
Emerging carbon shell-encapsulated metal nanocatalysts for fuel cells and water electrolysis
Jue-Hyuk
Jang†
a,
A. Anto
Jeffery†
b,
Jiho
Min
b,
Namgee
Jung
 *b and
Sung Jong
Yoo
*acd
*b and
Sung Jong
Yoo
*acd
aCenter for Hydrogen·Fuel Cell Research, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea
bGraduate School of Energy Science and Technology (GEST), Chungnam National University, 99 Daehak-ro, Yuseong-gu, Daejeon, 34134, Republic of Korea
cDivision of Energy & Environmental Technology, KIST school, University of Science and Technology (UST), Daejeon 34113, Republic of Korea
dKHU-KIST Department of Converging Science and Technology, Kyung Hee University, Seoul 02447, Republic of Korea
First published on 19th August 2021
Abstract
The development of low-cost, high-efficiency electrocatalysts is of primary importance for hydrogen energy technology. Noble metal-based catalysts have been extensively studied for decades; however, activity and durability issues still remain a challenge. In recent years, carbon shell-encapsulated metal (M@C) catalysts have drawn great attention as novel materials for water electrolysis and fuel cell applications. These electrochemical reactions are governed mainly by interfacial charge transfer between the core metal and the outer carbon shell, which alters the electronic structure of the catalyst surface. Furthermore, the rationally designed and fine-tuned carbon shell plays a very interesting role as a protective layer or molecular sieve layer to improve the performance and durability of energy conversion systems. Herein, we review recent advances in the use of M@C type nanocatalysts for extensive applications in fuel cells and water electrolysis with a focus on the structural design and electronic structure modulation of carbon shell-encapsulated metal/alloys. Finally, we highlight the current challenges and future perspectives of these catalytic materials and related technologies in this field.
 Jue-Hyuk Jang | Jue-Hyuk Jang received his PhD degree in Renewable Energy from KU-KIST Green School, Graduate School of Energy and Environment, Korea University in 2020. He has been working as a post-doc researcher at the Center for Hydrogen·Fuel cell Research of the Korea Institute of Science and Technology. His current research interests include nanomaterial-based electrocatalysts and membrane-electrode assemblies (MEAs) for electrochemistry. |
 A. Anto Jeffery | A. Anto Jeffery received his PhD in chemistry from Bangalore University, India in 2017. He has been working as a postdoctoral associate at the Graduate School of Energy Science and Technology in Chungnam National University, Korea under the supervision of Prof. Namgee Jung, since 2019. His research interests focus on the synthesis of novel electrocatalysts for fuel cells and water electrolysis. |
 Jiho Min | Jiho Min received his Bachelor's degree from Hanbat University in 2018 and pursued his Master's degree from Graduate School of Energy Science and Technology, Chungnam National University, Korea in 2020 under the supervision of Prof. Namgee Jung. He is currently pursuing his PhD studies at the Graduate School of Energy Science and Technology in Chungnam National University under the supervision of Prof. Namgee Jung. His main research interests focus on the synthesis/design of novel electrocatalysts for fuel cell applications. |
 Namgee Jung | Namgee Jung received his Ph.D. degree in the Department of Chemical and Biological Engineering from Seoul National University in 2012. He completed postdoctoral studies in Korea Institute of Science and Technology (2015). He moved to Chungnam National University in 2015 and became associate professor of the Graduate School of Energy Science and Technology in 2019. His research interests include novel electrocatalysts for electrochemical energy conversion systems such as fuel cells and water electrolysis. |
 Sung Jong Yoo | Sung Jong Yoo obtained his Ph.D. degree from the School of Chemical and Biological Engineering at Seoul National University in 2009 and moved to KIST (Korea Institute of Science and Technology) for postdoctoral research. He began his independent career in 2012 as a senior research scientist in the Fuel Cell Research Center at KIST. He is currently a principal scientist at the Center for Hydrogen & Fuel Cell Research of KIST. His current research interests include: (i) the influence of surface chemistry of metal nanoparticles on catalytic activities and stability, (ii) the design principles for oxygen reduction activity of oxide catalysts for fuel cells, and (iii) the catalytic activity trends of the oxygen reduction reaction for alkaline anion exchange membrane fuel cells. |
1. Introduction
Over the past decades, combustion-based energy technologies have continued to raise serious global concerns, having a significant impact on environmental pollution and climate change caused by tremendous fossil fuel consumption.1–4 Nevertheless, the global demand for energy is exponentially increasing for a better human life. To simultaneously solve the climate change issues and energy crisis, fuel cells have been intensively developed as one of the promising energy conversion devices. Fuel cells provide clean and efficient ways to convert chemical energy into electrical energy without causing environmental issues, while water electrolysis bridges the gap between fuel cell and energy storage technologies.5–7 Hydrogen energy conversion technologies based on fuel cells and water electrolysis are getting a lot of attention since they can be widely applied in various industries such as transportation and power generation.8–11In fuel cells, hydrogen gas is oxidized via the hydrogen oxidation reaction (HOR) at the anode, and oxygen gas is reduced via the oxygen reduction reaction (ORR) at the cathode, producing electricity and water.12–15 In contrast to this, in water electrolysis using an external power supply, pure hydrogen and oxygen gases can be electrochemically produced from water by hydrogen and oxygen evolution reactions, respectively, without emitting carbonaceous byproducts.16–18 Therefore, the water electrolysis reaction has the reverse mechanism of the fuel cell reaction.
In the past, hydrogen-based technologies have been used only for special and limited purposes, such as power sources for space shuttles due to their high cost.19,20 However, in recent years, fuel cell systems have become commercially available for applications in fuel cell vehicles (FCVs), residential power supplies, and power plants.21–23 Accordingly, the hydrogen production technology for fuel cell operation has been extensively studied.3,24–26 Despite this, to guarantee the performance of fuel cells and water electrolyzers, it is still necessary to develop a highly efficient and durable electrocatalyst, one of the key materials.
Precious metal-based nanoparticles (NPs) such as Pt and Ir have been commonly used in the electrochemical reactions in fuel cells and water electrolyzers.27–30 However, along with the high cost problems and the lack of noble metal reserves, the challenging goal of further improving their catalytic activity and durability remains. In addition, the development of non-precious metal-based catalysts has been urged to replace the Pt and Ir-based catalysts. For instance, Pt-based alloys have been studied to enhance the HER/ORR and HER performance.31–33 In addition, Ir- and Ru-based electrocatalysts have been used to reduce the overpotential for the OER in water electrolysis.34,35 However, precious metal-based alloy catalysts still suffer from dissolution and detachment of metal NPs during the electrochemical reactions.
Meanwhile, to solve the cost problem of noble metals, 3d transition metal (TM)- and carbon-based electrocatalysts such as metal–nitrogen–carbon (M–N–C) and heteroatom-doped graphene catalysts have been developed.36–40 Non-precious metal catalysts exhibit improved activity and durability; however, not only do they lack the number of active sites, but they also have non-ideal catalyst structures that are not suitable for practical application in compact electrochemical cells.41–43 In particular, a highly porous electrode structure is required for diffusion of reactants and products, but graphene-based M–N–C catalysts can be easily stacked on the electrode, thereby forming a high-density electrode structure.44–46
In recent years, to overcome the limitations of typical catalyst structures, carbon shell-encapsulated metal (M@C) NPs have been proposed as promising catalysts that can be practically used in electrodes.47–56 Metal NPs are encapsulated by a thin carbon shell through various carbon coating technologies such as chemical vapor deposition (CVD), the polymer coating method, the solvothermal method, and the high-temperature pyrolytic method. However, in M@C NPs, carbon shells play different roles or provide multi-functionality depending on the purpose. For instance, electrochemically active metal NPs such as Pt can be protected by a carbon shell, showing the high durability of the catalyst. On the other hand, the carbon shell directly acts as an active site due to the electronic structure modulated by the core metal NP. In some cases, M@C NPs exhibit high activity simultaneously against a variety of electrochemical reactions (e.g., HER and OER), demonstrating a bi-functional catalyst. Furthermore, the carbon shell with a finely tuned pore structure can be utilized as a molecular sieve layer with selective H2/O2 permeability under shut-down/start-up conditions during fuel cell operation.
In this review, starting with the classification of efficient carbon capsulation strategies, the important effects of the carbon shell on the physicochemical properties of M@C NPs are systematically described. Furthermore, the scientific achievements in the application of M@C NPs to water electrolysis and fuel cell reactions are introduced, and the effects of the carbon encapsulation on the electrochemical reactions are rationally explained. The most recent advances in M@C catalyst structures for water electrolysis and fuel cell applications are expected to have a significant impact on future work towards the development of new types of electrocatalysts.
2. Fabrication of carbon shell-encapsulated metal (M@C) nanocatalysts
2.1 Chemical vapour deposition (CVD)
CVD is one of the most common and reliable methods for synthesizing TM-based single and bimetallic M@C core–shell structured materials.57–66 A typical CVD method involves passing volatile carbon precursors on a metal-coated substrate. The carbon precursors are decomposed on the metal surface at high temperature, providing carbon sources to form a thin carbon shell at a controlled gas flow rate.67 The physicochemical property and morphology of M@C materials fabricated via CVD methods depend on several factors such as the gas flow rate, temperature, chamber pressure, carrier–gas type, and precursor material.68,69 For instance, in the fabrication of N-doped carbon shell-encapsulated NiMo NPs on 3D N-doped graphene using a two-step CVD process for the HER,70 pyridine was used as the carbon and nitrogen sources to form an N-doped carbon shell on NiMo NPs (Fig. 1a). Through the 1st CVD process, annealing NiMoO4 nanofiber under an Ar/H2 atmosphere along with pyridine followed by etching NiMoO4 yielded a porous 3D N-doped graphene substrate. In the 2nd CVD after Ni/Mo deposition on the substrate, well-dispersed NiMo@C NPs on the conductive graphene substrate were obtained.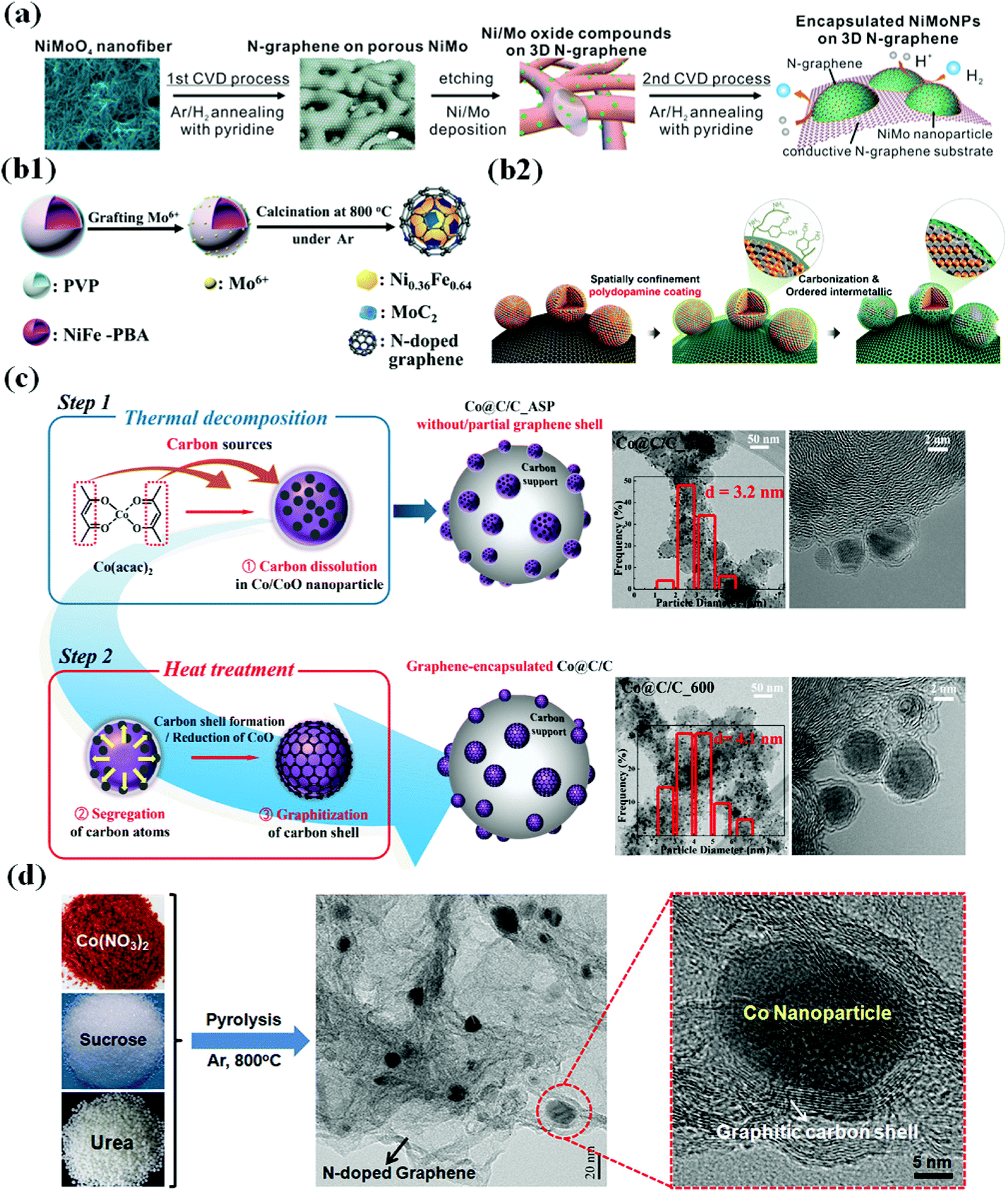 | ||
| Fig. 1 Schematic illustration of the synthesis of the metal/metal alloy encapsulated carbon shell by various strategies. (a) Preparation of N-doped graphene-encapsulated NiMo NPs via a two-step CVD approach, adapted from ref. 70 with permission from ACS, copyright 2018. (b1) Synthesis of MoC2-doped NiFe alloy NPs encapsulated within N-doped graphene via a polymer coating method, adapted from ref. 72 with permission from Elsevier, copyright 2018. (b2) Synthesis of carbon-supported and N-doped carbon-coated ordered intermetallic fct PtFe/C, reproduced with permission from ref. 51, ACS, copyright 2018. (c) TEM images of Co@G/C_ASP and Co@G/C_600 showing partial and complete encapsulation of Co NPs, reproduced with permission from ref. 47, RSC publications, copyright 2019. (d) Schematic illustration of the preparation of Co/N–C hybrids, adapted from ref. 92 with permission from Elsevier, copyright 2016. | ||
2.2 Polymer coating coupled thermal treatment
Polymer coating methods have been extensively developed particularly for synthesizing M@C materials composed of Earth-abundant metals.71–76 This method involves direct introduction of polymers (e.g., polyvinylpyrrolidone (PVP), polydopamine (PDA), polypyrrole, etc.) to the metal precursor solution at ambient temperature or at slightly elevated temperature, and the polymer-blended precursor solution is then subjected to pyrolysis or calcination/annealing at a desired temperature to yield carbon shell-coated metal NPs.68,69,71–77 The role of the polymers is to provide carbon sources for carbon shell formation on metal NPs. In addition, N-containing polymers can facilitate N doping on the outer carbon shell as well as carbon supports (CNTs, CNF, etc.), as depicted in Fig. 1b1.72 Alternatively, Sung et al. fabricated intermetallic ordered face-centered tetragonal (fct)-PtFe NPs with tunable N-doped carbon shell layers by thermal treatment of PDA-coated carbon-supported PtFe (PtFe/C) (Fig. 1b2). By controlling PDA coating time, the carbon shell thickness can be modified, and an appropriate carbon coating layer (<1 nm thickness) prevents aggregation, coalescence, or dissolution of PtFe NPs during fuel cell operation while simultaneously retaining high activity.512.3 Solvothermal coupled thermal treatment
Solvothermal methods to prepare M@C NPs have been of intense interest due to numerous advantages including a low-temperature process (<300 °C), morphology tuning, scale-up, less-time consumption, carbon shell engineering, and so on.47,48,78–83 Jung et al. have pioneered the synthesis of various TMs encapsulated by carbon shells including Earth-abundant TMs and noble metal/alloy-based catalysts such as Pt, Pd, Co, PtRu, PtFe, and PdAu,47,48,78–83 which involves decomposing metal precursors (e.g., metal acetylacetonates) in organic solvents with surfactants under inert atmospheres. The role of surfactants is to minimize particle agglomeration and control the size distribution and in some cases they are used as N-dopants.47,48,83 The decomposition temperature is kept below 300 °C, after which the products are processed and subjected to annealing under different gas conditions to yield different M@C structures. For example, Fig. 1c depicts the preparation of carbon supported graphene-encapsulated Co NPs (Co@G/C) for the ORR through a solvothermal route.472.4 High-temperature pyrolytic methods
A high-temperature pyrolytic method is also one of the prevalent methods to fabricate M@C core–shell materials.52,54,56,84–102 Pyrolysis methods involve the heat treatment of precursor compounds at high-temperatures (>500 °C) under inert gas atmospheres (e.g., N2 or Ar) wherein the precursor containing metal organic frameworks (MOFs) decompose to release simple volatile molecules (CO2, nitrates, sulphur oxides, NH3, volatile organic vapors, etc.), leaving a carbon material as a support (or frame) with carbon-encapsulated metals or alloys.56,68,69,84–88 In particular, pyrolysis is widely used for the synthesis of metal NPs with heteroatom-doped carbon framework catalysts,52,63,86,92,94,100 for example, calcination of carbon precursors containing metal species with hetero-dopant sources (melamine, H2S, urea, sodium hydrogen phosphate, etc.) (Fig. 1d).68,69,91,92 Inspired by the pioneering work of Jasinski's in 1964, wherein metal-phthalocyanines as metal macrocycles were converted to metal/N-doped carbon (M–N–C, M = Fe, Co, Ni) catalysts upon pyrolysis under inert conditions which were reported to show enhanced ORR performance,89 thereafter, the synthetic protocols of fabricating M–N–C based electrocatalysts were widely adopted and extended to fabricate various inexpensive M–N–C electrocatalysts, reported as the best alternatives to Pt-based catalysts.68,101,1023. Effects of the carbon shell and metal core on physicochemical properties of M@C catalysts
3.1 Electronic effects
The effect of core metals on the electronic structure of carbon shells is shown in Fig. 2. First of all, Kelly et al. reported the result of density functional theory (DFT) calculations for single graphene layer-coated metal surfaces.103 A graphene formed on TM substrates exhibits an n- or p-type character depending on the nature of the metal. For instance, graphene that strongly interacted with a Co, Ni, Pd, or Ti substrate has an n-type character while the work function of graphene on Pt or Au can change to a p-type due to weak interaction between them. Based on these results, for M@C NPs, the work function of the carbon shell is expected to be controlled depending on the type of core metal. Inspired by the previous studies, Jung et al. fabricated Pd@C and Co@C NPs with finely tuned carbon shells for the ORR in alkaline solutions.47,48 It has been clearly observed that electrons are transferred from the core metal to the carbon shell, forming an n-type graphene catalyst. The charge transfer between the core metal and carbon shell can be facilitated due to the large electronegativity difference and the short distance (strong interaction) between them. As a result, the work function of the carbon shell is reduced through strong interaction with the electron-donating metal NP, resulting in an electronic structure suitable for the ORR. The catalytic activity of the carbon shell can be further improved through doping of heteroatoms, mainly nitrogen.49,50,52,53 Chung et al. prepared N-doped carbon shell-coated PtFe NPs via polymer coating methods using N-containing polymers such as PDA followed by heat treatment for carbonization. The N-doped carbon shell provided additional active sites along with PtFe alloy NPs. By X-ray photoelectron spectroscopy (XPS), it was revealed that the structure of N-doped carbons (e.g., pyridinic N, graphitic N, pyrrolic N, etc.) has an effect on the activity of the carbon shell.51 However, it has been reported that the doping level is limited to less than 5 wt% since the doping process requires a high-temperature annealing enough to break the N–C bonds.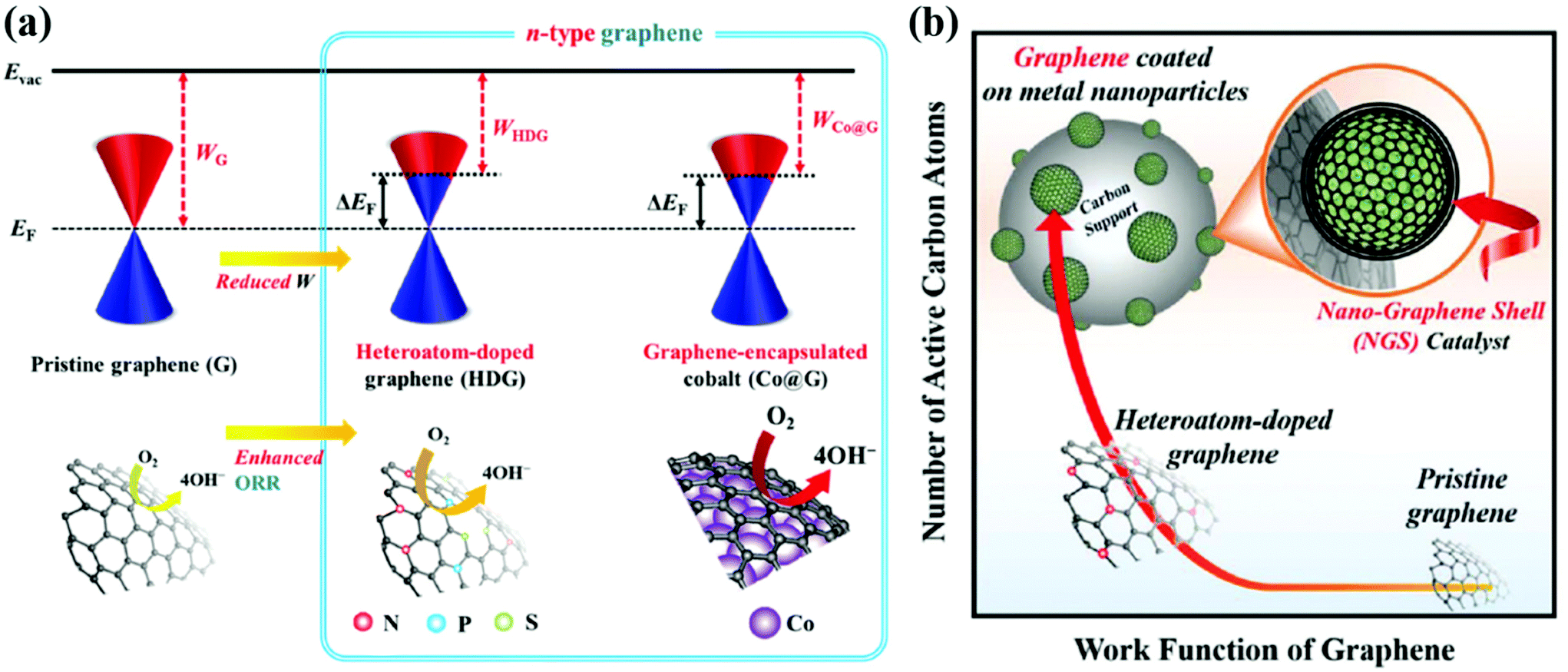 | ||
| Fig. 2 Schematic illustration of the electronic effects between the carbon shell and metal nanoparticles (a) Design concept to modify the work function of the carbon shell via Co encapsulation, adapted from ref. 47 with permission from RSC publications, copyright 2019. (b) Advantages of carbon shell encapsulated Pd nanoparticle catalysts, adapted from ref. 48 with permission from RSC publications, copyright 2019. | ||
3.2 Structural effects
It is well known that the thickness and porosity of the carbon shell are very important factors affecting the performance of M@C NPs for electrochemical catalysis. In addition, the stability of M@C NPs is strongly affected by the crystallinity of the carbon shell. During electrochemical reactions, the thicker the carbon shell and the more crystalline, the more durable, but less active. Chung et al. demonstrated the effects of the thickness of carbon shells on the ORR activity of Pt-based NPs. Highly porous carbon shells can also provide a sufficient number of active metal surfaces, but metal NPs inevitably suffer from dissolution of 3d TMs, causing performance degradation. If the carbon shell is the main active site, not the core metal, the carbon shell thickness should be very thin, less than ∼3 layers, since the charge transfer from the metal core to the carbon shell decreases exponentially as the thickness increases.51However, the finely controlled pore structure of the carbon shell improves reaction selectivity since it can provide a molecular sieve effect. For instance, a carbon shell with extremely small pores selectively allows the permeation of H2 gas rather than O2 due to the difference in the kinetic diameter. Using the molecular sieve effect by the carbon shell, Jung et al. developed HOR-selective Pt@C/C catalysts with high durability under shut-down-start-up conditions during fuel cell operation.80 Accordingly, fine control of the carbon shell structure is the most important technique for developing electrocatalysts suitable for each purpose (Fig. 3).
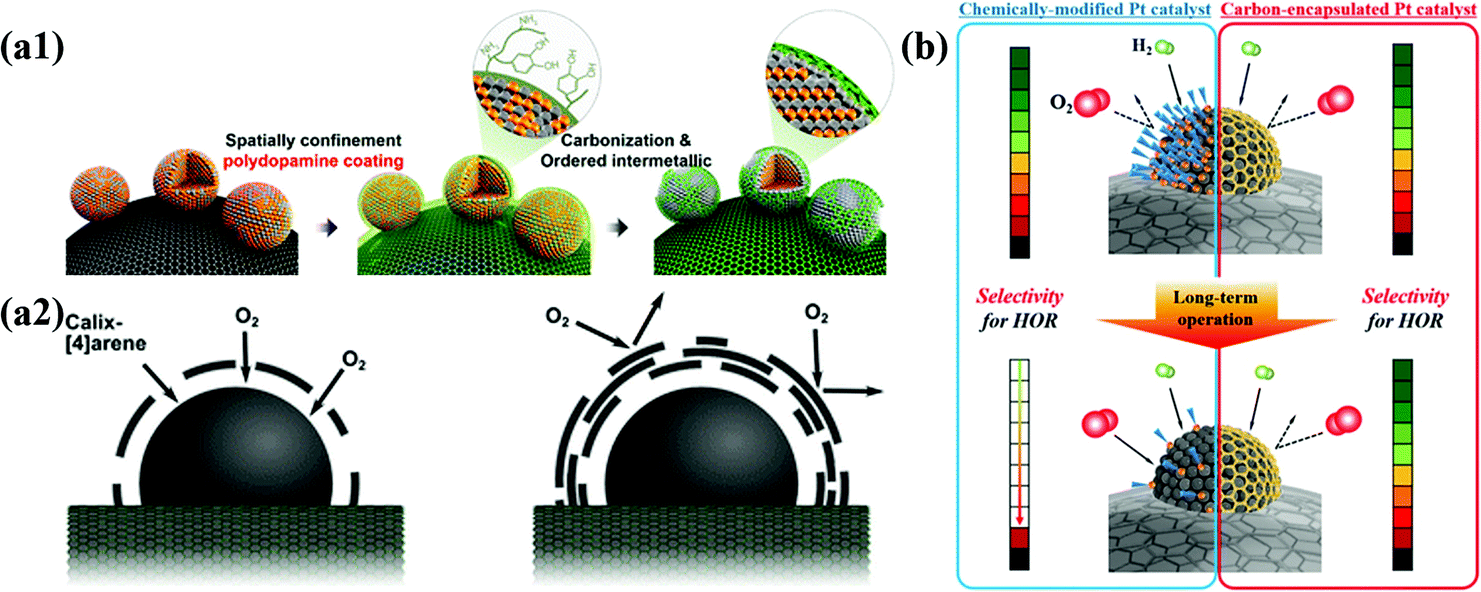 | ||
| Fig. 3 Schematic illustration of structural effects for carbon shell on metal nanoparticles (a1) Synthesis of N-doped carbon shell-coated ordered PtFe nanoparticles (a2) Effect of O2 permeability via carbon shell thickness for electrochemical activity, adapted from ref. 51 with permission from ACS, copyright 2015. (b) Schematic illustration of HOR selectivity change of Pt catalyst and carbon encapsulated Pt catalyst during PEMFC operation, with permission from ref. 80, ACS copyright 2019. | ||
4. Applications of metal@carbon core–shell catalysts to water electrolysis and fuel cell reactions
4.1 Direct utilization of carbon shells as active sites
Therefore, complete metal encapsulation by a carbon shell protects the TM NPs from harsh conditions because the embedded metal NP is not directly exposed to the electrolyte solutions. Furthermore, as supported by many experimental and theoretical calculation results,57,68,69,105–108 the electrochemical HER can be simultaneously triggered on the carbon surface due to the modification of the electronic structure by electron transfer from the core metal to the carbon shell. However, as mentioned earlier, the catalytic activity of M@C NPs strongly depends on the composition of the core metal and the thickness and crystallinity of the carbon shell.57,107 The HER performances of various carbon shell-encapsulated metal/alloys are summarized in Table 1.
| Catalysts | Synthetic method | Electrolyte | Overpotential (mV)@10 mA cm−2 | Tafel slope (mV dec−1) | Stability (cycle or hours) | Ref. |
|---|---|---|---|---|---|---|
| Co@NC | Hydrothermal-CVD | 0.5 M H2SO4 | 265 | 98 | 1000 | 57 |
| NiCu@C | Sol–gel/CVD | 0.5 M H2SO4 | 48 | 63 | 2000 | 61 |
| Fe@single shell | CVD | 0.5 M H2SO4 | 77 | 40 | 1000 | 66 |
| NiMo-G | CVD | 0.5 M H2SO4 | 56 | 49 | 24 h | 70 |
| Co@BCN | Hydrothermal-annealing | 0.5 M H2SO4 | 96 | 63.7 | 2000 | 71 |
| MoC2-doped carbon | Calcination | 1.0 M KOH | 320 | 31 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
72 |
| 3D Cu@N/CNTs | Pyrolysis | 1.0 M KOH | 123 | 63 | 10 h | 73 |
| FeCo@N-graphene | CVD | 0.5 M H2SO4 | 262 | 74 | 10000 |
76 |
| Co@N-doped carbon | Pyrolysis | 1.0 M HClO4 | 200 | 100 | 10000 |
86 |
| NiRu@N–C | Pyrolysis | 0.5 M H2SO4 | 50 | 36 | 1000 | 90 |
| FeCo@N-graphene | Pyrolysis | 1.0 M KOH | 233 | 102 | 1000 | 91 |
| IrNi@OC | Pyrolysis | 1.0 M KOH | 27 | 50 | 12 h | 96 |
| CoNi@NC | Hydrothermal-annealing | 0.5 M H2SO4 | 142 | 104 | 1000 | 113 |
| Ni2P@PCG | Solvothermal-annealing | 0.5 M H2SO4 | 110 | 59 | 5000 | 115 |
| PdCo@N-graphene | Pyrolysis | 0.5 M H2SO4 | 80 | 31 | 10000 |
117 |
| FeCo@Graphene | Pyrolysis | 1.0 M KOH | 149 | 77 | 10000 |
119 |
| Co@N-CNTs | Pyrolysis | 0.5 M H2SO4 | 260 | 69 | 8.5 h | 146 |
| Co@C | Hydrothermal-annealing | 1.0 M KOH | 90 | 91 | 5000 | 177 |
| CoFe@N-graphene | Pyrolysis | 1.0 M KOH | 272 | 96 | 1000 | 178 |
| FeCo@N–C | Pyrolysis | 0.5 M H2SO4 | 230 | 92 | 5000 | 179 |
| CoP/CO2P@NC | Pyrolysis | 0.5 M H2SO4 | 126 | 79 | 1000 | 192 |
| Ni@porous carbon | Carbonization | 0.5 M H2SO4 | 105 | 72 | 500 | 193 |
| FeCoNi@NC | Lyophilization-pyrolysis | 1.0 M KOH | 175 | 168 | 1000 | 194 |
| NiCo@N–C | Pyrolysis | 1.0 M KOH | 68 | 180 | 2000 | 195 |
| IrCo | Hydrothermal-annealing | 0.5 M H2SO4 | 24 | 23 | 10000 |
196 |
Laasonen et al. demonstrated this phenomenon in a chainmail type catalyst composed of Fe NPs completely encapsulated in graphene single shell layers (SCEIN) supported on single walled carbon nanotubes (SWCNTs) (Fig. 4a1), exhibiting high HER electrochemical activity on the graphene surface influenced by encapsulated Fe NPs with good stability in an acidic solution. It was revealed that the catalyst showed an onset potential of 0 V vs. RHE, which is close to the thermodynamic potential of the HER, and a low overpotential of 77 mV to reach j = 10 mA cm−2 almost similar to that of Pt/C (Fig. 4a2).66
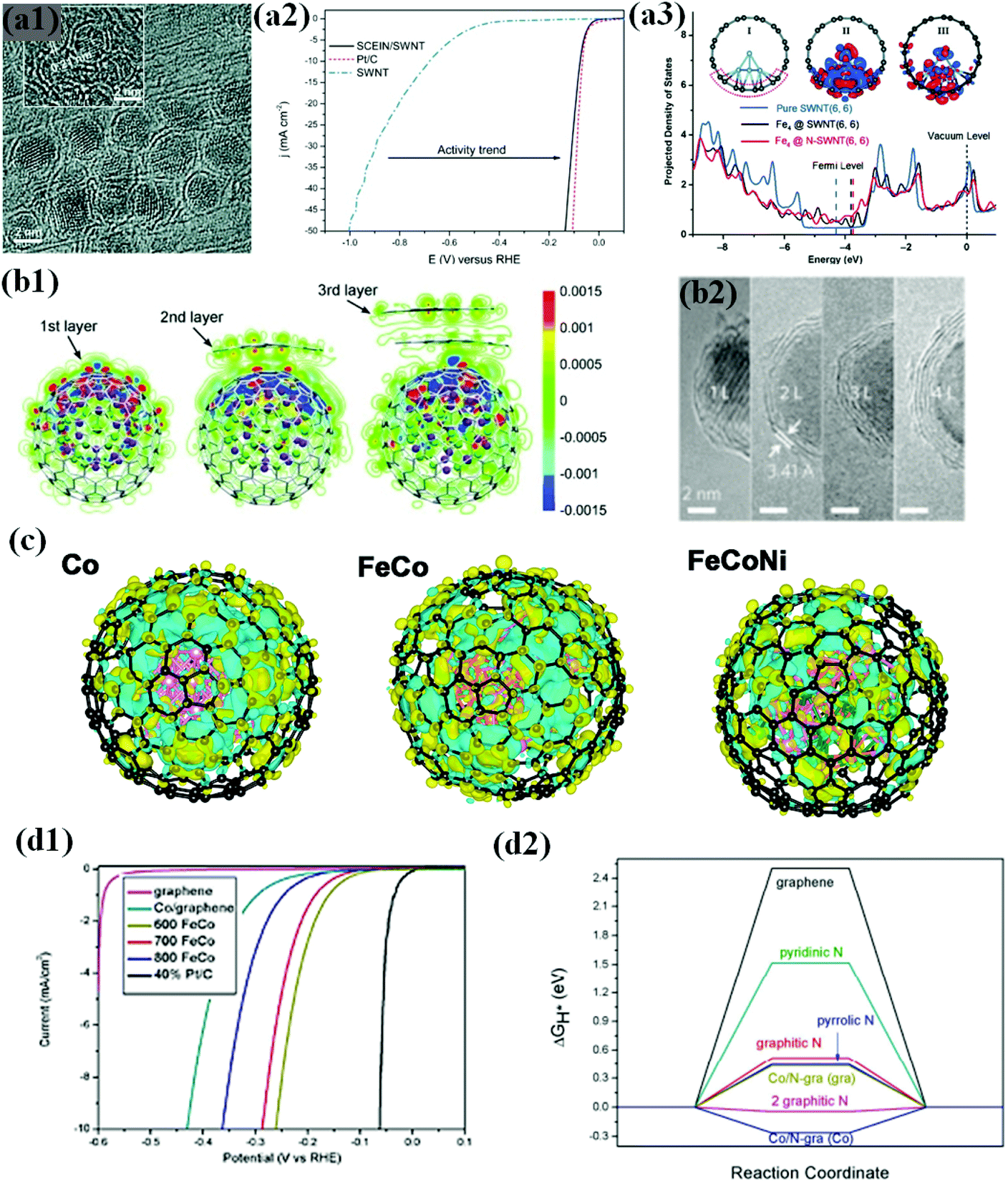 | ||
| Fig. 4 (a1) HRTEM image of the SCEIN/SWNT sample, the inset indicates the (110) lattice plane of the Fe particles in Science. (a2) Polarization curves of SWNT (blue), SCEIN/SWNT (black), and Pt/C (red). The polarization curves were measured at a scan rate of 50 mV s−1 in 0.5 M H2SO4, reproduced with permission from ref. 66, Wiley publications, copyright 2015. (a3) Projected DOS of the p orbitals of C atoms bonded to Fe4 in Fe4@SWNT and Fe4@N-SWNT compared with that in pure SWNT. Inserted plot I and plot II show the optimized structure of Fe4@SWNT and its difference charge density, and plot III corresponds to that of Fe4@N-SWNT. The red and blue regions in plots II and III indicate a charge increase and decrease, respectively, reproduced with permission from ref. 109, Wiley publications, copyright 2018. (b1) HRTEM images of CoNi@NC, showing the graphene shells and encapsulated metal NPs. Inset: Crystal (111) plane of the CoNi alloy. (b2) Redistribution of the electron densities after the CoNi clusters are covered by one to three layers of graphene. The red and blue regions are regions of increased and decreased electron density, respectively, reproduced with permission from ref. 113, Wiley publications, copyright 2015. (b3) High-resolution transmission electron microscopy (HRTEM) images of FeCo NPs covered by one to four layers of graphene (1–4 L), reproduced with permission from ref. 114, RSC publications, copyright 2013. (c) Calculated charge density differences of various models. The isosurface value of the color region is 0.01 e Å−3. The yellow and cyan regions refer to increased and decreased charge distributions, respectively. FeCoNi (Fe24Co24Ni7) refers to ternary alloys with lower and higher Ni contents, respectively, reproduced with permission from ref. 119, ACS publications, copyright 2017. (d1) Polarization curves of different FeCo annealed samples measured at a scan rate of 50 mV s−1 in 0.5 M H2SO4 solution. (d2) Calculated ΔGH* diagram of some models; the words in the bracket mean the H* adsorbed on whether graphene or Co4 side, reproduced with permission from ref. 76, RSC publications, copyright 2015. | ||
Experimental and theoretical investigations prove electron transfer from Fe to the graphene surface due to the difference in the work function, which increases the density of states (DOS) near the Fermi level of graphene and decreases the local work function of graphene (Fig. 4a3). Consequently, n-type graphene promotes the electrocatalytic reaction on the graphene surface.109–112 The electronic modulation of the graphene shell by inner metals/metal alloys has been realized in many catalytic systems ranging from Earth-abundant TMs to noble metal-like groups.69,90,113 Of note, there are several factors that can affect the electronic communication between metal and graphene, including graphene layer thickness, doped heteroatom, metal or alloy composition, and nanostructure of metal encapsulation.65
In particular, numerous studies have shown that the carbon shell thickness is tunable by optimising the preparation conditions, which in turn affects the number of electrons transferred and subsequent electronic alteration.61,104,105,107 To improve the HER activity of carbon-encapsulated NPs, the carbon shell thickness should not be more than 3 to 4 layers, i.e., electron penetration efficiency increases with decreasing layer thickness (Fig. 4b1 and b2).114 The best HER activity could be achieved by confining the NP in single shell encapsulation for efficient electron transfer.66,113 However, the single carbon shell might not provide high robustness and slowly tend to decline in activity, resulting in low stability during a long-term operation, while some studies indicate the formation of a partially oxidized state of the metal surface.
A number of DFT studies suggest that charge transfer takes place between the inner core and outer carbon shell, and the charge transfer can be further modified by alloying with heteroatoms caused by ligand or strain effects,71,115–118 which in turn affects the charge densities in the carbon network by the difference in the number of electrons transferred. Since electrocatalytic reactions occur at the carbon shell surface, the change in the electronic structure of the carbon shells can be evaluated by Bader charge analysis and several studies support the evidence that single metal, binary, and ternary alloy core materials exhibit different charge densities.59,61,68,76 Accordingly, the electronic structure of the outmost carbon shells also varies, affecting the binding energy of the reaction intermediates. For instance, Fig. 6c shows different charge densities of Co, FeCo, and FeCoNi encapsulated by the carbon shell. Among them, carbon shell-coated FeCoNi exhibits superior HER activity (η10: 149 mV vs. RHE) to their binary and single counterparts.119
One can also tailor the catalytic activity by changing the structure of the carbon shell. For example, doping of heteroatoms such as N, P, F, S, and B has proven to be effective in further tuning the electronic property and catalytic performance.71,75,76,115 Importantly, among the dopants, N doping in a carbon matrix is prevalent among core–shell structured materials due to numerous advantages.68,107,108
Doping N into a graphene matrix increases the DOS near the Fermi level and improves HER performance by decreasing ΔGH* close to zero which is the key indicator to probe HER activity.76 Intriguingly, increasing the N dopant level further maximizes HER performance by optimizing the electronic properties of graphene.76,104,105,107 Chen et al. found that an appropriate annealing temperature for the FeCo-MOF precursor resulted in a high doping level of N (8.2 at%) in carbon shell-coated bimetallic FeCo alloy NPs, exhibiting much enhanced HER activity with a low η10 of 262 mV vs. RHE (Fig. 4d1). Their DFT calculations indicated that an appropriate increase in N doping increases the H adsorption sites and decreases ΔGH*, thereby showing superior HER performance (Fig. 4d2).76 In addition, a few studies have observed a significant increase in HER activity by co-doping N and B in the carbon shell, leading to a more optimized electronic structure of the carbon surface and synergistically coupling with the encapsulated metal NPs to promote high HER activity.68,71,106
Utilizing carbon shells as active sites is not restricted to non-noble metals, but has been also studied in various carbon-coated noble metals and their alloy systems. Ultralow loading of noble metals with other non-noble metals can maintain intrinsic high activity and reduce material cost.98,116–118,120 In this regard, coupling noble metals like Ru with 3d TMs to form bimetallic alloys has been a promising way to fabricate highly active bimetallic catalysts, and it was revealed that the surface properties, charge distribution, strain and ligand effects can be also modified similarly to non-noble metal-based M@C catalysts.98,116–118
Su et al. reported high HER activity of RuCo alloy catalysts exceeding the activity of Pt/C, showing that they are economically advantageous over other Pt group metals. Their DFT calculations suggest that alloying Co with a small amount of Ru can enhance the HER activity comparable to a pure Ru catalyst through more electron transfer from the RuCo alloy core to the outer carbon shells and enhanced C–H bonds, thereby lowering ΔGH* for efficient catalytic activity.197 Similarly, Xu and co-workers fabricated NiRu alloys encapsulated by N-doped carbon, exhibiting superior HER activity close to Pt/C in acid solutions.100 Besides, Pd has a strong affinity for H adsorption; thus, Pd-based materials are widely used as primary catalysts for electrocatalytic HER applications. Therefore, Chen et al. designed N-doped graphene-encapsulated PdCo systems and reported that high conductivity of the graphene layer and N-doping enhanced the charge transfer and the number of active sites.117,121,122
| Catalysts | Synthetic method | Electrolyte | Overpotential (mV)@10 mA cm−2 | Tafel slope (mV dec−1) | Stability (cycle or hours) | Ref. |
|---|---|---|---|---|---|---|
| Porous CoNi@NC | Solvothermal | 0.1 M KOH | 500 | 66 | 1000 | 54 |
| FeCo@NC/NCNTs | Calcination | 0.1 M KOH | 355 | 57 | 60 h | 58 |
| FeNi@NC | CVD | 1.0 M NaOH | 280 | 70 | 10000 |
65 |
| MoC2-doped NiFe@C | Calcination | 1.0 M KOH | 320 | 31 | 10000 |
72 |
| FeCo@N-graphene | Pyrolysis | 1.0 M KOH | 380 | 99 | 1000 | 91 |
| Ni@N-doped C | Precipitation | 1.0 M KOH | 280 | 45 | 1000 | 94 |
| FeCoNi@graphene | Pyrolysis | 1.0 M KOH | 288 | 60 | 10000 |
119 |
| NiFe–N/C | Template-free pyrolysis | 0.1 M KOH | 320 | 44 | 2000 | 131 |
| NiFe@C | Electrical explosion | 1.0 M KOH | 274 | 57 | 5000 | 133 |
| NiCo@BC | Ultrasonic stripping | 1.0 M KOH | 309 | 62 | 1000 | 147 |
| NiFe@NCNFs | Electrospinning | 1.0 M KOH | 294 | 52 | 2000 | 148 |
| Co/FeC@NC | Polymer coating | 1.0 M KOH | 380 | 70 | 1000 | 151 |
| FeCo–N/PC | Solution-impregnation | 0.1 M KOH | 395 | 68 | 3000 | 171 |
| NiFe-G | Sol–gel/carbothermal | 1.0 M KOH | 280 | 43 | 10 h | 176 |
| Co@NC | Pyrolysis | 1.0 M KOH | 400 | 108 | 10000 |
186 |
| FeNi@C/NG | Calcination | 0.1 M KOH | 430 | 105 | 2000 | 190 |
| FeCo@NC | Ball milling | 0.1 M KOH | 440 | 90 | 1000 | 191 |
| FeCoNi@NC | Lyophilization-pyrolysis | 1.0 M KOH | 270 | 60 | 1000 | 194 |
| FeCo@NC | Electrospinning | 1.0 M KOH | 283 | 38 | 53 h | 198 |
| 3D NiCo@NCNTs | Pyrolysis | 0.1 M KOH | 41 | 177 | 1000 | 199 |
| FeCo@NG/NCNT | Ball milling | 1.0 M KOH | 450 | 77 | 5.5 h | 200 |
| FeCoNi@NC | Pyrolysis | 0.5 M KOH | 400 | 72 | 10 h | 201 |
Bao and co-workers attempted to find reliable evidence for the electronic interaction between carbon shells and encapsulated metal NPs through direct chemical imaging and spectral analysis using scanning transmission X-ray microscopy (STXM). Furthermore, they used X-ray absorption near edge structure (XANES) and computational simulation study to support the STXM results.128–130 In their study, the STXM chemical image (Fig. 5a1) shows Fe NP encapsulated pod-like carbon nanotubes (Pod-Fe) consisting of three different regions: Fe particle regions, thick CNT regions, and thin CNT regions, marked by red, green, and blue, respectively. In STXM XANES spectra for the three different regions, changes in the C 1s graphitic domains of π* (285.2 eV) and σ* (∼292 eV) were observed (Fig. 5a2). The increase in the intensity of the C 1s π* signal (285.2 eV) in the Fe particle region (red) implies that Fe NPs might tune the electronic structure of the carbon shells. As shown in the simulated XAS spectra (Fig. 5a4), it was revealed that the increase in the intensity of π* bonds of single-walled CNTs (SWCNTs) with encapsulated Fe was due to a decrease in the number of electrons in the anti-bonding orbital by electron transfer from Fe NPs to carbon shells. Furthermore, the electron transfer enhances the pre-edge signals at ∼283.5 eV of the C 1s peak (Fig. 5a2), and the corresponding mapping image (Fig. 5a3) shows white dots at the same location as the Fe NPs marked in red in Fig. 5a1. The analysis results clearly demonstrated the electronic interaction between Fe and the carbon shell which improves the adsorption of oxygen containing species (O, OH, OO, and OOH) as reaction intermediates for the OER.
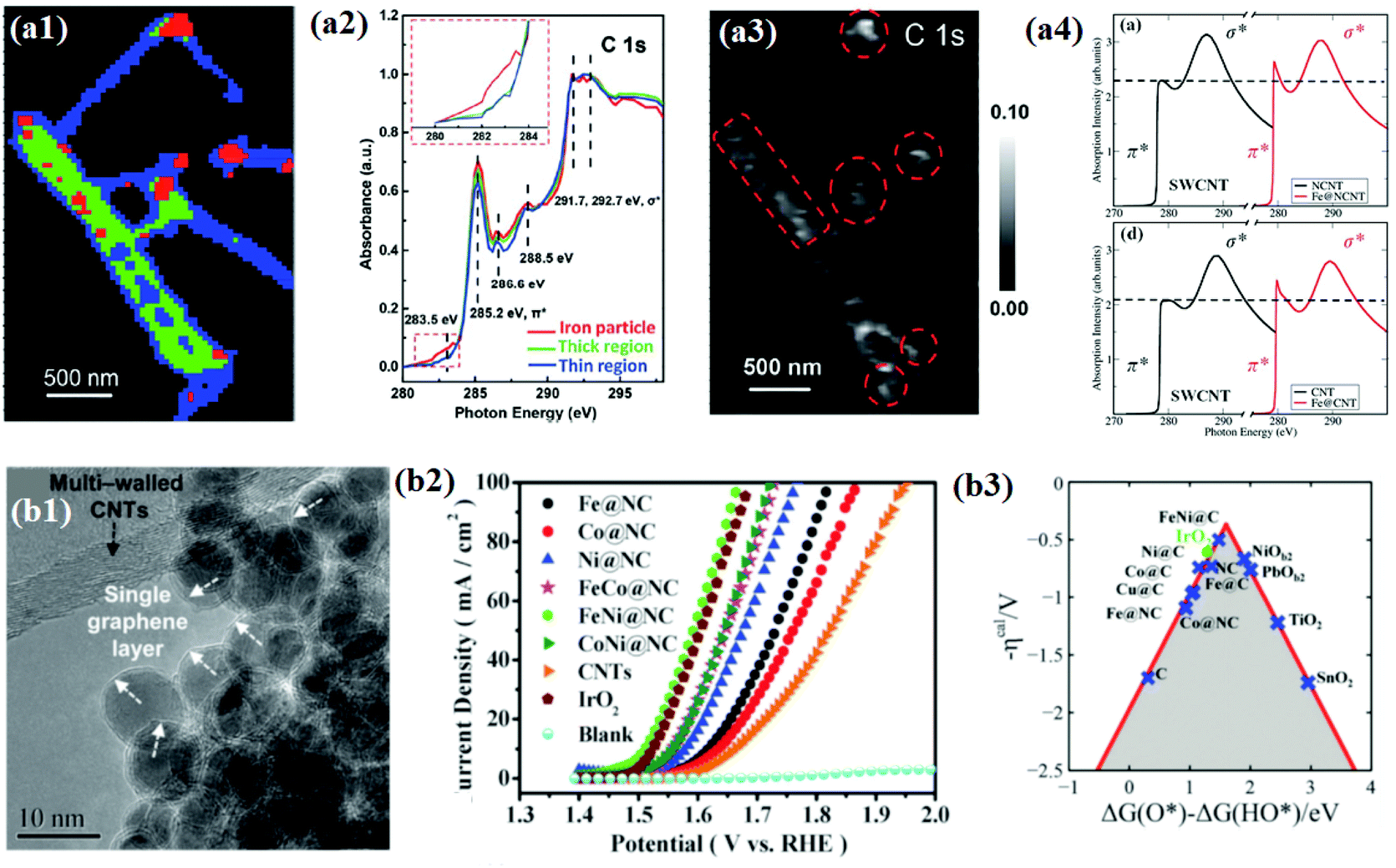 | ||
| Fig. 5 (a1) STXM chemical images of the pod-like carbon nanotube with encapsulated iron (Pod-Fe). Color composite regions, red: iron particle regions; green: thick CNT regions; blue: thin CNT regions. (a2) STXM XANES spectra and mappings of C 1s on Pod-Fe. Selected regions on the sample to extract normalized C 1s XANES spectra from the STXM stack (normalization range from the lowest absorbance to the highest), the inset shows the magnified image of the dashed red rectangle. (a3) Pre-edge peak map at 283.5 eV, using the pre-edge peak averaged images (from 282.5 to 283.5 eV) subtracting the pre-edge averaged images (from 280 to 281 eV). The red dashed circles indicate the main iron particles’ position. The scale bars on the right represent the absorbance intensity. (a4) Calculated X-ray absorption spectra of carbon atoms at the K-edge on CNTs with and without the Fe4 cluster with the N dopant and undoped SWCNT, reproduced with permission from ref. 128, RSC publications copyright 2015. (b1) Morphology of metal@NCs and the white arrows in the inset show the single layer graphene, and the black arrow in the inset shows the multi-walled carbon nanotubes. (b2) Electrocatalytic OER performance test of M@NCs in O2-saturated 1 M NaOH solution at 25 °C OER polarization curves for metal@NCs in comparison with CNTs and IrO2 with the same mass loading. (b3) Calculated negative overpotential (ηcal) against the universal descriptor ΔG(O*) − ΔG(HO*) on different catalysts, reproduced with permission from ref. 65, RSC publications copyright 2016. | ||
The understanding of the electronic interaction and the identification of the main active sites have led to the development of various 3d TMs and their alloys with core–shell structured materials for OER applications.65,105,107 The same group pioneered the synthesis of N-doped single layer graphene-encapsulated 3d TM NPs (M@NC) such as Fe, Co, Ni, and their alloys using a CVD process (Fig. 5b1). Electrochemical measurements of different types of M@NC catalysts revealed the OER activity trend in the order of FeNi@NC > CoNi@NC > FeCo@NC > Ni@NC > Co@NC > Fe@NC. In addition, for 10000 cycles, the optimized FeNi@NC catalyst exhibited superior stability compared to a commercial IrO2 catalyst in alkaline solutions (Fig. 5b2).65 Finally, the DFT calculation showed that by tuning the metal compositions, the OER activity descriptor, ΔG(O*) − ΔG(HO*), could be optimized and consequently the highest activity of the graphene layer could be found (Fig. 5b3). These observations conclude that the electronic transformation of the inert graphene shell also facilitates oxygen electrocatalysis, as observed for the HER.
Other research groups demonstrated a similar catalytic phenomenon,63,94,131–133 and some recent studies reported that apart from the graphene shell as active sites via modification of electronic structures, the in situ formed oxidized metal species synergistically contribute to OER activity.63,94,131,133
In recent years, M@C catalysts with an appropriate carbon shell structure have been proposed to improve the activity and durability of non-precious metal-based materials and to make a novel catalyst structure applicable to unit cells. Researchers have initially studied TM NPs encapsulated by an N-doped carbon shell and tried to highlight the improved ORR activities of the novel catalyst structure. Wang et al. fabricated carbon-encapsulated Co NPs (Co@G) deposited on N-doped graphitic carbon nanosheets (N-GCN), which exhibited a half-wave potential of 0.94 V comparable to that of Pt/C due to the optimized balance of graphitic N and pyridinic N sites in both the nanosheet and carbon shell.50 Similarly, Ning et al. synthesized CoNi@N–C via incorporating CoNi into ZIF followed by a high-temperature carbonization process.51 The enhanced ORR activity of CoNi@N–C was elucidated by the synergistic interaction between the N-doped carbon shell and the CoNi surface making active sites, CoNiNx.54 However, at that time, very few studies understood the carbon shell as the main active site since most researchers believed that carbon materials do not have catalytic activity for electrochemical reactions.
For the first time, to clearly identify the main active site of M@C catalysts, Gewirth et al. investigated the change in the electrochemical properties of a catalyst containing both Fe@C NPs and Fe–N–C sites depending on the annealing gas atmosphere (Cl2 and H2).87 Through the Cl2 treatment, the metallic Fe (Fe@C) and FeN4 (Fe–N–C) species are converted into FeCl3·xH2O that is reformed into reduced Fe species (Fe@C) by the H2 treatment. Interestingly, the ORR activity decreased significantly after the Cl2 treatment, but it recovered back to its initial state after the H2 treatment. Therefore, it was concluded that the FeN4 species do not affect the ORR activity, while the Fe@C NPs serve as the main active materials for the ORR. Furthermore, Jiang et al. synthesized N-doped carbon-encapsulated Fe3C–Co NPs, and conducted DFT calculations to unravel the mechanism for improved ORR activity of M@C catalysts.55 Combining with Fe3C–Co, the π-bonds between N–C and C–C of N-doped carbon shell are weakened, which makes the OOH* bond to the carbon shell stronger and leads to easier OOH formation.
In recent years, the importance of controlling the work function of the carbon shell through charge transfer between the core metal and the carbon shell has emerged. As shown in Fig. 6a, Jung et al. developed carbon-supported Pd@C (Pd@C/C) catalysts without doping of heteroatoms and revealed that the ORR can be promoted when the work function of the carbon shell is reduced by charge transfer from the core metal NP to the carbon shell.48 In addition, Yoo et al. reported a highly active and durable carbon-supported Co@C (Co@C/C) catalyst for the ORR (Fig. 6b), showing excellent AEMFC performance comparable to that of unit cells using Pt/C catalysts.47 The improved ORR activity of the Co@C/C catalyst was attributed mainly to the reduced work function of the carbon shell (n-type graphene shells) and the 3D catalyst structure with Co@C NPs highly dispersed on a carbon support, maximizing the electrochemically active surface area even in a unit cell. In the previous studies, complete encapsulation of Pd NPs by carbon shells accelerates the charge transfer, which has been clearly demonstrated in in-depth XPS studies. Fig. 7a also shows that the main active site of the Pd@C/C catalyst is only the carbon shell, not the Pd surface, via the Pd poisoning test using organic molecules with thiol groups. For the Co@C/C catalyst, the CN− adsorption/desorption (poisoning/washing) tests clearly prove that the active sites of the Co@C/C catalyst are the carbon shells, not the Co surface (Fig. 7b). Furthermore, it is demonstrated that the carbon shell can play a role of a protective layer to prevent dissolution of the core Co NPs during fuel cell operation (Fig. 6b2).
