
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light-mediated, additive-free, and open-to-air controlled radical polymerization of acrylates and acrylamides†
Jessica R.
Lamb
 ,
K. Peter
Qin
and
Jeremiah A.
Johnson
*
,
K. Peter
Qin
and
Jeremiah A.
Johnson
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: jaj2109@mit.edu
First published on 8th February 2019
Abstract
Oxygen tolerance in controlled radical polymerizations has been an active field of study in recent years. Herein, we report a photocontrolled, additive-free iniferter polymerization that operates in completely open vials utilizing the “polymerizing through oxygen” mechanism. Trithiocarbonates are directly activated with high intensity 450 nm light to produce narrowly dispersed (Mw/Mn = 1.1–1.6) polyacrylates and polyacrylamides. Living behavior is demonstrated through chain extension, block copolymer synthesis, and control over molecular weight through varying the monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :iniferter ratio. A slight increase in induction period is observed for the open vial polymerization compared to the air-free reaction, but polymers with similar Mn and Mw/Mn values are produced after 30–60 minutes of irradiation. This system will provide a convenient platform for living additive manufacturing because of its fast reaction time, air tolerance, wide monomer scope, and lack of any additives beyond the monomer, iniferter, and DMSO solvent.
:iniferter ratio. A slight increase in induction period is observed for the open vial polymerization compared to the air-free reaction, but polymers with similar Mn and Mw/Mn values are produced after 30–60 minutes of irradiation. This system will provide a convenient platform for living additive manufacturing because of its fast reaction time, air tolerance, wide monomer scope, and lack of any additives beyond the monomer, iniferter, and DMSO solvent.
Introduction
Controlled radical polymerization (CRP) techniques have transformed macromolecular synthesis by enabling access to polymers with controlled molecular weights, narrow molecular weight distributions, and well-defined architectures.1–3 Photocontrolled CRP (photo-CRP) affords the additional benefits of spatiotemporal control by using light as a low cost external stimulus. Light-induced free radical polymerizations have already found commercial use in photolithography, additive manufacturing, and for the synthesis of coatings, adhesives, and resins; however, most of these systems do not have “living” characteristics, such as chain-end fidelity and the ability to reinitiate over several cycles.4Our group recently combined the concepts of light-induced additive manufacturing5 and photo-CRP6 to achieve “living additive manufacturing” (LAM) in which a dormant parent polymer network can be subsequently altered to produce complex, responsive daughter materials.7 One downside of our reported LAM system – and most radical polymerizations – is oxygen inhibition due to trapping of propagating carbon-centered radicals by oxygen, which produces peroxy radicals.8,9 This deactivation is generally avoided by performing the polymerizations under inert conditions; however, degassing techniques such as freeze–pump–thaw cycles, sparging, or performing experiments in a glovebox can be impractical for certain applications.
We set out to develop a second-generation LAM system that did not require any degassing procedures and could ideally be done fully open to air (to mimic a benchtop 3D printer) on a reasonably fast time scale. Before we can achieve this goal, an efficient oxygen tolerant photo-CRP is required. Oxygen tolerant CRP reactions have garnered significant interest in recent years (Fig. 1A–C).10 In many cases, a sacrificial compound is added to scrub the oxygen or regenerate a catalyst that can react with and effectively remove oxygen.11,12 In other cases, no specific scrubbing mechanism is employed; oxygen simply reacts with excess radicals in the system, referred to as “polymerizing through oxygen” (Fig. 2, red path).13,14
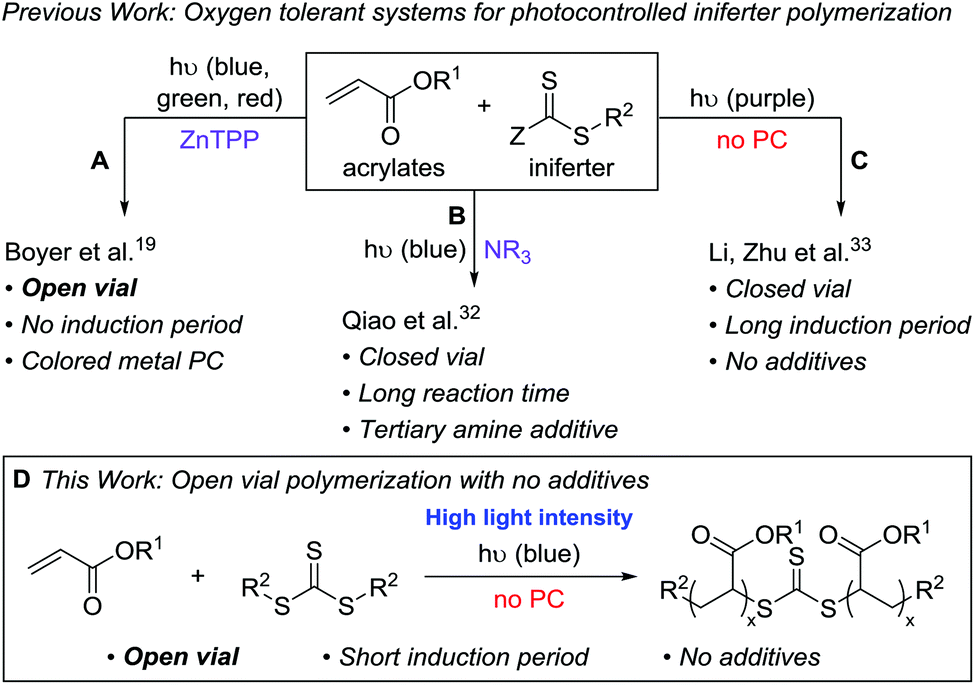 | ||
| Fig. 1 Progression of oxygen tolerant CRP using visible light and thiocarbonylthio iniferters: (A) Photocatalyst-mediated CRP in a completely open vial using a metal catalyst. (B) Tertiary amines as sacrificial reductants in photocatalyst-free CRP. (C) Additive-free CRP in a closed vial. (D) Additive-free, open vial CRP utilizing high light intensity. | ||
 | ||
| Fig. 2 Mechanism of trithiocarbonates (TTC)-mediated iniferter polymerization and structures of iniferters used in this study. | ||
For photo-CRP utilizing common RAFT agents as iniferters, the current best systems for achieving oxygen tolerance involve photoredox catalysts – such as Ir(ppy)3,15,16 Ru(bpy)3Cl2,17 eosin y,18 and zinc tetraphenylporphyrin (ZnTPP)19,20 – converting triplet oxygen into a reactive species (such as singlet oxygen) that can be trapped. These techniques rely on highly colored dyes or metal catalysts, which can be difficult to remove from the final materials. In addition, most of these systems still operate in a closed vial such that the amount of oxygen is finite; it is generally proposed that the oxygen in both the solution and the headspace is scrubbed during the long induction period before photopolymerization can begin. A few reports describe exceptions where polymerizations in completely open vials are possible, including methyl acrylate polymerization in DMSO using ZnTPP19 (Fig. 1A) and acrylic acid polymerization in water using a combination of fluorescein and ascorbic acid.21
To avoid additional purification to remove highly colored and/or metal photocatalysts, significant work has been done to directly photolyze thiocarbonylthio compounds, such as xanthates and trithiocarbonates (TTCs), for photo-CRP (Fig. 2, black path).22–24 Initially, UV light was utilized due to the large π to π* transition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond. This approach can lead to irreversible decomposition of the iniferter and the self-initiation of monomers.22,25,26 Recently, increased control over such iniferter polymerizations has been gained by using low intensity long wavelength UV/violet light27,28 and visible (blue or green) light,29–31 the latter operating via the spin-forbidden n to π* pathway. While these systems resulted in well-controlled polymerizations, many are quite slow due to the low extinction coefficient of the n to π* transition and most were done under inert atmosphere.
S bond. This approach can lead to irreversible decomposition of the iniferter and the self-initiation of monomers.22,25,26 Recently, increased control over such iniferter polymerizations has been gained by using low intensity long wavelength UV/violet light27,28 and visible (blue or green) light,29–31 the latter operating via the spin-forbidden n to π* pathway. While these systems resulted in well-controlled polymerizations, many are quite slow due to the low extinction coefficient of the n to π* transition and most were done under inert atmosphere.
Qiao and coworkers achieved oxygen tolerance without a photocatalyst by using tertiary amines as sacrificial reductants for both oxygen scrubbing and photoredox polymerization (Fig. 1B).32 While this system demonstrated excellent control over molecular weight and was effective for the synthesis of block copolymers through chain extension, it was performed in a closed vial and required multiple hours to days of irradiation to yield high molecular weight polymers. Li, Zhu, and coworkers reported a very fast (5–20 min) polymerization of n-butyl acrylate using 390 nm light under air-free conditions; however, when performed in a non-degassed, sealed vessel, the polymerization had a 110 min induction period (Fig. 1C).33
Despite these recent advances, our goals for LAM would be ideally served by a fast photo-CRP that displays living characteristics and can operate under open conditions without highly colored photocatalysts. Here, we report an open-vial polymerization with direct iniferter activation using blue (450 nm) light on a reasonably fast timescale (1 h). This additive-free photo-CRP provides polymers with narrow molecular weight distributions, linear molecular weight growth with conversion, and chain end fidelity, which allows for chain extension (Fig. 1D).
Results and discussion
Optimization and monomer scope
Inspired by the open vial polymerization achieved by Boyer and coworkers using ZnTPP,19 we began to pursue open to air polymerizations using symmetrical TTC 1 (Fig. 2) with various photocatalysts in our custom-built photoreactor (see ESI,† for photoreactor details).34 A serendipitous discovery was made when we ran control experiments for the open vial polymerization under red (625 nm), green (530 nm) and blue (450 nm) light (Table 1, entries 1–3): under 450 nm light in the absence of a photocatalyst, methyl acrylate (MA) could be polymerized in a completely open vial in 1 hour (entry 3). The unstirred polymerization in DMSO reached high conversion and yielded polymer products with a narrow molar mass distribution. In contrast to Boyer's system, in which the oxygen tolerance is proposed to originate from the ZnTPP,20 we hypothesized that our system relies on a polymerizing through oxygen mechanism, which could explain the presence of a small low molecular weight tail in the GPC trace (Fig. S7,† see the mechanism section for further discussion on the origin of oxygen tolerance in this system).| Entry | λ (nm) | MA:DMSO (v:v, μl) |
Conv.b (%) | M n,GPC (kg mol−1) | M w,GPC (kg mol−1) | M w/Mn |
|---|---|---|---|---|---|---|
|
a Conditions: MA:TTC (200:1), 0.5 ml scale, 60 min (entries 1–3), 45 min (entries 4–9), uncapped 1 dram vial, no stirring.
b Conversion is uncorrected for monomer evaporation.
|
||||||
| 1 | 625 | 250:250 |
<1 | — | — | — |
| 2 | 530 | 250:250 |
<1 | — | — | — |
| 3 | 450 | 250:250 |
98 | 13.25 | 15.03 | 1.13 |
| 4 | 450 | 500:0 |
55 | 7.45 | 9.30 | 1.25 |
| 5 | 450 | 400:100 |
74 | 12.78 | 15.23 | 1.19 |
| 6 | 450 | 325:175 |
85 | 11.95 | 14.78 | 1.24 |
| 7 | 450 | 250:250 |
83 | 13.75 | 15.85 | 1.15 |
| 8 | 450 | 175:325 |
81 | 12.37 | 15.51 | 1.26 |
| 9 | 450 | 100:400 |
31 | 5.84 | 8.58 | 1.47 |
We began to probe this reaction by varying the monomer concentration at a fixed MA:TTC ratio of 200:1 (Table 1, entries 4–9). Not only does the concentration affect the rate of polymerization, but the iniferter concentration also dictates the radical flux, which is known to require a certain threshold to successfully employ the polymerizing through oxygen strategy.10,13 Bulk polymerization (entry 4) and dilute polymerization (∼2.2 M, entry 9) both resulted in lower molar masses and bimodal molar mass distributions. All of the intermediate concentrations (entries 5–8) resulted in similar Mw values, with the Mn and dispersity differences arising from the slight changes in the low molecular weight tail (see Fig. S8† for GPC traces). A 1:1 ratio between monomer and solvent was used going forward. Other solvents were tested (Table S2,† entries 10–14), but toluene, ethyl acetate, acetone, and acetonitrile all resulted in no polymerization under standard conditions. N,N-Dimethylformamide did yield some polymer (Mw/Mn = 1.19) after 1 h of irradiation, but the molar mass was significantly reduced (Mn = 3.77 kg mol−1, Table S2,† entry 13). Solvent mixtures between DMSO and MeCN (Table S2,† entries 15–17) could be used successfully, but the polymer molecular weight quickly drops if the DMSO fraction is below ∼80%.
The reaction was then run on different scales (0.2–0.6 ml, Table S2, entries 18–22 and Fig. S9†) in a 1 dram vial which revealed that a reaction volume of at least 0.4–0.5 ml is needed for this setup to achieve well-controlled and high molecular weight polymers (vide infra). We chose 0.5 ml as the standard reaction volume for MA. Next, the effect of the iniferter was probed by using the commercially available TTCs 2 and 3 (Table S2,† entries 23 and 24). 2 is similarly symmetrical to 1, but is terminated by two carboxylic acids instead of benzyl esters. Under standard conditions, 2 yields polymer with slightly lower molar mass (Mn = 12.22 kg mol−1) and higher dispersity (1.26) (entry 23). The unsymmetrical TTC 3 was less effective, providing 6.51 kg mol−1 polymer with a dispersity of 1.36 in 1 hour (entry 24). These data indicate that further optimization will be needed when changing the iniferter for open vial polymerizations, a potential drawback when compared to oxygen-tolerant systems utilizing a photocatalyst.19
Finally, the efficacy of the polymerization was shown to be strongly dependent on the use of high light intensity (Fig. 3A and Table S2,† entries 25–29); however, the effect was somewhat dependent on the monomer investigated (Fig. S10†).34 We hypothesize that the high light intensity is necessary both to achieve high radical flux for the polymerizing through oxygen mechanism13 and for the accelerated polymerization rate35 compared to many previous systems29,30 that have directly activated the n to π* transition of the iniferter.
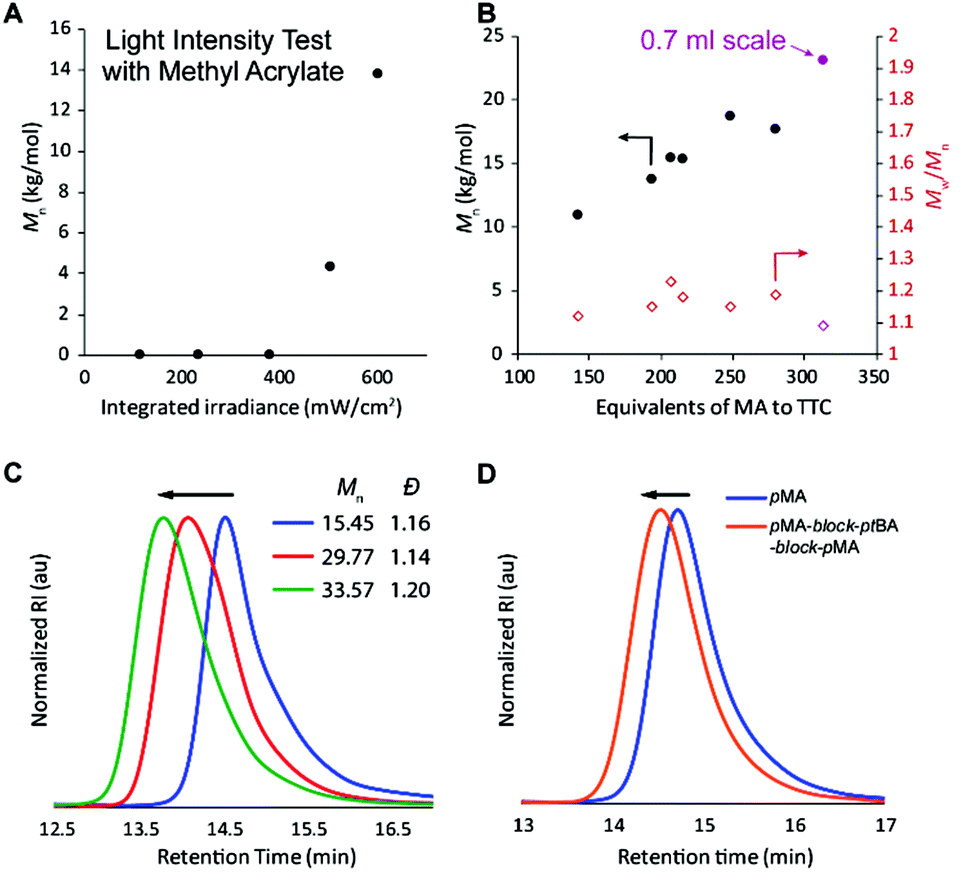 | ||
| Fig. 3 (A) Molecular weight dependence on light intensity. (B) Molecular weight and dispersity changes with MA:TTC ratio. (C) GPC traces of chain extension from precipitated pMA to form pseudoblocks. (D) GPC traces of block copolymer synthesis. | ||
After understanding which variables affect the polymerization, different monomer:TTC ratios were used to demonstrate that molar mass increases approximately linearly until ca. 250:1, at which point the molar mass achieved in 1 hour levels off (Fig. 3B and Table S3,† entries 1–6). We initially thought that perhaps the polymerization rate was limiting the molar mass that could be achieved within 60 minutes of irradiation, but increasing the polymerization time to 90 or 120 minutes resulted in bimodal molar mass distributions and slightly lower Mn (Table S3,† entries 7 and 8). We attribute this degradation to chains near the solution-air interface reacting with oxygen (red path, Fig. 2), resulting in chains with half of the molar mass compared to the original polymer. Increasing the reaction volume to 0.7 ml led to an increased molar mass (purple point in Fig. 3B and Table S3,† entry 9), though all reactions with high monomer:iniferter ratios (∼300:1) were fairly inconsistent and sometimes resulted in bimodal distributions. The inconsistency is likely due to the low TTC concentration, which is known to reduce control in TTC-mediated iniferter polymerizations.27
A variety of other acrylate and acrylamide monomers can be polymerized in fully open vials under these conditions (Table 2). n-Butyl acrylate (nBA) required a slightly larger reaction volume, but proceeded with similar control as MA (entry 1). t-Butyl acrylate (tBA) resulted in polymer with slight high-molecular-weight tailing but also showed good control over molar mass by varying the equivalents of monomer to iniferter (entries 2–4 and Fig. S12†). Poly(tBA) is of particular interest because it can be deprotected to form poly(acrylic acid) (PAA), a polyelectrolyte that is commonly used as a super-absorbing polymer and as a coating for biomedical applications.36–38 A sample of poly(tBA) was deprotected to PAA with trifluoroacetic acid as a proof of concept.34 The solubility properties and 1H NMR analysis (Fig. S3 versus Fig. S5†) indicated clean conversion to PAA. This completely metal-free synthesis of PAA avoids additional purification steps to remove any catalysts or additives that could contaminate the material.
| Entry | Monomer | Monomer:TTCb |
Monomer:DMSO (v:v, μl) |
Conv.c (%) | M n,Theo (kg mol−1) | M n,GPC (kg mol−1) | M w,GPC (kg mol−1) | M w/Mn |
|---|---|---|---|---|---|---|---|---|
|
a Conditions: Monomer:TTC (200:1), 60 min, uncapped 1 dram vial, no stirring.
b Molar ratio.
c Conversion is corrected for monomer evaporation.
d 30 min.
e Conversion not corrected for monomer evaporation.
f Solid monomer so concentration listed as weight (mg):volume (μl).
|
||||||||
| 1 | n-Butyl-acrylate | 200:1 |
350:350 |
65 | 17.23 | 20.40 | 24.18 | 1.19 |
| 2 | t-Butyl acrylate | 150:1 |
300:300 |
68 | 13.25 | 15.21 | 18.79 | 1.24 |
| 3 | t-Butyl acrylate | 200:1 |
300:300 |
62 | 16.05 | 17.97 | 21.96 | 1.22 |
| 4 | t-Butyl acrylate | 250:1 |
300:300 |
66 | 21.32 | 20.53 | 26.37 | 1.29 |
| 5 | 2-(2-Ethoxyethoxy)ethyl acrylate | 200:1 |
300:300 |
59 | 21.78 | 38.01 | 58.16 | 1.53 |
| 6d | Methyl methacrylate | 200:1 |
325:175 |
33e | 6.66 | 28.00 | 42.93 | 1.53 |
| 7 | N,N-Dimethyl acrylamide | 200:1 |
400:300 |
41e | 8.40 | 16.11 | 27.25 | 1.69 |
| 8 | N-Isopropyl acrylamide | 200:1 |
440:350f |
53 | 12.26 | 13.16 | 20.82 | 1.58 |
| 9 | Acrylamide | 200:1 |
274:400f |
55 | 8.07 | 14.18 | 16.23 | 1.15 |
2-(2-Ethoxyethoxy)ethyl acrylate (DEGA), methyl methacrylate (MMA), N,N-dimethylacrylamide (DMAA), and N-isopropyl acrylamide (NiPAAm) were all successfully polymerized in the open vial setup, but with less control under standard conditions (entries 5–8 and Fig. S13†). DEGA and NiPAAm resulted in generally broader molar mass distributions, while DMAA had a narrow main distribution with a pronounced low molecular weight tail. MMA reached very high molar mass at low conversion, which could be due to fast propagation relative to initiation or slower chain transfer. Acrylamide was polymerized with a narrow dispersity, similar to MA (entry 9). All molecular weights were larger than the theoretical values, presumably due to partial degradation of the iniferter through reactions with oxygen (red path, Fig. 2).
To prove that the iniferter was still active at the end of the polymerization, more monomer was added followed by an additional hour of irradiation, resulting in chain extension without a loss of control (Fig. S15†). If no extra monomer was added, the molecular weight did not change between 1 and 2 hours of irradiation. Additionally, pMA macro-iniferter was isolated by precipitation into cold hexanes before performing additional growth experiments. Over two reinitiation cycles, the GPC traces maintained a narrow, monomodal distribution and clearly shifted to shorter retention times (Fig. 3C). Following a similar procedure, a triblock copolymer of pMA-block-ptBA-block-pMA was prepared with excellent control (Fig. 3D).
Mechanistic investigations/origin of oxygen tolerance
To confirm that the polymerization was due to the visible light photolysis of the TTC, a series of control experiments were performed. A dry ice-acetone bath was used to continually cool the reactor during the polymerization, but the reaction solutions do reach 60 °C due to the exotherm of the polymerization. While this is still an elevated temperature, compared to the thermally-initiated, open-to-air RAFT polymerization,13 temperatures above 100 °C are not required to generate the requisite high radical flux needed to achieve oxygen tolerance. Notably, control reactions heated to 40, 60, and 80 °C in the dark resulted in no conversion (Table S4,† entries 1–3). These results are also supported by our initial finding that, of the visible wavelengths tested, only blue light is able to activate the TTC (Table 1, entries 1–3) since TTC 1 has a λmax,n→π* = 452 nm. During the initialization period when the first monomers are inserted into the iniferter C–S bonds, the n to π* transition slightly blue shifts to λmax,n→π* = 428 nm, but still overlaps sufficiently with the emission of the LED to induce photolysis (see Fig. S28†). If no TTC was added to the reaction, no conversion was observed (Table S4,† entry 4), corroborating Qiao's observation that visible light does not induce self-polymerization of the monomer.29We hypothesize that our system displays oxygen tolerance through a combination of high viscosity, low oxygen solubility, high irradiance, bottom irradiation, no stirring, and large reaction scale. These factors synergize so that the little oxygen that is present in the solution can be removed through the polymerizing through oxygen mechanism and oxygen mass transport from the air is limited. This proposal is supported by our optimization reactions and a variety of other control experiments. We hypothesize that DMSO is the best solvent due to its high viscosity39 and low solubility of oxygen40 compared to the other solvents. The reaction volume screen (vide supra, Table S2,† entries 18–22) indicated that there is a minimum reaction volume required to achieve controlled polymerization. Reaction volume affects oxygen diffusion by changing the surface-area/volume ratio41 and by spatially separating the oxygen in the air from the propagating radicals near the light source. If the reaction is stirred, which increases the mass transport of oxygen, no polymerization is observed (Table S4,† entry 5). It is worthy to note that the polymerization proceeds with excellent control under optimized conditions despite any potential inhomogeneities caused by lack of stirring. This control can possibly be attributed to the degenerative chain transfer of the RAFT process. This finding will be important for the application of this method to LAM because stirring is not employed when forming crosslinked polymer networks, but according to these data, living behavior should be maintained. The penetration depth of the light will also affect the maximum depth of the reaction, such that scaling up the reaction must be done by increasing volume laterally. This hypothesis was tested by running a 2 ml reaction in an uncapped 20 ml scintillation vial (Table S4,† entry 6). Comparable molar mass (Mn = 15.09 kg mol−1) and dispersity (1.16) values were obtained as on the smaller scale.
Many oxygen tolerant systems require a scrubbing mechanism in which all of the oxygen in a closed vial, both in the solution and in the headspace, is removed before the polymerization can proceed, often resulting in long induction periods.15,17,33 In an open vial system, the headspace is effectively infinite, such that all of the oxygen can never be purged. Therefore, for solutions that have not been degassed, the polymerization should not be drastically affected by a cap, besides the amount of monomer evaporation. This hypothesis was tested by running parallel reactions with different headspaces: one uncapped vial, one with a closed cap and vent needle, and one closed vial (Table S4,† entries 7–9). All were similarly well controlled, though the closed vial reactions went to higher conversion and molecular weight, presumably due to lower monomer evaporation. As expected, positive air pressure significantly affects the reaction; when a balloon of air was attached to the top of the reaction vial, the polymerization was inhibited (Mn = 3.58 kg mol−1 after 1 h) and less controlled (Mw/Mn = 1.35, Table S4,† entry 10).
We then sought to compare the kinetics of the open vial polymerization to the corresponding air-free reaction (Fig. 4). A conversion correction was applied to the open vial polymerizations based on an estimation of monomer evaporation (see Section D in ESI†). The air-free reaction had a 5 min induction period before displaying first order kinetics to 79% conversion in 1 hour. The open vial polymerization kinetics were run in triplicate to check for inconsistencies arising from oxygen scavenging. Indeed, in the first 20 minutes, there was variability in the GPC traces, from tailing to bimodality. There seemed to be a slight increase in induction period to around 10 minutes, though low molecular weight polymer was observed in 10 minutes in one of the trials. By 30 minutes, all traces had coalesced to monomodal, narrow peaks (Fig. S20†), and the molar masses matched very well with those from the air-free reaction (Fig. 4A).
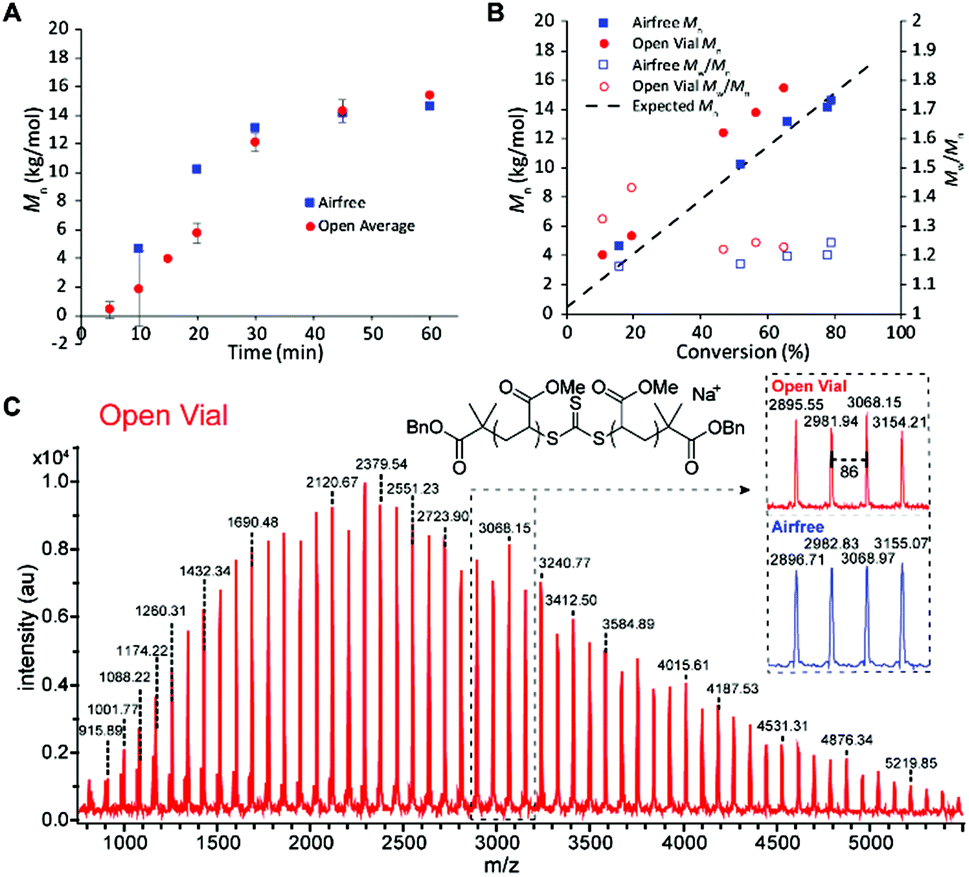 | ||
| Fig. 4 Comparing the open vial polymerization to the analogous air-free reaction: (A) Mn increase over time (open vial polymerization average of three runs). (B) Mnversus conversion to show linear increase (note only one open vial trial shown, conversions for the open vial are estimated due to monomer evaporation). (C) MALDI-TOF analysis of low molecular weight samples to verify the TTC moiety and end groups remain intact for both cases. | ||
The molar masses for both the air-free and open polymerizations increased approximately linearly with conversion (Fig. 4B). The air-free Mn values match very closely with the theoretical molar mass, whereas the open vial Mn values appear to be slightly higher. This observation could be due to some iniferter degradation at the beginning of the reaction as radicals react with dissolved oxygen, an error in the conversion correction, or both. MALDI-TOF mass spectrometry analysis of low molar mass samples verifies the expected polymer structure with the TTC moiety and two benzyl ester end groups (Na+ adduct observed) and confirms the polymer structure is identical between the air-free and open vial polymerizations (Fig. 4C).
Conclusions
In this study, we report an iniferter photo-CRP that proceeds in completely open vials without any photocatalysts or additives. The polymerization is well controlled for acrylate monomers and displays living behavior through control over the molecular weight by adjusting the monomer:TTC ratio and chain extension over multiple cycles. Acrylamide monomers could also be polymerized in an uncapped vial, but with slightly less control. Oxygen tolerance is proposed to arise via the polymerizing through oxygen mechanism and careful experimental setup to limit the transport of oxygen in the air to the site of polymerization. After understanding the parameters that affect oxygen tolerance, the polymerization was able to be scaled up 4× to produce identical material to that of the smaller scale. Compared to the air-free reaction, the induction period may have increased from 5 to 10 minutes, but is still relatively short in comparison to the total reaction time of 60 minutes. Polymer of the same molar mass was produced in 1 hour for both the air-free and open vial cases and MALDI-TOF confirms that both have identical structures with the preservation of the TTC moiety and benzyl ester end groups. This system will help advance the understanding of oxygen tolerance in photocatalyst-free CRP and presents a very convenient platform to perform reversible deactivation radical polymerizations under ambient conditions for LAM in the future.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank C. Perkinson and Prof. M. Bawendi for their help with the absolute irradiance measurements. We acknowledge the National Science Foundation (NSF CHE-1629358) for support of this work. This work was supported in part by the MRSEC Program of the NSF under award DMR 14-19807. J. R. L. gratefully acknowledges the National Institutes of Health for a postdoctoral fellowship (1F32GM126913-01A1).Notes and references
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed
.
- E. M. Pelegri-O'Day and H. D. Maynard, Acc. Chem. Res., 2016, 49, 1777–1785 CrossRef PubMed
- D. F. Grishin and I. D. Grishin, Russ. J. Appl. Chem., 2011, 84, 2021–2028 CrossRef CAS
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed
- J.-Y. Lee, J. An and C. K. Chua, Appl. Mater. Today, 2017, 7, 120–133 CrossRef
- M. Chen, M. J. MacLeod and J. A. Johnson, ACS Macro Lett., 2015, 4, 566–569 CrossRef CAS
- M. Chen, Y. Gu, A. Singh, M. Zhong, A. M. Jordan, S. Biswas, L. T. J. Korley, A. C. Balazs and J. A. Johnson, ACS Cent. Sci., 2017, 3, 124–134 CrossRef CAS PubMed
- V. A. Bhanu and K. Kishore, Chem. Rev., 1991, 91, 99–117 CrossRef CAS
- S. C. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557–589 CrossRef CAS PubMed
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC
- K. Matyjaszewski, S. Coca, S. G. Gaynor, M. Wei and B. E. Woodworth, Macromolecules, 1998, 31, 5967–5969 CrossRef CAS
- S. R. Zavada, T. Battsengel and T. F. Scott, Int. J. Mol. Sci., 2016, 17, 195–221 CrossRef PubMed
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC
- F. M. Calitz, M. P. Tonge and R. D. Sanderson, Macromolecules, 2003, 36, 5–8 CrossRef CAS
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed
- S. Shanmugam, J. Xu and C. Boyer, Macromolecules, 2014, 47, 4930–4942 CrossRef CAS
- J. Xu, K. Jung and C. Boyer, Macromolecules, 2014, 47, 4217–4229 CrossRef CAS
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed
- N. Corrigan, D. Rosli, J. W. J. Jones, J. Xu and C. Boyer, Macromolecules, 2016, 49, 6779–6789 CrossRef CAS
- L. Yu, Y. Wei, Y. Tu, S. Lin, Z. Huang, J. Hu, Y. Chen, H. Qiao and W. Zou, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2437–2444 CrossRef CAS
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS
- S. Muthukrishnan, E. H. Pan, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis, D. Lewis and L. Barner, Macromolecules, 2007, 40, 2978–2980 CrossRef CAS
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 3, 1500394 CrossRef PubMed
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed
- L. Lu, H. Zhang, N. Yang and Y. Cai, Macromolecules, 2006, 39, 3770–3776 CrossRef CAS
- H. Wang, Q. Li, J. Dai, F. Du, H. Zheng and R. Bai, Macromolecules, 2013, 46, 2576–2582 CrossRef CAS
- H. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS PubMed
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS
-
J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, in Controlled Radical Polymerization: Mechanisms, American Chemical Society, 2015, vol. 1187, pp. 247–267 Search PubMed
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558–564 CrossRef CAS
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519–1526 RSC
- J. Li, C. Ding, Z. Zhang, X. Pan, N. Li, J. Zhu and X. Zhu, Macromol. Rapid Commun., 2017, 38, 1600482 CrossRef PubMed
- See ESI† for additional information.
- R. W. Lewis, R. A. Evans, N. Malic, K. Saito and N. R. Cameron, Polym. Chem., 2018, 9, 60–68 RSC
- R. Bitar, P. Cools, N. De Geyter and R. Morent, Appl. Surf. Sci., 2018, 448, 168–185 CrossRef CAS
- M. Donegan and D. P. Dowling, Surf. Coat. Technol., 2013, 234, 53–59 CrossRef CAS
-
F. L. Buchholz, in Industrial Polymers Handbook, 2001, vol. 1, pp. 565–585 Search PubMed
-
J. R. Rumble, Handbook of Chemistry and Physics. 2017–2018, CRC Press, Boca Raton, 98th edn, 2017 Search PubMed
- R. Battino, T. R. Rettich and T. Tominaga, J. Phys. Chem. Ref. Data, 1983, 12, 163–178 CrossRef CAS
- A. J. Gormley, J. Yeow, G. Ng, Ó. Conway, C. Boyer and R. Chapman, Angew. Chem., Int. Ed., 2018, 57, 1557–1562 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental procedures, characterization methods, expanded data tables and figures. See DOI: 10.1039/c9py00022d |
| This journal is © The Royal Society of Chemistry 2019 |