliphatic side chain or without it. Starting from steroidal alcohols 87 and 91, they prepared different intermediate compounds via DDQ-assisted oxidation (Scheme 38).63
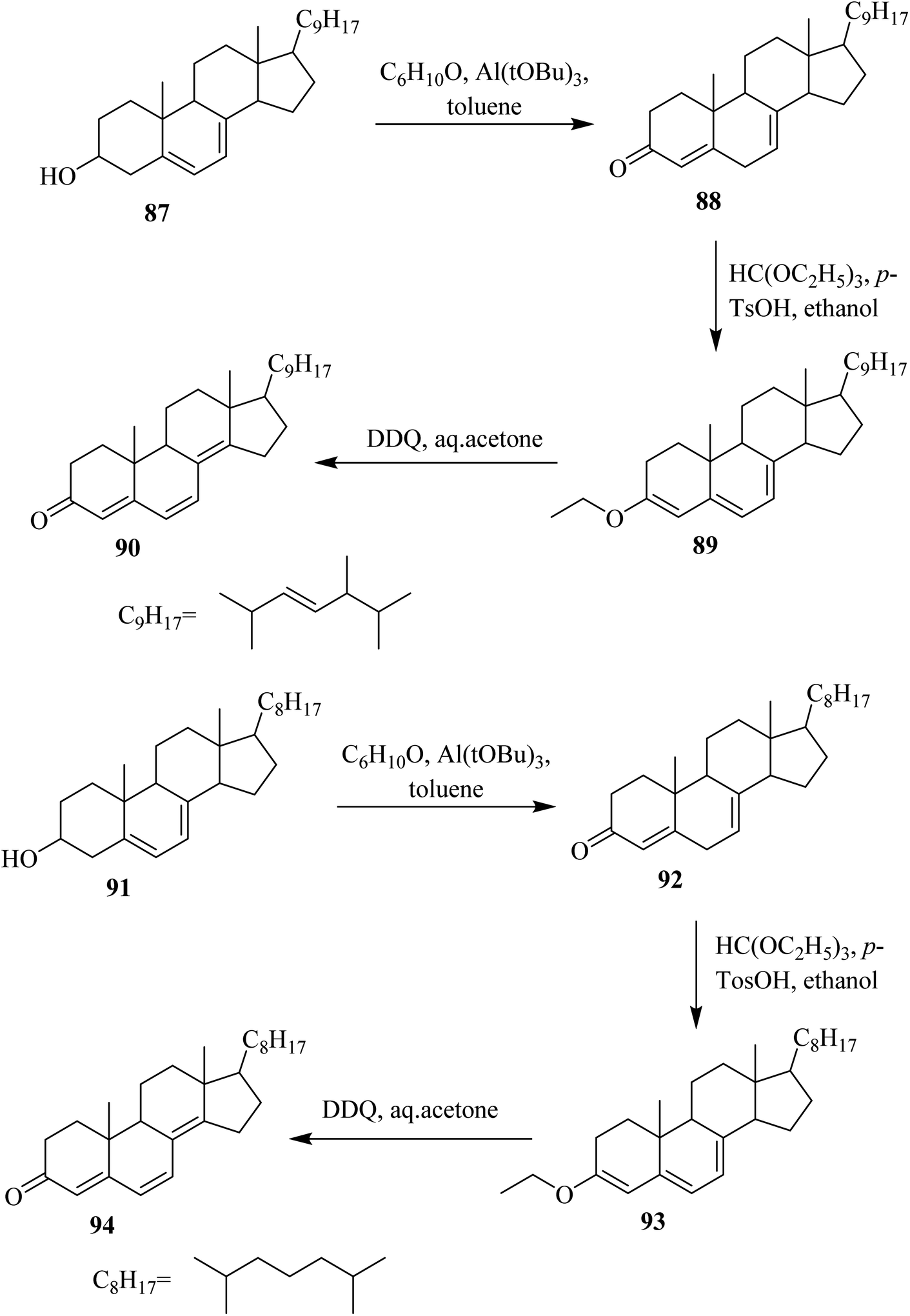 | ||
| Scheme 38 Synthesis of various steroids structures via DDQ-initiated oxidation. | ||
In continuation, Xu and co-workers (2010) explored the synthesis of keto-steroids 96 and 98 through the dehydrogenation of the reactants 95 and 97 by employing DDQ as an oxidant in the presence of tert-butyldimethylchlorosilane (TBDMSCl) at ordinary temperature (Scheme 39). From these results, they concluded that the addition of chlorosilane reagent could enhance the regioselectivity and reactivity in the synthesis of desired compounds under these oxidative conditions.64
 | ||
| Scheme 39 Dehydrogenation of ketosteroids by DDQ. | ||
Later on, Shen and co-workers (2015) performed great work on the oxidation of sensitive hydroxysteroids 99 and 101 to their respective ketosteroids 100 and 102 by utilizing a mixture of DDQ and a catalytic amount of tetramethyl piperidinyl-1-oxyl (TEMPO) reagent as an effective oxidative system (Scheme 40). These oxidative conditions proved a high yielding. This procedure was most effective for the preparation of 4,6-diene-3-one from the corresponding hydroxysteroids.65
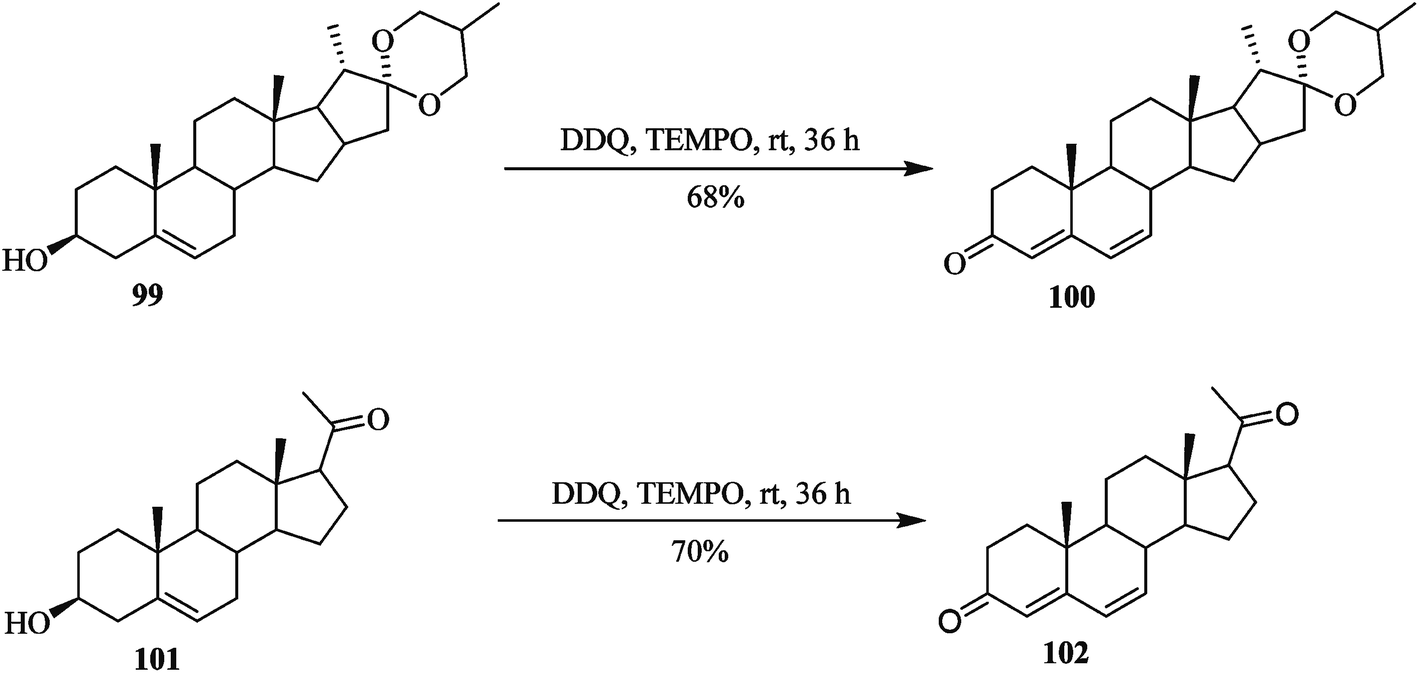 | ||
| Scheme 40 Oxidation of hydroxysteroids by DDQ. | ||
Enzymatic dehydrogenation of ketosteroids was quite popular method but it had two disadvantages associated with it. Firstly, it was not much stable and secondly it was difficult to handle. To replace this tedious and less stable process, DDQ is used as an oxidant for facile and quick oxidation of ketosteroids. Moreover, it controlled the selectivity of the reaction as well. The reaction proceeds rapidly at room temperature under acidic conditions with high chemo- and regioselectivity. Thus, the main advantages here are short reaction time, high yield, selectivity (chemo and regio) and stability with easy-to-handle methodology.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds to extend conjugation in α,β-unsaturated ketones in order to furnish dienones as well as extend aromatic conjugation (Scheme 41).67
C double bonds to extend conjugation in α,β-unsaturated ketones in order to furnish dienones as well as extend aromatic conjugation (Scheme 41).67
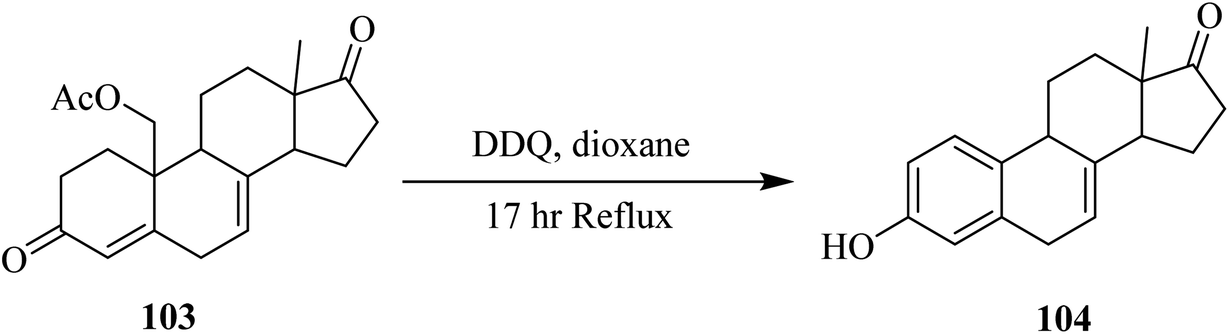 | ||
| Scheme 41 Dehydrogenation of acetoxy ketones. | ||
In 1960, Braude reported various examples to exemplify the process of aromatization. The possibilities of previously mentioned reactions might have occurred in the second step during the process of aromatization. For instance, an earlier and a facile example was the preparation of 1,2,3,5-tetramethylbenzene 106 from 1,5,5-trimethyl-3-methylenecyclohexene 105 by the use of DDQ (Scheme 42).68
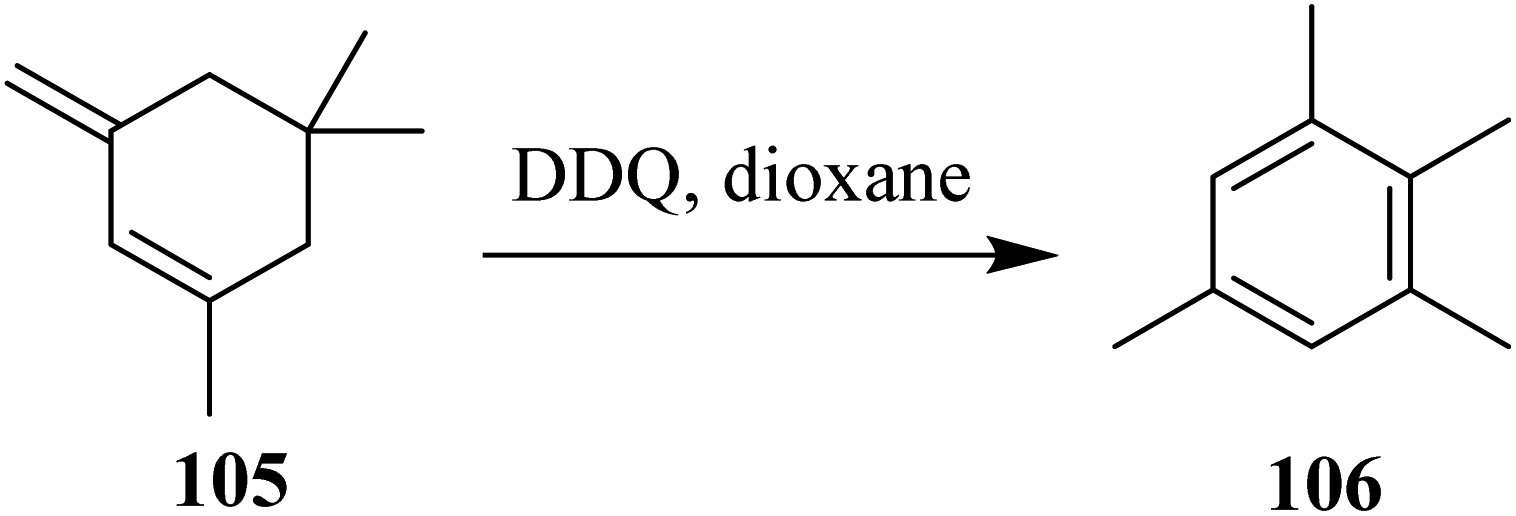 | ||
| Scheme 42 Aromatization induced by DDQ. | ||
In the following years (1963 and 1964), Gaudry and co-workers presented a facile route for the dehydrogenation of 19-acetoxyketone 107 by using DDQ to form the product equilin 108.69 The estrogen dehydrogenation process was highly dependent on the nature of the carbon-17 substituent and the DDQ reagent. Therefore, estrone methyl ether 109 dehydrogenation was performed with chloranil in the presence of dioxane and t-butanol to produce the product 110 and 111 which emerged from the cleavage of steroidal ring D (Fig. 1).70
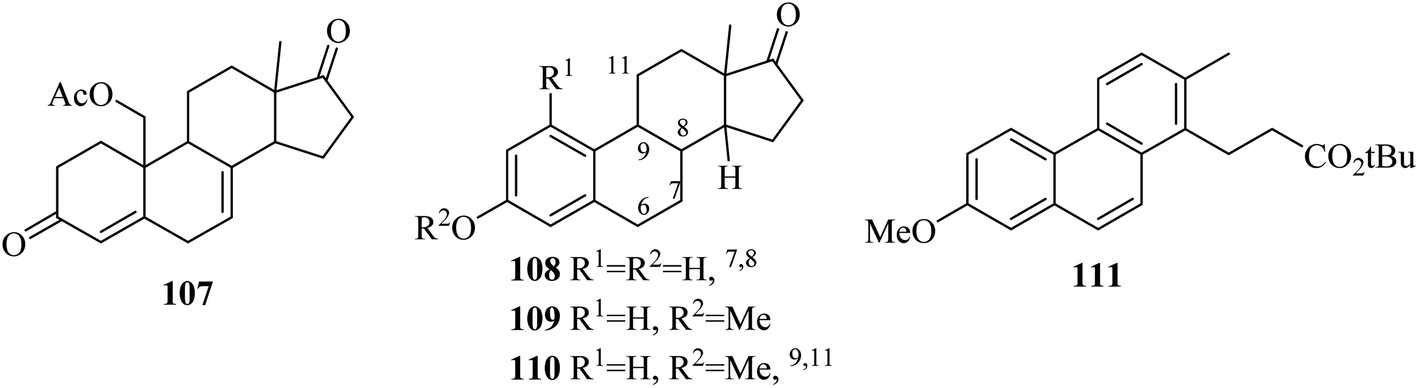 | ||
| Fig. 1 Aromatization using quinone-based oxidants. | ||
Another complex example was given in 1970 by Dannenberg in which the preparation of three hydrocarbons 112, 113, and 114 was possible from cholesterol 115 dehydrogenation using chloranil and DDQ oxidants (Fig. 2).71–74
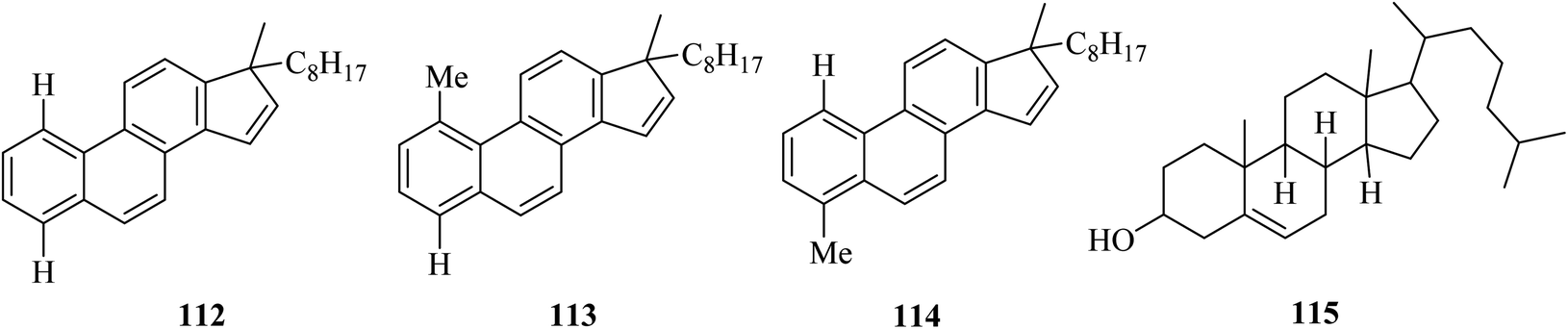 | ||
| Fig. 2 Hydrocarbons prepared by the dehydrogenation of cholesterol. | ||
Findlay and Turner (1971) reported that the oxidation of phenol l16 by DDQ leads to the formation of ketone 117.75 In addition, when methyl abietate 118 dehydrogenation took place with DDQ, an unexpected intermediate 119 was obtained.76 Menthofuran 120 oxidation by DDQ generated the lactone 121 instead of a p-cymene analog (Fig. 3).77
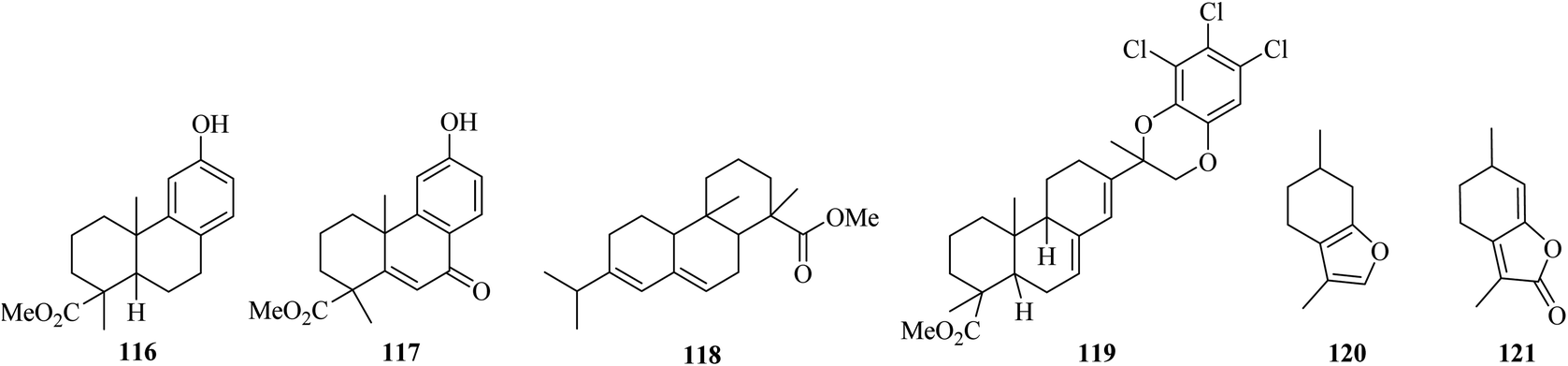 | ||
| Fig. 3 Aromatization promoted by DDQ. | ||
To avoid the prolonged treatment and by product formation during aromatization of steroids, DDQ is preferred as oxidant. It is considered as highly superior reagent for aromatization of steroids in neutral and weakly acidic conditions at a faster rate. DDQ is quite useful in case of sterically hindered molecules. The work we have described here has already referred to several useful aspects of DDQ as oxidant.
3.7. Oxidative cyclization
In 1960, Creighton and Jackman reported an interesting reaction involving an oxidative coupling process catalyzed by DDQ (Scheme 43).78 These types of coupling reactions (C–O bond formation) involve the formation of a carbocation intermediate, the stability of which is highly dependent upon the nature of attached substituents.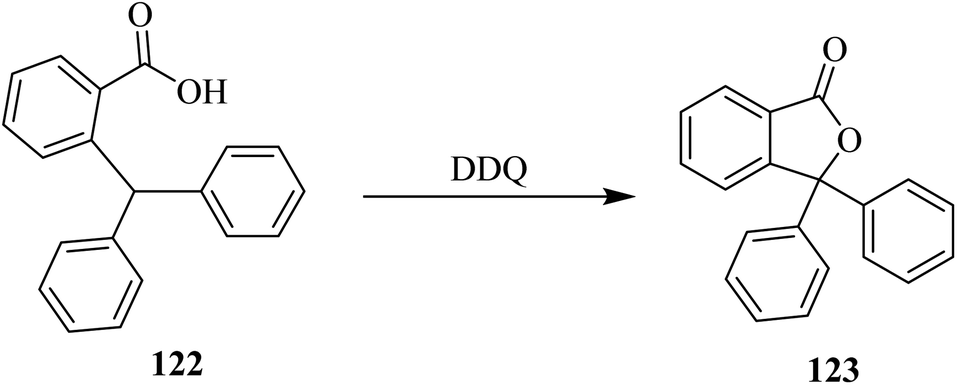 | ||
| Scheme 43 Oxidative coupling by DDQ. | ||
Thereafter, in 1965, chemists suggested that intermolecular coupling could be possible if enolizable ketones and phenols could not undergo α,β-dehydrogenation. However, oxidative dimerization was detected for a few starting materials, for instance, 2,6-dimethoxy-phenol.56
DuPont Pharm chemists synthesized Efavirenz (SUSTIVATM: anti-HIV) drug in which oxidative cyclization of butynol derivative 124 was achieved using DDQ in toluene to give the benzoxazine derivative 125 (Scheme 44).79
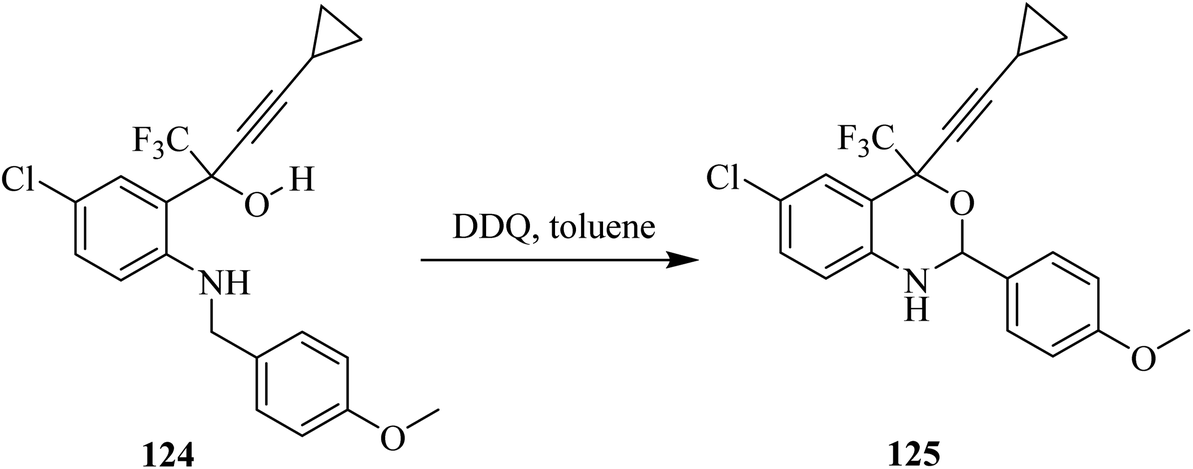 | ||
| Scheme 44 Synthesis of Efavirenz by DDQ-mediated oxidation. | ||
In recent years, Cho and Scott (2015) proposed an efficient way to synthesize triply-fused benzene rings system 127 (Scheme 45). This intermolecular oxidative cyclotrimerization reaction involves highly reactive benzyne intermediates generated, in situ, in the presence of DDQ and trifluoromethanesulfonic acid (TfOH) in 1,2-dichloroethane (DCE). This protocol is a convenient and advantageous metal-free reagent system as compared to the literature reported metal-based Scholl-type oxidants since it eradicates the probability of aromatic halogenation as a side reaction.80
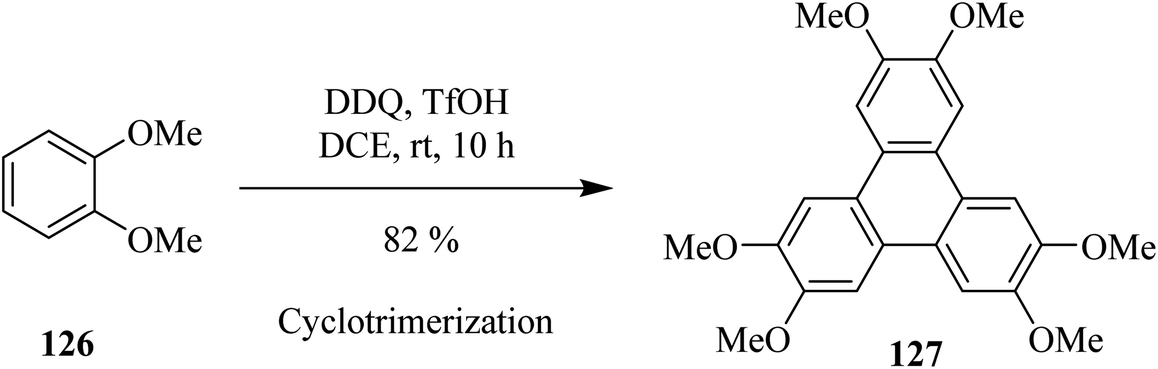 | ||
| Scheme 45 Oxidative cyclotrimerization of 1,2-dimethoxybenzene induced by DDQ. | ||
Several methods were reported in the literature for the oxidative cyclization of aromatic compounds. But low yields and low chemical purity were always the main problems faced by the synthetic chemists. DDQ reduced these problems by improving the yield and purity of products obtained in oxidative cyclization reactions. Furthermore, it also eliminated the risk of environmental pollution by promoting metal-free synthesis.
3.8. Role of DDQ in flavonoids synthesis
Flavonoids constitute an interesting class of secondary metabolites with plant origin. Researchers have paid special attention to the preparation of synthetic analogs in the laboratory. For instance, Kagei and colleagues (1978) described a unique procedure to prepare flavone 129a in excellent yield from the corresponding flavanone 128a by treating the latter with DDQ (Scheme 46).81 Subsequently, in 1983 Shanker and team members applied these oxidative conditions to other substrates such as 128b to synthesize flavone derivative 129b under dehydrogenation conditions. Prior to this work, dehydrogenation of chromanones and flavanones to chromones and flavones, respectively, by DDQ had not been investigated yet. Therefore, a various number of chromanones and flavanones were dehydrogenated by using DDQ at reflux temperature.82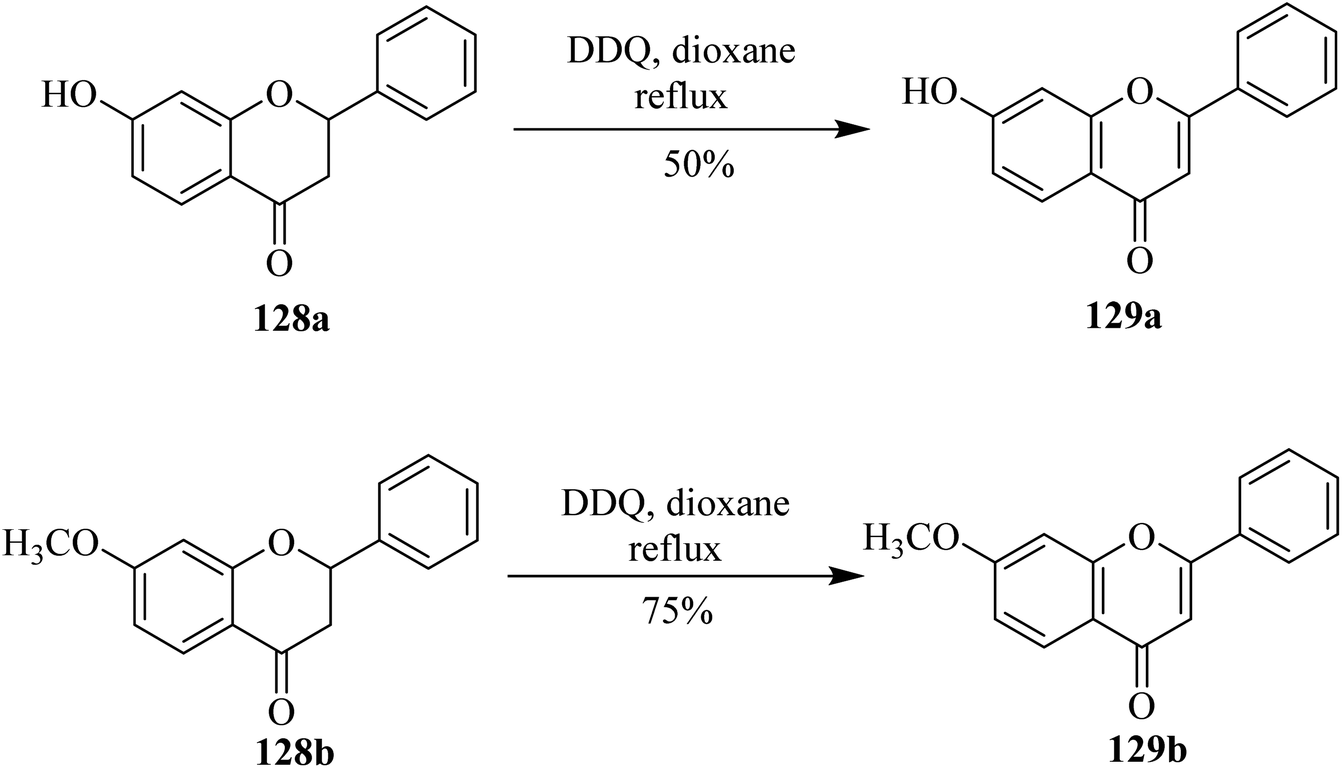 | ||
| Scheme 46 Dehydrogenation of 7-hydroxyflavanone (128a) and 7-methoxyflavanone (128b). | ||
Later on, Hoshino and Takeno (1987) determined the extent of the dehydrogenation of flavanones 130 and 132 with DDQ through HPLC analyses (Scheme 47). While substituents residing in the ortho position of the phenyl ring (ring B) of flavanones retarded the reaction, a corresponding study to explain the ortho effect by employing the linear combination model concluded that the inductive effect was more critical than the resonance and steric effects.83
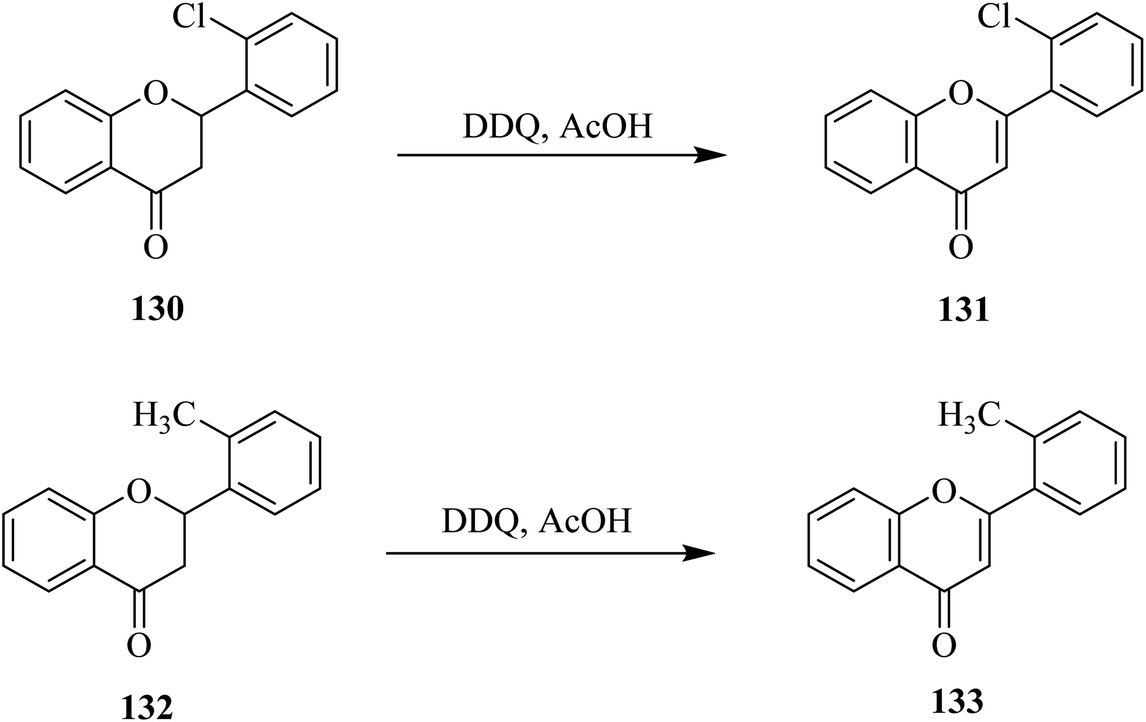 | ||
| Scheme 47 Dehydrogenation of flavanones with DDQ. | ||
In 1999, Somogyi's group reported a simple and efficient single-step dehydrogenation of 1-thioflavanone 134 into 1-thioflavone 135 in excellent yield by employing DDQ (Scheme 48). They applied the same conditions on the conversion of their oxidized and halogenated derivatives as well. For example, the bromo derivative 136 was successfully oxidized to form flavone 137 in high yield by treating the former with DDQ. Noteworthy, the same chemical transformation of flavanones 134 and 136 into flavones 135 and 137 failed when o-chloranil oxidant was used (Scheme 48).84
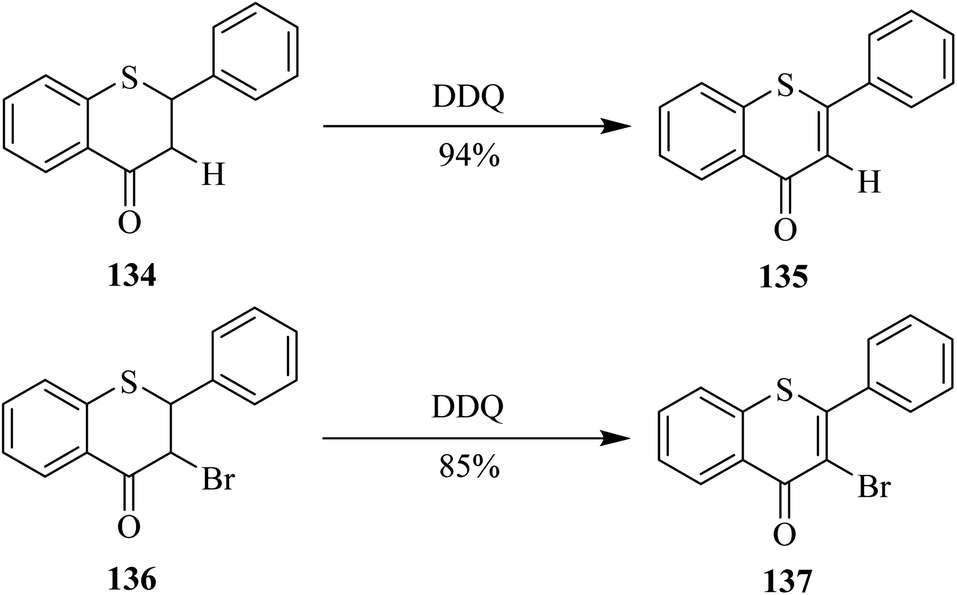 | ||
| Scheme 48 Dehydrogenation of 1-thioflavanone and 3-bromo analog by DDQ. | ||
Vercauteren and co-workers (2001) worked on the gram-scale preparation of 13C-labelled procyanidin 139 from anthocyanidin analog 138 by treating the latter with DDQ in an acidic medium (Scheme 49). This oxidation procedure was fairly stereoselective and exclusively produced one diastereomer. Higher yields (up to 76%) were achieved as compared to low-scale reactions performed in preliminary studies.85
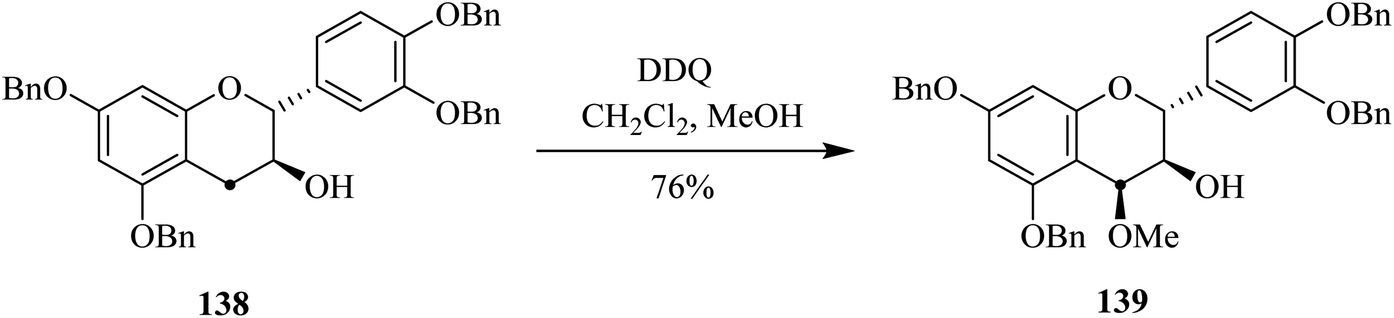 | ||
| Scheme 49 Conversion of catechin to flavanol analog by DDQ. | ||
In 2005, Tzeng and co-workers designed a method to synthesize flavone 141 and flavanone 142 from the requisite chalcone 140 by oxidizing the latter with DDQ at 110 °C. The reaction outcome depended highly on temperature and the group demonstrated the prospects of getting different products by gradually manipulating the reaction conditions (Scheme 50).86
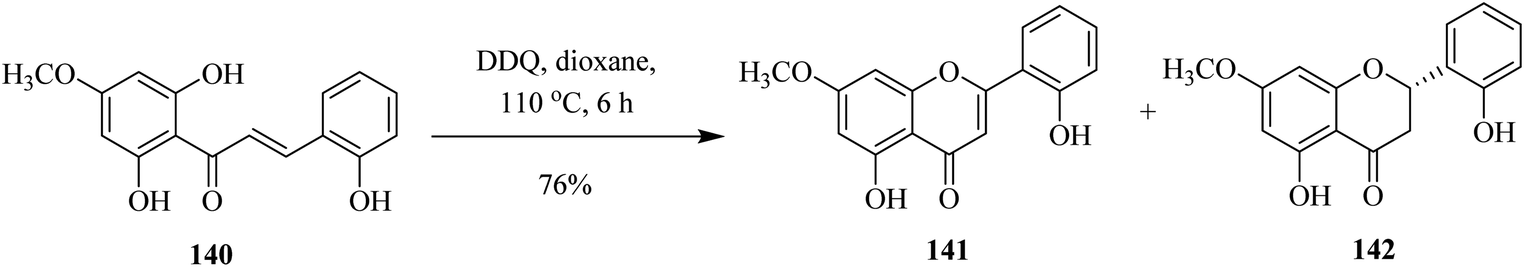 | ||
| Scheme 50 Synthesis of 5,2′-dihydroxy-7-methoxyflavone and flavanone. | ||
Although flavonoids may be oxidized by a number of oxidants, the applicability of these reagents is often limited due to several demerits. The sensitivity of substituents, side reactions, and certain substitution pattern of flavonoid molecules limits the use of these typical oxidants. To avoid these limitations, the mild and selective oxidant DDQ has been used as an alternative as discussed above. It also avoids tedious purification and provides higher yields. Moreover, it promotes one-step reaction that minimizes the risk of impurities.
3.9. Dehydrogenation by DDQ
DDQ plays a vital role in the dehydrogenation of aliphatic hydrocarbons to simple aromatic compounds. In this regard, Müller (1973) disclosed a method for the dehydrogenation of 1,4-cyclohexadiene 143 as well as cis- and trans-3,6-dimethyl-l,4-cyclohexadiene by employing a mixture of DDQ and tritylium tetrafluoroborate as an oxidizing agent (Scheme 51). The mechanism of dehydrogenation using tritylium tetrafluoroborate along with DDQ was investigated and proposed on the basis of kinetic isotopic effects studies they conducted and the products obtained.87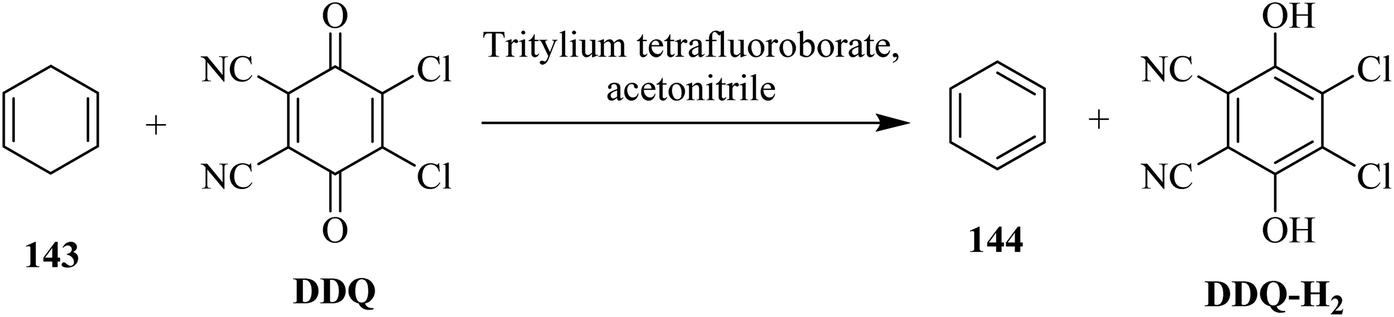 | ||
| Scheme 51 Dehydrogenation of 1,4-cyclohexadiene by DDQ. | ||
In 2008, Xu and co-workers extended this work and reported a method of oxidative dehydrogenation of dihydroarenes like 145 by DDQ and NaNO2 under an open atmosphere to furnish the aromatic system 146 (Scheme 52). The amalgamation of DDQ and NaNO2 offered better capability and selectivity than other benzoquinone and anthraquinone-based oxidants.88
 | ||
| Scheme 52 Dehydrogenation of dihydroarenes by DDQ. | ||
In continuation, Wang and co-workers (2010) developed a DDQ controlled oxidation–dehydrogenation protocol. In this process, they synthesized 2-aroylbenzopyran-4-one (148) from 2-aroyl-3,4-dihydro-2H-benzopyran (147) in the presence of a stoichiometric quantity of DDQ and acetic acid (Scheme 53). Firstly, DDQ and acetic acid generate DDQH+ which abstracts one of the benzylic hydrogens of 147 to generate a benzylic cation intermediate. As DDQH+ carries a positive charge, it behaves as a stronger electrophile compared to neutral DDQ. Therefore, the deprotonation process of chroman at the C-4 position is enhanced. After this dehydrogenation step, oxidation was carried out to introduce CO in the chroman moiety. Then, two DDQ equivalents were used sequentially for dehydrogenation to introduce CC in chroman ring to afford 2-aroylbenzopyran-4-one (148).89
 | ||
| Scheme 53 DDQ controlled oxidation-dehydrogenation of dihydrobenzopyrans. | ||
In 2011, Crabtree and co-workers90 introduced a DDQ catalyzed oxidative dehydrogenation of secondary amines which served as a model system for simulated storage of hydrogen.
The group conducted a computational to provide mechanistic information about a chemical reaction. They effectively performed dehydrogenation of secondary amine like 149 in the presence of a metal-free organocatalyst and produced 150 (Scheme 54). Additional work is still required to optimize the reported procedure.90
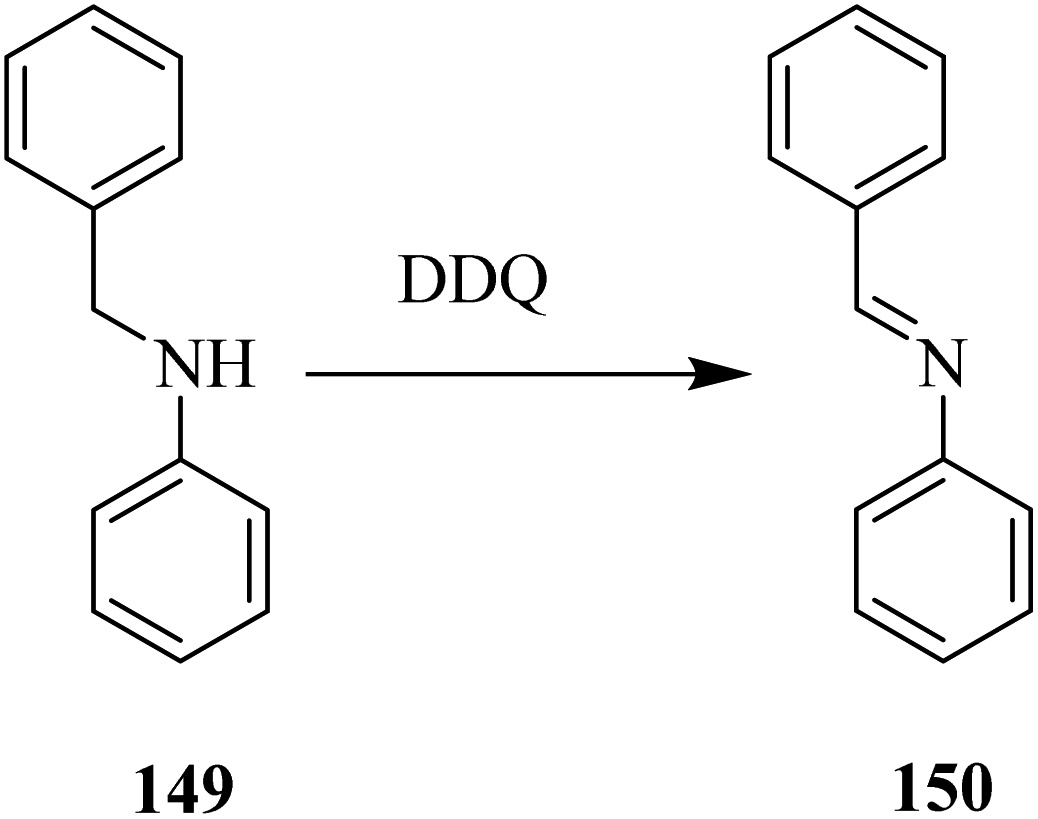 | ||
| Scheme 54 Secondary amine dehydrogenation by DDQ. | ||
Furthermore, Li and co-workers (2013) devised a mild procedure for thiazolines and oxazolines 151 oxidation into thiazoles and oxazoles 152 with the aid of DDQ (Scheme 55). The applied method showed several benefits over previously reported similar methods. For instance, this process offered a feasible, metal-free, and high functional group tolerance for the formation of heteroaryl compounds. To acquire an improved understanding of the reaction mechanism, they performed the natural population analysis.91
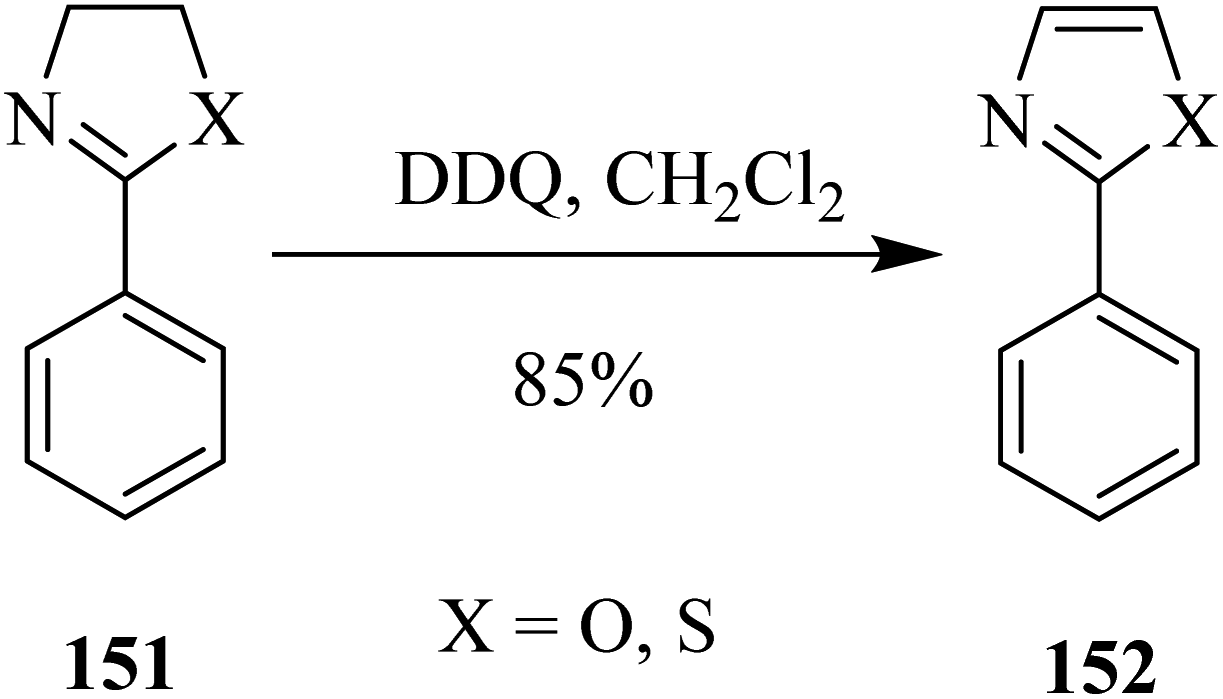 | ||
| Scheme 55 DDQ-induced dehydrogenation of saturated heterocycles. | ||
DDQ proved highly selective oxidation system for the dehydrogenation of different organic compounds like aliphatic hydrocarbons. It is the main replacement to the classical oxidative dehydrogenating agents and provides greater yields. The rapid reaction rate avoids the slow H2 evolution and the reagent may can be used in smaller amounts as mixture with other reagents. It is also easy to remove at the completion of the reaction. Further, it is metal free oxidation system that eliminates the risk of pollution.
3.10. DDQ-induced condensation of polyphenylenes into nanographenes
Over the past decade, DDQ-catalyzed condensation of polyphenylene hydrocarbons has gained increased attention in the scientific community involved in the synthesis of π-electrons rich aromatics. In this context, Rathore and co-workers (2009),92 probably for the first time, explored the use of DDQ in Scholl's reaction. They reported that Scholl's precursors could be subjected to oxidative cyclization with DDQ in the presence of a readily available methanesulfonic acid (MeSO3H) under the standard conditions to afford the corresponding cyclodehydrogenated products in excellent yields (Fig. 4). Remarkably, the Scholl reaction with the DDQ/CH3SO3H oxidation system was equally effective with substrates undergoing both intramolecular and intermolecular aryl–aryl C–C bond formations.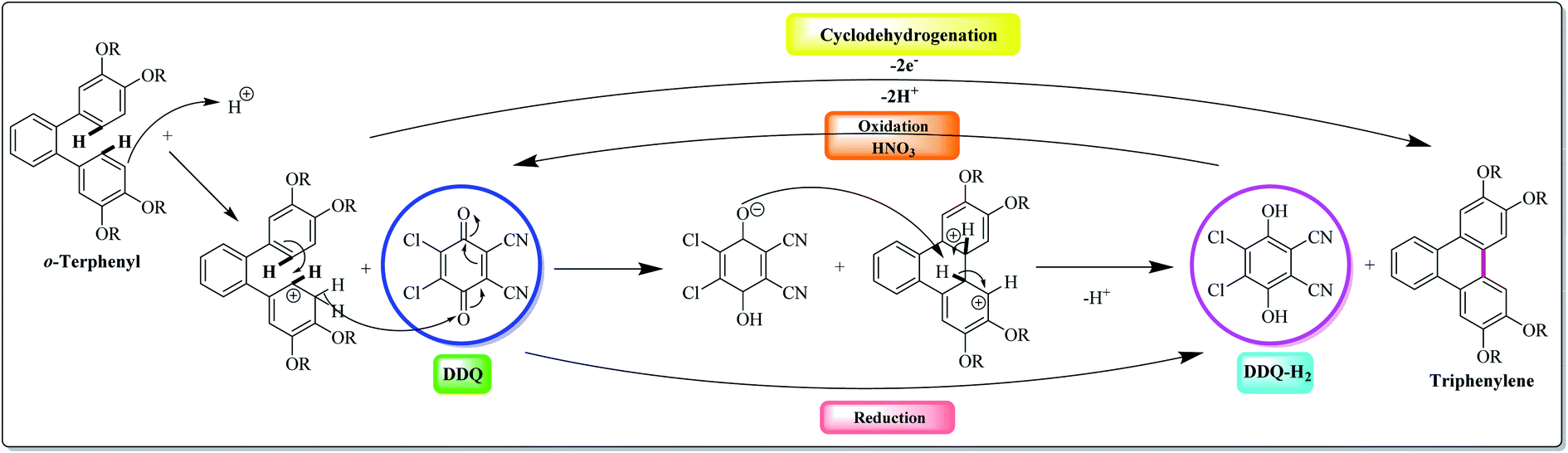 | ||
| Fig. 4 Possible mechanistic pathways for the C–H abstraction from Scholl's precursors by DDQ. | ||
Further, the DDQ/acid system (which readily oxidizes aromatics to the corresponding cation radicals) has been shown that it can be employed for the Scholl reaction. Mechanistically, the DDQ/H+ system probably proceeds through cationic intermediate as depicted in Fig. 4. Here, a high-potential quinone is used in conjunction with an aprotic organic acid to affect dehydrogenative biaryl formation. Although a full mechanistic understanding of this reaction remains elusive, it has been suggested that the formation of a radical cation or arenium cation precedes the crucial biaryl C–C bond-forming step. Noteworthy, acid is necessary with DDQ to work effectively. However, mostly other oxidants don't require Brønsted acid, because they are strong enough to oxidize the parent arene system themselves. The isolation of cyclized products and the recovery of the reduced DDQ–H2, which can be readily recycled into DDQ by oxidation with nitric acid, are easy.
Moreover, Rathore and his team also reported that the DDQ/H+ method can be efficiently employed for oxidative biaryl synthesis (or C–C bond formation) in polycyclic aromatic hydrocarbons (PAHs). The biaryl synthesis has been particularly useful for the oxidative cyclodehydrogenation of a variety of substituted hexaarylbenzene and o-terphenyls to produce the corresponding planar PAHs, for example, hexa-peri-hexabenzocoronenes (HBC's) and triphenylenes, respectively. The usefulness of the DDQ-acid system for the oxidative cyclodehydrogenation in the Scholl reaction was further validated by the generation of soluble HBC's [154a, b, c] from hexakis(4-isoalkylphenyl)benzenes [153a, b, c], where six new C–C bonds were formed in one step to give graphitic products in excellent yields and high purity (Scheme 56). Later, the DDQ/H+ oxidative system was applied in the oxidization of a variety of aromatic donors, with oxidation potential as high as ∼1.7 V, to the corresponding radical cations and can be utilized for the synthesis of a variety of PAHs including graphitic hexa-peri-hexabenzocoronenes.92
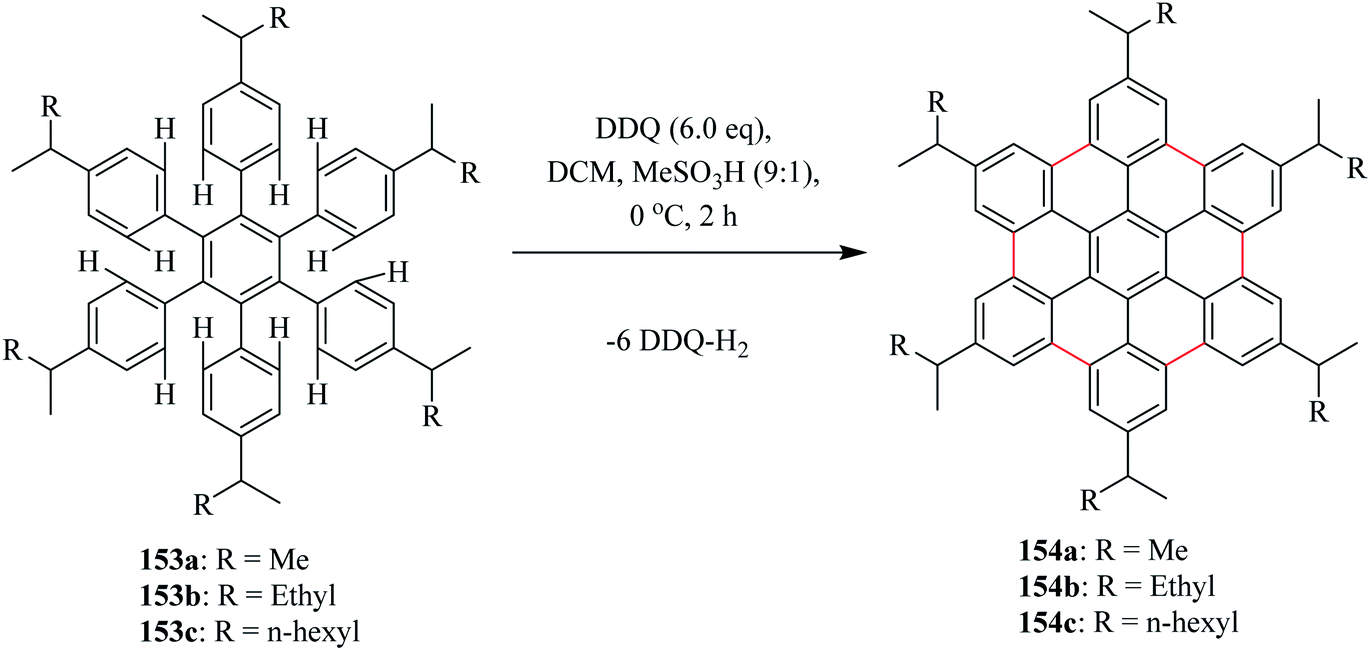 | ||
| Scheme 56 Synthesis of soluble HBCs from hexaarylbenzene precursors. | ||
Wong and co-workers (2012) developed a new set of reaction conditions for the oxidative cyclodehydrogenation of electron-poor polyphenylenes with various electron-withdrawing groups like bromo and fluoro, among others. Their coupling into planar nanographenes was achieved using the DDQ–CF3SO3H oxidative system.93 Bromo- and fluoro-HBC derivatives 158a–c were prepared from hexaphenylbenzene precursors 157a–c by treatment with a combination of DDQ and CF3SO3H. It is noteworthy that there are two proposed reaction mechanisms for the oxidative cyclodehydrogenation (Scholl) reaction, proceeding through either an arenium-ion or a radical-cation intermediates. In both pathways, acids play a vital role in the progress of the reaction. The hexaphenylbenzene precursors 157a–c were synthesized from the Diels–Alder cycloaddition reaction of acetylenes 155a–c to cyclo-pentadienones 156a–c followed by CO extrusion and aromatization (Scheme 57).
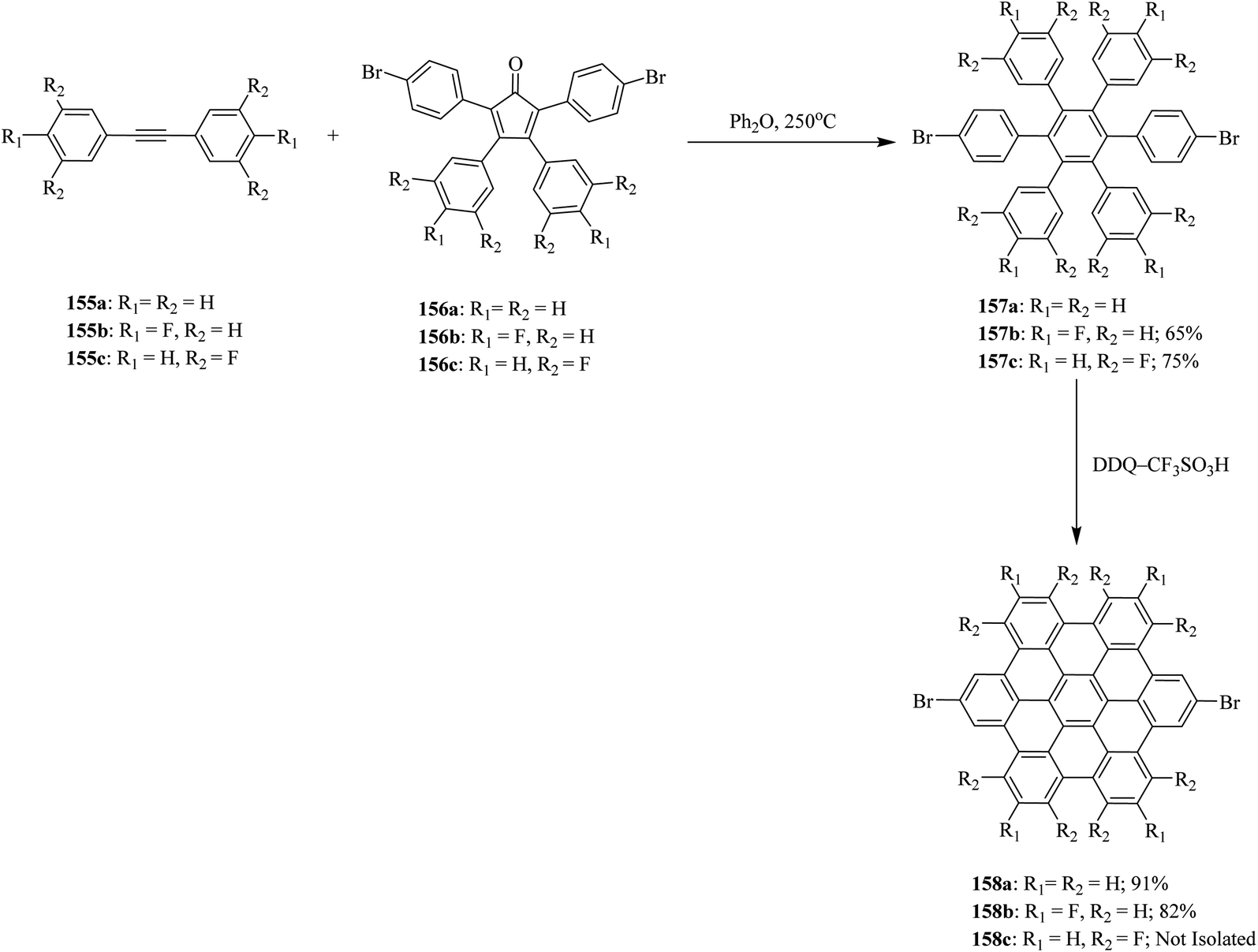 | ||
| Scheme 57 Synthesis of a series of HBC derivatives containing EWGs via DDQ. | ||
In 2012, Miao and co-workers also reported the synthesis of slightly non-planar nanographene molecules with overcrowded fjord regions like derivatives of hexabenzoperylene (HBP) 163 and 164 that led to the formation of both chiral twisted and anti-folded conformers. The oxidative cyclization step of the precursors 161 and 162 involves the use of a DDQ/CH3SO3H oxidizing system to give the PAHs 163 and 164 excellent yields without any side products (Scheme 58). Interestingly, introducing alkoxyl groups at the o- or p-positions next to the reaction sites was essential for successful and optimal cyclodehydrogenation process.94
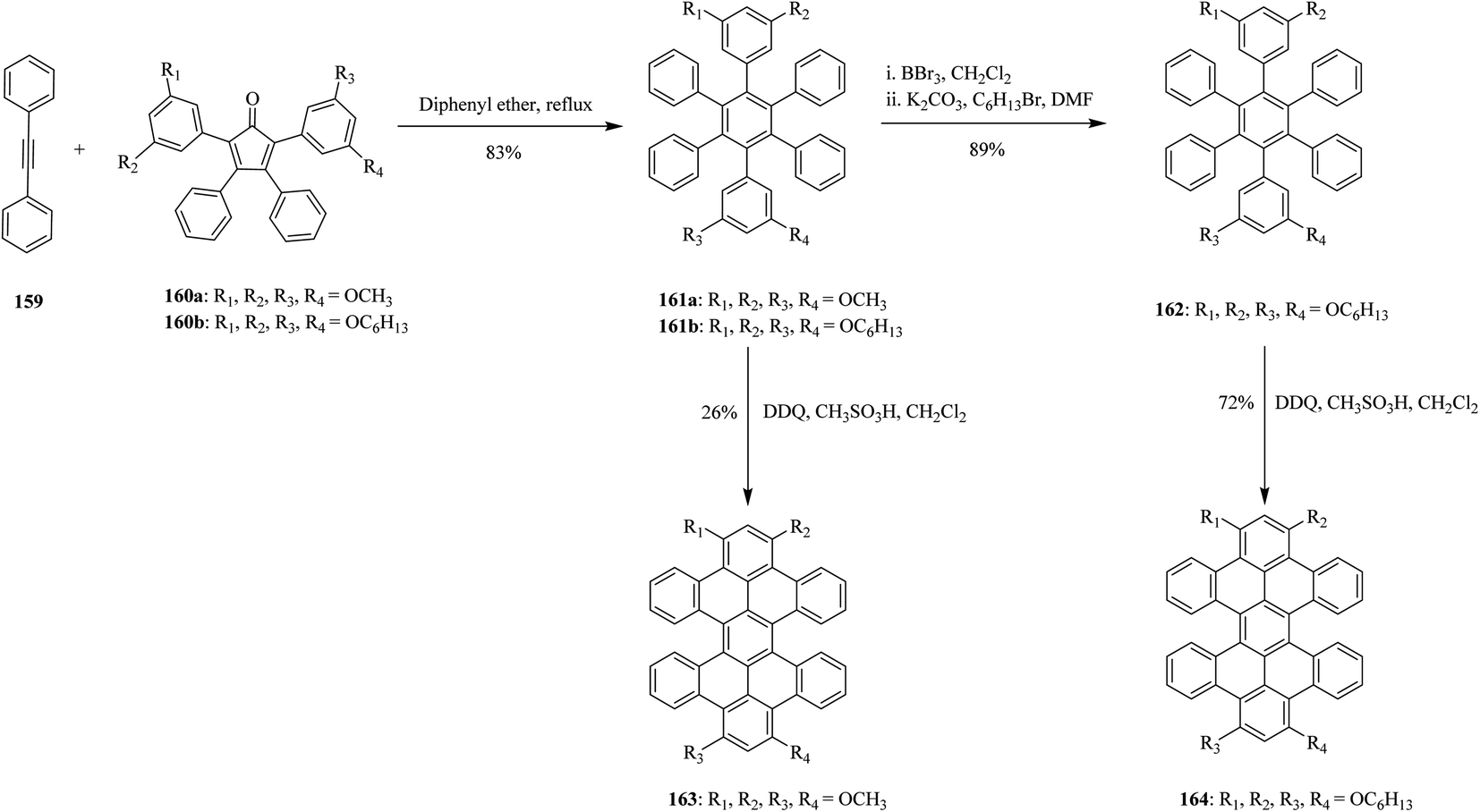 | ||
| Scheme 58 Synthesis of hexabenzoperylene via DDQ. | ||
Up till now, we have only discussed the use of DDQ in constructing a six-membered ring in nanographenes.
However, Scott and co-workers (2013) made remarkable progress in introducing a challenging two-step synthesis of completely warped nanographene molecule embedded with five seven-membered rings. Surprisingly, the intramolecular cyclization of the regioisomeric precursors 166 effectively afforded curved graphene molecules 167 in good yields, overcoming the high steric demand to produce five distorted seven-membered rings. The C–H arylation of corannulene 165 was successfully conducted in moderate yield. Subsequently, the oxidative cyclodehydrogenation of the precursor 166 was successfully achieved by utilizing a DDQ/trifluoromethane sulphonic acid (TfOH) oxidant mixture to furnish a distinctive convex–concave nanographene 167 containing five seven-membered rings (Scheme 59).95 The above-mentioned studies proved the success of the current protocol, thus paving the way to synthesize the challenging heptagon containing non-planar PAHs from polyphenylenes after condensing with DDQ/H+ oxidant system.
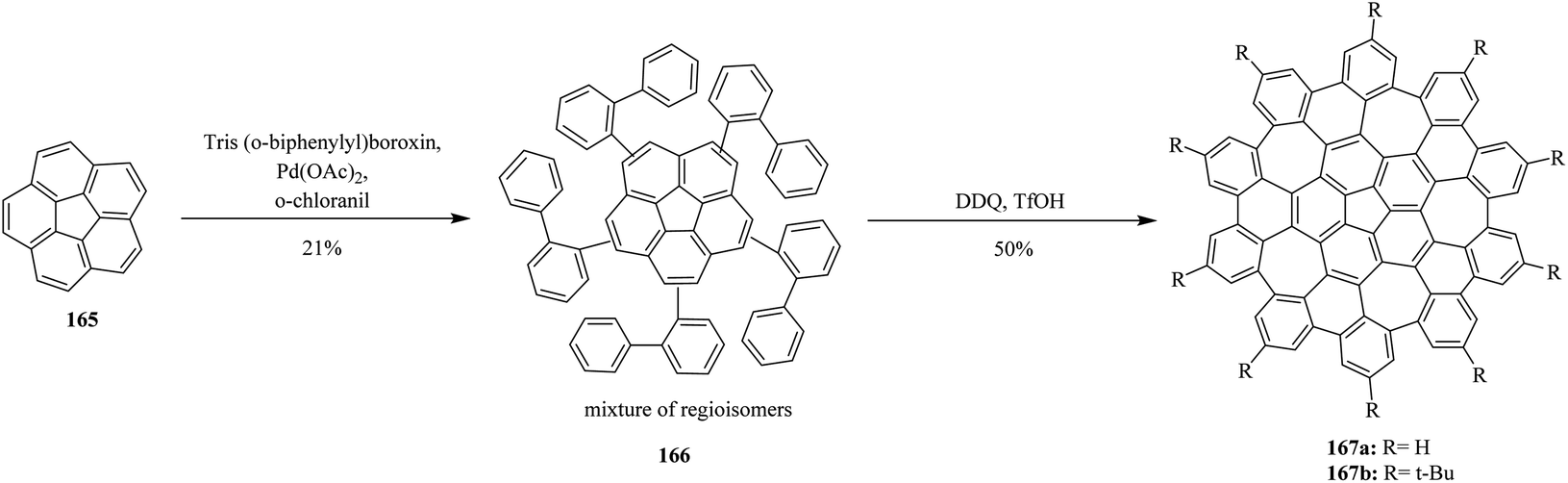 | ||
| Scheme 59 DDQ-induced synthesis of grossly warped graphene molecules. | ||
Inspired by previous findings, Müllen, and co-workers (2014) explained the synthesis of biphenylene-based condensed PAH 169 merged with two 8-membered and one 4-membered rings by cyclodehydrogenation of the precursor octaarylbiphenylene 168 using DDQ/MeSO3H mixture (Scheme 60). The cyclodehydrogenation did not proceed any further, possibly due to the greater steric hindrance and selectively, exclusively producing nanographene 169 in 90% yield.96 No doubt, this was also a breakthrough in showcasing the significance of DDQ as an oxidant in the field of nanographene syntheses. Additionally, the authors concluded that the isomeric graphene nanostructures were prepared to show a combination of four-, six- and eight-membered rings in a single structure.
 | ||
| Scheme 60 Synthesis of biphenylene-based PAH by DDQ. | ||
In follow-up, Müllen and co-workers (2015) further demonstrated recent progress in their work by illustrating the synthesis of nanographene by newly devised methods. These methods immensely broadened the scope of already available nanographene molecules. A great range of configurations, symmetries, embedded more than six-membered rings, edge functionalization, and heteroatom doping are included in these graphene molecules. They prepared these nanographenes by using various oxidants, and one of them was DDQ. For instance, they utilized DDQ in the chemical transformation of precursor 170 to the PAH 171 through consequent cyclodehydrogenation in 89% yield (Scheme 61).97
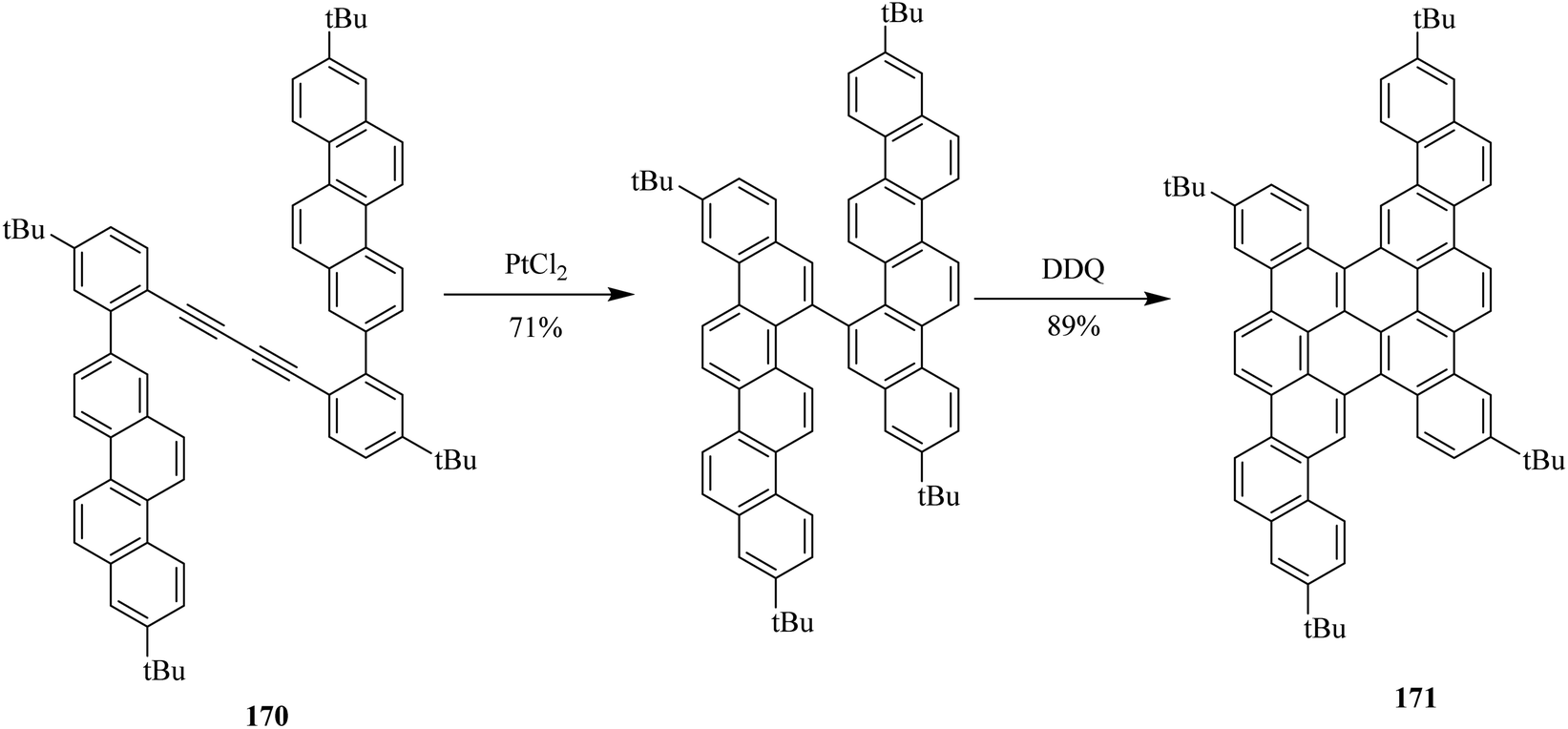 | ||
| Scheme 61 Synthesis of PAHs by DDQ. | ||
In 2015, Miao and co-workers synthesized two new saddle-shaped polycyclic arenes containing two heptagons from saddle-shaped diketone 172a, b (Scheme 62). The synthesis of two novel saddle-shaped PAHs 174a, b and 176a, b with two entrenched heptagons was conducted via the oxidative cyclodehydrogenation of the precursors 173a, b and 175a, b by employing DDQ/CF3SO3H initiated oxidation.98 These findings further strengthened the importance of the DDQ/H+ system in the insertion of a seven-membered ring in nanographenes structures, which could offer exciting electronic properties for applications in organic electronics.
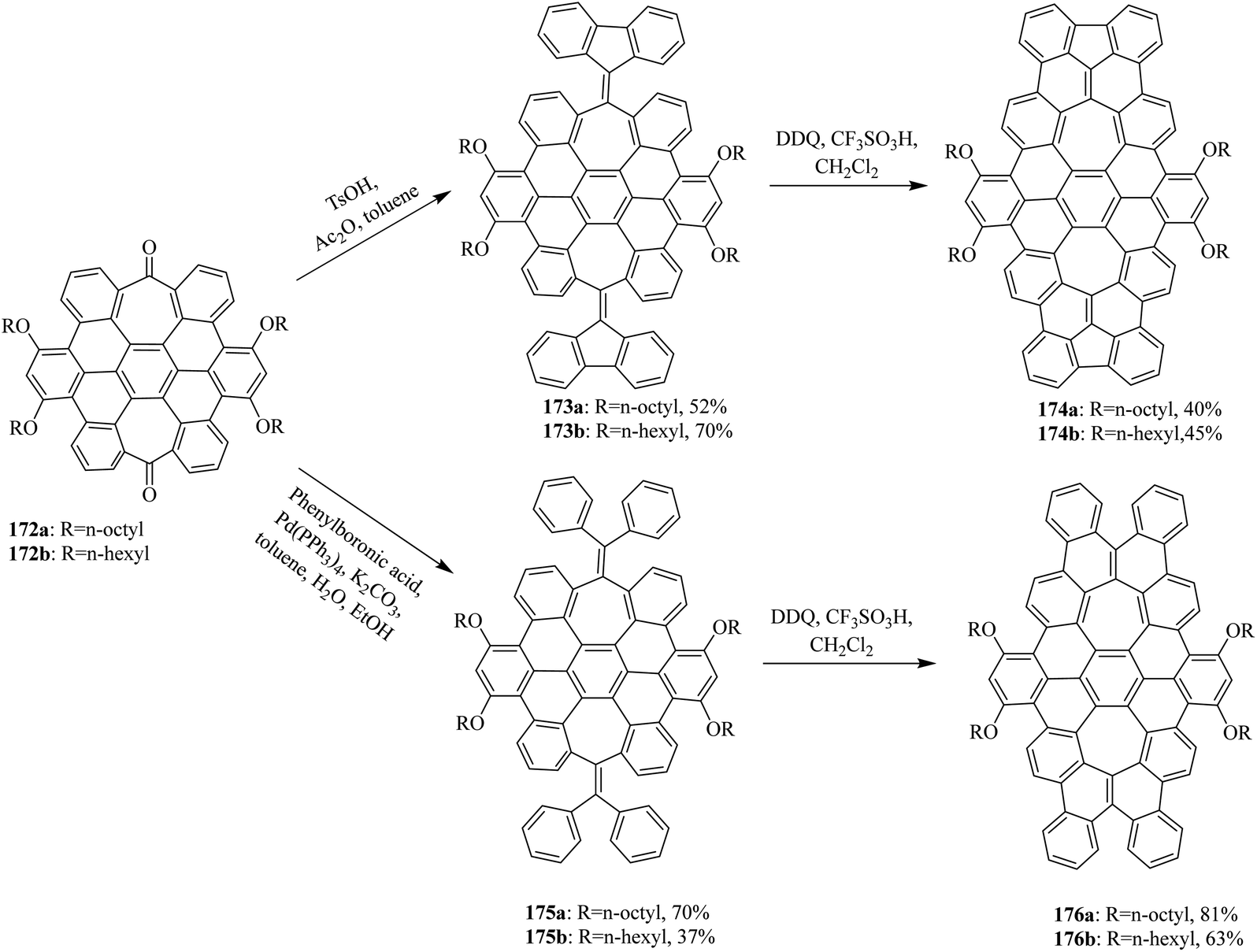 | ||
| Scheme 62 Graphene syntheses with an embedded 7-membered ring through cyclodehydrogenation. | ||
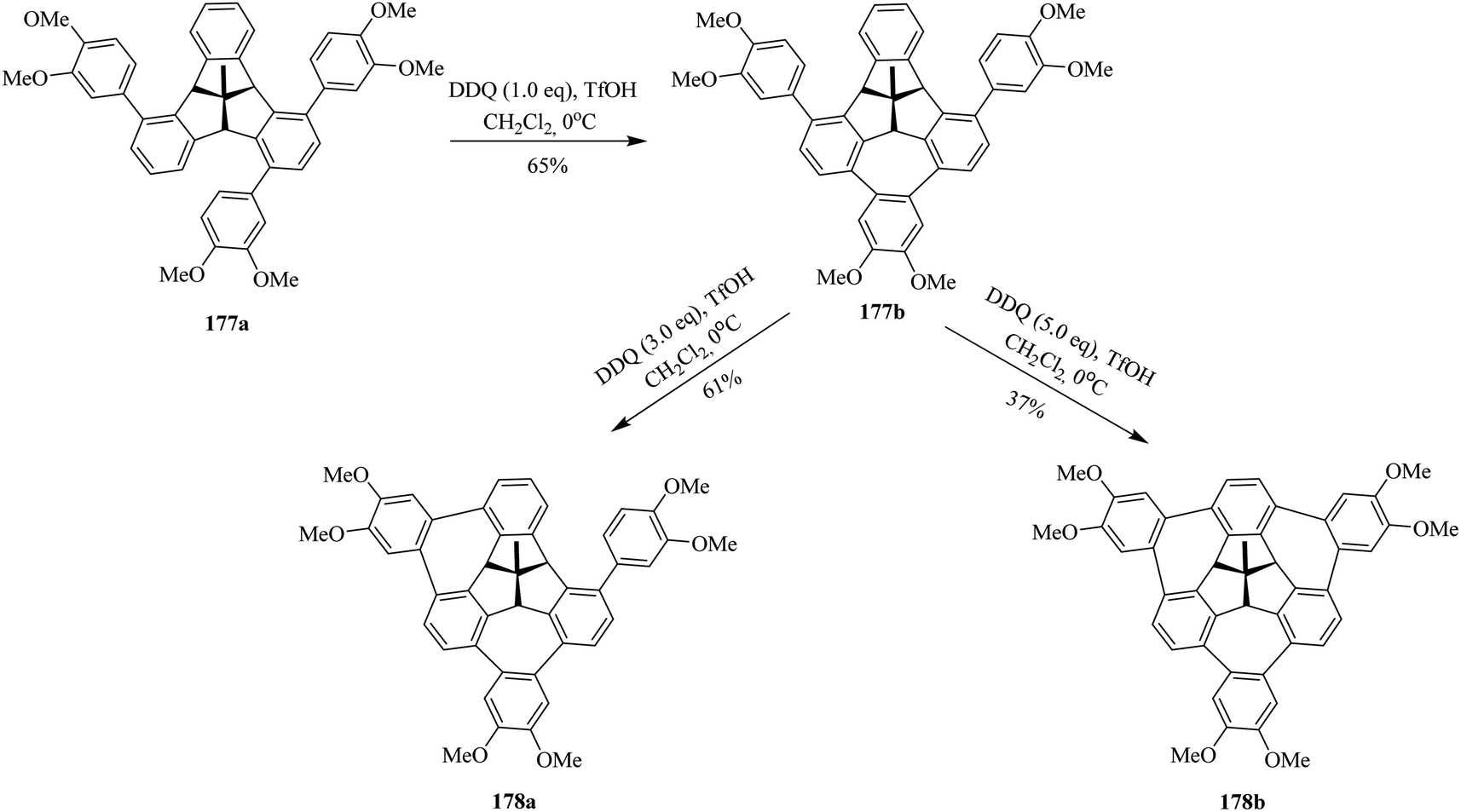
Encouraged by the findings, Kuck and co-workers (2016) published, for the first time, the challenging approach for the attachment of three o-phenylene units at the bay regions of C3v-symmetrical hat-shaped polycyclic aromatic hydrocarbon called tribenzotriquinacene (TBTQ) 177a. The key step in their synthetic sequence includes three Scholl-type cycloheptatriene rings formation around the TBTQ core. This has been a longstanding dream of Kuck to incorporate three 7-membered rings at the bay regions of the TBTQ carbon framework. Over the past years, they attempted several oxidants to promote these oxidative cyclization reactions and ultimately were successful in fulfilling their goals by utilizing DDQ/TfOH oxidant system. In this regard, they treated the hydrocarbon 177a with stoichiometric amounts of DDQ and TfOH in dichloromethane as displayed in Scheme 63. These oxidative conditions produced intermediate product 177b with incomplete cyclodehydrogenation in good yield. To get multiple functionalization around the peripheral arene positions of TBTQ is itself quite challenging. Anyhow, upon increasing the amount of DDQ to 3.0 equivalents, they observed the attachment of two 1,2-arylene units around the TBTQ core in the form of an unwanted product 178a (Scheme 63) produced in moderate yield. Nevertheless, the desired target compound 178b could be obtained in 37% yield by using 5.0 equivalents of DDQ along with TfOH. Despite the relatively lower yield, the TBTQ merged with three cycloheptatriene units represents a promising key intermediate for the construction of nonplanar nanographene molecules bearing a TBTQ at the central core.99 These intriguing results opened the door for others to design and prepare organic functional materials based on the TBTQ motif.
 | ||
| Scheme 63 DDQ promoted oxidative cycloheptatriene ring formation around the TBTQ core. | ||
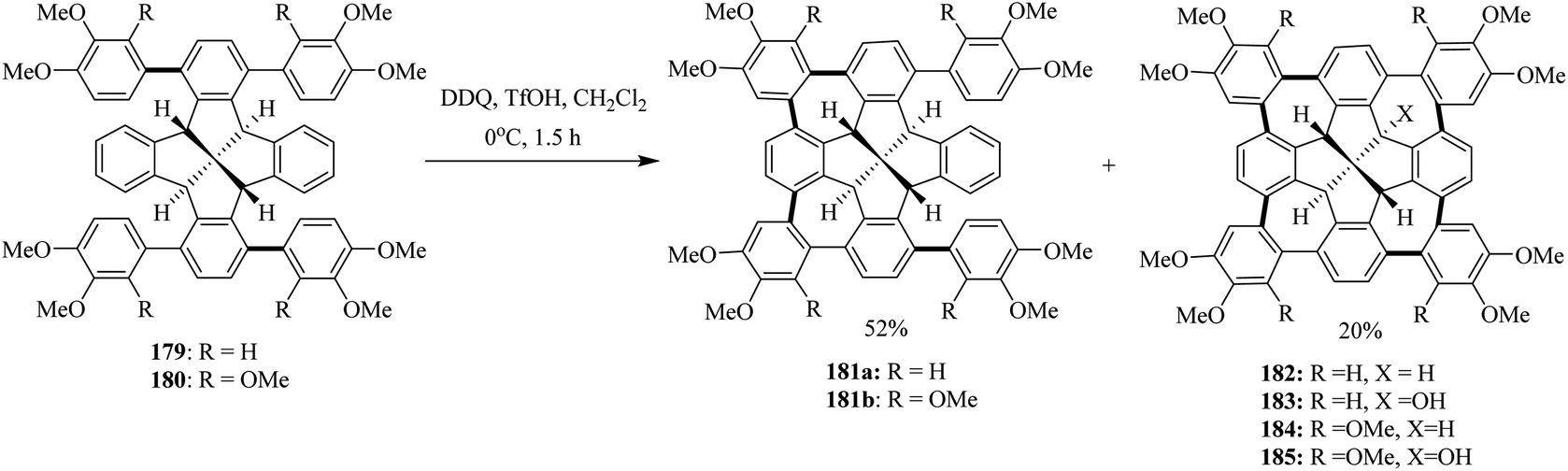
In the following years, Kuck and co-workers (2017) extended their research ventures to apply the previously reported oxidative conditions (DDQ/TfOH) in the synthesis of cycloheptatriene merged fenestrindane derivatives as shown in Scheme 64. Promoted by DDQ/TfOH, the key step featured four Scholl-type cycloheptatriene formation steps starting from the corresponding tetraarylfenestrindane derivatives 179 and 180 bearing electron-rich substituents. Consequently, the precursor hydrocarbons 179 and 180 were reacted with DDQ/TfOH in slight excess to produce a mixture of twofold-and fourfold–cyclization reactions in order to afford the products 181a (52%) and 182 (20%), respectively (Scheme 64), in moderate yields. In other attempts, neither products of single nor triple cyclization were observed. With the peripheral –OCH3 groups (184, 185) being available for further functionalization and C–C cross-coupling reactions, these molecules should provide access to various novel π-extended saddle-shaped nanographenes.100
 | ||
| Scheme 64 DDQ-mediated Scholl-type oxidative cycloheptatriene formation; a fourfold bridging of the fenestrindanes core with electron-rich aryl units. | ||
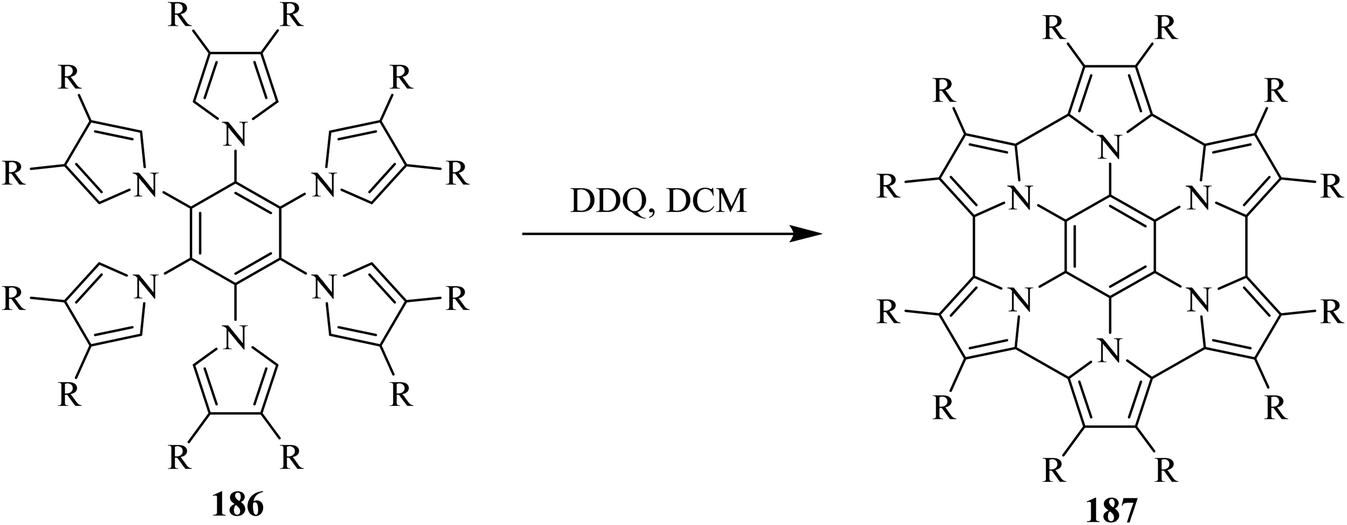
In 2017, a review by Stepien and co-workers discussed the extended form of two-dimensional polycyclic heteroaromatics like 187 which is also known as heterocyclic nanographene. These molecules were identified as an extremely versatile class of organic materials. The authors focused on the synthesis, properties, and applications of large π-extended heteroatom-doped nanographenes, and thus highlighted the use of DDQ as a privileged oxidant for their synthesis.101 A representative example of such heteroatom-doped planar nanographene molecule is illustrated in Scheme 65.
 | ||
| Scheme 65 Synthetic route to the synthesis of hexapyrrolohexaazacoronenes by DDQ. | ||
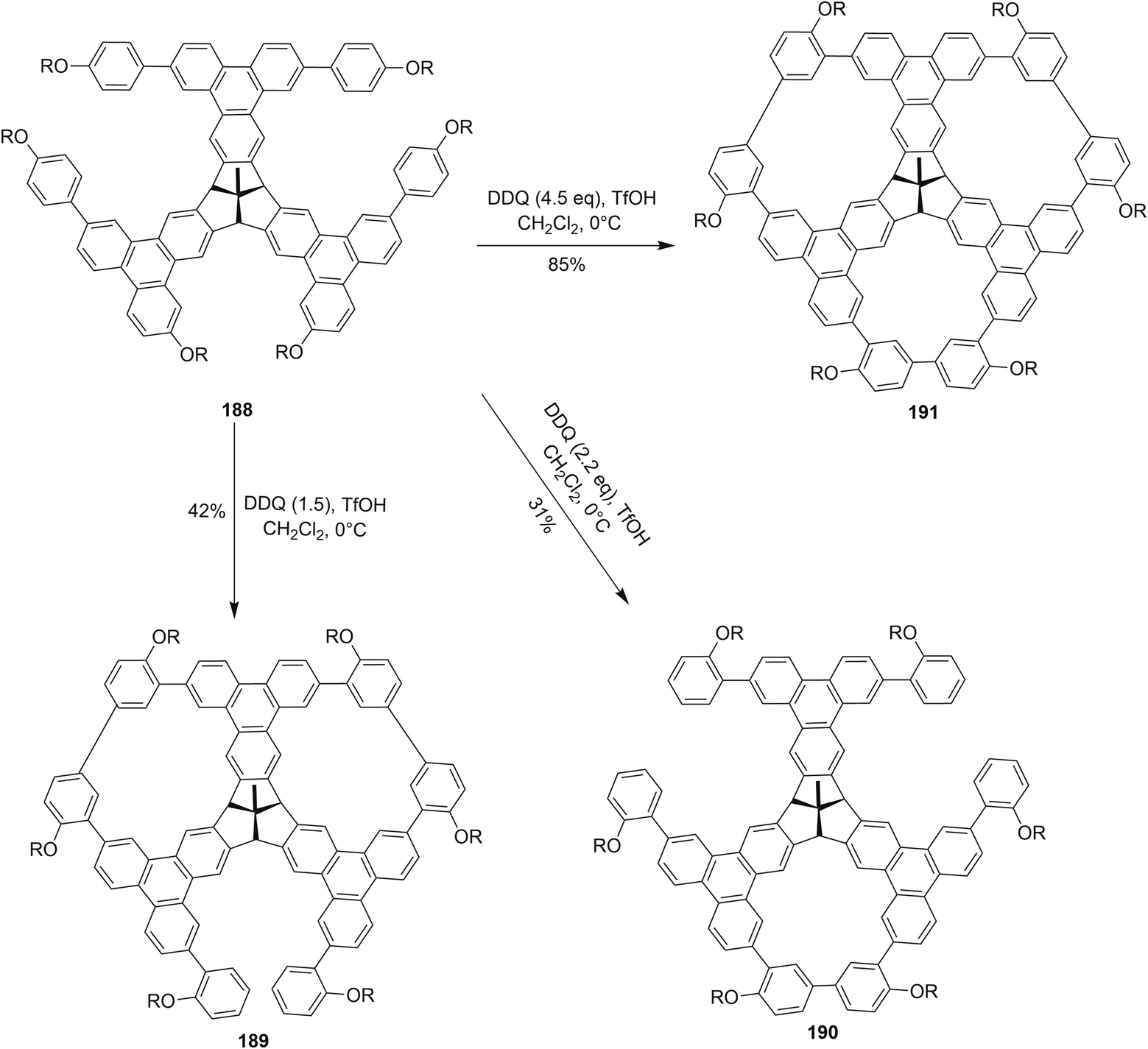
Recently, Kuck and coworkers (2018) described the formation of porous and complex bowl-shaped TBTQ-based nanographenes (Scheme 66). The precursor 188 was synthesized over several steps from easily accessible starting materials.102 The compound 188 was treated with different equivalents of DDQ–TfOH in order to promote Scholl type macrocyclization to produce the porous nanographene molecules 189–191 in different yields (Scheme 66). However, they concluded that in order to achieve threefold oxidative cyclization, 4.5 equivalents of DDQ/TfOH mixture were needed. The further extension of this cyclic porous nanographene 191 by Scholl's macrocyclization using DDQ/TfOH is still underway by the researchers to prepare a number of different novel PAHs.102
 | ||
| Scheme 66 DDQ–TfOH-promoted Scholl macrocyclization for porous nanographene syntheses. | ||
In 2020, Bonifazi and co-workers exploited the Pummerer oxidative annulation reaction to increase π-extension of the structure of 192 through an intramolecular C–O bond formation catalyzed by the use of appropriate oxidants (Scheme 67). The peripheral topology of the precursor 192 was used to regulate the formation of 5, 6, and 7-membered O-containing rings. They used DDQ as a key oxidant for the preparation of PAH 193 in good yield.103
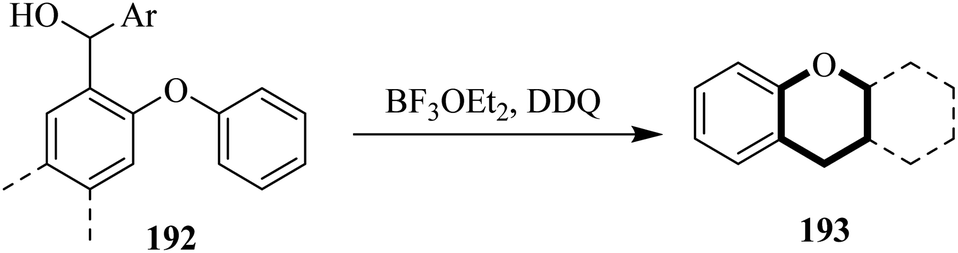 | ||
| Scheme 67 Synthesis of O-annulated PAHs by DDQ. | ||
In continuation, Gryko and co-workers (2020) demonstrated that oxidative aromatic coupling is central to the contemporary field of aromatic compounds. This type of coupling can be used for the construction of large and puzzling architectures from precursor-like indole 194. Substantial effort was also dedicated to the implementations of Scholl's reaction for the formation of these nanographene-containing natural products and chiral bisphenols like 195 (Scheme 68). The researchers used DDQ as a primary oxidant to promote this kind of aromatic coupling in order to furnish the valuable target compound 195.104
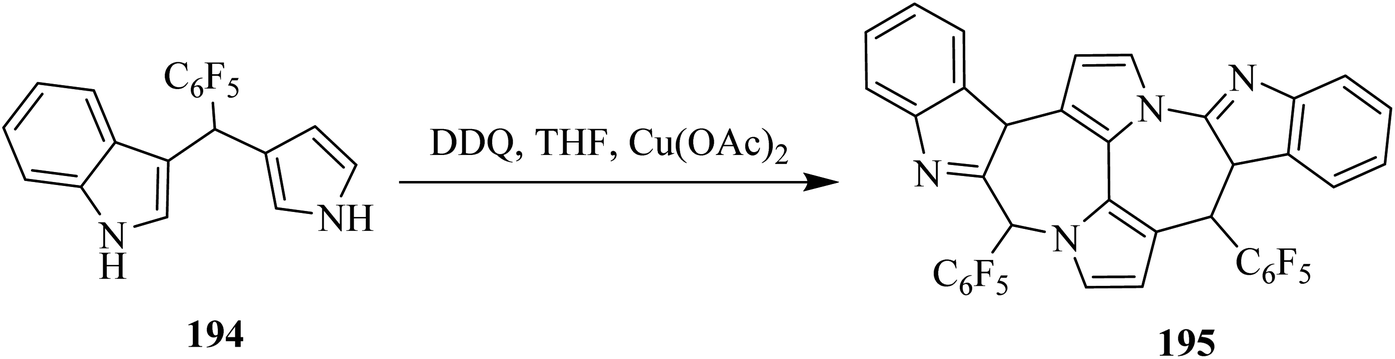 | ||
| Scheme 68 Oxidative aromatic coupling by DDQ. | ||
Very recently, Miao and co-workers (2020) devised a protocol for the synthesis of novel aromatic saddle-shaped nanographenes by employing Scholl's reaction conditions. In this context, different equivalents of DDQ were used throughout the experiment. It is worth mentioning that the stoichiometric amount of DDQ furnished incomplete oxidative cyclodehydrogenation as depicted in Scheme 69. Nevertheless, to overcome this problem and push the reaction towards complete oxidation, they used DDQ in excess (11.0 eq.). The nanographenes 199 and 200 were prepared from the reactant hydrocarbon 198 using DDQ along with TfOH in moderate to good yields. Thus, the team successfully synthesized novel aromatic strained non-planner nanographenes with embedded octagon by employing DDQ/TfOH oxidizing mixture. Moreover, this oxidant system helped avert the problems of skeletal rearrangement and incomplete cyclization.105
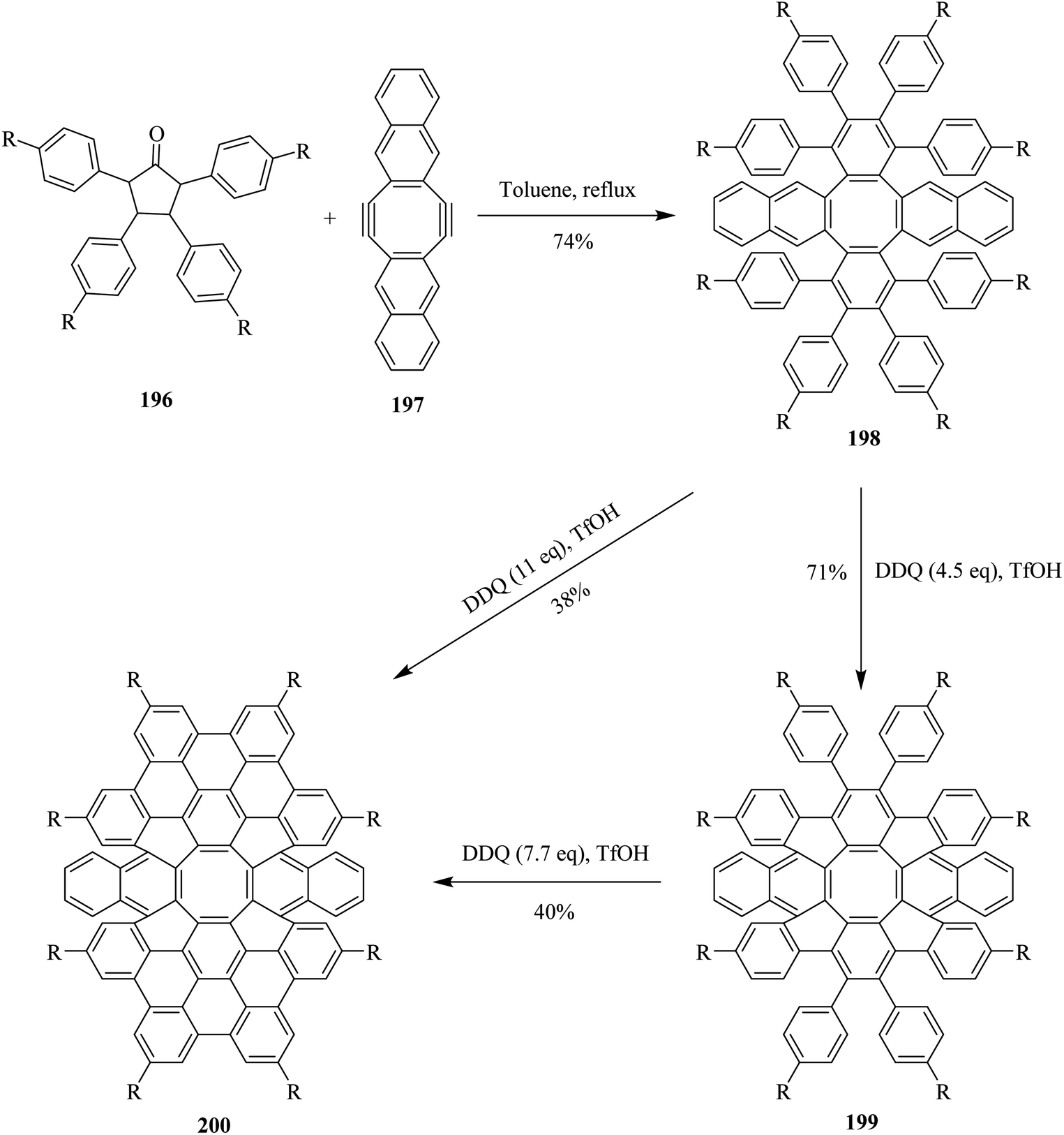 | ||
| Scheme 69 Synthesis of the aromatic saddle-shaped nanographenes by DDQ/TfOH system. | ||
This year (2021), Kuck and co-workers disclosed the synthesis of π-extended TBTQ-based wizard hat-shaped nanographene 202 from the precursor polycyclic aromatic compound 201 through Scholl type dehydrocyclization (Scheme 70). The key step involves Scholl-type cyclodehydrogenation of precursor 201 upon treatment with the DDQ-TfOH/CH2Cl2 oxidative system to afford the unique nonplanar PAH 202 in a 38% isolated yield. This DDQ-induced methodology has paved the way to design and synthesize π-electrons rich conjugated aromatics with a combination of five-, six-, and eight-membered carbocycles having improved optical and electronic properties.106
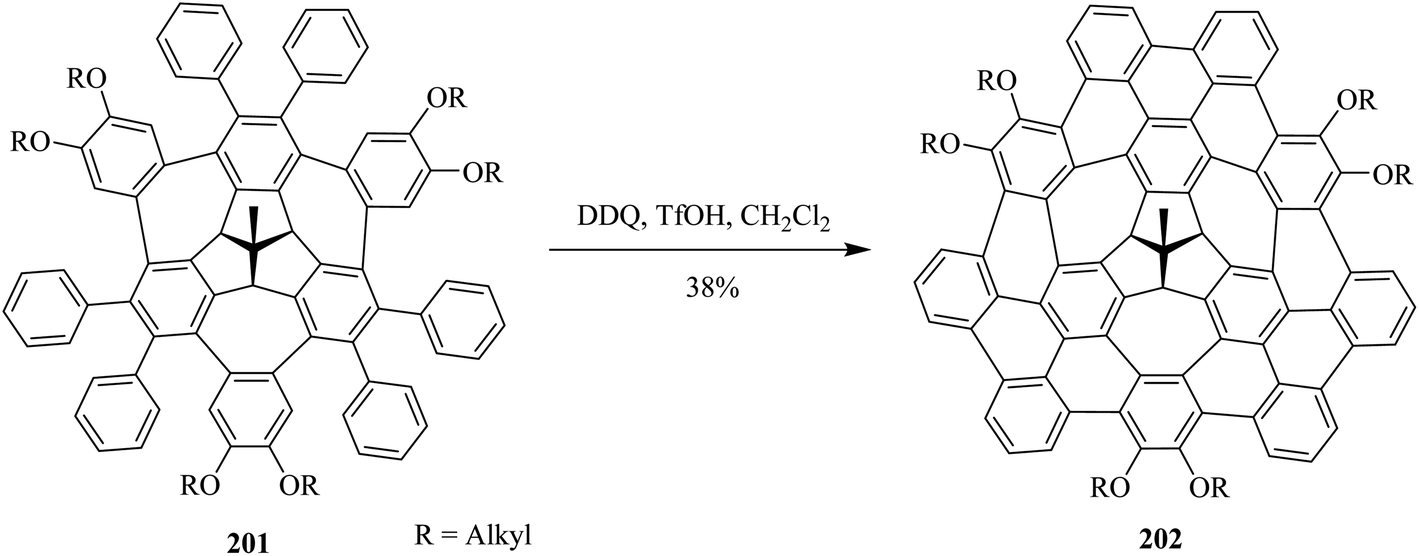 | ||
| Scheme 70 Synthesis of the π-extended nanographene from the aryloxy-substituted TBTQ via DDQ. | ||
The main challenge in the condensation of polyphenylenes into planar- and non-planar nanographenes lies in the complete removal of hydrogens in order to make multiple C–C bonds. In this context, several oxidant systems including iron chloride, vanadium chloride, and copper chloride have been used to achieve this goal. However, mostly these oxidants result in complex mixture of products. To circumvent these drawbacks, DDQ was used as an efficient, ecofriendly and easily recyclable alternative oxidant. It provided high yields and selective products under optimized conditions. The planar, non-planar, and porous nanographenes with extensive structures can be synthesized quite easily in less time using DDQ. It also promotes intramolecular multiple Scholl reactions. Other oxidants are metallic in nature, and thus hazardous for the environment and also lead to the formation of incomplete reaction. Therefore, DDQ is an effective and stable alternative oxidant to synthesize nanographenes from their precursors.
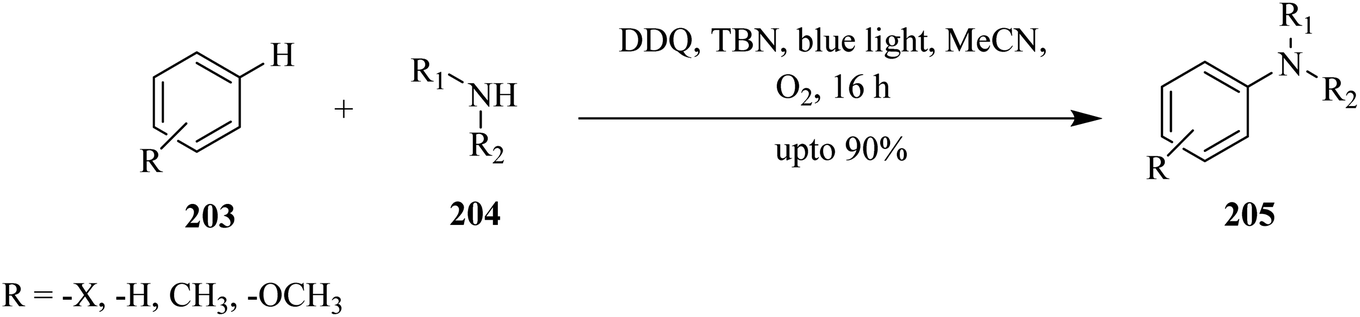 | ||
| Scheme 71 Amination of aromatic ring via DDQ under visible light irradiation. | ||
In 2017, Yuan and co-workers reported that DDQ acts as an effective catalyst for C–C bond formation reactions under visible-light irradiation. They described a DDQ-photocatalyzed direct trifluoromethylation of (hetero)arenes 206 using CF3SO2Na 207 as the trifluoromethyl 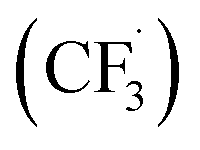 radical source under visible-light irradiation (Scheme 72). In this method, different benzoquinone derivatives including benzoquinone, methyl-p-benzoquinone, 2,6-dimethyl-p-benzoquinone and DDQ were tested. A wide variety of (hereto)arenes 206 with EWGs or EDGs gave the expected trifluoromethyl products 208 in moderate to good yields (Scheme 71).108
radical source under visible-light irradiation (Scheme 72). In this method, different benzoquinone derivatives including benzoquinone, methyl-p-benzoquinone, 2,6-dimethyl-p-benzoquinone and DDQ were tested. A wide variety of (hereto)arenes 206 with EWGs or EDGs gave the expected trifluoromethyl products 208 in moderate to good yields (Scheme 71).108
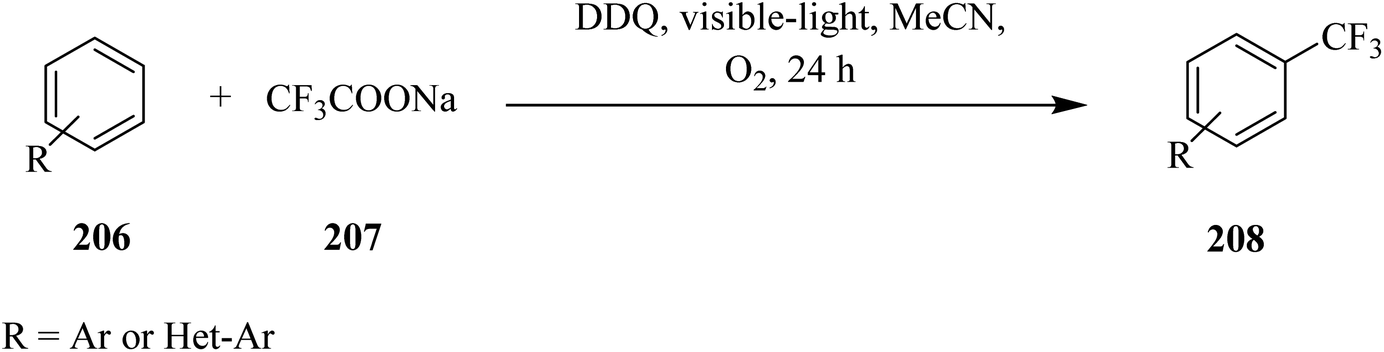 | ||
| Scheme 72 Radical trifluoromethylation of six-membered aromatics. | ||
In 2017, Song and co-workers reported a DDQ-photocatalyzed methodology for the direct C2–H amination of thiophene 209 by azoles 210 under visible-light irradiation (Scheme 72). This protocol has been found applicable to a series of thiophenes containing either EDGs (such as alkyl, trimethylsilyl or dioxolan) or EWGs (including chloride or bromide). They were selectively transformed into their corresponding amination products 211 in good yield. Moreover, various nitrogen sources like pyrazole, triazole, benzotriazole and their derivatives have shown to be tolerated and afforded the expected product in moderate to good yields (Scheme 73).109
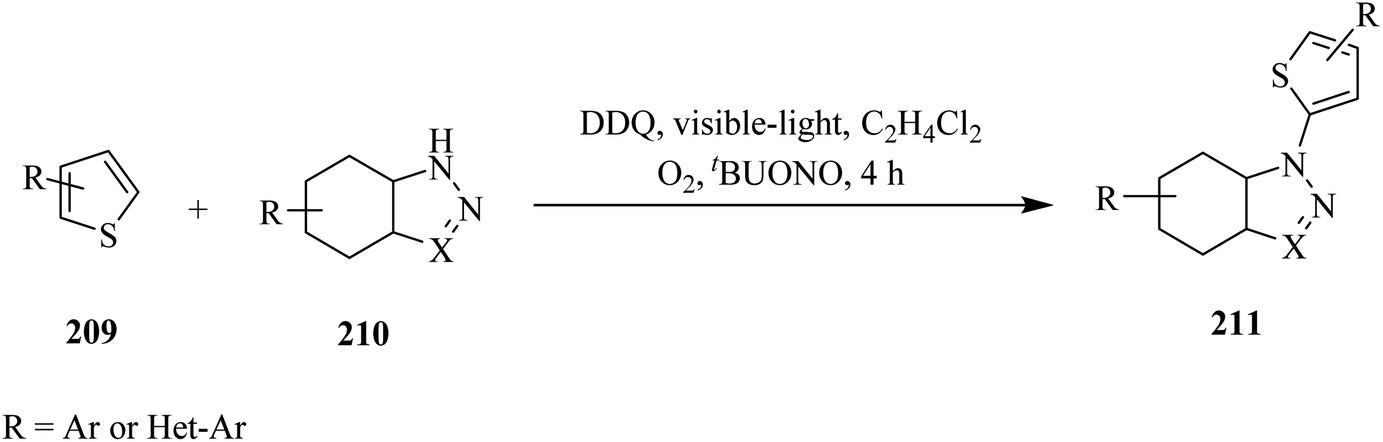 | ||
| Scheme 73 DDQ-photocatalyzed direct C2–H amination of thiophene under visible-light irradiation. | ||
In 2018, Natarajan and co-workers devised a unique protocol for the homocoupling reaction of readily available mono- and disubstituted olefins to afford symmetrical di- and tetra-substituted 1,3-butadienes in the presence of catalytic amounts of DDQ under visible-light-irradiation. The reaction works for a wide range of reactants giving 1,3-butadienes in moderate to good yields. Herein, they propose a reaction mechanism involving direct oxidative radical coupling followed by proton and hydrogen atom elimination. Primarily, a stoichiometric amount of DDQ was used as an oxidizing agent. Accordingly, a solution of ethene–1,1-diyldibenzene 212 (0.5 mmol) and DDQ (0.5 mmol) in anhyd. MeCN (5 mL) under a N2 atmosphere was irradiated with a 5 W blue LED at room temperature. After 10 hours, the desired product 1,1,4,4-tetraphenylbuta-1,3-diene 213 was obtained in moderate to good yield (Scheme 74).110
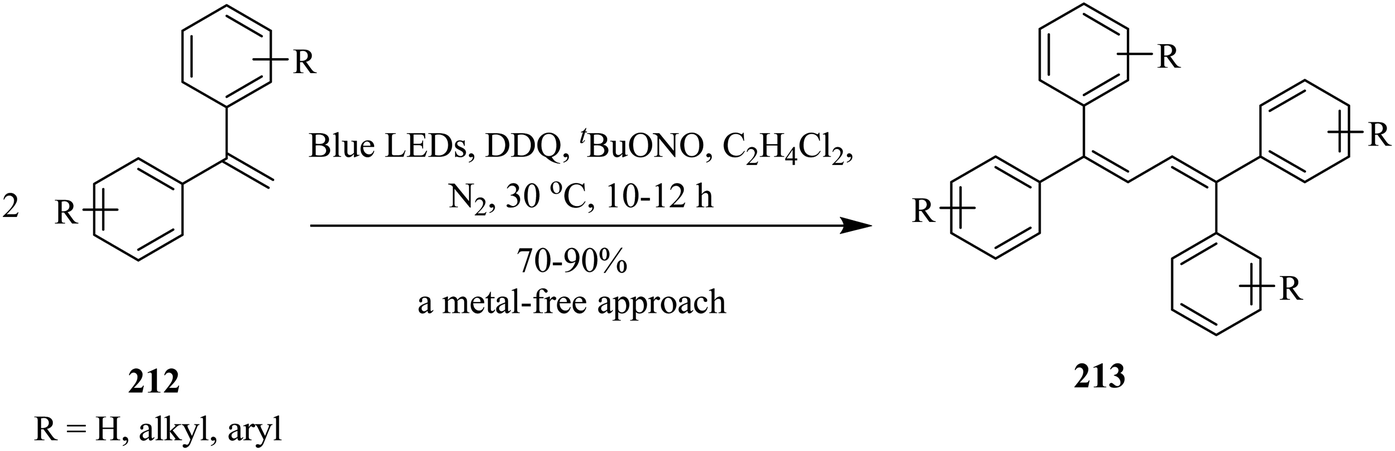 | ||
| Scheme 74 DDQ-catalyzed transformation of varyingly substituted 1,3-butadienes under visible-light-irradiation. | ||
In 2020, Sheridan and co-workers reported a protocol using DDQ for the C–F amination of fluoroarenes 214 with pyrazoles 215 under visible-light irradiation (Scheme 75). In this method, the authors used either trifluoroethanol (TFE) or a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of dichloroethane (DCE) and trifluoroethanol (TFE) as a reaction medium due to its ability to stabilize the radical cation intermediate of the substrate. Moreover, to avoid the direct reaction of pyrazole with DDQ, the authors added azole slowly throughout the reaction. However, it is likely that the DDQ*-assisted one-electron oxidation of fluoroarene to the corresponding radical cation intermediate followed by nucleophilic pyrazole attack and rearrangement gave the reaction products 216.111
:1 mixture of dichloroethane (DCE) and trifluoroethanol (TFE) as a reaction medium due to its ability to stabilize the radical cation intermediate of the substrate. Moreover, to avoid the direct reaction of pyrazole with DDQ, the authors added azole slowly throughout the reaction. However, it is likely that the DDQ*-assisted one-electron oxidation of fluoroarene to the corresponding radical cation intermediate followed by nucleophilic pyrazole attack and rearrangement gave the reaction products 216.111
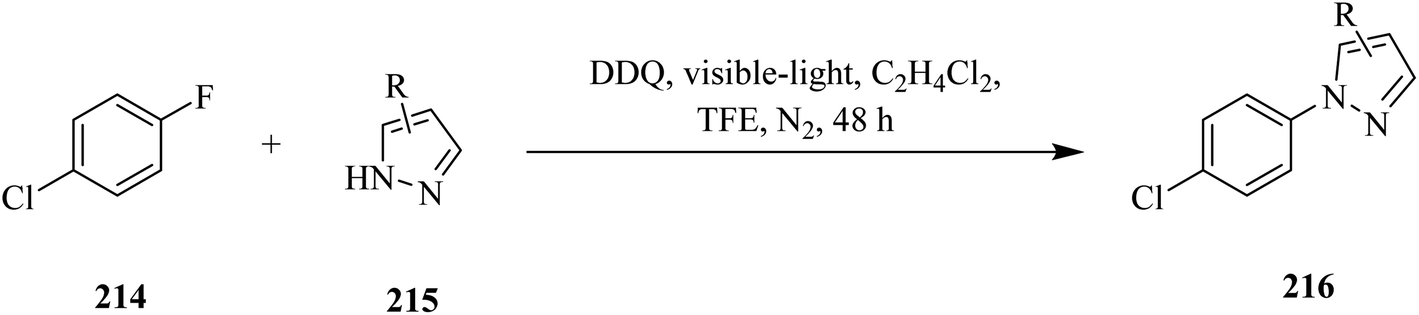 | ||
| Scheme 75 DDQ-assisted direct C–F amination of chlorofluoroarenes by pyrazoles under visible-light irradiation. | ||
In 2020, Cavedon and co-workers described a DDQ-assisted protocol for the oxidative debenzylation of benzyl ethers 217 under visible-light irradiation (Scheme 76). This protocol is applicable to a variety of substrates containing acetyl, benzoyl and isopropylidine protecting groups and smoothly deprotected them in less than 4 h in good yield. The authors pointed out that hydroxyethers 218, and several common protecting groups (that do not survive in a hydrogenolysis or Birch reduction) such as fluorenylmethoxy, carbonyl, levulinic ester, allyl carbonate, and propargyl carbonate, and benzylidene were tolerated by the photooxidative benzyl ether cleavage. Notably, green light irradiation was reported to be superior to blue light in suppressing the formation of side products during reactions. The control studies confirmed that light and DDQ are necessary for the reaction.112
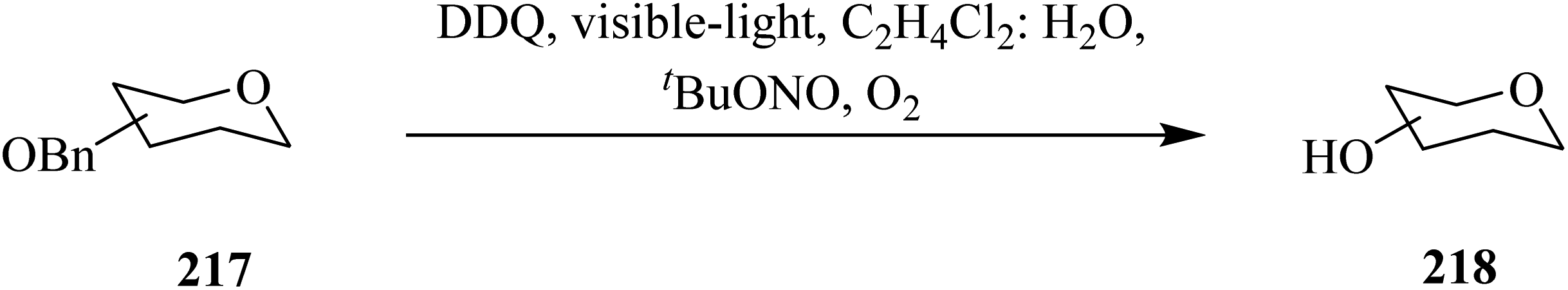 | ||
| Scheme 76 DDQ-assisted oxidative debenzylation of benzyl ethers under visible-light irradiation. | ||
DDQ, in combination with visible light, catalyzes several useful organic transformations like the photooxidative benzyl ether cleavage mentioned earlier. The ready availability, cheap price, lower toxicity, and good solubility in various solvents renders DDQ easy to handle and often leads to high product yields. The reagent enables functionalization of fluoroarenes of poor electronic nature and hydrocarbons. It is easier to oxidize and use it as photocatalyst. Owing to all these properties, DDQ is used in many synthetic pathways for small- and large-scale applications.
4. Conclusions & future perspectives
The commercially available DDQ plays an important role as an oxidant in numerous organic reactions. Throughout this review, we have showcased the facile and effective applications of DDQ in a wide range of organic transformations like oxidation of alcohols and ethers, oxidative cyclization and aromatization, oxidative protection and deprotection reactions, oxidative carbon–carbon bond formation reactions, dehydrogenation reactions, oxidation of phenols, amines, steroid ketones, and other such reactions. Additionally, we have focused on its uses in the condensation of polyphenylenes into planar and nonplanar nanographenes. Furthermore, we have also discussed the scope of DDQ oxidant in a vast range of organic chemical conversions. Most of the reactions known for C–H bond functionalization involve the use of aromatic compounds as starting material and only two examples used benzylic compounds and olefins. The use of aliphatic compounds as starting substrates in the DDQ-mediated synthesis of organic compounds awaits studies. DDQ, as a powerful oxidant, permits an effective metal-free oxidation. The methodology is quite simple and strong oxidant precludes the need of expensive transition-metal catalysts. It also minimizes the risk of environmental pollution by replacing poisonous metals. Its methodology is easy to handle and reactions often proceed at room temperature. Being a mild oxidizing agent, it promotes selective hydride-abstraction with good to excellent yields. That favours the synthesis of highly pure compounds and avoids the formation of side-products. DDQ is one of the most stable oxidants in acidic conditions. It is easily separable from the reaction mixture after the completion of reaction via ion-exchange separation technique. It can operate at room temperature, so no extra energy consumption is much needed. It is soluble in many solvents, such as DCM, dioxane, toluene, and acetic acid, facilitating its reactions. Noteworthy, polar solvents accelerate the rate of reaction with DDQ. We have seen so far how DDQ has broken all the taboos set by conventional oxidants. We anticipate that DDQ will be further explored and used in novel and selective transformations to design novel and highly selective complex organic compounds. Moreover, asymmetric reactions, improved reaction selectivity and larger-scale application are future challenges in DDQ catalysis.Abbreviations
| APEX | Annulative p-extension |
| Bn | Benzyl |
| BME | Benzyl methyl ethers |
| CBz | N4-Bz-cytidine |
| CDCs | Cross-dehydrogenative couplings |
| DCM | Dichloromethane |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| DMPM | 3,4-Dimethoxybenzyl |
| EOM | Ethoxy methyl |
| H-NMR | Proton nuclear magnetic resonance |
| HPLC | High-performance liquid chromatography |
| MeCN | Methyl cyanide |
| DMPM | 3,4-Dimethoxy benzyl |
| PAH | Polycyclic aromatic hydrocarbon |
| p-HBC | p-Hexabenzocoronene |
| PMB | p-Methoxy benzyl |
| TBDMSCl | tert-Butyldimethylchlorosilane |
| TBTQ | Tribenzotriquinacene |
| TEMPO | Tetramethyl piperidinyl 1-oxyl |
| TfOH | Triflic acid |
| UV-Vis | Ultraviolet-visible |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to the Deanship of Scientific Research at Umm Al-Qura University for financial support under grant code 19-SCI-1-01-0008. The authors are also highly grateful to the Higher Education Commission of Pakistan (HEC) for providing financial assistance under Project No. (NRPU-6484).References
- M. G. Clerici and P. Ingallina, Catal. Today, 1998, 41, 351–364 CrossRef CAS.
- Z. Shen, J. Dai, J. Xiong, X. He, W. Mo, B. Hu, N. Sun and X. Hu, Adv. Synth. Catal., 2011, 353, 3031–3038 CrossRef CAS.
- S. B. Bharate, Synlett, 2006, 205, 0496–0497 CrossRef.
- (a) J. J. V. Eynde, F. Delfosse, P. Lor and Y. Van Haverbeke, Tetrahedron, 1995, 51, 5813–5818 CrossRef; (b) J. Thiele and F. Günther, Justus Liebigs Ann. Chem., 1906, 349, 45–66 CrossRef CAS.
- D. Walker and T. D. Waugh, J. Org. Chem., 1965, 30, 3240 CrossRef CAS.
- M. Bouquet, A. Guy, M. Lemaire and J. Guette, Synth. Commun., 1985, 15, 1153–1157 CrossRef CAS.
- Q. Huang, B.-Z. Zheng and Q. Long, J. Chem. Sci., 2010, 122, 203–207 CrossRef CAS.
- T. Tomakinian, R. Guillot, C. Kouklovsky and G. Vincent, Angew. Chem., 2014, 126, 12075–12079 CrossRef.
- Y. Ding, Z. Huang, J. Yin, Y. Lai, S. Zhang, Z. Zhang, L. Fang, S. Peng and Y. Zhang, Chem. Commun., 2011, 47, 9495–9497 RSC.
- Y. Cui and P. E. Floreancig, Org. Lett., 2012, 14, 1720–1723 CrossRef CAS PubMed.
- A. Ali, F. Cheng, W.-H. Wen, X. Ying, J. Kandhadi, H. Wang, H.-Y. Liu and C.-K. Chang, Chin. Chem. Lett., 2018, 29, 1888–1892 CrossRef CAS.
- A. G. Slater, Y. Hu, L. Yang, S. P. Argent, W. Lewis, M. O. Blunt and N. R. Champness, Chem. Sci., 2015, 6, 1562–1569 RSC.
- E. S. Lewis, J. M. Perry and R. H. Grinstein, J. Am. Chem. Soc., 1970, 92, 899–905 CrossRef CAS.
- K. Tanemura, Y. Nishida and T. Suzuki, Bull. Nippon Dental Univ., 2011, 40, 31–34 CAS.
- W. Brown and A. Turner, J. Chem. Soc. C, 1971, 2566–2572 RSC.
- D. R. Buckle, S. J. Collier and M. D. McLaws, Encyclopedia of Reagents for Organic Synthesis, 2001 Search PubMed.
- P. Mitchell, Can. J. Chem., 1963, 41, 550–553 CrossRef CAS.
- J. Ma, Z. Hu, M. Li, W. Zhao, X. Hu, W. Mo, B. Hu, N. Sun and Z. Shen, Tetrahedron, 2015, 71, 6733–6739 CrossRef CAS.
- N. Iranpoor and I. M. Baltork, Tetrahedron Lett., 1990, 31, 735–738 CrossRef CAS.
- I. Paterson, C. J. Cowden, V. S. Rahn and M. D. Woodrow, Synlett, 1998, 98, 915–917 CrossRef.
- Z.-L. Wang, H.-L. Li, L.-S. Ge, X.-L. An, Z.-G. Zhang, X. Luo, J. S. Fossey and W.-P. Deng, J. Org. Chem., 2014, 79, 1156–1165 CrossRef CAS PubMed.
- P. Röse, S. Emge, C. A. König and G. Hilt, Adv. Synth. Catal., 2017, 359, 1359–1372 CrossRef.
- B. Bortolotti, R. Leardini, D. Nanni and G. Zanardi, Tetrahedron, 1993, 49, 10157–10174 CrossRef CAS.
- V. S. Batista, R. H. Crabtree, S. J. Konezny, O. R. Luca and J. M. Praetorius, New J. Chem., 2012, 36, 1141–1144 RSC.
- L. Zhai, R. Shukla and R. Rathore, Org. Lett., 2009, 11, 3474–3477 CrossRef CAS PubMed.
- (a) Y. Zhang and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 4242–4243 CrossRef CAS PubMed; (b) P. Natarajan and B. König, Eur. J. Org Chem., 2021, 1–18 Search PubMed.
- H. Lee and R. G. Harvey, J. Org. Chem., 1983, 48, 749–751 CrossRef CAS.
- T. R. Kasturi, S. K. Jayaram, J. A. Sattigeri, P. V. Pragnacharyulu, T. N. G. Row, K. Renuka, K. Venkatesan, N. S. Begum and N. Munirathinam, Tetrahedron, 1993, 49, 7145–7158 CrossRef CAS.
- K. Peng, F. Chen, X. She, C. Yang, Y. Cui and X. Pan, Tetrahedron Lett., 2005, 46, 1217–1220 CrossRef CAS.
- C. C. Cosner, P. J. Cabrera, K. M. Byrd, A. M. A. Thomas and P. Helquist, Org. Lett., 2011, 13, 2071–2073 CrossRef CAS PubMed.
- L. Wang, J. Li, H. Yang, Y. Lv and S. Gao, J. Org. Chem., 2012, 77, 790–794 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., 2015, 127, 260–264 CrossRef.
- Y. Hu, L. Chen and B. Li, Catal. Commun., 2018, 103, 42–46 CrossRef CAS.
- T. Katsina, L. Clavier, J.-F. Giffard, N. Macedo Portela da Silva, J. Fournier, R. Tamion, C. Copin, S. Arseniyadis and A. Jean, Org. Process Res. Dev., 2020, 24, 856–860 CrossRef CAS.
- A. Bhattacharya, L. M. DiMichele, U. H. Dolling, E. J. Grabowski and V. J. Grenda, J. Org. Chem., 1989, 54, 6118–6120 CrossRef CAS.
- E. Lee-Ruff and F. Ablenas, Can. J. Chem., 1989, 67, 699–702 CrossRef CAS.
- W. Wang, T. Li and G. Attardo, J. Org. Chem., 1997, 62, 6598–6602 CrossRef CAS.
- R. J. Fradette, M. Kang and F. West, Angew. Chem., Int. Ed., 2017, 56, 6335–6338 CrossRef CAS PubMed.
- H. Jo, A. H. Hassan, S. Y. Jung, J. K. Lee, Y. S. Cho and S.-J. Min, Org. Lett., 2018, 20, 1175–1178 CrossRef CAS PubMed.
- A. S.-K. Tsang, P. Jensen, J. M. Hook, A. S. K. Hashmi and M. H. Todd, Pure Appl. Chem., 2011, 83, 655–665 CAS.
- D. Waghray, C. de Vet, K. Karypidou and W. Dehaen, J. Org. Chem., 2013, 78, 11147–11154 CrossRef CAS PubMed.
- S. Nobusue, K. Fujita and Y. Tobe, Org. Lett., 2017, 19, 3227–3230 CrossRef CAS PubMed.
- M. Kawamura, E. Tsurumaki and S. Toyota, Synthesis, 2018, 50, 134–138 CrossRef CAS.
- A. Kar, B. Chakraborty, S. Kundal, G. Rana and U. Jana, Organic & Biomolecular Chemistry 2021 Search PubMed.
- Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 885–888 CrossRef CAS.
- Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 889–892 CrossRef CAS.
- Y. Oikawa, T. Tanaka, K. Horita, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1984, 25, 5393–5396 CrossRef CAS.
- K. Horita, T. Yoshioka, T. Tanaka, Y. Oikawa and O. Yonemitsu, Tetrahedron, 1986, 42, 3021–3028 CrossRef CAS.
- C. E. McDonald, L. E. Nice and K. E. Kennedy, Tetrahedron Lett., 1994, 35, 57–60 CrossRef CAS.
- P. B. Sampson and J. F. Honek, Org. Lett., 1999, 1, 1395–1397 CrossRef CAS.
- G. Sharma, Tetrahedron Lett., 2001, 42, 5571–5573 CrossRef CAS.
- J. Chang, K. Zhao and S. Pan, Tetrahedron Lett., 2002, 43, 951–954 CrossRef CAS.
- M. A. Rahim, S. Matsumura and K. Toshima, Tetrahedron Lett., 2005, 46, 7307–7309 CrossRef CAS.
- M. Guiso, C. Marra and F. Piccioni, Nat. Prod. Res., 2010, 24, 331–340 CrossRef CAS PubMed.
- Z. Shen, L. Sheng, X. Zhang, W. Mo, B. Hu, N. Sun and X. Hu, Tetrahedron Lett., 2013, 54, 1579–1583 CrossRef CAS.
- P. Kumar, S. K. Cherian, R. Jain and K. Show, Tetrahedron Lett., 2014, 55, 7172–7176 CrossRef CAS.
- K. Walsh, H. F. Sneddon and C. J. Moody, Tetrahedron, 2014, 70, 7380–7387 CrossRef CAS.
- P. Srishylam, A. R. Reddy, S. Banerjee, S. Penta and Y. S. Sanghvi, Tetrahedron Lett., 2017, 58, 2588–2591 CrossRef CAS.
- H.-D. Becker, J. Org. Chem., 1965, 30, 982–989 CrossRef CAS.
- H. D. Becker, J. Org. Chem., 1969, 34, 1203–1210 CrossRef CAS.
- K. Schofield, R. Ward and A. Choudhury, J. Chem. Soc. C, 1971, 2834–2837 RSC.
- S. Pradhan and H. J. Ringold, J. Org. Chem., 1964, 29, 601–604 CrossRef CAS.
- R. M. Böhme and M. A. Kempfle, Steroids, 1994, 59, 265–269 CrossRef.
- K. Chen, C. Liu, L. Deng and G. Xu, Steroids, 2010, 75, 513–516 CrossRef CAS PubMed.
- W. Zhang, D. Pan, A. Wu and L. Shen, Steroids, 2015, 96, 16–20 CrossRef CAS PubMed.
- E. Braude and R. Linstead, J. Chem. Soc., 1954, 3544–3547 RSC.
- D. Walker and J. D. Hiebert, Chem. Rev., 1967, 67, 153–195 CrossRef CAS PubMed.
- E. Braude, J. Chem. Soc., 1960, 3123 RSC.
- J. Bagli, P. Morand, K. Wiesner and R. Gaudry, Tetrahedron Lett., 1964, 5, 387–389 CrossRef.
- A. Cross, H. Carpio and P. Crabbé, J. Chem. Soc., 1963, 5539–5544 RSC.
- H. Dannenberg, Synthesis, 1970, 309, 74–81 CrossRef.
- S. G. Boots and W. S. Johnson, J. Org. Chem., 1966, 31, 1285–1287 CrossRef CAS.
- R. Cambie and V. F. Carlisle, J. Chem. Soc. C, 1970, 1706–1710 RSC.
- A. Bodenberger and H. Dannenberg, Chem. Ber., 1971, 104, 2389–2426 CrossRef CAS PubMed.
- J. Findlay and A. Turner, J. Chem. Soc. C, 1971, 23–29 RSC.
- T. Kasturi, E. Raghavan, S. Dev and D. Banerjee, Tetrahedron, 1966, 22, 745–752 CrossRef CAS.
- M. De Lucia, F. Mainieri, L. Verotta, M. Maffei, L. Panzella, O. Crescenzi, A. Napolitano, V. Barone, G. Appendino and M. d'Ischia, J. Org. Chem., 2007, 72, 10123–10129 CrossRef CAS PubMed.
- A. Creighton and L. Jackman, J. Chem. Soc., 1960, 3138–3144 RSC.
- H. Y. Meltzer, Neuropsychopharmacology, 1999, 21, 106–115 CrossRef.
- H. Y. Cho and L. T. Scott, Tetrahedron Lett., 2015, 56, 3458–3462 CrossRef CAS.
- S. Matsuura, M. Iinuma, K. Ishikawa and K. Kagei, Chem. Pharm. Bull., 1978, 26, 305–306 CrossRef CAS.
- C. G. Shanker, B. V. Mallaiah and G. Srimannarayana, Synthesis, 1983, 985, 310–311 CrossRef.
- Y. Hoshino and N. Takeno, Bull. Chem. Soc. Jpn., 1987, 60, 4468–4470 CrossRef CAS.
- L. Somogyi, Synth. Commun., 1999, 29, 1857–1872 CrossRef CAS.
- V. Arnaudinaud, B. Nay, S. Vergé, A. Nuhrich, G. Deffieux, J.-M. Mérillon, J.-P. Monti and J. Vercauteren, Tetrahedron Lett., 2001, 42, 5669–5671 CrossRef CAS.
- Y. K. Rao, S.-H. Fang and Y.-M. Tzeng, Bioorg. Med. Chem., 2005, 13, 6850–6855 CrossRef CAS PubMed.
- P. Müller, Helv. Chim. Acta, 1973, 56, 1243–1251 CrossRef.
- W. Zhang, H. Ma, L. Zhou, Z. Sun, Z. Du, H. Miao and J. Xu, Molecules, 2008, 13, 3236–3245 CrossRef CAS PubMed.
- L.-Y. Chen, S.-R. Li, P.-Y. Chen, H.-C. Chang, T.-P. Wang, I.-L. Tsai and E.-C. Wanga, Arkivoc, 2010, 11, 64–76 Search PubMed.
- O. R. Luca, T. Wang, S. J. Konezny, V. S. Batista and R. H. Crabtree, New J. Chem., 2011, 35, 998–999 RSC.
- X. Li, C. Li, B. Yin, C. Li, P. Liu, J. Li and Z. Shi, Chem. - Asian J., 2013, 8, 1408–1411 CrossRef CAS PubMed.
- L. Zhai, R. Shukla and R. Rathore, Org. Lett., 2009, 11, 3474–3477 CrossRef CAS PubMed.
- D. J. Jones, B. Purushothaman, S. Ji, A. B. Holmes and W. W. Wong, Chem. Commun., 2012, 48, 8066–8068 RSC.
- J. Luo, X. Xu, R. Mao and Q. Miao, J. Am. Chem. Soc., 2012, 134, 13796–13803 CrossRef CAS PubMed.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- F. Schlütter, T. Nishiuchi, V. Enkelmann and K. Müllen, Angew. Chem., 2014, 126, 1564–1568 CrossRef.
- A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- K. Y. Cheung, X. Xu and Q. Miao, J. Am. Chem. Soc., 2015, 137, 3910–3914 CrossRef CAS PubMed.
- H.-W. Ip, C.-F. Ng, H.-F. Chow and D. Kuck, J. Am. Chem. Soc., 2016, 138, 13778–13781 CrossRef CAS PubMed.
- W. S. Wong, C. F. Ng, D. Kuck and H. F. Chow, Angew. Chem., Int. Ed., 2017, 56, 12356–12360 CrossRef CAS PubMed.
- M. Stepien, E. Gonka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef CAS PubMed.
- L. He, C. F. Ng, Y. Li, Z. Liu, D. Kuck and H. F. Chow, Angew. Chem., Int. Ed., 2018, 57, 13635–13639 CrossRef CAS PubMed.
- L. Đorđević, D. Milano, N. Demitri and D. Bonifazi, Org. Lett., 2020, 22, 4283–4288 CrossRef PubMed.
- M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef CAS PubMed.
- S. H. Pun, C. K. Chan, Z. Liu and Q. Miao, Organic Materials, 2020, 2, 248–252 CrossRef CAS.
- H.-W. Ip, Y. Li, D. Kuck and H.-F. Chow, J. Org. Chem., 2021, 86, 5546–5551 CrossRef CAS PubMed.
- S. Das, P. Palani Natarajan and B. König, Chemistry, 2017, 23, 18161 CrossRef CAS PubMed.
- B. Chang, H. Shao, P. Yan, W. Qiu, Z. Weng and R. Yuan, ACS Sustainable Chem. Eng., 2017, 5, 334–341 CrossRef CAS.
- C. Song, H. Yi, B. Dou, Y. Li, A. K. Singh and A. Lei, Chem. Commun., 2017, 53, 3689–3692 RSC.
- D. Chuskit, R. Chaudhary, P. Venugopalan, B. König and P. Natarajan, Org. Chem. Front., 2018, 5, 3553–3556 RSC.
- T. Sheridan, H. G. Yayla, Y. Lian, J. Genovino, N. Monck and J. W. Burton, Eur. J. Org Chem., 2020, 20, 2766–2770 CrossRef.
- C. Cavedon, E. T. Sletten, A. Madani, O. Niemeyer, P. H. Seeberger and B. Pieber, Org. Lett., 2021, 23, 514–518 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |