
Immunostimulatory silica nanoparticle boosts innate immunity in brain tumors†
Peter A.
Bielecki‡
ab,
Morgan E.
Lorkowski‡
ab,
Wyatt M.
Becicka‡
a,
Prabhani U.
Atukorale
ab,
Taylor J.
Moon
a,
Yahan
Zhang
a,
Michelle
Wiese
a,
Gil
Covarrubias
ab,
Shruthi
Ravichandran
c and
Efstathios
Karathanasis
 *ab
*ab
aDepartment of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio, USA. E-mail: stathis@case.edu
bCase Comprehensive Cancer Center, Case Western Reserve University, Cleveland, Ohio, USA
cHathaway Brown School, Shaker Heights, Ohio, USA
First published on 23rd December 2020
Abstract
The high mortality associated with glioblastoma multiforme (GBM) is attributed to its invasive nature, hypoxic core, resistant cell subpopulations and a highly immunosuppressive tumor microenvironment (TME). To support adaptive immune function and establish a more robust antitumor immune response, we boosted the local innate immune compartment of GBM using an immunostimulatory mesoporous silica nanoparticle, termed immuno-MSN. The immuno-MSN was specifically designed for systemic and proficient delivery of a potent innate immune agonist to dysfunctional antigen-presenting cells (APCs) in the brain TME. The cargo of the immuno-MSN was cyclic diguanylate monophosphate (cdGMP), a Stimulator of Interferon Gene (STING) agonist. Studies showed the immuno-MSN promoted the uptake of STING agonist by APCs in vitro and the subsequent release of the pro-inflammatory cytokine interferon β, 6-fold greater than free agonist. In an orthotopic GBM mouse model, systemically administered immuno-MSN particles were taken up by APCs in the near-perivascular regions of the brain tumor with striking efficiency. The immuno-MSNs facilitated the recruitment of dendritic cells and macrophages to the TME while sparing healthy brain tissue and peripheral organs, resulting in elevated circulating CD8+ T cell activity (2.5-fold) and delayed GBM tumor growth. We show that an engineered immunostimulatory nanoparticle can support pro-inflammatory innate immune function in GBM and subsequently augment current immunotherapeutic interventions and improve their therapeutic outcome.
New conceptsCancer immunotherapies that leverage adaptive immunity often fall short in solid tumor masses and aggressive cancers like glioblastoma multiforme (GBM). This stems mainly from the inability to overcome the profound immunosuppression in the tumor microenvironment (TME), which is enriched with dysfunctional antigen-presenting cells (APCs). To drive a more robust anti-tumor response, we engineered an immunostimulatory mesoporous silica nanoparticle, termed immuno-MSN, to specifically boost the dysfunctional innate immune compartment of GBM from within the tumor itself. The highly versatile and tunable immuno-MSN particle offered a set of valuable features including (1) efficient delivery of a potent innate immune agonist to the site of disease, (2) protection of its cargo while in circulation and diminished systemic toxicities, (3) direct uptake by the dysfunctional tumor-resident APCs, and (4) proficient intracellular presentation of the immune agonist. While conventional approaches focus on local delivery, systemic delivery enabled immuno-MSNs to efficiently use the entire microvasculature and readily deposit into the APC-rich perivascular areas of the tumor itself, leading to predominant uptake by tumor-resident APCs. Due to its design, the immuno-MSN resulted in remarkable activation and expansion of APCs in the TME and improved therapeutic outcomes. |
1. Introduction
Glioblastoma multiforme (GBM) is resilient to current clinical treatment strategies like surgical resection, chemotherapy, and radiation therapy, resulting in more than 95% recurrence and a grim survival.1 The high mortality of GBM is primarily attributed to its invasive peripheral growth, hypoxic core, and highly resistant cell subpopulations.2,3 On the other hand, immunotherapies like CAR T cells and immune checkpoint blockade (ICB) have shown promise against certain hard-to-treat cancers by facilitating immune-recognition and T cell-mediated killing of tumor cells.4,5 However, patient responses vary widely, with only a small cohort of patients responding favorably to current immunotherapies.6–8 One major hurdle in immunotherapy is to overcome the profound immunosuppression within the tumor microenvironment (TME) of GBM.9 While GBM was once thought to be immune-privileged, overwhelming evidence now suggests that the TME of GBM is crowded with dysfunctional immune cells including antigen-presenting cells (APCs), such as macrophages and dendritic cells (DCs), and myeloid-derived suppressor cells (MDSCs) that drive immunosuppression and tumor progression.10An effective approach in GBM immunotherapy therefore is to alter the local innate immune compartment of the TME by reprograming inhibitory APCs into properly activated APCs that stimulate tumor antigen-specific T cells. We designed an immunostimulatory nanoparticle that systemically delivers a Stimulator of Interferon Gene (STING) agonist to the TME of GBM to achieve local and robust immunostimulation. More specifically, the cargo of the nanoparticle was cyclic diguanylate monophosphate (cdGMP), a cyclic dinucleotide that activates APCs by inducing pro-inflammatory Type I interferon (IFN) secretion. By targeting host pattern recognition receptors (PRRs), cdGMP has gained significant attention in recent years as a potent immunogenic molecule that triggers the body's natural defense mechanism to foreign DNA.11–14 Importantly, the compromised blood–brain barrier in GBM gives circulating nanoparticles direct access to the near-perivascular regions of the tumor,15–17 which is populated by dysfunctional resident immune cells (Fig. 1A). By using systemic administration, the immunostimulatory nanoparticles can selectively deposit into the APC-rich perivascular regions of the tumor, leading to the uptake of nanoparticles by the desirable subset of cells.18 When properly activated, DCs and macrophages recruit more immune cells to the local TME, process tumor-associated antigens that are shed from tumor cells, and can cross-present these antigens to prime T cells. In addition to the challenges associated with local intratumoral injections in GBM, systemic delivery enables circulating nanoparticles to access the majority of the tumor microvasculature and trigger widespread IFN-β-mediated danger signaling.
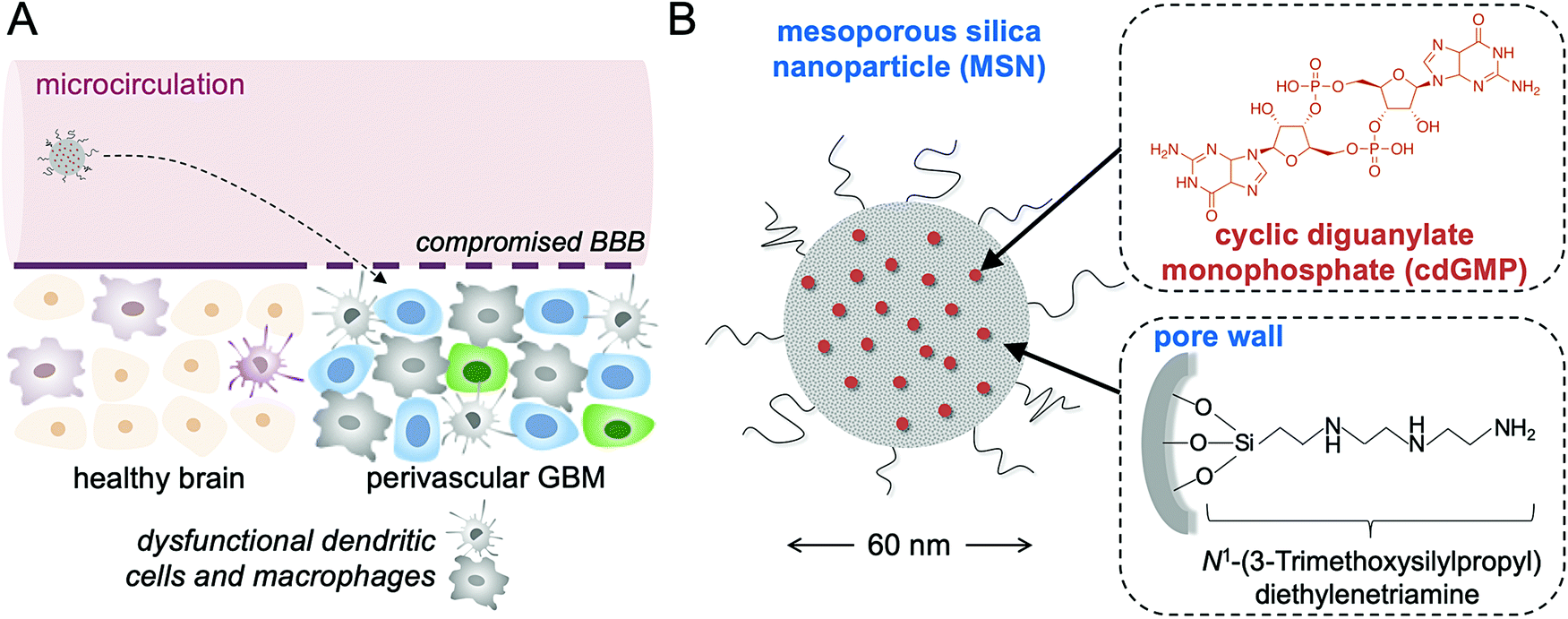 | ||
| Fig. 1 Schematic of (A) the systemic delivery of the immuno-MSN to the APC-rich space of GBM, and (B) the ∼60 nm immuno-MSN based on a mesoporous silica nanoparticle loaded with a potent STING agonist (cdGMP). | ||
The immunostimulatory nanoparticle used here was a 60 nm mesoporous silica nanoparticle stably loaded with high amounts of cdGMP (abbreviated as immuno-MSN), which diminished systemic toxicities and facilitated direct uptake by APCs in GBM (Fig. 1B). The mesoporous silica surface of the immuno-MSN was functionalized with a very high content of protonatable primary and secondary amines. With a pKa close to endosomal and lysosomal pH, these amines facilitated endosomal escape and the release of cdGMP in the cytosol. Considering the limitations of systemically delivered ‘free’ cdGMP associated with its hydrophilicity and negative charge, the immuno-MSN effectively presented the STING agonist to its binding site in the cytosol of APCs in GBM.
2. Experimental section
2.1 Mesoporous silica nanoparticle (MSN) synthesis and characterization
5.7 mL of 25% cetyltrimethylammonium bromide (CTAB, Sigma-Aldrich) was added to H2O heated at 75–80 °C for a total volume of 20 mL and mixed for 15 min. While continuing to heat and mix the solution, 0.8 mL of freshly prepared 10% triethylamine (TEA) was added. After 15 min, 1.5 mL of silica precursor TEOS (tetraethylorthosilicate, Sigma-Aldrich) was added dropwise to the mixing solution at a rate of approximately 75 μL per minute. The solution was stirred vigorously for 1 h at 80 °C to form silica particles of diameter near 60 nm. The solution was collected and washed several times with EtOH via centrifugation at ≥2000 rpm. To remove the CTAB surfactant, particles were suspended in 100 mL methanol and 3.8 mL HCl (12 M) and mixed for 24 h at room temperature. The solution was collected and washed several times with EtOH via centrifugation at ≥2000 rpm. Prior to removal of the surfactant, particles were resuspended in H2O to a volume corresponding to approximately 20 mL H2O per 250 mg of particles. The solution was adjusted to pH 8.0–9.0 using NH4OH and heated to 70 °C under stirring. 50 μL of N1-(3-trimethoxysilylpropyl) diethylenetriamine (Santa Cruz Biotechnology) was added in 10 μL increments and left for 3 h. For flow cytometry studies using fluorescently labeled MSNs, particles were conjugated with Alexa Fluor 750 NHS ester (Thermo Fisher Scientific). For PEGylation, MSN-NH2 particles were first resuspended in H2O to a volume corresponding to approximately 1 mL H2O per 10 mg of particles. The pH of the solution was kept near neutral (pH 7.0–7.5). A ratio of 20 μg of mPEG succinic acid NHS (2 kDa) (NANOCS) was suspended in DMSO and added to 1 mg of MSN-NH2 particles, and allowed to react for 2 h under mixing at room temperature. For loading into MSNs, particles were sonicated in an ultrasonic bath and washed several times in H2O adjusted to pH 10.2 using NH4OH. Loading was performed by combining MSNs in pH 10.2 H2O with cdGMP (InvivoGen) at a ratio of 62.5 μg cdGMP per mg MSNs. The mixture was briefly sonicated and placed on a shaker for 12 h. CdGMP encapsulation was measured by reading the loading supernatant on a spectrophotometer at an absorbance wavelength of 284 nm (Tecan Infinite 200). Stability studies of the immuno-MSN particles were performed by measuring cdGMP release across a 20k MWCO mini dialysis unit (Thermo Fisher Scientific) in PBS (pH 7.4 or 5.5) at 25 °C. MSN size diameter and surface charge were measured via dynamic light scattering (DLS) and zeta potential, respectively (90Plus, Brookhaven Instruments). MSN size diameter was also analyzed in ImageJ using images acquired via transmission electron microscopy.2.2 Institutional animal care and use committee statement
All animal procedures were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Case Western Reserve University (CWRU). CWRU follows the Guide for the Care and Use of Laboratory Animals, which is required by the United States Public Health Service Policy (PHS) on humane care and use of laboratory animals.2.3 Cell lines and animal models
Murine RAW 264.7 macrophages (ATCC) and murine GL261 cells expressing tdTomato and luciferase (a gift from the Jeremy Rich Laboratory at USCD) were cultured in DMEM (Gibco) containing 10% FBS (HyClone). All cell lines were authenticated using short tandem repeat (STR) profile and routinely tested for Mycoplasma contamination. All cells were grown at 37 °C with 5% carbon dioxide.For flank GL261 studies, 6–10 week-old, female C57BL/6 albino mice (Jackson Laboratories) were subcutaneously injected with 1 × 106 GL261 cells in 100 μL PBS. After tumor implantation, mice were randomized into groups for subsequent studies. Flank GL261 models were treated by intravenous administration on days 7, 8, and 9 after flank inoculation when tumors became palpable and measurable with calipers (∼70 mm3). Tumors were monitored by bioluminescence imaging (IVIS Spectrum, PerkinElmer) 10 min after intraperitoneal administration of 200 μL of D-luciferin (12.5 mg mL−1). Tumors were measured with calipers at least twice a week and tumor volume calculated using the following formula: volume = 0.5 × length × width2.
For orthotopic GL261 studies, 6–10 week-old, female C57BL/6 albino mice (Jackson Laboratories) were anesthetized and fitted into a stereotaxic rodent frame. A 10 μL Hamilton syringe was used to inoculate 2 × 105 GL261 cells AP = +0.5 and ML = −2.0 mm from the bregma at a rate of 1 μL min−1 in the right striatum and a depth of −3 mm from the dura. After tumor implantation, mice were randomized into groups for subsequent studies. Orthotopic brain tumor models were treated by intravenous administration beginning on day 7 after inoculation. 2.5 mg kg−1 of α-TGF-βR1 (Galunisertib, MedChemExpress) was administered by intraperitoneal injection for five consecutive days per week beginning on day 3 after inoculation. Tumors were monitored by bioluminescence imaging (IVIS Spectrum, PerkinElmer) 10 min after intraperitoneal administration of 200 μL of D-luciferin (12.5 mg mL−1).
2.4 Transmission electron microscopy
MSNs were diluted in EtOH, applied to 3 nm thick carbon film grids (Ted Pella), and left to dry. Imaging was performed using a FEI Tecnai F30 300 keV Transmission Electron Microscope.2.5 Immuno-MSN cell uptake and confocal microscopy
2 M RAW 264.7 macrophage cells were plated in glass bottom cell culture dishes and allowed to adhere for 24 h. Cells were then exposed to either 30 μg of cGMP (8-[Fluo]-cGMP, BIOLOG Life Science Institute) loaded into MSNs or free cGMP alone. Cells were quickly washed with PBS a fixed in 2% paraformaldehyde (in PBS) after incubation. Cells were mounted with VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories) and No. 1.5 glass coverslips and imaged using a Leica TCS SP8 gated STED Confocal Microscope (Leica Microsystems). Confocal images were analyzed on ImageJ software to quantify cGMP fluorescence signal per cell from immuno-MSN treatment compared to free cGMP alone.2.6 ELISA assay of IFN-β
6 M RAW 264.7 macrophage cells were plated in a 24-well plate. Cells were then treated in triplicate with 20 μg mL−1 cdGMP loaded into MSNs or an equivalent amount of cdGMP, either free or loaded into liposomes per previously established lab protocols.18 Cell culture supernatants were harvested 24 h later, centrifuged at 4 °C, and analyzed for mIFN-β per the manufacturer's protocols using LumiKine Xpress Bioluminescent Cytokine ELISA Kits (InvivoGen). Luminescence was measured using a Tecan Infinite 200 spectrophotometer.2.7 Immunostaining and confocal microscopy
Mice bearing orthotopic GBM tumors were injected i.v. with immuno-MSNs (10 μg of cdGMP) conjugated with Alexa Fluor 647 NHS ester (Thermo Fisher Scientific) on days 7 and 8 post tumor inoculation with GL261 cells. Mice were perfused on day 9 with PBS and PBS containing 4% paraformaldehyde (Alfa Aesar). Brains were harvested in 4% paraformaldehyde in PBS, transferred to 30% sucrose in PBS, and finally embedded in optimum cutting temperature gel (OCT, Thermo Fisher Scientific) at −80 °C. Primary (anti-CD31 and anti-CD11c) and Alexa Fluor 488 secondary antibodies were purchased from Thermo Fisher Scientific. Frozen sections were sectioned at a 10 μm thickness using a Leica Cryostat and stained with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50–1:100 dilution of anti-mouse primary antibodies overnight at 4 °C. Tissue sections were then stained with Alexa Fluor 488 secondary antibodies at a 1:150 dilution for 30 min at 25 °C before being mounted with a No. 1.5 glass coverslip and VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories). Images were collected using a Leica TCS SP8 gated STED Confocal Microscope (Leica Microsystems).
:50–1:100 dilution of anti-mouse primary antibodies overnight at 4 °C. Tissue sections were then stained with Alexa Fluor 488 secondary antibodies at a 1:150 dilution for 30 min at 25 °C before being mounted with a No. 1.5 glass coverslip and VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories). Images were collected using a Leica TCS SP8 gated STED Confocal Microscope (Leica Microsystems).
2.8 Flow cytometry
DAPI, anti-mouse CD16/32 (2.4G2), CD45 (30-F11), CD3e (145-2C11), CD11b (M1/70), CD11c (HL3), CD19 (1D3), CD49b (DX5), and F4/80 (T45-2342) dye-conjugated flow cytometry antibodies were purchased from BD Biosciences. Anti-mouse CD4 (GK1.5), CD8a (53-6.7), CD25 (3C7), CD80 (16-10A1), CD206 (C068C2), Ly-6C (HK1.5), and Ly-6G (1A8) antibodies were purchased from Biolegend. For flow cytometry studies using fluorescently labeled MSNs, particles were conjugated with Alexa Fluor 750 NHS ester (Thermo Fisher Scientific). Flow cytometry analysis was typically performed 24 h after two consecutive days of treatment. After blood collection via retro-orbital bleeding, mice were immediately euthanized followed by harvesting of brains, spleens, and livers. After removing the cerebellum, brains were separated into left (healthy control) and right (tumor-bearing) hemispheres prior to digestion for 15 min in trypsin–EDTA (25%). Single-cell suspensions were obtained by gently homogenizing organs and passing the homogenates through 70 μm filters. Myelin was separated from brain cell suspensions via a 30%/70% Percoll gradient (Sigma-Aldrich). ACK lysis buffer was used to remove residual red blood cells in blood and organ cell suspensions. In survival studies of orthotopic GL261 models, flow cytometry was performed weekly following retro-orbital bleeding. Cells were blocked with anti-mouse CD16/CD32 and stained to identify immune cell populations. Samples were analyzed using a BD FACS LSR II Flow Cytometer (Becton Dickinson) and FlowJo software. Threshold gating for AF750/MSN+ cells was determined using untreated (MSN−) cells as a background. This strategy was confirmed by comparing AF750 signals between CD45+ immune cells and CD45− non-immune cells. The AF750/MSN+ gate was the same for all immune cell subsets. For analysis, immune cell numbers were normalized by 105 viable (DAPI−) cells.In a separate study, 6–10 week-old, female C57BL/6 albino mice received intravascular administration of either immuno-MSNs or free cdGMP alone (10 μg cdGMP). 24 h later, mice were bled retro-orbitally and cell blood counts were measured using a HemaVet 950 (Drew Scientific).
2.9 In vivo imaging of the biodistribution of immuno-MSN
Mice with orthotopic GL261 tumors received i.v. injections of either empty MSNs or immuno-MSNs conjugated with Alexa Fluor 750 NHS ester (Thermo Fisher Scientific) on days 7 and 8 after tumor inoculation. Animals were imaged at various time points (0.5, 6, 24, and 44 hours after the first injection) via IVIS Spectrum (PerkinElmer, 124262). Blood was collected by retro-orbital bleeding, after which the mice were euthanized and organs were harvested (brain, spleen, liver, lungs, kidneys, heart). The organs were then imaged on the IVIS Spectrum.2.10 Statistical analysis
All statistical analyses were performed using Prism 8 (GraphPad) and are detailed in the figure legends. Data was analyzed by unpaired t-test (two-tailed) or either one- or two-way ANOVA with Tukey or Sidak post-test. Statistical significance was determined using P-values less than 0.05. All values are comprised of at least three independent biological replicates and are reported as the mean ± standard deviation, unless otherwise noted. In animal studies, each treatment group consisted of at least 5 mice unless otherwise noted.3. Results and discussion
3.1 Synthesis and characterization of the immuno-MSN
Immuno-MSNs were synthesized in a reproducible process that yielded approximately 500 mg of monodispersed nanoparticles per batch. Dynamic light scattering (DLS) measurements and transmission electron microscopy (TEM) images indicated MSN size distributions of 87.2 ± 5.0 nm and 59.1 ± 11.0, respectively (Fig. 2A and B). TEM imaging confirmed the mesoporous structure of the MSN, formed from tetraethylorthosilicate (TEOS) particle nucleation with cetyltrimethylammonium bromide (CTAB). The mesoporous structure creates a high surface area that is more than 15 times greater than that of similarly sized solid silica nanospheres.19 Amine functionalization with N1-(3-trimethoxysilylpropyl) diethylenetriamine was verified by zeta potential measurements where aminated MSNs (MSN-NH2) recorded a distinct positive surface charge (34.7 ± 6.2 mV) compared to unfunctionalized MSNs (MSN-OH) (−2.7 ± 7.1 mV) (Fig. 2C). Amine functionalization facilitates two major functions of the MSN: high cdGMP loading into the particle and effective release of cdGMP in acidic conditions as in endosomes upon internalization. For cdGMP loading, surface amines on the porous MSN-NH2 become deprotonated in basic conditions near pH 10.2 and attract negatively charged cdGMP molecules with high efficiency. MSN-NH2 particles loaded 99.2% of the available cdGMP compared to just 9.4% observed in unfunctionalized MSNs (Fig. 2D). This equates to roughly 60 μg of loaded cdGMP per mg of MSN-NH2 (Fig. 2E). The neutralization of surface charge during loading was reflected by a measured drop in zeta potential to −8.5 ± 24.1 mV (Fig. 2C). Further, PEGylation of the MSNs did not hinder the loading capacity of the particle under the same loading conditions. The immuno-MSN particles demonstrated good stability at physiological pH, releasing just 8.6% (5.2 μg) of the cdGMP payload over 5 h in phosphate buffer solution (PBS) (Fig. 2F).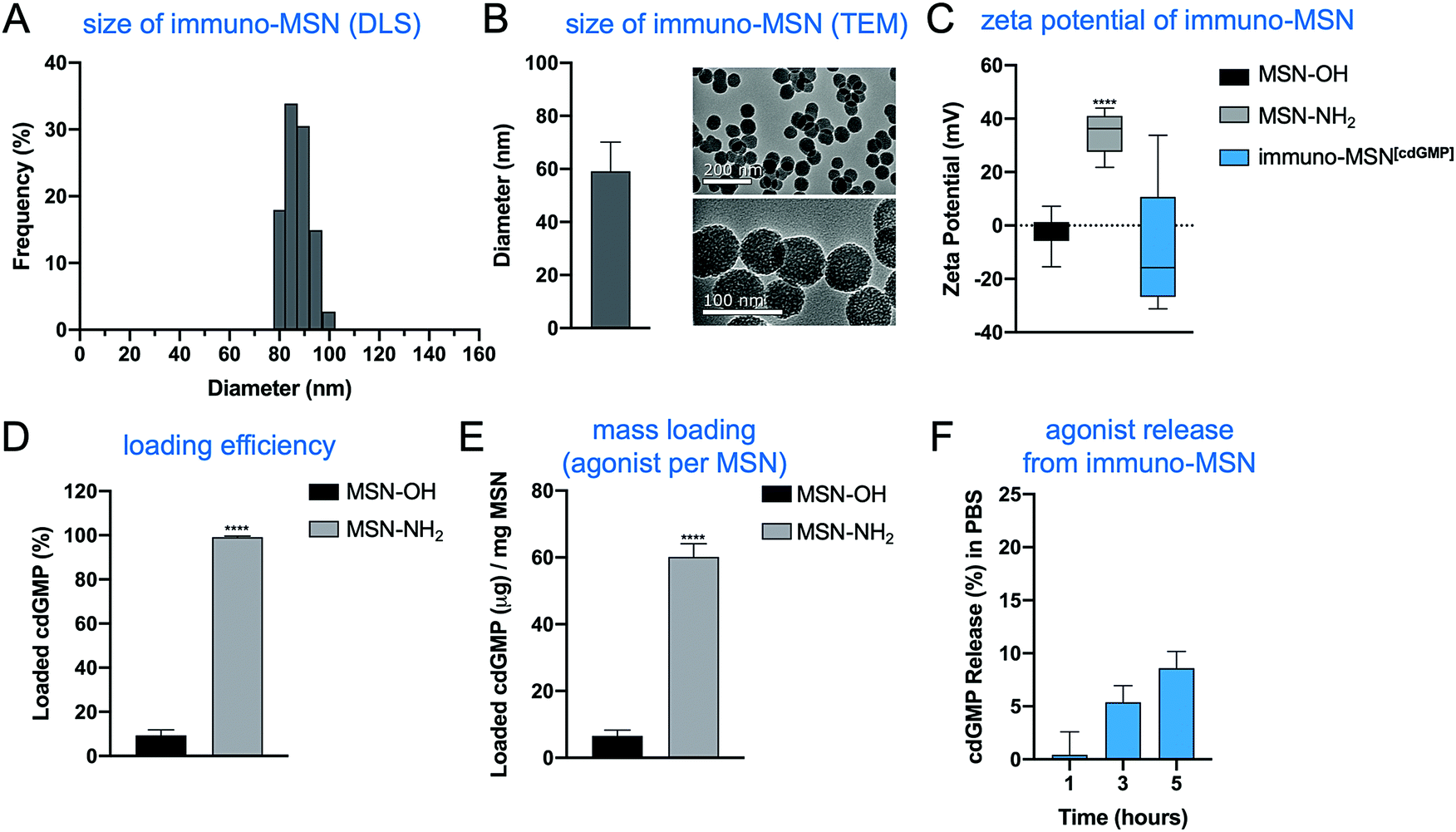 | ||
| Fig. 2 Effective loading and stability of STING agonist in monodispersed immuno-MSN particles after amine functionalization. MSN characterization with (A) dynamic light scattering (representative sample run) and (B) transmission electron microscopy to determine particle size distributions (nm) and confirm the mesoporous structure. The effects of MSN amine functionalization evaluated by (C) MSN zeta potential measurements (mV), (D) MSN loading efficiency of cdGMP (%), and (E) MSN mass loading of cdGMP per mg of MSN particles. (F) Stability study of cdGMP release from the immuno-MSN particle in PBS. All samples were run at least in triplicate. Statistical significance in zeta potential was conducted by one-way ANOVA with Tukey's post-test (****P < 0.0001). Statistical significance in cdGMP loading was conducted by unpaired t-test (two-tailed) (****P < 0.0001). | ||
3.2 Immuno-MSN facilitates efficient cdGMP cell uptake and IFN-β secretion from innate immune cells
To validate the ability of the immuno-MSN to facilitate intracellular delivery of cdGMP, RAW 264.7 murine macrophages were incubated with immuno-MSNs. A fluorescein variant of cdGMP (8-[Fluo]-cGMP) was loaded into the MSNs to directly visualize the uptake of the agonist by cells. Macrophage cells were exposed to either cGMP-loaded MSNs or an equivalent amount of free cGMP before imaging with confocal microscopy. Confocal microscopy showed that free STING agonist exhibited poor uptake by APCs with very low amounts reaching the cytosol (Fig. 3A). On the other hand, immuno-MSNs shuttled cdGMP rapidly into the cytosol of macrophages. After just 5 min of incubation, the immuno-MSN exhibited a 10-fold higher cGMP uptake than the ‘free’ agonist condition. Uptake became even more pronounced after 6 h where MSNs shuttled cGMP into macrophages with striking efficiency compared to free cGMP controls. Quantitative image analysis of fluorescence showed a 12.6-fold increase in cGMP signal per macrophage when cGMP was delivered with the MSN particles (Fig. 3B). The effective intracellular delivery of the agonist to its binding partner STING in the cytosol was mediated by the MSN and stems from the protonatable primary and secondary amines on the particle, which provide high pH buffering capability. Nanoparticles can escape endosomes and transition to the cytosol via the ‘proton sponge’ mechanism because of the particle's buffering capability.20 Through a titration study, the immuno-MSN showed strong pH buffering for a wide range of pH (5–7.4). In fact, the proton buffering property of the immuno-MSN was nearly 20-fold greater than the pH-buffering capacity of the well-established cationic polymer polyethylenimine, which is often used in proton sponge applications. At the same time, the intracellular pH is below the pKa of cdGMP, causing protonation of its phosphate groups, which eliminates the electrostatic attraction with the silica surface and results in the efficient release of cdGMP from the particle (Fig. 3C). Rapid and continuous release of cdGMP was observed at pH 5.5 (Fig. 3D).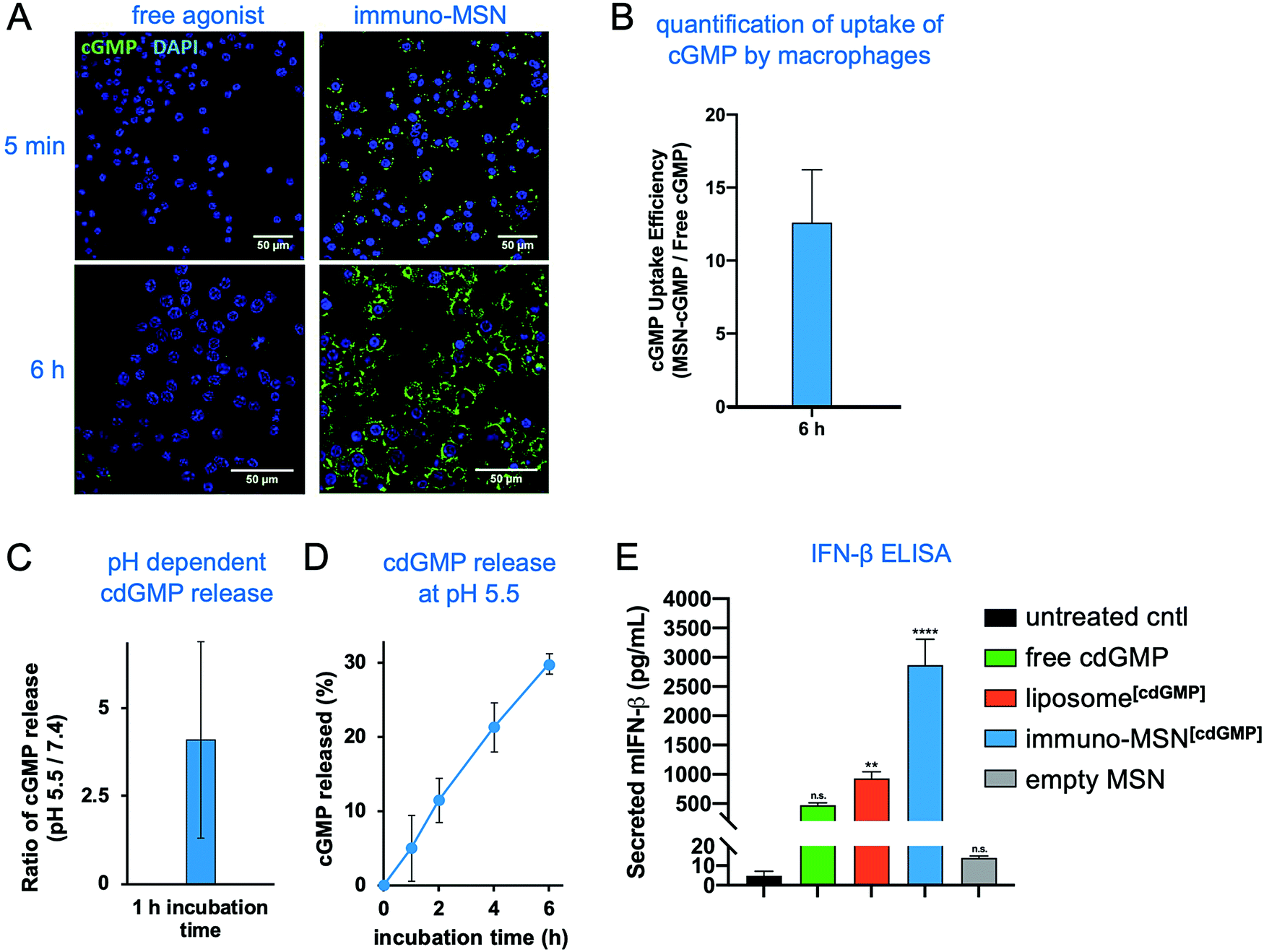 | ||
| Fig. 3 Immuno-MSN particles facilitate efficient uptake of STING agonist and subsequent release of IFN-β from RAW 264.7 murine macrophages in vitro. (A) Representative confocal images (40× – top row, 63× – bottom row) depicting the cell uptake of fluorescent STING agonist after incubation with macrophages in vitro. Fluorescein-cGMP (green), DAPI-stained nuclei (blue). (B) ImageJ analysis of confocal images evaluating the fold-increase of fluorescent STING agonist signal as a result of MSN delivery compared to free STING agonist alone. STING agonist signal (a.u.) was normalized per cell. (C and D) pH-Dependent release of cdGMP from the immuno-MSN particle. (E) ELISA analysis of in vitro IFN-β secretion (pg mL−1) from macrophage cells after a 24 h incubation with the tested formulations. A total of 30 μg cdGMP was tested per condition. All samples were run in triplicate. Statistical significance to the untreated control was conducted by one-way ANOVA with Tukey's post-test (**P < 0.01, ****P < 0.0001, n.s. = not significant). | ||
Proficient delivery to the cytosol is important for cdGMP since the STING machinery is located in the cell cytosol. This was directly assessed by measuring IFN-β cytokine secretion from macrophages. RAW 264.7 macrophages were incubated for 24 h with either free cdGMP, immuno-MSN, or empty MSN before sampling cell supernatants for enzyme-linked immunosorbent assay (ELISA) analysis. The immuno-MSN was also compared to a commonly used liposomal formulation of cdGMP.12,21 Treatment with immuno-MSNs elicited a 6.1-fold increase in IFN-β compared to equivalent amounts of free cdGMP (Fig. 3E). The empty MSN vehicle control resulted in negligible IFN-β secretion similar to the untreated control. Considering that liposomes have been widely used to deliver immunomodulators including STING agonists, the superior presentation of the STING agonist using the immuno-MSN was highlighted by comparing it to the liposome variant, which induced a 3.1-fold lower IFN-β production.
3.3 Immuno-MSN drives tumor reduction in a flank GBM model
First, the therapeutic potential of the immuno-MSN treatment was tested in mice bearing flank GBM tumors.22 The objective was to directly assess the antitumor immune response in the absence of the complexity associated with intracranial GBM and the blood–brain barrier. GL261 glioma cells (1 × 106 cells) were inoculated subcutaneously into C57BL/6 albino immunocompetent mice to form palpable tumors and treated for three consecutive days with either immuno-MSN or empty MSN-NH2 vehicle control (Fig. S1A, ESI†). Tumor progression was monitored with bioluminescent imaging (Fig. S1B, ESI†) and caliper measurements of tumor size. While tumors treated with the empty MSN-NH2 vehicle continued to grow, complete tumor clearance was achieved in 50% of mice treated with immuno-MSNs (Fig. S1C and D, ESI†). Complete tumor remission in the good responders was sustained through the duration of the 110 day study (Fig. S1E, ESI†). Meanwhile, the entire group treated with empty MSN-NH2 was euthanized by day 72 due to excessive tumor burden. The mouse weight progression is shown in Fig. S2A (ESI†). Both groups exhibited an initial mild weight loss, which was transient. The immuno-MSN-treated mice regained weight 3 days after treatment. In a separate study, a single dose of immuno-MSN or free agonist (10 μg cdGMP) was intravenously administered into healthy mice. A complete blood count was performed after 24 h to evaluate short-term safety and detect signs of acute adverse systemic inflammation caused by immune-potentiating agents. Within 24 h after treatment, mice treated with free cdGMP had significantly higher levels of leukocytes and lymphocytes (3.6 and 4.5-fold, respectively) compared to the immuno-MSN treatment group (Fig. S2B, ESI†). In a previous study,18 serum clinical chemistry, clinical observations, and body weight monitoring was performed in mice treated with systemically administered immunostimulatory nanoparticles to assess the safety profile over a period of one month after systemic administration. Analysis of serum chemistry revealed mild and transient elevation in liver enzyme levels at day 1 post-treatment, which returned to baseline at day 4 post-treatment.3.4 Microdistribution of immuno-MSNs in the tumor immune microenvironment of an intracranial GBM model
We next sought to evaluate the uptake of the MSN particles by key innate immune cells in the TME of an orthotopic GBM model. GL261 brain tumor cells were inoculated into the right striatum of immunocompetent mice. To first understand the immune landscape of the intracranial GL261 brain tumor, brains were excised 9 days after tumor inoculation. Cells were isolated from the brain and resident immune cell populations were analyzed using flow cytometry. The two hemispheres of the brain were processed independently in order to decipher immune cell populations in the tumor-burdened hemisphere against the healthy brain tissue of the contralateral hemisphere. Significantly elevated natural killer (NK) cells (5.4-fold) and monocytes (38.2-fold) were found in the brain tumor tissue compared to healthy brain, as well as heightened levels of macrophages (6.7-fold) and DCs (1.8-fold) (Fig. 4A). These findings are in agreement with reports that the brain tumor is not as immune-privileged as previously thought, and it is, in fact, occupied by innate and adaptive immune cells that can facilitate tumor progression.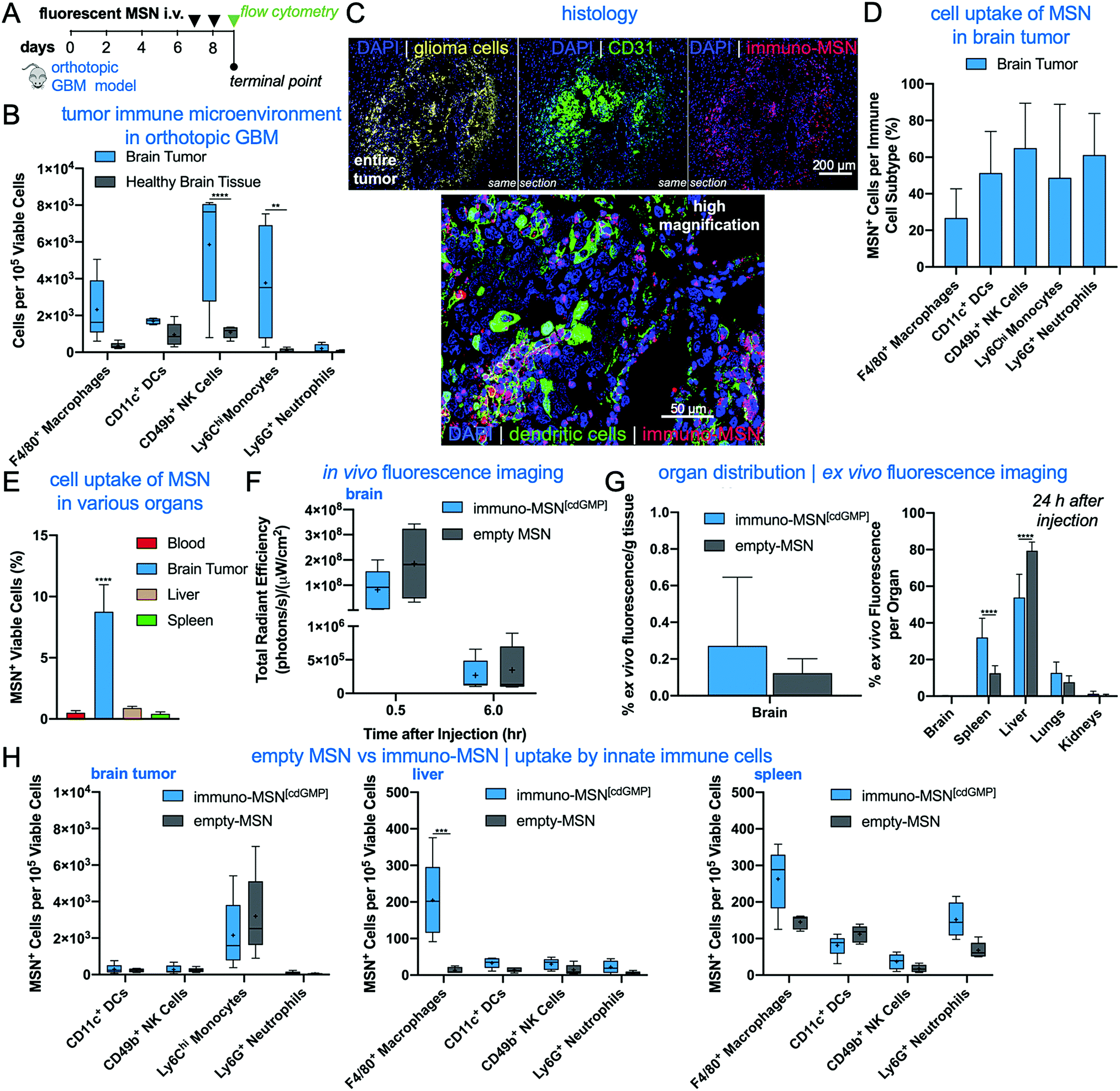 | ||
| Fig. 4 Immuno-MSN particles target APCs and innate immune cells in the TME with high efficiency in an orthotopic GBM model. (A) Flow cytometry analysis revealed heightened levels of resident immune cells in the brain tumor microenvironment. Statistical significance in the box and whisker plot (5–95 percentile, “+” mean) was conducted by two-way ANOVA with Sidak's post-test (**P < 0.01, ****P < 0.0001). (B) Treatment regimen of intravascularly administered fluorescent MSN particles. (C) Representative confocal images depicting the microdistribution of immuno-MSN particles in orthotopic GBM tumor sections. Fluorescent immuno-MSNs accumulate in near-perivascular regions rich with APCs. Alexa Fluor 647 tagged immuno-MSNs (red), tdTomato-expressing GL261 tumor cells (yellow), DAPI-stained nuclei (blue), CD31 (green, top panel), DCs (green, bottom). (D) Flow cytometry analysis of fluorescent MSN particle uptake by APCs and innate cells in the brain tumor (DCs and NK cells, n = 4). (E) Flow cytometry analysis of cell uptake of fluorescent MSNs in the brain TME, blood, liver, and spleen. (F) Live-animal spectrum imaging for fluorescently labeled MSN was performed longitudinally. The fluorescent immuno-MSN and the fluorescent MSN (empty, no cdGMP cargo) were compared following the administration schedule showed in (A). (F) Organs were collected 24 h after the second injection. Nanoparticle deposition was quantified ex vivo by measuring fluorescence signal from organs. The total signal from all organs was considered 100%. (H) Flow cytometry analysis of cell uptake of fluorescent immuno-MSN and fluorescent MSN in the brain (left panel), liver (middle panel), and spleen (right panel). Statistical significance was conducted by one-way ANOVA with Tukey's post-test (****P < 0.0001). All cell count data from flow cytometry analysis was collected from a sample size of n = 5, unless otherwise noted, and normalized to 105 viable cells. | ||
MSN particles were then fluorescently tagged with Alexa Fluor 750 to track their uptake by innate immune cells in the brain tumor and other major organs. Fluorescent MSNs were intravenously delivered on days 7 and 8 after orthotopic brain tumor inoculations and analyzed by histology and flow cytometry on day 9 (Fig. 4B). Histological analysis indicates that the intratumoral microdistribution of systemically administered MSNs was predominantly in the perivascular regions of GBM, which coincided with locations harboring high levels of APCs (Fig. 4C). Flow cytometry analysis showed that the uptake of MSNs by key innate immune cell subsets in brain tumors was remarkably high (Fig. 4D). A representative example of threshold gating for MSN+ cells is shown in Fig. S1A (ESI†). Regarding uptake of the particle by APCs, MSNs were found in roughly 50% of DCs and 27% of macrophages. Evaluating cell uptake in the reticuloendothelial (RES) organs, we found that less than 1% of cells in the liver and spleen were positive for the immuno-MSNs compared to 8.7% of cells in the glioma TME of the brain (Fig. 4E).10
Although the GBM vasculature is not as leaky as the angiogenic endothelium of other solid tumors,23–26 the BBB of brain tumors is partially breached,27 allowing for the intratumoral accumulation of nanoparticles in glioma patients.28 These nanoparticles typically exhibit a near-perivascular microdistribution and a limited penetration into the tumor interstitium.29 We previously showed that mesoporous silica nanoparticles of similar size to the immuno-MSN particles accumulate in the near-perivascular space in various orthotopic GBM models.30–32 While the near-perivascular accumulation of nanoparticles limits the effective delivery of cytotoxic drugs to the majority of glioma cells, the APC-rich perivascular space of GBMs is an ideal target as a deposition space for immunostimulatory nanoparticles. With systemic delivery, immuno-MSN particles circulate through the entire tumor microvasculature and readily gain access to these perivascular regions to promote their uptake by local APCs, which recruit antitumor immune cells.
Using IVIS Spectrum imaging, longitudinal in vivo imaging provided the overall organ distribution of fluorescent MSN (empty, no agonist cargo) and fluorescent immuno-MSN (Fig. 4F and Fig. S1B, C, ESI†). Quantification of NP fluorescence obtained ex vivo from the main organs indicated the fluorescent MSN and fluorescent immuno-MSN exhibited similar organ distribution with the majority of the particles being cleared by the reticuloendothelial organs (Fig. 4G). It should be noted that fluorescent labeling of the MSN gave the particle a similar zeta potential to the immuno-MSN. Overall, the MSN accumulation in the brain tumor was consistent to previous reports.30–32 Flow cytometry showed the uptake of fluorescent MSN and fluorescent immuno-MSN by innate immune cells was similar in the brain tumor, liver and spleen (Fig. 4H).
3.5 Immuno-MSN mediates activation and expansion of DCs and macrophages in brain tumors
To assess the cellular response in the glioma TME after treatment with immuno-MSNs, orthotopic GBM mice were treated for two consecutive days with immuno-MSNs and the innate immune cell content was analyzed after 24 h using flow cytometry (Fig. 5A). Significantly elevated levels of macrophages in the brain tumor (>3.6-fold) were measured compared to either the liver or spleen, as well as high levels of DCs, NK cells, and monocytes (>1.5-fold, >1.5-fold, and >6.2-fold, respectively) (Fig. 5B). Compared to untreated controls, significant increases in both macrophages (9.6-fold) and DCs (6.6-fold) were observed in the brain tumor (Fig. 5C). Importantly, healthy brain tissue was largely unaffected by the immuno-MSN treatment, seeing only a modest increase in macrophages, which was significantly lower than that of the brain tumor (Fig. 5C).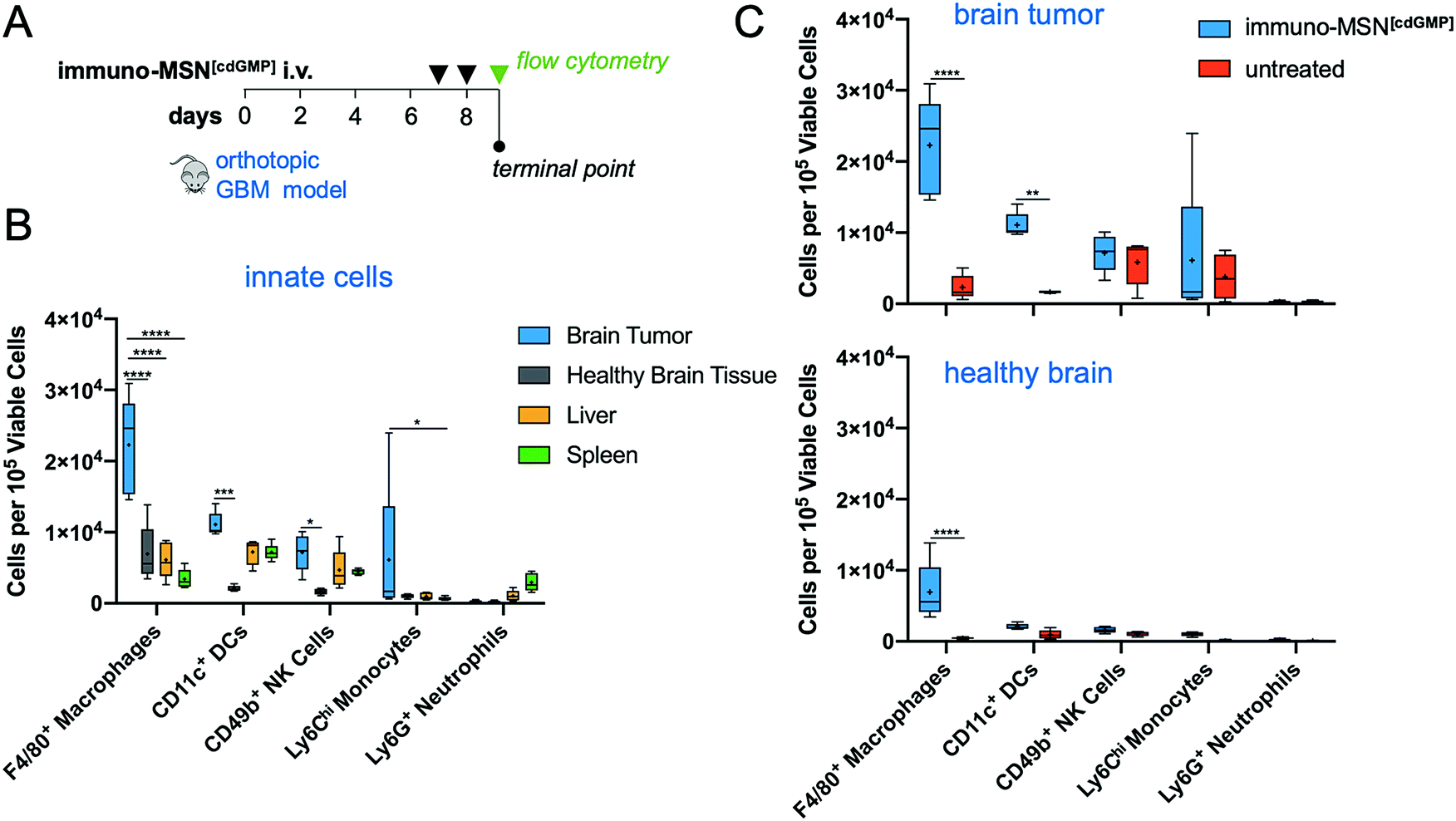 | ||
| Fig. 5 Heightened APC recruitment to the GBM microenvironment after treatment with immuno-MSN particles, sparing healthy brain tissue and peripheral organs. (A) Treatment regimen of intravascularly administered immuno-MSN particles delivering 10 μg of cdGMP per dose. (B) Flow cytometry analysis of excised organ tissue in mice receiving immuno-MSN treatment. Statistical significance in the box and whisker plot (5–95 percentile, “+” mean) was conducted by two-way ANOVA with Tukey's post-test (*P < 0.05, ***P < 0.001, ****P < 0.0001). (C) Flow cytometry analysis of the brain tumor microenvironment (top) and healthy brain tissue (bottom) in mice treated with immuno-MSNs compared to untreated controls. Statistical significance was conducted by two-way ANOVA with Sidak's post-test (**P < 0.01, ****P < 0.0001). All cell count data from flow cytometry analysis was collected from a sample size of n = 5 and normalized to 105 viable cells. | ||
3.6 Immuno-MSN delays GBM tumor growth and elevates peripheral CD8+ T cells
The therapeutic efficacy of the immuno-MSN and its ability to elicit CD8+ T cell activity was assessed in a long-term therapeutic study. After orthotopic inoculation of GL261 glioma cells, mice were treated with immuno-MSNs on days 7, 8 and 14 (Fig. 6A). Strikingly, BLI data showed delayed GBM tumor growth after the first week of treatment with the immuno-MSN particles compared to the untreated tumor controls (Fig. 6B). This finding highlights the importance of recruiting innate immunity to the TME for the treatment of immunosuppressive brain tumors. Additionally, weekly blood samples were analyzed for circulating DCs and CD8+ T cells using flow cytometry. Within one week after treatment, significantly elevated circulating DCs (3.7-fold) were present in the blood (Fig. 6C). Similarly, the immuno-MSN group had a significant 2.5-fold more CD8+ T cells in the blood than the untreated mice, indicating the immuno-MSN treatment successfully drove CD8+ T cell priming (Fig. 6D). It is also evident that the dose and schedule of administration of immuno-MSNs need to be refined to obtain a sustained antitumor immune response as the later data points indicate. It is important to note that the primary objective of the immuno-MSN treatment is the activation of pro-inflammatory innate immunity in the tumor itself, which is a pivotal step to enable consistent and robust outcomes of cancer immunotherapies. An inflamed “hot” brain tumor that is rich in activated APCs can be significantly more receptive to additional immunotherapeutic intervention.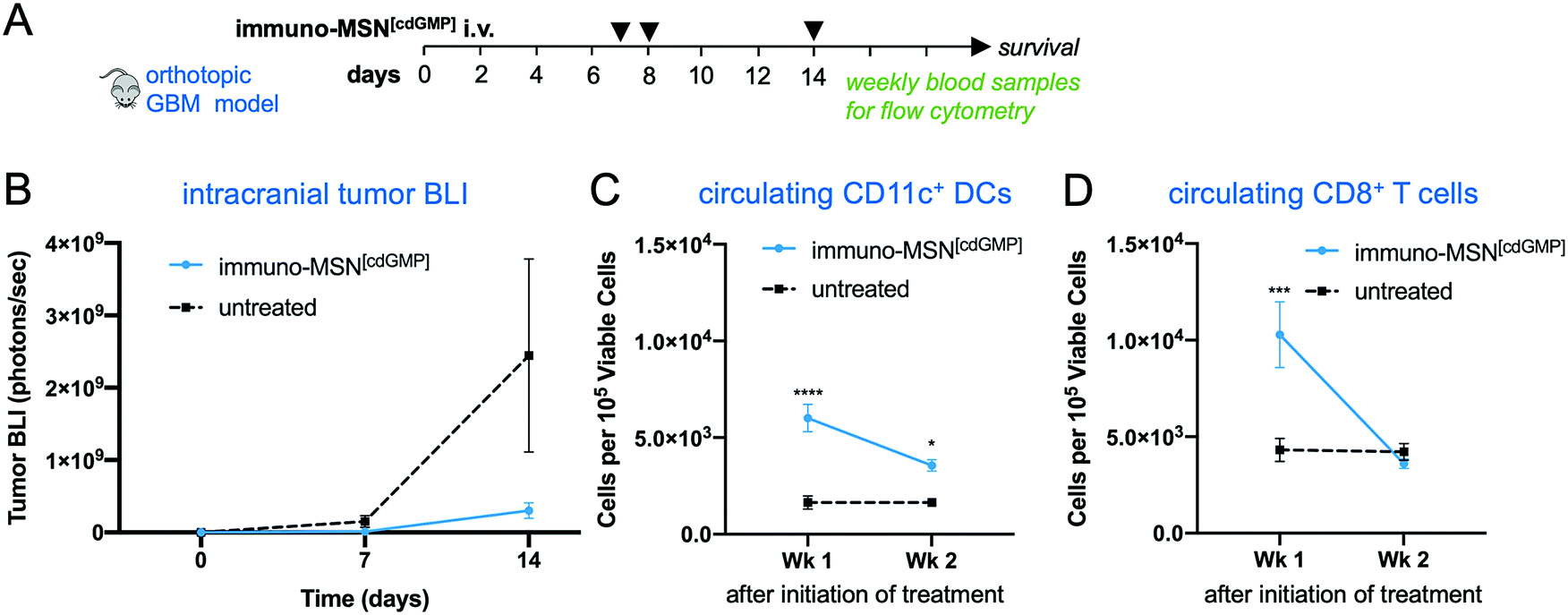 | ||
| Fig. 6 Immuno-MSN particles delay GBM tumor growth and elevate circulating CD11c+ DCs and CD8+ T cells in orthotopic GBM studies. (A) Treatment regimen of intravascularly administered immuno-MSN particles delivering 10 μg of cdGMP per dose. (B) BLI quantification of GBM tumor cell luminescence. Tumor signal is represented as mean ± standard error (n ≥ 5). Statistical significance was conducted by unpaired t-test (two-tailed). Flow cytometry analysis from Wk 1 and Wk 2 blood draws measuring levels of (C) CD11c+ DCs, and (D) CD8+ T cells after the start of immuno-MSN treatment compared to untreated controls. All cell count data from flow cytometry analysis is represented as mean ± standard error and was normalized to 105 viable cells (Wk 1 and Wk 2: n ≥ 5 for immuno-MSN and untreated). Statistical significance was conducted by two-way ANOVA with Sidak's post-test (*P < 0.05, ***P < 0.001, ****P < 0.0001). | ||
For example, we augmented the immuno-MSN treatment with an inhibitor of Transforming Growth Factor beta (TGF-β). Elevated TGF-β is accompanied by both a tumor-intrinsic effect on tumor antigenicity as well as an increase in immune suppressive regulatory T cells,33–38 MDSCs and TAMs.39–42 These immunosuppressive cells are known to depend on TGF-β either for their recruitment into the TME or as a mediator of their immune suppression.43–46 In this context, a survival study was conducted in the orthotopic GL261 model using a combination of immuno-MSNs and Galunisertib, a small molecule inhibitor of TGF-β receptor 1 (TGF-βR1). The combination treatment significantly prolonged median survival compared to the untreated tumor controls (Fig. S3, ESI†).
In the last decade, scientific efforts revealed the complex immune landscape of GBM, dismissing the long-standing dogma of an immune-privileged brain tumor. The GBM TME is comprised of a dynamic assortment of cells that includes dysfunctional innate and adaptive immune cells, MDSCs, and glioma stem-like cells.10 Notably, a system of lymphatic vessels was recently detected in the central nervous system that allows for immune cell trafficking to deep cervical lymph nodes.47 As the framework of the GBM microenvironment continues to evolve, redundant mechanisms of immune escape are becoming better understood.48 More combination therapies are being tested to overcome these immunosuppressive barriers that are not addressed by current standard of care treatments. While clinical trials are underway to concurrently block PD-1 suppression, VEGF signaling, and/or IDO1 suppression, there are no trials that aim to directly stimulate innate immune activity in the TME of GBM (source: ClinicalTrials.gov).
Here, we demonstrated the ability of the immuno-MSN particles to stimulate innate immune activity and IFN-β secretion by systemically delivering STING agonist to the brain TME. Activation and expansion of the innate immune arm within a brain tumor can further augment other immunotherapies with the ultimate objective being improved infiltration and function of effector immune cells. For example, many tumors, including GBM, display an ‘immune excluded’ phenotype, which is largely driven by the cytokine TGF-β.49–51 As previously described, TGF-β is a potent immunosuppressive cytokine that diminishes functionality and tumor infiltration of effector CD8+ T cells and NK cells.50–52 Specifically, elevated TGF-β potently suppresses MHC expression.53–57 To reverse the TGF-β-mediated immune suppression of effector immune cells in GBM, we augmented the immuno-MSN treatment with a small molecule inhibitor of TGF-βR1.58–63 Our studies found that the combination of the immuno-MSN treatment with the anti-TGF-β inhibitor improved the outcome compared to the immuno-MSN treatment alone and significantly prolonged GBM survival compared to the untreated tumor controls. The antitumor potential of the immuno-MSN system may also enhance standard of care treatments like temozolomide chemotherapy and radiotherapy in GBM. Future work includes optimizing the therapeutic potential of the immuno-MSN particles by identifying the minimal effective dose, treatment regimen and experimenting with combination treatment strategies.
4. Conclusions
We engineered an immuno-MSN system that systemically delivered a STING agonist to APCs in the brain TME to reverse immunosuppression. Immuno-MSN particles facilitated the recruitment of DCs and macrophages to the TME while sparing healthy brain tissue and peripheral organs, resulting in elevated circulating CD8+ T cell activity and delayed tumor growth. The immuno-MSN system seeks to address limitations in cancer immunotherapies by boosting innate immunity for a more robust antitumor immune response. The systemic design of the immuno-MSN complex may also translate well for metastatic lesions as well as more accessible cancers like breast cancer and melanoma.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Cancer Institute (R01CA253627, U01CA198892), Case Comprehensive Cancer Center GI SPORE 2P50CA150964-07A1, the Case Comprehensive Cancer Center Support Grant (P30CA043703) and the Shiverick Family Fund, the Clinical Translational Science Collaborative of Cleveland (UL1TR002548), and the Alex's Lemonade Stand Foundation (E. K.). P. B. was supported by the NSF graduate research fellowships program. M. L. was supported by a fellowship from the NIH Interdisciplinary Biomedical Imaging Training Program (T32EB007509) administered by the Department of Biomedical Engineering, Case Western Reserve University. We acknowledge the Case Center for Imaging Research, Case Comprehensive Cancer Center Flow Cytometry Core, and the Case School of Medicine Light Microscopy Core.References
- R. Stupp, M. E. Hegi, W. P. Mason, M. J. van den Bent, M. J. Taphoorn, R. C. Janzer, S. K. Ludwin, A. Allgeier, B. Fisher, K. Belanger, P. Hau, A. A. Brandes, J. Gijtenbeek, C. Marosi, C. J. Vecht, K. Mokhtari, P. Wesseling, S. Villa, E. Eisenhauer, T. Gorlia, M. Weller, D. Lacombe, J. G. Cairncross and R. O. Mirimanoff, Lancet Oncol., 2009, 10, 459–466 CrossRef CAS.
- Z. Li, S. Bao, Q. Wu, H. Wang, C. Eyler, S. Sathornsumetee, Q. Shi, Y. Cao, J. Lathia, R. E. McLendon, A. B. Hjelmeland and J. N. Rich, Cancer Cell, 2009, 15, 501–513 CrossRef CAS.
- C. E. Eyler, Q. Wu, K. Yan, J. M. MacSwords, D. Chandler-Militello, K. L. Misuraca, J. D. Lathia, M. T. Forrester, J. Lee, J. S. Stamler, S. A. Goldman, M. Bredel, R. E. McLendon, A. E. Sloan, A. B. Hjelmeland and J. N. Rich, Cell, 2011, 146, 53–66 CrossRef CAS.
- C. H. June, R. S. O’Connor, O. U. Kawalekar, S. Ghassemi and M. C. Milone, Science, 2018, 359, 1361–1365 CrossRef CAS.
- J. M. Michot, C. Bigenwald, S. Champiat, M. Collins, F. Carbonnel, S. Postel-Vinay, A. Berdelou, A. Varga, R. Bahleda, A. Hollebecque, C. Massard, A. Fuerea, V. Ribrag, A. Gazzah, J. P. Armand, N. Amellal, E. Angevin, N. Noel, C. Boutros, C. Mateus, C. Robert, J. C. Soria, A. Marabelle and O. Lambotte, Eur. J. Cancer, 2016, 54, 139–148 CrossRef CAS.
- D. Migliorini, P.-Y. Dietrich, R. Stupp, G. P. Linette, A. D. Posey and C. H. June, Clin. Cancer Res., 2018, 24(3), 535–540 CrossRef CAS.
- M. Preusser, M. Lim, D. A. Hafler, D. A. Reardon and J. H. Sampson, Nat. Rev. Neurol., 2015, 11, 504–514 CrossRef CAS.
- M. A. Postow, R. Sidlow and M. D. Hellmann, N. Engl. J. Med., 2018, 378, 158–168 CrossRef CAS.
- E. K. Nduom, M. Weller and A. B. Heimberger, Neuro. Oncol., 2015, 17(Suppl 7), vii9–vii14 CrossRef CAS.
- A. R. P. Antunes, I. Scheyltjens, J. Duerinck, B. Neyns, K. Movahedi and J. A. Van Ginderachter, eLife, 2020, 9, e52176 CrossRef CAS.
- D. K. R. Karaolis, T. K. Means, D. Yang, M. Takahashi, T. Yoshimura, E. Muraille, D. Philpott, J. T. Schroeder, M. Hyodo, Y. Hayakawa, B. G. Talbot, E. Brouillette and F. Malouin, J. Immunol., 2007, 178, 2171–2181 CrossRef CAS.
- N. Cheng, R. Watkins-Schulz, R. D. Junkins, C. N. David, B. M. Johnson, S. A. Montgomery, K. J. Peine, D. B. Darr, H. Yuan, K. P. McKinnon, Q. Liu, L. Miao, L. Huang, E. M. Bachelder, K. M. Ainslie and J. P. Y. Ting, JCI Insight, 2018, 3(22), e120638 CrossRef.
- T. Ohkuri, A. Ghosh, A. Kosaka, J. Zhu, M. Ikeura, M. David, S. C. Watkins, S. N. Sarkar and H. Okada, Cancer Immunol. Res., 2014, 2(12), 1199–1208 CrossRef CAS.
- D. Shae, K. W. Becker, P. Christov, D. S. Yun, A. K. R. Lytton-Jean, S. Sevimli, M. Ascano, M. Kelley, D. B. Johnson, J. M. Balko and J. T. Wilson, Nat. Nanotechnol., 2019, 14, 269–278 CrossRef CAS.
- I. Bechmann, J. Priller, A. Kovac, M. Böntert, T. Wehner, F. F. Klett, J. Bohsung, M. Stuschke, U. Dirnagl and R. Nitsch, Eur. J. Neurosci., 2001, 14, 1651–1658 CrossRef CAS.
- S. Watkins, S. Robel, I. F. Kimbrough, S. M. Robert, G. Ellis-Davies and H. Sontheimer, Nat. Commun., 2014, 5, 1–15 Search PubMed.
- P. Domingues, M. González-Tablas, Á. Otero, D. Pascual, D. Miranda, L. Ruiz, P. Sousa, J. Ciudad, J. M. Gonçalves, M. C. Lopes, A. Orfao and M. D. Tabernero, Brain, Behav., Immun., 2016, 53, 1–15 CrossRef CAS.
- P. U. Atukorale, S. Raghunathan, V. Raguveer, T. J. Moon, C. Zhang, P. Bielecki, M. L. Wiese, A. L. Goldberg, G. Covarrubias, C. J. Hoimes and E. Karathanasis, Cancer Res., 2019, 79, 5394–5406 CrossRef CAS.
- H. Meng, M. Wang, H. Liu, X. Liu, A. Situ, B. Wu, Z. Ji, C. H. Chang and A. E. Nel, ACS Nano, 2015, 9, 3540–3557 CrossRef CAS.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS.
- M. C. Hanson, M. P. Crespo, W. Abraham, K. D. Moynihan, G. L. Szeto, S. H. Chen, M. B. Melo, S. Mueller and D. J. Irvine, J. Clin. Invest., 2015, 125, 2532–2546 CrossRef.
- T. Oh, S. Fakurnejad, E. T. Sayegh, A. J. Clark, M. E. Ivan, M. Z. Sun, M. Safaee, O. Bloch, C. D. James and A. T. Parsa, J. Transl. Med., 2014, 12 Search PubMed.
- P. R. Lockman, R. K. Mittapalli, K. S. Taskar, V. Rudraraju, B. Gril, K. A. Bohn, C. E. Adkins, A. Roberts, H. R. Thorsheim, J. A. Gaasch, S. Huang, D. Palmieri, P. S. Steeg and Q. R. Smith, Clin. Cancer Res., 2010, 16, 5664–5678 CrossRef CAS.
- F. M. Boyle, S. L. Eller and S. A. Grossman, Neurooncology, 2004, 6, 300–306 CAS.
- M. Blanchette and D. Fortin, Methods Mol. Biol., 2011, 686, 447–463 CrossRef CAS.
- S. Sato, T. Kawase, S. Harada, H. Takayama and S. Suga, Acta Neurochir., 1998, 140, 1135–1142 CrossRef CAS.
- S. Liebner, A. Fischmann, G. Rascher, F. Duffner, E. H. Grote, H. Kalbacher and H. Wolburg, Acta Neuropathol., 2000, 100, 323–331 CrossRef CAS.
- M. I. Koukourakis, S. Koukouraki, I. Fezoulidis, N. Kelekis, G. Kyrias, S. Archimandritis and N. Karkavitsas, Br. J. Cancer, 2000, 83, 1281–1286 CrossRef CAS.
- B. C. Baumann, G. D. Kao, A. Mahmud, T. Harada, J. Swift, C. Chapman, X. Xu, D. E. Discher and J. F. Dorsey, OncoTargets Ther., 2013, 4, 64–79 CrossRef.
- O. Turan, P. Bielecki, V. Perera, M. Lorkowski, G. Covarrubias, K. Tong, A. Yun, A. Rahmy, T. Ouyang, S. Raghunathan, R. Gopalakrishnan, M. A. Griswold, K. B. Ghaghada, P. M. Peiris and E. Karathanasis, Nanoscale, 2019, 11, 11910–11921 RSC.
- O. Turan, P. A. Bielecki, V. Perera, M. Lorkowski, G. Covarrubias, K. Tong, A. Yun, G. Loutrianakis, S. Raghunathan, Y. Park, T. Moon, S. Cooley, D. Dixit, M. A. Griswold, K. B. Ghaghada, P. M. Peiris, J. N. Rich and E. Karathanasis, Adv. Ther., 2019, 2, 1900118 CrossRef CAS.
- O. Turan, P. Bielecki, K. Tong, G. Covarrubias, T. Moon, A. Rahmy, S. Cooley, Y. Park, P. M. Peiris, K. B. Ghaghada and E. Karathanasis, Mol. Pharm., 2019, 16, 4352–4360 CrossRef CAS.
- H. P. Kim, B. G. Kim, J. Letterio and W. J. Leonard, J. Biol. Chem., 2005, 280, 34042–34047 CrossRef CAS.
- M. C. Kullberg, V. Hay, A. W. Cheever, M. Mamura, A. Sher, J. J. Letterio, E. M. Shevach and C. A. Piccirillo, Eur. J. Immunol., 2005, 35, 2886–2895 CrossRef CAS.
- J. J. Letterio, Oncogene, 2005, 24, 5701–5712 CrossRef CAS.
- M. Mamura, W. K. Lee, T. J. Sullivan, A. Felici, A. L. Sowers, J. P. Allison and J. J. Letterio, Blood, 2004, 103, 4594–4601 CrossRef CAS.
- J. C. Marie, J. J. Letterio, M. Gavin and A. Y. Rudensky, J. Exp. Med., 2005, 201, 1061–1067 CrossRef CAS.
- I.-K. Park, L. D. Shultz, J. J. Letterio and J. D. Gorham, J. Immunol., 2005, 175, 5666–5674 CrossRef CAS.
- X. Chen, L. Wang, P. Li, M. Song, G. Qin, Q. Gao, Z. Zhang, D. Yue, D. Wang, S. Nan, Y. Qi, F. Li, L. Yang, L. Huang, M. Zhang, B. Zhang, Y. Gao and Y. Zhang, Int. J. Cancer, 2018, 143, 2561–2574 CrossRef CAS.
- B. Bierie and H. L. Moses, Cytokine Growth Factor Rev., 2010, 21, 49–59 CrossRef CAS.
- X. Xiang, A. Poliakov, C. Liu, Y. Liu, Z. Deng, J. Wang, Z. Cheng, S. V. Shah, G.-J. Wang, L. Zhang, W. E. Grizzle, J. Mobley and H.-G. Zhang, Int. J. Cancer, 2009, 124, 2621–2633 CrossRef CAS.
- S. V. Ryzhov, M. W. Pickup, A. Chytil, A. E. Gorska, Q. Zhang, P. Owens, I. Feoktistov, H. L. Moses and S. V. Novitskiy, J. Immunol., 2014, 193, 3155–3164 CrossRef CAS.
- D. Llopiz, J. Dotor, N. Casares, J. Bezunartea, N. Díaz-Valdés, M. Ruiz, F. Aranda, P. Berraondo, J. Prieto, J. J. Lasarte, F. Borrás-Cuesta and P. Sarobe, Int. J. Cancer, 2009, 125, 2614–2623 CrossRef CAS.
- H. Li, Y. Han, Q. Guo, M. Zhang and X. Cao, J. Immunol., 2009, 182, 240–249 CrossRef CAS.
- T. Cooks, I. S. Pateras, L. M. Jenkins, K. M. Patel, A. I. Robles, J. Morris, T. Forshew, E. Appella, V. G. Gorgoulis and C. C. Harris, Nat. Commun., 2018, 9, 1–15 CrossRef CAS.
- X. Ye, S. Xu, Y. Xin, S. Yu, Y. Ping, L. Chen, H. Xiao, B. Wang, L. Yi, Q. Wang, X. Jiang, L. Yang, P. Zhang, C. Qian, Y. Cui, X. Zhang and X. Bian, J. Immunol., 2012, 189, 444–453 CrossRef CAS.
- A. Louveau, I. Smirnov, T. J. Keyes, J. D. Eccles, S. J. Rouhani, J. D. Peske, N. C. Derecki, D. Castle, J. W. Mandell, K. S. Lee, T. H. Harris and J. Kipnis, Nature, 2015, 523, 337–341 CrossRef CAS.
- J. Adhikaree, J. Moreno-Vicente, A. P. Kaur, A. M. Jackson and P. M. Patel, Cells, 2020, 9, 263 CrossRef CAS.
- J. V. Joseph, V. Balasubramaniyan, A. Walenkamp and F. A. E. Kruyt, Biochem. Pharmacol., 2013, 85, 478–485 CrossRef CAS.
- H. Mi Lee, K. S. Kim and J. Kim, Cell. Immunol., 2014, 290, 52–61 CrossRef.
- R. Castriconi, A. Dondero, F. Bellora, L. Moretta, A. Castellano, F. Locatelli, M. V. Corrias, A. Moretta and C. Bottino, J. Immunol., 2013, 190, 5321–5328 CrossRef CAS.
- L. Yang, Y. Pang and H. L. Moses, Trends Immunol., 2010, 31, 220–227 CrossRef CAS.
- Y. Dong, L. Tang, J. J. Letterio and E. N. Benveniste, J. Immunol., 2001, 167, 311–319 CrossRef CAS.
- S. Kobayashi, K. Yoshida, J. M. Ward, J. J. Letterio, G. Longenecker, L. Yaswen, B. Mittleman, E. Mozes, A. B. Roberts, S. Karlsson and A. B. Kulkarni, J. Immunol., 1999, 163(7), 4013–4019 CAS.
- T. Nakabayashi, J. J. Letterio, A. G. Geiser, L. Kong, N. Ogawa, W. Zhao, T. Koike, G. Fernandes, H. Dang and N. Talal, J. Immunol., 1997, 158(11), 5527–5535 CAS.
- J. J. Letterio, A. G. Geiser, A. B. Kulkarni, H. Dang, L. Kong, T. Nakabayashi, C. L. Mackall, R. E. Gress and A. B. Roberts, J. Clin. Invest., 1996, 98, 2109–2119 CrossRef CAS.
- A. G. Geiser, J. J. Letterio, A. B. Kulkarni, S. Karlsson, A. B. Roberts and M. B. Sporn, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9944–9948 CrossRef CAS.
- R. Ueda, M. Fujita, X. Zhu, K. Sasaki, E. R. Kastenhuber, G. Kohanbash, H. A. McDonald, J. Harper, S. Lonning and H. Okada, Clin. Cancer Res., 2009, 15, 6551–6559 CrossRef CAS.
- P. Neviani, P. M. Wise, M. Murtadha, C. W. Liu, C. H. Wu, A. Y. Jong, R. C. Seeger and M. Fabbri, Cancer Res., 2019, 79, 1151–1164 CrossRef CAS.
- H. C. Tran, Z. Wan, M. A. Sheard, J. Sun, J. R. Jackson, J. Malvar, Y. Xu, L. Wang, R. Sposto, E. S. Kim, S. Asgharzadeh and R. C. Seeger, Clin. Cancer Res., 2017, 23, 804–813 CrossRef CAS.
- S. Regis, F. Caliendo, A. Dondero, B. Casu, F. Romano, F. Loiacono, A. Moretta, C. Bottino and R. Castriconi, Front. Immunol., 2017, 8, 868 CrossRef.
- J. M. Yingling, W. T. McMillen, L. Yan, H. Huang, J. S. Sawyer, J. Graff, D. K. Clawson, K. S. Britt, B. D. Anderson, D. W. Beight, D. Desaiah, M. M. Lahn, K. A. Benhadji, M. J. Lallena, R. B. Holmgaard, X. Xu, F. Zhang, J. R. Manro, P. W. Iversen, C. V. Iyer, R. A. Brekken, M. D. Kalos and K. E. Driscoll, OncoTargets Ther., 2018, 9, 6659–6677 CrossRef.
- K. Ganesh and J. Massagué, Immunity, 2018, 48, 626–628 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00446d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |