
Recent progress in the catalytic transformation of carbon dioxide into biosourced organic carbonates
Vatcharaporn
Aomchad
 ab,
Àlex
Cristòfol
a,
Francesco
Della Monica
*a,
Bart
Limburg
a,
Valerio
D'Elia
*b and
Arjan W.
Kleij
*ac
ab,
Àlex
Cristòfol
a,
Francesco
Della Monica
*a,
Bart
Limburg
a,
Valerio
D'Elia
*b and
Arjan W.
Kleij
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science & Technology, Av. Països Catalans 16, 43007 – Tarragona, Spain. E-mail: akleij@iciq.es; fdellamonica@iciq.es
bDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), 555 Moo 1, 21210, Payupnai, WangChan, Rayong, Thailand. E-mail: valerio.delia@vistec.ac.th
cCatalan Institute of Research and Advanced Studies (ICREA), Pg. Lluis Companys 23, 08010 – Barcelona, Spain
First published on 21st January 2021
Abstract
Cyclic organic carbonates are among the most widely studied targets in the nonreductive conversion of carbon dioxide using oxiranes as the common reaction partners. Apart from using fossil fuel based precursors, recent developments have shown that biomass related feedstock can also serve as coupling partner for CO2 allowing the preparation of more functional and complex types of carbonate architectures. This tutorial review places this latter development in the current context of new and more sustainable material designs, and highlights the main types of biomass that have been examined using primarily homogeneous catalysis approaches.
1. Introduction
Cyclic organic carbonates, more typically denoted as cyclic carbonates (CCs), have become a major target in the area of catalytic valorization of (waste) carbon dioxide.1–8 The most common way to prepare such carbonates is through the coupling of epoxides and CO2,9–11 a reaction that has reached a high level of sophistication thanks to the development of improved catalysts,12–16 and new concepts.17–19 A more recent trend shows a shift towards the use of biosourced feedstock in the synthesis of organic carbonates, i.e., so-called bio-carbonates. These biosourced carbonates are believed to give new impetus for the development of a variety of new applications such as their use as drop-in monomers for sustainable polymers,20,21 the creation of isocyanate-free polyurethanes (NIPUs),22,23 new types of plasticizers,24 green and biodegradable solvents and surfactants,25 and functionalized building blocks for organic synthesis.26,27With this context in mind, we decided to capture the most recent and important advances in this area in this review. The focal point will be on selected biosourced feedstock that has been utilized to prepare cyclic carbonate structures including glycerol, fatty acids, terpenes and carbohydrates (Fig. 1).
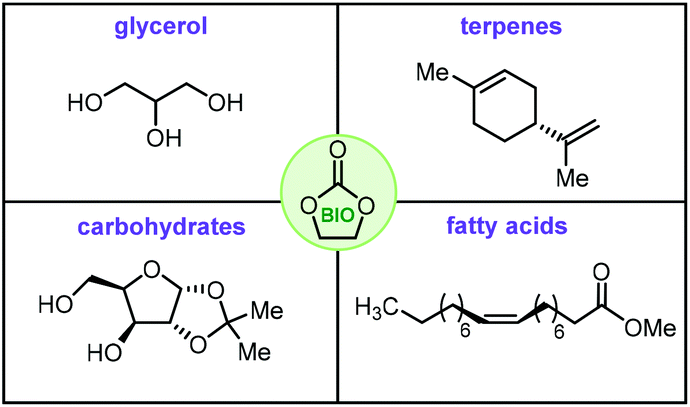 | ||
| Fig. 1 Biobased feedstock used to prepare bio-carbonates. Apart from glycerol, exemplary cases of the other categories are shown. | ||
Here, biosourced feedstock is defined as those starting materials produced by the growth of microorganisms, plants or animals or derived thereof.28,29 Each of these specific categories of biocarbonates is shortly introduced followed by a detailed description of the state-of-the-art. Mechanistic details on the formation of CCs from CO2 and epoxides are provided where necessary, since this topic has been recently and extensively reviewed.30 This article thus summarizes the most frequently used feedstock to build biobased organic carbonates serving as an inspiring stepping stone towards a greener development of CO2-derived heterocyclic building blocks.
2. Glycerol carbonate
The utilization of glycerol to produce valuable chemicals is an inspiring goal for chemists. Glycerol is the main byproduct derived from the production of biodiesel (i.e., through the transesterification of triglycerides, present in vegetable oils, with alcohols such as methanol), and is produced at a higher rate than it is consumed. Therefore, it is important to find ways that transform low-value glycerol into high-value products. Regarding the chemical transformations of glycerol, one of the most attractive conversion processes is its conversion into glycerol carbonate (GC).31–39 This cyclic organic carbonate finds interesting use in cosmetics and lubricants, coating materials and as a polar solvent.40 In addition, GC can also be used as building block in organic synthesis through ring-opening, decarboxylation, esterification or polymerization reactions.41–43The most common way to produce GC is by transesterification of glycerol with dimethyl carbonate (DMC) or urea with the aid of a catalyst.44–48 The most desirable sustainable and straightforward route to produce GC would be from glycerol and carbon dioxide combining two waste molecules. However, the direct coupling of CO2 and glycerol is an equilibrium-limited reaction making it difficult to achieve high conversions and yields unless sacrificial dehydrating agent are used (vide infra). Despite significant advances that have been achieved in the direct glycerol-to-GC conversion, an alternative route towards GC utilizes glycidol and CO2. Glycidol has much higher reactivity towards CO2 and, moreover, it can also be produced from biomass. The subsequent sections discuss the pros and cons of both formation routes.
2.1. Glycerol carbonate from glycerol and CO2
The first attempt towards the synthesis of GC from glycerol and CO2 was reported by Mouloungui in 1998.49 Unfortunately, under supercritical CO2 conditions, the reaction did not occur. It was not until 2006 when Dibenedetto successfully reported that Sn-based catalysts of the formula (n-Bu)2SnO or (n-Bu)2Sn(OMe)2 were able to catalyze the formation of GC at 5 MPa of CO2 pressure and 180 °C to obtain a maximum of 5.7% isolated yield of GC under solvent-free conditions using molecular sieves to remove water from the reaction mixture.50 The Sn-based catalyst activates glycerol by forming an active Sn-glycerate intermediate that allows for CO2 insertion. This process was greatly improved by Munshi in 2009 using MeOH as medium, which increased the yield of GC to 35% (Scheme 1).51 The addition of MeOH is believed to help preventing catalyst deactivation by avoiding oligomerization of the mononuclear catalytic species.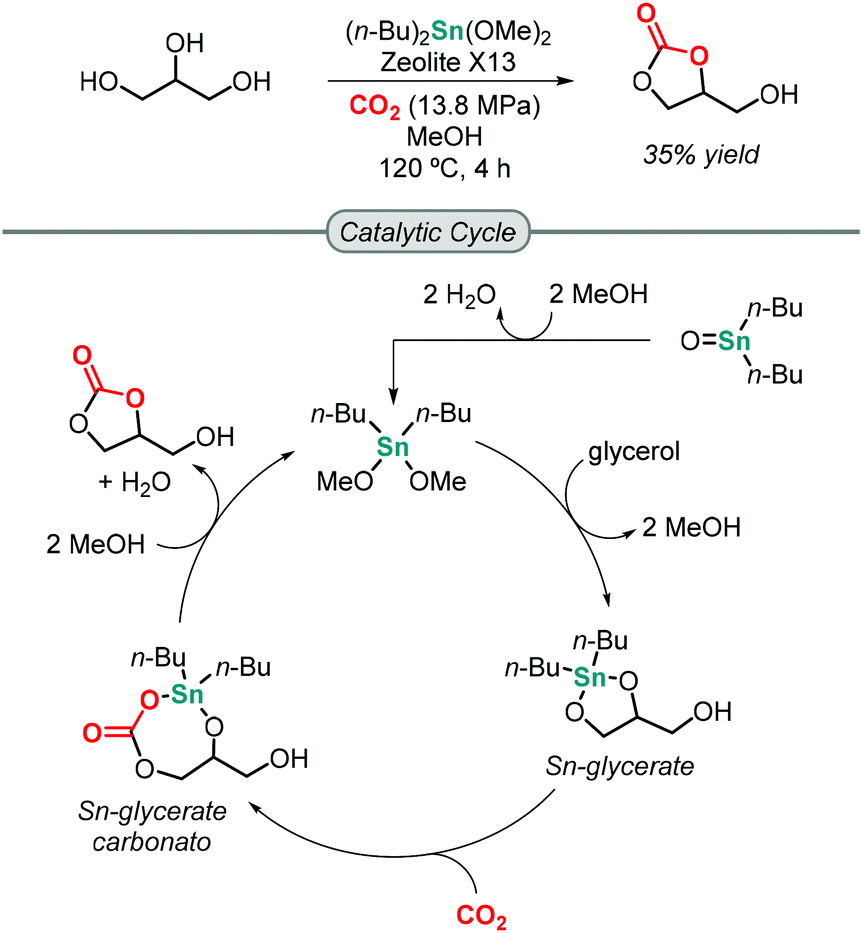 | ||
| Scheme 1 Formation of GC from glycerol using (n-Bu)2Sn(OMe)2 as catalyst. | ||
Up to now, many catalytic systems have been reported that are based on metal oxides/complexes,52–57 supported metal catalysts,58,59 or modified zeolites or hydrotalcites,60–62 in combination with a dehydrating reagent. Most of the reported heterogeneous catalysts typically require harsh reaction conditions for GC synthesis, i.e. a CO2 pressure above 40 bar and temperatures above 150 °C.
Another notable feature of these processes is the introduction of a dehydrating reagent, which was found to be more effective than molecular sieves, but on the other hand lowered in some cases the chemoselectivity towards GC (Scheme 2).57 In most reported approaches, acetonitrile is selected as an inexpensive dehydrating agent, which upon reaction with the in situ generated water forms acetamide. This in turn may further react with a second molecule of water to generate acetic acid. The latter can lower the selectivity towards GC through mono- and di-acetylation of glycerol.
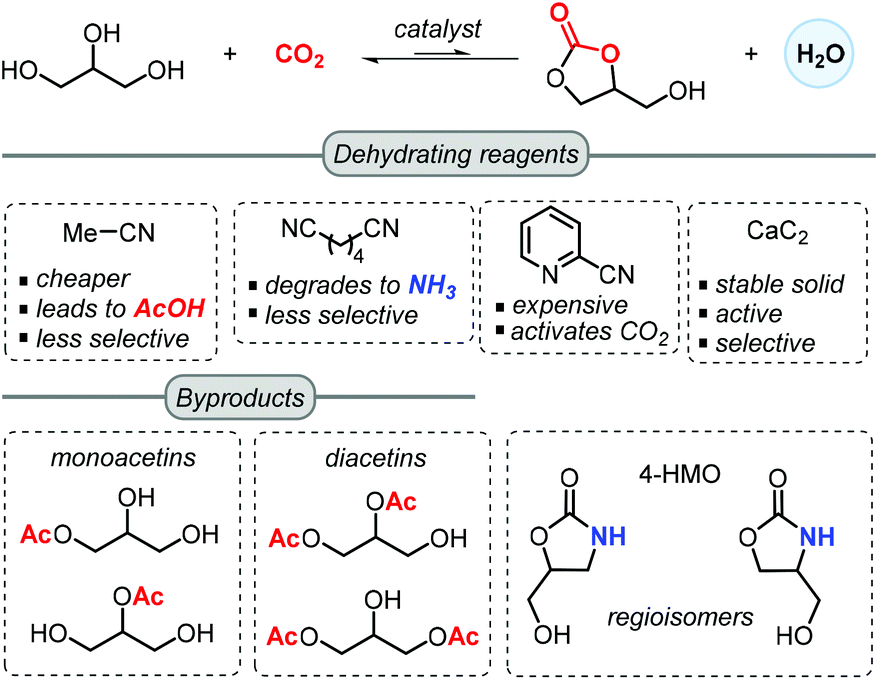 | ||
| Scheme 2 Comparison of different dehydrating agents in the formation of GC from glycerol and CO2. | ||
Adiponitrile has been used as a dehydrating agent by McGregor,57 who found that under the applied reaction conditions, adiponitrile degraded to NH3 that upon reaction with glycerol produces 4-(hydroxymethyl)oxazolidin-2-one (4-HMO) as a mixture of two regioisomers. Apart from acetonitrile and adiponitrile, He and co-workers reported in 2016 the use of 2-cyanopyridine as a superior dehydrating reagent. In the presence of a CeO2 based catalyst, the yield of GC is boosted up to 79% when using 3 equiv. of 2-cyanopyridine at 4 MPa of CO2 and 150 °C in DMF.63 Moreover, the CeO2 catalyst could be recycled 5 times through a calcination process at 400 °C. Recently, Choi et al. demonstrated the advantage of using CaC2 as a dehydrating agent for the synthesis of GC, which in combination of Zn(OTf)2/phen (1,10-phenanthroline) in NMP (N-methyl-2-pyrrolidone) could achieve 88% isolated yield at 50 bar of CO2 pressure at 180 °C for 24 h.55
One year later, Zhao et al. demonstrated that the formation of GC (via 2-cyanopyridine) also occurs in the absence of a metal catalyst achieving a 19% yield at 15 MPa of CO2 and 180 °C (Scheme 3).64 The authors confirmed by FTIR that 2-cyanopyridine activates CO2 to form a five-membered ring which then reacts with glycerol to produce GC. Interestingly, switching to 3-cyanopyridine, 4-cyanopyridine or acetonitrile did not provide such potential. Theoretical calculations pointed indeed to the formation of a five-membered heterocycle as the “activated” form of CO2, which is subsequently involved in a transesterification reaction of glycerol.
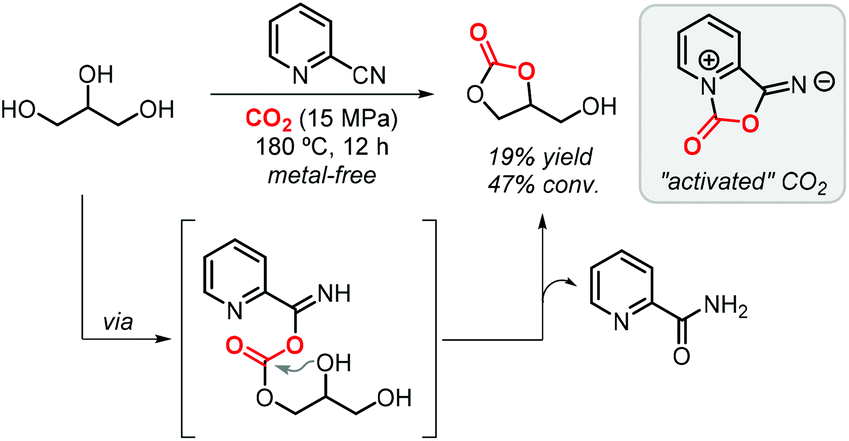 | ||
| Scheme 3 Metal-free formation of GC using 2-cyanopyridine. | ||
2.2. Glycerol carbonate using coupling agents
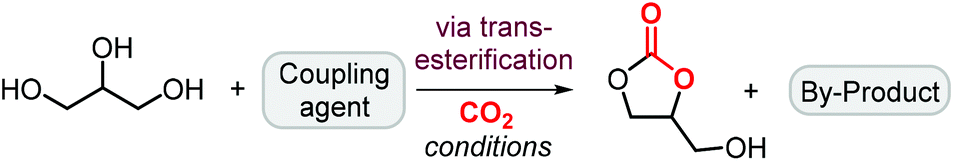
Glycerol carbonate can also be successfully prepared by reaction of glycerol and carbon dioxide in the presence of a coupling agent, which is used to produce a cyclic carbonate intermediate in situ allowing for transesterification of glycerol to form glycerol carbonate and a by-product (Scheme 4). The involvement of a third reactant allows to overcome the intrinsic thermodynamic limitation for the direct reaction between glycerol and CO2, whereas it also produces a new byproduct derived from this additional component.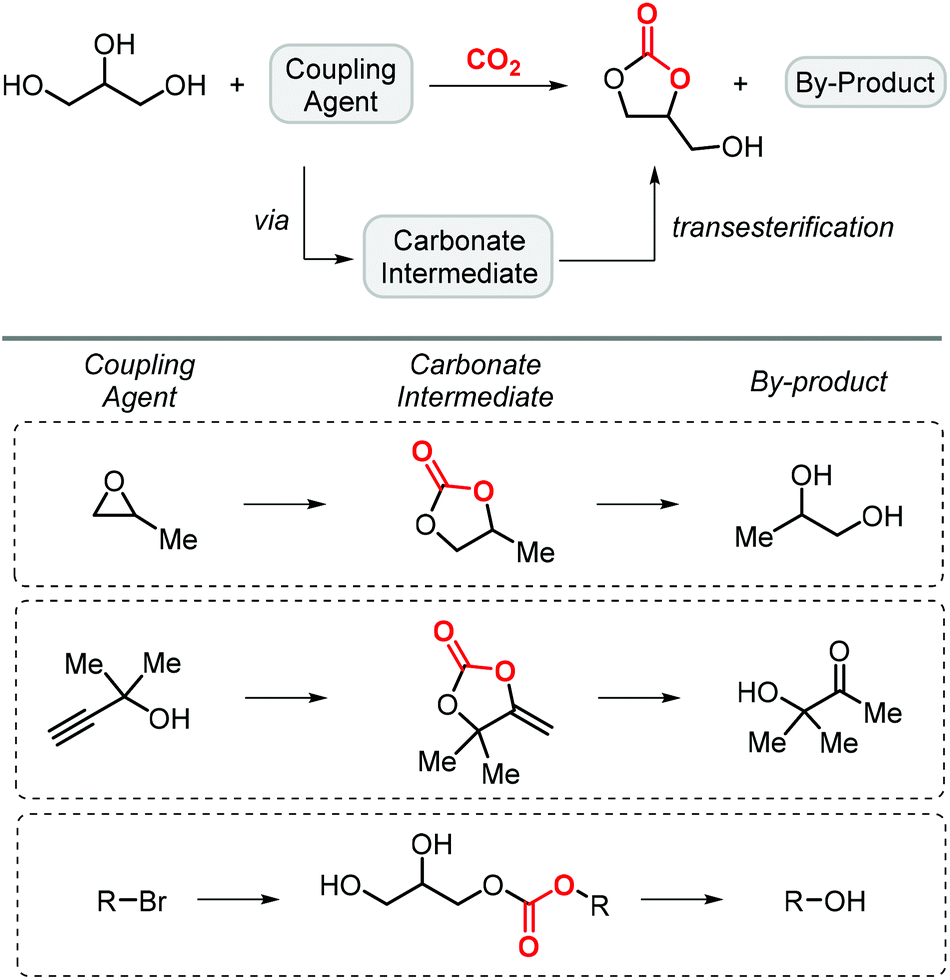 | ||
| Scheme 4 General reaction of glycerol with CO2 in the presence of a coupling agent. | ||
In this context, propylene oxide (PO) was the first additive to be reported by Han and co-workers in 2012 (entry 1, Table 1).65 Although PO constitutes an inexpensive precursor, the process would not be entirely biobased since PO is primarily produced from fossil fuel feedstock. The reaction uses KI as a catalyst and is carried out at 2 MPa of CO2 pressure and 115 °C affording GC in 75% yield. The catalyst promotes the formation of propylene carbonate (PC), which then engages in a transesterification of glycerol producing both GC and propylene glycol. In 2018, Xiao et al. improved the process by introducing a bromide-based heterogeneous catalyst obtained through the polymerization of divinyl benzene and 1-vinyl-3-butylimidazolium bromide (entry 2, Table 1).66 Importantly, not only the yield of GC could be improved to 81%, but also the catalyst could be recycled up to 5 times while maintaining the same level of activity.
|
|
||||
|---|---|---|---|---|
| Entrya | Coupling agent | Conditions | Yield of GC (%) | Yield of byproduct (%) |
| a Corresponding reference in parentheses. b 59% yield of propylene carbonate. c 76% yield of propylene carbonate. | ||||
| 1 [65] |
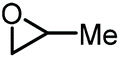
|
CO2 (2 MPa), KI 115 °C, 1.5 h | 77 | 39b |
| 2 [66] |
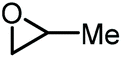
|
CO2 (2 MPa) P-DVB-(vIm-BuBr) 100 °C, 4 h | 81 | 20c |
| 3 [67] |
|
CO2 (1 MPa) Ag2CO3, XantPhos MeCN, 80 °C, 16 h | 82 | 83 |
| 4 [68] |
|
CO2 (1 MPa) Ag sulfadiazene, Et4NBr 80 °C, 24 h | 56 | 69 |
| 5 [69] |
|
1. CO2 (0.1 MPa) Amidine-CO2 25 °C, 24 h 2. MTBD MeCN, 25 °C, 24 h | 85 | NR |
| 6 [70] |
|
CO2 (3 MPa), DBU DMF, 120 °C, 10 h | 97 | 63 |
| 7 [71] | CH2Br2 | CO2 (1 MPa) DBU, BmimPF6 70 °C, 18 h | 86 | NR |
| 8 [72] | n-BuBr | CO2 (0.1 MPa) Guanidine cat. NMP, 50 °C, 4 h | 74 | NR |
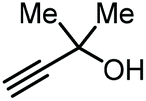
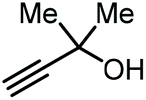
Apart from the use of PO, dimethylethynyl carbinol (i.e., a propargylic alcohol) has also been applied as a reagent to generate GC in high yield, along with 3-hydroxy-3-methyl-2-butanone as the byproduct. The reaction proceeds through the intermediacy of a cyclic alkenyl carbonate, which allows for the transesterification of glycerol. Since 3-hydroxy-3-methyl-2-butanone contains a tertiary alcohol group, it is less nucleophilic than propylene glycol, and thus does not readily react with the formed GC to return the carbonate intermediate. Initially, the group of He reported in 2017 the use of a Ag2CO3/Xantphos as a catalyst promoting this transformation in MeCN at 80 °C under 1 MPa of CO2 pressure to give GC in 82% yield (entry 3, Table 1).67 A solvent-free version was later reported by Song and Zhang using a silver sulfadiazine/Et4NBr catalytic system, although a lower yield for GC was obtained (entry 4, Table 1).68
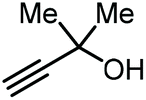
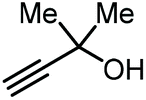
On the other hand, the groups of Lu,69 and Liu,70 independently reported an organocatalytic approach to prepare GC. In the case of Lu, a one-pot approach was applied by formation of the intermediate carbonate using a catalytic amount of an amidine-CO2 adduct, followed by the transesterification with glycerol catalyzed by MTBD (entry 5, Table 1; MTBD = methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene). In this way, 85% yield of GC was obtained under very mild reaction conditions. The system of Liu and co-workers was based on DBU (8-diazabicyclo[5.4.0]undec-7-ene) and was also found to promote both steps to deliver GC in 97% yield albeit under harsher reaction conditions (entry 6, Table 1).
The last example of this strategy illustrates that alkyl halides are also effective reactants. Compared to the other methods, the use of halogenated reagents implies the generation of halogen-containing waste, which is not ideal from a sustainable point of view. Initially, Jang reported in 2014 one example using BmimPF6 (an ionic liquid), DBU as a homogeneous base and CH2Br2 as both the reactant and solvent to afford GC in 86% isolated yield (entry 7, Table 1).71 An intermediary linear carbonate salt attacks the CH2Br2 reagent following ring-closure leading to the CC. The authors propose that the ionic liquid helps to improve the solubility of CO2 although under basic conditions, it is possible that an NHC-carbene forms able to capture/activate CO2, which can then react with glycidol. In another approach, Mihara et al. used n-BuBr as the additional component, together with a guanidine-based catalyst (entry 8, Table 1).72 By adjusting the reaction conditions, it was possible to selectively form GC in 74% yield at 50 °C under 0.1 MPa of CO2 pressure and using NMP (N-methyl pyrrolidone) as solvent.
2.3. Glycerol carbonate from glycidol
Apart from the conversion of bio-glycerol into glycerol carbonate, an alternative and popular route starts from its epoxy alcohol derivative, viz. glycidol (Gly). This precursor can be derived from glycerol present in industrial waste produced in the synthesis of epichlorohydrin, thus providing an opportunity for recycling it into valuable glycerol carbonate in two steps.73 This route based on Gly features excellent atom-economy and, additionally, since many highly efficient catalytic systems have been developed over the years for the coupling of oxiranes and CO2, glycidol has often been included in the substrate scope.Gly has distinct reactivity compared to other common epoxides such as PO or styrene oxide. The non-innocent hydroxyl group can actively participate in the reaction in two distinct ways. Capacchione and co-workers found that ring-opening of glycidol by an external nucleophile was faster than with other epoxides (Table 2).74 By means of NMR and DFT studies, they concluded that glycidol dimers were present under the catalytic reaction conditions. Intermolecular hydrogen bonds between the Gly molecules are important to explain the activation the oxirane ring of Gly towards nucleophilic ring-opening by bromide (Fig. 3). Hence, using simple TBAB (tetrabutylammonium bromide, 5 mol%) at 60 °C and 1 MPa of CO2 pressure it was possible to convert Gly in >99% conversion and selectivity producing GC (entry 1, Table 2). Lowering the temperature to 40 °C or reducing the loading of TBAB to 1 mol% while heating at 80 °C for 1 h reduced the conversion to 87% and 85%, respectively (entries 2 and 3, Table 2). Lowering the pressure to 0.1 MPa had a negative impact on the Gly conversion, which reached 52% after stirring for 24 h at 40 °C. The authors also examined the conversion of PO at the optimized reaction conditions but noted only 4% of conversion. This Gly/TBAB derived binary catalyst was then successfully applied to other epoxide/CO2 combinations, and significantly better substrate conversion levels were achieved compared to the reactions performed without Gly.

 | ||
| Fig. 2 Examples of naturally occurring terpenoid carbonates. The cyclic carbonate rings are highlighted in red. | ||
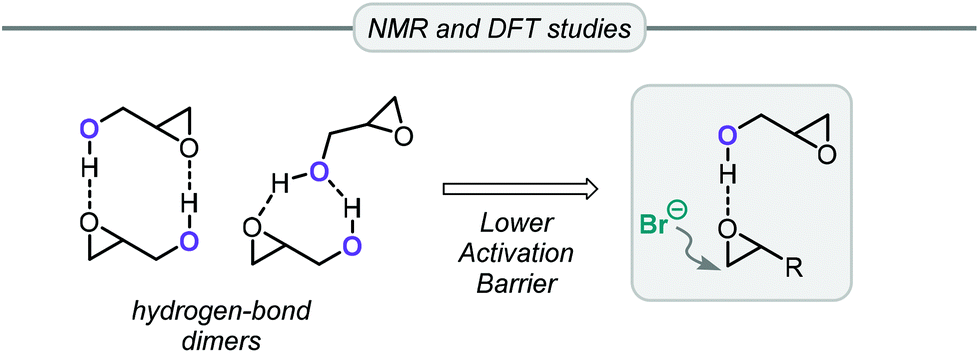 | ||
| Fig. 3 Lower part: Hydrogen bonding in Gly and implications for the overall reaction. | ||
The group of Kleij discovered that the hydroxyl group of Gly can be involved in the activation of CO2 by forming a transient hemi-carbonate species (Scheme 5) that acts as an intramolecular nucleophile able to ring-open the oxirane ring under mild conditions: as such, no external nucleophilic additive is required facilitating thus a halide-free route.17 The use of the aminotriphenolate aluminum catalyst 1tBu (1 mol%) at 75 °C and 1 MPa of CO2 pressure in methylethyl ketone (MEK) as solvent enabled to obtain >99% Gly conversion in 2 h and provided GC in 93% isolated yield. The mechanism of the reaction was studied in detail by ATR-IR, kinetic studies, DFT analysis and X-ray crystallography, which provided proof that a hemi-carbonate intermediate forms under the reaction conditions aiding the intramolecular ring-opening of the oxirane ring of Gly and being facilitated by a hydrogen bond network between the catalyst, Gly and co-catalytic H2O.75
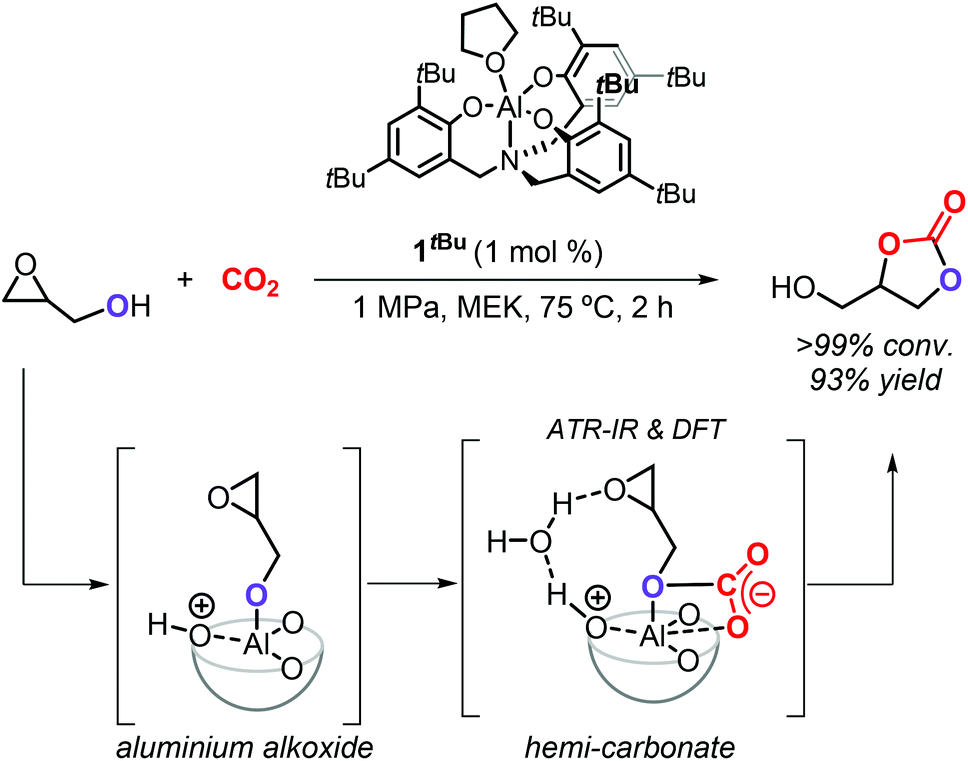 | ||
| Scheme 5 Halide-free carboxylation of glycerol using the aluminum aminotriphenolate catalyst 1tBu. | ||
While more than 140 publications have been reported that discuss the conversion of Gly into GC, not all the catalyst systems proved to be superior to simple TBAB. However, some catalyst systems provide other advantages, such as recyclability, reduced catalyst loading and/or milder reaction conditions among others. For example, the use of heterogeneous catalysts (Scheme 6) allows for easy catalyst recovery after some postsynthetic treatment such as filtration or product extraction. Most of these heterogeneous systems for GC synthesis from Gly are derived from ionic liquids (ILs),76–82 which accelerate the reaction in two ways.
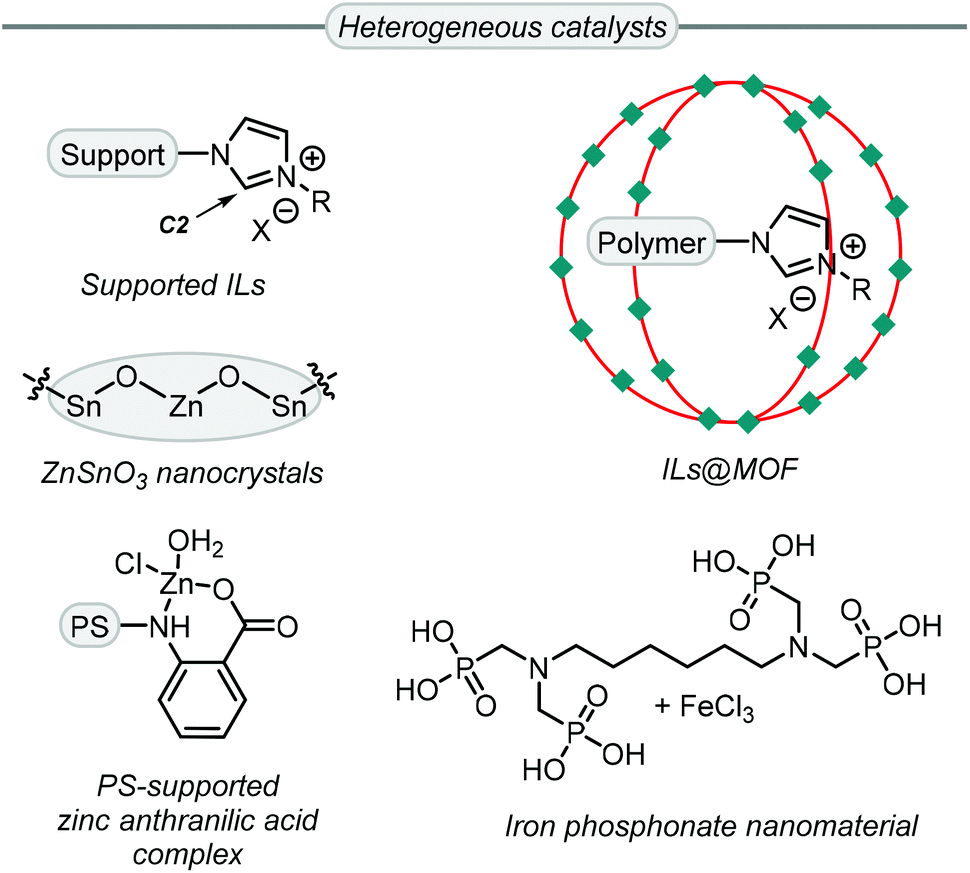 | ||
| Scheme 6 Highly efficient heterogeneous catalysts for the synthesis of GC from Gly. ILs = ionic liquids, MOF = metal–organic framework, PS = polystyrene. | ||
First, a nucleophilic anion helps to ring-open the oxirane ring and, second, an appropriate IL can establish hydrogen bonds between the oxirane ring and the acidic C2 proton (Scheme 6) of the imidazolium ring. The group of Jiang reported an efficient supported ionic liquid catalyst which was confined inside a metal–organic framework (MOF), and this multi-component system displayed synergistic effects between the CO2 capturing capability of the MOF, the Lewis acidic sites of the MOF and the basic sites of the poly-IL.83 Islam et al. reported three examples of active heterogeneous catalysts that are not based on ILs. An iron-phosphonate nanomaterial,84 and a polystyrene supported zinc catalyst,85 showed good activity as Lewis acidic materials with TBAB used as a co-catalyst. Conversely, the catalyst based on ZnSnO3,86 did not require any additive.
