
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA thermoresponsive crosslinker for reversible micelle stabilization†
Mark Q.
Tolentino
and
Jessica L.
Rouge
 *
*
University of Connecticut, Department of Chemistry, 55 North Eagleville Road, Storrs, CT 06269, USA. E-mail: Jessica.rouge@uconn.edu
First published on 16th March 2021
Abstract
Herein we report a thermoresponsive crosslinker that can be utilized to stabilize the surface of micelles through a “click” reaction and to trigger the disassembly via a retro Diels Alder (rDA) mechanism. The micelles generated were 100 nm in size and monodispersed. Above 60 °C, rDA dissociation of crosslinker occurs resulting to the degradation of the micelles and release of hydrophobic cargo.
Owing to their significant impact on nanomedicine and materials science, tremendous effort has been directed towards the development of stimuli-responsive or “smart” nanomaterials.1 These synthetic constructs have the ability to respond to external stimuli (e.g. light, heat, pH, enzymes) by conformational change, aggregation, disassembly or induction of motion.2–4 Of the various types of nanoparticles, micelles have attracted significant attention due to their straightforward synthesis, compatibility in aqueous systems, and their ability to solubilize hydrophobic molecules in water.5 Additionally, micelles can be built to react to biological (e.g. pH6 and enzymes7–9) and external (e.g. temperature10) stimuli, affording controlled cargo delivery and release.11 This can be achieved by either functionalizing the surfactant12 or introducing an additional assembly component with stimuli-responsive moeities.7–9,13 Despite their accessible and modular fabrication, the use of micelles as nanocarriers for therapeutic cargo is hampered by their low thermodynamic stability and extremely dynamic nature of assembly.14 To address this issue, covalent crosslinking of surfactants is usually employed.14–16 In doing so, surfactant molecules are locked in to their assembled state, leading to an intact and stable micelle structure.15 Such micelles have been termed surface crosslinked micelles (SCMs) and have been explored for applications ranging from catalysis17,18 to biology.19
SCMs such as those first developed by Zhao et al. often possess surfactant head groups that are covalently tethered using crosslinkers reactive with functionalities presented by the surfactants.20 Chemical groups receptive to a specific stimuli are integrated into the crosslinker, generating stimuli-responsive SCMs.7–9,11 Recently, our group designed an alkyne-terminated surfactant which self assembles in water for the purpose of drug delivery upon encountering cellular esterases.7 To do so we designed a unique diazido ester crosslinker which reacted with the surfactant via an azide–alkyne click reaction to facilitate the crosslinking. While relying on such biochemical triggers is an attractive avenue for controlled cargo release, their therapeutic efficacy may still be limited by poor target specificity and slow degradation kinetics.1,21 In contrast, an external stimulus such as temperature provides a manual way to control nanoparticle degradation, irrespective of its chemical environment and targeting ability.1,21 For biological applications this ensures cargo release to be more likely to occur at a desired site, minimizing potential side effects.1 For these reasons, we developed a novel thermoresponsive crosslinker to create a temperature-sensitive surfactant assembly or thermo-SCM. Prior to this study, there has been no reported thermoresponsive crosslinked micelles built from a small molecule surfactant, as all are polymer-based.22
The thermally responsive crosslinker, which we refer to hear as thermoXlinker, was synthesized to consist of a furan-maleimide DA adduct as the temperature-responsive domain and two triethyleneglycol (EG)3-azide arms as the click-reactive handles (Fig. 1A). Such structural composition was formulated for three reasons. First, the Diels–Alder [4+2] cycloaddition reaction between the maleimide and furan is thermally reversible and highly efficient without requiring any catalyst.23 The adduct formation typically occurs at moderate temperatures (25–60 °C) while the dissociation, wherein maleimide and furan moieties are regenerated, occurs at elevated temperatures (above 60 °C).23 Second, the presence of PEG units will aid in the solubility of the thermoXlinker in aqueous solution. Third, the azido ends are intended to participate in a crosslinking reaction with the alkyne-modified surfactant via Cu(I)-catalyzed 1,3-dipolar cycloaddition. This click reaction can be conducted in aqueous solution at room temperature conditions with very high yields.24
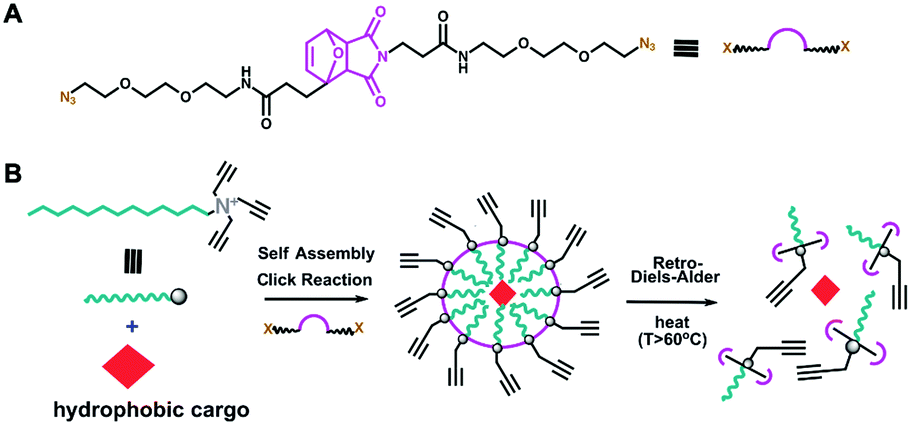 | ||
| Fig. 1 (A) Chemical structure of the novel thermoresponsive crosslinker. (B) Assembly and breakdown of temperature-responsive surface crosslinked micelle (thermo SCM). In water, the trialkyl-functionalized surfactant 2 shown at the left, readily self-assembles into a micelle structure as it encapsulates a hydrophobic cargo. Heating above 60 °C initiates the retro Diels Alder (rDA) reaction within the crosslinker, and the consequent breakdown of SCM releases the internal cargo. | ||
Successful synthesis and structure validation of the thermoXlinker was confirmed by 1H-NMR, 13C-NMR and mass spectrometry (Fig. S4–S6 and Table S2, ESI†). Prior to SCM synthesis, the thermoresponsive behavior of the thermoXlinker as a small molecule was evaluated by 1H-NMR at a temperature of 70 °C (Fig. 2A). This particular temperature was selected over 60 °C to promote a faster rDA and dissociation of the thermoXlinker (Fig. S7, ESI†). Fig. 2B indicates substantial rDA and dissociation (86%) of the thermoXlinker within two hours, as evidenced by the prominent and diminished intensity of protons unique to the DA adduct (a–e), and the appearance and amplified intensity of protons attributed to the regenerated maleimide (a′ and b′) and furan (c′–e′) moieties. After 24 hours, DA adduct protons are hardly noticeable signifying an almost complete rDA conversion and dissociation (98%) of the thermoXlinker. To further support these results, mass spectrometry was performed to detect the decomposition products after 24 hours of heating at 70 °C. The mass spectrum obtained confirms the presence of the two compounds produced from the thermoXlinker decomposition (Fig. S8 and Table S3, ESI†). Altogether, these results demonstrate the temperature-responsive behavior of the synthesized thermoXlinker.
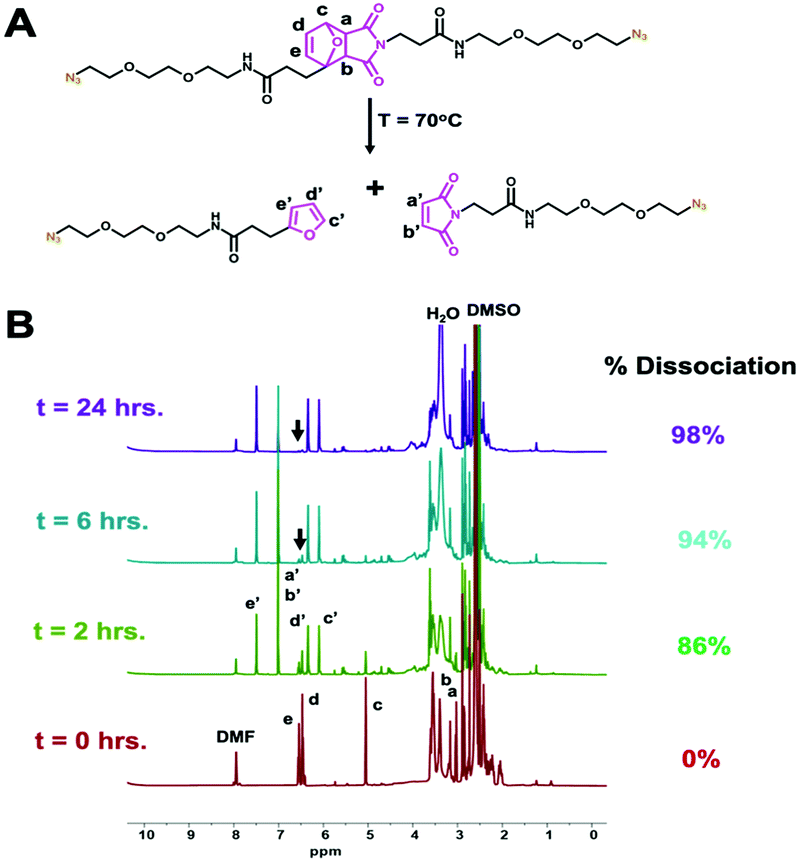 | ||
| Fig. 2 (A) Schematic representation of the thermoXlinker undergoing retro-Diels–Alder (rDA) at 70 °C and yielding the decomposition products. (B) 1H NMR spectral monitoring of the rDA reaction indicating the percent dissociation of thermoXlinker (cinitial = 1.5 mM) over time. Black arrows point to protons d and e. | ||
Using the thermoXlinker, empty SCMs were synthesized and their temperature response in solution investigated. As a control, a non-thermoresponsive PEG crosslinker (Scheme S1, ESI†) was utilized. Dynamic light scattering (DLS) measurements indicated that the thermo SCMs are less than 100 nm in size and exhibited a uniform size distribution (Fig. 3A). TEM analysis (Fig. 3B) corroborates the size and dispersity data from DLS. Additionally, the SCMs displayed a positive surface charge (+39.2 ± 0.8 mV) (Fig. S9, ESI†) ascribed to the quaternary amine head group of the surfactant. Next, the particle size of the PEG and thermo SCMs were monitored at different temperatures to examine their thermal responsive nature in aqueous solution. To prevent the total disintegration of thermo SCMs, the heating time was limited to 30 minutes. This time was also enough for 50% of the thermoXlinker to dissociate (Fig. S7, ESI†) and for the disintegration of the thermo SCMs surface crosslinkages, while partially maintaining the micelle structure (Fig. S11B, ESI†). Fig. 3C reflects a prominent decrease in the particle size of the thermo SCMs starting at 60 °C, indicating the dissociation of the thermoXlinker. An almost complete degradation of thermo SCMs is observed as shown by the absence of intact micelles, achieved after 6 hours of heating at 70 °C (Fig. S11C, ESI†). Minimal changes in particle size are seen at temperatures below 50 °C where rDA and dissociation of the thermoXlinker are not expected. Likewise, PEG SCMs exhibited little to no change in particle size with increasing temperature.
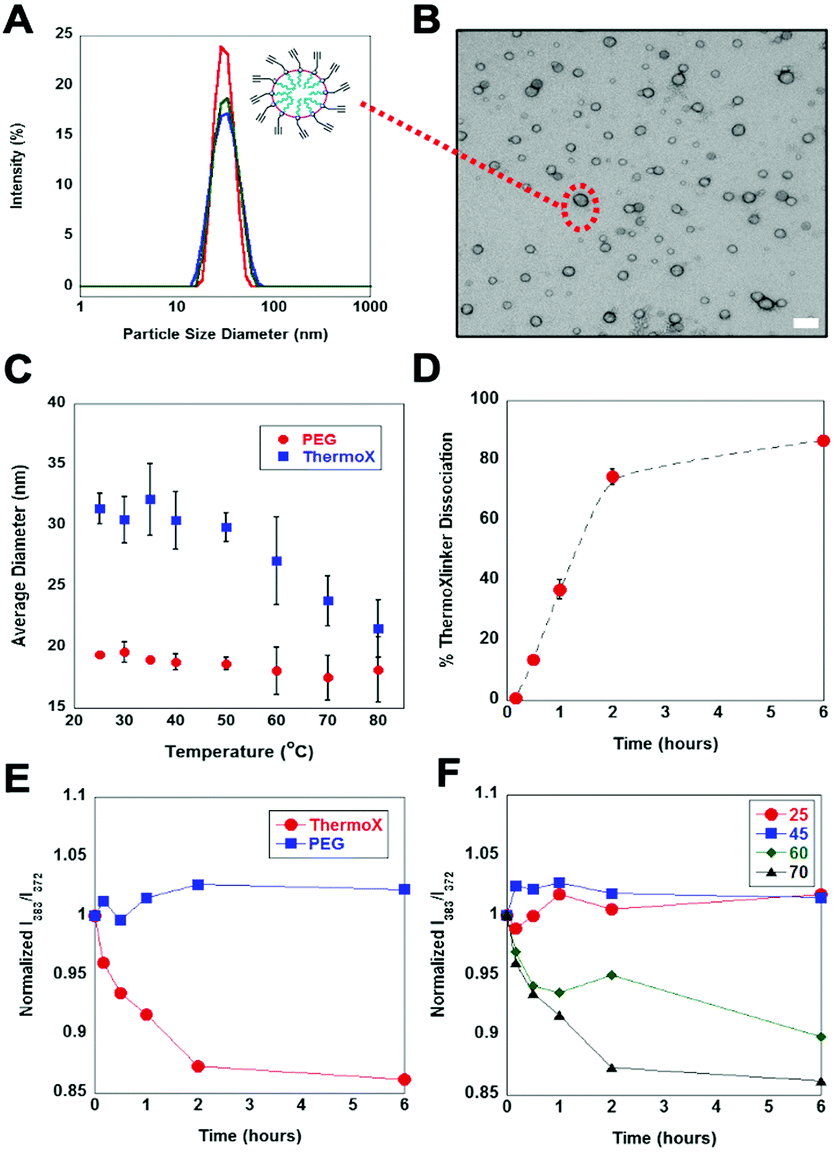 | ||
| Fig. 3 Characterization and temperature responsive behavior of thermo SCMs. (A) Representative particle size measurement of SCMs (B) TEM micrograph showing the average size of thermo SCMs stained with 0.5% uranyl acetate. Scale bar is 100 nm. (C) Particle size monitoring (n = 3) of empty thermo SCMs indicating the disintegration of the micelles at temperatures above 60 °C. No evident change in particle size was observed for PEG SCMs. (D) Kinetics of the thermoXlinker dissociation during thermo SCMs decomposition. Empty thermo SCM solutions were heated at 70 °C and liberated maleimides quantified by fluorescence assay (n = 3). (E) Pyrene I383/I372 ratio as a function of heating time (T = 70 °C) revealing the change in the solution environment of pyrene as a result of thermo SCM breakdown and its release. No significant change was observed for PEG SCMs. (F) Pyrene I383/I372 ratio examined at different temperatures indicating pyrene release occurring only above 60 °C. | ||
To probe the effect of elevated temperatures on the particle size uniformity of SCMs, the polydispersity index (PDI) was measured at 25 °C and 80 °C, and the obtained values were compared (Fig. S10, ESI†). For thermo SCMs, the PDI at 80 °C (0.269) was found to be four times as great as the PDI at 25 °C (0.067), suggesting the presence of non-micellar structures such as smaller modified surfactant aggregates, which could arise from SCM disintegration (Table S4, ESI†). In contrast, the PDI of PEG SCMs at 25 °C and 80 °C appeared to be comparable, indicating that heating at a higher temperature did not affect the particle size distribution.
To gain additional insight into the rDA-induced degradation process, we quantified the amount of maleimide regenerated from thermoXlinker dissociation within the SCM structure using a commercially available maleimide quantification kit. We chose to heat the thermo SCMs at 70 °C for six hours in order to make a rational comparison with the 1H-NMR results. The percent dissociation was calculated by relating the computed maleimide concentration to the initial thermoXlinker concentration utilized in SCM synthesis (Fig. S12, ESI†). Within two hours, the thermoXlinker dissociation reached 75% conversion and 86% after six hours (Fig. 3D). While these values are lower compared to those acquired from the 1H-NMR study (Fig. 2B), both results indicate that extensive thermoXlinker dissociation can occur within two hours of heating at 70 °C. The lesser percent dissociation analyzed from SCM degradation may be associated with the hydrolysis of the maleimide over the course of heating and incomplete “click” reaction during SCM synthesis.
Having shown the thermoresponsive behavior of thermo SCMs, we then encapsulated pyrene as a mock hydrophobic drug cargo and examined its thermally induced release. Pyrene has been widely used as a fluorescence probe for biophysical studies of micelles and membranes.25 The ratio between the intensities of the third (∼383 nm) and first (∼372) vibronic peaks (I383/I372) from the monomer emission spectrum, is strongly sensitive to the polarity of its solution environment.25 Specifically, I383/I372 decreases as the solvent polarity increases.25 Thermo and PEG SCMs loaded with pyrene exhibited uniform particle size and positive surface charge (Fig. S13, ESI†). The amount of encapsulated pyrene in the SCMs was determined by constructing a calibration curve (Fig. S14, ESI†). Based on the initial [surfactant]/[pyrene] ∼8 and our approximate micelle aggregation number (∼60),26 roughly one pyrene molecule is expected to be contained in each micelle. This deduction is attested by the absence of pyrene excimer peak at 480 nm in the fluorescence emission spectra (Fig. S15, ESI†). Importantly, the entrapment of pyrene in the SCM was confirmed by comparing the I383/I372 of the stock (10 mM surfactant) and diluted (1 mM surfactant) SCM solutions. For both thermo and PEG SCMs, the I383/I372 values remain unchanged between 0.89–0.91 despite being below the critical micelle concentration (10 mM). This suggests that the pyrene is physically confined and trapped within the micelle. For the succeeding fluorescence readings, SCM loaded with pyrene contained 0.075 mM surfactant to ensure that no pyrene would be released from a micelle, even uncrosslinked assemblies of surfactants.
SCM solutions were first heated at 70 °C for six hours and fluorescence emission spectra were recorded at specific time points. Looking at Fig. 3E the steep decrease in I383/I372 for thermo SCMs signifies the pyrene being exposed to an aqueous environment as a result of SCM degradation. The magnitude of decrease is comparable to the previously reported pyrene SCM system.14 Notably, most of the pyrene was released after two hours which is consistent with the results from the SCM degradation study (see Fig. 3D). In contrast, no apparent decrease in I383/I372 values was observed for PEG SCMs suggesting that the pyrene remains inside the hydrophobic core. We then studied the pyrene release from thermo SCMs over a range of temperatures (Fig. 3F). At 25 °C and 45 °C, where the thermoXlinkers dissociation and SCM disintegration are not anticipated to happen, no evident decrease in I383/I372 values was observed. This confirms the lack of pyrene release. Conversely, a pronounced decrease in the I383/I372 is observed at 60 °C when compared to 70 °C, denoting a more gradual pyrene release at 60 °C than at 70 °C. This result supports the earlier 1H-NMR finding that rDA and the thermoXlinker dissociation is slower at 60 °C than at 70 °C.
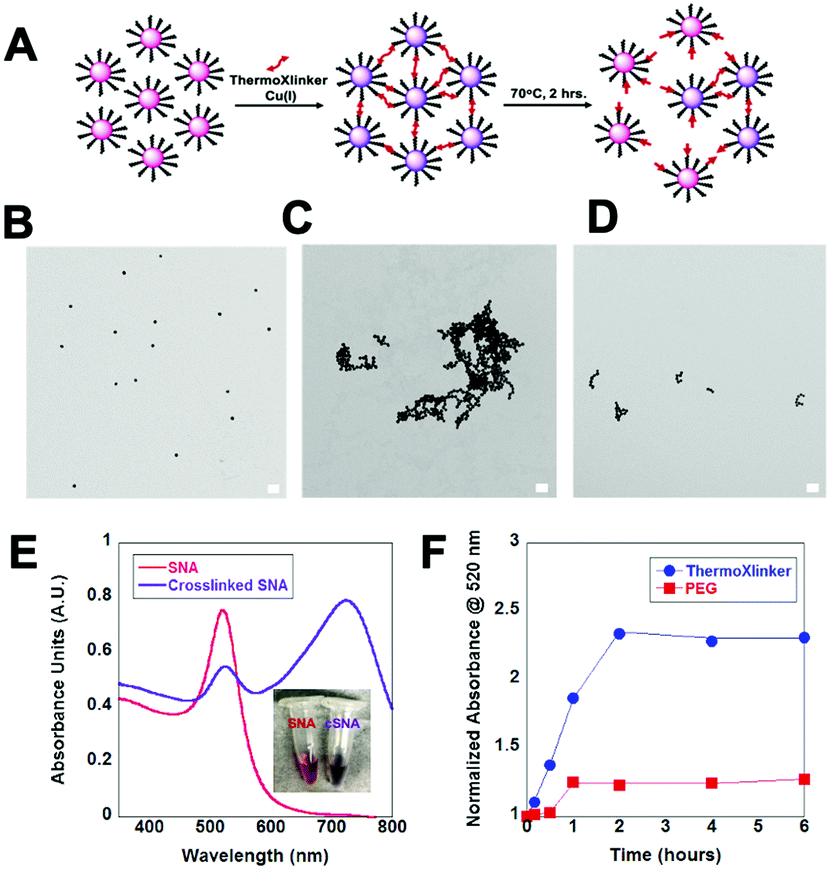 | ||
| Fig. 4 Crosslinking of spherical nucleic acids. (A) Schematic representation of Cu(I) catalyzed SNA crosslinking using the thermoXlinker and the subsequent thermal dissociation. (B) TEM micrograph of SNAs prior to crosslinking (C) TEM after crosslinking with thermoXlinker (D) TEM after heating at 70 °C for 2 hours. Scale bars are 100 nm. (E) UV-VIS spectrum change as a result of SNA crosslinking. (F) Monitoring the absorbance at 520 nm of crosslinked SNAs in response to heating as a function of time (T = 70 °C). | ||
After the successful application of the thermoXlinker in SCM synthesis, we investigated its use as a crosslinker for DNA-modified gold nanoparticles (Au NPs) to further demonstrate the utility of the temperature response of the crosslinker outside a micellar system. We explored whether the thermoXlinker could be used to initiate gold nanoparticle aggregation via a Cu(I)-catalyzed click reaction and be reversibly disassembled by rDA-induced heating. To accomplish this, a 30 nm gold NP was functionalized with thiolated-polyT20 DNA presenting a terminal 5′-hexynyl, to build alkyne modified spherical nucleic acids (SNAs). The synthesis of the SNA structure was verified by an increase in particle size (42 nm) (see Fig. S17A, ESI†) and the enhanced electrophoretic mobility of the Au NPs (see Fig. S17B, ESI†). Afterwards, a Cu(I)-catalyzed click reaction was performed between the SNAs and the thermoXlinker, wherein equimolar concentrations of the crosslinker and DNA were utilized to reach a high degree of crosslinking (see Fig. 4A). To characterize the temperature-response of the thermoXlinker, the crosslinked SNAs were heated at 70 °C for two hours to allow the rDA-driven disassembly. This phenomenon was observed using TEM. Fig. 4C–E display the aggregation of SNAs induced through crosslinking and the subsequent breakdown after heating. SNAs crosslinked with a PEG crosslinker remained in an aggregated state after subjecting them to equivalent heating conditions (see Fig. S18, ESI†). In addition to TEM, the solution behavior of the crosslinked SNAs was studied using UV-VIS spectroscopy. Au NPs in aqueous solution possess a unique absorption peak at 520 nm arising from surface plasmon absorption thus giving the solution an intense red color.27 This peak is shifted to a longer wavelength when aggregation occurs.27 The expected red-shift as a result SNA crosslinking was observed in Fig. 4E after 30 minutes of running the Cu(I) click reaction. The heat-dependent disassembly of crosslinked SNA was also observed by monitoring the absorbance of crosslinked SNAs at 520 nm at 70 °C over six hours (see Fig. 4F).
The increase in absorbance at 520 nm can be correlated to the disassembly of crosslinked SNAs. For SNAs crosslinked with the thermoXlinker, an increase in absorbance was apparent in the first 2 hours, whereas no appreciable increase was observed for PEG-crosslinked SNAs. These results reflect the temperature-responsive behavior of the thermoXlinker observed in the 1H-NMR and pyrene release experiments.
In summary, we have provided a comprehensive analysis of a new temperature responsive crosslinker for creating thermoresponsive SCMs shown through a combination of DLS, TEM microscopy and fluorescence studies, indicating that SCMs crosslinked with the thermoXlinker are less than 100 nm, uniform in size and controllably degrade at temperatures above 60 °C. Although the temperatures ranges studied here are high for biological systems, this can be circumvented by tuning the construct to respond to physiologically relevant temperatures via localized heating using iron oxide nanoparticles.28
Moreover, the thermoresponsive behavior of the thermoXlinker is applied to reversible crosslinking studies of Au NPs. The facile synthesis and the controllable degradation of the micelles enabled by the crosslinker make it a versatile conjugation strategy for a variety of micelle-based applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work under NSF grant 1847869 is gratefully acknowledged.References
- A. P. Blum, J. K. Kammeyer, A. M. Rush, C. E. Callmann, M. E. Hahn and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137(6), 2140 CrossRef CAS PubMed.
- Z. Dai, H. M. Leung and P. K. Lo, Small, 2017, 13, 1602881 CrossRef PubMed.
- L. Xu, M. Zhao, Y. Yang, Y. Liang, C. Sun, W. Gao, S. Li, B. He and Y. Pu, J. Mater. Chem. B, 2017, 5, 9157 RSC.
- Q. Pan, B. Zhang, X. Peng, S. Wan, K. Luo, W. Gao, Y. Pu and B. He, Chem. Commun., 2019, 55, 13896 RSC.
- N. A. N. Hanafy, M. El-Kemary and S. Leporatti, Cancers, 2018, 10(7), 238 CrossRef PubMed.
- W. Hong, S. Hong, M. Qiao, Z. Zhang, W. Yang, L. Dong, F. Xie, C. Zhao and L. Kang, Sci. Rep., 2017, 7, 42465 CrossRef CAS PubMed.
- J. K. Awino, S. Gudipati, A. K. Hartmann, J. J. Santiana, D. F. Cairns-Gibson, N. Gomez and J. L. Rouge, J. Am. Chem. Soc., 2017, 139(18), 6278 CrossRef CAS PubMed.
- J. J. Santiana, B. Sui, N. Gomez and J. L. Rouge, Bioconjugate Chem., 2017, 28(12), 2910 CrossRef CAS PubMed.
- A. K. Hartmann, D. F. Cairns-Gibson, J. J. Santiana, M. Tolentino, H. Barber and J. L. Rouge, ChemBioChem, 2018, 19, 1734 CrossRef CAS PubMed.
- J. Akimoto, M. Nakayama and T. Okano, J. Controlled Release, 2014, 193, 2 CrossRef CAS PubMed.
- M. Q. Tolentino, A. K. Hartmann, D. Loe and J. L. Rouge, J. Mater. Chem. B, 2020, 8, 5627 RSC.
- H. Feng, N. A. L. Verstappen, A. J. C. Kuehne and J. Sprakel, Polym. Chem., 2013, 4, 1842 RSC.
- J. Guo, G. Chen, X. Ning, M. A. Wolfert, X. Li, B. Xu and G.-J. Boons, Chem. – Eur. J., 2010, 16, 13360 CrossRef CAS PubMed.
- S. Zhang and Y. Zhao, J. Am. Chem. Soc., 2010, 132, 10642 CrossRef CAS PubMed.
- R. K. O’Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- G. Chadha and Y. Zhao, J. Colloid Interface Sci., 2013, 390(1), 151 CrossRef CAS PubMed.
- M. Arifuzzaman and Y. Zhao, ACS Catal., 2018, 8(9), 8154 CrossRef CAS.
- L. Hu, M. Arifuzzaman and Y. Zhao, ACS Catal., 2019, 9(6), 5019 CrossRef CAS.
- Y.-Z. Chen, P.-Z. Chen, H.-Q. Peng, Y. Zhao, H.-Y. Ding, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Commun., 2013, 49, 5877 RSC.
- S. Zhang and Y. Zhao, Macromolecules, 2010, 43(9), 4020 CrossRef CAS.
- D. Cui, P. Li, X. Zhen, J. Li, Y. Jiang, A. Yu, X. Hu and K. Pu, Adv. Funct. Mater., 2019, 29, 1903461 CrossRef.
- Y. Li, K. Xiao, W. Zhu, W. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2014, 66, 58 CrossRef CAS PubMed.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS PubMed.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99(7), 2039 CrossRef CAS.
- S. P. Moulik, M. E. Haque, P. K. Jana and A. R. Das, J. Phys. Chem., 1996, 100, 701–708 CrossRef CAS.
- C. Hua, W. H. Zhang, S. R. M. De Almeida, S. Ciampi, D. Gloria, G. Liu, J. B. H. Harper and J. J. Gooding, Analyst, 2012, 137, 82 RSC.
- T. T. T. N’Guyen, H. T. T. Duong, J. Basuki, V. Montembault, S. Pascual, C. Guibert, J. Fresnais, C. Boyer, M. R. Whittaker, T. P. Davis and L. Fontaine, Angew. Chem., Int. Ed., 2013, 52, 14512 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00089f |
| This journal is © The Royal Society of Chemistry 2021 |