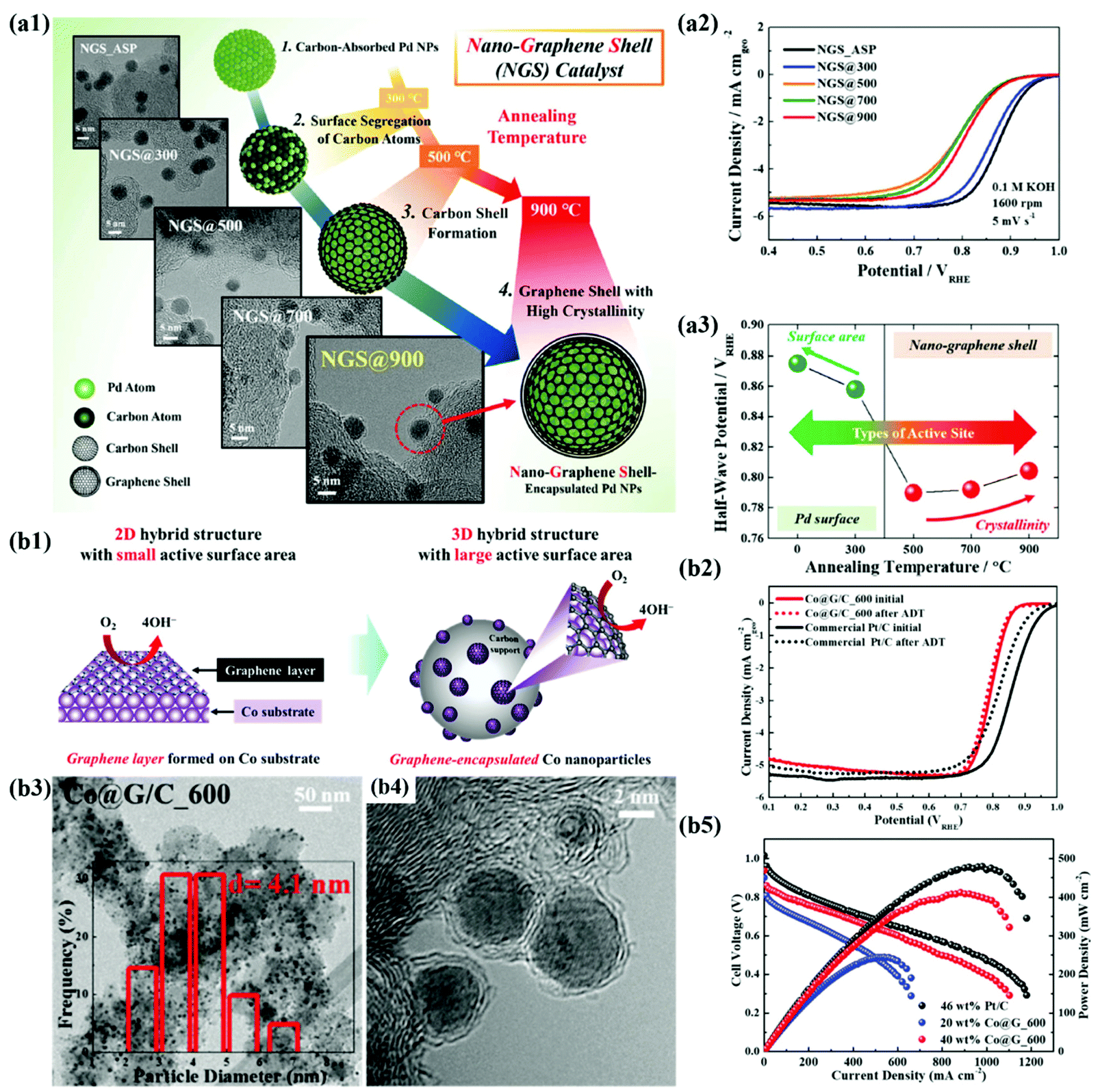 | ||
| Fig. 6 (a1) Structural changes of NGS catalysts based on TEM images at different annealing temperatures (300–900 °C). (a2) ORR polarization curves of NGS catalysts. (a3) Correlation between half-wave potential and heat treatment temperature, reproduced with permission from ref. 48, RSC publications copyright 2019. (b1) Schematic diagram of the maximized active site of Co@G/C via the 3D structure of the carbon shell (b2) ORR polarization curves of Co@G/C. (b3, 4) TEM images of Co@G/C_600. (b5) MEA performances of Co@G/C_600 in AEMFC, reproduced with permission from ref. 47, RSC publications copyright 2019. | ||
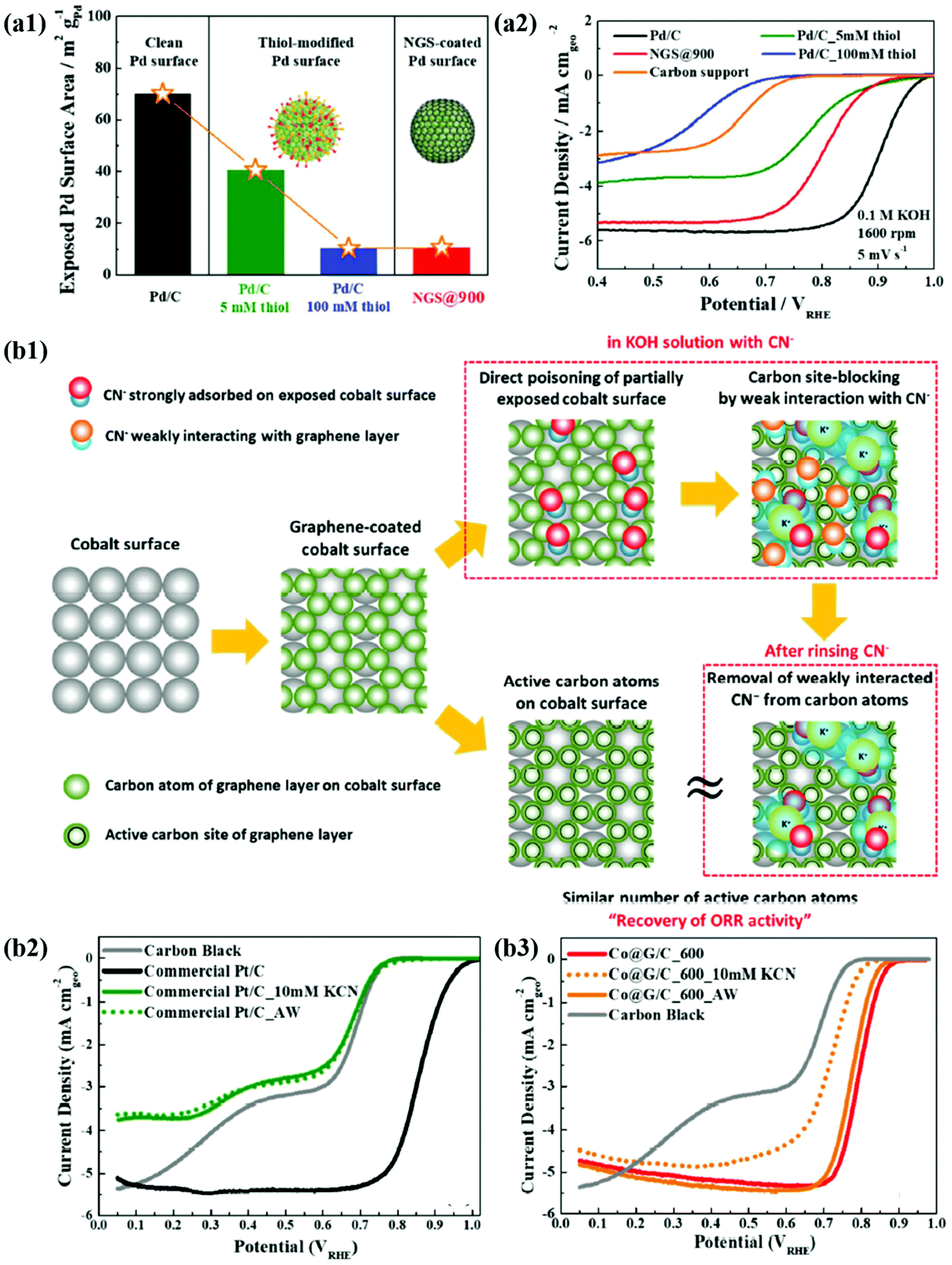 | ||
| Fig. 7 (a1) The exposed Pd surface area of Pd@C. (a2) ORR polarization curves of Pd/C, Pd/C-5 mM thiol, NGS@900, Pd/C_100 mM thiol, and carbon support, reproduced with permission from ref. 48, RSC publications copyright 2019. (b1) Schematic illustration of the mechanism for CN− effect on Co@G/C. (b2) ORR polarization curves of commercial Pt/C. (b3) Co@G/C in 0.1 M KOH with 10 mM KCN and after cleaning in pure 0.1 M KOH, reproduced with permission from ref. 47, RSC publications copyright 2019. | ||
Accordingly, M@C catalysts with highly active and durable carbon shells are considered promising ORR catalysts to overcome the disadvantages of M–N–C catalysts. Table 3 shows the recently reported electrochemical properties (ORR activity and durability) of M@C catalysts, mainly focusing on AEMFC applications.
| Catalysts | Synthetic method | Electrolyte | Overpotential (mV)@10 mA cm−2 | Tafel slope (mV dec−1) | Stability (cycle or hours) | Ref. |
|---|---|---|---|---|---|---|
| Co@G/C_600 | Solvothermal | 0.1 M KOH | 0.80 | — | 5000 | 47 |
| NGS@900 | Solvothermal | 0.1 M KOH | 0.87 | — | 5000 | 48 |
| O–Pd–Fe@NC/C | Pyrolysis | 0.1 M KOH/0.1 M HClO4 | 0.91/0.88 | — | 30000/10000 |
49 |
| Co@G/N-GCNs | Pyrolysis | 0.1 M KOH | 0.84 | 70 | 2000/10 h | 50 |
| Fct–PtFe/C | Solvothermal | 0.1 M HClO4 | 0.93 | — | 10000 |
51 |
| PdCo@NPNCs | Pyrolysis | 0.1 M KOH | 0.90 | 49 | 5.6 h | 52 |
| Pd-PANI-K@500 | Pyrolysis | 0.1 M HClO4 | 0.79 | — | 300 | 53 |
| Co2Ni1@N–C | Calcination | KOH | 0.82 | 63 | 4.2 h | 54 |
| Fe3C–Co/NC | Pyrolysis | 1 M KOH | 0.885 | — | 16.7 h | 55 |
| NFC@Fe/Fe3C-8 | Ball milling | 0.1 M KOH/0.1 M HClO4 | 0.87/0.73 | 80/44 | 50000/30000 |
75 |
| Fe/C_H2-treated | Solvothermal | 0.1 M HClO4 | 0.63 | — | — | 87 |
| Fe@N–C-700 | Pyrolysis | 0.1 M KOH | 0.83 | — | 30000 |
127 |
| UsPtCu@C | Pyrolysis | 0.1 M KOH | 0.87 | — | 5000 | 160 |
| Co3O4@C–N | Solvothermal | 0.1 M HClO4 | 0.37 (Ag/AgCl) | — | 1000 | 163 |
| CoNi/NG | Pyrolysis | 0.1 M KOH | 0.85 | 51 | 12 h | 202 |
| Ni@NCNTs-800 | Pyrolysis | 0.1 M KOH | 0.83 | 84 | 2000 | 203 |
| Fe30@N/HCSs | Pyrolysis | 0.1 M KOH | 0.85 | 62 | 8.3 h | 204 |
| 3C-900 | Pyrolysis | 0.1 M KOH | 0.82 | — | 4.4 h | 205 |
4.2 Highly durable catalysts encapsulated by carbon shells as protective layers
Some previous studies demonstrate that chainmail catalysts composed of non-precious earth-abundant metals/alloys supported on CNTs trigger electrochemical reactions on the outer carbon shell surface during water electrolysis, while carbon layers protect the metals against acid leaching/dissolution, thus enhancing durability in an acidic medium.109,113,114 In the same context, Deng et al. developed highly active and durable Fe, Co, and FeCo alloy NPs encapsulated in CNTs through a CVD method (Fig. 8a1).59 In their work, the optimized FeCo NPs encapsulated in N-doped CNTs (FeCo@NCNTs) show high HER activity even in H2SO4 solution (Fig. 8a2). The FeCo NPs completely coated by rigid graphitic carbon shells are protected against surface oxidation and particle migration during a long-term durability test of 10000 cycles, without any noticeable decline in HER activity (Fig. 8a3). Yang et al. observed similar results for FeCo@NC prepared by direct carbonization of the MOF precursor, Fe3[Co(CN)6]2.76 These studies provide insights into the electrochemical stability of Earth-abundant M@C type catalysts under acidic conditions during long-term operation.
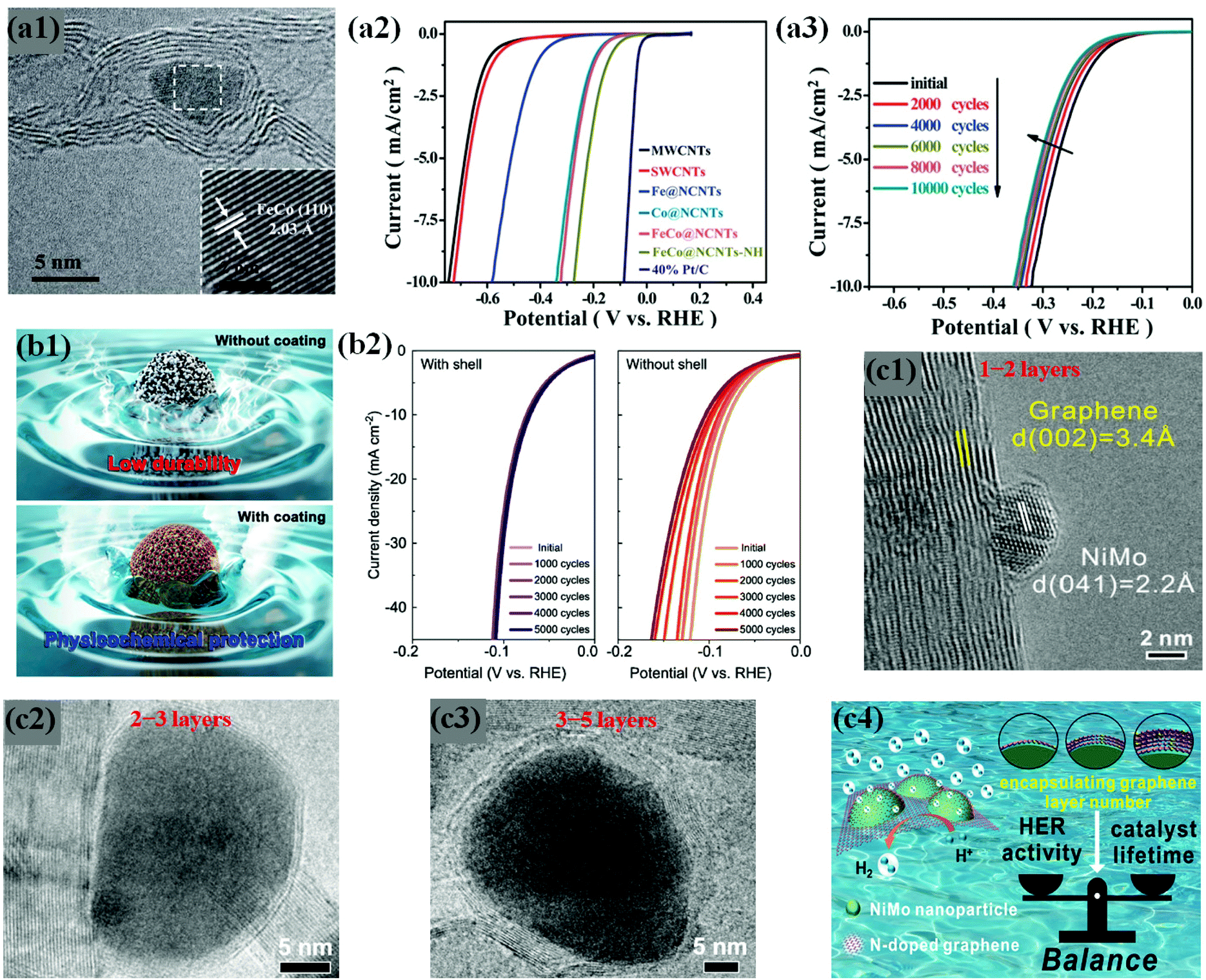 | ||
| Fig. 8 (a1) HRTEM image of FeCo@NCNTs with the inset showing the (110) crystal plane of the FeCo nanoparticle. (a2) Polarization curves of Fe@NCNTs, Co@NCNTs, FeCo@NCNTs, and FeCo@NCNTs–NH along with MWCNTs, SWCNTs and 40% Pt/C for comparison. (a3) Durability measurement of FeCo@NCNTs: polarization curves recorded initially and after every 2000 CV sweeps between +0.77 and _0.18 V (vs. RHE) at 100 mV s−1. All the polarization curves were recorded in 0.1 M H2SO4 at a scan rate of 2 mV s−1, reproduced with permission from ref. 59, RSC publications copyright 2014. (b1) Schematic illustrations of FeP NPs with and without carbon shell coating. (b2) Polarization curves for FeP/C with and without a carbon shell and commercial Pt/C electrocatalysts. The corresponding Tafel plots are shown in the inset. Data from control experiments with the samples with no NPs (“C with shell”) and iron oxide NPs instead of FeP (“FeOx with shell”) are shown together. Electrochemical performance for the HER is measured in 0.5 M H2SO4 solution with a rotating ring disk electrode at a rate of 2000 rpm. (b3) Long-term durability test of FeP/C electrocatalysts showing polarization curves for 5000 cycle tests of FeP NPs with (left) and without (right) a carbon shell, adapted with permission from ref. 74, ACS publications, copyright 2017. (c1) Typical HRTEM image of the NiMo NP encapsulated by a monolayer graphene sheet on 3DNG. (c2) High-resolution TEM images of 3DNG supported NiMoNPs encapsulated by 2–3 layers. (c3) High-resolution TEM images of 3DNG supported NiMoNPs encapsulated by 3–5 layers. (c4) Graphics depicting the importance of appropriate carbon shell layers, which provides the balance between HER activity and catalyst lifetime, adapted with permission from ref. 70, ACS publications, copyright 2018. | ||
Using different types of active materials, Chung and co-workers have revealed the importance of the carbon shell to enhance the durability by both theoretical and experimental works. In their work, carbon shell (<1 nm thickness)-coated FeP NPs are supported on a carbon support (FeP/C) by single step heating of polydopamine (PDA)-coated iron oxide with NaH2PO2, which exhibited high HER activity comparable to that of FeP/C without the carbon shell (Fig. 8b1). The durability tests for FeP/C with the carbon shell in acid media proved no significant activity loss for up to 10000 potential cycles, while FeP/C without the carbon shell indicated an increase in overpotential to ∼100 mV even after 5000 cycles. Therefore, the role of the carbon shells in protecting FeP NPs from activity loss is clearly demonstrated (Fig. 8b2).74
Based on a number of recent studies, single-layer encapsulation of NPs affords excellent activity but less chemical stability. In contrast, a thick carbon shell (>15 layers) may act as a complete protective layer but impede the efficient adsorption of the reactants to the active material, resulting in poor activity.71,76,114,115,117 Therefore, it is very essential to simultaneously achieve excellent activity and durability for sustainable electrochemical hydrogen production.
Hu et al. have attempted to overcome this problem by controlling the carbon shell thickness to balance the HER performance and durability. They fabricated NiMo bimetallic alloy NPs encapsulated by N-doped graphene layers supported on a 3D N-doped porous graphene network (NiMoNPs/3DNG) through a CVD process. As expected, the HER activity and chemical stability of NiMoNPs/3DNG depended on the number of graphene layers, showing that 1–2 and 2–3 graphene layers exhibited better HER activity but poor stability, while 3–5 layers showed slightly lower HER activity but better durability against metal dissolution under acidic conditions for 24 hours (Fig. 8c1–4).70 Interestingly, the concept of carbon shell protection has been pronounced in abundant studies, regardless of the nature of metal core NPs.61,71,106,113–115,117,146 However, the core metal/alloy composition is a crucial parameter for the development of highly efficient and durable HER catalysts, which requires deep understanding of physicochemical and electronic properties through experimental and theoretical studies. Several HER stability studies of carbon shell-encapsulated metals/alloys are listed in Table 1.
In view of the above challenges, L. Du et al. attempted to synthesize a highly durable N-doped carbon shell encapsulated NiFe OER catalyst through pyrolysis of NiFe-MOFs.63 The optimized catalyst NiIIFeIII@NC had spherical particles shielded by 6–12 carbon layers with a high degree of graphitization (DOG). OER activity of alloyed NiIIFeIII@NC and NiIIFeII@NC catalysts with different Ni/Fe ratios is superior in alkaline solution to that of individual counterparts (Fig. 9a1). XANES and EXAFS studies of NiFe@NC spectra reveal that the encapsulated Ni and Fe have separate roles that boost the activity. Ni was found to tune the local and electronic structure through the strain effect and triggers inert graphite, whereas Fe contributes to electron transfer.
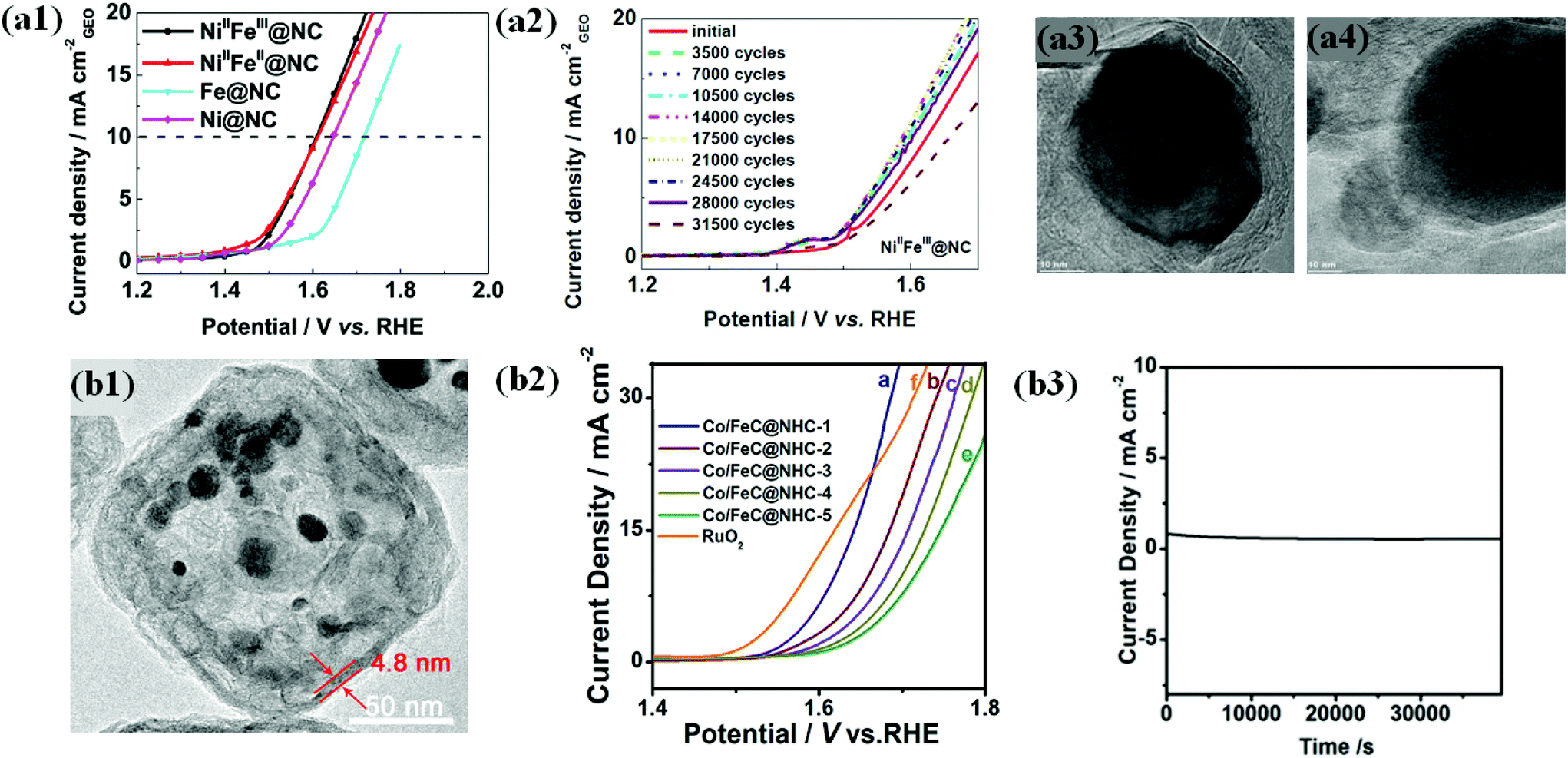 | ||
| Fig. 9 (a1) LSV curves (10 mV s–1@900 rpm) of NiIIFeIII@NC, NiIIFeII@NC, FeIII4[FeII(CN)6]3 derived Fe@NC and NiII[NiII(CN)4] derived Ni@NC catalysts in oxygen-saturated 0.1 M KOH solutions. (a2) LSV curves of NiIIFeIII@NC after a certain number of ADT cycles between 1.2 and 1.9 V at 500 mV s−1. TEM images of (a3) NiIIFeIII@NC and (a4) NiIIFeII@NC catalysts after ADT, reproduced with permission from ref. 63, Elsevier copyright 2017. (b1) TEM images of (A) Co/FeC@NHC-1 (b2) OER polarization curves of Co/FeC@NHC-1, Co/FeC@NHC-2, Co/FeC@NHC-3, Co/FeC@NHC-4, Co/FeC@NHC-5, and RuO2 in 1 M KOH. (b3) Potentiostatic response (current density versus time) under a constant potential of 1.54 V vs. RHE, reproduced with permission from ref. 151, Elsevier copyright 2020. | ||
Durability studies under harsh alkaline conditions (1 M KOH) demonstrated that NiIIFeIII@NC with a high DOG exhibited robust activity without significant activity loss upon up to 25000 accelerated degradation test (ADT) cycles between 1.2 and 1.9 (Fig. 9a2). The TEM image of NiIIFeIII@NC after the ADT shows no carbon shell corrosion (Fig. 9a3). In contrast, NiIIFeII@NC with a poor DOG shows a significantly decreased activity under the same conditions, indicating severe carbon corrosion (Fig. 9a4).63 Therefore, the encapsulation strategy using a highly graphitized carbon shell layer is very effective in improving the durability of OER catalysts and provides insights into industrial applications. Similarly, recent studies demonstrated that the carbon shell modified with heteroatom dopants (B, N, P, and S) ensures efficient OER catalysis along with durability.71,131,147,150
Nevertheless, the doping level in the carbon shell layer of M@C catalysts should be carefully controlled since there are several factors that balance OER activity and chemical stability.
In another way, Zhang and co-workers proposed the fabrication of highly durable Co/FeC NPs embedded in a cubic N-doped hollow carbon matrix (Co/FeC@NHC) with tunable thickness from the MOF precursor (Fig. 9b1).151 The OER activity of the Co/FeC@NHC catalyst increased with decreasing the thickness of the carbon matrix (Fig. 9b2). The outstanding durability of Co/FeC@NHC is attributed mainly to the protection of the embedded core Co/FeC NPs by the carbon shell from harsh alkaline environments, showing extremely stable performance during chronoamperometry at a constant potential of 1.54 V vs. RHE (Fig. 9b3). However, maintaining high durability under wide pH conditions is still technically challenging. Recently reported OER stability studies of carbon shell-encapsulated metals/alloys are summarized in Table 2.
In this regard, M@C type catalysts encapsulated in carbon shells with appropriate thickness and porosity have been intensively developed. The concept of M@C catalysts can be applied to both noble and non-precious metal-based NPs. As shown in Fig. 10, the TEM image shows the typical morphology of M@C NPs, and the change in the polarization curves before and after ADTs indicates extremely high durability of the M@C catalyst. Regardless of the type of carbon support, carbon-encapsulated Pt- and Pd-based NPs have been mainly investigated.49,51–53,99,160,161
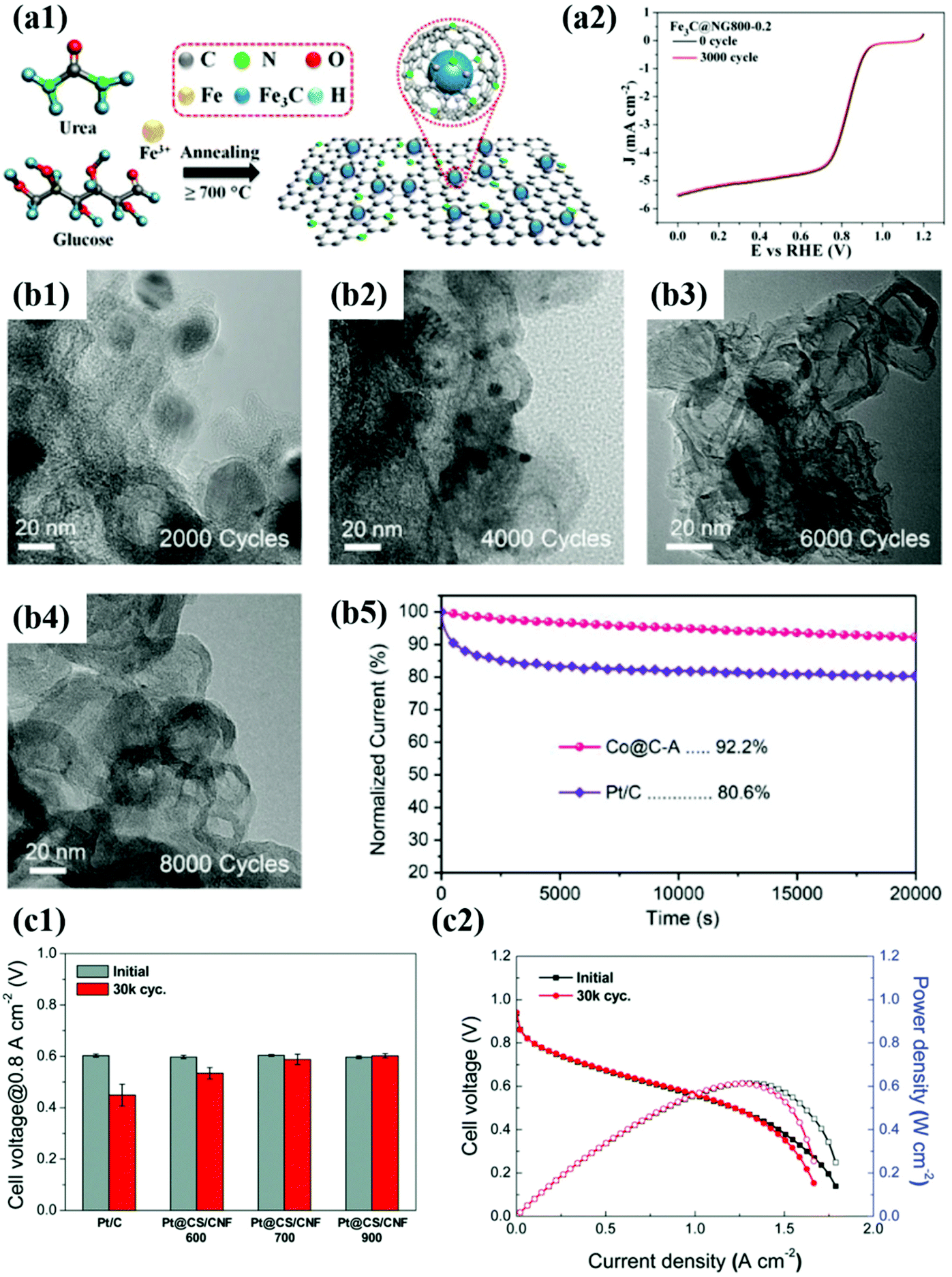 | ||
| Fig. 10 (a1) Schematic illustration of synthesis of Fe3C@NG (a2) ORR curves of the Fe3C@NG800 before and after 3000 cycles in O2-saturated 0.1 M KOH solution, reproduced with permission from ref. 164, ACS copyright 2015. (b1–4) TEM images of Co@C-A before and after ADT. (b5) Chronoamperometry of Co@C-A for 20000 s compared to commercial Pt/C in O2-saturated 0.1 M KOH, reproduced with permission from ref. 97, Wiley copyright 2019. (c1) Cell voltages measured at 0.1 A cm−2 of MEAs before and after ADT. (c2) Polarization curves of Pt@CS/CNF900 before and after ADT, reproduced with permission from ref. 162, RSC copyright 2019. | ||
For Pt@C type catalysts, Pt@CS/CNF, which is prepared with the N-containing polymer and Pt precursor, exhibited high activity and durability not only under half-cell conditions but also in unit cell tests, satisfying the DOE target of 2025 (voltage loss@0.8 A cm−2 <30 mV, ECSA loss <40%) after ADTs using the DOE protocol.162 In the case of Pt-based alloy catalysts, it is very important to prevent the dissolution of 3d TMs that change the Pt electronic structure by strain and electronic effects. Accordingly, Sung et al. synthesized highly durable PtFe NPs with a face-centered-tetragonal (fct) structure through polydopamine (PDA) coating followed by a high-temperature annealing process.51 The thickness of the carbon shell was rationally controlled by changing the polymer coating time, and the fct–PtFe alloy structure was achieved by using the carbon shell as a hard template during heat treatment. In the half-cell test, the half-wave potential (E1/2) of the fct–PtFe/C catalyst decreased by less than 10 mV after ADT (for Pt/C, E1/2: decreased by more than 100 mV), exhibiting excellent stability. Particularly, in the unit cell test, the maximum power density decreased only by 3.4% (for Pt/C, the maximum power density loss: 27%).
Pd-Based NPs have been extensively studied as ORR catalysts since Pd has an electronic structure similar to Pt. As expected, Pd shows high ORR activity in both acid and alkaline solutions; however, the durability cannot be guaranteed, especially in acidic environments. Therefore, to improve the durability of Pd-based catalysts, Kwon et al. proposed N-doped carbon shell-encapsulated Pd NPs (Pd-PANI-K), which are fabricated using aniline as the carbon and nitrogen sources. Wang et al. developed Pd-based alloy (PdFe) NPs encapsulated by a graphitic carbon layer via a one-step thermal annealing method.49 In their study, the O–Pd–Fe@NC/C catalyst annealed at 800 °C showed 74.5% MA and 57.2% SA losses after 10000 cycles in an acid solution, while Pt/C indicated a loss in MA of 83.8% and SA of 88.2% under the same conditions. In particular, in alkaline solution, E1/2 of the O–Pd–Fe@NC/C catalyst decreased by only 2 mV even after 30000 cycles.
Recently, non-precious metal-based catalysts such as Fe- and Co-based materials have attracted much more attention to replace Pt- and Pd-based catalysts. Although carbon-encapsulated TM NPs have been reported since 2000,47,97,163–165 in contrast to precious metal catalysts, both catalytic activity and durability issues remain challenging. For instance, N-doped graphene-supported Fe3C@C showed a negligible decrease in E1/2 after 3000 cycles in O2 saturated 0.1 M KOH.75 However, the number of potential cycles for ADT was not sufficient to evaluate the protective effect of the carbon shell.164 However, a few studies on Co-based catalysts such as Co9C8@C and Co@C-MOF adopted harsh test conditions with 5000–8000 potential cycles in alkaline solution, showing much enhanced durability compared to Pt/C.163,164 However, Co-based NPs still suffered from metal dissolution and detachment from carbon supports during the ADTs.
In contrast, Yoo et al. suggested highly stable Co@C/C catalysts synthesized by a bottom-up approach to the carbon shell formation.47 In this fabrication method, the Co precursor containing acetylacetonates as carbon sources is thermally decomposed and then provides the carbon atoms to reduced Co NPs. The carbons incorporated into the Co NPs are diffused to the metal surface and simultaneously graphitized during a high-temperature annealing process, forming a robust thin carbon shell (less than 1 nm) at the sub-nm level. The ORR activity of the synthesized Co@C/C catalyst hardly changed even after 5000 potential cycles, exhibiting extremely high durability compared to Pt/C. In addition, the Co@C NPs maintained the initial structure even after the ADTs due to the protective effect of the graphitic carbon shell. Therefore, it is believed that the interfacial bonding strength between the carbon shell and the core metal NP can be affected by the fabrication method. Nevertheless, it is necessary to develop non-precious metal-based M@C catalysts that exhibit high stability under both acid and alkaline conditions.
In addition, Gao et al. synthesized Ni@C NPs as non-precious metal-based catalysts by heat treatment at 500, 600, and 700 °C, respectively (Fig. 11).56 Despite the low HOR activity, after 10000 cycles in alkaline solutions, Ni@C catalysts showed negligible performance degradation, as follows: 20% (500 °C), 8% (600 °C), and 0% (700 °C), respectively. From the XPS analysis, it was confirmed that the number of C–C bonds increases and the interaction between the metallic Ni NP and the carbon shell becomes stronger as the annealing temperature increases. In addition, the enhanced crystallinity of the carbon shell was clearly identified by Raman spectra showing the change in the D/G ratio.80 Unfortunately, few studies have been conducted on M@C catalysts for the HOR; however, in contrast, it can be thought that there is plenty of room for new research.
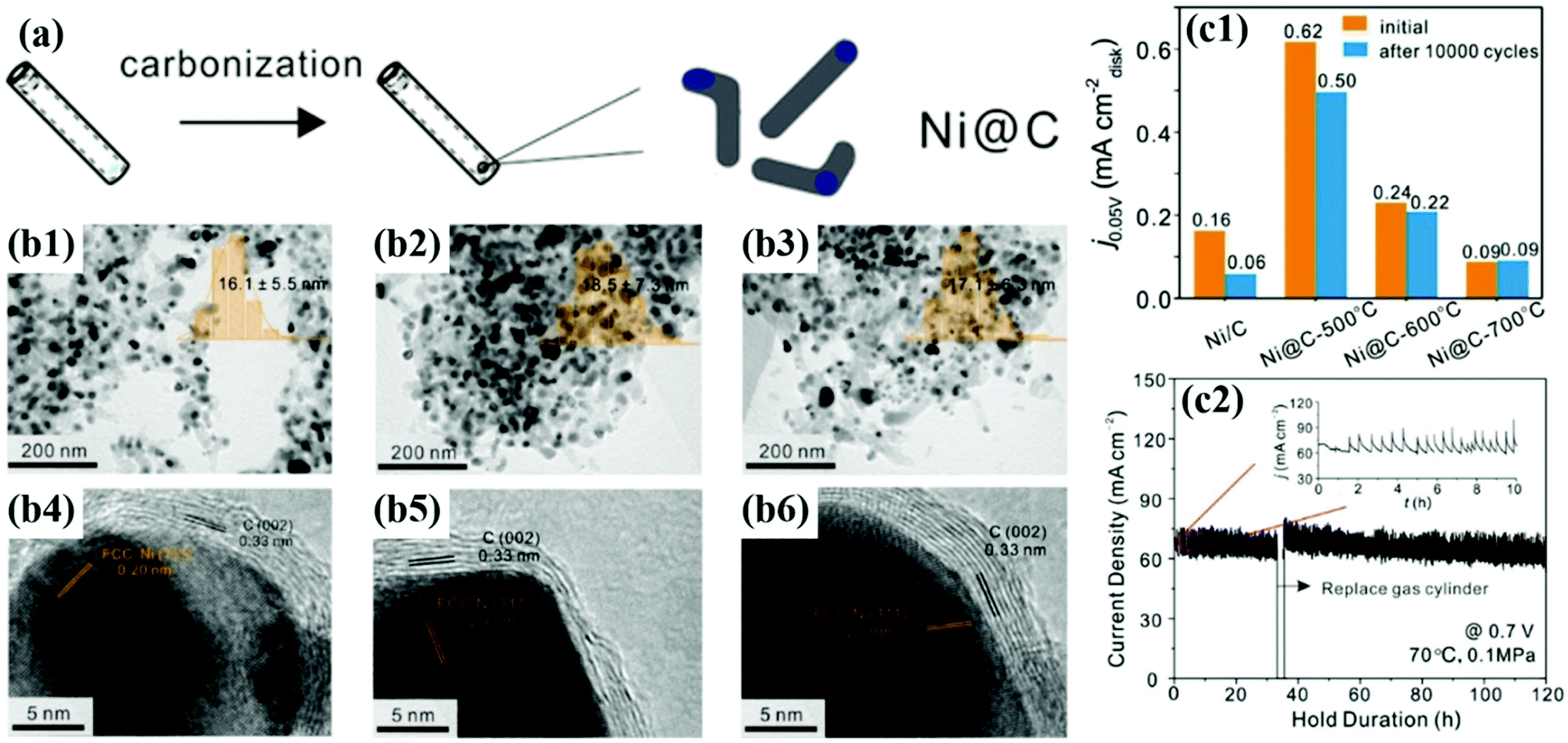 | ||
| Fig. 11 (a) Synthesis diagram of the Ni@C catalyst. (b1–3) TEM images of Ni@C with the inset showing the particle sized distribution histogram. (b1–6) HR-TEM images of Ni@C showing carbon shell thickness for Ni@C-500, Ni@C-600, and Ni@C-700, respectively. (c1) Current densities of Ni@C catalysts before and after ADT (10000 cycles). (c2) Stability test at 0.7 V for 120 h under single cell conditions at 70 °C with fully humidified H2 and O2 fed at the anode and cathode, respectively, with permission from ref. 56, ACS copyright 2020. | ||
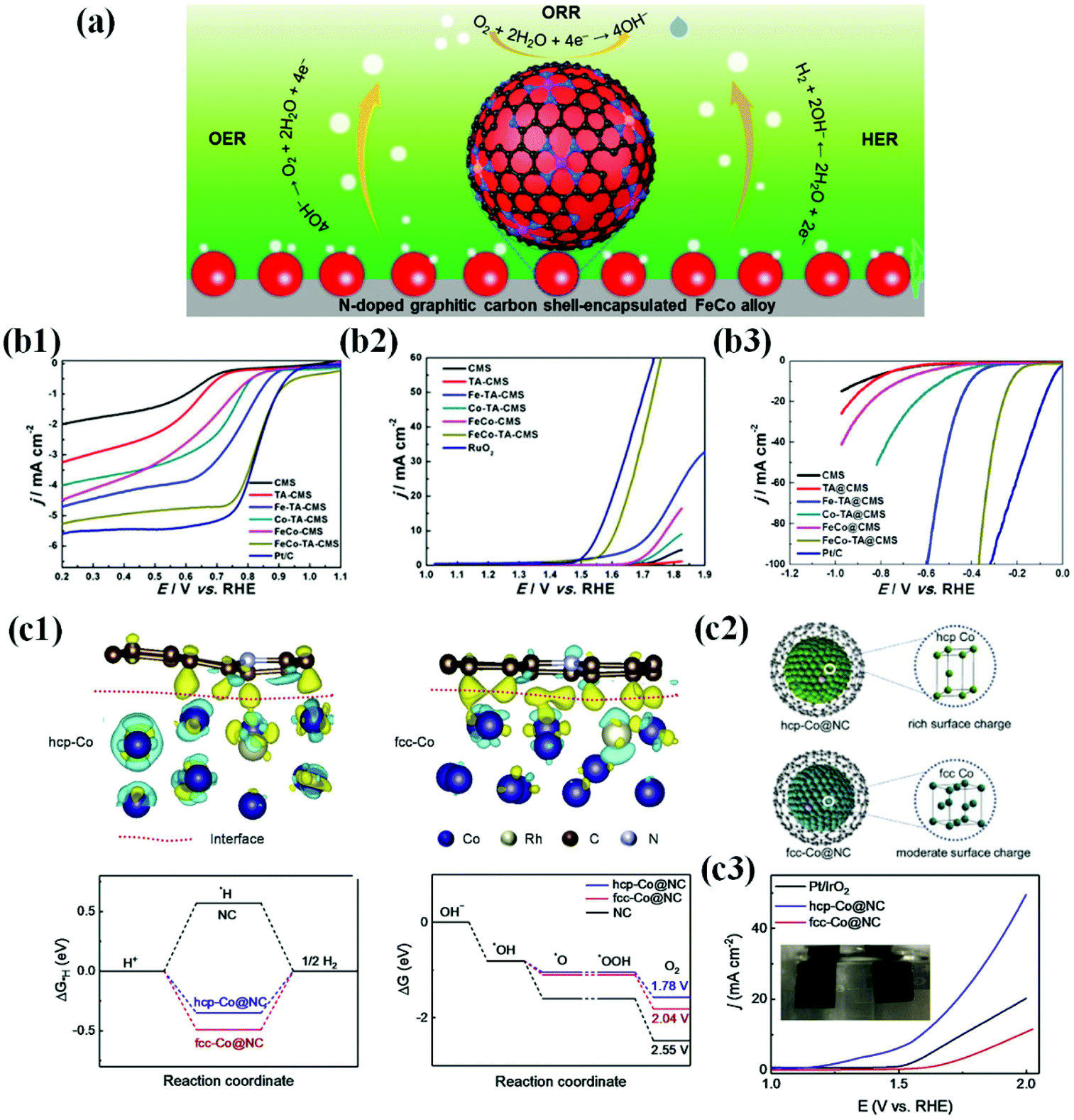 | ||
| Fig. 12 (a) Multifunctional activity of the N-doped carbon shell coated FeCo alloy for the ORR, OER and HER. (b1) Electrocatalytic performances of all as-prepared catalysts and Pt/C for the ORR. LSV curves and the corresponding half wave potential (E1/2) of FeCo-TA@CMS, FeCo@CMS, Fe-TA@CMS, Co-TA@CMS, TA@CMS, CMS, and Pt/C in O2-saturated 0.1 M KOH at room temperature. (b2) LSV curves of FeCo-TA@CMS, FeCo@CMS, Fe-TA@CMS, Co-TA@CMS, TA@CMS, CMS, and RuO2. (b3) LSV curves of FeCo-TA@CMS, FeCo@CMS, Fe-TA@CMS, Co-TA@CMS, TA@CMS, CMS and Pt/C, reproduced with permission from ref. 91, Elsevier publications, copyright 2021. (c1) TEM micrographs of G-Co0.5Fe0.5 (before and after electrochemical activation. (c2) Electrochemical OER activities of graphite-encapsulated 3d TMs and their alloys in 1 M KOH solution scanned at 10 mV s−1. (c3) Electrochemical HER activities of graphite-encapsulated 3d TMs and their alloys in 0.1 M KOH solution scanned at 10 mV s−1, reproduced with permission from ref. 177, Elsevier publications, copyright 2020. | ||
However, in recent years, numerous efforts have been devoted to the development of novel metal encapsulated carbon shell materials as promising multifunctional or bifunctional electrocatalysts because the concept of metal–carbon hybrid interaction and their electronic structure modification are expected to manifest multifunctional activity.54,94,119,127,171–173 In particular, first-row TMs (Mn, Co, Ni, and Fe) and their alloys have been well explored due to their improved intrinsic activity by electronic modification of enveloping carbon layers. Importantly, metal alloy-based catalysts such as FeCo, NiCo, and FeNi exhibit superior bifunctional or multifunctional activity to their individual counterparts since the meta-metal bonding enhances intrinsic polarity and facilitates charge transfer.59,113,174,175 Liu et al. proposed the multifunctional ability of FeCo alloy NPs encapsulated in the N-doped carbon shell prepared by a simple pyrolysis process and identified that the FeCo catalysts exhibit superior activity to individual metal species for the ORR, OER, and HER (Fig. 12b1–3).91
Recently, Goodenough and co-workers developed a series of trifunctional electrocatalysts based on binary and ternary alloys of Fe, Co, and Ni NPs encapsulated by graphitic carbon shells. The initially synthesized catalysts had a thick graphitic shell (∼20–30 layers) that was not catalytically active due to the suppression of charge penetration efficiency. However, this situation is circumvented when the catalysts undergo electrochemical exfoliation through the surface activation process by potential cycling between 1.2 and 2.0 V vs. RHE in an alkaline solution. Consequently, this electrochemical exfoliation transformed the inert thick graphite layer into a thin, highly active graphene shell (1–3 layers). Screening of the activated catalysts for the ORR, OER, and HER in alkaline solutions indicated that the binary Ni–Fe system showed the best activity for the OER, and the Co–Fe system was promising for the HER and ORR.176 As the catalytic reactions occur on the surface, the strong metal–carbon electronic interactions at the interface are significant to tailor the surface charge density and to improve physicochemical properties of the outer carbon shell towards their multifunctional characteristics. However, this depends on the electronic properties and charge injection capability of metal/alloy cores.72,86,119 For instance, Li et al. demonstrated that the hcp-Co@NC catalyst composed of hcp-Co NPs encapsulated by the N-doped carbon shell triggers delocalization of interfacial charge density and strong electronic coupling at the interface, while the fcc-Co@NC catalyst is not possible. Consequently, when hcp-Co@NC NPs are applied to a water electrolyzer, superior reaction activity and good stability are realized, requiring only 1.58 V to attain 10 mA cm−2 (Fig. 12c).177 Interestingly, it was reported that tuning of metal/alloy composition can also have a strong influence on the electronic structure of graphene shells, tailoring the catalytic activity.178
Several factors such as appropriate metal composition, carbon shell thickness, and dopant level can affect the multifunctional performance of M@C type catalysts. According to the design of the catalyst structure, the activity and durability can be dramatically modulated. This scenario was exemplified by Noh et al. when they fabricated a series of TM alloys encapsulated in an N-doped carbon shell (M@N–C, M = Fe, Co, Ni, Cu, or Fe alloys) supported on Ketjen black (KB) as bifunctional electrocatalysts. Based on the screening result of different transition metals, FeCo@N–C/KB was found to achieve excellent bifunctional activity toward the HER as well as ORR with high durability in acidic solutions. By DFT and experimental investigations on the number of factors (N-doping level, active N sites, thickness of shell layers, and alloying elements), a quantitative method is proposed to tune the ORR activity by carefully controlling the aforementioned factors (Fig. 13a1–3).179
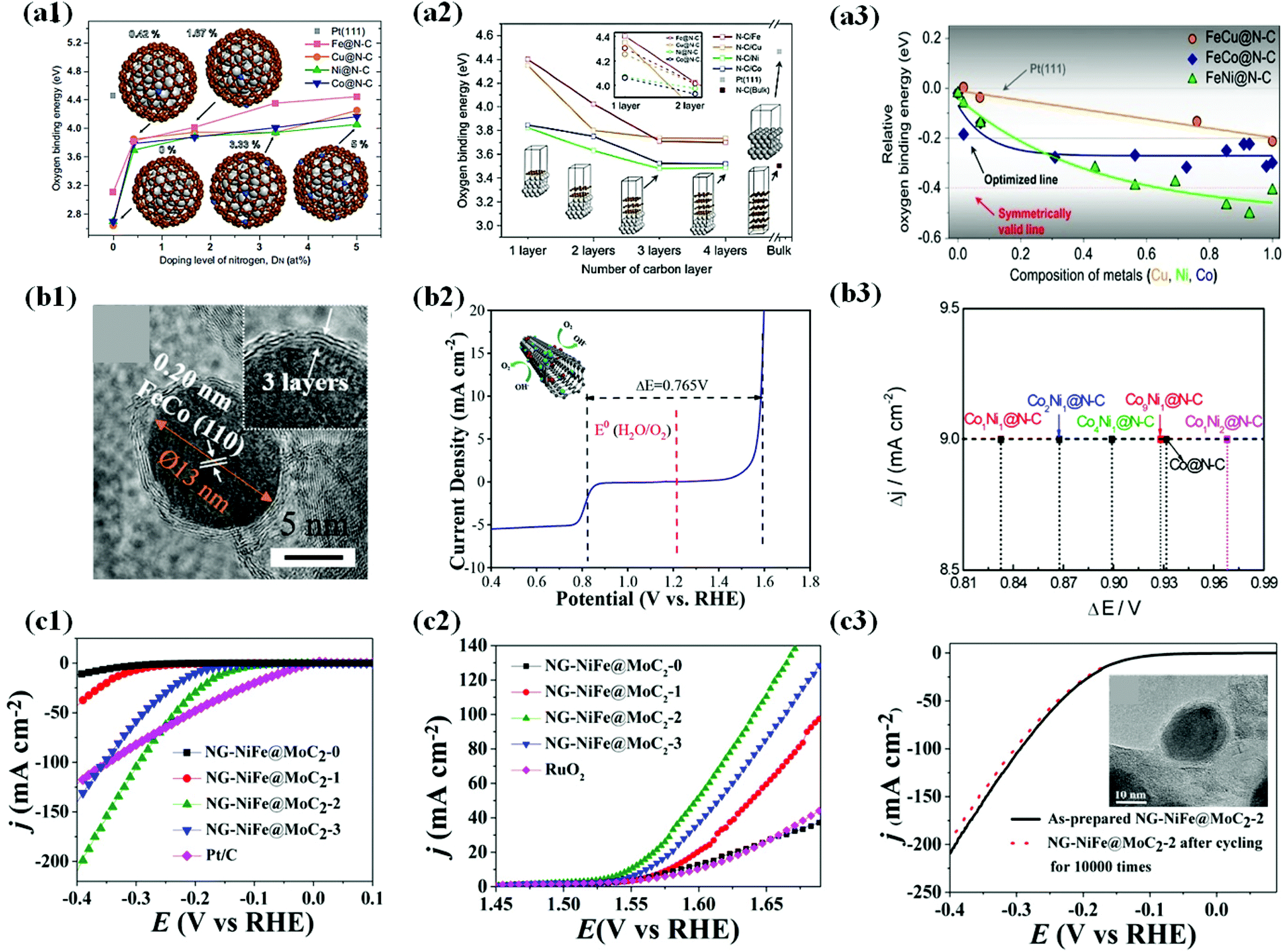 | ||
| Fig. 13 (a1) Oxygen binding energies as a function of the doping level of nitrogen on M@N–C (M = Fe, Cu, Ni and Co) as a function of DN, typically 0, 0.42, 1.67, 3.33 and 5 at%. The oxygen binding energy (4.46 eV) on Pt(111) is marked for comparison with those on M@N–C. (a2) Oxygen binding energy as a function of the number of carbon layers for N–C/M (bulk) and M@N–C (nanoparticle, 1.5 nm) as a function of the number of carbon layers, including Pt(111) and N–C (bulk). (a3) Relative oxygen binding energies on FeM@N–C NPs as a function of the composition of second metal (M). Oxygen binding energies are represented as E(FeM@N–C) E(Pt(111)), where E(FeM@N–C) and E(Pt(111) are oxygen binding energies on FeM@N–C and Pt(111), respectively, reproduced with permission from ref. 179, Nature group publications, copyright 2019. (b1) HRTEM images of the Fe1.2Co@NC/NCNTs. (b2) Overall polarization curve of the Fe1.2Co@NC/NCNTs in the entire ORR and OER region, reproduced with permission from ref. 58, ACS publications, copyright 2018. (b3) Potential differences (E@6.0 mA cm−2 − E@−3.0 mA cm−2) on CoxNiy@N–C, reproduced with permission from ref. 54, ACS publications, copyright 2018. (c1) Steady-state polarization curves of NGNiFe@MoC2-0, NG-NiFe@MoC2-1, NGNiFe@MoC2-2, NG-NiFe@MoC2-3 and Pt/C electrocatalysts for HER. (c2) Steady-state polarization curves of NGNiFe@MoC2-0, NG-NiFe@MoC2-1, NGNiFe@MoC2-2, NG-NiFe@MoC2-3 and RuO2 electrocatalysts for OER. (c3) HER polarization of NG-NiFe@MoC2-2 before and after cycling for 10000 times. Inset shows HRTEM image of NG-NiFe@MoC2-2 after 10000 cycles, reproduced with permission from ref. 72, Elsevier publications, copyright 2018. | ||
Recently, Li et al. developed an efficient bifunctional electrocatalyst composed of FeCo alloy NPs coated by an ultrathin N-doped carbon layer (∼1–3) encapsulated in N-doped carbon nanotubes (FeCo@NC/NCNTs) (Fig. 13b1). Electrochemical measurements show that the optimized catalyst, Fe1.2Co@NC/NCNTs, with a low Fe/Co content exhibits high ORR activity, while OER activity of the same exhibited superior activity among all the catalysts in an alkaline medium. The potential difference (ΔE = Ej = 10 − E1/2) between the ORR and OER for Fe1.2Co@NC/NCNTs is 0.765 V (Fig. 13b2), which was lower than that of Pt/C and Ir/C. The outstanding ORR and OER activity of the catalyst was found to be due to the following reasons: (1) synergy between Fe and Co in the metal alloy, (2) electronic interaction between FeCo NPs and the N-doped carbon layer, (3) formation of Fe–Nx and Co–Nx bonds generated more active sites for the OER, and (4) enhanced conductivity.58 The potential difference (ΔE) as an important descriptor of bifunctional (ORR/OER) activity can be further manipulated by tuning the metal composition ratios (Fig. 13b3).54
In addition, co-doping other heteroatoms (B, S, and P) with nitrogen in graphene is proven to potentially improve bifunctional activity as well.179–181 Zhang et al. studied bifunctional water splitting reactions on Co encapsulated B/N co-doped nanocarbon by carbonization of the Co-MOF precursor. The optimized catalyst, Co/NBC-900, in their work displayed efficient bifunctional activity and stability toward the HER and OER in alkaline media.182
In some instances, coupling two different metal species to form an alloy alters the lattice and bond length of the crystals and changes the adsorption energies of reactive intermediates for optimal catalytic activity toward bifunctional or multifunctional electrocatalytic activity.76,183 For instance, incorporating Mo into bimetallic NiFe-based catalysts may emerge as a new type of ternary metal electrocatalyst and can potentially serve as bifunctional electrocatalysts toward the overall water splitting process owing to improved metal–hydrogen (M–H) strength and enhanced affinity toward OH− ions.184–186 Hu et al. in their work demonstrated the above concept by doping MoC2 in NiFe NPs encapsulated in N-doped graphene layers (∼3–11 layers) for water electrolysis. The electrochemical tests and structural analysis proved that the MoC2-doped NiFe cores were the active sites for the HER/OER but not the N-doped carbon shell (Fig. 13c1 and 2), which took the role to provide high conductivity and chemical protection against corrosion/oxidation during long-term durability tests. However, OER stability tests indicated inevitable in situ formed oxidized species (i.e., Ni2+, Fe2+, and Mo3+), which acted as additional active sites for the OER (Fig. 13c3).72
4.3 Reaction-selective catalysts via fine-control of the carbon shell structure
First of all, we introduce the development of alcohol-tolerant M@C catalysts for the ORR in fuel cells.49,50,75,187,188 Direct alcohol fuel cells (DAFCs) have been extensively studied due to their high energy density and safety of fuel storage. In DAFCs, along with the alcohol oxidation reaction (AOR) at the anode, the sluggish ORR kinetics at the cathode is a crucial factor in decreasing fuel cell performance. In addition, it is well known that the alcohol crossover from the anode to the cathode impedes the ORR by forming a mixed potential, causing serious performance degradation. Accordingly, in the past decades, to improve the cathode performance, Pt-based alloys NPs such as PtPdCo have been developed as alcohol-tolerant cathode catalysts.189 However, it is very difficult to completely prevent the formation of a mixed potential since the surface of metal alloy NPs is still exposed to the crossovered alcohols.189Accordingly, Wang et al. demonstrated that a thick carbon shell perfectly encapsulating the PdFe alloy surface can significantly reduce the alcohol crossover effect.50 As shown in Fig. 14a, the CV and ORR polarization curves of the O–Pd–Fe@NC/C catalyst hardly changed even after injecting 0.5 M methanol into the electrolyte, resulting in much enhanced alcohol tolerance compared to Pt/C. In addition, the Fe/Fe3C@C catalyst maintained the ORR current for 1000 s after adding the methanol solution to both acidic and alkaline solutions (Fig. 14a).188 Cheng et al. also encapsulated Pt NPs in a 3D-structured N-doped carbon nanocage (Pt@NCNC) through a vacuum filling process, showing high methanol tolerance without change in ORR activity.187 Therefore, it is clearly confirmed that the blocking effect by the carbon shell coated on the metal NPs is superior to the electronic effect by simple metal alloying. However, to guarantee high ORR activity, it is necessary to develop an alcohol-tolerant M@C type catalyst with a carbon shell that has an appropriate thickness and pores.
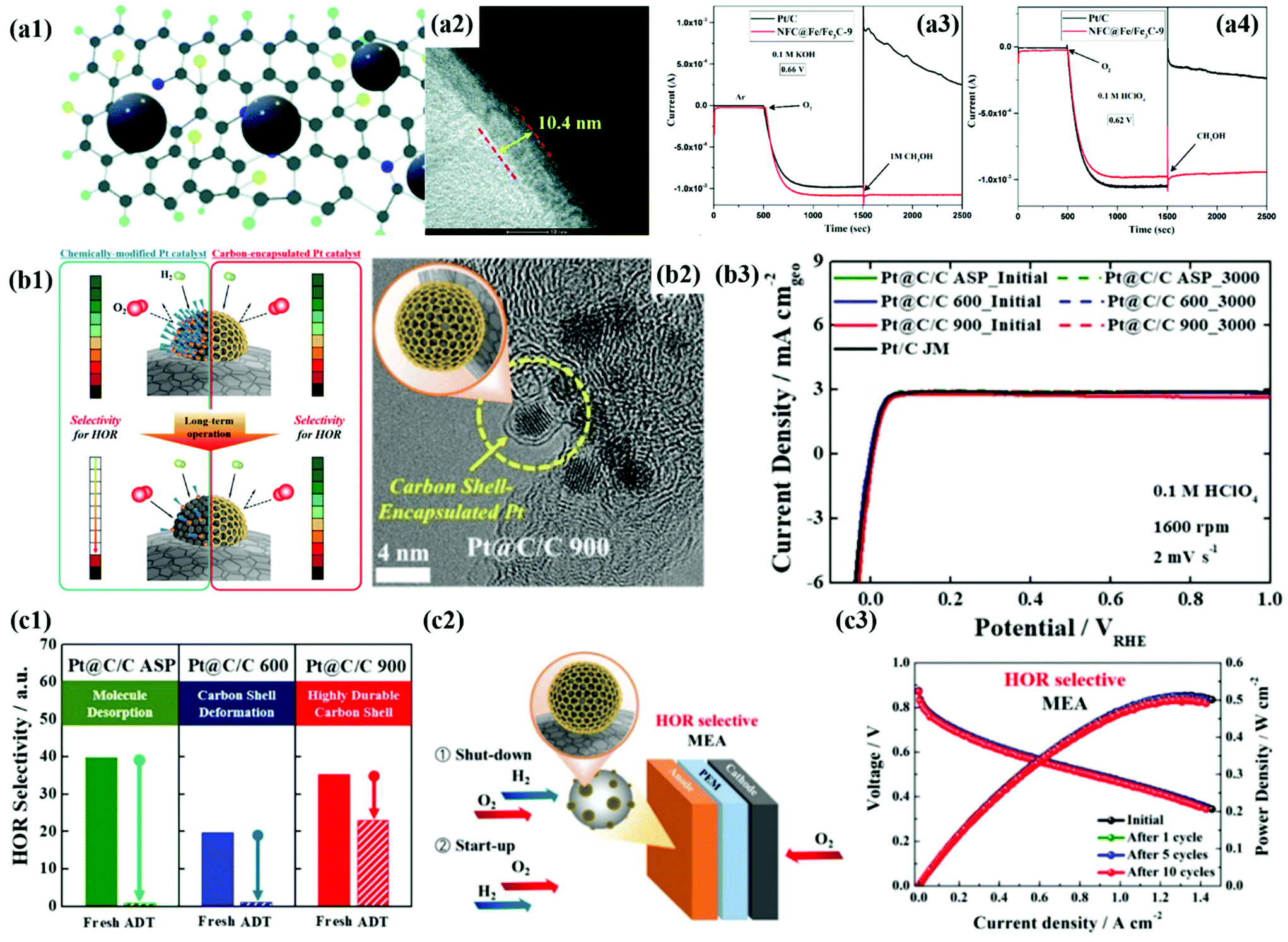 | ||
| Fig. 14 (a1) Schematic illustration of N and F co-doped carbon (NFC) encapsulated Fe/Fe3C. (a2) HR-TEM images of NFC@Fe/Fe3C-9 showing carbon shell thickness. (a3–4) Chronoamperometry of NFC@Fe/Fe3C-9 for methanol tolerance 0.1 M KOH (a3) and 0.1 M HClO4 (a4), with permission from ref. 75, RSC copyright 2020. (b1) Schematic illustration of HOR selectivity change of the Pt catalyst and carbon encapsulated Pt catalyst during PEMFC operation. (b2) TEM images of Pt@C/C 900 (b3) HOR polarization curves of Pt@C/C catalysts before and after 3000 cycles (b4) exposed Pt surface areas of Pt@C/C catalysts before and after 3000 cycles. (b5) Schematic diagram of the start-up/shut-down cycle test of the single cell system using Pt@C/C catalysts. (b6) i–V polarization curves of Pt@C/C catalysts during the start-up/shut-down test in the single cell system, with permission from ref. 80, ACS copyright 2019. | ||
Second, during fuel cell operation, a reverse current can occur by the undesired ORR due to the formation of H2/O2 boundaries at the anode under shut-down/start-up conditions.80 It is well known that the reverse current causes serious deterioration of fuel cell performance since a potential of ∼1.5 V or higher is applied to the cathode, resulting in catalyst dissolution and carbon corrosion. To avoid this problem, Yoo et al. fabricated a Pt@C/C anode catalyst that has high HOR selectivity along with fully subdued ORR activity when H2/O2 boundaries are formed in the anode under shut-down/start-up conditions. The finely-tuned carbon shell as a molecular sieve layer rationally induced the selective adsorption/reaction of H2 rather than O2 due to the different kinetic diameters of the gases. In particular, the Pt@C/C catalyst heat-treated at 900 °C had a robust carbon shell with high HOR selectivity and exhibited superior chemical stability compared to the catalyst annealed at 600 °C. In unit cell tests, the membrane electrode assembly (MEA) with a HOR-selective anode catalyst showed excellent durability even after shut-down/start-up cycles, while the performance of MEA with a typical Pt/C catalyst significantly decreased (50% reduction) due to serious deterioration of the cathode catalyst layer. Similarly, Choi et al. synthesized Pt@C/C catalysts with hydrogen peroxide (H2O2) production selectivity against the ORR by controlled coating of Pt NPs, effectively suppressing the 4-electron pathway in the course of the ORR. The carbon shell induced the end-on adsorption of O2 on the Pt surface, remarkably enhancing the H2O2 selectivity up to 41%.60 Therefore, the M@C type catalyst with reaction selectivity is expected to be a key material that can be applied not only to fuel cells, but also to various electrochemical energy conversion reactions.
5. Summary and outlook
Carbon-encapsulated metal (M@C) type catalysts have been extensively studied as promising electrocatalysts for fuel cell and water electrolysis reactions. In this review, we introduced the M@C NPs composed of Earth-abundant TMs and noble metals that can promote the advancement of energy conversion technologies and bridge the gap to the development of energy storage technologies. In particular, the technical achievements by the development of M@C type catalysts trigger the electrocatalytic reactions of the inert carbon shell, ensure the durability of the active material, and facilitate the selective electrochemical reactions by tuning the electronic structure and physicochemical properties of the carbon shell. Furthermore, the N-dopant induces excellent properties of the carbon shell through the modification of the electronic structure and the chemical bonding character between the metal and the outer N-doped carbon than other dopants (B, S, and P). Now, beyond the experimental studies of water electrolysis and fuel cells, an in-depth theory must be established to fully understand interesting phenomena in electrochemical reactions. In particular, in water electrolysis, it is necessary to rationally describe the physical changes of metal NPs encapsulated by the carbon shell during the reaction, and to clearly investigate the role of the carbon shell. Further advances in computational modelling will provide insight into the novel design of M@C type catalysts. Nevertheless, simple and reliable experimental strategies to fabricate a well-defined and fine-tuned carbon shell are always essential to realizing potential applications that will mark great breakthroughs in this field.Author contributions
S. J. Yoo and N. Jung conceived the idea of the work. J.-H. Jang and A. Anto Jeffry wrote the manuscript. J. Min investigated the paper and research trends. N. Jung and S. J. Yoo guided the work and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the KIST Institutional Program (2E31002) and a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. 2021R1A2C2012685, 2018M1A2A2061975, and 2021M3H4A1A02042948). This work was also supported by the New & Renewable Energy Core Technology Program of KETEP (20203020030010) in Korea.Notes and references
- R. O'hayre, S.-W. Cha, W. Colella and F. B. Prinz, Fuel cell fundamentals, John Wiley & Sons, 2016 Search PubMed.
- H. A. Gasteiger and N. M. Marković, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- O. Z. Sharaf and M. F. Orhan, Renewable Sustainable Energy Rev., 2014, 32, 810–853 CrossRef CAS.
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS.
- G. Shen, J. Liu, H. B. Wu, P. Xu, F. Liu, C. Tongsh, K. Jiao, J. Li, M. Liu and M. Cai, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- J. Yang, S. Yang, Y. Chung and Y. Kwon, Korean J. Chem. Eng., 2020, 37, 176–183 CrossRef CAS.
- S. Miyanishi and T. Yamaguchi, Polym. Chem., 2020, 11, 3812–3820 RSC.
- J.-H. Wee, Renewable Sustainable Energy Rev., 2007, 11, 1720–1738 CrossRef CAS.
- S. Khilari, S. Pandit, M. Ghangrekar, D. Das and D. Pradhan, RSC Adv., 2013, 3, 7902–7911 RSC.
- J.-J. Hwang, Renewable Sustainable Energy Rev., 2013, 19, 220–229 CrossRef CAS.
- C. Tang, H.-F. Wang and Q. Zhang, Acc. Chem. Res., 2018, 51(4), 881–889 CrossRef CAS PubMed.
- C. M. Zalitis, D. Kramer and A. R. Kucernak, Phys. Chem. Chem. Phys., 2013, 15, 4329–4340 RSC.
- A. Roy, M. R. Talarposhti, S. J. Normile, I. V. Zenyuk, V. De Andrade, K. Artyushkova, A. Serov and P. Atanassov, Sustainable Energy Fuels, 2018, 2, 2268–2275 RSC.
- E. Antolini, Energy Environ. Sci., 2009, 2, 915–931 RSC.
- B. P. Setzler, Z. Zhuang, J. A. Wittkopf and Y. Yan, Nat. Nanotechnol., 2016, 11, 1020–1025 CrossRef CAS PubMed.
- K. Karthick, A. B. M. Basha, A. Sivakumaran and S. Kundu, Catal. Sci. Technol., 2020, 10, 3681–3693 RSC.
- Y. P. Zhu, T. Y. Ma, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 1324–1328 CrossRef CAS PubMed.
- P. Ganesan, A. Sivanantham and S. Shanmugam, J. Mater. Chem. A, 2016, 4, 16394–16402 RSC.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS.
- M. Conte, A. Iacobazzi, M. Ronchetti and R. Vellone, J. Power Sources, 2001, 100, 171–187 CrossRef CAS.
- V. Das, S. Padmanaban, K. Venkitusamy, R. Selvamuthukumaran, F. Blaabjerg and P. Siano, Renewable Sustainable Energy Rev., 2017, 73, 10–18 CrossRef CAS.
- M. Ehsani, Y. Gao, S. Longo and K. Ebrahimi, Modern electric, hybrid electric, and fuel cell vehicles, CRC press, 2018 Search PubMed.
- J. M. Ogden, M. M. Steinbugler and T. G. Kreutz, J. Power Sources, 1999, 79, 143–168 CrossRef CAS.
- R. Van den Hoed, J. Cleaner Prod., 2007, 15, 1014–1021 CrossRef.
- Y. Chen, A. Lavacchi, H. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC.
- S. Park, Y. Shao, J. Liu and Y. Wang, Energy Environ. Sci., 2012, 5, 9331–9344 RSC.
- L. Xiao, S. Zhang, J. Pan, C. Yang, M. He, L. Zhuang and J. Lu, Energy Environ. Sci., 2012, 5, 7869–7871 RSC.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS.
- T. Schuler, T. Kimura, T. J. Schmidt and F. N. Büchi, Energy Environ. Sci., 2020, 13, 2153–2166 RSC.
- T. Asset, R. Chattot, M. Fontana, B. Mercier-Guyon, N. Job, L. Dubau and F. Maillard, ChemPhysChem, 2018, 19, 1552–1567 CrossRef CAS PubMed.
- N. Jung, D. Y. Chung, J. Ryu, S. J. Yoo and Y.-E. Sung, Nano Today, 2014, 9, 433–456 CrossRef CAS.
- A. Oh, Y. J. Sa, H. Hwang, H. Baik, J. Kim, B. Kim, S. H. Joo and K. Lee, Nanoscale, 2016, 8, 16379–16386 RSC.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- L. Huo, B. Liu, G. Zhang, R. Si, J. Liu and J. Zhang, J. Mater. Chem. A, 2017, 5, 4868–4878 RSC.
- J. Li, M. Chen, D. A. Cullen, S. Hwang, M. Wang, B. Li, K. Liu, S. Karakalos, M. Lucero and H. Zhang, Nat. Catal., 2018, 1, 935–945 CrossRef CAS.
- C. Li, H. Liu and Z. Yu, Appl. Catal., B, 2019, 241, 95–103 CrossRef CAS.
- C.-X. Zhao, J.-N. Liu, B.-Q. Li, D. Ren, X. Chen, J. Yu and Q. Zhang, Adv. Funct. Mater., 2020, 30, 2003619 CrossRef CAS.
- L. Wang, X. Wan, S. Liu, L. Xu and J. Shui, J. Energy Chem., 2019, 39, 77–87 CrossRef.
- H. Wu, J. Wang, J. Yan, Z. Wu and W. Jin, Nanoscale, 2019, 11, 20144–20150 RSC.
- K. Im, D. Kim, J.-H. Jang, J. Kim and S. J. Yoo, Appl. Catal., B, 2020, 260, 118192 CrossRef CAS.
- L. Liu, B. Wang, R. Gao, D. Zhang, W. Xu, L. Chen, X. Yan and Y. Li, RSC Adv., 2020, 10, 10689–10694 RSC.
- E. Antolini, Appl. Catal., B, 2012, 123, 52–68 CrossRef.
- X. Zhou, J. Qiao, L. Yang and J. Zhang, Adv. Energy Mater., 2014, 4, 1301523 CrossRef.
- S. H. Hur and J. N. Park, Asia-Pac. J. Chem. Eng., 2013, 8, 218–233 CrossRef CAS.
- M. Sharma, J.-H. Jang, D. Y. Shin, J. A. Kwon, D.-H. Lim, D. Choi, H. Sung, J. Jang, S.-Y. Lee, K. Y. Lee, H.-Y. Park, N. Jung and S. J. Yoo, Energy Environ. Sci., 2019, 12, 2200–2211 RSC.
- H. Sung, M. Sharma, J. Jang, S.-Y. Lee, M.-G. Choi, K. Lee and N. Jung, Nanoscale, 2019, 11, 5038–5047 RSC.
- Y. Hu, Y. Lu, X. Zhao, T. Shen, T. Zhao, M. Gong, K. Chen, C. Lai, J. Zhang, H. L. Xin and D. Wang, Nano Res., 2020, 13, 2365–2370 CrossRef CAS.
- H.-J. Niu, L. Zhang, J.-J. Feng, Q.-L. Zhang, H. Huang and A.-J. Wang, J. Colloid Interface Sci., 2019, 552, 744–751 CrossRef CAS PubMed.
- D. Y. Chung, S. W. Jun, G. Yoon, S. G. Kwon, D. Y. Shin, P. Seo, J. M. Yoo, H. Shin, Y.-H. Chung, H. Kim, B. S. Mun, K.-S. Lee, N.-S. Lee, S. J. Yoo, D.-H. Lim, K. Kang, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2015, 137, 15478–15485 CrossRef CAS PubMed.
- Z. Zhang, S. Liu, X. Tian, J. Wang, P. Xu, F. Xiao and S. Wang, J. Mater. Chem. A, 2017, 5, 10876–10884 RSC.
- J. Hwang, Y. Kim, M. Karuppnan, T. Lim and O. J. Kwon, Electrocatalysis, 2020, 11, 77–85 CrossRef CAS.
- H. Ning, G. Li, Y. Chen, K. Zhang, Z. Gong, R. Nie, W. Hu and Q. Xia, ACS Appl. Mater. Interfaces, 2018, 11, 1957–1968 CrossRef.
- C. C. Yang, S. F. Zai, Y. T. Zhou, L. Du and Q. Jiang, Adv. Funct. Mater., 2019, 29, 1901949 CrossRef.
- Y. Gao, H. Peng, Y. Wang, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2020, 12, 31575–31581 CrossRef CAS PubMed.
- H. Fei, Y. Yang, Z. Peng, G. Ruan, Q. Zhong, L. Li, E. L. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2015, 7, 8083–8087 CrossRef CAS.
- S. Li, W. Chen, H. Pan, Y. Cao, Z. Jiang, X. Tian, X. Hao, T. Maiyalagan and Z.-J. Jiang, ACS Sustainable Chem. Eng., 2019, 7, 8530–8541 CrossRef CAS.
- J. Deng, P. Ren, D. Deng, L. Yu, F. Yang and X. Bao, Energy Environ. Sci., 2014, 7, 1919–1923 RSC.
- C. H. Choi, H. C. Kwon, S. Yook, H. Shin, H. Kim and M. Choi, J. Phys. Chem. C, 2014, 118, 30063–30070 CrossRef CAS.
- Y. Shen, Y. Zhou, D. Wang, X. Wu, J. Li and J. Xi, Adv. Energy Mater., 2018, 8, 1701759 CrossRef.
- C. Wang, H. Yang, Y. Zhang and Q. Wang, Angew. Chem., Int. Ed., 2019, 58, 6099–6103 CrossRef CAS.
- L. Du, L. Luo, Z. Feng, M. Engelhard, X. Xie, B. Han, J. Sun, J. Zhang, G. Yin and C. Wang, Nano Energy, 2017, 39, 245–252 CrossRef CAS.
- H. Kim, A. W. Robertson, S. O. Kim, J. M. Kim and J. H. Warner, ACS Nano, 2015, 9, 5947–5957 CrossRef CAS.
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123–129 RSC.
- M. Tavakkoli, T. Kallio, O. Reynaud, A. G. Nasibulin, C. Johans, J. Sainio, H. Jiang, E. I. Kauppinen and K. Laasonen, Angew. Chem., 2015, 127, 4618–4621 CrossRef.
- R. S. Bhoria, in Graphene and Its Derivatives-Synthesis and Applications, IntechOpen, 2019 Search PubMed.
- J. Wang, J. Kim, S. Choi, H. Wang and J. Lim, Small Methods, 2020, 4, 2000621 CrossRef CAS.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2020, 121, 934 Search PubMed.
- K. Hu, T. Ohto, L. Chen, J. Han, M. Wakisaka, Y. Nagata, J.-I. Fujita and Y. Ito, ACS Energy Lett., 2018, 3, 1539–1544 CrossRef CAS.
- H. Zhang, Z. Ma, J. Duan, H. Liu, G. Liu, T. Wang, K. Chang, M. Li, L. Shi and X. Meng, ACS Nano, 2016, 10, 684–694 CrossRef CAS PubMed.
- Q. Hu, X. Liu, B. Zhu, L. Fan, X. Chai, Q. Zhang, J. Liu, C. He and Z. Lin, Nano Energy, 2018, 50, 212–219 CrossRef CAS.
- Y. Zhang, Y. Ma, Y.-Y. Chen, L. Zhao, L.-B. Huang, H. Luo, W.-J. Jiang, X. Zhang, S. Niu and D. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 36857–36864 CrossRef CAS.
- D. Y. Chung, S. W. Jun, G. Yoon, H. Kim, J. M. Yoo, K.-S. Lee, T. Kim, H. Shin, A. K. Sinha and S. G. Kwon, J. Am. Chem. Soc., 2017, 139, 6669–6674 CrossRef CAS PubMed.
- M. Karuppannan, J. E. Park, H. E. Bae, Y.-H. Cho and O. J. Kwon, Nanoscale, 2020, 12, 2542–2554 RSC.
- Y. Yang, Z. Lun, G. Xia, F. Zheng, M. He and Q. Chen, Energy Environ. Sci., 2015, 8, 3563–3571 RSC.
- L. Li, B. Yan, L. Zhang, Y. Tian and H. Zeng, Chem. Commun., 2015, 51, 15780–15783 RSC.
- Y. Kim, A. A. Jeffery, J. Min and N. Jung, J. Nanomater., 2019, 9, 1491 CrossRef CAS PubMed.
- J. Min, A. A. Jeffery, Y. Kim and N. Jung, J. Nanomater., 2019, 9, 1425 CrossRef CAS PubMed.
- J. Jang, M. Sharma, D. Choi, Y. S. Kang, Y. Kim, J. Min, H. Sung, N. Jung and S. J. Yoo, ACS Appl. Mater. Interfaces, 2019, 11, 27735–27742 CrossRef CAS.
- J. Jang, M. Sharma, H. Sung, S. Kim and N. Jung, Korean J. Mater. Res., 2018, 28, 646–652 CrossRef.
- H. Sung, M. Sharma, J. Jang and N. Jung, Korean J. Mater. Res., 2018, 28, 633–639 CrossRef.
- M. Sharma, N. Jung and S. J. Yoo, Chem. Mater., 2018, 30, 2–24 CrossRef CAS.
- S.-F. Xue, W.-Y. Wu, X. Bian, Z.-F. Wang and Y.-F. Wu, Green Process. Synth., 2018, 7, 241–247 CAS.
- C. Lv, B. Liang, K. Li, Y. Zhao and H. Sun, Biosens. Bioelectron., 2018, 117, 802–809 CrossRef CAS PubMed.
- J. Wang, D. Gao, G. Wang, S. Miao, H. Wu, J. Li and X. Bao, J. Mater. Chem. A, 2014, 2, 20067–20074 RSC.
- J. A. Varnell, C. Edmund, C. E. Schulz, T. T. Fister, R. T. Haasch, J. Timoshenko, A. I. Frenkel and A. A. Gewirth, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- M. X. Chen, M. Zhu, M. Zuo, S. Q. Chu, J. Zhang, Y. Wu, H. W. Liang and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 1627–1633 CrossRef CAS PubMed.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- Y. Xu, S. Yin, C. Li, K. Deng, H. Xue, X. Li, H. Wang and L. Wang, J. Mater. Chem. A, 2018, 6, 1376–1381 RSC.
- H. Liu, D.-H. Yang, X.-Y. Wang, J. Zhang and B.-H. Han, J. Colloid Interface Sci., 2021, 581, 362–373 CrossRef CAS PubMed.
- G. Zhang, W. Lu, F. Cao, Z. Xiao and X. Zheng, J. Power Sources, 2016, 302, 114–125 CrossRef CAS.
- L. Zhang, C. Qi, A. Zhao, G. Xu, J. Xu, L. Zhang, C. Zhang and D. Jia, Appl. Surf. Sci., 2018, 445, 462–470 CrossRef CAS.
- Y. Xu, W. Tu, B. Zhang, S. Yin, Y. Huang, M. Kraft and R. Xu, Adv. Mater., 2017, 29, 1605957 CrossRef PubMed.
- H. Lee, Y.-E. Sung, I. Choi, T. Lim and O. J. Kwon, J. Power Sources, 2017, 362, 228–235 CrossRef CAS.
- S. Gong, C. Wang, P. Jiang, K. Yang, J. Lu, M. Huang, S. Chen, J. Wang and Q. Chen, J. Mater. Chem. A, 2019, 7, 15079–15088 RSC.
- X. Yan, C. L. Dong, Y. C. Huang, Y. Jia, L. Zhang, S. Shen, J. Chen and X. Yao, Small Methods, 2019, 3, 1800439 CrossRef.
- M. Gobbi, S. Bonacchi, J. X. Lian, Y. Liu, X.-Y. Wang, M.-A. Stoeckel, M. A. Squillaci, G. D'avino, A. Narita and K. Müllen, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- J. Liu, W. Li, R. Cheng, Q. Wu, J. Zhao, D. He and S. Mu, Langmuir, 2019, 35, 2580–2586 CrossRef CAS.
- Y. Xu, Y. Li, S. Yin, H. Yu, H. Xue, X. Li, H. Wang and L. Wang, Nanotechnology, 2018, 29, 225403 CrossRef PubMed.
- Y. He, Q. Tan, L. Lu, J. Sokolowski and G. Wu, Electrochem. Energy Rev., 2019, 2, 231–251 CrossRef CAS.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 CAS.
- G. Giovannetti, P. A. Khomyakov, G. Brocks, V. V. Karpan, J. van den Brink and P. J. Kelly, Phys. Rev. Lett., 2008, 101, 026803 CrossRef CAS PubMed.
- F. Yang, D. Deng, X. Pan, Q. Fu and X. Bao, Natl. Sci. Rev., 2015, 2, 183–201 CrossRef CAS.
- D. Deng, K. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- Y. Peng and S. Chen, Green Energy Environ., 2018, 3, 335–351 CrossRef.
- J. Deng, D. Deng and X. Bao, Adv. Mater., 2017, 29, 1606967 CrossRef PubMed.
- W. Zhou, J. Jia, J. Lu, L. Yang, D. Hou, G. Li and S. Chen, Nano Energy, 2016, 28, 29–43 CrossRef CAS.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., 2013, 125, 389–393 CrossRef.
- L. Liang, J. Wang, W. Lin, B. G. Sumpter, V. Meunier and M. Pan, Nano Lett., 2014, 14, 6400–6406 CrossRef CAS PubMed.
- A. Castro-Neto and F. Guinea, Rev. Mod. Phys., 2009, 81, 109 CrossRef CAS.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- J. Deng, L. Yu, D. Deng, X. Chen, F. Yang and X. Bao, J. Mater. Chem. A, 2013, 1, 14868–14873 RSC.
- M. Miao, R. Hou, Z. Liang, R. Qi, T. He, Y. Yan, K. Qi, H. Liu, G. Feng and B. Y. Xia, J. Mater. Chem. A, 2018, 6, 24107–24113 RSC.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS.
- J. Chen, G. Xia, P. Jiang, Y. Yang, R. Li, R. Shi, J. Su and Q. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 13378–13383 CrossRef CAS PubMed.
- A. F. Pedersen, E. T. Ulrikkeholm, M. Escudero-Escribano, T. P. Johansson, P. Malacrida, C. M. Pedersen, M. H. Hansen, K. D. Jensen, J. Rossmeisl and D. Friebel, Nano Energy, 2016, 29, 249–260 CrossRef CAS.
- Y. Yang, Z. Lin, S. Gao, J. Su, Z. Lun, G. Xia, J. Chen, R. Zhang and Q. Chen, ACS Catal., 2017, 7, 469–479 CrossRef CAS.
- W. Zhou, T. Xiong, C. Shi, J. Zhou, K. Zhou, N. Zhu, L. Li, Z. Tang and S. Chen, Angew. Chem., Int. Ed., 2016, 55, 8416–8420 CrossRef CAS PubMed.
- A. Chen and C. Ostrom, Chem. Rev., 2015, 115, 11999–12044 CrossRef CAS PubMed.
- W. Ju, T. Brülle, M. Favaro, L. Perini, C. Durante, O. Schneider and U. Stimming, ChemElectroChem, 2015, 2, 547–558 CrossRef CAS.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk, I. Katsounaros and K. J. Mayrhofer, ChemCatChem, 2014, 6, 2219–2223 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach and A. Ludwig, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- G. C. Dismukes, R. Brimblecombe, G. A. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935–1943 CrossRef CAS PubMed.
- J. Wang, H. Wu, D. Gao, S. Miao, G. Wang and X. Bao, Nano Energy, 2015, 13, 387–396 CrossRef CAS.
- X. Chen, J. Xiao, J. Wang, D. Deng, Y. Hu, J. Zhou, L. Yu, T. Heine, X. Pan and X. Bao, Chem. Sci., 2015, 6, 3262–3267 RSC.
- B. J. Schultz, C. Jaye, P. S. Lysaght, D. A. Fischer, D. Prendergast and S. Banerjee, Chem. Sci., 2013, 4, 494–502 RSC.
- X. Liu, T. Pichler, M. Knupfer and J. Fink, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 245435 CrossRef.
- Y. Feng, X.-Y. Yu and U. Paik, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- J. Wang and F. Ciucci, Small, 2017, 13, 1604103 CrossRef PubMed.
- S.-W. Park, I. Kim, S.-I. Oh, J.-C. Kim and D.-W. Kim, J. Catal., 2018, 366, 266–274 CrossRef CAS.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- F. Hasché, M. Oezaslan and P. Strasser, ChemCatChem, 2011, 3, 1805–1813 Search PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund and H. Ogasawara, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- K. Mohanraju and L. Cindrella, RSC Adv., 2014, 4, 11939–11947 RSC.
- M. Oezaslan and P. Strasser, J. Power Sources, 2011, 196, 5240–5249 CrossRef CAS.
- R. Sgarbi, K. Kumar, F. Jaouen, A. Zitolo, E. A. Ticianelli and F. Maillard, J. Solid State Electrochem., 2019, 1–12 Search PubMed.
- M. M. Hossen, K. Artyushkova, P. Atanassov and A. Serov, J. Power Sources, 2018, 375, 214–221 CrossRef CAS.
- P. G. Santori, F. D. Speck, S. Cherevko, H. A. Firouzjaie, X. Peng, W. E. Mustain and F. Jaouen, J. Electrochem. Soc., 2020, 167, 134505 CrossRef CAS.
- H. Kishi, T. Sakamoto, K. Asazawa, S. Yamaguchi, T. Kato, B. Zulevi, A. Serov, K. Artyushkova, P. Atanassov and D. Matsumura, J. Nanomater., 2018, 8, 965 CrossRef PubMed.
- S. G. Peera, H. J. Kwon, T. G. Lee, J. Balamurugan and A. M. Hussain, Novel Catalyst Materials for Bioelectrochemical Systems: Fundamentals and Applications, 2020, pp. 231–278 Search PubMed.
- H. Lee, M. J. Kim, T. Lim, Y.-E. Sung, H.-J. Kim, H.-N. Lee, O. J. Kwon and Y.-H. Cho, Sci. Rep., 2017, 7, 1–8 CrossRef PubMed.
- H.-C. Huang, Y.-C. Lin, S.-T. Chang, C.-C. Liu, K.-C. Wang, H.-P. Jhong, J.-F. Lee and C.-H. Wang, J. Mater. Chem. A, 2017, 5, 19790–19799 RSC.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmeková and T. Asefa, Angew. Chem., 2014, 126, 4461–4465 CrossRef.
- C. Shuai, Z. Mo, X. Niu, Y. Du, Q. Gao, J. Liu, N. Liu and R. Guo, Appl. Surf. Sci., 2020, 532, 147381 CrossRef CAS.
- P. Wei, X. Sun, Q. Liang, X. Li, Z. He, X. Hu, J. Zhang, M. Wang, Q. Li and H. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 31503–31513 CrossRef CAS PubMed.
- C.-H. Liu, Y.-J. Tang, X.-L. Wang, W. Huang, S.-L. Li, L.-Z. Dong and Y.-Q. Lan, J. Mater. Chem. A, 2016, 4, 18100–18106 RSC.
- Z. Wang, S. Peng, Y. Hu, L. Li, T. Yan, G. Yang, D. Ji, M. Srinivasan, Z. Pan and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 4949–4961 RSC.
- Q. Zhang, M. Fu, G. Ning, Y. Sun, H. Wang, X. Fan, H. Lu, Y. Zhang and H. Wang, J. Colloid Interface Sci., 2020, 580, 794–802 CrossRef CAS PubMed.
- Y. Zhou, K. Neyerlin, T. S. Olson, S. Pylypenko, J. Bult, H. N. Dinh, T. Gennett, Z. Shao and R. O'Hayre, Energy Environ. Sci., 2010, 3, 1437–1446 RSC.
- H. R. Colón-Mercado and B. N. Popov, J. Power Sources, 2006, 155, 253–263 CrossRef.
- S. Rudi, D. Teschner, V. Beermann, W. Hetaba, L. Gan, C. Cui, M. Gliech, R. Schlögl and P. Strasser, ACS Catal., 2017, 7, 6376–6384 CrossRef CAS.
- A. S. Haile, W. Yohannes and Y. S. Mekonnen, RSC Adv., 2020, 10, 27346–27356 RSC.
- G. Wang, B. Huang, L. Xiao, Z. Ren, H. Chen, D. Wang, H. D. Abruña, J. Lu and L. Zhuang, J. Am. Chem. Soc., 2014, 136, 9643–9649 CrossRef CAS PubMed.
- M. Bele, P. Jovanovič, A. Pavlišič, B. Jozinović, M. Zorko, A. Rečnik, E. Chernyshova, S. Hočevar, N. Hodnik and M. Gaberšček, Chem. Commun., 2014, 50, 13124–13126 RSC.
- T. P. Johansson, E. T. Ulrikkeholm, P. Hernandez-Fernandez, P. Malacrida, H. Hansen, A. Bandarenka, J. Nørskov, J. Rossmeisl, I. Stephens and I. Chorkendorff, Top. Catal., 2014, 57, 245–254 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- J. Gao, M. Mao, P. Li, R. Liu, H. Song, K. Sun and S. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 6298–6308 CrossRef CAS PubMed.
- C. Galeano, C. Baldizzone, H. Bongard, B. Spliethoff, C. Weidenthaler, J. C. Meier, K. J. Mayrhofer and F. Schüth, Adv. Funct. Mater., 2014, 24, 220–232 CrossRef CAS.
- M. Karuppannan, Y. Kim, S. Gok, E. Lee, J. Y. Hwang, J.-H. Jang, Y.-H. Cho, T. Lim, Y.-E. Sung and O. J. Kwon, Energy Environ. Sci., 2019, 12, 2820–2829 RSC.
- K. Wang, R. Wang, H. Li, H. Wang, X. Mao, V. Linkov and S. Ji, Int. J. Hydrogen Energy, 2015, 40, 3875–3882 CrossRef CAS.
- H. Jiang, Y. Yao, Y. Zhu, Y. Liu, Y. Su, X. Yang and C. Li, ACS Appl. Mater. Interfaces, 2015, 7, 21511–21520 CrossRef CAS PubMed.
- L. Li, L. Song, H. Guo, W. Xia, C. Jiang, B. Gao, C. Wu, T. Wang and J. He, Nanoscale, 2019, 11, 901–907 RSC.
- M. Zeng, Y. Liu, F. Zhao, K. Nie, N. Han, X. Wang, W. Huang, X. Song, J. Zhong and Y. Li, Adv. Funct. Mater., 2016, 26, 4397–4404 CrossRef CAS.
- F. Aftab, H. Duran, K. Kirchhoff, M. Zaheer, B. Iqbal, M. Saleem and S. N. Arshad, Welcome to ICONN-2019, 2020, p. 35 Search PubMed.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grützke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- T. Reier, M. Oezaslan, P. Strasser and A. Catal, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef PubMed.
- M. Li, T. Liu, L. Fan, X. Bo and L. Guo, J. Alloys Compd., 2016, 686, 467–478 CrossRef CAS.
- Y. Wu, J. Meng, Q. Li, C. Niu, X. Wang, W. Yang, W. Li and L. Mai, Nano Res., 2017, 10, 2364–2376 CrossRef CAS.
- L. Zhang, J. Xiao, H. Wang and M. Shao, ACS Catal., 2017, 7, 7855–7865 CrossRef CAS.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- X. Zhang, H. Xu, X. Li, Y. Li, T. Yang and Y. Liang, ACS Catal., 2016, 6, 580–588 CrossRef CAS.
- H. Khani, N. S. Grundish, D. O. Wipf and J. B. Goodenough, Adv. Energy Mater., 2020, 10, 1903215 CrossRef CAS.
- N. Li, H. Tan, X. Ding, H. Duan, W. Hu, G. Li, Q. Ji, Y. Lu, Y. Wang and F. Hu, Appl. Catal., B, 2020, 266, 118621 CrossRef CAS.
- B. K. Barman and K. K. Nanda, ACS Sustainable Chem. Eng., 2018, 6, 12736–12745 CrossRef CAS.
- S. H. Noh, M. H. Seo, J. Kang, T. Okajima, B. Han and T. Ohsaka, NPG Asia Mater., 2016, 8, e312–e312 CrossRef CAS.
- M. Wang, C. Zhang, T. Meng, Z. Pu, H. Jin, D. He, J. Zhang and S. Mu, J. Power Sources, 2019, 413, 367–375 CrossRef CAS.
- Y. B. Adegbemiga, N. Ullah, M. Xie, S. Hussain, C. J. Oluigbo, W. Yaseen, A. J. Kumar, Y. Xu and J. Xie, J. Alloys Compd., 2020, 835, 155267 CrossRef CAS.
- M. R. Liu, Q. L. Hong, Q. H. Li, Y. Du, H. X. Zhang, S. Chen, T. Zhou and J. Zhang, Adv. Funct. Mater., 2018, 28, 1801136 CrossRef.
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef CAS PubMed.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- Z.-Y. Yu, Y. Duan, M.-R. Gao, C.-C. Lang, Y.-R. Zheng and S.-H. Yu, Chem. Sci., 2017, 8, 968–973 RSC.
- X. Long, J. Li, S. Xiao, K. Yan, Z. Wang, H. Chen and S. Yang, Angew. Chem., 2014, 126, 7714–7718 CrossRef.
- W. Ren, W. Zang, H. Zhang, J. Bian, Z. Chen, C. Guan and C. Cheng, Carbon, 2019, 142, 206–216 CrossRef CAS.
- L. Shen, T. Sun, O. Zhuo, R. Che, D. Li, Y. Ji, Y. Bu, Q. Wu, L. Yang and Q. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 16664–16669 CrossRef CAS PubMed.
- Q. Wang, F. Chen, Y. Liu, T. T. Gebremariam, J. Wang, L. An and R. L. Johnston, J. Power Sources, 2018, 404, 106–117 CrossRef CAS.
- G.-L. Li, B.-B. Yang, X.-C. Xu, S. Cao, Y. Shi, Y. Yan, X. Song and C. Hao, Chem. – Eur. J., 2020, 26, 2890–2896 CrossRef CAS PubMed.
- N. Wu, Y. Lei, Q. Wang, B. Wang, C. Han and Y. Wang, Nano Res., 2017, 10, 2332–2343 CrossRef CAS.
- X. Lv, J. Ren, Y. Wang, Y. Liu and Z.-Y. Yuan, ACS Sustainable Chem. Eng., 2019, 7, 8993–9001 CrossRef CAS.
- M. A. Ahsan, A. R. P. Santiago, M. F. Sanad and J. M. Weller, J. Colloid Interface Sci., 2021, 581, 905–918 CrossRef CAS PubMed.
- M. Khalid, A. M. B. Honorato, G. T. Filho and H. Varela, J. Mater. Chem. A, 2020, 8, 9021–9031 RSC.
- H. Guo, N. Youliwasi, L. Zhao, Y. Chai and C. Liu, Appl. Surf. Sci., 2018, 435, 237–246 CrossRef CAS.
- P. Jiang, J. Chen, C. Wang, K. Yang, S. Gong, S. Liu, Z. Lin, M. Li, G. Xia, Y. Yang, J. Su and Q. Chen, Adv. Mater., 2018, 30, 1705324 CrossRef PubMed.
- J. Su, Y. Yang, G. Xia, J. Chen, P. Jiang and Q. Chen, Nat. Commun., 2017, 8, 14969 CrossRef PubMed.
- F. Aftab, H. Duran, K. Kirchhoff, M. Zaheer, B. Iqbal, M. Saleem and S. N. Arshad, ChemCatChem, 2020, 12, 932–943 CrossRef CAS.
- J. Yu, Y. Zhong, W. Zhou and Z. Shao, J. Power Sources, 2017, 338, 26–33 CrossRef CAS.
- B. Du, Q.-T. Meng, J.-Q. Sha and J.-S. Li, New J. Chem., 2018, 42, 3409–3414 RSC.
- S. Saha and A. K. Ganguli, ChemistrySelect, 2017, 2, 1630–1636 CrossRef CAS.
- L. Yang, D. Wang, Y. Lv and D. Cao, Carbon, 2019, 144, 8–14 CrossRef CAS.
- G. Zhong, S. Li, S. Xu, W. Liao, X. Fu and F. Peng, ACS Sustainable Chem. Eng., 2018, 6, 15108–15118 CrossRef CAS.
- B. Wang, Y. Ye, L. Xu, Y. Quan, W. Wei, W. Zhu, H. Li and J. Xia, Adv. Funct. Mater., 2020, 2005834 CrossRef CAS.
- T. Li, Y. Lu, S. Zhao, Z.-D. Gao and Y.-Y. Song, J. Mater. Chem. A, 2018, 6, 3730 RSC.
Footnote |
| † These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |