
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn advanced cathode material for high-power Li-ion storage full cells with a long lifespan†
Haijian
Huang
 a,
Long
Pan
a,
Xi
Chen
a,
Elena
Tervoort
a,
Alla
Sologubenko
b and
Markus
Niederberger
*a
a,
Long
Pan
a,
Xi
Chen
a,
Elena
Tervoort
a,
Alla
Sologubenko
b and
Markus
Niederberger
*a
aLaboratory for Multifunctional Materials, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zürich, Switzerland. E-mail: markus.niederberger@mat.ethz.ch
bLaboratory for Nanometallurgy, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, Zürich, 8093, Switzerland
First published on 24th September 2019
Abstract
Compared to the progress made in high-rate anodes, the exploration of cathode materials with comparable performance remains a challenge in the fabrication of high power Li-ion storage devices. Here a cathode material with fast Li-ion storage is reported, which is composed of Ni0.25V2O5·nH2O nanobelts with a layered structure and reduced graphene oxide. Operando X-ray diffraction and ex situ X-ray photoelectron spectroscopy results reveal an electrochemical process with high structural reversibility during charging/discharging. Kinetics analysis based on cyclic voltammetry measurements demonstrates fast Li-ion storage in the cathode material. These positive properties lead to high rate capability and cycling stability of the composite. Even at a high mass loading of 11.3 mg cm−2, the material still offers high-performance Li-ion storage, indicating its great promise for practical applications. Furthermore, we report for the first time the combination of such a high-rate cathode with a surface redox pseudocapacitive anode made of N-doped reduced graphene oxide foam for a fast-charging Li-ion storage full-cell device with a long lifespan. The high performance of the full cell suggests that the concept of using materials with fast Li-ion storage in both the positive and negative electrodes represents a promising strategy to develop high-power and high-energy Li-ion storage devices.
1 Introduction
The impressive development of consumer electronics, electric vehicles and smart grid storage technology motivates the battery research community to strive for breakthroughs in high-performance Li-ion energy storage devices, not only providing high energy density, but also with a high power output.1–5 Inspired by this trend, the development of high-rate Li-ion battery (LIB) anode materials has progressed at an unprecedentedly high speed over the last decade.6–14 However, the research on high-power cathodes, as the pairing counterpart of the anodes, is significantly lagging behind.One of the challenges for the development of high-rate cathodes lies in discovering materials with fast Li+ diffusion kinetics during operation. Conventional faradaic LIB cathodes possess Li+ intercalation channels, whereas the slow diffusion rate will lead to prolonged charging time and thus limits the power density.15–18 On the other hand, non-faradaic cathodes, e.g., activated carbon (AC), intrinsically offer fast charge/discharge capability, because the capacitive charge storage in this type of cathode is based on fast physical adsorption/desorption processes.19–22 The high rate capability of AC as the cathode resulted in the emergence of high-power supercapacitors (SCs) and Li-ion hybrid supercapacitors (Li-HSCs).11,23–30 As the capacitive storage mechanism is a surface effect, the energy storage capability of AC is limited by its accessible surface area. Therefore, the high power density of SCs and HSCs comes at the cost of low energy density. In this context, developing high-energy and high-power Li-ion storage devices is still challenging.
Layered metal vanadates have been proven to show good electrochemical performance in rechargeable batteries due to their open-framework crystal structure, which leads to fast ion diffusion kinetics.31–35 In a recent report, we demonstrated that fast Na-ion insertion/extraction in layered metal vanadates leads to high-rate Na-ion storage performance.36 However, so far this type of material has rarely been studied as high-power LIB cathodes. Here we report the synthesis of Ni0.25V2O5·nH2O/reduced graphene oxide (referred to as NVO@rGO) as a new type of LIB cathode with high rate capability for LIBs. The layered crystal structure of Ni0.25V2O5·nH2O consists of V2O5 layers pillared by NiO6 octahedra in between. Ni0.25V2O5·nH2O has a large interlayer spacing (d001 = 10.3 Å), which is able to provide spacious channels for fast Li+ intercalation/extraction. Ni0.25V2O5·nH2O was further integrated with reduced graphene oxide (rGO) sheets, forming a conducting network to facilitate electron transport. The synergetic effects between the two constituents, one offering channels for fast Li+ storage and the other one good electronic conductivity, lead to the high rate capability of the composite material when working as the LIB cathode within the voltage range of 2.0–4.3 V. Moreover, it is worth noting that for practical applications, the electrodes usually have about 10 mg cm−2 of mass loading. However, in most of the studies on Li-ion battery cathodes, the electrochemical performances were tested at low mass loadings,37–41 which results in low areal capacities. In our study, due to the favourable Li-ion storage kinetics and the conducting network, the composite material is able to deliver high-performance Li-ion storage even at a high mass loading, indicative of its large application perspective.
Furthermore, conventional strategies for improving the ionic diffusion kinetics of electrode materials usually involve nanoscaling and complex nanostructural design to shorten the diffusion distance,11,42–48 which often complicates large-scale manufacturing. Even without downsizing the particles, anode materials of niobium oxide49 and niobium tungsten oxides50 have been reported to possess fast ion storage kinetics mainly related to the crystal structure rather than to the diffusion length. However, analogous properties of cathode materials have been overlooked. In our study, it is found that NVO@rGO shows fast Li+ intercalation kinetics even when the diffusion length is of the order of micrometers, indicating its great promise in practical applications.
To produce a high-power and high-energy LIB full cell, the NVO@rGO cathode is combined with an anode composed of N-doped reduced graphene oxide (N-rGO) foam. Cyclic voltammetry (CV) analysis indicates surface redox pseudocapacitance in the N-rGO foam, which results in a high rate performance of the anode material in half-cell tests. Prototype LIB full cells were fabricated from NVO@rGO as the cathode and N-rGO as the anode. To the best of our knowledge, this is the first time that a surface redox pseudocapacitive anode is combined with a cathode with fast Li-ion storage in a full cell configuration. The fast Li+ storage on both sides is expected to improve the rate capability of the full cells without compromising the energy density.
2 Results and discussion
2.1. Synthesis and physical characterization
Pristine NVO and NVO@rGO were both prepared by a rapid one-pot microwave approach (the detailed procedures are described in the Experimental section, see ESI†). For the synthesis of NVO@rGO, nickel acetate tetrahydrate, vanadium oxide and graphene oxide were used as the precursors (Scheme 1). Due to the fast Li-ion intercalation/de-intercalation into/from the layer-structured NVO and the efficient electron transport within the composite structure, the material is expected to show high rate performance as a LIB cathode, which will be discussed in detail below.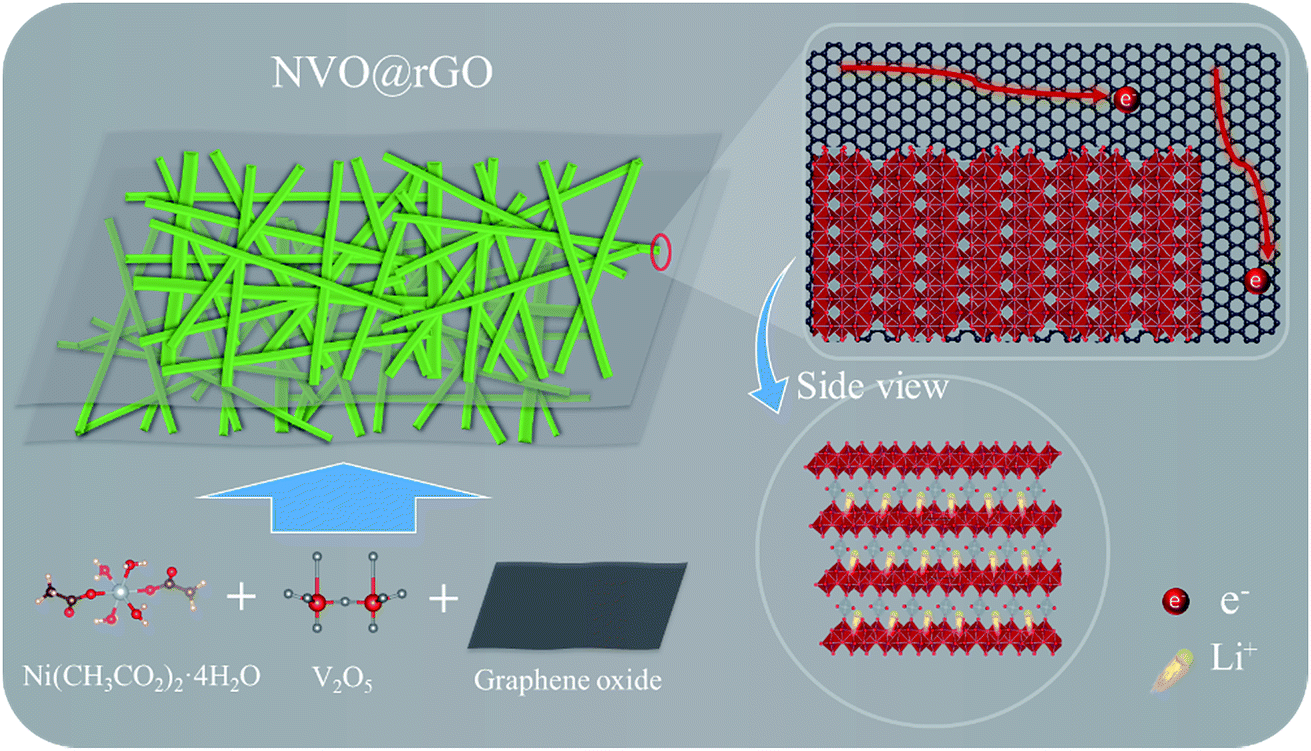 | ||
| Scheme 1 Schematic of the preparation of NVO@rGO and its advantages as a high-rate Li-ion battery cathode. | ||
Interestingly, GO was partially reduced under microwave radiation during the reaction. As shown in the X-ray photoelectron spectroscopy (XPS) data (Fig. S1a and b†), a significant decrease in the C–O/C–C (C1s) signal ratio was observed for NVO@rGO compared with pristine GO. Such reduced GO sheets with the restoration of the sp2 hybridization of carbon facilitate the formation of a conductive network between the NVO nanobelts, enabling enhanced rate capability as discussed below. Thermogravimetric analysis (TGA) (Fig. S2†) reveals ∼0.9 molecules of lattice water per formula unit in NVO. In addition, the weight loss difference between NVO@rGO and NVO indicates a carbon content of 7% in the NVO@rGO composite. The X-ray diffraction (XRD) results of NVO and NVO@rGO (Fig. 1a) show that there is no phase change with the introduction of reduced graphene oxide in the composite. The XRD peaks of both samples can be indexed to monoclinic Ni0.25V2O5·nH2O51 (ICSD PDF no. 04-009-5930) although the pattern of our sample is dominated by (00l) reflections due to the highly anisotropic shape of the nanobelts. The same observation was reported for Zn0.25V2O5·nH2O with a similar structure.33,52Fig. 1b shows the crystal structure of NVO, where NiO6 octahedra connect the V2O5 layers. The open framework of the structure creates spacious and stable pathways for fast Li+ diffusion in the ab plane along the [0![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] and [010] directions, which will be discussed in detail below.
0] and [010] directions, which will be discussed in detail below.
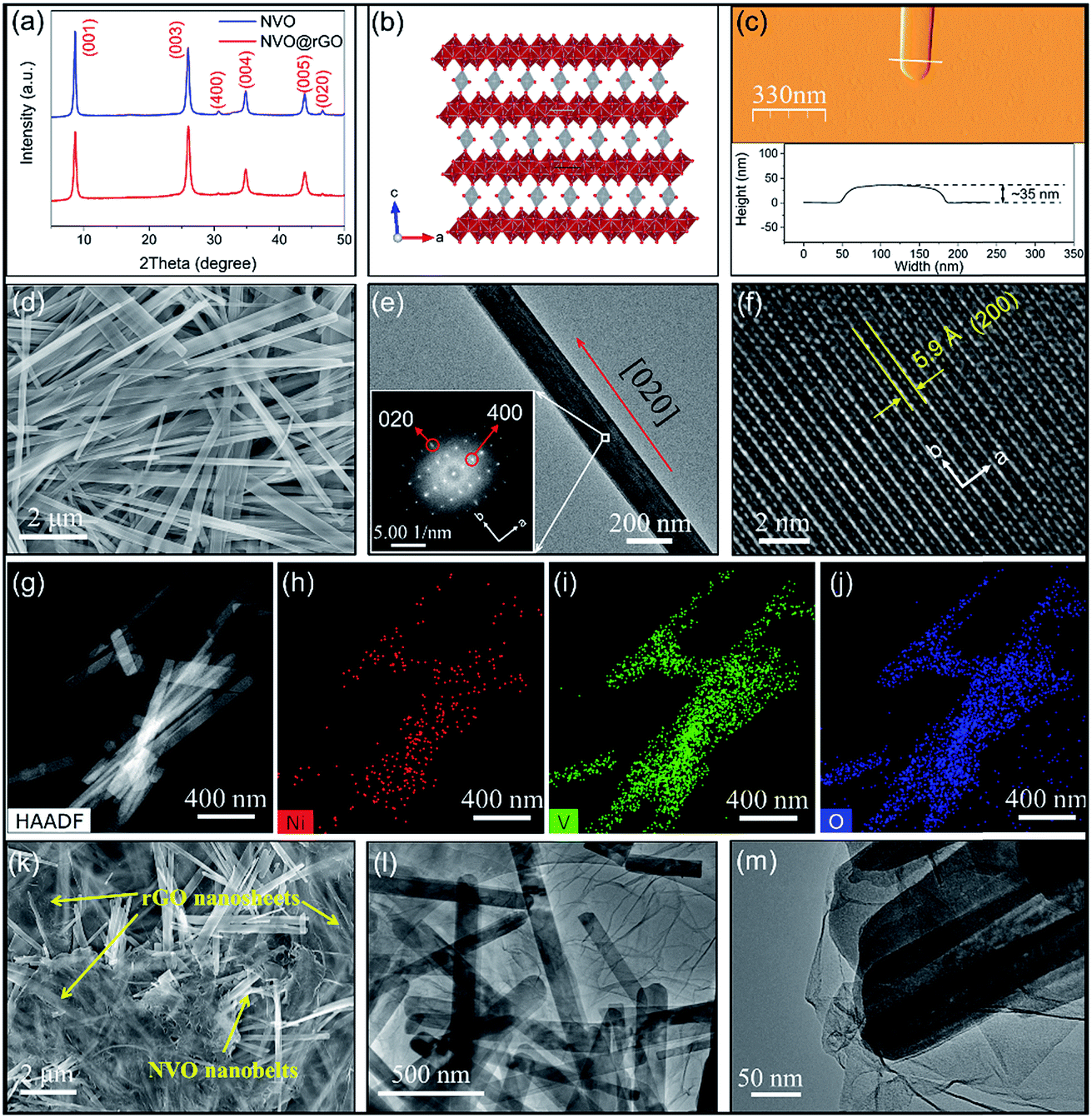 | ||
| Fig. 1 (a) XRD patterns of NVO and NVO@rGO. (b) Crystal structure of NVO viewed along the b axis; the red polyhedra represent the VOx units in the V2O5 layers, while the interlayer NiO6 octahedra are indicated in grey. The V atoms in the V2O5 polyhedra and the Ni atoms in the NiO6 octahedra are indicated in blue and grey, respectively. The isolated O atoms bonded to the Ni ions belong to water molecules (hydrogen atoms not shown). (c) AFM image and the corresponding height information of the NVO nanobelt. (d–f) SEM and TEM images of NVO nanobelts. The inset in (e) shows the corresponding FFT pattern. (g–j) HAADF image and the corresponding element distribution maps of the NVO nanobelts. (k–m) SEM and TEM images of the NVO@rGO composite. | ||
The morphology of NVO and NVO@rGO was studied by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM overview image in Fig. 1d reveals the 1-dimensional morphology of NVO with anisotropic particles that are tens of micrometers long and 100–500 nm wide. The structures are nearly transparent to the electron beam, indicating a thin nanobelt-like shape. The thickness of the NVO nanobelts is determined to be ∼35 nm by atomic force microscopy (AFM) measurements (Fig. 1c), further corroborating the belt-like morphology with a high width to thickness aspect ratio. Fig. 1e shows a representative TEM image of a typical nanobelt. The high-resolution TEM (HRTEM) image of a selected area in the nanobelt is presented in Fig. S3a.† The corresponding fast Fourier transform (FFT) acquired from Fig. S3a† (inset in Fig. 1e) reveals well-resolved spots, typical for a single-crystalline material. The evaluation of the FFTs (Fig. S3b†) confirms the monoclinic C12/m1 structure of the Ni0.25V2O5·nH2O nanobelts51 and reveals that their growth direction is [020]. The enlarged HRTEM image in Fig. 1f reveals well-defined lattice fringes with a spacing of 0.59 nm corresponding to the (200) planes, further confirming that the nanobelts grow along the b direction. It is interesting to note that, as discussed above, Li+ diffuses into the structure along the [00] and [010] directions, i.e., along the growth direction of the nanobelts, which results in long diffusion paths of several micrometers. Nevertheless, the Li+ intercalation rates are still high due to the spacious structure of the NVO host lattice, as discussed below. In addition, energy dispersive spectroscopy (EDS) analyses using an atomic number sensitive high-angle annular dark-field detector (HAADF) confirm the co-existence of Ni, V, and O inside the NVO nanobelts (Fig. 1g–j). The resulting atomic ratio of Ni to V is about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 (Table S1†), which is in line with the structural formula of Ni0.25V2O5·nH2O. The SEM image of NVO@rGO (Fig. 1k) reveals a composite architecture with NVO nanobelts embedded in the rGO sheets. TEM images at low (Fig. 1l) and at higher magnification (Fig. 1m) prove the interconnected composite structure of NVO@rGO with the nanobelts embedded in the rGO sheets. Although not all NVO nanobelts are similarly well connected with the rGO nanosheets (a less connected example is shown in Fig. S4†) due to the low content of rGO sheets, the rGO in the composite is able to create a conductive network and the interaction between these two components facilitates fast and efficient electron transfer in the composite nanostructure. This feature is especially beneficial for achieving high-rate capability, as discussed below.
:8 (Table S1†), which is in line with the structural formula of Ni0.25V2O5·nH2O. The SEM image of NVO@rGO (Fig. 1k) reveals a composite architecture with NVO nanobelts embedded in the rGO sheets. TEM images at low (Fig. 1l) and at higher magnification (Fig. 1m) prove the interconnected composite structure of NVO@rGO with the nanobelts embedded in the rGO sheets. Although not all NVO nanobelts are similarly well connected with the rGO nanosheets (a less connected example is shown in Fig. S4†) due to the low content of rGO sheets, the rGO in the composite is able to create a conductive network and the interaction between these two components facilitates fast and efficient electron transfer in the composite nanostructure. This feature is especially beneficial for achieving high-rate capability, as discussed below.
The N-rGO foam was prepared by a hydrothermal process using urea as the source for the nitrogen dopant. A typical photograph of monolithic N-rGO foam on a dandelion flower is presented in Fig. S5,† illustrating the low density of the foam. High-resolution C1s XPS spectra in Fig. S6† reveal the significantly decreased intensity of the C–O signal in N-rGO compared with pristine GO, indicating the reduction of GO in the N-rGO foam. Furthermore, the N1s XPS spectrum confirms the successful doping of N in the rGO sheets. As shown in Fig. S7,† the envelope of the N1s peak can be deconvoluted into three components that are assigned to pyridinic N (398.7 eV), pyrrolic N (400.0 eV) and graphitic N (401.5 eV), respectively.53,54 The reduction of GO together with N doping is well-known means to optimize the electronic properties of such materials.55,56 N2 gas sorption analysis demonstrates the coexistence of mesoporosity and macroporosity with pore sizes between 5 and 75 nm (Fig. S8†). SEM images (Fig. S9†) clearly show the hierarchical porous structure of the foam. The high porosity with a surface area of 154 m2 g−1 (Fig. S10†) leads to more surface active sites on the sheet-like structure of N-rGO (Fig. S11†), which is especially important for improving the rate capability as discussed below.
2.2. Half-cell performance and study of the Li-ion storage mechanism
The electrochemical properties of NVO and NVO@rGO are first evaluated separately in half-cell configurations using lithium foil as both counter and reference electrodes. Fig. 2a shows the discharge rate capability of NVO@rGO. The rate performance of the reference sample, NVO, is also shown for comparison. As expected, with increasing current densities, much higher specific capacities are retained in NVO@rGO compared to NVO. The enhanced rate capability can be attributed to the improved conductivity induced by rGO, as confirmed by electrochemical impedance spectroscopy (EIS) measurements (Fig. S12†). Through simulation of the EIS results, the charge transfer resistance Rct for NVO@rGO is calculated to be 34 Ω, which is much lower than 85 Ω for NVO, suggesting that the reduced graphene oxide in G-ZVO successfully decreases the charge transfer resistance at the electrode/electrolyte interface.57,58 Interestingly, it is observed that the reference cells based on NVO already fail at 30C (1C corresponds to 150 mA g−1 in this study), whereas at the same current rate the NVO@rGO samples still deliver a capacity of 108 mA h g−1, equivalent to ∼65% of the initial capacity achieved at 1C (Fig. 2a). Similar to other studies,9,59,60 a capacity increase is observed at this stage. The reason for this observation is not clear yet, but it could be due to the improved wetting of the electrodes with the electrolyte during the measurement.59 Capacities of 90 and 74 mA h g−1 are achieved at 50C and 70C, respectively. Even at an extremely high current rate of 100C, a capacity of 52 mA h g−1 still remains for NVO@rGO, demonstrating its high rate performance. Note that the capacity recovers to 181 mA h g−1, when the current density returns to 1C, indicating its high structural stability. Fig. 2b shows the galvanostatic charge–discharge curves for the first 5 cycles of NVO@rGO at a current rate of 15C and 50C. The charge–discharge profiles almost overlap with each other during cycling, underlying the high electrochemical reversibility. Finally, the long-term cycling performance of the NVO@rGO composite was evaluated. As shown in Fig. 2d, NVO@rGO offers a high capacity of 108 mA h g−1 at 15C after 500 cycles, with an average coulombic efficiency close to 100% (Fig. S13†). At a much higher current density of 50C, the composite still achieves a capacity of 90 mA h g−1 after 500 cycles (Fig. 2e), proving the high rate capability and the good cycling stability of the material in half cells. To investigate the practical application potential, the cycling performance of the electrode material at high mass loading was also tested and is shown in Fig. 2c. With 11.3 mg cm−2 of the active material, the composite still exhibits good cyclability, delivering a high reversible areal capacity of 1.3 mA h cm−2 at 2.8 mA cm−2 after 100 cycles.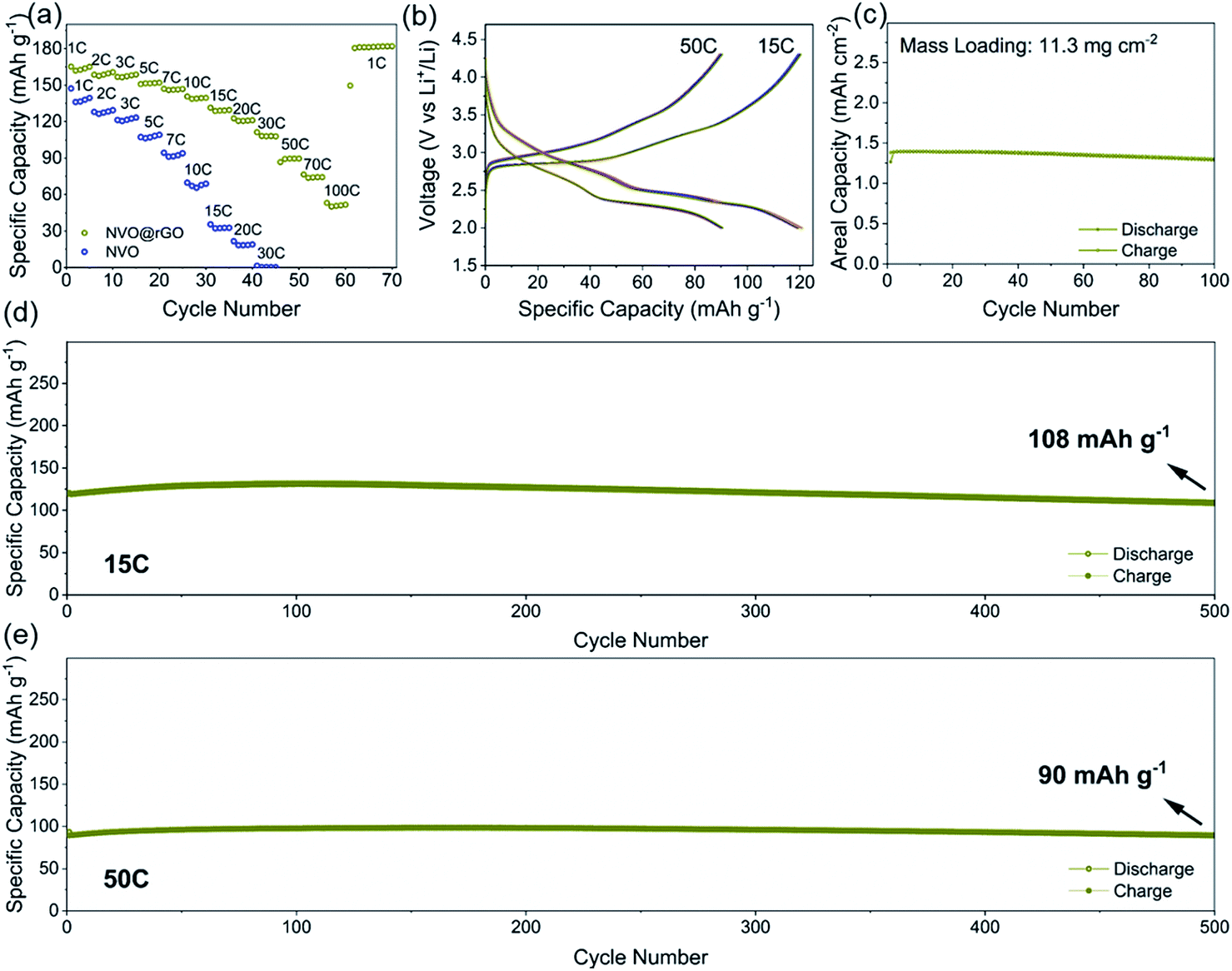 | ||
| Fig. 2 (a) Rate performance of NVO@rGO in comparison to NVO. (b) Galvanostatic charge/discharge profiles for the first 5 cycles of NVO@rGO at 15C and 50C, respectively. (c) Cycling performance of NVO@rGO at 2.8 mA cm−2 with a mass loading of 11.3 mg cm−2. (d) Cycling performance of NVO@rGO at 15C and (e) 50C. | ||
To gain better insight into the electrochemical mechanism of the NVO@rGO electrodes, operando XRD investigation, ex situ high-resolution XPS analysis and CV measurements were performed. The operando XRD results are illustrated in Fig. 3a–d. A broad peak centered around 25°, which stems from the operando cell as confirmed by the XRD pattern of the blank cell (Fig. S14†), is found for all patterns. Apart from this, three dominating reflections, which are indexed to the (001), (003) and (004) crystal planes of NVO, are observed. The exclusive (00l) set of reflections provides detailed information on the evolution of the a–b interlayer spacing, i.e., on the c parameter, during Li+ intercalation/extraction. Specifically, upon discharging to 2.0 V, a slight shift of the (00l) reflections to higher 2θ positions is observed, which corresponds to a decrease of the interplanar spacing from 10.3 to 9.8 Å. The small contraction of the NVO lattice is likely due to the reinforced electrostatic attraction between the inserted Li+ and the VOx layers. A continuous intensity decrease of the (00l) is also observed, which might be the result of an uneven distribution of Li-ions between the layers, leading to the broadening and weakening of the peaks. During charging back to 4.3 V, the structural evolution follows the reverse process of discharge. The intensity and the peak positions of the charged electrodes are almost the same as those of the pristine electrodes, indicating the good structural reversibility of the cathode. The high reversibility can be attributed to the NiO6 octahedra, which work as mechanically stable pillars between the VOx layers, efficiently stabilizing the layered crystal structure during charging/discharging. In addition, the high structural stability makes it possible to maintain the morphology of the NVO nanobelts in the composite for 250 cycles at 15C, as confirmed by the ex situ SEM measurements (Fig. S15†).
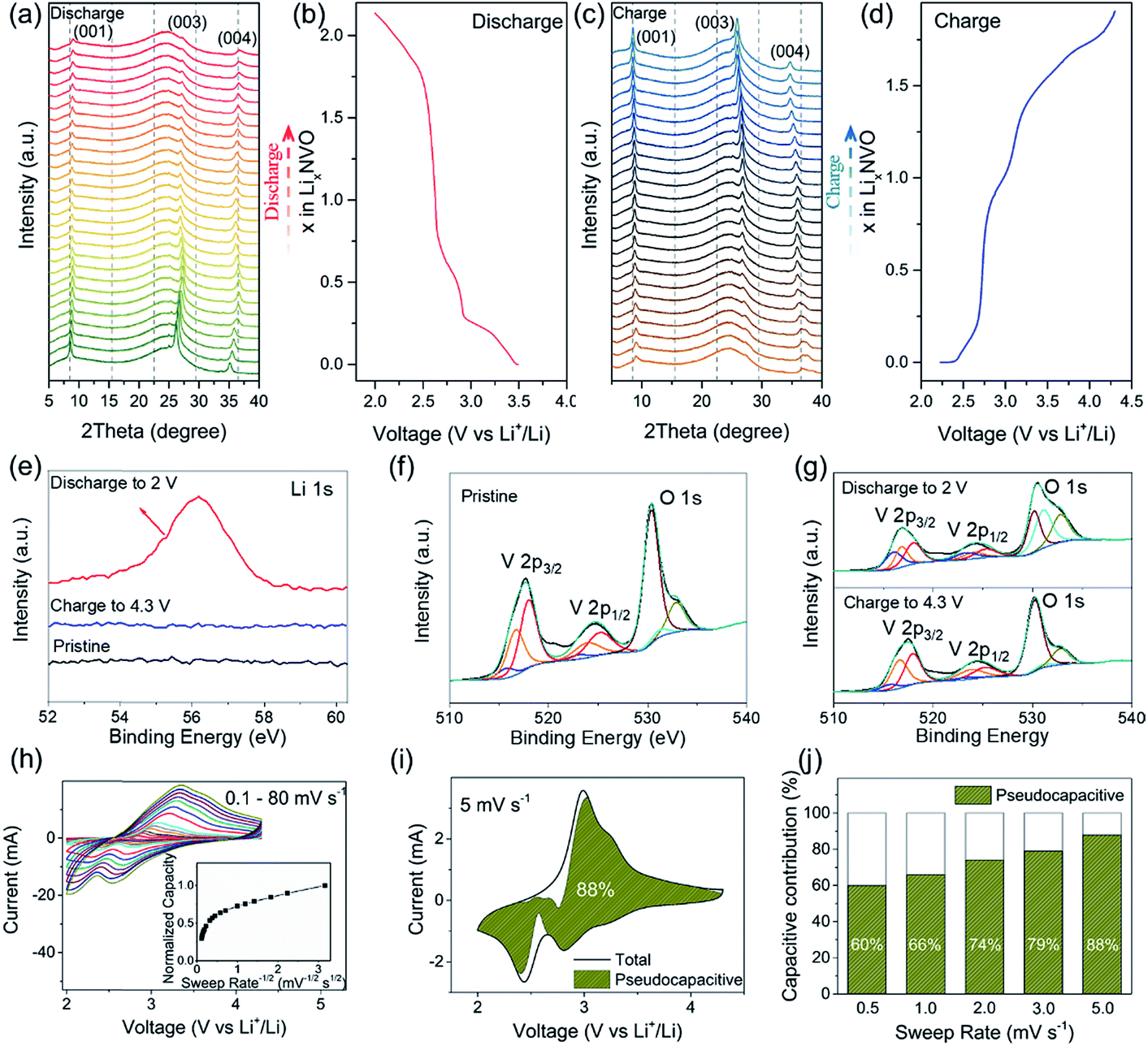 | ||
| Fig. 3 Operando XRD patterns collected for the NVO@rGO electrodes during (a) the first discharge and (c) the first charge, and the corresponding galvanostatic voltage profiles for the (b) discharge and (d) charge processes. (e) High-resolution XPS spectra of Li1s for the discharged, the charged and the pristine NVO@rGO electrodes. V2p and O1s regions of the XPS spectra of (f) the pristine, (g) the discharged and the charged NVO@rGO electrodes. (h) CV curves of NVO@rGO at 0.1–80 mV s−1 (inset: the plot of normalized capacity vs. sweep rate−1/2). (i) Voltammetric response of NVO@rGO separated into the total current (solid line) and the current contributed by pseudocapacitance (shaded area) at a sweep rate of 5 mV s−1. (j) Contributions of the non-diffusion limited processes to the total capacity of NVO@rGO at various sweep rates. | ||
To further explore the Li+ storage mechanism in NVO@rGO, ex situ high-resolution XPS spectra of the pristine electrodes, the discharged electrodes, and the charged electrodes were collected. As shown in Fig. 3e, no Li signal is observed in the pristine NVO cathode as expected. However, the Li1s peak emerges when the NVO electrodes were discharged to 2 V, demonstrating the intercalation of Li+ into the NVO structure. After charging back to 4.3 V, the Li signal nearly disappears again, clearly indicating that most of the intercalated Li ions are extracted during charging. The V2p spectra of the discharged and charged electrodes are shown in Fig. 3g. Relative to the original V2p state of the pristine electrodes (see Fig. 3f), the intensity of V3+ strongly increased upon discharging, reflecting the reduction of V during Li+ intercalation. After recharging to 4.3 V, the V3+ signal intensity decreases again and the pristine V2p spectrum is almost completely regenerated, indicating that the initial chemical state of V is restored upon extraction of Li+. The XPS results also underline the reversibility of the electrochemical processes, which is in good agreement with the high coulombic efficiency of the cathode as discussed above.
Cyclic voltammetry (CV) measurements at various sweep rates were carried out (Fig. 3h) to evaluate the electrochemical kinetics of the cathode material during charging/discharging. In the inset of Fig. 3h, the plot of normalized capacity (Q) vs. sweep rate−1/2 (v−1/2) shows that the capacity does not vary significantly with the sweep rates increasing from 0.1 to 5 mV s−1, indicating that the non-diffusion limited processes are independent of the scan rate. The linear decrease of Q with v−1/2 observed at a sweep rate >10 mV s−1 reflects a rate-limited diffusion process. Similar observations were reported for other intercalation pseudocapacitive materials, e.g., T-Nb2O5 (ref. 49) and partially bonded graphene–TiO2.61 Possible reasons for the rate-limited process at high sweep rates are the increase of the ohmic contribution and/or the diffusion constraints/limitations.49 The total contribution of the non-diffusion limited processes at a certain sweep rate can be quantified based on eqn (1):
| i = k1v + k2v1/2 | (1) |
The electrochemical performance of N-doped reduced graphene oxide (N-rGO) foam is also first tested in the half-cell configuration within the voltage range of 0.01–2 V. As shown in Fig. S16,† the N-rGO foam shows a high rate capability with a capacity decrease of only 29.1% at 4.5 A g−1 compared with that at 1.05 A g−1. In the long-cycling tests, a capacity of 160 mA h g−1 is achieved at 4.5 A g−1 after 500 cycles, with an average coulombic efficiency of close to 100% (Fig. S17†). When cycled at an even higher current rate of 7.5 A g−1, the N-rGO anode still delivers a capacity of 131 mA h g−1 after 500 cycles (see Fig. S18†), further supporting its high rate capability and cycling stability. The Li+ storage kinetics of the N-rGO is also investigated by CV measurements. Fig. S19† shows the CV curves at various sweep rates ranging from 0.1–5 mV s−1. Based on eqn (1), the relative contributions from pseudocapacitive processes are accessible for different sweep rates. For instance, as shown in Fig. S20a,† 85% of the total stored charge can be attributed to surface redox pseudocapacitive effects at 5 mV s−1. The pseudocapacitive contributions at various rates are illustrated in Fig. S20b.† The results clearly indicate the dominant pseudocapacitive charge storage mechanism in the N-rGO anode.
2.3. Full-cell performance
Based on the high performance of the NVO@rGO with fast Li-ion storage and the surface redox pseudocapacitive N-rGO in half cells, prototype Li-ion storage full cells were also fabricated by paring them as the cathode and the anode, respectively (see the schematic in Fig. 4a). Note that to match the capacity between the anode and the cathode and to improve the working voltage, both the electrodes went through an activation procedure during which the anode and the cathode were pre-cycled for 5 times at 1C and then discharged to 0.01 V and charged to 4.3 V, respectively. Fig. 4b presents the typical galvanostatic charge/discharge profiles of the NVO@rGO‖N-rGO full cells at various current rates. The curves exhibit a nearly linear correlation between voltage and time at high current densities, indicating the dominating capacitive behaviour of the full cells during fast charging/discharging. The deviation from the ideal linear slope at low current rates is due to a combined faradaic and non-faradaic charge storage mechanism.62,63 As shown in Fig. 4c, the energy density of the NVO@rGO‖N-rGO full cells reaches 168 W h kg−1 at a specific current of 0.1 A g−1. With the current density increased to 5 A g−1, the full cells still deliver a remarkable energy density of 63 W h kg−1. Even at an extremely high rate of 10 A g−1, an energy density of 32 W h kg−1 is still obtained, indicating the outstanding rate performance of the as-developed full cells. Besides the rate capability, the NVO@rGO‖N-rGO full cells also exhibit good cycling stability. Fig. 4d presents the cycling performance of the full cells tested at a current rate of 5 A g−1. When cycled at such a high current density, the full cell still achieves 55 W h kg−1 with an energy retention of 87% over 5000 cycles. The remarkable cycling performance of the full cells can be ascribed to an ideally balanced kinetics between NVO@rGO and the N-rGO foam. To the best of our knowledge, such a lifespan is among the best ever reported for Li-ion storage full cells (Table S2†).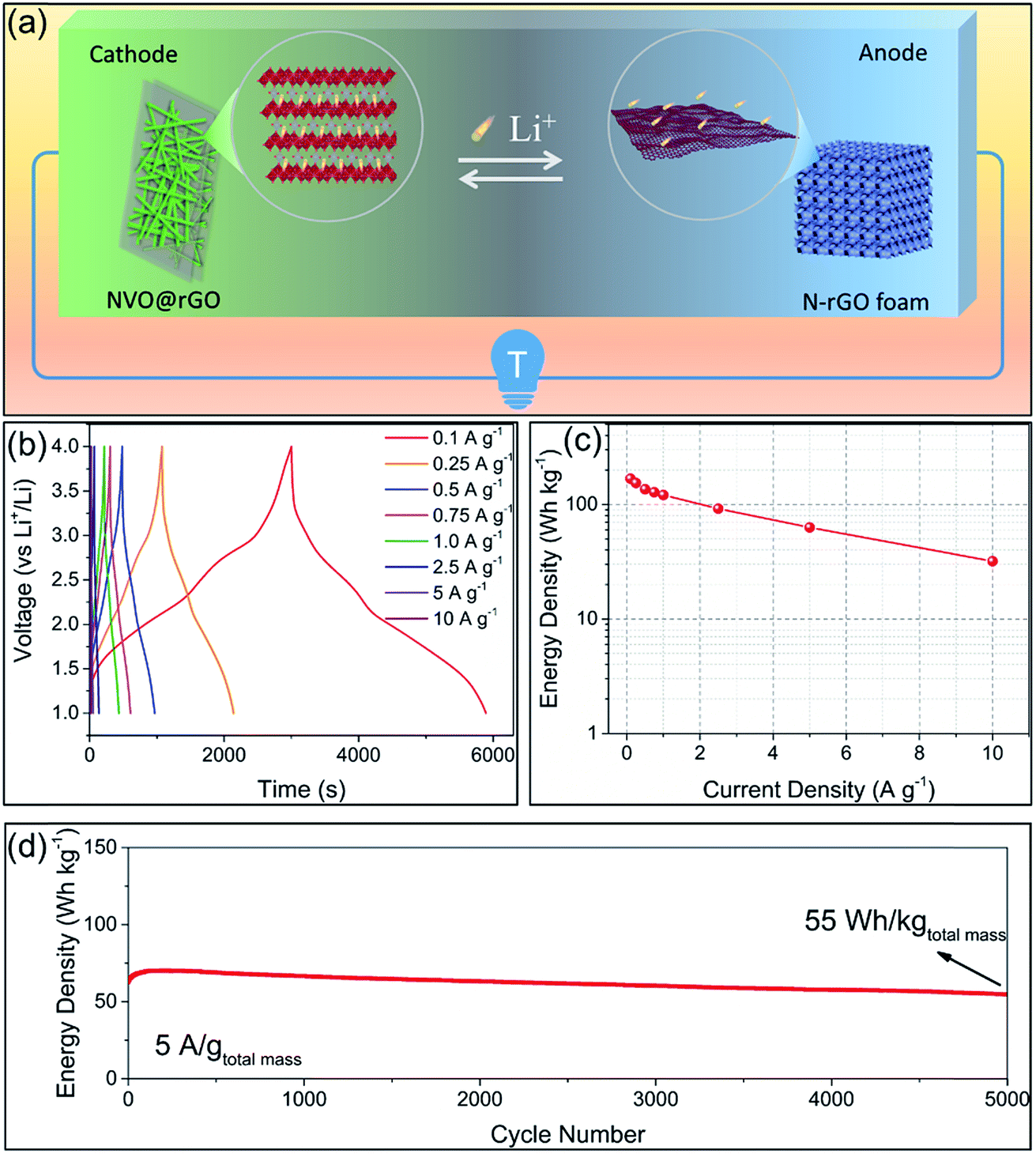 | ||
| Fig. 4 (a) Schematic of the LIB full cell combining the fast-Li-ion-storage cathode of NVO@rGO with the surface redox pseudocapacitive anode of the N-rGO foam. (b) Typical charge/discharge voltage profiles of the NVO@rGO‖N-rGO full cells as a function of time. (c) Energy density vs. rate plot of the NVO@rGO‖N-rGO full cell devices. (d) Long-term cycling performance of the NVO@rGO‖N-rGO full cells at a current rate of 5 A g−1. The energy densities and the current rates are all based on the total mass of active materials in the cathode and the anode. | ||
The performance of our full cell is compared with that of some reported high-energy LIB full cells and high-power Li-HSCs, which represent the two typical Li-ion storage devices. As shown in the Ragone plots in Fig. 5, the NVO@rGO‖N-rGO full cells are not only able to deliver an energy density (168 W h kg−1 at 209 W kg−1) comparable to that of high-performance LIB full cells (e.g., graphene‖LiFePO4,64 Sn–C‖Li[Ni0.45Co0.1Mn1.45]O4,65 Si/graphene‖LiNi1/3Mn1/3Co1/3O2,66 graphene‖LiNi0.5Mn1.5O4,67 and Li4Ti5O12–Li2Ti3O7‖LiFePO4 (ref. 68)), but additionally our full cell device exhibits an impressively high power-output (17194 W kg−1 with 32 W h kg−1 retained), which outperforms some recently reported most advanced Li-HSCs (e.g., pillared Ti3C2 MXene‖AC,25 Li3VO4‖AC,69 NixFeyOz@rGO aerogel‖AC,11 m-Nb2O5/C‖AC,70 and TiO2@rGO‖AC62). The successful combination of high energy density and high power density within one device can be ascribed to the fast Li-ion storage of the two materials used as the cathode and anode, both able to drive high rate and charge storage capability. The well-balanced kinetics between the two electrodes thus leads to the high power output of the full cells. Compared with AC, the cathode of NVO@rGO shows much higher charge storage capability, which finally provides the high energy density of the full cells.
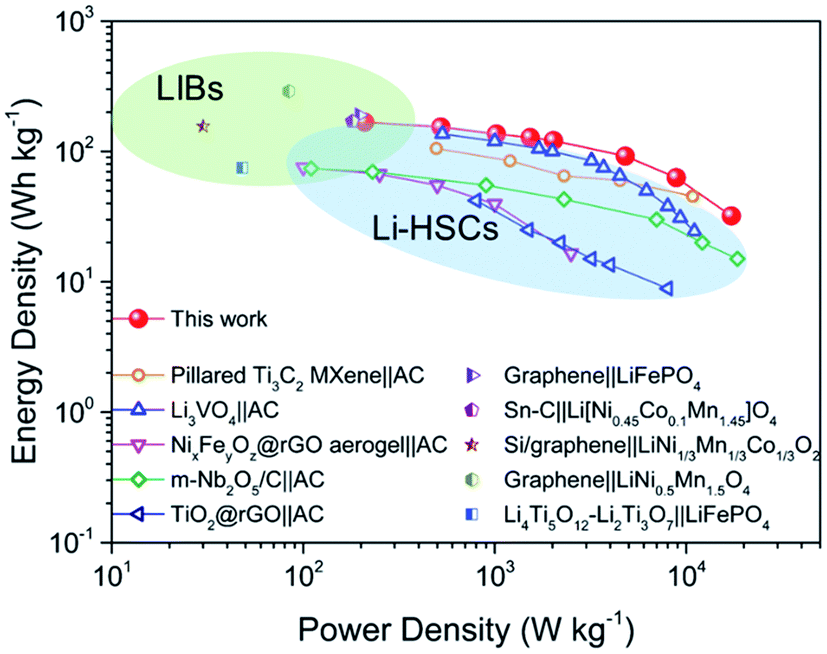 | ||
| Fig. 5 Ragone plots (energy density vs. power density) of NVO@rGO‖N-rGO LIB full cells developed in this work in comparison to some advanced Li-HSCs and LIB full cells reported in the literature. | ||
3 Conclusions
In this study, we synthesized Ni0.25V2O5·nH2O/reduced graphene oxide, which can be used as a cathode material with fast Li-ion storage in LIBs. Operando XRD, ex situ XPS and CV analysis shed light on the electrochemical mechanism, offering possible reasons for the excellent performance of this material in half-cell configurations. To take full advantage of its promising electrochemical properties, the cathode with fast Li-ion storage was paired with a surface redox pseudocapacitive anode composed of N-rGO foam, leading to a Li-ion storage full cell with high rate capability (63 W h kg−1 at 5 A g−1 and 32 W h kg−1 at 10 A g−1), long-term cyclability (an energy retention of 87% over 5000 cycles at 5 A g−1), and high energy density (168 W h kg−1 at 209 W kg−1). This study introduced a new type of LIB cathode with no diffusion limitations, which may promote the development of fast-charging Li-ion storage devices. Furthermore, the investigation of full cells in this work also provides an efficient method to achieve a high power output without sacrificing the high energy density, i.e., it offers a concept to bridge the gap between LIBs and Li-HSCs, or even Na-ion/K-ion batteries and Na-ion/K-ion hybrid supercapacitors, through combining materials with fast Li-ion storage in both positive and negative electrodes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge ETH Zurich (ETH Research Grant ETH-13 16-1) and the China Scholarship Council for financial support. L. Pan acknowledges a fellowship from the Office of China Postdoctoral Council (No. 32 Document of OCPC, 2017). We thank the following colleagues for giving us access to their research equipment: G. Cossu and Prof. A. Rossi for XPS, Prof. A. Studart for SEM, Prof. P. Gambardella for AFM (and J. Feng for the help with the measurements) and Prof. J. Vermant for TGA. The authors also thank the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zurich for providing the electron microscopy facilities.Notes and references
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed.
- W. Lv, Z. Li, Y. Deng, Q.-H. Yang and F. Kang, Energy Storage Materials, 2016, 2, 107–138 CrossRef.
- S. Lou, X. Cheng, Y. Zhao, A. Lushington, J. Gao, Q. Li, P. Zuo, B. Wang, Y. Gao and Y. Ma, Nano Energy, 2017, 34, 15–25 CrossRef CAS.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- B. Wang, W. Al Abdulla, D. Wang and X. Zhao, Energy Environ. Sci., 2015, 8, 869–875 RSC.
- Y. T. Liu, P. Zhang, N. Sun, B. Anasori, Q. Z. Zhu, H. Liu, Y. Gogotsi and B. Xu, Adv. Mater., 2018, 30, 1707334 CrossRef PubMed.
- H. Li, L. Jiang, Q. Feng, Z. Huang, H. Zhou, Y. Gong, Z. Hou, W. Yang, C. Fu and Y. Kuang, Energy Storage Materials, 2019, 17, 157–166 CrossRef.
- T. Yang, Y. Liu, D. Yang, B. Deng, Z. Huang, C. D. Ling, H. Liu, G. Wang, Z. Guo and R. Zheng, Energy Storage Materials, 2019, 17, 374–384 CrossRef.
- L. Pan, X. D. Zhu, X. M. Xie and Y. T. Liu, Adv. Funct. Mater., 2015, 25, 3341–3350 CrossRef CAS.
- H. Sun, L. Mei, J. Liang, Z. Zhao, C. Lee, H. Fei, M. Ding, J. Lau, M. Li and C. Wang, Science, 2017, 356, 599–604 CrossRef CAS PubMed.
- H. Huang, X. Wang, E. Tervoort, G. Zeng, T. Liu, X. Chen, A. Sologubenko and M. Niederberger, ACS Nano, 2018, 12, 2753–2763 CrossRef CAS PubMed.
- H. Liu, W. Li, D. Shen, D. Zhao and G. Wang, J. Am. Chem. Soc., 2015, 137, 13161–13166 CrossRef CAS PubMed.
- L. Lin, X. Xu, C. Chu, M. K. Majeed and J. Yang, Angew. Chem., Int. Ed., 2016, 55, 14063–14066 CrossRef CAS PubMed.
- B.-S. Lee, H.-S. Yang, K. H. Lee, S. Han and W.-R. Yu, Energy Storage Materials, 2019, 17, 62–69 CrossRef.
- M. Gu, I. Belharouak, J. Zheng, H. Wu, J. Xiao, A. Genc, K. Amine, S. Thevuthasan, D. R. Baer and J.-G. Zhang, ACS Nano, 2012, 7, 760–767 CrossRef PubMed.
- S. K. Jung, H. Gwon, J. Hong, K. Y. Park, D. H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2014, 4, 1300787 CrossRef.
- J. Kim, H. Ma, H. Cha, H. Lee, J. Sung, M. Seo, P. Oh, M. Park and J. Cho, Energy Environ. Sci., 2018, 11, 1449–1459 RSC.
- P. Yan, J. Zheng, J.-G. Zhang and C. Wang, Nano Lett., 2017, 17, 3946–3951 CrossRef CAS PubMed.
- G. G. Amatucci, F. Badway, A. Du Pasquier and T. Zheng, J. Electrochem. Soc., 2001, 148, A930–A939 CrossRef CAS.
- Y.-G. Wang and Y.-Y. Xia, Electrochem. Commun., 2005, 7, 1138–1142 CrossRef CAS.
- S. Nohara, T. Asahina, H. Wada, N. Furukawa, H. Inoue, N. Sugoh, H. Iwasaki and C. Iwakura, J. Power Sources, 2006, 157, 605–609 CrossRef CAS.
- V. Khomenko, E. Raymundo-Piñero and F. Béguin, J. Power Sources, 2008, 177, 643–651 CrossRef CAS.
- Y. Ma, H. Chang, M. Zhang and Y. Chen, Adv. Mater., 2015, 27, 5296–5308 CrossRef CAS PubMed.
- E. Lim, C. Jo, H. Kim, M.-H. Kim, Y. Mun, J. Chun, Y. Ye, J. Hwang, K.-S. Ha and K. C. Roh, ACS Nano, 2015, 9, 7497–7505 CrossRef CAS PubMed.
- J. Luo, W. Zhang, H. Yuan, C. Jin, L. Zhang, H. Huang, C. Liang, Y. Xia, J. Zhang and Y. Gan, ACS Nano, 2017, 11, 2459–2469 CrossRef CAS PubMed.
- Z. Chen, V. Augustyn, J. Wen, Y. Zhang, M. Shen, B. Dunn and Y. Lu, Adv. Mater., 2011, 23, 791–795 CrossRef CAS PubMed.
- X. Wang, G. Li, Z. Chen, V. Augustyn, X. Ma, G. Wang, B. Dunn and Y. Lu, Adv. Energy Mater., 2011, 1, 1089–1093 CrossRef CAS.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS.
- J. Gamby, P. Taberna, P. Simon, J. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109–116 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz and M. Thommes, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- P. Hu, T. Zhu, X. Wang, X. Wei, M. Yan, J. Li, W. Luo, W. Yang, W. Zhang and L. Zhou, Nano Lett., 2018, 18, 1758–1763 CrossRef CAS PubMed.
- C. Zhang, H. Song, C. Liu, Y. Liu, C. Zhang, X. Nan and G. Cao, Adv. Funct. Mater., 2015, 25, 3497–3504 CrossRef CAS.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed.
- H. Huang, D. Kundu, R. Yan, E. Tervoort, X. Chen, L. Pan, M. Oschatz, M. Antonietti and M. Niederberger, Adv. Energy Mater., 2018, 8, 1802800 CrossRef.
- H. Huang, T. Tian, L. Pan, X. Chen, E. Tervoort, C.-J. Shih and M. Niederberger, J. Mater. Chem. A, 2019, 7, 16109–16116 RSC.
- S.-I. Nishimura, M. Nakamura, R. Natsui and A. Yamada, J. Am. Chem. Soc., 2010, 132, 13596–13597 CrossRef CAS PubMed.
- J. Ahn, J. H. Kim, B. W. Cho, K. Y. Chung, S. Kim, J. W. Choi and S. H. Oh, Nano Lett., 2017, 17, 7869–7877 CrossRef CAS PubMed.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- Y. Li, C. Wu, Y. Bai, L. Liu, H. Wang, F. Wu, N. Zhang and Y. Zou, ACS Appl. Mater. Interfaces, 2016, 8, 18832–18840 CrossRef CAS PubMed.
- C. Zhang, S.-H. Park, S. E. O'brien, A. Seral-Ascaso, M. Liang, D. Hanlon, D. Krishnan, A. Crossley, N. Mcevoy, J. N. Coleman and V. Nicolosi, Nano Energy, 2017, 39, 151–161 CrossRef CAS.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. Tarascon, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. Chen, Chem. Soc. Rev., 2015, 44, 5926–5940 RSC.
- L. Jin, G. Zeng, H. Wu, M. Niederberger and M. Morbidelli, J. Mater. Chem. A, 2016, 4, 11481–11490 RSC.
- W. Cheng, F. Rechberger, D. Primc and M. Niederberger, Nanoscale, 2015, 7, 13898–13906 RSC.
- L. Pan, P. Gao, E. Tervoort, A. M. Tartakovsky and M. Niederberger, J. Mater. Chem. A, 2018, 6, 18551–18560 RSC.
- L. Pan, H. Huang, M. Zhong and M. Niederberger, Energy Storage Materials, 2019, 16, 519–526 CrossRef.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- K. J. Griffith, K. M. Wiaderek, G. Cibin, L. E. Marbella and C. P. Grey, Nature, 2018, 559, 556–563 CrossRef CAS PubMed.
- Y. Oka, T. Yao and N. Yamamoto, J. Solid State Chem., 1997, 132, 323–329 CrossRef CAS.
- Y. Oka, O. Tamada, T. Yao and N. Yamamoto, J. Solid State Chem., 1996, 126, 65–73 CrossRef CAS.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- C. Zhang, H. Song, C. Liu, Y. Liu, C. Zhang, X. Nan and G. Cao, Adv. Funct. Mater., 2015, 25, 3497–3504 CrossRef CAS.
- S. Zhang, K. Xu and T. Jow, Electrochim. Acta, 2004, 49, 1057–1061 CrossRef CAS.
- Z. Le, F. Liu, P. Nie, X. Li, X. Liu, Z. Bian, G. Chen, H. B. Wu and Y. Lu, ACS Nano, 2017, 11, 2952–2960 CrossRef CAS PubMed.
- G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS.
- C. Chen, Y. Wen, X. Hu, X. Ji, M. Yan, L. Mai, P. Hu, B. Shan and Y. Huang, Nat. Commun., 2015, 6, 6929 CrossRef CAS PubMed.
- H. Kim, M. Y. Cho, M. H. Kim, K. Y. Park, H. Gwon, Y. Lee, K. C. Roh and K. Kang, Adv. Energy Mater., 2013, 3, 1500–1506 CrossRef CAS.
- E. Lim, C. Jo, M. S. Kim, M. H. Kim, J. Chun, H. Kim, J. Park, K. C. Roh, K. Kang and S. Yoon, Adv. Funct. Mater., 2016, 26, 3711–3719 CrossRef CAS.
- J. Hassoun, F. Bonaccorso, M. Agostini, M. Angelucci, M. G. Betti, R. Cingolani, M. Gemmi, C. Mariani, S. Panero and V. Pellegrini, Nano Lett., 2014, 14, 4901–4906 CrossRef CAS PubMed.
- J. Hassoun, K.-S. Lee, Y.-K. Sun and B. Scrosati, J. Am. Chem. Soc., 2011, 133, 3139–3143 CrossRef CAS PubMed.
- L. Ji, H. Zheng, A. Ismach, Z. Tan, S. Xun, E. Lin, V. Battaglia, V. Srinivasan and Y. Zhang, Nano Energy, 2012, 1, 164–171 CrossRef CAS.
- O. Vargas, A. Caballero, J. Morales, G. A. Elia, B. Scrosati and J. Hassoun, Phys. Chem. Chem. Phys., 2013, 15, 20444–20446 RSC.
- G. N. Zhu, L. Chen, Y. G. Wang, C. X. Wang, R. C. Che and Y. Y. Xia, Adv. Funct. Mater., 2013, 23, 640–647 CrossRef CAS.
- L. Shen, H. Lv, S. Chen, P. Kopold, P. A. Van Aken, X. Wu, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1700142 CrossRef PubMed.
- E. Lim, H. Kim, C. Jo, J. Chun, K. Ku, S. Kim, H. I. Lee, I.-S. Nam, S. Yoon and K. Kang, ACS Nano, 2014, 8, 8968–8978 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Fig. S1–S21, and Tables S1 and S2. See DOI: 10.1039/c9ta08000g |
| This journal is © The Royal Society of Chemistry 2019 |