
Exosome-mediated delivery of gene vectors for gene therapy
Li
Duan
a,
Limei
Xu
a,
Xiao
Xu
a,
Zhuan
Qin
a,
Xiaoying
Zhou
a,
Yin
Xiao
 b,
Yujie
Liang
*c and
Jiang
Xia
*d
b,
Yujie
Liang
*c and
Jiang
Xia
*d
aDepartment of Orthopedics, Shenzhen Intelligent Orthopaedics and Biomedical Innovation Platform, Guangdong Artificial Intelligence Biomedical Innovation Platform, Shenzhen Second People's Hospital, the First Affiliated Hospital of Shenzhen University Health Science Center, Shenzhen, 518035, China
bInstitute of Health and Biomedical Innovation, Faculty of Science and Engineering, Queensland University of Technology, Kelvin Grove Campus, Brisbane, QLD 4059, Australia
cDepartment of Child and Adolescent Psychiatry, Shenzhen Kangning Hospital, Shenzhen Mental Health Center, Shenzhen Key Laboratory for Psychological Healthcare and Shenzhen Institute of Mental Health, Shenzhen, Guangdong, China. E-mail: liangyjie@126.com
dDepartment of Chemistry, and Center for Cell & Developmental Biology, School of Life Sciences, the Chinese University of Hong Kong, Shatin, Hong Kong SAR, China. E-mail: jiangxia@cuhk.edu.hk
First published on 8th December 2020
Abstract
Gene vectors are nucleic acids that carry genetic materials or gene editing devices into cells to exert the sustained production of therapeutic proteins or to correct erroneous genes of the cells. However, the cell membrane sets a barrier for the entry of nucleic acid molecules, and nucleic acids are easily degraded or neutralized when they are externally administered into the body. Carriers to encapsulate, protect and deliver nucleic acid molecules therefore are essential for clinical applications of gene therapy. The secreted organelles, exosomes, which naturally mediate the communications between cells, have been engineered to encapsulate and deliver nucleic acids to the desired tissues and cells. The fusion of exosomes with liposomes can increase the loading capacity and also retain the targeting capability of exosomes. Altogether, this review summarizes the most recent designs of exosome-based applications for gene delivery and their future perspectives in gene therapy.
 Li Duan | Dr Li Duan received her master degree from Kunming Medical University in 2004, and her PhD degree from the School of Stomatology, Wuhan University, in 2007. Now she is a full Research Professor in Guangdong Artificial Intelligence Biomedical Innovation Platform at Shenzhen Second People's Hospital, the First Affiliated Hospital of Shenzhen University Health Science Center. Her research focuses on biomaterials and osteoarthritis treatment. |
 Yujie Liang | Dr Yujie Liang received a PhD degree in Chemistry from The Chinese University of Hong Kong in 2020 under the supervision of Prof. Jiang Xia. He is currently a Principal Investigator at the Department of Child and Adolescent Psychiatry, Shenzhen Kangning Hospital and Shenzhen Mental Health Center. Currently, his interest is to engineer exosomes for tissue repair and gene therapy of neurological disorders. |
 Jiang Xia | Dr Jiang Xia received a PhD degree from Stanford University, USA, in 2006. Now he is an Associate Professor in the Department of Chemistry, the Chinese University of Hong Kong. His research interest lies in chemical biology and synthetic biology, with a focus on subcellular structures, such as organelles and protein coacervates, and their biocatalytic and biomedical applications. |
1. Introduction
Gene therapy is a form of therapy that treats genetic diseases by introducing genetic materials such as the expression system of a therapeutic protein or an editing tool to correct the erroneous gene in the diseased cells.1,2 The advancement of this field has brought hope to the treatment of previously incurable diseases, such as genetic disorders,3 cancers,4 and autoimmune diseases.5 To date, at least 943 clinical trials of gene therapy have been undertaken worldwide.Delivery is essential for gene therapy. The cell membrane sets an impermeable barrier to the negatively charged nucleic acids. Also, genetic materials are fragile in the human body, so they must be protected to take effect. mRNAs and viral or nonviral plasmid DNAs are commonly the genetic materials used in gene therapy. For example, the adeno-associated virus (AAV) vector is popularly used.6 However, the AAV vector induces a host immune response, which results in the rapid degradation or neutralization of the vector and seriously compromises its efficiency for gene therapy.7,8 Encapsulation in nanostructures, such as exosomes, protects the AAV vectors from the host immune system and delivers them into cells across the plasma membrane.9,10 This review describes the development of exosomes as a carrier for encapsulation and delivery of gene vectors to cells and the related disease treatment (Fig. 1). The pros and cons of exosomes as carriers for various genetic materials, strategies to engineer exosomes for increased loading and delivery, and methods to maximize the production of gene-loaded exosomes, together with the future perspectives on exosome-mediated targeted gene therapy, will be discussed.
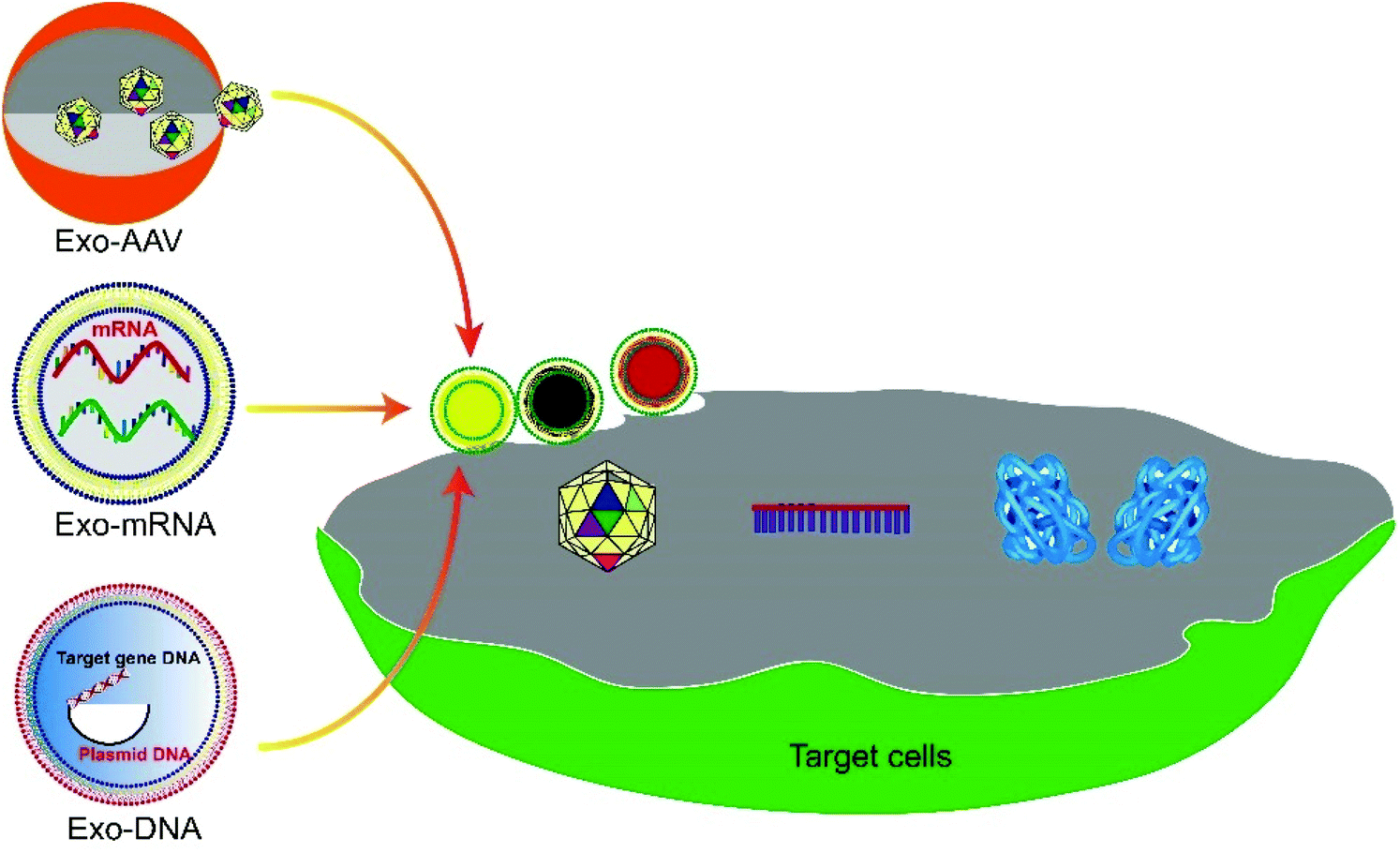 | ||
| Fig. 1 Schematic illustration of exosome-mediated gene delivery strategies. Functionalized exosome encapsulated gene vectors, such as AAV, DNA and mRNA, deliver to target cells and exert functional therapies by translating into proteins, RNA interference, genome editing, etc. | ||
2. Gene therapy
The history of gene therapy dated back to the 1960s, when Joshua Lederberg first presented the concept and laid the foundation for treating diseases with genes.11 In 1972, gene therapy was formally proposed and accepted as a therapeutic method for genetic diseases in humans.12 In 1989, the National Institutes of Health approved the first clinical trial of gene therapy which used retrovirus labeling to detect tumour homing in vivo.13 This study demonstrated that human cells can be genetically engineered in vitro and then be returned safely to the patient's body.13 The first successful clinical study of gene therapy in vivo was reported in 1995.14 Autologous leukocytes from patients with severe combined immune deficiency syndrome (SCID) were corrected by retrovirus reinfusion. However, further development of gene therapy experienced setbacks due to adverse effects caused by the adenovirus-induced lethal immune response and malignancy. The advent of viral vectors (such as lentivirus and AAV) and gene-editing tools has overcome the drawbacks and broadened the applications of gene therapy since then. To date, 943 clinical trials of gene therapy have been launched worldwide. Initially used only for genetic disorders, now gene therapies have extended in the treatment of cancers, infectious diseases, cardiovascular diseases, and autoimmune diseases.15–17 Since the China Food and Drug Administration (CFDA) approved the first-ever gene therapy product Gendicine (recombinant human p53 adenovirus) to treat head and neck squamous cell carcinoma (HNSCC) in 2003, nine products have been approved by the European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA) (Table 1).18–29| Products | Approval Organization | Approval time | Diseases | Working principles | Bottleneck |
|---|---|---|---|---|---|
| N/A, not available. | |||||
| Gendicine | CFDA | 2003 | Head and neck squamous-cell carcinoma (HNSCC) | Recombinant human p53 adenovirus18 | High immunogenicity29 |
| Glybera | EMA | 2012 | Lipoprotein lipase deficiency (LPLD) | Adeno-associated virus (AAV) carries the functional LPL gene19 | 1. Immune response |
| 2. Low packaging capacity | |||||
| 3. Random vector genome insertion28 | |||||
| Imlygic | FDA | 2015 | Melanoma | Using Oncolytic, engineered herpes virus to anticancer20 | Immune response29 |
| Zalmoxis | EMA | 2016 | Leukemia and lymphoma | Treatment with donor T cells were genetically modified to express the herpes simplex thymidine kinase (HSV-TK) suicide gene21 | Immune response21 |
| Strimvelis | EMA | 2016 | ADA-SCID | Using retroviral vector to insert a functional copy of the gene ADA to CD34 + cells22 | 1. Immune response |
| 2. Random vector genome insertion28 | |||||
| Spinraza | EMA | 2016 | Spinal muscular atrophy | Using an antisense oligonucleotide to alter the splicing of the survival motor neuron (SMN) gene23 | Unstable molecules23 |
| Kymriah | FDA | 2017 | Non-Hodgkin's lymphoma (NHL) | Chimeric antigen receptor T-cell immunotherapy targets CD19 antigen24 | 1. Immune response |
| 2. Lack of targetable tumour-specific surface antigens | |||||
| 3. Tumour cell heterogeneity29 | |||||
| Luxturna | FDA | 2017 | Leber congenital amaurosis | Using AAV to deliver normal human RPE65 gene to retinal cells25 | 1. Immune response |
| 2. Low packaging capacity | |||||
| 3. Random vector genome insertion28 | |||||
| Yescarta | FDA | 2017 | Diffuse large B-cell lymphoma (DLBCL); primary mediastinal large B-cell lymphoma (PMBCL) | Chimeric antigen receptor T-cell immunotherapy targets CD19 antigen26 | 1. Immune response |
| EMA | 2018 | 2. Lack of targetable tumour-specific surface antigens | |||
| 3. Tumour cell heterogeneity29 | |||||
| Zynteglo | EMA | 2019 | Trans-fusion-dependent β thalassemia | Autologous CD34 + cells encoding βA-T87Q-globin gene in vitro using a lentiviral vector27 | 1. Immune response |
| 2. Random vector genome insertion28 | |||||
| CTX001 | FDA | 2019 | β-thalassemia and sickle anemia | Using the CRISPR gene-editing technology to increase the production of fetal hemoglobin in patients’ red blood cells | N/A |
| EDIT-101 | FDA | 2017 | Leber congenital amaurosis 10 | Eliminating a mutation in the CEP290 gene using CRISPR | N/A |
The development of CRISPR-Cas9 gene editing technology, known as the “magic scissors” of DNA, has markedly spurred the development of gene therapy.30–34 Scientists can now efficiently and accurately change, delete, or replace the genes of plants,35,36 animals,37,38 and even humans.39 Two CRISPR-based therapeutics have entered clinical trials: CTX001 to treat rare genetic diseases, β-thalassemia and sickle anemia, and EDIT-101 to treat Leber congenital amaurosis 10, a rare inherited eye disease that causes blindness.40
3. Exosome-associated AAV vectors for gene therapy
Based on the source, gene delivery carriers can be categorized as viral or non-viral systems (Fig. 2). Virus-mediated gene delivery systems use modified viruses that are replication-deficient but can deliver DNA for expression, including adenoviruses, retroviruses, and lentiviruses.41 Traditional non-viral vehicles include micelles,42,43 liposomes, dendrimers,44,45 and carbon nanotubes.46 Recently, extracellular vesicles secreted by cells, especially exosomes, have been used as non-viral vehicles for gene delivery and show features including high biocompatibility, low clearance rate, and amenability for cell-targeted delivery.47–50 Hybrid gene delivery technologies, such as the exosome–AAV hybrid, combine the virus vectors with non-viral vehicles and show significantly reduced immunogenicity.51,52 In this review, we focus on gene delivery carriers based on viral vesicles and exosomes.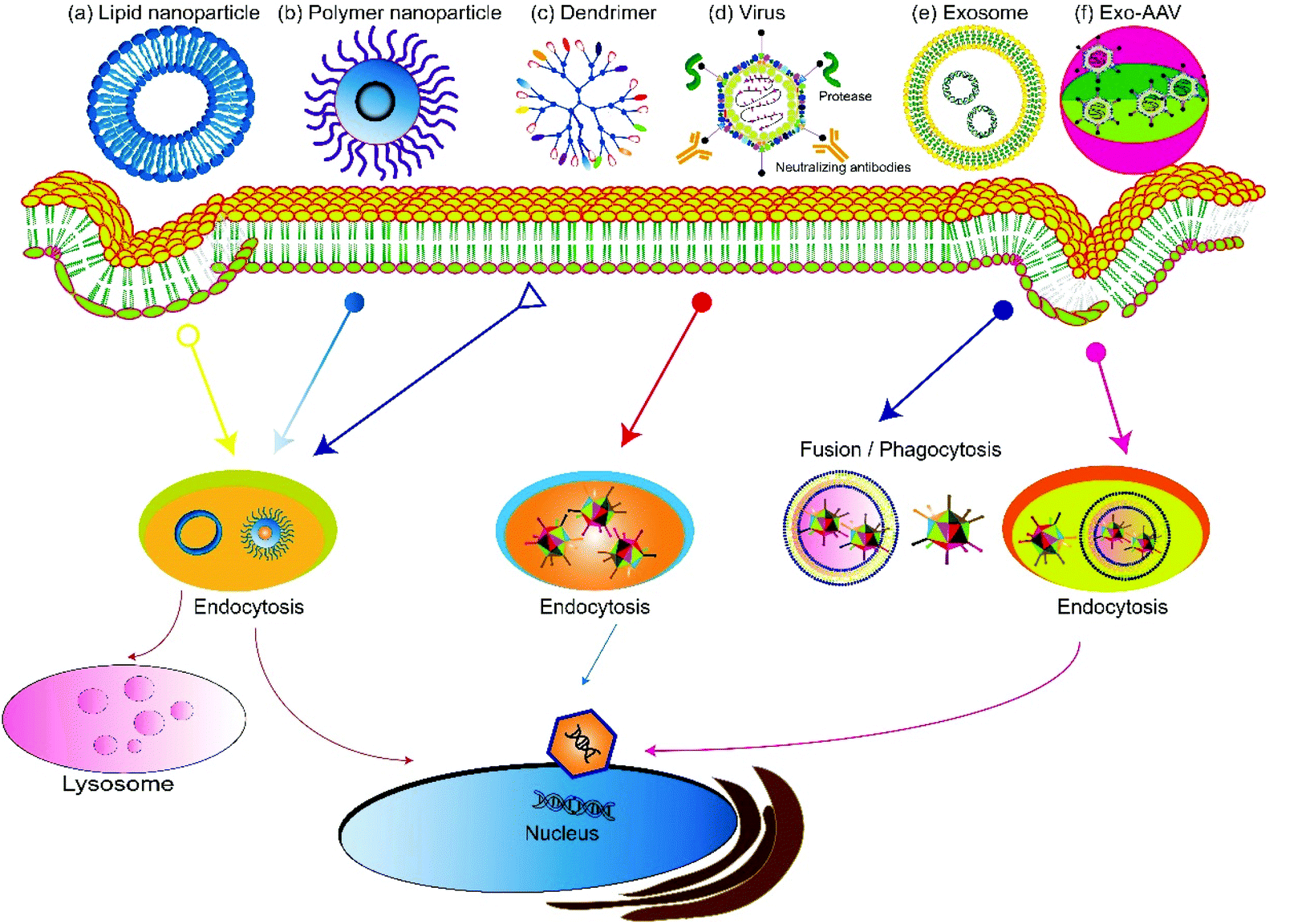 | ||
| Fig. 2 A summary of gene delivery carriers. Current gene delivery carriers contain three major categories: non-viral vectors, viral-vectors, and combined hybrid vectors. Commonly used non-viral vectors include synthetic vectors, such as lipid nanoparticles, polymer nanoparticles, and dendrimers, and biological vectors, such as exosomes. A hybrid gene delivery system based on exosome-associated AAV (Exo-AAV). Most synthetic vectors mainly enter endosomes and later lysosomes. Exosomes and Exo-AAVs enter cells via multiple pathways, such as membrane fusion and various types of endocytosis pathways such as phagocytosis, macropinocytosis, and lipid raft-dependent and clathrin-/caveolin-mediated endocytosis. | ||
3.1 AAV vectors
Adeno-associated viruses (AAV) are small viruses that infect humans and some other primate species. A single-stranded AAV DNA with a genome size of 4.7 kb encoding three genes, Rep (Replication), Cap (Capsid), and aap (Assembly), is packed as an icosahedron. The two ends of the genes are inverted terminal repeats (ITRs). In recombinant AAV (rAAV) used as a gene vector, all the viral protein genes have been deleted, and only the terminal ITR sequences associated with viral replication and packaging are retained. Therapeutic transgenes and regulatory elements are inserted between the ITRs. Deleting the viral protein gene maximizes the transgene capacity of AAV and reduces immunogenicity and cytotoxicity. Because the rAAV vector does not express the REP protein, its ability to integrate into the host genome is significantly reduced. Furthermore, the rAAV genome may form a long-standing circular tandem in the nucleus, thus realizing long-term transgene expression. Unlike lentiviral vectors, adenoviral vectors, retrovirus vectors, and other commonly used viral vectors, AAV vectors do not produce harmful infections without a helper virus. Therefore, AAV has become the primary vector in these applications because of its low immunogenicity, low gene toxicity, diverse capsid proteins, long duration of action, and ease of production.3.2. Exosomes for gene delivery
Exosomes are lipid bilayer-enclosed vesicles of 40–160 nm in diameter that circulate in the extracellular environment. They originate from the internal budding of the late endosomal membrane and are released by all cell types. Therefore, exosomes are natural carriers for cell–cell communication. This feature then allured researchers to develop exosome-based drug delivery systems. Compared with other gene delivery systems, exosomes are advantageous due to the following reasons. First, exosomes are versatile carriers. A variety of biological cargos such as small RNAs, mRNAs, and proteins can be encapsulated and delivered by exosomes. Second, exosomes possess the natural capacity of traversing biological barriers, for example the blood–brain barrier. They can also migrate into the tissues or areas with no blood supply, e.g. the dense cartilage matrix. Furthermore, after arriving at the target tissue, exosomes can remain there for a long period of time; the low clearance rate can be ascribed to the biocompatibility of exosomes. Another important feature is that exosomes are also genetically engineerable. Multiple purposes can be achieved by engineering the surface proteins of exosomes, for example, cell or tissue-targeting peptides can be appended on the surface of exosomes to realize selective targeting to specific tissues and avoid unwanted accumulation in other organs, thus decreasing systemic toxicity. Studies have demonstrated that exosome-based targeted gene therapies can enhance both therapeutic efficacy and safety. For example, we engineered an exosome-based chondrocyte-targeted miRNA delivery system for cartilage defect repair.53 In this work, a chondrocyte-affinity peptide (CAP) was expressed on the surface of exosomes, and miR-140 was loaded inside the exosome. Intra-articular injection of the CAP-exosome/miRNA composites showed selective and long-term enrichment in the cartilage and a significant increase of therapeutic efficacy toward cartilage defect as compared to miRNA encapsulated in exosomes without surface modification.3.3 Exosome-associated AAV for gene therapy
Association with exosomes can alleviate the toxicity and mitigate the rapid clearance of AAV vectors. AAV vectors suffer from drawbacks such as rapid clearance by neutralizing anti-AAV antibodies in the body and off-target delivery (often to the liver). Maguire et al. demonstrated in 2012 that a fraction of AAV vectors is associated with microvesicles/exosomes.54 AAV capsids were found to associate on the surface and in the interior of microvesicles under an electron microscope. Exosome-associated AAVs (Exo-AAVs) purified from cell medium (Fig. 3) outperformed conventionally purified AAV vectors in transduction efficiency. One marked problem of the AAV vector is that it induces a potent long-term humoral response, including neutralizing antibodies (NAbs), which bind with the AAV vectors and inhibit the transduction, and non-Nabs, which flag the virus. Therefore, NAbs adversely affect the efficacy of AAV-mediated gene therapy and the AAV vector distribution and safety.55 In contrast, exo-AAVs are more resistant to neutralizing anti-AAV antibodies as compared to the conventional AAVs. This has been experimentally proven. Exosome-associated AAV vectors were much more resistant to NAbs than AAV both in vitro and in vivo and AAV9Exo-SERCA2a outperformed the conventional AAV vectors in preserving cardiac functions both in the presence and absence of Nabs.56 Exo-AAV8 gene delivery also decreased the therapeutic dose of the vector and achieved efficient transduction even in the presence of moderate NAb titers.57 Exo-AAVs also showed higher transduction efficacy in the central nervous system and auditory and vestibular systems.51,54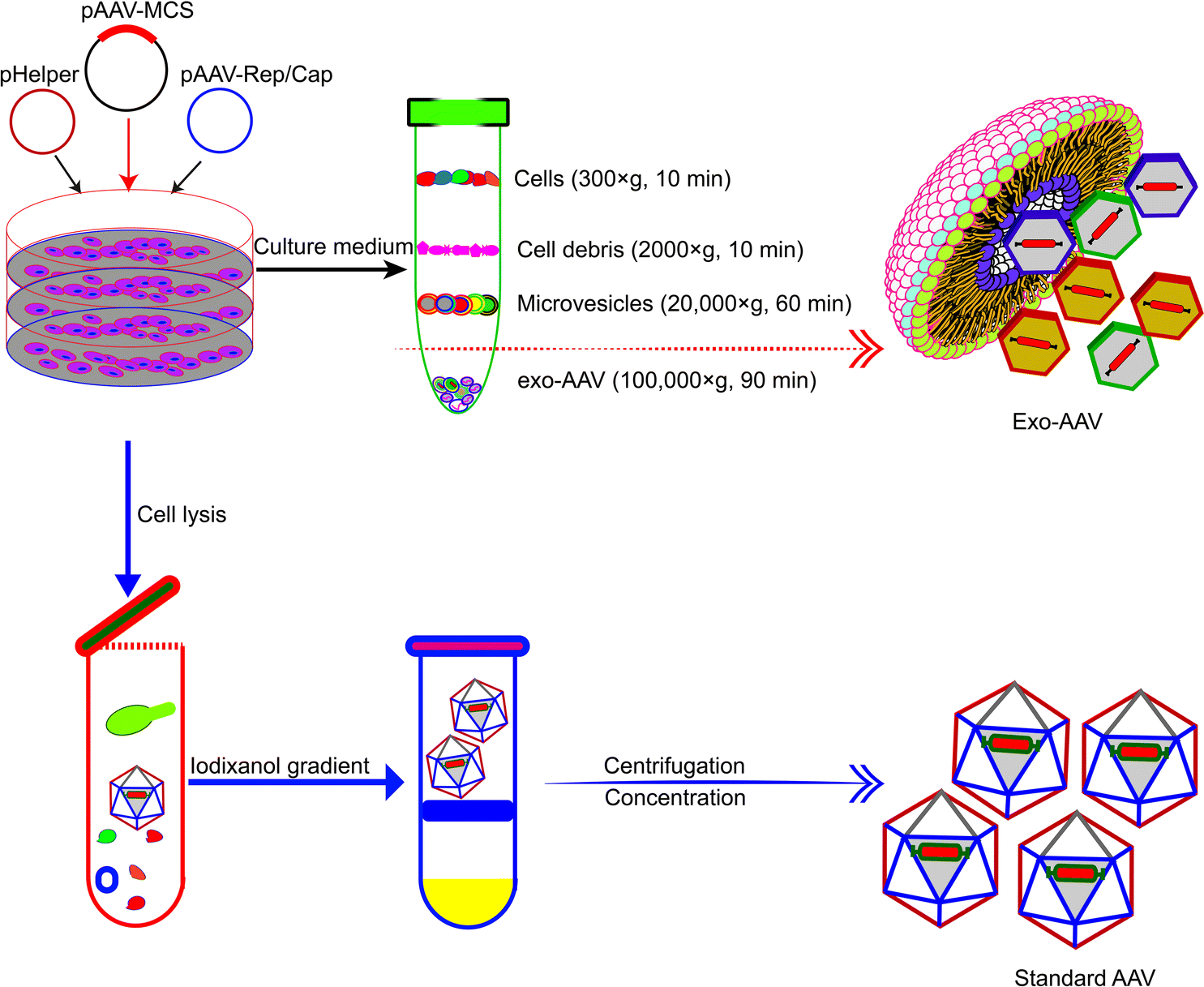 | ||
| Fig. 3 Schematic diagram showing the different procedures for the production and purification of Exo-AAVs (upper) and AAVs alone (lower), respectively.54 Exo-AAVs are harvested from cell medium and purified by differential centrifugation, whereas AAVs are purified from cell lysates by iodixanol density gradient and ultra-centrifugation. | ||
The association of AAV with exosomes also achieves targeted delivery. In one example, magnetic targeting can be realized by expressing a biotin acceptor peptide with a transmembrane receptor (BAP-TM) on their surface by transducing 293T cells with a lentivirus encoding BAP-TM. The vector–exosome complexes, termed vexosomes by the authors, were then biotinylated by a biotin ligase enzyme BirA. The biotinylated vector–exosome complexes then bind with streptavidin-functionalized magnetic beads, and can be guided by a magnetic field.58 The genetic fusion of a rabies virus glycoprotein (RVG) peptide with the platelet-derived growth factor receptor transmembrane domain results in Exo-AAVs that can deliver cargoes to the brain more efficiently than untagged EV-AAVs.59 Similar strategies can be used to develop Exo-AAVs for myocardial gene delivery.56,59,60
The production of Exo-AAVs is also less costly. Compared with the complex purification protocol of AAVs, the purification of Exo-AAVs can be simply done by pelleting using ultra-centrifugation (Fig. 3).61 In addition, it has also been shown that overexpression of CD9 can enhance the production of Exo-AAV which increased the hope to further reduce the cost of Exo-AAVs for clinical trials.62 These benefits make Exo-AAV a promising platform for gene therapy. Here we summarize in Table 2 the reported Exo-AAV systems for gene therapy.51,52,54,56–61,63–65
| Exo-AAV constructs | Delivery target | Advantages |
|---|---|---|
| N/A, not available. | ||
| Exo-AAV6-GFP, Exo-AAV9-GFP | Brain | Efficient gene delivery tool in brain tissue51,52 |
| Exo-AAV1-Fluc, Exo-AAV2-GFP | Cultured cells | Improved gene delivery and magnetic targeting of exosomes54 |
| Exo-AAV1-GFP | Inner-ear hair cells | Efficient gene delivery tool and no toxicity58 |
| Exo-AAV2-GFP | Murine retina | Convenient and powerful tool for intravitreal gene transfer in vivo61 |
| Exo-AAV5-hF.IX, Exo-AAV8-hF.IX | Liver | Enhanced gene transfer and liver-targeted efficacy in vivo57 |
| Exo-AAV9-SERCA2a | Cardiomyocytes | High-efficient myocardial gene delivery, minimized susceptibility to neutralizing antibodies56 |
| Exo-AAV6-mCherry, Exo-AAV9-FLuc | Cardiovascular | Neutralization of anti-AAV antibodies and more efficient gene transfer for cardiovascular therapeutics60 |
| Exo-AAV9-FLuc | Brain | Brain-targeted delivery and evasion of neutralizing antibodies59 |
| Exo-AAV6-iCasp9 | Hepatocellular carcinoma | Enhanced delivery efficiency of a suicide gene for tumour suppression63 |
| Exo-AAV8-GFP | Lymphocytes | In vivo gene modification of immune cells64 |
| Exosome-associated SUMOylation mutant AAV2 | Ocular | Enhanced permeation ability across the retinal layers65 |
4. Exosome-mediated delivery of CRISPR-Cas9
Guided by the CRISPR sequence, the Cas9 enzyme can edit genomic DNA with unprecedented precision, efficiency, and flexibility in the cells. However, the CRSPR-Cas9 system designed ex vivo needs to be delivered into the targeted cell and organisms for gene therapy. The delivery of the CRISPR-Cas9 system can be enabled by physical interaction, chemical modification, and biological carriers. The CRISPR-Cas9 system can be delivered into the cell through a transient gap in the lipid bilayer caused by physical methods such as electroporation, hydrodynamic injection, membrane deformation, and others. Although simple and easy to perform, irreversible physiological damage can be caused to the cell membrane. Chemical modification of CAS9-sgRNA will also enable its delivery to the cells or biological carriers such as bacteriophages, viruses, and extracellular vesicles can mediate the delivery of CRISPR-Cas systems into the cells (Table 3).| Exosome | Lipid nanoparticle | Polymer nanoparticle | Dendrimer | Virus | |
|---|---|---|---|---|---|
| Biocompatibility | Excellent | Low | May accumulate undesirably in the liver, spleen etc. | Non-toxic | Poor |
| Loading efficiency | High | N/A | N/A | N/A | High |
| Targeting engineerability | Yes | Yes | Yes | Yes | Yes |
4.1 Exosomes as carriers for CRISPR-Cas9 plasmids
Compared to viral carriers, exosomes from certain cell sources can selectively deliver the cargo to tumor tissue.66 Kim et al. reported that cancer cell-derived exosomes could transport the CRISPR-Cas9 system to target tumors.67 The CRISPR-Cas9 system targeting poly (ADP-ribose) polymerase-1 (PARP-1) was electroporated into exosomes from the ovarian cancer cell line SKOV3 and the epithelial cell line HEK293 and delivered into SKOV3 cancer cells and HEK293. Compared with epithelial cell-derived exosomes, cancer-derived exosomes can selectively accumulate in the ovarian cancer tumors of SKOV3 xenograft mice, enhance the chemosensitivity to cisplatin and enable the synergistic killing of tumor cells.67 This study shows that cell tropism can guide the tumor-origin exosomes to cancer cells, suggesting a targeting effect of tumor-specific proteins on the surface of exosomes. Despite the tumor-targeting effect, tumor-origin exosomes carry intracellular materials that may promote tumor growth and progression. This then highlights the balance of pros and cons in choosing exosomes from different cell types for cargo carrying and delivery.4.2 Engineering exosomes for CRISPR-Cas9 delivery
Natural exosomes are not designed to deliver giant molecules such as plasmids; therefore, the loading of plasmids in exosomes is low. Exosomes then need to be engineered to increase the efficiency and capacity of plasmids.68 Engineering exosomes also endows these delivery nanoparticles with the targeting capability to deliver the cargo to specific cells or tissues. CD63 and CD9, marker proteins expressed on the surface of exosomes, are often chosen as the target proteins for exosome engineering. For example, Ye et al. produced engineered exosomes (M-CRISPR-Cas9 exosomes) that can efficiently load CRISPR-Cas9 components through protein–protein interactions. The authors fused GFP with CD63, and a GFP nanobody with the Cas9 protein. The interaction between GFP and the GFP nanobody guides the CRISPR-Cas9 system into the exosomes and increases the loading capacity.69 The increase of gene delivery was proven in a reporter cell line that the authors engineered. In another example, Li et al. fused CD9 with an RNA binding protein HuR, which is known to bind RNA molecules with AU rich elements (AREs) such as miR-155. Engineered exosomes containing CD9-HuR enriched miR-155 into exosomes and delivered the cargo into the recipient cells. The CD9-HuR exosome can also enrich CRISPR-dCas9 that is engineered with AU rich elements. 3× AREs were cloned downstream of the dCas9 stop codon, and this significantly enhanced the encapsulation efficiency of dCas9 mRNA into CD9-HuR exosomes.70 The CRISPR-dCas9 RNA was successfully delivered and exerted its function both in vitro and in vivo. Recently, Gee et al. have developed a transient delivery system for therapeutic genome editing by utilizing two homing mechanisms: Cas9 protein was recruited to the vesicles by chemical induced dimerization, and sgRNA was introduced to the vesicles by a viral RNA packaging signal and two self-cleaving riboswitches. Efficient genome editing was then achieved in various hard-to-transfect cell types, including human induced pluripotent stem (iPS) cells, neurons, and myoblasts.714.3 Exosome–liposome hybrids for gene delivery
The nanoscale size of exosomes (40–160 nm) makes them qualified vehicles for small therapeutic agents, such as chemical compounds, siRNAs, and miRNAs. For large molecules, such as CRISPR-Cas9 expressing plasmids with a minimal size of 5–6 kb, the capacity of native exosomes is still relatively low.72 Liposomes, on the other hand, are capable of encapsulating and delivering large plasmids, but demonstrate relatively high cytotoxicity due to the unnatural nature of the lipids. The fusion of the lipid bilayers of the exosomal membrane and the liposome forms an exosome–liposome hybrid, which enables the encapsulation and delivery of large DNA molecules, such as the CRISPR-Cas9 expression plasmids, and also mitigates the toxicity problem of liposomes (Fig. 4).73 Therefore, the exosome–liposome hybrid expands the application scope of exosomes in drug delivery. For example, CRISPR-Cas9-sgRUNX2 was encapsulated in exosome–liposome hybrid vesicles, and delivered to human bone marrow mesenchymal stem cells which cannot be transfected by the liposome alone.71 In another example, Lv and co-workers developed genetically engineered exosome–thermosensitive liposome hybrid nanoparticles (gETL NPs) to deliver granulocyte-macrophage colony-stimulating factor (GM-CSF) and docetaxel (a chemoimmunotherapy drug) to large tumor nodules that cause metastatic peritoneal carcinoma, a deadly disease without effective treatment. The authors showed that combining the locoregional delivery of hyperthermic intraperitoneal chemotherapy (HIPEC) and systemic delivery of chemoimmunotherapy via gETL NPs could significantly improve the disease treatment.74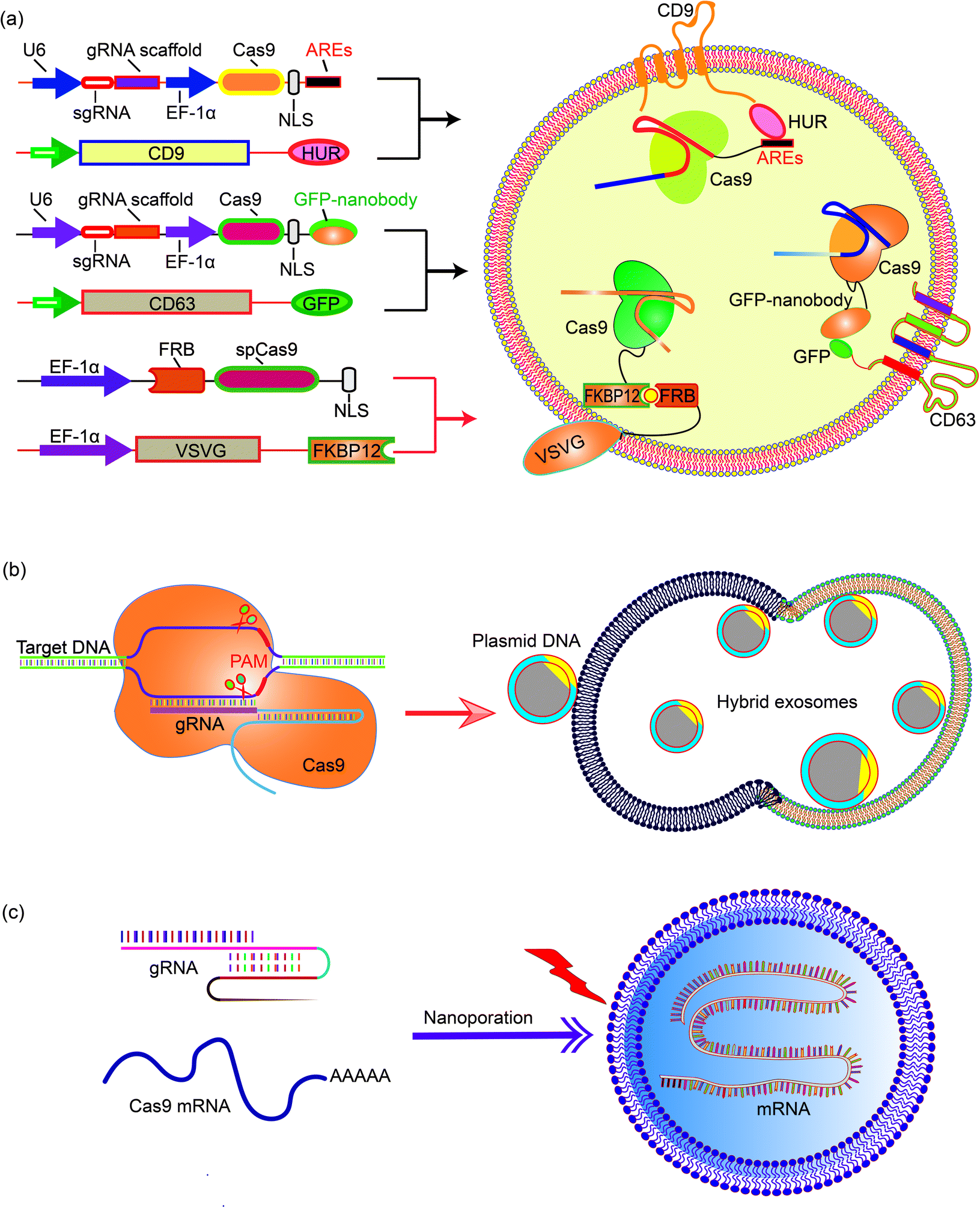 | ||
| Fig. 4 Encapsulating large nucleic acids in exosome or exosome-derived vesicles. Strategies to realize efficient encapsulation include (a) interaction mediated loading of the Cas9 protein in exosomes,69–72 (b) enriching CRISPR-Cas9 plasmid in the exosome–liposome hybrid,71 and (c) nanoporation to deliver mRNA molecules into exosomes.76 | ||
5 Exosome-mediated mRNA delivery for gene editing
Encapsulation of large mRNA transcripts to exosomes is a challenging task. Although native secreted exosomes contain multiple RNAs, including miRNAs, ncRNAs and fragments, most of the fragments are small.75 Large mRNA molecules can be transferred to exosomes from erythrocytes by electroporation, but this process is still inefficient.50 In view of this obstacle, Yang et al. presented an approach to generate large-scale functional mRNA-encapsulating exosomes.76 In this work, a cellular nanoporation biochip with nanochannels of about 500 nm diameter was developed. The authors transfected cells with plasmid DNAs and stimulated the transfected cells with a focal and transient electrical stimulus. This nanoporation procedure promoted the release of exosomes carrying transcribed mRNAs and targeting peptides from cells, leading to up to 50-fold more exosomes and a more than 103-fold increase in exosomal mRNA transcripts, reaching estimably 2–10 intact, functional mRNAs per exosome.6 Conclusion and perspectives
Tissue or cell-specific delivery of the gene vectors by designed carriers enables an advanced version of gene therapy with reduced toxicity or risk. The carriers from cell sources, exosomes, can mediate the delivery of various gene therapy molecules or vectors. In particular, genetic or chemical engineering of exosomes allows cell-specific recognition sequences to be installed on the exterior of exosomes to guide the targeted delivery of the cargos or exosome marker proteins can be engineered to enrich target proteins through a designed protein–protein interaction. Besides, engineering exosomes with nucleic acid-binding properties also will help the encapsulation of the plasmids or vectors and enhance the loading efficiency. Fusing exosomes with liposomes will create vesicles that can encapsulate large plasmids or mRNA transcripts while retaining the advantages of exosomes such as biocompatibility and targeting capability. As an important step towards clinical trials, progress has also been made to realize the large-scale production of exosomes or drug-loaded exosomes. Breakthroughs in instruments and procedures such as nanoporation also solve the bottleneck of loading and production. Taken together, with the advancement and optimization of gene editing tools, the continuous development of exosome-based carrier systems will power the targeted gene therapies as accessible treatments for difficult diseases in the future, through both the in vitro and in vivo routes (Fig. 5).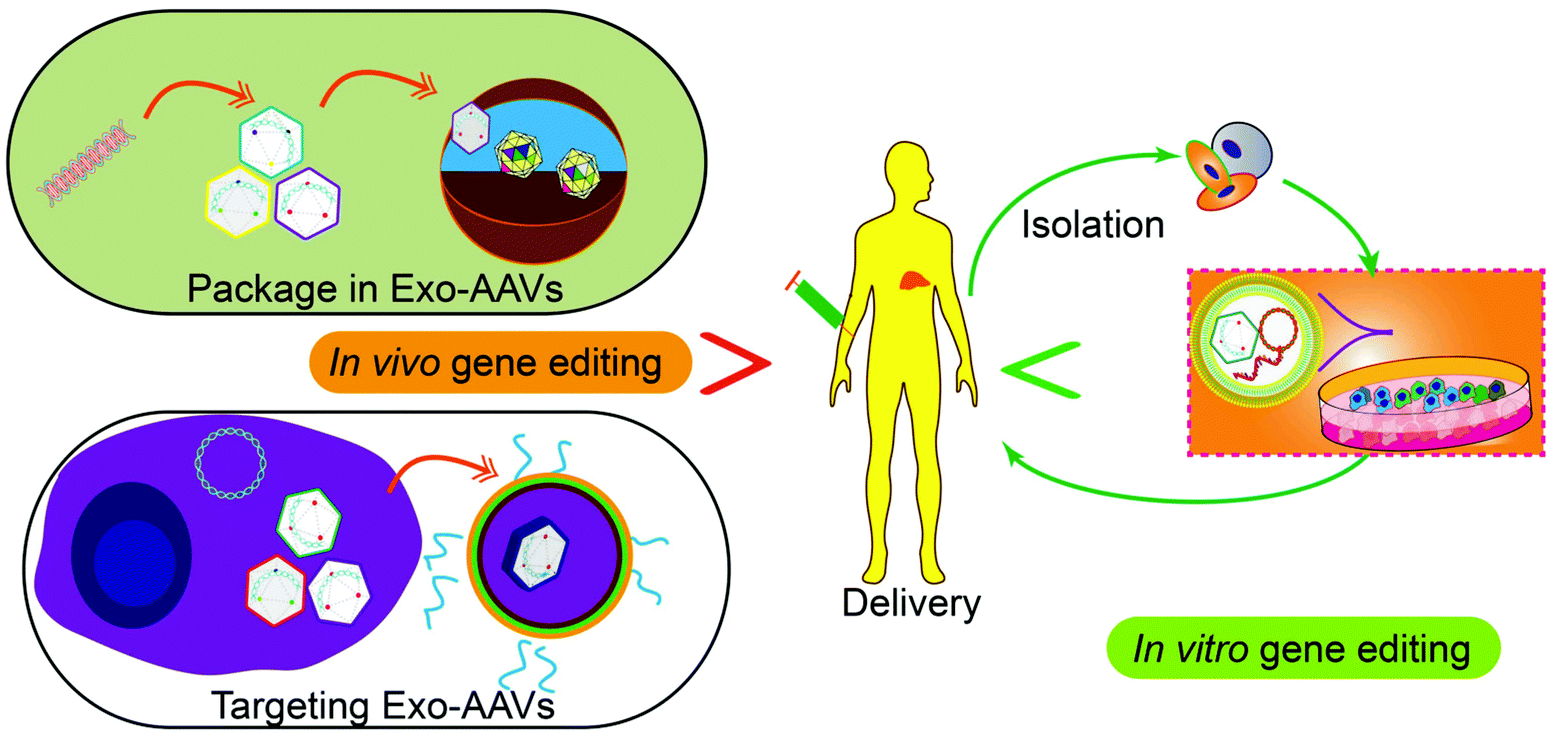 | ||
| Fig. 5 Potential future applications of gene therapy. Nucleic acid-based therapeutics can be packaged in Exo-AAVs, or Exo-AAVs engineered with targeting capability can be used for gene delivery into human bodies for gene editing in vivo. Alternatively, the cells isolated from patients can be edited by Exo-AAVs carrying gene-editing tools in vitro and then returned to the patients for disease treatment. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. L., Y. J., and J. X designed and wrote the paper. This work was supported by the National Natural Science Foundation of China (81772394 and 81972116), the Natural Science Foundation of Guangdong Province (2018B0303110003 and 2020A1515011581), the Shenzhen Science and Technology Projects (KQTD20170331100838136, JCYJ20180306170922163, JCYJ20200109150700942, JCYJ20170413161649437, and JCYJ20170817172023838), the Shenzhen Fund for Guangdong Provincial High level Clinical Key Specialties (SZGSP007 and SZGSP013), the Shenzhen Key Medical Discipline Construction Fund (SZXK042 and SZXK049), the University Grants Committee of Hong Kong (GRF Grants 14306317, N_CUHK422/18, 14307218, and AoE/M-09/12), and a CUHK RSFS grant.Notes and references
- O. Shalem, N. E. Sanjana, E. Hartenian, X. Shi, D. A. Scott, T. Mikkelson, D. Heckl, B. L. Ebert, D. E. Root, J. G. Doench and F. Zhang, Science, 2014, 343, 84–87 CrossRef CAS.
- S. DeWeerdt, Nature, 2018, 564, S6–S8 CrossRef CAS.
- D. R. Hampson, A. W. M. Hooper and Y. Niibori, Brain Sci., 2019, 9, 32 CrossRef CAS.
- A. G. Souza, V. A. F. Bastos, I. B. B. Silva, K. Marangoni and V. A. Goulart, Curr. Gene Ther., 2016, 16, 287–291 CrossRef.
- C. Greenhill, Nat. Rev. Endocrinol., 2018, 14, 127 CrossRef.
- J. Sanmiguel, G. Gao and L. H. Vandenberghe, Methods Mol. Biol., 2019, 1950, 51–83 CrossRef CAS.
- P. Colella, G. Ronzitti and F. Mingozzi, Mol. Ther.—Methods Clin. Dev., 2018, 8, 87–104 CrossRef CAS.
- J. Rabinowitz, Y. K. Chan and R. J. Samulski, Viruses, 2019, 11, 102 CrossRef CAS.
- P. Pofali, A. Mondal and V. Londhe, Curr. Cancer Drug Targets, 2020, 20, 821–830 CrossRef.
- H. Zhang, J. Wu, J. Wu, Q. Fan, J. Zhou, J. Wu, S. Liu, J. Zang, J. Ye, M. Xiao, T. Tian and J. Gao, J. Nanobiotechnol., 2019, 17, 29 CrossRef.
- J. Lederberg, Nature, 1963, 198, 428–429 CrossRef CAS.
- T. Friedmann and R. Roblin, Science, 1972, 175, 949–955 CrossRef CAS.
- S. A. Rosenberg, P. Aebersold, K. Cornetta, A. Kasid, R. A. Morgan, R. Moen, E. M. Karson, M. T. Lotze, J. C. Yang and S. L. Topalian, et al. , N. Engl. J. Med., 1990, 323, 570–578 CrossRef CAS.
- R. M. Blaese, K. W. Culver, A. D. Miller, C. S. Carter, T. Fleisher, M. Clerici, G. Shearer, L. Chang, Y. Chiang, P. Tolstoshev, J. J. Greenblatt, S. A. Rosenberg, H. Klein, M. Berger, C. A. Mullen, W. J. Ramsey, L. Muul, R. A. Morgan and W. F. Anderson, Science, 1995, 270, 475–480 CrossRef CAS.
- S. Ylä-Herttuala and A. H. Baker, Mol. Ther., 2017, 25, 1095–1106 CrossRef.
- K. Ishikawa, T. Weber and R. J. Hajjar, Circ. Res., 2018, 123, 601–613 CrossRef CAS.
- J. Lähteenvuo and S. Ylä-Herttuala, Hum. Gene Ther., 2017, 28, 1024–1032 CrossRef.
- S. W. Zhang, S. W. Xiao, C. Q. Liu, Y. Sun, X. Su, D. M. Li, G. Xu, G. Y. Zhu and B. Xu, Zhonghua Zhongliu Zazhi, 2005, 27, 426–428 Search PubMed.
- H. Büning, EMBO Mol. Med., 2013, 5, 1–3 CrossRef.
- A. Gangi and J. S. Zager, Expert Opin. Drug Saf., 2017, 16, 265–269 CAS.
- R. Greco, G. Oliveira, M. T. Stanghellini, L. Vago, A. Bondanza, J. Peccatori, N. Cieri, S. Marktel, S. Mastaglio, C. Bordignon, C. Bonini and F. Ciceri, Front. Pharmacol., 2015, 6, 95 Search PubMed.
- J. Hoggatt, Cell, 2016, 166, 263 CrossRef CAS.
- L. Wan and G. Dreyfuss, Cell, 2017, 170, 5 CrossRef CAS.
- V. Date and S. Nair, Expert Opin. Biol. Ther., 2020, 1–16 Search PubMed.
- D. A. Prado, M. Acosta-Acero and R. S. Maldonado, Curr. Opin. Ophthalmol., 2020, 31, 147–154 CrossRef.
- I. Papadouli, J. Mueller-Berghaus, C. Beuneu, S. Ali, B. Hofner, F. Petavy, K. Tzogani, A. Miermont, K. Norga, O. Kholmanskikh, T. Leest, M. Schuessler-Lenz, T. Salmonson, C. Gisselbrecht, J. L. Garcia and F. Pignatti, Oncologist, 2020, 2019–0646 Search PubMed.
- M. Schuessler-Lenz, H. Enzmann and S. Vamvakas, Clin. Pharmacol. Ther., 2020, 107, 492–494 CrossRef.
- T. V. Mashel, Y. V. Tarakanchikova, A. R. Muslimov, M. V. Zyuzin, A. S. Timin, K. V. Lepik and B. Fehse, Biomaterials, 2020, 258, 120282 CrossRef CAS.
- G. V. Kochneva, G. F. Sivolobova, A. V. Tkacheva, A. A. Gorchakov and S. V. Kulemzin, Mol. Biol., 2020, 54, 3–16 CrossRef CAS.
- F. Hille, H. Richter, S. P. Wong, M. Bratovič, S. Ressel and E. Charpentier, Cell, 2018, 172, 1239–1259 CrossRef CAS.
- E. Charpentier, EMBO Mol. Med., 2015, 7, 363–365 CrossRef CAS.
- J. A. Doudna and E. Charpentier, Science, 2014, 346, 1258096 CrossRef.
- P. K. Jain, J. H. Lo, S. Rananaware, M. Downing, A. Panda, M. Tai, S. Raghavan, H. E. Fleming and S. N. Bhatia, Nanoscale, 2019, 11, 21317–21323 RSC.
- F. Ding, X. Huang, X. Gao, M. Xie, G. Pan, Q. Li, J. Song, X. Zhu and C. Zhang, Nanoscale, 2019, 11, 17211–17215 RSC.
- H. Kim, S. T. Kim, J. Ryu, B. C. Kang, J. S. Kim and S. G. Kim, Nat. Commun., 2017, 8, 14406 CrossRef CAS.
- A. Schindele, A. Dorn and H. Puchta, Curr. Opin. Biotechnol, 2020, 61, 7–14 CrossRef CAS.
- J. Cohen, Science, 2019, 365, 426–429 CrossRef CAS.
- D. Du, A. Roguev, D. E. Gordon, M. Chen, S. H. Chen, M. Shales, J. P. Shen, T. Ideker, P. Mali, L. S. Qi and N. J. Krogan, Nat. Methods, 2017, 14, 577–580 CrossRef CAS.
- K. Baumann, Nat. Rev. Mol. Cell Biol., 2017, 18, 591 CrossRef CAS.
- E. V. Grant, Food Drug Law J., 2016, 71, 608–633 Search PubMed.
- D. Escors and K. Breckpot, Arch. Immunol. Ther. Exp., 2010, 58, 107–119 CrossRef CAS.
- Y. Lu, L. Zhong, Z. Jiang, H. Pan, Y. Zhang, G. Zhu, L. Bai, R. Tong, J. Shi and X. Duan, Nanoscale Res. Lett., 2019, 14, 193 CrossRef CAS.
- M. M. Yousefpour and K. A. Yari, Cancer Chemother. Pharmacol., 2017, 79, 637–649 CrossRef.
- Y. Dong, T. Yu, L. Ding, E. Laurini, Y. Huang, M. Zhang, Y. Weng, S. Lin, P. Chen, D. Marson, Y. Jiang, S. Giorgio, S. Pricl, X. Liu, P. Rocchi and L. Peng, J. Am. Chem. Soc., 2018, 140, 16264–16274 CrossRef CAS.
- F. Abedi-Gaballu, G. Dehghan, M. Ghaffari, R. Yekta, S. Abbaspour-Ravasjani, B. Baradaran, J. E. N. Dolatabadi and M. R. Hamblin, Appl. Mater. Today, 2018, 12, 177–190 CrossRef.
- A. Singh, H. M. Hua, N. Gupta, P. Khanra, P. Kumar, V. V. Prakash and M. Kapoor, ChemPlusChem, 2020, 85, 466–475 CrossRef CAS.
- S. Kamerkar, V. S. LeBleu, H. Sugimoto, S. Yang, C. F. Ruivo, S. A. Melo, J. J. Lee and R. Kalluri, Nature, 2017, 546, 498–503 CrossRef CAS.
- S. Li, Y. Wu, F. Ding, J. Yang, J. Li, X. Gao, C. Zhang and J. Feng, Nanoscale, 2020, 12, 10854–10862 RSC.
- J. Wang, W. Li, Z. Lu, L. Zhang, Y. Hu, Q. Li, W. Du, X. Feng, H. Jia and B. F. Liu, Nanoscale, 2017, 9, 15598–15605 RSC.
- W. M. Usman, T. C. Pham, Y. Y. Kwok, L. T. Vu, V. Ma, B. Peng, Y. S. Chan, L. Wei, S. M. Chin, A. Azad, A. B. He, A. Y. H. Leung, M. Yang, N. Shyh-Chang, W. C. Cho, J. Shi and M. T. N. Le, Nat. Commun., 2018, 9, 2359 CrossRef.
- E. Hudry, C. Martin, S. Gandhi, B. György, D. I. Scheffer, D. Mu, S. F. Merkel, F. Mingozzi, Z. Fitzpatrick, H. Dimant, M. Masek, T. Ragan, S. Tan, A. R. Brisson, S. H. Ramirez, B. T. Hyman and C. A. Maguire, Gene Ther., 2016, 23, 380–392 CrossRef CAS.
- N. S. Orefice, B. Souchet, J. Braudeau, S. Alves, F. Piguet, F. Collaud, G. Ronzitti, S. Tada, P. Hantraye, F. Mingozzi, F. Ducongé and N. Cartier, Mol. Ther.—Methods Clin. Dev., 2019, 14, 237–251 CrossRef CAS.
- Y. Liang, X. Xu, X. Li, J. Xiong, B. Li, L. Duan, D. Wang and J. Xia, ACS Appl. Mater. Interfaces, 2020, 12, 36938–36947 CrossRef CAS.
- B. György, C. Sage, A. A. Indzhykulian, D. I. Scheffer, A. R. Brisson, S. Tan, X. Wu, A. Volak, D. Mu, P. I. Tamvakologos, Y. Li, Z. Fitzpatrick, M. Ericsson, X. O. Breakefield, D. P. Corey and C. A. Maguire, Mol. Ther., 2017, 25, 379–391 CrossRef.
- R. Calcedo and J. M. Wilson, Front. Immunol., 2013, 4, 341 Search PubMed.
- Y. Liang, P. Mathiyalagan, E. Kohlbrenner, E. Chepurko, D. Jeong, D. Ceholski, N. Dubois, R. Hajjar and S. Sahoo, Circulation, 2017, 136, A15439 Search PubMed.
- A. Meliani, F. Boisgerault, Z. Fitzpatrick, S. Marmier, C. Leborgne, F. Collaud, S. M. Simon, S. Charles, G. Ronzitti, A. Vignaud, L. van Wittenberghe, B. Marolleau, F. Jouen, S. Tan, O. Boyer, O. Christophe, A. R. Brisson, C. A. Maguire and F. Mingozzi, Blood Adv., 2017, 1, 2019–2031 CrossRef CAS.
- C. A. Maguire, L. Balaj, S. Sivaraman, M. H. Crommentuijn, M. Ericsson, L. Mincheva-Nilsson, V. Baranov, D. Gianni, B. A. Tannous, M. Sena-Esteves, X. O. Breakefield and J. Skog, Mol. Ther., 2012, 20, 960–971 CrossRef CAS.
- B. György, Z. Fitzpatrick, M. H. Crommentuijn, D. Mu and C. A. Maguire, Biomaterials, 2014, 35, 7598–7609 CrossRef.
- C. M. Ambrosi, G. Sadananda, J. L. Han and E. Entcheva, Front. Physiol., 2019, 10, 168 CrossRef.
- S. J. Wassmer, L. S. Carvalho, B. György, L. H. Vandenberghe and C. A. Maguire, Sci. Rep., 2017, 7, 45329 CrossRef CAS.
- L. T. Schiller, N. Lemus-Diaz, F. R. Rinaldi, K. O. Böker and J. Gruber, Mol. Ther.—Methods Clin. Dev., 2018, 9, 278–287 CrossRef CAS.
- N. Khan, S. Maurya, S. Bammidi and G. R. Jayandharan, Mol. Ther.—Methods Clin. Dev., 2020, 17, 497–504 CrossRef CAS.
- C. B. Breuer, K. S. Hanlon, J. S. Natasan, A. Volak, A. Meliani, F. Mingozzi, B. P. Kleinstiver, J. J. Moon and C. A. Maguire, Sci. Rep., 2020, 10, 4544 CrossRef CAS.
- S. Maurya and G. R. Jayandharan, Virus Res., 2020, 283, 197966 CrossRef CAS.
- D. J. Palmer, D. L. Turner and P. Ng, Mol. Ther.—Methods Clin. Dev., 2020, 17, 441–447 CrossRef CAS.
- S. M. Kim, Y. Yang, S. J. Oh, Y. Hong, M. Seo and M. Jang, J. Controlled Release, 2017, 266, 8–16 CrossRef CAS.
- M. Lu and Y. Huang, Biomaterials, 2020, 242, 119925 CrossRef CAS.
- Y. Ye, X. Zhang, F. Xie, B. Xu, P. Xie, T. Yang, Q. Shi, C. Y. Zhang, Y. Zhang, J. Chen, X. Jiang and J. Li, Biomater. Sci., 2020, 8, 2966–2976 RSC.
- Z. Li, X. Zhou, M. Wei, X. Gao, L. Zhao, R. Shi, W. Sun, Y. Duan, G. Yang and L. Yuan, Nano Lett., 2019, 19, 19–28 CrossRef CAS.
- P. Gee, M. S. Y. Lung, Y. Okuzaki, N. Sasakawa, T. Iguchi, Y. Makita, H. Hozumi, Y. Miura, L. F. Yang, M. Iwasaki, X. H. Wang, M. A. Waller, N. Shirai, Y. O. Abe, Y. Fujita, K. Watanabe, A. Kagita, K. A. Iwabuchi, M. Yasuda, H. Xu, T. Noda, J. Komano, H. Sakurai, N. Inukai and A. Hotta, Nat. Commun., 2020, 11(1), 1334 CrossRef CAS.
- Y. Lin, J. Wu, W. Gu, Y. Huang, Z. Tong, L. Huang and J. Tan, Adv. Sci., 2018, 5, 1700611 CrossRef.
- Y. T. Sato, K. Umezaki, S. Sawada, S. A. Mukai, Y. Sasaki, N. Harada, H. Shiku and K. Akiyoshi, Sci. Rep., 2016, 6, 21933 CrossRef CAS.
- Q. Lv, L. Cheng, Y. Lu, X. Zhang, Y. Wang, J. Deng, J. Zhou, B. Liu and J. Liu, Adv. Sci., 2020, 7, 2000515 CrossRef.
- A. O. Batagov and I. V. Kurochkin, Biol. Direct, 2013, 8, 12 CrossRef CAS.
- Z. Yang, J. Shi, J. Xie, Y. Wang, J. Sun, T. Liu, Y. Zhao, X. Zhao, X. Wang, Y. Ma, V. Malkoc, C. Chiang, W. Deng, Y. Chen, Y. Fu, K. J. Kwak, Y. Fan, C. Kang, C. Yin, J. Rhee, P. Bertani, J. Otero, W. Lu, K. Yun, A. S. Lee, W. Jiang, L. Teng, B. Y. S. Kim and L. J. Lee, Nat. Biomed. Eng., 2020, 4, 69–83 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |