
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tin(IV) fluoride complexes with neutral phosphine coordination and comparisons with hard N- and O-donor ligands†
Rhys P.
King
a,
Madeleine S.
Woodward
 a,
Julian
Grigg
b,
Graeme
McRobbie
b,
William
Levason
a and
Gillian
Reid
*a
a,
Julian
Grigg
b,
Graeme
McRobbie
b,
William
Levason
a and
Gillian
Reid
*a
aSchool of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
bGE Healthcare, Pollards Wood, Nightingales Lane, Chalfont St Giles, Bucks, HP8 4SP, UK
First published on 15th September 2021
Abstract
The reactions of trans-[SnF4(PMe3)2] with one, two or three equivalents of Me3SiO3SCF3 (TMSOTF), respectively, in anhydrous CH2Cl2 form six-coordinate [SnF4−n(PMe3)2(OTf)n] (n = 1–3), which have been characterised by microanalysis, IR and multinuclear NMR (1H, 19F{1H}, 31P{1H} and 119Sn) spectroscopy. The crystal structure of [SnF3(PMe3)2(OTf)] reveals the three fluorines are in a mer-arrangement with mutually trans PMe3 ligands. The multinuclear NMR spectra confirm this structure is retained in solution, and show that [SnF2(PMe3)2(OTf)2] has trans-phosphines, while [SnF(PMe3)2(OTf)3] has trans PMe3 groups and hence mer-triflate ligands. The [SnF4−n(PMe3)2(OTf)n] are unstable in solution and the decomposition products include [Me3PF]+ and the tin(II) complexes [Sn(PMe3)2(OTf)2] and [Sn3F5(OTf)], both of the latter identified by their crystal structures. The reaction of trans-[SnF4(PiPr3)2] containing the bulkier phosphine, with one and two equivalents of TMSOTf produced unstable mono- and bis-triflates, which the NMR data also suggest contain weakly coordinated triflate, [SnF3(PiPr3)2(OTf)] and [SnF2(PiPr3)2(OTf)2], again with axial phosphines, although some OTf dissociation from the former to give [SnF3(PiPr3)2]+ may occur in solution at room temperature. The new phosphine complexes of SnF4, trans-[SnF4(PiPr3)2] and (cis) [SnF4(κ2-triphos)] (triphos = CH3C(CH2PPh2)3) have also been fully characterised, including the crystal structure of [SnF4(κ2-triphos)]. Attempts to promote P3-coordination by further treatment of this complex with TMSOTf were unsuccessful. The [SnF4(L)2] (L = dmso, py, pyNO, DMF, OPPh3) complexes, which exist as mixtures of cis and trans isomers, react with one equivalent of TMSOTf, followed by addition of one equivalent of L, to form the ionic [SnF3(L)3][OTf] complexes, which were characterised by microanalysis, IR and multinuclear NMR spectroscopy. In nitromethane solution they are a mixture of mer and fac isomers based upon multinuclear NMR data (1H, 19F{1H}, 119Sn). Reaction of [SnF4(OPPh3)2] with two equivalents of TMSOTf and further OPPh3 produced [SnF2(OPPh3)4][OTf]2, which is a mixture of cis and trans isomers in solution. The crystal structure of [SnF2(OPPh3)4][OTf]2 confirms the trans isomer in the solid state, with the triflate ionic. These complexes are rare examples of fluorotin(IV) cations with neutral monodentate ligands.
Introduction
In addition to their intrinsic interest, coordination complexes of the main group elements have attracted considerable interest in recent years for a variety of applications, including Lewis acid based and metal free catalysis, development of precursors for the chemical vapour deposition of semiconductor thin films and, particularly in the case of main group fluoride complexes, towards new types of 18F carriers for positron emission tomography imaging in medicine.1The tin(IV) halides, SnX4 (X = Cl, Br or I) are tetrahedral monomers that are widely used as Lewis acids and as synthons for tin(IV) complexes.2–5 The complexes are generally formed by adding two neutral donor groups to form six-coordinate cis- or trans-[SnX4(L)2] or cis-[SnX4(bidentate)]. Only very rarely do neutral ligands displace a coordinated halide to form Sn(IV) cations; exceptions are provided by the macrocycles 1,4,7-trimethyl-1,4,7-triazacyclononane, 1,3,5-trimethyl-1,3,5-triazacyclohexane and 1,4,7-trithiacyclononane (L3), which generate the ‘self-ionisation’ complexes, [SnX3(L3)]2[SnX6] (X = Cl or Br),6,7 although more flexible acyclic polydentate ligands, such as MeC(CH2AsMe2)3, behave as bidentate ligands, as in [SnX4{κ2-MeC(CH2AsMe2)3}].8 Formation of tin(IV) cations can be achieved by using halide abstractors such as AlCl3, CF3SO3SiMe3 (TMSOTf) or Na[BArF] (BArF = [B{3,5-(CF3)2(C6H3)}4]−).9,10
Tin(IV) fluoride contains six-coordinate tin in vertex sharing octahedra11 and although readily hydrolysed, is unreactive towards many neutral ligands. As a result of the lack of commercial availability and this limited reactivity, its coordination chemistry was little explored until recently; only one crystal structure was reported (for the complex [SnF4(2,2′-bipy)]) and spectroscopic data were limited.12–17 A key development was the synthesis of [SnF4(MeCN)2] from SnF2 and I2 in MeCN by Tudela.18 The other product, SnI4, does not form a stable MeCN adduct and is readily removed by washing with CS2. The MeCN is easily displaced from [SnF4(MeCN)2] by neutral ligands, and hence this is a valuable synthon for developing wider coordination chemistry of SnF4 with a range of ligands. Detailed studies of the reactions of [SnF4(MeCN)2] with phosphine and arsine oxides produced a series of complexes [SnF4(R3EO)2] (R = Me, Ph; E = P, As) and [SnF4(L-L)] (L-L = (L-L = o-C6H4(P(O)Ph2)2, o-C6H4(P(O)Me2)2 or Ph2P(O)CH2P(O)Ph2),19 and comparison of spectroscopic and structural data on these and their heavier SnX4 (X = Cl, Br, I) analogues confirmed SnF4 as the strongest Lewis acid of the four. Subsequently, these studies were extended to tertiary phosphine complexes, although arsine ligands did not form isolable complexes with SnF4 (in contrast to their behaviour with other SnX4).8 Further studies of N- and O-donor complexes of SnF4 were also included, but none of the ligands showed any tendency to displace fluoride from SnF4.8 Fluorotin cations were obtained with the tetradentate N-donor ligand, tris(1-ethyl-benzoimidazol-2-ylmethyl)amine (BIMEt3) by reaction of SnF4, BIMEt3 and appropriate amounts of TMSOTf, when [SnF4−x(BIMe3)][OTf]x (x = 1, 2, 3) were isolated.20 Other fluorotin(IV) cations appear unknown.12
Here we report the reactions of phosphine complexes of tin(IV) tetrafluoride with some halide abstractors, and several cationic N- and O-donor ligand complexes, [SnF4−xL2+x]x+, prepared by abstraction of fluoride from [SnF4L2] with TMSOTf in the presence of added L.
Experimental
All complex syntheses were carried out using standard Schlenk and vacuum line techniques. Samples were handled and stored in a glove box under a dry dinitrogen atmosphere to exclude moisture. TMSOTf was obtained from Sigma-Aldrich and distilled before use. Tin(II) fluoride was obtained from Alfa Aesar and used as received. Phosphine ligands, OPPh3 and pyNO were obtained from Sigma-Aldrich or Strem and used as received. Na[BArF] was made as described.10 CH2Cl2, DMF, dmso and MeCN were dried by distillation from CaH2, py, diethyl ether and n-hexane from Na. [SnF4(MeCN)2] was made from SnF2 and I2 in MeCN as described by Tudela.18 [SnF4(PMe3)2],8 [SnF4(Py)2]8 and [SnF4(OPPh3)2]19 were made by literature methods.Infrared spectra were recorded as Nujol mulls between CsI plates using a Perkin Elmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H, 19F{1H}, 31P{1H}, and 119Sn NMR spectra were recorded from CH2Cl2/CD2Cl2 or CH3NO2/CD3NO2 solutions using a Bruker AV400 spectrometer and referenced to TMS via the residual solvent resonance, CFCl3, 85% H3PO4, or neat SnMe4, as appropriate. Typically, [Cr(acac)3] was added as a relaxation agent when recording the 119Sn spectra. Microanalyses were undertaken by London Metropolitan University or Medac.
[SnF3(PMe3)2(OTf)]
To a solution of [SnF4(PMe3)2] (0.100 g, 0.288 mmol) in CH2Cl2 (2 mL) a solution of TMSOTf (0.064 g, 0.288 mmol) in CH2Cl2 (2 mL) was added dropwise to form a clear solution. The reaction was stirred for 2 h, the volatiles were then removed in vacuo to leave a solid, which was washed with hexane (3 × 10 mL) and dried in vacuo to form a white powder. Crystals suitable for single crystal X-ray diffraction were grown by layering a CH2Cl2 solution of the complex with hexane. Yield 0.083 g, 60%. Required for C7H18F6P2S2Sn (476.91): C, 17.6; H, 3.8. Found: C, 17.5; H, 4.2%. IR (Nujol/cm−1): ν = 517m, 546m, 573m (Sn–F), 1156 (–OSO2), 1224, 1261 (CF3). 1H NMR (CD2Cl2, 298 K): δ = 1.70 (d, 2JPH = 12 Hz, CH3). 19F{1H} NMR (CD2Cl2, 298 K): δ = −78.8 (s, OTf), −126.9 (br s, Sn–F), −149.2 (br s, Sn–F); (CD2Cl2, 183 K): δ = −78.5 (s, [3F], OTf), −127.9 (td, [2F], 2JPF(cis-OTf) = 153 Hz, 2JFF = 41 Hz, 1J119SnF = 3253 Hz, 1J117SnF = 3108 Hz, Sn–Fcis-OTf), −151.4 (tt, [F], 2JPF(trans-OTf) = 121 Hz, 2JFF = 41, 1J119SnF = 3013 Hz, 1J117SnF = 2893 Hz, Sn–Ftrans-OTf). 31P{1H} NMR (CH2Cl2, 298 K): δ = −6.01 (s); (CH2Cl2, 183 K): δ = −3.2 (td, 2JPF(cis-OTf) = 153 Hz; 2JPF(trans-OTf) = 121 Hz; 1J119SnP = 3412 Hz; 1J117SnP = 3263). 119Sn NMR (CH2Cl2, 183 K): δ = −599.9 (m).[SnF2(PMe3)2(OTf)2]
To a solution of [SnF4(PMe3)2] (0.100 g, 0.288 mmol) in CH2Cl2 (2 mL) a solution of TMSOTf (0.128 g, 0.576 mmol) in CH2Cl2 (2 mL) was added dropwise to form a clear solution. The reaction was stirred for 2 h. The volatiles were then removed in vacuo to leave a solid, which was washed with hexane (3 × 10 mL) and dried in vacuo to form a white powder. Yield 0.103, 59%. Required for C8H18F8O6P2S2Sn (606.98): C, 15.8; H, 3.0. Found: C, 15.7; H, 3.6%. IR (Nujol/cm−1): ν = 531 (s) (Sn–F). 1H NMR (CD2Cl2, 298 K): δ = 1.88 (d, 2JPH = 12 Hz, CH3). 19F{1H} NMR (CD2Cl2, 298 K): δ = −78.0 (s, [6F], OTf), −140 (br s, [2F], Sn–F); (CD2Cl2, 183 K): δ = −77.8, −78.4 (OTf−), −142.7 (t, 2JP− = 107 Hz, 1J119SnF = 3393 Hz, 1J117SnF = 3232, Sn–F). 31P{1H} NMR (CH2Cl2, 298 K): δ = 7.3 (br); (CH2Cl2, 183 K): δ = 10.2 (t, 2JPF 107 Hz; 1J119SnP = 3654 Hz; 1J117SnP = 3484). 119Sn NMR (CH2Cl2, 183 K): δ = −609 (tt, 1J119SnP = 3654 Hz, 1J119SnF = 3393 Hz).[SnF(PMe3)2(OTf)3]
To a solution of [SnF4(PMe3)2] (0.100 g, 0.288 mmol) in CH2Cl2 (2 mL) a solution of TMSOTf (0.192 g, 0.864 mmol) in CH2Cl2 (2 mL) was added dropwise to form a clear solution. The reaction was stirred for 10 min, the volatiles were then removed in vacuo to leave a solid, which was washed with hexane (3 × 10 mL) and dried in vacuo to form a white powder. Yield 0.137 g (64%). Required for C9H18F10O9P2S3Sn (737.07): C, 14.7; H, 2.6. Found: C, 14.3; H, 3.7%. IR (Nujol/cm−1): ν = 510m (Sn–F), 1158m, 1164m (–OSO2), 1191m, 1202m, 1237m, 1243m (CF3). 1H NMR (CD2Cl2, 298 K): δ = 2.0 (d, 2JPH = 13 Hz, CH3). 19F{1H} NMR (CD2Cl2, 298 K): δ = −77.6 (s, [9F], OTf), −132.9 (t, 2JPF = 88 Hz, 1J119SnF = 3557 Hz, 1J117SnF = 3396 Hz), [F], (Sn–F). 31P{1H} NMR (CD2Cl2, 298 K): δ = 18.3 (d, 2JPF = 88 Hz, 1J119SnP = 3740 Hz; 1J117SnP = 3568 Hz). 119Sn NMR (CH2Cl2, 183 K): δ = −617 (m).[SnF4(PiPr3)2]
To a suspension of [SnF4(MeCN)2] (0.648 g, 2.34 mmol) in CH2Cl2 (5 mL) a solution of PiPr3 (0.750 g, 4.68 mmol) in CH2Cl2 was added to form a slightly cloudy solution. The reaction was stirred for 1 h, then the solution was filtered and the filtrate was concentrated in vacuo to yield a white solid, which was washed with hexane (3 × 10 mL) and dried in vacuo. Yield: 0.862 g (71%). C18H42F4P2Sn·3/4CH2Cl2 (578.84): C, 38.9; H, 7.6. Found: C, 38.8 H, 8.1%. IR (Nujol/cm−1): ν = 535s (Sn–F) 1H NMR (CD2Cl2, 298 K): δ = 1.41 (dd, 3JPH = 15Hz, 3JHH = 7 Hz, [18H], CH3), 2.55 (septet of d, 3JHH = 7 Hz, 2JPH = 1 Hz, [3H], CH), 19F{1H} NMR (CD2Cl2, 298 K): δ = −99.1 (t, 2JPF = 124 Hz, 1J119SnF = 2477 Hz, 1J117SnF = 2363 Hz), 31P{1H} NMR (CD2Cl2, 298 K): δ = 30.6 (quint, 2JPF = 124, 1J119SnP = 2920 Hz, 1J117SnP = 2792 Hz). 119Sn NMR (CH2Cl2, 183 K): δ = −622.0 (quin of t, 1J119SnF = 2920 Hz, 1J117SnP = 2477 Hz).[SnF3(PiPr3)2(OTf)]
To a solution of [SnF4(PiPr3)2] (0.100 g, 0.194 mmol) in CH2Cl2 (2 mL) a solution of TMSOTf (0.043 g, 0.194 mmol) was added dropwise to form a clear solution. The reaction was stirred for 1 h, volatiles were then removed in vacuo to yield a white solid, which was washed with hexane (3 × 10 mL) and dried in vacuo. Yield: 0.095 g (76%). Required for C19H42F6O3P2S·1/2CH2Cl2 (718.68): C, 32.6; H, 6.0. Found: C, 32.1; H, 6.7%. IR (Nujol/cm−1): ν = 540m, 558m, 571m (Sn–F). 1150m (–OSO2) 1223, 1260 (CF3). 1H NMR (CD2Cl2, 298 K): δ = 1.45 (dd, 3JPH = 15Hz, 3JHH = 7 Hz, [18H], CH3), 2.77 (septet of d, 3JHH = 7 Hz, 2JPH = 2 Hz, CH), 19F{1H} NMR (CD2Cl2, 298 K): δ = −73.5 (br s, 1JSnF = 3518 Hz), −78.5 (s, OTf), −116.9 (br s, 1JSnF = 3244 Hz). 31P{1H} NMR (CH2Cl2, 298 K): δ = 38.2 (q, 2JPF = 121 Hz, 1J119SnP = 2827 Hz, 1J117SnP = 2702 Hz), 119Sn NMR (CH2Cl2, 183 K): −611.1 (tdt, 1J119SnF = 3558, 3200 Hz, 1J119SnP = 2856 Hz.[SnF2(PiPr3)2(OTf)2]
To a solution of [SnF4(PiPr3)2] (0.200 g, 0.388 mmol) in CH2Cl2 (2 mL) a solution of TMSOTf (0.173 g, 0.778 mmol) was added dropwise to form a clear solution. The reaction was stirred for 1 h, volatiles were removed in vacuo to yield a white solid which was washed with hexane (3 × 10 mL) and dried in vacuo. Yield: 0.231 g (77%). Required for C20H42F8O6P2S2Sn·CH2Cl2 (860.20): C, 29.3; H, 5.2. Found: C, 29.1; H, 5.8%. IR (Nujol/cm−1): ν = 519w, 533w (Sn–F), 1150 (OSO2), 1234w, 1261w (CF3). 1H NMR (CD2Cl2, 298 K): 1.49 (dd, 3JPH = 16Hz [8H], CH3), 3.05 (septet of d, 3JHH = 8 Hz, 2JPH = 3 Hz, [3H], CH). 19F{1H} NMR (CD2Cl2, 298 K): −56.9 (br, 1JSnF = 4082 Hz), −77.9 (s, OTf). 31P{1H} NMR (CH2Cl2, 298 K): 49.4 (t, 2JPF = 117 Hz, 1J119SnP = 2971 Hz, 1J117SnP = 2839 Hz), 119Sn NMR (CH2Cl2, 183 K): −630 (tt, 1J119SnF = 4013, 1J119SnP = 2941).[SnF4(triphos)]
To a suspension of [SnF4(MeCN)2] (0.200 g, 0.722 mmol) in CH2Cl2 (2 mL), triphos (0.451 g, 0.722 mmol) was added as a solid and the resulting solution was stirred for 2 h to yield a cloudy solution, which was then filtered. Volatiles were removed from the filtrate in vacuo to yield a white solid, which was washed with hexane (3 × 10 mL) and dried in vacuo to yield a white solid. Crystals suitable for single crystal X-ray diffraction were grown by layering a CH2Cl2 solution of the complex with hexane. Yield: 0.326 g (55%). Required for C41H39F4P3Sn·1/2CH2Cl2 (861.78): C, 57.8; H, 4.7. Found: C, 58.2 H, 4.8%. IR (Nujol/cm−1): ν = 516m, 552m, 564m (Sn–F). 1H NMR (CD2Cl2, 298 K): δ = 0.87 (s, [3H], CH3), 2.28 (s, [2H]), 2.91–2.95 (m, [2H]), 3.11 (m, [2H]), 7.1–8.1 (m, [30H], Ar–H). 19F{1H} NMR (CD2Cl2, 298 K): δ = −144.2 (tt, 2JPF = 98 Hz, 2JFF = 45, [2F]), −109.7 (m, [2F]). 31P{1H} NMR (CD2Cl2, 298 K): δ = −6.9 (dtd, 2JPF = 121, 98, 51, [2P]), −27.1 (s, [P]). 119Sn NMR (CH2Cl2, 183 K): δ = −681.4 (m).[SnF4(dmso)2]
[SnF4(MeCN)2] (0.20 g, 0.72 mmol) was suspended in excess dmso and stirred for 5 min. The solvent was removed, and the white powder was dried in vacuo. Yield 0.120 g, 47%. Required for C4H12F4O2S2Sn (351.0): C, 13.7; H, 3.5%. Found C, 13.1; H, 3.5%. IR (Nujol/cm−1): ν = 936s, 908sh (SO), 573vs, 552sh, 522m (SnF). 1H NMR (CD3NO2, 298 K): δ = 3.1 (s), 3.0 (s). 19F{1H} NMR (CD3NO2, 298 K): δ = −161.6 (t, 1J119SnF = 2030, 2JFF = 52 Hz), −149.3 (t, 1J119SnF = 2020, 2JFF = 52 Hz), −149.0 (s, 1J119SnF = 2466 Hz). 119Sn NMR (CH3NO2, 253 K): δ = −778.4.[SnF3(dmso)3][OTf]
[SnF4(dmso)2] (0.15 g, 0.43 mmol) was suspended in CH2Cl2 (10 mL). To this TMSOTf (0.035 g, 0.43 mmol) in CH2Cl2 (5 mL) was added and the solution stirred for 2 h. Dmso (0.04 g, 0.43 mmol) in MeCN (5 mL) was then added and the solution was again stirred for 2 h. The solvent and volatiles were removed and dried in vacuo, which yielded a colourless, gel-like solid which was recrystalised from MeCN/Et2O to a colourless powder. Yield 0.11 g, 46%. Required for C7H18F6O6S4Sn (559.2): C, 15.0; H, 3.2. Found C, 14.9; H, 3.2%. IR (Nujol/cm−1): ν = 912 vbr (SO), 574br, 519br (SnF). 1H NMR (CD3NO2, 298 K): δ = 3.11 (s), 3.10 (s), 3.08 (s). 19F{1H} NMR (CD3NO2, 298 K): δ = −155.2 (s, 1J119SnF = 2063 Hz), −153.7 (t, 1J117SnF = 1852, 1J119SnF = 2181 Hz), −145.8 (d, 1J117SnF = 1847, 1J119SnF = 2063 Hz, 2JFF = 59 Hz), −79.5 (s, OTf). 119Sn NMR (CH3NO2, 253 K): δ = −744, −735.[SnF4(pyNO)2]
[SnF4(MeCN)2] (0.15 g, 0.54 mmol) was dissolved in CH2Cl2 and pyNO (0.10 gm, 1.08 mmol) was added. The solution was stirred for 2 h. The white powder was filtered, washed in hexane (3 × 5 mL) and dried in vacuo. Yield 0.125 g, 60%. Required for C10H10F4N2O2Sn (384.9): C, 31.2; H, 2.6; N, 7.3. Found: C, 31.4; H, 2.6; N, 7.1%. IR (Nujol/cm−1): ν = 1202m (NO), 573br (Sn–F). 1H NMR (CD3NO2, 298 K): δ = 8.7 (m, [2H]), 8.2 (m, [1H]), 7.9 (m, [2H]). 19F{1H} NMR (CD3NO2, 298 K): δ = −168.7 (t, 1J119SnF = 2030, 2JFF = 51 Hz), −166.5 (t, 1J119SnF = 2081, 2JFF = 51 Hz), −164.2 (s, 1J119SnF = 1951). 119Sn NMR (CH3NO2, 253 K): δ = −778.5.[SnF3(pyNO)3][OTf]
[SnF4(pyNO)2] (0.30 g, 0.78 mmol) was suspended in CH2Cl2. To this TMSOTf (0.17 g, 0.78 mmol) was added in MeCN (5 mL). The solution was stirred for 2 h. PyNO (0.074 g, 0.78 mmol) was added and the solution was stirred for a further 2 h. The solvent was then removed in vacuo which yielded a colourless, gel-like solid which was recrystallised from MeCN/Et2O to give a white powder. Yield: 0.22 g, 46%. Required for C16H15F6N3O6Sn·1/2CH2Cl2 (652.54): C, 30.4; H, 2.5; N, 6.4. Found: C, 30.3; H, 2.9; N, 6.6%. IR (Nujol/cm−1): ν = 1225m (NO), 574br,s, 517m (Sn–F). 1H NMR (CD3CN, 298 K): δ = 8.8 (m, [2H]), 8.2 (m, [1H]), 7.9 (m, [2H]). 19F{1H} NMR (CD3CN, 298 K): δ = −172.4 (s, 1J119SnF = 2159 Hz), −171.6 (t, 1J119SnF = 2268 Hz, 2JFF = 59 Hz), −169.9 (d, 1J119SnF = 2244 Hz, 2JFF = 59 Hz), −78.7 (s, OTf). 119Sn NMR (CH3NO2, 253 K): δ ∼−770 (multiplets for the two isomers are almost coincident).[SnF4(DMF)2]
[SnF4(MeCN)2] (0.20 g, 0.72 mmol) was added to an excess of DMF (5 mL). The suspension was stirred for 2 h. The white precipitate was filtered, washed in hexane (3 × 3 mL) and dried in vacuo. Yield 0.195 g, 80%. Required for SnF4C6H14N2O2·CH2Cl2 (425.8): C, 19.7; H, 3.8; N, 6.6. Found C, 19.5; H, 3.8; N, 7.4%. IR (Nujol/cm−1): ν = 1669 (CO), 585s (Sn–F). 1H NMR (CD3NO2, 298 K): δ = 8.16 (s), 7.91 (s) (H), 3.31 (s), 3.28 (s), 3.15 (s), 3.12 (s) CH3. 19F{1H} NMR (CD3NO2, 298 K): δ = −169.5 (t, 1J119SnF = 2015 Hz, 2JFF = 50 Hz), −161.9 (t, 1J119SnF = 1907 Hz, 2JFF = 50 Hz), −161.8 (s). 119Sn NMR (CH3NO2, 253 K): n.o.[SnF3(DMF)3][OTf]
[SnF4(DMF)2] (0.31 g, 0.91 mmol) was suspended in CH2Cl2. To this TMSOTf (0.20 g, 0.91 mmol) was added in CH2Cl2. The solution was stirred for 2 h. DMF (0.067 g, 0.91 mmol) in MeCN was then added and the solution was stirred for 2 h. Addition of n-hexane formed a viscous oil from which the solvent was decanted and the residue was washed with further hexane and dried in vacuo, leaving a colourless glassy solid. Yield 0.085 g, 17%. Microanalytical data were not obtained due to the glassy nature of the product. IR (Nujol/cm−1): ν = 1666 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 582s (Sn–F), 518s (Sn–F). 1H NMR (CD3NO2, 298 K): δ = 8.27 (br s), 8.21 (s), 3.34 (m), 3.18 (m). 19F{1H} NMR (CD3NO2, 298 K): δ = −169.2 (t, 1J119SnF = 2143 Hz, 2JFF = 55 Hz), −168.8 (s, 1J119SnF = 2150 Hz), −164.0 (d, 1J119SnF = 1985 Hz, 2JFF = 55 Hz), −79.5 (s, OTf). 119Sn NMR (CH3NO2, 253 K): n.o.
O), 582s (Sn–F), 518s (Sn–F). 1H NMR (CD3NO2, 298 K): δ = 8.27 (br s), 8.21 (s), 3.34 (m), 3.18 (m). 19F{1H} NMR (CD3NO2, 298 K): δ = −169.2 (t, 1J119SnF = 2143 Hz, 2JFF = 55 Hz), −168.8 (s, 1J119SnF = 2150 Hz), −164.0 (d, 1J119SnF = 1985 Hz, 2JFF = 55 Hz), −79.5 (s, OTf). 119Sn NMR (CH3NO2, 253 K): n.o.
[SnF3(py)3][OTf]
[SnF4(py)2] (0.10 g, 0.28 mmol) was suspended in CH2Cl2 (10 mL). To this TMSOTf (0.063 g, 0.28 mmol) was added in CH2Cl2 (5 mL). The solution was stirred for 2 h. Py (0.022 g, 0.28 mmol) was added and the solution was stirred for a further 2 h. The solvent was then removed in vacuo which yielded a colourless, gel-like solid which was recrystallised from MeCN/Et2O to a white powder. Although we were unable to obtain satisfactory microanalytical data for this complex, the spectroscopic data are in accord with the formulation above. IR (Nujol/cm−1): ν = 568br,s (Sn–F). 1H NMR (CD3NO2, 298 K): δ = 8.9 (m), 8.8 (m), 8.4 (m), 8.3 (m), 7.9 (m), 7.8 (m). 19F{1H} NMR (CD3NO2, 298 K): δ = −165.8 (m, 1J117SnF = 1637 Hz, 1J119SnF = 1717 Hz, 2JFF = 40 Hz), −158.8 (d, 1J117SnF = 1658 Hz, 1J119SnF = 1737 Hz, 2JFF = 40 Hz), −157.2 (s, 1J117SnF = 1515 Hz, 1J119SnF = 1588 Hz), −79.9 (s, OTf). 119Sn NMR (CH3NO2, 253 K): n.o.[SnF3(OPPh3)3][OTf]
[SnF4(OPPh3)2] (0.073 g, 0.097 mmol) was dissolved in CH2Cl2 (10 mL). To this TMSOTf (0.20 g, 0.91 mmol) was added in CH2Cl2 (5 mL). The solution was stirred for 2 h. To this OPPh3 (0.027 g, 0.097 mmol) then added and the solution was stirred for 2 h. The solution was concentrated in vacuo and excess hexane was added, the solvent was removed, and the resulting white powder was dried in vacuo. Yield 0.070 g, 63%. IR (Nujol/cm−1): ν = 1145sh, 1059 (PO), 554m, 537m (Sn–F). 1H NMR (CD2Cl2, 298 K): δ = 7.9–7.3(m). 19F{1H} NMR (CD2Cl2, 298 K): δ = −142.1 (s), −141.2 (t, 2JFF = 61 Hz), −134.5(d, 2JFF = 61 Hz), −79.1 (s, OTf). 31P{1H} NMR (CD2Cl2, 298 K): δ = 46.0 (s), 43.9 (s), 41.7 (s). 119Sn NMR (CH3NO2, 253 K): n.o.
[SnF2(OPPh3)4][OTf]2
[SnF4(OPPh3)2] (0.085 g, 0.11 mmol) was dissolved in CH2Cl2 (5 mL). To this TMSOTf (0.050 g, 0.23 mmol) was added in CH2Cl2 (5 mL). The suspension was stirred for 2 h and then OPPh3 (0.063 g, 0.23 mmol) was added and the solution was stirred for a further 2 h. The solution was concentrated in vacuo and excess hexane was added, the solvent was decanted, and the resulting white powder was dried in vacuo. Yield 0.08 g, 47%. Required for C74H60F8O10P4S2·CH2Cl2 (1652.9): C, 54.5; H, 3.8. Found: C, 54.6; H, 3.8%. IR (Nujol/cm−1): ν = 1150(sh), 1060 (PO), 550s, 537s, 517m (Sn–F). 1H NMR (CD3NO2, 298 K): δ = 7.3–7.9 (m). 19F{1H} NMR (CD3NO2, 298 K): δ = −123.1 (s, 1J117SnF = 1730, 1J119SnF = 1812 Hz), −122.4 (s, 1J117SnF = 1977, 1J119SnF = 2069 Hz), −79.8 (s, OTf). 31P{1H} NMR (CD3NO2, 298 K): δ = 50.5 (s, 1JSnP = 78 Hz), 48.4 (s, 1JSnP = 78 Hz), 47.1 (s, 1JSnP = 96 Hz) average 117Sn/119Sn; separate couplings were not resolved. 119Sn NMR (CH3NO2, 253 K): n.o.
X-Ray experimental
Crystals of the phosphine complexes were grown by layering CH2Cl2 solutions with hexane, while those for the N- and O-donor complexes were obtained by slow evaporation from MeNO2 solutions in the glovebox. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 μm focus) with the crystal held at 100 K. Structure solution and refinement were performed using SHELX(S/L)97, SHELX-2013, or SHELX-2014/7.41.21,22 H atoms bonded to C were placed in calculated positions using the default C–H distance and refined using a riding model. The structure of [SnF4(py)2] showed some twinning, which we were not able to fully resolve, hence the final structure quality if less good than for the other complexes reported. Details of the crystallographic parameters are given in Table S1 (ESI†). CCDC reference numbers for the crystallographic information files in cif format are CCDC 2104490 ([Sn(PMe3)2(OTf)2]), 2104491 (Sn3F5OTf), 2104492 ([SnF3(PMe3)2(OTf)]), 2104493 ([SnF4(κ2-triphos)]), 2104976 ([SnF2(OPPh3)4)][OTf]2), 2106812 ([SnF4(py)2] and 2104984 ([SnF4(pyNO)2]).†Results and discussion
The neutral tin(IV) fluoride complex trans-[SnF4(PMe3)2] was synthesised following the literature method7 and two new complexes [SnF4(PiPr3)2] and [SnF4(κ2-triphos)] (triphos = CH3C(CH2PPh2)3) were made similarly. The presence of a quintet in the 31P{1H} and a triplet in the 19F{1H} NMR spectrum of [SnF4(PiPr3)2] shows this exists only as the trans isomer as found for other tertiary phosphine complexes of SnF4.7 The reaction of [SnF4(MeCN)2] with triphos leads to the formation of the complex [SnF4(κ2-triphos)]. The 31P{1H} NMR spectrum of this complex at 298 K in CH2Cl2 solution is a doublet of triplets of doublets at δ = −6.9 ppm and a singlet at δ = −27.1 ppm with integrals in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, consistent with a κ2-coordinated triphos in solution, and with no evidence of other isomers present. The crystal structure of the complex (Fig. 1) confirms this, and there is no evidence that the free arm can spontaneously displace a fluoride from the metal centre, a result similar to that found in the heavier tin(IV) halide complexes.8 The geometry at the tin is distorted octahedral and the Sn–F and Sn–P bond lengths are much as expected.8
:1 ratio, consistent with a κ2-coordinated triphos in solution, and with no evidence of other isomers present. The crystal structure of the complex (Fig. 1) confirms this, and there is no evidence that the free arm can spontaneously displace a fluoride from the metal centre, a result similar to that found in the heavier tin(IV) halide complexes.8 The geometry at the tin is distorted octahedral and the Sn–F and Sn–P bond lengths are much as expected.8
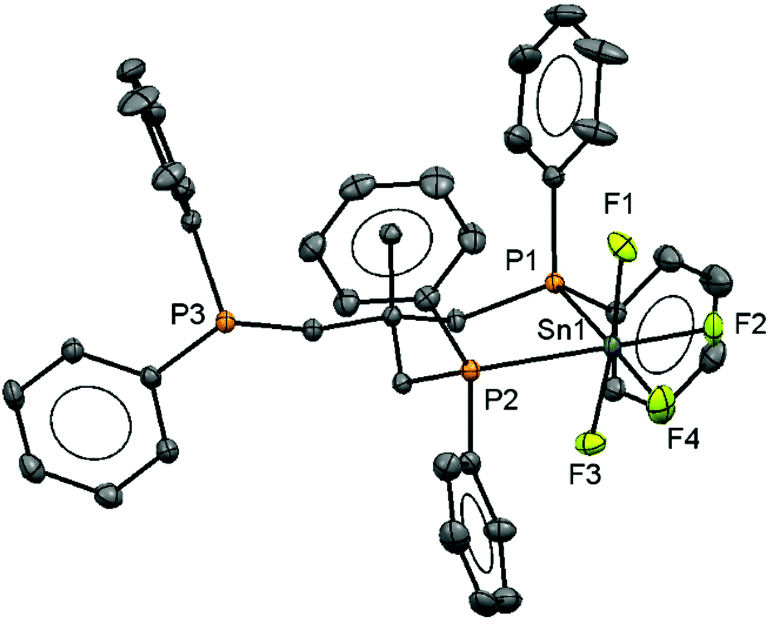 | ||
| Fig. 1 Crystal structure of [SnF4(κ2-triphos)] showing the atom labelling scheme. The ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Sn1–P1 = 2.6302(5), Sn1–P2 = 2.6314(5) Sn1–F1 = 1.9670(12), Sn1–F2 = 1.9488(12), Sn1–F3 = 1.9791(12), Sn1–F4 = 1.9307(12), P1–Sn1–P2 = 91.245(14), P1–Sn1–F4 = 172.46(5), P2–Sn1–F2 = 174.74(4), F1–Sn1–F3 = 173.44(5). | ||
Fluoride abstraction from [SnF4(PR3)2] complexes
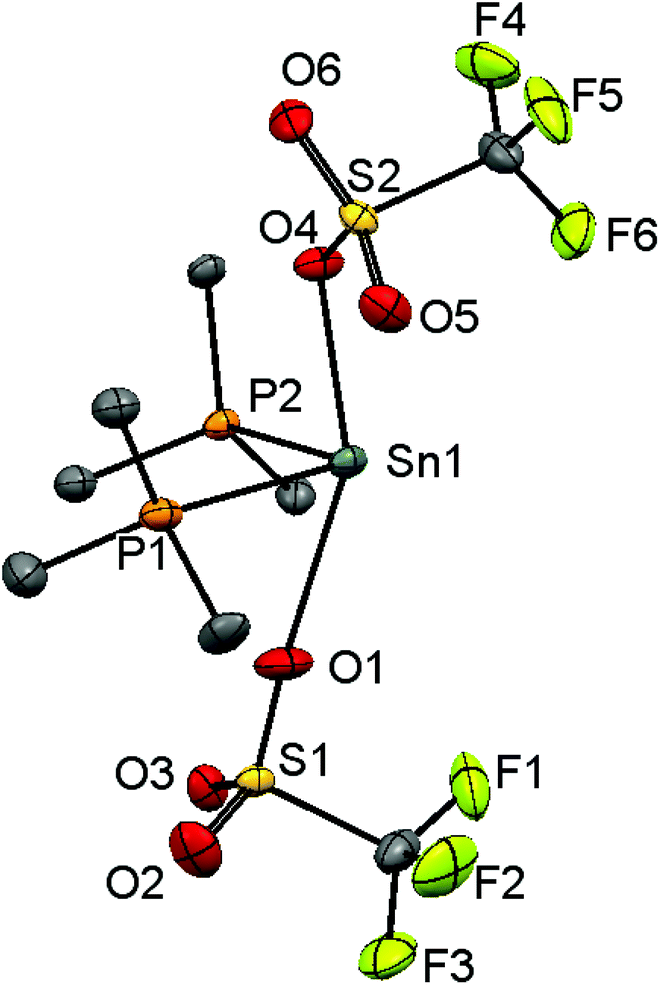
Since neutral phosphine ligands do not displace fluoride from the tin centre directly, the reactions with the potential fluoride abstraction reagents9 Na[BArF] and Me3SiO3SCF3 (TMSOTf) were explored. Some reaction was evident with Na[BArF], but did not go to completion (cf. ref. 10) and hence studies were focussed on TMSOTf (Scheme 1). The reaction of trans-[SnF4(PMe3)2] with one equivalent of TMSOTf in CH2Cl2 leads to the formation of [SnF3(PMe3)2(OTf)]. The crystal structure of this complex (Fig. 2) shows that in the solid state the three fluorines are in a mer-arrangement with OTf trans-F and with mutually trans-PMe3 ligands. The same isomer is present in [SnCl3(PMe3)2(OTf)],10 and comparison of the structures shows the d(Sn–P) and d(Sn–O) are only marginally shorter in the fluoride complex. | ||
| Scheme 1 | ||
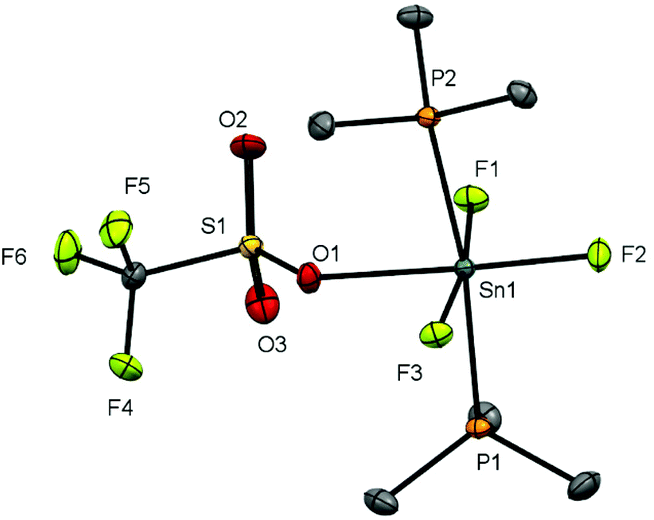 | ||
| Fig. 2 Crystal structure of [SnF3(PMe3)2(OTf)] showing the atom labelling scheme. The ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Sn1–P1 = 2.5278(4), Sn1–P2 = 2.5405(4), Sn1–F1 = 1.9761(9), Sn1–F2 = 1.9595(10), Sn1–F3 = 1.9871(9), Sn1–O1 = 2.2445(12), P1–Sn1–P2 = 171.615(13), F1–Sn1–F3 = 171.00(4), O1–Sn1–F3 = 176.66(4). | ||
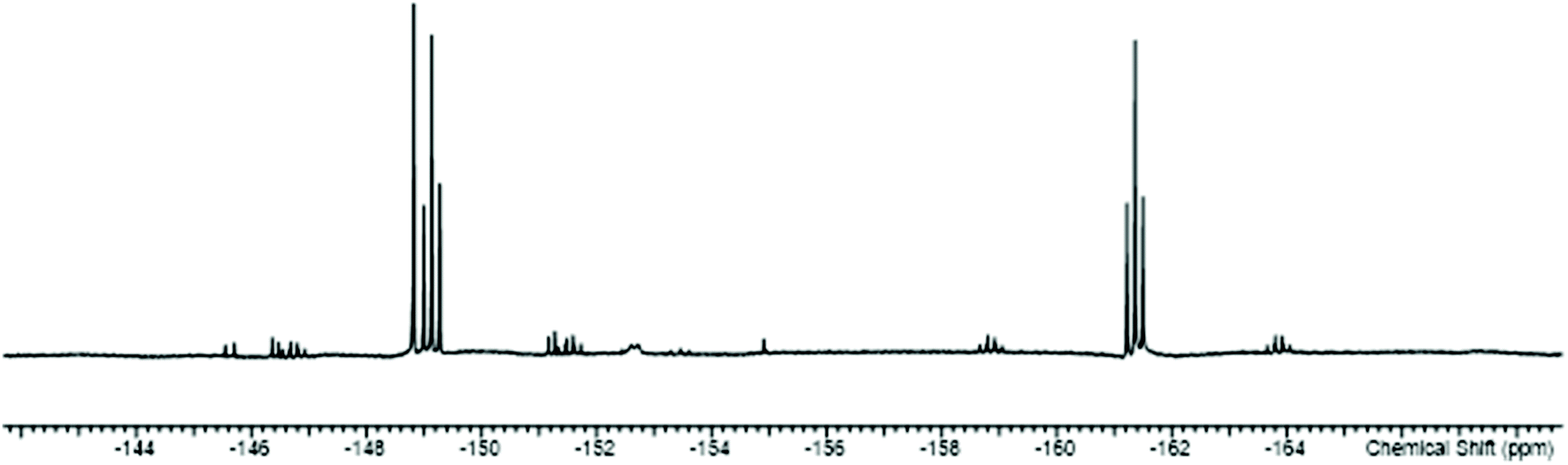
In general the triflate complexes are less robust than the [SnF4(PR3)2] and resonances of decomposition products, which include [R3PF]+, build up quite rapidly in solution, especially for the mono-fluoride species. In solution at 298 K the [SnF3(PMe3)2(OTf)] exhibits broad 19F{1H} and 31P{1H} NMR spectra, probably due to some reversible OTf dissociation. However, cooling the solution to 183 K sharpened these resonances; in the 31P{1H} NMR spectrum there is a triplet of doublets and in the 19F{1H} NMR spectrum a triplet of triplets and a triplet of doublets are seen in a 1:2 ratio (Table 1 and Fig. 3), as expected if the structure in Fig. 2 was retained in solution. Obtaining 119Sn NMR spectra proved to be difficult from the triflate complexes, even at 183 K, the full couplings were often unclear, although the chemical shifts were identified.
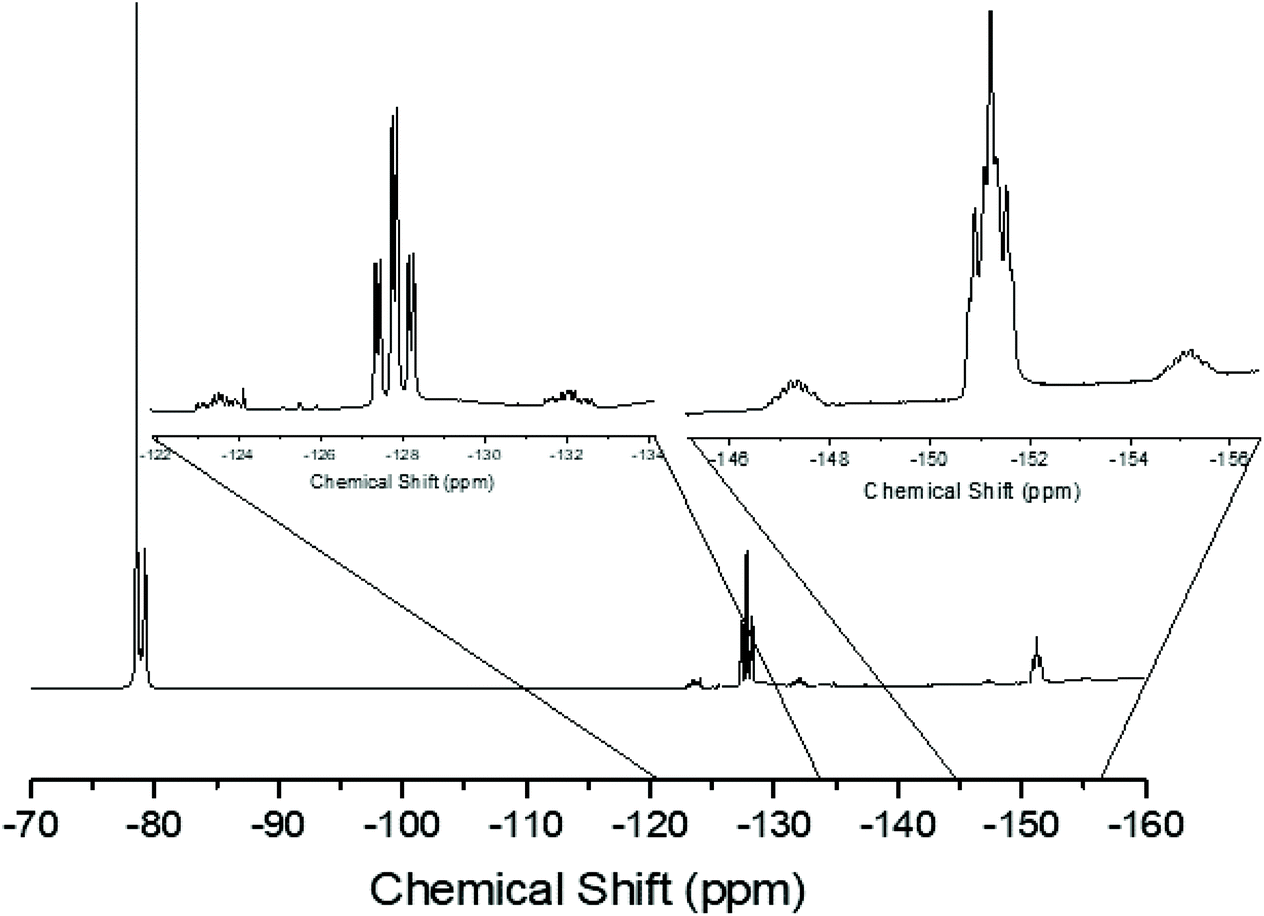 | ||
| Fig. 3 The 19F{1H} NMR spectrum of [SnF3(PMe3)2(OTf)] (CD2Cl2, 183 K). | ||
| Compound | δ(31P{1H})/ppm | δ(19F{1H})c/ppm | δ(119Sn)/ppm | 2 J(31P–19F)/Hz | 2 J(19F–19F)/Hz | 1 J(119Sn–31P)/Hz | 1 J(119Sn–19F)/Hz |
|---|---|---|---|---|---|---|---|
| a NMR data from CH2Cl2 (phosphine complexes) or CH3NO2 (N- and O-donor ligands) solution at 298 K. b Full data are in the Experimental section. c Non triflate resonances. d Data from ref. 20. e Data from ref. 18. f Not clear due to near coincident spectra for the two isomers; n.o. = not observed. g Averaged 117/119Sn couplings since separate resonances not resolved. | |||||||
| trans-[SnF4(PMe3)2]d | −19.1 (quin) | −132.8 (t) | −628.0 (t of quin) | 155 | 2975 | 2745 | |
| [SnF3(PMe3)2(OTf)] | −3.2 (td) | −127.9 (td) | −599.9 (m) | 153 | 41 | 3412 | 3253 |
| −151.4 (tt) | 121 | 3013 | |||||
| [SnF2(PMe3)2(OTf)2] | 10.2 (t) | −142.7 (t) | −609 (tt) | 107 | 3654 | 3393 | |
| [SnF(PMe3)2(OTf)3] | 18.3 (d) | −132.9(t) | −617 (m) | 88 | 3740 | 3557 | |
| trans-[SnF4(PiPr3)2] | 30.6 (quin) | −99.1 (t) | −622.0 (quin of t) | 124 | 2477 | 2920 | |
| [SnF3(PiPr3)2(OTf)] | 38.2 (q) | −73.5 (br) | −611.1 (tdt) | 121 | 2856 | 3518 | |
| −116.9 (br) | 3244 | ||||||
| [SnF2(PiPr3)2(OTf)2] | 49.4 (t) | −56.9 (br) | −630 (tt) | 117 | — | 2971 | 4082 |
| [SnF4(dmso)2] cis | −161.6 (t), −149.3 (t) | n.o. | 52, 52 | 2030, 2020 | |||
| trans | −149.0 (s) | −778.4 (quin) | 2466 | ||||
| [SnF3(dmso)3][OTf] mer | −153.7 (t), −145.8 (d) | −744 (m) | 59, 59 | 2063. 2181 | |||
| fac | −155.2 (s) | −735 (q) | 2063 | ||||
| [SnF4(pyNO)2] cis | −168.7 (t), −166.5 (t) | n.o. | 51, 51 | 2030, 2081 | |||
| trans | −164.2 (s) | −778.5 (quin) | 1951 | ||||
| [SnF3(pyNO)3][OTf] mer | −171.6 (t), −169.9 (d) | ∼−770 | 59, 59 | 2268, 2244 | |||
| fac | −172.4 (s) | ∼−770 | 2159 | ||||
| [SnF4(DMF)2] cis | −169.5 (t), −161.9 (t) | n.o. | 50, 50 | 2015. 1907 | |||
| trans | −161.8 (s) | n.o. | unclear | ||||
| [SnF3(DMF)3][OTf] mer | −169.2 (t), −164.0 (d) | n.o. | 55, 55 | 2143, 1985 | |||
| fac | −168.8 (s) | n.o | 2150 | ||||
| trans-[SnF4(py)2]d | −163.8 (s) | −670.8 (quin) | 1983 | ||||
| [SnF3(py)3][OTf] mer | −165.8 (t), −158.8 (d) | −771 (m) | 40, 40 | 1717, 1737 | |||
| fac | −157.2 (s) | −763 (q) | 1588 | ||||
| [SnF4(OPPh3)2]ecis | 42.3 | −159.8 (t), −146.2 (t) | −775.1 (ttt) | 53, 53 | 22f | 1850, 1730 | |
| Trans | 42.5 | −149.8 (s) | −770.0 (quin) | 20f | 1704 | ||
| [SnF3(OPPh3)3][OTf] mer | 46.0, 43.9 | −141.2 (t), −134.5 (d) | n.o. | 61, 61 | 1811, 2000 | ||
| fac | 41.7 | −142.1 (s) | 1878 | ||||
| [SnF2(OPPh3)4][OTf]2cis | 47.1, 48.4 | −122.4 (s) | n.o. | 96g, 78g | 2069 | ||
| trans | 50.6 | −123.1 (s) | n.o. | 78g | 1812 | ||
The reaction of trans-[SnF4(PMe3)2] with two equivalents of TMSOTf forms the complex [SnF2(PMe3)2(OTf)2] as identified by the microanalysis. Here the low temperature 19F{1H} and 31P{1H} NMR spectra are triplets which suggests that the complex has trans phosphines, with the fluorides either cis or trans. At 183 K both the 19F{1H} and 31P{1H} NMR spectra (ESI S3.2 and S3.3†) show, in addition to resonances of [SnF2(PMe3)2(OTf)2], some minor species which may be [SnF2(PMe3)2(OTf)2−n][OTf]n. Attempts to grow crystals for an X-ray study to confirm the geometry of [SnF2(PMe3)2(OTf)2] failed due to limited solution stability, and instead gave some crystals of the tin(II) complex, [Sn(PMe3)2(OTf)2] (see below). The reaction of trans-[SnF4(PMe3)2] with three equivalents of TMSOTf leads to the formation of the complex [SnF(PMe3)2(OTf)3]. The 19F{1H} and 31P{1H} spectra of this complex are sharp at room temperature; the 31P{1H} NMR spectrum shows a doublet resonance indicating equivalent phosphines (mutually trans), whilst the 19F{1H} NMR spectrum has two resonances, a singlet corresponding to triflate and a triplet corresponding to the tin bound fluoride, in a 9:1 ratio. This complex is not stable in solution and over a number of hours these resonances decrease in intensity and new resonances consistent with the formation of [PMe3F]+ appear.23 A few crystals deposited from the solution were revealed by structure solution to be a phosphine free tin(II) polymer, Sn3F5(OTf) (see below).
The sequential abstraction of fluoride from [SnF4(PMe3)2] leads to a high frequency shift in the phosphorus resonance and an increase in 1J119SnP and 1J119SnF, which is consistent with an increase in charge at the tin centre. There is a decrease in 2JPF as the series is traversed. The triflate has a trans influence on the fluorides, best demonstrated in the monotriflate complex where both the 2JPF and 1J119SnF couplings are smaller for the trans fluorine than for the cis. The 119Sn NMR spectra are expected to be complex, and in some the couplings are not all clearly resolved even at 183 K (Table 1 and ESI†).
In an attempt to generate cations, the reaction of TMSOTf with trans-[SnF4(PiPr3)2] was explored. The PiPr3 has a Tolman cone angle24 of 160° which compares with 118° in PMe3. Whilst it was possible to sequentially remove one or two fluorines from the tin, the resulting complexes had very limited stability and attempts to obtain X-ray crystallographic data were unsuccessful. However, based upon the microanalysis and multinuclear NMR data they were formulated as [SnF3(PiPr3)2(OTf)] and [SnF2(PiPr3)2(OTf)2], with the NMR data suggesting the triflates are most likely coordinated to the tin in solution at low temperature, like the trimethylphosphine complexes, although dissociated at room temperature. The reaction of [SnF4(PiPr3)2] with one equivalent of TMSOTf produces the complex [SnF3(PiPr3)2(OTf)]. The room temperature 31P{1H} NMR spectrum of which is a quartet, indicating the three Sn-bound F− groups are equivalent, whilst the 19F{1H} NMR spectrum has broad resonances flanked by tin satellites (as well as a triflate resonance), which sharpen as the temperature is lowered to 183 K. The room temperature data support the tentative assignment of the tin species as a trans-trigonal bipyramid, although cation-OTf interactions most likely occur at 183 K and in the solid state.
The reaction of [SnF4(PiPr3)2] with two equivalents of TMSOTf leads to the formation of [SnF2(PiPr3)2(OTf)2]. The 31P{1H} NMR spectrum of this complex is a triplet, whilst the 19F{1H} spectrum is a broad resonance at δ = −57.0, which is flanked by tin satellites. It is likely that weak interactions with the triflate anions may be present in solution, and the solution decomposes quickly. Curiously, the reaction of TMSOTf with [SnCl4(ER3)2] (E = P or As) only generated [SnCl3(ER3)2OTf]; further halides were not removed even with the addition of excess TMSOTf.10
As described above, decomposition of some of the triflate complexes in solution produced crystals of tin(II) compounds. From [SnF2(PMe3)2(OTf)2] one decomposition product was [Sn(PMe3)2(OTf)2] (Fig. 4), which has a four-coordinate primary coordination sphere around the Sn(II) centre, based upon a trigonal bipyramid with equatorial phosphines, axial triflates and a vacant equatorial vertex. There are weak long-range contacts between neighbouring molecules through a Sn-triflate interaction (3.044 Å) to yield a 1D polymer (ESI Fig. S8†). The Sn–P bond distances in this species are ca. 0.2 Å longer and the Sn-OTf ca. 0.3 Å shorter than those in the Sn(IV) complex, [SnF3(PMe3)2(OTf)] (above).
 | ||
| Fig. 4 Crystal structure of [Sn(PMe3)2(OTf)2] showing the atom labelling scheme. The ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Sn1–P1 = 2.7796(10), Sn1–P2 = 2.6610(8), Sn1–O1 = 2.523(3), Sn1–O4 = 2.342(3), P1–Sn1–P2 = 96.27(3), O1–Sn1–O4 = 154.39(10). | ||
One product from the decomposition of [SnF(PMe3)2(OTf)3] in solution is the Sn(II) phosphine-free compound, Sn3F5(OTf), which has an extended 2D sheet structure formed mainly through Sn–F bridges (see ESI. Fig. S9†).
Fluoro-tin(IV) cations with hard N- and O-donor ligands
The work described above shows that fluoride can be abstracted sequentially from tetrafluorotin(IV) phosphine complexes, to form tri-, di- and mono-fluoride complexes in which OTf coordination replaces the fluorides removed. However, these complexes mostly have limited stability, and attempts to introduce further phosphine into the coordination sphere, for example by reacting TMSOTf with [SnF4(κ2-triphos)] (bearing a pendant –PPh2 function) does not lead to formation of the intended [SnF3(κ3-triphos)][OTf]. Similarly, adding PMe3 to [SnF3(PMe3)2(OTf)] did not lead to clean substitution of the OTf. Hence, in this section we explore fluoride abstraction from tetrafluorotin(IV) complexes containing hard, neutral nitrogen and oxygen donor ligands in the presence of further neutral ligand. The neutral complexes [SnF4(L)2] (L = dmso, pyNO, OPPh3, py, DMF) have been reported in the earlier literature,8,13,14,19 but most with limited data. The key spectroscopic data are given in Table 1 and the Experimental section. The structures of trans-[SnF4(OPR3)2] (R = Me, Ph)8,19 have been determined previously and those of trans-[SnF4(pyNO)2] and trans-[SnF4(py)2] are shown in Fig. 5. The NMR spectra of most of the [SnF4(L)2] complexes show both cis and trans isomers present in solution, with the relative amounts varying significantly with L (Table 1) (Scheme 2).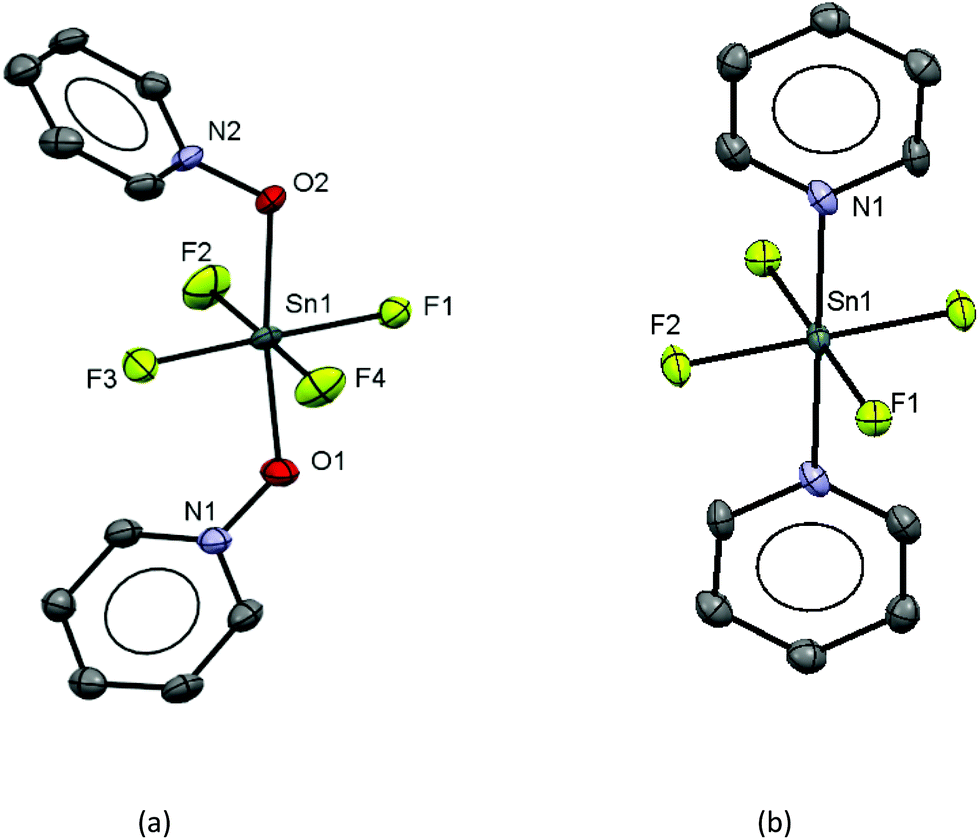 | ||
| Fig. 5 Crystal structures of (a) [SnF4(pyNO)2]·CH2Cl2 and (b) the centrosymmetric [SnF4(py)2] showing the atom labelling schemes. The ellipsoids are drawn at the 50% probability level and H atoms and lattice CH2Cl2 are omitted for clarity. There are two crystallographically independent [SnF4(pyNO)2]·CH2Cl2 moieties in asymmetric unit and only one is shown. Selected bond lengths (Å) and angles (°) are: (a) Sn1–F1 = 1.945(3), Sn1–F2 = 1.945(4), Sn1–F3 = 1.943(4), Sn1–F4 = 1.953(3), Sn1–O1 = 2.081(3), Sn1–O2 = 2.081(3), N1–O1 = 1.364(5), N1–O2 = 1.359(5), O1–Sn1–O2 = 172.29(15), F1–Sn1–F3 = 177.59(16), F2–Sn1–F4 = 177.20(16); (b) Sn1–F1 = 1.954(7), Sn1–F2 = 1.956(7), Sn1–N1 = 2.175(10), F1–Sn1–F2 = 90.1(3), F1–Sn1–N1 = 90.0(3), F2–Sn1–N1 = 90.0(3). | ||
 | ||
| Scheme 2 | ||
The complexes were mostly poorly soluble in CH2Cl2 and generally solution data were acquired from CH3NO2/CD3NO2 solutions, with stronger donor solvents being avoided since these may displace the neutral ligands from the tin. The high melting point of MeNO2 (244 K) limited low temperature NMR studies. The isomers present are readily identified from the 19F{1H} NMR spectra with the trans showing a sharp singlet resonance and the cis, two triplets, in both cases with accompanying 117/119Sn satellites. The relative amounts of the two isomers are solvent dependent, although we have not explored this in any detail. The 1JSnF couplings are large and usually the separate couplings to the two tin isotopes were resolved, despite their similar values (the magnetogyric ratio 119Sn/117Sn is 1.046) (Table 1). A typical example is shown in Fig. 6.
 | ||
| Fig. 6 The 19F{1H} NMR spectrum of [SnF4(dmso)2] (298 K) showing the resonances of the two isomers with 117/119Sn satellites. | ||
The reactions of the [SnF4(L)2] complexes with one equivalent of TMSOTf, followed by addition of one equivalent of L, afforded [SnF3(L)3][OTf] (L = dmso, py, pyNO, DMF, OPPh3). The 19F{1H} NMR spectra show that in solution both the mer- and fac-isomers are present, the former with doublet and triplet resonances due to 2JFF, and the latter a singlet, again all with 117/119Sn isotope satellites. A typical example is shown in Fig. 7.
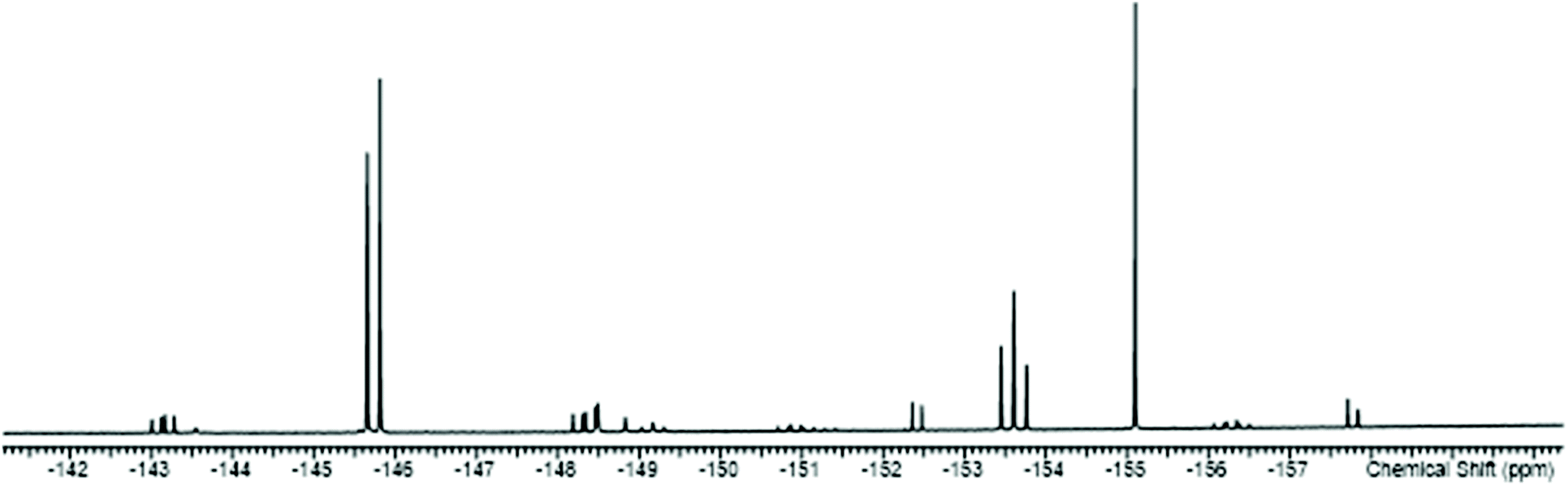 | ||
| Fig. 7 The 19F{1H} NMR spectrum of [SnF3(dmso)3][OTf] (298 K) showing the resonances of the two isomers with 117/119Sn satellites (the OTf resonance is not shown). | ||
Observing the 119Sn spectra proved more challenging due to a combination of factors, including the presence of two isomers, the complex multiplet patterns which often overlapped, and the modest receptivity of 119Sn (Dc = 26). Typically, the central lines of the multiplets were identified, allowing δ(119Sn) be determined, but the weaker outer lines of the multiplets were often less clear. Several complexes, including [SnF4(DMF)2], [SnF3(DMF)3][OTf] and [SnF3(OPPh3)3][OTf], failed to exhibit a 119Sn resonance (in CH3NO2 at 253 K), presumably due to reversible neutral ligand dissociation on the tin NMR timescale. Similar behaviour has been reported for [SnF4(MeCN)2].8 Attempts to isolate pure samples of difluorotin(IV) cations [SnF2(L)4][OTf]2 from [SnF4(L)2], with two equivalents of TMSOTf and excess L, proved more challenging. Typically, mixtures of [SnF2(L)4][OTf]2 and [SnF3(L)3]OTf were identified in situ by the 19F{1H} NMR spectra. It did prove possible to isolate an analytically pure sample of [SnF2(OPPh3)4][OTf]2. The 19F{1H} and 31P{1H} NMR spectra of this complex confirmed that both cis and trans isomers of the phosphine oxide complex were present in CH3NO2 solution, with the cis form the more abundant. However, we did not observe a 119Sn NMR spectrum from this complex even at 253 K (the lower limit for CH3NO2). The X-ray crystal structure of [SnF2(OPPh3)4][OTf]2 showed it to be of the trans isomer (Fig. 8).
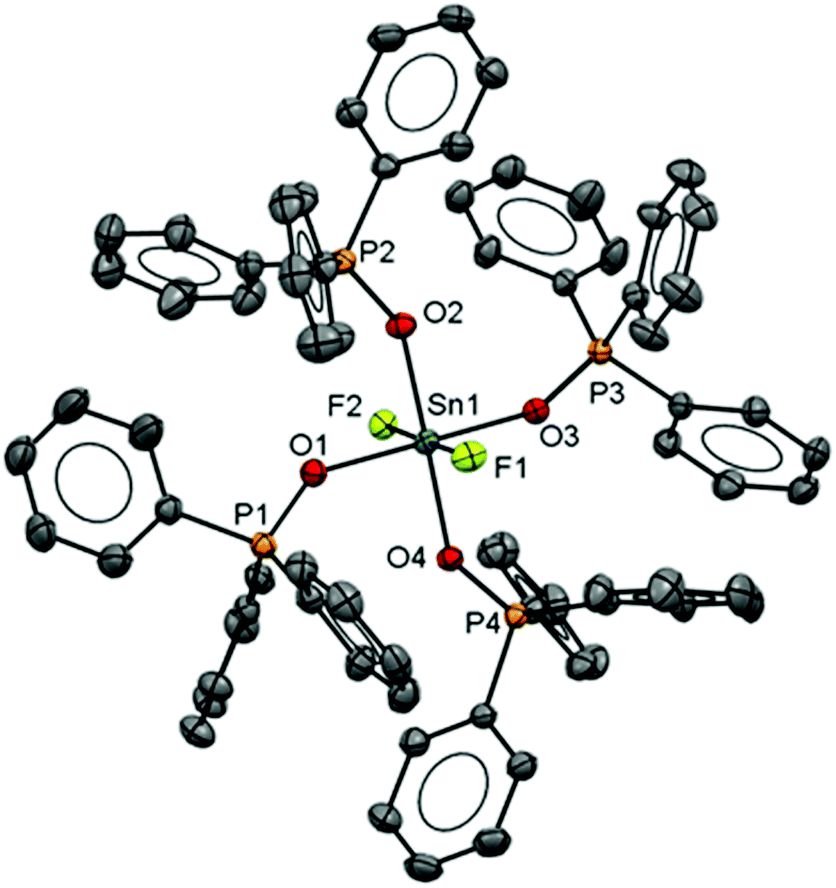 | ||
| Fig. 8 Structure of the cation in [SnF2(OPPh3)4][OTf]2 showing the atom labelling scheme. The ellipsoids are drawn at the 50% probability level and H atoms and OTf anions are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Sn1–F2 = 1.9314(16), Sn1–F1 = 1.9164(17), Sn1–O3 = 2.040(2), Sn1–O4 = 2.0447(19), Sn1–O2 = 2.040(2), Sn1–O1 = 2.057(2), F1–Sn1–F2 = 178.78(7), O3–Sn1–O1 = 176.24(8), O–2Sn1–O4 = 178.85(8). | ||
The tin environment is very close to a regular octahedron and the d(Sn–F) are rather shorter than in the neutral [SnF4(L)2] (L = pyNO, py) complexes above, although the d(Sn–O) are not significantly different to those in the neutral parent complex trans-[SnF4(OPPh3)2].8
The [SnF3(L)3][OTf] complexes are less robust in solution than the neutral [SnF4(L)2], and upon standing, resonances corresponding to varying amounts of decomposition/disproportionation products, including [SnF4(L)2], were observed in some systems, especially for L = OPPh3 and DMF. It is notable that the crystal used to determine the structure of [SnF4(py)2] (above) was grown from a sample of [SnF3(py)3][OTf], and our inability to isolate pure [SnF2(L)4][OTf]2 with other N- or O-donor ligands suggest some ligand redistribution occurs in solution.
Conclusions
A series of new triflate complexes of tin(IV) fluoride with neutral donor co-ligands have been synthesised. Variable temperature multinuclear NMR spectroscopic data show that soft phosphine ligands lead to neutral tri- and (unstable) di- and mono-fluoride complexes with two trans phosphines and one, two or three κ1-OTf groups, respectively. The triflate is not displaced by added phosphine (even using the potentially tridentate triphos ligand). In contrast, harder O- and N-donor ligands form the six-coordinate mer- and fac-[SnF3L3]+ (L = dmso, dmf, OPPh3, pyNO and py) monocations, with further TMSOTf producing cis- and trans-[SnF2(OPPh3)4]2+ dications, the latter being confirmed as the trans isomer in the solid state by X-ray analysis.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank EPSRC for funding via the ADEPT Programme grant (EP/N035437/1), a studentship to R. P. K. (EP/N509747/1) and GE Healthcare and EPSRC for a CASE studentship to M. S. W. (EP/R513325/1).References
-
(a) S. Aldridge and C. Jones, Chem. Soc. Rev., 2016, 45, 763 RSC
and references therein; (b) K. Chansaenpak, B. Vabre and F. P. Gabbaı, Chem. Soc. Rev., 2016, 45, 954 RSC
-
P. G. Harrison, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, J. A. McCleverty and R. D. Gillard, Pergamon, Oxford, 1988, vol. 3, p. 183 Search PubMed
-
J. Parr, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 3, p. 545 Search PubMed
-
The Chemistry of Tin, ed. P. J. Smith, Chapman & Hall, London, 1998 Search PubMed
-
P. G. Harrison, The Chemistry of Tin, Blackie, London, 1989 Search PubMed
- G. R. Willey, T. J. Woodman, U. Somasundaram, D. R. Aris and W. Errington, J. Chem. Soc., Dalton Trans., 1998, 2575 Search PubMed
- G. R. Willey, A. Jarvis, J. Palin and W. Errington, J. Chem. Soc., Dalton Trans., 1994, 255 RSC
- M. F. Davis, M. Clarke, W. Levason, G. Reid and M. Webster, Eur. J. Inorg. Chem., 2006, 2773 CrossRef CAS
- E. MacDonald, L. Doyle, S. S. Chitnis, U. Werner-Zwanziger, N. Burford and A. Decken, Chem. Commun., 2012, 48, 7922 RSC
- V. K. Greenacre, R. P. King, W. Levason and G. Reid, Dalton Trans., 2019, 48, 17097 RSC
- M. Bork and R. Hoppe, Z. Anorg. Allg. Chem., 1996, 622, 1557 CrossRef CAS
-
(a) S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460 RSC
- C. E. Michelson, D. S. Dyer and R. O. Ragsdale, J. Inorg. Nucl. Chem., 1970, 32, 833 CrossRef CAS
- C. J. Wilkins and H. M. Haendler, J. Chem. Soc., 1965, 3174 RSC
- S. H. Hunter, V. M. Langford, G. A. Rodley and C. J. Wilkins, J. Chem. Soc. A, 1968, 305 RSC
- A. D. Adley, P. H. Bird, A. R. Fraser and M. Onyszchuk, Inorg. Chem., 1972, 11, 1402 CrossRef CAS
- J. P. Clark, V. M. Langford and C. J. Wilkins, J. Chem. Soc. A, 1967, 792 RSC
-
(a) D. Tudela and F. Rey, Z. Anorg. Allg. Chem., 1989, 575, 202 CrossRef CAS
- M. F. Davis, W. Levason, G. Reid and M. Webster, Polyhedron, 2006, 25, 930 CrossRef CAS
- R. Suter, A. Swidan, C. L. B. Macdonald and N. Burford, Chem. Commun., 2018, 54, 4140 RSC
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS
- A. M. Forster and A. J. Downs, Polyhedron, 1985, 4, 1625 CrossRef CAS
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Multinuclear NMR and IR spectra associated with each of the new compounds described, together with the crystal structure of Sn3F5(OTf), the X-ray crystallographic parameters. CCDC 2104490–2104493, 2104976, 2104984 and 2106812. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02948g |
| This journal is © The Royal Society of Chemistry 2021 |