
The structural fate of lipid nanoparticles in the extracellular matrix†
Sarith R.
Bandara
 a,
Thomas G.
Molley
b,
Hojun
Kim
c,
Priyalini A.
Bharath
a,
Kristopher A.
Kilian
*b and
Cecilia
Leal
*a
a,
Thomas G.
Molley
b,
Hojun
Kim
c,
Priyalini A.
Bharath
a,
Kristopher A.
Kilian
*b and
Cecilia
Leal
*a
aDepartment of Materials Science and Engineering, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: cecilial@illinois.edu
bSchool of Chemistry, School of Materials Science and Engineering, Australian Centre for Nanomedicine, University of New South Wales, Sydney NSW 2052, Australia. E-mail: k.kilian@unsw.edu.au
cCenter for Biomaterials, Biomedical Research Institute, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea
First published on 31st July 2019
Abstract
Drug-loaded liposomes are the most successful nanomedicine to date, with multiple FDA-approved systems for a myriad of diseases. While liposome circulation time in blood and retention in tissues have been studied in detail, the structural fate of liposomes—and nanoparticles in general—in the body has not been extensively investigated. Here, we explore the interactions of liposomes with synthetic and natural hydrogel materials to understand how the natural extracellular matrix influences liposome structural characteristics. Small angle X-ray scattering, confocal microscopy, and cryogenic transmission electron microscopy data demonstrate that poly(ethylene glycol) (PEG), gelatin, alginate, and Matrigel® hydrogels cause 200 nm liposomes of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) to transform into micrometer-sized aggregates. These aggregates are composed of multilamellar vesicles around 100 nm in diameter with a mean interlamellar separation of 5.5 nm. Protecting the liposomes with a corona of PEG damps this restructuring effect, making the multilamellar vesicles less stable. We attribute this unilamellar to multilamellar transition to an osmotic driving force from the hydrogel environment. This lipid restructuring has broad ramifications in the design and use of nanomedicines, and in understanding the fate and function of natural lipid-based materials within the tissue microenvironment.
New conceptsUsing robust structural characterization techniques (X-ray scattering and Cryo-EM), we have demonstrated that lipid nanoparticles get significantly reconfigured when exposed to the materials of the extracellular milieu. The study and design of nanoparticles for the delivery of cargo to cells mostly focus on their biochemical interactions with cells and cell organelles. This new concept highlighting the structural fate of nanoparticles even before they reach cells will critically impact the design of soft materials, lipid nanoparticles in particular, for the delivery of cargo to cells. The majority of drug-delivery nanomedicines being used in the clinic are some form of a lipid vesicle. Future systems in therapeutics and diagnosis will go beyond synthetic vesicles as recent discoveries demonstrate that natural extracellular vesicles are secreted to biological fluids and mark for a variety of diseases. We show that extracellular hydrogels dehydrate and significantly restructure lipid vesicles. Specifically, they impose a phase transformation from single-membrane vesicles to multilamellar bodies, which we attribute to an osmotic stress effect. We argue that such structural transformations might not be a mere consequence of biomolecular interactions but have a regulatory function in living systems, and can be exploited to design more efficient therapeutic and diagnosis materials. |
Introduction
Nanomedicine has gained the spotlight in the last two decades as a means to more effectively treat disease,1 particularly cancer. In the realm of drug delivery, small molecule drugs already known to be effective against cancer have been formulated into nanoparticles to render them more potent by increasing their time in circulation and by making them specifically target the desired location, concomitantly lowering toxic effects to healthy cells. For a nanoparticle to reach a tumor site, it must evade the body's mononuclear phagocytic system, which predominantly clears particles larger than a few hundred nanometers in the liver and spleen,2 and renal clearance, which clears particles smaller than around five nanometers in the kidneys.3–5 Evidently, nanoparticles between 5 and 100 nm in size are tailored for this purpose. Once a nanoparticle survives the liver, the spleen, and the kidneys, it must also persist in the extracellular matrix (ECM) of the tumor cells before it can finally reach the tumor. The enhanced permeability and retention (EPR) effect—where leaky vasculature in the tumor allows larger nanoparticles to enter and compromised lymphatic drainage hinders the nanoparticles from leaving—increases the likelihood that the nanoparticle enters and remains in the tumor.6 Even though the EPR effect has been exploited widely to target tumors, how effective it actually is on nanoparticle retention has recently been questioned.7 Many studies have focused on nanoparticle retention in tissues. Yet the structural fate of nanoparticles in the body remains largely unexplored.A distinction has been made between the synthetic identity and the biological identity of a nanoparticle, where the biological identity refers to the structure of the nanoparticle in the body, which can be different from its synthesized structure.8–10 Proteins binding to nanoparticle surfaces result in a protein “corona” that imparts the nanoparticle with physiological responses different from those of the original protein-devoid nanoparticle.8–10 Interactions of proteins with nanoparticles made of various materials such as gold, copolymers, quantum dots, and lipids have been investigated.8,9,11 Doxil®, a liposomal formulation of the cancer drug doxorubicin, is one of few non-targeted nanoparticle formulations approved for clinical use.12,13 Since lipids, the major component of cell membranes, are ubiquitous in nature, they are associated with fewer toxic effects than other nanomaterials in drug delivery applications. Indeed, of the nanomedicine drugs approved by the Federal Drug Administration (FDA), lipid-based systems constitute the vast majority and are the leading material in future drug development systems.14,15 In addition, lipids exist in numerous complex structures within the ECM including exosomes and several other lipid-based extracellular vesicles,16 which have been implicated in ECM modulation within several physiological and pathological contexts.17 To prevent protein adsorption onto nanoparticles, poly(ethylene glycol) (PEG) has been incorporated onto the surfaces of nanoparticles via the inclusion of a lipid that is covalently linked to PEG, effectively “cloaking” the particles to evade protein interactions, thus prolonging their circulation life-time.18 Even though the fate of liposomes after endocytosis has been extensively investigated, with liposomes observed to form aggregates in the micrometer scale,19,20 the structural fate of liposomes after extravasation into tissue and transport through the ECM prior to engaging the cell membrane remains a mystery.
The ECM is a natural hydrogel comprising proteoglycans, polysaccharides, proteins, and water.21 A bulk of the prior work with synthetic hydrogels and liposomes has been conducted in the context of drug delivery, where liposomes are encapsulated within hydrogel networks to enhance biocompatibility, biodegradability, and controlled release.22 The osmotic pressure of proteoglycans, a major component of the ECM, is evaluated to range from 30 to 350 kPa.23,24 Under these conditions, significant structural transformations have been shown to occur in a myriad of soft materials in vivo. Examples include dehydration and conformation change of DNA molecules,25 cytoskeleton filamentous protein bundle formation,26 and DNA ejection from phage viruses.27 Similarly, we can expect the osmotic stress generated in the ECM to have the power to influence the structure of lipid-based particles.
Synthetic hydrogels—which have been used increasingly as 3D matrices in tissue engineering—have numerous advantages over natural materials when exploring ECM structure–function relationships, including control over molecular weight, cross-link density and architecture, diffusion, swelling, degradation, and osmotic pressure.28–30 One synthetic hydrogel material commonly used in biomedical applications is a network of PEG chains formed through addition polymerization or condensation reactions between multiarmed-PEG macromolecules.31 As a model hydrogel, PEG is arguably the most widely explored synthetic matrix for tissue engineering32 and has previously been used as a platform to study the diffusion of proteins33 and nanomaterials.34 The osmotic pressure of an aqueous solution of 20 kDa, 15 wt% PEG is 260 kPa, and this osmotic pressure can be controlled by changing the molecular weight or the weight percentage of PEG.25 Since the osmotic pressure of hydrophilic PEG hydrogels is well within the osmotic pressure range of the ECM—with tunability through control of molecular weight and crosslinking—PEG is a well-suited synthetic matrix to model the ECM.
This work elucidates in detail the structures of liposomes in synthetic and natural hydrogel systems at several length scales, with the aim of understanding the structural fate of liposomes in the ECM before they even reach their intended cellular destination. Small angle X-ray scattering (SAXS), confocal microscopy, and cryogenic transmission electron microscopy (cryo-EM) results demonstrate that poly(ethylene glycol), gelatin, alginate, and Matrigel® hydrogels cause 200 nm liposomes of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) to transform into micrometer-sized aggregates composed of multilamellar vesicles around 100 nm in diameter. Incorporating PEG-lipids in the liposomes using 8 mol% of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DOPE-PEG2000) attenuates this restructuring effect, making the multilamellar vesicles less stable. We attribute this restructuring effect to an osmotic driving force from the hydrogel on the liposomes.
Results and discussion
Lipids assemble hierarchically into a variety of structures, including single bilayers, multilamellar stacks, 2D hexagonal phases, and even cubic phases.35–39 The stability of a lipid mesophase is dictated by molecular packing, membrane elasticity, and environmental factors. For example, a single bilayer vesicle will adopt a multilamellar stack architecture as it incorporates DNA.40 To determine how single bilayers enclosed in a lipid vesicle are affected by immersion into a continuous matrix made of hydrated polymer chains (hydrogels), we used a combination of techniques including optical microscopy, electron microscopy, and SAXS.Structural changes of DOPC liposomes in polyethylene glycol
Fig. 1A shows SAXS data of DOPC liposomes that were added to uncrosslinked PEG. Three peaks were observed at q = 0.109 Å−1, 0.214 Å−1, and 0.327 Å−1. The ratio between the q positions of these peaks is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3, which is known to correspond to a multilamellar structure. The repeat spacing d (thickness of water plus lipid layers) can be obtained by SAXS as d001 = 2π/q001 = 5.8 nm. The blue SAXS curve was produced by uncrosslinked PEG with no liposomes. When liposomes are analyzed in the absence of hydrogel (Fig. S1, ESI†), no multilamellar peaks are detected, and a SAXS profile characteristic of the form factor of a single lipid bilayer is observed instead. These data suggest that uncrosslinked PEG causes unilamellar liposomes to transform into multilamellar structures. The domain size of the multilamellar structures is determined to be 56 nm using the full-width at half maximum of the first peak in the Scherrer formula.41 Even though it is not possible to determine the fraction of the liposomes that become multilamellar, since SAXS is a bulk average method, the presence of intense multilamellar peaks suggests that a significant fraction has assumed multilamellar structure. We expect the multilamellar structures to be interspersed among the PEG network, which would not be much of a barrier to liposome motion since the uncrosslinked PEG chains are pliable. To the right of the SAXS curves is a blown up (not to scale) illustration of a quartz capillary with uncrosslinked PEG and multilamellar lipid constructs. Based on just the SAXS data, the lipid morphologies could be multilamellar sheets, vesicles, or a hybrid of the two.
:2:3, which is known to correspond to a multilamellar structure. The repeat spacing d (thickness of water plus lipid layers) can be obtained by SAXS as d001 = 2π/q001 = 5.8 nm. The blue SAXS curve was produced by uncrosslinked PEG with no liposomes. When liposomes are analyzed in the absence of hydrogel (Fig. S1, ESI†), no multilamellar peaks are detected, and a SAXS profile characteristic of the form factor of a single lipid bilayer is observed instead. These data suggest that uncrosslinked PEG causes unilamellar liposomes to transform into multilamellar structures. The domain size of the multilamellar structures is determined to be 56 nm using the full-width at half maximum of the first peak in the Scherrer formula.41 Even though it is not possible to determine the fraction of the liposomes that become multilamellar, since SAXS is a bulk average method, the presence of intense multilamellar peaks suggests that a significant fraction has assumed multilamellar structure. We expect the multilamellar structures to be interspersed among the PEG network, which would not be much of a barrier to liposome motion since the uncrosslinked PEG chains are pliable. To the right of the SAXS curves is a blown up (not to scale) illustration of a quartz capillary with uncrosslinked PEG and multilamellar lipid constructs. Based on just the SAXS data, the lipid morphologies could be multilamellar sheets, vesicles, or a hybrid of the two.
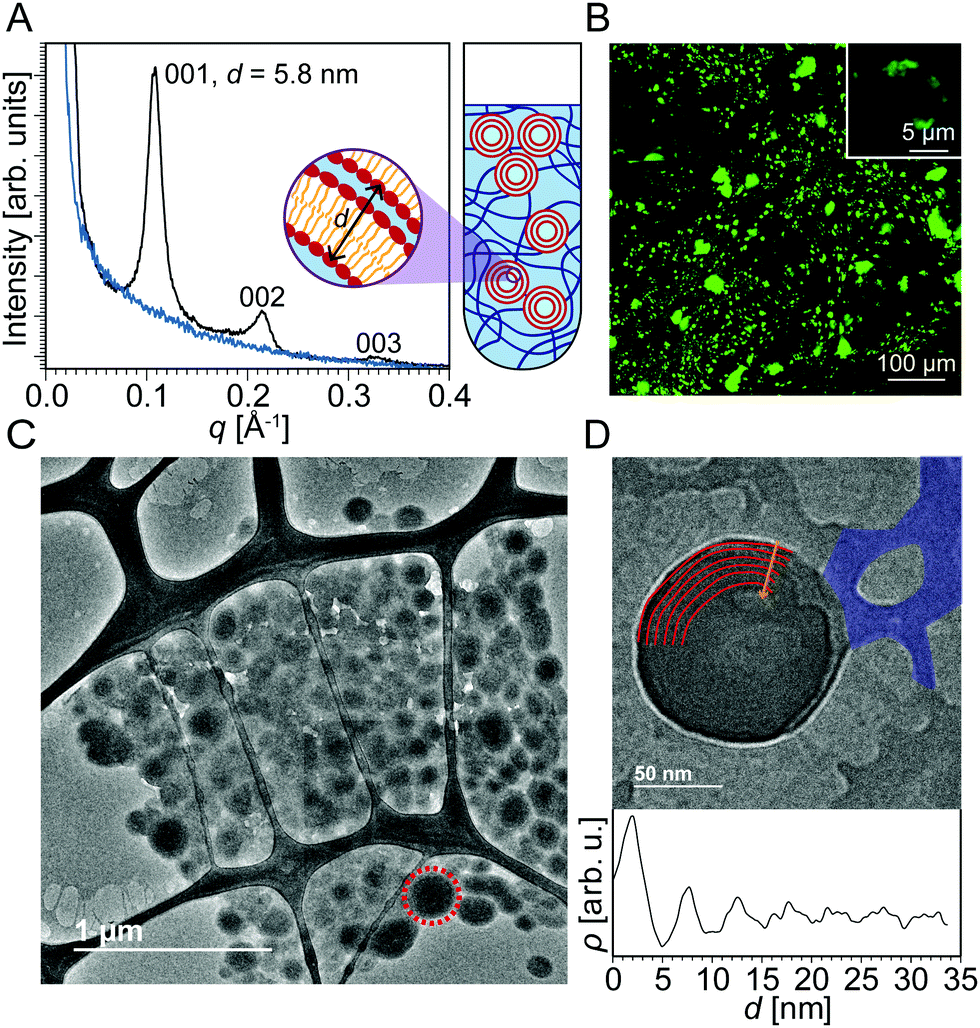 | ||
| Fig. 1 (A) SAXS data of DOPC liposomes added to uncrosslinked PEG (black) display three peaks at q = 0.109 Å−1, 0.214 Å−1, and 0.327 Å−1. SAXS data of uncrosslinked PEG only (blue) show a steady decay in intensity with no peaks. A schematic representation to the right of the SAXS data depicts multilamellar lipid aggregates and the uncrosslinked PEG network in a quartz capillary. The interlamellar repeat spacing d = 5.8 nm. (B) Confocal microscopy of the uncrosslinked PEG/liposome composite displays aggregates several micrometers in size. The inset is at a higher magnification. (C) A cryo-EM image of the PEG/liposome composite displays micrometer-sized aggregates of multilamellar vesicles a few hundred nanometers in diameter. (D) A cryo-EM image of one multilamellar vesicle. An arc of each layer of the multilamellar wall of the vesicle is outlined in red. At the bottom is a plot of image intensity versus distance along the yellow arrow. A series of high-low contrast patterns indicates a mean interlamellar spacing of 5.2 nm. A section of the PEG network has been shaded in blue. | ||
To support the SAXS data, we employed confocal microscopy to image the lipid structures in both the presence and absence of hydrogels. Liposomes were imaged with confocal microscopy (Fig. 1B) by doping the lipid aggregates with 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (NBD-PE), an amphiphilic dye that integrates well within the liposome bilayers. When liposomes are imaged in the absence of a hydrogel network (Fig. S1, ESI†), only a halo of green is observed because the resolution of the microscope is just around the size of individual liposomes. In contrast, for liposomes integrated within the PEG network, large green aggregates several micrometers in size are revealed. This observation also suggests that the hydrogel is destabilizing the liposomes and inducing some form of aggregation.
To probe the identity of these supramolecular aggregates at higher resolution, we employed cryo-EM. Cryo-EM images of individual liposomes and hydrogels are shown in Fig. S1 and S2 of ESI.† When hydrogels and liposomes are combined, very different morphologies are observed (Fig. 1C). The network characteristic of the hydrogel is still present in addition to several multilamellar vesicles a few hundred nanometers in diameter, most of which locally aggregate into clusters. These morphologies are in sharp contrast to the unilamellar liposome structures observed in the absence of a hydrogel network. This result corroborates the data obtained by confocal microscopy where aggregates several micrometers in size are observed. Fig. 1D displays a thinner sample section where one multilamellar vesicle is discerned within the surrounding hydrogel network. An arc of each layer of the multilamellar wall of the vesicle is outlined in Fig. 1D. A plot of image intensity versus distance demonstrates a series of high-low contrast patterns that indicate that the mean interlamellar spacing of the multilamellar aggregate is 5.2 nm, which is in close agreement with the interlamellar spacing of 5.8 nm obtained by SAXS. A possible mechanism for this unilamellar to multilamellar transformation could be a phase separation effect that causes the liposomes to aggregate together, minimizing interactions with the hydrogel. The surface-area-to-volume ratio of the multilamellar vesicles is 6.3 times higher than that of the liposomes, suggesting better drug-loading capability.
Multilamellar restructuring of DOPC liposomes in crosslinked polyethylene glycol hydrogels
The result that PEG restructures liposomes is surprising because we anticipated that hydrogels offer a very fluid environment with sufficient water for liposomes to integrate unperturbed. However, our results indicate that the fluid environment may facilitate aggregation. To investigate this phase separation conjecture, we tested the effect of crosslinking PEG, which would result in a less fluid environment that restricts liposome mobility, but would still be representative of the crowded cellular environment with limited free water.42 We synthesized PEG with distal acryloyl moieties for radical polymerization as described previously43 using chemical crosslinking via ammonium persulfate (APS) and tetramethylethylenediamine (TEMED). Fig. 2A shows the SAXS results of that sample where a broad peak was observed at q = 0.044 Å−1 in addition to the peaks at the same q as in Fig. 1A corresponding to multilamellar structures. Waters et al. have previously used SAXS to characterize crosslinked PEG and have attributed the broad peak to a correlation length between densely crosslinked junctions in PEG.44 In the PEG/liposome composite sample, where the PEG was crosslinked in the presence of liposomes, the broad peak at q = 0.044 Å−1 corresponds to a correlation length of 14 nm. Fig. S2 of ESI† shows the SAXS profile of crosslinked PEG chains without any liposomes. The end-to-end distance r0 of the PEG network can be calculated using the equation r0 = bNν, where b is the Kuhn length for PEG (0.76 nm), N is the number of Kuhn segments (130 for 10 kDa PEG), and ν is the scaling exponent (depends on the model used).45 The correlation length almost matches the end-to-end distance of a 3D self-avoiding random walk model, for which ν = 0.588.46 Since the SAXS pattern obtained by scanning through the whole volume of the quartz capillary showed the PEG correlation length in addition to the peaks from the multilamellar lipids (Fig. 2A), it can be inferred that the multilamellar lipid constructs are well-integrated within the PEG hydrogel network. | ||
| Fig. 2 (A) SAXS data of PEG crosslinked in the presence of DOPC liposomes (black) display a broad peak at q = 0.044 Å−1 and two peaks at q = 0.109 Å−1 and 0.214 Å−1. SAXS data of just crosslinked PEG (blue) show only the broad peak at q = 0.044 Å−1. A schematic representation to the right of the SAXS data depicts multilamellar lipid aggregates and the crosslinked PEG network in a quartz capillary. The interlamellar repeat spacing d = 5.8 nm. The correlation length of crosslinked PEG *L = 14 nm. (B) Confocal microscopy of the crosslinked PEG/liposome composite displays aggregates several micrometers in size. The inset is at a higher magnification. (C) DOPC liposomes added to already crosslinked PEG yields two layers (schematic representation on right). SAXS data of the lipid layer (red) display peaks at q = 0.109 Å−1, 0.214 Å−1, and 0.327 Å−1. SAXS data of the PEG layer (blue) display a broad peak at q = 0.044 Å−1. The interlamellar repeat spacing d = 5.9 nm. The correlation length of crosslinked PEG *L = 14 nm. 2D SAXS scans of the two layers are shown next to the schematic. | ||
Waters et al. used free-radical photopolymerization reactions to crosslink PEG, whereas we used free-radical chemical polymerization. Even though Waters et al. state that the polymerization method could influence crosslink functionality and nanoscale morphology, both methods—photo- and chemical polymerization—produced correlation peaks in SAXS, though this result does not necessarily imply that the two differently crosslinked hydrogels have identical morphologies. Importantly, these results for crosslinked PEG demonstrate that a gel network with smaller, less pliable pores does little to prevent liposome restructuring. We conclude that the liposome restructuring process is unlikely due to phase separation. Fig. 2B shows a confocal microscope image of crosslinked PEG in the presence of liposomes revealing large lipid aggregates analogous to what was observed in the uncrosslinked PEG/liposome sample.
To further investigate liposome integration within hydrogels, an aqueous solution of liposomes was added to an already crosslinked PEG hydrogel. In this case, by scanning the whole volume of the quartz capillary, two distinct SAXS patterns were detected in different regions of the sample tube. Fig. 2C shows the SAXS pattern obtained at the bottom of the sample tube that unsurprisingly depicted the characteristic peak of crosslinked PEG. Interestingly, the top of the sample tube yields a SAXS pattern that is consistent with lipid layers arranged in a multilamellar structure showing three structure-factor peaks: (001), (002), and (003). SAXS is a bulk average method, so for these intense multilamellar peaks to appear, a significantly high number of multilamellar structures should be present in the lipid region. These data demonstrate that regardless of the extent of physical integration of liposomes within the hydrogel network, hydrogels still induce liposome restructuring. In all three scenarios—DOPC liposomes integrated within uncrosslinked PEG, DOPC liposomes integrated within uncrosslinked PEG followed by PEG crosslinking, and DOPC liposomes added to already crosslinked PEG—liposomes underwent transformation into multilamellar aggregates. PEG is known to generate high osmotic stress environments and has been used in a technique to infer intermolecular interactions of soft materials dispersed in water.25,47 An aqueous solution of 20 kDa, 15 wt% PEG can exert a pressure of 260 kPa while an aqueous solution of 20 kDa, 60 wt% PEG can exert a pressure as high as 11 MPa.25 One could argue that physiological salinity yields similar osmotic pressure to that of PEG. However, in contrast to long PEG chains, small ions move freely within aqueous interstitials. Consequently, the driving force to pull water by a concentrated PEG solution is much more prominent than that of a salt solution of the same nominal osmotic pressure. We conjecture that in the systems investigated here, PEG polymer chains of the hydrogel are too bulky to penetrate small water gaps, pulling water from liposomes and inducing their fusion into multilamellar aggregates.
Hydrogel physical properties influence liposome restructuring
To test this hypothesis, we investigated different hydrogels—gelatin, alginate, poly(acrylamide) (PAM), and Matrigel® in addition to PEG—that display distinct crosslinking mechanisms and swelling behavior. Hydrogels are formed in gelatin through interstrand H-bonds that stabilize triple helices,48 alginate through divalent cations,49 PAM through radical-initiated monomer polymerization, and PEG through end-functionalized radical cross-linking. Matrigel®, a reconstituted basement membrane preparation comprising several ECM proteins extracted from murine tumors,50,51 is an actual ECM hydrogel. Livney et al. demonstrated that there is a linear correlation between the swelling ratio and osmotic pressure of dextran in salt solutions.52 Since hydrogels absorb water via an osmotic effect, the higher the osmotic pressure, the higher the swelling. To rank the above hydrogels in order of osmotic strength, we therefore carried out swelling ratio tests (Fig. 3B). The hydrogels ranked as PAM, PEG, alginate, and gelatin in ascending order of swelling ratio. Since gelatin, alginate, and PEG have different crosslinking mechanisms, it is difficult to relate their relative swelling ratios to crosslinking mechanisms. However, the difference in swelling between PEG and PAM, both of which were crosslinked through free radical means, can be easily justified. The covalent crosslinks in PEG hydrogels are constrained to form at the chain ends, whereas in PAM hydrogels the crosslinks are formed in situ during polymerization via the incorporation of bis-acrylamide at different ratios as described by Tse and Engler.53 We expect these differences in crosslinking mechanism to lead to distinct morphologies and mesh-size.54,55 | ||
| Fig. 3 (A) SAXS data of DOPC liposomes in gelatin (green), alginate (blue), PEG (pink), PAM (yellow), and Matrigel® (black). Peaks are observed for gelatin, alginate, PEG, and Matrigel®. For PAM, there is a steady decay of SAXS intensity with no peaks. (B) Swelling ratios of gelatin (n = 6), alginate (n = 4), PEG (n = 8), and PAM (n = 6) are 19.6, 17.7, 16.6, and 7.3, respectively (standard deviations shown). (C) Confocal microscopy displays aggregates for DOPC liposomes in gelatin, alginate, and PEG. For DOPC liposomes in PAM, confocal microscopy displays a green haze, very similar to that seen with liposomes only. | ||
SAXS data of liposomes mixed within these five different hydrogels are presented in Fig. 3A. The SAXS line of DOPC liposomes mixed with PAM shows a decay of the SAXS intensity without any structure factor peaks indicating scattering from an unstructured sample. In this case, PAM hydrogels are unable to restructure DOPC liposomes. It is noteworthy that of the different gels, PAM has the lowest osmotic stress capability. In contrast, PEG, alginate, gelatin, and Matrigel® all induce the transformation of liposomes into multilamellar vesicles. Confocal microscopy (Fig. 3C) displays signs of liposome aggregation seen by multiple micrometer-scale patterns of lipid dye. Confocal images of liposomes in PAM are nearly identical to confocal images of liposomes alone (Fig. S1, ESI†).
Lipid PEGylation attenuates restructuring in model hydrogels
PEG has been used in many drug delivery applications, often covalently linked to lipid molecules (PEGylated lipids), to prolong the blood circulation of liposomal drugs as well as to hinder protein adsorption and aggregation in vivo.18 Doxil® is a famous example of a liposomal formulation of the cancer drug doxorubicin that has been coated with PEG to enhance its circulation properties. Complements are specific proteins in the immune system that are known to attack liposomal membranes and help ingest liposomes.56,57 Bradley et al. investigated how different lipid systems could be incorporated with PEG to prolong their circulation half-life by inhibiting complement activation.58 They discovered that the optimal conditions to completely block complement activation by anionic liposomes was the incorporation of 5–10 mol% of DSPE-PEG2000 within the liposomal membrane. Leal et al. also found that incorporation of more than 10 mol% of PEGylated lipid results in demixing and formation of PEGylated lipid micelles in coexistence with liposomes.59 The ECM—composed of proteoglycans, polysaccharides, proteins, and water—is a natural hydrogel.21 During blood circulation, drug-loaded nanoparticles constantly encounter the ECM. Since PEGylated lipids ensure longer circulation lifetimes and prevent liposome aggregation, we anticipated that adding PEGylated lipids to DOPC liposomes could attenuate the restructuring effects of the hydrogels.We synthesized liposomes of DOPC incorporating 8 mol% of DOPE-PEG2000. We selected DOPE-PEG2000 to ensure that both the lipids and PEGylated lipids had identical hydrophobic tails. Compared to the DOPC liposomes, these liposomes have an external PEG corona, which can potentially shield the liposomes from aggregation due to osmotic stress induced by the surrounding PEG hydrogel. SAXS data (Fig. 4A) showed that multilamellar structures are still present with the characteristic three equally-spaced peaks at q = 0.046 Å−1, 0.093 Å−1, and 0.138 Å−1. Interestingly, the d-spacing measured is d001 = 2π/q001 = 14 nm. This spacing is three times larger than the interlamellar spacing obtained with DOPC liposomes. The significantly increased d-spacing suggests that PEG is indeed incorporated within the multilamellar aggregates. It is noteworthy that the structure factor peaks obtained for the multilamellar aggregates of the PEGylated liposomes are considerably broader and of lower intensity compared to those from DOPC-only liposomes. This difference indicates a loss in order or a smaller domain size of the multilamellar aggregates that are formed by osmotic action of the PEG hydrogels.
 | ||
| Fig. 4 (A) SAXS data of DOPC + DOPE-PEG2000 liposomes added to uncrosslinked PEG (black) display broad peaks at q = 0.046 Å−1, 0.093 Å−1, 0.138 Å−1, and 0.184 Å−1. SAXS data of the non-PEGylated lipids are shown by a dotted red curve for comparison. SAXS data of uncrosslinked PEG only (blue) show a steady decay in intensity with no peaks. A schematic representation to the right of the SAXS data depicts multilamellar PEGylated lipid aggregates and the uncrosslinked PEG network in a quartz capillary. The interlamellar repeat spacing d = 14 nm. (B) Confocal microscopy of the uncrosslinked PEG/DOPC + DOPE-PEG2000 composite displays aggregates slightly smaller than those formed with DOPC liposomes. (C) SAXS data of DOPC liposomes in PEG display no change in the interlamellar spacing when the temperature is increased to 70 °C and returned to 20 °C. (D) SAXS data of DOPC + DOPE-PEG2000 liposomes in PEG display a decrease in peak intensity with increasing temperature, with the peaks disappearing between 60 and 70 °C (the curves have been vertically shifted to better discern differences). | ||
Confocal microscopy predictably displayed the existence of lipid aggregates (Fig. 4B). To determine the stability of the multilamellar structures formed, we examined the effect of temperature on the PEG/liposome composite. The temperature was increased up to 90 °C and decreased back to 20 °C at 10 °C intervals. With the PEG/DOPC system, this change in temperature caused no significant change to the interlamellar spacing or peak width and intensity (Fig. 4C). However, with the PEG/DOPC + DOPE-PEG2000 system, there was a significant temperature effect. Increasing the temperature caused the multilamellar peaks to decrease in intensity and eventually vanish between 60 and 70 °C (Fig. 4D). Decreasing the temperature did not cause the multilamellar peaks to return. This observation—that the multilamellar structures formed with the PEGylated lipids were not as stable to temperature changes as the lipids without a PEG corona—is consistent with the notion that PEGylation of liposomes damps the restructuring ability of PEG hydrogels. In other words, even though restructuring of liposomes into multilamellar aggregates still occurs, the aggregates are considerably less ordered and are thermally unstable.
Most of the evidence point to the hydrogels exerting an osmotic pressure on the liposomes, causing them to transform into multilamellar lipid aggregates. Fig. 5 depicts a plausible mechanism underlying liposome restructuring when they are embedded in the hydrogels. Gibbs free energy minimization dictates that the activity of the water inside and outside the liposomes be equal. An osmotic pressure (Π) is produced because of this difference in water activity, which is the driving force for the water within the liposomes to permeate the hydrogel network. Depending on the molecular weight and the weight percentage of PEG, an osmotic pressure ranging from 102 to 104 kPa can be exerted on the liposomes, with lower molecular weights and higher weight percentages serving to increase the osmotic pressure. The hydrophilic hydrogel osmotically absorbs water from the liposomes, inducing their deswelling and concomitant membrane-fusion into multilamellar structures.
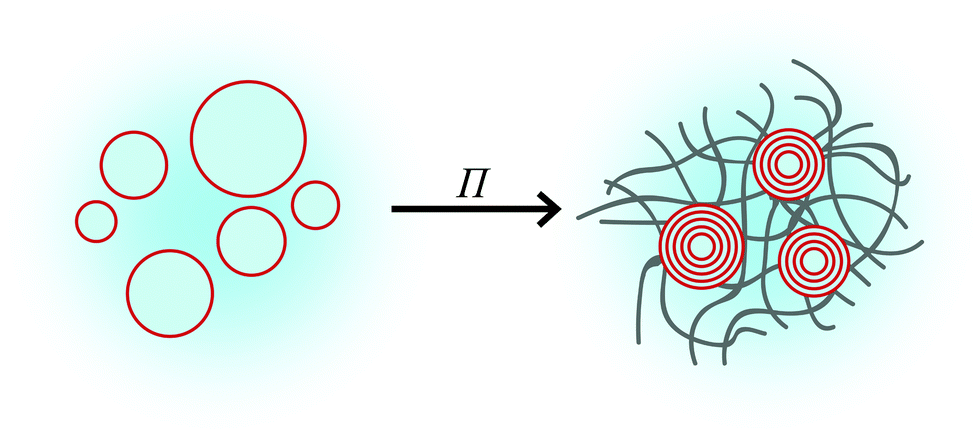 | ||
| Fig. 5 A schematic of a plausible mechanism for the restructuring effect on liposomes by hydrogels. The hydrogel exerts an osmotic pressure Π on the liposomes, transforming them into multilamellar vesicles with small interlamellar spacing. | ||
Conclusions
In this work, we have demonstrated that natural and synthetic hydrogels cause unilamellar liposomes to transform into aggregates of multilamellar structures. We attribute this restructuring effect—which is much less pronounced when a PEG corona surrounds the DOPC liposomes—to an osmotic driving force from the hydrogel. This observation has multiple implications in both liposomal drug delivery and in the mechanism of action of extracellular vesicles such as exosomes in ECM modulation. Currently, PEG is used to envelop liposomal formulations for drug delivery to increase circulation half-life and prevent an immune response. Encasing the liposomes in PEG may have the added benefit of stabilizing the liposomes. On the other hand, liposomes that do not have the protection garnered by a PEG corona and, therefore, transform into multilamellar structures in a hydrogel environment may have better drug-loading and -release capabilities arising from a significantly higher surface-area-to-volume ratio compared to that of liposomes. It is possible that extracellular vesicles, which are similar in structure to liposomes, meet a similar fate when they encounter the hydrogel environment of the ECM.Methods
Liposome preparation
DOPC, DOPE-PEG2000, and NBD-PE lipids were purchased from Avanti Polar Lipids (AL, USA). All lipids were used without further purification. A stock solution of lipids—either DOPC or 92 mol% DOPC + 8 mol% DOPE-PEG2000—dissolved in chloroform was dried under a stream of nitrogen in a glass vial and then vacuum-dried overnight. Milli-Q water was added to the glass vial to achieve the required lipid concentration (62.5 mM) and the vial was vortexed. After at least 30 min, the solution was extruded 11 times with a mini-extruder using a polycarbonate membrane of 200 nm pore size. For experiments with confocal microscopy, 0.2 mol% of NBD-PE was also mixed in before drying the chloroform. The concentration of lipids in the final liposome solutions used was 12.5 mM.PEG diacrylate synthesis and gelation
10 kDa and 20 kDa PEG (Sigma-Aldrich, MO, USA) were diacrylated by following the procedure used by Aydin et al.60 but omitting the filtration step over a plug of alumina. The synthesized PEG diacrylate was dissolved in DI water to achieve a 15 wt% solution. This solution was degassed under argon for 10 min. To crosslink 1 mL of PEG diacrylate, 20 μL of 0.438 M (10% w/v) aqueous APS (Sigma-Aldrich, MO, USA) and 4 μL of TEMED (Sigma-Aldrich, MO, USA) were added, and the mixture was either pipetted up and down multiple times or sonicated for a few seconds. The PEG diacrylate was left to crosslink for at least half an hour.PAM synthesis and gelation
Acrylamide (Amresco, OH, USA) was dissolved in DI water to achieve a 40% w/v solution. Bis-acrylamide (Amresco, OH, USA) was dissolved in DI water to achieve a 2% w/v solution. The two solutions were combined with DI water at different ratios to achieve PAM that would crosslink to different stiffnesses as described by Tse and Engler.53 To crosslink 1 mL of PAM, 10 μL of 0.438 M aqueous APS and 1 μL of TEMED were added, and the mixture was either pipetted up and down multiple times or sonicated for a few seconds. The PAM solution was left to crosslink for at least half an hour. PAM gels of 3 kPa, 4 kPa, 7 kPa, 11 kPa, 20 kPa and 40 kPa Young's moduli were synthesized.Gelatin gelation
Gelatin (Sigma-Aldrich, MO, USA) was dissolved in DI water to achieve a 10 wt% solution. The solution was heated in a water bath at around 65 °C and stirred at 1200 rpm using a stir bar for one hour or until all gelatin particles dissolved. 2 mL of heated gelatin solution was poured into a cast with 37.5 mm × 2 mm cross-section and allowed to cool to room temperature and gel.Alginate gelation
Sodium alginate (Sigma-Aldrich, MO, USA) was dissolved in DI water to achieve a 5 wt% solution. The solution was heated in a water bath at around 65 °C and stirred at 1200 rpm using a stir bar for one hour or until all alginate particles dissolved. To crosslink the alginate, 2 mL of the alginate solution was poured into a cast with 37.5 mm × 2 mm cross-section and 1.1 wt% calcium chloride solution was poured onto the alginate solution. The solution was left to gel at room temperature for an hour.Liposome incorporation into hydrogels
In all following, the final liposome concentration was 12.5 mM (by 5× dilution of 62.5 mM liposomes in the hydrogel). For PEG, PAM, and sodium alginate: (i) liposomes were incorporated into uncrosslinked hydrogel (15 wt%); (ii) liposomes were added to uncrosslinked hydrogel, and crosslinking agents APS and TEMED were added after; and (iii) hydrogel was crosslinked first, and then the liposomes were added. For gelatin: (i) liposomes were incorporated into warmed (uncrosslinked) gelatin, and then the gel was allowed to physically crosslink by bringing to room temperature; and (ii) liposomes were added to already physically crosslinked gelatin. For Matrigel® (Corning, NY, USA): liposomes were mixed with liquid Matrigel® at 4 °C, which polymerized at higher temperatures.SAXS
SAXS experiments were carried out at Beamlines 12-ID-B and 12-ID-C at the Advanced Photon Source at Argonne National Laboratory. An average photon energy of 13.3 keV was used at Beamline 12-ID-B. An average photon energy of 12 or 18 keV was used at Beamline 12-ID-C. 2D scattering data were radially averaged upon acquisition. The q-calibrant was silver behenate. Fit2D and IGOR Pro were used to integrate the 2D plots. Data analysis was carried out using OriginPro.Confocal microscopy
Confocal microscope images were captured at the LSM700 at the Institute for Genomic Biology and at the LSM800 at the Leal Lab at UIUC. An excitation wavelength of 488 nm was used. NBD-PE was used to fluorescently label the lipids (see Liposome preparation in ESI†).Cryo-EM
Liposome and hydrogel-liposome complex samples for cryo-EM (JEOL 2100 Cryo-TEM at 200 kV) were prepared on a 200-mesh sized lacey carbon-coated copper grid (Structure Probe Incorporation, PA) using a semi-automated Vitrobot (Vitrobot Mark II, FEI). The liposome sample solution was casted on the carbon coated side of the grid at 4 °C and 100% relative humidity. After blotting twice with one-second blotting time, the grid was quenched in liquid ethane to vitrify the liposomes with less than 500 nm ice thickness. For uncrosslinked liposome-hydrogel samples, the procedure was identical except for the blotting condition (blotted thrice with two-second blotting time). The images were obtained at a defocus of ∼4000 nm.Swelling ratio measurements
Gels were dried in a vacuum desiccator and the dry weight (W0) was measured. They were then immersed in DI water till they reached maximum swelling. The surface water was then removed, and the saturated weight (Ws) was measured. The swelling ratio was calculated using:| Swelling ratio = (Ws − W0)/W0. |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Institutes of Health, grant number: 1DP2EB024377 (structure of lipid particles) and by the Office of Naval Research (lipid/hydrogel composites and fluorescence imaging), grant numbers: N000141612886 and N000141812087 (DURIP – Defense University Research Instrumentation Program). SAXS experiments were carried out at beamlines 12-ID-B and 12-ID-C at the Advanced Photon Source (APS), Argonne National Laboratory. Use of APS was supported by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under contract number DE-AC02-06CH11357. The LSM700 was used at the Carl R. Woese Institute for Genomic Biology at the University of Illinois at Urbana-Champaign for confocal microscopy. Cryogenic transmission electron microscopy was performed at the Materials Research Laboratory at the University of Illinois at Urbana-Champaign.References
- K. Kostarelos, The Emergence of Nanomedicine: A Field in the Making, Nanomedicine, 2006, 1(1), 1–3, DOI:10.2217/17435889.1.1.1.
- M. Yu and J. Zheng, Clearance Pathways and Tumor Targeting of Imaging Nanoparticles, ACS Nano, 2015, 9(7), 6655–6674, DOI:10.1021/acsnano.5b01320.
- H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Renal Clearance of Quantum Dots, Nat. Biotechnol., 2007, 25(10), 1165–1170, DOI:10.1038/nbt1340.
- J. Liu, M. Yu, X. Ning, C. Zhou, S. Yang and J. Zheng, PEGylation and Zwitterionization: Pros and Cons in the Renal Clearance and Tumor Targeting of Near-IR-emitting Gold Nanoparticles, Angew. Chem., Int. Ed., 2013, 52, 12572–12576, DOI:10.1021/nl061786n.Core-Shell.
- J. Liu, M. Yu, C. Zhou, S. Yang, X. Ning and J. Zheng, Passive Tumor Targeting of Renal-Clearable Luminescent Gold Nanoparticles: Long Tumor Retention and Fast Normal Tissue Clearance, J. Am. Chem. Soc., 2013, 135(13), 4978–4981, DOI:10.1021/ja401612x.
- Y. Matsumura and H. Maeda, A New Concept for Macromolecular Therapeutics in Cnacer Chemotherapy: Mechanism of Tumoritropic Accumulatio of Proteins and the Antitumor Agents Smancs, Cancer Res., 1986, 46(12Pt1), 6387–6392, DOI:10.1021/bc100070g.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Analysis of Nanoparticle Delivery to Tumours, Nat. Rev. Mater., 2016, 1(1), 1–138 Search PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Understanding the Nanoparticle-Protein Corona Using Methods to Quantify Exchange Rates and Affinities of Proteins for Nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(7), 2050–2055, DOI:10.1073/pnas.0608582104.
- C. D. Walkey and W. C. W. Chan, Understanding and Controlling the Interaction of Nanomaterials with Proteins in a Physiological Environment, Chem. Soc. Rev., 2012, 41(7), 2780–2799, 10.1039/c1cs15233e.
- A. Albanese, C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, Secreted Biomolecules Alter the Biological Identity and Cellular Interactions of Nanoparticles, ACS Nano, 2014, 8(6), 5515–5526, DOI:10.1021/nn4061012.
- S. H. De Paoli Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, a. Karim and J. F. Douglas, Interaction of Gold Nanoparticles with Common Human Blood Proteins, ACS Nano, 2010, 4(1), 365–379, DOI:10.1021/nn9011187.
- C. M. Dawidczyk, C. Kim, J. H. Park, L. M. Russell, K. H. Lee, M. G. Pomper and P. C. Searson, State-of-the-Art in Design Rules for Drug Delivery Platforms: Lessons Learned from FDA-Approved Nanomedicines, J. Controlled Release, 2014, 187, 133–144, DOI:10.1016/j.jconrel.2014.05.036.
- T. M. Allen and P. R. Cullis, Liposomal Drug Delivery Systems: From Concept to Clinical Applications, Adv. Drug Delivery Rev., 2013, 65(1), 36–48, DOI:10.1016/j.addr.2012.09.037.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Liposomal Formulations in Clinical Use: An Updated Review, Pharmaceutics, 2017, 9(2), 1–33, DOI:10.3390/pharmaceutics9020012.
- R. Savla, J. Browne, V. Plassat, K. M. Wasan and E. K. Wasan, Review and Analysis of FDA Approved Drugs Using Lipid-Based Formulations, Drug Dev. Ind. Pharm., 2017, 43(11), 1743–1758, DOI:10.1080/03639045.2017.1342654.
- R. Shah, T. Patel and J. E. Freedman, Circulating Extracellular Vesicles in Human Disease, N. Engl. J. Med., 2018, 379(10), 958–966, DOI:10.1056/NEJMra1704286.
- W. Mu, S. Rana and M. Zöller, Host Matrix Modulation by Tumor Exosomes Promotes Motility and Invasiveness, Neoplasia, 2013, 15(8), 875–887, DOI:10.1593/neo.13786.
- M. L. Immordino, F. Dosio and L. Cattel, Stealth Liposomes: Review of the Basic Science, Rationale, and Clinical Applications, Existing and Potential, Int. J. Nanomed., 2006, 1(3), 297–315 CrossRef CAS PubMed.
- G. Bozzuto and A. Molinari, Liposomes as Nanomedical Devices, Int. J. Nanomed., 2015, 10, 975–999, DOI:10.2147/IJN.S68861.
- C. Matthäus, A. Kale, T. CHernenko, V. Torchilin and M. Diem, New Ways of Imaging Uptake and Intracellular Fate of Liposomal Drug Carrier Systems inside Individual Cells, Based on Raman Microscopy, Mol. Pharmacol., 2008, 5(2), 287–293, DOI:10.1021/mp7001158.New.
- C. Frantz, K. M. Stewart and V. M. Weaver, The Extracellular Matrix at a Glance, J. Cell Sci., 2010, 123(24), 4195–4200, DOI:10.1242/jcs.023820.
- S. Grijalvo, J. Mayr, R. Eritja and D. D. Díaz, Biodegradable Liposome-Encapsulated Hydrogels for Biomedical Applications: A Marriage of Convenience, Biomater. Sci., 2016, 555–574, 10.1039/C5BM00481K.
- J. P. G. Urban, A. Maroudas, M. T. Bayliss and J. Dillon, Swelling Pressures of Proteoglycans at the Concentrations Found in Cartilaginous Tissues, Biorheology, 1979, 16(6), 447–464, DOI:10.3233/BIR-1979-16609.
- S. Ehrlich, N. Wolff, R. Schneiderman, A. Maroudas, K. H. Parker and C. P. Winlove, The Osmotic Pressure of Chondroitin Sulphate Solutions: Experimental Measurements and Theoretical Analysis, Biorheology, 1999, 35(6), 383–397, DOI:10.1016/S0006-355X(99)80018-3.
- V. Parsegian, R. Rand, N. Fuller and D. Rau, Osmotic Stress for the Direct Measurement of Intermolecular Forces, Methods Enzymol., 1986, 127, 400–416, DOI:10.1016/0076-6879(86)27032-9.
- M. Kornreich, E. Malka-Gibor, B. Zuker, A. Laser-Azogui and R. Beck, Neurofilaments Function as Shock Absorbers: Compression Response Arising from Disordered Proteins, Phys. Rev. Lett., 2016, 117(14), 1–5, DOI:10.1103/PhysRevLett.117.148101.
- A. Evilevitch, M. Castelnovo, C. M. Knobler and W. M. Gelbart, Measuring the Force Ejecting DNA from Phage, J. Phys. Chem. B, 2004, 108, 6838–6843, DOI:10.1021/jp0376525.
- A. A. Abdeen, J. Lee and K. A. Kilian, Capturing Extracellular Matrix Properties in Vitro: Microengineering Materials to Decipher Cell and Tissue Level Processes, Exp. Biol. Med., 2016, 241(9), 930–938, DOI:10.1177/1535370216644532.
- G. A. Hudalla and W. L. Murphy, Mimicking the Extracellular Matrix: The Intersection of Matrix Biology and Biomaterials, Royal Society of Chemistry, Great Britain, 2016 Search PubMed.
- L. Shang, Y. Cheng and Y. Zhao, Emerging Droplet Microfluidics, Chem. Rev., 2017, 117, 7964–8040, DOI:10.1021/acs.chemrev.6b00848.
- C.-C. Lin and K. S. Anseth, PEG Hydrogels for the Controlled Release of Biomolecules in Regenerative Medicine, Pharm. Res., 2009, 26(3), 631–643, DOI:10.1007/s11095-008-9801-2.
- B. D. Ratner, Biomaterials Science, 3rd edn, 2013, pp. xli–liii, DOI:10.1016/B978-0-08-087780-8.00154-6.
- K. Engberg and C. W. Frank, Protein Diffusion in Photopolymerized Poly(Ethylene Glycol) Hydrogel Networks, Biomed. Mater., 2011, 6(5), 055006, DOI:10.1088/1748-6041/6/5/055006.
- N. Ziębacz, S. A. Wieczorek, T. Kalwarczyk, M. Fiałkowski and R. Hołyst, Crossover Regime for the Diffusion of Nanoparticles in Polyethylene Glycol Solutions: Influence of the Depletion Layer, Soft Matter, 2011, 7(16), 7181, 10.1039/c0sm01357a.
- D. F. Evans and H. Wennerström, The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet, Wiley-VCH, 1999 Search PubMed.
- H. Kim, Z. Song and C. Leal, Super-Swelled Lyotropic Single Crystals, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(41), 10834–10839, DOI:10.1073/pnas.1710774114.
- S. S. W. Leung and C. Leal, The Stabilization of Primitive Bicontinuous Cubic Phases with Tunable Swelling over a Wide Composition Range, Soft Matter, 2019, 15(6), 1269–1277, 10.1039/C8SM02059K.
- D. Steer, S. S. W. Leung, H. Meiselman, D. Topgaard and C. Leal, Structure of Lung-Mimetic Multilamellar Bodies with Lipid Compositions Relevant in Pneumonia, Langmuir, 2018, 34(25), 7561–7574, DOI:10.1021/acs.langmuir.8b01359.
- M. Kang, G. Huang and C. Leal, Role of Lipid Polymorphism in Acoustically Sensitive Liposomes, Soft Matter, 2014, 10(44), 8846–8854, 10.1039/C4SM01431F.
- J. O. Rädler, I. Koltover, T. Salditt and C. R. Safinya, Structure of DNA-Cationic Liposome Complexes: DNA Intercalation in Multilamellar Membranes in Distinct Interhelical Packing Regimes, Science, 1997, 275(5301), 810–814 CrossRef.
- A. L. Patterson, The Scherrer Formula for X-Ray Particle Size Determination, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- S. Mittal, R. K. Chowhan and L. R. Singh, Macromolecular Crowding: Macromolecules Friend or Foe, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850(9), 1822–1831, DOI:10.1016/j.bbagen.2015.05.002.
- A. A. Abdeen, J. Lee, S. H. Mo and K. A. Kilian, Spatially Defined Stem Cell-Laden Hydrogel Islands for Directing Endothelial Tubulogenesis, J. Mater. Chem. B, 2015, 3(40), 7896–7898, 10.1039/C5TB01294E.
- D. J. Waters, K. Engberg, R. Parke-Houben, L. Hartmann, C. N. Ta, M. F. Toney and C. W. Frank, Morphology of Photopolymerized End-Linked Poly(Ethylene Glycol) Hydrogels by Small-Angle X-Ray Scattering, Macromolecules, 2010, 43(16), 6861–6870, DOI:10.1021/ma101070s.
- J. E. Mark and P. J. Flory, The Configuration of the Polyoxyethylene Chain, J. Am. Chem. Soc., 1965, 87(7), 1415–1423, DOI:10.1021/ja01085a001.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, New York, 2003, p. 443 Search PubMed.
- D. C. Rau and V. A. Parsegian, Direct Measurement of the Intermolecular Forces between Counterion-Condensed DNA Double Helices. Evidence for Long Range Attractive Hydration Forces, Biophys. J., 1992, 61, 246–259, DOI:10.1016/S0006-3495(92)81831-3.
- M. Djabourov, J. Leblond and P. Papon, Gelation of Aqueous Gelatin Solutions. I. Structural Investigation, J. Phys., 1988, 49(2), 319–332, DOI:10.1051/jphys:01988004902031900.
- R. Kohn, Ion Binding on Polyuronates-Alginate and Pectin, Pure Appl. Chem., 1975, 42(3), 371–397 CAS.
- H. K. Kleinman and G. R. Martin, Matrigel: Basement Membrane Matrix with Biological Activity, Semin. Cancer Biol., 2005, 15(5 SPEC. ISS.), 378–386, DOI:10.1016/j.semcancer.2005.05.004.
- C. S. Hughes, L. M. Postovit and G. A. Lajoie, Matrigel: A Complex Protein Mixture Required for Optimal Growth of Cell Culture, Proteomics, 2010, 10(9), 1886–1890, DOI:10.1002/pmic.200900758.
- Y. D. Livney, O. R. Y. Ramon, E. Kesselman, U. R. I. Cogan, S. Mizrahi and Y. Cohen, Swelling of Dextran Gel and Osmotic Pressure of Soluble Dextran in the Presence of Salts, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2740–2750 CrossRef CAS.
- J. R. Tse and A. J. Engler, Preparation of Hydrogel Substrates with Tunable Mechanical Properties, Curr. Protoc. Cell Biol., 2010,(suppl. 47), 1–16, DOI:10.1002/0471143030.cb1016s47.
- V. Calvino-Casilda, A. J. López-Peinado, E. Vaganova, S. Yitzchaik, I. E. Pacios and I. F. Piérola, Porosity Inherent to Chemically Crosslinked Polymers. Poly(N-Vinylimidazole) Hydrogels, J. Phys. Chem. B, 2008, 112(10), 2809–2817, DOI:10.1021/jp7106473.
- S. Lee, X. Tong and F. Yang, Effects of the Poly(Ethylene Glycol) Hydrogel Crosslinking Mechanism on Protein Release, Biomater. Sci., 2016, 4(3), 405–411, 10.1039/C5BM00256G.
- C. A. Janeway, Jr, P. Travers, M. Walport and M. J. Shlomchik, Immunobiology: The Immune System in Health and Disease, 5th Edition, Garland Science, New York, 5th edn, 2001 Search PubMed.
- D. V. Devine, K. Wong, K. Serrano, A. Chonn and P. R. Cullis, Liposome-Complement Interactions in Rat Serum: Implications for Liposome Survival Studies, Biochim. Biophys. Acta, Biomembr., 1994, 1191(1), 43–51, DOI:10.1016/0005-2736(94)90231-3.
- A. J. Bradley, D. V. Devine, S. M. Ansell, J. Janzen, D. E. Brooks and B. E. T. Al, Inhibition of Liposome-Induced Complement Activation by Incorporated Poly(Ethylene Glycol)-Lipids, Arch. Biochem. Biophys., 1998, 357(2), 185–194 CrossRef CAS.
- C. Leal, S. Rögnvaldsson, S. Fossheim, E. A. Nilssen and D. Topgaard, Dynamic and Structural Aspects of PEGylated Liposomes Monitored by NMR, J. Colloid Interface Sci., 2008, 325(2), 485–493, DOI:10.1016/j.jcis.2008.05.051.
- D. Aydin, I. Louban, N. Perschmann, J. Blümmel, T. Lohmüller, E. A. Cavalcanti-Adam, T. L. Haas, H. Walczak, H. Kessler and R. Fiammengo, et al., Polymeric Substrates with Tunable Elasticity and Nanoscopically Controlled Biomolecule Presentation, Langmuir, 2010, 26(19), 15472–15480, DOI:10.1021/la103065x.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mh00835g |
| This journal is © The Royal Society of Chemistry 2020 |