Compared to heterogeneous catalysts, homogeneous catalysts derived from earth-abundant metals show superior activity and, thus, reduced catalyst loadings are typically utilized. Moreover, bifunctional catalysts, which have the halide anion incorporated within the catalyst structure, have demonstrated good activity and selectivity towards the formation of GC. For instance, aluminum scorpionate complexes (Scheme 7) reported by Lara-Sánchez and co-workers can achieve an excellent GC yield using 0.25–0.5 mol% of catalyst loading at 70–85 °C.87–89 Similar results were obtained by the group of He, who reported a bifunctional zinc salen-like complex that shows appreciable activity at 0.3 mol% catalyst loading and 0.1 MPa of CO2 pressure, though heating to 100 °C was necessary.90 Another type of bifunctional catalyst was reported by North et al. showing that a bimetallic aluminum salen complex displays high activity in the synthesis of GC from Gly, achieving full conversion in 3 h at only 27 °C and 0.1 MPa.91
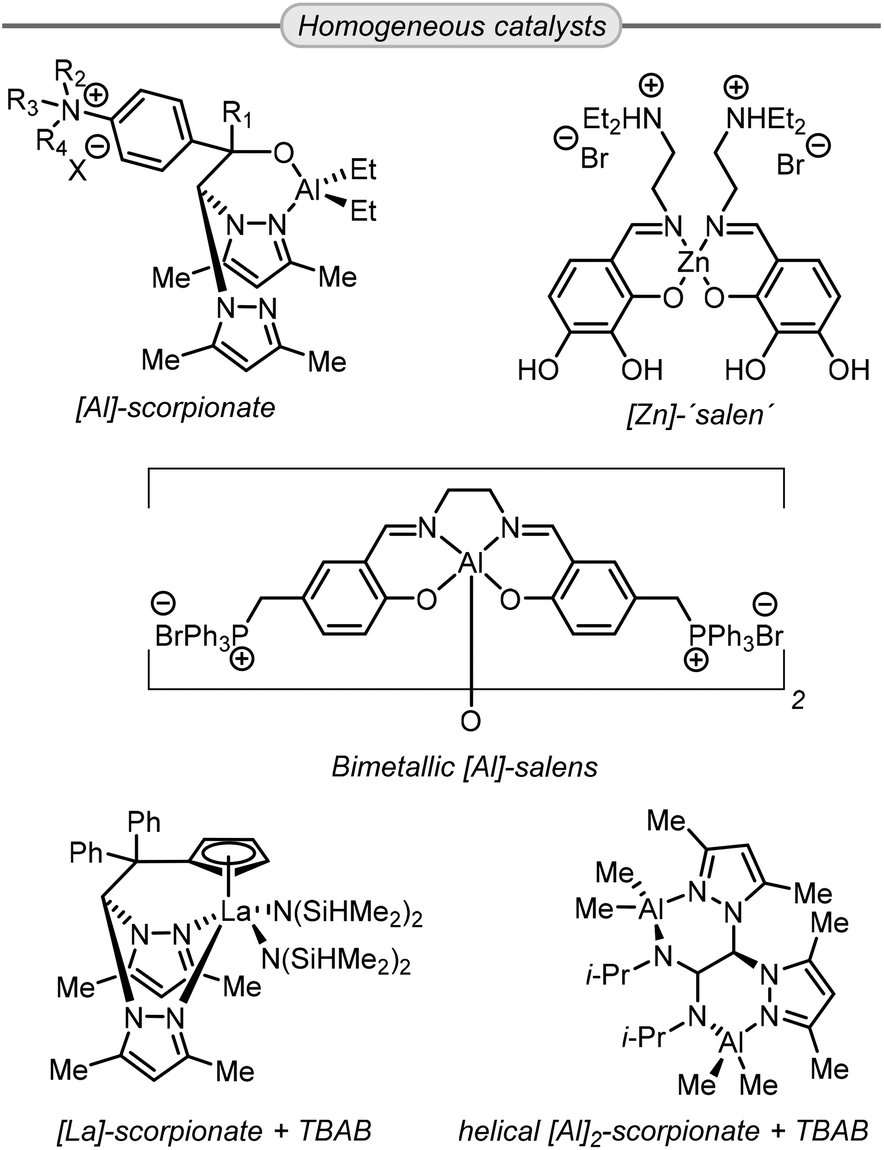 | ||
| Scheme 7 Highly efficient homogeneous catalysts for the synthesis of GC from Gly. | ||
Other homogeneous metal catalysts combined with a co-catalytic amount of halide also exhibit high activity such as the aluminum,92 and lanthanum,93 based scorpionate catalysts reported by Otero, Lara-Sánchez et al. In particular the La-based catalyst shows good activity at remarkably low catalyst loading (0.05 mol%) achieving 98% yield of GC at 70 °C and 1 MPa CO2 pressure in 4 h.
Lastly, some organocatalysts have also been demonstrated as competitive systems for metal-based catalysts. Most of these organocatalysts effectively activate the oxirane ring by establishing hydrogen bonds that facilitate the ring-opening by a halide nucleophile. For instance, Cokoja,94 and Lara-Sánchez,95 reported recyclable organocatalysts that contain imidazolium rings featuring halide counter anions (Scheme 8). Similarly, the group of Kim published a scorpionate type organocatalyst comprising of an aminodiphenol scaffold which acts as a hydrogen-bond activator, and additionally contains a quaternary ammonium salt in its structure.96
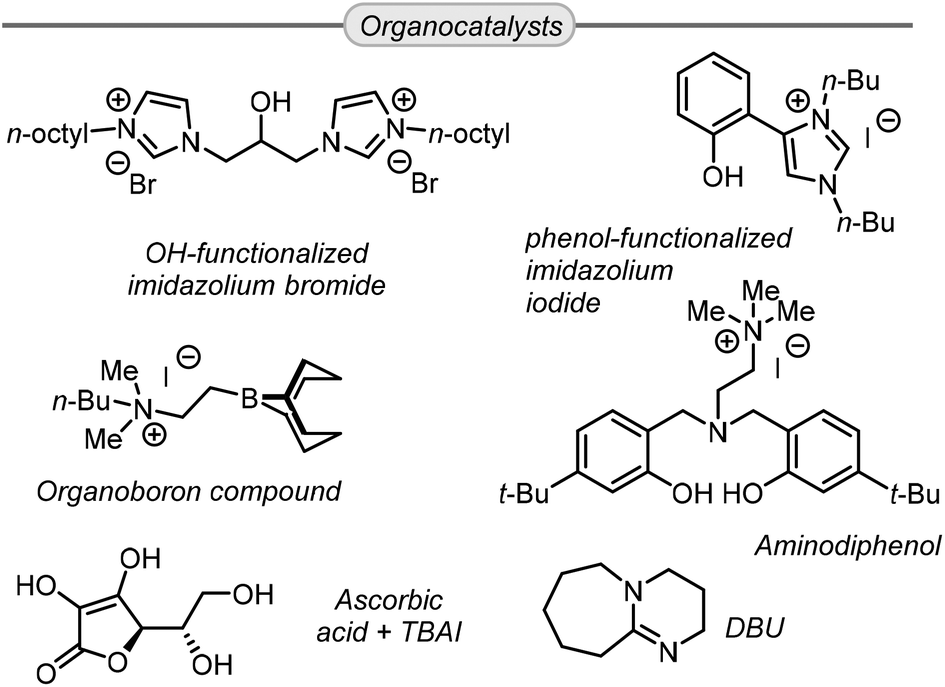 | ||
| Scheme 8 Efficient reported organocatalysts for the conversion of Gly into GC. | ||
D'Elia reported that ascorbic acid is an efficient hydrogen bond donor which, in combination with a co-catalytic amount of TBAI, could achieve high yield of GC at room temperature and 0.1 MPa of CO2.97 On the other hand, the group of Wu reported a bifunctional organoboron organocatalyst able to achieve full conversion and excellent yield of GC at remarkably low (0.02 mol%) catalyst loading though under somewhat harsher reaction conditions (120 °C, 20 MPa CO2).98 In the latter case, mechanistic studies indicated that the boron acts as a Lewis acid and activates the oxirane ring near a closely positioned ammonium iodide unit (Scheme 8). Lastly, Kleij et al. showed that DBU can promote the formation of GC from Gly and other epoxy alcohols through the formation of a hemi-carbonate intermediate under mild reaction conditions (45 °C, 1 MPa CO2).99
3. Terpene based carbonates
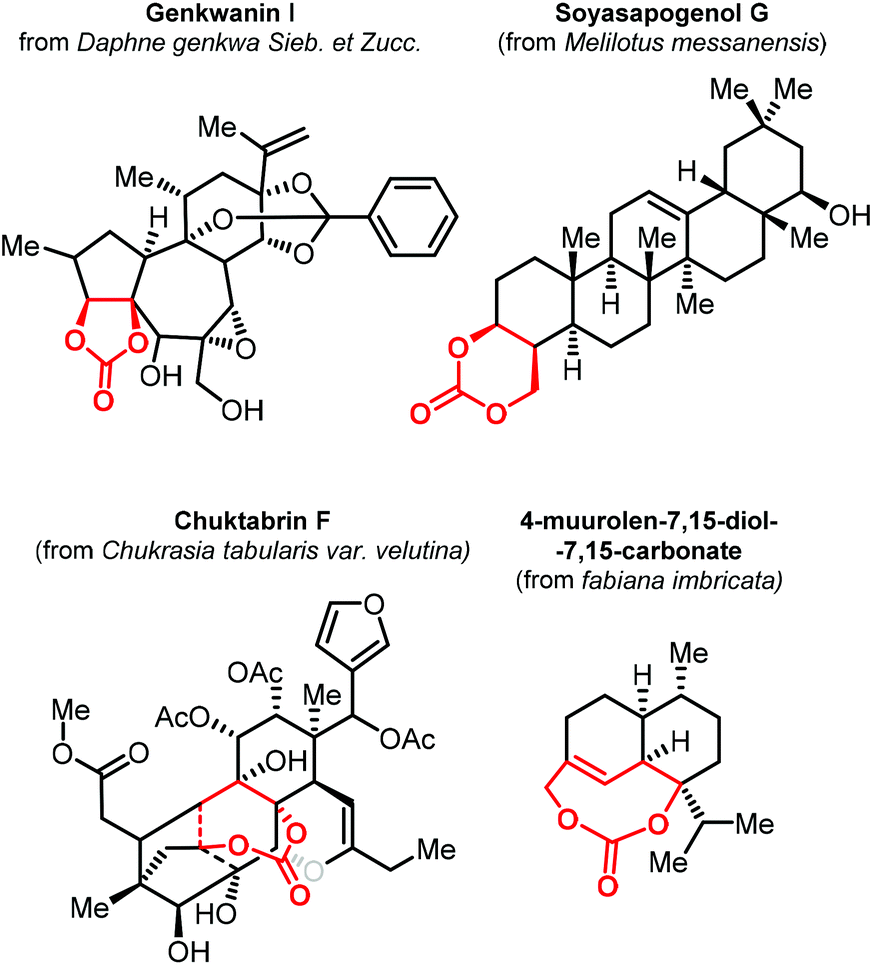
As stated before, the synthesis of CCs from CO2 has become a mature research field. In most contributions, there has been a primary focus on the preparation of relatively simple five- and six-membered CCs with a low degree of substitution/functionality. To explore new applications of CCs, the preparation of structurally more complex products is currently an emerging topic in the area of CO2 utilization. In this regard, terpenes represent attracting starting materials for the preparation of new, more complex and partially biobased CCs.The isolation of 4-muurolen-7,15-diol-7,15-carbonate in 1994 demonstrated that there are naturally occurring terpenoid carbonates (Fig. 2).100 Since then, several other examples of terpene carbonates have been reported,101,102 ranging from compounds having five- to eight-membered CC moieties within their structure with some of them showing biological activity such as Genkwanin I,103 Soyasapogenol G,104 and Chuktabrin F (Fig. 2).105 These findings triggered the effort of several research groups to use terpene scaffolds for the preparation of new types of CCs. The high-structural modularity of terpenes and the almost ubiquitous presence of double bonds that can be easily oxidized and further functionalized, offer an ideal starting point for the preparation of CCs using CO2.106,107
In addition to these attractive structural features, it must be noted that terpenes can be isolated from natural sources, rendering these molecules an attracting alternative to fossil fuel-based raw materials. In particular, the cyclic terpene limonene (both enantiomers) can be conveniently isolated from natural sources such as citrus fruit and fir cone oil (Scheme 9).108,109 It is likely for this reason that limonene has been the most studied terpene in the coupling reaction with CO2, and especially in the synthesis of polycarbonates.110–116
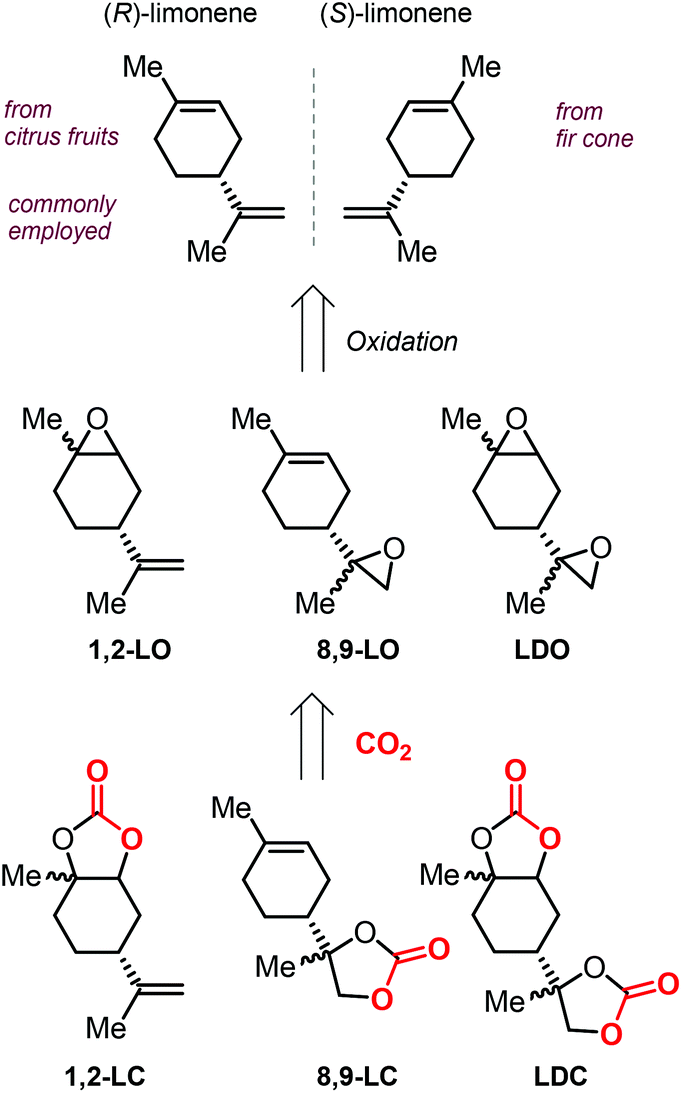 | ||
| Scheme 9 Structures of both limonene enantiomers, limonene-based oxides and limonene-based cyclic carbonates, and their abbreviations. | ||
The extraction of (R)-limonene from orange peel has resulted as economically feasible, and thus the use of this enantiomer is predominant in literature. The structure of limonene shows the presence of two different double bonds, that can be selectively epoxidized to afford either 1,2-limonene oxide (1,2-LO), 8,9-limonene oxide (8,9-LO) and limonene dioxide (LDO, being a mixtures of cis/trans isomers), and their coupling with CO2 leads to the formation of the corresponding 1,2-limonene carbonate (1,2-LC), 8,9-limonene carbonate (8,9-LC) and limonene dicarbonate (LDC) (Scheme 9).
Due to the higher reactivity of the endocyclic double bond and hence the easier synthetic access to 1,2-LO, this particular limonene oxide has been investigated preferentially. A summary of metal-based catalytic systems reported to promote the coupling of 1,2-LO with CO2 is given in Fig. 4.92,93,117–121 With respect to other epoxides commonly used for the preparation of CCs, sterically demanding 1,2-LO shows substantially lower reactivity during its coupling with CO2. This results in the typical use of relatively high temperatures (75–100 °C) and pressures (10–30 bar) and relatively long reaction times (16–66 hours); therefore, achieving full substrate conversion remains a challenge.
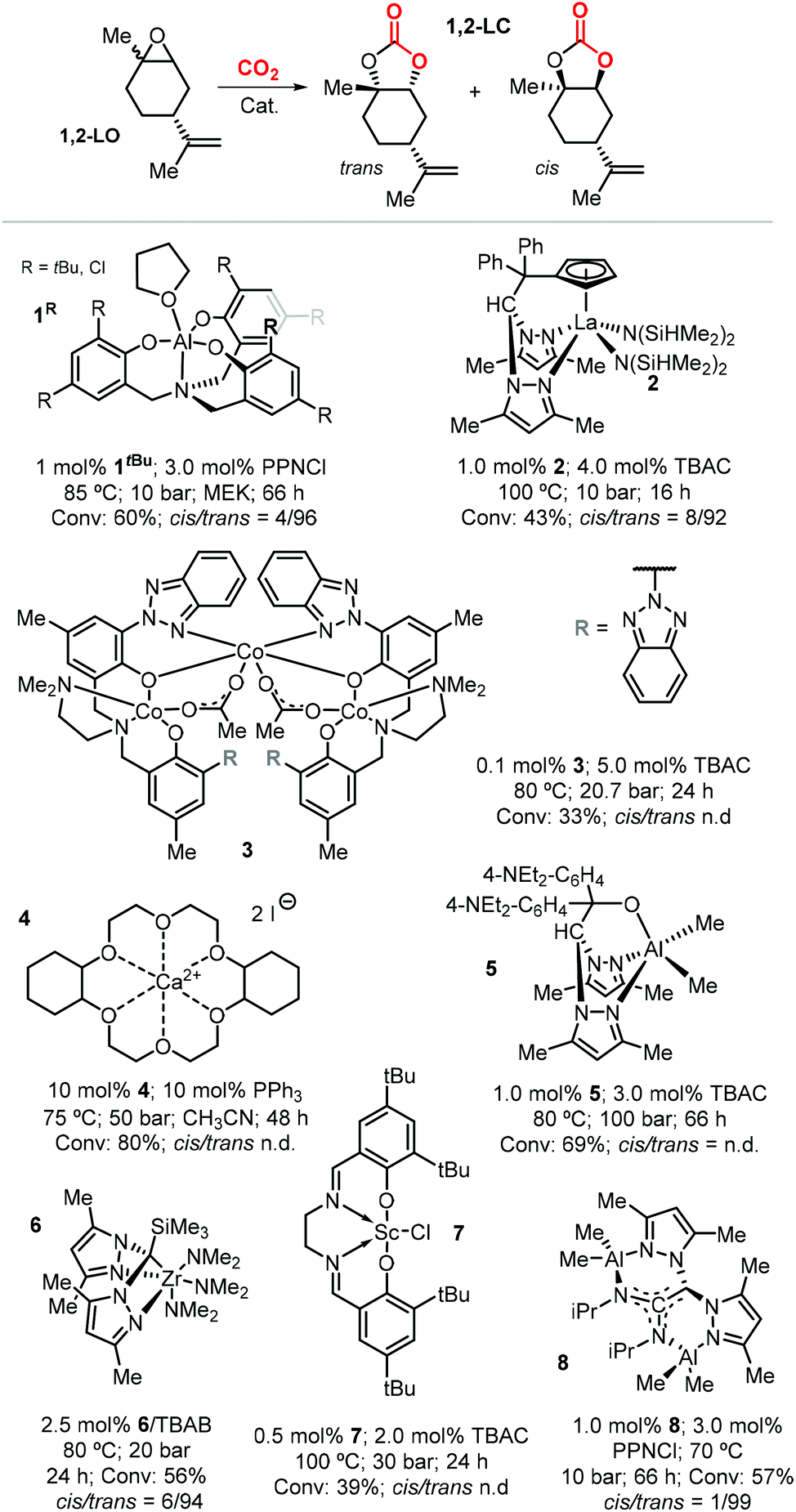 | ||
| Fig. 4 Metal-based catalytic systems reported for the preparation of 1,2-LC, comparative reaction conditions and product stereochemistry. N.d. stands for not determined. | ||
Selective formation of trans-1,2-LC has been observed in several cases such as with catalysts based on 1tBu, 2, 6 and 8 (Fig. 4). This suggests that the cis and trans isomers of 1,2-LO exhibit different reactivity as previously described in the preparation of poly(limonene carbonate).114,115 Indeed, reactions conducted with the binary system 1tBu/PPNCl [bis(triphenyl-phosphine)iminium chloride] using either the pure trans-1,2-LO or cis-1,2-LO resulted in higher conversion (73%) and stereoselectivity (cis/trans > 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :99), and only very low conversion (4%), respectively.116 The formation of trans-1,2-LC was confirmed by X-ray analysis, and more recently the same reactivity difference was observed with the binary catalyst 8/PPNCl.93
:99), and only very low conversion (4%), respectively.116 The formation of trans-1,2-LC was confirmed by X-ray analysis, and more recently the same reactivity difference was observed with the binary catalyst 8/PPNCl.93
In 2016, Fiorani et al. extended the use of the binary systems 1R/PPNCl to the synthesis of other CCs from CO2 and both bicyclic and acyclic terpene epoxides (Scheme 10).116 Under the optimized conditions, the conversion of these bicyclic substrates typically proceeds with high chemoselectivity and moderate isolated yields between 45–52% providing thus access to carvone (c1), limonene dioxide (c3) and menthene (c4) based CCs. Attempts to produce limonene dicarbonate c2 were less successful because of the competitive formation of a polyether (PE) product. The crystal structures of cis-c1, trans-c2 and c3 were elucidated by X-ray analysis for the first time, and confirmed the assigned stereochemistry on the basis of NMR spectroscopic studies.
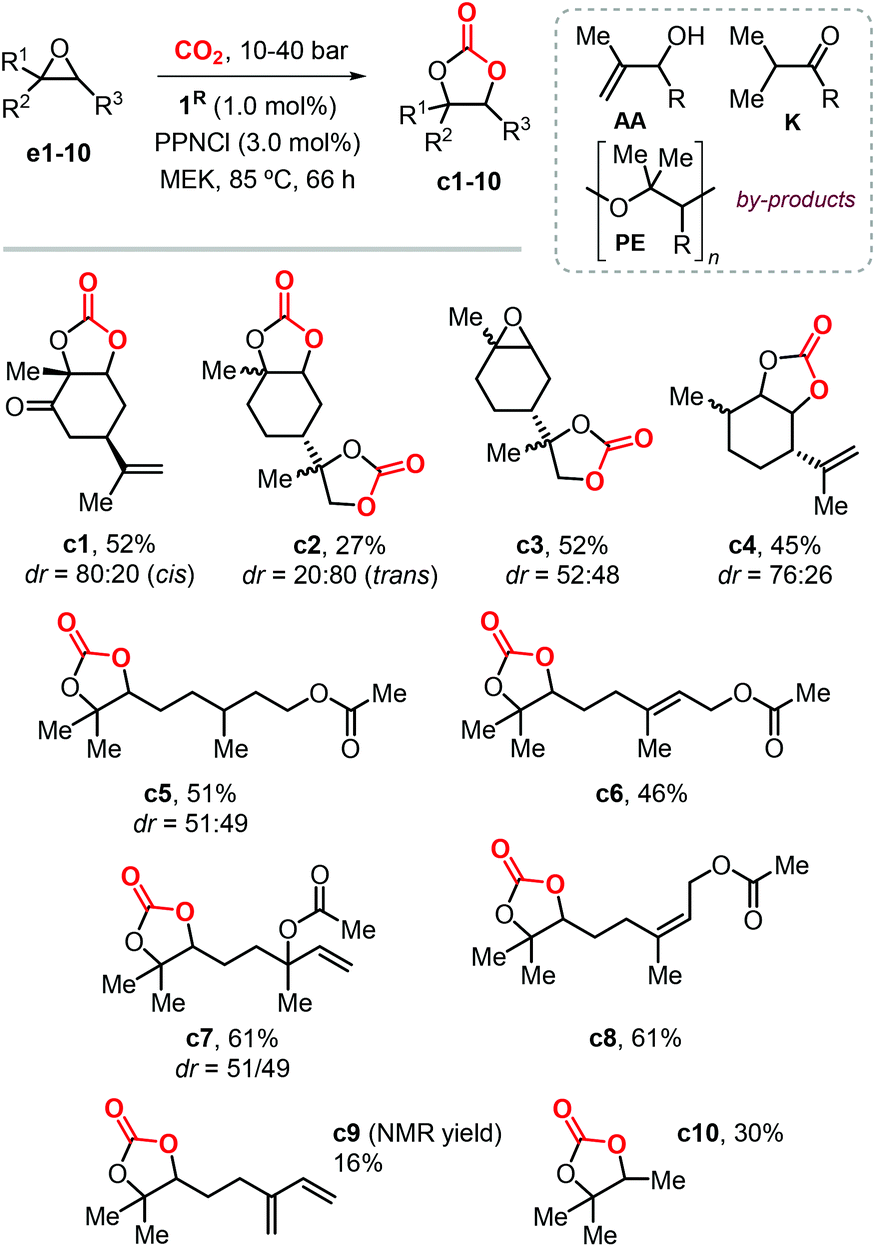 | ||
| Scheme 10 Terpene-based CCs obtained with binary catalysts 1R/PPNCl. | ||
The conversion of acyclic substrates proceeded with lower chemoselectivities, and generally higher pressure of CO2 (4.0 MPa) was necessary to obtain appreciable yields. In the conversion of these acyclic terpene oxide substrates apart from the formation of a PE, the formation of allylic alcohol (AA) and ketone (K) side-products was also detected (Scheme 10). The formation of AA and K by-products was attributed to the phenolate-assisted,122,123 and the Lewis-acid induced Meinwald-type rearrangements of the starting epoxide,124,125 respectively.
The preparation of model compound c10 resulted into a similar product distribution, thus ruling out that by-product formation depends on the nature of the substrate. Despite the issue with the overall chemoselectivity, the synthesis of terpene CCs derived from citronellyl acetate (c5), geranyl acetate (c6), linalyl acetate (c7) and neryl acetate (c8) was possible in moderately high yields. The conversion of myrcene gave a complex reaction mixture, resulting in low yield of CC c9 due to additional side-reactions likely involving the conjugated double-bond.
Following this study, Werner et al. reported the preparation of carbonate c2 (78%) and c4 (81%) in high yields in the presence of 10 mol% of the catalyst 4/PPh3 (Fig. 4) and at 50 bar of CO2 and 75 °C.118 Under the same reaction conditions, the preparation of c5 (19%) and c6 (23%) was less efficient and provided only low yields. In 2019, Lara-Sánchez et al. reported the preparation and characterization of several new terpene-based CCs obtained through the use of binary catalyst 5/TBAC (Fig. 4).119 Carvone-based carbonates c11–14 (Scheme 11) were obtained in good to high yields.
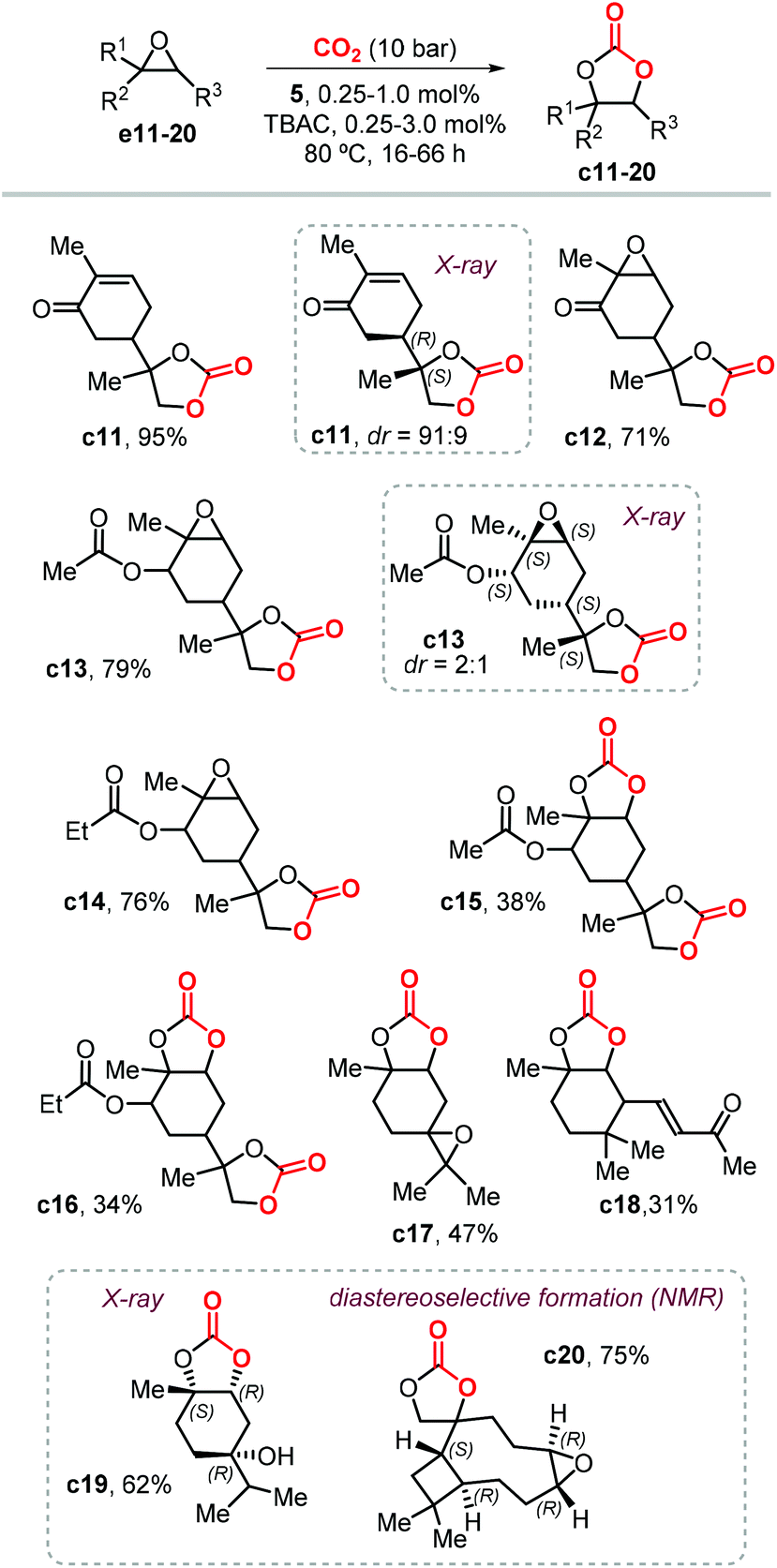 | ||
| Scheme 11 New terpene-based CCs obtained with the binary catalyst 5/TBAC. | ||
Diastereo-enriched samples of c11 and c13 were obtained after crystallization, and their atom connectivities were revealed by X-ray analysis. Carbonates c15 and c16 were only isolated in low yield because of the low stability of the bicyclic carbonates derived from the endo epoxide. These latter products undergo a decarboxylative decomposition toward the formation of the corresponding syn diols. The challenging carbonates c17 and c18, derived from terpinolene and ionone were obtained in moderate yields, whereas the preparation of terpinene-4-ol and caryophellene carbonates c19 and c20 were found to proceed in a diastereo-selective fashion as presented in the bottom part of Scheme 11.
In 2016, Kleij et al. described a new protocol for the synthesis of highly substituted CCs using epoxy alcohols as substrates via a “substrate-directed” mechanism promoted by complex 1R (Fig. 4).17,126 In this context, the authors reported the diastereoselective synthesis of 1,2-geraniol carbonate from 2,3-epoxy geraniol and CO2 (Scheme 12). Notably, this regio-divergent method achieved the conversion of a sterically demanding terpene-based substrate under mild conditions.
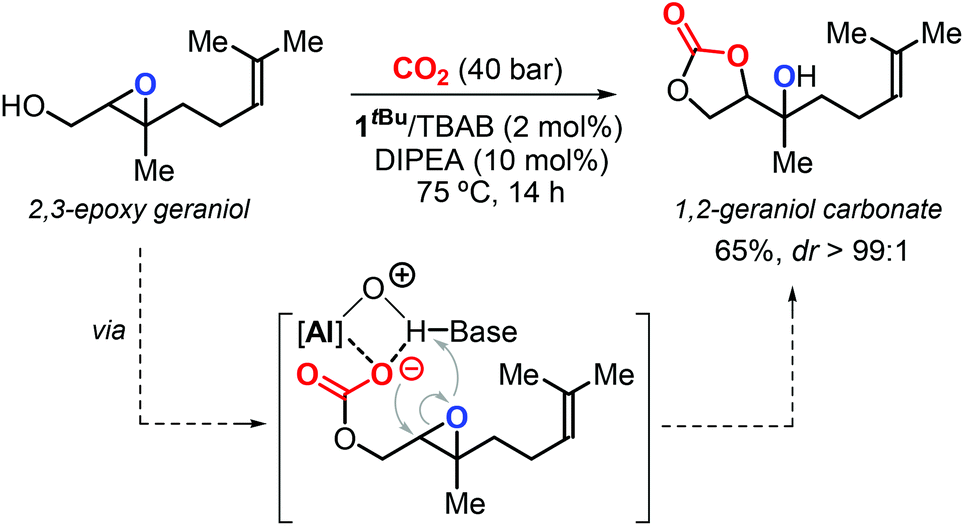 | ||
| Scheme 12 Diastereoselective synthesis of 1,2-geraniol carbonate via a substrate-directed CO2 activation in the presence of 1tBu as catalyst. | ||
Metal-based catalysts may offer several advantages such as the use of milder reaction conditions, shorter reaction times and higher stereocontrol. At the same time, drawbacks such as high(er) cost, multistep synthesis and air/moisture sensitivity create incentives to select more simple catalysts of commercial interest and a practical point of view. In 2018, Morikawa et al. described the reaction of 1,2-LO with CO2 in the presence of TBAC, TBAB or TBAI as catalyst.127 In the presence of 10 mol% of halide salt, the reaction at 100 °C and 30 bar proceeds more efficiently with TBAC. Due to the steric impediment of LO, the smaller radius chloride anion gives the best trade-off in terms of nucleophilicity, leaving group ability and size features. Under these conditions, the reactions using pure trans-1,2-LO and cis-1,2-LO provided higher (76%) and lower (19%) conversions, respectively, compared with the commercial cis/trans mixture (51%). This finding is in line with the different reactivity reported for both LO stereoisomers in the case of metal-based catalysts.93,116 Interestingly, pure cis-1,2-LC was isolated for the first time and characterized by NMR spectroscopy. In addition, the relative configuration of the chiral centers in cis-1,2-LC was confirmed by X-ray analysis of the corresponding diol obtained by reduction with LiAlH4 (Scheme 13).
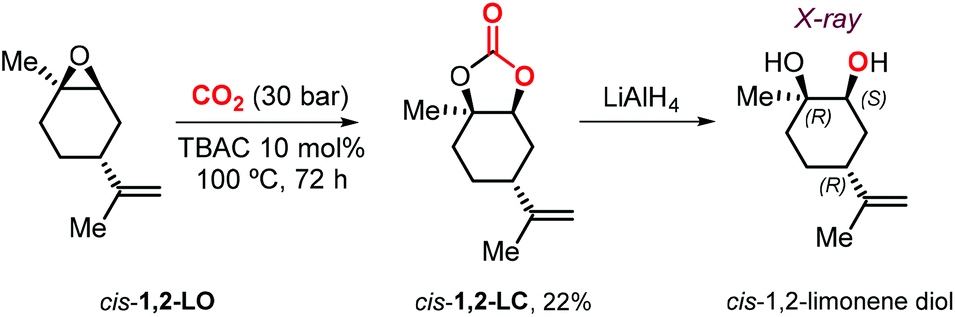 | ||
| Scheme 13 Synthesis of cis-1,2-LO and reduction towards the corresponding syn diol. | ||
For comparative reasons only, the same research group reported, for the first time the synthesis of two 1,2-LC diastereoisomers in which the oxygen atoms of the carbonate ring are in a trans configuration by treatment of 1,2-diols stereoisomers with triphosgene rather than using a more preferred CO2/epoxide coupling strategy (Scheme 14).128
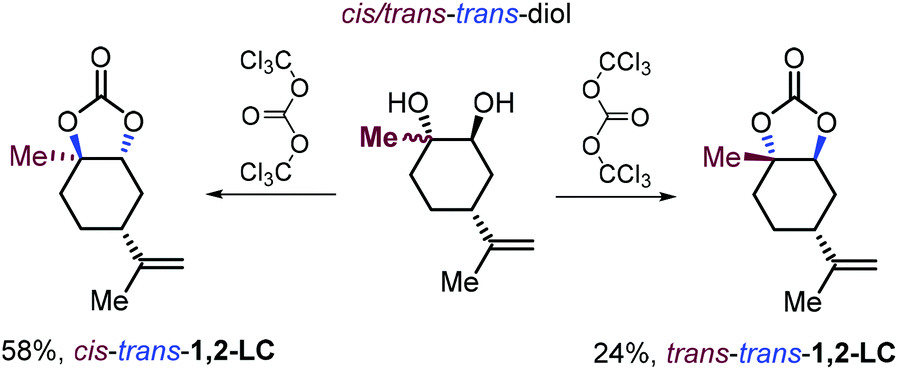 | ||
| Scheme 14 Synthesis of 1,2-LC diastereoisomers having a trans configured carbonate ring. | ||
Recently, Rehman et al. reported a detailed kinetic study of the 1,2-LO/CO2 coupling reaction in the presence of TBAC.129 This investigation confirmed the higher reactivity of the trans isomer in the formation of 1,2-LC. It was found that the reaction kinetics show a first-order dependence with respect to all the reaction components (1,2-LO, CO2 and TBAC). In addition, the thermodynamic parameters of this conversion were determined using the Eyring equation, providing activation enthalpy and entropy values of 60.6 kJ mol−1 and −103.6 J (mol K)−1, respectively.
In the last decade, the use of CCs have emerged as an attracting, more sustainable alternative for the production of the so-called non-isocyanate based polyurethanes (NIPUs), and the preparation of CCs from renewable feedstock is of importance to develop more sustainable materials.22,130,131 In this respect, the research group of Mülhaupt investigated the use of limonene dicarbonate (LDC) for the synthesis of new types of NIPUs.132,133 They investigated the synthesis of LDC from LDO and CO2 catalyzed by TBAB on a kilogram scale. First, the reaction parameters (CO2 pressure, temperature and catalyst loading) where optimized, after which full LDO conversion could be achieved using 3 mol% of TBAB in less than 50 h at 140 °C and 30 bar of CO2. A brownish oil was is obtained this way, and it was initially directly used (without purification) for the preparation of NIPUs preparation.132 Afterwards, a more detailed analysis of the reaction products based on NMR and mass spectrometry was carried out revealing the formation of several by-products (B1–4, Scheme 15).133 The authors proposed that the formation of these products occurs by bromide elimination after initial bromide-assisted epoxide ring-opening being essentially the first step of the catalytic cycle leading to LDC. Therefore, in order to obtain a pure compound, LDC was crystallized obtaining a mixture of cis and trans isomers in a 2:3 ratio. Further crystallization led to the isolation of pure trans-LDC as confirmed by X-ray analysis.
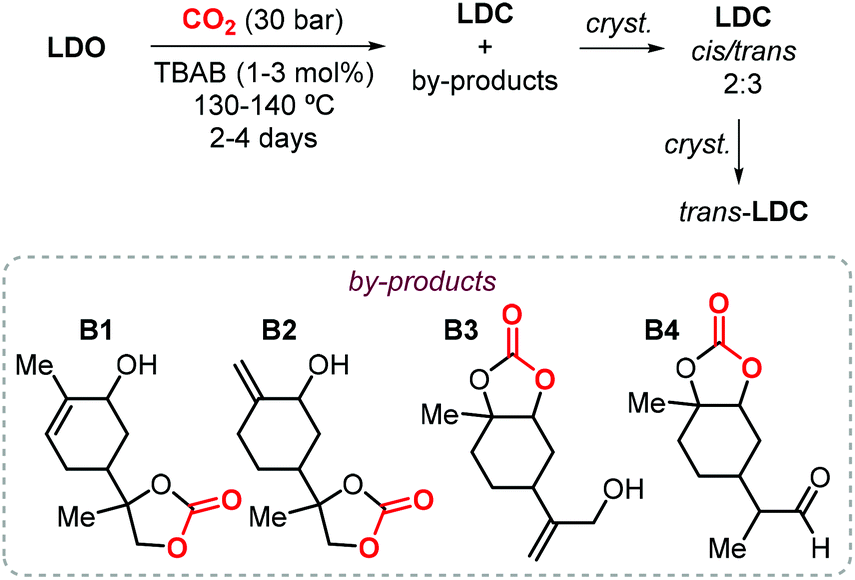 | ||
| Scheme 15 Synthesis of LDC catalyzed by TBAB followed by crystallization of trans-LDC, and structures of reaction by-products B1–B4. | ||
The use of limonene-based CCs in the preparations of NIPUs was also investigated by Hintermair et al.134 The authors reported the preparation of 8,9-LC by reaction of 8,9-LO and CO2 mediated by TBAB. The key step for the synthesis of the desired carbonate was the selective epoxidation of (R)-limonene at the less hindered terminal alkene. This reaction was performed using perchloric acid as the oxidant in the presence of a bulky polyoxometalate (POM) catalyst previously reported by Mizuno and coworkers (Scheme 16).135 Because of a lower degree of steric hindrance, the formation of 8,9-LC occurs comparatively much faster than the formation of 1,2-LC under the same reaction conditions reaching 80% conversion in 2.5 hours. Remarkably, the two different 8,9-LO diastereoisomers generated during the epoxidation reaction show similar reactivity in their coupling reaction with CO2, which is in contrast to the behaviour typically observed for cis and trans1,2-LO.
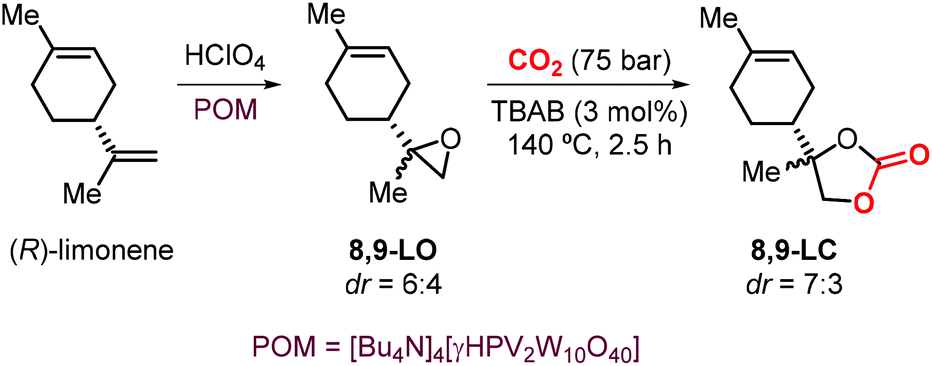 | ||
| Scheme 16 Selective epoxidation of (R)-limonene and synthesis of 8,9-LC. | ||
4. Carbohydrate derived carbonates
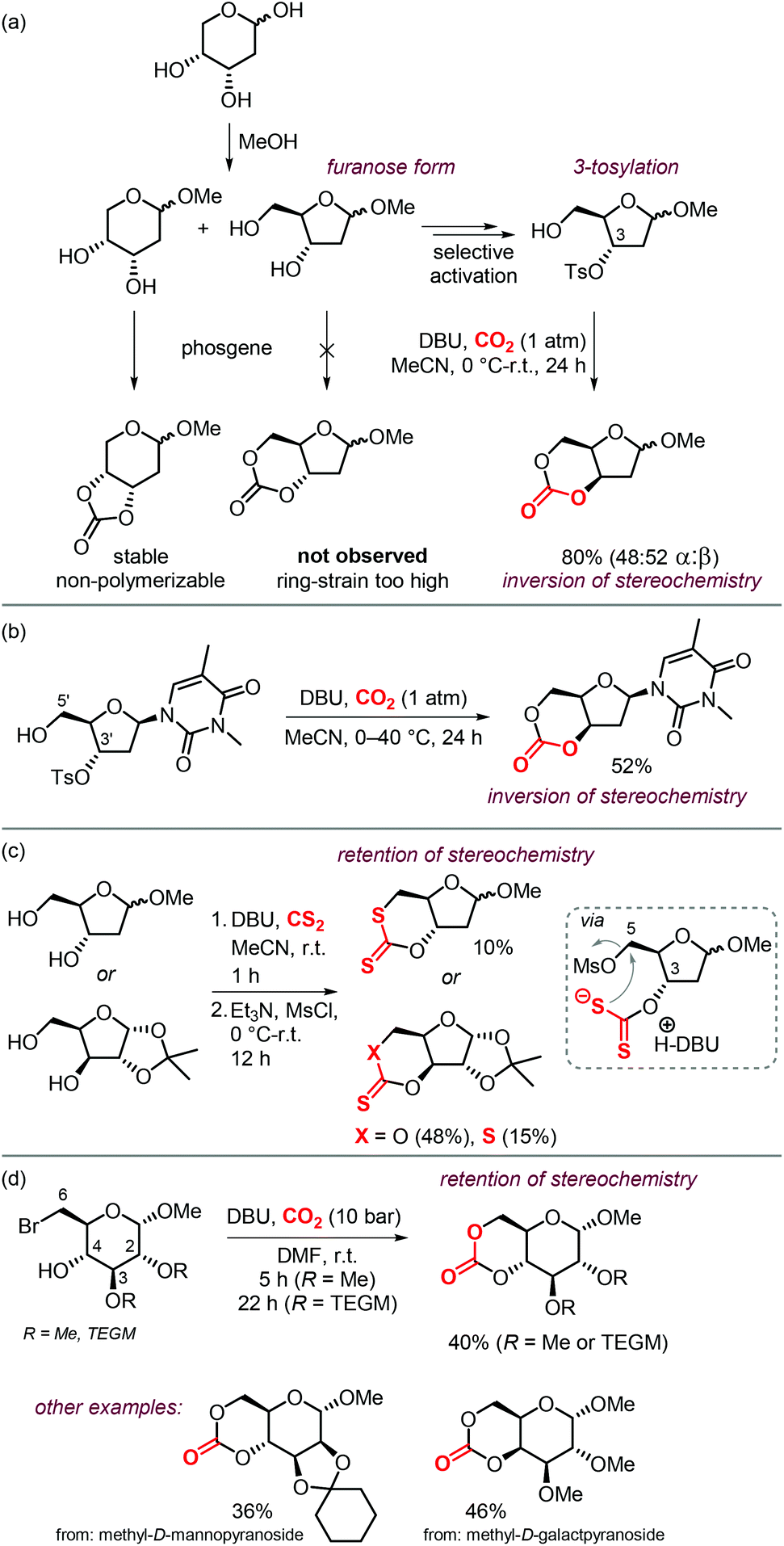
Sugars are another interesting and ubiquitous source for CCs. Different from terpenes or fatty acids, these compounds bear multiple alcoholic functionalities, and therefore the preparation of cyclic carbonates on sugars is generally performed using phosgene-derived reagents.136–139 Well-defined, non-toxic and biodegradable poly-glycocarbonates with a narrow distribution of molecular weights can be prepared from sugar-based monomers bearing a six-membered carbonate,136,137,140–142 or a trans-positioned five-membered carbonate,138,143 showing the huge potential of functional polymers from such substrates. In addition, the sugar molecules can first be converted into simpler compounds, which can then be converted into polymerizable monomers.144–147Recently, a protocol to replace phosgene-derivatives was developed (Scheme 17).148 In this process, CO2 and an organic base (DBU) form an ionic hemi-carbonate intermediate, after which tosyl-chloride is added to yield a cyclic carbonate. 1H NMR spectroscopy was used to monitor the formation of the ionic intermediate of 1,3-butanediol, showing that under optimized conditions, 49% of the in situ product is carbonated on the primary alcohol, 24% on the secondary alcohol, and 5% is bis-carbonated. Subsequent tosylation rapidly led to cyclic carbonate formation, without the observation of any intermediates. To discriminate between two mechanistic possibilities where either the hemi-carbonate or the remaining free alcohol is tosylated, enantiopure (R)-1,3-butanediol or (R,R)-2,4-pentanediol were employed. Interestingly, the stereochemistry was preserved during the reaction, indicating that the hemi-carbonate group is likely tosylated instead of the remaining free alcohol (Scheme 17a, top pathway).148 The proposed mechanism was further supported by DFT calculations. The reaction proved to be capable of providing the 6-membered cyclic carbonate derivative of D-xylose (Scheme 17b), albeit in low yield, showing the potential of the overall transformation.
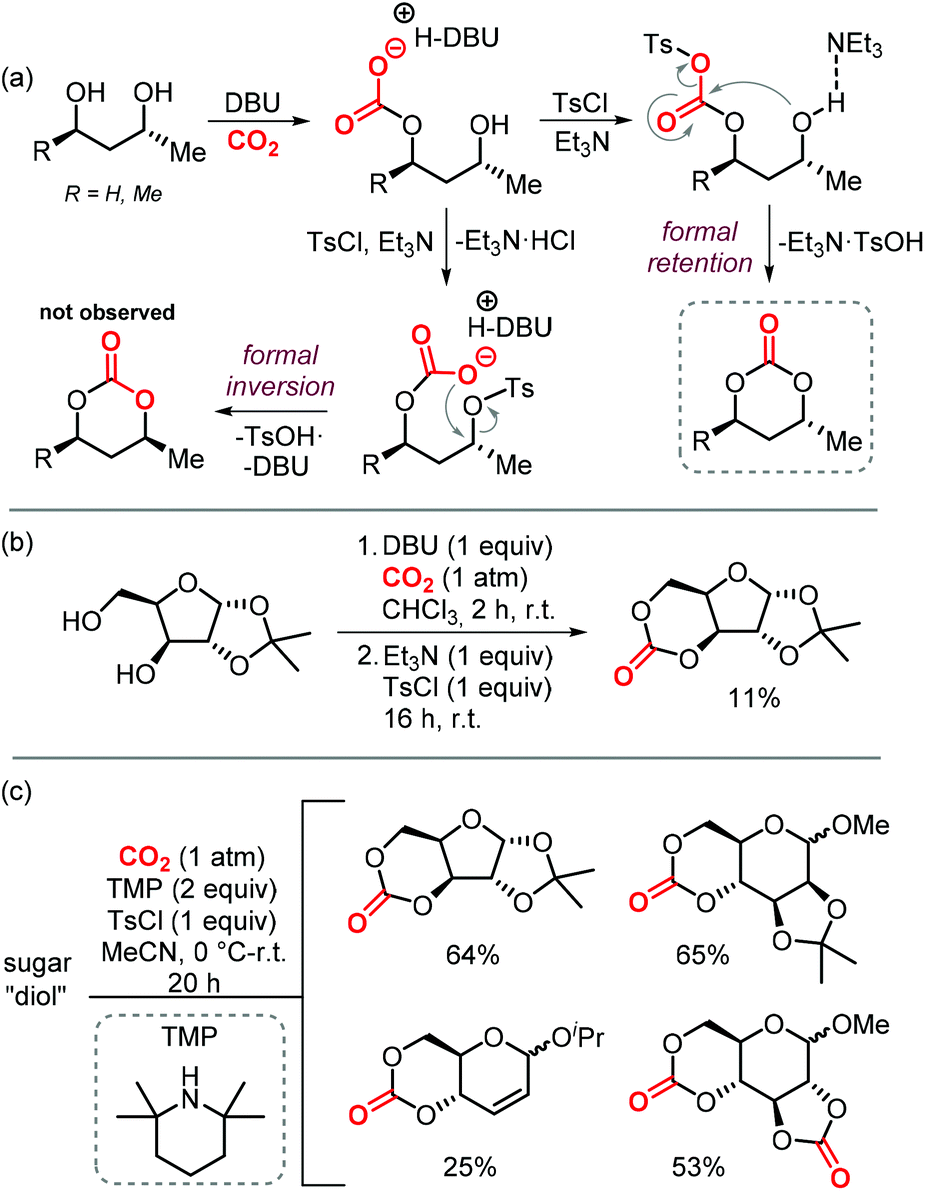 | ||
| Scheme 17 (a) and (b): Conversion of 1,3-diols to 6-membered cyclic carbonates by a stepwise, 1-pot procedure. (c) 1-step procedure to form cyclic carbonates from sugars. | ||
Later on, the process was improved significantly as to allow the carbonation to occur in one step, and higher yields of the cyclic carbonate versus byproducts such as oligomeric species or tosylation of the alcohols were reported.149 The use of a weaker base such as Et3N or 2,2,6,6-tetramethylpiperidine (TMP) proved to be essential for the overall chemoselectivity. Even though the initial formation of the hemi-carbonate is strongly disfavored in the presence of weaker bases (2% or 4% for TMP or Et3N, respectively vs. 85% for DBU), the selective tosylation of the hemi-carbonate over the alcohol and the ring-closure step are strongly favored energetically as calculated by DFT. A comparison between the mechanism using Et3N,149vs. DBU,148 shows that the barriers for formation of the tosylated carbonate are 18.4 and 23.8 kcal mol−1, and the ring-closure step is subject to a barrier of 15.4 and 18.9 kcal mol−1, respectively. Using this new strategy, the authors synthesized a series of 5- to 8-membered cyclic carbonates, amongst which four sugar-based CCs (Scheme 17c) in good yields comparable to or exceeding the yields obtained using phosgene-reagent based syntheses.
The possibility to convert diols to carbonates by this method rapidly led to further development of CO2- and sugar-based CCs. A first report describes the synthesis and ring-opening polymerization (ROP) of a cyclic carbonate-functionalized mannose derivative.150 1-O-Methyl-α-D-mannose was protected at position 2 and 3 by an isopropylidene acetal. Subsequently, a six-membered cyclic carbonate was formed by subjecting the compound to DBU and CO2 followed by tosylation with TsCl and Et3N (Scheme 18). The stereochemistry was retained, which suggests that the mechanism discussed above is operative. The yield of the product (57%) was higher than in similar syntheses for D-glucose (36%) and D-xylose (41%) derived CCs mediated by phosgene-reagents. An X-ray molecular structure was obtained, confirming the trans-positioning of the carbonate on the mannose ring. These types of bicyclic carbonates are easily polymerized through ROP. Indeed, the authors showed that a controlled polymerization of the obtained cyclic carbonate is feasible under organocatalytic conditions using TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene) and 4-methylbenzyl alcohol as initiator.
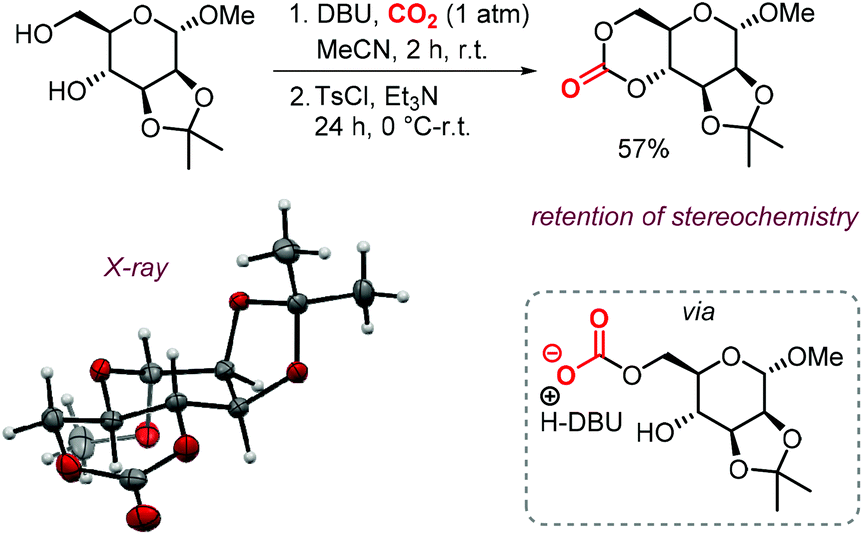 | ||
| Scheme 18 Synthesis of a 6-membered carbonate based on D-mannose. X-ray ellipsoids at 50% probability. | ||
In addition to pyranose-sugars, there has been interest in the use of furanoses due to their stiffness in the backbone for potential polymers based on it. An interesting candidate is 2-deoxy-D-ribose. Naturally, this sugar exists in its pyranose form, which exposes a cis-diol, which can be converted into a CC through phosgene-based pathways. cis-5-Membered CCs, however, do not readily undergo ROP.151 Therefore, this sugar was treated with MeOH in acidic conditions to transform it to its furanose-form (Scheme 19a). The furanose-form has a trans 1,3-diol that cannot be carbonated using phosgene-derived methods,152 likely due to the high ring strain. Therefore, an alternative route was envisioned, based on selective preactivation of the secondary alcohol at the 3-position using tosyl chloride.153
 | ||
| Scheme 19 (a) Synthesis of sugar-derived CCs through a DBU mediated insertion of CO2 through SN2 displacement of pre-activated alcohols or halides. (b) synthesis of a thymidine-based cyclic carbonate. (c) preparation of thiobased CCs. (d) synthesis of a D-glucose-based cyclic carbonate (TEGM = –(CH2CH2O)3CH3, a D-mannose-based cyclic carbonate and a D-galactose-based cyclic carbonate. | ||
Using the activated sugar, the protocol based on DBU and CO2 led to a 6-membered CC in one step as a mixture of anomers that could be separated by column chromatography. By reversing the order of tosylation and hemi-carbonate formation, the stereochemistry of the reaction can therefore be controlled. However, it is necessary to first isolate the tosylate, and the regioselectivity had to be forced by protection of the 5-alcohol. The reason for the successful synthesis lies in the fact that the stereochemistry is formally inverted in this process leading to a cis-configured carbonate of 2-deoxy-D-xylose that is less strained than its trans-analogue. ROP was attempted for both anomers, and it was shown that the β-anomer could not be polymerized. In contrast, the α-anomer was converted into a high-molecular weight polymer using trimethylene carbonate (TMC) as a comonomer in the presence of TBD as catalyst and benzyl alcohol as initiator.153
The same research group set out to investigate the possibility of generating CCs of thymidine, one of the bases of DNA containing the same 2-deoxy-D-ribose sugar backbone.154 Both phosgene-reagent and DBU-CO2 mediated syntheses were unsuccessful likely due to high ring strain of trans-fused CC units in furanose-sugars. In order to relieve the ring-strain but retain polymerization potential, the secondary alcohol at position 3′ of the thymidine was tosylated. Subsequent hemi-carbonate formation at alcohol 5′ and ring-closure led to a 6-membered CC with the stereochemistry at the 3′-position inverted (Scheme 19b). In addition, the free NH-group of thymidine required methylation as not to inhibit carbonate formation. As in other comparable cases, the cyclic carbonate monomer could be polymerized with a high control over the molecular weight of the resultant polycarbonate.
Later on, the ribose substrate was investigated for the formation of thiocarbonates or xanthates using CS2 as a replacement for CO2 (Scheme 19c).155 The authors hypothesized that the larger C–S bond distance could better accommodate a trans-fused ring on the sugar. Reacting 1-O-methyl-2-deoxy-D-ribose with CS2 and DBU, followed by MsCl (mesyl chloride) and Et3N led to a single product in low yield (10%). The product was proven to contain a xanthate ring fused trans to the ribofuranose ring. When using a substrate where the diol is positioned cis (such as in 1,2-protected xylose) the xanthate product was obtained in similar yield (15%) but in addition the expected thiocarbonate product was formed in moderate yield (48%). The authors explain these results by proposing that the alcohol at the 5-position is mesylated, and the hemi-xanthate intermediate is formed at position 3, formed by ring-closure. Indeed, when the mesylated compound is prepared prior to reacting with DBU and CS2, the same product is formed in higher yield.
The group of Gnanou published a similar strategy to obtaining carbonates of glucose by inclusion of CO2.156 By a protection-group strategy, they produced a fully protected glucopyranose with a benzylidene-acetal on alcohols 4 and 6, methylated anomeric alcohol and either a methyl or a methyl triethyleneglycol group on alcohols 2 and 3. Bromination of the acetal-group and hydrolysis of the resulting benzoyl-group on position 4 led to a halo-alcohol derivative. Under slightly elevated pressure of CO2 (10 bar) and in the presence of DBU, this compound was carbonated to give the glucose-based cyclic carbonate with retention of stereochemistry (Scheme 19d). Although inversion of stereochemistry would be expected on position 6, this carbon center is achiral. The authors were able to generate hydrophilic or hydrophobic polymers by ROP of the cyclic-carbonate monomers, with R = Me, or TEGM, respectively or amphiphilic polymers by using both monomers. The new synthetic method proves a greener alternative to the method described by Wooley et al., using phosgene derivatives to generate the carbonate.136–138 Two more derivatives were later synthesized through the same method, i.e., selective protection and bromination of the 6-position followed by reaction with DBU and CO2 in DMF.157 By this method, the authors could synthesize cyclic carbonates from a D-mannose and a D-galactose, and polymerize them through ROP.
In the same article, the authors report a procedure to generate 5-membered cyclic carbonates regioselectively from galactose and mannose that were only methylated at the anomeric position (Scheme 20a).157 The simple one-step reaction involves CH2Br2 to generate a productive leaving group in the hemi-carbonate fragment that allows for subsequent ring-closure to yield the cyclic carbonate (Scheme 20b).158 The procedure is selective for the formation of cis-5-membered carbonates, no trans-cyclic carbonates or 6-membered (even if they are cis in the case of galactose) were observed. Although not polymerizable, the authors show that the carbonates can react with amines to afford linear carbamates, which holds promise for their use in the production of isocyanate-free hydroxypolyurethanes.157
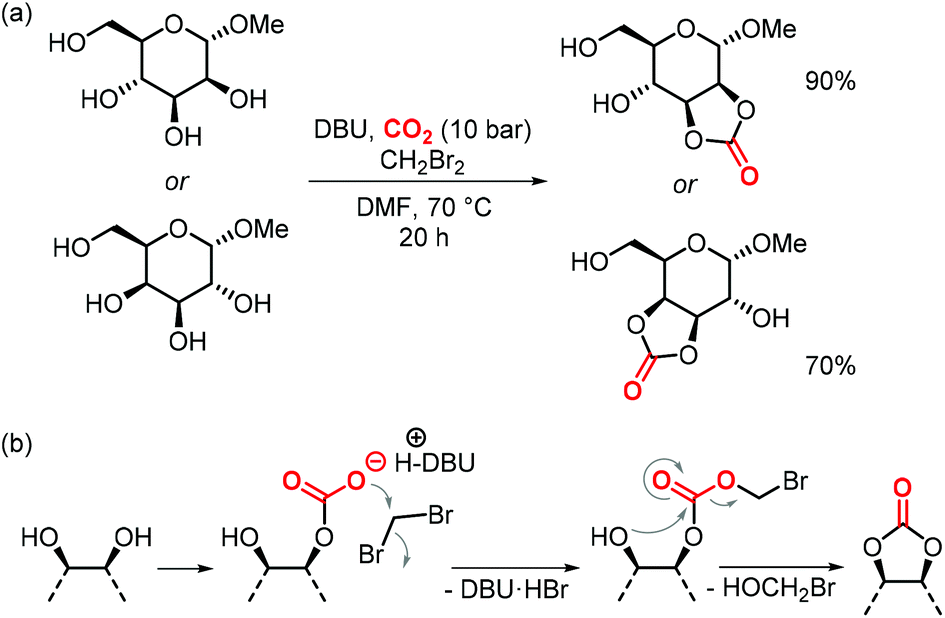 | ||
| Scheme 20 (a) one-step synthesis to form cis-5-membered CCs from a D-mannose and a D-galactose.(b) Proposed mechanism of the reaction. | ||
5. Fatty acid based carbonates
As can be judged from the preceding sections, the partial replacement of fossil fuels-based chemicals with compounds sourced from bio-based, renewable sources is an attractive and rewarding target in chemical research in the pursuit of increased sustainability.159–162 It is clear that the realization of such a target requires the use of feedstock that are available in large volumes such as biogenic and food waste materials,163–165 or those derivable from mass production crops.166,167In this context, vegetable oils (VOs, Fig. 5), with a global production of over 200 Mt per year,168,169 represent a valuable feedstock for the production of several chemicals.170 The transesterification of VOs leads to fatty acid methyl esters (FAMEs, Fig. 5) that find wide application as commodity chemicals,171 biodiesel fuel,172 and as intermediates for further chemical diversification.173–177 In particular, epoxidized fatty acid esters (EFAs, Fig. 5) can be easily obtained and display potential for applications such as lubricants, additives and plasticizers.178 Similarly, triglycerides in vegetables oils can be epoxidized to afford epoxidized vegetable oils (EVOs, Fig. 5), that find application as green materials for the preparation of PVC [poly(vinyl chloride)], plasticizers,179 elastomers,180 coatings,181 epoxy resins and blends.182 Finally, both EFAs and EVOs can be carbonated via catalytic cycloaddition chemistry using CO2,1,2,4,181,183 leading to carbonated fatty acids and carbonated vegetable oils (CFAs and CVOs, Fig. 5). These latter compounds can serve as plasticizers for PVC,24 and as building blocks for the synthesis of NIPUs, respectively.185,186 Importantly, the latter processes have received increasing attention in recent years,118,187,188 as they enable the integration between highly sought-after recycling of CO2 into chemicals,189–191 and the use of renewable substrates as building blocks for commodity chemicals with a low(er) carbon footprint.24,192,193
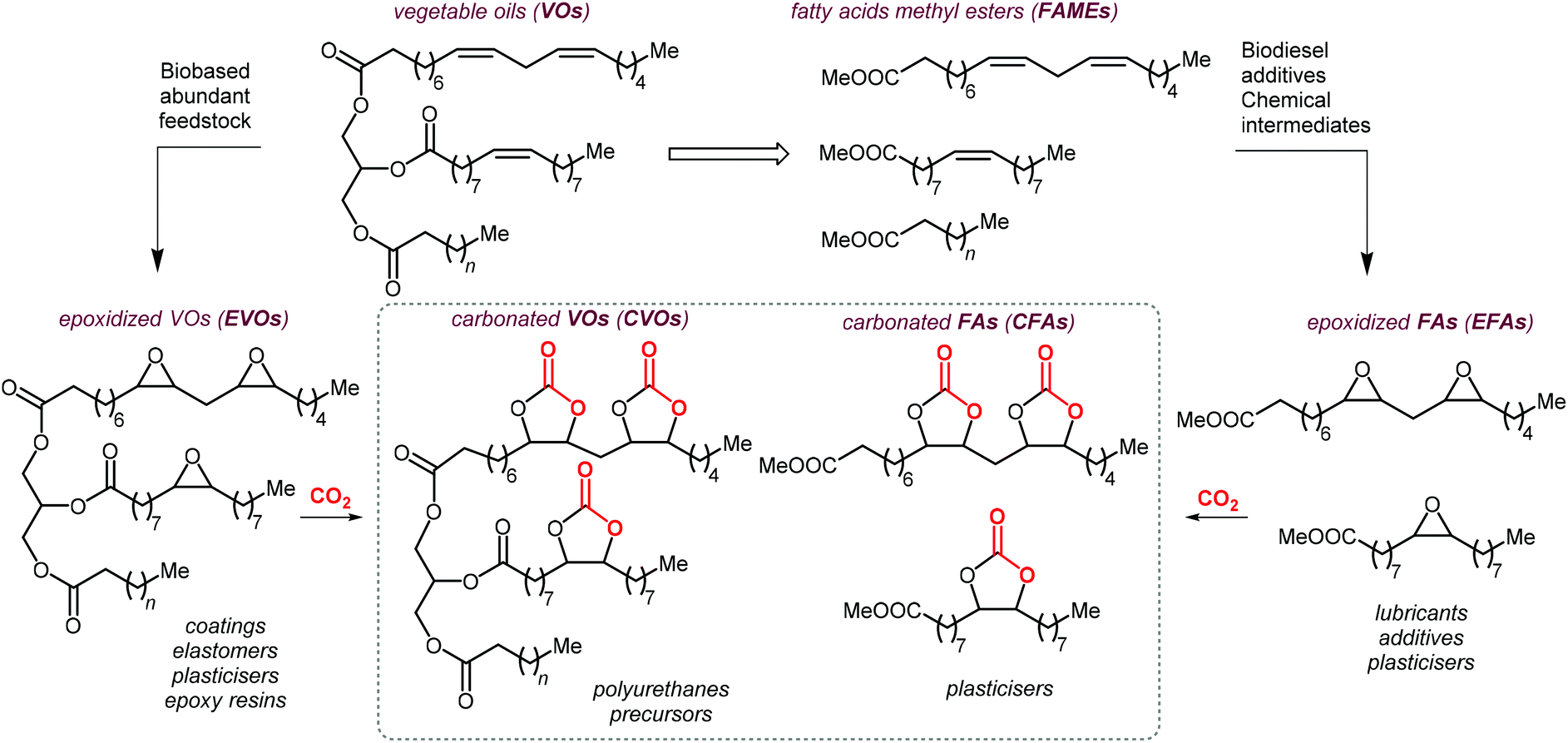 | ||
| Fig. 5 Products from transesterification and functionalization of vegetables oils depicted as triglycerides of saturated, mono- and poly-unsaturated fatty acids. | ||
In this section, we review the catalytic processes that have been developed (mostly in the last decade) for the cycloaddition of CO2 to EFAs and EVOs providing oleochemical carbonates. For the sake of clarity, terminal cyclic carbonates prepared from epoxidized fatty acid derivatives are initially discussed whereas the carbonation of internal epoxides present in fatty acids and vegetable oils is discussed separately.
5.1 Terminal carbonates from EFA derivatives
A variety of terminal carbonates can be generated from methyl 10-undecenoate, a terminal fatty acid (tEFA) that in turn can be obtained from the pyrolysis of methyl ricinoleate, which is a main fatty acid component of renewable castor oil.194 In Scheme 21, several possible synthetic routes are illustrated leading to (terminal) mono- and bis-carbonates. Terminal carbonate tc1 can be obtained from the carbonation of epoxidized methyl 10-undecenoate (route a, Scheme 21). In recent years, the synthesis of tc1 was studied by Werner et al. by employing various organocatalysts derived from phosphonium salts, or calcium-based catalysts (9, Scheme 22).118,195–197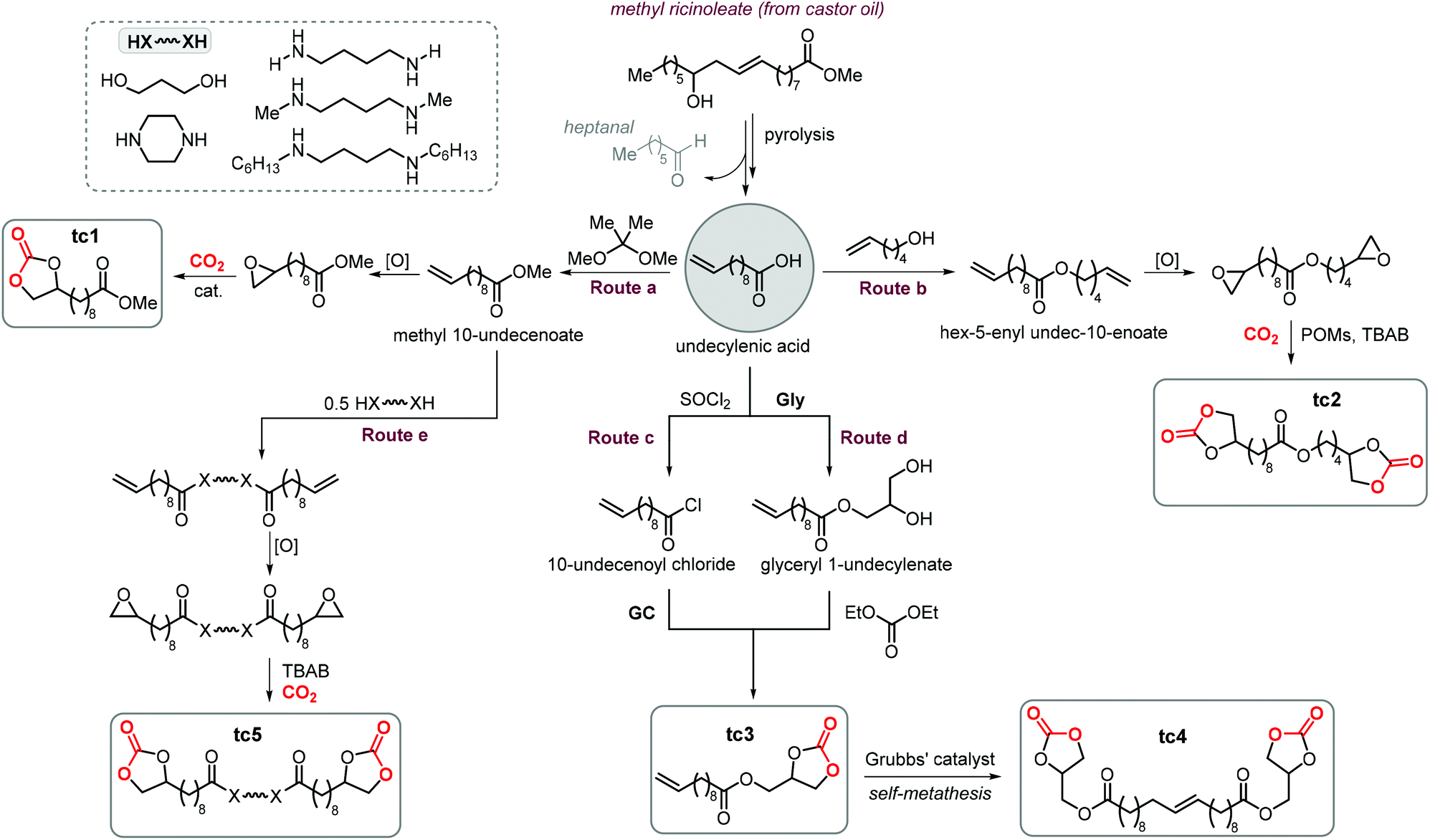 | ||
| Scheme 21 Five-membered CC diversity derived from castor oil. | ||
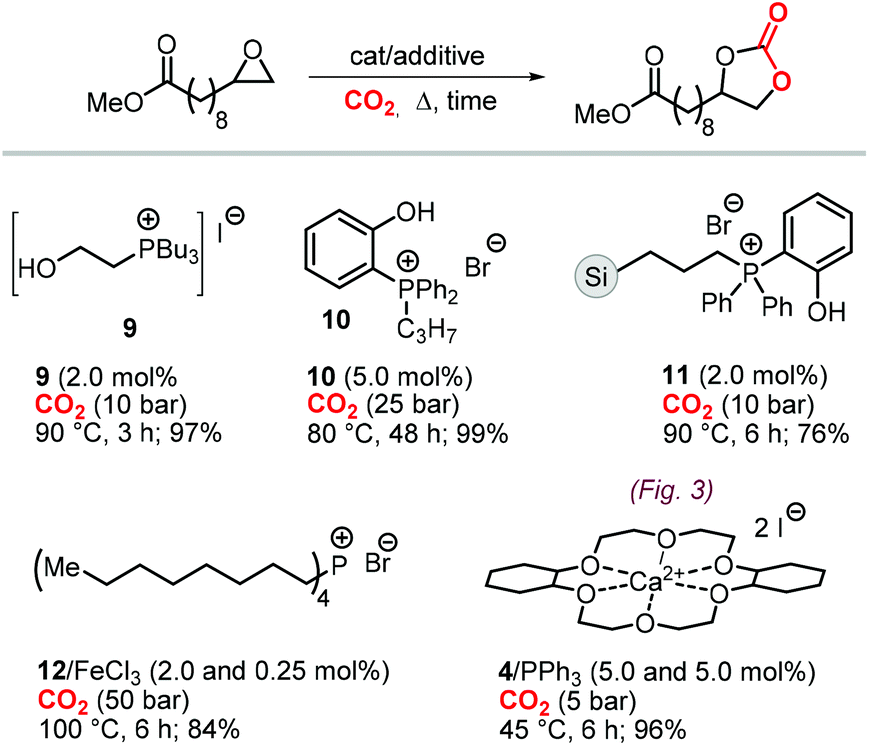 | ||
| Scheme 22 Catalysts for the formation of fatty acid based CC tc1. | ||
The first attempt to prepare tc1 was carried out by using bifunctional single-component organocatalysts based on tetra-alkylphosphonium salts bearing an alcoholic moiety acting as hydrogen bond donor (HBD) for the activation of the epoxide (9, Scheme 22).195 The latter class of organocatalysts is structurally tunable and accessible through the simple reaction between trialkylphosphines and halogenated alcohols under ambient conditions. Using catalyst 9, the coupling of the epoxy precursor and CO2 was carried out in the temperature range 45–90 °C and at 10 bar of CO2 pressure. A different single-component organocatalyst for the carbonation of this epoxidized fatty acid is the triphenylphosphine derivative 10 bearing an ortho-hydroxy functionality (Scheme 22).198 Importantly, due to an optimal pKa, phenolic hydroxyl groups have been found among the best H-bonding activating moieties in the conversion of epoxides into CCs.199 The presence and the position of the hydroxy substituent in 10 was found to be crucial for its catalytic performance likely because it allows activation of the epoxide in the proximity of the bromide anion associated to the phosphonium group, with the halide acting as nucleophile for the ring-opening of the epoxide. The latter aspect allowed the use of 10 also as organocatalyst for the carbonation of more challenging internal bio-based epoxides (vide infra). However, in comparison to the previously discussed single-component organocatalyst 9, the carbonation of epoxidized methyl 10-undecenoate using 10 required higher pressure (25 bar) and longer reaction times.
In order to avoid the limitations related to the use of homogeneous catalysts in terms of cost and product purification, immobilization of 10 onto polystyrene and a silica based support was carried out to ease separation from the reaction products and to allow for its recycling (11, Scheme 22) with the silica-supported catalyst 11 found to perform better than the polystyrene-supported one.197 Catalyst 11 proved to be even more active than its homogeneous counterpart achieving a satisfactory yield in the carbonation of the same epoxide in 6 h at 10 bar of CO2 pressure. The increased activity of the supported catalyst was attributed to the presence of abundant Si–OH moieties on thermally untreated silica likely acting as additional hydrogen-bonding moieties.200,201 Catalyst 11 could be recovered and reused for over ten catalytic cycles, albeit some leaching and deactivation was noted.
As an alternative to the application of organocatalysts, inexpensive and readily available coordination compounds in combination with ammonium and phosphonium salts have frequently found to act as highly active catalysts for the cycloaddition of CO2 to epoxides.202–204 Additionally, Lewis acids based on earth-abundant iron-derived complexes are particularly attractive.2,7,205,206 On the other hand, catalysts based on metal halides are potentially corrosive and unlikely to find application in industrial reactors.207,208 In this context, within a study on the application of iron-based coordination compounds as Lewis acids for the carbonation of several bio-based epoxides, Werner et al. investigated the catalytic performance of the binary catalyst tetra-n-octylphosphonium bromide 12/FeCl3 (Scheme 22) for the carbonation of epoxidized methyl 10-undecenoate.196 The complete carbonation of this substrate was achieved in 6 h albeit under harsh reaction conditions (100 °C, 50 bar of CO2).
More recently, the in situ complexation of calcium halides by crown ethers allows to prepare soluble calcium-based catalysts (4, Scheme 22 and Fig. 4).209 This system was able to convert several internal and terminal epoxides to their corresponding CCs under ambient or very mild conditions without the need for additional quaternary salts. Later on, 4/PPh3 was applied for the conversion of several bio-based epoxides including epoxidized methyl 10-undecenoate (Scheme 22).118 Whereas the calcium based complex 4, formed by the reaction of CaI2 with dicyclohexyl-functionalized 18-crown-6 ether (DCFCE), performed well in the carbonation of a benchmark substrate (i.e., methyl oleate), the addition of triphenylphosphine (PPh3, 5 mol%) allowed for reducing the CO2 pressure from 20 to 5 bar and the reaction temperature from 60 to 45 °C. Under such conditions, epoxidized methyl 10-undecenoate was efficiently converted into tc1 in 6 h.
Bis-carbonates containing two terminal cyclic carbonate moieties are useful synthons for the preparation of non-isocyanate based polyurethanes (NIPUs) by step-growth polymerization using diamine reagents.185,186,210,211 Leitner et al. studied the carbonation of bis-epoxidized hex-5-enyl undec-10-enoate (route b, Scheme 21) to afford tc2, and this process was carried out under relatively harsh conditions using supercritical CO2 (scCO2) at 100 °C.212 In this case, the authors chose tetraheptylammonium silicotungstate containing chromium (a polyoxometalate, POM, abbreviated as THA-Cr-Si-POM) in combination with TBAB as the catalyst. The authors proposed that the POM is capable of activating CO2 by binding it to the catalyst surface. However, this aspect was not experimentally proven and it should be considered that the metal atoms (W, Cr) on the surface of the POM could, alternatively, accelerate the cycloaddition reaction by acting as Lewis acids. This could facilitate the ring-opening of the epoxide in a similar way as observed in MOFs,213,214 and metalated porous polymers.215,216 Despite the harsh reaction conditions, the advantage of the binary, heterogeneous system THA-Cr-Si-POM/TBAB was its simple separation from the products and the potential implementation of a flow process.
Cramail et al. carried out the coupling between GC, a versatile biobased building block,217,218 with 10-undecenoyl chloride leading to the preparation of CC tc3 (route c, Scheme 21) bearing both terminal carbonate and alkene moieties.219 The same CC (tc3) was also prepared by Plasseraud et al. using a different strategy that involves the ring opening of glycidol (Gly) by undecylenic acid followed by carbonation of the obtained diol with diethyl carbonate (route d, Scheme 21).220 Dimerization of tc3via self-metathesis using Grubbs catalyst gave access to bis-carbonate tc4 that could be used to produce NIPUs by treatment with diamines. Cramail et al. also utilized 6-membered bis CCs derived from undecylenic acid using a procedure similar to route d giving other types of NIPUs precursors.221
A different strategy to advance the synthesis of cyclic bis-carbonates for NIPUs using undecylenic acid derivatives was followed by Cramail and coworkers (route e, Scheme 21). This strategy consists of bridging two undecylenic acid units by flexible “diamino or diol” linkers via transamidation or transesterification reactions.222 Following epoxidation of the terminal alkenes to afford a bis-epoxide precursor, the bis-carbonate product (tc5) was produced quantitatively under harsh reaction conditions (80–140 °C, 50–60 bar of CO2) using TBAB as the catalyst.
Finally, terminal cyclic bis-carbonates were prepared also from the methyl ester of oleic acid (MO, Scheme 23). The ethenolysis of the latter is known to produce useful synthons such as 9-decanoic acid methyl ester (9-DAME) and 1-decene.173 Cramail et al. coupled two molecules of 9-DAME by transesterification with pentanediol catalyzed by Zn(OAc)2 at 140 °C. Alternatively, parent MO was directly transesterified with pentanediol to afford an intermediate with internal double bonds.223 Epoxidation of both compounds led to bis-epoxides serving as precursors for their respective CCs, which was carried out under close-to-supercritical or supercritical conditions using TBAB as a catalyst. Terminal bis-carbonate tc6 was found to be more reactive than the bis-internal one tc7 despite the latter being more soluble in the liquid CO2 phase.
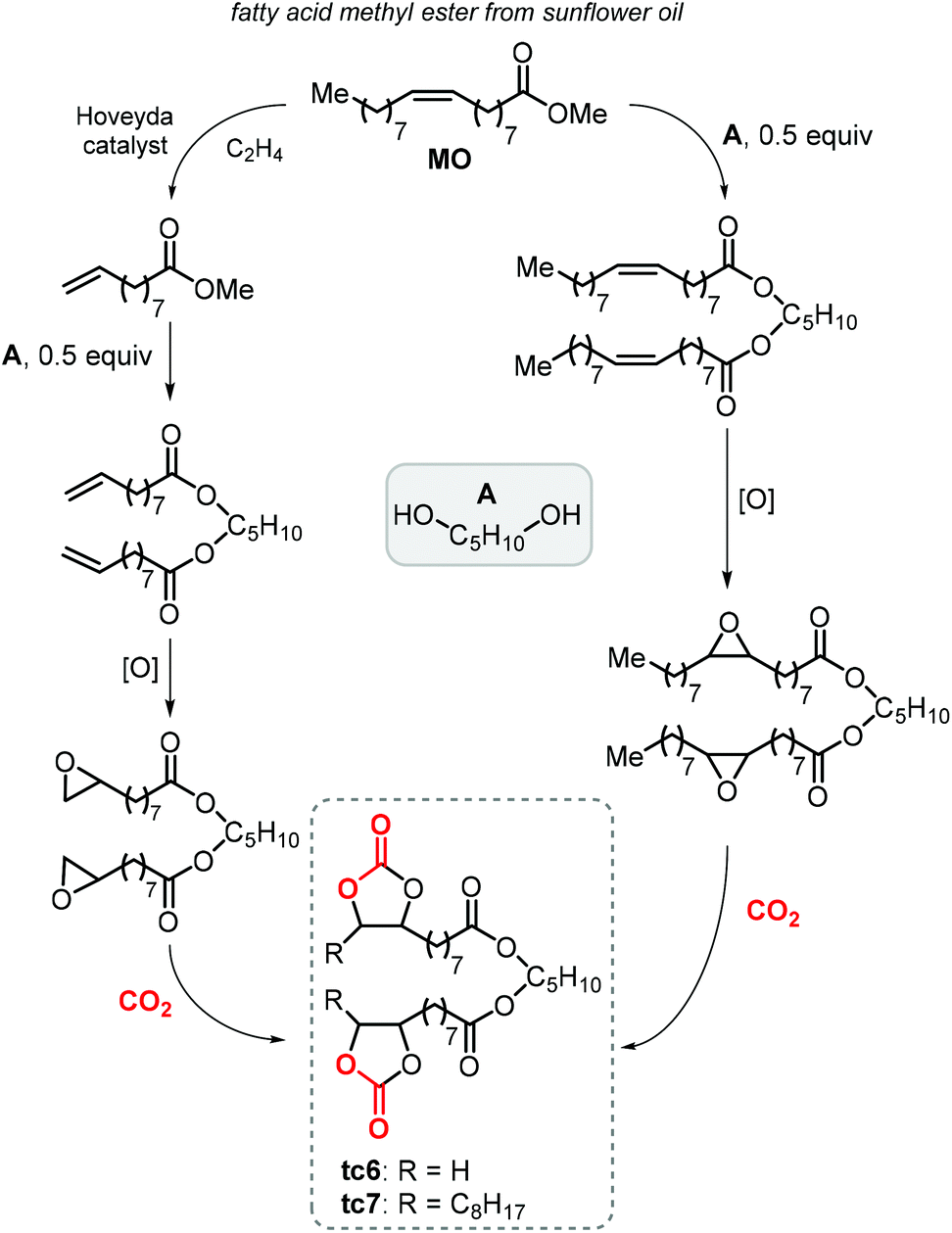 | ||
| Scheme 23 Synthesis of terminal carbonated fatty acid diester (tc6) and internal carbonated fatty acid diester (tc7). | ||
5.2 Internal carbonates from EFA derivatives
The synthesis of cyclic carbonates from epoxidized fatty acids with internal epoxy groups (iEFAs) is more complex than from terminal ones (tEFAs) because of the increased steric hindrance around the epoxy groups and the occurrence of rearrangement and/or isomerization processes compromising the selectivity and thus the yield of the targeted product. In the case of iEFAs, the internal epoxides are generally available in the stereochemically pure cis-configuration, and an attractive challenge is to perform the carbonation reaction with control over the stereoselectivity to obtain the fatty acid based cyclic carbonates as pure cis- or trans-isomers.A general mechanism along with competitive side-reactions is shown in Scheme 24 for a reaction catalyzed by a binary catalyst comprising of a Lewis acid [M] and a nucleophilic halide(X).224 Starting from a pure cis-epoxide, both cis and trans configured CCs can be formed after an initial SN2-type epoxide ring-opening step mediated by the halide that affords a metal alkoxide.225,226 In the subsequent step, a hemicarbonate intermediate is formed after CO2 insertion into the metal–alkoxide bond. This linear carbonate species undergoes a second, intramolecular nucleophilic substitution having either SN1 or SN2 character that strongly depends on the choice of the halide nucleophiles.227–229 Halides possessing excellent leaving group ability (i.e., Br and I) may provide under suitable reaction conditions SN1 type chemistry with the intermediacy of a carbocation. In this case, ring-closure of the carbocation affords either the cis- or trans-carbonates with the latter being typically the thermodynamically favored product. Conversely, an anion with reduced leaving group ability (i.e., Cl) is less likely to provide an in situ generated carbocation. In this case, a second SN2 at the same carbon atom of the metallo-hemicarbonate will undergo ring-closure while restoring the initial configuration leading thus to a cis CC product.
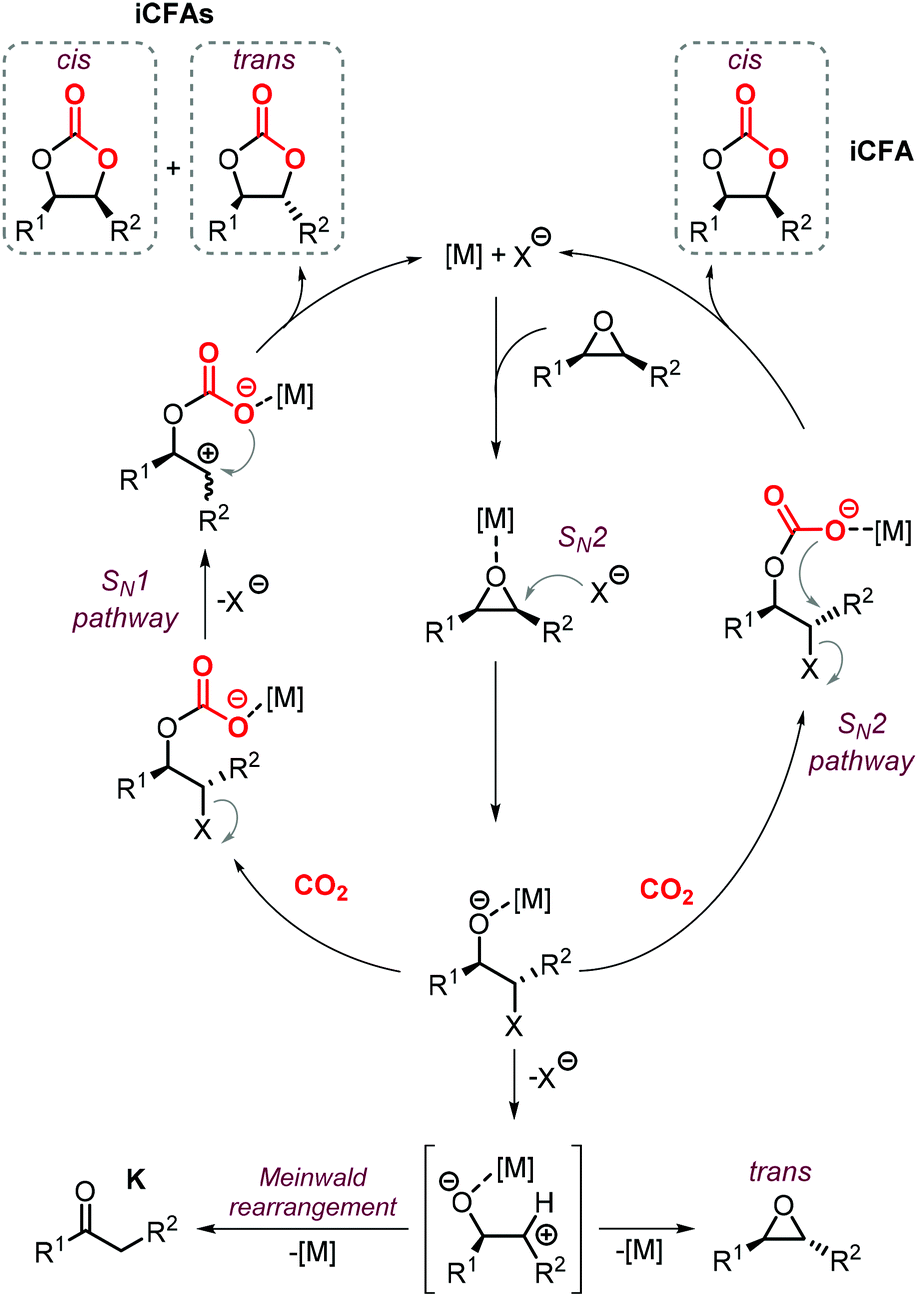 | ||
| Scheme 24 General mechanism of the coupling reaction between a fatty acid containing internal epoxide units and CO2 catalyzed by a binary system [M]/X. X is a halide source such as an ammonium or phosphonium salt. iCFA stands for internal carbonated fatty acid, K for a ketone byproduct. | ||
To further substantiate these concepts, the catalytic performance of several binary/bifunctional catalysts (1Cl and 13–15) incorporating different nucleophilic halides(X) is compared in Table 3. For all catalysts featuring chloride nucleophiles, the cis-iCFA1 was observed as the main product (entries 1–4). Furthermore, the use of 13/TBAI allowed the formation of trans-iCFA1 with nearly complete diastereoselectivity.230 In agreement with the mechanistic picture of Scheme 24, the use of bromide or iodide as the nucleophilic anion should lead to a decreased selectivity for the cis-isomer and thus increased selectivity for the trans isomer by partially favoring the SN1 pathway (entries 1–4), and this appears to be generally the case.
|
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | iEFA1 | Cat. | I | Br | Cl | Ref. | ||||||
| Conv. | Selectivity | Conv. | Selectivity | Conv. | Selectivity | |||||||
| iCFA1 |
cis:transb |
iCFA1 |
cis:transb |
iCFA1 |
cis:transb |
|||||||
|
a Determined by 1H NMR from the integration of the peaks corresponding to the carbonates (cisiCFA1 + transiCFA1) and ketone (K) by-product.
b
Cis:trans ratio determined by 1H NMR by integration of the corresponding signals of cis and trans carbonate products.
c Isolated yield.
d Not reported.
|
||||||||||||
| 1 | cis | 13 | 91c | <1:99 |
85c | 15:85 |
72c | 76:24 |
230 | |||
| 2 | cis | 1Cl | 94 | >99 | 76:24 |
>99 | >99 | 95:5 |
187 | |||
| 3 | cis | 14 | >99 | 19 | 17:83 |
98 | 74 | 24:76 |
69 | >99 | 85:15 |
231 |
| 4 | cis | 15 | 89 | 79 | 39:61 |
95 | 91 | 64:36 |
64 | 92 | 94:6 |
224 |
| 5 | trans | 1Cl | 65 | 99 | <1:99 |
187 | ||||||
| 6 | trans | 13 | 78c | 17:83 |
81c | 11:89 |
79c | 7:93 |
230 | |||
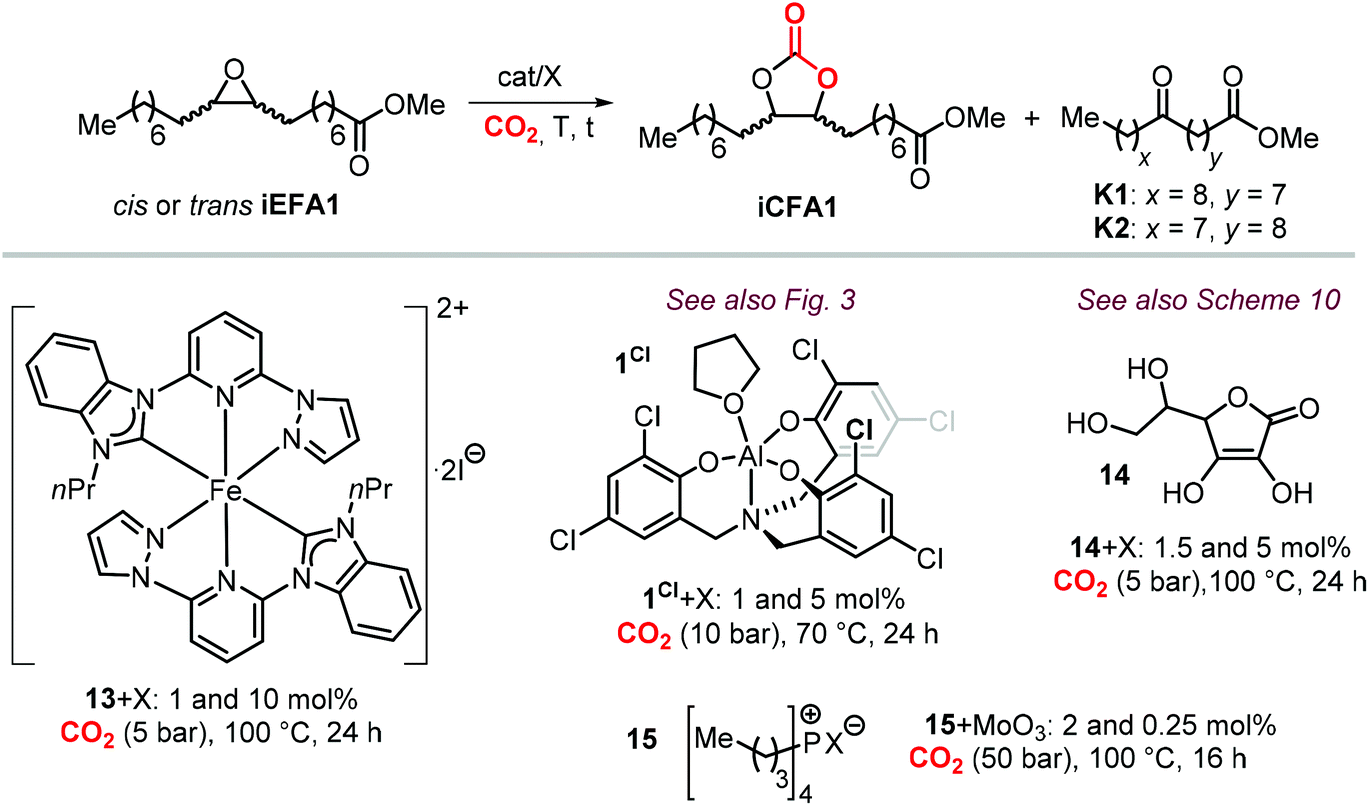
Among the catalysts, simple and readily available L-ascorbic acid 14/TBAC allowed the synthesis of the cis-iCFA1 from cis-iEFA1 with high diastereoselectivity (Cis:trans = 85:15).231 The system 14/TBAC showed (expectedly) lower substrate conversion than 14/TBAB and 14/TBAI but higher chemoselectivity towards cis-iCFA1 (>99%), whereas in the other two cases a significant amount of the side products were formed (entry 3). When 13/TBAI or 14/TBAI were selected as catalysts, trans-iCFA1 was observed as the main carbonate product (entries 1 and 3), whereas iCFA1 was preferentially formed when utilizing bifunctional catalyst 15 regardless of the nature of the halide (entry 4) though with the lowest stereocontrol towards cis-iCFA1 in the presence of iodide. These results suggest that the use of iodide based catalysts have higher preference for the trans-carbonate product by favoring an SN1 pathway.229,230
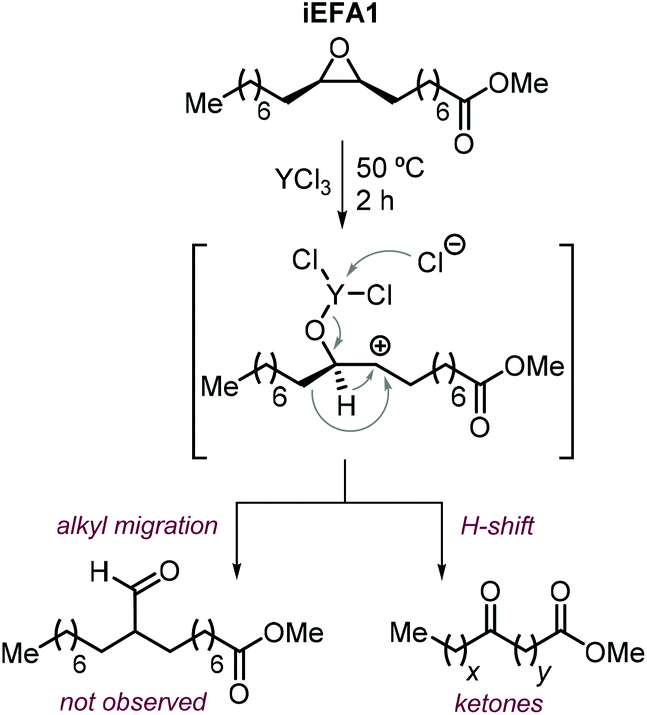
The nature of the halide has thus a clear impact on the overall selectivity of the process, and ketones K are typical by-products in the formation of iCFAs.24,116,118,196,212,224,231,232 Ketone formation is attributed to Meinwald rearrangement via a 1,2-hydride shift in the presence of Lewis- or Brønsted acids (Scheme 24, below).124,233 For example, the formation mechanism of K proposed for YCl3 is shown in Scheme 25.234–236 In the case of CO2 cycloaddition to internal epoxides catalyzed by Lewis acids in the presence of halides as nucleophiles, the carbocation species (precursor of the ketone via hydride shift) may be formed by dissociation of the halide from the alkoxide intermediate similar to what discussed for the SN1 mechanism. Therefore, catalysts with halides that can serve as good leaving groups (Br and I) are expected to favor the formation of a ketone by-product (K), whereas catalysts delivering a chloride nucleophile should suppress this side-product formation.
 | ||
| Scheme 25 Plausible formation of ketone side-products via Meinwald rearrangement in the presence of YCl3.234 | ||
This is in agreement with the observations when using catalysts 14,231 and 15,224 with the selectivity for the carbonate product cis-iCFA1 progressively decreasing in the series Cl > Br > I (entries 3 and 4) in line with the ability of Cl-derived catalysts to suppress the occurrence of undesired Meinwald rearrangement.
Interestingly, the use of Lewis acid complex 1Cl led preferentially to carbonate cis-iCFA1 as the major product independent of the use of chloride or bromide anions as nucleophiles (entry 2). Finally, the formation of a carbocation from the crucial alkoxide intermediate can also lead to cis-to-trans isomerization of the epoxide (Scheme 24, below). Indeed, trans-iEFA1 is often observed as minor by-product along with the formation of trans-iCFA1 and ketones K.224
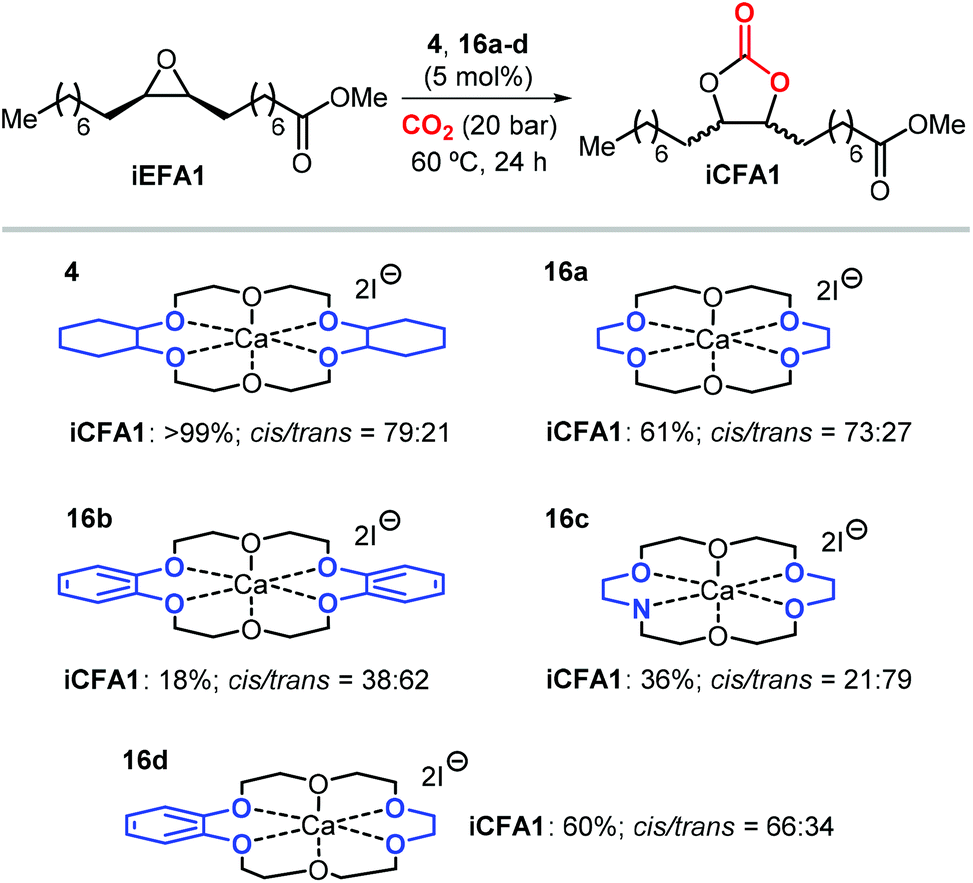
Apart from the nature of the nucleophilic halide anion, modifications of the (Lewis acid) structure can also play a role in controlling the stereoselectivity and kinetics of the carbonation reaction. In the case of calcium-based crown ether complexes, the use of complexes 4 and 16a (Scheme 26) containing fully aliphatic crown ether ligands and iodide as co-catalyst principally led to carbonate product iCFA1 with high cis-selectivity, which is somehow different from the results obtained using catalysts 13 and 14 in the presence of TBAI where the principal product was the trans isomer of iCFA1.118 When part of the bridging groups were aromatic (16b), the selectivity switched from mostly cis to trans product though the overall yield (18%) of iCFA1 was low, likely due to the low solubility of 16c in the reaction mixture. Similarly, by replacing one oxygen for a nitrogen atom (16c) largely the formation of trans-iCFA1 was noted. Finally, compound 16d with a single aromatic bridging unit displayed a catalytic performance similar to that observed for 16a. All these results combined indicate that, beside the choice of the nucleophilic halide anion, other structural factors can contribute to the overall efficacy and stereo-outcome of the process.
 | ||
| Scheme 26 Dependency of catalytic activity and stereocontrol on the macrocyclic ligand structure in calcium-based crown ether complexes in the cycloaddition of CO2 to iEFA1 to afford iCFA1.118 | ||
Some authors examined the cycloaddition of CO2 to trans-iEFA1 (Table 3) as a way to confirm the occurrence of a double inversion (or: double SN2) pathway in the formation of iCFAs. The binary catalytic system 1Cl/PPNCl, that converts cis-iEFA1 mostly to its corresponding carbonate cis-iCFA1 (entry 2, Table 3), led to almost total diastereoselective formation of trans-iCFA1 from trans-iEFA1 (entry 5, Table 3) in agreement with a double inversion pathway. Interestingly, the application of complex 13 for the carbonation of trans-iEFA1 led principally to trans-iCFA1 as the thermodynamically most stable isomer regardless of the type of halide employed (entry 6). This observation is somewhat in contrast with that observed for the carbonation of cis-iEFA1 where the use of iodide and bromide as nucleophiles led to substantial degrees of inversion of configuration via a pseudo-SN1 mechanism (entry 1). These results suggest that the outcome of the cycloaddition process may be subject to a more complex set of interactions between the catalyst components and the substrate.
5.3 Catalytic performances in the cycloaddition of CO2 to various iEFAs and EVOs: the role of quaternary salts
Initial attempts to carry out the carbonation of EFAs where carried out using quaternary ammonium, phosphonium or other salts in the absence of Lewis acidic catalysts or HBDs. An overview of quaternary salts applied under various reaction conditions for the carbonation of iEFAs1–3 derived from mono-unsaturated MO (iEFA1), bis-unsaturated methyl linoleate (iEFA2) and, more rarely, tris-unsaturated methyl linolenate (iEFA3) is given in Table 4.223|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | iEFA | cat. (mol%) | Reaction conditions T (°C), CO2 (bar), time (h) | Conversiona (%) | Selectivity for iCFA | Selectivity (Cis:trans)b |
Ref. |
|
a Conversion determined by titration and/or 1H NMR.
b
Cis:trans ratio determined by 1H NMR by integration of the corresponding signals of cis and trans carbonate products.
c Isolated yield.
d Not reported.
e (C14mim)Br = 1-n-tetradecyl-3-methylimidazolium bromide.
|
|||||||
| 1 | 1 | TBAB (5) | 100, 103, 15 | 93c | 238 | ||
| 2 | 1 | TBAF (5) | 100, 117, 24 | 62 | 0 | 212 | |
| 3 | 1 | TBAC (5) | 100, 117, 24 | 21 | 95 | 77:23 |
212 |
| 4 | 1 | TBAB (5) | 100, 117, 24 | 97 | ≥99 | 72:28 |
212 |
| 5 | 1 | TBAI (5) | 100, 117, 17 | 80 | 92 | 20:80 |
212 |
| 6 | 1 | NH4Br | 100, 117, 24 | 4 | 75 | 212 | |
| 7 | 1 | ((n-C7H15)4N)Br | 100, 117, 24 | 99 | ≥99 | 70:30 |
212 |
| 8 | 1 | (C14mim)Br e | 100, 117, 24 | 97 | 96 | 74:26 |
212 |
| 9 | 1 | ((n-C14H29)(n-C6H13)3P)Br | 100, 117, 24 | 97 | 97 | 69:31 |
212 |
| 10 | 2 | TBAB (5) | 100, 117, 24 | 71 | 95 | 212 | |
| 11 | 3 | TBAB (5) | 100, 117, 24 | 68 | 89 | 212 | |
| 12 | 1 | TBAB (2) | 100, 50, 16 | 39 | 82 | 46:56 |
224 |
| 13 | 1 | 15-Br (2) | 100, 50, 16 | 49 | 94 | 71:29 |
224 |
| 14 | 1 | 15-Cl (2) | 100, 50, 16 | 39 | 99 | 90:10 |
224 |
| 15 | 1 | 15-I (2) | 100, 50, 16 | 35 | 71 | 57:43 |
224 |
| 16 | 1 | TBAB (7) | 130, 30, 8 | 99 | 239 | ||
| 17 | 1 | TBAB (5) | 70, 10, 24 | >99 | >99 | 51:49 |
187 |
| 18 | 1 | TBAC (5) | 70, 10, 24 | 6 | >99 | >99:1 |
187 |
| 19 | 1 | PPNCl (5) | 70, 10, 24 | 53 | >99 | 96:4 |
187 |
| 20 | 2 | PPNCl (5) | 85, 10, 24 | 95 | >99 | 95:5 |
187 |
| 21 | 3 | PPNCl (5) | 70, 10, 24 | 75 | >99 | 90:10 |
187 |
| 22 | 1 | TBAI (5) | 100, 5, 24 | 70 | 59 | 22:78 |
231 |
| 23 | 1 | TBAB (5) | 100, 5, 24 | 83 | 87 | 36:64 |
231 |
| 24 | 1 | TBAC (5) | 100, 5, 24 | 44 | >99 | 90:10 |
231 |
The obvious advantage of using quaternary salts as catalysts is that they are metal-free, generally inexpensive, and commercially available. In addition, Doll and Erhan found that TBAB can be conveniently removed from the product mixture by thermal breakdown into volatile compounds at 190 °C by Hofmann elimination.237 In different studies, the use of halide salts often required harsh reaction conditions such as the use of scCO2 (entries 1–11, Table 4) or temperatures above 100 °C (entry 16) to convert iEFA1–3 to their corresponding carbonates iCFA1–3 in reasonable to high yields.
Based on the earlier report by Doll and Erhan highlighting the catalytic competence of TBAB in the carbonation of iEFA1 under supercritical conditions (entry 1),238 Leitner et al. showed that the employment of several quaternary ammonium halides for the cycloaddition of CO2 to iEFA1 under scCO2 conditions generally provided the target carbonate with good conversion rates and selectivities (entries 2–9). Exceptions in this series were NH4Br and tetra-n-butylammonium fluoride (TBAF) because of the poor leaving group character of the fluoride anion (see entries 2 and 6) and the tighter ion pair when using ammonium cations.212 In line with earlier results discussed in section 5, all salts employed provided cis-iEFA1 as the main stereoisomer with the exception of TBAI for which the trans-isomer was found to be the main product (entry 5). The use of ammonium or phosphonium salts bearing longer alkyl chains compared to TBAB did not lead to any significant improvement of the catalytic activity (entries 7–9). Therefore, TBAB was selected as catalyst for the conversion of other substrates such as cis-iEFA2 and cis-iEFA3, obtaining satisfactory performances despite a slight drop in conversion and carbonate selectivity (entries 10 and 11).
Similar results under comparable reaction conditions were found for substrate cis-iEFA2 in a later study by Buchholz et al.24 However, Werner and coworkers demonstrated that the catalytic performance is strongly reduced together with some loss of selectivity for iCFA1 when using TBAB for the carbonation of cis-iEFA1 at 100 °C but lower CO2 pressure (50 bar) and lower catalyst loading (2 mol%; cf., entries 4 and 12).224 In the same study it was observed that the use of tetrabutylphosphonium halides, in particular 15-Br and 15-Cl, leads to slight improvement in terms of epoxide conversion and iCFA selectivity compared to TBAB under identical conditions (cf., entry 12 and 13–15). In addition, Leveneur et al. showed that the use of a slightly higher TBAB loading (7 mol%) at 130 °C allowed complete conversion of cis-iEFA1 under 30 bar CO2 pressure as an alternative for supercritical conditions, though the selectivity towards iCFA1 was not reported (entry 16).239
Recently, the group of Kleij studied the performance of various halide salts in an attempt to develop catalysts able to operate under milder conditions.187 They found that TBAB converts cis-iEFA1 quantitatively into iCFA1 at 70 °C and 10 bar of CO2, although without any observable stereocontrol (entry 17). The use of TBAC, however, provided cis-iCFA1 selectively but only at very low epoxide conversion, while the use of PPNCl as chloride source led to higher conversion of cis-iEFA1 maintaining very high selectivity for cis-iCFA1 (cf., entries 18 and 19). PPNCl served also as an efficient and stereoselective catalyst for carbonation of cis-iEFA2 and cis-iEFA3 at 70–85 °C and 10 bar of CO2 (entries 20 and 21).
Recently, it was shown that quaternary ammonium salts give appreciable conversion levels of cis-iEFA1 at 5 bar of CO2 and 100 °C. Under these conditions, the stereocontrol exerted by TBAI in the preparation of cis-iCFA1 is moderately high, and similar results are obtained switching to TBAB though with significantly higher chemoselectivity for iCFA1. The use of TBAC led to moderate epoxide conversion but with very high overall selectivity for cis-iCFA1 (cf., entries 22–24).
A further expansion of the portfolio of fatty acid-derived CCs can be realized by using epoxidized estolides as starting point. This class of compounds is generated by the reaction between a carboxylic acid of a fatty acid with a double bond or hydroxyl moiety of another one.232,240 The estolide compounds are attractive functional fluids when compared to standard vegetable oils because of a higher stability towards oxidation and lower pour points.241 The epoxidation and carbonation of estolides can be used to further tune crucial properties of these compounds such as viscosity.242
In an initial study, Isbell et al. used 2-ethylhexyl estolide esters of oleic acids constituted by a complex mixture of oligomers generated by the addition of the carboxylic acid of oleic acid to the double bond of other oleate chains.242 Following epoxidation using in situ generated performic acid, the epoxidized estolide was successfully carbonated using TBAB as the catalyst under scCO2 conditions at ∼100 bar of CO2 and 100 °C. The viscosity of the carbonated estolide was substantially higher than that of the parent epoxidized estolide. The same group developed the synthesis of estolide esters obtained through the reaction between oleic acid and the hydroxyl group of saturated and unsaturated alkyl esters of castor oil (Scheme 27).232 The resulting compounds were epoxidized and carbonated using TBAB as catalyst under supercritical conditions as described in the previous example. The resulting carbonated estolides displayed increased viscosity and higher oxidation stability (oxidation onset around 200 °C) compared to the non-functionalized estolides and its epoxy derivatives, thus offering good candidates for application as industrial fluids.
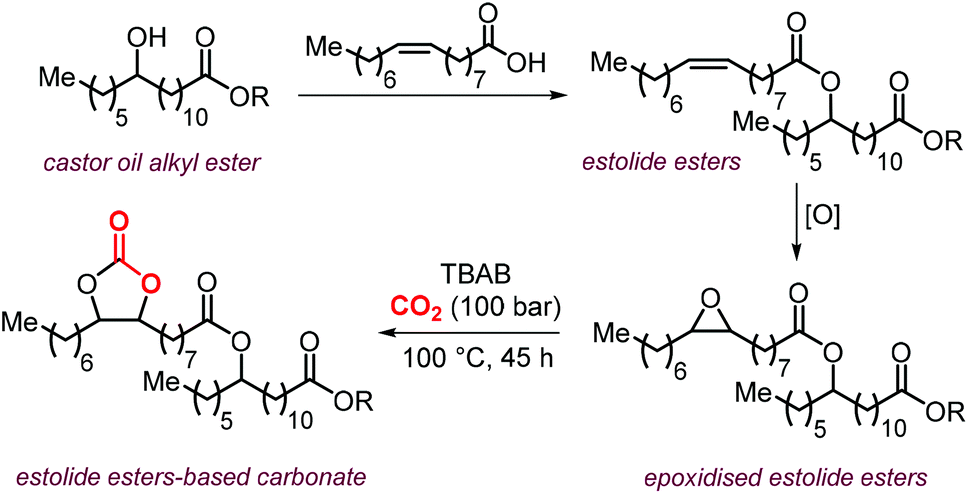 | ||
| Scheme 27 The synthesis of estolides from oleic acid and castor oil alkyl esters. | ||
Beside the case of EFAs, the carbonation of EVOs (Table 5) such as epoxidized vernonia, castor, soybean, linseed, sunflower, cottonseed and olive oils has been extensively studied.243 The resulting CVOs (Table 5) are attractive synthons for the synthesis of NIPUs by reaction with diamines,23,185,211,244–251 and find applications in paints, coatings, and bio-based materials.252,253 Remarkably, some properties such as the thermal stability and oxidative stability of polyurethanes (PUs) derived from soybean oil were found to rival those of PUs derived from poly(propylene oxide).254
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | EVO | TBAB (mol%) | Reaction conditions T (°C), CO2 (bar), time (h) | Conversiona (%) | Selectivitya for CVOs | Ref. |
|
a Conversion/selectivity determined by titration and/or 1H NMR.
b Weight percent (wt%) loading.
c Weight percent (wt%), epoxide conversion determined by epoxy titration according to ASTM D 1652.
d Not Reported.
e Conversion determined by FTIR.
f TBAI loading in mol%.
g Conversion determined by FTIR analysis of the reaction mixture at intervals of 24 h until completion.
h Addition of water, with a molar ratio H2O:EVO of 1:3.
i Weight percent.
|
||||||
| 1 | Soybean | 5b | 140, 152, 18 | 96c | 263 | |
| 2 | Sucrose soyate | 5b | 140, 131, 20 | 70c | 263 | |
| 3 | Soybean | 16.6 | 100, 138, 46 | 96 | 264 | |
| 4 | Soybean | 4.5 | 140, 124, 54 | 99 | 265 | |
| 5 | Soybean | 5 | 100, 117, 24 | 47 | 73 | 212 |
| 6 | Soybean | 2.7i | 120, 100, 9 | 100 | 266 | |
| 7 | Soybean | 5 | 100, 100, 20 | 94 | 237 | |
| 8 | Soybean | 0.18 | 140, 56.5, 22 | 98 | 267 | |
| 9 | Cottonseed | 3.5 | 130, 50, 7 | 94 | 268 | |
| 10 | Sunflower | 3.5 | 120, 50, 12 | 86 | 269 | |
| 11 | Cottonseed | 5 | 140, 30, 24 | 99.9 | 270 | |
| 12 | Linseed | 3 | 140, 30, 20 | 100 | 271 | |
| 13 | Soybean | 3.4i | 140, 10, 23 | 97 | 272 | |
| 14 | Castor | 5 | 130, 5, 8 | 93 | 273 | |
| 15 | Soybean | 5 | 110, 4, 12 | 100e | 274 | |
| 16 | Soybean | 5f | 110, 1, 70 | 100 | 255 | |
| 17 | Linseed | 5 | 100, 1, 72 | 100g | 256 | |
| 18 | Soybeanh | 5 | 120, 1, 70 | 77 | 89 | 257 |
| 19 | Soybeanh | 5 | 120, 1, 40 | 80 | 92 | 259 |
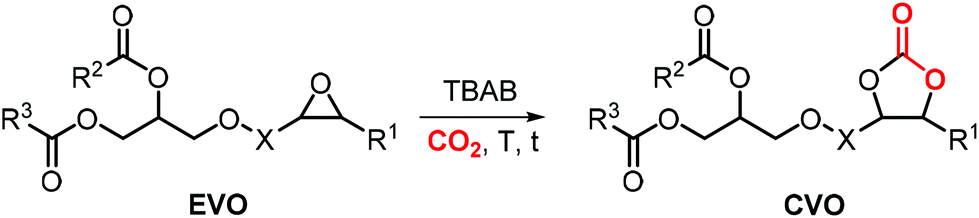
The synthesis of CVOs has been principally carried out on epoxidized linseed, sunflower and soybean oils due to the relatively high epoxy content that can be introduced in the fatty acid alkyl chains. The cycloaddition reaction of CO2 to such EVOs has often been carried out using TBAB as the catalyst (Table 5). As in the case of EFAs (see entries Table 4), these reactions are conveniently performed at high pressure (CO2 ≥ 50 bar) or under sc-CO2 conditions (entries 1–10, Table 5) often in combination with high reaction temperatures (120–140 °C). However, the CO2 pressure can be reduced to 4 bar when the carbonation reaction is carried out at ≥110 °C (entries 11–15). Under these conditions, high to nearly quantitative conversion of the epoxide groups was generally observed, however the selectivities for the carbonate products have seldom been reported. Leitner et al. observed a relatively low selectivity (73%) for carbonate formation in the cycloaddition of CO2 to epoxidized soybean oil (ESBO) under supercritical conditions (entry 5).212 Further attempts were carried out for the carbonation of EVOs under atmospheric pressure at T ≥ 110 °C using TBAB or TBAI as catalyst by extending the reaction time to 40–70 h (entries 16–19). In some of these cases, despite the disappearance of epoxide and the appearance of IR-bands for the carbonate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching were observed, the actual chemoselectivity for the CC was not reported.255,256
O stretching were observed, the actual chemoselectivity for the CC was not reported.255,256
Mazo and Rios observed that the addition of water (about 33 mol%) significantly accelerates the carbonation of ESBO thus obtaining the corresponding CVO under atmospheric pressure with good conversion and selectivity in 70 h (entry 18).257 This result is in line with the known ability of water to serve as a HBD in the cycloaddition of CO2 to epoxides.258 Additionally, the same authors have shown that a further acceleration of the reaction rate can be achieved by combining the addition of water to the use of a microwave reactor resulting in a reduction of the reaction time to 40 h without affecting the selectivity for the carbonated product (entry 19).259
Finally, several examples of comprehensive physicochemical studies on the carbonation of EVOs in the presence of TBAB have been carried out. These studies concern reaction kinetics modelling, the role of mass transfer, CO2 solubility, substrate viscosity, the differences between EFAs and EVOs and the effect of microwave irradiation on the process kinetics, and were carried out by Leveneur et al.260–262 These important, but rather technical studies are not described in detail in this section.
5.4 Catalytic performance of binary catalytic systems in the cycloaddition of CO2 to iEFA1: metal salts and coordination compounds
The vast majority of catalytic systems for the cycloaddition of CO2 to epoxides are binary systems. These systems typically involve one component (Lewis acid or HBD) coordinating or better activating the epoxide substrate, and a nucleophilic component that serves to ring-open the activated epoxide.30,183,184 These bicomponent catalysts often allow for the cycloaddition reactions to take place under relatively mild or even ambient conditions when compared to the exclusive use of (quaternary) halide salts.1In recent years, several examples of binary catalytic systems suitable for the mild and stereoselective carbonation of EFAs and EVOs have been developed and are discussed in this and following sections, by taking iEFA1 as a model substrate, according to the kind of epoxide-activator including metal coordination compounds, metal–organic catalysts and organocatalysts.
Coordination compounds and metal salts represent readily available and inexpensive compounds for the cycloaddition of CO2 to epoxides.202,203,229 They are usually commercially available and can be easily immobilized onto silica supports.225,275,276 However, in the case of metal halide salts, their large-scale application could be limited by their relatively low moisture stability and by the risk of reactor corrosion.207
Polyoxometalates (POMs) are clusters of metal atoms connected by oxo-bridges and terminated by anionic MO moieties. Such compounds are able to activate CO2/epoxides via interaction with the basic oxygen atoms and Lewis acidic metal centers of the POM.277 Based on this dual activation ability, the halogen-free cycloaddition of CO2 to epoxides catalyzed by transition-metal-substituted silicotungstates ([n-C7H15)4N]6[α-SiW11O39M(II)], with M = Mn, Co) was demonstrated by Sakakura et al. already in 2005.278 The sustainable character of this approach was limited by the harsh reaction conditions (150 °C, 35 bar of CO2) and the need for a reaction solvent.
Later in 2013, Leitner et al. employed [(n-C7H15)4N]5[α-SiW11O39Cr(III)] (THA-Cr-Si-POM) for the carbonation of iEFAs in scCO2.212 As expected, under harsh conditions (100 °C, ∼130 bar of CO2), THA-Cr-Si-POM (2 mol%) was able to catalyze the quantitative conversion of iEFA1 to cis-iCFA1 in 20 h. However, the authors also observed that the addition of equimolar, catalytic amounts of TBAB could accelerate the reaction that was complete in 6 h (entry 1, Table 6).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat/add. (mol%) | T, p, t (°C, bar, h) | Yield iCFA1 (%) | Sel.aiCFA1 (%) | Sel.acis vs trans | Ref. |
|
a
Cis:trans ratios and selectivity towards iCFA1 determined by 1H NMR from the integration of the corresponding signals of cis and trans carbonate products.
b PEG DME 500: oligo(ethyleneglycol) dimethyl ether with Mn ∼ 400 g mol−1.
c Not reported.
d Conversion determined by GC analysis. THA = tetra-n-heptyl ammonium, p is the partial pressure of CO2 and TEA is triethanolamine.
|
||||||
| 1 | THA-Cr-Si-POM (2.0) | 100, 130, 6 | 95 | 98 | 96:4 |
212 |
| TBAB (2.0) | ||||||
| 2 | 15-Br (2.0) | 100, 50 | 98 | 98 | 77:23 |
224 |
| MoO3 (0.25) | 20 | |||||
| 3 | 12 (2.0) | 100, 50 | 96 | 96 | 68:32 |
196 |
| FeCl3 (0.25) | 24 | |||||
| 4 | CaI2 | 90, 50 | 91 | 53:47 |
280 | |
| PEG-DME 500 (5.0)b | 48 | |||||
| 5 | 4 (5.0) | 45, 5 | 86 | 84:16 |
118 | |
| Ph3P (5.0) | 24 | |||||
| 6 | KI (2.0) | 100, 50 | 8d | 281 | ||
| TEA (2/2) | 16 | |||||
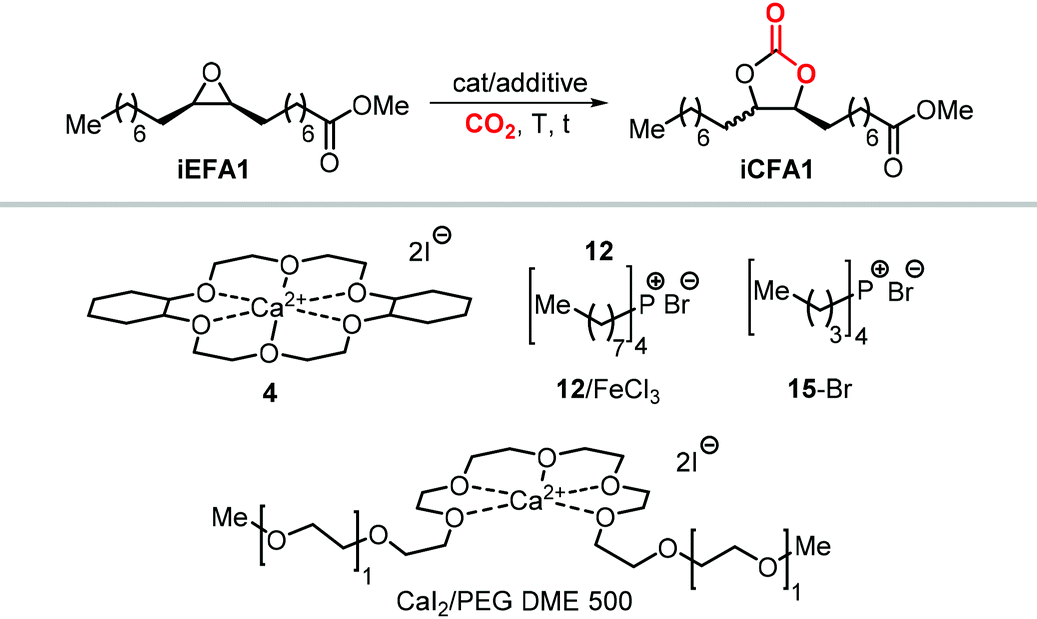
More recent, the use of coordination compounds and metal salts for the carbonation of fatty acids has led to the discovery of binary systems operating under milder conditions than the THA-Cr-Si-POM reported by Leitner. Werner et al. extensively studied the application of coordination compounds as additives for the cycloaddition of CO2 to iEFA1. Initial studies involved the application of phosphonium salts 12 and 15 in combination with commercially available coordination metal compounds of transition metals such as MoO3 and FeCl3 under relatively harsh conditions (100 °C, 50 bar of CO2; entries 2 and 3 in Table 6).196,224 The presence of MoO3 appeared as a more convenient choice for iEFA1 conversion and carbonate selectivity in a wider screening of metal salts (mostly Al- and Mo-based) in the presence of 15-Br (see also Table 3) as the nucleophile.
Interestingly, compared to the exclusive use of phosphonium salts as catalysts, the addition of coordination compounds accelerate iEFA1 conversion at the cost of a slight drop in carbonate selectivity.224 Under the same reaction conditions, the use of FeCl3 in the presence of phosphonium salts (12 or 15-Br) led to a similar result as MoO3.196 The use of iron-based catalysts is considered advantageous, since Fe is an earth-abundant, non-toxic and non-endangered metal.2,7,205,279
Similarly, the application of calcium may be seen as more sustainable compared to the use of transition metals in catalyst systems. Whereas most coordination compounds of calcium are generally insoluble in most reaction media, Werner et al. showed that the use of chelating ligands (crown ethers) in combination with calcium halides leads to soluble and highly active Lewis acids for CO2/epoxide cycloaddition.118,209,280–282 The in situ complexation of CaI2 by poly(ethylene glycol)dimethyl ether (PEG-DME-500) further promotes the nucleophilicity of the iodide anion leading to a system able to mediate the cycloaddition of CO2 to terminal and internal epoxides including iEFA1 (entry 4, Table 6).280 Alternatively, crown ethers are effective complexing agents for CaI2 leading to highly active calcium catalysts such as 4 for the cycloaddition of CO2 to terminal epoxides under ambient conditions.209 Catalyst 4 can be successfully applied for the carbonation of iEFA1 under relatively mild conditions (60 °C, 20 bar of CO2). In addition, it was found to work under even milder conditions (45 °C, 5 bar of CO2, entry 5) when used in the presence of a relatively high loading (5 mol%) of PPh3 obtaining iCFA1 mostly as the cis-isomer.118 In a previous study, the same group showed that KI, a frequently used source of nucleophilic iodide,283 forms an efficient catalyst for the carbonation of terminal epoxides when combined with triethanolamine as HBD, although this catalyst was virtually inactive for the conversion of iEFA1 (entry 6),281 justifying the design of more sophisticated binary catalyst architectures.
5.5 Catalytic performance of binary catalytic systems in the CO2 cycloaddition to iEFA1: metal complexes
Metal complexes based on Schiff-base ligands are among the most studied metal–organic compounds often displaying remarkable catalytic activity for the cycloaddition of CO2 to terminal epoxides.121,284,285 Thus, it is not surprising that these complexes have also been studied for the carbonation of EFAs.Masdeu-Bultó et al. prepared NN′O- and N2O2-type Schiff-base ligands that were used for the complexation of earth-abundant metals such as zinc,286 and aluminum, respectively (complexes 17 and 18, Table 7).287 These complexes were found to be active Lewis acidic catalysts for the cycloaddition of CO2 to terminal epoxides, but their application for the carbonation of iEFA1 required harsh reaction conditions (100 °C, 100 bar of CO2) to afford only moderate yields of iCFA1 (entries 1 and 2). Under these conditions, catalyst 18/TBAB proved to be much faster with reaction times as short as 30 min.287
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat/add. (mol%) | T, p, t (°C, bar, h) | Yield/sel.a for iCFA1 (%) | Sel.aCis:trans |
Ref. |
|
a
Cis:trans ratio and selectivity for iCFA1 determined by 1H NMR from the integration of the corresponding signals of cis and trans carbonate products, and substrate/side-products.
b Not reported.
c Using epoxidized methyl ricinoleate (cis) as the substrate. Note that p is the partial pressure of CO2.
d Conversion, determined by 1H NMR.
|
|||||
| 1 | 17 (2.0) | 100, 100, 24 | 53, b | 95:5 |
286 |
| TBAB (2.0) | |||||
| 2 | 18 (2.0) | 100, 100, 0.5 | 63, b | 52:48 |
287 |
| TBAB (2.0) | |||||
| 3 | 19 (0.5) | 85, 10, 18 | 46, >99 | >99:1 |
291 |
| TBAB (5.0) | |||||
| 4 | 1Cl (0.5) | 70, 10, 24 | 99d, >99 | 97:3 |
187 |
| PPNCl (3.0) | |||||
| 5c | 1Cl (1.0) | 70, 10, 24 | 99 d, 99 | < 1:99 |
187 |
| 6 | 13 (1.0) | 100, 5, | 91, b | < 1:99 |
230 |
| TBAI (10) | 24 | ||||
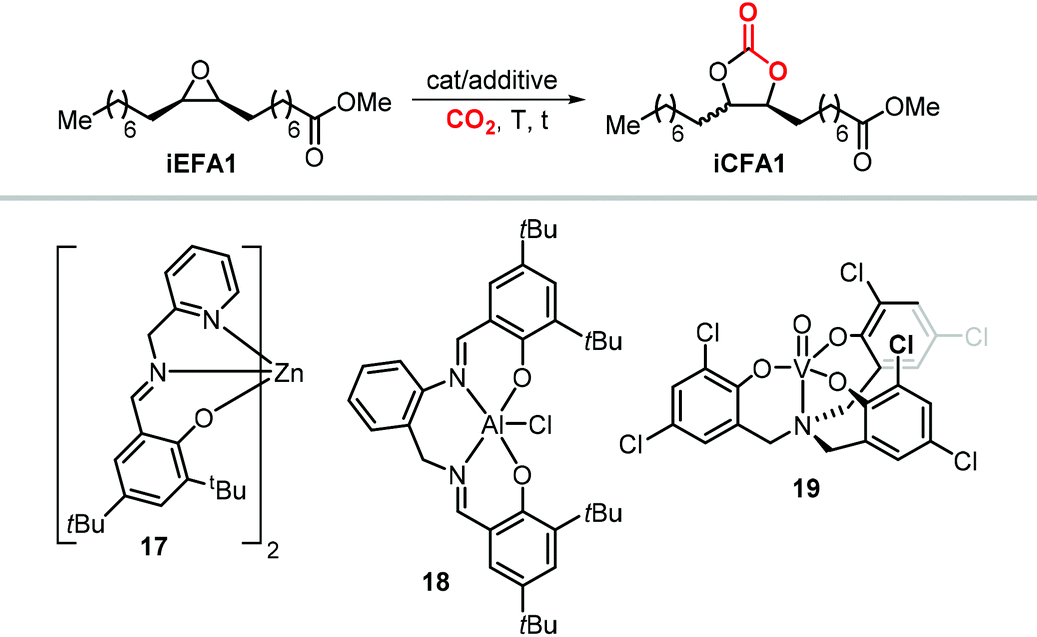
Aminotriphenolates (TPA) complexes, extensively studied by the group of Kleij,206,288–291 are a different class of highly efficient and strongly Lewis acidic metal complexes for the cycloaddition of CO2 to internal epoxides. In particular, TPA complexes derived from aluminum (1R, Table 3) and vanadium (19, Table 7) are highly chemo- and stereoselective Lewis acids for the carbonation of iEFA1 under comparatively mild conditions (75–80 °C, 10 bar of CO2) in the presence of TBAB, obtaining cis-iCFA1 (entries 3 and 4), with the V-based binary catalyst 19/TBAB only providing moderate conversion.
One peculiar feature of Al-aminotriphenolate complexes is their capability to catalyze the conversion of CO2 and epoxy alcohols such as glycidol and its derivative, to cyclic carbonates in the absence of nucleophilic additives. The conversion of epoxy alcohols has been proposed to take place by formation of a carbonic acid hemi-ester stabilized by the metal–organic Lewis acid.17,18,75 A similar mechanism was proposed when epoxidized methyl ricinoleate was combined with CO2 in the presence of 1Cl.187 Despite the initial cis-configuration of the epoxide precursor, in the absence of halide nucleophiles the trans-isomer of carbonate product was obtained in high yield and selectivity indicating effective inversion of configuration (entry 5, Table 7). To explain this observation and in line with their previous findings,17,75 the authors postulated a mechanism in which an Al-stabilized carbonic acid-like intermediate, formed by reaction of the substrate with CO2, first evolves into a six-membered carbonate intermediate by nucleophilic attack on the nearest epoxide carbon (Scheme 28). A subsequent nucleophilic attack of the produced oxyanion on the electrophilic carbon center of the latter carbonate would then lead to formation of the final trans-carbonate with stereo-inversion.
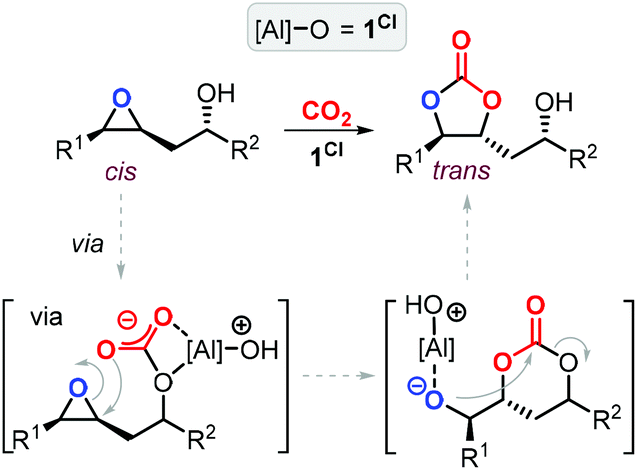 | ||
| Scheme 28 Proposed mechanism for the conversion of CO2 to epoxidized methyl ricinoleate catalyzed by Al-complex 1Cl. | ||
Recently, Liu and coworkers reported the cycloaddition of CO2 to iEFAs catalyzed by the iron bis-pincer complex 13 (Table 3).230 By using a large amount of TBAI (10 mol%, entry 6, Table 7) at 100 °C and low CO2 pressure (5 bar), iEFA1 was fully converted to trans-iCFA1 (entry 6, Table 7) according to an SN1 pathway described in Scheme 24. Independent from the substrate, the authors managed to control the stereochemistry of the final carbonate iCFA1 by proper selection of a suitable nucleophilic halide as described in section 5.2.
Based on previous investigations, the authors proposed that 13 acts as a precatalyst in this cycloaddition process by generating an iron-based Lewis acid (LA) and a (free) N-heterocyclic carbene (NHC), see Scheme 29.292
 | ||
| Scheme 29 Proposed catalytically competent species generated from 13 under the applied reaction conditions. Tetra-n-butylammonium cations (TBA) are omitted for clarity. | ||
In the presence of TBAB, the Lewis acid is supposed to evolve into hexa-coordinated tri-halide –ate complexes of type [Fe(CNN)X3] (structures A–C in Scheme 29). According to DFT calculations, compound C is the most stable one. After dissociation of one of the halide anions, a coordination site on the Fe-center becomes available for the coordination (activation) of the epoxide prior to nucleophilic attack by the liberated halide. In principle, the free NHC fragment stemming from 13 can also play a role in the reaction by capturing and activating CO2,293 but in this specific case this was not discussed or proven.
5.6 Catalytic performance of organocatalysts in the cycloaddition of CO2 to iEFA1
Organocatalysts have received considerable attention as relatively nontoxic, readily available, and in most cases moisture-insensitive compounds.294–297 In the cycloaddition of CO2 to epoxides, organocatalysts are often applied as single-component nucleophilic species, such as DBU, TBD and 4-dimethylamino-pyridine (DMAP), or as HBDs in the presence of nucleophilic halide sources.1,5,31,298 Thus far, the organocatalyzed carbonation of fatty acids has been generally carried out using both binary and bifunctional catalytic systems, with the second category embedding halide nucleophiles and HBD moieties within the same molecule.The Werner group developed two families of bifunctional phosphonium halides (such as 9, Scheme 22),195 and ammonium halides (such as 20, Table 8),299 bearing hydroxyl groups for the activation of epoxides. Such catalysts performed efficiently in the carbonation of terminal epoxides under mild conditions (45–90 °C, 5–10 bar of CO2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat/add. (mol%) | T, p, t (°C, bar, h) | Yield/sel. of iCFA1a (%) | Sel.aCis:trans |
Ref. |
|
a
Cis:trans ratio determined by 1H NMR by integration of the corresponding signals of cis and trans carbonate products, isolated yields of iCFA1 are given.
b GC yield.
c Not reported.
d Yield determined by 1H NMR using mesitylene as the internal standard. Note that p is the partial pressure of CO2.
|
|||||
| 1 | 9 (5.0) | 100, 50, 24 | 65, >99 | 40:60 |
195 |
| 2 | 20 (2.0) | 100, 50, 16 | 20b, >99 | 48:52 |
299 |
| 3 | 10 (5.0) | 80, 25, 24 | 98, c | 54:46 |
198 |
| 4 | 21 (1.0) | 90, 10, 24 | 30, c | 28:72 |
300 |
| 5 | 11 (2.0) | 90, 10, 24 | 26d,c | 43:57 |
197 |
| 6 | 22 (4.0) | 100, 5, 24 | 51, c | 19:81 |
301 |
| TBAI (12) | |||||
| 7 | 14 (1.5) | 100, 5, 48 | 90, >99 | 77:23 |
231 |
| TBAC (5.0) | |||||
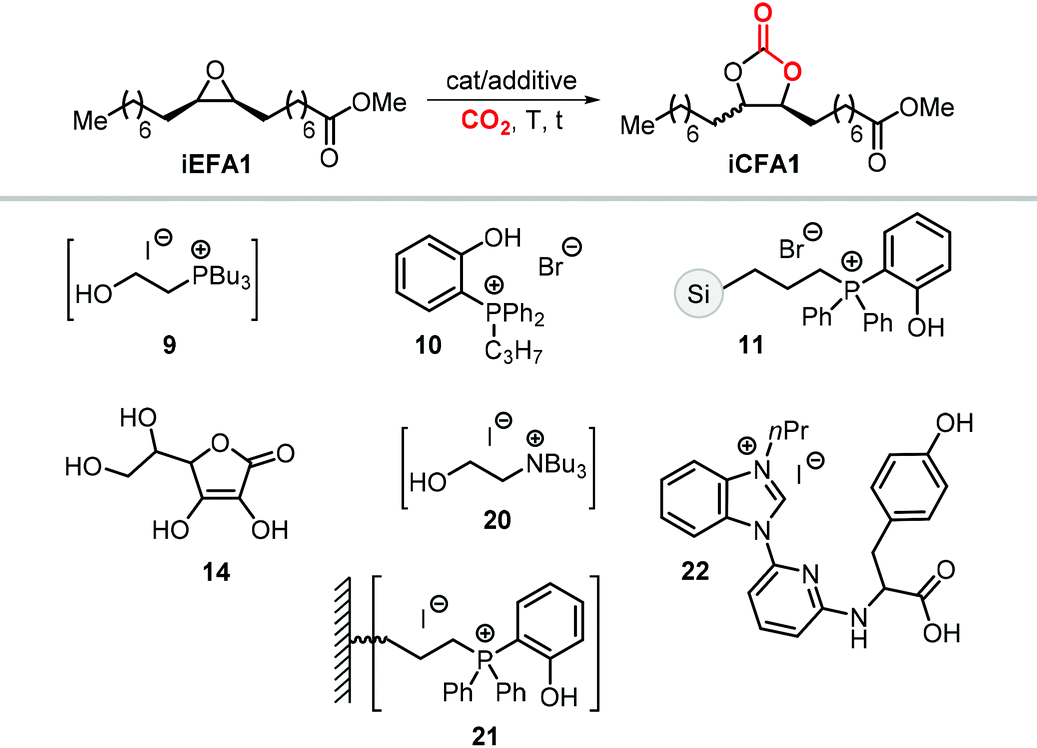
When using iEFA1 as the substrate, 9 promotes its chemoselective carbonation in good yields under demanding reaction conditions (100 °C, 50 bar of CO2), whereas 20 was less efficient under similar conditions providing only low yields of iCFA1 (cf., entries 1 and 2, Table 8). In both cases, the carbonate product was obtained as a mixture of cis and trans stereoisomers. Due to their higher acidity, phenolic hydroxyls are more active HBDs in the cycloaddition of CO2 to epoxides than aliphatic hydroxyls.199 Werner prepared bifunctional phenolic phosphonium salts (such as 10; see also Scheme 22) and applied them for the cycloaddition of CO2 to fatty acids. The carbonation of iEFA1 proceed quantitatively under significantly milder conditions than for aliphatic HBDs (entry 3) though without any stereocontrol.198 The same group reported the immobilization of 10 either on a traditional support such as silica (i.e., catalyst structure 11; see also Scheme 22),197 or on amorphous hydrogenated carbon coating through a plasma-assisted method leading to recyclable catalysts 21.300 The application of these heterogeneous compounds for the carbonation of iEFA1 at lower CO2 pressure compared to the homogeneous catalyst 10 led to low yields of iCFA1 (entries 4 and 5).
Dai et al. reported a multifunctional pincer-type organocatalyst (22) for the cycloaddition of CO2 to iEFA1. This catalyst bears several active functionalities such as a nucleophilic imidazolium iodide, a pyridine moiety, –NH and –OH (carboxylic and phenolic) HBDs groups.301 Higher loadings of 22 and TBAI were required to produce moderate yields of iCFA1 mostly as the trans-isomer.
Despite its excellent catalytic activity, compound 10 was prepared from the reaction between expensive and toxic (2-hydroxyphenyl)-diphenylphosphine (hazard: GSH07) and carcinogenic 1-bromopropane (hazard: GSH08). Similarly, the synthesis of 22 required 2,6-dibromopyridine as the starting material and a two-step CuI-mediated coupling process.301 In order to better exploit the generally assumed benefits of organocatalysis (lower catalyst cost and toxicity), the application of ubiquitous biobased compounds such as amino acids,302,303 peptides,304–306 sugars,307 and vitamins,308 would be highly attractive.309
In this context, L-ascorbic acid (14), a well-established bio-based HBD for the cycloaddition of heterocumulenes to epoxides under ambient or mild conditions,310,311 was applied in combination with TBAC for the cycloaddition of CO2 to iEFAs.231 Ascorbic acid was found to accelerate the quaternary ammonium salt-catalyzed cycloaddition of CO2 to iEFA1, with the binary combination 14/TBAC showing the best performance in terms of iCFA1 selectivity (see also Table 3). Under similar reaction conditions as reported for 22/TBAI apart from the longer reaction time (48 h), the use of a lower loading of both catalyst components of 14/TBAC led to quantitative iEFA1 conversion at 5 bar CO2 pressure at 100 °C (entry 7).
5.7 Catalytic performance of binary catalysts in the cycloaddition of CO2 to polyunsaturated EFAs
Several of the catalytic systems discussed before were also applied for the carbonation of bis-unsaturated epoxidized linoleic acid methyl ester iEFA2 (entries 1–10, Table 9). Generally, the reaction conditions optimized for the carbonation of iEFA1 highlighted in the previous sections could be successfully applied to the conversion of iEFA2, therefore there will not be any detailed discussion of each example. For systems operating under supercritical conditions (entries 1 and 2), good epoxide conversion rates and iCFA2 selectivities were obtained using alkali metals halides in combination with crown ethers.24 The best results were obtained using KI/[18]crown-6 (entry 2). Whereas other catalytic systems could afford the carbonation of iEFA2 under subcritical conditions (entries 3 and 4) or even at moderate CO2 pressures (entries 5–10), the mildest conditions were reported for the in situ generated complex between CaI2 and [18]crown-6 (4) combined with PPh3 (entry 9).118 It is worth noting that the combination of complex 1Cl and PPNCl performed also well in the carbonation of iEFA2 at slightly higher temperature providing a shorter reaction time and with high (chemo)selectivity for cis-10b (entry 7).187|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | iEFA | Cat/additivea (mol%) | T, p, t (°C, bar, h) | Conv.b (%) | Sel. For iCFAb | Yield (%) | Sel. (Cis:trans)c |
Ref. |
|
a The loading value in brackets is relative to the total amount of epoxides unit.
b Conversion and selectivity determined by 1H NMR.
c Overall Cis:trans ratio determined by 1H NMR from the integration of the corresponding signals of cis and trans carbonate units.
d THA = tetra-n-heptylammonium.
e Not reported.
f Conversion determined by 1H NMR and GC. Note that p is the partial pressure of CO2, 18-C-6 is short for 18-crown-6.
|
||||||||
| 1 | iEFA2 | THA-Cr-Si-POM (2.0)d | 100, 130, 6 | 85 | 50 | 212 | ||
| TBAB (2.0) | ||||||||
| 2 | iEFA2 | KI (5.0 wt%) | 100, 100, 17 | 90f | 97 | 24 | ||
| 18-C-6 (3.5 wt%) | ||||||||
| 3 | iEFA2 | 12 (1.0) | 100, 50, 48 | 99 | 85 | 196 | ||
| FeCl3 (0.13) | ||||||||
| 4 | iEFA2 | 15-Br (1.0) | 100, 50, 40 | 88 | 93 | 82 | 70:30 |
224 |
| MoO3 (0.13) | ||||||||
| 5 | iEFA2 | 10 (2.5) | 80, 25, 48 | >99 | >99 | 99 | 198 | |
| 6 | iEFA2 | 14 (0.75) | 100, 10, 48 | >99 | 94 | 85 | 91:9 |
231 |
| TBAC (1.5) | ||||||||
| 7 | iEFA2 | 1 Cl (0.30) | 70, 10, 24 | >99 | >99 | 97:3 |
187 | |
| PPNCl (5.0) | ||||||||
| 8 | iEFA2 | 22 (4.0) | 100, 5, 48 | 43 | 301 | |||
| TBAI (12) | ||||||||
| 9 | iEFA2 | 4 (10) | 45, 5, 48 | 90 | 118 | |||
| PPh3 (10) | ||||||||
| 10 | iEFA2 | 13 (1.0) | 100, 5, 24 | 84 | 16:84 |
230 | ||
| TBAI (10) | ||||||||
| 11 | iEFA3 | THA-Cr-Si-POM (2.0)d | 100, 130, 6 | 71 | 12 | 212 | ||
| TBAB (2.0) | ||||||||
| 12 | iEFA3 | 1Cl (0.20) | 70, 10, 24 | >99 | >99 | 68:32 |
187 | |
| PPNCl (5.0) | ||||||||
| 13 | iEFA3 | 1tBu (1.0) | 70, 10, 24 | 92 | >99 | 96:4 |
187 | |
| PPNCl (5.0) | ||||||||
| 14 | iEFA3 | 14 (1.5) | 80, 10, 48 | 94 | 93 | 75 | 93:7 |
231 |
| TBAC (5.0) | ||||||||
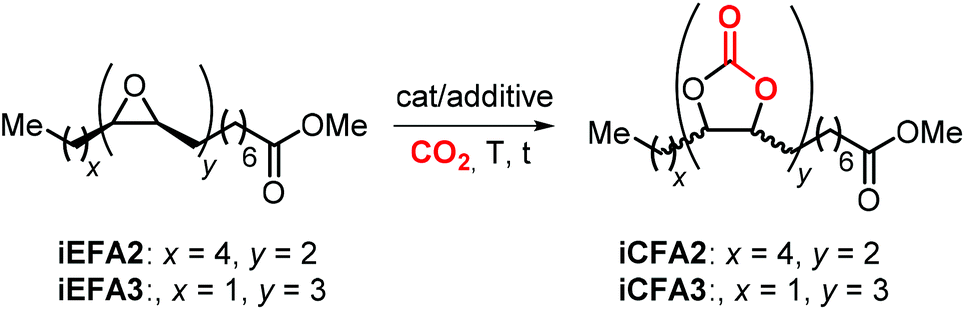
Finally, organocatalysts 10 (entry 5),198 and 14/TBAC (entry 6),231 also performed well under different reaction conditions, with 14 achieving a selectivity for cis-iCFA2 close to that observed with Al-based 1Cl.
Few catalytic systems have been reported to date that are able to mediate the cycloaddition of CO2 to tris-epoxide iEFA3 derived from triply unsaturated linolenic acid (entries 11–14). To note, under scCO2 conditions, the binary system THA-Cr-Si-POM/TBAB afforded only poor yields of iCFA3 because of a low chemoselectivity for the tris-carbonate product (entry 11).212 Under considerably milder conditions, the use of 1Cl/PPNCl provided iCFA3 with excellent conversion and carbonate selectivity with an approximate and overall 2:1 cis/trans isomer ratio (entry 12).187 However, excellent selectivity for all-cisiCFA3 in appreciable yield was achieved by replacing the chlorine for tert-butyl substituents in complex 1 (entry 13). The organocatalytic pair 14/TBAC achieved comparable results by applying similar reaction conditions but using a longer reaction time (48 h versus 24 h, entry 14).231
5.8 Catalytic performance of binary catalytic systems in the cycloaddition of CO2 to EVOs
Whereas the cycloaddition of CO2 to EVOs produced from several vegetable oils (soybean, linseed, olive, sunflower etc.) has been reported, here we mostly focus on the carbonation of ESBO as a benchmark substrate. The catalytic performance of various catalyst systems for the carbonation of EVOs is summarized and discussed in Table 10.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | EVO | Cat/additive (mol%) | T, p, t (°C, bar, h) | Yield (%) | Sel.a for CVO | Ref. |
| a Selectivity determined by 1H NMR. b THA: tetra-n-heptylammonium. c Conversion determined by 1H NMR. d Not reported. e Conversion determined by standard titration. f PEG DME 500: poly(ethyleneglycol) dimethyl ether with Mn ∼ 400 g mol−1. g Yield determined by 1H NMR. | ||||||
| 1 | Soybean | THA-Cr-Si-POM (2.0)b | 100, 130, 24 | 41c | 60 | 212 |
| TBAB (2.0) | ||||||
| 2 | Soybean | 23 (1.0) | 100, 100, 10 | 100 | 312 | |
| TBAB (1.0) | ||||||
| 3 | Soybean | KI (2.0) | 130, 60, 120 | 98e | 314 | |
| [18]crown-6 (1.0) | ||||||
| 4 | Soybean | 12 (2.0) | 100, 50, 24 | 94 | >99 | 196 |
| FeCl3 (0.25) | ||||||
| 5 | Soybean | 15-Br (2.0) | 100, 50, 20 | 89 | 90 | 224 |
| MoO3 (0.25) | ||||||
| 6 | Sunflower | CaI2 (5.0) | 90, 50, 120 | 83 | 282 | |
| PEG-DME-500 (5.0)f | ||||||
| 7 | Soybean | CaCl2 (5.0) | 140, 40, 40 | 98 | 315 | |
| TBAB (2.5) | ||||||
| 8 | Soybean | SnCl4·5H2O (1.0) | 140, 15, 30 | 99e | 316 | |
| TBAB (3.0) | ||||||
| 9 | Soybean | 10 (5.0) | 80, 25, 24 | 77 | 198 | |
| 10 | Soybean | 14 (1.5) | 100, 5, 48 | 81 | 89 | 231 |
| TBAC (5.0) | ||||||
| 11 | Soybean | 4 (5.0) | 45, 5, 24 | 81 | >99 | 118 |
| PPh3 (5.0) | ||||||
| 12 | Sunflower | 24a (4.0) | 100, 1, 30 | 99g | 320 | |
| TBAB (4.0) | ||||||
| 13 | Sunflower | 24b (4.0) | 100, 1, 30 | 8g | 320 | |
| TBAB (4.0) | ||||||
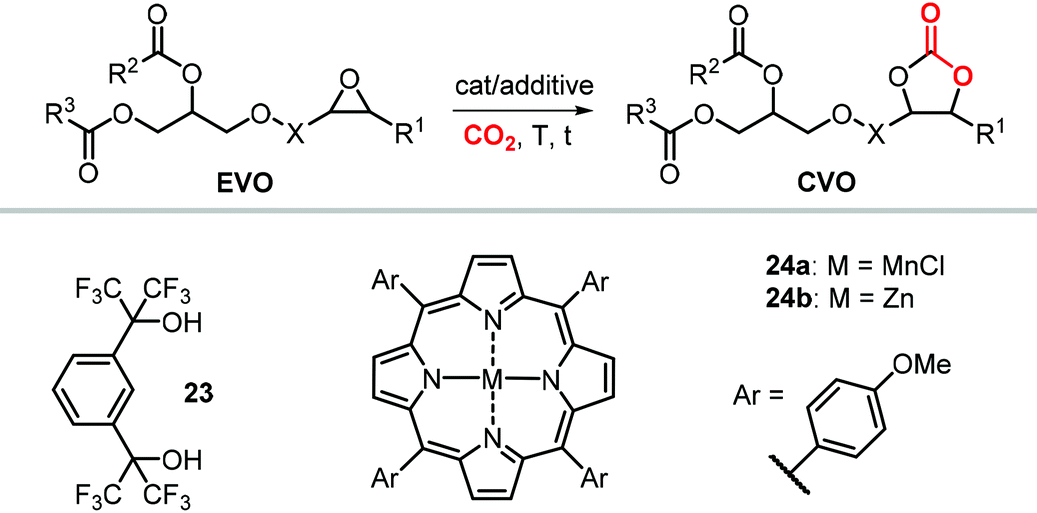
Several catalytic systems already highlighted in Tables 7–9 could be successfully applied, under optimized conditions, for the conversion of EVOs into CVOs (entries 1, 4–6, 9–11), although in the case of THA-Cr-Si-POM/TBAB only a relatively modest yield of carbonated product was observed (entry 1).212 Therefore, these examples will not be discussed in detail to avoid overlap with previous sections. A different system able to efficiently carbonate ESBO under supercritical conditions was developed by Jerome, Detrembleur et al. in a study targeting the preparation of NIPU foams from CVOs.312 The authors used TBAB in the presence of fluorinated HBD [1,3-bis(2-hydroxyhexafluoroisopropyl)benzene, 23],313 providing quantitative carbonation of ESBO in 10 h (entry 2).
Rokicki et al. reported the carbonation of ESBO at lower CO2 pressure than the previous example but a higher reaction temperature and longer reaction time (5 d) were needed employing KI/18-crown-6 as a catalyst. In this case, the CVO was obtained in high yield in the form of cis- and trans mixture (entry 3).314
Previously discussed coordination compounds (see also Table 6) displayed good performance in the carbonation of EVOs under relatively high pressure (entries 4–6, Table 10). A simple alkali salt (CaCl2) displayed a catalytic activity comparable to these aforementioned Lewis acid based systems for the carbonation of ESBO in the presence of TBAB albeit at a significantly higher reaction temperature (entry 7).315 In a related contribution, the same authors showed that CaCl2/TBAB can be used for the synthesis of carbonated soybean oil (CSBO) under a flow of atmospheric CO2 at 110 °C, although at the expense of the reaction time.254
A different, readily available and highly Lewis acidic compound, SnCl4·5H2O, used in combination with TBAB allowed for quantitative conversion of ESBO into CSBO under moderate CO2 pressure but at a high temperature (entry 8).316 Previously discussed organocatalytic systems 10 and 14/TBAC (entries 9 and 10) and coordination complex 4 combined with PPh3 (entry 11) had attractive catalytic activity for the synthesis of CSBO under milder temperatures and/or CO2 pressures with performances comparable to those observed for fatty acids (see Tables 6 and 8). Thus, it appears that the type of ester and the presence of saturated fatty acid chains in the starting material has a negligible impact on the reactivity of the epoxide moieties. As a testament for this hypothesis, 14/TBAC proved to be an efficient catalyst for the carbonation of epoxidized FAME.231
Finally, Lewis acidic metal complexes were tested for the carbonation of epoxidized sunflower oil (ESFO) under atmospheric pressure in the presence of TBAB. Metalloporphyrins are known as efficient catalysts for the cycloaddition of CO2 to epoxides under ambient pressure.1,317–319 Safari et al. showed that the Mn(II) metalloporphyrin complex 24a in the presence of TBAB permits full carbonation of ESFO under atmospheric pressure at 100 °C in 30 h, whereas the analogous Zn(II) complex 24b is inactive under identical conditions (entries 12 and 13).320
5.9 Recyclable catalytic systems for the carbonation of EFAs and EVOs
Whereas most of the highlighted catalytic systems are homogeneous, the development of recoverable and recyclable heterogeneous catalysts is crucial for easier purification of the products and for the sake of cost and sustainability, especially in the context of large-scale application. The development and communication of recyclable catalysts for the carbonation of fatty acids is rare.In the case of TBAB as homogeneous catalyst for the carbonation of ESBO, Doll and Erhan were able to recover TBAB from the products mixture by liquid–liquid extraction using water followed by freeze-drying (96% recovery rate). However, they did not report the catalyst reuse for the same reaction.237 Similarly, D'Elia et al. attempted to recover 14/TBAC from the crude reaction mixture containing iCFA1 by extraction with water.231 In this case, the recovered catalyst showed poor reusability as it converted only 38% of iEFA1 into iCFA1 without altering the chemoselectivity for the carbonate product.
Some homogeneous catalysts highlighted in the previous sections such as 20,29924a,320 CaI2/PEG-DME-500,282 and KI/hydroxyl-functionalized imidazoles,281 can be recovered after the synthesis of the respective CCs by methods such as column chromatography, product distillation and liquid–liquid extraction, and reused for the same cycloaddition reaction. However, their recyclability for the synthesis of CFAs and CVOs has not been specifically addressed. Catalyst separation protocols that require evaporation of large volumes of water or distillation of high-boiling carbonates are likely not convenient or sustainable for commercial exploitation. Along similar lines, Werner et al. prepared some heterogeneous, reusable catalysts such as 11,197 and 21,300 but their recyclability was not demonstrated for processes that focus on carbonated oleochemicals.
Bähr and Mülhaupt reported the application of silica-supported 4-pyrrolidinopyridinium iodide (previously developed by Motokura)321 as a heterogeneous catalyst for the carbonation of ESBO and epoxidized linseed oils (ELSO) with full conversion of the respective EVO realized at high temperature and moderate pressure (140 °C, 30 bar of CO2, 45 h).271 Whereas the catalyst could be easily recovered by simple filtration after the reaction and thus avoid product purification by liquid–liquid extraction, the performance of the recycled catalyst for a new run of carbonation was not reported.
As a rare example of a recyclable catalyst for the coupling of CO2 and epoxidized oleochemicals, Wang et al. reported a ZrO2-supported heteropolyacid (H3PW12O40/ZrO2) that was successfully applied for the carbonation of ESBO at high temperature (150 °C) and moderate CO2 pressure (10 bar) in the presence of DMF.322 Its catalytic performance was attributed to the synergy between the strong acidic metal centers of the heteropolyacid and the basic zirconia surface providing sites for CO2 adsorption and activation. Nonetheless, H3PW12O40/ZrO2 showed poor reuse features due to the strong adsorption of bulky reaction by-products at the active sites of the catalyst surface that could not be efficiently regenerated even after calcination.
To improve the regeneration of an active catalyst, the catalyst was modified by doping it with platinum (5%) via a co-impregnation approach. As expected, the addition of platinum increased the ability of the material to oxidize hydrocarbons, and the undesired adsorbed organic molecules could be removed below 300 °C.323 At the same time, the presence of platinum did not affect the efficiency of the carbonation reaction. The Pt-doped H3PW12O40/ZrO2 displayed significantly better recyclability despite the gradual decrease in catalytic activity upon reuse with the ESBO conversion dropping from 93 to 78% after four catalytic cycles. An obvious drawback of this catalytic system is the need for expensive noble metal dopant to achieve reusability.
A different way to generate internal carbonates from fatty acids was reported by Cádiz et al. by using heptanal, a product of the thermal cracking of castor oil, as a novel precursor for NIPU synthesis.324 The Horner–Wadsworth–Emmons reaction between heptanal and trimethyl phosphonoacetate yielded methyl 2-nonenoate that was further oxidized to achieve the corresponding 2,3-epoxynonanoic methyl ester (see Scheme 30) as a short-chain epoxidized fatty acid derivative.
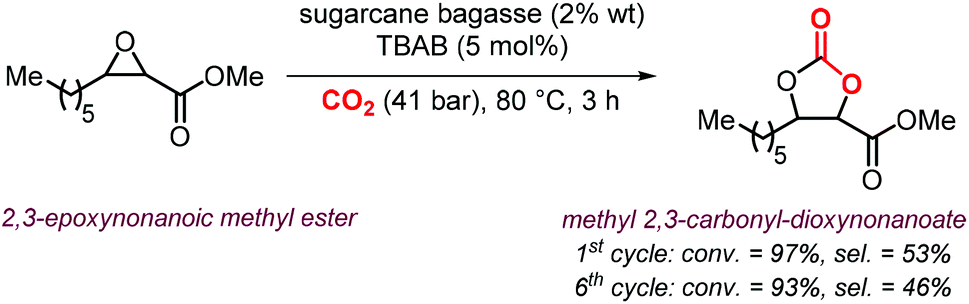 | ||
| Scheme 30 The coupling of 2,3-epoxynonanoic methyl ester with CO2 catalyzed by a recyclable sugarcane bagasse/TBAB binary catalyst. | ||
Interestingly, the carbonation of the latter compound can be performed by employing sugarcane bagasse as a heterogeneous HBD in combination with TBAB at 80 °C and about 41 bar of CO2 pressure (Scheme 30). The insolubility of sugarcane bagasse allows for easy separation from the crude reaction mixture by simple filtration. This catalyst component can be reused for at least six runs with only slight loss of activity. However, the chemoselectivity toward carbonate for this system was only moderate due to secondary reactions (such as hydrolysis) taking place involving the epoxy groups. Additionally, fresh TBAB, likely the most expensive catalyst component, had to be added in each consecutive cycle.
6. Conclusions and perspectives
This tutorial review demonstrates that the area of biobased carbonate synthesis has tremendously advanced over the last decade. Key to the success has been and will continue to be the development of suitable catalysts that, according to the principles of green chemistry and commercial applications, should preferentially be low-cost, readily available, scalable and sustainable in terms of their components. The incorporation of carbon dioxide in biobased feedstock such as terpenes, sugar-derived architectures, glycerol and fatty acids (including vegetable oils) offers a way to increase the application potential of low-value materials into high-value functional additives, solvents and polymer precursors for, inter alia, NIPUs.There are several aspects that still deserve attention. Most of the catalytic processes developed to date are operated with purified and single-component substrates, whereas larger scale commercially available feedstock are often mixtures containing impurities that may affect the stability, activity and reuse of the involved catalyst system. Therefore, it is important to further develop catalysts that are not only able to combine high (chemo)selectivity and sufficient activity, but are also compatible with less defined mixtures of waste streams such as the case for fatty acids that are available from the biodiesel industry. As catalyst cost is paramount for scale up, cheap(er) catalyst design is a crucial aspect and particularly when bulk chemical applications are foreseen for biocarbonates attained by integration of CO2 into biomolecules.
In the following sections we present a summary of key advances and future perspectives for each class of compounds.
6.1 Glycerol carbonate
Glycerol carbonate is a promising outlet for the valorization of waste glycerol from saponification reactions, and it is expected to represent a valuable bio-refinery product.325 The ideal route to prepare GC, i.e. the combination of two renewable substrates such as glycerol and CO2, is affected by thermodynamic limitations that lead to low GC yields under typically harsh reaction conditions. Such limitations can be partially eased by the use of dehydrating agents or by the presence of additional reaction components that, however, unavoidably negatively affect the overall sustainability, cost, and purification requirements of the product. In this context, the atom-economic cycloaddition of CO2 to glycidol appears as a convenient approach as it can be carried out under relatively mild conditions using molecular catalysts based on readily available organic compounds such as ascorbic acid. Moreover, recent advances in the synthesis of GC from glycidol and CO2via Payne rearrangement chemistry rather than the traditional cycloaddition mechanism,99 demonstrate that this reaction can be carried out using single-component and halogen-free systems. Therefore, the development of bio-based homogeneous and heterogeneous catalysts exploiting such a reaction manifold for the synthesis of GC is highly promising. To note, the sustainability of the glycidol-based approach can be increased by the implementation of green routes to produce the latter compound; for instance using 2-chloro-1,3-propanediol, a waste product of the Epicerol process, as the substrate,326 or from glycerol deoxydehydration affording allyl alcohol.3276.2 Terpene-derived carbonates
Terpene compounds have been used for a long time for the preparation of fragrances, flavors and pharmaceuticals. Some terpenes, such as pinene, carvone, myrcene and limonene are currently obtained from turpentine oil, paper pulping process and extraction from citrus fruits.328 Such terpenes are produced on million tons per year, and have been also proposed for the production of biofuels.329 Interesting, terpene-based cyclic carbonates have been isolated from natural sources with some of them showing biological activity. In comparison with other biobased feedstock, terpenes present an impressive structural diversity, which offers the possibility to produce complex cyclic carbonate structures by relatively easy transformations (i.e., oxidation followed by coupling with CO2). Consequently, the synthesis of terpene-based CCs has gained momentum, and future investigations could lead to the discovery of compounds with promising pharmaceutical activity. However, most of the current reports focus on limonene- and pinene-derived carbonates. Thus, future investigations are required to expand the portfolio of terpene-based carbonates. To date, different catalytic methodologies for the coupling of CO2 with terpene oxides have been reported, with in several cases significant byproduct formation when more complex substrates were employed. This clearly calls for further development of more efficient and selective catalytic systems.In recent years, the use of terpene oxides in the field of sustainable polymer chemistry has also emerged.110 In particular, polycarbonates such as poly(limonene carbonate) and poly(menth-2-ene carbonate) have been obtained by direct coupling with CO2.111,330 Unfortunately, up to now these reactions can only be promoted by two types of catalytic systems. Alternatively, polycarbonates can be obtained via ROP of reactive cyclic carbonates in the presence of simple catalytic systems.331,332 We believe that further investigations should focus on the discovery of terpene-based cyclic carbonates that will serve as monomers for the production of polycarbonates by easy-to-tune ROP protocols. This will offer an alternative strategy for the production of polymeric materials with attractive properties.
6.3 Sugar-derived carbonates
Thanks to the ubiquity of sugars in nature, this class of compounds has been recognized as an attractive sustainable feedstock. However, in order to avoid competition with the food processing industry, a lot of research is still needed that should focus on sugar-based biofuels, chemicals and polymers obtained from widely-available cellulose and lignocellulose.333–335 Sugar structures are characterized by the presence of hydroxyl groups and, consequently, their transformation into cyclic carbonates has been mainly conducted by well-known stoichiometric procedures in the presence of non-ideal reactants such as phosgene. During the last few years, alternative methodologies have been reported for the carbonation of sugars under milder conditions. In general, these procedures are based on the reaction of low pressure carbon dioxide (1–10 bar) with an in situ formed activated alcohol species, followed by cyclization promoted by elimination of a suitable leaving group (e.g., OTs and OMs). These reactions do avoid the use of phosgene-related compounds, though they still require the presence of stoichiometric amounts of base (e.g., DBU and Et3N) and halogenated reactants (e.g., TsCl and CH2Br2). Future development of catalytic methodologies are likely needed to improve the potential application and scale up of this synthetic process. Interestingly, the possibility to control the stereochemistry during the synthesis of sugar-based CCs has also been demonstrated. This aspect is highly relevant, especially with respect to the use of such carbonates as monomers in polycarbonate synthesis. Indeed, the stereochemistry of bicyclic carbonates strongly influences their reactivity during the polymerization process, and the possibility to compare compounds with different stereochemistry will offer the possibility to further investigate structure–reactivity relationships. In addition, phosgene-free routes for sugar-based carbonates render these molecules as an attracting platform for the synthesis of biobased NIPUs.6.4 Fatty acid and vegetable oil-based carbonates
The attractive aspects of these feedstock is that they are available in large volumes either as-produced or from the recovery of spent cooking oils. Carbonated vegetable oils are versatile substrates for the preparation of NIPUs via aminolysis reactions (i.e., amine-promoted ring-opening of the cyclic carbonate). With NIPUs being regarded as ideal green polymers for the replacement of phosgene-based PUs, it is expected that the demand for these types of biocarbonates will considerably increase in the near future when current process issues such as low molecular weights, low aminolysis rates and side reactions can be adequately solved.336 Additionally we foresee that carbonated fatty acids could be suitable substrates for the preparation of additives to improve the anti-wear properties of group II base oils.337In the last decade, the synthesis of fatty acid/vegetable oil based CCs has been characterized by a remarkable development of several binary homogeneous systems able to catalyze the carbonation reactions of oils under practical reaction conditions (45–100 °C, 5–10 bar). This is especially relevant when comparing to the exclusive use of quaternary ammonium or phosphonium salts and, often, binary systems provide better/improved control of the stereochemistry of the final products. As a drawback, and due to the low reactivity of the substrates, long reaction times (24–48 h) are generally required to reach high degrees of conversion (>80%). Therefore, substantial improvement of the performance of homogeneous (binary) catalysts for the carbonation of vegetable oil-derived epoxides is a future requisite. It can be anticipated that a substantial increase in the demand for carbonated vegetable oils should be met by the development of heterogeneous, or at least, recyclable catalysts with powerful reactivity profiles. As discussed in section 5.8, there is limited knowledge on the development of heterogeneous catalysts for the carbonation of epoxidized fatty acids, and up to now no viable catalyst has been developed for application in large-scale production. This may be related to the more challenging nature of internal epoxide conversion in the presence of CO2 mediated by heterogeneous catalysts, making the latter much less efficient than their homogeneous counterparts.338 A different and promising approach to the development of recoverable and reusable catalysts for the carbonation of epoxidized fatty acids was recently proposed by Duguet et al. by using a thermomorphic polyethylene-supported catalyst.339 In this latter example, the polymeric catalyst is soluble in the reaction mixture (T = 100 °C) but insoluble in the product at room temperature. Therefore, it can essentially be recovered by filtration and its recyclability for the carbonation of epoxidized methyl oleate was indeed demonstrated. Whereas such polymeric catalysts are still limited by relatively modest molecular weights (∼1000 g mol−1) and low product stereoselectivity, the concept of using a catalyst that can be separated from the carbonated fatty acid after reaction is a promising molecular approach and could be further developed in the future.
An interesting direction for all the categories of biobased substrates presented in this account could be to merge flow catalysis approaches with biocarbonate synthesis, with catalyst recycling and a continuous operation mode helping to increase the overall sustainability. Parallel flow catalysis may help to scale up the preparation of any desired target, but market demands will likely determine which type of process will be preferred to create critical amounts of the biocarbonate for eventual (commercial) use.
From the diversity and functionality of biobased structures that can be accessed via catalytic process, and the prospect of biocarbonates in various academic and commercial applications, a bright future is ahead of these CO2 based cyclic carbonates.
Conflicts of interest
There are no conflicts to declare by the authors.Acknowledgements
AWK thanks the CERCA Program/Generalitat de Catalunya, ICREA, the Spanish MINECO (CTQ2017-88920-P) and AGAUR (2017-SGR-232) for financial support. We also thank the Ministerio de Ciencia e Innovación for support through Severo Ochoa Excellence Accreditation 2020–2023 (CEX2019-000925-S, MIC/AEI). BL and FDM thank the European Community for Marie Curie individual fellowships (PHOTOCARBOX grant agreement 889754, and SUPREME grant agreement 840557). V. D. E. thanks the Thailand Research Fund (Grant No. RSA6080059) for funding.Notes and references
- R. Rajjak Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- T. K. Pal, D. De and P. K. Bharadwaj, Coord. Chem. Rev., 2020, 408, 213173 CrossRef CAS.
- F. Della Monica, A. Buonerba and C. Capacchione, Adv. Synth. Catal., 2019, 361, 265–282 CrossRef CAS.
- L. Tao, T. S. Choksi, W. Liu and J. Pérez-Ramírez, ChemSusChem, 2020, 13, 6066–6089 CAS.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- D. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed.
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489–4491 RSC.
- Y. Qin, H. Guo, X. Sheng, X. Wang and F. Wang, Green Chem., 2015, 17, 2853–2858 RSC.
- W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem. – Eur. J., 2010, 16, 6828–6843 CrossRef CAS PubMed.
- F. Della Monica, S. V. C. Vummaleti, A. Buonerba, A. De Nisi, M. Monari, S. Milione, A. Grassi, L. Cavallo and C. Capacchione, Adv. Synth. Catal., 2016, 358, 3231–3243 CrossRef.
- J. Rintjema, R. Epping, G. Fiorani, E. Martín, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2016, 55, 3972–3976 CrossRef CAS PubMed.
- B. Limburg, À. Cristòfol, F. Della Monica and A. W. Kleij, ChemSusChem, 2020, 13, 6056–6065 CAS.
- C. Qiao, A. Villar-Yanez, J. Sprachmann, B. Limburg, C. Bo and A. W. Kleij, Angew. Chem., Int. Ed., 2020, 59, 18446–18451 CrossRef CAS PubMed.
- P. Furtwengler and L. Avérous, Sci. Rep., 2018, 8, 9134 CrossRef PubMed.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- R. P. Tiger and E. M. Gotlib, Polym. Sci., Ser. D, 2017, 10, 9–12 CrossRef CAS.
- B. Schäffner, M. Blug, D. Kruse, M. Polyakov, A. Köckritz, A. Martin, P. Rajagopalan, U. Bentrup, A. Brückner, S. Jung, D. Agar, B. Rüngeler, A. Pfennig, K. Müller, W. Arlt, B. Woldt, M. Graß and S. Buchholz, ChemSusChem, 2014, 7, 1133–1139 CrossRef PubMed.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- W. Guo, J. E. Gómez, À. Cristòfol, J. Xie and A. W. Kleij, Angew. Chem., Int. Ed., 2018, 57, 13735–13747 CrossRef CAS PubMed.
- B. D. W. Allen, C. P. Lakeland and J. P. A. Harrity, Chem. – Eur. J., 2017, 23, 13830–13857 CrossRef CAS PubMed.
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”), Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 1997 Search PubMed.
- Review of the EU Bioeconomy Strategy and its Action Plan, Chair: Alice Newton, European Commission, Brussels, 2017.
- F. Della Monica and A. W. Kleij, Catal. Sci. Technol., 2020, 10, 3483–3501 RSC.
- J. A. Kenar, Lipid Technol., 2007, 19, 249–253 CrossRef CAS.
- C.-H. C. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. M. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- F. Jérôme and J. Barrault, in Green Polymerization Methods, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 57–87 Search PubMed.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, C. Ramírez-López and M. Belsué, Org. Process Res. Dev., 2012, 16, 389–399 CrossRef.
- M. O. Sonnati, S. Amigoni, E. P. T. de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- A. Galadima and O. Muraza, Waste Biomass Valorization, 2016, 8, 141–152 CrossRef.
- N. Kindermann, T. Jose and A. W. Kleij, Top. Curr. Chem., 2017, 375, 1–28 CrossRef CAS PubMed.
- G. M. Lari, G. Pastore, M. Haus, Y. Ding, S. Papadokonstantakis, C. Mondelli and J. Pérez-Ramírez, Energy Environ. Sci., 2018, 11, 1012–1029 RSC.
- P. de Caro, M. Bandres, M. Urrutigoïty, C. Cecutti and S. Thiebaud-Roux, Front. Chem., 2019, 7, 1–13 CrossRef PubMed.
- S. Christy, A. Noschese, M. Lomelí-Rodriguez, N. Greeves and J. A. Lopez-Sanchez, Curr. Opin. Green Sust., 2018, 14, 99–107 CrossRef.
- G. Fiorani, A. Perosa and M. Selva, Green Chem., 2018, 20, 288–322 RSC.
- M. Szőri, B. R. Giri, Z. Wang, A. E. Dawood, B. Viskolcz and A. Farooq, Sustainable Energy Fuels, 2018, 2, 2171–2178 RSC.
- P. G. Parzuchowski, A. Świderska, M. Roguszewska, T. Frączkowski and M. Tryznowski, Polymer, 2018, 151, 250–260 CrossRef CAS.
- W. K. Teng, G. C. Ngoh, R. Yusoff and M. K. Aroua, Energy Convers. Manage., 2014, 88, 484–497 CrossRef CAS.
- K. Shukla and V. C. Srivastava, Catal. Rev., 2017, 59, 1–43 CrossRef CAS.
- K. Kim and E. Lee, Energies, 2017, 10(1790), 1–15 Search PubMed.
- H. Zhang, H. Li, A. Wang, C. C. Xu and S. Yang, Adv. Polym. Technol., 2020, 2020, 1–17 Search PubMed.
- A. O. Esan, A. D. Adeyemi and S. Ganesan, J. Cleaner Prod., 2020, 257, 1–21 CrossRef.
- C. Vieville, J. Yoo, S. Pelet and Z. Mouloungui, Catal. Lett., 1998, 56, 245–247 CrossRef CAS.
- M. Aresta, A. Dibenedetto, F. Nocito and C. Pastore, J. Mol. Catal. A: Chem., 2006, 257, 149–153 CrossRef CAS.
- J. George, Y. Patel, S. M. Pillai and P. Munshi, J. Mol. Catal. A: Chem., 2009, 304, 1–7 CrossRef CAS.
- A. Dibenedetto, A. Angelini, M. Aresta, J. Ethiraj, C. Fragale and F. Nocito, Tetrahedron, 2011, 67, 1308–1313 CrossRef CAS.
- H. Li, D. Gao, P. Gao, F. Wang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Sci. Technol., 2013, 3, 2801–2809 RSC.
- L. P. Ozorio and C. J. A. Mota, ChemPhysChem, 2017, 18, 3260–3265 CrossRef CAS PubMed.
- Q. Zhang, H.-Y. Yuan, X.-T. Lin, N. Fukaya, T. Fujitani, K. Sato and J.-C. Choi, Green Chem., 2020, 22, 4231–4239 RSC.
- J. Liu, Y. Li, H. Liu and D. He, Biomass Bioenergy, 2018, 118, 74–83 CrossRef CAS.
- N. A. Razali, M. Conte and J. McGregor, Catal. Lett., 2019, 149, 1403–1414 CrossRef CAS.
- J. Zhang and D. He, J. Colloid Interface Sci., 2014, 419, 31–38 CrossRef CAS PubMed.
- J. Liu, Y. Li, H. Liu and D. He, Appl. Catal., B, 2019, 244, 836–843 CrossRef CAS.
- L. P. Ozorio, R. Pianzolli, L. da Cruz Machado, J. L. Miranda, C. C. Turci, A. C. Guerra, E. F. Souza-Aguiar and C. J. Mota, Appl. Catal., A, 2015, 504, 187–191 CrossRef CAS.
- H. Li, X. Jiao, L. Li, N. Zhao, F. Xiao, W. Wei, Y. Sun and B. Zhang, Catal. Sci. Technol., 2015, 5, 989–1005 RSC.
- H. Li, C. Xin, X. Jiao, N. Zhao, F. Xiao, L. Li, W. Wei and Y. Sun, J. Mol. Catal. A: Chem., 2015, 402, 71–78 CrossRef CAS.
- J. Liu, Y. Li, J. Zhang and D. He, Appl. Catal., A, 2016, 513, 9–18 CrossRef CAS.
- X. Su, W. Lin, H. Cheng, C. Zhang, Y. Wang, X. Yu, Z. Wu and F. Zhao, Green Chem., 2017, 19, 1775–1781 RSC.
- J. Ma, J. Song, H. Liu, J. Liu, Z. Zhang, T. Jiang, H. Fan and B. Han, Green Chem., 2012, 14, 1743–1748 RSC.
- X. Song, Y. Wu, D. Pan, J. Zhang, S. Xu, L. Gao, R. Wei and G. Xiao, J. CO2 Util., 2018, 28, 326–334 CrossRef CAS.
- Z.-H. Zhou, Q.-W. Song and L.-N. He, ACS Omega, 2017, 2, 337–345 CrossRef CAS PubMed.
- J.-Y. Li, L.-H. Han, Q.-C. Xu, Q.-W. Song, P. Liu and K. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 3378–3388 CrossRef CAS.
- H. Zhou, H. Zhang, S. Mu, W.-Z. Zhang, W.-M. Ren and X.-B. Lu, Green Chem., 2019, 21, 6335–6341 RSC.
- L.-H. Han, J.-Y. Li, Q.-W. Song, K. Zhang, Q.-X. Zhang, X.-F. Sun and P. Liu, Chin. Chem. Lett., 2020, 31, 341–344 CrossRef CAS.
- Y. N. Lim, C. Lee and H.-Y. Jang, Eur. J. Org. Chem., 2014, 1823–1826 CrossRef CAS.
- M. Mihara, K. Moroga, T. Iwasawa, T. Nakai, T. Ito, T. Ohno and T. Mizuno, Synlett, 2018, 1759–1764 CrossRef CAS.
- D. Cespi, R. Cucciniello, M. Ricciardi, C. Capacchione, I. Vassura, F. Passarini and A. Proto, Green Chem., 2016, 18, 4559–4570 RSC.
- F. Della Monica, A. Buonerba, A. Grassi, C. Capacchione and S. Milione, ChemSusChem, 2016, 9, 3457–3464 CrossRef CAS PubMed.
- R. Huang, J. Rintjema, J. González-Fabra, E. Martín, E. C. Escudero-Adán, C. Bo, A. Urakawa and A. W. Kleij, Nat. Catal., 2018, 2, 62–70 CrossRef.
- U. R. Seo and Y. K. Chung, Adv. Synth. Catal., 2014, 356, 1955–1961 CrossRef CAS.
- S. M. Sadeghzadeh, Green Chem., 2015, 17, 3059–3066 RSC.
- S. M. Sadeghzadeh, Res. Chem. Intermed., 2015, 42, 2317–2328 CrossRef.
- A. H. Jadhav, G. M. Thorat, K. Lee, A. C. Lim, H. Kang and J. G. Seo, Catal. Today, 2016, 265, 56–67 CrossRef CAS.
- T. Jin, F. Dong, Y. Liu and Y. L. Hu, New J. Chem., 2019, 43, 2583–2590 RSC.
- P. Zhang and R. Zhiani, Catal. Lett., 2020, 150, 2254–2266 CrossRef CAS.
- Y. Zhang, G. Chen, L. Wu, K. Liu, H. Zhong, Z. Long, M. Tong, Z. Yang and S. Dai, Chem. Commun., 2020, 56, 3309–3312 RSC.
- M. Ding and H.-L. Jiang, ACS Catal., 2018, 8, 3194–3201 CrossRef CAS.
- S. Ghosh, P. Bhanja, N. Salam, R. Khatun, A. Bhaumik and S. M. Islam, Catal. Today, 2018, 309, 253–262 CrossRef CAS.
- S. Ghosh, P. Mondal, D. Das, K. Tuhina and S. M. Islam, J. Organomet. Chem., 2018, 866, 1–12 CrossRef CAS.
- S. Roy, B. Banerjee, A. Bhaumik and S. M. Islam, RSC Adv., 2016, 6, 31153–31160 RSC.
- J. Martínez, J. A. Castro-Osma, C. Alonso-Moreno, A. Rodríguez-Diéguez, M. North, A. Otero and A. Lara-Sánchez, ChemSusChem, 2016, 10, 1175–1185 CrossRef PubMed.
- F. de la Cruz-Martínez, J. Martínez, M. A. Gaona, J. Fernández-Baeza, L. F. Sánchez-Barba, A. M. Rodríguez, J. A. Castro-Osma, A. Otero and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2018, 6, 5322–5332 CrossRef.
- J. Martínez, F. de la Cruz-Martínez, M. A. Gaona, E. Pinilla-Peñalver, J. Fernández-Baeza, A. M. Rodríguez, J. A. Castro-Osma, A. Otero and A. Lara-Sánchez, Inorg. Chem., 2019, 58, 3396–3408 CrossRef PubMed.
- X.-D. Lang, Y.-C. Yu and L.-N. He, J. Mol. Catal. A: Chem., 2016, 420, 208–215 CrossRef CAS.
- M. North, P. Villuendas and C. Young, Tetrahedron Lett., 2012, 53, 2736–2740 CrossRef CAS.
- J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, J. A. Castro-Osma, A. Lara-Sánchez and A. Otero, ChemSusChem, 2017, 10, 2886–2890 CrossRef PubMed.
- M. Navarro, L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, I. Fernández, A. Lara-Sánchez and A. M. Rodríguez, Catal. Sci. Technol., 2020, 10, 3265–3278 RSC.
- M. H. Anthofer, M. E. Wilhelm, M. Cokoja, M. Drees, W. A. Herrmann and F. E. Kühn, ChemCatChem, 2014, 7, 94–98 CrossRef.
- J. A. Castro-Osma, J. Martínez, F. de la Cruz-Martínez, M. P. Caballero, J. Fernández-Baeza, J. Rodríguez-López, A. Otero, A. Lara-Sánchez and J. Tejeda, Catal. Sci. Technol., 2018, 8, 1981–1987 RSC.
- M. Hong, Y. Kim, H. Kim, H. J. Cho, M.-H. Baik and Y. Kim, J. Org. Chem., 2018, 83, 9370–9380 CrossRef CAS PubMed.
- N. Yadav, F. Seidi, S. Del Gobbo, V. D'Elia and D. Crespy, Polym. Chem., 2019, 10, 3571–3584 RSC.
- Y.-Y. Zhang, G.-W. Yang, R. Xie, L. Yang, B. Li and G.-P. Wu, Angew. Chem., Int. Ed., 2020, 59, 23291–23298 CrossRef CAS PubMed.
- S. Sopeña, M. Cozzolino, C. Maquilón, E. C. Escudero-Adán, M. Martínez Belmonte and A. W. Kleij, Angew. Chem., Int. Ed., 2018, 57, 11203–11207 CrossRef PubMed.
- G. D. Brown, Phytochemistry, 1994, 35, 425–433 CrossRef CAS.
- H. Zhang, H.-B. Liu and J.-M. Yue, Chem. Rev., 2014, 114, 883–898 CrossRef CAS PubMed.
- Q. Shi, S. Lu, D. Li, J. Lu, L. Zhou and M. Qiu, Fitoterapia, 2020, 145, 104635 CrossRef CAS PubMed.
- L. Z. Li, P. Y. Gao, Y. Peng, L. H. Wang and S. Song, Chem. Nat. Compd., 2010, 46, 380–382 CrossRef CAS.
- F. A. Macias, A. M. Simonet and J. C. G. Galindo, J. Chem. Ecol., 1997, 23, 1781–1803 CrossRef CAS.
- J. Luo, Y. Li, J. S. Wang, J. Lu, X. B. Wang, J. G. Luo and L. Y. Kong, Chem. Pharm. Bull., 2012, 60, 195–204 CrossRef CAS PubMed.
- Terpenes: Flavors, Fragrances, Pharmaca, Pheromones, ed. E. Breitmaier, Wiley-VCH, Weinheim, 2006 Search PubMed.
- M. Touaibia, C. Boutekedjiret, S. Perino and F. Chemat, Natural Terpenes as Building Blocks for Green Chemistry, in Plant Based “Green Chemistry 2.0”. Green Chemistry and Sustainable Technology, ed. Y. Li and F. Chemat, Springer, Singapore, 2019 Search PubMed.
- G. Paggiola, S. Van Stempvoort, J. Bustamante, J. M. Vega Barbero, A. J. Hunt and J. H. Clark, Biofuels, Bioprod. Biorefin., 2016, 10, 686–698 CrossRef CAS.
- R. Ciriminna, M. Lomeli-Rodriguez, P. Demma Carà, J. A. Lopez-Sanchez and M. Pagliaro, Chem. Commun., 2014, 50, 15288–15296 RSC.
- F. Della Monica and A. W. Kleij, Polym. Chem., 2020, 11, 5109–5127 RSC.
- N. Kindermann, À. Cristòfol and A. W. Kleij, ACS Catal., 2017, 7, 3860–3863 CrossRef CAS.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed.
- C. Li, R. J. Sablong and C. E. Koning, Angew. Chem., Int. Ed., 2016, 55, 11572–15576 CrossRef CAS PubMed.
- L. Peña Carrodeguas, J. González-Fabra, F. Castro-Gómez, C. Bo and A. W. Kleij, Chem. – Eur. J., 2015, 21, 6115–6122 CrossRef PubMed.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- G. Fiorani, M. Stuck, C. Martín, M. Martínez Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, ChemSusChem, 2016, 9, 1304–1311 CrossRef CAS PubMed.
- C.-Y. Li, Y.-C. Su, C.-H. Lin, H.-Y. Huang, C.-Y. Tsai, T.-Y. Lee and B.-T. Ko, Dalton Trans., 2017, 46, 15399–15406 RSC.
- L. Longwitz, J. Steinbauer, A. Spannenberg and T. Werner, ACS Catal., 2018, 8, 665–672 CrossRef CAS.
- F. de la Cruz-Martínez, M. Martínez, de S. Buchaca, J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Rodríguez-Diéguez, J. A. Castro-Osma and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2019, 7, 20126–20138 CrossRef.
- J. Fernández-Baeza, L. F. Sánchez-Barba, A. Lara-Sánchez, S. Sobrino, J. Martínez-Ferrer, A. Garcés, M. Navarro and A. M. Rodríguez, Inorg. Chem., 2020, 59, 12422–12430 CrossRef PubMed.
- V. Aomchad, S. Del Gobbo, P. Yingcharoen, A. Poater and V. D'Elia, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.01.021.
- R. P. Thummel and B. Rickborn, J. Org. Chem., 1971, 36, 1360–1365 CrossRef.
- M. J. Södergren, S. K. Bertilsson and P. G. Andersson, J. Am. Chem. Soc., 2000, 122, 6610–6618 CrossRef.
- J. R. Lamb, Y. Jung and G. W. Coates, Org. Chem. Front., 2015, 2, 346–349 RSC.
- J. R. Lamb, M. Mulzer, A. M. LaPointe and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 15049–15054 CrossRef CAS PubMed.
- C. Maquilón, B. Limburg, V. Laserna, D. Garay-Ruiz, J. González-Fabra, C. Bo., M. Martínez Belmonte, E. C. Escudero-Adán and A. W. Kleij, Organometallics, 2020, 39, 1642–1651 CrossRef.
- H. Morikawa, M. Minamoto, Y. Gorou, J.-I. Yamaguchi, H. Morinaga and S. Motokucho, Bull. Chem. Soc. Jpn., 2018, 91, 92–94 CrossRef CAS.
- H. Morikawa, J.-i. Yamaguchi, S.-I. Sugimura, M. Minamoto, Y. Gorou, H. Morinaga and S. Motokucho, Beilstein J. Org. Chem., 2019, 15, 130–136 CrossRef CAS PubMed.
- A. Rehman, A. M. López Fernández, M. F. M. Gunam Resul and A. Harvey, J. CO2 Util., 2019, 29, 126–133 CrossRef CAS.
- J. Guan, Y. Song, Y. Lin, X. Yin, M. Zuo, Y. Zhao, X. Tao and Q. Zheng, Ind. Eng. Chem. Res., 2011, 50, 6517–6527 CrossRef CAS.
- S. Wendels and L. Avérous, Bioact. Mater., 2021, 6, 1083–1106 CrossRef CAS PubMed.
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- V. Schimpf, B. S. Ritter, P. Weis, K. Parison and R. Mülhaupt, Macromolecules, 2017, 50, 944–955 CrossRef CAS.
- K. A. Maltby, M. Hutchby, P. Plucinski, M. G. Davidson and U. Hintermair, Chem. – Eur. J., 2020, 26, 7405–7415 CrossRef CAS.
- K. Kamata, K. Sugahara, K. Yonehara, R. Ishimoto and N. Mizuno, Chem. – Eur. J., 2011, 17, 7549–7559 CrossRef CAS PubMed.
- A. T. Lonnecker, Y. H. Lim and K. L. Wooley, ACS Macro Lett., 2017, 6, 748–753 CrossRef CAS.
- Y. Song, X. Ji, M. Dong, R. Li, Y. N. Lin, H. Wang and K. L. Wooley, J. Am. Chem. Soc., 2018, 140, 16053–16057 CrossRef CAS PubMed.
- S. E. Felder, M. J. Redding, A. Noel, S. M. Grayson and K. L. Wooley, Macromolecules, 2018, 51, 1787–1797 CrossRef CAS.
- W. M. Doane, B. S. Shasha, E. I. Stout, C. R. Russell and C. E. Rist, Carbohydr. Res., 1967, 4, 445–451 CrossRef CAS.
- Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 2799–2802 CrossRef CAS.
- R. Kumar, W. Gao and R. A. Gross, Macromolecules, 2002, 35, 6835–6844 CrossRef CAS.
- K. Mikami, A. T. Lonnecker, T. P. Gustafson, N. F. Zinnel, P. J. Pai, D. H. Russell and K. L. Wooley, J. Am. Chem. Soc., 2013, 135, 6826–6829 CrossRef CAS PubMed.
- O. Haba, H. Tomizuka and T. Endo, Macromolecules, 2005, 38, 3562–3563 CrossRef CAS.
- G. L. Gregory, E. M. Lopez-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC.
- J. A. Stewart, R. Drexel, B. Arstad, E. Reubsaet, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2016, 18, 1605–1618 RSC.
- P. Brignou, M. Priebe Gil, O. Casagrande, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2010, 43, 8007–8017 CrossRef CAS.
- D. M. Pearson, N. R. Conley and R. M. Waymouth, Adv. Synth. Catal., 2011, 353, 3007–3013 CrossRef CAS.
- G. L. Gregory, M. Ulmann and A. Buchard, RSC Adv., 2015, 5, 39404–39408 RSC.
- T. M. McGuire, E. M. López-Vidal, G. L. Gregory and A. Buchard, J. CO2 Util., 2018, 27, 283–288 CrossRef CAS.
- G. L. Gregory, L. M. Jenisch, B. Charles, G. Kociok-Köhn and A. Buchard, Macromolecules, 2016, 49, 7165–7169 CrossRef CAS.
- K. Tezuka, K. Koda, H. Katagiri and O. Haba, Polym. Bull., 2015, 72, 615–626 CrossRef CAS.
- M. Suzuki, T. Sekido, S. I. Matsuoka and K. Takagi, Biomacromolecules, 2011, 12, 1449–1459 CrossRef CAS.
- G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2017, 8, 2093–2104 RSC.
- G. L. Gregory, E. M. Hierons, G. Kociok-Köhn, R. I. Sharma and A. Buchard, Polym. Chem., 2017, 8, 1714–1721 RSC.
- E. M. López-Vidal, G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2018, 9, 1577–1582 RSC.
- D. Pati, X. Feng, N. Hadjichristidis and Y. Gnanou, Macromolecules, 2017, 50, 1362–1370 CrossRef CAS.
- D. Pati, X. Feng, N. Hadjichristidis and Y. Gnanou, J. CO2 Util., 2018, 24, 564–571 CrossRef CAS.
- D. Pati, Z. Chen, X. Feng, N. Hadjichristidis and Y. Gnanou, Polym. Chem., 2017, 8, 2640–2646 RSC.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- Ó. Ögmundarson, M. J. Herrgård, J. Forster, M. Z. Hauschild and P. Fantke, Nat. Sustainable, 2020, 3, 167–174 CrossRef.
- F. Schipfer, L. Kranzl, D. Leclère, L. Sylvain, N. Forsell and H. Valin, Biomass Bioenergy, 2017, 96, 19–27 CrossRef.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- X. Xiong, I. K. M. Yu, D. C. W. Tsang, N. S. Bolan, Y. Sik Ok, A. D. Igalavithana, M. B. Kirkham, K.-H. Kim and K. Vikrant, Chem. Eng. J., 2019, 375, 121983 CrossRef CAS.
- M. Yabushita, H. Kobayashi and A. Fukuoka, Appl. Catal., B, 2014, 145, 1–9 CrossRef CAS.
- J. Goldemberg, Science, 2007, 315, 808–810 CrossRef CAS PubMed.
- G. Paggiola, S. V. Stempvoort, J. Bustamante, J. M. V. Barbero, A. J. Hunt and J. H. Clark, Biofuels, Bioprod. Biorefin., 2016, 10, 686–698 CrossRef CAS.
- S. M. Danov, O. A. Kazantsev, A. L. Esipovich, A. S. Belousov, A. E. Rogozhin and E. A. Kanakov, Catal. Sci. Technol., 2017, 7, 3659–3675 RSC.
- Oilseeds and oilseed products, in OECD-FAO Agricultural Outlook 2018–2027, OECD Publishing, Paris/Food and Agriculture Organization of the United Nations, Rome, 2018 Search PubMed.
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- J. A. Kenar, B. R. Moser and G. R. List, Naturally Occurring Fatty Acids: Source, chemistry, and uses, in Fatty Acids: Chemistry, Synthesis, and Applications, ed. M. U. Ahmad, Elsevier, Amsterdam, 2017 Search PubMed.
- J. F. Sierra-Cantor and C. A. Guerrero-Fajardo, Renewable Sustainable Energy Rev., 2017, 72, 774–790 CrossRef CAS.
- P. Rouge, K. C. Szeto, Y. Bouhoute, N. Merle, A. De Mallmann, L. Delevoye, R. M. Gauvin and M. Taoufik, Organometallics, 2020, 39, 1105–1111 CrossRef CAS.
- N. Herrmann, K. Köhnke and T. Seidensticker, ACS Sustainable Chem. Eng., 2020, 8, 10633–10638 CAS.
- M. von Czapiewski, M. Rhein and M. A. R. Meier, ACS Sustainable Chem. Eng., 2018, 6, 15170–15179 CrossRef CAS.
- Y. Liu and S. Mecking, Angew. Chem., Int. Ed., 2019, 58, 3346–3350 CrossRef CAS PubMed.
- X. Sun, J. Chen and T. Ritter, Nat. Chem., 2018, 10, 1229–1233 CrossRef CAS PubMed.
- W. He, Z. Fang, Q. Tian, D. Ji, K. Zhang and K. Guo, Chem. Eng. Process., 2015, 96, 39–43 CrossRef CAS.
- H. Hosney, B. Nadiem, I. Ashour, I. Mustafa and A. El-Shibiny, J. Appl. Polym. Sci., 2018, 135, 46270 CrossRef.
- A. J. Clark and S. S. Hoong, Polym. Chem., 2014, 5, 3238–3244 RSC.
- M. Alam, D. Akram, E. Sharmin, F. Zafar and S. Ahmad, Arabian J. Chem., 2014, 7, 469–479 CrossRef CAS.
- S. G. Tan and W. S. Chow, Polym. – Plast. Technol. Eng., 2010, 49, 1581–1590 CrossRef CAS.
- G. Karmakar, P. Ghosh, K. Kohli, B. K. Sharma and S. Z. Erhan, Chemicals from Vegetable Oils, Fatty Derivatives, and Plant Biomass, in Innovative Uses of Agricultural Products and Byproducts, ed. M. H. Tunick and L. S. Liu, ACS Symposium Series; American Chemical Society, Washington, DC, 2020 Search PubMed.
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- N. Yadav, F. Seidi, D. Crespy and V. D'Elia, ChemSusChem, 2019, 12, 724–754 CrossRef CAS PubMed.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, ChemSusChem, 2019, 12, 3410–3430 CrossRef PubMed.
- L. Peña Carrodeguas, À. Cristòfol, J. M. Fraile, J. A. Mayoral, V. Dorado, C. I. Herrerías and A. W. Kleij, Green Chem., 2017, 19, 3535–3541 RSC.
- M. Alves, B. Grignard, S. Gennen, C. Detrembleur, C. Jerome and T. Tassaing, RSC Adv., 2015, 5, 53629–53636 RSC.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2017, 118, 434–504 CrossRef PubMed.
- S. Dabral and T. Schaub, Adv. Synth. Catal., 2019, 361, 223–246 CrossRef CAS.
- N. A. Tappe, R. M. Reich, V. D'Elia and F. E. Kühn, Dalton Trans., 2018, 47, 13281–13313 RSC.
- H. Büttner, C. Kohrt, C. Wulf, B. Schäffner, K. Groenke, Y. Hu, D. Kruse and T. Werner, ChemSusChem, 2019, 12, 2701–2707 CrossRef PubMed.
- N. von der Assen, J. Jung and A. Bardow, Energy Environ. Sci., 2013, 6, 2721–2734 RSC.
- H. Guobin, L. Zuyu, Y. Suling and Y. Rufeng, J. Am. Oil Chem. Soc., 1996, 73, 1109–1112 CrossRef CAS.
- H. Büttner, J. Steinbauer and T. Werner, ChemSusChem, 2015, 8, 2655–2669 CrossRef PubMed.
- H. Büttner, C. Grimmer, J. Steinbauer and T. Werner, ACS Sustainable Chem. Eng., 2016, 4, 4805–4814 CrossRef.
- J. Steinbauer, L. Longwitz, M. Frank, J. Epping, U. Kragl and T. Werner, Green Chem., 2017, 19, 4435–4445 RSC.
- H. Büttner, J. Steinbauer, C. Wulf, M. Dindaroglu, H.-G. Schmalz and T. Werner, ChemSusChem, 2017, 10, 1076–1079 CrossRef PubMed.
- P. Yingcharoen, C. Kongtes, S. Arayachukiat, K. Suvarnapunya, S. V. C. Vummaleti, S. Wannakao, L. Cavallo, A. Poater and V. D′Elia, Adv. Synth. Catal., 2019, 361, 366–373 CrossRef CAS.
- A. M. Hardman-Baldwin and A. E. Mattson, ChemSusChem, 2014, 7, 3275–3278 CrossRef CAS PubMed.
- N. Maity, S. Barman, E. Abou-Hamad, V. D'Elia and J.-M. Basset, Dalton Trans., 2018, 47, 4301–4306 RSC.
- H. Kisch, R. Millini and I.-J. Wang, Chem. Ber., 1986, 119, 1090–1094 CrossRef CAS.
- A. Monassier, V. D'Elia, M. Cokoja, H. Dong, J. D. A. Pelletier, J.-M. Basset and F. E. Kühn, ChemCatChem, 2013, 5, 1321–1324 CrossRef CAS.
- A. Barthel, Y. Saih, M. Gimenez, J. D. A. Pelletier, F. E. Kühn, V. D'Elia and J.-M. Basset, Green Chem., 2016, 18, 3116–3123 RSC.
- F. Della Monica, M. Leone, A. Buonerba, A. Grassi, S. Milione and C. Capacchione, Mol. Catal., 2018, 460, 46–52 CrossRef CAS.
- C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef CAS.
- A. W. Kleij, Curr. Opin. Green Sustainable., 2020, 24, 72–81 CrossRef.
- I. D. V. Ingram, M. North and X. Wu, Halide-Free Synthesis of Cyclic and Polycarbonates, in Chemistry Beyond Chlorine, ed. P. Tundo, L. N. He, E. Lokteva and C. Mota, Springer, Cham, 2016 Search PubMed.
- J. Steinbauer, A. Spannenberg and T. Werner, Green Chem., 2017, 19, 3769–3779 RSC.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnama and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS.
- J. Langanke, L. Greiner and W. Leitner, Green Chem., 2013, 15, 1173–1182 RSC.
- M. H. Beyzavi, C. J. Stephenson, Y. Liu, O. Karagiaridi, J. T. Hupp and O. K. Farha, Front. Energy Res., 2015, 2, 63 Search PubMed.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef PubMed.
- M. H. Alkordi, Ł. J. Weseliński, V. D'Elia, S. Barman, A. Cadiau, M. N. Hedhili, A. J. Cairns, R. G. AbdulHalim, J.-M. Basset and M. Eddaoudi, J. Mater. Chem. A, 2016, 4, 7453–7460 RSC.
- A.-C. Simao, B. Lynikaite-Pukleviciene, C. Rousseau, A. Tatibouet, S. Cassel, A. Sackus, A. P. Rauter and P. Rollin, Lett. Org. Chem., 2006, 3, 744–748 CrossRef CAS.
- Z. Zhang, D. W. Rackemann, W. O. S. Doherty and I. M. O'Hara, Biotechnol. Biofuels, 2013, 6, 153 CrossRef CAS PubMed.
- O. Lamarzelle, P.-L. Durand, A.-L. Wirotius, G. Chollet, E. Grau and H. Cramail, Polym. Chem., 2016, 7, 1439–1451 RSC.
- S. Bigot, M. Daghrir, A. Mhanna, G. Boni, S. Pourchet, L. Lecamp and L. Plasseraud, Eur. Polym. J., 2016, 74, 26–37 CrossRef CAS.
- L. Maisonneuve, A.-L. Wirotius, C. Alfos, E. Grau and H. Cramail, Polym. Chem., 2014, 5, 6142–6147 RSC.
- L. Maisonneuve, A. S. More, S. Foltran, C. Alfos, F. Robert, Y. Landais, T. Tassaing, E. Grau and H. Cramail, RSC Adv., 2014, 4, 25795–25803 RSC.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205–2213 RSC.
- N. Tenhumberg, H. Büttner, B. Schäffner, D. Kruse, M. Blumenstein and T. Werner, Green Chem., 2016, 18, 3775–3788 RSC.
- O. Sodpiban, S. Del Gobbo, S. Barman, V. Aomchad, P. Kidkhunthod, S. Ould-Chikh, A. Poater, V. D'Elia and J.-M. Basset, Catal. Sci. Technol., 2019, 9, 6152–6165 RSC.
- F. Castro-Gómez, G. Salassa, A. W. Kleij and C. Bo, Chem. – Eur. J., 2013, 19, 6289–6298 CrossRef PubMed.
- V. D'Elia, A. A. Ghani, A. Monassier, J. Sofack-Kreutzer, J. D. A. Pelletier, M. Drees, S. V. C. Vummaleti, A. Poater, L. Cavallo, M. Cokoja, J.-M. Basset and F. E. Kühn, Chem. – Eur. J., 2014, 20, 11870–11882 CrossRef PubMed.
- B. Dutta, J. Sofack-Kreutzer, A. A. Ghani, V. D'Elia, J. D. A. Pelletier, M. Cokoja, F. E. Kühn and J.-M. Basset, Catal. Sci. Technol., 2014, 4, 1534–1538 RSC.
- C. J. Whiteoak, E. Martin, E. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2233–2239 CrossRef CAS.
- F. Chen, Q.-C. Zhang, D. Wei, Q. Bu, B. Dai and N. Liu, J. Org. Chem., 2019, 84, 11407–11416 CrossRef CAS PubMed.
- W. Natongchai, S. Pornpraprom and V. D′Elia, Asian J. Org. Chem., 2020, 9, 801–810 CrossRef CAS.
- K. M. Doll, S. C. Cermak, J. A. Kenar, E. L. Walter and T. A. Isbell, Ind. Crops Prod., 2017, 104, 269–277 CrossRef CAS.
- V. L. Mamedova and G. Z. Khikmatova, Chem. Heterocycl. Compd., 2017, 53, 976–978 CrossRef CAS.
- R. R. Shaikh, R. A. Khan and A. Alsalme, J. Ind. Eng. Chem., 2018, 64, 446–452 CrossRef CAS.
- L. A. Rios, B. A. Llano and W. F. Hoelderich, Appl. Catal., A, 2012, 445–446, 346–350 CrossRef CAS.
- V. Dorado, L. Gil, J. A. Mayoral, C. I. Herrerías and J. M. Fraile, Catal. Sci. Technol., 2020, 10, 1789–1795 RSC.
- K. M. Doll and S. Z. Erhan, Green Chem., 2005, 7, 849–854 RSC.
- K. M. Doll and S. Z. Erhan, J. Agric. Food Chem., 2005, 53, 9608–9614 CrossRef CAS PubMed.
- W. Y. Pérez-Sena, X. Cai, N. Kebir, L. Vernières-Hassimi, C. Serra, T. Salmi and S. Leveneur, Chem. Eng. J., 2018, 346, 271–280 CrossRef.
- S. C. Cermak and T. A. Isbell, J. Am. Oil Chem. Soc., 2001, 78, 557–565 CrossRef CAS.
- S. C. Cermak, K. B. Brandon and T. A. Isbell, Ind. Crops Prod., 2006, 23, 54–64 CrossRef CAS.
- K. M. Doll, S. C. Cermak, J. A. Kenar and T. A. Isbell, J. Am. Oil Chem. Soc., 2016, 93, 1149–1155 CrossRef CAS.
- D. Miloslavskiy, E. Gotlib, O. Figovsky and D. Pashin, Int. Lett. Chem. Phys. Astron., 2014, 27, 20–29 Search PubMed.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- D. J. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC.
- L. Maisonneuve, T. Lebarbé, E. Grau and H. Cramail, Polym. Chem., 2013, 4, 5472–5517 RSC.
- J. Datta and M. Włoch, Polym. Bull., 2016, 73, 1459–1496 CrossRef CAS.
- A. Cornille, R. Auvergne, O. Figovsky, B. Boutevin and S. Caillol, Eur. Polym. J., 2017, 87, 535–552 CrossRef CAS.
- C. Zhang, T. F. Garrison, S. A. Madbouly and M. R. Kessler, Prog. Polym. Sci., 2017, 71, 91–143 CrossRef CAS.
- M. Ghasemlou, F. Daver, E. P. Ivanova and B. Adhikari, Eur. Polym. J., 2019, 118, 668–684 CrossRef CAS.
- V. Sharma and P. P. Kundu, Prog. Polym. Sci., 2006, 31, 983–1008 CrossRef CAS.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- M. Jalilian, H. Yeganeh and M. N. Haghighi, Polym. Int., 2008, 57, 1385–1394 CrossRef CAS.
- S. Hu, X. Chen and J. M. Torkelson, ACS Sustainable Chem. Eng., 2019, 7, 10025–10034 CrossRef CAS.
- A. R. Mahendran, N. Aust, G. Wuzella, U. Müller and A. Kandelbauer, J. Polym. Environ., 2012, 20, 926–931 CrossRef CAS.
- P. Mazo and L. Rios, J. Am. Oil Chem. Soc., 2013, 90, 725–730 CrossRef CAS.
- Y. A. Alassmy and P. P. Pescarmona, ChemSusChem, 2019, 12, 3856–3863 CrossRef CAS PubMed.
- P. C. Mazo and L. A. Rios, Chem. Eng. J., 2012, 210, 333–338 CrossRef CAS.
- X. Cai, J. L. Zheng, J. Wärnå, T. Salmi, B. Taouk and S. Leveneur, Chem. Eng. J., 2017, 313, 1168–1183 CrossRef CAS.
- X. Cai, M. Matos and S. Leveneur, Ind. Eng. Chem. Res., 2019, 58, 1548–1560 CrossRef CAS.
- J. L. Zheng, P. Tolvanen, B. Taouk, K. Eränen, S. Leveneur and T. Salmi, Chem. Eng. Res. Des., 2018, 132, 9–18 CrossRef CAS.
- A. Z. Yu, R. A. Setien, J. M. Sahouani, J. Docken and D. C. Webster, J. Coat. Technol. Res., 2019, 16, 41–57 CrossRef CAS.
- N. Mann, S. K. Mendon, J. W. Rawlins and S. F. Thames, J. Am. Oil Chem. Soc., 2008, 85, 791–796 CrossRef CAS.
- S. Samanta, S. Selvakumar, J. Bahr, D. S. Wickramaratne, M. Sibi and B. J. Chisholm, ACS Sustainable Chem. Eng., 2016, 4, 6551–6561 CrossRef CAS.
- L. Poussard, J. Mariage, B. Grignard, C. Detrembleur, C. Jérôme, C. Calberg, B. Heinrichs, J. De Winter, P. Gerbaux, J. M. Raquez, L. Bonnaud and P. Dubois, Macromolecules, 2016, 49, 2162–2171 CrossRef CAS.
- I. Javni, D. P. Hong and Z. S. Petrović, J. Appl. Polym. Sci., 2013, 128, 566–571 CrossRef CAS.
- J. L. Zheng, F. Burel, T. Salmi, B. Taouk and S. Leveneur, Ind. Eng. Chem. Res., 2015, 54, 10935–10944 CrossRef CAS.
- S. Doley and S. K. Dolui, Eur. Polym. J., 2018, 102, 161–168 CrossRef CAS.
- L. Zhang, Y. Luo, Z. Hou, Z. He and W. Eli, J. Am. Oil Chem. Soc., 2014, 91, 143–150 CrossRef CAS.
- M. Bähr and R. Mülhaupt, Green Chem., 2012, 14, 483–489 RSC.
- I. Javni, D. P. Hong and Z. S. Petrović, J. Appl. Polym. Sci., 2008, 108, 3867–3875 CrossRef CAS.
- A. F. Guzmán, D. A. Echeverri and L. A. Rios, J. Chem. Technol. Biotechnol., 2017, 92, 1104–1110 CrossRef.
- O. Türünç, N. Kayaman-Apohan, M. V. Kahraman, Y. Menceloğlu and A. Güngör, J. Sol-Gel Sci. Technol., 2008, 47, 290–299 CrossRef.
- G. Bresciani, F. Marchetti, G. Rizzi, A. Gabbani, F. Pineider and G. Pampaloni, J. CO2 Util., 2018, 28, 168–173 CrossRef CAS.
- M. J. Kelly, A. Barthel, C. Maheu, O. Sodpiban, F.-B. Dega, S. V. C. Vummaleti, E. Abou-Hamad, J. D. A. Pelletier, L. Cavallo, V. D'Elia and J.-M. Basset, J. CO2 Util., 2017, 20, 243–252 CrossRef CAS.
- B. Yu, B. Zou and C.-W. Hu, J. CO2 Util., 2018, 26, 314–322 CrossRef CAS.
- H. Yasuda, L. He, T. Sakakura and C. Hu, J. Catal., 2005, 233, 119–122 CrossRef CAS.
- F. Della Monica, M. Ricciardi, A. Proto, R. Cucciniello and C. Capacchione, ChemSusChem, 2019, 12, 3448–3452 CrossRef PubMed.
- J. Steinbauer and T. Werner, ChemSusChem, 2017, 10, 3025–3029 CrossRef CAS PubMed.
- T. Werner, N. Tenhumberg and H. Büttner, ChemCatChem, 2014, 6, 3493–3500 CrossRef CAS.
- Y. Hu, J. Steinbauer, V. Stefanow, A. Spannenberg and T. Werner, ACS Sustainable Chem. Eng., 2019, 7, 13257–13269 CrossRef CAS.
- S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- L. Cuesta-Aluja, A. Campos-Carrasco, J. Castilla, M. Reguero, A. M. Masdeu-Bultó and A. Aghmiz, J. CO2 Util., 2016, 14, 10–22 CrossRef CAS.
- L. Cuesta-Aluja, J. Castilla and A. M. Masdeu-Bultó, Dalton Trans., 2016, 45, 14658–14667 RSC.
- C. J. Whiteoak, B. Gjoka, E. Martin, M. M. Belmonte, E. C. Escudero-Adán, C. Zonta, G. Licini and A. W. Kleij, Inorg. Chem., 2012, 51, 10639–10649 CrossRef CAS PubMed.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083–3090 RSC.
- J. Rintjema, W. Guo, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem. – Eur. J., 2015, 21, 10754–10762 CrossRef CAS.
- C. Miceli, J. Rintjema, E. Martin, E. C. Escudero-Adán, C. Zonta, G. Licini and A. W. Kleij, ACS Catal., 2017, 7, 2367–2373 CrossRef CAS.
- F. Chen, N. Liu and B. Dai, ACS Sustainable Chem. Eng., 2017, 5, 9065–9075 CrossRef CAS.
- W. Desens and T. Werner, Adv. Synth. Catal., 2016, 358, 622–630 CrossRef CAS.
- M. P. van der Helm, B. Klemm and R. Eelkema, Nat. Rev. Chem., 2019, 3, 491–508 CrossRef CAS.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed.
- S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178–2189 RSC.
- M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed.
- H. Büttner, K. Lau, A. Spannenberg and T. Werner, ChemCatChem, 2015, 7, 459–467 CrossRef.
- Y. Hu, S. Peglow, L. Longwitz, M. Frank, J. D. Epping, V. Brüser and T. Werner, ChemSusChem, 2020, 13, 1825–1833 CrossRef CAS.
- N. Liu, Y.-F. Xie, C. Wang, S.-J. Li, D. Wei, M. Li and B. Dai, ACS Catal., 2018, 8, 9945–9957 CrossRef CAS.
- L.-W. Xu and Y. Lu, Org. Biomol. Chem., 2008, 6, 2047–2053 RSC.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- E. R. Jarvo and S. J. Miller, Tetrahedron, 2002, 58, 2481–2495 CrossRef CAS.
- V. D'Elia, H. Zwicknagl and O. Reiser, J. Org. Chem., 2008, 73, 3262–3265 CrossRef PubMed.
- M. B. Schmid, M. Fleischmann, V. D'Elia, O. Reiser, W. Gronwald and R. M. Gschwind, ChemBioChem, 2009, 10, 440–444 CrossRef CAS.
- M. Toda, A. Takagaki, M. Okamura, J. N. Kondo, S. Hayashi, K. Domen and M. Hara, Nature, 2005, 438, 178–178 CrossRef CAS PubMed.
- T. Morack, J. B. Metternich and R. Gilmour, Org. Lett., 2018, 20, 1316–1319 CrossRef CAS PubMed.
- S. Meninno, ChemSusChem, 2019, 13, 439–468 CrossRef PubMed.
- S. Arayachukiat, C. Kongtes, A. Barthel, S. V. C. Vummaleti, A. Poater, S. Wannakao, L. Cavallo and V. D'Elia, ACS Sustainable Chem. Eng., 2017, 5, 6392–6397 CrossRef CAS.
- P. Yingcharoen, W. Natongchai, A. Poater and V. D′Elia, Catal. Sci. Technol., 2020, 10, 5544–5558 RSC.
- B. Grignard, J. M. Thomassin, S. Gennen, L. Poussard, L. Bonnaud, J. M. Raquez, P. Dubois, M. P. Tran, C. B. Park, C. Jerome and C. Detrembleur, Green Chem., 2016, 18, 2206–2215 RSC.
- M. Alves, B. Grignard, S. Gennen, R. Mereau, C. Detrembleur, C. Jerome and T. Tassaing, Catal. Sci. Technol., 2015, 5, 4636–4643 RSC.
- P. G. Parzuchowski, M. Jurczyk-Kowalska, J. Ryszkowska and G. Rokicki, J. Appl. Polym. Sci., 2006, 102, 2904–2914 CrossRef CAS.
- M. Jalilian, H. Yeganeh and M. N. Haghighi, Polym. Adv. Technol., 2010, 21, 118–127 CrossRef CAS.
- Z. Li, Y. Zhao, S. Yan, X. Wang, M. Kang, J. Wang and H. Xiang, Catal. Lett., 2008, 123, 246–251 CrossRef CAS.
- C. Maeda, J. Shimonishi, R. Miyazaki, J.-Y. Hasegawa and T. Ema, Chem. – Eur. J., 2016, 22, 6556–6563 CrossRef CAS PubMed.
- Y. Chen, R. Luo, Q. Xu, W. Zhang, X. Zhou and H. Ji, ChemCatChem, 2017, 9, 767–773 CrossRef CAS.
- Y. Chen, R. Luo, Q. Xu, J. Jiang, X. Zhou and H. Ji, ChemSusChem, 2017, 10, 2534–2541 CrossRef CAS PubMed.
- A. Farhadian, M. B. G. Afshani, A. B. Miyardan, M. R. Nabid and N. Safari, ChemistrySelect, 2017, 2, 1431–1435 CrossRef CAS.
- K. Motokura, S. Itagaki, Y. Iwasawa, A. Miyaji and T. Baba, Green Chem., 2009, 11, 1876–1880 RSC.
- J. Wang, Y. Zhao, Q. Li, N. Yin, Y. Feng, M. Kang and X. Wang, J. Appl. Polym. Sci., 2012, 124, 4298–4306 CrossRef CAS.
- L. He, Y. Fan, J. Bellettre, J. Yue and L. Luo, Renewable Sustainable Energy Rev., 2020, 119, 109589 CrossRef CAS.
- L. Ruiz, A. Aghmiz, A. M. Masdeu-Bultó, G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Polymer, 2017, 124, 226–234 CrossRef CAS.
- S. C. D'Angelo, A. Dall'Ara, C. Mondelli, J. Pérez-Ramírez and S. Papadokonstantakis, ACS Sustainable Chem. Eng., 2018, 6, 16563–16572 CrossRef.
- M. Ricciardi, F. Passarini, C. Capacchione, A. Proto, J. Barrault, R. Cucciniello and D. Cespi, ChemSusChem, 2018, 11, 1829–1837 CrossRef CAS.
- S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 6393–6397 CrossRef CAS.
- A. E. Harman-Ware, Conversion of Terpenes to Chemicals and Related Products, in Chemical Catalysts for Biomass Upgrading, ed. M. Crocker and E. Santillan–Jimenez, Wiley–VCH Verlag GmbH, 2020 Search PubMed.
- R. Mewalal, D. K. Rai, D. Kainer, F. Chen, C. Külheim, G. F. Peter and G. A. Tuskan, Trends Biotechnol., 2017, 35, 227–240 CrossRef CAS.
- A. Wambach, S. Agarwal and A. Greiner, ACS Sustainable Chem. Eng., 2020, 8, 14690–14693 CrossRef CAS.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- T. M. McGuire, C. Pérale, R. Castaing, G. Kociok-Köhn and A. Buchard, J. Am. Chem. Soc., 2019, 141, 13301–13305 CrossRef CAS.
- H. Kobayashi and A. Fukuoka, Green Chem., 2013, 15, 1740–1763 RSC.
- G. Diamond, A. Hagemeyer, V. Murphy and V. Sokolovskii, Comb. Chem. High Throughput Screening, 2018, 21, 616–630 CrossRef CAS PubMed.
- J. A. Galbis, M. de Gracia García-Martín, M. Violante de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS.
- Y. Ecochard and S. Caillol, Eur. Polym. J., 2020, 137, 109915 CrossRef CAS.
- M. R. Ortega Vega, J. Ercolani, S. Mattedi, C. Aguzzoli, C. A. Ferreira, A. S. Rocha and C. F. Malfatti, Ind. Eng. Chem. Res., 2018, 57, 12386–12396 CrossRef CAS.
- T. Jose, S. Cañellas, M. A. Pericàs and A. W. Kleij, Green Chem., 2017, 19, 5488–5493 RSC.
- A. Akhdar, K. Onida, N. D. Vu, K. Grollier, S. Norsic, C. Boisson, F. D'Agosto and N. Duguet, Adv. Sustainable Syst., 2020, 2000218, DOI:10.1002/adsu.202000218.
| This journal is © The Royal Society of Chemistry 2021 |