
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold nanoclusters for biomedical applications: toward in vivo studies
Estelle
Porret
,
Xavier
Le Guével
* and
Jean-Luc
Coll
 *
*
Université Grenoble Alpes – INSERM U1209 – CNRS UMR 5309, 38000 Grenoble, France. E-mail: Estelle.porret@univ-grenoble-alpes.fr; Xavier.le-guevel@univ-grenoble-alpes.fr; Jean-luc.coll@univ-grenoble-alpes.fr
First published on 18th February 2020
Abstract
In parallel with the rapidly growing and widespread use of nanomedicine in the clinic, we are also witnessing the development of so-called theranostic agents that combine diagnostic and therapeutic properties. Among them, ultra-small gold nanoclusters (Au NCs) show promising potential due to their optical properties and activatable therapeutic activities under irradiation. Furthermore, due to their hydrodynamic diameter of smaller than 6 nm and unique biophysical properties, they also present intriguing behaviors in biological and physio-pathological environments. In this review, we aim to present the latest research studies published on such nanoparticles in animals. We also propose guidelines to identify the main physico-chemical parameters that govern the behaviour of Au NCs after administration in small animals, notably concerning their renal elimination and their ability to accumulate in tumors. Then, we present recent advances in their use as theranostic agents putting them in parallel with other contrast agents.
 Estelle Porret | Dr Estelle Porret received her Chemistry and Physical Engineering degree, with specialization in nano- and microtechnology, from Graduate School of Chemistry, Biology and Physics (ENSCBP) and a Master's level degree in physical chemistry of materials from Bordeaux University in 2016. She joined Dr Jean-Luc Coll's group at the Institute for Advanced Biosciences the same year to obtain her PhD, focusing on the development of novel gold nanoclusters for cancer diagnosis and therapy. |
 Xavier Le Guével | Dr Xavier Le Guevel obtained his PhD in 2006 in Chemistry from the University of Tours (France). Following this, he did some post-doctoral fellowships in physics (Rome, Italy), biophotonics (Dublin, Ireland) and biotechnology (Saarbruecken, Germany) between 2006 and 2010 before starting as an independent researcher at the Nanomedicine Centre Bionand in Malaga (Spain). Since 2016, XLG has been a CNRS researcher at the Institute for Advanced Biosciences in Grenoble (France). His work focuses on the development of new nanomaterials such as metal nanoclusters and optical instruments for medical applications and more specifically for cancer treatment and diagnosis from fundamental research to pre-clinical application. |
 Jean-Luc Coll | Dr Jean-Luc Coll is the Director of Research at INSERM (Institut National de la Santé et de la Recherche Médicale) in France. He is in charge of the team “Cancer Targets and Experimental Therapeutics” in the Institute for Advanced Biosciences in Grenoble. JLC had an initial training in molecular biology (thesis in microbial genetics on E. coli), and then focused on cancer, first as a postdoc at the Burnham Institute (La Jolla USA) and then at the Cancer Research Center of Lyon (CRCL – Centre Léon Bérard). For the last 20 years, he has been working at the interfaces between biology, chemistry, physics and medicine with a clinical (veterinary and human) and industrial (2 Start-up) vision. JLC is focused on the use of near-infrared labeled nanoparticles to target tumors, guide surgery and enhance radiotherapy, phototherapy or innovative therapies. In addition to the developments of nanovectors, he is also deeply involved in the generation of innovative adapted medical devices. |
1 Introduction
In the past few years, a new class of photoluminescent ultra-small size metal nanoparticles (NPs) with a core size between 0.2 and 3 nm and usually referred to in the literature as nanoclusters (NCs)1 have gained growing interest in biomedical applications. They are composed of an assembly of tens to hundreds of metallic atoms (gold, silver, platinum, zinc, copper, etc.)2–6 stabilized by ligands (organic thiolate molecules,7–15 dendrimers,16–18 DNA,19,20 amino acids,21–23 peptides,24–26 or proteins27–29). NCs can be viewed as the missing link between metal–ligand complexes and plasmonic metal NPs.Their optical and physicochemical properties are suitable for biomedical applications since:
• Their ultra-small size should favor renal elimination, but increase passive accumulation in the tumor micro-environment compared to small molecules.30
• They can be detected in vivo by multimodal imaging techniques owing to their tunable photoluminescence (PL), from the ultra-violet (UV) to near-infrared (NIR) region17 and by X-ray CT31,32 or, as recently detected, by photoacoustic imaging33 owing to their metallic composition.
• They can be used as radio-sensitizers34–36 due to the electronic properties of metal NCs.
• They can be used as delivery systems.37–39
Gold is often preferred over other metals for biomedical applications because of its biocompatibility and inertness. Therefore, we mainly focus this review on the in vivo studies of gold NCs (Au NCs).
2 Design of Au NCs
2.1 Synthesis parameters
Au NCs can be synthesized in solution by the classical “bottom-up” approach using metal precursors. Depending on the nature of the ligands and the synthesis parameters such as the metal:ligand:reducing agent proportion, pH, temperature, or strength of reducing agents, a large library of Au NCs have been synthesized and used for in vivo studies.40 They can be divided into two main categories.The first one corresponds to Au NCs with a discrete composition at the atomic level synthesized with thiolated molecules. These clusters are represented by their formula Mn(L)m, where n and m are respectively the number of metal atoms (M) and ligands (L = thiolate ligand SR31 or poly(amino-amide) PAMAM17).
Using this “bottom-up” approach, it was difficult to control the growth of single-size stable Au NCs and the final products were often composed of mixtures of Au NCs. Indeed, in 2004, the work of Tsukuda et al.24,41 on the characterization of gold NCs stabilized with glutathione (SG) (AunSGm), by polyacrylamide gel electrophoresis and electrospray ionization mass spectrometry, revealed the presence of nine species (Au10SG10, Au15SG13, Au18SG14, Au22SG16, Au22SG17, Au25SG18, Au29SG20, Au33–35SG22, and Au38–39SG22). Furthermore, it was challenging to synthesize gram-scale monodispersed Au NCs. In 2007, a so-called etching method was then proposed42 based on excess ligands with increasing temperature to obtain stable and single-size Au NCs. This process was first used to produce Au25SG18 on a large scale,43 prior to adopting a size-focusing methodology with the aim of extending the obtention of large-scale monodispersed Au NCs to other sizes such as Au10SG10, Au15SG13, Au18SR13 (SR = SG, S-c-C6H11), Au38SR24 and Au144SR60, (SR = SC2H4Ph, SC12H25).1,44–46 Recent reviews have summarized the advances in synthesis of Au NCs and their properties.44,47–49 Size focusing still has some limitations to obtain atomically precise Au NCs with some ligands. Au NCs stabilized with polyethylene glycol (PEG),15,50 zwitterions (Zw)8,33 or proteins (“bovine serum albumin” BSA or transferrin)27,29 as ligands exhibit some polydispersity and correspond to the second category of Au NCs: subnanometer-scaled NPs with a core size between 1 and 3 nm.51
Metal NPs tend to aggregate in solutions over time due to ionic and electrostatic interactions (van der Waals forces).52 To reduce this effect, NPs can be stabilized with ligands in order to generate electrostatic or steric repulsions between the NPs. Thiolate ligands are usually used due to the strong bond between the sulfur group and the gold surface (40 kcal mol−1).53 A large library of biocompatible ligands such as proteins,27 DNA,19 peptides,24 dendrimers,17 polymers16 or small organic molecules following the previous criteria are suitable for stabilizing NCs.7
Lipoic acid (LA)10,11,37,54 is an example of a bidentate thiol ligand with strong anchors onto the metal surface, and its carboxylic groups that are negatively charged at physiological pH are known to generate electrostatic repulsion between NCs. Following this, a large panel of zwitterions based on lipoic acid sulfobetaine (LA-sulfobetaine) for stabilizing Au NCs have been designed, which exhibited remarkable colloidal stability in various media with high antifouling effects.12,13,55 Indeed, F. Aldeek et al.8 demonstrated that Au NCs stabilized by LA-sulfobetaine with a zwitterion moiety remained stable for at least 3 months even in acidic phosphate buffer (pH 2) or in the presence of 0.25 M antioxidant biomolecules. In contrast, Au NCs stabilized only by LA aggregated with time, which confirms the enhanced colloidal stability of zwitterionic functions. Another zwitterionic ligand widely used in NC synthesis is SG.26 This tripeptide is naturally present in eukaryotic cells to maintain their intracellular redox homeostasis.3 SG was used to synthesize atomically precise NC AunSGm, negatively charged at physiological pH, initiating the metal growth from the cysteine end of the peptide.41,42 Single amino acids, such as histidine23 and cysteine,21,22 which are positively charged and neutral at physiological pH, respectively, have also been used to stabilize NCs. The use of longer sequences of amino acid will induce steric repulsion between the NCs. The first protein employed to grow NCs is the BSA.27 This 66.5 kDa protein is composed of tyrosines that could reduce gold atoms under alkaline conditions (pH ∼ 12),56 and then be trapped by the thiol group of cysteines. Several studies reported the in vivo applications of AuBSA NCs.34,39,57–62 Following the same process, it was possible to grow Au NCs within transferrin,63 a protein that transports iron with receptors overexpressed at the surface of some cancer cells.64 NCs stabilized by transferrin and conjugated with graphene oxide have already been used in vivo.65 Finally, PEG is often used as a biocompatible capping agent to form a hydrating layer and generate steric hindrance between the NCs.8,13–15
2.2 Specificities of the Au NCs for in vivo applications
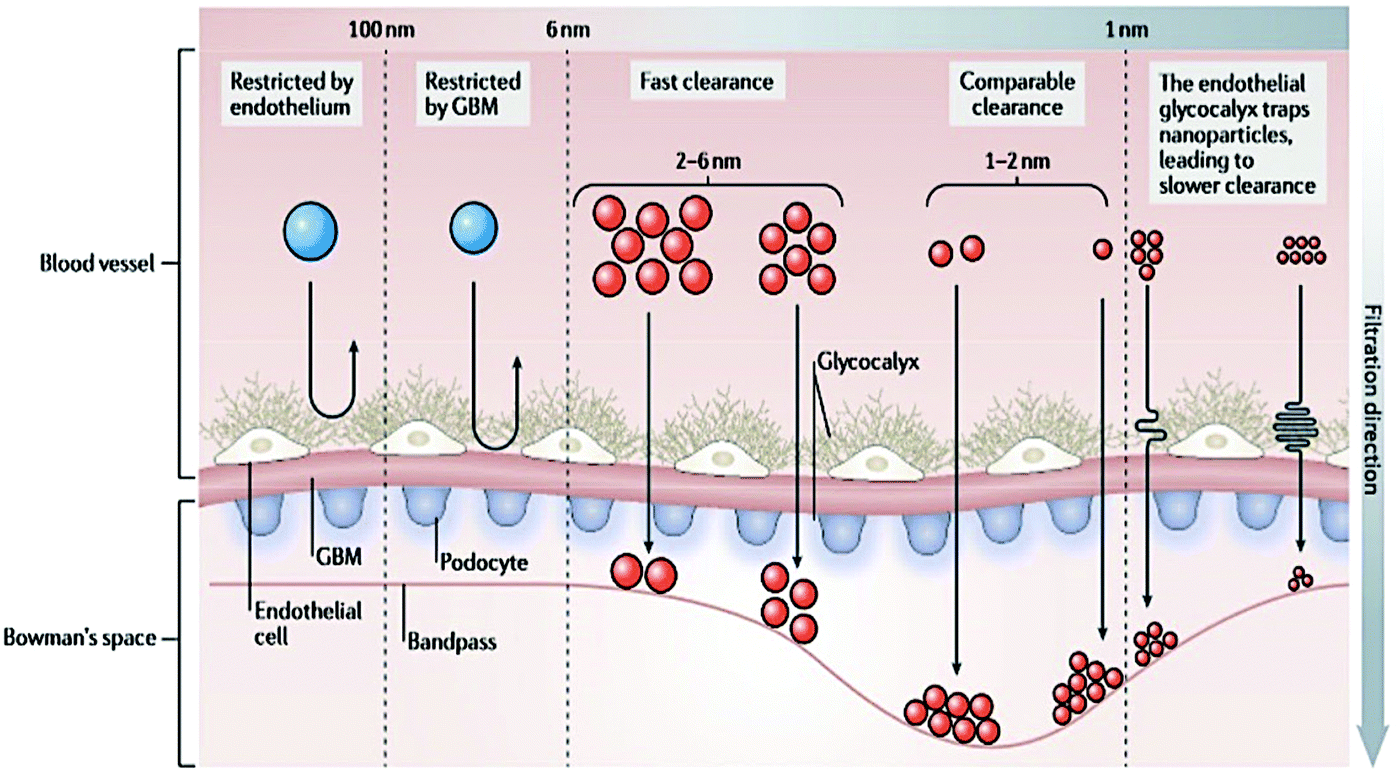 | ||
| Fig. 1 Layer composition of the glomerular filtration barrier. The filtration takes place through the endothelial fenestrae, across the glomerular basement membrane and the pores between podocytes. The combination of these three layers defines the size of the compounds that could cross the glomerular filtration barrier. Reproduced from ref. 66 with permission from Springer Nature publishing group. | ||
A study was conducted to follow the biodistribution of Au and Ag NPs with sizes ranging between 1.4 and 250 nm after intravenous administration in mice. It confirmed the size-dependent toxicity of NPs. If small NPs were widely spread into the organs, larger ones were mainly found in the liver and spleen.70 The accumulation of metal NPs in different organs is considered a major issue due to their poor degradation that could induce acute toxicity.69,71 This size effect could be extended to other inorganic nanomaterials such as quantum dots,72 lanthanide NPs,73 and carbon nanotubes.74
A deeper toxicology study75 was carried out on Au NPs with sizes between 3 and 100 nm after their intraperitoneal injection in mice. Surprisingly, the smallest and largest NPs (3, 5, 50, and 100 nm) did not show any harmful effects in mice, while those with intermediate sizes (8, 12, 17, and 37 nm) induced severe sickness that can lead to premature death in mice. These studies revealed the potential toxicity of Au NPs larger than 6 nm and advantages of using ultra-small Au NCs.
Several Au NCs exhibit PL in the NIR window between 600 and 850 nm.14,21,77 Very recently, the spectral window has been extended to the shortwave infrared (SWIR) region (900–1700 nm).78 Developing biocompatible optical probes in the NIR/SWIR optical window presents an advantage for in vivo imaging in deep tissues due to reduced auto-fluorescence, exponential decrease in light scattering and improved penetration of light into the tissues, offering potentially a higher spatial resolution.79 Au NCs also present large Stokes shift in comparison to most of the organic fluorophores (<20–30 nm80), which can exceed 100 nm.81,82 This reduces the scattering and absorption of light and improves fluorescence detection.
The main limitation of atomically precise Au NCs as optical probes is related to their low quantum yield (QY) one order of magnitude lower than those of quantum dots,83 lanthanide NPs,84 and organic dyes85 especially in the NIR/SWIR windows. Most of the Au NCs exhibit a QY < 1% with the well-characterized Au25SG18 exhibiting a QY ∼ 0.3%,86 which is 107 times higher than that of bulk gold.87 Fortunately, a few AunSGm exhibit higher QYs, such as Au18SG14 (λexc./em. = 590 nm/745 nm, QY ∼ 5.3%)88 or Au22SG18 (λexc./em. = 520 nm/665 nm, QY ∼ 8%).89 Different strategies have been reported to improve the brightness of Au NCs based on (i) doping with another metal,90–92 (ii) using ligands that can delocalize their electronic density to the gold core,93,94 or (iii) rigidification of the ligand shell surrounding the metal core.77,95,96
Therefore, it is crucial to develop new strategies to improve their QY. A first approach to enhance the brightness involves doping Au NCs with another metal. As an example, Rongchao Jin et al. obtained a QY of around 40% by doping Au NCs with 13 silver atoms90 (Fig. 2A). We also obtained a QY of around 15% by doping AuSG with silver.91 Similar results were obtained when doping Au NCs with 2% of different metals (Ag, Cu, Pt, Zn, and Cd).68 A second approach is based on the use of ligands that can delocalize their electronic density to the gold core. Replacing hexyl (C6H13) by dodecyl (C12H25) to stabilize Au25, or mixing Au25SG18 with a peptide nucleic acid (PNA) containing electron-rich atoms (e.g. O, N) induced a 2–3 fold enhancement of the PL93 (Fig. 2B). The exchange of some ligands of Au38(SPhC2)24 by HSPhNO2 rather SPhOCH3 also induced an increase of the PL.94
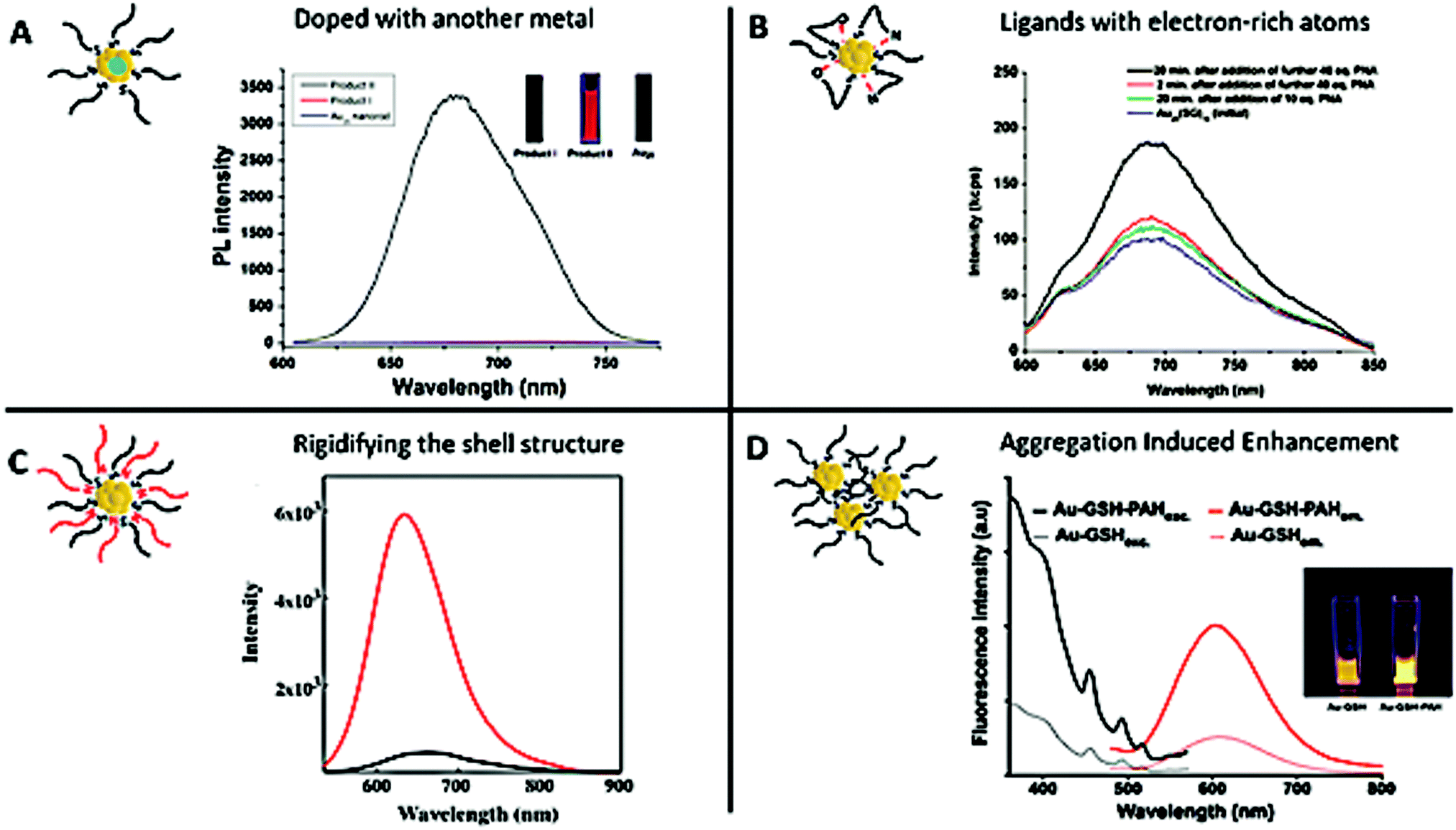 | ||
Fig. 2 Strategies to enhance the PL of the metal NCs. (A) Silver doping: Product I was obtained by reacting Au NPs with an AgI thiolate complex, which gave rise to AgxAu25−x NCs with a maximum of 12 Ag atoms. Product II was obtained by reacting Au11 NCs with the AgI thiolate complex, which gave rise to Ag13Au12 NCs. The PL spectra of Au25 NCs (bottom, blue line), Product I (middle, red line), and Product II (top, black line) show that the number of silver atoms has a strong impact on the optical properties of the NCs. The inset presents the fluorescence emission of the solution of the corresponding products under UV illumination (λexc. = 365 nm). (B) LMCT: luminescence spectra of Au25SG18 (blue) and after the addition of different equivalents of an electron-rich atom, PNA (initial concentration of Au25SG18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 μM, OD614 ∼ 0.025, λexc. = 514 nm). (C) Rigidification of the shell: luminescence spectra of Au22SG18 (black) and after the addition of tetraoctylammonium cations (red) into toluene to rigidify the shell of ligands (OD514 ∼ 0.025, λexc. = 514 nm). (D) AIE effect: excitation and emission spectra at similar Au NC concentrations of AuSG (dashed lines) and after the addition of PAH to promote electrostatic cross-linking between Au NCs (PAH-AuSG, solid lines). The inset presents the fluorescence emission of the solution of the corresponding products under UV illumination (λexc. = 366 nm). Reproduced with permission from John Wiley and Sons for (A) ref. 90, and American Chemical Society for (B) ref. 93 and 94, (C) ref. 95, and (D) ref. 97. :1.1 μM, OD614 ∼ 0.025, λexc. = 514 nm). (C) Rigidification of the shell: luminescence spectra of Au22SG18 (black) and after the addition of tetraoctylammonium cations (red) into toluene to rigidify the shell of ligands (OD514 ∼ 0.025, λexc. = 514 nm). (D) AIE effect: excitation and emission spectra at similar Au NC concentrations of AuSG (dashed lines) and after the addition of PAH to promote electrostatic cross-linking between Au NCs (PAH-AuSG, solid lines). The inset presents the fluorescence emission of the solution of the corresponding products under UV illumination (λexc. = 366 nm). Reproduced with permission from John Wiley and Sons for (A) ref. 90, and American Chemical Society for (B) ref. 93 and 94, (C) ref. 95, and (D) ref. 97. | ||
A third strategy is based on blocking the loss of energy caused by intramolecular rotation of the ligand, by rigidifying the shell structure. Very bright fluorescent Au22SG18 (QY ∼ 62% in toluene, λem. = 630 nm) was obtained after adding tetraoctylammonium cations while the initial NCs presented a QY ∼ 7% (in water, λem = 665 nm)95 (Fig. 2C). A rigidification of the shell structure of Au NCs stabilized with 6-aza-2thiothymine (ATT) was also obtained by adding L-arginine (QY ∼ 65%, versus QY ∼ 1.8% for the NCs alone; λem. = 530 nm).96 It is also possible to rigidify the shell by creating a second layer and a PL enhancement of 300 was observed when the ratio Au:Zw passed from 1:1 to 1:40.77
Finally, the aggregation-induced enhancement effect98 can also be seen as a strategy to boost Au NC PL. Indeed, aggregation will create stronger intra- and inter-complex aurophilic Au(I)⋯Au(I) interactions that could enhance the PL signal. At the same time, the intramolecular rotation and vibration of the ligands are reduced and the Au core is better protected from solvent molecules, thereby reducing the probability of nonradiative relaxation of the excited states.47,99 These two phenomena induce an enhancement of the emission of the NCs. By generating self-assembled AuSG using cationic polyelectrolytes such as poly(allylamine hydrochloride) (PAH), it was possible to control the distance between Au NCs by adjusting the pH and to increase the QY from 7 to almost 25% using the AIE effect97 (Fig. 2D).
For example, zwitterions based on lipoic acid sulfobetaine (LA-sulfobetaine), a bidentate thiol ligand with strong anchors to the metal surface, present remarkable colloidal stability in various media with high antifouling effects,8,13,100 while PEGylated ligands are often used as a biocompatible capping agent to form a hydrating layer and generate steric hindrance between the NCs.8,13–15
Other small organic molecules and biomolecules are also commonly used for functionalizing the NC surface:
— fluorophores to shift the optical properties of Au NCs toward the NIR region;23,62,101 drugs, photo or radiosensitizers for cancer therapy;
— targeting molecules to specifically interact with receptors overexpressed at the surface of tumor cells, such as:
• Folic acid (FA) that can recognize the folate receptors (FRs), overexpressed in many human cancer cells (ovary, breast, colon, kidneys, liver, testes, brain, lungs, and blood),54,61,101–103
• Luteinizing hormone-releasing hormone (LHRH) overexpressed in several types of cancer cells (ovary, breast, prostate, lungs, and liver),104
• Hyaluronic acid (HA) that can bind to CD44 receptors overexpressed at the surface of different cancer cells, in particular cancer stem cells,61
• AMD3100 that can interact with CXCR4, an up-regulating receptor, in particular in leukemia or breast cancer cells,105
• Cyclic RGD (cRGD), a zwitterionic compound capable of interacting with αvβ3, α5β1, and αvβ5 integrins highly present in neoangiogenic endothelial cells and several solid tumors.106–108
Two main techniques can be used to functionalize the Au NCs with specific molecules. An efficient approach involves directly synthesizing Au NCs with the molecule of interest usually with a terminal thiol group that could bind to the metal surface.8,9 A second approach is based on the post-functionalization of Au NCs. Click chemistry15 and succinimidyl ester105,109,110 reactions have been used to covalently bind molecules of interest to the ligand stabilizing the Au NCs,15,54,101,107 or ligand exchange could be used if the molecule of interest has a thiol group.110
3. Physico-chemical parameters affecting the biodistribution of Au NCs
C. Zhou et al.111 studied the biodistribution of AuSG with NIR emission. The highest uptake of the AuSG in the kidneys (21% of injected dose per gram of tissue, ID per g) was obtained at 5 min pi (post injection) and decreased almost by 3 fold (7.5 ID per g) after urinary excretion. These observations were consistent with their short circulation time in blood (half-life, t1/2α ∼ 5 min) and indicated that AuSG were mainly eliminated via the urinary system. The accumulation of AuSG in the liver and spleen was lower than that in the kidneys (respectively 4 and 2.4% ID per g) and remained constant over time. After 48 h, more than 50% of the injected AuSG were eliminated.We obtained comparable results with AuZw, which presented a short blood circulation time (t1/2α ∼ 6 min).112 Using laser-induced breakdown spectroscopy (LIBS), we detected a strong Au signal in the medulla of the kidneys 30 min pi that was almost undetectable at 1 h (Fig. 3A). In contrast, a low but constant Au signal was observed in the liver and spleen during 24 h (Fig. 3B and C, respectively). These results were confirmed ex vivo by fluorescence and inductively coupled plasma mass spectrometry measurements.
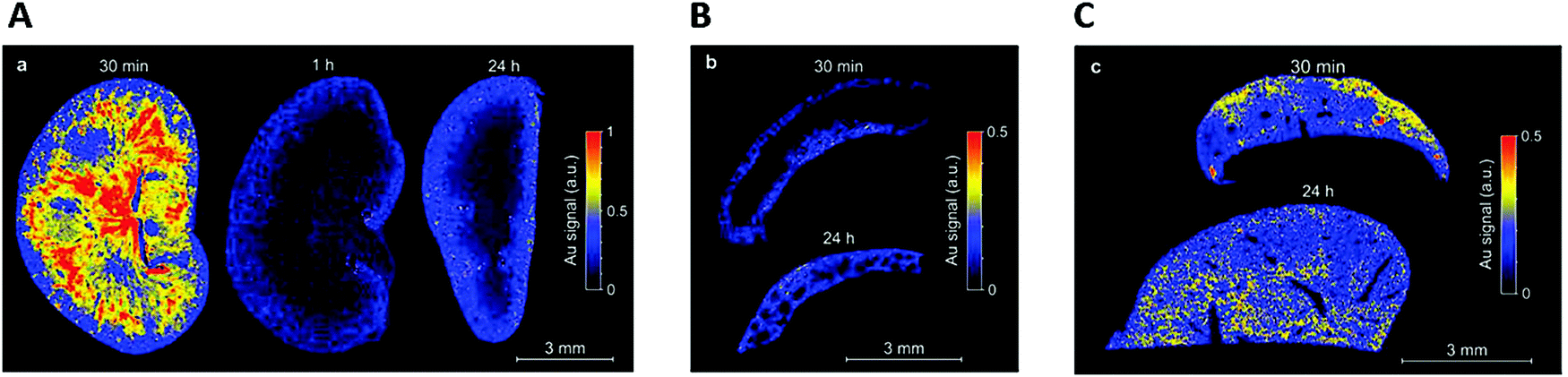 | ||
| Fig. 3 After the intravenous injection of AuZwMe2 (600 μM; 200 μL) into NMRI (Naval Medical Research Institute) nude mice, LIBS detection was carried out for: (A) kidney slices 30 min, 1 h, and 24 h pi, (B) spleen slices 30 min and 24 h, and (C) liver slices 30 min and 24 h. Reproduced from ref. 112 with permission from The Royal Society of Chemistry publishing group. | ||
These two studies demonstrated that ultra-small size Au NCs have a very rapid renal clearance, but it is important to better understand the different physicochemical parameters that may affect the kinetics and completeness of this renal elimination.
3.1 Length of the ligand
Due to the high surface reactivity of NCs, small changes in the ligand have huge impacts on their biodistribution. For example, the liver-to-blood and the kidney-to-blood ratios are decreased 22 and 1.9 times, respectively, when a glycine moiety is added to a cysteine ligand.21X. D. Zhang et al.59 synthesized Au NCs with the same core size, but stabilized by different ligands such as SG or BSA. AuSG showed a very efficient renal clearance with 36% of the NCs excreted into the urine 24 h pi and only 6% were remaining in the mouse 28 days pi. In contrast, more than 95% of AuBSA were retained 28 days pi, with elevated concentrations in the liver and spleen (Fig. 4A). This difference in terms of pharmacokinetics and biodistribution was attributed to the size of the ligand that changed the HD of the Au NCs. Indeed, the AuSG with its HD of 2 nm could be taken up by the kidneys, but not the HD of 7 nm of AuBSA.34,113
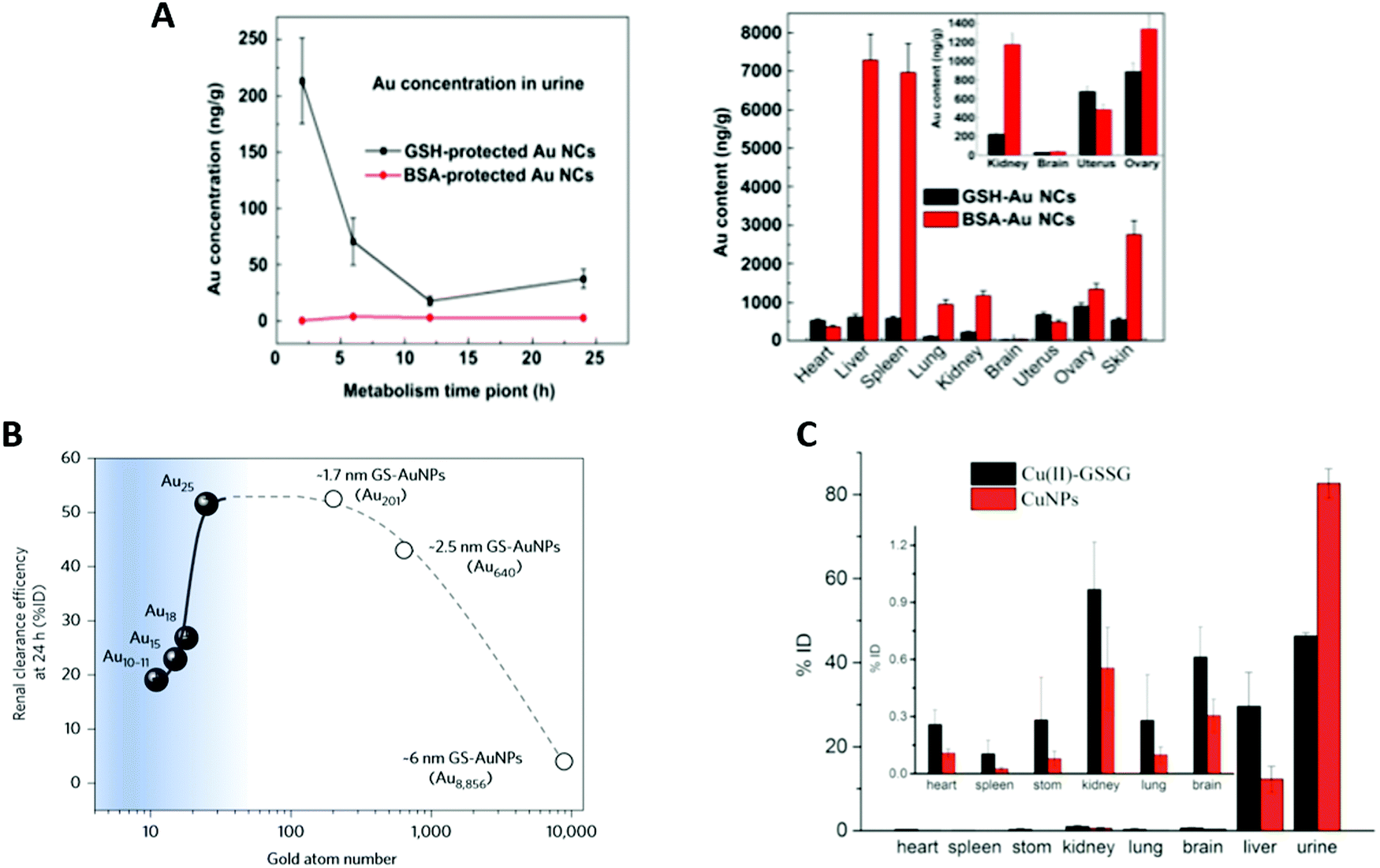 | ||
| Fig. 4 (A) Renal elimination and biodistribution in mice intravenously injected (7550 μg kg−1, 151 μg mL−1) with AuSG and AuBSA NCs: renal elimination 24 h pi (top left) and biodistribution in the main organs 28 days pi (top right). AuBSA NCs have 10 times higher distribution in the liver and spleen than AuSG NCs. (B) Effect of the sub-nanometer size of the NCs on their renal elimination. Renal elimination efficiencies of different sizes of Au NCs, intravenously injected (∼100 μM, 100 μL) in BALB/c mice, versus the number of gold atoms at 24 h pi. The renal elimination increased with the number of gold atoms up to 25 (Au25SG18) and reached a plateau followed by a decrease for larger sizes. (C) Biodistribution in the main organs 24 h after intravenous injections of Cu(II)-GSSG complexes (300 μL, 0.33 mg mL−1) or luminescent CuSG NCs (300 μL, 0.83 mg mL−1) in BALB/c mice (N = 6). Reproduced with permission from Elsevier for (A) ref. 59, Springer Nature for (B) ref. 46 and American Chemical Society for (C) ref. 6. | ||
3.2 Size of the metal core
AuSG NPs with increasing metallic core sizes, leading to an increase in their HD (2, 6, and 13 nm) were administered intravenously114 and, as could be expected, the results indicated that the larger the size, the stronger the liver uptake and the lower the kidney elimination. After one day pi, 50% of the injected dose (ID) of AuSG with an HD of 2 nm was eliminated in the urine, while only 4% or 0.5% ID for the 6 or 13 nm ones was observed. On the other hand, 3.7% ID, 27.1% ID, and 40.5% ID were respectively blocked in the liver. This tendency was also confirmed in a study that compared the clearance efficiency of 3.4 and 18.4 nm large Au NPs stabilized by SG.115 However, a reverse size dependency has been observed recently when a sub-nanometer size was reached. Au25SG18 were filtered faster than Au18SG14 or Au10–11SG10–1146 (Fig. 4B). In the latter case, a very small reduction in the number of gold atoms (from 18 to 10–11), and thus a very small reduction in their HD dramatically reduced their renal elimination because of an augmented binding of the clusters on the glomerular glycocalyx.Accordingly, biodegradable NCs that dissociate into small fragments should also be cleared from the body. CuSG are 2 nm large NCs (core diameter and 2.7 nm HD) that gradually degrade in Cu(II)-SG and disulfide (Cu(II)-GSSG) under physiological conditions.6 Surprisingly, 90% of the Cu collected in the urine 2 h pi was still in the form of CuSG, while 60% of Cu in the liver was in the form of Cu(II)-GSSG-complexes bound to serum proteins. Indeed, CuSG had a higher resistance to serum protein adsorption than Cu(II)-GSSG. This explains why these NCs were quickly eliminated and less accumulated in the liver than the degradation product (Fig. 4C).
3.3 Protein corona formation
Proteins can be adsorbed onto the surface of NPs forming the protein corona.113,116 The protein coating is a dynamic system with competition between different proteins until an equilibrium is reached. The first layer of proteins is rapidly adsorbed onto the surface of the particles to form the soft corona. In the second step, proteins that have a stronger affinity to the surface of the particles slowly remove the first layer of proteins to generate a stable layer: the hard corona. This means that the composition and thickness of the protein corona evolve with time.117 The affinity and exchange rate of the protein corona depend on the size, charge, composition, and shape of the NPs, the incubation conditions (temperature, concentration, and time), the type of proteins, and their stability.116,117The formation of this protein layer will have an impact on cell internalization, bio-distribution, and toxicity of the NPs,118,119 and NCs.120
Concerning NCs, it was established that incubation of Au NCs with an increasing concentration of human serum albumin (HSA) improved their fluorescence intensity by 6 at high protein concentrations (3 μM).121 This was probably due to the adsorption of proteins that decreased the non-radiative recombination by forming a rigid protection layer or inducing new metal–ligand interactions.
The surface charge strongly affects the amount and composition of the protein corona.109 Indeed, negative NCs interact preferentially with Apolipoprotein (Pi > 5.5), whereas positive NCs prefer albumin (Pi < 5.5), and as a result, positive NCs accumulate in the spleen, lungs, heart, and kidneys, while negatively charged NCs will rather end up in the liver and testis.109
Neutral surfaces obtained with PEGylated ligands or with zwitterionic molecules (SG or BSA) do not bind proteins. However, the non-fouling properties observed with zwitterionic systems are pH and particle curvature dependent.14,55,115
3.4 Density
The size of a nanoparticle is important, but its density (i.e. mass/volume) may also have a strong impact. Increasing the density of a NC will augment its interaction with the surface of the blood vessels. NCs stabilized by SG but with different metal cores (Au, Ag, and Au/Ag alloy) were synthesized.122 As compared to high density AuSG, the low-density AgSG presented low affinity for the blood vessels, faster clearance, and shorter retention time.3.5 Electrical charge
The charge of the NCs also determines the affinity of the NCs for the blood vessels. AuSG NCs functionalized with ethanediamine or ethanedioic acid to generate positively and negatively charged surfaces were more slowly excreted than neutral NCs over a 90 day period.109 Just 24 h after intraperitoneal administration, the amount of positively charged NCs in the kidneys was two times higher than those of neutral NCs. Various studies have demonstrated that, after glomerular filtration, positively charged compounds bind through their cationic sites to megalin receptors overexpressed on negatively charged proximal tubule epithelial cells.66,67,123 This re-absorption phenomenon could explain that small positively charged compounds were slowly excreted by the kidneys.3.6 Influence of the concentration of injected Au NCs
Finally, the concentration of the injected Au NCs also influences their transport by the blood flow.124 An interesting two-compartment kidney elimination process was observed for AuSG intravenously injected into mice at nine different concentrations (from 0.15 to 1 059 mg kg−1 of body weight). At low concentrations (<15 mg kg−1), kidney elimination was constant at 35% ID. Then, the renal elimination linearly increased with the concentration of NCs. One explanation is that, for small doses, the AuSG could easily cross the blood vessel wall and enter the extravascular space. The reduction in their blood concentration slows down the renal process. At higher concentrations, the Au NCs are strictly confined to the blood vessels, more rapidly transported by the blood flow, and more efficiently eliminated through the kidneys.4 Tumor targeting of Au NCs
If the renal clearance of the NCs is reckoned as highly relevant to reduce their toxicity, these species should still have enough time to circulate in the body in order to accumulate passively and/or specifically in the tumor microenvironment. The growth of a tumor generally induces the formation of new blood vessels with pore sizes between 300 and 1200 nm and a perturbation of the lymphatic drainage. This allows NP accumulation in cancer tissues at higher concentrations and for a longer time than in normal tissues. This phenomenon is called the Enhanced Permeability and Retention (EPR) effect. Maeda et al.125 were one of the first groups who studied the EPR effect in 1986. They discovered that NPs with a size larger than the KFT can circulate in the blood at high concentrations and for longer times (at least 6 h) to allow a passive uptake.4.1 Passive accumulation of renal-clearable Au NCs by the EPR effect
Au NCs combine a high renal clearance with surprisingly elevated passive uptake in MCF-7 tumors.30 As can be seen in Fig. 5A, AuSG and IRDye 800CW present a rapid whole-body distribution and a maximum tumor accumulation 40 min pi. However, 24 h pi, the concentration of NCs in the tumor was 10 times higher than that of IRDye 800CW (Fig. 5B). This augmented EPR effect probably occurred because AuSG NCs have a longer blood elimination half-life (t1/2β) than IRDye (Fig. 5C).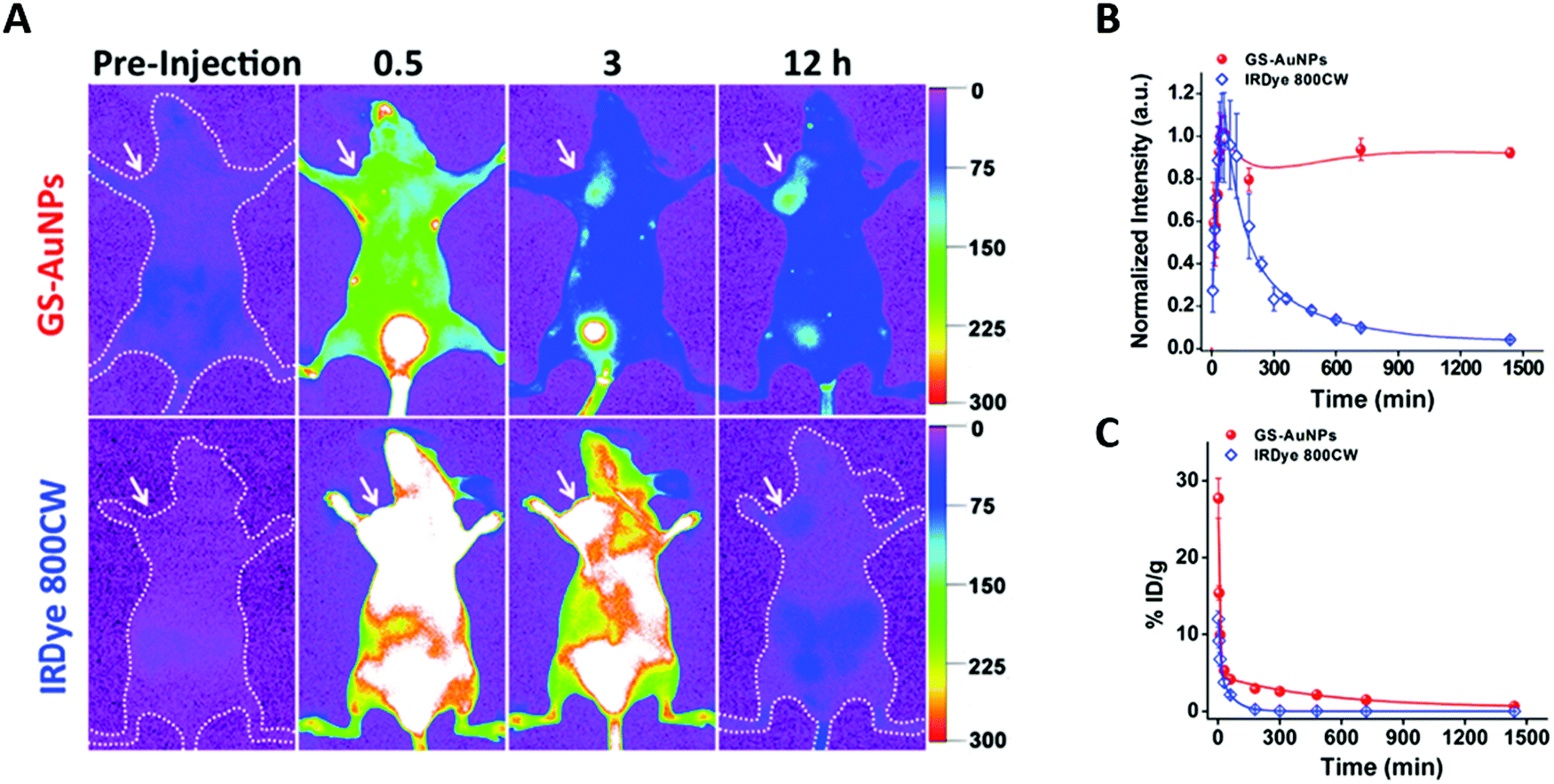 | ||
| Fig. 5 Tumor uptake and renal elimination in mice intravenously injected with AuSG NCs and IRDye 800CW. (A) In vivo NIR fluorescence images of MCF-7 tumor-bearing mice 0.5, 3, and 12 h after intravenous injection of AuSG NCs and IRDye 800CW (200 μL at 20 mg mL−1 and 10 μM, respectively). The tumor areas are indicated by arrows. (B) MCF-7 tumor uptake of AuSG NCs and IRDye 800CW 24 h pi. (C) Renal elimination of AuSG and IRDye 800CW, intravenously injected (200 μL at 7 mg mL−1 and 10 mM, respectively) at 24 h pi. The curves were fitted to a biexponential function with R2 values of 0.9711 and 0.9838, respectively. The distribution half-life (t1/2α) values are 5.4 ± 1.2 and 6.3 ± 2.5 min, respectively, and the t1/2β values are 8.5 ± 2.1 and 0.98 ± 0.08 h, respectively, for AuSG NCs and IRDye 800CW (n = 3). Reproduced from ref. 30 with permission from the American Chemical Society. | ||
Compared to larger Au NPs that are not cleared by the kidneys, AuSG NCs present a two times higher tumor accumulation. This could be attributed to the 10 times lower accumulation of AuSG in the RES compared with Au NPs, which also maintained the highest NC concentration in the blood during the first hours. This implies that NCs escaping RES uptake and remaining long enough in circulation in the blood stream will have a better chance to accumulate in tumors via the EPR effect.
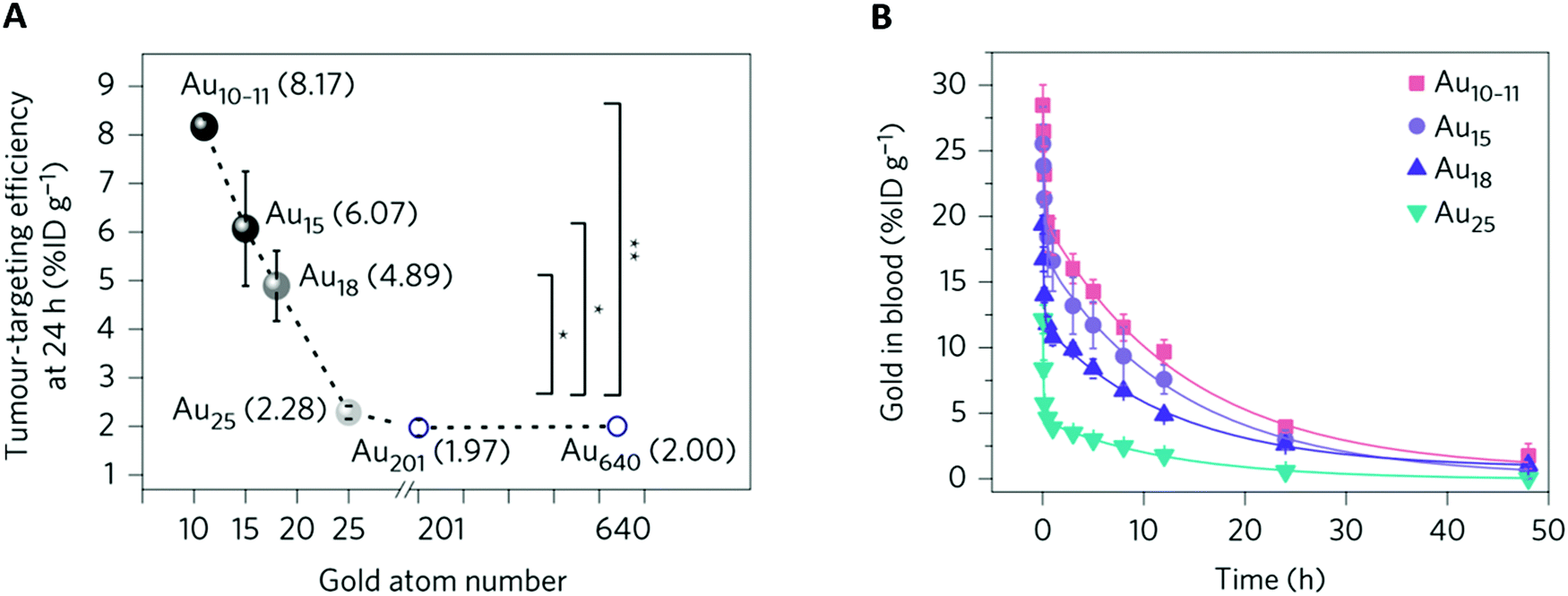 | ||
| Fig. 6 Effect of the subnanometer size of the NCs on tumor uptake: (A) MCF-7 tumor accumulation efficiency of different-sized Au NCs versus the number of gold atoms at 24 h p.i. For NCs smaller than Au25SG18 (Au10–11SG10–11, Au15SG13, Au18SG14), the tumor accumulation decreased with increasing number of gold atoms. For NCs larger than Au25SG18 (Au201 and Au640), tumor uptake was constant. *P < 0.05, **P < 0.005, Student's t-test. (B) Blood pharmacokinetics of Au10–11SG10–11, Au15SG13, Au18SG14, and Au25SG18 show two-compartment pharmacokinetics with different distributions and t1/2β between the NCs. BALB/c mice (n = 3) were intravenously injected (∼100 μM, 100 μL). Reproduced from ref. 46 with permission from Springer Nature publishing group. | ||
Overall, this confirms that increasing the circulation time of the NCs favors their passive uptake by the tumor.
4.2 Active tumor targeting
The addition of a targeting agent, usually a small (bio)molecule that could specifically interact with receptors overexpressed at the surface of tumor cells is expected to augment the specific tumor accumulation. Cell surface proteins such as integrins, transferrin, and folate receptors (FRs) are commonly targeted because they are overexpressed in tumors.54,61,64,65,101–103,106,108,128AuBSA functionalized with folic acid (FA)51 provided maximum tumor/normal tissue ratios (T/N) in mice with FR-positive HCT116 tumors two times higher than for negative A549 tumor. The functionalization of AuBSA with hyaluronic acid (HA)61 (Fig. 7) or Luteinizing hormone-releasing hormone (LHRH)83 allowed increasing the tumor accumulation by three compared to the AuBSA.
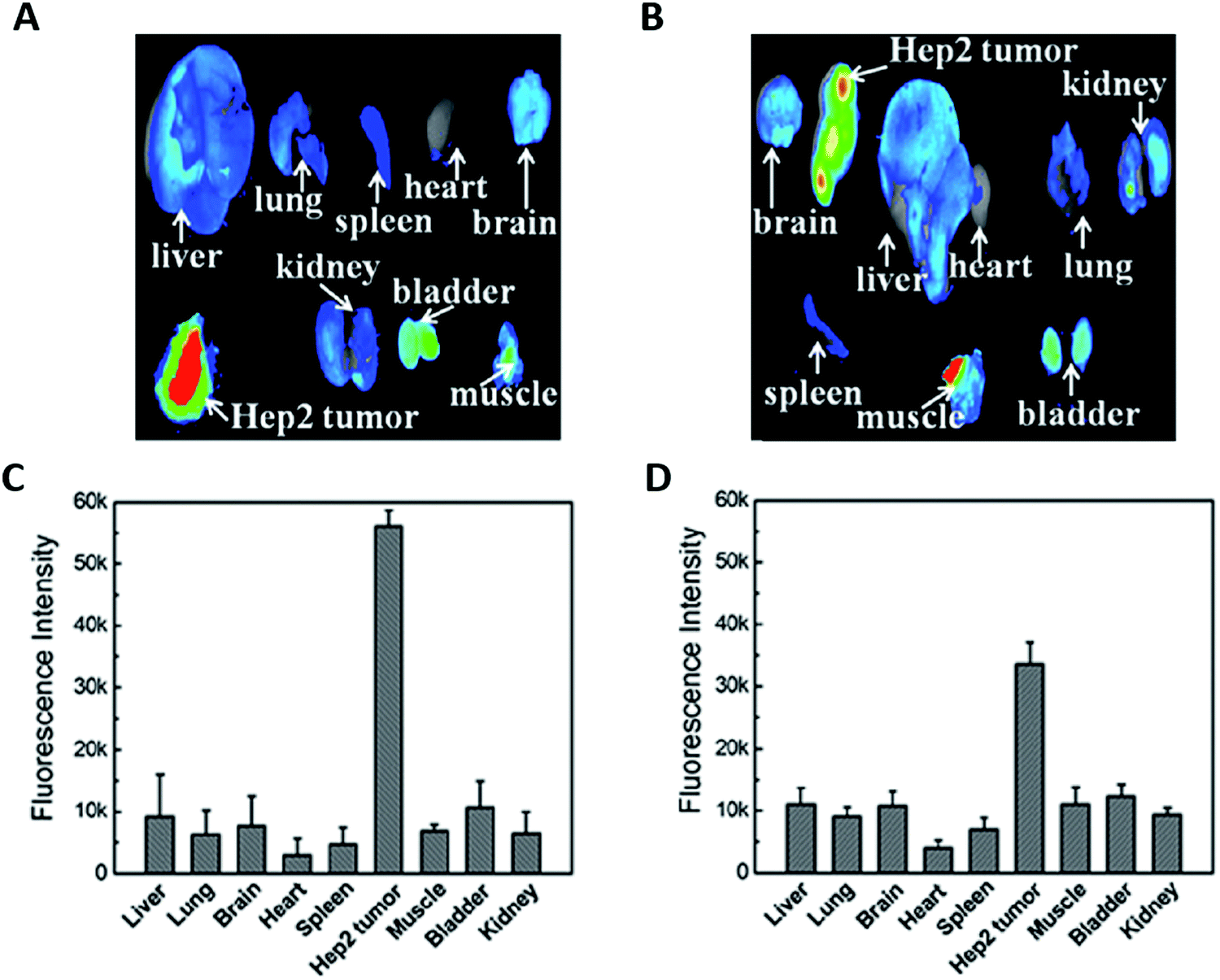 | ||
| Fig. 7 Ex vivo fluorescence images of the tumor and of the major organs in mice 7 h p.i. of HA-AuBSA NCs (A) or free HA and AuBSA NCs (B) with the corresponding average fluorescence intensity analysis (C and D). The addition of the targeting ligand HA grafted onto the Au NC surface improves the tumor uptake by two times. Reproduced from ref. 61 with permission from The Royal Society of Chemistry publishing group. | ||
Grafting an RGD-containing peptide on AuBSA can improve the targeting of human glioblastoma U87MG tumors engrafted in mice.86 U87MG cells express integrin ανβ3, a cell surface receptor recognized by RGD. If, 1 h after the intravenous injection of AuBSA, the tumor was detected due to the EPR effect, the signal dramatically decreased after 2 h. In contrast, the NIR fluorescence of the RGD–Au in tumors was still visible after 24 h. Thus, functionalized Au NCs with a targeting molecule could improve the tumor accumulation of the Au NCs and prolong their retention time.
In another study, a dual targeting was tested. A cRGD peptide and a 26-base G-rich DNA oligonucleotide that functions as a nucleolin-binding aptamer (AS1411) were added to Au NCs to induce a dual targeting.38 The nucleolin receptors are overexpressed on the cell membrane of most of the cancer cells. The tumor signal disappeared 8 h pi when naked Au NCs were used. In contrast, it was still detectable 48 h later when only one targeting molecule was added. This confirmed the interest of using targeted Au NCs to augment their retention time in the tumor. If both ligands were co-presented by the Au NCs, the T/N fluorescence ratio was increased to a greater extent (7.2) than when the Au NCs were labeled with only cRGD (5.4).
If the addition of a targeting agent on the Au NC surface favors higher and longer tumor accumulation,38,103–105,107 it also increases the size and changes the charge of the Au NCs, which could be at the expense of their renal elimination.
5 Au NCs: promising theranostic agents
Au NCs are useful for multimodal cancer diagnosis and therapy.5.1 Multimodal diagnostic applications
Many imaging techniques are used daily in hospitals for diagnosis, image-guided surgery, and follow-up of treatment efficacy. We will present only imaging techniques that have already been tested with Au NCs in animal models.However, OI also presents drawbacks and in particular suffers from a weak penetration of light in biological tissues, especially because hemoglobin absorbs light below 650 nm while water will absorb wavelengths above 900 nm.129,132 In addition, in the visible range (±400–700 nm), auto-fluorescence of some tissues (skin especially) leads to a high background noise that reduces the optical contrast. For all these reasons, it is particularly interesting to work in the NIR wavelength range between 700 and 900 nm, called the “transparent imaging window.”79,129
Au NCs, with their water solubility, biocompatibility, tunable PL from the UV to the NIR, and resistance to photobleaching in contrast to organic fluorophores, are potentially good optical probes. In the earliest works, the low emission wavelength at 480 nm of Au NCs stabilized with histidine (His)23 limited their use to superficial tissues or cell imaging. A hydrophilic indocyanine green fluorophore needed to be added to have an emission at 800 nm that can be followed in vivo. In the case of AuBSA, even though the 680 nm emission58,61,133 enabled their detection under few millimeters of tissues, the conjugation with an indocyanine green fluorophore also permitted a shift of the emission in the NIR and facilitated their detection.62 As mentioned in the first part, several strategies exist to shift the emission to higher wavelengths and to increase the QY of the NCs for improving their limit of detection. In the meantime, other NCs such as AuSG14,30 and AuPEG14 with intrinsic NIR fluorescence were employed.
Seminal works by H. Dai79,132,134 have demonstrated the benefit in terms of spatial and temporal resolutions to move from the NIR to a SWIR spectral window with the parallel development of new contrast agents and camera.79 LA-sulfobetaine-capped Au NCs with relatively good fluorescence in the NIR/SWIR region (QY of 0.6% at 1000 nm and 3.8% at 900 nm) and broad emission were produced.78 After their intravenous injection, images of the blood vessels were obtained with higher contrast and spatial resolution in the SWIR using a long-pass filter at 1250 nm than in the NIR.
Au NCs thus appeared as new promising SWIR (Fig. 8A) and NIR (Fig. 8B) contrast agents.
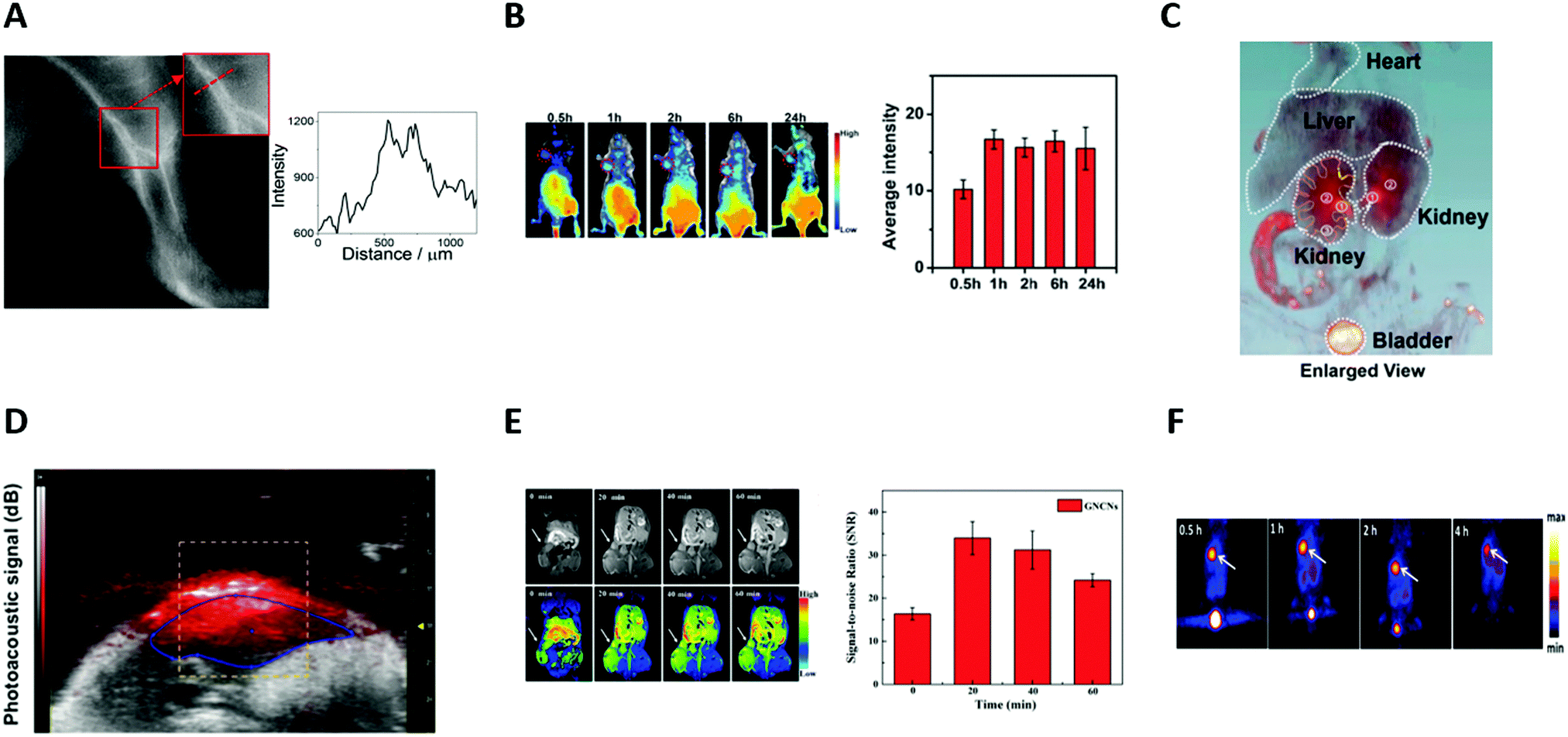 | ||
| Fig. 8 (A) Image of the left leg of a wild-type CS7BL/6 mouse taken using an InGaAs camera equipped with a 1250 LP filter after the intravenous injection of LA-sulfobetaine-capped Au NCs (0.5 mg of Au). Signal intensity across a line of interest drawn in the inset images showed the spatial resolution of the vessels. (B) In vivo fluorescence images of a HeLa tumor-bearing nude mouse at different time points (30 min, 1 h, 2 h, 6 h, and 24 h) after intravenous injection of cRGD-Au NCs (200 μL, 8.0 mg mL−1). Red circles indicate the tumor. Quantification of the signal intensity of the tumor side (n = 3). (C) In vivo 3D CT images of the heart, liver, kidneys, and bladder (excluding bone) in mice 2 h after intravenous injection of AuBSA NCs (200 μL, 9.5 mgAu per mL). The ureter, renal pelvis, and the major calyx are marked in orange and yellow dashed curves. (D) Noninvasive in vivo photoacoustic imaging of the belly of a NMRI nude mouse, 3 h after intravenous injection of AuZw NCs (200 μL, 600 μM). (E) In vivo T1-weighted MR images of A549 tumor in a BALB/c-nude mouse at different time points (20, 30, and 40 min) after intratumoral injection of Gd-AuSG NCs (50 μL, 0.11 M). White arrows indicate the position of the tumor. The graph represents the signal-to-noise ratio around the tumor (n = 3). (F) Representative PET images of coronal single slices of an orthotopic A549 lung tumor-bearing mouse at different time points (0.5, 1, 2, and 4 h), after intravenous injection of 6.7 MBq of [64Cu]CuNC@BSA-LHRH. White arrows indicate the position of the tumor. Reproduced with permission from American Chemical Society for (A) ref. 78, (C) ref. 31 and (F) ref. 104; The Royal Society of Chemistry for (B) ref. 106, AIP for (D) ref. 33 and Elsevier for (E) ref. 135. | ||
AuBSA were thus tested in mice, allowing an improved visualization of the structure of major organs (heart, liver, kidneys, bladder, and intestine) and a clear delineation of the calyces, pelvis, ureters, and bladder (Fig. 8C) making AuBSA NC a promising candidate to study renal excretion.31 Other NCs such as AuSG were also successfully tested.32
Because the Au NCs are also fluorescent, they can serve as bi-modal imaging agents.60 X-ray CT images provide anatomical information, but can also be used to confirm the results obtained by optical imaging, such as tumor uptake139 or renal elimination of AuSG.32,35,36,114
The use of Au NCs decorated with Gd3+ ions compared to traditional Gd chelates (diethylenetriamine penta-acetic acid, DTPA) improved the performance of Gd as a MRI contrast agent itself and prolonged its detection time three-fold135 (Fig. 8E). These Au NCs were still visible by X-ray CT or optical imaging techniques which permit visualization of the organs, where MRI had a weak signal-to-noise ratio, making Gd-functionalized Au NCs promising tools for tri-modal imaging: MIR/X-ray CT/optical imaging.
64Cu, a well-known β+ radioisotope, is generally complexed with bifunctional chelates, such as DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) and TETA (1,4,8,11-tetraazacyclododecane1,4,8,11-tetraacetic acid). However, these complexes suffer from low stability, and the released copper ions will be complexed with proteins to be finally stored in the liver. This could induce toxic effects.104
The direct incorporation of 64Cu in NCs may overcome these limitations and the ligand shell could favor the radiolabeling stability (Fig. 8F). These NCs keep their initial properties, which means renal excretion and the ability to escape the RES.6,104
For example, CuSG NCs are progressively degraded into Cu(II)-GSSG.6 Due to their interaction with serum proteins, they were then retained in the liver. After intravenous injection in mice, CuSG or Cu(II)-GSSG showed t1/2β of 3.2 and 4.9 h, respectively. The successful addition of 64Cu, with its half-time of decay of 12.7 h, helped in following the elimination of CuSG with precision. Indeed, the strong signal from the kidneys and the bladder just 1.5 min pi confirmed the fast renal elimination of [64Cu]CuSG. The signal from the bladder still visible 4 h pi attested the constant elimination of the NCs from the body. Finally, at 4 h pi, the signal detected in the liver confirmed the dissociation of CuSG into Cu(II)-GSSG complexes.
Radioactive NCs can thus be used for dual imaging. PET offers more sensitive and quantitative follow-up, which will be an addition to the benefits of optical imaging.104,144
5.2 Therapy
In addition to their use in diagnostics, Au NCs can be used as therapeutic agents and delivery systems.Zhang et al.35 first studied the potential of a Au10–12SG10–12 or of a saline solution after intraperitoneal administration (0.2 mL, 3 mM) in nude mice engrafted with a human cervical U14 tumor. When the tumor uptake reached a maximum (24 pi), the mice were either irradiated (under 137Cs gamma radiation of 3600 Ci at 5 Gy) or not. Regarding the controls, no decrease in tumor size was observed in mice treated with only the Au NCs over a period of 23 days. In contrast, the tumor volume decreased by 8% upon irradiation and 65% with the combination of Au NCs plus irradiation.
The same experiment was carried out with Au29–43SG27–37.36 Compared with the control group, the tumor volume in mice treated with only radiation and mice treated with both Au29–43SG27–37 and radiation decreased by 10% and 76%, respectively. No effects were observed in mice injected only with Au NCs (Fig. 9A). In both cases, Au NCs thus generated a toxic effect only if they were irradiated. Similar results were obtained with AuBSA.34
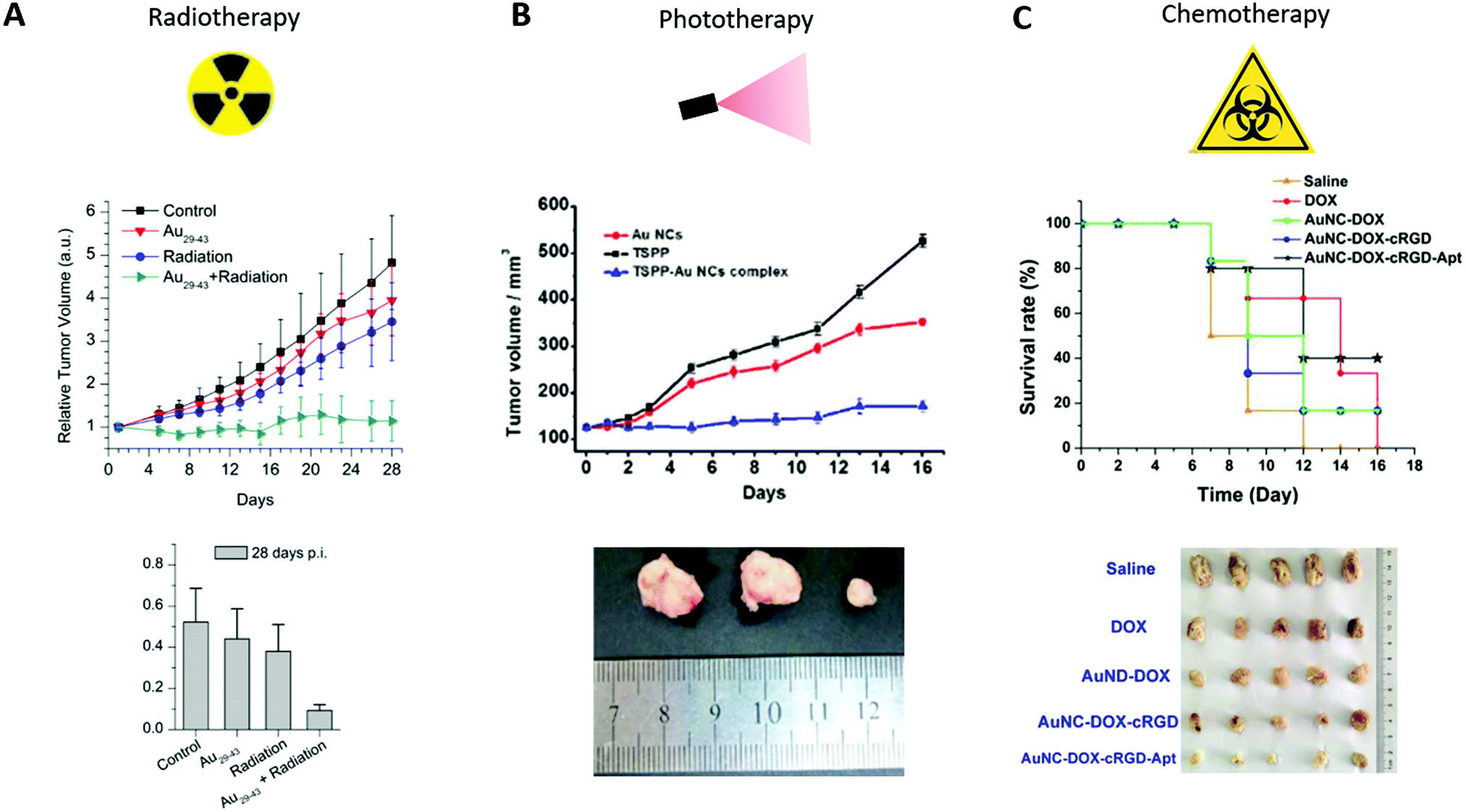 | ||
| Fig. 9 (A) Time-course studies of the U14 tumor volumes until 28 days pi (top) and measurement of the U14 tumor weights at 28 days pi (bottom) of mice intraperitoneally injected with saline solution (control) or with Au29–43SG27–37 (5.9 mgAu per kg of body), nude mice treated with only radiation (under 137Cs gamma radiation of 3600 Ci at 5 Gy, 24 h pi), and nude mice injected with Au29–43SG27–37 followed by irradiation (n = 8). (B) Time-course studies of U87 tumor volumes until 16 days pi (top) of mice intraperitoneally injected with Au NCs (0.1 mL, 5 mM), TSPP (0.1 mL, 0.1 mM) and TSPP-Au NCs (equivalent 0.1 mL, 0.1 mM TSPP) (n = 3) and corresponding images isolated from tumor-bearing mice after 16 days of treatment (Au NCs, TSPP and TSPP-Au NCs from left to right, bottom). (C) Survival rate curves of U87MG tumor-bearing mice intravenously injected with free DOX, DOX-Au NCs, cRGD-DOX-Au NCs, and Apt-cRGD-DOX-Au NCs (all at 5.0 μg kg−1 equivalent DOX) and corresponding images isolated from tumor-bearing mice after 14 days of treatment (bottom) Reproduced from ref. 36, 38 and 133 with permission from Scientific Reports, The Royal Society of Chemistry and Elsevier publishing group. | ||
Concerning PDT, Au25SG18 can generate singlet oxygen species under 650 and 808 nm irradiation in solution145 but most of the current studies use Au NCs combined with photosensitizer molecules such as chlorine6 (Ce6)37,39,146,147 or porphyrin derivatives.37,133
Most of the PDT compounds present unwanted cutaneous photosensitivity, and inadequate selectivity that can be ameliorated when combined with NCs. A four-time decrease in the size of C6 tumors was obtained 8 days after the intravenous injection of Au NCs grafted with protoporphyrin IX, followed by laser treatment 3 h later (532 nm, 15 min, 1.5 W cm−2)37 compared to the control treated with only radiation. Moreover, U87 tumor growth can be stopped, after intratumoral injection of Au NCs conjugated with TSPP (meso-tetra(4-sulfonatophenyl)porphine dihydrochloride), a photosensitizer, in U87 tumors, and light excitation for 30 min per day with IR light indicating that Au NCs can enhance the PDT effect of the photosensitizer alone133 (Fig. 9B).
In order to study whether this synergistic effect could be generalized, C. Zhang et al.39 intravenously injected Ce6–Au25SG18, free Ce6, or PBS into mice with human gastric MGC-803 tumors of 150 mm.3 The tumors were illuminated (633 nm, 10 min, 100 mW cm−2) 8 h and 24 h later. After 21 days, the volumes of the tumors for the mice injected with PBS, Ce6, and Ce6–Au25SG18 without laser treatment were similar and close to 1200 mm3. However, the tumor growth slowed down and reached a volume of around 650 mm3 or 350 mm3, in mice injected with free Ce6 or Ce6–Au25SG18 followed by illumination. This thus confirmed that Au NCs can enhance the radiotherapeutic activity of a co-injected radiosensitizer.
Based on this observation, it appeared interesting to generate a core–shell structure using silica146 or liposomes147 to increase the amount of Ce6 in contact with the Au NCs. The compounds were intravenously injected into mice with melanoma MDA-MB-435 or breast MDA-MB-361 tumors for compounds with silica or liposome shells, respectively. The tumors were irradiated for 10 min using laser (671 nm, 100 mW cm−2) for the silica model and 3 times every 3 days for 25 min (660 nm, 150 mW cm−2) for the liposome model. In both tumor models, regarding the control, the injection of free Ce6 slowed down the growth of the tumors almost 2 times, while the core–shell structure composed of Ce6 and Au NCs totally inhibited the growth of the tumors. These studies confirmed that Au NCs can enhance the effect of the Ce6 radiosensitizer even after encapsulation in silica or liposomes.
More complex systems combining multimodal imaging and therapeutic compounds were generated146 combining Au NCs functionalized with Ce6 carbon dots. These theranostic agents were usable for fluorescence, MRI and PDT cancer treatments.
To conclude, Au NCs are promising multimodal imaging probes. Due to their intrinsic properties, toxic effects can be generated in the microenvironment of the tumors upon activation by an external stimulus like X-ray radiation or illumination, with reduced damage to healthy tissues. They can be used as therapeutic compounds alone or to enhance the effects of sensitizer agents.
The multimodal properties of the Au NCs make these theranostic agents highly promising in oncology.
6 Conclusions and perspectives
In this review, we described recent works performed on metal NCs, mainly gold, for in vivo applications. Progress in synthesis methods has enabled the development of effective strategies to produce photoluminescent atomically precise NCs on a large scale, with high purity and different functional groups.The unique physicochemical properties and good biocompatibility of metal NCs have facilitated their use in biomedical applications. The PL and metallic composition of NCs allow their use as optical or X-ray probes, or radiosensitizers. By integrating Au NCs with other imaging or therapeutic agents, complex systems could be achieved. These combinations result in powerful image-guided therapy that takes advantage of each technique. Finally, their fast renal elimination decreases their nonspecific accumulation in different organs, and thus, their toxicity. These optical features combined with their low toxicity and rather simple synthesis method make them very good candidates as contrast agents with respect to other nanomaterials such as Quantum Dots, organic fluorophores, lanthanide NPs, and carbon nanotubes.
Different parameters such as size, length, and charge of the ligands or density affect the formation of the protein corona, optical properties, biodistribution, targeting potential, renal elimination, tumor uptake, and biodegradability of metal NCs. Each of these parameters must be carefully addressed when designing NCs for specific biomedical applications and their possible clinical translation.
Au NCs have already been applied in vivo on mice and monkeys.124 They have shown promising results as theranostic agents, even in parts of the body difficult to access. NCs are not limited to cancer treatments and can be used for diagnosis of other diseases such as renal dysfunctions,31,148,149 and Alzheimer's disease.150,151
However, some of their properties such as their tumor accumulation and retention or their luminescent properties in the NIR and SWIR regions for in vivo imaging still need to be improved. Further studies are necessary to confirm their radio and photosensitizing effects and more broadly, to get a deeper understanding of the relationships among structure, stability, and luminescence of Au NCs.
Precise and early diagnosis of tumors and the development of targeted therapies are two major lines of research in oncology. In this context, we believe that photoluminescent Au NCs are promising candidates. Their biomedical potential will go beyond cancer research and toward a wide range of (biomedical) applications (such as fluorophores for fluorescence resonance energy transfer applications,152,153 biosensors, and optoelectronics).
Once their pharmacokinetic and luminescence properties, combined with their proven theranostic potential under light or X-ray irradiation, are clearly demonstrated at the preclinical level, we can expect a rapid translation of such ultrasmall Au NCs in clinical trials because of their method of preparation, low toxicity and powerful elimination. In addition, they will benefit from the fact that the Food and Drug Administration has already approved different gold nanoparticles for diagnostics and therapy.
Abbreviations
| BSA | Bovine serum albumin |
| CI | Contrast index |
| cRGD | Cyclic RGD |
| DTPA | Diethylenetriamine penta-acetic acid |
| EPR | Enhancement permeability and retention |
| FA | Folic acid |
| FR | Folate receptor |
| HA | Hyaluronic acid |
| HD | Hydrodynamic diameter |
| H | Hour |
| ID | Injected dose |
| ID per g | Percentage of injected dose per gram of tissue |
| KFT | Kidney filtration threshold |
| LA | Lipoic acid |
| LHRH | Luteinizing hormone-releasing hormone |
| LIBS | Laser induced breakdown spectroscopy |
| LMCT | Ligand-to-metal charge transfer |
| LMMCT | Ligand-to-metal–metal charge transfer |
| MRI | Magnetic resonance imaging |
| NCs | Nanoclusters |
| NIR | Near-infrared |
| NPs | Nanoparticles |
| PDT | Photodynamic therapy |
| PEG | Polyethylene glycol |
| PET | Positron emission tomography |
| pi | Post injection |
| PL | Photoluminescence |
| PNA | Peptide nucleic acid |
| PTT | Photothermal therapy |
| QY | Quantum yield |
| ROS | Reactive oxygen species |
| RES | Reticuloendothelial system |
| SG | Glutathione |
| SWIR | Shortwave infrared |
| t 1/2α | Distribution half-life |
| t 1/2β | Elimination half-life |
| UV | Ultraviolet |
| X-ray CT | X-ray computed tomography |
| Zw | Zwitterion molecules |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Rhône-Alpes region for the financial support of EP (ARC1 Santé fellowship 2016). We thank Cancéropôle Lyon Auvergne Rhône-Alpes (CLARA), Plan Cancer (C18038CS) and ARC (R17157CC) for financial support. All authors declare that they have no financial/commercial Conflict of Interest.References
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- J. Wang, J. Ye, H. Jiang, S. Gao, W. Ge, Y. Chen, C. Liu, C. Amatore and X. Wang, RSC Adv., 2014, 4, 37790 RSC.
- S. Gao, D. Chen, Q. Li, J. Ye, H. Jiang, C. Amatore and X. Wang, Sci. Rep., 2014, 4, 4384–4390 CrossRef PubMed.
- D. Chen, C. Zhao, J. Ye, Q. Li, X. Liu, M. Su, H. Jiang, C. Amatore, M. Selke and X. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 18163–18169 CrossRef CAS PubMed.
- C. Zhao, L. Lai, F. U. Rehman, C. Qian, G. Teng, H. Jiang and X. Wang, RSC Adv., 2016, 6, 110525–110534 RSC.
- S. Yang, S. Sun, C. Zhou, G. Hao, J. Liu, S. Ramezani, M. Yu, X. Sun and J. Zheng, Bioconjugate Chem., 2015, 26, 511–519 CrossRef CAS PubMed.
- C. J. Ackerson, P. D. Jadzinsky and R. D. Kornberg, J. Am. Chem. Soc., 2005, 127, 6550–6551 CrossRef CAS PubMed.
- F. Aldeek, M. A. H. Muhammed, G. Palui, N. Zhan and H. Mattoussi, ACS Nano, 2013, 7, 2509–2521 CrossRef CAS PubMed.
- X. Yuan, B. Zhang, Z. Luo, Q. Yao, D. T. Leong, N. Yan and J. Xie, Angew. Chem., Int. Ed., 2014, 53, 4623–4627 CrossRef CAS PubMed.
- H.-H. Wang, C.-A. J. Lin, C.-H. Lee, Y.-C. Lin, Y.-M. Tseng, C.-L. Hsieh, C.-H. Chen, C.-H. Tsai, C.-T. Hsieh, J.-L. Shen, W.-H. Chan, W. H. Chang and H.-I. Yeh, ACS Nano, 2011, 5, 4337–4344 CrossRef CAS PubMed.
- L. Shang, N. Azadfar, F. Stockmar, W. Send, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Small, 2011, 7, 2614–2620 CrossRef CAS PubMed.
- E. Porret, L. Sancey, A. Martín-Serrano, M. I. Montañez, R. Seeman, A. Yahia-Ammar, H. Okuno, F. Gomez, A. Ariza, N. Hildebrandt, J.-B. Fleury, J.-L. Coll and X. Le Guével, Chem. Mater., 2017, 29, 7497–7506 CrossRef CAS.
- T. D. Fernández, J. R. Pearson, M. P. Leal, M. J. Torres, M. Blanca, C. Mayorga and X. Le Guével, Biomaterials, 2015, 43, 1–12 CrossRef PubMed.
- J. Liu, M. Yu, X. Ning, C. Zhou, S. Yang and J. Zheng, Angew. Chem., Int. Ed., 2013, 52, 12572–12576 CrossRef CAS PubMed.
- E. Oh, F. K. Fatemi, M. Currie, J. B. Delehanty, T. Pons, A. Fragola, S. Lévêque-Fort, R. Goswami, K. Susumu, A. L. Huston and I. L. Medintz, Part. Part. Syst. Charact., 2013, 30, 453–466 CrossRef CAS.
- H. Duan and S. Nie, J. Am. Chem. Soc., 2007, 129, 2412–2413 CrossRef CAS PubMed.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402–077406 CrossRef PubMed.
- F. Qu, N. B. Li and H. Q. Luo, Anal. Chem., 2012, 84, 10373–10379 CrossRef CAS PubMed.
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS PubMed.
- G. Liu, Y. Shao, K. Ma, Q. Cui, F. Wu and S. Xu, Gold Bull., 2012, 45, 69–74 CrossRef CAS.
- X. Ning, C. Peng, E. S. Li, J. Xu, R. D. Vinluan, M. Yu and J. Zheng, APL Mater., 2017, 5, 053406 CrossRef PubMed.
- Y. Yu, Z. Luo, Y. Yu, J. Y. Lee and J. Xie, ACS Nano, 2012, 6, 7920–7927 CrossRef CAS PubMed.
- H. Chen, B. Li, C. Wang, X. Zhang, Z. Cheng, X. Dai, R. Zhu and Y. Gu, Nanotechnology, 2013, 24, 055704 CrossRef PubMed.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- Q. Wen, Y. Gu, L.-J. Tang, R.-Q. Yu and J.-H. Jiang, Anal. Chem., 2013, 85, 11681–11685 CrossRef CAS PubMed.
- R. D. Vinluan, J. Liu, C. Zhou, M. Yu, S. Yang, A. Kumar, S. Sun, A. Dean, X. Sun and J. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 11829–11833 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- A. Baksi, P. L. Xavier, K. Chaudhari, N. Goswami, S. K. Pal and T. Pradeep, Nanoscale, 2013, 5, 2009 RSC.
- X. L. Guével, N. Daum and M. Schneider, Nanotechnology, 2011, 22, 275103 CrossRef PubMed.
- J. Liu, M. Yu, C. Zhou, S. Yang, X. Ning and J. Zheng, J. Am. Chem. Soc., 2013, 135, 4978–4981 CrossRef CAS PubMed.
- Y. Wang, C. Xu, J. Zhai, F. Gao, R. Liu, L. Gao, Y. Zhao, Z. Chai and X. Gao, Anal. Chem., 2015, 87, 343–345 CrossRef CAS PubMed.
- J. Xu, M. Yu, P. Carter, E. Hernandez, A. Dang, P. Kapur, J.-T. Hsieh and J. Zheng, Angew. Chem., Int. Ed., 2017, 56, 13356–13360 CrossRef CAS PubMed.
- D. Shen, M. Henry, V. Trouillet, C. Comby-Zerbino, F. Bertorelle, L. Sancey, R. Antoine, J.-L. Coll, V. Josserand and X. Le Guével, APL Mater., 2017, 5, 053404 CrossRef.
- X. D. Zhang, J. Chen, Z. Luo, D. Wu, X. Shen, S. S. Song, Y. M. Sun, P. X. Liu, J. Zhao, S. Huo, S. Fan, F. Fan, X. J. Liang and J. Xie, Adv. Healthcare Mater., 2014, 3, 133–141 CrossRef CAS PubMed.
- X. D. Zhang, Z. Luo, J. Chen, X. Shen, S. Song, Y. Sun, S. Fan, F. Fan, D. T. Leong and J. Xie, Adv. Mater., 2014, 26, 4565–4568 CrossRef CAS PubMed.
- X. D. Zhang, Z. Luo, J. Chen, S. Song, X. Yuan, X. Shen, H. Wang, Y. Sun, K. Gao, L. Zhang, S. Fan, D. T. Leong, M. Guo and J. Xie, Sci. Rep., 2015, 5, 8669 CrossRef CAS PubMed.
- L. V. Nair, S. S. Nazeer, R. S. Jayasree and A. Ajayaghosh, ACS Nano, 2015, 9, 5825–5832 CrossRef CAS PubMed.
- D. Chen, B. Li, S. Cai, P. Wang, S. Peng, Y. Sheng, Y. He, Y. Gu and H. Chen, Biomaterials, 2016, 100, 1–16 CrossRef CAS PubMed.
- C. Zhang, C. Li, Y. Liu, J. Zhang, C. Bao, S. Liang, Q. Wang, Y. Yang, H. Fu, K. Wang and D. Cui, Adv. Funct. Mater., 2015, 25, 1314–1325 CrossRef CAS.
- X.-R. Song, N. Goswami, H.-H. Yang and J. Xie, Analyst, 2016, 141, 3126–3140 RSC.
- Y. Negishi, Y. Takasugi, S. Sato, H. Yao, K. Kimura and T. Tsukuda, J. Am. Chem. Soc., 2004, 126, 6518–6519 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS PubMed.
- M. A. Habeeb Muhammed and T. Pradeep, Chem. Phys. Lett., 2007, 449, 186–190 CrossRef CAS.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- N. Goswami, Q. Yao, T. Chen and J. Xie, Coord. Chem. Rev., 2016, 329, 1–15 CrossRef CAS.
- B. Du, X. Jiang, A. Das, Q. Zhou, M. Yu, R. Jin and J. Zheng, Nat. Nanotechnol., 2017, 12, 1096–1102 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- M. A. H. Muhammed, F. Aldeek, G. Palui, L. Trapiella-Alfonso and H. Mattoussi, ACS Nano, 2012, 6, 8950–8961 CrossRef CAS PubMed.
- J. Zheng, C. Zhou, M. Yu and J. Liu, Nanoscale, 2012, 4, 4073 RSC.
- J. T. Au, G. Craig, V. Longo, P. Zanzonico, M. Mason, Y. Fong and P. J. Allen, Am. J. Roentgenol., 2013, 200, 1347–1351 CrossRef PubMed.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed.
- Y. Wang, C. Dai and X.-P. Yan, Chem. Commun., 2014, 50, 14341–14344 RSC.
- D. F. Moyano, K. Saha, G. Prakash, B. Yan, H. Kong, M. Yazdani and V. M. Rotello, ACS Nano, 2014, 8, 6748–6755 CrossRef CAS PubMed.
- S. Si, R. R. Bhattacharjee, A. Banerjee and T. K. Mandal, Chem. – Eur. J., 2006, 12, 1256–1265 CrossRef CAS PubMed.
- W. Zhang, J. Ye, Y. Zhang, Q. Li, X. Dong, H. Jiang and X. Wang, RSC Adv., 2015, 5, 63821–63826 RSC.
- X. Wu, X. He, K. Wang, C. Xie, B. Zhou and Z. Qing, Nanoscale, 2010, 2, 2244 RSC.
- X. D. Zhang, D. Wu, X. Shen, P. X. Liu, F. Y. Fan and S. J. Fan, Biomaterials, 2012, 33, 4628–4638 CrossRef CAS PubMed.
- A. Zhang, Y. Tu, S. Qin, Y. Li, J. Zhou, N. Chen, Q. Lu and B. Zhang, J. Colloid Interface Sci., 2012, 372, 239–244 CrossRef CAS.
- P. Zhang, X. X. Yang, Y. Wang, N. W. Zhao, Z. H. Xiong and C. Z. Huang, Nanoscale, 2014, 6, 2261–2269 RSC.
- H. Chen, B. Li, X. Ren, S. Li, Y. Ma, S. Cui and Y. Gu, Biomaterials, 2012, 33, 8461–8476 CrossRef CAS PubMed.
- X. Le Guével, V. Trouillet, C. Spies, G. Jung and M. Schneider, J. Phys. Chem. C, 2012, 116, 6047–6051 CrossRef.
- C. Sun, H. Yang, Y. Yuan, X. Tian, L. Wang, Y. Guo, L. Xu, J. Lei, N. Gao, G. J. Anderson, X.-J. Liang, C. Chen, Y. Zhao and G. Nie, J. Am. Chem. Soc., 2011, 133, 8617–8624 CrossRef CAS PubMed.
- Y. Wang, J.-T. Chen and X.-P. Yan, Anal. Chem., 2013, 85, 2529–2535 CrossRef CAS.
- B. Du, M. Yu and J. Zheng, Nat. Rev. Mater., 2018, 3, 358–374 CrossRef.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- L. Yang, H. Kuang, W. Zhang, Z. P. Aguilar, H. Wei and H. Xu, Sci. Rep., 2017, 7, 3303–3315 CrossRef PubMed.
- E. B. Ehlerding, F. Chen and W. Cai, Adv. Sci., 2016, 3, 1500223 CrossRef.
- T. S. Hauck, R. E. Anderson, H. C. Fischer, S. Newbigging and W. C. Chan, Small, 2010, 6, 138–144 CrossRef CAS PubMed.
- Y. Sun, W. Feng, P. Yang, C. Huang and F. Li, Chem. Soc. Rev., 2015, 44, 1509–1525 RSC.
- K. Kostarelos, Nat. Biotechnol., 2008, 26, 774–776 CrossRef CAS PubMed.
- Y.-S. Chen, Y.-C. Hung, I. Liau and G. S. Huang, Nanoscale Res. Lett., 2009, 4, 858–864 CrossRef CAS PubMed.
- Y. Huang, L. Fuksman and J. Zheng, Dalton Trans., 2018, 47, 6267–6273 RSC.
- X. L. Guevel, O. Tagit, C. E. Rodríguez, V. Trouillet, M. Pernia Leal and N. Hildebrandt, Nanoscale, 2014, 6, 8091–8099 RSC.
- Y. Chen, D. M. Montana, H. Wei, J. M. Cordero, M. Schneider, X. Le Guével, O. Chen, O. T. Bruns and M. G. Bawendi, Nano Lett., 2017, 17, 6330–6334 CrossRef CAS PubMed.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS.
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455–462 CrossRef.
- B. Santiago-González, C. Vázquez-Vázquez, M. C. Blanco-Varela, J. M. Gaspar Martinho, J. M. Ramallo-López, F. G. Requejo and M. A. López-Quintela, J. Colloid Interface Sci., 2015, 455, 154–162 CrossRef PubMed.
- Y.-C. Shiang, C.-C. Huang, W.-Y. Chen, P.-C. Chen and H.-T. Chang, J. Mater. Chem., 2012, 22, 12972 RSC.
- Y. Ma, Y. Zhang and W. W. Yu, J. Mater. Chem. C, 2019, 7, 13662–13679 RSC.
- J.-C. Bünzli, Trends Chem., 2019, 1, 751–762 CrossRef.
- D. Wu, L. Chen, W. Lee, G. Ko, J. Yin and J. Yoon, Coord. Chem. Rev., 2018, 354, 74–97 CrossRef CAS.
- S. Link, A. Beeby, S. FitzGerald, M. A. El-Sayed, T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2002, 106, 3410–3415 CrossRef CAS.
- A. Mooradian, Phys. Rev. Lett., 1969, 22, 185–187 CrossRef CAS.
- A. Ghosh, T. Udayabhaskararao and T. Pradeep, J. Phys. Chem. Lett., 2012, 3, 1997–2002 CrossRef CAS.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS PubMed.
- X. Le Guével, V. Trouillet, C. Spies, K. Li, T. Laaksonen, D. Auerbach, G. Jung and M. Schneider, Nanoscale, 2012, 4, 7624 RSC.
- E. Oh, J. B. Delehanty, L. D. Field, A. J. Mäkinen, R. Goswami, A. L. Huston and I. L. Medintz, Chem. Mater., 2016, 28, 8676–8688 CrossRef CAS.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- G. Wang, R. Guo, G. Kalyuzhny, J.-P. Choi and R. W. Murray, J. Phys. Chem. B, 2006, 110, 20282–20289 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- H.-H. Deng, X.-Q. Shi, F.-F. Wang, H.-P. Peng, A.-L. Liu, X.-H. Xia and W. Chen, Chem. Mater., 2017, 29, 1362–1369 CrossRef CAS.
- A. Yahia-Ammar, D. Sierra, F. Mérola, N. Hildebrandt and X. Le Guével, ACS Nano, 2016, 10, 2591–2599 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- C. Colombé, X. Le Guével, A. Martin-Serrano, M. Henry, E. Porret, C. Comby-Zerbino, R. Antoine, I. Atallah, B. Busser, J.-L. Coll, C. A. Righini and L. Sancey, Nanomedicine, 2019, 20, 102011 CrossRef PubMed.
- H. Chen, S. Li, B. Li, X. Ren, S. Li, D. M. Mahounga, S. Cui, Y. Gu and S. Achilefu, Nanoscale, 2012, 4, 6050 RSC.
- K. Pyo, N. H. Ly, S. Y. Yoon, Y. Shen, S. Y. Choi, S. Y. Lee, S.-W. Joo and D. Lee, Adv. Healthcare Mater., 2017, 1700203, DOI:10.1002/adhm.201700203.
- D. Chen, Z. Luo, N. Li, J. Y. Lee, J. Xie and J. Lu, Adv. Funct. Mater., 2013, 23, 4324–4331 CrossRef CAS.
- F. Gao, P. Cai, W. Yang, J. Xue, L. Gao, R. Liu, Y. Wang, Y. Zhao, X. He, L. Zhao, G. Huang, F. Wu, Y. Zhao, Z. Chai and X. Gao, ACS Nano, 2015, 9, 4976–4986 CrossRef CAS PubMed.
- Y. Zhao, L. Detering, D. Sultan, M. L. Cooper, M. You, S. Cho, S. L. Meier, H. Luehmann, G. Sun, M. Rettig, F. Dehdashti, K. L. Wooley, J. F. DiPersio and Y. Liu, ACS Nano, 2016, 10, 5959–5970 CrossRef CAS PubMed.
- X. Wang, H. He, Y. Wang, J. Wang, X. Sun, H. Xu, W. M. Nau, X. Zhang and F. Huang, Chem. Commun., 2016, 52, 9232–9235 RSC.
- S.-K. Sun, L.-X. Dong, Y. Cao, H.-R. Sun and X.-P. Yan, Anal. Chem., 2013, 85, 8436–8441 CrossRef CAS PubMed.
- S. Lucie, G. Elisabeth, F. Stéphanie, S. Guy, H. Amandine, A.-R. Corinne, B. Didier, S. Catherine, G. Alexeï, D. Pascal and C. Jean-Luc, Mol. Ther., 2009, 17, 837–843 CrossRef PubMed.
- J. Y. Wang, J. Chen, J. Yang, H. Wang, X. Shen, Y. M. Sun, M. Guo and X. D. Zhang, Int. J. Nanomed., 2016, 11, 3475–3485 CrossRef CAS PubMed.
- E. S. Shibu, M. A. H. Muhammed, T. Tsukuda and T. Pradeep, J. Phys. Chem. C, 2008, 112, 12168–12176 CrossRef CAS.
- C. Zhou, G. Hao, P. Thomas, J. Liu, M. Yu, S. Sun, O. K. Öz, X. Sun and J. Zheng, Angew. Chem., 2012, 124, 10265–10269 CrossRef.
- X. Le Guével, M. Henry, V. Motto-Ros, E. Longo, M. I. Montañez, F. Pelascini, O. de La Rochefoucauld, P. Zeitoun, J.-L. Coll, V. Josserand and L. Sancey, Nanoscale, 2018, 10, 18657–18664 RSC.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- C. Zhou, M. Long, Y. Qin, X. Sun and J. Zheng, Angew. Chem., Int. Ed., 2011, 50, 3168–3172 CrossRef CAS PubMed.
- C. Peng, X. Gao, J. Xu, B. Du, X. Ning, S. Tang, R. M. Bachoo, M. Yu, W.-P. Ge and J. Zheng, Nano Res., 2017, 10, 1366–1376 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- P. d. Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Mater. Horiz., 2014, 1, 301–313 RSC.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- R. D. Vinluan and J. Zheng, Nanomedicine, 2015, 10, 2781–2794 CrossRef CAS PubMed.
- L. Shang and G. U. Nienhaus, Int. J. Biochem. Cell Biol., 2016, 75, 175–179 CrossRef CAS PubMed.
- L. Shang, S. Brandholt, F. Stockmar, V. Trouillet, M. Bruns and G. U. Nienhaus, Small, 2012, 8, 661–665 CrossRef CAS PubMed.
- S. Tang, C. Peng, J. Xu, B. Du, Q. Wang, R. D. Vinluan, M. Yu, M. J. Kim and J. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16039–16043 CrossRef CAS PubMed.
- A. Briat, C. H. F. Wenk, M. Ahmadi, M. Claron, D. Boturyn, V. Josserand, P. Dumy, D. Fagret, J.-L. Coll, C. Ghezzi, L. Sancey and J.-P. Vuillez, Cancer Sci., 2012, 103, 1105–1110 CrossRef CAS PubMed.
- J. Xu, M. Yu, C. Peng, P. Carter, J. Tian, X. Ning, Q. Zhou, Q. Tu, G. Zhang, A. Dao, X. Jiang, P. Kapur, J.-T. Hsieh, X. Zhao, P. Liu and J. Zheng, Angew. Chem., 2018, 130, 272–277 CrossRef.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- E. Koren and V. P. Torchilin, Trends Mol. Med., 2012, 18, 385–393 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmaceutics, 2008, 5, 505–515 CrossRef CAS PubMed.
- G. Liang, X. Jin, S. Zhang and D. Xing, Biomaterials, 2017, 144, 95–104 CrossRef CAS PubMed.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- S. Wang, N. Li, W. Pan and B. Tang, TrAC, Trends Anal. Chem., 2012, 39, 3–37 CrossRef CAS.
- S. Lee and X. Chen, Mol. Imaging, 2009, 8, 7290 CrossRef.
- G. Hong, S. Diao, J. Chang, A. L. Antaris, C. Chen, B. Zhang, S. Zhao, D. N. Atochin, P. L. Huang, K. I. Andreasson, C. J. Kuo and H. Dai, Nat. Photonics, 2014, 8, 723–730 CrossRef CAS PubMed.
- Y. Zhang, J. Li, H. Jiang, C. Zhao and X. Wang, RSC Adv., 2016, 6, 63331–63337 RSC.
- G. Hong, J. C. Lee, J. T. Robinson, U. Raaz, L. Xie, N. F. Huang, J. P. Cooke and H. Dai, Nat. Med., 2012, 18, 1841–1846 CrossRef CAS PubMed.
- W. Hou, F. Xia, G. Alfranca, H. Yan, X. Zhi, Y. Liu, C. Peng, C. Zhang, J. M. de la Fuente and D. Cui, Biomaterials, 2017, 120, 103–114 CrossRef CAS PubMed.
- L. W. Goldman, J. Nucl. Med. Technol., 2007, 35, 115–128 CrossRef PubMed.
- M. F. Kircher and J. K. Willmann, Radiology, 2012, 263, 633–643 CrossRef PubMed.
- J. F. Hainfeld, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Br. J. Radiol., 2006, 79, 248–253 CrossRef CAS PubMed.
- C. Zhang, Z. Zhou, Q. Qian, G. Gao, C. Li, L. Feng, Q. Wang and D. Cui, J. Mater. Chem. B, 2013, 1, 5045 RSC.
- H. F. Zhang, K. Maslov, G. Stoica and L. V. Wang, Nat. Biotechnol., 2006, 24, 848–851 CrossRef CAS PubMed.
- J.-H. Lee, Y.-M. Huh, Y.-w. Jun, J.-w. Seo, J.-t. Jang, H.-T. Song, S. Kim, E.-J. Cho, H.-G. Yoon, J.-S. Suh and J. Cheon, Nat. Med., 2007, 13, 95–99 CrossRef CAS PubMed.
- G. Liang, D. Ye, X. Zhang, F. Dong, H. Chen, S. Zhang, J. Li, X. Shen and J. Kong, J. Mater. Chem. B, 2013, 1, 3545 RSC.
- S. S. Gambhir, Nat. Rev. Cancer, 2002, 2, 683–693 CrossRef CAS PubMed.
- F. Chen, S. Goel, R. Hernandez, S. A. Graves, S. Shi, R. J. Nickles and W. Cai, Small, 2016, 12, 2775–2782 CrossRef CAS PubMed.
- H. Kawasaki, S. Kumar, G. Li, C. Zeng, D. R. Kauffman, J. Yoshimoto, Y. Iwasaki and R. Jin, Chem. Mater., 2014, 26, 2777–2788 CrossRef CAS.
- P. Huang, J. Lin, S. Wang, Z. Zhou, Z. Li, Z. Wang, C. Zhang, X. Yue, G. Niu, M. Yang, D. Cui and X. Chen, Biomaterials, 2013, 34, 4643–4654 CrossRef CAS PubMed.
- F. Gao, W. Zheng, L. Gao, P. Cai, R. Liu, Y. Wang, Q. Yuan, Y. Zhao and X. Gao, Adv. Healthcare Mater., 2017, 6, 1601453 CrossRef PubMed.
- M. Zhou, C. Zeng, Y. Chen, S. Zhao, M. Y. Sfeir, M. Zhu and R. Jin, Nat. Commun., 2016, 7, 13240 CrossRef CAS PubMed.
- M. Yu, J. Zhou, B. Du, X. Ning, C. Authement, L. Gandee, P. Kapur, J.-T. Hsieh and J. Zheng, Angew. Chem., Int. Ed., 2016, 55, 2787–2791 CrossRef CAS PubMed.
- L. Lai, C. Zhao, X. Li, X. Liu, H. Jiang, M. Selke and X. Wang, RSC Adv., 2016, 6, 30081–30088 RSC.
- L. Lai, X. Jiang, S. Han, C. Zhao, T. Du, F. U. Rehman, Y. Zheng, X. Li, X. Liu, H. Jiang and X. Wang, Langmuir, 2017, 33, 9018–9024 CrossRef CAS PubMed.
- M. A. H. Muhammed, A. K. Shaw, S. K. Pal and T. Pradeep, J. Phys. Chem. C, 2008, 112, 14324–14330 CrossRef CAS.
- E. Oh, A. L. Huston, A. Shabaev, A. Efros, M. Currie, K. Susumu, K. Bussmann, R. Goswami, F. K. Fatemi and I. L. Medintz, Sci. Rep., 2016, 6, 35538–35555 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |