
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme catalysis powered micro/nanomotors for biomedical applications
Motilal
Mathesh†
,
Jiawei
Sun†
and
Daniela A.
Wilson
 *
*
Institute of Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands. E-mail: d.wilson@science.ru.nl
First published on 22nd July 2020
Abstract
With recent developments in the field of autonomous motion for artificial systems, many researchers are focusing on their biomedical application for active and targeted delivery. In this context, enzyme powered motors are at the forefront since they can utilize physiologically relevant fuels as their substrate and carry out catalytic reactions to power motion under in vivo conditions. This review focuses on the design and fabrication of enzyme powered motors together with their propulsion mechanism by using fuels present in biological environments. In addition, the recent advances in the field of enzyme powered motors for biomedical applications have been discussed together with the parameters that need to be considered for designing such systems. We believe that this review will provide insights and better understanding for the development of next generation biomedical technologies based on enzyme powered motors.
 Motilal Mathesh | Motilal Mathesh received his PhD degree (2016) from Deakin University (Australia), in the field of bio-nanotechnology. During his PhD studies, he worked on enzyme architectonics on graphene oxides. He joined Prof. Wilson's group in 2017 as a postdoctoral fellow and was awarded the Marie Curie Individual fellowship in 2018. Recently, he was awarded the Alfred Deakin Postdoctoral Fellowship and joined as a Research Fellow at Deakin University, Australia. His current research interest focuses on the design and fabrication of supramolecular nanomotors for biomedical applications and light driven systems. |
 Jiawei Sun | Jiawei Sun graduated from Sichuan University, Chengdu (China) in 2017 with her master's degree in (MSc) Pharmaceutical Science. Currently, she is a PhD candidate in the Department of Systems Chemistry at Radboud University Nijmegen, in the Netherlands. Her research focuses on developing self-propelled assembled nano-motors with the enzyme system as a fuel, and observing the potential for biomedical application, such as drug delivery and biosensing. |
 Daniela A. Wilson | Daniela A. Wilson received her PhD degree (2007) “summa cum laude” from “Gh. Asachi” Technical University of Iasi, Romania. She is currently a full professor and chair of the systems Chemistry Department at the Institute for Molecules and Materials, Radboud University, Nijmegen, in the Netherlands. She is also a theme leader for nanomedicine in the Radboud Institute for Molecular Life sciences. Her research interests focus on the design of intelligent, self-propelled, and self-guided supramolecular assemblies and their communication and interaction as next generation nanoengineered delivery systems. |
1. Introduction
Nanotechnology has played a crucial role over the past few decades in the development of nanomedicine, from targeted drug delivery, therapeutics, and diagnostic agents to the fabrication of biosensors and medical devices for maximizing their positive responses while minimizing side effects.1 In the past decade, micro/nanomotors (MNMs) have been proved to be a potential tool for addressing complicated in vivo problems, such as drug delivery, cell sensing, imaging, wound healing and tissue uptake. Compared to passive diffusion, active motion is powerful and shows faster cargo delivery and overcomes drawbacks such as systemic toxicity and reliance on passive diffusion for transport.2 The idea of fabricating artificial motors is inspired from biological motors in living systems that are ubiquitous and capable of performing precise tasks like intracellular transport, cell division, and cell locomotion by converting chemical energy into mechanical force.3 One of the typical examples is kinesin, a motor protein, that is able to walk along microtubules by unbinding and rebinding to the filaments through adenosine triphosphate (ATP) hydrolysis.4,5 In past decades, tremendous progress has been made in developing hybrid (biological and non-biological) devices with ATP-dependent motor proteins such as kinesin, myosin, and dynein. Back in 2001, Vogel et al. developed a molecular shuttle with kinesin protein motors, capable of moving cargo along an engineered pathway using ATP as a fuel.6 Whitesides et al. designed a synthetic motor powered by hydrogen peroxide (H2O2) in which platinum (Pt) was introduced on the surface of the motor as a catalyst to decompose H2O2, resulting in an impulse of oxygen bubbles for autonomous motion.7 Since then mimicking the behavior of bio-motors and bio-organisms by synthetic motors with catalysts has attracted considerable attention, together with intensive studies towards the realization of fuel dependent self-propulsion of synthetic MNMs.8 H2O2 has been widely used as a fuel because of the production of oxygen, while Pt has also become a popular catalyst. Besides, H2O2, hydrazine,9 water,10 acid,11 base,12 and bromine (iodine)13 have also been used as fuels for propelling the motors, with iridium (Ir), aluminum (Al), zinc (Zn), Al/palladium (Pd) and Pt being used as catalysts.In spite of great progress in this field, we are still far from using motors for in vivo tasks due to the bottlenecks present. The main limitations are their ability to move in biological fluids, the size and the poor biocompatibility of both materials used for fabrication and the fuels used for active motion.14 For biomedical applications, enzymes are ideal candidates as catalysts that can power the motors because of their biocompatibility, high turnover numbers and great selectivity under physiological conditions.15,16 Back in 2005, Heller et al. reported the propulsion of carbon microfibers at an air–glucose interface by the ion flow produced by a catalytic reaction, using glucose oxidase (GOx) and bilirubin oxidase (BOD) as catalysts.17 From then on, more and more enzymes have been introduced in the intelligent systems as biocatalysts for developing enzyme-powered motors (EPMs), such as catalase,18,19 urease,20–22 atpase,23 lipase24etc. Besides biocatalysts, the use of biocompatible substrates is also essential, in particular, the ones present in living systems are considered as good candidates. Motors with proof of concept studies relying on toxic fuels limit their use for biomedical application, since it is not possible to use them in in vivo scenarios.25 Moreover, the external addition of substrates to the biological system seems unrealistic, with their safety issues that need to be evaluated. To have a better view of this field, several reviews have been published recently. The mimicking of natural motility behaviour using synthetic soft materials has been discussed by Samuel Sanchez et al.26 The biocompatibility of propulsion techniques has been discussed by Halder and Sun.27 Several reviews towards biomedical applications have also been published. In 2018, Guan et al. discussed the use of micro/nanorobots for active drug delivery.28 Besides, Soto et al. discussed the use of micro/nanomotors for medical applications and emphasized the current and foreseeable perspectives of their commercialization.29 Moreover, the importance of biocompatibility of motors for biomedical applications has also been discussed by Peng et al.30 As a promising catalyst candidate, using enzyme to power micro- and nanoswimmers has been discussed by Sanchez et al.31 and Sen et al.15 An overview of most recent advances of micro/nanomotors has been discussed by Städler et al.32 However, of all the reviews, using enzyme motors for biomedical applications has not been discussed. The importance of biofuels and the accessibility of biofuels for in vivo powering motors are also essential for the future perspectives of enzyme powered motors.
Our discussion in this review will first focus on the biological fuels available in biological systems and will highlight several possibilities of using them for motion, as they are not adequately reported in the literature. Besides, several popular biocatalysts that can carry out catalytic reactions using these biological fuels as substrates are provided. As already described before, MNMs equipped with inorganic catalysts will not be discussed here as it is not the scope of this review.33,34 The chemotaxis behavior of EPMs is also discussed, as they are essential for further biomedical applications. Finally, the recent progress on EPMs as well as their biomedical applications based on the material used for fabrication has been addressed. At the end, an outlook of current challenges and future prospects to enable them for in vivo applications is discussed. We believe that this review will lay a platform for the design and fabrication of autonomous, intelligent and stimuli responsive systems for next generation in vivo focused MNMs.
2. Fuels in biological environments
Using enzyme-powered micro/nanomotors for biomedical applications holds great promise for many medical challenges, such as targeted drug delivery, bio-sensing and imaging. Designing such a system has quite some restrictions, with primary challenges such as the fuels that can be used, whether the fuels are available in a biological system and whether the fuel concentration (conc.) can support the motion of motors, therefore warranting the urgent search for a “green fuel” present in the biological system. Furthermore, the biological microenvironment with pH and temperature changes also needs to be considered, and also whether the addition of fuels to the system could change the microenvironment or if the product from the catalytic reactions could induce a toxic response in our body should be studied.33,35 In the next section, we are going to look at the possible fuels for MNMs that can be used for biomedical applications and discuss their possible associated risks.2.1 Hydrogen peroxide (H2O2)
As a reactive oxygen species (ROS), H2O2 is involved in a wide variety of physiological functions, from cytokine, insulin and AP-1 signaling to redox regulation.36–40 H2O2 is present in all aerobic metabolism, and it is generated through a variety of extracellular and intracellular actions. The biochemical pathways for the generation and metabolism of H2O2 have been reported by Hua Cai40 (Fig. 1a) showing that the mitochondrial electron transport chain, the arachidonic acid metabolizing enzymes lipoxygenase and cycloxygenase, the cytochrome P450s, xanthine oxidase, NAD(P)H oxidases, uncoupled nitric oxide synthase (NOS) and peroxidases can all be possible ROS enzymatic sources, which is also supported by other research studies.39–43 However, it is noteworthy that H2O2 is also considered to be a cytotoxic agent, and it can be harmful to cells when it reaches certain conc. (∼50 μM), resulting in the oxidation of DNA, lipids and proteins.44,45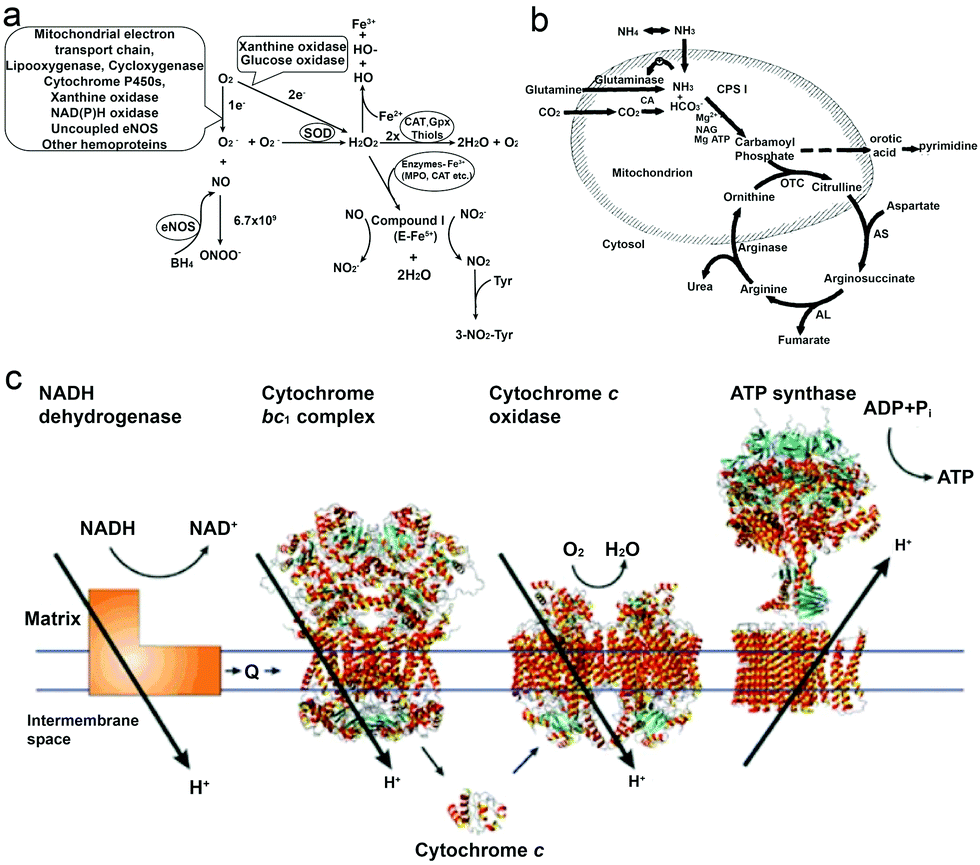 | ||
| Fig. 1 (a) Biochemical pathways of H2O2 generation and metabolism. Molecular oxygen undergoes one or two-electron reduction (1 or 2 e−) to form superoxide (O2−) or H2O2, respectively. The degradation of H2O2 involves intracellular catalase (CAT), extracellular glutathione peroxidase (GPX) or small molecules like thiols.40 (b) Formation of urea from carbon dioxide and ammonia within the hepatocyte. Bicarbonate is formed from carbon dioxide by the action of carbonic anhydrase (CA).54 (c) Electrons are transferred from NADH dehydrogenase to cytochrome c oxidase by coenzyme Q (Q), the cytochrome bc1 complex and cytochrome c. The established proton gradient across the inner mitochondrial membrane drives the proton flow in ATP synthase that accompanies ATP synthesis.55 Adapted with permission from ref. 40. Copyright 2002 Oxford University Press. Adapted with permission from ref. 54. Copyright 1994 Wiley. Adapted with permission from ref. 55. Copyright 2001 Springer Nature. | ||
Recent progress in MNMs has made H2O2 an important fuel in powering micro/nanomotors, since they can be decomposed into water (H2O) and oxygen (O2) bubbles (2H2O2 → 2H2O + O2), in which oxygen bubbles provide the thrust to propel motors.46 The propulsion mechanism associated with these systems is the so-called bubble propulsion, in which the fuel can be decomposed by a motor equipped with catalysts to form micron sized gas bubbles and released from the motor surface, thus driving the motor directionally away from the catalyst.35 The most widely used biocatalysts for the decomposition of H2O2 are Pt and catalase (EC 1.11.1.6),16 of which catalase being one of the most efficient enzymes reported is suggested for biomedical applications because of its biocompatibility and high turnover rate (kcat = 2.12 × 105 s−1).47,48 Moreover, research studies have also shown that human tumor cells produce more H2O2 than normal cells,49,50 as high conc. of ROS is required for promoting cell cycle progression and differentiation.51–53 The difference in conc. between normal cells and tumor cells may create a gradient of H2O2, which helps motors to perform chemotaxis behavior for achieving targeted delivery.56 However, whether H2O2 present in tumor tissues would promote the motion of motors still needs to be investigated, since most of the studies rely on the addition of fuels externally with almost no in vivo effect reported to the best of our knowledge. Moreover, the amount of fuel needed for the motor sustained motion is variable, and the differences in size and materials can also influence the motion behavior.
2.2 Urea
Urea (CO(NH2)2) is an essential metabolite containing nitrogen in the human body, produced by the metabolic transformations and catabolism of amino acids, to eliminate the generated nitrogen as the waste in our body. It is mainly expressed in the liver by the so-called urea cycle54 (Fig. 1b), in which five enzymes are involved: carbamoyl phosphate synthetase-I (CPS-I), ornithine transcarbamylase, argininosuccinate synthetase (ASS), argininosuccinate lyase (ASL), and arginase.57,58 After being produced in the liver, urea is then transported from the blood and excreted mostly through urine by the kidneys, with some being hydrolyzed to ammonia in the lumen of the large intestine by urease-producing bacteria.59,60 The blood urea conc. in the human body was reported by MacKay et al. in 1927,61 to be 29.3 mg of urea in 100 cc of blood (≈5 mM) on average, and studies from other groups have also reported similar results.62 Furthermore, the conc. in urine can vary but it is much higher than in plasma, and Bailly et al.63 reported a conc. of 1.91 g L−1 (≈30 mM) while Raftery et al.62 recently reported the conc. To be around 126 mM. Moreover, the increase of urea conc. in plasma might relate to the imbalance between production and excretion, which can indicate several diseases, such as chronic-persistent hepatitis (CPH), chronic-active hepatitis (CAH) and liver cirrhosis.64,65Urea has become a popular fuel in powering motors in recent years, and it can be hydrolyzed by urease into ammonium ions (NH4+) and bicarbonate anions (HCO3−). Interestingly, a study by Butler et al. has shown that during hydrolysis, the diffusion coefficient of urease is increased, and the change is considered to be a result of the formation of the local electric field generated by the faster diffusion of NH4+ ions.66 Furthermore, when urease is introduced on the surface of a particle, the biocatalytic conversion of urea can result in a diffusive motion of the particles.67 However, to power urease motors, the conc. required for the fuel is quite high, even though urease is relatively robust and has a high turnover rate (kurease = 2.34 × 104 s−1) compared to other enzymes.68,69 As reported by researchers, 50 mM conc. is necessary for the particles to move and show an increased diffusion coefficient,15,31,70 which is hard to be achieved in normal biological fluids, except urine. However, if the motors can be used in organs where there is higher urea conc., they would be advantageous for biomedical application, since particles will not show motion before reaching the targeted area. For example, in the treatment of bladder cancer or in intravascular drug/gene delivery, passing through the bladder permeability barrier is considered to be tougher than passing through the blood–brain barrier (BBB),71,72 and in such a scenario, active particles might be helpful for the delivery of drugs benefitting from the enhanced penetration efficiency and a recent study by Sánchez et al.67 has already proved the possibility of this strategy. However, the risk of introducing urease into our body also needs to be considered.73
2.3 Glucose
Glucose is an essential nutrient for the human body and a major energy source for cells delivered by the bloodstream. The circulation of glucose in the bloodstream is mainly due to exogenous nutrients (food) and endogenous glucose production. The liver plays a major role in controlling the conc. of blood glucose by balancing the uptake and storage and the release of glucose via several metabolic pathways like glycogenesis, glycogenolysis and gluconeogenesis. In these procedures, β-cells act as sensors and secret insulin for the regulation of glucose.74 The normal conc. of glucose in the blood stream is between 3.9 and 7.1 mM with the level varying throughout the day.75 It is worth noting that the liver glucose conc. can be influenced by the plasma glucose conc. in an opposite way, i.e. when the plasma conc. increases, the liver shows a lower conc. and vice versa.76 Moreover, in many solid tumor tissues, the glucose level is lower than normal tissues (less than 1 mM), because of the disorganized vasculature system and the inefficient capillary bed.77–79 Glucose 1-oxidase (EC 1.1.3.4) is a FAD-dependent enzyme consisting of two identical 80 kDa subunits, widely used to catalyze the oxidation of β-D-glucose (C6H12O6) to D-gluconolactone (C6H10O6) and H2O2 in the presence of molecular O2.80 Besides glucose oxidase (GOX), three other types of enzyme can also oxidize glucose: glucose dehydrogenases, quinoprotein glucose dehydrogenases and glucose 2-oxidases. Although glucose dehydrogenases require a soluble cofactor, quinoprotein glucose dehydrogenase is not so stable and glucose 2-oxidases lack specificity; these drawbacks of other enzymes make GOX a better choice for applications.81 GOX was first discovered from Aspergillus niger, and it can also be produced naturally in some fungi and insects. As it is considered to be safe, GOX has been used as an additive in food processing for decades, and also for the fabrication of glucose biosensors.82 It has been coupled together with catalase or other inorganic catalysts for cascade catalytic reactions, to power MNMs. Recently, Zhang et al. reported a motor system for tumor therapy, in which glucose oxidase (GOD) and manganese dioxide (MnO2) were used as pair enzymes to consume glucose in solid tumor, resulting in glucose depletion and significant tumor suppression.83 Further research is required for biomedical applications using glucose powered motors.2.4 Adenosine 5′-triphosphate (ATP)
It is well known that ATP is an essential cellular energy. It plays an important role in many biological processes such as muscle contraction, synthesis and degradation of biological molecules and intracellular or extracellular signaling.84 ATP is synthesized from adenosine diphosphate (ADP) and inorganic phosphate (Pi) by F1F0–ATP synthase in an energy-requiring reaction. In F1F0–ATP synthase, the F0 portion of the ATP synthase synthesises ATP in biological systems and the F1 portion of ATP is embedded in the membrane and catalyzes ion translocation, while an electrochemical proton (or Na+) gradient provides energy for ATP synthesis.55,85 The human plasma ATP conc. has been measured, around 1 μmol L−1,86 while the intracellular conc. is maintained between 1 and 10 mmol L−1.87,88 However, it is noteworthy that the addition of extracellular adenosine (Ade) is proved to be toxic to cells.89ATP has been used as a fuel for many motor proteins found in eukaryotic cells, for example ATP transports kinesins along the microtubule filaments based on ATP binding, hydrolysis and ADP release.90 ATP synthase itself has been known to be a ubiquitous biological nanomotor, because of the rotation motion during ATP synthesis.23,91 This biological nanomotor has also been used in developing devices with multiple functions in terms of monitoring, diagnosing and curing diseases. Kinesin, as one of the ATP motor proteins, has been used for making molecular shuttles for constructing nanoscale assembly lines,6 and transporting and stretching DNA molecules across a surface.92 However, the use of ATP might cause the accumulation of ADP which inhibits the activity of these biomotors,93 and hence the balance between enzymes and substrates still needs to be addressed.
2.5 Other fuels
In the quest for physiologically relevant fuels, other chemicals present in the biological system were also studied by researchers as fuels for powering motors; for instance, a major extracellular matrix (ECM) constituent, collagen, has been used as a fuel for collagenase powered superparamagnetic nanoparticles, and demonstrated enhanced tissue penetration.94 Triglyceride, a major constituent of body fat in humans, which is also present in blood for the bidirectional transference of adipose fat and blood glucose from the liver,95 was recently used as a fuel for powering motors based on lipase, and the study shows that lipase motors can destroy triglyceride droplets while consuming triglyceride.96 Apart from them, a plethora of fuels still needs to be investigated for powering MNMs.3. Enzyme chemotaxis behavior
In nature, microorganisms control the directionality of their motion by sensing the environment. One of the important behavioural responses is called chemotaxis, meaning that microorganisms can sense and move along the gradient of chemical species, to find and locate a region with better living conditions.97,98For example, bacteria have a molecular memory system to help them move accordingly in a chemical gradient. In recent years, the motion of enzymes has been widely studied and reported to be due to binding or unbinding the substrate or by chemotaxis behavior. In 2009, Schwartz et al. reported a complex molecular motor driven by transcription substrates—nucleoside triphosphates (NTPs) that undergo a biased movement along a conc. gradient of the substrate for NTPs.102 Sen et al. also reported the chemotaxis behavior in many models, in which experiments used a microfluidic device with two inlets and one outlet channel, with a fluorescently labeled enzyme in one inlet and the substrate solution passing through the other as shown in Fig. 2a. Using this device, the chemotaxis behavior of catalase, urease and DNA polymerase was observed, and reported to move toward a higher substrate conc. area.99 A study by Shelley D. Minteer et al. of mitochondrial malate dehydrogenase and citrate synthase using similar techniques also proved the chemotaxis behavior among enzymes.103 For the study of enzymatic catalysis of enzymes involved in cascade reactions, devices with multiple inlets were used, where enzymes involved in cascade reactions were observed to move independently along their own specific substrate gradient, while the chemotactic assembly of enzymes was also observed under cytosolic crowding conditions.104 In another research study, the chemotactic behavior of enzymes has been used for the separation of active and inactive enzymes with a two-inlet five-outlet microfluidic device.68 Furthermore, the anti-chemotaxis behavior of enzymes has also been reported both experimentally and theoretically, showing that enzymes move away from their substrate,105,106 which might be a result of different experimental conditions, the observation time and the conc. difference allowing them to be closer to the shape of the ultimate steady state distribution of the enzyme, leading to a contrary result.107 For an in-depth mechanistic understanding of the chemotaxis behaviour, several studies have analysed it in more detail. The first theory for chemotaxis was proposed by Schurr et al.108 They suggest that a thermodynamic force due to ligand binding might drive the chemotaxis behaviour of enzymes, which has been proved experimentally by Zhao et al.109 Recently, the potential mechanisms of enzyme chemotaxis have been discussed in the review by Gilson et al.,110 in which very detailed discussions were given. First, a theoretical framework for mechanisms was discussed based on the Fokker–Planck (FP) equation. Second, the mechanisms based on the force-induced drift and position-dependent diffusion coefficient studied in recent years were summarized. Here these theories will not be discussed, however, the mechanism of chemotactic behaviour of EPMs could be essential in EPM based research.
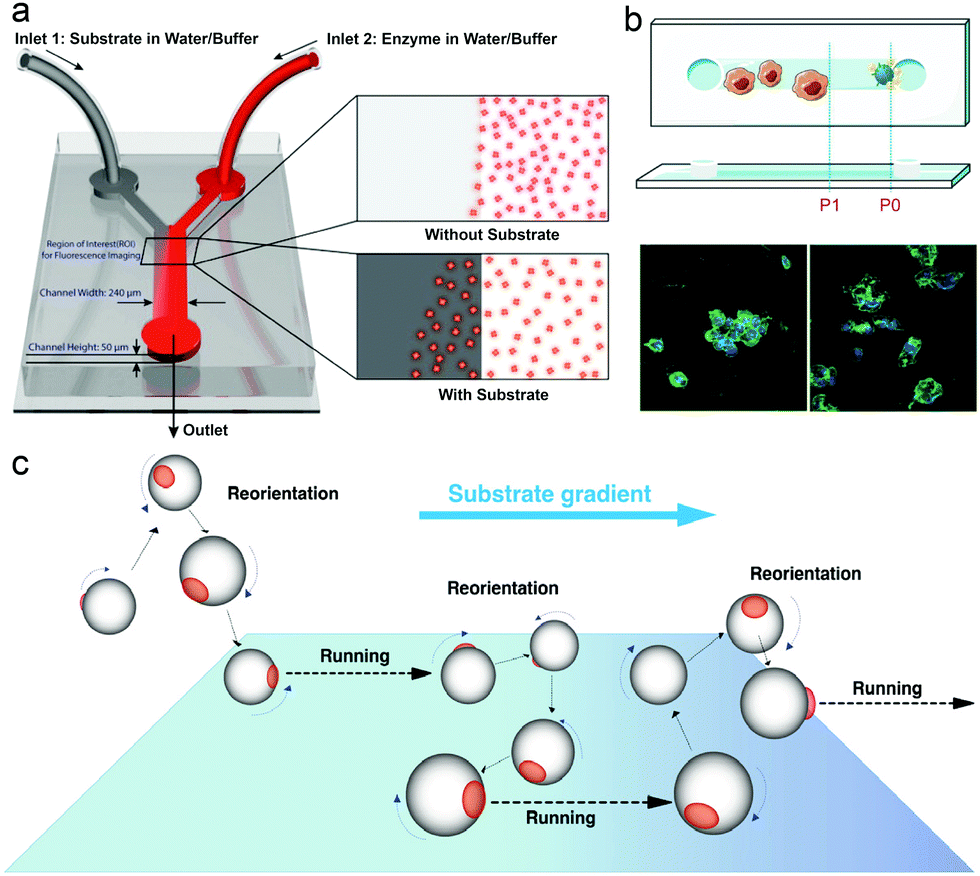 | ||
| Fig. 2 (a) Schematic representation of the Y-shaped microfluidic channel used for chemotactic studies of ensembles of enzyme molecules.99 (b) Schematic overview of the chemotactic model. Raw 264.7 cells before and after being triggered by PMA (green: actin cytoskeleton, blue: cell nuclei).100 (c) Schematics of the proposed mechanisms of asymmetric polymersome chemotaxis, which consist of alternative running and reorientation events.101 Adapted with permission from ref. 99. Copyright 2013 American Chemical Society. Adapted with permission from ref. 100. Copyright 2020 Wiley. Adapted with permission from ref. 101. Copyright 2017 American Association for the Advancement of Science. | ||
With the development of EPM systems, the effect of chemotaxis behavior of enzymes on EPM systems needs to be addressed. Recently, Battaglia et al. reported asymmetric polymersomes powered by glucose oxidase and catalase, and in this system, motors were observed in response to an external gradient of glucose and move toward regions with higher conc. of substrate glucose according to the superdiffusional behaviors in the presence of a gradient (Fig. 2c).101 Similar behavior was also reported by Sen et al.69 In liposome based enzyme driven motors, the same group observed that the motion of the motor is related to the enzyme-catalysis-induced positive chemotaxis and solute–phospholipid-based negative chemotaxis of enzymes attached to the liposome model.111 In this paper, the propulsion mechanism of this EPM was hypothesized and proved to be a result of enzyme-catalysis-induced positive chemotaxis and solute–phospholipid-based negative chemotaxis. Recently, work from our group has made use of chemotaxis behavior as a strategy for motors to deliver doxycycline for the treatment of periodontal disease, and the study showed that the enzyme powered micromotor can move with controlled directionality because of the substrate gradient produced by phagocytes (Fig. 2b).100 We believe that the use of chemotaxis behavior for enzyme powered MNMs can be an ideal approach for developing the next generation of nanocarriers for biomedical applications, however, mechanistic studies based on EPMs are necessary for further understanding of these motors in particular when placed in biological environments.
4. Enzyme powered motors (EPMs)
The first example of enzyme powered artificial systems was demonstrated by Mano and Heller by binding glucose oxidase and bilirubin oxidase on either side of a carbon fiber at the water–O2 interface.17 Later in 2007, Feringa et al. fabricated carbon nanotubes covalently functionalized with glucose oxidase and catalase enzymes, reaching speeds up to 0.8 cm s−1 in the presence of glucose and oxygen.112 These demonstrations laid the platform for the fabrication of micro/nanomotors (MNMs) powered by enzymes by various research groups (Fig. 3). As discussed in the previous section, there is a plethora of fuels available in the biological environment that can be exploited as fuels to power micro/nanomotors (MNMs) with the help of enzymes, using them as substrates for carrying out catalytic reactions and thus providing the propulsion force. This section will showcase the enzymes utilized to power MNMs to date together with the main focus on using biologically relevant fuels together with their fabrication and propulsion mechanism.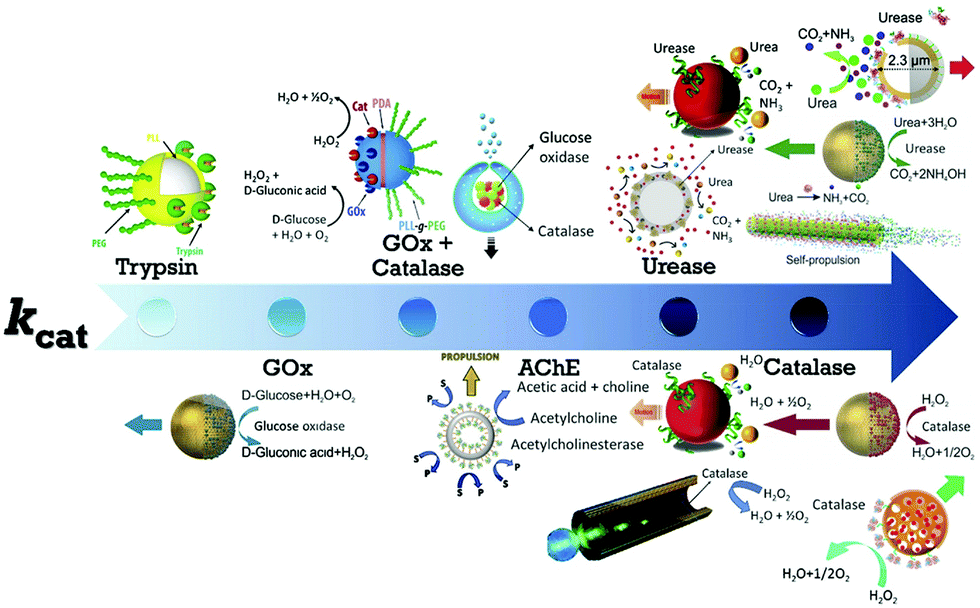 | ||
| Fig. 3 Schematic of enzymes reported to power micro/nanomotors (MNMs) arranged in an ascending order of turnover number (kcat): trypsin,113 glucose oxidase (GOx),114 GOx coupled with catalase,115,116 acetylcholinesterase (AChE),117 urease,20,70,114,118 and catalase.18,69,114,119 Adapted with permission from ref. 20, 69, 113–116 and 118 and 119. Copyrights 2010, 2015, 2016, and 2017 American Chemical Society. Adapted with permission from ref. 117. Copyright 2019 Springer Nature. Adapted with permission from ref. 70. Copyright 2018 Wiley. Adapted with permission from ref. 18. Copyright 2017 Elsevier. | ||
4.1 Catalase based EPMs
Catalase decomposes H2O2 to produce oxygen that can power MNMs based on the bubble propulsion mechanism. The first report of using catalase as a sole enzyme to drive micromotors was demonstrated by Sanchez et al., with the help of a roll up technique. The fabrication was carried out using thin Au/Ti films rolled up with a covalently bound catalase enzyme that could reach speeds up to 8 body-lengths per s, much higher than the corresponding Pt-based micromotors.120 Later, the same group used mesoporous silica clusters (MSC) to fabricate micromotors coated with catalase enzymes by EDC–NHS functionalization and Ni by electron beam deposition on either side to provide them with directionality using magnetic guidance (Fig. 4a).121 Furthermore, they fabricated nanomotors with hollow mesoporous silica nanoparticles (HMSNPs) due to their superior biocompatibility on account of the glutaraldehyde linkage of catalase on one side of the HMSNPs. These nanomotors with a size of around 390 nm were observed to self-propel due to the chemo-phoretic mechanism arising due to a product conc. gradient around the surface owing to the asymmetric bio-catalytic reaction.114 In order to reduce the size of nanomotors for biomedical applications Sanchez et al. also fabricated motors of a size of around 100 nm powered by a similar mechanism.18 Sen et al., in 2015, used polystyrene beads to anchor the catalase enzyme by biotin–streptavidin linkage and showed an enhanced diffusivity of the beads and the directional migration along the substrate gradient by enzyme catalysis (Fig. 4b).69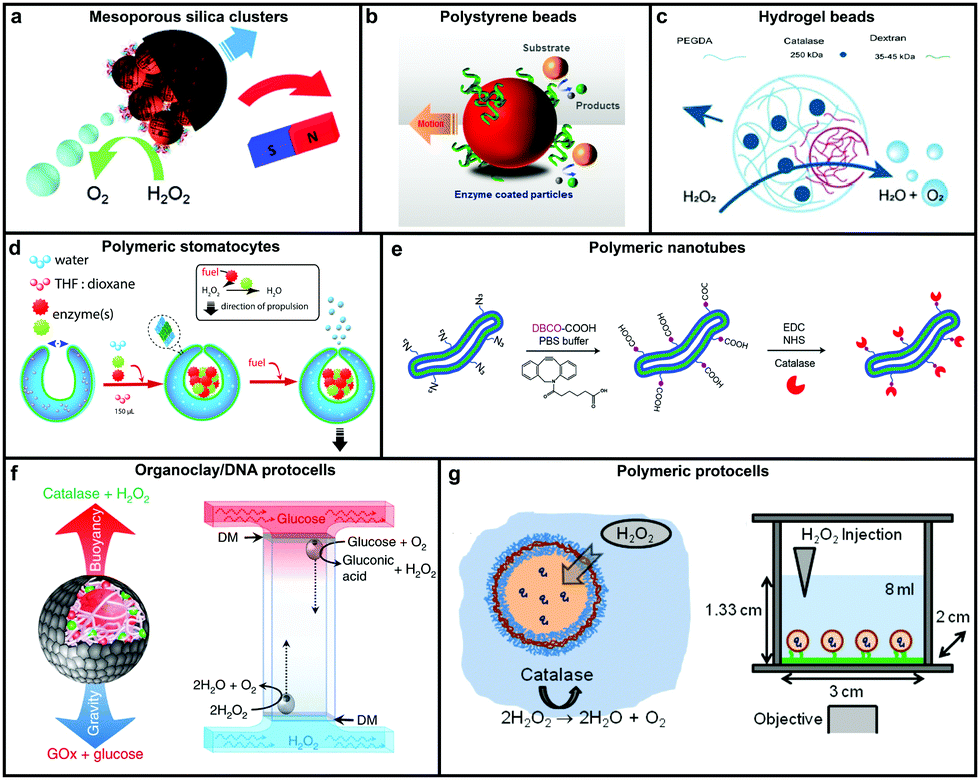 | ||
| Fig. 4 Designs for catalase powered MNMs. (a) Mesoporous silica nanoclusters with magnetic control bound to catalase on one side,121 (b) polystyrene beads coated with catalase by biotin–streptavidin linkage,69 (c) biocompatible hydrogel fabricated by PEGDA and dextran,124 (d) polymeric (PEG-b-PS) stomatocytes encapsulated with enzymes,115 (e) biodegradable polymeric (PEG-b-PDLLA) nanotubes coated with catalase,125 (f) organoclay/DNA protocells encapsulated with enzymes,126 and (g) polymeric protocells encapsulated with catalase for autonomous motion.127 Adapted with permission from ref. 121 and 125. Copyrights 2015, 2018 Royal Society of Chemistry. Adapted with permission from ref. 69 and 115. Copyrights 2015 and 2016 American Chemical Society. Adapted with permission from ref. 124 and 127. Copyrights 2018 Wiley. Adapted with permission from ref. 126. Copyrights 2018 Springer Nature. | ||
In this regard, Wilson et al. fabricated polymer based nanomotors using the bottom-up approach involving supramolecular chemistry called stomatocytes.122 The bowl shaped stomatocytes were formed by the controlled shape transformation of polymeric vesicles.46 These structures have an opening on one side providing the necessary asymmetry for nanomotor application and can be encapsulated with enzymes to power them (Fig. 4d).123 The polymeric structures provide a soft interface to cells, do not contain hard metal surfaces, provide easy functionalization and contain polyethylene glycol (PEG) that is highly biocompatible.128 The stomatocytes were encapsulated with catalase in the stomach that could propel in the presence of H2O2 with speeds up to 60 μm s−1 due to the expulsion of oxygen bubbles produced through the opening, providing forward thrust. In the same study, stomatocytes were encapsulated with a two enzyme system comprising GOx and catalase that participates in a cascade reaction in the presence of glucose as a substrate and propels due to self-diffusiophoresis.115 In order to have a completely biodegradable polymer, a poly(ethylene glycol)-block-poly(D,L-lactide) (PEG-b-PDLLA) diblock copolymer was used to fabricate tubular structures functionalized with catalase, showing enhanced diffusion (Fig. 4e).125 PEG–PDLLA was also used to fabricate stomatocytes with spatial control for functionalization by providing two different functional groups on the inside and outside of stomatocytes. The stomach of the stomatocytes was functionalized with GOx and catalase, showing propelled motion with speeds reaching up to 15.8 μm s−1 in the presence of glucose.129 The same group also fabricated asymmetric hydrogel based micromotors composed of dextran and poly(ethylene glycol) diacrylate (PEGDA) with entrapped catalase (Fig. 4c). The low molecular weight dextran can diffuse into hydrogels upon UV polymerization forming an opening for bubble propulsion, when H2O2 is decomposed by catalase in the PEGDA phase reaching speeds up to 100 μm s−1.124 Furthermore, control over the speed of these micromotors was achieved by controlling the surface of the opening.130
Protocells have also been fabricated by encapsulating catalase in the lumen of a biotinylated polymersome. In the presence of H2O2 the force generated by the enzymatic reaction on the surface of protocells was enough to break adhesive contacts between the polymersome and the surface, to drive their autonomous motion, mimicking the formation and breakage of adhesive contacts as showcased by mammalian cells (Fig. 4g).127 In another study, catalase containing organoclay/DNA semipermeable microcapsules were fabricated that displayed oxygen gas bubble dependent buoyancy (Fig. 4f).126 When the same system was co-encapsulated with GOx and catalase a sustained vertical oscillatory movement was established due to antagonistic bubble generation and depletion. Such systems could be used for the flotation of macroscopic objects, self-sorting of mixed protocell communities and delivery of a biocatalyst from an inert to chemically active environment.126
4.2 Urease based EPMs
In order to overcome the use of non-biocompatible fuels such as H2O2 required for catalase driven systems and the bubble propulsion mechanism, researchers have exploited the urease enzyme to power motion through the use of biocompatible fuels. Sen et al. used gold (Au) nanoparticles anchored with urease enzymes and used them as pumps that could move tracer particles in the presence of their substrate urea.131 The observed migration of tracer particles was due to fluid pumping caused by the enzymatic reactions. Sanchez et al., later fabricated nanomotors using HMSNPs covalently bound to urease enzymes on one side to create a Janus structure (Fig. 5a). The HMSNP nanomotors were observed to have an enhanced diffusion coefficient due to the chemophoretic mechanism arising from the catalytic reaction of urease in the presence of urea.114 In addition, the control over urease powered HMSNPs reaching speeds up to 5 body-lengths s−1 was achieved by the addition of inhibitors such as Ag+ or Hg2+ for modulating the enzymatic activity (Fig. 5b).20 The modulation of the enzyme activity resulted in manipulating the mechanical output on the motors, thereby gaining control over its speed. The inhibition process was reversible upon the addition of dithiothreitol (DTT) due to competitive binding, resulting in control over the start and stop motion of the micromotors. Furthermore, the incorporation of magnetic materials helped in gaining control over the directionality of the as fabricated micromotors. Ultrasmall tubular silica nanojets with 220 nm diameter and lengths ranging from 3 to 40 μm were also fabricated by binding urease through glutaraldehyde linkage molecules (Fig. 5c).118 The decomposition of urea caused an internal flow that extended to an external fluid through the opening, thus giving rise to self-propulsion. The speed of the motors reached up to 10 μm s−1 and was observed to be dependent on the conc. of the fuel, fitting well with Michaelis–Menten enzymatic kinetics. Interestingly, for this system when the enzyme was functionalized on the outside of the nanotubes, only Brownian motion was observed showcasing the need for internal flows for propulsion.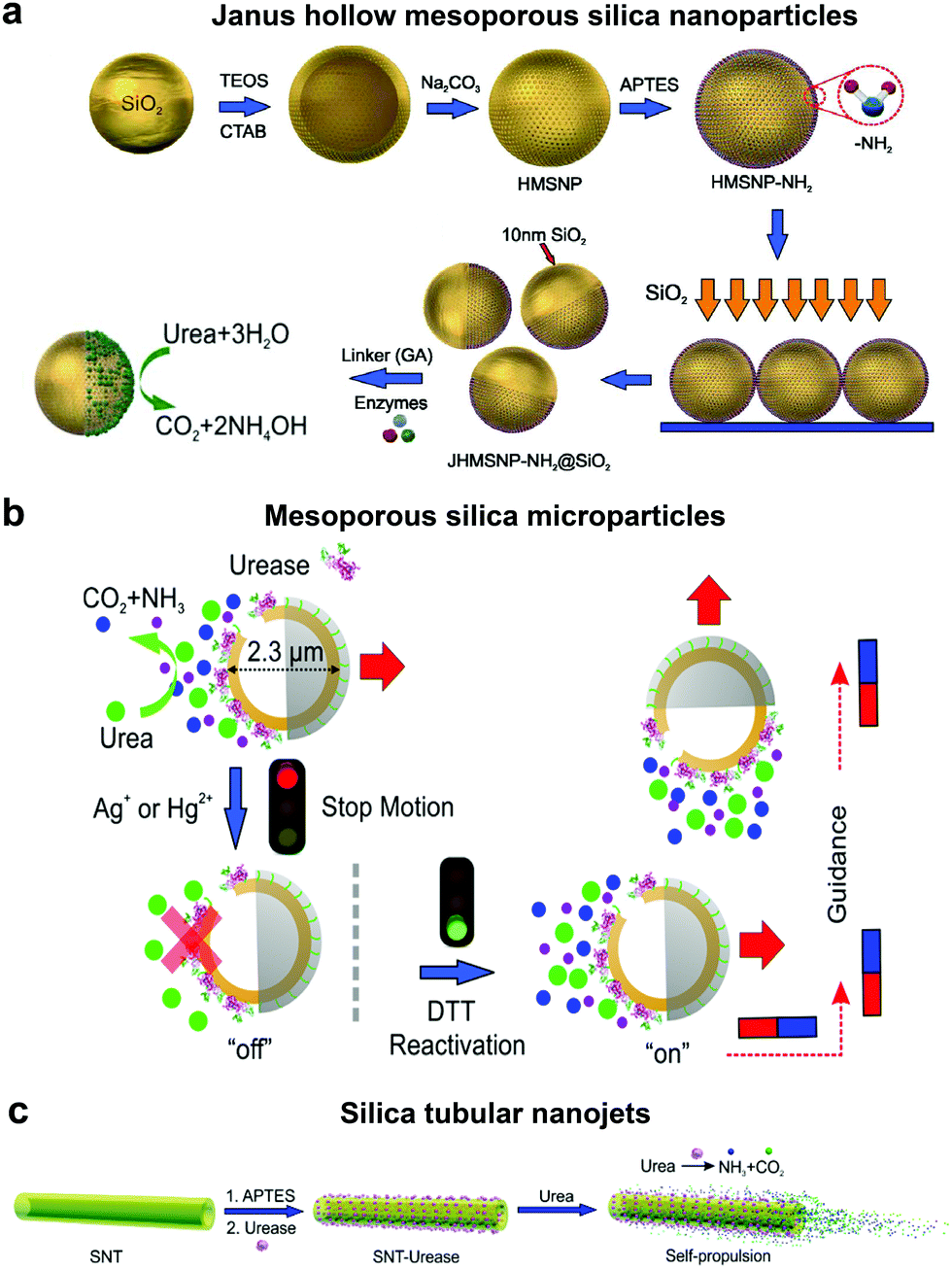 | ||
| Fig. 5 Urease powered motors. (a) JHMSNP nanomotors powered by urease due to the chemo-phoretic mechanism arising from the conc. gradient obtained from the asymmetric catalytic reaction,114 (b) mesoporous silica microparticles powered by urease with control over motion and directionality,20 and (c) silica tubular nanojets fabricated by covalently binding urease.118 Adapted with permission from ref. 20, 114 and 118. Copyrights 2015 and 2016 American Chemical Society. | ||
4.3 Glucose oxidase (GOx) based EPMs
Glucose oxidase catalyzes the oxidation of glucose to D-glucono-1,5-lactone and H2O2 that can be used in tandem with enzymes such as catalase for powering nanomotors. The first example of using GOx in tandem with bilirubin oxidase (BOD) to power artificial systems was shown by Mano and Heller in 2005.17 In this system, one side of the fibre was decorated with GOx carrying out the oxidation of glucose and another side by BOD carrying out the reduction of oxygen (Fig. 6a). This reaction results in the travelling of generated protons in the electrical double layer of the fibre, dragging water molecules with them and thus propelling the nanomotor in the opposite direction. Since then, there have been many MNMs fabricated by using GOx and catalase to power motion as discussed in catalase powered systems. Here, we will discuss a few more examples of MNMs that are driven by the self-diffusiophoresis mechanism. Stadler et al. fabricated micromotors featuring a Janus architecture with a poly(dopamine) (PDA)-coated silica (SiO2/PDA) particle as the core, followed by PEGylation with poly(L-lysine)-grafted-poly-(ethylene glycol) (PLL-g-PEG) on one side and immobilizing GOx and catalase on the other side. The micromotors were observed to have enhanced diffusion dependent on glucose conc. The locomotion was based on self-diffusiophoresis due to the generation of a conc. gradient of molecules accomplished by the conversion of one molecule into two.116 Later, a similar system was modified with Pt nanoparticles instead of catalase enzymes that showcased a 100% increase in diffusion properties due to a higher density of GOx immobilization and no net loss in enzyme activity for metallic nanoparticles. The system was also co-immobilized with trypsin that cleaves peptide bonds at the carboxylic acid sites of amino acids coated with superparamagnetic manganese ferrite nanoparticles (MF-NPs) (Fig. 6b). The microswimmer, could thus be propelled by two fuel sources and could be guided remotely with the help of a magnet, hence, giving rise to directionality.113 He et al. fabricated carbonaceous nanoflasks (CNF) that could self-propel in the presence of glucose and the directionality could be premeditated depending on the surface wettability of the nanoflasks (Fig. 6c). The hydrophilic CNF exhibited backward movement dominated by a local glucose gradient and hydrophobic CNF exhibited forward movement by the generated local glucose acid gradient.132 Simulation results revealed that the hydrophilic nanoflask motor generates a puller-like flow field, whereas the hydrophobic motor creates a pusher-like flow field resulting in backward and forward movement, respectively. In the same year, his group also fabricated nanomotors by grafting polymer brushes on one side and GOx on the other side of Au nanoparticles (Fig. 6d). These particles were observed to have positive chemotaxis towards the glucose gradient with speeds reaching up to 120 body-lengths s−1. The grafted polymer brush provided the motor with improved translational diffusion resulting in bacteria with swarming behavior.133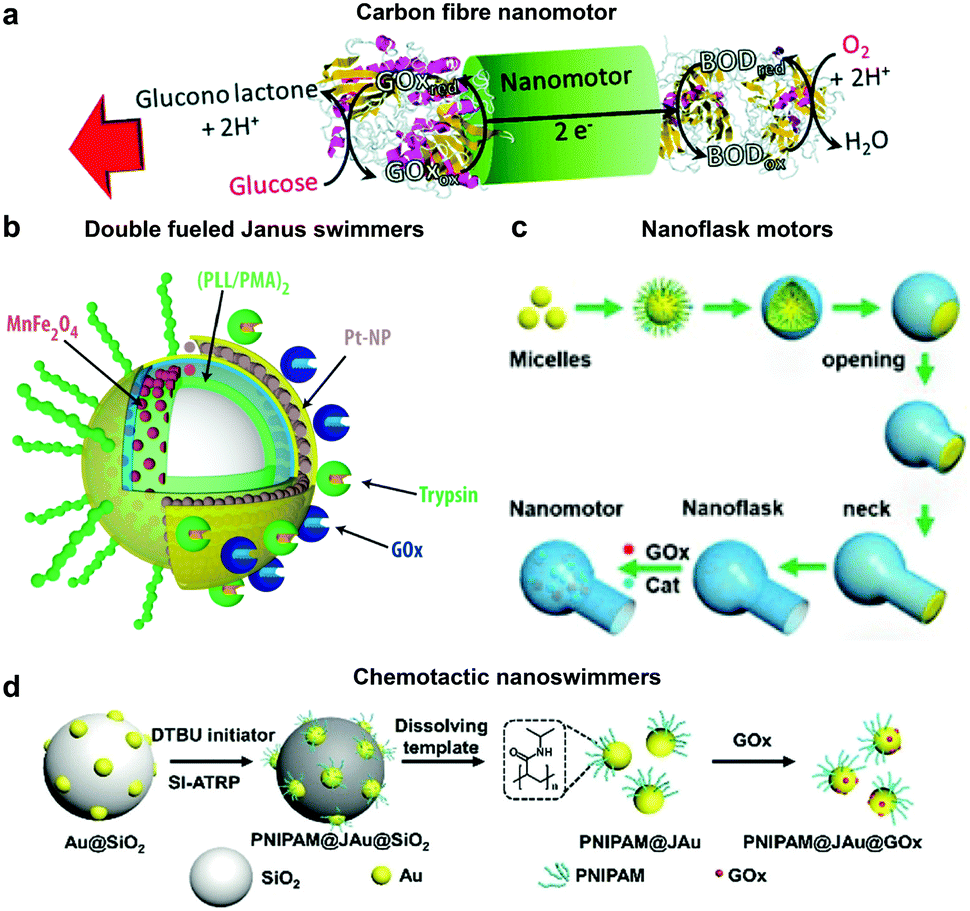 | ||
| Fig. 6 Glucose oxidase powered motors. (a) Carbon fibre decorated with GOx on one side and BOD on other side resulting in bioelectrochemical propulsion,16 (b) double fueled Janus swimmers decorated with GOx, trypsin, Pt and Mn–Fe nanoparticles with magnetic guidance,113 (c) surface wettability directed carbonaceous nanoflasks with GOx and catalase,132 and (d) polymer brush functionalized Au nanoswimmers with swarming chemotactic behavior.133 Adapted with permission from ref. 16. Copyrights 2014 Royal Society of Chemistry. Adapted with permission from ref. 113 and 132. Copyrights 2017 and 2019 American Chemical Society. Adapted with permission from ref. 133. Copyrights 2019 Wiley. | ||
4.4 Other EPMs
Although the majority of research is focused on the above-mentioned enzymatic systems, researchers have used other enzymes for powering motion. One such example are lipases, an industrially important enzyme that hydrolyze carboxylic ester bonds in hydrophobic compounds such as triglycerides.134 Recently, lipase was immobilized covalently on MSNPs and were observed to have enhanced Brownian motion in the presence of dissolvable triglyceride solution (Fig. 7a). The lipase enzyme had two functions; (a) powering the motor and (b) to degrade triglyceride (tributyrin).24 At the same time, another group also reported four different types of micromotor fabricated with poly(glycidylmethacrylate)/polystyrene (PGMA/PS) particles and using hydrophobic interactions to bind lipase (Fig. 7b).135 The micromotors revealed substrate conc. dependent enhanced diffusion and facilitated enzymatic reactions. The propulsion mechanism of both the above-mentioned motors was based on a product gradient due to the degradation of triglyceride molecules.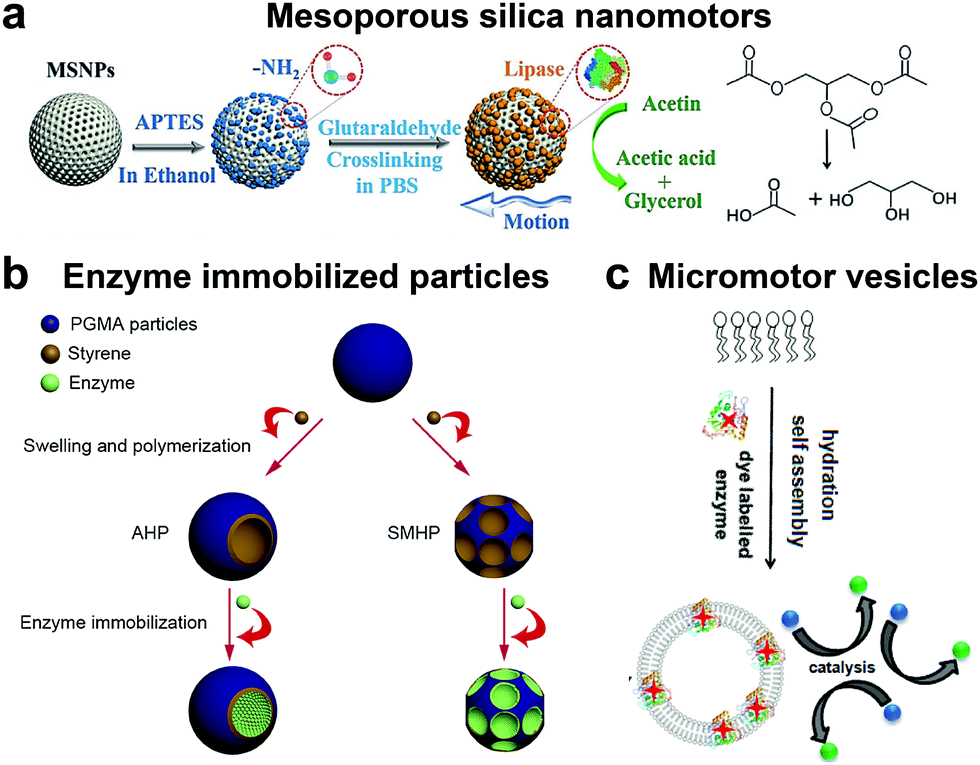 | ||
| Fig. 7 Other enzyme powered motors. (a) Mesoporous silica nanoparticles covalently immobilized with lipase for nanomotor application,24 (b) enzyme immobilized particles showcasing different micromotor configurations immobilized with lipase,135 and (c) ATPase and AP as membrane bound enzymes for enzyme powered vesicles.136 Adapted with permission from ref. 24. Copyrights 2019 Wiley. Adapted with permission from ref. 135. Copyrights 2019 Elsevier. Adapted with permission from ref. 136. Copyrights 2019 American Chemical Society. | ||
Apart from lipase, adenosine 5′-triphosphatase has also been studied by Sen et al. to fabricate active phospholipid vesicles using the hydration assembly of L-α-phosphatidylcholine (EPC) in the presence of ATPase (Fig. 7c).136 These biocompatible phospholipid vesicles exhibited enhanced diffusion in the presence of their substrate ATP by hydrolyzing the phosphate bonds. In the same study, similar results were also obtained for acid phosphatase (AP) enzymes that were bound to vesicles using a biotin–streptavidin linkage.136
4.5 Biologically relevant fluid driven motors
Until now we have discussed EPMs using biocompatible fuels such as glucose, physiologically relevant urea and H2O2; herein MNMs powered by another important biologically relevant fuel i.e. acidic conditions prevalent in stomach and tumor cells together with water will be discussed. The first acidic environment based on tubular micromotors was fabricated using polyaniline with the zinc (Zn) coating on the outer surface and inner surface of the tube, respectively (Fig. 8a). A spontaneous redox reaction was carried out on the Zn surface, thereby generating hydrogen (H2), and the as-produced H2 bubbles provided forward thrust to the tube and hence propelling them at a speed of 100 body-lengths s−1.11 Later the system was modified into double conical micromotors fabricated only with Zn for higher capacity, combinatorial delivery of cargoes, autonomous release of encapsulated payloads, and self-destruction.137 Simultaneously, magnesium (Mg) based micromotors were also fabricated that could propel in the presence of H2O and acid. Wang et al. fabricated Mg based Janus micromotors that self-propelled in seawater without an external fuel due to the oxidation of the Mg surface which reduces water to H2 bubbles (Fig. 8c). The micromotor was coated with Ni for magnetic guidance and Au for increasing the propulsion efficiency associated with the microgalvanic corrosion mechanism.138 The system was further modified to fabricate tubular micromotors that could propel in the gastrointestinal tract at a speed of 60 μm s−1 due to H2 bubbles produced from the water-based reaction (Fig. 8b). Furthermore, Mg based micromotors were also demonstrated to propel under acidic conditions by a spontaneous reaction between the Mg microsphere and the surrounding protons generating H2 bubbles providing efficient micromotor thrust (Fig. 8d).139 With respect to acidic conditions, calcium carbonate (CaCO3) Janus micromotors were fabricated to achieve motion under slightly acidic conditions found in the tumor microenvironment with velocities of 1.8 μm s−1 (Fig. 8e).140 The propulsion mechanism was based on self-diffusiophoresis due to the controlled decomposition of CaCO3 into Ca2+, HCO3−, OH− and H+ ions, thereby inducing diffusioosmotic flows generated by the difference in diffusion coefficients of the ions produced.141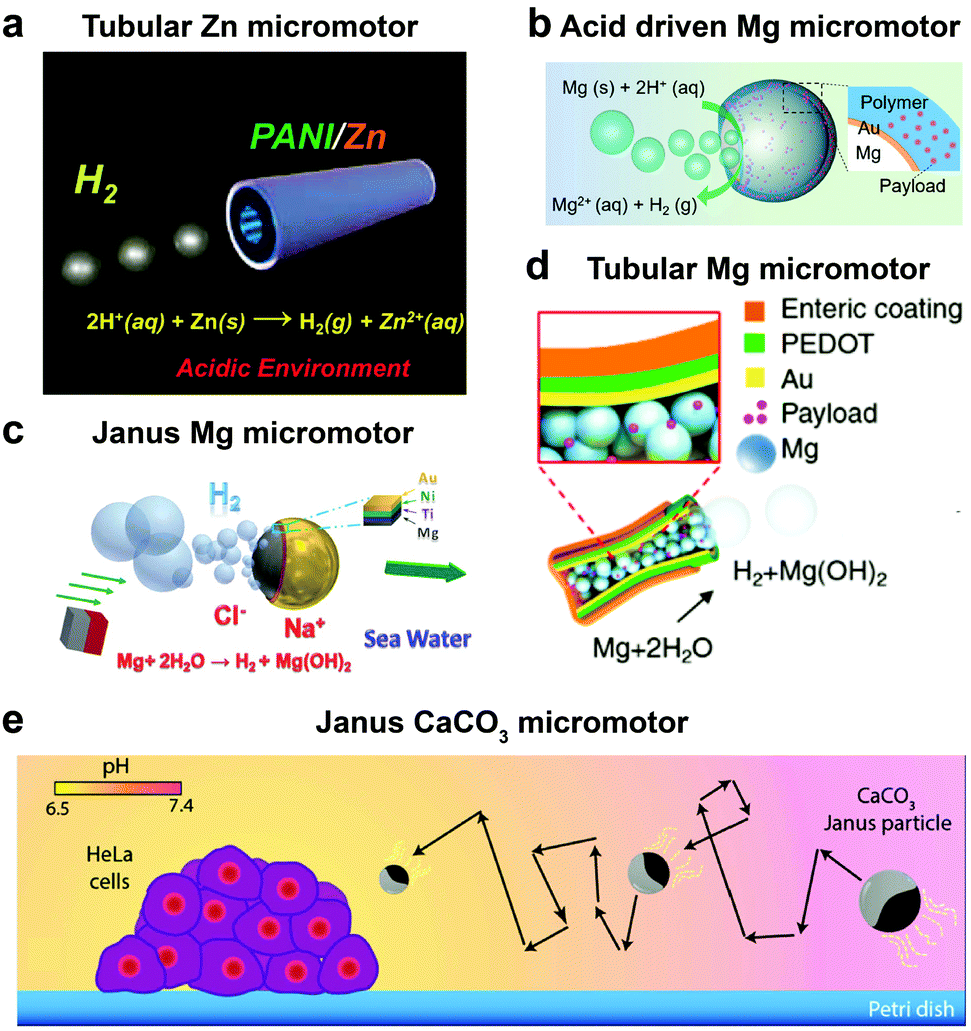 | ||
| Fig. 8 Biologically relevant fluid driven systems. Acid driven micromotor systems based on (a) Zn11 and (b) Mg;139 water driven systems (c) Janus Mg,138 (d) tubular Mg base micromotors147 and (e) calcium carbonate based Janus micromotors driven by acidic conditions in the tumour microenvironment.140 Adapted with permission from ref. 11 and 147. Copyrights 2012 and 2016 American Chemical Society. Adapted with permission from ref. 138. Copyrights 2013 Royal Society of Chemistry. Adapted with permission from ref. 139. Copyrights 2017 Wiley. Adapted with permission from ref. 140. Copyrights 2016 Springer Nature. | ||
5. Biomedical applications of EPMs
Enzymatically powered motors can propel themselves through an aqueous solution by self-generated gradients and gas bubbles. With recent advancements, MNMs have been observed to have active motion using fuels present in bodily fluids or physiologically relevant molecules that could be used by enzymes as substrates. In this section, we will discuss examples for EPMs based on materials used for fabrication and discuss their relevant biomedical applications.5.1 Mesoporous silica based EPMs
Mesoporous silica offers various advantages in terms of easy synthesis, high loading capacity, large surface area and good biocompatibility.142 They are easy to functionalize with the possibility of various surface chemistry modifications, high potential for the physical entrapment of particles and have been clinically approved by FDA for biomedical applications.143 In 2012, MSNs were used for the fabrication of nanomotors for capture and cargo transport that was powered by the catalase enzyme. The asymmetry to the nanomotor was achieved by coating with Au on one side that was used for functionalizing single-stranded DNA and the other side was functionalized with catalase that provided the driving force in the presence of a low conc. of H2O2.144 The system was able to capture cargo (DNA functionalized particle) with the help of analyte DNA strands whose ends are complimentary to the DNA strands on nanomotors and cargoes, thereby acting as a bridge due to the high selectivity of the DNA hybridization process.144 The system was envisioned to be integrated in lab-on-chip devices for biomedical diagnostic applications. Wang et al. used MSNs to fabricate acoustically propelled nanomotors that could release insulin in the presence of glucose.145 Even though the system was not powered by enzymes, they were used to fabricate glucose responsive nanovalves based on phenylboronic acid (PBA)–GOx supramolecular nanostructures. The MSN particles were loaded with insulin gated with (PBA)–GOx nanovalves to prevent the unloading of insulin. GOx present in the nanovalves produces H2O2 in the presence of glucose, thereby decreasing the pH and cleaving the C–B bond and thus releasing the loaded insulin. The use of active propelled motion by ultrasound helped in increasing the insulin release efficiently.145 Sanchez et al. coated MSNs with urease enzymes for powering the DOX loaded nanobots in the presence of urea (Fig. 9a). The nanobots could self-propel in ionic media and showed improved effect on HeLa cells in the presence of urea when compared to its passive counterparts, due to a synergistic effect between improved drug release kinetics and ammonia production by the catalytic decomposition of urea.70 In another study, the above system was modified to the target by specific interaction and to reduce the proliferation of 3D bladder cancer spheroids found in the urinary bladder (Fig. 9b).146 For this, the MSN was modified with urease for powering the system and bound with an anti-FGFR3 antibody. The enhanced diffusion of the nanomotors at biologically relevant conc. of urea allowed them to explore a greater area in comparison to passive diffusion, thus, improving the chances of interaction between the antigen and the antibody and decreasing the proliferation of the spheroids.146 Furthermore, the urease powered MSN was further used for the delivery of DOX to HeLa cells using a pH gated system (Fig. 9c). The nanomotors were loaded with DOX grafted with benzimidazole groups on the outer surface, and capped by the formation of inclusion complexes between benzimidazole and cyclodextrin-modified urease (CD-U).148 Benzimidazole CD-U nanovalves prevented the release of DOX at physiological pH, but in the presence of acidic pH prevalent in the tumor microenvironment the protonation of benzimidazole causes the subsequent dethreading of inclusion complexes thereby releasing the cargo. Cellular studies revealed that the presence of active nanomotors enhanced both tumor internalization and intracellular cargo delivery.148 Ramón Martínez-Máñez et al. modified the above system by powering the motors with the catalase enzyme and glutathione (GSH) responsive valves for cargo delivery (Fig. 9d). Glutathione is a tripeptide that carries out redox reactions and is present in high conc. in intracellular compartments.150 In brief, MSN particles were attached with Au nanoparticles on one side that were further covalently functionalized with catalase. Later, the MSN scaffold was loaded with DOX and capped with disulfide linked chains.149 In the presence of H2O2 the MSN nanomotors showed enhanced diffusion resulting in higher internalization in HeLa cells and due to the presence of GSH, the disulfide bonds are broken thereby releasing DOX molecules. The latter two systems could sense the environment and release the cargo molecules in the presence of a stimulus thus displaying features essential for biomedical applications.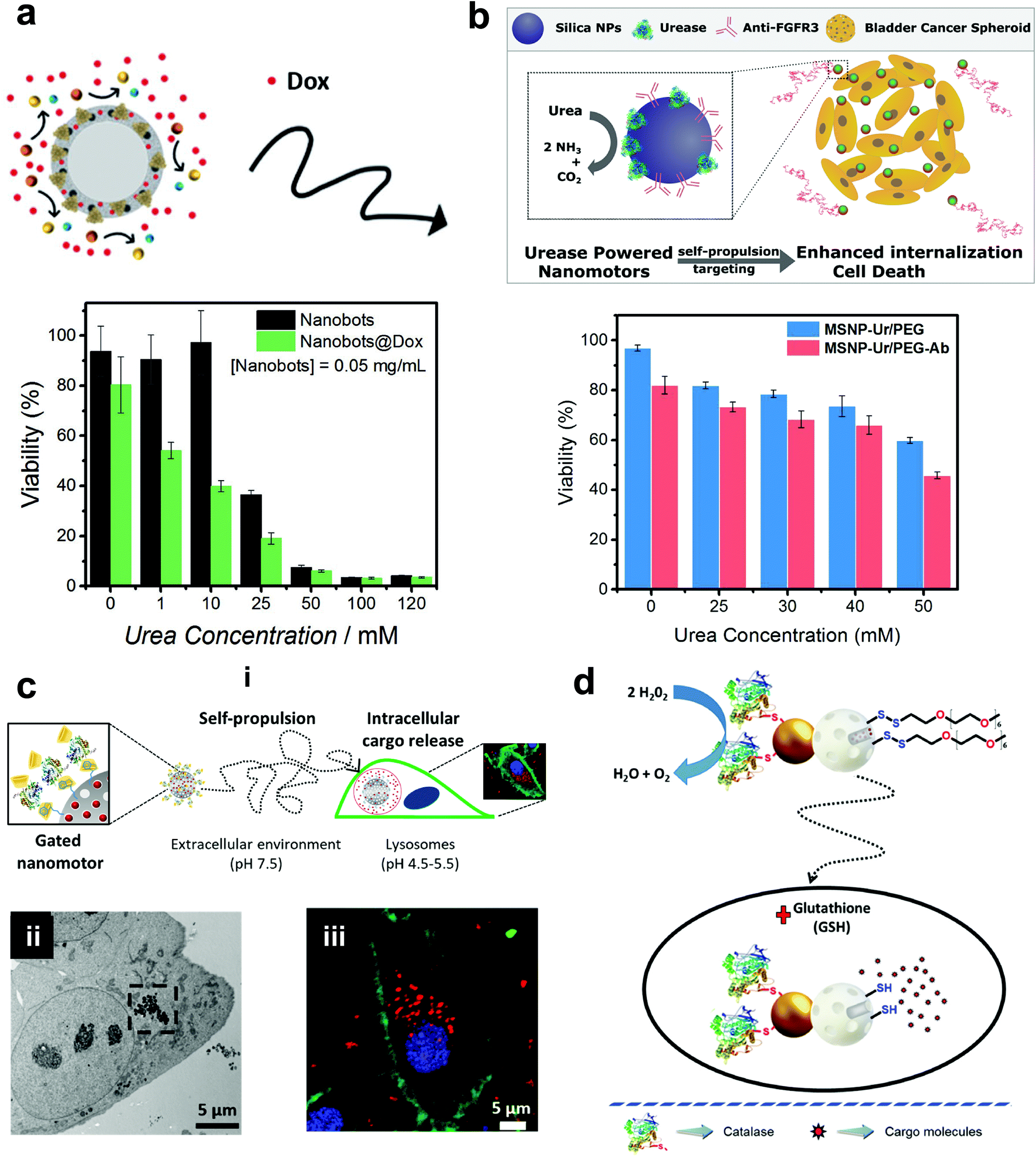 | ||
| Fig. 9 Mesoporous silica based EPMs. (a) Urease powered motors for DOX delivery showing a decrease in cell viability with increasing urea conc.70 (b) Urease powered motors with antibodies against 3D bladder cancer spheroids showing decreased cell viability corresponding to urea conc.146 (c) Schematic representation of benzimidazole and cyclodextrin-modified urease powered nanomotors (i), showing internalization into HeLa cells via TEM (ii) and high resolution confocal images with different signals corresponding to the WGA membrane marker (green), DNA-marker Hoechst 33342 (blue), and DOX (red).148 (d) Schematic representation of the performance of nanobots with self-propulsion and glutathione-responsive cargo delivery capabilities inside a cell.149 Adapted with permission from ref. 70. Copyrights 2017 Wiley. Adapted with permission from ref. 146 and 148. Copyrights 2018 and 2019 American Chemical Society. Adapted with permission from ref. 149. Copyrights 2019 Royal Society of Chemistry. | ||
5.2 Polymer based EPMs
Recently, polymers have attracted attention for the fabrication of MNMs due to easy functionalization, ease of scaling up, and appropriate physicochemical properties for biomedical applications with good biocompatibility.151 In addition, they can be self-assembled into supramolecular structures that can provide a soft interface to cells in comparison to hard metal surface based MNMs together with good biodegradability. He et al. fabricated Janus capsules loaded with DOX by template-assisted polyelectrolyte layer-by-layer (LBL) deposition using poly(styrenesulfonate) sodium salt (PSS) and poly(allylamine hydrochloride) (PAH).152 This was followed by the deposition of Au, Cr and Ni, with Au coated with catalase enzymes that powered the motors by the biocatalytic decomposition of H2O2. The Janus capsule was observed to swim at 25 μm s−1 and was directed towards HeLa cells using an external magnetic field and upon application of near-infra red (NIR) light, it induced the shell breakage of Au particles thereby releasing DOX molecules. Such systems provide self-driving with navigation capabilities and perform drug loading and targeted transportation together with remotely controlled release in the vicinity of cells.152 Using a similar technique, biodegradable poly-L-lysine hydrochloride/bovine serum albumin multilayered microtubes were fabricated with the integration of a thermal-sensitive gelatin hydrogel containing Au nanoparticles, DOX and catalase enzymes.153 The catalase reaction propelled the motors, with Au increasing the local temperature upon illumination with NIR light, thereby releasing DOX molecules. In addition to this, the protein based structure provided high biodegradability in the presence of α-chymotrypsin and hence satisfying all the requirements for biomedical applications.153Battaglia et al. fabricated chemotactic vesicles based on polymersomes encapsulated with GOx alone or in combination with catalase exhibiting self-propulsion in response to a glucose gradient. The polymersomes upon further functionalization with low-density lipoprotein receptor-related protein 1 (LRP-1) showed a four-fold increase in blood brain barrier crossing (Fig. 10a).101 The asymmetric polymerosome propulsion velocity was directly proportional to the glucose gradient and larger particles were observed to have better binding affinity to the vessels due to minimized rotational diffusion.101 In 2019, Stadler et al. fabricated biocompatible self-propelled swimmers functionalized with collagenase enzymes for using collagen as a fuel and supermagnetic nanoparticles that can impart heat generation and used calcium for triggering the motion (Fig. 10b).154 Collagenase enzymatically breaks collagen in the extracellular matrix and the asymmetry required for self-diffusiophoretic motion is provided by the calcium gradient that also enhanced the enzymatic activity. The microswimmers enhanced penetration into cancer cell spheroids and further resulted in a decreased fraction of live cells when exposed to an alternate magnetic field due to heat generation.154 With respect to tumor cell penetration, Wilson et al. fabricated both small and ultra-small stomatocytes encapsulated with the catalase enzyme in the size ranging around 150 nm (Fig. 10c).155 The smaller size in comparison to normal stomatocytes resulted in increased cellular uptake in the presence of H2O2 as a fuel due to the enhanced permeation and retention (EPR) effect. These systems can be further extrapolated as cargo delivery vehicles or for targeted delivery of drugs. In this context, poly(lactic-co-glycolic acid) microspheres were fabricated for the delivery of the doxycycline drug, used for the treatment of periodontal disease to prevent bacterial infection. The micromotors were powered with catalase enzymes that could induce motion and provide directional motion in the presence of a H2O2 gradient produced by macrophage cells incubated with phorbol-12-myristate-13-acetate.100 All the above examples showcase the advantages of polymer based MNMs powered by enzymes and warrant further investigation for their biomedical application.
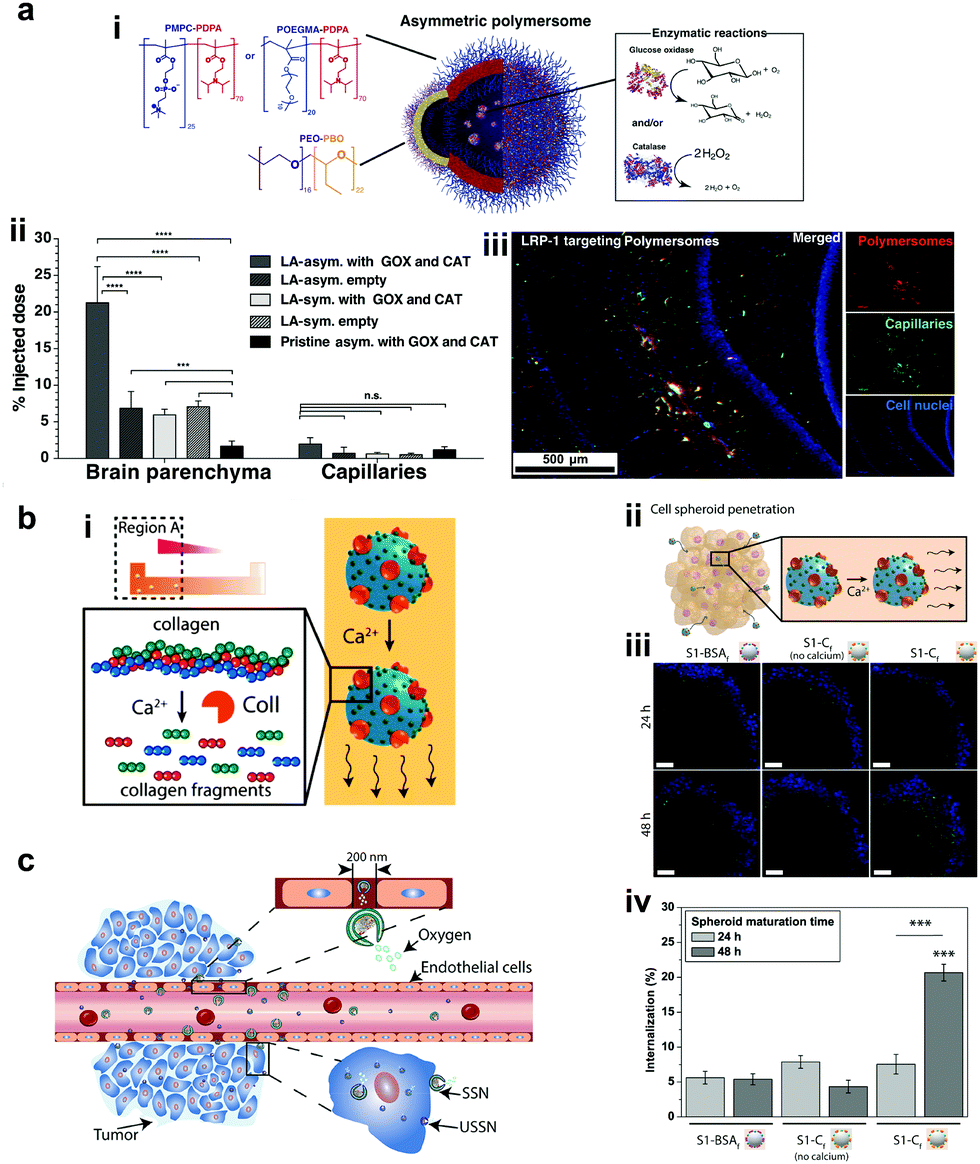 | ||
| Fig. 10 Polymer based EPMs. (a) Schematic representation of a chemotactic polymersome using a combination of copolymers. The polymersomes encapsulate glucose oxidase and/or catalase enzymes (i), percentage of the injected dose found in the rat brain parenchyma and the capillary fraction 10 min after the carotid artery in situ perfusion of polymersomes loaded with Gox + Cat and empty, as well as pristine asymmetric polymersomes loaded with Gox and Cat (ii) and (iii) immunofluorescence histologies of rat hippocampus sections of animals treated with asymmetric polymersomes loaded with Gox + Cat.101 (b) Schematic representation of collagenase loaded polymeric swimmers (i) and their penetration into spheroids (ii), CLSM images of the spheroids aged over 24 h and 48 h, upon addition of polymeric nanomotors for 24 h. The blue channel represents the stained spheroids, and the green channel represents the swimmers (iii) and swimmers located inside the spheroid (represented as % of internalization), determined as the percentage of green pixels in the spheroid area of images (iv).154 (c) Schematic representation of enhanced cellular uptake of ultra-small stomatocytes due to the EPR effect.155 Reproduced with permission from ref. 101. Copyrights 2017 American Association for the Advancement of Science. Reproduced with permission from ref. 154 and 155. Copyrights 2019 American Chemical Society. | ||
5.3 Inorganic oxides and MOF based EPMs
Silica materials are well studied and are reported to have negligible cytotoxicity towards cell proliferation with good biocompatibility at adequate doses.156 Fischer et al. fabricated micropropellers using SiO2 particles coated with nickel and functionalized with the urease enzyme. The micropropellers were able to penetrate the mucin gels by increasing the pH locally due to the biocatalytic reaction of urease enzymes which liquefies the mucus and also provides the driving force.160 The micropropellers were directed by an external magnetic field and could possibly be used for drug delivery applications by overcoming the limitations of acidic conditions present in the stomach.160 CaCO3 is another example for inorganic oxide that is highly biocompatible, has low costs, is highly reproducible and can be easily loaded with drug molecules that could be released under acidic pH conditions in the tumor microenvironment. With regards to micromotors, CaCO3 has been studied to power microspheres loaded with enzyme creatine phosphate kinase (CPK) that produce ATP. The microspheres once bound to microtubules were able to glide with an average velocity of 92 ± 7 nm s−1 on a surface coated with kinesin and used for cargo delivery in the presence of ADP and CP.161 In another example, CaCO3 cores modified with the urease enzyme on one side for powering motors, hyaluronic acid on another side for targeting and loaded with camptothecin for the antitumor effect (Fig. 11a).157 The nanomotors showed enhanced penetration and cellular uptake into tumor cells. Upon reaching the tumor cells the disintegration of the core due to the presence of acidic pH conditions aids in localized delivery to CPT molecules thereby resulting in increased necrosis. Using such nanomotors is a feasible strategy to boost the antitumor efficacy by promoting the local accumulation, deep tumor penetration, tumor cell capture and the intracellular release of chemotherapeutic drugs.157 Recently, metal organic frameworks (MOF) have gained interest due to their porous structure and disintegration based on pH stimuli.151 Ma et al. fabricated MOF based nanomotors by encapsulating them with photosensitizers to generate cytotoxic 1O2 for efficient photodynamic therapy (PDT), powered by GOx and catalase that caused starvation of cells due to the utilization of glucose (Fig. 11b).158 The catalase enzyme also decomposed H2O2 present in the cells to produce 3O2 molecules that help in the generation of 1O2 molecules. Overall, the active system increased the cellular uptake by increasing the diffusivity by 27% and the synergistic effects of PDT and starvation therapy resulted in decreased cell viability of HeLa cells accounting to 73%, far better than individual therapies used in the study.158 In another study, the pH regulated delivery of DOX molecules to HeLa cells was carried out using a MOF based system. The micromotors consisted of succinylated β-lactoglobulin and the catalase enzyme superassembled onto MOF particles (Fig. 11c).159 In the presence of neutral pH, succinylated β-lactoglobulin is permeable through which H2O2 can reach the catalase enzyme to power the nanomotors. Once acidic pH conditions were encountered in tumor cells, succinylated β-lactoglobulin undergoes reversible gelation that seizes the motion of micromotors and due to degradation it releases the encapsulated DOX molecules to HeLa cells as observed via deconvolution microscopy.159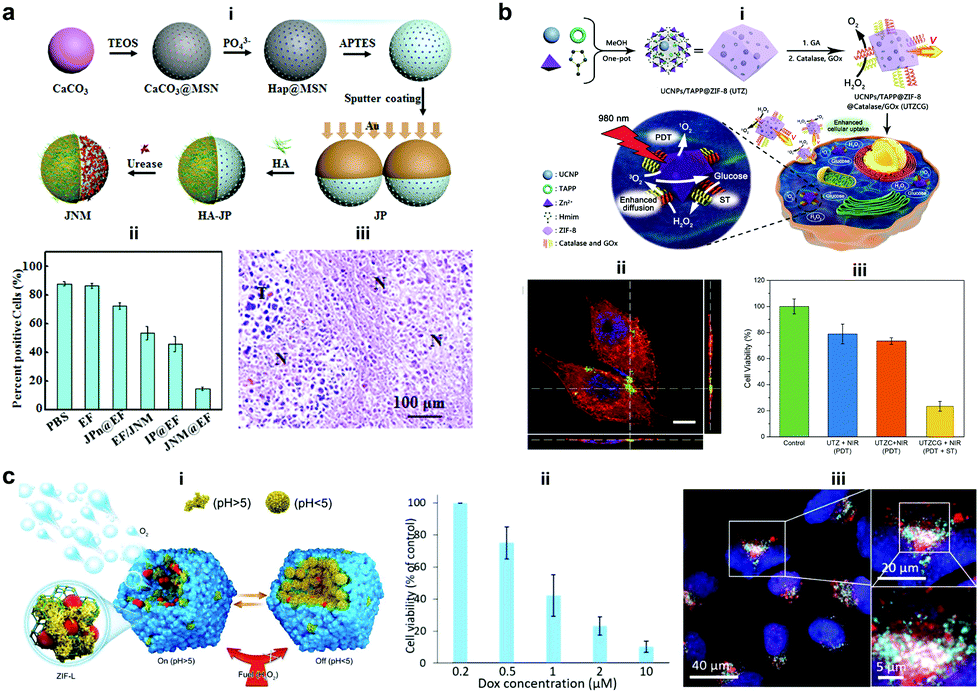 | ||
| Fig. 11 Inorganic and MOF based EPMs. (a) Fabrication process of JNMs obtained by separately grafting HA and urease on the opposite sides of JPs as targeting moieties and power sources, respectively (i), the number of ki-67-positive cells compared with the total number of cells in IHC staining images of tumors retrieved on day 21 after intratumoral administration (ii), and H&E staining images (“N” represents necrotic area, “T” represents tumor mass) (iii).157 (b) Schematic illustration for MOF based nanomotors and synergetic photodynamic-starvation therapy involving self-accelerated cascade reactions (i), CLSM image of HeLa cells after incubation with nanomotors (green). The cytoskeleton and cell nucleus were stained with Phalloidin-TRITC (red) and DAPI (blue), respectively (ii) and cell viability in different nanoparticle solutions of 100 g mL−1 under 980 nm laser irradiation (iii).158 (c) Schematic representation of the assembly of micromotors with pH-responsive on/off motion (i), Cell viability of HeLa cells in the presence of DOX-loaded cat-β@ZIF micromotor particles (ii), and CLSM overlay of DOX (red), lysosome staining using lysosomal-associated membrane protein 1 (LAMP1) antibody (cyan) and cell nuclei (blue).159 Reproduced with permission from ref. 157 and 158. Copyrights 2019 Elsevier. Reproduced with permission from ref. 159. Copyrights 2019 Wiley. | ||
To the best of our knowledge we have discussed all the well-known enzyme powered motors and their biomedical applications. Apart from these, there are few examples of MNMs based on Zn and Mg metals studied by Wang et al. that can power motion in the presence of acidic pH conditions present in the stomach and utilized for biomedical applications.162–165 However, these MNMs are not the focus of this review and are hence only briefly discussed here, to showcase the other possibilities.
6. Outlook and future perspectives
Enormous progress has been made in the field of EPMs with ample proof-of-concept studies exhibiting their biomedical applications. To realize the full potential of such systems a number of factors require to be addressed. First, the use of biocompatible and biodegradable components that comprise the body parts of the EPM is to be optimized. Even though the use of stimuli responsive polymers can be an ideal solution, their specific interaction with tissues and cells needs to be studied. Second, there is a need of exploring fuels present under physiologically relevant conditions; until now only a few fuels have been studied and there is a plethora of fuels available that has not yet been explored. Furthermore, currently, many studies have been carried out using a relevant conc. of physiologically present fuels by externally supplying them. It is the need of the hour to test EPMs using endogenously produced fuels that can act as substrates for enzymes to power them. Third, for taking EPMs towards more application-oriented studies, a better understanding is required of their propulsion mechanism and they need to be engineered for enhanced stability in the bodily fluids as current systems exhibit lower efficiency. Fourth, it is required to throw light and gain deep insights and knowledge on the shape and geometry of EPMs, which is best suited for the proposed biomedical applications together with control over motion and directionality for targeted therapy. Fifth, a thorough investigation of the bioavailability, retention, toxicity, and therapeutic efficacy of EPMs and their tracking in real world clinical applications is required for true in vivo biomedical applications.The current advancements only form the tip of the iceberg and all the above issues warrant immediate research to understand the role of EPMs in complex biological environments to make them truly applicable for biomedical applications. To achieve this, a close collaboration is required between researchers in the field of medicine, materials science, robotics, biology, nanotechnology and chemistry to expand the use of EPMs in real world applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. M. would like to acknowledge funding from the European Union's Horizon 2020 framework programme under the Marie Skłodowska-Curie Individual Fellowship Grant Agreement No. 794657. D. A. Wilson and J. S. acknowledge the support from the Ministry of Education, Culture and Science (Gravity Program 024.001.035).References
- S. M. Moghimi, A. C. Hunter and J. C. Murray, FASEB J., 2005, 19, 311–330 CrossRef CAS PubMed.
- F. Peng, Y. Tu and D. A. Wilson, Chem. Soc. Rev., 2017, 46, 5289–5310 RSC.
- R. Lipowsky, J. Beeg, R. Dimova, S. Klumpp, S. Liepelt, M. J. I. Muller and A. Valleriani, Biophys. Rev. Lett., 2009, 4, 77–137 CrossRef CAS.
- K. Svoboda, C. F. Schmidt, B. J. Schnapp and S. M. Block, Nature, 1993, 365, 721–727 CrossRef CAS PubMed.
- A. Yildiz, M. Tomishige, R. D. Vale and P. R. Selvin, Science, 2004, 303, 676 CrossRef CAS PubMed.
- H. Hess, J. Clemmens, D. Qin, J. Howard and V. Vogel, Nano Lett., 2001, 1, 235–239 CrossRef CAS.
- R. F. Ismagilov, A. Schwartz, N. Bowden and G. M. Whitesides, Angew. Chem., Int. Ed., 2002, 41, 652–654 CrossRef CAS.
- Z. Wu, X. Lin, T. Si and Q. He, Small, 2016, 12, 3080–3093 CrossRef CAS PubMed.
- W. Gao, A. Pei, R. Dong and J. Wang, J. Am. Chem. Soc., 2014, 136, 2276–2279 CrossRef CAS PubMed.
- W. Gao, A. Pei and J. Wang, ACS Nano, 2012, 6, 8432–8438 CrossRef CAS PubMed.
- W. Gao, A. Uygun and J. Wang, J. Am. Chem. Soc., 2012, 134, 897–900 CrossRef CAS PubMed.
- W. Gao, M. D'Agostino, V. Garcia-Gradilla, J. Orozco and J. Wang, Small, 2013, 9, 467–471 CrossRef CAS PubMed.
- R. Liu and A. Sen, J. Am. Chem. Soc., 2011, 133, 20064–20067 CrossRef CAS PubMed.
- D. Patra, S. Sengupta, W. Duan, H. Zhang, R. Pavlick and A. Sen, Nanoscale, 2013, 5, 1273–1283 RSC.
- X. Zhao, K. Gentile, F. Mohajerani and A. Sen, Acc. Chem. Res., 2018, 51, 2373–2381 CrossRef CAS PubMed.
- S. Gáspár, Nanoscale, 2014, 6, 7757–7763 RSC.
- N. Mano and A. Heller, J. Am. Chem. Soc., 2005, 127, 11574–11575 CrossRef CAS PubMed.
- X. Ma and S. Sánchez, Tetrahedron, 2017, 73, 4883–4886 CrossRef CAS.
- J. Simmchen, A. Baeza, D. Ruiz-Molina and M. Vallet-Regí, Nanoscale, 2014, 6, 8907–8913 RSC.
- X. Ma, X. Wang, K. Hahn and S. Sánchez, ACS Nano, 2016, 10, 3597–3605 CrossRef CAS PubMed.
- T. Patiño, N. Feiner-Gracia, X. Arqué, A. Miguel-López, A. Jannasch, T. Stumpp, E. Schäffer, L. Albertazzi and S. Sánchez, J. Am. Chem. Soc., 2018, 140, 7896–7903 CrossRef PubMed.
- M. Luo, S. Li, J. Wan, C. Yang, B. Chen and J. Guan, Langmuir, 2020, 36, 7005–7013 CAS.
- T. Hornung, J. Martin, D. Spetzler, R. Ishmukhametov and W. D. Frasch, in Single Molecule Enzymology, Springer, 2011, pp. 273–289 Search PubMed.
- L. Wang, A. C. Hortelão, X. Huang and S. Sánchez, Angew. Chem. Int. Ed., 2019, 58, 7992–7996 CrossRef CAS PubMed.
- X. Ma, A. C. Hortelão, T. Patiño and S. Sánchez, ACS Nano, 2016, 10, 9111–9122 CrossRef CAS PubMed.
- L. Wang, S. Song, J. van Hest, L. K. E. A. Abdelmohsen, X. Huang and S. Sánchez, Small, 2020, 1907680 CrossRef CAS PubMed.
- A. Halder and Y. Sun, Biosensors Bioelectron., 2019, 139, 111334 CrossRef CAS PubMed.
- M. Luo, Y. Feng, T. Wang and J. Guan, Adv. Funct. Mater., 2018, 28, 1706100 CrossRef.
- F. Soto and R. Chrostowski, Front. Bioeng. Biotechnol., 2018, 6, 170 CrossRef PubMed.
- J. Ou, K. Liu, J. Jiang, D. A. Wilson, L. Liu, F. Wang, S. Wang, Y. Tu and F. Peng, Small, 2020, 1906184 CrossRef CAS PubMed.
- T. Patiño, X. Arqué, R. Mestre, L. Palacios and S. Sánchez, Acc. Chem. Res., 2018, 51, 2662–2671 CrossRef PubMed.
- M. Fernández-Medina, M. A. Ramos-Docampo, O. Hovorka, V. Salgueiriño and B. Städler, Adv. Funct. Mater., 2020, 30, 1908283 CrossRef.
- J. Wang and W. Gao, ACS Nano, 2012, 6, 5745–5751 CrossRef CAS PubMed.
- K. K. Dey, F. Wong, A. Altemose and A. Sen, Curr. Opin. Colloid Interface Sci., 2016, 21, 4–13 CrossRef CAS.
- L. K. Abdelmohsen, F. Peng, Y. Tu and D. A. Wilson, J. Mater. Chem. B, 2014, 2, 2395–2408 RSC.
- H. Zhang, A. M. Gomez, X. Wang, Y. Yan, M. Zheng and H. Cheng, Cardiovasc. Res., 2013, 98, 248–258 CrossRef CAS PubMed.
- T. Finkel, Curr. Opin. Cell Biol., 1998, 10, 248–253 CrossRef CAS PubMed.
- D. B. Zorov, M. Juhaszova and S. J. Sollott, Physiol. Rev., 2014, 94, 909–950 CrossRef CAS PubMed.
- E. Veal and A. Day, Antioxid. Redox Signaling, 2011, 15, 147–151 CrossRef CAS PubMed.
- H. Cai, Cardiovasc. Res., 2005, 68, 26–36 CrossRef CAS PubMed.
- B. Commoner, J. Townsend and G. E. Pake, Nature, 1954, 174, 689–691 CrossRef CAS PubMed.
- R. K. Root and J. A. Metcalf, J. Clin. Invest., 1977, 60, 1266–1279 CrossRef CAS PubMed.
- S. Miwa, J. St-Pierre, L. Partridge and M. D. Brand, Free Radical Biol. Med., 2003, 35, 938–948 CrossRef CAS PubMed.
- B. Halliwell, M. V. Clement, J. Ramalingam and L. H. Long, IUBMB Life, 2000, 50, 251–257 CrossRef CAS PubMed.
- F. Lacy, T. Kailasam Mala, T. O’Connor Daniel, W. Schmid-Schönbein Geert and J. Parmer Robert, Hypertension, 2000, 36, 878–884 CrossRef CAS PubMed.
- D. A. Wilson, R. J. M. Nolte and J. C. M. van Hest, Nat. Chem., 2012, 4, 268–274 CrossRef CAS PubMed.
- B. K. Vainshtein, W. R. Melik-Adamyan, V. V. Barynin, A. A. Vagin and A. I. Grebenko, Nature, 1981, 293, 411–412 CrossRef CAS PubMed.
- A. Deisseroth and A. L. Dounce, Physiol. Rev., 1970, 50, 319–375 CrossRef CAS PubMed.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794 CAS.
- S. Toyokuni, K. Okamoto, J. Yodoi and H. Hiai, FEBS Lett., 1995, 358, 1–3 CrossRef CAS PubMed.
- J. Boonstra and J. A. Post, Gene, 2004, 337, 1–13 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS PubMed.
- D. S. Dimski, J. Vet. Intern. Med., 1994, 8, 73–78 CrossRef CAS PubMed.
- M. Yoshida, E. Muneyuki and T. Hisabori, Nat. Rev. Mol. Cell Biol., 2001, 2, 669–677 CrossRef CAS PubMed.
- F. Peng, Y. Tu, J. C. M. van Hest and D. A. Wilson, Angew. Chem. Int. Ed., 2015, 54, 11662–11665 CrossRef CAS PubMed.
- H. Kamin and P. Handler, Annu. Rev. Biochem., 1957, 26, 419–490 CrossRef CAS PubMed.
- S. M. Morris Jr, Annu. Rev. Nutr., 2002, 22, 87–105 CrossRef PubMed.
- A. J. Cooper, Cirrhosis, Hyperammonemia, and Hepatic Encephalopathy, Springer, 1993, pp. 21–37 Search PubMed.
- J. Huizenga, C. Gips and A. Tangerman, Ann. Clin. Biochem., 1996, 33, 23–30 CrossRef PubMed.
- E. M. MacKay and L. L. MacKay, J. Clin. Invest., 1927, 4, 295–306 CrossRef CAS PubMed.
- A. Taylor and P. Vadgama, Ann. Clin. Biochem., 1992, 29, 245–264 CrossRef CAS PubMed.
- J. P. Bretaudiere, H. T. Phung and M. Bailly, Clin. Chem., 1976, 22, 1614–1617 CrossRef CAS.
- K. P. Maier, H. Talke and W. Gerok, Klin. Wochenschr., 1979, 57, 661–665 CrossRef CAS.
- B. K. Burton, Clin. Liver Dis., 2000, 4, 815–830 CrossRef CAS.
- H. S. Muddana, S. Sengupta, T. E. Mallouk, A. Sen and P. J. Butler, J. Am. Chem. Soc., 2010, 132, 2110–2111 CrossRef CAS.
- A. C. Hortelão, R. Carrascosa, N. Murillo-Cremaes, T. Patiño and S. Sánchez, ACS Nano, 2019, 13, 429–439 CrossRef PubMed.
- K. K. Dey, S. Das, M. F. Poyton, S. Sengupta, P. J. Butler, P. S. Cremer and A. Sen, ACS Nano, 2014, 8, 11941–11949 CrossRef CAS PubMed.
- K. K. Dey, X. Zhao, B. M. Tansi, W. J. Méndez-Ortiz, U. M. Córdova-Figueroa, R. Golestanian and A. Sen, Nano Lett., 2015, 15, 8311–8315 CrossRef CAS PubMed.
- A. C. Hortelão, T. Patiño, A. Perez-Jiménez, À. Blanco and S. Sánchez, Adv. Funct. Mater., 2018, 28, 1705086 CrossRef.
- M. D. Shelley, M. D. Mason and H. Kynaston, Cancer Treat. Rev., 2010, 36, 195–205 CrossRef PubMed.
- P. Tyagi, P.-C. Wu, M. Chancellor, N. Yoshimura and L. Huang, Mol. Pharmaceutics, 2006, 3, 369–379 CrossRef CAS PubMed.
- D. P. Griffith, D. M. Musher and C. Itin, Invest. Urol., 1976, 13, 346–350 CAS.
- B. R. Landau, J. R. Leonards and F. M. Barry, Am. J. Physiol., 1961, 201, 41–46 CrossRef CAS PubMed.
- J. W. Appelboom, W. A. Brodsky and W. S. Rehm, J. Gen. Physiol., 1959, 43, 467–479 CrossRef CAS PubMed.
- G. F. Cahill, J. Ashmore, A. S. Earle and S. Zottu, Am. J. Physiol., 1958, 192, 491–496 CrossRef CAS PubMed.
- G. Bergers and L. E. Benjamin, Nat. Rev. Cancer, 2003, 3, 401–410 CrossRef CAS PubMed.
- A. Hirayama, K. Kami, M. Sugimoto, M. Sugawara, N. Toki, H. Onozuka, T. Kinoshita, N. Saito, A. Ochiai and M. Tomita, Cancer Res., 2009, 69, 4918–4925 CrossRef CAS PubMed.
- S. A. Skinner, P. J. Tutton and P. E. O'Brien, Cancer Res., 1990, 50, 2411–2417 CAS.
- H. J. Hecht, H. M. Kalisz, J. Hendle, R. D. Schmid and D. Schomburg, J. Mol. Biol., 1993, 229, 153–172 CrossRef CAS PubMed.
- R. Wilson and A. P. F. Turner, Biosens. Bioelectron., 1992, 7, 165–185 CrossRef CAS.
- C. M. Wong, K. H. Wong and X. D. Chen, Appl. Microbiol. Biotechnol., 2008, 78, 927–938 CrossRef CAS PubMed.
- Y.-H. Zhang, W.-X. Qiu, M. Zhang, L. Zhang and X.-Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 15030–15039 CrossRef CAS PubMed.
- H. Imamura, K. P. Huynh Nhat, H. Togawa, K. Saito, R. Iino, Y. Kato-Yamada, T. Nagai and H. Noji, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15651 CrossRef CAS PubMed.
- C. von Ballmoos, A. Wiedenmann and P. Dimroth, Annu. Rev. Biochem., 2009, 78, 649–672 CrossRef CAS PubMed.
- M. W. Gorman, E. O. Feigl and C. W. Buffington, Clin. Chem., 2007, 53, 318–325 CrossRef CAS PubMed.
- M. L. Casem, in Case Studies in Cell Biology, ed. M. L. Casem, Academic Press, Boston, 2016, pp. 263–281 Search PubMed.
- F. M. Gribble, G. Loussouarn, S. J. Tucker, C. Zhao, C. G. Nichols and F. M. Ashcroft, J. Biol. Chem., 2000, 275, 30046–30049 CrossRef CAS PubMed.
- M. Leist, B. Single, A. F. Castoldi, S. Kühnle and P. Nicotera, J. Exp. Med., 1997, 185, 1481–1486 CrossRef CAS PubMed.
- M. J. Schnitzer and S. M. Block, Nature, 1997, 388, 386–390 CrossRef CAS PubMed.
- H. Noji, R. Yasuda, M. Yoshida and K. Kinosita, Nature, 1997, 386, 299–302 CrossRef CAS PubMed.
- S. Diez, C. Reuther, C. Dinu, R. Seidel, M. Mertig, W. Pompe and J. Howard, Nano Lett., 2003, 3, 1251–1254 CrossRef CAS.
- W. R. Schief, R. H. Clark, A. H. Crevenna and J. Howard, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1183 CrossRef CAS PubMed.
- M. A. Ramos-Docampo, M. Fernández-Medina, E. Taipaleenmäki, O. Hovorka, V. N. Salgueiriño and B. Städler, ACS Nano, 2019, 13, 12192–12205 CrossRef CAS PubMed.
- V. Truong, S. Huang, J. Dennis, M. Lemire, N. Zwingerman, D. Aïssi, I. Kassam, C. Perret, P. Wells, P.-E. Morange, M. Wilson, D.-A. Trégouët and F. Gagnon, Sci. Rep., 2017, 7, 11207 CrossRef PubMed.
- L. Wang, A. C. Hortelão, X. Huang and S. Sanchez, Angew. Chem., 2019, 131, 8076–8080 CrossRef.
- H. C. Berg and D. A. Brown, Nature, 1972, 239, 500–504 CrossRef CAS PubMed.
- A. C. Grimm and C. S. Harwood, Appl. Environ. Microbiol., 1997, 63, 4111–4115 CrossRef CAS PubMed.
- S. Sengupta, K. K. Dey, H. S. Muddana, T. Tabouillot, M. E. Ibele, P. J. Butler and A. Sen, J. Am. Chem. Soc., 2013, 135, 1406–1414 CrossRef CAS PubMed.
- J. Wang, B. J. Toebes, A. S. Plachokova, Q. Liu, D. Deng, J. A. Jansen, F. Yang and D. A. Wilson, Adv. Healtcare Mater., 2020, 9, 1901710 CrossRef CAS PubMed.
- A. Joseph, C. Contini, D. Cecchin, S. Nyberg, L. Ruiz-Perez, J. Gaitzsch, G. Fullstone, X. Tian, J. Azizi, J. Preston, G. Volpe and G. Battaglia, Sci. Adv., 2017, 3, e1700362 CrossRef PubMed.
- H. Yu, K. Jo, K. L. Kounovsky, J. J. D. Pablo and D. C. Schwartz, J. Am. Chem. Soc., 2009, 131, 5722–5723 CrossRef CAS.
- F. Wu, L. N. Pelster and S. D. Minteer, Chem. Commun., 2015, 51, 1244–1247 RSC.
- X. Zhao, H. Palacci, V. Yadav, M. M. Spiering, M. K. Gilson, P. J. Butler, H. Hess, S. J. Benkovic and A. Sen, Nat. Chem., 2018, 10, 311–317 CrossRef CAS PubMed.
- J. Agudo-Canalejo, P. Illien and R. Golestanian, Nano Lett., 2018, 18, 2711–2717 CrossRef CAS PubMed.
- A.-Y. Jee, S. Dutta, Y.-K. Cho, T. Tlusty and S. Granick, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 14–18 CrossRef CAS PubMed.
- M. Feng and M. K. Gilson, Annu. Rev. Biophys., 2020, 49, 87–105 CrossRef CAS PubMed.
- J. M. Schurr, B. S. Fujimoto, L. Huynh and D. T. Chiu, J. Phys. Chem. B, 2013, 117, 7626–7652 CrossRef CAS PubMed.
- X. Zhao, H. Palacci, V. Yadav, M. M. Spiering, M. K. Gilson, P. J. Butler, H. Hess, S. J. Benkovic and A. Sen, Nat. Chem., 2018, 10, 311 CrossRef CAS PubMed.
- M. Feng and M. K. Gilson, Annu. Rev. Biophys., 2020, 49, 87–105 CrossRef CAS PubMed.
- A. Somasundar, S. Ghosh, F. Mohajerani, L. N. Massenburg, T. Yang, P. S. Cremer, D. Velegol and A. Sen, Nat. Nanotechnol., 2019, 14, 1129–1134 CrossRef CAS.
- D. Pantarotto, W. R. Browne and B. L. Feringa, Chem. Commun., 2008, 1533–1535 RSC.
- P. S. Schattling, M. A. Ramos-Docampo, V. Salgueiriño and B. Städler, ACS Nano, 2017, 11, 3973–3983 CrossRef CAS PubMed.
- X. Ma, A. Jannasch, U.-R. Albrecht, K. Hahn, A. Miguel-López, E. Schäffer and S. Sánchez, Nano Lett., 2015, 15, 7043–7050 CrossRef PubMed.
- L. K. E. A. Abdelmohsen, M. Nijemeisland, G. M. Pawar, G.-J. A. Janssen, R. J. M. Nolte, J. C. M. van Hest and D. A. Wilson, ACS Nano, 2016, 10, 2652–2660 CrossRef CAS PubMed.
- P. Schattling, B. Thingholm and B. Städler, Chem. Mater., 2015, 27, 7412–7418 CrossRef CAS.
- X. Arqué, A. Romero-Rivera, F. Feixas, T. Patiño, S. Osuna and S. Sánchez, Nat. Commun., 2019, 10, 2826 CrossRef PubMed.
- X. Ma, A. C. Hortelao, A. Miguel-López and S. Sánchez, J. Am. Chem. Soc., 2016, 138, 13782–13785 CrossRef CAS PubMed.
- S. Sanchez, A. A. Solovev, Y. Mei and O. G. Schmidt, J. Am. Chem. Soc., 2010, 132, 13144–13145 CrossRef CAS PubMed.
- A. A. Solovev, S. Sanchez, M. Pumera, Y. F. Mei and O. G. Schmidt, Adv. Funct. Mater., 2010, 20, 2430–2435 CrossRef CAS.
- X. Ma and S. Sanchez, Chem. Commun., 2015, 51, 5467–5470 RSC.
- I. Ortiz-Rivera, M. Mathesh and D. A. Wilson, Acc. Chem. Res., 2018, 51, 1891–1900 CrossRef CAS PubMed.
- M. Nijemeisland, L. K. E. A. Abdelmohsen, W. T. S. Huck, D. A. Wilson and J. C. M. van Hest, ACS Cent. Sci., 2016, 2, 843–849 CrossRef CAS PubMed.
- S. Keller, S. P. Teora, G. X. Hu, M. Nijemeisland and D. A. Wilson, Angew. Chem. Int. Ed., 2018, 57, 9814–9817 CrossRef CAS PubMed.
- B. J. Toebes, L. K. E. A. Abdelmohsen and D. A. Wilson, Polym. Chem., 2018, 9, 3190–3194 RSC.
- B. V. V. S. P. Kumar, A. J. Patil and S. Mann, Nat. Chem., 2018, 10, 1154–1163 CrossRef CAS PubMed.
- W.-S. Jang, H. J. Kim, C. Gao, D. Lee and D. A. Hammer, Small, 2018, 14, 1801715 CrossRef PubMed.
- M. Mathesh and D. A. Wilson, Adv. Intell. Sys., 2020, 2, 2000028 CrossRef.
- B. J. Toebes, F. Cao and D. A. Wilson, Nat. Commun., 2019, 10, 5308 CrossRef PubMed.
- S. Keller, G. X. Hu, M. I. Gherghina-Tudor, S. P. Teora and D. A. Wilson, Adv. Funct. Mater., 2019, 29, 1904889 CrossRef CAS.
- S. Sengupta, D. Patra, I. Ortiz-Rivera, A. Agrawal, S. Shklyaev, K. K. Dey, U. Córdova-Figueroa, T. E. Mallouk and A. Sen, Nat. Chem., 2014, 6, 415–422 CrossRef CAS PubMed.
- C. Gao, C. Zhou, Z. Lin, M. Yang and Q. He, ACS Nano, 2019, 13, 12758–12766 CrossRef CAS PubMed.
- Y. Ji, X. Lin, Z. Wu, Y. Wu, W. Gao and Q. He, Angew. Chem. Int. Ed., 2019, 58, 12200–12205 CrossRef CAS.
- M. Mathesh, B. Luan, T. O. Akanbi, J. K. Weber, J. Liu, C. J. Barrow, R. Zhou and W. Yang, ACS Catal., 2016, 6, 4760–4768 CrossRef CAS.
- Y. Hu and Y. Sun, Biochem. Eng. J., 2019, 149, 107242 CrossRef CAS.
- S. Ghosh, F. Mohajerani, S. Son, D. Velegol, P. J. Butler and A. Sen, Nano Lett., 2019, 19, 6019–6026 CrossRef CAS PubMed.
- S. Sattayasamitsathit, H. Kou, W. Gao, W. Thavarajah, K. Kaufmann, L. Zhang and J. Wang, Small, 2014, 10, 2830–2833 CrossRef CAS PubMed.
- W. Gao, X. Feng, A. Pei, Y. Gu, J. Li and J. Wang, Nanoscale, 2013, 5, 4696–4700 RSC.
- J. Li, P. Angsantikul, W. Liu, B. Esteban-Fernández de Ávila, S. Thamphiwatana, M. Xu, E. Sandraz, X. Wang, J. Delezuk, W. Gao, L. Zhang and J. Wang, Angew. Chem. Int. Ed., 2017, 56, 2156–2161 CrossRef CAS PubMed.
- M. Guix, A. K. Meyer, B. Koch and O. G. Schmidt, Sci. Rep., 2016, 6, 1–7 CrossRef.
- J. J. McDermott, A. Kar, M. Daher, S. Klara, G. Wang, A. Sen and D. Velegol, Langmuir, 2012, 28, 15491–15497 CrossRef CAS PubMed.
- J. G. Croissant, Y. Fatieiev, A. Almalik and N. M. Khashab, Adv. Healtcare Mater., 2018, 7, 1700831 CrossRef PubMed.
- F. H. Ma Xing, L. Chunyan, L. Xiaojia, Z. Fanyu and W. Yong, J. Mater. Sci. Technol., 2017, 33, 1067–1074 CrossRef.
- J. Simmchen, A. Baeza, D. Ruiz, M. J. Esplandiu and M. Vallet-Regí, Small, 2012, 8, 2053–2059 CrossRef CAS PubMed.
- P. Díez, B. Esteban-Fernández de Ávila, D. E. Ramírez-Herrera, R. Villalonga and J. Wang, Nanoscale, 2017, 9, 14307–14311 RSC.
- A. C. Hortelão, R. Carrascosa, N. Murillo-Cremaes, T. Patiño and S. Sánchez, ACS Nano, 2018, 13, 429–439 CrossRef PubMed.
- J. Li, S. Thamphiwatana, W. Liu, B. Esteban-Fernández de Ávila, P. Angsantikul, E. Sandraz, J. Wang, T. Xu, F. Soto, V. Ramez, X. Wang, W. Gao, L. Zhang and J. Wang, ACS Nano, 2016, 10, 9536–9542 CrossRef CAS PubMed.
- A. Llopis-Lorente, A. García-Fernández, N. Murillo-Cremaes, A. C. Hortelão, T. Patiño, R. Villalonga, F. Sancenón, R. Martinez-Manez and S. Sanchez, ACS Nano, 2019, 13, 12171–12183 CrossRef CAS PubMed.
- A. Llopis-Lorente, A. García-Fernández, E. Lucena-Sánchez, P. Díez, F. Sancenón, R. Villalonga, D. A. Wilson and R. Martínez-Máñez, Chem. Commun., 2019, 55, 13164–13167 RSC.
- D. Montero, C. Tachibana, J. Rahr Winther and C. Appenzeller-Herzog, Redox Biol., 2013, 1, 508–513 CrossRef CAS PubMed.
- S. Wang, X. Liu, Y. Wang, D. Xu, C. Liang, J. Guo and X. Ma, Nanoscale, 2019, 11, 14099–14112 RSC.
- Y. Wu, X. Lin, Z. Wu, H. Möhwald and Q. He, ACS Appl. Mater. Interfaces, 2014, 6, 10476–10481 CrossRef CAS PubMed.
- Z. Wu, X. Lin, X. Zou, J. Sun and Q. He, ACS Appl. Mater. Interfaces, 2015, 7, 250–255 CrossRef CAS PubMed.
- M. A. Ramos-Docampo, M. Fernández-Medina, E. Taipaleenmäki, O. Hovorka, V. Salgueiriño and B. Städler, ACS Nano, 2019, 13, 12192–12205 CrossRef CAS PubMed.
- J. Sun, M. Mathesh, W. Li and D. A. Wilson, ACS Nano, 2019, 13, 10191–10200 CrossRef CAS PubMed.
- Q. He and J. Shi, J. Mater. Chem., 2011, 21, 5845–5855 RSC.
- Z. Chen, T. Xia, Z. Zhang, S. Xie, T. Wang and X. Li, Chem. Eng. J., 2019, 375, 122109 CrossRef CAS.
- Y. You, D. Xu, X. Pan and X. Ma, Appl. Mater. Today, 2019, 16, 508–517 CrossRef.
- S. Gao, J. Hou, J. Zeng, J. J. Richardson, Z. Gu, X. Gao, D. Li, M. Gao, D.-W. Wang, P. Chen, V. Chen, K. Liang, D. Zhao and B. Kong, Adv. Funct. Mater., 2019, 29, 1808900 CrossRef.
- D. Walker, B. T. Käsdorf, H.-H. Jeong, O. Lieleg and P. Fischer, Sci. Adv., 2015, 1, e1500501 CrossRef PubMed.
- Y. Jia, W. Dong, X. Feng, J. Li and J. Li, Nanoscale, 2015, 7, 82–85 RSC.
- W. Gao, R. Dong, S. Thamphiwatana, J. Li, W. Gao, L. Zhang and J. Wang, ACS Nano, 2015, 9, 117–123 CrossRef CAS PubMed.
- X. Wei, M. Beltrán-Gastélum, E. Karshalev, B. Esteban-Fernández de Ávila, J. Zhou, D. Ran, P. Angsantikul, R. H. Fang, J. Wang and L. Zhang, Nano Lett., 2019, 19, 1914–1921 CrossRef CAS PubMed.
- M. Zhou, T. Hou, J. Li, S. Yu, Z. Xu, M. Yin, J. Wang and X. Wang, ACS Nano, 2019, 13, 1324–1332 CAS.
- E. Karshalev, Y. Zhang, B. Esteban-Fernández de Ávila, M. Beltrán-Gastélum, Y. Chen, R. Mundaca-Uribe, F. Zhang, B. Nguyen, Y. Tong, R. H. Fang, L. Zhang and J. Wang, Nano Lett., 2019, 19, 7816–7826 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |