
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precisely tailored shell thickness and Ln3+ content to produce multicolor emission from Nd3+-sensitized Gd3+-based core/shell/shell UCNPs through bi-directional energy transfer†
York E.
Serge Correales‡
a,
Chanchal
Hazra‡
*a,
Sajjad
Ullah
 b,
Laís R.
Lima
a and
Sidney J. L.
Ribeiro
*a
b,
Laís R.
Lima
a and
Sidney J. L.
Ribeiro
*a
aInstitute of Chemistry, São Paulo State University, UNESP, 14800-060, Araraquara, SP, Brazil. E-mail: sidney.jl.ribeiro@unesp.br; sjlribeiro@gmail.com; chanchalhazra007@gmail.com
bInstitute of Chemical Sciences, University of Peshawar, 25120, Peshawar, Pakistan
First published on 21st March 2019
Abstract
Lanthanide (Ln3+)-doped upconversion nanoparticles (UCNPs) have been paid great attention as multiplexing agents due to their numerous uses in biological and clinical applications such as bioimaging and magnetic resonance imaging (MRI), to name a few. To achieve efficient multicolor emission from UCNPs under single 808 nm excitation and avoid detrimental cross-relaxations between the Ln3+ activator ions (positioned in either the core and/or shell in the core/shell), it is essential to design an adequate nanoparticle architecture. Herein, we demonstrate the tailoring of multicolor upconversion luminescence (UCL) from Nd3+-sensitized Gd3+-based core/shell/shell UCNPs with an architecture represented as NaGdF4:Tm3+(0.75)/Yb3+(40)/Ca2+(7)/Nd3+(1)@NaGdF4:Ca2+(7)/Nd3+(30)@NaGdF4:Yb3+(40)/Ca2+(7)/Nd3+(1)/Er3+(X = 1, 2, 3, 5, 7) [hereafter named CSS (Er3+ = 1, 2, 3, 5 and 7 mol%)]. Such UCNPs can be excited at a single wavelength (∼808 nm) without generation of any local heat. Incorporation of substantial Nd3+-sensitizers with an appropriate concentration in the middle layer allows efficient harvesting of excitation light which migrates bi-directionally across the core/shell interfaces in sync to produce blue emission from Tm3+ (activator) ions in the core as well as green and red emission from Er3+ (activator) ions in the outermost shell. Introduction of Ca2+ lowers the local crystal field symmetry around Ln3+ ions and subsequently affects their intra 4f–4f transition probability, thus enhancing the upconversion efficiency of the UCNPs. By simple and precise control of the shell thickness along with tuning the content of Ln3+ ions in each domain, multicolor UCL can be produced, ranging from blue to white. We envision that our sub-20 nm sized Nd3+-sensitized Gd3+-based UCNPs are not only potential candidates for a variety of multiplexed biological applications (without impediment of any heating effect), but also can act as MRI contrast agents in clinical diagnosis.
Introduction
In view of the recent research upsurge, multicolor luminescence from photoluminescent probes is of great interest due to its widespread use in chemical and biological sciences.1–7 The multicolor luminescence provides remarkable insight into molecular biological processes, and, using a number of color-encoded probes, makes a large number of biochemical entities feasible for high-throughput sequencing technologies together with brief sequencing chemistry.8–10 Along with multicolor luminescence, multiplexing experiments are adequately used in human genome decoding, and biological and clinical applications.11–13 To date, there are several reports available on photoluminescent probes that have been used as multiplexing agents in bio-system analysis and therapy applications.14–18 Among them, Ln3+-doped UCNPs hold promise because of their unique anti-Stokes (known as upconversion) optical properties.19–24 Typically, these Ln3+-doped nanoparticles are composed of sensitizers (e.g., Yb3+/Nd3+) and activators (Er3+/Tm3+/Ho3+) which are spatially distributed in an appropriate host matrix or in a dilute host–guest system. Successive absorption of near infrared (NIR) photons and/or energy transfer (ET) generally promote the activators to higher excited states and concurrently lead to the emission of higher energy radiation spanning over a wide region, from the ultraviolet (UV) to NIR spectral regions.25–29 The relatively high penetration depth of NIR light in biological structures causes a minimal autofluorescence background signal and low biotoxicity, which again make UCNPs ideal candidates for a wide range of biological applications such as sensing, imaging and therapy, among others.30–38 Moreover, compact, inexpensive continuous-wave NIR diode lasers can be used to excite UCNPs, leading to the generation of multiphoton UCL which is an important additive to conventional multiphoton microscopy. This characteristic physiochemical property of UCNPs also allows their extensive use in biological domains.39–41Doping different types of activators (e.g., Er3+/Tm3+/Ho3+) together with sensitizers (e.g., Yb3+) with a precise concentration in a suitable host matrix can produce multicolor UCL under single NIR wavelength excitation at 980 nm, which corresponds to the absorption of Yb3+ ions.42,43 However, this strategy can lead to cross-relaxation processes between the different types of activators, resulting in lower UCL efficiency from the UCNPs.44–46 Moreover, in the Yb3+-sensitized upconversion process, the excitation wavelength (980 nm) is strongly absorbed by water molecules in biological environment which causes local heating problems in the vicinity of cells and tissues, thus damaging them.42,47 To address this problem, recent research has been focused on the extension of the upconversion excitation spectrum to shorter wavelengths (especially, in the vicinity of 808 nm), where water molecules do not absorb significantly.48–52 Nevertheless, efficient multicolor UCL output from Nd3+-sensitized UCNPs remains vague until this point.
Furthermore, the multicolor UCL from Nd3+-sensitized UCNPs, obtained through a combination of different types of activators (such as Er3+ and Tm3+) in a single domain, can introduce deleterious cross-relaxation quenching processes, leading to low UCL efficiency from UCNPs. Therefore, it is necessary to separate different types of activators at a nanoscale distance. Moreover, to date, Nd3+-sensitized core/shell UCNPs have typically been exploited to transfer the energy from a Nd3+-doped light-harvesting layer across the core/shell interface to a Yb3+/X3+ (X = Er, Ho or Tm) co-doped layer in one single direction. This uni-directional ET process provides no plausible scope to sensitize two different types of activators in separated layers which again leads to the decrease in UCL efficiency, knowing that the efficiency depends on the distance between the Nd3+-doped layer and the activator-doped layer. It is worth mentioning that the efficiency of Nd3+ → Yb3+ non-radiative ET is directly proportional to the number of Yb3+ ions per Nd3+ ion in the doped region. In fact, the ability to harvest light most efficiently by UCNPs at 808 nm is also directed by the total number of doped Nd3+ ions. Thus, to achieve efficient multicolor UCL, it is essential to optimize the concentration of Yb3+ ions and Nd3+ ions in their defined domains. Recently, Hao et al. reported that the Tm3+ and Er3+ ions doped into sandwich-structured UCNPs of core/shell/shell NaYbF4:Tm3+@CaF2:Nd3+@CaF2:Er3+ effectively produce multicolour UCL via a bi-directional ET process.53 However, to our knowledge, there is no report available on the use of a bi-directional ET process along with precise control of the shell thickness and proper tuning of the content of Ln3+ ions in each domain to produce bright multicolour emission from Nd3+-sensitized Gd3+-based core/shell/shell UCNPs. Such strategies could be a potential tool for bio-imaging and MRI dual modal applications in the near future.
In this context, we report Nd3+-sensitized Gd3+-based core/shell/shell (CSS) UCNPs to achieve multicolor UCL under single wavelength excitation at 808 nm. In particular, NaGdF4 has been selected as a host matrix (both as the core and the shell layer) as it allows the formation of small sized nanoparticles. In small sized UCNPs, the surface-to-volume ratio is very high and a large portion of dopant ions are located at the surface. Moreover, with proper functionalization, Gd3+-based core/shell/shell UCNPs could effectively serve as MRI contrast agents in medical diagnosis. Ca2+ was chosen as it has lower valence than Gd3+ ions. Upon replacement of some Gd3+ ions by Ca2+ ions, a charge imbalance in NaGdF4 occurs which lowers the local crystal field symmetry around Yb3+, Er3+ and Tm3+ ions and subsequently affects the intra 4f–4f transition probability of the Ln3+ ions, thus enhancing the upconversion efficiency.34,54–56 The Nd3+ sensitizer is doped at a high concentration (30 mol%) in the middle layer to effectively and efficiently absorb the excitation light in this region and simultaneously conveys it to core and outermost shell in a bi-directional manner to emit efficient multicolor emissions from the nanoparticles. Tm3+ and Er3+ activators are separated into the inner core and the outermost shell, respectively, to prevent deleterious cross-relaxation processes between them. Moreover, the core/shell/shell nanoconfiguration allows simultaneous non-radiative ET from the middle layer to both the Tm3+-doped core and Er3+-doped outermost shell. Since high concentrations of Yb3+ ions (40 mol%) are incorporated in both the outermost shell and core domain, efficient core/shell interface energy migration takes place from the Nd3+ ions to the Yb3+ ions, which then sensitizes the activator ions (both Er3+ and Tm3+) to produce bright UCL. A lower doping of Nd3+ (1 mol%) in the outermost shell and in the core region is expected to assist the energy migration across the core/shell interfaces through a resonant ET process between Nd3+ ions. We demonstrated that the CSS (Er3+ = 1, 2, 3, 5 and 7 mol%) UCNPs are able to emit efficient multicolor UCL, ranging from blue to white under laser excitation at 808 nm. Tailoring the shell thickness, along with a precise control of the concentration of sensitizers and activators in their defined domains, allows us to achieve such tunable multicolor UCL from our designed sub-20 nm sized core/shell/shell UCNPs.
Experimental
Materials
Gd2O3 (99.99%, Sigma-Aldrich), Yb2O3 (99.99%, Sigma-Aldrich), Er2O3 (99.99%, Sigma-Aldrich), Nd2O3 (99.99%, Sigma-Aldrich), Tm2O3 (99.99%, Sigma-Aldrich), CaCl2 (Synth), NaOH (Synth), NH4F (Merck), methanol (CH3OH, Synth), 1-octadecene (ODE, Aldrich), oleic acid (OA, Aldrich), cyclohexane (Synth, Brazil), and ethanol (C2H5OH, Synth, Brazil) were used in this work. All chemicals were used without further purification.Methods
Characterization
Results and discussion
Structural characterization of materials
Nd3+-sensitized Gd3+-based CSS UCNPs were successfully synthesized following a modified seed-mediated layer-by-layer growth method according to a literature report57 (Scheme 1). Phase analysis of the core, CS (30 mol% Nd3+) and CSS (5 mol% Er3+) UCNPs was carried out using powder X-ray diffraction (XRD) measurements (Fig. 1a). All diffraction peaks perfectly match with the standard diffraction pattern of hexagonal NaGdF4 crystals (JCPDS no: 27-0699). The shape and size of the nanoparticles were examined by TEM. TEM images indicate the formation of uniform and monodisperse nanoparticles with an average diameter of 7 nm, 12 nm and 19 nm for core (NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7)), CS (30 mol% Nd3+) and CSS (5 mol% Er3+) UCNPs, respectively (Fig. 1b, c and d). The shell thickness of the CS and CSS UCNPs is thus estimated to be around 2.5 and 3.5 nm, respectively. Monodispersity in the size of these core, CS and CSS UCNPs is evident from the size distribution histograms shown in Fig. S1.† Along with the size increase (core → CS → CSS), there is hardly any change in the morphology observed, indicating the homogeneous nature of shell growth over core and CS UCNPs. The high resolution TEM results further supported the formation of high quality hexagonal phase core, CS and CSS UCNPs (Fig. S2†). The qualitative chemical composition of these UCNPs was studied by energy-dispersive X-ray spectroscopy (EDS) (Fig. S3†). The EDS spectra of the samples show the presence of Gd, Nd, Ca, Yb, and Tm, Gd, Nd, Ca, Yb, and Tm and Gd, Nd, Ca, Yb, Tm, and Er for core, CS and CSS UCNPs, respectively, implying the layer-by-layer growth of shells on core nanoparticles. The capping of core, CS and CSS UCNPs with oleic acid onto the surface of the same is confirmed by the appearance of strong carbonyl stretching vibration near 1500 cm−1 (Fig. S4†). This frequency is much lower than that observed for the free oleic acid molecules (1700 cm−1), indicating strong binding of the ligands (oleic acid) onto the surface of the nanoparticles.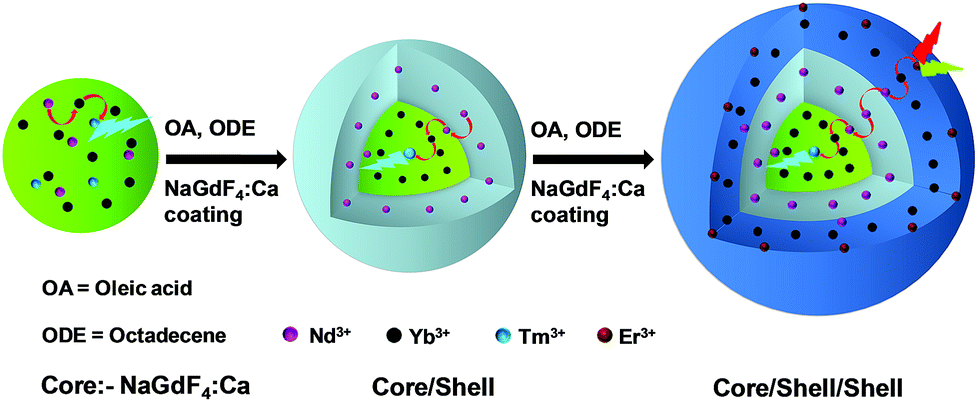 | ||
| Scheme 1 Schematic illustration of the synthesis of CSS UCNPs following a seed-mediated layer-by-layer growth method. | ||
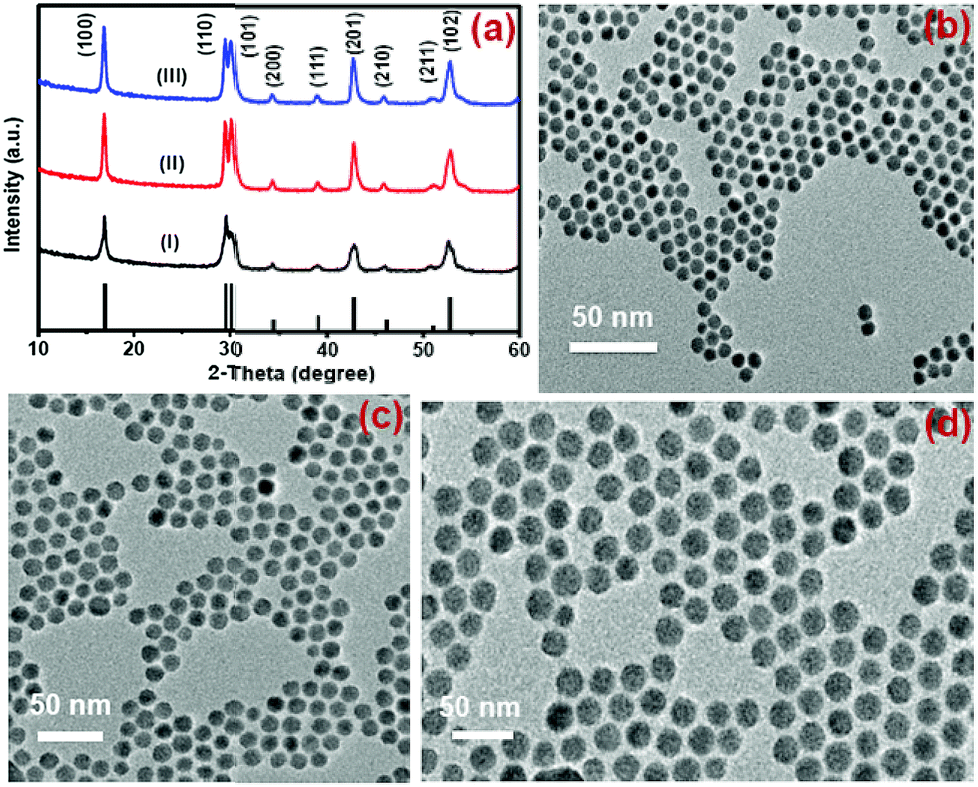 | ||
| Fig. 1 (a) XRD patterns of the NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7) core (i), CS (30 mol% Nd3+) (ii) and CSS (5 mol% Er3+) (iii) UCNPs. (b, c, and d) present the TEM images of these core, CS and CSS UCNPs, respectively. | ||
Study of optical properties
The presence of Yb3+ ions (along with 1 mol% Nd3+ ions) in core, (Yb3+ + Nd3+) ions in CS (30 mol% Nd3+) and (Yb3+ + Nd3+) ions in CSS (5 mol% Er3+) UCNPs is confirmed by the presence of their characteristic peaks in the electronic absorption spectra of the samples (Fig. S5†). In comparison to core UCNPs, the absorption intensity of the characteristic peaks of Nd3+ ions increases with increase in the Nd3+ concentration in the CS and CSS UCNPs, suggesting efficient incorporation of Nd3+ ions in the shell of CS and in the middle layer of CSS UCNPs. To justify the importance of a Nd3+-enriched shell to efficiently harvest light at 808 nm for simultaneous sensitization of both Tm3+ and Er3+ activators to produce bright multicolor UCL, we first prepared the core-NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7), CS (30 mol% Nd3+) and CSS (5 mol% Er3+) UCNPs. Fig. 2 shows the upconversion emission spectra for these core and CS UCNPs obtained using 808 nm laser excitation. The UCL peaks centered around 451 and 477 nm are assigned to the 1D2 → 3F4 and 1G4 → 3H6 transitions of Tm3+ ions, respectively. The calculated integrated UCL intensity of the blue (442–505 nm) emission of CS UCNPs is 77 times higher than that of core UCNPs. The reason behind this observation might be explained on the basis of two factors. First, surface related harsh quenching of the core NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7) is diminished by coating the NaGdF4:Nd3+(30)/Ca2+(7) shell over the former. Secondly, the high concentration of Nd3+ ions (30 mol%) in the shell of the core/shell UCNPs might harvest the light at 808 nm more efficiently and subsequently transfer it to the core region to sensitize Yb3+ ions and simultaneously the Tm3+ ions (activator) to produce blue emission.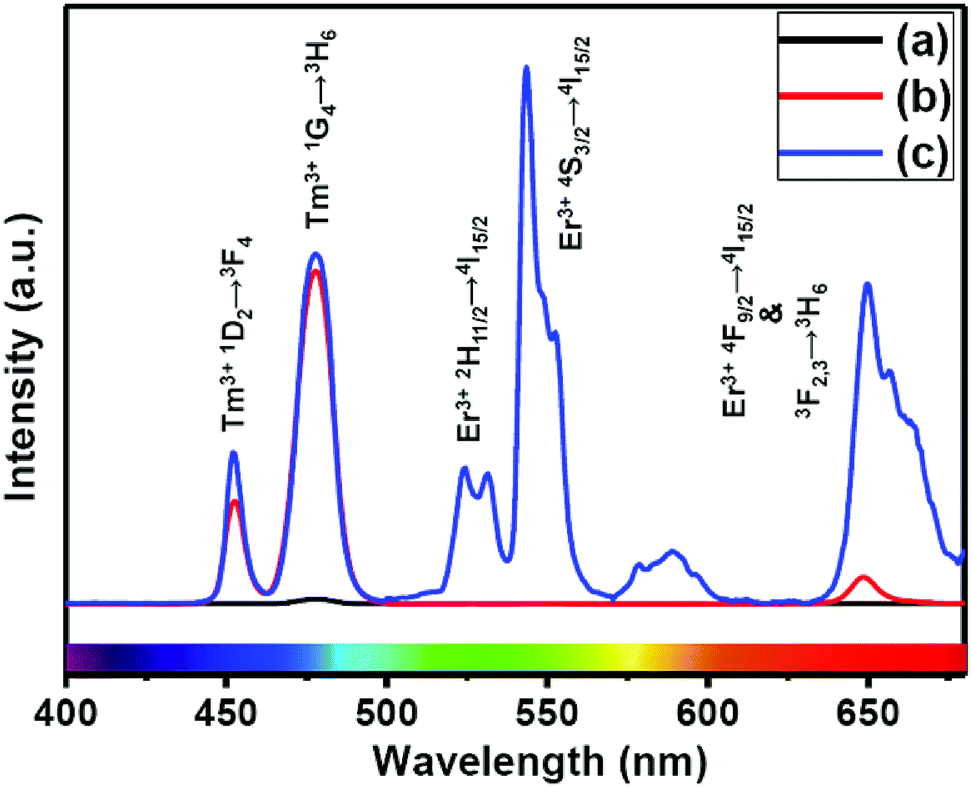 | ||
| Fig. 2 UCL spectra of the (a) core NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7), (b) CS (30 mol% Nd3+) and (c) CSS (5 mol% Er3+) UCNPs. | ||
To understand the effect of shell thickness on the UCL intensity from CS (30 mol% Nd3+), the core particles were coated with a shell of variable thickness (i.e., 1.3, 1.8, 2.5, 3.9 and 4.8 nm) using epitaxial growth (Fig. 3C and D). As shown in Fig. 3A, the luminescence intensity first increases, reaching a maximum for a shell thickness of 2.5 nm, and then decreases. From Fig. 3B, it could be noted that the calculated integrated intensity of the blue (462–505 nm) emission of CS UCNPs with 2.5 nm shell thickness is 7.6 and 3.5 times higher than that of CS nanoparticles with 1.3 and 4.8 nm shell thickness, respectively. This result clearly suggests that shell thickness plays a pivotal role in the UCL intensity of CS UCNPs. Thinner shells (i.e., shell thickness < 2.5 nm) allow the activator (Tm3+, 0.75 mol% in the core) and the sensitizer (Nd3+, 30 mol% in the shell) to come closer in proximity. Hence, a good overlap between emission of Tm3+ ions and the absorption of Nd3+ ions could be expected which assures an efficient back ET from Tm3+ ions to Nd3+ ions, leading to a decrease in blue UCL from the CS UCNPs. On the other hand, with an increase in the shell thickness (>2.5 nm), though the shell with 30 mol% Nd3+ efficiently harvests excitation light, it does not allow it to migrate effectively to Yb3+ ions (in core) across the core/shell interfaces to produce efficient blue emission from Tm3+ (activator) ions in the core. In fact, such CS UCNPs with a thicker shell (>2.5 nm) enable excitation energy to travel a long Nd3+–Yb3+ distance, which again leads to the decrease in the UCL intensity from the nanoparticles.
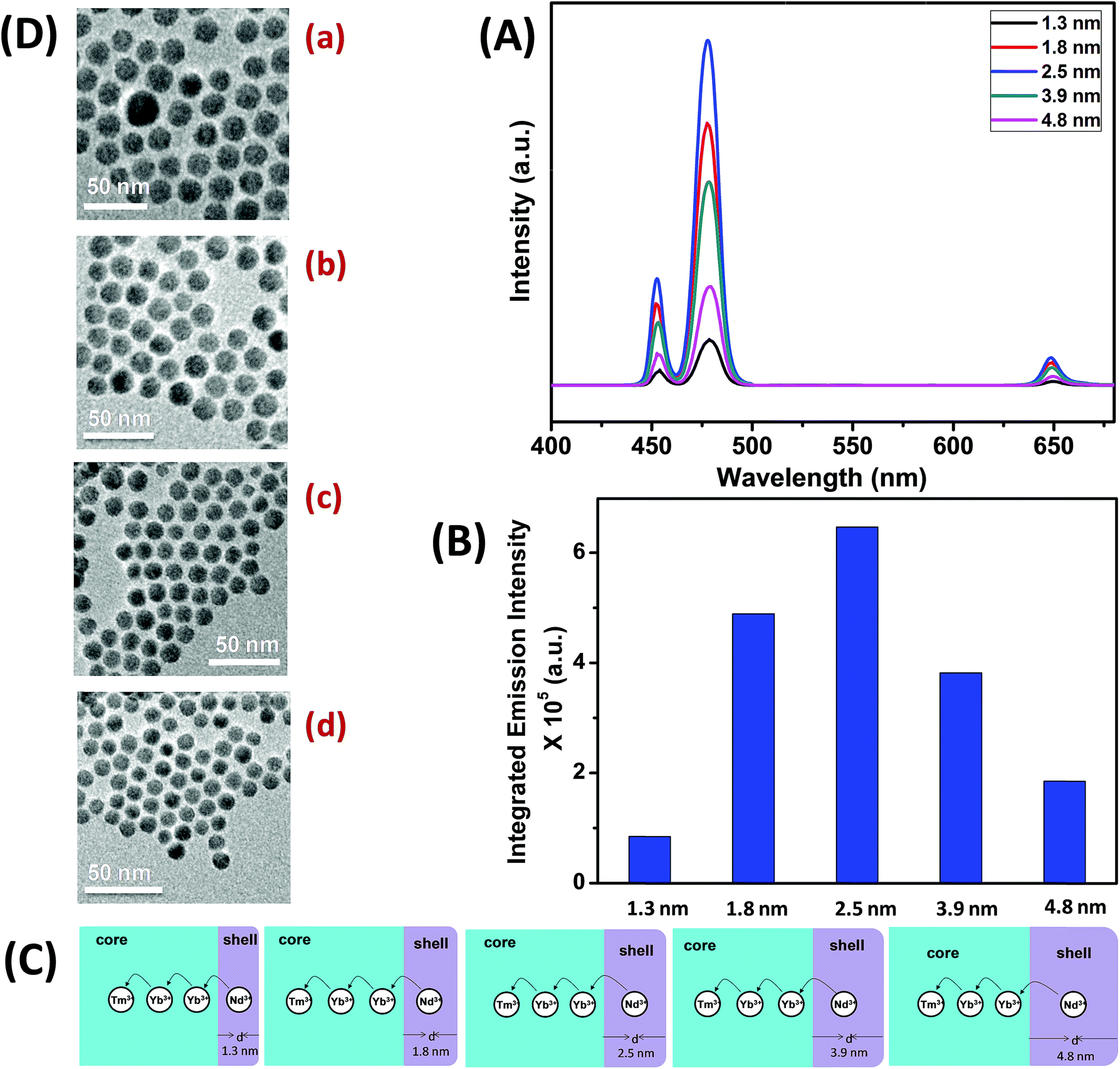 | ||
| Fig. 3 (A) UCL spectra of CS (30 mol% Nd3+) UCNPs with different thickness of the NaGdF4:Ca2+(7)/Nd3+(30) shell (d = 1.3–4.8 nm). (B) Integrated intensity of the blue (462–505 nm) emission of CS (30 mol% Nd3+) UCNPs with different shell thickness (d = 1.3–4.8 nm). (C) Mechanistic investigation of interfacial ET from the Nd3+-containing shell to the Tm3+ containing core in different shell thickness controlled (d = 1.3–4.8 nm) CS (30 mol% Nd3+) UCNPs. (D) TEM images of CS (30 mol% Nd3+) UCNPs with different shell thickness ((a) 1.3 nm, (b) 1.8 nm, (c) 3.9 nm and (d) 4.8 nm). | ||
Upon growth of the second shell (i.e., NaGdF4:Yb3+(40)/Nd3+(1)/Ca2+(7)/Er3+(5) onto the surface of CS (30 mol% Nd3+) UCNPs, the UCL from both activators (Er3+ and Tm3+) was observed together (Fig. 2, spectrum c). Along with the 1D2 → 3F4 and 1G4 → 3H6 transitions of Tm3+ ions, the green emission peak centered at 544 nm (4S3/2 → 4I15/2) and the red emission peak centered at 650 nm (4F9/2 → 4I15/2) from Er3+ ions are also observed. This result substantiates that the Nd3+ enriched shell can harvest excitation light more efficiently and allows migration of energy across the core/shell interface to both Tm3+ ions in the core and Er3+ in the outermost shell to produce multicolor UCL from CSS UCNPs.
Similar to CS (30 mol% Nd3+), to examine the effect of shell thickness on the UCL intensity from CSS (5 mol% Er3+), CS (30 mol% Nd3+) was used as a seed for the epitaxial growth of outermost shells with different thickness (1.2, 2.5, 3.5, 5 and 5.7 nm) (Fig. 4C and D). From Fig. 4A, a shell thickness of 3.5 nm results in the highest UCL. From Fig. 4B, it can be clearly observed that the calculated integrated intensity of the green (536–570 nm) emission of these CSS UCNPs with 3.5 nm shell thickness is 2.3 and 4.5 times higher than that of CSS nanoparticles with 1.2 and 5.7 nm outermost shell thicknesses, respectively. A similar trend was observed for red (630–680 nm) emission too. Thinner outermost shells (i.e., shell thickness 1.2 and 2.5 nm) allow the activator (Er3+, 5 mol% in the outermost shell) and the sensitizer (Nd3+, 30 mol% in the middle shell layer) to come closer in proximity. Therefore, in this case too, a good overlap between emission of Er3+ ions and the absorption of Nd3+ ions can be expected which manifests itself in the form of an efficient back ET from Er3+ ions to Nd3+ ions, leading to the decrease in UCL (green + red) from the CSS UCNPs. On the other hand, with an increase in the outermost shell thickness (>3.5 nm), the shell with 30 mol% Nd3+, despite efficiently harvesting excitation light, does not allow it to migrate efficiently to Yb3+ ions (40 mol%, outermost shell) concurrently to produce green and red emissions from Er3+ (activator) ions in the outermost shell.
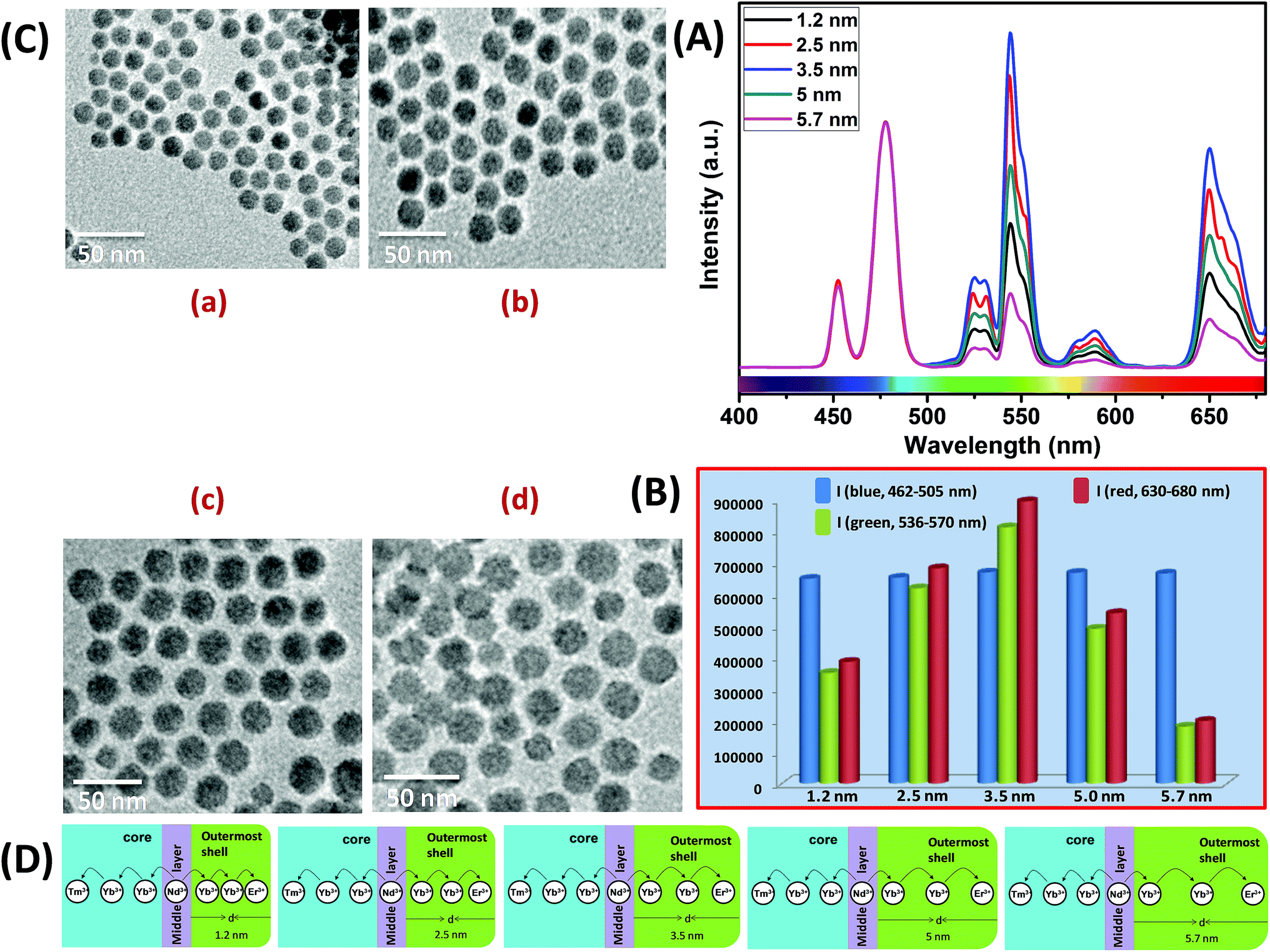 | ||
| Fig. 4 (A) UCL spectra of CSS UCNPs (5 mol% Er3+) with different thickness of the outermost shell over CS (30 mol% Nd3+) UCNPs (d = 1.2–5.7 nm). (B) Integrated intensity of blue (Tm3+, 462–505 nm), green (Er3+, 536–570 nm) and red (Er3+, 630–680 nm) emissions of CSS UCNPs with different outermost shell thickness (d = 1.2–5.7 nm). Note that, the UCL spectra and integrated emissions in (A) and (B), respectively, are normalized to the blue (462–505 nm) emission. (C) TEM images of CSS (5 mol% Er3+) UCNPs with different outermost shell thickness ((a) 1.2 nm, (b) 2.5 nm, (c) 5.0 nm and (d) 5.7 nm). (D) Mechanistic investigation of interfacial ET from the Nd3+-containing middle shell layer to the Tm3+ containing core (with defined shell thickness d = 2.5 nm) and Er3+ containing outermost shell (with different shell thickness d = 1.2–5.7 nm) in CSS (5 mol% Er3+) UCNPs. | ||
Moreover, like the CS UCNPs, the CSS UCNPs with a thicker shell (>3.5 nm) make the excitation energy travel a long Nd3+–Yb3+ distance, which again leads to a decrease in the green and red UCL intensity from the nanoparticles. The UCL spectra and integrated emissions in Fig. 4A and B, respectively, are normalized to the blue (462–505 nm) emission. Actually, to prepare CSS nanoparticles via the epitaxial growth mechanism, we used CS nanoparticles with similar size in each reaction. We only varied the amount of outermost shell precursor to deposit an outermost shell (with different thickness) over CS nanoparticles. All other synthesis parameters (time, temperature etc.) were kept constant. Hence, there was no remarkable change in the blue emission intensity as the energy transfer efficiency of Nd → Yb → Tm remained unaltered in each case. Though we acquired all the spectra under identical experimental conditions, still we observed a slight change in the blue emission. To further clarify the effect of outermost shell thickness on the green and red emission of Er3+ ions (doped in outermost shell), we normalized the whole spectra and intensity with respect to the blue emission. The original spectra and intensity (without normalization) are shown in Fig. S6 and Table S1,† respectively.
The CSS (5 mol% Er3+) nanostructure thus plays an important role in separating two activators (Tm3+ and Er3+) to mitigate deleterious cross-relaxation processes between them. Fig. 5a depicts a schematic route of nonradiative ET processes of Tm3+ ← Yb3+ ← Nd3+ → Yb3+ → Er3+ in the CSS UCNPs, upon 808 nm excitation. The schematic illustration of ET pathways to produce bright UCL from both the core and the outermost shell in the designed CSS UCNPs (upon 808 nm excitation) is shown in Fig. 5b. Moreover, to better understand, we recorded the UCL spectra of three different NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Ca2+(7)/Nd3+(30), NaGdF4:Er3+(2)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Ca2+(7)/Nd3+(30) and NaGdF4:Tm3+(0.75)/Er3+(2)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Ca2+(7)/Nd3+(30) core/shell UCNPs to verify the adverse cross-relaxation processes between the activator ions (Tm3+ and Er3+). From Fig. 6A, it is clearly observed that the UCL intensity is significantly reduced when Tm3+ and Er3+ ions are doped together in the core region (spectrum c), indicating the unwanted cross-relaxation process between them. As a reasonable explanation of our observation, a simplified energy level diagram depicting the cross-relaxation quenching mechanisms between Tm3+ and Er3+ (under 808 nm excitation) in Nd3+-sensitized Gd3+-based core/shell UCNPs is shown in Fig. 6B. In order to achieve efficient multicolor UCL from CSS (5 mol% Er3+) UCNPs, it is essential to first optimize the concentration of Tm3+ ions in the core region of CS (30 mol% Nd3+) nanoparticles. To achieve the utmost blue emission (808 nm excitation), the optimal concentration of Tm3+ ions in CS nanoparticles (against 40 mol% Yb3+ ions) was found to be 0.75 mol% (Fig. S7†). The reason behind the use of such high mol% of Yb3+ ions has also been explained in this article (vide infra). We also investigated the effect of Nd3+ concentration in the middle layer of CS UCNPs and the optimum concentration of Nd3+ ion was determined to be 30% (Fig. S8†). The gradual increase of Nd3+ concentration in the middle layer is consistent with the overall increase in absorption of excitation energy, leading to the utmost photon harvesting efficiency and, concurrently, the strongest UCL intensity from the UCNPs. However, at a Nd3+ concentration higher than 30 mol%, the mutual interaction between Nd3+ ions increases in the shell. This enhanced interaction consumes the excitation energy, leading to less efficient energy migration across the core/shell interface to the Tm3+-doped core. We believe that a similar phenomenon might occur in the Er3+-doped outermost shell in CSS UCNPs. Thus, multicolor UCL intensity from the UCNPs is greatly reduced. Moreover, a good overlap between the emission of Tm3+ and Er3+ ions with the absorption of Nd3+ ions ensures an efficient back ET from Tm3+ or Er3+ ions to Nd3+ ions which again leads to a decrease in UCL from the nanoparticles. Upon variation in the concentration of Nd3+ ions in the shell, the crystalline phase and size of the CS UCNPs remain intact (Fig. S9†). It is worth mentioning that the coating of at least one inert shell over CS UCNPs is necessary to preserve excitation energy and simultaneously achieve strong luminescence from the nanoparticles. In this work, we did not follow the growth of any inert shell over CS (30 mol% Nd3+) UCNPs as our goal was to allow the excitation energy to migrate along the core/shell interface to both the core and the outermost shell to produce efficient multicolor UCL. Though Nd3+–Nd3+ energy migration is not reported in the literature, using lower doping of Nd3+ ions (1 mol%) in the core and outermost shell is due to two reasons: (i) to systematically follow the UCL spectra of UCNPs upon growth of shells in a layer-by-layer fashion, under single 808 nm excitation and (ii) to help the energy transfer (Nd3+–Yb3+) across the core/shell interfaces through a resonant ET process between Nd3+ ions.
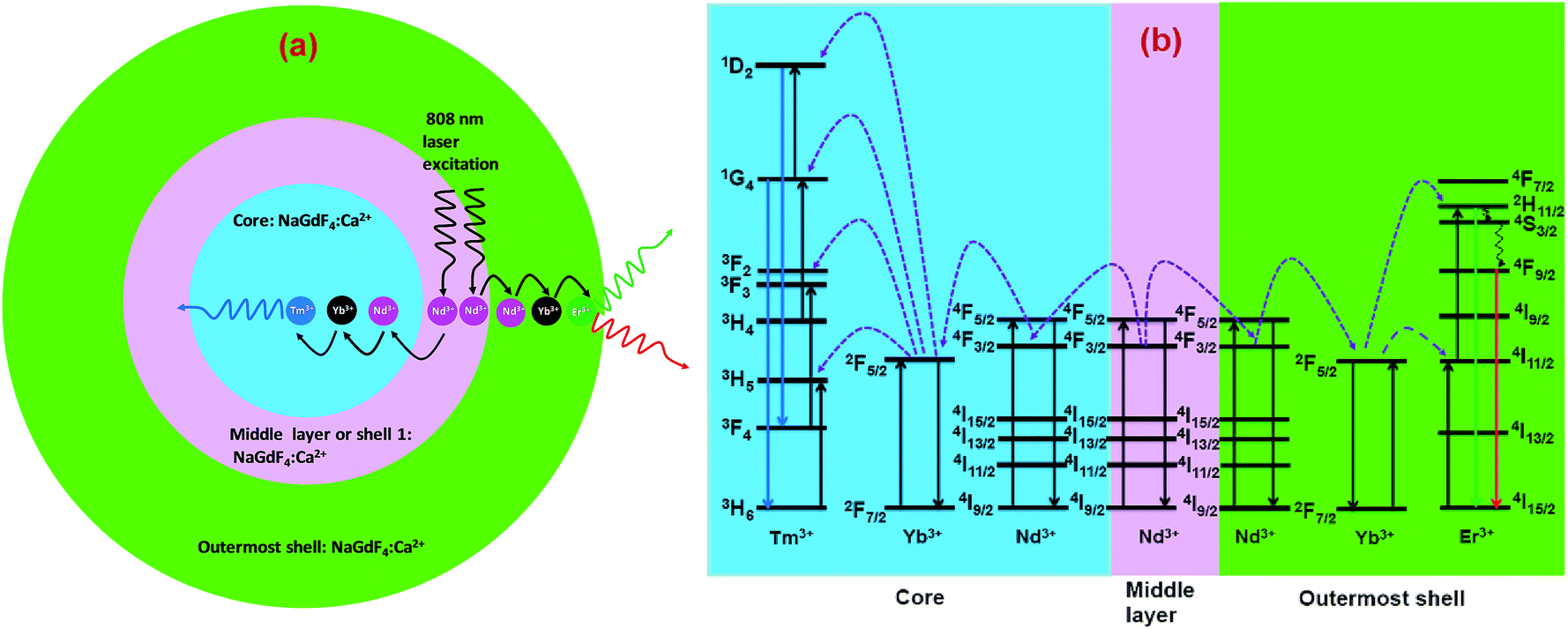 | ||
| Fig. 5 (a) Schematic illustration of the non-radiative ET mechanism of Tm3+ ← Yb3+ ← Nd3+ → Yb3+ → Er3+ in CSS (5 mol% Er3+) UCNPs (808 nm laser excitation). (b) Schematic illustration of ET pathways to produce bright UCL from both the core (blue, Tm3+) and the outermost shell (red and green, Er3+) in the same CSS UCNPs upon 808 nm excitation. | ||
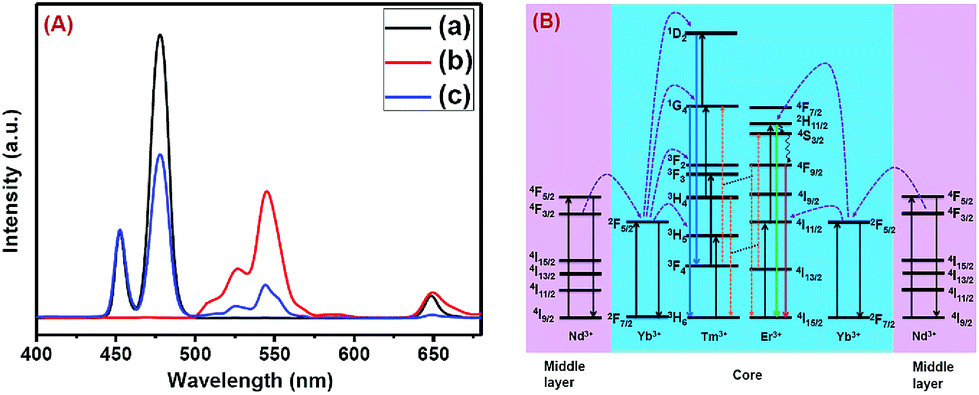 | ||
| Fig. 6 (A) Comparison of UCL spectra of (a) NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Ca2+(7)/Nd3+(30), (b) NaGdF4:Er3+(2)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Ca2+(7)/Nd3+(30) and (c) NaGdF4:Tm3+(0.75)/Er3+(2)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Ca2+(7)/Nd3+(30) core/shell UCNPs, upon 808 nm laser excitation, to verify the cross-relaxation between Tm3+ and Er3+ ions. (B) Energy level diagram indicating the cross-relaxation mechanism between Tm3+ and Er3+ ions in NaGdF4:Tm3+(0.75)/Er3+(2)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Ca2+(7)/Nd3+(30) core/shell UCNPs, upon 808 nm laser excitation. | ||
It is interesting to note that, in the core and outermost shell, a relatively high concentration (40 mol%) of Yb3+ ions was used in our designed CSS (5 mol% Er3+) UCNPs. To examine the impact of Yb3+ concentration in the outermost shell on the complete UCL spectrum of these CSS UCNPs, we varied the concentration of Yb3+ ions in the outermost shell, while the Yb3+ concentration (40 mol%) in the core was kept fixed. As shown in Fig. 7A, maximum green and red emission intensities are observed with an increase in Yb3+ ions concentration (in the outermost shell) up to 40 mol%, whereas the intensity of blue emissions remains almost unchanged. From Fig. 7B, it is clear that the calculated integrated intensity of the green (536–570 nm) emission of CSS UCNPs with 40 mol% Yb3+ concentration in the outermost shell is 4.4 and 1.2 times higher than that of CSS nanoparticles with 10 and 50 mol% Yb3+ concentration in the outermost shell, respectively. The change in the calculated integrated intensity of red (630–680 nm) emission was similar to that of green emission.
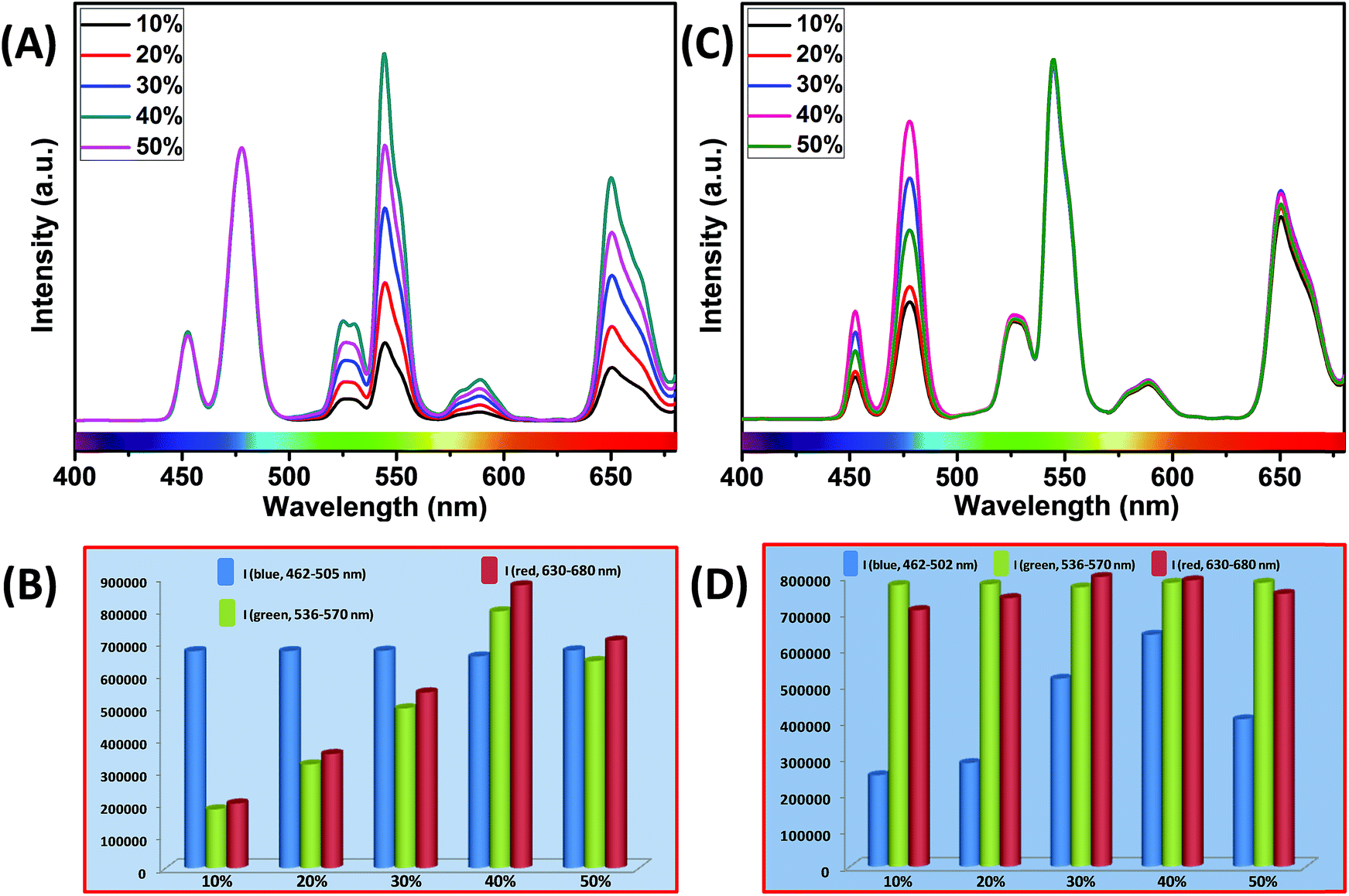 | ||
| Fig. 7 (A) UCL spectra of CSS (5 mol% Er3+) UCNPs with different Yb3+ concentrations in the outermost shell, where the Yb3+ concentration in the core remains unchanged at 40 mol%. Note that the UCL spectra are normalized to the blue (Tm3+, 462–505 nm) emission. (B) Integrated intensity of blue (Tm3+, 462–505 nm), green (Er3+, 536–570 nm) and red (Er3+, 630–680 nm) emissions of CSS UCNPs with different Yb3+ concentrations in the outermost shell. Note that integrated emissions are normalized to the blue (Tm3+, 462–505 nm) emission. (C) UCL spectra of CSS (5 mol% Er3+) UCNPs with different Yb3+ concentrations in the core, where the Yb3+ concentration in the outermost shell remains intact at 40 mol%. Note that the UCL spectra are normalized to the green (Er3+, 536–570 nm) emission. (D) Integrated intensity of blue (Tm3+, 462–505 nm), green (Er3+, 536–570 nm) and red (Er3+, 630–680 nm) emissions of CSS UCNPs with different Yb3+ concentrations in the core. Note that integrated emissions are normalized to the green (Er3+, 536–570 nm) emission. | ||
Furthermore, we examined the impact of Yb3+ ions in the core on the complete UCL spectrum of our designed CSS UCNPs. We varied the concentration of Yb3+ ions in the core while fixing its concentration at 40 mol% in the outermost shell. As shown in Fig. 7C, as the concentration of Yb3+ ions is increased, the UCL intensity of blue emission increased concomitantly, reaching a maximum at 40 mol% concentration of Yb3+ ions, while hardly any change in the green and red emission intensity was observed.
From Fig. 7D, it is quite clear that the calculated integrated intensity of the blue (462–502 nm) emission of CSS UCNPs with 40 mol% Yb3+ concentration in the core is 2.5 and 1.6 times higher than that of CSS nanoparticles with 10 and 50 mol% Yb3+ concentrations in the core, respectively. In fact, the use of a high concentration of Yb3+ ions is expected to strongly affect the UCL intensity of the nanoparticles as Yb3+-mediated sub-lattice enables excitation energy migration from the primary sensitizer to the activator. Upon increase in Yb3+concentration, the total absorption cross-section of the same increases which again leads to the enhanced efficiency of ET from Nd3+ (middle layer domain) to Tm3+ (core region) and Er3+ (outermost shell). Moreover, a higher concentration of Yb3+ ions resulted in shorter Yb3+–Yb3+ interionic distance within the sublattice ensuring an efficient ET from Nd3+ to both the Tm3+-doped core and Er3+-doped outermost shell in CSS UCNPs. To our knowledge, literature reports suggest that the ET rate is proportional to r−6 (where, r is the average donor–acceptor distance) within the dipole interaction.58,59 Based on this fundamental, we assume that a suitable shell thickness (2.5 nm for CS and 3.5 nm for CSS, in our work) and high concentration of Nd3+ ions in the middle layer along with a high concentration of Yb3+ ions in both the core and outermost shell not only allow our designed UCNPs to migrate excitation energy directionally across the core/shell interface but also prevent the excitation energy from travelling long Nd3+–Nd3+, Yb3+–Yb3+ and Nd3+–Yb3+ distances. However, when the Yb3+–Yb3+ interionic distance exceeds the optimal value, back ET might occur from Tm3+ or Er3+ to Yb3+ ions, leading to the reduction of the UCL intensity from the nanoparticles. In this point of view, a high concentration of Yb3+ ions played a pivotal role in limiting the possibility of producing unwanted cross-relaxation pathways, while also affecting the process of bridging efficient ET from Nd3+ (middle layer) to Tm3+ (core region) and Er3+ (outermost shell) to produce efficient multicolor UCL from CSS UCNPs. Now, to check whether the use of a higher concentration of Yb3+ ions leads to the formation of bigger sized nanoparticles and simultaneously affect the UCL intensity of the same, we performed TEM measurements of core nanoparticles where the Yb3+ concentration was varied from 20 to 50 mol% while the concentration of other ions was kept unchanged. Almost no change in the size of the core nanoparticles was observed (Fig. S10†). Hence, from Fig. 7, we believe that the change in the UCL intensity of CSS (5 mol% Er3+) UCNPs is not related to the variation in size of the core but with the variation of Yb3+ ions in the core as well as in the outermost shell.
In addition, in our work, the Ca2+ ion has been chosen as dopant in each domain as the UCL intensity (438–503, blue emission) of the CS (30 mol% Nd3+) UCNPs with 7 mol% Ca2+ (in both core and shell) showed an around 1.4 times enhancement as compared to the same sample prepared without Ca2+ (Fig. S11†).
Very small sized NaGdF4 UCNPs possess a very high surface-to-volume ratio and, at the same time, a large portion of dopant ions occupy positions at the surface of the nanoparticles. Thus, the excitation energy is easily quenched either by surface defects or surface capping ligands. Therefore, it is essential to modify the host lattice and this is possible while Ca2+ ions are introduced into the NaGdF4 core (and even into the shell). Due to the lower valence, when Ca2+ is introduced to replace Gd3+, a charge imbalance occurs in NaGdF4. In addition, F− vacancies play an important role in charge compensation inside the crystal lattice. This induces the formation of transient electric dipoles with positive poles pointing outward on the grain surfaces. These transient electric dipoles accelerate the diffusion of ions for the crystal growth, needing F− ions from the solution to the grain and thus promoting the growth of NaGdF4. In this case, the growth of NaGdF4 nanoparticles is indeed a diffusion-controlled process, resulting in better size-uniformity and fewer surface defects which lead to the enhancement of the UCL intensity of the UCNPs. Moreover, the formation of F− vacancies induced by the substitution of Gd3+ with Ca2+ in the host lattice lowers the local crystal field symmetry around Yb3+, Nd3+, Er3+ and Tm3+ ions, which in turn affects the intra 4f–4f transition possibility of Ln3+ ions and subsequently enhances the UCL intensity from the CSS UCNPs.
Furthermore, to shed light on the position of the Nd3+-enriched harvesting layer (i.e., first shell or middle layer) in the CSS (5 mol% Er3+) UCNPs, we synthesized NaGdF4:Nd3+(30)/Ca2+(7)@NaGdF4:Nd3+(1)/Ca2+(7)/Tm3+(0.75)/Yb3+(40)@NaGdF4:Yb3+(40)/Nd3+(1)/Ca2+(7)/Er3+(5) CSS and NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Nd3+(1)/Ca2+(7)/Yb3+(40)/Er3+(5)@NaGdF4:Nd3+(30)/Ca2+(7) CSS UCNPs, whereas the Nd3+-enriched domain was placed in the core and outermost shell, respectively, and the energy transfer process (Nd3+ → Yb3+ → Er3+/Tm3+) was uni-directional. Fig. 8 clearly depicts that the UCL [blue or (red + green)] is only generated from Tm3+ or Er3+ ions from those CSS UCNPs. On the other hand, the Nd3+-enriched middle shell along with separation of Tm3+ (in core) and Er3+ ions (in outermost shell) enable the CSS UCNPs to produce efficient multicolor UCL (Fig. 8c). These results suggest that the ET in a bi-directional manner dominates the uni-directional one to achieve efficient multicolor UCL without implication of any deleterious cross-relaxation mechanism between the activator ions.
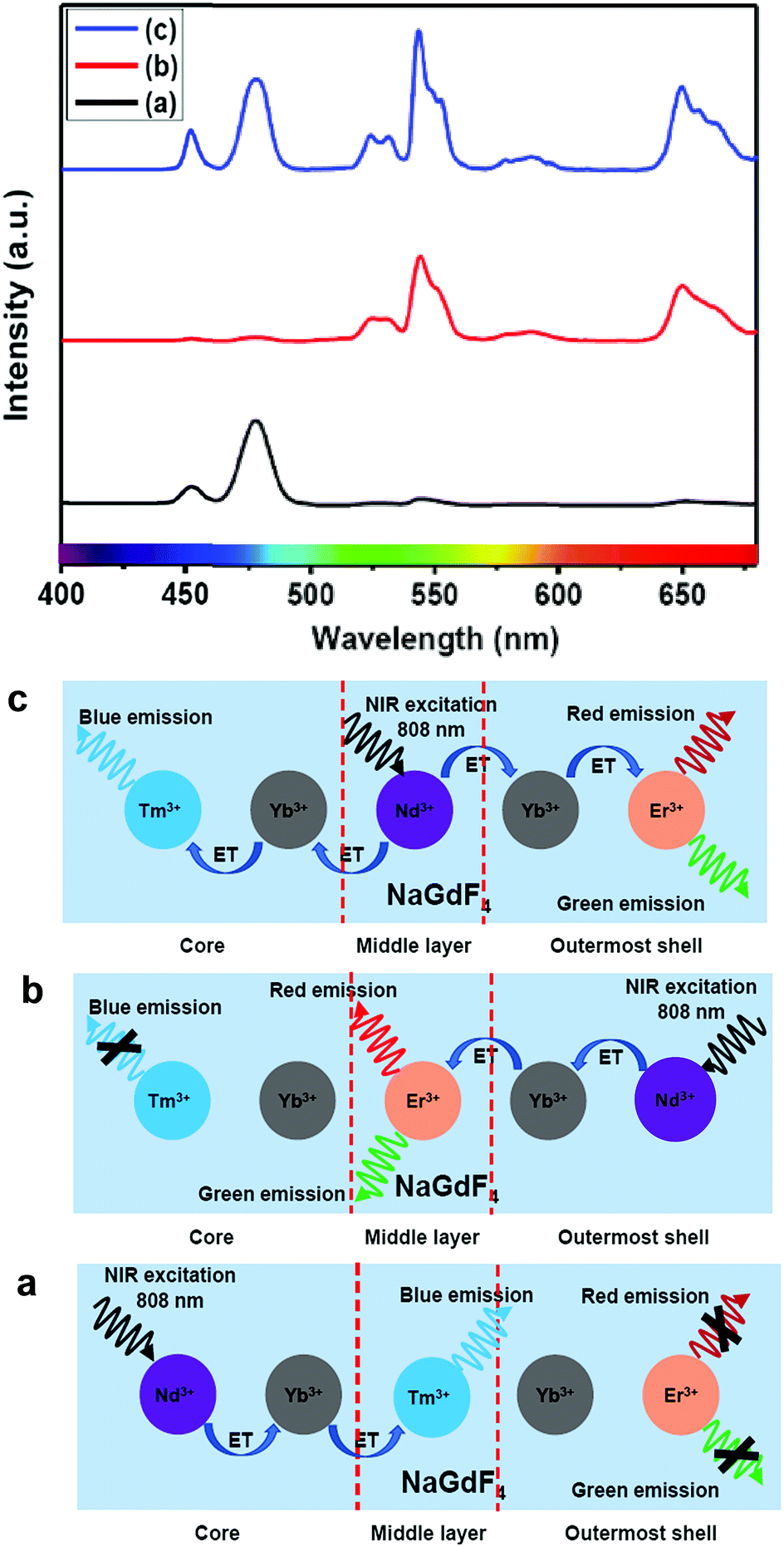 | ||
| Fig. 8 UCL spectra of (a) NaGdF4:Nd3+(30)/Ca2+(7)@NaGdF4:Nd3+(1)/Ca2+(7)/Tm3+(0.75)/Yb3+(40)@NaGdF4:Yb3+(40)/Nd3+(1)/Ca2+(7)/Er3+(5), (b) NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Nd3+(1)/Ca2+(7)/Yb3+(40)/Er3+(5)@NaGdF4:Nd3+(30)/Ca2+(7) and (c) NaGdF4:Tm3+(0.75)/Yb3+(40)/Nd3+(1)/Ca2+(7)@NaGdF4:Nd3+(30)/Ca2+(7)@NaGdF4:Yb3+(40)/Nd3+(1)/Ca2+(7)/Er3+(5) core/shell/shell UCNPs (808 nm laser excitation). Schematic illustration of the switching of the Nd3+− enriched domain to emit multicolor UCL from corresponding core/shell/shell UCNPs. | ||
Precise control of the concentration of Er3+ (activator) ions in the outermost shell of CSS UCNPs makes them potential candidates to produce efficient multicolor UCL output. There is hardly any change in the size (Fig. S12†) and size distribution (Fig. S13†) observed for CSS UCNPs with varying Er3+ concentration in the outermost shell. Moreover, it is evident from Fig. 9A that the appropriate tuning of the concentration of Er3+ ions in the outermost shell plays a vital role in the change in relative intensities of the green (4S3/2 → 4I15/2) and red (4F9/2 → 4I15/2) emissions of Er3+ ions, resulting in the continuous production of UCL colors ranging from blue to white. Such spectral changes could also be observed by the naked eye (Fig. 9B, right side). Furthermore, the calculated chromaticity coordinates (Fig. 9B) reveal a similar trend of the emission colors as perceived through spectroscopic study and naked eye visualization. It is worth mentioning that control in the use of each activator (e.g., Tm3+ or Er3+) in the CSS UCNPs allows manipulating and producing different UCL spectra with different colors. From Fig. S14,† it is quite clear that the core/shell/shell UCNPs (analogues to CSS and without Er3+ in the outermost shell) show pure blue emission at 451 and 477 nm from Tm3+ ions positioned in the core. However, core/shell/shell UCNPs (analogues to CSS and without Tm3+ in the core) exhibit both green (540 nm) and red (650 nm) emissions, which originate from Er3+ ions present in the outermost shell. On the other hand, our designed CSS (5 mol% Er3+) shows a three primary color (blue, green and red) blended spectrum responsible for generating white UCL light.
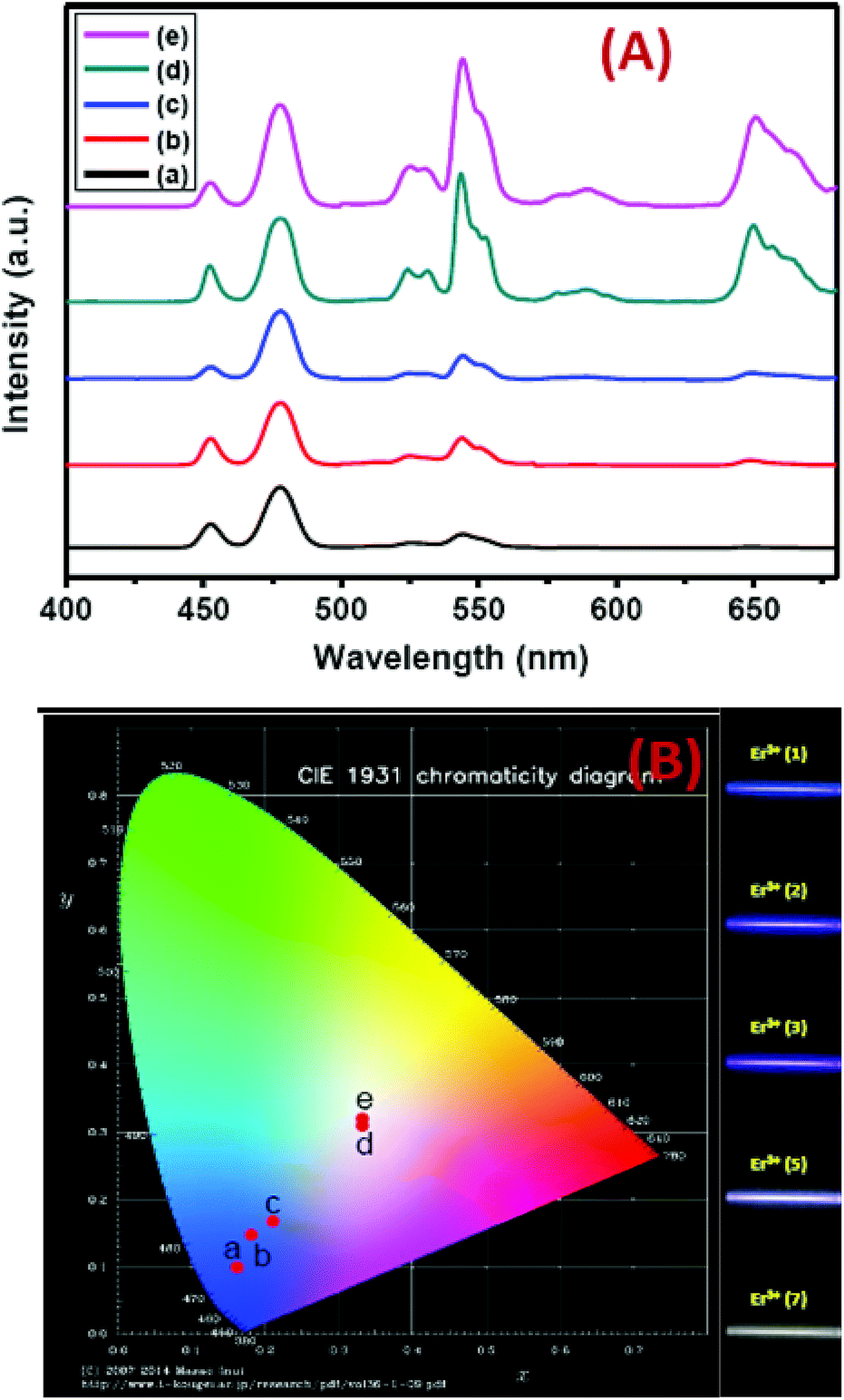 | ||
| Fig. 9 (A) UCL spectra (B) corresponding ‘Commission Internationale de l’Eclairage’ (CIE) and digital images of CSS UCNPs. a, b, c, d and e in the CIE diagram indicate the corresponding UCNPs, having Er3+ concentrations 1, 2, 3, 5 and 7 mol%, respectively, in the outermost shell. Digital images are taken using an 808 nm laser, 10 W cm−2 (without using any filter). | ||
To understand whether the middle shell layer-to-core (Nd3+ to Tm3+) ET or the middle shell layer-to-outermost shell (Nd3+ to Er3+) ET has higher contribution to the overall UCL to produce multicolor emission, as a control experiment, Tm3+ ions in the core and Er3+ ions in the shell of the CSS (5 mol% Er3+) were subjected to the corresponding direct excitations at 357 and 488 nm, respectively. As shown in Fig. S15,† the integrated PL emission intensity of Tm3+ ions (λexi = 357 nm) was slightly higher (1.3 times) than that of Er3+ ions (λexi = 488 nm). As the absorption coefficients of Tm3+ and Er3+ ions are lower and similar (∼2–10 M−1 cm−1),60 we may conclude that Tm3+ excited states favour a radiative decay pathway slightly more compared to that of Er3+ ions. However, the ratio between the integrated area of green + red (510–570 nm + 630–680 nm, Er3+) and blue (436–500 nm, Tm3+) regions in the UCL spectrum of the same CSS UCNPs was found to be 2.4 indicating higher contribution from Er3+. This is only possible if the ET from Nd3+ to Er3+ is more compared to Nd3+ to Tm3+ in the UCNPs.
Furthermore, to investigate the number of photons involved to produce white UCL, the power dependence of UCL was measured from CSS (5 mol% Er3+) UCNPs. Fig. S16† shows that the number of photons involved in multicolor UCL is 3 (slope, 3.01), 2 (slope, 1.98) and 2 (slope, 1.76) for blue, green and red emissions, respectively. These results are in good agreement with the literature reports.
Conclusion
In summary, we have successfully illustrated multicolor UCL from Nd3+-sensitized Gd3+-based core/shell/shell UCNPs under biologically relevant, single wavelength excitation (808 nm) which, unlike 980 nm excitation, does not cause biological heating problems that could damage cells and tissues. The separation of Tm3+ (in the core) and Er3+ (in the outermost shell layer) in the core/shell/shell UCNPs effectively alleviates the deleterious cross-relaxation processes between them. The doping of a Nd3+ sensitizer (30 mol%) in the middle layer, on the other hand, enables efficient harvesting of excitation light and concurrent migration of the energy across the core/shell interfaces to both Tm3+ in the core and Er3+ in the outermost shell layer. Precise control of the shell thickness and judicious manipulation of Ln3+ ions (activators and sensitizers) along with their spatial distribution in their defined layer domain provide a broad range of multicolor UCL (from blue to white) through a bi-directional ET process. Such tuneable multicolor UCL makes these sub-20 nm sized Nd3+-sensitized Gd3+-based core/shell/shell UCNPs potential candidates for bioimaging and MRI dual modal applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Brazilian agencies CNPq, CAPES and FAPESP for financial support. C. Hazra, S. Ullah and L. R. Lima acknowledge financial support from the São Paulo Research Foundation (FAPESP, Brazil) under fellowship grant numbers 2015/18733-0, 2015/22875-4 and 2014/05948-5, respectively. Y. E. S. Correales thanks FAPESP for PhD fellowship (grant no. 2018/15791-7) and CNPq for master degree fellowship (grant no. 133105/2017-2). The authors thank N. A. F. Perruci for the help in XRD measurements.Notes and references
- Y. Li, X. Yu and T. Yu, J. Mater. Chem. C, 2017, 5, 5411–5419 RSC.
- H. Nie, M. Li, Q. Li, S. Liang, Y. Tan, L. Sheng, W. Shi and X.-A. A. Zhang, Chem. Mater., 2014, 26, 3104–3112 CrossRef CAS.
- M. X. Zhao, Y. Li, E. Z. Zeng and C. J. Wang, Chem.–Asian J., 2014, 9, 1349–1355 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldmani and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- D. Qu, M. Zheng, J. Li, Z. Xie and Z. Sun, Light: Sci. Appl., 2015, 4, 364–371 CrossRef.
- Z. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765–4769 CrossRef CAS.
- H. Kim, Y. Park, S. Beack, S. Han, D. Jung, H. J. Cha, W. Kwon and S. K. Hahn, Adv. Sci., 2017, 4, 1700325 CrossRef PubMed.
- J. Chen, R. V. Dalal, A. N. Petrov, A. Tsai, S. E. O'Leary, K. Chapin, J. Cheng, M. Ewan, P.-L. Hsiung, P. Lundquist, S. W. Turner, D. R. Hsu and J. D. Puglisi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 664–669 CrossRef CAS PubMed.
- Y.-P. Ho, M. C. Kung, S. Yang and T.-H. Wang, Nano Lett., 2005, 5, 1693–1697 CrossRef CAS PubMed.
- Y. Leng, K. Sun, X. Chen and W. Li, Chem. Soc. Rev., 2015, 44, 5552–5595 RSC.
- N. Rohland and D. Reich, Genome Res., 2012, 22, 939–946 CrossRef CAS PubMed.
- J. Zoll, J. Rahamat-Langendoen, I. Ahout, M. I. de Jonge, J. Jans, M. A. Huijnen, G. Ferwerda, A. Warris and W. J. Melchers, J. Clin. Virol., 2015, 66, 6–11 CrossRef CAS PubMed.
- Q. Fu, F. S. Schoenhoff, W. J. Savage, P. Zhang and J. E. Van Eyk, Proteomics: Clin. Appl., 2010, 4, 271–284 CAS.
- Y. I. Park, K. T. Lee, Y. D. Suh and T. Hyeon, Chem. Soc. Rev., 2015, 44, 1302–1317 RSC.
- L. Zhou, R. Wang, C. Yao, X. Li, C. Wang, C. Zhang, C. Xu, A. Zeng, D. Zhao and F. Zhang, Nat. Commun., 2015, 6, 6938 CrossRef CAS PubMed.
- L. Cheng, K. Yang, S. Zhang, M. Shao, S. Lee and Z. Liu, Nano Res., 2010, 3, 722–732 CrossRef CAS.
- H. H. Gorris, R. Ali, S. M. Saleh and O. S. Wolfbeis, Adv. Mater., 2011, 23, 1652–1655 CrossRef CAS PubMed.
- F. Zhang, Q. Shi, Y. Zhang, Y. Shi, K. Ding, D. Zhao and G. D. Stucky, Adv. Mater., 2011, 23, 3775–3779 CAS.
- S. Ullah, C. Hazra, E. P. Ferreira-Neto, T. C. Silva, U. P. Rodrigues-Filho and S. J. L. Ribeiro, CrystEngComm, 2017, 19, 3465–3475 RSC.
- Z. Li, Y. Zhang, H. La, R. Zhu, G. El-Banna, Y. Wei and G. Han, Nanomaterials, 2015, 5, 2148–2168 CrossRef CAS PubMed.
- J. Zhang, Y. Huang, L. Jin, F. Rosei, F. Vetrone and J. P. Claverie, ACS Appl. Mater. Interfaces, 2017, 9, 8142–8150 CrossRef CAS PubMed.
- C. Hazra, V. N. K. B. Adusumalli and V. Mahalingam, ACS Appl. Mater. Interfaces, 2014, 6, 7833–7839 CrossRef CAS PubMed.
- P. A. Rojas-Gutiérrez, C. E. DeWolf and J. A. Capobianco, Part. Part. Syst. Charact., 2016, 33, 865–870 CrossRef.
- Q. Chen, X. Xie, B. Huang, L. Liang, S. Han, Z. Yi, Y. Wang, Y. Li, D. Fan, L. Huang and X. Liu, Angew. Chem., 2017, 129, 7713–7717 CrossRef.
- C. Hazra, S. Ullah, Y. E. Serge-Correales, L. G. Caetano and S. J. L. Ribeiro, J. Mater. Chem. C, 2018, 6, 4777–4785 RSC.
- J. C. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- C. Dong and F. C. J. M. van Veggel, ACS Nano, 2009, 3, 123–130 CrossRef CAS PubMed.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- C. D. S. Brites, X. Xie, M. L. Debasu, X. Qin, R. Chen, W. Huang, J. Rocha, X. Liu and L. D. Carlos, Nat. Nanotechnol., 2016, 11, 851–856 CrossRef CAS PubMed.
- K. Nigoghossian, S. Ouellet, J. Plain, Y. Messaddeq, D. Boudreau and S. J. L. Ribeiro, J. Mater. Chem. B, 2017, 5, 7109–7117 RSC.
- G. Jalani, R. Naccache, D. H. Rosenzweig, L. Haglund, F. Vetrone and M. Cerruti, J. Am. Chem. Soc., 2016, 138, 1078–1083 CrossRef CAS PubMed.
- S. Hao, G. Chen and C. Yang, Theranostics, 2013, 3, 331–345 CrossRef PubMed.
- Y. Zhang, Z. Yu, J. Li, Y. Ao, J. Xue, Z. Zeng, X. Yang and T. T. Y. Tan, ACS Nano, 2017, 11, 2846–2857 CrossRef CAS PubMed.
- J. Peng, W. Xu, C. L. Teoh, S. Han, B. Kim, A. Samanta, J. C. Er, L. Wang, L. Yuan, X. Liu and Y. T. Chang, J. Am. Chem. Soc., 2015, 137, 2336–2342 CrossRef CAS PubMed.
- X. Ai, C. J. Ho, J. Aw, A. B. Attia, J. Mu, Y. Wang, X. Wang, Y. Wang, X. Liu, H. Chen, M. Gao, X. Chen, E. K. Yeow, G. Liu, M. Olivo and B. Xing, Nat. Commun., 2016, 7, 10432 CrossRef CAS PubMed.
- Y. Huang, A. Skripka, L. Labrador-Páez, F. Sanz-Rodríguez, P. Haro-González, D. Jaque, F. Rosei and F. Vetrone, Nanoscale, 2018, 10, 791–799 RSC.
- P. Cortelletti, A. Skripka, C. Facciotti, M. Pedroni, G. Caputo, N. Pinna, M. Quintanilla, A. Benayas, F. Vetrone and A. Speghini, Nanoscale, 2018, 10, 2568–2576 RSC.
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- J. Xu, W. Han, Z. Cheng, P. Yang, H. Bi, D. Yang, N. Niu, F. He, S. Gai and J. Lin, Chem. Sci., 2018, 9, 3233–3247 RSC.
- H. Li, R. Wei, G.-H. Yan, J. Sun, C. Li, H. Wang and L. Shi, ACS Appl. Mater. Interfaces, 2018, 10, 4910–4920 CrossRef CAS PubMed.
- X. Xie and X. Liu, Nat. Mater., 2012, 11, 842–843 CrossRef CAS PubMed.
- X. Xie, N. Gao, R. Deng, Q. Sun, Q. H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 135, 12608–12611 CrossRef CAS PubMed.
- Q. Duo, N. M. Idris and Y. Zhang, Biomaterials, 2013, 34, 1722–1731 CrossRef PubMed.
- F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924–2929 CrossRef CAS.
- H. S. Qian and Y. Zhang, Langmuir, 2008, 24, 12123–12125 CrossRef CAS PubMed.
- Y. F. Wang, G. Y. Liu, L. D. Sun, J. W. Xiao, J. C. Zhou and C. H. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed.
- X. Li, Z. Guo, T. Zhao, Y. Lu, L. Zhou, D. Zhao and F. Zhang, Angew. Chem., 2016, 128, 2510–2515 CrossRef.
- G. Chen, J. Damasco, H. Qiu, W. Shao, T. Y. Ohulchanskyy, R. R. Valiev, X. Wu, G. Han, Y. Wang, C. Yang, H. Ågren and P. N. Prasad, Nano Lett., 2015, 15, 7400–7407 CrossRef CAS PubMed.
- D. Chen, L. Liu, P. Huang, M. Ding, J. Zhong and Z. Ji, J. Phys. Chem. Lett., 2015, 6, 2833–2840 CrossRef CAS PubMed.
- J. Xu, P. Yang, M. Sun, H. Bi, B. Liu, D. Yang, S. Gai, F. He and J. Lin, ACS Nano, 2017, 11, 4133–4144 CrossRef CAS PubMed.
- F. Liégard, J. L. Doualan, R. Moncorgé and M. Bettinelli, Appl. Phys. B, 2005, 80, 985–991 CrossRef.
- S. Hao, G. Chen, C. Yang, W. Shao, W. Wei, Y. Liu and P. N. Prasad, Nanoscale, 2017, 9, 10633–10638 RSC.
- C. Zhao, X. Kong, X. Liu, L. Tu, F. Wu, Y. Zhang, K. Liu, Q. Zeng and H. Zhang, Nanoscale, 2013, 5, 8084–8089 RSC.
- L. Lei, D. Chen, J. Xu, R. Zhang and Y. Wang, Chem.–Asian J., 2014, 9, 728–733 CrossRef CAS PubMed.
- G. Chen, C. Yang and P. N. Prasad, Acc. Chem. Res., 2013, 46, 1474–1486 CrossRef CAS PubMed.
- F. Wang, R. Deng and X. Liu, Nat. Protoc., 2014, 9, 1634–1644 CrossRef CAS PubMed.
- B. Zhou, W. Yang, S. Han, Q. Sun and X. Liu, Adv. Mater., 2015, 27, 6208–6212 CrossRef CAS PubMed.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- T. Samanta, A. E. Praveen and V. Mahalingam, J. Mater. Chem. C, 2018, 6, 4878–4886 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Histograms, TEM, infrared spectra, EDS, absorption spectra, XRD, UCL spectra, PL emission spectra, and calculation of the number of photons. See DOI: 10.1039/c9na00006b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |