
Functionalized tungsten disulfide nanotubes for dopamine and catechol detection in a tyrosinase-based amperometric biosensor design
Quentin
Palomar
 a,
Chantal
Gondran
*a,
Jean-Paul
Lellouche
b,
Serge
Cosnier
a and
Michael
Holzinger
*a
a,
Chantal
Gondran
*a,
Jean-Paul
Lellouche
b,
Serge
Cosnier
a and
Michael
Holzinger
*a
aUniv. Grenoble Alpes, CNRS, DCM UMR 5250, F 38000, Grenoble, France. E-mail: chantal.gondran@univ-grenoble-alpes.fr; michael.holzinger@univ-grenoble-alpes.fr
bBar Ilan University, Ramat Gan, 5290002, Israel
First published on 21st November 2019
Abstract
WS2 nanotubes functionalized with carboxylic acid functions (WS2-COOH) were used for improved immobilization of the enzyme tyrosinase in order to form an electrochemical biosensor towards catechol and dopamine. The nanotubes were deposited on glassy carbon electrodes using a dispersion-filtration-transfer procedure to assure the reproducibility of the deposits. After the electrochemical and morphological characterization of these WS2-COOH nanotube deposits, the formed biosensors showed very satisfying performance towards catechol detection with a linear range of 0.6–70 μmol L−1 and a sensitivity of 10.7 ± 0.2 mA L mol−1. The apparent Michaelis Menten constant of this system is slightly lower than the KM value of tyrosinase in solution, reflecting an excellent accessibility of the active site of the enzyme combined with a good mass transport of the target molecule through the deposit. For dopamine detection, we observed an accumulation of this substrate due to electrostatic interactions between the amine function of dopamine and the carboxylic acid groups of the nanotubes. This led to improved signal capture at low dopamine concentrations. With linear ranges of 0.5–10 μmol L−1 and 10–40 μmol L−1, and respective sensitivities of 6.2 ± 0.7 mA L mol−1 and 3.4 ± 0.4 mA L mol−1, the overall sensor performance is within the average of comparable results using carbon nanotubes. Nonetheless, the simplified handling of these nanotubes and their reduced environmental impact make these WS2-COOH nanotubes a promising nanomaterial for biosensing applications.
Introduction
Nanomaterials are nowadays essential tools in biosensor research not only due to the enhanced surface area but also due to a variety of particular phenomena related to their nanosized structures.1–3 Within the vast availability of different nanomaterials, carbon nanotubes4,5 and later graphene6,7 became prominent candidates for biosensing applications because of their beneficial electronic and optical properties for enhanced signal capture of the biological recognition event. More recently, 2D transition metal dichalcogenides such as WS2 and MoS2 showed promising properties in biosensor designs,8 which are principally based on the layered form of these metal dichalcogenides. The detection of the target is commonly transduced via optical detection9 or in a field effect transistor design,10 but such dichalcogenides have also shown their appropriateness in electrochemical biosensor setups.11 The synthesis of WS2 and MoS2 can also form differently shaped materials like nanotubes or nanorods12,13 with improved surface activities and might be more appropriate for biosensor applications.14 Raichman et al. reported an efficient functionalization method for tungsten disulfide inorganic nanotubes using a highly electrophilic acidic Vilsmeier–Haack reagent for homogeneous covalent carboxylation of the WS2 walls. The carboxylic acid functions lead to improved dispersions in aqueous media and represent efficient anchor groups for the immobilization of bioreceptor units.15,16 Furthermore, the clearly reduced cytotoxicity of such transition-metal dichalcogenides compared to carbon nanomaterials represents an advantage when combined with biological entities.17,18 In this work, we used such carboxylated WS2 (WS2-COOH) nanotubes for the immobilization of the tyrosinase enzyme as a protein model and evaluated this sensor for the detection of catechol and dopamine.Catechol (CA) is a toxic compound19 but that serves as a starting material for the synthesis of many different chemicals and therein a wide variety of pesticides.20 This compound therefore contributes to the pollution of soils and groundwater. It further has a low degradation rate that worsens its environmental impact.
Dopamine (DA) is a neurotransmitter and plays an important role as an extracellular chemical messenger in the cardiovascular, renal, hormonal and nervous systems. It is at the centre of cell signal transmission and therefore has a strong influence on human behaviour. Abnormal concentrations of DA in biological fluids are an indicator of several diseases such as schizophrenia, Huntington's disease or Parkinson's disease.21,22 An efficient filtration and transfer technique23 to form identical WS2-COOH nanotube deposits was adjusted for the electro-enzymatic detection of these two compounds. As illustrated in Scheme 1, the carboxylic acid groups served as an anchor for the immobilization of tyrosinase, an enzyme that oxidises specifically catechol and dopamine to its respective quinones. These quinones are then monitored at cathodic potentials via electrochemical reduction to the initial hydroquinones. The WS2-COOH nanotubes serve here principally as porous nanostructures on the working electrodes to immobilize and enhance the amount of tyrosinase enzymes.
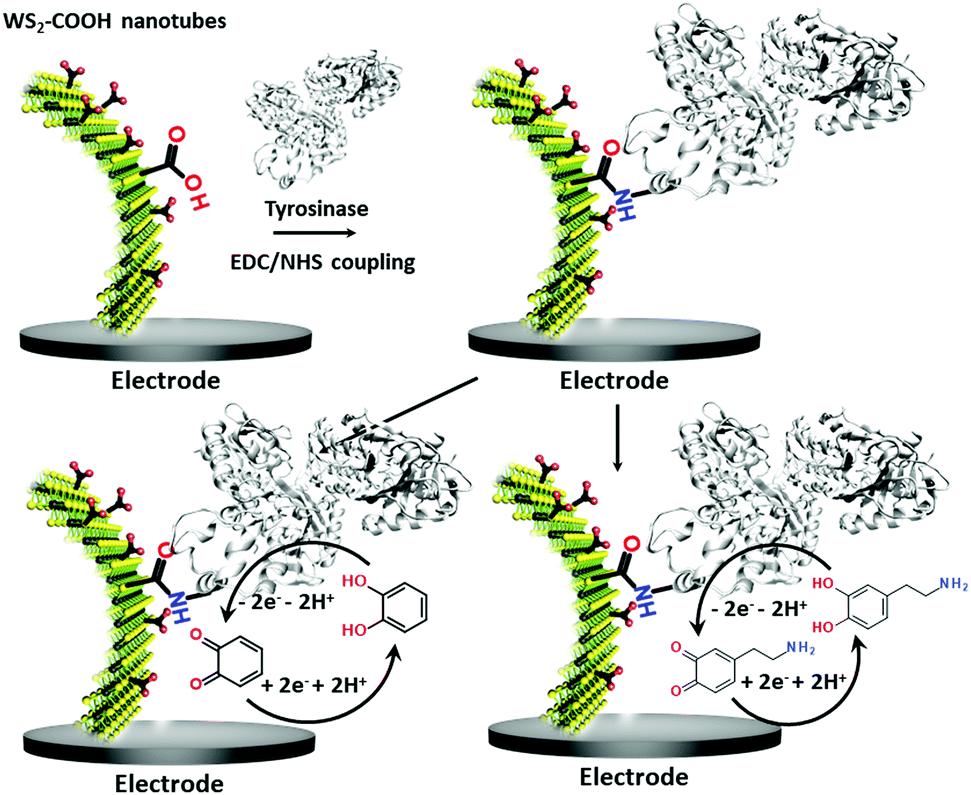 | ||
| Scheme 1 Sketch of the functionalization of WS2 modified glassy carbon electrodes with the enzyme tyrosinase via a standard EDC/NHS coupling reaction. These modified bioelectrodes served in the detection of catechol (bottom left) and dopamine (bottom right) at −0.2 V vs. Ag+/Ag. | ||
Results and discussion
Characterization of the WS2-COOH nanotubes
Before the construction of the biosensor, the morphology and electrochemical behaviour of the WS2-COOH nanotubes were characterized. Fig. 1 shows the scanning electron microscopy images of these as synthesized nanotubes (Fig. 1A and B) and after film formation and transfer onto a GCE (Fig. 1C and D). At the same magnification (Fig. 1A and C), the as synthesized WS2-COOH nanotubes and the formed films do not show significant differences and demonstrate that the stirring-dispersion, filtration and transfer procedures do not affect these nanotubes. | ||
Fig. 1 SEM images of (A and B) as synthesized WS2-COOH powder at magnifications of (A) ×20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and (B) ×50000. SEM images of (C and D) thin films of WS2-COOH nanotubes at magnifications of (C) ×20000 and (D) ×5000. 000 and (B) ×50000. SEM images of (C and D) thin films of WS2-COOH nanotubes at magnifications of (C) ×20000 and (D) ×5000. | ||
The length of these nanotubes comprises between 2 and 10 microns for a diameter of about 100 to 200 nm. It also seems that the WS2-COOH nanotubes are formed of rolled-up WS2 sheets. This lamellar structure is particularly visible at higher magnification, as shown in Fig. 1B. Fig. 1D shows the deposits at lower magnification and reveals an assembly of randomly oriented nanotubes forming a highly porous structure.
To determine the thickness and roughness of the formed WS2-COOH films, a confocal laser microscope was used. This technique allows mapping the height and the morphology of the films by profilometry. Fig. 2 shows images taken at the boundary between the gold substrate and the WS2-COOH films.
 | ||
| Fig. 2 Height mapping images and 3D views realized with a confocal laser microscope at a magnification of ×50 of WS2-COOH film at the cut edge (A and B) with the film on the left and the substrate on the right. Microscope images of the center of the film (C and D). | ||
A thickness of 6.2 μm was determined for a roughness of ±0.6 μm. This relatively high roughness, 10% of the total thickness, can be explained by the highly porous nature of the formed films. Finally, it appears that thickness and roughness values for 3 different films exhibit a relative standard deviation of 6% and 3.6%, respectively, highlighting the reproducibility of the procedure.
The electrochemical behaviour of these films on GCEs was studied using cyclic voltammetry in organic media (Fig. 3). Within a scan range of −2.0 and +2.0 V vs. Ag+/Ag, the first cycle shows a strong and large irreversible anodic peak at 1.7 V that starts at 0.6 V. This peak almost disappears during the second scan. According to the studies about the inherent electrochemistry of transition-metal dichalcogenides by Bonde et al.,24 this peak can be assigned to the irreversible oxidation of WS2 on surface forming WO42−. The potential shift in the second scan is related to the oxidation of remaining WS2 in a WO42− environment. At the reverse scan towards negative potentials, an irreversible cathodic peak was observed at −0.7 V, which also disappeared at the second scan. Here, it is more likely that loosely adsorbed species are released during the first scan or a reduction of the protons since WS2 can be used as electrocatalysts for hydrogen evolution reaction (HER).25
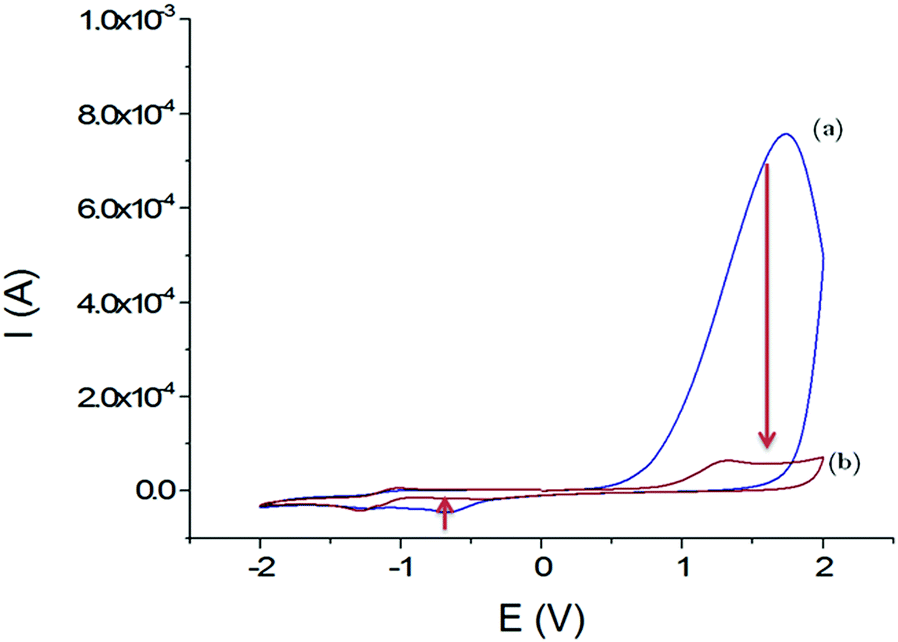 | ||
| Fig. 3 Voltammograms obtained in CH3CN + 0.1 mol L−1 TBAP; first cycle (a) and second cycle (b) with the GCE/WS2-COOH modified electrode between −2.0 and +2.0 V vs. Ag+/Ag; scan rate: 100 mV s−1. | ||
Most importantly, the WS2-COOH nanotubes are electrochemically silent in the potential range around −0.2 V where the amperometric detection of dopamine and catechol with the tyrosinase based biosensor should occur. Unfortunately, films of WS2-COOH are almost not conductive at this potential (1.6 ± 0.1 × 10−7 S cm−1, measured using a four-point probe) and can therefore not be considered as an extension of the electrode.
Detection of catechol
The detection of catechol using tyrosinase is based on the enzymatic oxidation of phenols and o-bisphenols like catechol to o-quinones. The product of the enzymatic reaction is then reduced at the electrode at −0.2 V vs. SCE. The resulting chronoamperometric current density is proportional to the analyte concentration. In addition, the target molecule is regenerated during the reduction at the electrode, which allows the amplification of the electrochemical signal and leads to increased sensitivity.26,27 The measurements were done by immersing the modified electrode in an electrochemical cell containing 20 mL of PBS (0.1 mol L−1, pH = 7.4).Fig. 4A shows the increase of steady-state current response of the bioelectrode upon successive additions of catechol and illustrates the fast response time (3 s). Fig. 4B shows the amperometric response of the bioelectrode as a function of catechol concentration in the range of 0.3–220 μmol L−1 resulting in a calibration curve that follows Michaelis–Menten kinetics.
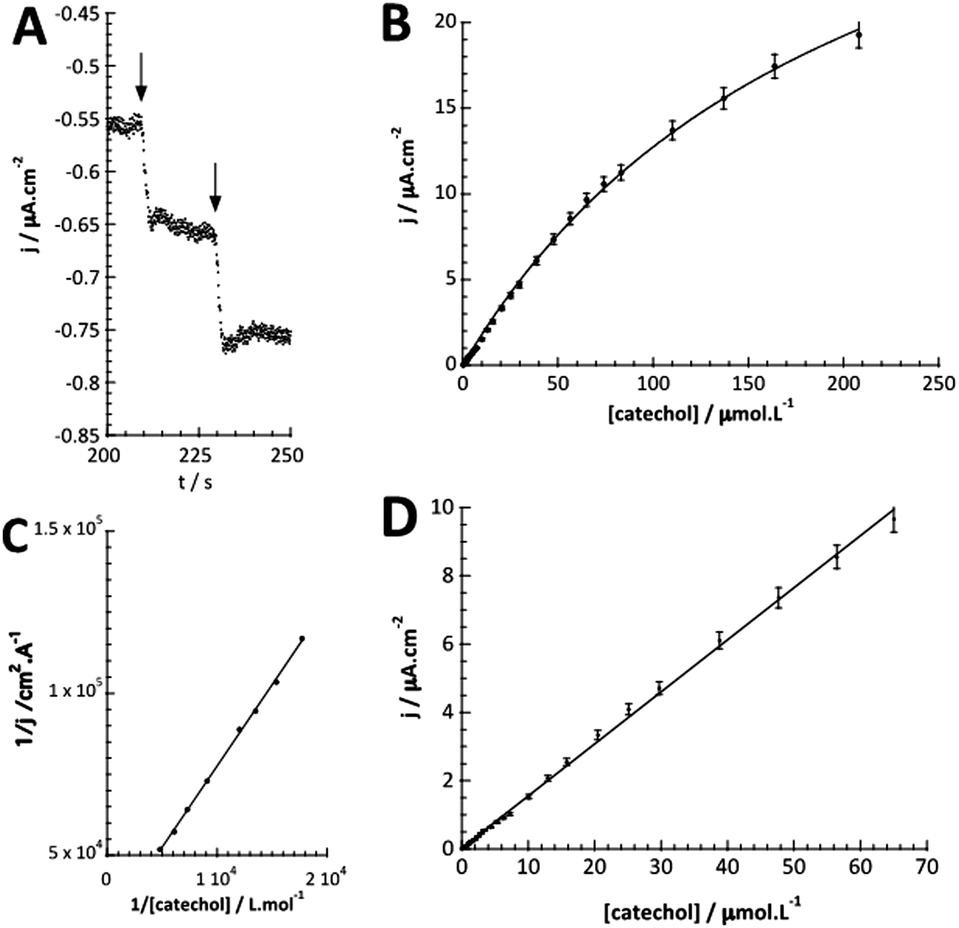 | ||
| Fig. 4 (A) Chronoamperometric response of the GC/WS2-COOH/tyrosinase electrode (E = −0.2 V vs. SCE; 0.1 mol L−1 PBS; pH 7.4) upon successive increase of catechol concentration from 0.3 μmol L−1 to 220 μmol L−1. (B) Plot of the current density vs. catechol concentration on tyrosinase/WS2-COOH/GCE. (C) Lineweaver–Burk plot of the corresponding data. (D) Plot of the linear part of the current density vs. increasing catechol concentrations. | ||
The maximum current density, jmax, is related to the amount of immobilized enzymes on the electrode surface and to the affinity of the enzyme for its target and thus to the apparent Michaelis–Menten constant (KappM). The linear fit equation (R2 = 0.99897) obtained with the Lineweaver–Burk representation leads to 36 μA cm−2 and 183 μmol L−1 for jmax and KappM, respectively (Fig. 4C):
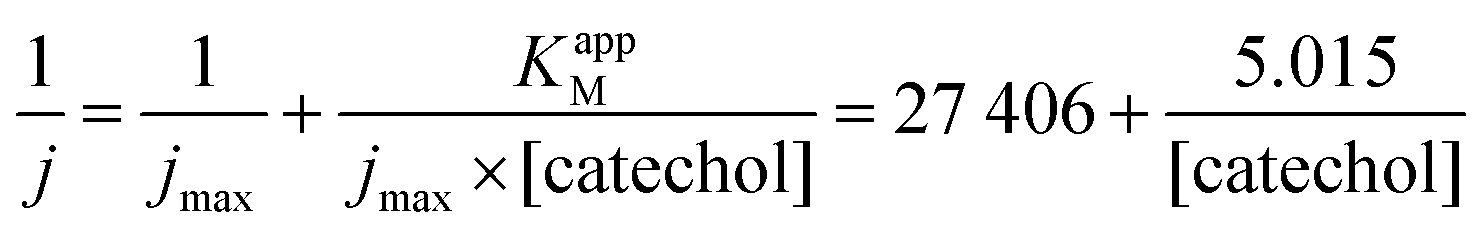
where k1 represents the kinetics of the enzyme–substrate complex formation and k2 represents the transformation of enzyme–substrate complex into the product of the enzymatic reaction and the regenerated enzyme. This formula for KM does not take into account the co-substrate (oxygen) concentration of the enzymatic reaction but this is actually the case since tyrosinase reduces oxygen to water to regenerate itself. By integrating the co-substrate concentration in the equation for
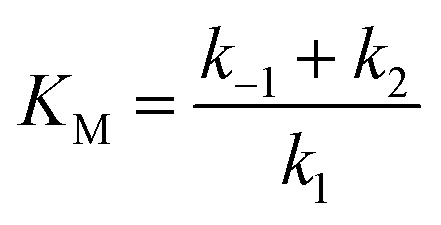
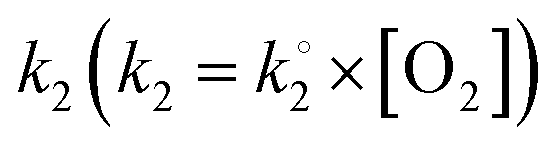 , KappM can be calculated:
, KappM can be calculated:Thus, the value of KappM is directly related to the oxygen concentration. However, the use of nanomaterials considerably increases the quantity of immobilized enzymes and the co-substrate amount is rapidly consumed. Since the oxygen supply is limited by diffusion, a concentration gradient of oxygen can thus be established between the solution and the enzyme. The co-substrate concentration in the enzyme's environment is then lower than in solution and results in a decrease of KappM since the value of k2 is lower than expected. Assuming that the enzyme is here in a favorable environment, it may then be assumed that the oxygen concentration decreases to 40%, close to that of the enzyme compared to the rest of the solution.
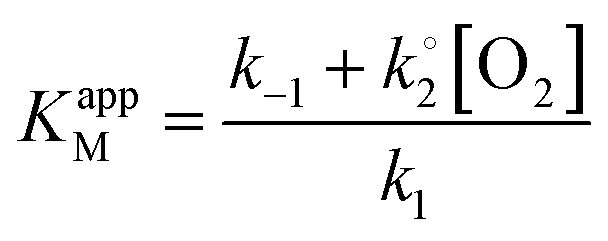
The linear part of the calibration curve within the concentration range of 0.6 μmol L−1 and 70 μmol L−1 reveals a sensitivity value of 152.5 mA L cm−2 mol−1 (R2 = 0.9986). It should be noted that the linearity range and the sensitivity are identic for the two electrodes. The electrochemical characteristics in terms of molecular architecture, KappM, maximum current density, linearity, and sensitivity are summarized in Table 1.
| System used | K appM/μmol L−1 | i max/μA | Linearity/μmol L−1 | Sensitivity/mA L mol−1 | |
|---|---|---|---|---|---|
| This work | WS2-COOH nanotube + GCE | 183 ± 20 | 2.5 ± 0.6 | 0.6–70 | 10.7 ± 0.2 |
| Karim et al.31 | Gold nanoparticle + screen printed carbon electrode | — | — | 0.01–80 | 13720 |
| Sethuraman et al.32 | PEDOT-rGO + GCE | 30.48 | 92 | 0.04–62 | 1600 |
| Zhou et al.33 | Mesoporous carbon nitride + GCE | 11.07 | 11.07 | 0.05–12.5 | 593 |
| Bujduveanu et al.34 | CNT + CaCO3 nanoparticles + GCE | 7 | 39.2 | 0–8 | 2500 |
| Mei et al.35 | rGO–PdCu nanocage + GCE (laccase) | — | — | 500–1155 | 12.65 |
| 1655–5155 | 5.51 |
The linearity range of our system is similar to those found in the literature.35 This can be explained by the presence of WS2-COOH nanotubes, which allow a high amount of immobilized enzymes on the electrode surface, as do other nanomaterials. However, the sensitivity remains relatively low compared to other reports.34 This may be due either to the rapid oxygen consumption and/or to reduced permeability of the WS2-COOH film but finally results most likely from the almost inexistent conductivity at this potential.
Detection of dopamine
As for catechol, tyrosinase also oxidizes dopamine to dopamine-o-quinone, which can be detected at the same potential (−0.2 V vs. SCE). The detection of dopamine was thus performed under identical conditions as described above. Fig. 5A illustrates the chronoamperometric response of the modified GC/WS2-COOH/tyrosinase electrodes as a function of the dopamine concentrations. It can be seen that the current stabilizes rapidly after dopamine injection, confirming the fast electronic transfer and the permeation of the substrate through the WS2-COOH nanotube deposit. The range of concentrations tested varies from 0.5 μmol L−1 to 137 μmol L−1. From the shape of the curve, it seems that the dopamine oxidation via tyrosinase is also governed by Michaelis–Menten kinetics. Fig. 5B shows the evolution of the current density as a function of the dopamine concentration. The observed maximum current density is clearly higher than for catechol detection.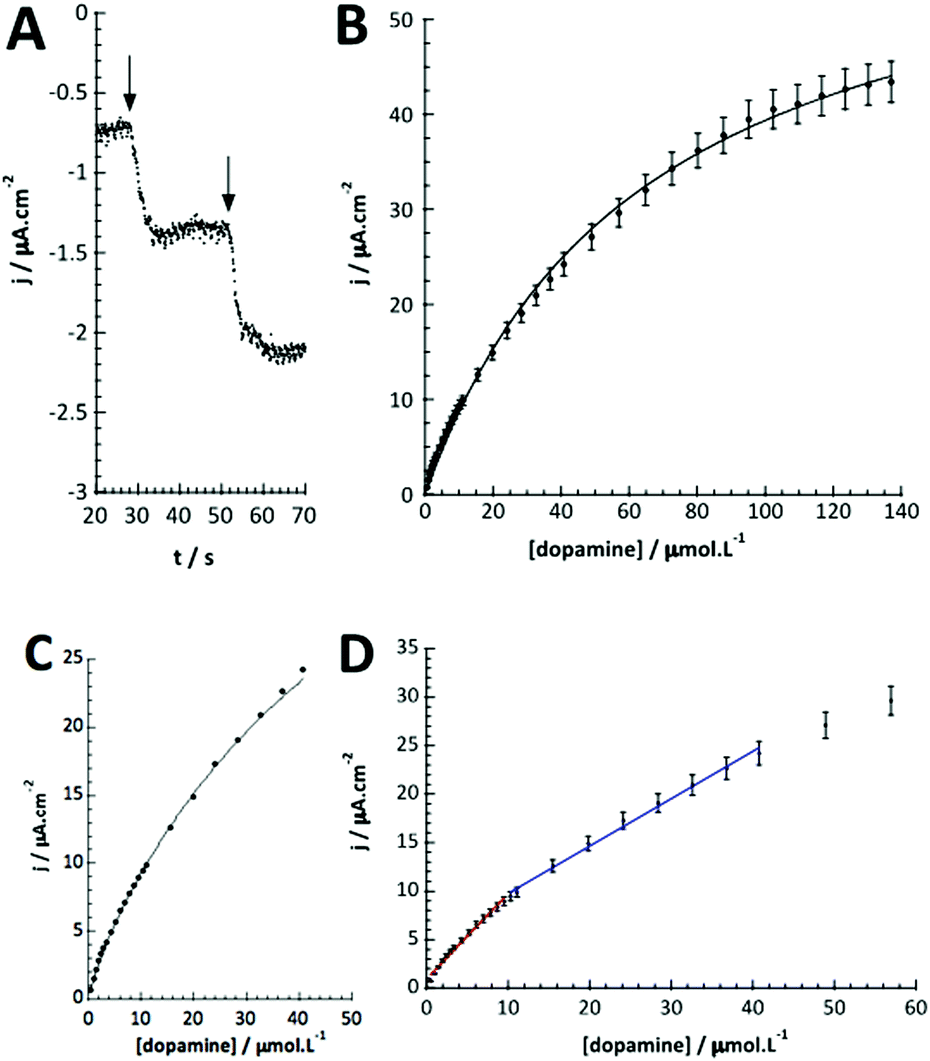 | ||
| Fig. 5 (A) Chronoamperometric response of the GC/WS2-COOH/tyrosinase electrodes (E = −0.2 V vs. SCE; 0.1 mol L−1 PBS; pH 7.4) for increasing dopamine concentrations from 0.5 μmol L−1 to 137 μmol L−1, (B) plot of the current density vs. dopamine concentration. (C) Fit of the calibration curve with the Michaelis–Menten equation taking into account the accumulation of dopamine in the biosensor matrix. (D) Plot of linear parts of the current density vs. increasing dopamine concentrations measured with GC/WS2-COOH/tyrosinase electrode. | ||
This may be due to the fact that the amine function of dopamine is partially positively charged at pH 7.4 and hence can display a faster and deeper penetration inside the WS2-COOH matrix, which is negatively charged. As a consequence, the calibration curve, which is obtained by plotting the current density as a function of the dopamine concentration in solution, does not necessarily reflect the dopamine concentration in the WS2-COOH matrix.
From the Lineweaver–Burk representation (not shown), a KappM of 41 μmol L−1 and jmax of 57 μA cm−2 could be determined following the linear fit equation (R2 = 0.99344):
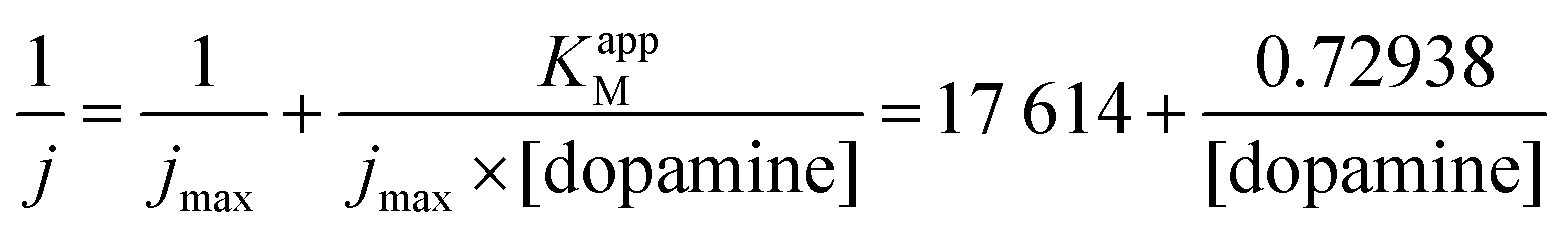
The experimental KappM value is, as for the catechol experiments, lower than the KM of the free enzyme (KM = 2.2 mmol L−1 for the tyrosinase towards dopamine),28 which is related to the limited oxygen concentration. It should be noted that the experimentally determined KappM is lower for dopamine (41 μmol L−1) than for catechol (183 μmol L−1). In solution, the KM value of tyrosinase for dopamine is higher (2.2 mmol L−1) compared to catechol (300 μmol L−1). This inversion may be related to an accumulation of dopamine in the WS2-COOH matrix related to electrostatic interactions. This can be deduced by the arc shaped (and not linear) calibration curve at low dopamine concentrations. In order to estimate this accumulation effect, we fitted this part of the calibration curve in Fig. 5C using an equation that takes the concentration change of dopamine into account:
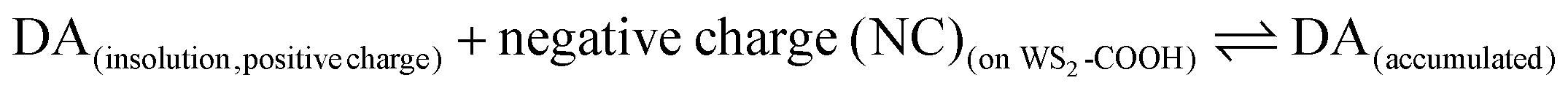
with Kd as the equilibrium dissociation constant. [DA], [NC] and [DA]acc are the dopamine concentrations in solution, negative charges on WS2-COOH nanotubes, and accumulated dopamine in the deposit, respectively. The unknown amount of negative charges on WS2-COOH can be substituted by the difference between the maximum accumulating dopamine concentration, [DA]maxacc and the actually accumulated dopamine concentration, [DA]acc:
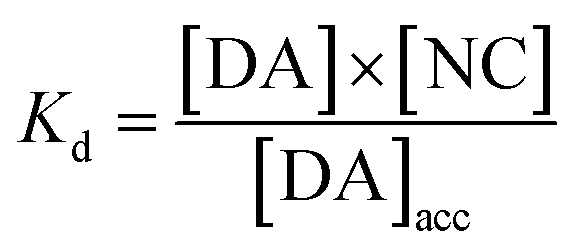
leading for [DA]ads to:
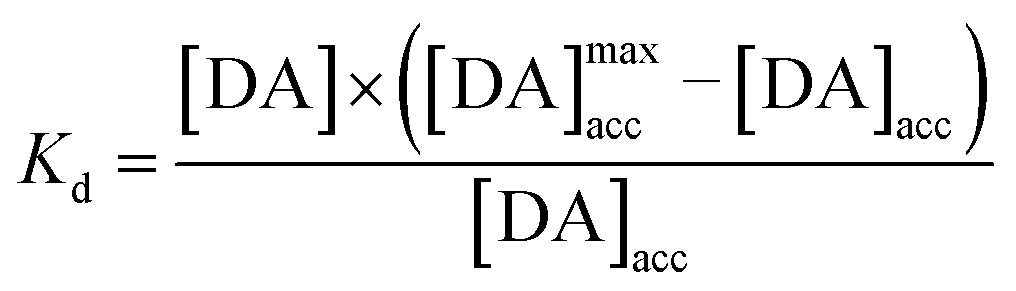
The integration of this equation inside the Michaelis–Menten equation gives:
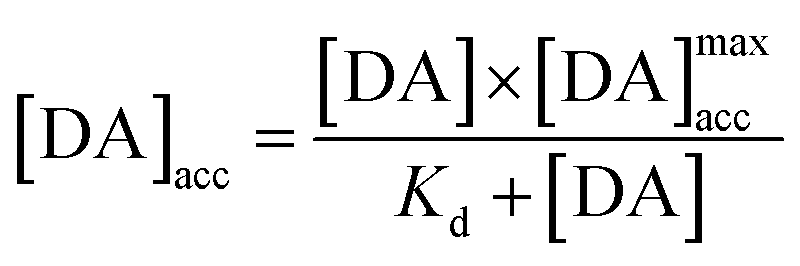
By fitting the first part of the calibration curve with this equation (Fig. 5C, R2 = 0.99728), the dissociation constant, Kd, is estimated at 5 × 10−5 and the maximum accumulating dopamine concentration, [DA]maxacc, is estimated at 35 μmol L−1. It can therefore be concluded that below the value of 35 μmol L−1, the dopamine concentration is greater around the enzymes than in solution. At higher values, the dopamine concentration is the same in the WS2-COOH matrix and in solution.
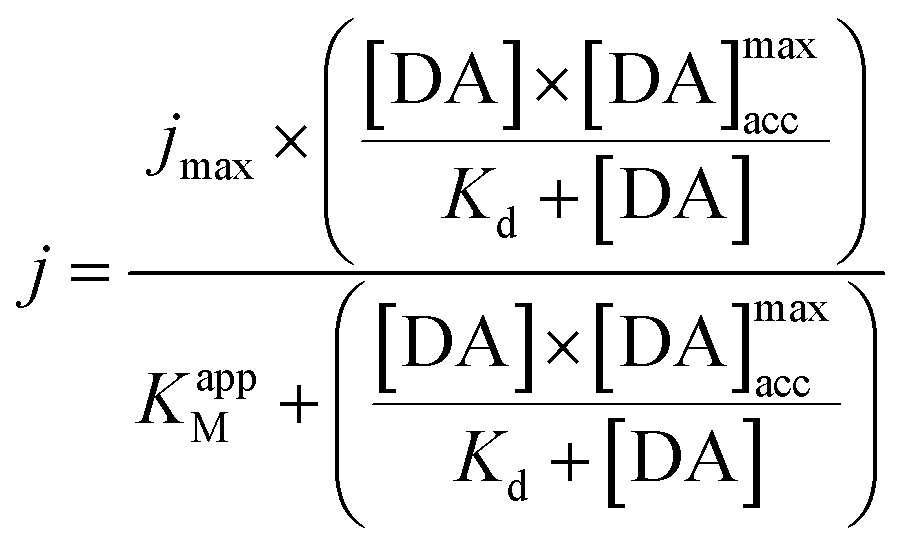
Fig. 5D shows that at low dopamine concentration, the calibration curve presents two successive linear parts, as in other examples reported in the literature for PPO biosensors.36–38 For a concentration range varying from 0.5 to 10 μmol L−1, the biosensor sensitivity for dopamine is 88.0 mA L cm−2 mol−1 (or 6.2 mA L mol−1) and 48.8 mA L cm−2 mol−1 (or 3.4 mA L mol−1) between 10 and 40 μmol L−1 dopamine. This setup and phenomena are quite good reproducible with a relative standard deviation of 4.8% for the three different electrodes.
Table 2 compares our findings with representative examples found in the literature for dopamine detection using tyrosinase combined with a nanomaterial focusing on various criteria such as sensitivity, linearity, the maximum current densities and the calculated KappM. The system presented here has slightly lower performance in terms of linearity range, sensitivity, and limit of detection compared to carbon nanotubes (CNTs), widely used for biosensing application, despite the good maximum current densities.
| System used | K appM/μmol L−1 | i max/μA | Linearity/μmol L−1 | Sensitivity/mA L mol−1 | |
|---|---|---|---|---|---|
| This work | WS2-COOH nanotube + GCE | 41 ± 2 | 4 ± 0.5 | 0.5–10 | 6.2 ± 0.7 |
| 10–40 | 3.4 ± 0.4 | ||||
| Bujduveanu et al.34 | CNT + CaCO3 nanoparticles + GCE | 30 | — | 0.015–30 | 250 |
| Wang et al.39 | SiO2 nanoparticle + acid phytic + GCE (laccase) | — | — | 0.99–103.1 | 1.9 |
| Lete et al.40 | CNT + glutaraldehyde + PEDOT + gold IDE | 11.1 | — | 100–500 | 14.1 |
| Zhuang et al.36 | Overoxidized polypyrrole/graphene + GCE | — | — | 0.5–10 | 94 ± 15 |
| 25–1000 |
We have shown that our WS2-COOH nanotube deposits are not conductive at appropriate potentials and therefore, in contrast to CNTs, they cannot contribute to the electrochemical signal capture. However, the increased surface area of the nanotubes for improved enzyme loading and the particular interaction with dopamine almost compensate this lack of conductivity.
Experimental
Chemicals
Dopamine hydrochloride 98%, 1,2-dihydroxybenzene (catechol), phosphate buffered saline (PBS) tablets, N-hydroxysuccinimide (NHS) 98%, 4-(dimethylamino)pyridine (DMAP) and tyrosinase from mushroom were purchased from Sigma-Aldrich. WS2 nanotubes were provided by NanoMaterials Ltd (Yavne, Israel). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) was purchased from Fluka. All chemicals and solvents were of analytical grade and were used as received, without further purification. WS2-COOH nanotubes were synthesized and characterized as described in ref. 15. The obtained material was characterized using FT-IR spectroscopy (Fig. 6), where the appearance of characteristic peaks, summarized in Table 3, indicates the successful grafting of carboxylic acid functions.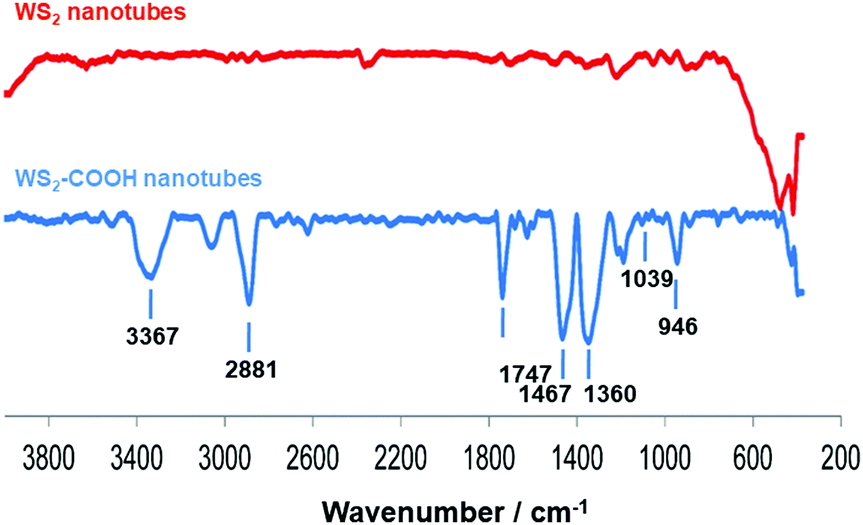 | ||
| Fig. 6 IR spectra of WS2 nanotubes before (top) and after (below) carboxylation. | ||
| Wavenumber (cm−1) | FT-IR vibrational modes |
|---|---|
| 3367 | O–H stretching |
| 2881 | C–H stretching |
| 1747 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carboxylic acid functionality O stretching of carboxylic acid functionality |
| 1467 | C–H bends |
| 1360 | C–H rocks |
| 1039 | C–S stretching |
| 946 | O–H stretching of carboxylic acid functions |
Moreover and as additional proof for COOH functionalization, characteristic fluorescence-based KAISER tests41 have been performed after EDC-based derivatization/activation of the polyCOOH shell by 1,3-diaminopropane and ninhydrin reactivity. Fluorescence measurements helped to obtain the optimized carboxylation degree of 0.5 mmol COOH group g−1 of the functional WS2 nanotubes.
Apparatus
FE-SEM images were recorded using an ULTRA 55 FESEM with a GEMINI FESEM column, beam booster (Nanotechnology Systems Division, Carl Zeiss NTS GmbH, Germany), and a tungsten gun with an accelerating voltage of 3 kV. The WS2-COOH deposits were studied in terms of surface roughness and thickness by using a Keyence Vega confocal laser microscope. For this study, WS2-COOH films were deposited onto a flat gold substrate. In order to determine a representative surface roughness, the images were recorded at low magnifications (×50).Cyclic voltammetry and chronoamperometric experiments were performed using an Autolab potentiostat 100 (Eco Chemie, Utrecht, The Netherlands). The data were recorded and operated using Nova software (version 2.1). Modified glassy carbon electrodes (ϕ = 3 mm) were used as the working electrode for all characterization experiments. Platinum wire was used as the counter electrode. All electrochemical experiments were carried out using a basic three-electrode cell. A 10−2 mol L−1 Ag+/Ag electrode was used as a reference electrode in organic media (CH3CN + 0.1 mol L−1 tetrabutylammonium perchlorate, TBAP), whereas a saturated calomel electrode (SCE) was used as a reference electrode in aqueous media.
Preparation of the WS2-COOH nanotube films
Prior to any modification, the surface of the glassy carbon electrodes (GCE, ϕ = 3 mm) was polished with a 2 μm diamond paste purchased from Presi (France) and rinsed successively with water, acetone, and ethanol under sonication (5 min). Afterwards, the GCEs were modified with the WS2-COOH nanotubes. These nanotubes were dispersed by mechanical stirring in distilled water (15 mg of WS2-COOH in 1 L of H2O). The solution was then allowed to settle for 60 min in order to sediment the larger nanotube clusters. The supernatant was then filtered over cellulose nitrate filter (Sartorius, 0.45 μm, ϕ 3.5 cm). The obtained film was finally transferred to a GCE surface by dissolution of the cellulose nitrate filter according to a procedure described in ref. 23. The same deposition technique was used during laser microscopy characterization, this time using a gold substrate.Enzyme immobilization on the WS2-COOH nanotube films
The WS2-COOH nanotube films with a controlled thickness of 6.2 μm, were then used as a support for the immobilization of tyrosinase. This reaction involves the activation of the carboxylic acid functional groups of the film in order to allow the coupling of the NHS function.For this, the electrode is incubated with PBS (75 μL) containing EDC (20 mmol L−1), NHS (10 mmol L−1) and DMAP (10 mmol L−1) for 12 h at 4 °C. EDC and DMAP serve as activation agents. Eventually, the GCE is incubated with a solution having the concentration of tyrosinase of 0.3 mg mL−1 in PBS 0.1 mol L−1 at pH 7.4 for 12 h at 4 °C in order to immobilize the enzyme. This enzyme concentration was chosen after a short optimization procedure using 0.1, 0.3, and 0.5 mg mL−1 tyrosinase in a cyclic voltammetric setup. The reduction peak currents at −0.1 V vs. SCE were recorded in 0.1 mol L−1 PBS; pH 7.4, containing 5 μmol L−1 catechol. Fig. 7 shows the obtained values revealing an optimal concentration of 0.3 mg mL−1.
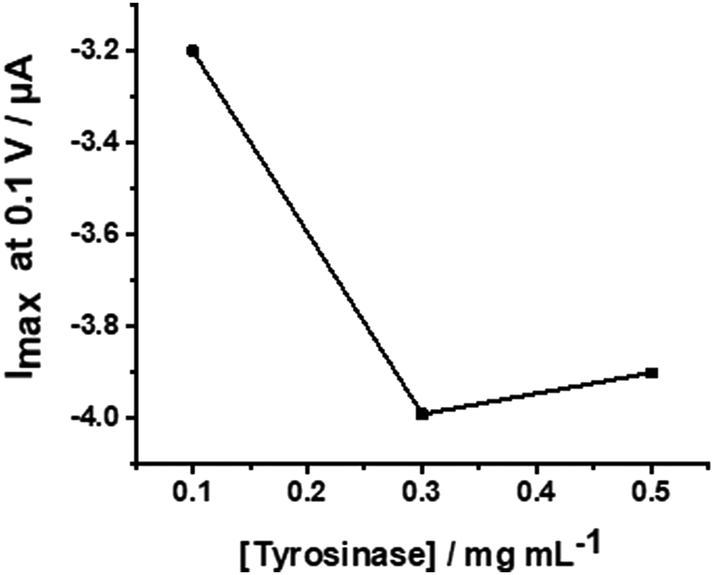 | ||
| Fig. 7 Plot of the peak currents at −0.1 V versus different tyrosinase concentrations from 0.1 to 0.5 mg mL−1. | ||
The resulting electrodes were then washed carefully several times with PBS solution.
Conclusions
COOH functionalized WS2 nanotubes were used as a platform for enhanced immobilization of the tyrosinase enzyme leading to a biosensor for catechol and dopamine, with very satisfying performance. Despite the lack of conductivity of this semiconductor material at appropriate redox potentials for amperometric biosensor applications, these tubes form highly porous deposits with improved enzyme wiring capacity. The determined KappM values of immobilized tyrosinase are close to the values for the enzyme in solution, which represents optimal accessibility of the target molecules to the active site of the enzyme. For dopamine detection, we observed an accumulation of the enzymatically produced dopamine o-quinone leading to enhanced signals at low analyte concentration. The possibility to functionalize these WS2 nanotubes with carboxylic acid functions makes this material easier to process and facilitate post functionalization. At the current state of research, these WS2-COOH nanotubes do not provide superior properties, compared to other described nanomaterials, for electrochemical biosensing applications, but they can become promising components in electrochemical biosensor setups as additives due to the shown beneficial properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the University Grenoble Alpes (Contrats Doctoraux thématiques UJF 2014) for the PhD fellowship funding of Quentin Palomar, and the CNRS PICS program 6344 France Israel for further support. The authors wish also to acknowledge the support from the platform Chimie NanoBio ICMG FR 2607 (PCN-ICMG), from the LabEx ARCANE (ANR-11-LABX-0003-01 and CBH-EUR-GS, ANR-17-EURE-0003), and from the Institut Carnot PolyNat (CARN 0007-01).Notes and references
- M. Holzinger, A. Le Goff and S. Cosnier, Sensors, 2017, 17, 1010 CrossRef.
- P. Mehrotra, J. Oral Biol. Craniofac. Res., 2016, 6, 153–159 CrossRef.
- X. Jia, S. Dong and E. Wang, Biosens. Bioelectron., 2016, 76, 80–90 CrossRef CAS.
- N. Yang, X. Chen, T. Ren, P. Zhang and D. Yang, Sens. Actuators, B, 2015, 207(Part A), 690–715 CrossRef CAS.
- C.-M. Tîlmaciu and M. C. Morris, Front. Chem., 2015, 3, 59 Search PubMed.
- E. Morales-Narváez, L. Baptista-Pires, A. Zamora-Gálvez and A. Merkoçi, Adv. Mater., 2017, 29, 1604905 CrossRef.
- Y. Song, Y. Luo, C. Zhu, H. Li, D. Du and Y. Lin, Biosens. Bioelectron., 2016, 76, 195–212 CrossRef CAS PubMed.
- M. Pumera and A. H. Loo, TrAC, Trends Anal. Chem., 2014, 61, 49–53 CrossRef CAS.
- X. Sun, J. Fan, C. Fu, L. Yao, S. Zhao, J. Wang and J. Xiao, Sci. Rep., 2017, 7, 10290 CrossRef.
- D. Sarkar, W. Liu, X. Xie, A. C. Anselmo, S. Mitragotri and K. Banerjee, ACS Nano, 2014, 8, 3992–4003 CrossRef CAS.
- N. Rohaizad, C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, ACS Appl. Mater. Interfaces, 2017, 9, 40697–40706 CrossRef CAS.
- Y. Q. Zhu, W. K. Hsu, N. Grobert, B. H. Chang, M. Terrones, H. Terrones, H. W. Kroto, D. R. M. Walton and B. Q. Wei, Chem. Mater., 2000, 12, 1190–1194 CrossRef CAS.
- M. Nath, A. Govindaraj and C. N. R. Rao, Adv. Mater., 2001, 13, 283–286 CrossRef CAS.
- S. Barua, H. S. Dutta, S. Gogoi, R. Devi and R. Khan, ACS Appl. Nano Mater., 2018, 1, 2–25 CrossRef CAS.
- D. Raichman, D. A. Strawser and J.-P. Lellouche, Nano Res., 2015, 8, 1454–1463 CrossRef CAS.
- D. Raichman, D. Strawser and J.-P. Lellouche, Inorganics, 2014, 2, 455 CrossRef CAS.
- W. Z. Teo, E. L. K. Chng, Z. Sofer and M. Pumera, Chem. – Eur. J., 2014, 20, 9627–9632 CrossRef CAS.
- J. Laloy, H. Haguet, L. Alpan, D. Raichman, J.-M. Dogné and J.-P. Lellouche, Nano Convergence, 2018, 5, 31 CrossRef.
- N. Schweigert, A. J. B. Zehnder and R. I. L. Eggen, Environ. Microbiol., 2001, 3, 81–91 CrossRef CAS.
- H. Fiege, H. Voges, T. Hamamoto, S. Umemura, T. Iwata, H. Miki, Y. Fujita, H. Buysch, D. Garbe and W. Paulus, Ullmann's Encyclopedia of Industrial Chemistry, 2000 DOI:10.1002/14356007.a19_313.
- G. Lunardi, S. Galati, D. Tropepi, V. Moschella, L. Brusa, M. Pierantozzi, A. Stefani, S. Rossi, F. Fornai, E. Fedele, P. Stanzione, A. H. Hainsworth and A. Pisani, Parkinsonism & Related Disorders, 2009, vol. 15, pp. 383–389 Search PubMed.
- Y. Tao, Y. Lin, J. Ren and X. Qu, Biosens. Bioelectron., 2013, 42, 41–46 CrossRef CAS.
- Q. Palomar, C. Gondran, M. Holzinger, R. Marks and S. Cosnier, Biosens. Bioelectron., 2017, 97, 177–183 CrossRef CAS.
- J. Bonde, P. G. Moses, T. F. Jaramillo, J. K. Nørskov and I. Chorkendorff, Faraday Discuss., 2009, 140, 219–231 RSC.
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef CAS.
- S. Cosnier, J. J. Fombon, P. Labbé and D. Limosin, Sens. Actuators, B, 1999, 59, 134–139 CrossRef CAS.
- S. Cosnier and I. C. Popescu, Anal. Chim. Acta, 1996, 319, 145–151 CrossRef CAS.
- J. C. Espín, R. Varón, L. G. Fenoll, M. A. Gilabert, P. A. García-Ruíz, J. Tudela and F. García-Cánovas, Eur. J. Biochem., 2000, 267, 1270–1279 CrossRef.
- D. Shan, M. Zhu, E. Han, H. Xue and S. Cosnier, Biosens. Bioelectron., 2007, 23, 648–654 CrossRef CAS.
- V. Carralero Sanz, M. L. Mena, A. González-Cortés, P. Yáñez-Sedeño and J. M. Pingarrón, Anal. Chim. Acta, 2005, 528, 1–8 CrossRef CAS.
- M. N. Karim, J. E. Lee and H. J. Lee, Biosens. Bioelectron., 2014, 61, 147–151 CrossRef CAS.
- V. Sethuraman, P. Muthuraja, J. Anandha Raj and P. Manisankar, Biosens. Bioelectron., 2016, 84, 112–119 CrossRef CAS.
- Y. Zhou, L. Tang, G. Zeng, J. Chen, Y. Cai, Y. Zhang, G. Yang, Y. Liu, C. Zhang and W. Tang, Biosens. Bioelectron., 2014, 61, 519–525 CrossRef CAS.
- M.-R. Bujduveanu, W. Yao, A. Le Goff, K. Gorgy, D. Shan, G.-W. Diao, E.-M. Ungureanu and S. Cosnier, Electroanalysis, 2013, 25, 613–619 CrossRef CAS.
- L.-P. Mei, J.-J. Feng, L. Wu, J.-Y. Zhou, J.-R. Chen and A.-J. Wang, Biosens. Bioelectron., 2015, 74, 347–352 CrossRef CAS.
- Z. Zhuang, J. Li, R. Xu and D. Xiao, Int. J. Electrochem. Sci., 2011, 6, 2149–2161 CAS.
- X. Liu, Y. Peng, X. Qu, S. Ai, R. Han and X. Zhu, J. Electroanal. Chem., 2011, 654, 72–78 CrossRef CAS.
- B. Rezaei, E. Havakeshian and A. A. Ensafi, Sens. Actuators, B, 2015, 213, 484–492 CrossRef CAS.
- K. Wang, P. Liu, Y. Ye, J. Li, W. Zhao and X. Huang, Sens. Actuators, B, 2014, 197, 292–299 CrossRef CAS.
- C. Lete, S. Lupu, B. Lakard, J.-Y. Hihn and F. J. del Campo, J. Electroanal. Chem., 2015, 744, 53–61 CrossRef CAS.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595–598 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |